top ten priorities for fda in 2017 - american bar ... ten priorities for fda in 2017 aba ad law...
TRANSCRIPT

TOP TEN PRIORITIES FOR FDAIN 2017
ABA AD LAW COMMITTEE ON FOOD & DRUG LAW
PROF. JAMES T. O’REILLY
PROF. KATHARINE VAN TASSEL
EDGAR J. ASEBEY, ESQ.

FDA’S SAFETY IMPORTANCE TO CONSUMERSIS A MISSION THAT DETERS POLITICAL FORCES• ADMINISTRATION POSITIONS TOWARD FDA REFLECT A WARINESS OF
APPEARING TO THE PUBLIC AS INCREASING SAFETY RISKS
• THIS INDUSTRY DOES NOT WANT “WILD WEST” REGULATORY VIEW
• TARIFF POLICY WILL CHANGE TO DISCOURAGE LOW-COST IMPORTS
• CHINA’S KEY SOURCE FOR DRUGS MAY FACTOR INTO TRUMP TRADE
• CONCERN ABOUT LESSENING HEALTHY FOOD & LESS SAFE DRUGS
• FDA COMMISSIONER JOB IS SENATE-CONFIRMED
• WATCH LOBBYISTS ASSIGNED TO HHS TRANSITION – NO POLICY PAPERS ON FDA ISSUES WERE AVAILABLE DURING CAMPAIGN

REAL “TOP TEN” DECIDED BELOW LEVEL OF TRUMP BY SELECTED HHS TEAM• LINGERING WOUND: REJECTION OF TRUMP DURING CAMPAIGN BY
OLD-LINE REPUBLICANS LEADING “ESTABLISHMENT”
• WILL THERE BE PAYBACK FOR KOCH FOODS’ INVESTMENTS?
• PHARMA ASSN & DEVICE MAKERS WILL HAVE WAY MORE POWER
• ACA REPEAL WILL KILL DEVICE TAX; UNCLEAR HOW SUBSIDIES FOR LOW-INCOME INSURC. NEEDS WILL BE FUNDED, OR JUST ELIMINATED
• WATCH THE HHS TRANSITION TO SEE WHO DISPLACES COM. CALIFF
• WATCH 115TH CONGRESS COMMITTEE CHAIR SELECTION BY RYAN AS INDICATORS OF WHICH REPEALS WILL MOVE FIRST

DON’T JUST WATCH FDA
• GIULIANI’S JUSTICE DEPT WILL SHIFT STAFF AWAY FROM REGULATORY ENFORCEMENT TEAMS TO “HARD NOSED” CRIME SUPPRESSION
• HENCE FEWER DoJ ENFORCEMENT CASES ACCEPTED FROM FDA
• OMB/OIRA WILL SHIFT INTO REVERSE, EXTINGUISHING PENDING SET OF OBAMA “MIDNIGHT RULES’
• HHS GRANT MAKING VIA NIH LIKELY TO BE CUT SHARPLY
• DEA: MORE RESOURCES SPENT ON BORDER, LESS LIKELY TO ACT ON REGULATORY DOCUMENTATION ISSUES
• CUSTOMS: MUCH MORE ACTIVITY IN SCREENING CHINA CARGOES

1. IMPORT SAFETY ISSUES
• EDGAR J. ASEBEY, ESQ., REGULATORY & BUSINESS LAW (POMPANO BEACH, FL)
• TRADE & JOBS FOCUS OF CAMPAIGN: TRANSLATE INTO NEW TARIFF LAW & REDUCTION IN REGULATORY OVERSIGHT OF FDA-REGULATED INDUSTRIES
• “JOB CREATION” PLEDGE IN FOOD: WHO WANTS TO BE PICKERS & CLEANERS?
• FOREIGN TRADE CONCERNS FACTORED INTO ELECTION CAMPAIGN (I.E. NAFTA REPEAL?)
• FSMA LAW PROTECTIONS OF SAFETY OF FOOD IMPORTS
• ANTI-IMPORT MOOD: FOODS FROM TROPICAL AREAS, UNLIKE SOME INDUSTRIAL PLANTS, CAN’T BE MOVED BACK TO USA; IMMIGRATION WALL THREATENS US FARMERS’ ACCESS TO FIELD LABORERS
• WTO CHALLENGE ALLEGING UNFAIR TRADE PRACTICES?
• PREFERENCE FOR IMPORTATION OF CHEAPER RX DRUGS VS. REPUBLICAN PHARMA LOBBY
• API & DRUGS GENERIC PRODUCTION 80% EX-USA, CLOSER TO RAW MATERIALS & TECHNOLOGIES FOR MANUFACTURING, LESS LIKELY TO BE RETURNED TO USA
• DEVICES INCREASINGLY TESTED & SOURCED OUTSIDE USA

Does Mandatory Listing of Novel Ingredients Violate the First
Amendment's Protection Against Forced Speech?
Katharine Van TasselRetired Professor of Law, Creighton University Law School
Adjunct Professor, Georgetown UniversityRegulatory Counsel, FDA Imports, Inc.

GM Salmon

GM Salmon





Lab Grown Meat

Lab Grown Meat or “Shmeat”

Nanoparticles

Mandatory Listing of Novel Ingredients and the First Amendment
For ideological, spiritual, or aesthetic reasons, consumers care about
– who produced
– how produced
– where produced
– what produced with
Is there a consumer “right to know” these facts about consumer products?

Mandatory Listing of Novel Ingredients and the First
Amendment
These preferences are real
– but, as we will see, they are unlikely to rise to a sufficiently substantial interest to justify compelling speech
As a public health agency, what position can the FDA take to support a listing requirement when it comes to novel ingredients that won’t run afoul of the Frist Amendment ?

Mandatory Listing of Novel Ingredients and the First Amendment
The Supreme Court in Zauderer held that
compelled commercial speech, and the compelled disclosure of factualinformation in particular, should be subject to less demanding scrutiny.
Further, the Court stated that
the constitutionally protected interest in not providing any particular factualinformation in advertising is minimal.

Mandatory Listing of Novel Ingredients and the First Amendment
The Zuaderer Court found that only reasonable basis review should apply, explaining that:
a requirement that a seller disclose factual information will be upheld so long as the requirement is not unduly burdensome and the requirement is “reasonably related” to a substantial governmental interest.

What could be a sufficiently substantial interest?
The novel nature of ingredients created using novel technologies suggests a need
To animate the public health product safety net
Post market tracking to protect public health

Public Health Product Safety Net
Ingredient Labeling= Post Market Surveillance
Report Injury to Physician for Proper
Treatment
Report to the FDA
Report to the CDC

Public Health Product Safety Net
Ingredient Labeling= Post Market Surveillance
Report Injury to Physician for Proper
Treatment
Report to the FDA
Report to the CDC Data Gathering
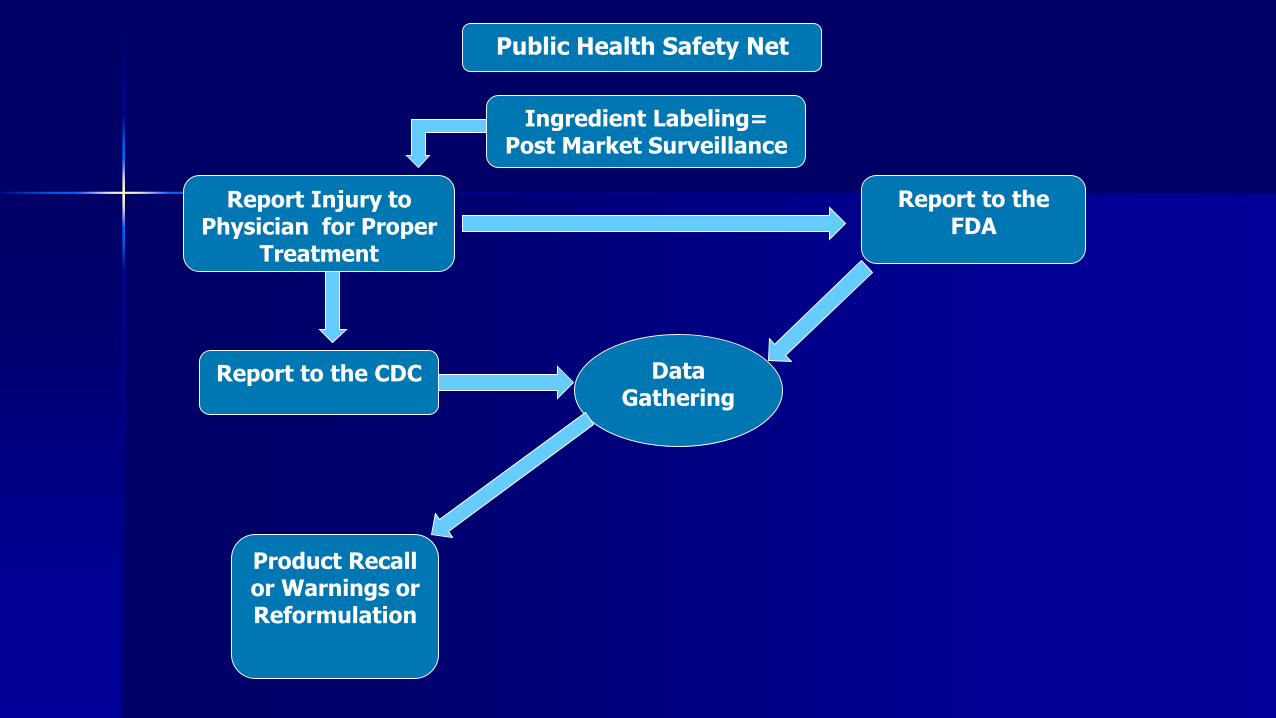
Public Health Safety Net
Ingredient Labeling= Post Market Surveillance
Report Injury to Physician for Proper
Treatment
Report to the FDA
Report to the CDC
Product Recall or Warnings or Reformulation
Data Gathering

Public Health Product Safety Net
Ingredient Labeling= Post Market Surveillance
Report Injury to Physician for Proper
Treatment
Report to the FDA
Report to the CDC
Product Recall or Warnings or Reformulation
Enables Tort
System to Assess
Liability
Data Gathering
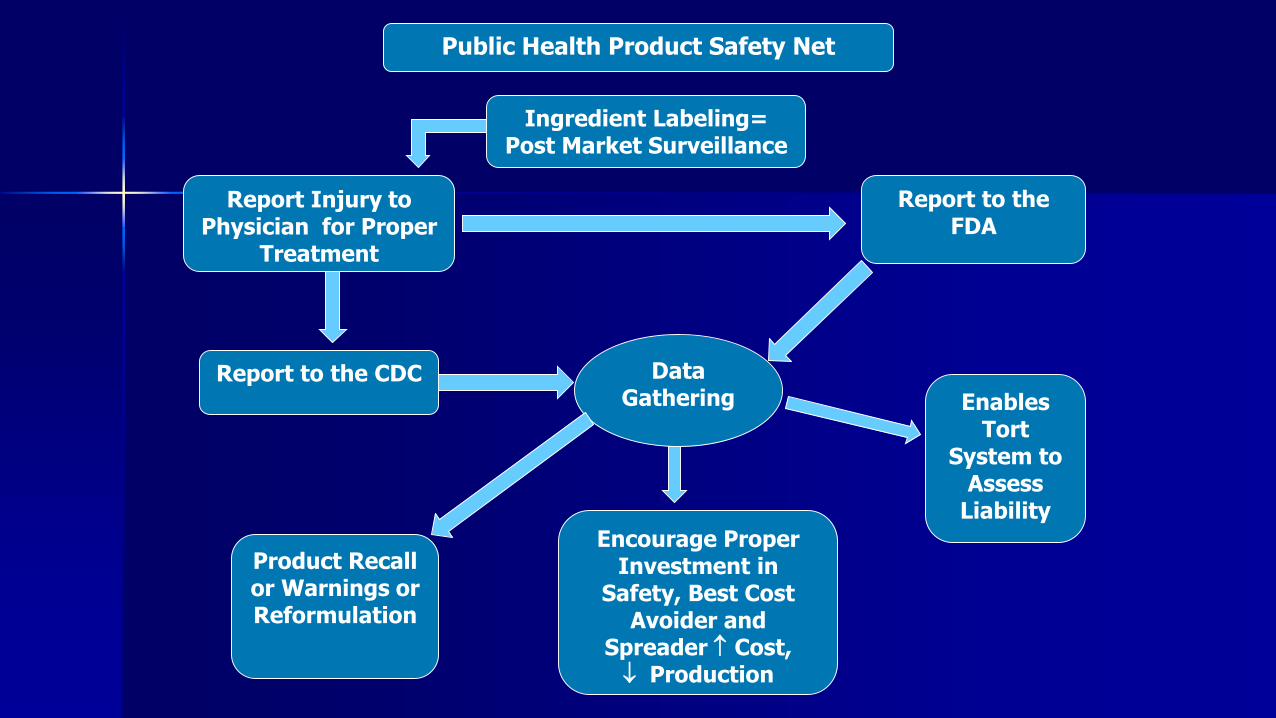
Public Health Product Safety Net
Ingredient Labeling= Post Market Surveillance
Report Injury to Physician for Proper
Treatment
Report to the FDA
Report to the CDC
Product Recall or Warnings or Reformulation
Enables Tort
System to Assess
Liability
Data Gathering
Encourage Proper Investment in
Safety, Best Cost Avoider and
Spreader Cost, Production

Public Health Product Safety Net
Ingredient Labeling= Post Market Surveillance
Report Injury to Physician for Proper
Treatment
Report to the FDA
Report to the CDC
Product Recall or Warnings or Reformulation
Enables Tort
System to Assess
Liability
Data Gathering
Encourage Proper Investment in
Safety, Best Cost Avoider and
Spreader Cost, Production


3. MARIJUANA LEGALIZATION, WHAT NEXT?
• PROF. JIM O’REILLY, UNIV. CINCINNATI
• DEA REGTY. PRIMACY SHAKEN BY MULTIPLE STATE LEGALIZATIONS
• WILL THIS NEW CONGRESS ALLOW “LIBERALIZATION” OF MARIJUANA?
• HEALTH EFFECTS RESEARCH VERY CONSTRAINED BY SCHED 1 STATUS
• IF DEA REDUCE SCHEDULE 1 STATUS: FDA & RESEARCH SAFETY ISSUES
• STATES NOW HAVE DOMINANT ROLE, BUT VERY FRACTURED
• WILL FDA WORK WITH STATES TO DEVELOP CLIN. TESTING OPTIONS?
• BETWEEN FDA’S DRUG ROLE AND FDA TOBACCO ROLE, SHOULD FDA WILLINGLY ACCEPT NEW REGULATORY POWERS TO CONTROL SAFETY OF FORMS OF MARKETED MARIJUANA

4. IMPLEMENTING FSMA
• EDGAR J. ASEBEY, ESQ., REGULATORY & BUSINESS LAW (POMPANO BEACH, FL)
• ROLL OUT OF LEGISLATION INTO RULES IS NOT LIKELY TO BE HALTED
• WILL NEW ADMINISTRATION LIMIT THE EFFECTIVENESS OF FSMA REGULATIONS BY LIMITING FUNDING FOR ENFORCEMENT?
• PRIORITIES FOR FDA ARE TOP 5 FSMA RULES:• PREVENTIVE CONTROLS FOR HUMAN FOOD• PREVENTIVE CONTROLS FOR ANIMAL FOOD• PRODUCE SAFETY• FOREIGN SUPPLIER VERIFICATION PROGRAM• ACCREDITATION OF 3RD PARTY AUDITORS
• HOW WILL THE NEW TRUMP BIAS AGAINST IMPORTS FACTOR INTO FINAL RULES?
• DOES NEW POLITICAL CLOUT OF FOOD CORP’S MEAN FDA RULES ON FSMA RETREAT FROM TOUGH OVERSIGHT OF FOOD PLANTS?

5. REORGANIZATION OF FDA’S FIELD FORCE
• PROF. JIM O’REILLY
• PRODUCTIVITY CHALLENGES AS FDA FIELD STAFF SHRINKS
• CHANGES IN “WHO IN FDA DOES WHAT, WHERE” ARE PROGRESSING
• OLD FDA GEOGRAPHICAL DISTRICTS GOING AWAY
• NEW PRODUCT-CENTERED FIELD FOCUS REPLACING IT
• MORE TRAVEL BY MORE FDA FIELD STAFFERS
• SIGNIFICANT ISSUE RE COST OF INTERNATIONAL INSPECTIONS FORCE
• WHAT ARE MAJOR EFFECTS ON MORALE? FEAR FACTOR? TRAVELS?
• HOW HAVE “REGRETTED DEPARTURES” OF FDA VETS INCREASED?

Katharine Van TasselRetired Professor of Law, Creighton University Law School
Adjunct Professor, Georgetown UniversityRegulatory Counsel, FDA Imports, Inc.
When Is the Use of the Term “Healthy” on Foods
Deceptive and Misleading?


“Healthy” Claims on Foods

“Healthy” Claims on Foods







Obesity Crisis
• More than one-third (36.5%) of U.S. adults have obesity
• Obesity-related conditions include heart disease, stroke, type 2 diabetes and certain types of cancer, some of the leading causes of preventable death.
• The estimated annual medical cost of obesity in the U.S. was $147 billion in 2008 U.S. dollars; the medical costs for people who are obese were $1,429 higher than those of normal weight.


FDA Guidance - focuses on “healthy” as implied nutrient content claim
• Healthy means
– foods that have a fat profile of predominantly mono and polyunsaturated fats
– but do not meet the regulatory definition of “low fat”
– or that contain at least 10 percent of the Daily Value per reference amount customarily consumed of potassium or vitamin D

FDA Solicits Comments
• For example, what current dietary recommendations should be reflected in the definition of “healthy”?
• What are the public health benefits of defining the term “healthy”?
• What do consumers expect of foods that carry a “healthy” claim?
• What factors and criteria should be used for the new definition of “healthy”?

“Healthy” Claims on Foods
• Down the rabbit hole… again
• public comments on ‘natural’ claims
• heavily they rely on words such as ‘chemicals,’ 'natural, ‘artificial’ and ‘processed,’ which themselves are not clearly defined in law.

“Healthy” Claims on Foods
• Healthy: Not just about nutrition?
• comments reflect a shift in the way consumers think about ‘healthy’
– from being narrowly focused on fat, sugar, calories or other nutrients
• to more wide ranging definitions – ‘artificial ingredients’
– GMOs
– Equating healthy means 'organic

“Healthy” Claims on Foods
• Kelie Cichoski:
Healthy should mean no chemicals, no preservatives that are not naturally occurring, no hormones, and no antibiotics.

“Healthy” Claims on Foods
• George Inashvili:
–By far and away the most important definition of healthy is that it's true organic. Healthy is nonGMO… Healthy is elimination of sugar… Healthy is a mostly vegan, organic diet full of dark leafy greens, green teas, and plant protein.


Exhibit 6-17 MISBRANDING CASE - DESCRIPTION OF CHARGE FROM A COMPLAINT - FDA Manual
• Labeling that is alleged to be false or misleading under 21 U.S.C. 352(a) (or 343(a))
– A misleading statement need not be false to violate the Act; it is enough that a statement has the capacity or tendency to deceive, by indirection, ambiguity, or by partial or half-truths.
– A statement can even be technically true in its entirety and still violate the Act.

Standard for “Deceptive and Misleading” Claims?
What standard that should be used when
determining whether a claim is deceptive and
misleading?
Should the relative vulnerability of the health
status of the targeted population be considered?

Standard for “Deceptive and Misleading” Claims?
On one end of the spectrum, courts state that the purpose of the FDCA is
"to protect the public, the vast multitude which includes the ignorant, the unthinking and the credulous who, when making a purchase, do not stop to analyze."
“The test is not the effect of the label on a 'reasonable consumer,' but upon 'the ignorant, the unthinking and the credulous' consumer.“

Standard for “Deceptive and Misleading” Claims?
On the other end of the spectrum, some courts have held that the test is that of "purchasers who are of normal capacity and use that capacity in a common sense way."

Standard for “Deceptive and Misleading” Claims?
2002 Guidance: FDA adopted
reasonable consumer standard

Standard for “Deceptive and Misleading” Claims?
The "reasonable person" standard
used to assign fault, and thereby liability, through tort law that governs conduct between manufacturers and consumers
Is imported into the public health context

Standard for “Deceptive and Misleading” Claims?
Reasonable person approach
ignores the role of the FDA to protect the health of all of the population



7. NOVEL: SENSORS, GENES & TELEMEDICINE
• PROF. JIM O’REILLY, UNIV. CINCINNATI
• FDA OFFICE OF COMBINATION PRODUCTS SEEKS TO RECONCILE INTERESTS
• INDUSTRY LIKELY TO CONTINUE WORK ON SENSORS FOR “PERSONALIZED MEDICINE” DIAGNOSTIC ABILITIES
• MUCH PROGRESS ON TELEMEDICINE WITH SENSORS
• TELEMED BATTLES ARE WITH STATE MEDICAL LICENSING BOARDS, UNCLEAR WHERE GIULIANI’S DoJ ANTITRUST DIVISION WILL ACT (OR NOT)
• PROGRESSIVE INTERACTIONS BY RX & DEVICE FIRMS NEEDED
• QUESTION OF IMPACTS OF LIKELY CUTS TO NIH AND HHS FUNDING
• WILL PVT SECTOR FUND ENOUGH GENE RESEARCH IF NIH GRANTS SHRINK

Vaccine Compensation Fund
And
Vaccine Court
Katharine Van TasselRetired Professor of Law, Creighton University Law School
Adjunct Professor, Georgetown UniversityRegulatory Counsel, FDA Imports, Inc.

Importance of Vaccines
The current routine childhood immunization schedule is estimated to:
–Prevent 42,000 deaths
–Prevent 20 million cases of disease
– Save $14 billion in direct medical costs
Per US birth cohort
The success of vaccines in reducing disease-associated mortality is second only to the introduction of safe drinking water



The National Vaccine Injury Compensation Program (VICP)
• To ensure individuals thought injured by routine childhood vaccines are provided with fair and efficient compensation
• To ensure a stable vaccine supply by limiting liability for manufacturers and administrators

The National Vaccine Injury Compensation Program (VICP)
• To ensure individuals thought injured by routine childhood vaccines are provided with fair and efficient compensation
• To ensure a stable vaccine supply by limiting liability for manufacturers and administrators

Vaccine Injury Compensation Trust Fund
• Derives from excise tax of $.75 on each dose of covered vaccine purchased
• Examples:
• DTaP Vaccine -- $2.25
• IPV -- $.75
• Provides payment of awards
• Current balance is $1.3 billion
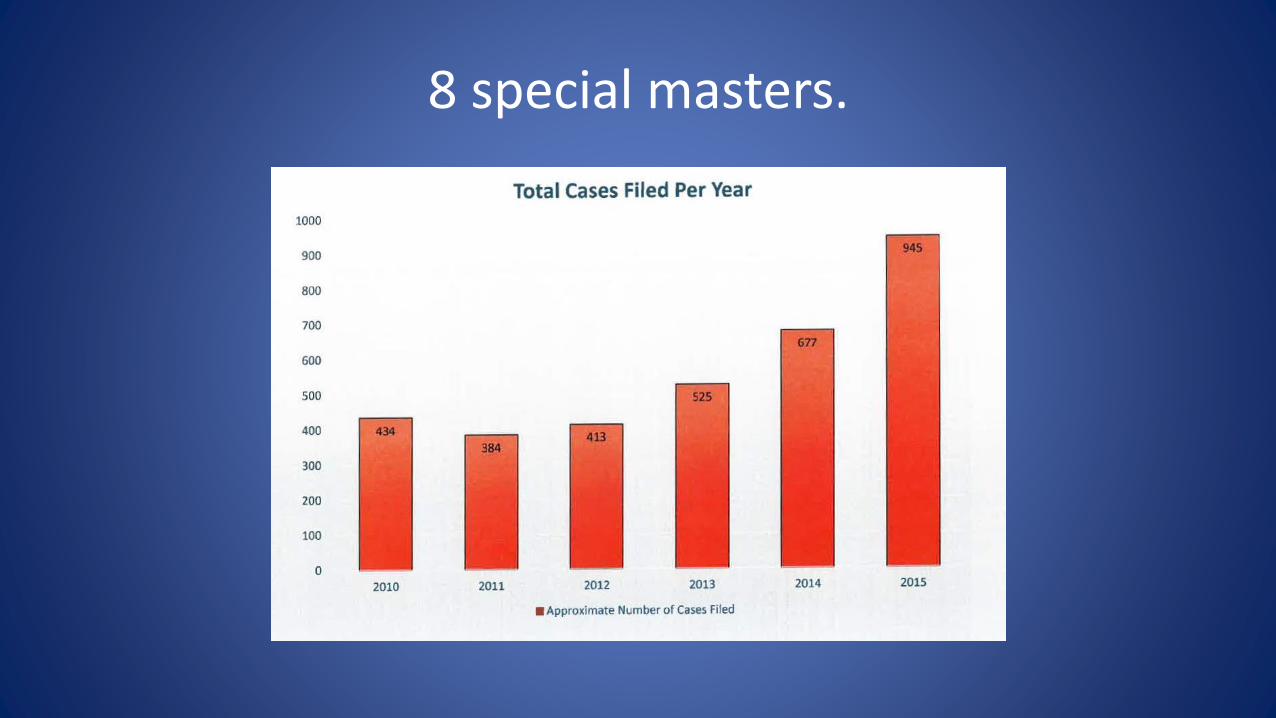
8 special masters.

Only 8 Special Masters

8 special masters.



Vaccine Compensation Fund
And
Vaccine Court
Katharine Van TasselRetired Professor of Law, Creighton University Law School
Adjunct Professor, Georgetown UniversityRegulatory Counsel, FDA Imports, Inc.

9. BACKLOGS OF GENERIC RX’S: HOW TO FIX?
• PROF. JIM O’REILLY, UNIV. OF CINCINNATI
• NOT LIKELY TRUMP COULD SIMPLY MASS-APPROVE OLDEST ANDA’S
• GDUFA MONEY VERY MUCH NEEDED TO STAFF UP, MOVE BACKLOGS
• VIEWS OF TRUMP RE “RED TAPE” IN DRUG APPROVALS ARE HOSTILE
• ANTI-IMPORT, ANTI-CHINA VIEWS MAY TIGHTEN SAMPLING AT PORT
• INSIDER LOBBYIST EFFORTS TO BLOCK MORE GENERICS WILL RISE
• INSIDER REPUBLICAN WASHINGTON PHARMA LOBBY MAY CLASH WITH “CUT COST” INCENTIVES FOR REDUCTION OF WAIT TIME FOR MARKETING OF NEW GENERICS

10. WHITHER RESEARCH ON DEMENTIA?
• PROF. JIM O’REILLY, UNIV. OF CINCINNATI
• SEE MORE EXTERNAL STRESSES TO “FAST TRACK” NEW BRAIN DRUGS
• DON’T FORGET FINDING CURE FOR ALZHEIMER’S…IF YOU STILL CAN
• MANY RX TRIALS UNDERWAY, NO SILVER BULLET DRUGS YET
• STATISTICS SHOW BIG INCREASE AMONG BOOMERS & POST-70’S
• FAST TRACKED DRUGS MAY REDUCE ASSURANCE OF SAFETY
• TRUMP: LOSS OF NIH FUNDING LIKELY, FEWER GRANTS AVAILABLE
• UNCERTAINTY HOLDS BACK INVESTOR FUNDS TO LONG-SHOT R&D

TOUGH TIME TO BE INSIDE FDA STAFF
• REDUCTIONS IN “RED TAPE” ARE NOW POLICY
• ELIMINATION OF RULES, STOPPING NEW RULES AT OMB/OIRA
• DECISIONS MORE LIKELY TO RUN AGAINST FDA INITIATIVES
• INDUSTRY’S ALUMNI LIKELY TO BE IN KEY POLICY SETTING ROLES
• RETURN TO POWER OF FORMER ADVOCATES FOR INDUSTRY
• CAREER REGULATORS: “LAST ONE OUT, TURN OUT THE LIGHTS”



FSMA RULEMAKING – on line at
http://www.fda.gov/Food/GuidanceRegulation/FSMA/default.htm
Implementation Activity Mitigation Strategies to Protect Food Against Intentional
Adulteration Final Rule Sanitary Transportation of Human and Animal Food Final
Rule Produce Safety Final Rule and Environmental Impact
Statement Foreign Supplier Verification Programs (FSVP) Final Rule Accredited Third-Party Certification Final Rule Preventive Controls for Human Food Final Rule Preventive Controls for Food for Animals Final Rule

FSMA Guidance Documents – on line at
http://www.fda.gov/Food/GuidanceRegulation/FSMA/default.htm
Guidance for Industry: FDA's Voluntary Qualified Importer Program
November 10, 2016
Draft Guidance for Industry: Questions and Answers Regarding Food Facility Registration (Seventh Edition)
November 7, 2016
Guidance for Industry: What You Need to Know About the FDA Regulation: Current Good Manufacturing Practice, Hazard Analysis, and Risk-Based Preventive Controls for Human Food; Small Entity Compliance Guide
October 31, 2016

PLEASE NOTE: PANEL WILL ALSO ADDRESS NEW FDA LEGISLATION IF ADOPTED BEFORE CONFERENCE
“21st Century Cures Act” – FDA Most Relevant Segments as of Nov. 25, 2016 – Final Text of Bill as Passed is on congress.gov
==============================================
HOUSE RULES COMMITTEE PRINT 114-67
TITLE III—DEVELOPMENT
Subtitle A—Patient-Focused Drug Development
Sec. 3001. Patient experience data.
Sec. 3002. Patient-focused drug development guidance.
Sec. 3003. Streamlining patient input.
Sec. 3004. Report on patient experience drug development.

Subtitle B—Advancing New Drug Therapies
Sec. 3011. Qualification of drug development tools.
Sec. 3012. Targeted drugs for rare diseases.
Sec. 3013. Reauthorization of program to encourage treatments for rare pediatric diseases.
Sec. 3014. GAO study of priority review voucher programs.
Sec. 3015. Amendments to the Orphan Drug grants.
Sec. 3016. Grants for studying continuous drug manufacturing.

Subtitle C—Modern Trial Design and Evidence Development
Sec. 3021. Novel clinical trial designs.
Sec. 3022. Real world evidence.
Sec. 3023. Protection of human research subjects.
Sec. 3024. Informed consent waiver or alteration for clinical investigations.

Subtitle D—Patient Access to Therapies and Information
Sec. 3031. Summary level review.
Sec. 3032. Expanded access policy.
Sec. 3033. Accelerated approval for regenerative advanced therapies.
Sec. 3034. Guidance regarding devices used in the recovery, isolation, or delivery of regenerative advanced therapies.
Sec. 3035. Report on regenerative advanced therapies.
Sec. 3036. Standards for regenerative medicine and regenerative advanced therapies.
Sec. 3037. Health care economic information.
Sec. 3038. Combination product innovation. ======================================


Subtitle E—Antimicrobial Innovation and Stewardship
Sec. 3041. Antimicrobial resistance monitoring.
Sec. 3042. Limited population pathway.
Sec. 3043. Prescribing authority.
Sec. 3044. Susceptibility test interpretive criteria for microorganisms; antimicrobial susceptibility testing devices.

Subtitle F—Medical Device Innovations
Sec. 3051. Breakthrough devices.
Sec. 3052. Humanitarian device exemption.
Sec. 3053. Recognition of standards.
Sec. 3054. Certain class I and class II devices.
Sec. 3055. Classification panels.
Sec. 3056. Institutional review board flexibility.
Sec. 3057. CLIA waiver improvements.
Sec. 3058. Least burdensome device review.
Sec. 3059. Cleaning instructions and validation data requirement.
Sec. 3060. Clarifying medical software regulation. ===============================

Subtitle G—Improving Scientific Expertise and Outreach at FDA
Sec. 3071. Silvio O. Conte Senior Biomedical Research and Biomedical Product Assessment Service.
Sec. 3072. Hiring authority for scientific, technical, and professional personnel.
Sec. 3073. Establishment of Food and Drug Administration Intercenter Institutes.
Sec. 3074. Scientific engagement.
Sec. 3075. Drug surveillance.
Sec. 3076. Reagan-Udall Foundation for the Food and Drug Administration.


Electronic copy available at: http://ssrn.com/abstract=2240034
VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433
Regulating in Uncertainty: Animating the
Public Health Product Safety Net to Capture
Consumer Products Regulated by the FDA
that Use Innovative Technologies, Including
Nanotechnologies, Genetic Modification,
Cloning, and Lab Grown Meat
Katharine Van Tassel†
I. INTRODUCTION
“For want of a nail, the war was lost.”1
The past several decades have seen the creation of
transformative new technologies that are being used to design
† Professor of Law, University of Akron School of Law, BSN, Case Western Reserve
University; JD, Case Western Reserve University School of Law; MPH, Harvard School
of Public Health.
1 Part of a proverbial rhyme showing that small actions can result in large
consequences:
For want of a nail the shoe was lost.
For want of a shoe the horse was lost.
For want of a horse the rider was lost.
For want of a rider the message was lost.
For want of a message the battle was lost.
For want of a battle the kingdom was lost.
And all for the want of a horseshoe nail.
Benjamin Franklin, Poor Richard’s Almanack 69 (1758). This Article will explain how
the powerful public health product safety net is activated when innovative ingredients
are identified on product labels. For example, nanotech ingredients can be identified
with just four simple letters, “nano,” in front of each ingredient that is nano-sized, or just
two letters, “GM,” can be placed in front of ingredients that are genetically modified.
With just the simple step of placing a couple of letters on the ingredient lists on product
labels, the public health product safety net will be activated, potentially averting a
public health crisis from exposures to novel, innovative ingredients. Thus, this small
action could avert a large consequence.

Electronic copy available at: http://ssrn.com/abstract=2240034
VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
434 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
innovative consumer product ingredients never before seen in
nature. Examples include the use of nanotechnology and genetic
modification, and, right around the corner, cloned animals used
for food, as well as lab-grown meat.2 These innovative, novel3
technologies are harbingers of more pioneering consumer
product ingredients to come. This remarkable pace in the
development of groundbreaking new technologies means that
the population is being steadily exposed to novel ingredients
with unknown health risks.
Optimally, the Food and Drug Administration (FDA) should
be regulating these innovative, novel ingredients in consumer
products to meet the twin goals of fostering innovation while
protecting public health. However, the current litmus test that
the FDA is using to trigger regulation to protect public health is
focused on hazard.4 Linking public health protections to the
degree of hazard when operating in scientific uncertainty is
outcome determinative—it means no regulation to protect public
health at all. This is because it is common for the development
of innovative, novel technologies to far outpace the development
of the science necessary to test for the health risks associated
with these technologies.5 This scientific lag time creates an
information void with regard to risks to human health that can
2 See generally Henry Fountain, Building a $350,000 Burger, NY Times (May 12,
2013), online at http: //www.nytimes.com/2013/05/14/science/engineering-the-325000-in-
vitro-burger.html?pagewanted=all&_r=0 (visited Sept 15, 2013) (describing a five ounce
hamburger being assembled from tiny bits of lab grown beef muscle tissue in a lab that
will be eaten at an event at London to show the world that lab grown meat is possible);
Zachary Schneider, Comment, In Vitro Meat: Space Travel, Cannibalism, and Federal
Regulation, 50 Houston L Rev 991 (2013) (describing why lab grown meat could ease
many of the environmental burdens of worldwide meat production).
3 These products are described as both innovative and novel. In technology, an
innovation “may be an improvement to something already existing.” Merriam-Webster,
“Innovation” (Merriam-Webster 2013), online at http: //www.merriam-webster.
com/dictionary/innovation (visited Sept 15, 2013). On the other hand, to be “novel”
means “new and not resembling something formerly known or used.” Merriam-Webster,
(Merriam-Webster 2013), online at http: //www.merriam-webster.com/dictionary/novel
(visited Sept 15, 2013). Thus, the descriptive “novel innovation” as used in this Article
means an improvement to an already existing product that involves the use of
ingredients that are new and that do not resemble anything formerly used or known.
Examples include nanotech particles, GM food, cloned animals used as food, or lab-grown
meat.
4 See notes 95–109 and accompanying text.
5 For a series of examples dealing with tributylin (“TBT”), polychlorinated
biphenyls (“PCBs”), diethylstilbestrol (“DES”), the Great Lakes pollution, thalidomide,
medical x-rays, benzene, asbestos, chlorofluorocarbons (“CFCs”), and DES, see Part
II.E.1.

Electronic copy available at: http://ssrn.com/abstract=2240034
VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 435
last for decades.6 This health risk information void cripples the
FDA’s ability to regulate to protect public health during this
often decades-long period of scientific uncertainty.7
Many propose that the proper way to deal with the
problems caused by this health risk information void is to adopt
some form of the Precautionary Principle. According to Professor
Cass Sunstein, “[s]imply put, the principle counsels that we
should avoid steps that will create a risk of harm; until safety is
established through clear evidence, we should be cautious. In a
catch-phrase: Better safe than sorry.”8 However, there are
downsides to this approach. Professor Sunstein points out that
the bar to the use of innovative technologies for the time period
it takes to prove them to be safe comes with its own harm—
keeping the potentially life-saving benefits of these innovative
technologies from reaching people in need.9 Professor Sunstein
gives the example of genetically modified food (“GM food”),
which may allow the production of food that is healthier,
cheaper, and easier to grow under difficult environmental
conditions.10 This GM food may provide great benefits to the
populations of developing countries, including the prevention of
many deaths.11 Following the Precautionary Principle means
refusing to allow the use of GM food until it is proven to be safe.
6 See notes 112–121 and accompanying text.
7 See notes 122–134 and accompanying text.
8 Cass R. Sunstein, The Paralyzing Principle, 25 Reg 32, 32 (2003). Professor
Sunstein explains that the Precautionary Principle has a wide range of definitions, from
weak to strong. An example of what he refers to as a weak version is that which was
adopted by the 1992 Rio Declaration that states “[w]here there are threats of serious or
irreversible damage, lack of full scientific certainty shall not be used as a reason for
postponing cost-effective measures to prevent environmental degradation.” Id at 33. An
example of a strong version of the Precautionary Principle that Professor Sunstein uses
is drawn from the
widely publicized Wingspread Declaration, from a meeting of
environmentalists in 1998 . . . “When an activity raises threats of harm to
human health or the environment, precautionary measures should be taken
even if some cause and effect relationships are not established scientifically. In
this context, the proponent of the activity, rather than the public, should bear
the burden of proof.”
Id.
9 Id.
10
Id.
11
Professor Sunstein clarifies that “the point is not that genetic modification will
definitely have those benefits, or that the benefits of genetic modification outweigh the
risks. The point is only that if the Precautionary Principle is taken literally, it is
offended by regulation and by non-regulation.” Id.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
436 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
This means that the potential benefits of GM food will be
withheld during the health risk information void. Thus, this
choice brings its own risks of harm.
How should the FDA regulate consumer products to protect
public health when operating in scientific uncertainty? This
Article suggests that, when it comes individual risk,12 such as
those risks faced by individual consumers when choosing and
using a particular product—in contrast to collective risks such as
those environmental risks faced by all individuals as a
collective13—neither of these “all or nothing” approaches fits the
bill.
This Article is the first to propose a unique, yet entirely
feasible, middle ground that uses what this Article calls the
public health product safety net. Instead of a focus on the
unknown, which is the degree of hazard associated with a new
and innovative technology, the lodestar of the FDA for
regulation in the context of consumer products should be on
novelty.14 Keying regulation to novelty allows for “strings” to be
attached to consumer products using novel technologies. These
strings take the form of a requirement that all ingredients
created through the use of novel technologies be listed in the
ingredient section of product labels. This requirement allows for
data collection and tracking to ensure that, if an innovative
technology is, in fact, harmful, public health officials can
identify this fact relatively quickly, pull the attached “strings” to
rapidly recall the consumer product, and avert, or at least
mitigate, a possible public health disaster. This Article describes
how a focus on novelty, coupled with an already-existing
provision in the Food, Drug, and Cosmetic Act (FDCA, or “the
Act”) requiring that all material information be provided on
product labels, triggers an ingredient-listing requirement.
Importantly, ingredient listing is the key to the public
health product safety net.15 In a nutshell, novel ingredient
12
For a discussion of the difference between individual risk and collective risk and
what this difference means to appropriate regulatory choices, see notes 189–193 and
accompanying text.
13
Id. Of note is that these approaches are also problematic when dealing with
collective risk. This topic is beyond the scope of this Article as the issues relating to
collective risk are, in many ways, very different from those arising in the context of
individual risks, including the appropriate role of government.
14
See notes 189–195 and accompanying text.
15
This term is different from what the popular literature refers to as the health

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 437
listing protects consumer safety through consumer self-
protection. If a consumer knows that she is being exposed to a
novel ingredient, she can use heightened vigilance for symptoms
of harm. It also allows for the appropriate treatment of injured
consumers by medical professionals who will, for the first time,
be able to identify novel ingredients as potential causative
agents through accurate exposure reporting by consumers.
Ingredient listing also allows for the proper reporting of injury-
causing agents to state and federal public health protection
agencies in charge of the early warning and product recall
systems. Finally, the data collected by these public health
protection agencies provides the evidence needed to actuate the
instrumental use of the tort system to encourage the proper
investment in product safety and to insulate against the overuse
and overconsumption of relatively risky products. Together,
these public and private actors join to form the public health
product safety net.16
While this system is far from perfect, it is likely to improve
exponentially as this country moves into the era of big data.17
The public health product safety net will use big data strategies
to take more traditionally created data generated by the state
and federal consumer product reporting systems and link it to
the information that is only recently being gathered from a
massive collection of electronic health records.18 Adding to this
care safety net that refers to health care providers who provide care to low-income
people. See, for example, Coalition of Community Health Clinics, What is the Health
Care Safety Net, online at http: //www.coalitionclinics.org /safety-net.html (visited Sept
15, 2013) (“Health care safety net clinics are community-based providers who offer
health services to low-income people, including those without insurance.”).
16
See notes 175–184 and accompanying text.
17
See Data, data everywhere, The Economist (Feb 25, 2010), online at
http: //www.economist.com/node/15557443 (visited Sept 15, 2013) (“[T]he world contains
an unimaginably vast amount of digital information which is getting ever vaster ever
more rapidly. This makes it possible to do many things that previously could not be done:
spot business trends, prevent diseases, combat crime and so on. Managed well, the data
can be used to unlock new sources of economic value, provide fresh insights into science
and hold governments to account.”); Dan Kusnetzky, What is “Big Data”?, Virtually
Speaking (ZDNet Feb 26, 2010), online at http: //www.zdnet.com/blog /virtualization/
what-is-big-data/1708 (visited Sept 15, 2013) (“In simplest terms, the phrase refers to the
tools, processes and procedures allowing an organization to create, manipulate, and
manage very large data sets and storage facilities.”).
18
Thomas L. Friedman, Obamacare’s Other Surprise, NY Times (May 25, 2013),
online at http: //www.nytimes.com/2013/05/26/opinion/sunday/friedman-obamacares-
other-surprise.html?emc=tnt&tntemail0=y (visited Sept 15, 2013) (detailing the
remarkable growth in conversion to electronic medical records and the use of data
mining to access information contained therein).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
438 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
data stream is an enormous amount of new information that is
being culled through the 2013 phenomenon of data mining,
consumer use of social media, such as tweets and on-line
discussion forums, as well as search engines such as those
created by Google, Microsoft, and Yahoo, in order to identify
defective products that cause physical harm. Just this year,
scientists announced that they were able to use the data from
these unique sources to identify harmful product defects
significantly before the FDA,19 and several apps have been
developed to track food-borne illnesses using data collected from
Twitter.20 All told, these integrated data streams will ultimately
create a highly sensitive, state-of-the-art, consumer product
safety surveillance system.21 This system will identify early
warnings of product safety problems associated with ingredients
created by innovative technologies to proactively mitigate their
effects through national product warning notifications and
recalls.
Adding to this picture, a focus on novelty fits well with the
overarching theme of the FDCA that titers the degree and kind
of regulation it uses to protect consumers from harm from third
parties to the level and type of vulnerability of those consumers.
It also tracks how the Act has historically dealt with regulating
novel ingredients when the health risks associated with these
ingredients are unknown.22
Finally, the result of this focus on novelty reflects the
appropriate role of the government when protecting its citizens
against the risks of harm by third parties. In the context of
consumer products and individual risk, the proper role of the
government is to protect individual choice with regard to the
amount and nature of the risk the consumer is willing to
encounter. This contrasts with the appropriate role of
government in the context of collective risk created by third
19
See notes 122–124 and accompanying text.
20
Beth Krietsch, Social Media Apps Use Twitter to Track Illness Outbreaks, Food
Safety News (Aug 19, 2013), online at http: //www.foodsafetynews.com/2013/08/social-
media-moves-to-help-detect-outbreaks/ (visited Sept 15, 2013); Tracking Twitter May
Enhance Monitoring of Food Safety at Restaurants, ScienceDaily (Aug 7, 2013), online at
http: //www.sciencedaily.com/releases/2013/08/130807134510.htm (visited Sept 15, 2013).
21
See note 20.
22
See notes 135–175 and accompanying text.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 439
parties when the individual has no ability to avoid the risk
through their own means.23
This Article will use nanotechnology as an example that
highlights how regulation based on novelty rather than hazard
achieves the proper balance between protecting public health
while encouraging innovation through the animation of the
public health product safety net. In Part II, this Article starts by
explaining what nanotechnology is and the remarkable growth
of its use in everyday consumer products. It then summarizes
the steadily increasing number of studies that suggest that
there are likely to be serious health risks associated with the
use of nanotech consumer products. Next, it explains how the
FDA is currently regulating these products by focusing on the
degree of hazard and the serious problems that arise from this
misguided focus. In Part III, the history of the FDCA is
summarized in order to explain how, and why, the FDCA came
to titer the degree and kind of regulation it uses to protect
consumers from harm from third parties to the level and type of
vulnerability of targeted consumers. Next, this Article builds on
this history to describe how the Act has historically dealt
specifically with regulating novel ingredients with unknown
health risks in order to deal with consumer vulnerabilities. This
history is then used to illuminate the reasons why a switch from
a focus on hazard to a focus on novelty is supported by precedent
and better fits the policies and goals of the FDCA while, at the
same time, achieving the proper balance between protecting
public health and encouraging innovation. This Article shows
how a simple provision that is already a part of the FDCA can be
relied upon to require ingredient listing based on novelty so that
additional legislation is not required. In Part IV, this Article
illustrates how this focus will operate to achieve the balance
between safety and innovation with other innovative, novel
technologies used as ingredients in consumer products such as
cloned animals, genetically modified plants and animals used for
food, as well as with lab-grown meat. Finally, in Part V, this
Article describes why the use of cost-benefit analysis by the
Office of Management and Budget to evaluate the change
23
Daniel Wickler, Persuasion and Coercion for Health: Ethical Issues in
Government Efforts to Change Life-Styles, 56 Health & Society 303–38 (1978), reprinted
in Rolf Sartorius, Paternalism (Minnesota 1984).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
440 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
suggested by this Article is inappropriate when dealing with
novel ingredients in the context of individual risk.
II. NANOTECHNOLOGY USED IN CONSUMER PRODUCTS
The benefits of nanotechnologies are varied. On one hand,
some claim that nanotechnology could provide the solution to
global challenges such as providing a cure for cancer, a source
for renewable energy, and the solution to the provision of clean
water.24 Others assert that nanotechnology could provide new
medical treatments, reduced use of limited resources, and
economic benefits.25 On the other hand, nanoparticles used in
consumer products offer far less important benefits. Nanotech
cosmetics may improve appearance,26 and nanotech sunscreens
are transparent instead of white and pasty.27 Some nanotech
dietary supplement manufacturers claim that their products are
better absorbed by the body.28 Nanotechnology used in foods
could provide more vivid colors and more potent flavors,
24
Fabio Salamanca-Buentello, et al, Nanotechnology and the Developing World, 2
PLOS Med 0383, 0385 (2005). According to a study by the Canadian Program on
Genomics and Global Health at the University of Toronto Joint Centre for Bioethics, the
top ten applications of nanotechnology most likely to benefit developing countries, and
which may contribute to the attainment of the United Nations Millennium Development
Goals (MDGs), are as follows: energy storage; production and conversion; agricultural
productivity enhancement; water treatment and remediation; disease diagnosis and
screening; drug delivery systems; food processing and storage; air pollution and
remediation; construction; health monitoring; vector and pest detection; and control. Id.
25
The Royal Society and The Royal Academy of Engineering, Nanoscience and
Nanotechnologies: Opportunities and Uncertainties *vii (July 2004), online at
http: //www.nanotec.org.uk/report /Nano%20report%202004%20fin.pdf (visited Sept 15,
2013).
26
Cristina Buzea, Ivan I. Pacheco Blandino, and Kevin Robbie, Nanomaterials and
Nanoparticles: Sources and Toxicity, 2 Biointerphases MR17, MR36 (2007) (discussing
how engineered nanomaterials in cosmetic products regenerate skin cells, help maintain
a youthful appearance of the skin, and hide wrinkles and creases).
27
Natasha Singer, New Products Bring Side Effect: Nanophobia, NY Times E1 (Dec
3, 2008).
28
William B. Schultz and Lisa Barclay, A Hard Pill to Swallow: Barriers to
Effective FDA Regulation of Nanotechnology-Based Dietary Supplements *9 (Woodrow
Wilson International Center for Scholars 2009), online at http: //www.nanotechproject.
org /process/assets/files/7056/pen17_final.pdf (visited Sept 15, 2013) (“Examples of
product claims that tout special properties due to the use of nanotechnology include:
increased effectiveness in a calcium/magnesium product; more rapid, uniform and
complete absorption of nutrients in a spray form; increased absorption of a B12 vitamin
spray; supplements that pass through membranes directly into human cells; and
increased absorption of gel supplements by transforming fat-soluble nutrients into
water-soluble ones.”).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 441
nutritional additives, and antibacterial ingredients for food
packaging.29
To take advantage of some of these less socially beneficial
improvements, engineered nanoparticles are pouring into
everyday consumer products in steadily increasing amounts,
resulting in a concomitant increase in consumer exposure to
these novel ingredients. These nanoparticles are engineered to
be only one to 100 nanometers in size.30 To give an idea of just
how small these nanoparticles are, a human hair is about 80,000
nanometers wide.31
Consumer products that use nanoscale materials can be
found almost everywhere. Nanotech ingredients are in
cosmetics, electronics, catalytics, magnetic and materials
applications, as well as in drug delivery and medical imaging
products.32 Just a small sampling of the specific products that
contain nanotech ingredients includes cell phones, stain-
29
Toxic Nanoparticles Might Be Entering Human Food Supply, ScienceDaily (Aug
22, 2013), online at http: //www.sciencedaily.com/releases/2013/08/130822194530.htm?
utm_source=feedburner&utm_medium=email&utm_campaign=Feed%3A+sciencedaily%
2Ftop_news%2Ftop_health+%28ScienceDaily%3A+Top+News+—+Top+Health%29
(visited Sept 15, 2013).
30
“Nanotechnology is the art and science of manipulating matter at the nanoscale
to create new and unique materials and products.” Schultz and Barclay, A Hard Pill to
Swallow at 20–23 (cited in note 28). The National Nanotechnology Initiative (“NNI”) is a
federal research and development initiative that manages and coordinates the nanoscale
research and technology endeavors of twenty-five different government agencies,
including the FDA. The NNI considers nanotechnology to include activities that involve
the following characteristics:
(1) research and technology development at the atomic, molecular or
macromolecular level, in the length scale of 1–100 nanometers; (2) creating and
using structures, devices and systems that have novel properties and functions
because of their small or intermediate size; and (3) ability to control or
manipulate on the atomic scale.
Id.
31
The Royal Society and The Royal Academy of Engineering, Nanoscience and
Nanotechnologies at viii–x (cited in note 25) (noting that a nanometer is one billionth of a
meter.); National Nanotechnology Initiative, What is Nanotechnology? (nano.gov), online
at http: //www.nano.gov/nanotech-101/what /definition (visited Sept 15, 2013). To attempt
to intellectualize just how small a nanoparticle is, compare nanoparticles to the
thickness of a sheet of paper, which is roughly 100,000 nanometers wide, or to the size of
a human hair, which is approximately 80,000 nanometers wide. National
Nanotechnology Initiative, Size of the Nanoscale (nano.gov), online at http: //www.
nano.gov/nanotech-101/what /nano-size (visited Sept 15, 2013).
32
See, for example, Centers For Disease Control and Prevention, Nanotechnology:
Frequently Asked Questions, online at http: //www.cdc.gov/niosh/topics/nanotech/faq.html
(visited Sept 15, 2013) (acknowledging the broad array of different categories of products
that contain nanoparticles).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
442 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
resistant clothing, sporting goods, digital cameras, eyeglasses,
paints, and computers.33
Importantly, some of these types of products use nanotech
ingredients that are “fixed” inside a solid matrix: for example,
cell phones, computers, and sporting goods, such as tennis
racquets and golf clubs. This makes these types of nanotech
ingredients less likely to move into the human body or the
environment.34 While nanotech ingredients that are
manufactured and then “fixed” in consumer products are of less
concern to consumers, they are of great concern to the workers
who make the products as they are exposed to large amounts of
these nanotech ingredients during the manufacturing process.35
33
See Hope Shand and Kathy Jo Wetter, Shrinking Science: An Introduction to
Nanotechnology, in Linda Starke, ed, State of the World 2006 78, 80–82 (2006) (providing
a list of various consumer products that containing nanotech ingredients); Project On
Emerging Nanotech, Nanotechnology Consumer Products Inventory, online at
http: //www.nanotechproject.org /inventories/consumer/browse/categories/ (visited
Apr 3, 2013) (acknowledging the broad array of different categories of products that
contain nanoparticles).
34
The industry opines that fixed nanoparticles are unlikely to be absorbed by the
human body or migrate into the environment. However, whether this is true is still being
studied. See, for example, Singer, New Products Bring Side Effect: Nanophobia, NY
Times at E1 (cited in note 27) (noting representatives of the cosmetic industry have
stated that there was no evidence that nanotech personal care products are a health
hazard). Dr. Andrew Maynard, the former chief science advisor to the Project on
Emerging Nanotechnologies, states that: “I would be very surprised if [fixed carbon
nanotubes are] dangerous to use, let us say, [in] a tennis racket or baseball bat[,] . . .
[b]ut I do not think it is OK to tell people that we think it is safe—we’ve got to have
evidence.” Ann Fernholm, Carbon Nanotubes May Be as Harmful as Asbestos, SF
Chronicle C-1 (May 21, 2008). The unresolved problem is what happens when fixed
nanoparticles are released as the result of natural wear and tear. For example, when a
product breaks or the surface of one of these products is rubbed against the ground. Id.
Dr. Maynard points out that the level of human exposure to asbestos as car brake pads
containing asbestos wore down and roads paved with asbestos-containing materials
deteriorated was high. See generally Agency for Toxic Substances and Disease Registry,
United States Department of Health and Human Services, Asbestos Toxicity, online at
http: //www.atsdr.cdc.gov/csem/asbestos/
docs/asbestos.pdf (visited Sept 15, 2013).
35
See generally Department of Health and Human Services, Current Intelligence
Bulletin 65: Occupational Exposure to Carbon Nanotubes and Nanofibers *1–2 (National
Institute for Occupational Safety and Health Apr 2013), online at
http: //www.cdc.gov/niosh/docs/2013-145/pdfs/2013-145.pdf (visited Sept 15, 2013)
(“NIOSH Bulletin”) (discussing the risks associated with exposure of workers to
nanoparticles during the manufacturing process); Arthur Miller, et al, Characterizing
Exposures to Airborne Metals and Nanoparticle Emissions in a Refinery, 54 Annals of
Occup Hygiene 504, 511–12 (2010); R.J. Aitken, K.S. Creely, and C.L. Tran,
Nanoparticles: An Occupational Hygiene Review 2–3 (Health and Safety Executive 2004),
online at http: //www.hse.gov.uk/research/rrpdf/rr274.pdf (visited Sept 15, 2013).
Importantly, higher exposure levels may occur during cleaning and maintenance of
production, research, and handling facilities. See NIOSH Bulletin at 1–2; Georgia Miller,

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 443
Products that contain predominantly “free” nanotech
particles, such as liquid products, should be of more concern to
consumers. Nanotech particles that are “free” are extremely
mobile,36 and the human body can absorb them through several
different routes of exposure.37 Nanoparticles can enter the blood
stream through the skin,38 the gastrointestinal tract,39 or the
lungs.40 When inhaled, nanoparticles can invade all areas of the
lungs.41 They can pass into the brain via the olfactory nerves42
et al, Nanomaterials, Sunscreens and Cosmetics: Small Ingredients, Big risks *10
(Friends of the Earth Report 2006), online at http: //libcloud.s3.amazonaws.com/93/
ce/0/633/Nanomaterials_sunscreens_and_cosmetics.pdf (visited Sept 15, 2013). As the
science on safe exposure levels and protective equipment in the work place is still
changing, little is known about the levels at which workers can safely be exposed to
nanoparticles while on the job. NIOSH Bulletin at 1–2. This is a pressing issue as it is
estimated that, by 2015, over two million workers worldwide will be directly employed by
industries using nanoparticles. Mihail C. Roco, Converging Science and Technology at
the Nanoscale: Opportunities for Education and Training, 21 Nature Biotech 1247, 1248
(2003). It is likely that the numbers of workers employed indirectly in the supply chain
will be significantly higher. Id (explaining that nanotechnology has the potential to
create five million related jobs by 2015).
36
David Rotman, Measuring the Risks of Nanotechnology, Tech Rev 71–73 (Apr
2003) (interviewing Dr. Vicki Colvin, Director of the Center for Biological and
Environmental Nanotechnology at Rice University, about possibly unique health and
environmental risks associated with nanotechnology); Buzea, Pacheco Blandino, and
Robbie, 2 Biointerphases at MR44–48, MR50–57 (cited in note 26).
37
Günter Oberdörster, et al, Principles for Characterizing the Potential Human
Health Effects from Exposure to Nanomaterials: Elements of a Screening Strategy, 2
Particle and Fibre Toxicology 1, 2, 4 (2005).
38
Cosmetics that use nanotech ingredients that are rubbed onto the skin contain
nano-size particles 1000 nm in size. At this size, they can be absorbed through intact
skin. Jillian Rouse and Jianzhong Yang, Repetitive Motion Speeds Nanoparticle Uptake:
‘Bucky Amino Acid’ Penetrates Faster, Deeper When Skin Is Flexed, Sciencedaily (Jan 9,
2007), online at http: //www.sciencedaily.com/releases/2007/01/070104144839.htm
(visited Sept 15, 2013) (noting that chemists and toxicologists find that uptake of
nanoparticles through the skin is sped up through repetitive movement); Sally S. Tinkle,
et al, Skin as a Route of Exposure and Sensitization in Chronic Beryllium Disease, 111
Envir Health Persp 1202, 1204–05 (2003) (presenting a study of the degree of
penetration of nano-size particles into human skin). Damage to the skin can occur
through sunburn, blemishes, shaving cuts, eczema, or other trauma. When skin is
traumatized, nano-sized particles up to 7000 nm are absorbed by the skin. Günter
Oberdörster, Eva Oberdörster, and Jan Oberdörster, Nanotoxicology: An Emerging
Discipline Evolving from Studies of Ultrafine Particles, 113 Envir Health Persp 823, 834
(2005). Importantly, many cosmetics and sunscreens containing nanotech ingredients
are especially formulated to be used on damaged skin.
39
Oberdörster, Oberdörster, and Oberdörster, 113 Envir Health Perspectives at
833–37 (cited in note 38); Peter H.M. Hoet, Irene Bruske-Hohlfeld, and Oleg V. Salata,
Nanoparticles: Known and Unknown Health Risks, 2 J Nanobiotech 1, 1, 2–10 (Dec
2004).
40
NIOSH Bulletin at 1–2 (cited in note 35).
41
Oberdörster, Oberdörster, and Oberdörster, 113 Envir Health Persp at 837 (cited
in note 38); Hoet, Bruske-Hohlfeld, and Salata, 2 J Nanobiotech at 1–4 (cited in note 39).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
444 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
and can also cross the formidable blood-brain barrier.43 In
contrast to macro-particles that are trapped and eradicated by
the body’s immune systems,44 once nanoparticles are absorbed
into the blood stream, they can travel unhindered into the
muscles, liver, bone marrow, and spleen, and can actually move
into cells.45 Amazingly, nanoparticles can move through the
cytoplasm and bind to cellular structures.46 This includes the
ability to get wedged in the mitochondria.47 Finally, if absorbed
by a pregnant woman, nanoparticles can cross the placenta,
enter the fetus, and invade all of the areas listed above.48
This use of free nanoparticles in food,49 drugs,50 cosmetics,51
dietary supplements,52 and sunscreens53 creates a large and
42
Buzea, Pacheco Blandino, and Robbie, 2 Biointerphases at MR50–51 (cited in
note 26); Alex Kirby, Tiny Particles ‘Threaten Brain’ (BBC News Jan 8, 2004), online at
http: //news.bbc.co.uk/2/hi/science/nature/3379759.stm (visited Sept 15, 2013); Annabelle
Hett, Nanotechnology: Small Matter, Many Unknowns *17 (Swiss Reins Co 2004), online
at http: //media.swissre.com/documents/nanotechnology_small_matter_many_unknowns_
en.pdf (visited Sept 15, 2013).
43
Rotman, Tech Rev at 73 (cited in note 36); Buzea, Pacheco Blandino, and Robbie,
2 Biointerphases at MR51 (cited in note 26); Kirby, Tiny Particles at 17 (cited in note 42).
44
Hett, Nanotechnology at 21 (cited in note 42).
45
Id at 22–23.
46
Karen Florini, et al, Nanotechnology: Getting It Right the First Time, 3 Nanotech
L and Bus 39, 42 (2006).
47
Dusica Maysinger, et al, Nanoparticles in Medicine, in 3 Oxford Handbook of
Nanoscience and Technology: Applications 503, 519 (A.V. Narlikar, et al, eds, 2010);
Buzea, Pacheco Blandino, and Robbie, 2 Biointerphases at MR48 (cited in note 26).
48
Karin S. Hougaard, et al, Effects of Prenatal Exposure to Surface-Coated
Nanosized Titanium Dioxide (UV-Titan): A Study in Mice, 7 Particle & Fibre Toxicology
16, 22 (2010).
49
See, for example, Georgia Miller and Rye Senjen, Out of the Laboratory and on to
our Plates: Nanotechnology in Food and Agriculture *9 (Friends of the Earth 2008),
online at http: //www.foeeurope.org /activities/nanotechnology/Documents/Nano_food_
report.pdf (visited Sept 15, 2013) (stating nano-size additives can be found in some soft
drinks, dairy products, sausages, beer, and other processed foods).
50
See, for example, Christopher Weldon, Bozhi Tian, and Daniel S. Kohane,
Nanotechnology for Surgeons, 3 Wiley Interdisciplinary Rev: Nanomed & Nanobiotech
223, 226 (2011) (discussing how the science of nanotechnology could create powerful new
tools greatly enhancing surgeons’ access to therapeutic and diagnostic measures); Kevin
O’Donnell and Robert O. Williams, Nanoparticulate Systems for Oral Drug Delivery to
the Colon, 8 Intl J Nanotech 4, 4–15 (2011) (arguing that by encapsulating a drug
molecule within a nano-size module, a highly effective tool for controlling drug delivery
to a specific target could be created, for example for the treatment of a cancerous tumor);
Dorothy Farrell, et al, Recent Advances from the National Cancer Institute Alliance for
Nanotechnology in Cancer, 4 ACS Nano 589 (2010) (noting nanotechnology holds great
promise for the treatment of cancer).
51
See, for example, Miller and Senjen, Out of the Laboratory at 4 (cited in note 49).
52
See, for example, Schultz and Barclay, A Hard Pill to Swallow at 8 (cited in note
28). Dietary supplements with nanotech ingredients have jumped in number from eleven

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 445
growing level of consumer exposure to novel ingredients never
before seen in nature. Nanotech ingredients are being used in
rapidly increasing amounts to create “more potent food
colourings, flavourings and nutritional additives, antibacterial
ingredients for food packaging, and more potent agrochemicals
and fertilisers.”54 Food analysts estimate that nanotech food
additives are being used in more than six hundred different food
products.55 Food packagers are also steadily increasing their use
of nanotech particles in food packaging. This creates the
opportunity for these particles to migrate into the food.56
The National Institute of Occupational Safety and Health
(“NIOSH”) reports that consumer products containing nanotech
ingredients are being introduced into the market at a frequency
of three to four per week.57 Consumer products that contain
nanotech ingredients number in the thousands,58 while
thousands of tons of nanoparticles are produced each year.59
Nanotech research and development is predicted to reach $3.1
trillion globally by 2015.60 In 2007, $147 billion in manufactured
in 2007 to forty-four in 2009. Id at 9.
53
Id.
54
Miller and Senjen, Out of the Laboratory at 4 (cited in note 49). Nanotech particle
additives can be found in sodas, margarine, dairy products, and sausages. Id at 9.
Another growing area is the use of nanotech content in food and beverage packaging, for
example “nanoclay composites—plastics to which nanoscale clay platelets have been
added.” Id at 4. These nanoclay materials are also used “in agriculture pipes and plastics
to allow controlled release of herbicides.” Id.
55
Stephen Daniells, Think Big, Think Nano (Foodnavigator.com Dec 19, 2007),
online at http: //www.foodnavigator.com/Science-Nutrition/Think-big-think-nano (visited
Sept 15, 2013).
56
Miller and Senjen, Out of the Laboratory at 4 (cited in note 49) (stating that
between four hundred and five hundred foods have nanotech packaging).
57
See, for example, Center For Disease Control and Prevention, Nanotechnology:
Frequently Asked Questions (Sept 22, 2010), online at http: //www.cdc.gov/niosh/topics/
nanotech/faq.html (visited Sept 15, 2013) (acknowledging the broad array of different
categories of products that contain nanoparticles).
58
See Schultz and Barclay, A Hard Pill to Swallow at 20–23 (cited in note 28)
(explaining that the increasing amounts of products integrating nanotechnology go
unregulated, due to the dated FDA regulatory scheme).
59
The Royal Society and The Royal Academy of Engineering, Nanoscience and
Nanotechnologies at 26–27 (cited in note 25).
60
Schultz and Barclay, A Hard Pill to Swallow at 8 (cited in note 28). See also
Buyer Beware: Product List Highlights Both Nanotech and Nano-marketing (Electro IQ
Mar 16, 2006), online at http: //www.electroiq.com/articles/stm/2006/03/buyer-beware-
product-list-highlights-both-nanotech-and-nano-marketing.html (visited Sept 15, 2013)
(detailing the growing number of products containing nanoparticles); Center for Disease
Control and Prevention, Nanotechnology: Frequently Asked Questions (cited in note 32).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
446 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
goods using nanotech ingredients were produced.61
Unfortunately, the financial resources invested into the
development, application, and production of nanoparticle
technology is far more than the amount of money invested in the
exploration of health and safety issues.62
A. Why Are Nanotech Ingredients Novel?
Nanotech particles are unique. Nanoparticles and normal
size particles have a whole range of fundamentally different
properties. There are remarkable differences between the two in
toxicity, bioaccumulation, persistence, chemical, magnetic,
electrical, explosiveness, and optical characteristics.63
Engineered nanoparticles differ significantly from their
normal size counterparts64 for two main reasons. First, the laws
61
Schultz and Barclay, A Hard Pill to Swallow at 8 (cited in note 28). See also
Buyer Beware: Product List Highlights Both Nanotech and Nano-marketing, Electro IQ
(Mar 16, 2006), http: //www.electroiq.com/articles/stm/2006/03/buyer-beware-product-list-
highlights-both-nanotech-and-nano-marketing.html (detailing the growing number of
products containing nanoparticles); Center for Disease Control and Prevention,
Nanotechnology: Frequently Asked Questions (cited in note 32).
62
Compared to the amount of funding for nanotech commercial applications, the
amount of money spent on health and environmental risks associated with nanotech
products is very small. For example, as of 2010, only approximately 5 percent of the
NNI’s budget is dedicated to the health and environmental implications of this new
technology. John F. Sargent Jr, Nanotechnology and Environmental, Health, and Safety:
Issues for Consideration *11 (Congressional Research Service Jan 20, 2011), online at
http: //www.fas.org /sgp/crs/misc /RL34614.pdf (visited Sept 15, 2013).
63
Buzea, Pacheco Blandino, and Robbie, 2 Biointerphases at MR17, MR23–25,
MR47, MR59 (cited in note 26). See also Ernie Hood, Nanotechnology: Looking as We
Leap, 112 Envir Health Persp A740, A741 (2004); The Royal Society and The Royal
Academy of Engineering, Nanoscience and Nanotechnologies: Opportunities and
Uncertainties at 5 (cited in note 25).
64
In order to conform to the nomenclature adopted by the literature, this Article
refers to particles that manifest these different properties as “nanoparticles” or
“nanoscale” or “nano-sized” materials or versions and will refer to larger scale particles
of the same chemical that do not have these unique properties as normal size materials
or bulk materials. Buzea, Pacheco Blandino, and Robbie, 2 Biointerphases at MR23–25
(cited in note 26); The Royal Society and The Royal Academy of Engineering,
Nanoscience and Nanotechnologies: Opportunities and Uncertainties at 7 (cited in note
25). For example, the list of FDA-approved active ingredients for use in sunscreens
includes titanium dioxide for use up to a 25 percent concentration. Food and Drug
Administration, Sunscreen Drug Products for Over-The-Counter Human Use: Final
Monograph, 64 Fed Reg 27666, 27672 (May 21, 1999), to be codified at 21 CFR § 352
(“Final Monograph”). The safety and effectiveness of titanium dioxide in sunscreens was
reviewed by the FDA prior to industry use of the engineered nanoparticle form of
titanium dioxide pursuant to the normal process for over-the-counter (“OTC”) approval.
Food and Drug Administration, Small Business Assistance: Frequently Asked Questions
on the Regulatory Process of Over-the-Counter (OTC) Drugs, online at
http: //www.fda.gov/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/ucm0

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 447
of classical physics apply to normal size particles.65
Comparatively, the laws of quantum mechanics apply to
nanoparticles.66 Second, nanoparticles enjoy an enormous
surface-to-volume ratio, which translates into a greater
proportion of atoms existing on the particle’s surface.67 As
chemical reactions occur on the surface of particles, a
nanoparticle has a greater potential for biological interaction.
This means that nanoparticles are far more bioreactive than
their normal size counterparts.68 It is critical to note, as a public
health matter, that this means that the inherent toxicity of any
given quantity of nano-sized particles is much greater than the
same quantity of their normal-sized counter parts.69
69917.htm (visited Sept 15, 2013) (describing the process for OTC approval). The FDA
stated that it is aware that sunscreen products use engineered nanoparticles but that it
“does not consider micronized titanium dioxide to be a new ingredient but it considers it
a specific grade of titanium dioxide originally reviewed by the Panel.” 64 Fed Reg at
27671–72.
65
Buzea, Pacheco Blandino, and Robbie, 2 Biointerphases at MR23 (cited in note
26) (stating the laws of classical physics don’t apply to particles that are smaller than
approximately 100 nanometers (nm)); The Royal Society and The Royal Academy of
Engineering, Nanoscience and Nanotechnologies: Opportunities and Uncertainties at 7
(cited in note 25).
66
Buzea, Pacheco Blandino, and Robbie, 2 Biointerphases at MR24–25 (cited in
note 26) (noting the laws of quantum mechanics apply to nanoparticles affecting the
magnetic, optical, and electric behavior of materials); The Royal Society and The Royal
Academy of Engineering, Nanoscience and Nanotechnologies: Opportunities and
Uncertainties at 7 (cited in note 25).
67
Andre Nel, et al, Toxic Potential of Materials at the Nanolevel, 311 Sci 622, 622
(2006). Compared to macro-particles,
[T]his provides a greater surface area per unit mass. In the size range of < 100
nm, the number of surface molecules (expressed as a % of the molecules in the
particle) is inversely related to particle size. For instance, in a particle of 30 nm
size, about 10% of its molecules are expressed on the surface, whereas at 10
and 3 nm size the ratios increase to 20% and 50%, respectively. Because the
number of atoms or molecules on the surface of the particle may determine the
material reactivity, this is key to defining the chemical and biological
properties of nanoparticles.
Id at 623 fig 1.
68
Id at 622.
69
Id; Scientific Committee on Emerging and Newly Identified Health Risks
(“SCENIHR”), Modified Opinion on the Appropriateness of Existing Methodologies to
Assess the Potential Risks Associated with Engineered and Adventitious Products of
Nanotechnologies 13, 13 (European Commission 2005), online at http: //ec.
europa.eu/health/ph_risk/committees/04_scenihr/docs/scenihr_o_003b.pdf (visited Sept
15, 2013).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
448 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
B. What Are the Health Risks Associated with Exposure to
Nanotech Ingredients?
Over the past several years, a number of scientific animal
studies have started to provide at least some ideas about the
type of negative health effects that can be expected to occur from
exposure to the unique properties of nanotech ingredients. Some
of the side effects could include a contribution to the
development of neurodegenerative processes, such as
Alzheimer’s disease70 or mesothelioma, the condition caused by
asbestos.71
Studies show that there are two main negative physical
effects that could cause these disorders.72 First, the increased
bioreactivity of nanoparticles discussed above can harm living
tissue.73 Once inside cells, this enhanced bioreactivity can
interfere with cell signaling, damaging the cell’s structure and
DNA.74 In the opposite effect of the saying “the poison is in the
dose,” the smaller the size of the particle, the more likely it is to
have a toxic effect because its bioreactivity increases.75 For
example, even if a material, like titanium dioxide, is harmless at
a normal size, when it shrinks to nano-size, pulmonary toxicity
increases.76 Second, the human body contains scavenger cells
70
Andre E. Nel, et al, Understanding Biophysicochemical Interactions at the Nano-
Bio Interface, 8 Nature Mat 543, 550 (2009).
71
Id at 546.
72
See Jelena Kolosnjaj, Henri Szwarc, and Fathi Moussa, Toxicity Studies of
Carbon Nanotubes, Bio-Apps of Nanoparticles 181 (Warren C.W. Chan ed 2007)
(containing an exceptional survey of toxicity studies). For a comprehensive summary of
the experimental mechanisms of nanotech toxicity, see Nel, et al, 8 Nature Mat at 551,
table 4 (cited in note 70).
73
Nel, et al, 8 Nature Mat at 543, table 4 (cited in note 70).
74
Jirasak Wong-Ekkabut, et al, Computer Simulation Study of Fullerene
Translocation Through Lipid Membranes, 3 Nature Nanotech 363, 363, 367 (2008); C.L.
Tran, et al, A Scoping Study to Identify Hazard Data Needs for Addressing the Risks
Presented by Nanoparticles and Nanotubes, Institution of Occupational Medicine 15, 25
(2005); Hisao Hidaka, et al, In Vitro Photochemical Damage to DNA, RNA and Their
Bases by an Inorganic Sunscreen Agent on Exposure to UVA and UVB Radiation, 111 J
Photochem & Photobio 205, 212 (1997); Rosemary Dunford, et al, Chemical Oxidation
and DNA Damage Catalysed by Inorganic Sunscreen Ingredients, 418 FEBS Letters 87,
87–90 (1997).
75
Qamar Rahman, et al, Evidence That Ultrafine Titanium Dioxide Induces
Micronuclei and Apoptosis in Syrian Hamster Embryo Fibroblasts, 110 Envir Health
Persp 797, 797, 799 (2002); T. Uchino, et al, Quantitative Determination of OH Radical
Generation and its Cytotoxicity Induced by TiO2-UVA Treatment, 16 Toxicology in Vitro
629, 634 (2002); Nel, et al, 311 Sci at 622 (cited in note 67).
76
Tran, et al, A Scoping Study, at 21–23 (cited in note 74); Nel, et al, 311 Sci at 622

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 449
called phagocytes that eliminate foreign substances. These
phagocytes can cease to function when they become clogged with
nanotech particles.77 This means that foreign particles,
including bacteria, can invade the body with impunity as the
phagocytes have been neutralized, causing an HIV-like effect.78
While unfortunate, it is no surprise that there are very few
animal studies of the health effects of exposure to nanoparticles
in light of the relative amounts of investment in product
development as compared to the investment in safety testing.79
One of the first well-known animal studies observes the impact
of exposures of nanoparticles called buckyballs80 on fish.81 The
result was that the brains of the fish, here largemouth bass,
developed a toxic side effect that manifested itself in significant
(cited in note 67).
77
Nel, et al, 8 Nature Mat at 550–52 (cited in note 70); Margot Lundborg, et al,
Human Alveolar Macrophage Phagocytic Function is Impaired by Aggregates of Ultrafine
Carbon Particles, 86 Envir Rsrch Sect A86 244, 252 (2001); Peter G. Barlow, et al,
Reduced Alveolar Macrophage Migration Induced by Acute Ambient Particle (PM10)
Exposure, 24 Cell Bio & Toxicology 243, 248–51 (2008); Buzea, Pacheco Blandino, and
Robbie, 2 Biointerphases at MR45–46 (cited in note 26).
78
Lundborg, et al, 86 Envir Rsrch Sect at 252 (cited in note 77); Barlow, et al, 24
Cell Bio & Toxicology at 251 (cited at 77); Buzea, Pacheco Blandino, and Robbie, 2
Biointerphases at MR45–46 (cited in note 26).
79
See Sargent, Nanotechnology and Environmental, Health, and Safety (cited in
note 62).
80
Buckminsterfullerene (C60), called “buckyballs,” was named for Richard
Buckminster Fuller, the famed engineer recognized for the creation of the geodesic dome.
Buckyballs Could Keep Water Systems Flowing, Sciencedaily (Mar 12, 2009), online at
http: //www.sciencedaily.com/releases/2009/03/090305080139.htm (visited Sept 15, 2013).
81
Eva Oberdörster, Manufactured Nanomaterials (Fullerenes, C60) Induce
Oxidative Stress in the Brain of Juvenile Largemouth Bass, 112 Envir Health Persp
1058, 1058 (2004). Buckyballs are soccer-ball-shaped, spherical molecules with sixty
carbon molecules. They are the smallest of the fullerene family. T. Csörgő, M. Gyulassi,
and D. Kharzeev, Letter to the Editor, Buckyballs and Gluon Junction Networks on the
Femtometre Scale, 30 J Physics G: Nuclear and Particle Physics L17, L17–18 (2004);
H.W. Kroto, et al, Letters to Nature, C60 Buckminsterfullerene, 318 NATURE 162, 162–
63 (1985). Fullerenes are molecules made entirely of carbon. Csörgő, Gyulassi, and
Kharzeev, 30 J Physics G: Nuclear and Particle Physics at L17.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
450 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
lipid peroxidation.82 Buckyballs are commonly used in
cosmetics,83 food packaging, and dietary supplements.84
Another startling set of recent, significant studies85 suggests
that another form of nanoparticle, called multi-walled carbon
nanotubes,86 could turn out to be as harmful as asbestos.87 It
82
Oberdörster, 112 Envir Health Persp at 1060 (cited in note 81). See also Emil
Venere, ‘Buckyballs’ Have a High Potential to Accumulate in Living Tissue, Purdue News
(Sept 18, 2008), online at http: //news.uns.purdue.edu/x/2008b/080918JafvertBuckyballs.
html (visited Sept 15, 2013) (interviewing author Chad T. Jafvert who explains that his
research suggests that buckyballs have a greater probability of partitioning into fatty
tissue than the banned pesticide DDT), referring to Chad T. Jafvert and Pradnya P.
Kulkarni, Buckminsterfullerene’s (C60) Octanol—Water Partition Coefficient (Kow) and
Aqueous Solubility, 42 Envir Sci & Tech 5945, 5946–49 (2008).
83
Bethany Halford, Fullerene for the Face: Cosmetics Containing C60 Nanoparticles
are Entering the Market even if Their Safety is Unclear, 84 Chem & Engineering News
47, 47 (2006); Miller, et al, 54 Annals of Occup Hygiene at 7 (cited in note 35).
84
Leslie Pray and Ann Yaktine, Nanotechnology in Food Products: Workshop
Summary, Inst of Med 739, 741 (2009), online at http: //www.nap.edu/catalog.php?
record_id=12633 (visited Sept 15, 2013); Schultz and Barclay, A Hard Pill to Swallow at
*9 (cited in note 28); Walter Derzko, Novel, Safe Natural Food Supplement, Hydrated
Fullerenes (C60-HyFn) or Water-Soluble Buckyballs Could Make 50–60% of the Riskier
Synthetic Drugs and Pharmaceuticals Obsolete by 2025–30, Smart Economy (May 17,
2010), online at http: //smarteconomy.typepad.com/smart_economy/2010/05/novel-safe-
natural-food-supplement-hydrated-fullerenes-c60hyfn-or-watersoluble-buckyballs-could-
make.html (visited Sept 15, 2013); Questioning Safety Of Nanotechnology in Your
Vitamins, Sciencedaily (Jan 15, 2009), online at http: //www.sciencedaily.com/releases/
2009/01/090114114936.htm (visited Sept 15, 2013); Nanoparticles in Dietary
Supplements Cause Health Concerns, Regulatory Challenges, Sciencedaily (Feb 10,
2009), online at http: //www.sciencedaily.com/releases/2009/02/090209075633.htm
(visited Sept 15, 2013). The buckyballs negatively impacted the water habitat by killing
all the water fleas and bacteria. Oberdörster, 112 Envir Health Persp at 1059 (cited in
note 81). Another separate study showed that nanoparticles in small amounts are toxic
to soil bacteria. J.D. Fortner, et al, C60 in Water: Nanocrystal Formation and Microbial
Response, 39 Envir Sci and Tech 4307, 4307 (2005); Buckyballs Could Keep Water
Systems Flowing, Sciencedaily (cited in note 80).
85
Vincent Castranova, et al, Persistent Pulmonary Fibrosis, Migration to the
Pleura, and Other Preliminary New Findings After Subchronic Exposure to Multi-Walled
Carbon Nanotubes, NIOSH Sci Blog (Mar 19, 2009, 10:24 AM), online at
http: //blogs.cdc.gov/niosh-science-blog /2009/03/nano-2/ (visited Sept 15, 2013) (“NIOSH
Study”), reprinted as abstract in 108 Toxicologist 457 (2009); Craig A. Poland, et al,
Letters, Carbon Nanotubes Introduced into the Abdominal Cavity of Mice Show Asbestos-
Like Pathogenicity in a Pilot Study, 3 Nature Nanotech 423, 423–28 (2008); Carbon
Nanotubes that Look like Asbestos, Behave like Asbestos, Could Lead to Asbestos-Related
Disease, Sciencedaily (May 22, 2008), online at http: //www.sciencedaily.com/
releases/2008/05/080520144004.htm (visited Sept 15, 2013).
86
“Discovered nearly 20 years ago, carbon nanotubes have been described as the
wonder material of the 21st Century. Light as plastic and stronger that [sic] steel, they
are being developed for use in new drugs, energy-efficient batteries and futuristic
electronics.” Carbon Nanotubes that Look Like Asbestos, Sciencedaily (cited in note 85).
Carbon nanotubes are atom-thick sheets of graphite formed into cylinders.
They may be formed from a single layer of graphite or they may consist of
multiple concentric layers of graphite, resulting in multi-walled carbon

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 451
appears that these nanotubes have the potential to cause
mesothelioma,88 a cancer of the lung lining that appears
following exposure to asbestos.89 A pair of studies released in
nanotubes. While the diameter of a nanotube can vary from a few nanometers
up to tens of nanometers, they can be hundreds or even thousands of
nanometers long. Carbon nanotubes come in many forms, with different
shapes, different atomic arrangements, and varying amounts and types of
added chemicals—all of which affect their properties and might influence their
impact on human health and the environment.
Id.
87 Asbestos fibers are harmful because they are thin enough to penetrate deep
into the lungs, but sufficiently long to confound the lungs’ built-in clearance
mechanisms for getting rid of particles. Widespread exposure to asbestos has
been described as the worst occupational health disaster in U.S. history and
the cost of asbestos-related disease is expected to exceed $200 billion.
Id.
The toll of asbestos-related cancer, first noticed in the 1950s and 1960s, is
likely to continue for several more decades even though usage reduced rapidly
some 25 years ago. While there are reasons to suppose that nanotubes can be
used safely, this will depend on appropriate steps being taken to prevent them
from being inhaled in the places they are manufactured, used and ultimately
disposed of. Such steps should be based on research into exposure and risk
prevention, leading to regulation of their use.
Id (quoting Anthony Seaton, professor emeritus at the University of Aberdeen, UK).
Similar to buckyballs, nanotubes are part of the fullerene family. They consist entirely of
carbon molecules. Unlike buckyballs, nanotubes are made up of carbon atoms bonded
into a tube shape, sometimes with a single wall, called single-wall carbon nanotubes
(“SWCN”), or multiple walls, called multi-wall carbon nanotubes (“MWCN”). Marking on
this similarity, carbon nanotubes are called “buckytubes” by some as their ends, when
closed, take on the spherical shape of buckyballs. Nanotubes and Buckyballs,
nanotechnologynow.com, online at http: //www.nanotech-now.com/nanotube-buckyball-
sites.htm (visited Sept 15, 2013).
88
Nanotubes and Buckyballs, nanotechnologynow.com (cited in note 87).
89
Id. In a 2008 study, nanoparticles were injected into the abdominal cavity of mice
because this is a good predictor of how long fibers will affect the lung lining. Based on
the study findings, it appears that long, thin, multi-walled carbon nanotubes that look
like asbestos fibers, actually behave like asbestos fibers. This study suggests that people
who breathe in nanotubes may develop cancer at some point after exposure. Poland, et
al, 3 Nature Nanotech at 426–27 (cited in note 85). In 2009, only a year later, NIOSH
took the Poland study one step further by examining the impact on mice of inhaling a
small drop of liquid containing the multi-walled carbon nanotubes. See Castranova, et al,
Persistent Pulmonary Fibrosis (cited in note 85). This study was the first to demonstrate
that multi-walled carbon nanotubes aspirated by laboratory mice can actually migrate
throughout even the tiniest area of the lungs and can actually migrate into the pleura.
Id. See also LM Sargent, et al, Induction of Aneuploidy by Single-Walled Carbon
Nanotubes, 50 Envir Molecular Mutagenesis 708, 713–15 (2009) (showing that single-
walled carbon nanotubes can cause genotoxicity and abnormal chromosome number
caused by interference with cell division (mitosis)); Atsuya Takagi, et al, Induction of
Mesothelioma in p53+/- Mouse by Intraperitoneal Application of Multi-Walled Carbon
Nanotube, 33 J Toxicology Sci 105, 110–14 (2008) (describing intraperitoneal injection of
multi-walled carbon nanotubes causes mesothelial tumors in mice after). The mice had

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
452 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
May of 2013, completed by a consortium of seventy researchers
from seven universities and the National Institute of
Occupational Health and Safety, confirms that some types of
nanoparticles are likely to cause physical harm.90 This
consortium was able to measure similar adverse health effects
after exposure to nanoparticles by replicating their results in
separate studies across multiple independent labs.91 In one
study, laboratories measured pulmonary inflammation in
rodents exposed to carbon nanotubes and nanotech titanium
dioxide.92 In the second study, researchers demonstrated
adverse reactions in cell cultures exposed to carbon nanotubes,
nanotech titanium dioxide and nanotech zinc oxide.93 Based on
these collective studies, it appears that it is likely that carbon
persistent inflammation and fibrosis (scarring) in their lungs after inhaling the particles.
Castranova, et al, Persistent Pulmonary Fibrosis, NIOSH Sci Blog (cited in note 85).
These are important findings because multi-walled carbon nanotubes have properties
that are similar to asbestos and a form of cancer called mesothelioma takes form in the
pleura after exposure to asbestos. Id. The still unanswered question is whether MWCN
will cause mesothelioma. Id. Answering this question, according to the authors of the
study,
is of considerable importance, because research and business communities
continue to invest heavily in carbon nanotubes for a wide range of products
under the assumption that they are no more hazardous than graphite. Our
results suggest the need for further research and great caution before
introducing such products into the market if long-term harm is to be avoided.
Poland, et al, 3 Nature Nanotech at 423 (cited in note 85) (footnote omitted). Dr. Andrew
Maynard, the co-author of the NIOSH study, opines that “[t]his study is exactly the kind
of strategic, highly focused research needed to ensure the safe and responsible
development of nanotechnology. . . . It looks at a specific nanoscale material expected to
have widespread commercial applications and asks specific questions about a specific
health hazard. Even though scientists have been raising concerns about the safety of
long, thin carbon nanotubes for over a decade, none of the research needs in the current
U.S. federal nanotechnology environment, health and safety risk research strategy
address this question.” Carbon Nanotubes that Look like Asbestos, Sciencedaily (cited in
note 85) (internal quotation marks omitted).
90
See generally Robert Iafolla, Groundbreaking Nanotechnology Studies Replicate
Result Across Independent Labs, 28 BNA Toxics Law Reporter 533 (May 9, 2013).
91
Id.
92
Id; Interlaboratory Evaluation of Rodent Pulmonary Responses to Engineered
Nanomaterials: The NIEHS NanoGo Consortium, Environmental Health Perspectives,
(May 6, 2013), online at http: //ehp.niehs.nih.gov/wp-content /uploads/121/5/ehp.1205693.
pdf (visited Sept 15, 2013).
93
Iafolla, 28 BNA Toxics Law Reporter 533 (cited in note 90); Intralaboratory
Evaluation of In Vitro Cytotoxicity and Inflammatory Responses to Engineered
Nanomaterials: The NIEHS NanoGo Consortium (Environmental Health Perspectives
May 6, 2013), online at http: //ehp.niehs.nih.gov/wp-content /uploads/121/5/ehp.1306561.
pdf (visited Sept 15, 2013).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 453
nanotubes, nanotech titanium dioxide, and nanotech zinc oxide
may pose a serious risk to human health.94
C. FDA Regulation of Nanotech Consumer Products or FDA
Regulations on Ingredient Labeling
Currently, the Food and Drug Administration (FDA) is
regulating nanotech ingredients the same way that it regulates
these ingredients’ bulk counterparts.95 If the macro-molecule
version of an ingredient has previously been found by the FDA
to be safe, for regulatory purposes, the FDA presumes that the
94
See, for example, Kolosnjaj, Toxicity Studies of Carbon Nanotubes, Bio-Apps Of
Nanoparticles at 181 (cited in 72) (opining that “available data clearly show that, under
some conditions, nanotubes can cross the membrane barriers and suggests that if raw
materials reach the organs they can induce harmful effects as inflammatory and fibrotic
reactions”); Alexandra E. Porter, et al, Direct Imaging of Single-Walled Carbon
Nanotubes in Cells, 2 Nature Nanotech 713, 713, 716 (2007) (showing that nanotubes
can penetrate into cell “cytoplasm and localize within the cell nucleus, causing cell
mortality in a dose-dependent manner”); Chiu-Wing Lam, et al, A Review of Carbon
Nanotube Toxicity and Assessment of Potential Occupational and Environmental Health
Risks, 36 Crit Rev in Toxicology 189, 207 (2006). See also A. Hubbs, et al, Persistent
Pulmonary Inflammation, Airway Mucous Metaplasia and Migration of Multi-Walled
Carbon Nanotubes from the Lung After Subchronic Exposure, 108 Toxicologist 457, 457
(2009) (describing how the fiber-like dimensions and durability of multi-walled carbon
nanotubes, as well as their ability to cause inflammation in the abdominal cavity, are
similar to asbestos); Elena Kisin, et al, Pulmonary Response, Oxidative Stress and
Genotoxicity Induced by Carbon Nanotubes, 114 Toxicologist A793, A793 (2010) (finding
acute inflammation and interstitial fibrosis in mice exposed to carbon nanofibers);
Robert R. Mercer, et al, Distribution and Persistence of Pleural Penetrations by Multi-
Walled Carbon Nanotubes, 7 Particle & Fibre Toxicology 1, 5–6 (2010) (demonstrating
the toxic effects of MWCNTs because they can invade both the alveollar epithelium and
visceral pleura); Jürgen Pauluhn, Subchronic 13-Week Inhalation Exposure of Rats to
Multi-Walled Carbon Nanotubes: Toxic Effects are Determined by Density of Agglomerate
Structures, not Fibrillan Structures, 113 Toxicology Sci 226, 226 (2010) (showing how
rats exposed to carbon nanotubes at low doses have lower lung clearance); Dale W.
Porter, et al, Mouse Pulmonary Dose—and Time Course—Responses Induced by
Exposure to Multi-Walled Carbon Nanotubes, 269 Toxicology 136, 136–47 (2010)
(observing the resemblance between asbestos fibers and the long and thin structures of
common carbon nanotubes and carbon nanofibres and how both can migrate from
pulmonary alveoli to pleural tissue, which is the same location where malignant
mesothelioma develops).
95
Katharine Van Tassel and Rose H. Goldman, The Growing Consumer Exposure to
Nanotechnology in Everyday Products: Regulating Innovative Technologies in Light of
Lessons from the Past, 44 U Conn 481, 503–13 (2010). Consistent with the nomenclature
adopted by the literature, this Article will refer to particles that manifest these different
properties as “nanoparticles” or “nanoscale” materials or versions and will refer to larger
scale particles of the same chemical that do not have these unique properties as normal
size materials or bulk materials. Buzea, Pacheco Blandino, and Robbie, 2 Biointerphases
at MR23-25 (cited in note 26); The Royal Society and The Royal Academy of Engineering,
Nanoscience and Nanotechnologies: Opportunities and Uncertainties at 7 (cited in note
25).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
454 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
nanotech version is also safe.96 Thus, the FDA is treating
nanotech ingredients as bioequivalent to their normal size
counterparts.97
Regulating nanotech products exactly like their non-
nanotech counterparts has serious consequences.98 First, as
manufacturers of food, dietary supplements, and cosmetics are
not required to test their products for safety and are not
required to obtain premarket approval from the FDA, the
nanotech versions of these products are also not subject to these
premarket steps.99 With regard to sunscreens, nano-sized
ingredients are deemed to be just as safe as their previously
approved macro-sized ingredient counterparts, so no testing of
these nano-ingredients is required.100
Second, this regulatory stance of bioequivalence means that
the listing of nanotech ingredients on product packaging is not
required by the FDA. The FDA opines that product ingredient
lists that refer to nanomaterial content by the same name as the
normal size material counterpart are not false and misleading
based on its presumption of bioquivalence. The FDA grounds
this conclusion on its finding that there is no scientific basis
on which to conclude that nanoscale materials as a class are
inherently more hazardous than nonnanoscale materials.101
Thus, the FDA has taken the position that the fact that a
consumer product contains nanotech ingredients is not
“material” and need not be disclosed on labels.102
96
Van Tassel and Goldman, 44 U Conn at 503–15 (cited in note 95).
97
Id.
98
Id.
99
Id.
100
See note 64.
101
Food and Drug Administration, Nanotechnology Task Force Report 2007, online
at http: //www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/UCM2006659.htm
(visited Sept 15, 2013).
102
Under the FDCA, a drug, device, food, dietary supplement, cosmetic, or sunscreen
is deemed misbranded if its labeling is “false or misleading in any particular.” 21 USC
§ 362(a) (2006). See also 21 CFR § 701.1 (2011).
If an article is alleged to be misbranded because the labeling or advertising is
misleading, then in determining whether the labeling or advertising is
misleading . . . there shall be taken into account (among other things) not only
representations made or suggested by statement, word, design, device, or any
combination thereof, but also the extent to which the labeling or advertising
fails to reveal facts material in the light of such representations or material
with respect to consequences which may result from the use of the article to
which the labeling or advertising relates under the conditions of use prescribed

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 455
It is important to note that, while the FDA has recently
acknowledged on its website that nanoparticles “can have
chemical, physical, and biological properties that differ from
those of their larger counterparts,”103 the acknowledgment of the
possibility that “nano can mean different” has yet to be given
voice in any of the FDA’s regulatory positions on safety.
Third, conditioning the use of regulatory power to protect
public health with the establishment of scientific evidence that
will support a finding of hazard, will, as a general matter, be
outcome determinative. This is because, as explained in the next
section, the development of the scientific evidence needed to
show a product is hazardous to health significantly lags behind
the creation of the innovative technology itself. This regulatory
posture means that there will be no regulatory protection of
public health for perhaps decades.
Fourth, with regard to nanotech food, dietary supplements,
sunscreens, and cosmetics, if the FDA does have concerns about
their safety, it must use its seizure or injunctive powers to
remove the product from the market.104 In these court actions,
the FDA has the burden of proving that the product is
adulterated.105 A product is adulterated if it “presents a
in the labeling or advertising relates under the conditions of use prescribed in
the labeling or advertising thereof or under such conditions of use as are
customary or usual.
21 USC § 321(n) (emphasis added). See also 21 USC § 331(a) (prohibiting the
introduction into commerce of any food, device, or cosmetic that is misbranded); 21 USC
§ 343(a) (stating that foods are misbranded if their labeling is “false or misleading in any
particular”); 21 USC § 352(a) (stating that drugs and devices are misbranded if their
labeling is “false or misleading in any particular”); 21 USC § 362(a) (stating that
cosmetics are misbranded if their labeling is “false or misleading in any particular”).
103
Food and Drug Administration, Nanotechnology, online at http: //www.fda.gov/
ScienceResearch/SpecialTopics/Nanotechnology/default.htm (visited Sept 15, 2013) (“The
U.S. Food and Drug Administration (FDA) regulates a wide range of products, including
foods, cosmetics, drugs, devices, veterinary products, and tobacco products, some of
which may utilize nanotechnology or contain nanomaterials. Nanotechnology allows
scientists to create, explore, and manipulate materials measured in nanometers
(billionths of a meter). Such materials can have chemical, physical, and biological
properties that differ from those of their larger counterparts.”).
104
Richard A. Merrill, Regulating Carcinogens in Food: A Legislator’s Guide to the
Food Safety Provisions of the Federal Food, Drug, and Cosmetic Act, 77 Mich L Rev 171,
186–90 (1978).
105
21 USC § 342(a)(I); Merrill, 77 Mich L Rev at 186–90 (cited in note 104). For a
naturally occurring substance found in the food product, the food product is rendered
adulterated if the substance is ordinarily injurious to health. 21 USC § 342(a)(I).
However, if the substance in the food product is “added,” the food product is adulterated
if the substance “may render” the food injurious to health. 21 USC § 342(a)(I).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
456 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
significant or unreasonable risk of illness or injury.”106 The FDA
interprets “the plain meaning of ‘unreasonable’ . . . [to] connote
[ ] comparison of the risks and benefit of the product.”107 Thus,
the FDA has the burden of “gathering data, soliciting comments
and conducting the risk-benefit analysis.”108
Once again, the FDA is likely to be unsuccessful using this
route for a long period of time, perhaps decades, unless a great
number of people have already been seriously injured. Even
then, as the case of Ephedra demonstrates, it is likely to take a
decade, or longer, for the FDA to be successful.109
106
21 USC § 342(f )(1).
107
Food and Drug Administration, Final Rule Declaring Dietary Supplements
Containing Ephedrine Alkaloids Adulterated Because They Present an Unreasonable
Risk, 69 Fed Reg 6788, 6823 (Feb 11, 2004) (“Final Rule”).
108
Nutraceutical Corp v Andrew Von Eschenbach, 459 F3d 1033, 1040 (10th Cir
2006) (“The plain meaning of ‘significant risk’ is a great danger.”).
109
Id at 1036. Ephedra was an ingredient that was commonly used in dietary
supplements. Id. While ephedrine alkaloids occur naturally in some plants, ephedra falls
into the same chemical category as the street drug called “speed.” Barry A. Palevitz,
Harmless Energizers or Dangerous Drugs?, The Scientist (Dec 9, 2002), online at
http: //www.the-scientist.com/?articles.view/articleNo/14399/title/Harmless-Energizers-
or-Dangerous-Drugs-/ (visited Sept 15, 2013) (“Ephedrine is a close relative of
amphetamine, sometimes called benzedrene. A little chemical tinkering creates the
street drugs methamphetamine and Ecstasy.”). Products containing ephedrine alkaloids
were marketed as dietary supplements for weight loss and to enhance sports
performance. See Nutraceutical Corp, 459 F3d at 1036. Over time, the FDA began
receiving adverse event reports (“AERs”) from consumers, which included numerous
complaints of heart attacks, strokes, seizures, and deaths associated with the
consumption of products containing ephedrine alkaloids. See id. One of the most highly
publicized cases of a fatal consequence from the use of ephedrine alkaloids in a dietary
supplement was the death of Steve Belcher, a twenty-three-year-old baseball player with
the Baltimore Orioles. See Fran Hawthorne, Inside the FDA: The Business and Politics
Behind the Drugs We Take and the Food We Eat 57 (John Wiley and Sons 2005). In order
to meet its burden of proof necessary to remove this ingredient from the market, the
FDA took seven years to gather sufficient evidence on the safety of ephedrine alkaloids.
The FDA compiled an administrative record of 130,000 pages, 19,000 AERs, and engaged
in extensive notice and comment before it passed a regulation banning the sale of
products containing ephedrine alkaloids in 2004. See 69 Fed Reg at 6788 (cited in note
107); Nutraceutical Corp, 459 F3d at 1036. In this final rule, the FDA stated that “[t]he
best clinical evidence for a benefit . . . supports only a modest short-term weight loss,
insufficient to positively affect cardiovascular risk factors or health conditions associated
with being overweight or obese.” Nutraceutical Corp, 459 F3d at 1036–37. Then, the FDA
had to spend several additional years in litigation until Ephedra was finally taken off the
market. The manufacturer of Ephedra filed suit, arguing that the FDA had failed to
meet its burden of proof of showing that products containing ephedrine alkaloids were
unsafe. Id at 1043–44. The district court found for the manufacturer; however, in 2006,
the FDA prevailed on appeal. Id at 1038–39. The total time and expense involved in this
process, including the cost of the harm suffered by consumers, was tremendous. 69 Fed
Reg at 6788 (cited in note 107). Not only did this proceeding take almost a decade, the
amount of time and money that was spent by the FDA, which it could have used on other
matters, was enormous. It is worth pointing out that the taxpayers foot the bill for this

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 457
D. Impact of the FDA’s Focus on Hazard to Trigger Public
Health Safety Regulation
The FDA’s reliance on hazard to trigger the use of its
regulatory power to protect public health when innovative
ingredients are used in consumer products short-circuits its
ability to act for sometimes-lengthy periods of time. This is
because a substantial lag time, likely a decade or more,
invariably exists between the production and distribution of the
innovative technology to the public and the development of the
science necessary to identify the risks associated with that
technology. This lag time occurs when a manufacturer is not
required by the FDCA to prove safety before being allowed to
market a novel, innovative product as there is, otherwise, little
incentive to invest in the testing process to prove safety. The
cost of testing for safety is, instead, born by third parties—
almost entirely the government and, thus, the taxpayers110—and
is performed after the product has already been introduced into
effort, not the manufacturers who reaped tremendous profits over several decades from
marketing Ephedra.
110
Ian Urbina, Think Those Chemicals Have Been Tested?, NY Times (April 13,
2013), online at http: //www.nytimes.com/2013/04/14/sunday-review/think-those-
chemicals-have-been-tested.html (visited Sept 15, 2013). “Many Americans assume that
the chemicals in their shampoos, detergents and other consumer products have been
thoroughly tested and proved to be safe. This assumption is wrong. Unlike
pharmaceuticals or pesticides, industrial chemicals do not have to be tested before they
are put on the market.” Id.
Companies have to alert the Environmental Protection Agency before
manufacturing or importing new chemicals. But then it is the E.P.A.’s job to
review academic or industry data, or use computer modeling, to determine
whether a new chemical poses risks. Companies are not required to provide
any safety data when they notify the agency about a new chemical, and they
rarely do it voluntarily, although the E.P.A. can later request data if it can
show there is a potential risk. If the E.P.A. does not take steps to block the new
chemical within 90 days or suspend review until a company provides any
requested data, the chemical is by default given a green light. The law puts
federal authorities in a bind. ‘It’s the worst kind of Catch-22,’ said Dr. Richard
Denison, senior scientist at the Environmental Defense Fund. ‘Under this law,
the E.P.A. can’t even require testing to determine whether a risk exists
without first showing a risk is likely.’ As a result, the overwhelming majority of
chemicals in use today have never been independently tested for safety. In its
history, the E.P.A. has mandated safety testing for only a small percentage of
the 85,000 industrial chemicals available for use today. And once chemicals are
in use, the burden on the E.P.A. is so high that it has succeeded in banning or
restricting only five substances, and often only in specific applications:
polychlorinated biphenyls, dioxin, hexavalent chromium, asbestos and
chlorofluorocarbons.
Id.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
458 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
the market. Thus, the focus on hazard means that the FDA
must wait to regulate to protect public health until the state of
the science on health risks catches up to the innovation itself.
During this scientific lag time, manufacturers are free to market
their products with no interference and no notice to
unsuspecting consumers.111 As explained in the next section,
this Article refers to this scientific lag time as the health risk
information void.
1. The health risk information void.
When a manufacturer is not required to prove safety before
being allowed to market a novel, innovative product, it is
common for the development of novel technologies to far outpace
the development of the science necessary to test for the health
risks associated with these technologies.112 This scientific lag
time creates a period during which there is an information void
with regard to risks to human health. As this information void is
slowly filled through scientific experimentation, the level of
uncertainty over health risks commonly progresses from
ignorance (where scientists don’t know what they don’t know) to
indeterminacy (where scientists know what they don’t know, but
can plan the scientific experiments necessary to find out) to,
finally, a tipping point in the state of knowledge when classic
probability analysis can be applied to predict, or quantify, risk
levels to human health.113 Thus, as health risks take time to
quantify, the result of the reliance on establishing hazard
through the use of risk/benefit analysis as a precondition to
regulation to protect public health is a foregone conclusion when
many new technologies first enter the market. The practical
result is that an unsafe product containing an innovative, novel
111
John M. Broder, New Alliance Emerges to Tighten Chemical Rules, NY Times
(May 24, 2013), online at http: //www.nytimes.com/2013/05/25/us/politics/lautenbergs-
chemical-safety-bill-gains-momentum.html?pagewanted=all (visited Sept 15, 2013) (“Of
roughly 85,000 chemicals registered for use in the United States, only 200 have been
tested by the Environmental Protection Agency and fewer than a dozen—including
polychlorinated biphenyls, dioxin and hexavalent chromium—have been restricted.”).
112
Nicholas A. Ashford, The Legacy of the Precautionary Principle in U.S. Law: The
Rise of Cost-Benefit Analysis and Risk Assessment as Undermining Factors in Health,
Safety and Environmental Protection, in Nicolas de Sadeleer, ed, Implementing The
Precautionary Principle: Approaches From The Nordic Countries, The EU and The
United States 356–61 (Routledge 2006).
113
Id.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 459
ingredient is likely to remain on the market for a long period of
time before the FDA can take action.
a) Examples of lag times with serious health
consequences. There are numerous examples114 of the negative
impact on public health of this lag time between the
introduction of new chemical compounds and innovative
technologies into the market and the development of the science
necessary to identify the associated hazard to human health.115
For example, it took decades to develop the science necessary to
establish the endocrine disruption hazard caused by novel
product ingredients such as polychlorinated biphenyls (“PCBs”),
tributylin (“TBT”), diethylstilbestrol (“DES”), Thalidomide, and
the Great Lakes pollution.116 Illustratively, some evidence
existed in the 1930s that PCBs were a serious hazard to human
health.117 However, it took four decades before the state of the
science evolved to the point of identifying the extent of the
hazard to human health. Sweden was the first country to ban
PCBs in the 1970s, with the European Union following suit in
1996.118
Other examples include those of medical x-rays, benzene,
and asbestos. The first scientific warnings over medical x-rays
occurred in 1896, those relating to benzene occurred in 1897,
and those relating to asbestos occurred in 1898. Yet it took
between thirty (for PCBs)119 and one hundred years (for x-
114
For a comprehensive analysis of fourteen case studies of these types of scenarios,
see European Environmental Agency, Late Lessons From Early Warnings: The
Precautionary Principle 1896–2000, 22 Envir Issue Rpt, 66–69 (2001).
115
Urbina, Think Those Chemicals Have Been Tested?, NY Times (cited in note 110).
116
David Gee, Late Lessons from Early Warnings: Toward Realism and Precaution
with Endocrine-Disrupting Substances, 114 Envir Health Persp 152, 156 (2006). See also
Katharine A. Van Tassel, Slaying the Hydra: The History of Quack Medicine, the Obesity
Epidemic and the FDA’s Battle to Regulate Dietary Supplements Marketed as Weight
Loss Aids, 6 Ind Health L Rev 203, 228–29 (2009) (discussing the case of Thalidomide).
117
European Environmental Agency, 22 Envir Issue Rpt at 66–69 (cited in note 114).
118
Id at 126. In 1962, the book Silent Spring warned of the negative health effects
on humans and wildlife of this exposure. The marketing of these novel chemicals
continued unabated in spite of this warning. And the full extent of the devastating
consequences to the Great Lakes area of a five decades-long period of human and
environmental exposure to pesticides with the novel organochlorine compound
ingredients is still unknown. See generally Rachel Carson, Silent Spring (Houghton
Mifflin Harcourt 1962).
119
Gee, 114 Envir Health Persp at 155 (cited in note 116). Another example is the
introduction of CFCs. CFCs have created the ozone hole, resulting in thousands of
additional skin cancer cases that will only peak in number in the middle of this century.
Id at 155–56.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
460 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
rays)120 for scientists to determine the extent of these hazards
and then for regulations to be promulgated to mitigate the
already extensive damage to public health caused by these
products.
b) Nanotech ingredients and the health risk information
void. While the use of nanoparticles in consumer products and
the resultant exposure of consumers are growing daily, the body
of science necessary to identify the health risks associated with
engineered nanoparticles is still in its infancy. When the FDA
first made its decisions on how to regulate nanotech products,
far less was known about the distinctive properties of
nanoparticles, their uniquely high level of bioreactivity, and the
resultant heightened potential for adverse health effects.
Scientists simply “did not know what they did not know” about
the health risks associated with nanotech particles. At that
point in time, the FDA made its regulatory choices in an
environment of ignorance, choosing to regulate based on what
scientists now know to be a false assumption of bioequivalence.
Over the past several years, a parade of major scientific
discoveries has shifted the nature of the uncertainty over the
public health risks of nanotech particles from ignorance to
indeterminacy.121 In other words, scientists have progressed
from not knowing what they do not know, to knowing what they
do not know. Scientists now understand that engineered
nanoparticles may create novel health risks caused by powerful
nano-bio interactions that have never before existed in nature.
In order to move from indeterminacy to classic risk analysis,
scientists must still determine the nature and extent of the
harm that occurs as a result of these nano-bio interactions.
Then, using classic risk analysis, scientists must quantify the
probability and degree of those harms. Thus, the new awareness
on the part of scientists that nanoparticles can cause serious
physical harm, and the identification of some of the potential
mechanisms for causation, opens the door to the ability to plan
out the systematic study of each new type of engineered
nanoparticle in order to eliminate or confirm the associated
health risks. In spite of the fact that the state of the science has
now moved into indeterminacy, the FDA’s reliance on
establishing hazard through the use of classic risk/benefit
120 European Environmental Agency, 22 Envir Issue Rpt at 66–69 (cited in note 114).
121 See Part II.B.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 461
analysis in making decisions over whether to regulate for safety
means that, until the science on the health risks associated with
nanoparticles has matured to the point that the risks can be
quantified, the FDA will proceed as if this growing body of
science did not exist.
As explained in the next section, switching the FDA’s
regulatory focus onto novelty, rather than hazard, enables the
public health product safety net, which can act to safeguard the
public while this ponderous cycle of scientific inquiry runs its
course.
2. The FDA’s focus on hazard disables the public health
product safety net.
The public health safety net is a powerful, interactive
network that involves consumers as well as the healthcare
system, the state and federal public health protection agencies,
and the tort system. Importantly, ingredient listing is essential
to the creation of this safety net—without this listing, the safety
net unravels into nothing. To summarize, novel ingredient
listing protects consumer safety through consumer self-
protection. If a consumer knows that she is being exposed to a
novel ingredient, she can use heightened vigilance to watch for
symptoms of harm. It also allows for the appropriate treatment
of injured consumers by medical professionals who can identify
novel ingredients as potential causative agents through accurate
exposure reporting by consumers. Ingredient listing is also
necessary to the proper reporting of injury-causing agents to
state and federal public health protection agencies that manage
the early warning and product recall systems. Finally, the data
collected by these public health protection agencies makes up
the evidence required to enable the instrumental use of the tort
system, which encourages the proper investment by
manufactures in product safety and protects against the overuse
and overconsumption of harmful products by consumers.
Together, these public and private actors join to form the public
health product safety net.
a) Negative impact on consumer self-protection, medical
treatment, and the early warning and product recall systems.
The FDA’s current focus on hazard means that it can’t act to
regulate nanotech ingredients until each separate variety of
nanoparticle is proved to be hazardous to human health. Recall

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
462 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
that the number of different varieties of nanoparticles is only
limited by the creativity and ingenuity of engineers and
scientists so that this may number in the tens of thousands.
Until each variety of nanoparticle is proven to be hazardous to
human health, nanotech products will continue to be regulated
like their macro-sized counterparts. This means that
nanoparticles will not be listed on ingredient labels. Thus, the
FDA’s focus on hazard to trigger its authority to require
ingredient listing means that consumers are unaware of the
extent of their exposure to novel nanotech substances. This lack
of knowledge bars the ability of public health officials to monitor
whether the heavy exposure of US consumers to nanotech
products is causing acute or latent toxic reactions.
Consequently, it is currently highly unlikely that any potential
health risks to the population from this exposure can be
identified and eliminated.
For example, if a consumer uses a nanotech product and has
a toxic reaction, the consumer will assume that the reaction is to
the product itself, not an exposure to the nanoparticle ingredient
because the new nanotech ingredient is not listed on the product
label. The only result is that the consumer will avoid that
particular product in the future. A mild reaction will not merit a
visit to a physician and will go unreported.
If the reaction is moderate to severe, a physician may be
consulted. If the physician reports the adverse reaction, it will
be incorrectly reported as a reaction to the particular host
product based on the inaccurate information given by the
patient. It will not be correctly reported as a reaction to the host
product’s nanotech ingredients.
This misinformation has a broader impact in light of the
new way scientists are using big data collected through social
media to proactively identify product safety problems. “Using
data drawn from queries entered into Google, Microsoft, and
Yahoo search engines, scientists at Microsoft, Stanford, and
Columbia University have for the first time been able to detect
evidence of unreported prescription drug side effects before they
were found by the Food and Drug Administration’s warning
system.”122 Another group of scientists are using data analysis
122
John Markoff, Unreported Side Effects of Drugs are Found Using Internet Search
Data, Study Finds, NY Times (March 6, 2013), online at http: //www.nytimes.com/
2013/03/07/science/unreported-side-effects-of-drugs-found-using-internet-data-study-

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 463
tools to analyze social media, including tweets and on-line
discussion forums, to identify adverse drug reactions.123 These
scientists
retroactively analyzed four types of public on-line media
(websites, blogs, Web forums and social networking sites)
posted from 2000 to early 2012 and were able to identify
hundreds and thousands of documents containing
adverse drug reaction-related information. The
preliminary results suggest that these documents can
accurately provide warnings earlier—in some cases years
earlier—than existing channels.124
In the context of food safety, several apps have been developed
to help public health officials track food-borne illnesses using
data collected from Twitter.125 These big data strategies are just
the start of muscular new additions to product safety
surveillance systems representing a fundamental shift from a
reactive to proactive monitoring systems. However, if consumers
are not provided accurate information on the innovative new
ingredients in consumer products, this revolutionary data
collection system can’t be used by scientists or public health
officials to monitor the safety of novel, innovative ingredients,
such as those using nanotechnology.
The bottom line is that the FDA’s decision that
manufacturers need not list nanotech ingredients on package
labels means that the public health system in place for collecting
data on the harms from exposures to novel ingredients had been
disabled. This means that the key elements of the public health
product safety net have been deactivated: (1) a consumer cannot
engage in self-protection and avoid the novel ingredient if it is
causing injury; (2) a consumer’s ability to obtain appropriate
medical treatment if injured has been sabotaged as they can’t
correctly inform their physicians what new ingredients they
have been exposed to; (3) physicians and consumers can’t
finds.html?_r=0 (visited Sept 15, 2013).
123
H. Brevy Cannon, Research to Sift Social Media for Early Signs of Adverse Drug
Reactions, UVA Today (Sept 20, 2012), online at http: //news.virginia.edu/content /
research-sift-social-media-early-signs-adverse-drug-reactions (visited Sept 15, 2013).
124
Id.
125
Krietsch, Social Media Apps (cited in note 20); Tracking Twitter May Enhance
Monitoring of Food Safety at Restaurants, ScienceDaily (cited in note 20).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
464 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
contribute to, or benefit from, the early warning system that
gives notice to consumers that a product is causing injuries to
some people so as to trigger heightened vigilance by all; and (4)
the product recall systems that all consumers rely upon to
protect against personal injury from unsafe products does not
work for novel ingredients.
b) Negative impact on the tort system. While the prime
objective of the tort system is to compensate innocent victims
harmed by defective products, shifting the cost of these injuries
onto the manufacturers is also instrumental to the proper
functioning of the public health product safety net. Forcing a
manufacturer to bear the costs of injuries incurred from its
products that are faulty in their manufacture or design may
deter future misconduct and may tacitly encourage more careful
behavior, such as increasingly diligent testing and product
design.
The cost of injuries from the use of a product may also then
be built into the price of the product and passed on to the
consumer. If these effects are realized, several additional goals
of the public health product safety net are furthered. First, the
cost of the risk will be borne by all of those using the product
generally through the increased price, instead of the innocent
victim alone. Second, the price of the product will reflect its true
social cost. This price will then mediate consumer choice, more
likely resulting in optimum levels of production and purchase.
As the price of the product increases as a result of
manufacturers’ internalizing the cost of injuries to product
users, the consumption of the product will likely decline as
consumers switch to less costly alternatives, resulting,
ultimately, in a decrease in injuries due to the fall in the use of
the product. Thus, the tort system is an important part of the
public health product safety net as it insulates against the
overuse and overconsumption of relatively risky products.
Moreover, as a part of the public health product safety net,
the tort system places the cost of injury avoidance on the least-
cost accident avoider. The manufacturer is often in the best
position to accurately access the various ways of avoiding costs
of injuries through redesign, quality control, and other safety
measures. As a result of its level of access, the manufacturer is
also often in the best position to insure against future injuries.
Internalizing all of these costs—as well as the costs associated
with injuries—into the price of the product may ultimately force

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 465
a manufacturer to consider the true cost of certain products
when making its choices of which products to produce. It is
hoped that the end result of this part of the public health
product safety net is a socially-efficient output.
However, this cascade of beneficial effects that can occur
from the proper use of the tort system is short-circuited when
the FDA focuses on hazard to regulate innovative products. The
principle reasons for this result are three-fold. First, as nanotech
ingredients are not required to be listed on product labels, the
consumer will not know that she was exposed to a nanotech
ingredient. Thus, the consumer is unlikely to suspect that her
injuries were caused by a novel substance.126 Second, if the
consumer learns that a nanotech ingredient was the cause of her
injury, she will be required to establish proximate cause under
tort law by showing that the manufacturer could have foreseen
the risk of harm.127 As with many new technologies, the rate of
the introduction of nanotech products into the market has far
outpaced the science needed to demonstrate its associated risks.
Under either the Daubert128 or Frye129 tests, this research lag
acts to insulate a manufacturer from liability based on a lack of
causation130 and foreseeability.131
The final hurdle arises in the context of both negligence and
strict liability claims. The lack of labeling means that the public
health protection agencies, such as the Center for Disease
Control (CDC) and the FDA, cannot collect the data that, over
time, could accumulate to the point of meeting the evidentiary
hurdles of causation and foreseeability. Over and above this
problem, unless the consumer can establish that she is a
126
Katharine Van Tassel, The Introduction of Biotech Foods to the Tort System:
Creating a New Duty to Identify, 72 U Cin L Rev 1645, 1681 (2004).
127
Id at 1683–84.
128
See Daubert v Merrell Dow Pharmceuticals, Inc, 509 US 579, 589 (1993).
129
See Frye v United States, 293 F 1013, 1014 (DC Cir 1923).
130
Under the Daubert and Frye standards, evidentiary principles are likely to bar
any introduction of scientific evidence to meet the burden of proof on causation as long as
there is scientific uncertainty. Daubert, 509 US at 589; Frye, 293 F at 1014. The Daubert
factors counsel judges to ask the following questions in making decisions on the
admissibility of scientific evidence: (1) Has the technique been tested in actual field
conditions (and not just in a laboratory)?; (2) Has the technique been subject to peer
review and publication?; (3) What is the known or potential rate of error? Is it zero, or
low enough to be close to zero?; (4) Do standards exist for the control of the technique’s
operation?; and, (5) Has the technique been generally accepted within the relevant
scientific community? Daubert, 509 US at 580.
131
Van Tassel, 72 U Cin L Rev at 1683–84 (cited in note 126).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
466 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
member of a substantial class of people who are at risk for the
same type of adverse reaction, the case is likely to be dismissed
under what is commonly referred to as the “idiosyncratic
plaintiff defense.”132 This “de minimus harm” liability threshold
can range from tens of thousands to millions of people.133 As
with causation, because nanotech ingredients in consumer
products are unlabeled, injured consumers are unlikely to
recognize what actually caused their injuries. As a result, these
injuries will go unreported. Without this data, a consumer will
be unable to establish that she is a member of a substantial
class, creating an almost impassable barrier to recovery.134
The bottom line is that the FDA’s no-labeling decision
means that another key element of the public health product
safety net, the tort system, has also been disabled.
III. HISTORY OF FDCA REGULATION OF NOVEL INGREDIENTS
As explained in the prior section, the current focus by the
FDA on hazard to trigger regulation of novel, innovative
ingredients in the context of scientific uncertainty fosters
innovation but does nothing to protect public health. In the past,
the FDA has avoided this problem by focusing on novelty rather
than on hazard. As described in the next sections, this focus had
a domino effect that created the public health product safety net
that managed the scientific uncertainty over possible negative
health effects of these novel ingredients. This focus on novelty
also fits with the overall scheme of the FDCA, which titers the
degree and kind of regulation it uses to protect consumers from
harm from third parties to the level and type of vulnerability of
a product’s targeted consumers. The overall result was that an
appropriate balance between public health protection and
innovation was achieved. A look back at this history explains
why, and how, this balance was realized in the past and how it
can be reached in the modern day context of rapidly emerging
innovative technologies.
132
Id at 1680, 1683–84. This defense is basically a contention that a reasonable
consumer would not have had the reaction and that the defect is in the consumer, not
the product. Id.
133
Id.
134
Id.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 467
A. The Pure Food Act of 1906
In the 1850s, a large majority of Americans lived and
worked on farms.135 Grain was milled at the local level by
thousands of mills and rural areas were peppered with hundreds
of small packing plants that packaged all of the locally produced
foods.136 By the turn of the century, the majority of the
population moved to the city, a far smaller number of mills did
the vast majority of the work, and packaging companies grew in
size, dwindled sharply in number, and moved to the cities.137
Food produced locally was shipped to these big city packing
companies, packaged in cans and jars and was then returned,
“watered down, preserved and cheap.”138 Soon, these products
were moving across great distances as horses were replaced with
cars, trucks, and trains. Food, clothing, and simple tools were no
longer made by people for their own use and the use of their
neighbors. “The modern estrangement between the people who
create goods and the people who consume them now emerged.”139
This “modern estrangement”
made adulteration and deception both easy and profitable
as manufacturers of food and medicines no longer had to
face their customers. Under laissez-faire regulation,
corruption and abuse were rampant. Food producers
scammed consumers by adding fillers to food to increase
weight (such as chalk, clay, or plaster of paris to flour
and ground up insect carcasses, commonly lice, to brown
sugar). Large amounts of untested chemicals (such as
formaldehyde, sulfites, borax, salicylic acid and benzoic
acid) were used liberally to preserve food for transport
and to disguise the taste and appearance of food that was
spoiled.140
135
Phillip J. Hilts, Protecting America’s Health: the FDA, Business, and One
Hundred Years of Regulation 11 (UNC 2004).
136
Id.
137
Id at 12.
138
Id.
139
Hilts, Protecting America’s Health at 12 (cited in note 135).
140
Van Tassel, 6 Ind Health L Rev at 230 (cited in note 116). See also Hilts,
Protecting America’s Health at 21–22 (cited in note 135) (“Copper sulfate can make faded
vegetables appear green again; sodium benzoate can prevent decayed tomatoes from
rotting altogether; stearins can stretch lard; borax can make odorous ham acceptable

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
468 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
Milk became one of the most adulterated products at the
turn of the century. It was frequently watered down or mixed
with formaldehyde, causing the deaths of numerous children.141
This was the state of affairs until Upton Sinclair’s book The
Jungle was published in 1905.142 The Jungle exposed the filthy
and appalling conditions for workers in meat packing
factories.143 An unintended consequence was to turn the
stomachs of the nation who read about men with tuberculosis
who spit globs of bloody mucous onto filthy, factory floors. The
workers then dragged dead, skinned carcasses along these same
filthy, contaminated floors to the cutting tables.144 In an attempt
to deal with these problems, Congress first passed the Pure Food
and Drug Act of 1906 (“1906 Act”), creating the first federal law
prohibiting the interstate transportation and sale of adulterated
food or drugs.145
B. The Food Drug and Cosmetic Act of 1938
Unfortunately, the 1906 Pure Food Act did not anticipate
two subsequent major developments: the processed food
revolution and a series of Supreme Court cases that eviscerated
the 1906 Act leaving unchecked the remarkable growth of quack
medicines.146
New food processing techniques triggered the processed food
revolution that swept across the country between the 1930s and
1950s. This new processed and packaged food created a black
box for consumers. For example, jelly had customarily been half
sugar and half fruit.147 But Strawberry BRED-SPRED did not
when canned.”); Food and Drug Administration, The Long Struggle for the 1906 Law
(June 1981), online at http: //www.fda.gov/AboutFDA/WhatWeDo/History/Centennial
ofFDA/TheLongStrugglefortheLaw/ (visited Sept 15, 2013).
141
Richard M. Cooper, The Struggle for the 1906 Act, in FDA: a Century of Consumer
Protection 28 (Wayne L. Pines, ed, 2006) (“Milk was one of the most adulterated products
in America at the turn of the century; it was frequently watered down and preserved
with formaldehyde.”).
142
Id.
143
Id.
144
Id.
145
Pure Food and Drug Act of 1906, Pub L No 59-384, 34 Stat 768 (1906).
146
Van Tassel, 6 Ind Health L Rev at 230 (cited in note 116).
147
Michelle Meadows, A Century of Ensuring Safe Foods and Cosmetics, FDA
Consumer Magazine, The Centennial Edition (Jan–Feb 2006), online at http: //www.fda.
gov/AboutFDA/WhatWeDo/History/FOrgsHistory/CFSAN/ucm083863.htm (visited Sept
15, 2013).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 469
contain a single strawberry.148 It was made of highly
carcinogenic coal tar, artificial chemical pectin, artificial
chemical flavorings, grass seeds, and other chemical
preservatives.149 Consumers had no way to learn of this poor
quality, as ingredients were not listed on labels.150 This
information asymmetry meant that consumers were not able to
engage in self-protection.
This period of time also saw a remarkable growth in the
marketing of sham products to treat and cure disease.151
During this long period in U.S. history, the curative
claims of predatory sham medicine salesmen were
limited only by the gullibility of their targets. In many
cases, the degree of gullibility was proportional to the
level of desperation of the individual for a cure. The more
dire the condition, the more vulnerable an individual was
to the “flim flam” of the greedy snake oil salesman. And
the more dire the condition, the greater the degree of
harm when the sham medicine did not work, causing
injury over and above the original illness and/or causing
a delay in seeking effective medical treatment.152
To address both of these problems, Congress passed the
1938 Food, Drug, and Cosmetic Act (“1938 Act”), which is still in
force today. The 1938 Act answered the question of the proper
regulatory balance between the protection of individual choice in
matters involving self-regarding behavior, such as choosing a
consumer product or medicine, and the need to protect
vulnerable consumers from harm from third parties. The 1938
Act created this balance by linking the level of product
regulation with the level of vulnerability of the product’s
targeted population. This linkage translates into the provision of
the greatest amount of regulatory protection when products are
targeted at vulnerable populations. This linkage also established
the proper balance between regulating to protect public health
while allowing innovation to flourish.
148
Id.
149
Id.
150
Id.
151
Van Tassel, 6 Ind Health L Rev at 219–24 (cited in note 116).
152
Id at 216–17.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
470 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
1. Regulating to protect vulnerability created by ill-health.
The FDCA of 1938 identifies two types of vulnerability. The
first is vulnerability that is created by ill health. Thus, products
claiming to aid in an individual’s struggle to return to normal
health fall into the categories of products that require the
greatest amount of regulation. Examples of products that fall
into this category include drugs and devices that require
premarket approval.153 This places the burden of proving both
safety and effectiveness on the manufacturer before these
products can be placed on the market.154 Thus, the 1938 FDCA
sharply curtailed the ability of the quack salesman to take
advantage of the desperation of those who suffer from ill health.
On the other hand, the FDCA requires far less regulatory
protection when products are targeted to healthy populations to
maintain or improve a normal state of health—for example,
dietary supplements or functional foods.155 Less regulation is
needed as this group of consumers is not blinded by desperate
desires to cure unbearable illnesses and return to good health.
Finally, the FDCA requires the least amount of regulation
to protect consumers in the context of traditional food. There is
little need to provide regulatory protection for consumers in this
context
as thousands of years of use of traditional food provides
consumers with the common knowledge, and thus the
ability, to protect themselves from the ordinary risks
associated with different traditional food products. . . .
This common knowledge and ability to self-protect
supports the presumption of safety that is granted to
traditional food under the FDCA.156
Therefore, traditional food products can be placed directly on the
market without undergoing any testing for safety.157
153
21 USC § 355(a) and 28 USC § 360e, respectively.
154
Van Tassel, 6 Ind Health L Rev at 236–41 (cited in note 116).
155
Id. The term “traditional food” is used to distinguish this category of product from
genetically modified food.
156
Id at 230.
157
Id at 230–31. Therefore, if a particular food poses a safety risk over and above
those which are normally associated with a food product, such as salmonella in peanut
butter, the FDA carries the burden of proving that the food is adulterated or misbranded
before it can be removed from the market. Id.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 471
With these second two categories, the FDCA assumes that
consumers can engage in self-protection based on common,
customary knowledge regarding the risks associated with these
products. Of course, with regard to dietary supplements, this
assumption is suspect as many contain biologically active
ingredients that are being newly introduced into US markets.
2. Regulating to protect vulnerability created by
information asymmetry.
In addition to recognizing and protecting consumer
vulnerabilities relating to ill-health, the 1938 Act also
recognized a second kind of vulnerability that occurs when there
is an information asymmetry between the manufacturer of a
product and the consumers that the product targets. For
example, in order to cure the asymmetry created by the
processed food revolution, the FDCA required that the
ingredients of a food product be listed on the label. A good
example of information asymmetry in the processed food context
is the black box that was created by the advent of canned stews
with unknown contents. The FDCA’s listing cure worked for
traditional food product ingredients that were contained in these
canned stews, such as tomatoes and potatoes, as the health risks
associated with these ingredients were common knowledge.158
With this ingredient listing, consumers who had a history of an
allergic or toxic reaction to a particular traditional ingredient
could engage in self-protection by avoiding the product.
Enabling consumers to engage in self-protection created the first
line of defense that makes up the first part of the public health
product safety net.
158
See id, explaining that “The 1938 Act eliminated the ‘distinctive name proviso’
and required instead that the label of a food ‘bear its common or usual name.’ The food
would be illegal or misbranded if it represented itself as a standardized food unless it
conformed to that standard.” Meadows, A Century of Ensuring Safe Foods and Cosmetics
(cited in note 147). The FDCA authorized three kinds of food standards—identity,
quality, and fill of container. Id. In 1939, the first food standards were issued for canned
tomatoes, tomato purée, and tomato paste. Id. The standards looked like a recipe of
listed ingredients. Id. The next standards were for jams and jellies. “By 1957, standards
had been set for many varieties of foods such as chocolate, flour, cereals, bakery
products, milk, cheese, juices, and eggs.” Id.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
472 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
C. Novel Ingredients and the Food Additives Amendments of
1958
Unfortunately, the 1938 Act did not anticipate the creation
of hundreds of novel chemicals for the use in processed foods
during the 1930s through the 1950s. Unlike the listing of
common traditional foods, such as tomatoes and potatoes, the
listing of these novel ingredients on food labels did nothing to
help consumers deal with these novel ingredients, as no common
knowledge existed regarding their health risks.159 By the 1950s,
the popular movement to regulate these chemical food additives
slowly gained traction and ultimately led to passage by Congress
of the Food Additives Amendment of 1958.160 The Food Additives
Amendment placed the burden on the chemical additive
industry to establish the safety of novel chemicals through
premarket testing for safety. Significantly, as the next two
subsections explain, this testing only decreased the uncertainty
over any possible health risks to a small degree for average
consumers and almost not at all for idiosyncratic consumers.161
The question then became how to deal with this excess
uncertainty. The answer, as explained in the next sections, was
the creation of what this Article calls the public health product
safety net.
159
In 1950, the Delaney Committee started a congressional investigation of the
safety of additives that laid the foundation for the Food Additives Amendment
and the Color Additive Amendments. Rep. James Delaney, D-NY, later
submitted a change to the bill proposing the Food Additives Amendment by
inserting the Delaney Clause, which prohibited the approval of any food
additive shown to induce cancer in humans or animals in studies with a
relevant route of exposure. Variations of the Delaney Clause were also
included in the Color Additive Amendments and animal drug provisions.
Id.
160
Id.
161
Id, explaining that
Enacted in 1958, the Food Additives Amendment required manufacturers of
new food additives to establish their safety to FDA’s satisfaction before
marketing. Food additives are substances that have no proven track record of
safety and that must be approved by the FDA before they can be used. A food
substance generally recognized by qualified experts as safe (GRAS) for its
intended use, based on publicly available information, is excluded from the
definition of food additive. Also in 1958, the FDA published the first list of
GRAS substances, which contained nearly 200 substances including ascorbic
acid, papain, and propylene glycol.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 473
1. Excess uncertainty regarding health risks: premarket
testing and predictability of health risks for average
consumers.
As a general matter, premarket safety testing required by
the FDCA involves studies that use very small sample sizes.162
These small sample sizes mean that the uncertainty over the
possibility of health risks from exposure to novel ingredients is
decreased to only a certain degree by premarket testing, leaving
a significant excess amount of uncertainty.
For example, similarly small sample sizes with relatively
low statistical power are relied upon to test safety and
effectiveness for drugs today.163 The FDA recognizes that these
drug studies are too small to predict a large portion of the health
risks associated with novel substances. However, as larger, more
statistically powerful studies are deemed too expensive, the FDA
regularly approves novel drugs with the understanding that the
only way to seriously decrease the excess uncertainty over safety
is to expose the drug to the genetic diversity of the general
population.164 The FDA then uses the post-market surveillance
system specially created for drugs and medical devices to collect
heath data over time relating to the long-term exposures of the
population to these novel chemicals to fully identify the
statistical probability of adverse reactions.165 Under this
premarket testing regime, to date, serious adverse effects were
not detected for approximately one-half of the drugs on the
market until after the drugs received regulatory approval and
were made available to the general population.166 Discovering
these serious adverse side-effects has led to many drugs and
medical devices being pulled from the market as the information
162
Shelby D. Reed, et al, Use of Larger Versus Smaller Drug-Safety Databases Before
Regulatory Approval: The Trade-Offs, 27 Health Aff 360, 360–61 (2008).
163
Id.
164
Id. See also Marshall S. Shapo, Experimenting with the Consumer: The Mass
Testing of Risky Products on the American Public (Praeger 2008) (providing an excellent
discussion across a wide range of products of how manufacturers of new products fail to
disclose potential risks—or uncertainties about danger—to consumers who then become
unwitting experimental subjects when products are first introduced into the market).
165
Kevin Bullis, Screening for Toxic Nanoparticles: Researchers Suggest a Strategy
That Could Weed Out Dangerous Nanoparticles, Tech Rev (Feb 7, 2006), online at
http: //www.technologyreview.com/news/405268/screening-for-toxic-nanoparticles/
(visited Sept 15, 2013).
166
Reed, 27 Health Aff at 366–67 (cited in note 162).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
474 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
gathered from post-market monitoring has demonstrated that
their actual risks outweigh their benefits. In the case of some
other drugs and medical devices, the discovery of serious side-
effects have led to the addition of safety measures, such as
warnings, when it is discovered over time that their actual risks,
though serious, are still outweighed by their potential benefits.
Thus, there is a regulatory recognition that premarket testing
will not detect many adverse reactions when novel substances
are distributed to the general population for use as drugs. The
bottom line is that post-market monitoring of novel substances
protects public health by managing the uncertainty over health
risks that premarket testing cannot significantly diminish.
For example, news of new and serious risks associated with
FDA-approved drugs is common.167 A readily recognized
example is the case of Vioxx.168 Subsequent to FDA approval,
distribution to over 2 million people in the United Sates, and
marketing to 80 different countries for total sales of 2.5 billion
dollars, it was discovered that Vioxx increased the risk of heart
attack and stroke by 50 percent.169 Another more recent case is
that of Omontys, a drug approved by the FDA in March of 2012
to treat adult patients on hemodialysis with anemia stemming
from chronic kidney disease.170 This approval was based upon a
relatively large premarket approval study, after which an FDA
advisory panel endorsed the drug by a vote of 15–1.171 The chair
of the panel remarked that the drug “doesn’t have any safety
signals.”172 Only one year later, in February of 2013, based on
data gathered from post-market surveillance, the FDA warned
of serious and fatal reactions to Omontys and counseled that
physicians should discontinue its use in all patients
immediately.173
167
Id.
168
Rita Rubin, How Did Vioxx Debacle Happen?, USA Today (Oct 12, 2004), online
at http: //usatoday30.usatoday.com/news/health/2004-10-12-vioxx-cover_x.htm# (visited
Sept 15, 2013).
169
Id.
170
John Gever, Fatal Reactions Prompt Omontys Recall, Medpage Today (Feb 24,
2012), online at http: //www.medpagetoday.com/HematologyOncology/Anemia/37509
(visited Sept 15, 2013).
171
Id.
172
Id.
173
Id.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 475
Just as is the current situation with drugs, chemical
preservatives and flavorings were tested under the Food
Additive Amendments Act of 1958 using small sample sizes with
the understanding that these studies could only identify a
portion of these novel chemical’s adverse effects. Those that
were approved were added to food and distributed to the general
population. As with novel drugs and the current post-market
surveillance system set up by the FDA, the public heath product
safety net came into play when novel chemical food additives
were approved based on premarket testing which only partially
decreased the uncertainty over the associated health risks. This
safety net was activated by the simple, but essential, step—the
requirement that novel ingredients be listed on a product’s
packaging.
Activating the safety net started with the FDA’s recognition
of the fact that the novelty of these chemicals meant that their
addition to food was “material” because neither the
manufacturers nor the FDA knew what the actual risks of
exposure to these novel ingredients would turn out to be, in spite
of premarket safety testing. Under the 1938 Act, the material
nature of the new ingredients meant that consumers were
required to be given notice so that they would not assume that
the nature of the risk was the same as the traditional food
products without the novel chemical additives.174 In order to give
consumers notice, these novel chemical ingredients were
required to be listed on the product labels. Listing activated the
first line of safety by allowing the consumer to engage in self-
protection. Then, if the consumer experienced an injury through
exposure to a product, that consumer could search the label and
identify the novel ingredient and avoid it in the future. If the
injury was serious enough to warrant a trip to the doctor,
because the consumer could identify the causative agent, proper
treatment could be provided, minimizing the cost of treatment,
the extent of the injury, and the time for recovery. The physician
could then accurately report the injury and its cause to the
public health officials of the appropriate state or federal agency.
If the cumulative data suggested that the product was unsafe,
the early warning and recall mechanisms could be used by the
public health officials of the state or federal agency to avert a
174
See notes 101 and 102.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
476 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
public health crisis. This data could also be used to meet the
evidentiary requirements of the tort system, allowing it to be
used instrumentally to increase product safety.
2. Excess uncertainly regarding health risks: premarket
testing and predictability of health risks for
idiosyncratic consumers.
More seriously, the initial studies performed during the
premarket approval process to decrease some uncertainty for the
average consumer, provide little to no assurance for those who
are referred to by the law as the “idiosyncratic consumers” or
non-average consumers.
The premarket studies required by the FDA are simply too
small to say much about safety for those who are not average.175
These consumers must rely 100 percent on self-protection by
watching ingredients listed on labels and stopping exposures to
novel ingredients as soon as they are symptomatic. Or, in
relation to long-term exposures, by hoping the public health
product safety net will be sensitive enough to pick up a surge in
injuries relating to novel products so that public health agencies
can issue a recall of that product. So, with novel ingredients, the
only way to significantly decrease uncertainty over health effects
for idiosyncratic consumers is to distribute the product to the
general population. Then, public health officials must “wait and
see,” while, at the same time, hoping to catch early warning
signs of a serious problem in time to minimize injuries through
early warnings and product recalls.
D. In the Past, a Focus on Novelty Created the Public Heath
Product Safety Net to Deal with Uncertainty Over Health
Risks
To recap, this history explains the overall FDCA regulatory
scheme that makes regulatory choices by balancing the
protection of individual choice in matters involving self-
regarding behavior, such as choosing a consumer product, with
the need to protect vulnerable consumers from harm from third
parties. This balance is created by linking the level of product
regulation with the nature and level of vulnerability of the
175
See Reed, 27 Health Aff at 366–67 (cited in note 162).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 477
product’s targeted population. This linkage translates into the
provision of the greatest amount of regulatory protection when
products are targeted at the most vulnerable populations.
Vulnerability can arise in two basics ways. First,
vulnerability can arise in situations where products are targeted
to those with ill health. The FDA ensures that product
manufacturers cannot take advantage of this vulnerability, as
was the case with predatory snake oil salesman, by requiring
premarket testing to ensure that the product is actually as
effective as claimed. On the other hand, for products that are
targeted to the healthy population, vulnerability can arise
because of information asymmetries between the manufacturer
of a product and the consumers that the product targets. The
FDA cures this second type of asymmetry by requiring that all of
the information that is material to a reasonable consumer is
listed on the label. History shows that one type of information
asymmetry that creates a consumer vulnerability is when novel
ingredients are not listed on labels. Without this information,
consumers can’t engage in self-protection—just like when
canned stews with novel chemical preservatives and flavorings
were first introduced.
The FDCA dealt with novel chemical food additives and
consumer vulnerability arising out of their unknown health
risks by requiring premarket testing to decrease some of the
uncertainty over health risks. However, this testing still left a
great deal of remaining uncertainty. The FDCA dealt with this
remaining uncertainty by acknowledging that this uncertainty
was material and, therefore, requiring ingredient listing in order
to enable the public health product safety net. This safety net
was, and still is, used to capture signals that indicate that a
product may cause injury to health in order to avoid, or at least
mitigate, harm to all consumers through early warnings and
product recalls
The next section describes how this precedent should be
used to regulate the novel, innovative ingredients of today and
tomorrow.

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
478 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
IV. ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET TO
CAPTURE CONSUMER PRODUCTS THAT USE INNOVATIVE
TECHNOLOGIES
As it did with novel chemicals in the 1950s, the focus on
novelty176 rather than hazard in the context of nanotech
ingredients takes the analysis out of the realm of scientific
uncertainty and empowers the FDA to regulate nanotech
consumer products to protect public health. A focus on novelty
acknowledges that the public health product safety net plays a
critical role in the identification of unsafe products in the
context of both products that have gone through premarket
testing and, importantly, those that have not.
A. Information Vulnerability and Novel Ingredients
The recognition that the vulnerability in the case of novel
ingredients, for example nanotech ingredients, is related to an
information asymmetry and that this asymmetry is material has
a domino effect that results in animating the powerful public
health product safety net. The only step the FDA needs to take
in order to engage the public health product safety net is to
recognize that the failure to list a novel ingredient, such as a
nanotech component, is a “material” omission under the FDCA.
In order for this omission to be “material,” it must be
“misleading.”177
176
One way to define novelty is to track the way this term is defined by the
European Union in the context of dietary supplements: “European Union novel foods
require any food or ingredient that can’t demonstrate a history of use prior to May 1997
to verify its safety and intended use.” Shane Sterling, Consultant: ‘No Need to Fear EU
Novel Food Laws’, NutraIngredients.com (William Reed Business Media Apr 29, 2013),
online at http: //www.nutraingredients.com/Regulation/Consultant-No-need-to-fear-EU-
novel-foods-laws (visited Sept 15, 2013). Another possibility is to link the definition to
the substances that are subject to a patent.
177
See text accompanying note 102 explaining that, under the FDCA, a drug, device,
food, dietary supplement, cosmetic, or sunscreen is deemed misbranded if its labeling is
“false or misleading in any particular.” 21 USC § 362(a) (2006). See also 21 CFR § 701.1
(2011).
[I]n determining whether the labeling or advertising is misleading . . . there
shall be taken into account (among other things) not only representations
made or suggested by statement, word, design, device, or any combination
thereof, but also the extent to which the labeling or advertising fails to reveal
facts material in the light of such representations or material with respect to
consequences which may result from the use of the article.
21 USC § 321(n) (2009).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 479
As described in the next section, there are several
compelling arguments that this information asymmetry created
through intentionally omitting to list novel, innovative
ingredients is misleading. If the omission is misleading, this
creates an information defect that renders the product
misbranded. If the product is misbranded, it cannot be
distributed to consumers.
1. Intentionally failing to list novel, innovative ingredients
is materially misleading, rendering the product
misbranded.
To put the analysis of “materiality” into a real world
context, the ingredient lists on nanotech products currently refer
to the host product’s nanotech content by the same name as the
ingredient’s normal size counterpart. For example, the use of
nanotech titanium dioxide is so ubiquitous in sunscreens as to
be almost unavoidable. Instead of this nanotech titanium
dioxide content being listed as “nano titanium dioxide,” as is
required by law in the European Union,178 manufacturers in the
United States just list this ingredient as they always did before
they started using the nanotech version of this chemical—it is
listed as “titanium dioxide.” Thus, manufacturers in the United
States are allowed to hide their products’ nanotech ingredient
content from consumers.
This practice is misleading for numerous reasons. First,
when consumers choose to use a new product for the first time,
and they are aware that they are using a new product, they
engage in self-protection by being more vigilant in watching for
any untoward effects. This is particularly true for parents of
small children, those who suffer from ill health, and those who
deal with multiple allergies. When manufacturers
misrepresent179 to consumers that the ingredient is a normal
178
Iris Eisnenberger, et al, On Voluntary and Obligatory Nano-Labelling, 31 Nano
Trust Dossiers 1 (July 2012), online at http: //epub.oeaw.ac.at /0xc1aa500d_
0x002c9d9a.pdf (visited Sept 15, 2013) (providing publication of the Institute of
Technology Assessment of the Austrian Academy of Sciences explaining the European
Union requirements for labeling nanotech cosmetics and food and summarizing the
status of the labeling movement among EU countries).
179
One who fraudulently makes a misrepresentation of fact, opinion, intention or
law for the purpose of inducing another to act or to refrain from action in

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
480 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
size ingredient, the consumer will likely have been exposed to
this normal size ingredient in the past. If this past exposure has
not caused any harm, the consumer will assume that the need
for self-protection through heightened vigilance is unnecessary.
Thus, the consumer’s natural self-defense mechanism will have
been disarmed by the manufacture’s misrepresentation.
The second reason that this practice is misleading is that, if
a consumer has a negative physical reaction to the nanotech
ingredient (which the current state of the science suggests is
possible, even likely),180 the consumer will be unaware that the
adverse reaction is to the novel ingredient. Instead, the
consumer will assume that the injury was caused by the host
product and will avoid that product in the future. If, for
example, the product is a sunscreen containing unidentified
nanotech titanium dioxide and nanotech zinc oxide, this incident
may cause the consumer to incorrectly believe that they can no
longer use sunscreens at all. This results in an unnecessary
avoidance of a product that could protect the consumer from
other serious health consequences such as skin cancers, a
category of diseases that includes malignant melanomas, which
have a rapidly increasing incidence rate over the past several
years as a result of current problems with the ozone layer.181
The third reason that this deceptive practice is misleading
is that, if a consumer has a negative physical reaction to the
nanotech ingredient, the consumer will rely upon the proper
listing of ingredients on product labels in order to provide their
physicians with an accurate list of exposures so that they can be
correctly diagnosed and treated. A simple but very common
example occurs when a consumer develops a rash. In
determining the proper treatment, one of the first questions that
a physician is likely to ask is whether the consumer/patient has
purchased any new products for regular use. In other words,
reliance upon it, is subject to liability to the other in deceit for pecuniary loss
caused to him by his justifiable reliance upon the misrepresentation.
Restatement (Second) of Torts § 525 (1977). “A misrepresentation is material if it would
be likely to induce a reasonable person to manifest his assent, or if the maker knows that
it would be likely to induce the recipient to do so.” Restatement (Second) of Torts § 162
(1965).
180
See notes 70–94 and accompanying text.
181
Doris Day, As Cancer Deaths Fall, Malignant-Melanoma Rates Rise, LiveScience
(June 3, 2012), online at http: //www.livescience.com/37124-skin-cancer-deaths-grow.html
(visited Sept 15, 2013).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 481
whether there are any new exposures that could be the cause for
the rash. A misleading label will thwart the physician’s ability
to determine the cause of the rash in order to properly treat the
condition.
The fourth reason this practice is misleading is that
individual consumers assume that the public health product
safety net is fully engaged and that, if the product is unsafe, the
early warning and product recall system will warn them to avoid
the product if it is causing harm. Parents with small children,
those who suffer from ill health, and those who deal with
multiple allergies are particularly reliant on these product
warning and recall systems. As both the consumer and the
physician have no idea that the consumer has been exposed to
the novel nanotech ingredient, neither can properly report the
injury to any to the state or federal agencies that collect and
analyze the data that is relied upon to alert the public of unsafe
products through early warning and product recall systems.
The fifth reason this practice is misleading is that
consumers purchase products with the customary
understanding that, if the product is unsafe and a consumer is
injured, the manufacturer will compensate them for these
injuries. As explained earlier, the failure to list nanotech
ingredients on product labels means that manufacturers of
nanotech ingredients are insulated from tort recovery.
Consumers have no notice of this special immunity and,
consequently, are not aware that the product carries a special
risk as the consumer won’t be compensated for any injuries the
product causes.
These arguments support a finding that novel, innovative
ingredient content in a consumer product is information that is
material to a consumer making a choice over whether to use a
consumer product. Failure to provide material information
renders a product misbranded, violating the FDCA.
B. Application to Future Technologies
This focus on novelty will engage the public health product
safety net to provide post-market surveillance for novel
ingredients created by the new technologies of the future. Thus,
as the food is created and placed on the market using meat from

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
482 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
cloned animals,182 or meat grown in the lab,183 or through the
genetic modification of animals,184 the FDA can regulate to
protect public health in a way that minimizes the roadblocks to
innovation. It is interesting to note that, if genetically modified
plants used for food had been identified as such pursuant to an
FDA focus on novelty, it is likely that much of the controversy
over its possible health effects would have abated as the public
health product safety net is likely to have picked up any short
term allergic or toxic reactions.185
182
Food and Drug Administration, Animal Cloning, online at http: //www.fda.gov/
animalveterinary/safetyhealth/animalcloning /default.htm (visited Sept 15, 2013).
183
People for the Ethical Treatment of Animals, PETA Offers $1 Million Reward to
First to Make In Vitro Meat, online at http: //www.peta.org /features/In-Vitro-Meat-
Contest.aspx (visited Sept 15, 2013); Julieanne Strachan, From a Petrie Dish to Your
Plate, ACT News (June 12, 2012), online at http: //www.canberratimes.com.au/act-
news/from-a-petrie-dish-to-your-plate-20120602-1zpau.html (visited Sept 15, 2013).
184
Food and Drug Administration, Genetically Engineered Animals, online at
http: //www.fda.gov/animalveterinary/developmentapprovalprocess/geneticengineering /ge
neticallyengineeredanimals/default.htm (visited Sept 15, 2013).
185
From the outset, food manufacturers using genetically modified (“GM”)
ingredients have declined to provide consumers notice of GM content. The FDA has not
mandated disclosure as it takes the position that the introduction of GMO ingredients
into food is not material. See generally Katharine Van Tassel, Genetically Modified Food,
Risk Assessment and Scientific Uncertainty Principles: Does the New Understanding of
the Networked Gene Trigger the Need for Post-Market Surveillance to Protect Public
Health?, 15 BU J Sci & Tech L 250 (2009); Stephanie Strom, Genetic Changes to Food
May Get Uniform Labeling, NY Times (Jan 31, 2013), online at http: //www.nytimes.com/
2013/02/01/business/food-companies-meet-to-weigh-federal-label-for-gene-engineered-
ingredients.html?pagewanted=all&_r=0 (visited Sept 15, 2013). This lack of
transparency resulted in consumer rights groups testing products for GMO use and
disclosing that use to consumers. Non GMO Project, GMO Facts: Frequently Asked
Questions (Non-GMO Project 2013), online at http: //www.nongmoproject.org /learn-more/
(visited Sept 15, 2013). As consumers have become aware of the extensive use of GMOs
in their food, a rising number have expressed the desire that these ingredients be
labeled. Rachel Hennessey, GMO Food Debate In The National Spotlight, Forbes, (Nov 3,
2012) online at http: //www.forbes.com/sites/rachelhennessey/2012/11/03/gmo-food-
debate-in-the-national-spotlight / (visited Sept 15, 2013) (“70% of items in America’s
grocery stores contain genetically modified organisms”). A recent ABC poll suggests that
93 percent of consumers now support mandatory disclosure of GMO content on labels.
Gary Langer, Poll: Skepticism of Genetically Modified Foods, ABC News (June 19, 2012),
online at http: //abcnews.go.com/Technology/story?id=97567&page=1 (visited Sept 15,
2013). When the industry ignored the consumer preference for listing of GM content, a
market was created for products that are “GMO-free.” Thus, the practice of “GMO-free”
labeling was born. The growing consumer labeling movement also triggered repeated
attempts to pass labeling laws. While these efforts have been unsuccessful to date, they
are gaining traction—for instance, it cost the industry 40 million dollars to block
California’s Proposition 37 calling for mandatory labeling last fall. Strom, Genetic
Changes to Food, NY Times (cited in note 185). With more legislative proposals cropping
up (a ballot initiative in Washington state and legislative proposals in Connecticut,
Vermont, New Mexico, and Missouri), a growing consumer boycott of some organic or
“natural” brands owned by major food companies and a recently introduced popular

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 483
V. DOES COST-BENEFIT ANALYSIS BAR THE LISTING OF NOVEL
INGREDIENTS ON PRODUCT LABELS?
Every president since Richard Nixon has established a
program to require review and clearance of agency regulatory
decisions before they are placed into effect.186 Usually, this
review is conducted by the Office of Information of Regulatory
Affairs (OIRA), which is part of the Office of Management &
Budget (OMB), using cost-benefit analysis.187 Cost-benefit
analysis
mobile app by Fooducate that allows consumers to check for GMO content in a growing
number of products, industry may be seeing the writing on the wall. Id; Hank Schultz,
Popular Mobile Food App Adds GMO Feature, FOODnavigator-usa.com (Oct 17, 2012),
online at http: //www.foodnavigator-usa.com/Business/Popular-mobile-food-app-adds-
GMO-feature (visited Sept 15, 2013). Just this year, Ben & Jerry’s Ice Cream has
decided to remove GMO ingredients from its supply chain. Hank Schultz, Research
Results Pointed Ben & Jerry’s Down Non-GMO Path, FOODnavigator-usa.com (Feb 1,
2013), online at http: //www.foodnavigator-usa.com/Business/Research-results-pointed-
Ben-Jerry-s-down-non-GMO-path (visited Sept 15, 2013). And the Meridian Institute,
which organizes discussion of major issues, convened a meeting in Washington just last
month that included executives from PepsiCo, ConAgra, and about 20 other major food
companies, as well as Wal-Mart and advocacy groups that favor labeling. Strom, Genetic
Changes to Food (cited in note 185). Just in March of 2013, three major grocery chains
announced that they will require food with genetically modified ingredients to list these
innovative ingredients on product labels. Stephanie Strom, Major Grocer to Label Foods
With Gene-Modified Content, NY Times (March 8, 2013), online at http: //www.
nytimes.com/2013/03/09/business/grocery-chain-to-require-labels-for-genetically-
modified-food.html?pagewanted=all (visited Sept 15, 2013). Many are predicting that
voluntary labeling may be right around the corner.
186
Dana D. Nelson, Bad for Democracy: How the Presidency Undermines the Power
of the People 152–53 (Minnesota 2008) (discussing the increasing presidential power
pursuant to the theory of the unitary executive that promises undivided presidential
control of the executive branch and its agencies as well as expanded unilateral powers).
187
See Office of Management and Budget, Circular A–4 (September 17, 2003), online
at http//:www.whitehouse.gov/omb/circulars_a004_a-4 (visited Sept 15, 2013):
In theory, cost-benefit analysis of a policy option enumerates all possible
consequences, both positive and negative; estimates the probability of each;
estimates the benefit or loss to society should each occur, expressed in
monetary terms; computes the expected social benefit or loss from each
consequence by multiplying the amount of the associated benefit or loss by its
probability of occurrence; and computes the net expected social benefit or loss
associated with the government policy by summing over the various possible
consequences. The reference point for these calculations is the state of the
economy in the absence of the government policy, termed the “baseline.”
Nicholas A. Ashford, The Legacy of the Precautionary Principle in US Law: The Rise of
Cost-Benefit Analysis and Risk Assessment as Undermining Factors in Health, Safety
and Environmental Protection, in Nicolas de Sadeleer, ed, Implementing the
Precautionary Principle: Approaches from the Nordic Countries, EU and USA 352, 366–
67 (Routledge 2007). Cost-benefit analysis attempts to describe the consequences of a
candidate regulation in monetary terms. This poses two problems. One is the difficulty,

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
484 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
is one of the primary tools used in regulatory analysis to
anticipate and evaluate the likely consequences of rules.
Although some regulatory benefits and costs are difficult
to quantify or monetize, those preparing those analyses
generally attempt to estimate the overall benefits that a
proposed or final rule would create as well as the
aggregate costs that it would impose on society, and then
determine whether the former justify the later.188
The first question becomes whether this cost-benefit tool should
be used to judge any FDA regulations requiring the listing of
novel ingredients on product labels? And, secondly, if this cost-
benefit tool is used, appropriately or not, will such a regulation
survive this process?
A. Is the Use of Cost-Benefit Analysis in the Context of
Consumer Products and Individual Risk Appropriate?
How people react to scientific evidence of risk is governed by
many factors, including how risk information is perceived and
communicated, how individuals react to social and cultural
influences, and how choices are structured.189 Examples abound
of situations where individuals’ risk perceptions lead them to act
in ways that appear contrary to their own interests, overreacting
even arbitrariness, of placing a monetary value on human life, health and safety, and a
healthy environment. Another is that by translating all of these consequences into
equivalent monetary units, discounting each to current value (since a US $/Euro
invested now is expected to earn interest over time), and aggregating them into a single
US$/Euro value intended to express the net social effect of the government policy, the
effects on the economy from investing now in future health, safety, and environmental
benefits are weighted far more heavily than those benefits that occur in the future,
including those to future generations.
188
Id. For an excellent critique of the role of OIRA, its use of cost-benefit analysis,
and suggestions for modification of OIRA’s role, see John S. Applegate, et al, Comments
Regarding Executive Order on OMB Regularity Review, Center for Progressive Reform
(Mar 16, 2009), online at http: //www.reginfo.gov/public /jsp/EO/fedRegReview/CPR_
Comments_New_EO_Reg_Rev_Final.pdf (visited Sept 15, 2013) (pointing out that the
use of cost-benefit analysis by OIRA is inconsistent with the law in most cases and
explaining how it is a failed tool of regulatory analysis).
189
See generally Cass R. Sunstein, Laws of Fear—Beyond the Precautionary
Principle (Cambridge 2005) (analyzing the relationship between “fear,” danger, and the
law); Richard A. Posner, Catastrophe—Risk and Response (Oxford 2004) (arguing that
“realism about science and scientists, innovative applications of cost-benefit analysis, a
scientifically literate legal profession, enhanced international cooperation, and a
pragmatic attitude toward civil liberties are among the keys to coping effectively with
catastrophic risks”).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 485
to or neglecting risks.190 Regardless of whether these choices are
rational or not, when it comes to individual risk, as a general
matter our society respects the choice of the individual to
encounter or avoid a particular risk in accord with that
individual’s personal values.
In contrast, environmental risks are those faced by the
collective population. In the context of collective risk, the
question becomes how much risk the collective is willing to
encounter, for example, in the air, water, and soil.191 Under our
democratic system, one might suppose that the desired level of
risk would be assigned by the voting public. Instead, the current
use of cost-benefit analysis as a proxy for the collective will is an
attempt to impose rationality into the decision-making
process.192 Cost-benefit analysis disregards the will of the
collective that may be driven by what some label as “irrational”
perceptions of risk in light of the mathematical probability of
that risk actually occurring.193
Regardless of the merits of this sanitizing approach when it
comes to collective risk,194 it has no role in the area of consumer
products and individual risk. With collective risk, the
government is fulfilling its proper role in protecting individuals
from the risks that arise from the activities of third parties
190
Sunstein, Laws of Fear at 89–106 (cited in note 189).
191
Many strongly argue that this choice should be made in a democratic fashion.
Nelson, Bad for Democracy at 150, 152 (cited in note 186) (noting that the inception of
the series of presidential executive orders requiring review and clearance of agency
regulatory decisions before they are placed into effect conducted by the Office of
Management & Budget using cost-benefit analysis was a “significant turning point in the
accumulation of presidential power, in the executive’s receptiveness to business
concerns, and in the diminished responsiveness of federal government to citizen
interest”).
192
Sunstein, Laws of Fear at 89–106 (cited in note 189).
193
Id; Posner, Catastrophe at 9–12 (cited in note 189). The question becomes
whether collective choices that are driven by the values of individuals created by their
life experiences should be pejoratively labeled as “irrational fear,” a loaded term in
American society that values “courage” and disparages the fearful. See John M. Kang,
The Burdens of Manliness, Harv J L & Gender 477, 487, 490–507 (2010) (A stereotype
exists that men, to be considered men, must prove that they are courageous. So
embedded is the assumption that “[c]ourage and notions of manhood [are] inseparable,
the very word for courage in many languages deriv[es] from the word for man.”). Should
these individual values be discounted or not counted at all through the use of cost-benefit
analysis?
194
For a collection of critiques of cost-benefit analysis from a wide variety of stellar
academics, see Thomas O. McGarity, Sidney A. Shapiro, and David Bollier, Sophisticated
Sabotage: The Intellectual Games Used to Subvert Responsible Regulation
(Environmental Law Institute 2004).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
486 THE UNIVERSITY OF CHICAGO LEGAL FORUM [2013
when the individual has no ability to avoid the risk through
their own initiative.195 In contrast, in the context of consumer
products and individual risk, the proper role of the government
is to protect individual choice with regard to the amount and
nature of the risk the consumer is willing to encounter.
B. What Result If Cost-Benefit Is Applied?
Even if the OMB inappropriately applies a cost-benefit
analysis to a requirement that novel ingredients be listed on
product labels, the benefits to public health far outweigh the
costs. Thus, this FDA regulation is likely to survive this last
OMB hurdle.
The cost of such a regulation will be de minimus. For
example, the cost of placing two to four letters (“GM” or “nano”)
in front of all novel, innovative ingredients listed on product
labels is small. With regard to GM food, the old arguments that
the majority of the cost lies in trying to maintain separate food
collection and distribution systems so that GM versus GM-free
food can be identified have become moot. The organic and GM-
free movements have set up certification systems that require
food producers and packagers to meet a set of strict
requirements in order to be allowed to use the label denoting
that their products are organic and/or GM free. Thus, the cost of
keeping the GM-free food and GM food separated has been
placed on the GM-free, natural food and is, thus, no longer
relevant to the cost of the listing of GM ingredients in food.
The benefits of novel ingredient listing, as explained earlier,
are the ability of a consumer to self-protect, to receive proper
treatment if injured, and to allow for data collection and
surveillance so that any uptick in injuries can be quickly
detected and a crisis averted or mitigated. Finally, ingredient
listing also allows for the data collection necessary for the tort
system to work appropriately, which allows for compensation of
injured parties and for the instrumental benefits of the tort
system to accrue.
195
Wickler, 56 Health & Society at 303–38 (cited in note 23).

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
433] ANIMATING THE PUBLIC HEALTH PRODUCT SAFETY NET 487
VI. CONCLUSION
Currently, the litmus test the FDA uses to trigger
regulation to protect public health when novel technologies are
used to create innovative ingredients in consumer products is
keyed to a finding of hazard. Linking public health protections
to the degree of hazard when operating in scientific uncertainty
is outcome determinative—it means no regulation at all. Thus, a
focus on hazard translates into the FDA ignoring its prime
mission, the protection of public health.
Instead, the FDA’s lodestar for triggering regulation should
be novelty. A focus on novelty results in the proper regulatory
balance between the twin goals of fostering innovation while
protecting public health. The results of this focus also fit well
with the overarching theme of the FDCA that titers the degree
and kind of regulation it uses to protect consumers from harm
from third parties to the level and type of vulnerability of those
consumers. It also tracks how the Act has historically dealt with
regulating novel ingredients with unknown health risks by
using what this Article calls the public health product safety
net.
This safety net is created when the healthcare system, the
state and federal public health protection agencies, and the tort
system are all networked together to protect public health. This
network is created through the simple expedient of novel
ingredient listing. This one small step empowers consumers’
abilities to self-protect, allows for appropriate medical treatment
by the correct identification of harmful causative ingredients,
and permits the proper reporting of products containing novel
ingredients to the state and federal public health protection
agencies in charge of the early warning and product recall
systems. This data can then be collected to allow for the
instrumental use of the tort system to encourage the proper
investment in product safety by manufacturers and insulate
against the overuse and overconsumption of relatively risky
products by consumers. While this network is far from perfect, it
is likely to improve exponentially as this country moves into the
era of big data and the collection of product safety information
reported directly by consumers in social media.
The proposed regulation of the use of nanotechnology in
consumer products set forth in this Article provides a good
example of how regulation based on novelty rather than hazard

VAN TASSEL PROOF F (DO NOT DELETE) 10/23/2013 1:13 AM
488 THE UNIVERSITY OF CHICAGO LEGAL FORUM
achieves the proper balance between protecting public health
while encouraging innovation through the animation of the
public health product safety net. This same regulatory strategy
should be applied to other novel technologies used as ingredients
in consumer products such as cloned animals used for food,
genetically modified plants and animals used for food, and lab
grown meat.


VACCINE INJURY COMPENSATION
Most Claims Took Multiple Years and Many Were Settled through Negotiation
Report to the Chairman, Committee on Oversight and Government Reform, House of Representatives
November 2014
GAO-15-142
United States Government Accountability Office

United States Government Accountability Office
Highlights of GAO-15-142, a report to the Chairman, Committee on Oversight and Government Reform, House of Representatives
November 2014
VACCINE INJURY COMPENSATION Most Claims Took Multiple Years and Many Were Settled through Negotiation
Why GAO Did This Study Vaccines save lives by preventing disease in the people who receive them. In some instances, however, a vaccine can have severe side effects, including death or an injury requiring lifetime medical care. VICP provides compensation to people for injuries and deaths associated with certain vaccines for medical and other costs. The program includes an injury table that lists the injuries that are presumed to be caused by vaccines covered by the program. The program may also compensate individuals for injuries not on the table; however, in those cases causation is not presumed. In both cases, medical and other records are required. VICP pays claims from a trust fund. Since the program began in 1988, it has awarded more than $2.8 billion in compensation.
GAO was asked to review the program. GAO examined (1) how long it has taken to adjudicate claims and how claims have been adjudicated, (2) the changes to the vaccine injury table, and (3) how the balance of and spending from the Vaccine Injury Compensation Trust Fund have changed, among other objectives.
GAO examined data and interviewed officials from HHS, DOJ, and USCFC, including data on claims filed since fiscal year 1999 and their status as of March 31, 2014; reviewed laws and agency documents; and reviewed Treasury data and agency data on compensation and obligations for other VICP-related expenses for fiscal years 2009 through 2013.
HHS and USCFC agreed with GAO’s findings, and HHS, USCFC, DOJ, and Treasury provided technical comments that were incorporated as appropriate.
What GAO Found Most of more than 9,800 claims filed with the National Vaccine Injury Compensation Program (VICP) since fiscal year 1999 have taken multiple years to adjudicate (see fig.). More than 1,000 (11 percent) of claims filed since fiscal year 1999 were still in process (pending) as of March 31, 2014; most of these were pending for 2 years or less. A greater percentage of the claims filed since fiscal year 2009 were resolved within 1 or 2 years. In all but 1 year since fiscal year 2009, the program has met the target for the average time to adjudicate claims (about 3.5 years) tracked by the Department of Health and Human Services (HHS), which administers the program. Officials from the U.S. Court of Federal Claims (USCFC), where VICP claims are adjudicated, report that delays may occur while petitioners gather evidence for their claims. Since 2006, about 80 percent of compensated claims have been resolved through a negotiated settlement.
Time to Adjudicate National Vaccine Injury Compensation Program Claims Filed Fiscal Years 1999-2014, as of March 31, 2014
Since fiscal year 1999, HHS has added six vaccines to the vaccine injury table, but it has not added covered injuries associated with these vaccines to the table. This means that while individuals may file VICP claims for these vaccines, each petitioner must demonstrate that the vaccine that was administered caused the alleged injury. HHS is considering adding covered injuries associated with these vaccines; but as of September 2014, it had not published any final rules to do so.
The balance of the Vaccine Injury Compensation Trust Fund, managed by the Department of the Treasury (Treasury) increased from $2.9 billion in fiscal year 2009 to nearly $3.3 billion at the end of fiscal year 2013 as the trust fund’s income (from net revenues from vaccine excise taxes and interest on investments) outpaced its disbursements to HHS, USCFC, and the Department of Justice (DOJ), which represents HHS in VICP proceedings. VICP compensation, funded by the trust fund, increased from less than $126 million in each of fiscal years 1999 to 2009 to over $254 million in fiscal year 2013.
View GAO-15-142. For more information, contact Marcia Crosse at (202) 512-7114 or [email protected].

Page i GAO-15-142 National Vaccine Injury Compensation Program
Letter 1
Background 4 Most Claims Took Multiple Years to Adjudicate and Many Were
Adjudicated by Settlement or Addressed by an Omnibus Proceeding 9
Vaccines Have Been Added to the Vaccine Injury Table since Fiscal Year 1999 without Covered Injuries, Resulting in More Off-Table Claims 16
Trust Fund Balance Has Increased since 2009, As Has Petitioner and Attorney Compensation and Other VICP-Related Spending 21
Information on VICP Petitioner Experience Is Limited; HHS Is Taking Steps to Address Criticism of Its Outreach Efforts 29
Concluding Observations 33 Agency Comments 34
Appendix I Vaccine Injury Table, September 2014 36
Appendix II Covered Vaccines and Injuries on the Vaccine Injury Table, Fiscal Years 1999-2014 38
Appendix III Compensation to Petitioners and Attorneys’ Fees and Costs, Fiscal Years 1999-2013 40
Appendix IV Comments from the Department of Health and Human Services 42
Appendix V Comments from the United States Court of Federal Claims 44
Appendix VI GAO Contact and Staff Acknowledgments 46
Contents

Page ii GAO-15-142 National Vaccine Injury Compensation Program
Tables
Table 1: Amounts Obligated by HRSA, DOJ, and USCFC for the National Vaccine Injury Compensation Program Administrative Expenses 28
Table 2: Compensation to Petitioners under the National Vaccine Injury Compensation Program, Fiscal Years 1999-2013 40
Table 3: Payments to Attorneys under the National Vaccine Injury Compensation Program, Fiscal Years 1999-2013 41
Figures
Figure 1: Time Taken to Adjudicate National Vaccine Injury Compensation Program Claims Filed Fiscal Years 1999-2014, as of March 31, 2014 10
Figure 2: Status of National Vaccine Injury Compensation Program Claims Filed Fiscal Years 2009-2014, as of March 31, 2014 11
Figure 3: Number of National Vaccine Injury Compensation Program Claims Filed Alleging Autism, Fiscal Years 1999-2014, as of March 31, 2014 16
Figure 4: Vaccine Injury Compensation Trust Fund Income, Disbursements, and Balance for Fiscal Years 2009–2013 23
Figure 5: Compensation to Petitioners under the National Vaccine Injury Compensation Program 25
Figure 6: Payments to Attorneys under the National Vaccine Injury Compensation Program 27

Page iii GAO-15-142 National Vaccine Injury Compensation Program
Abbreviations DOJ Department of Justice FTE full-time equivalent HHS Department of Health and Human Services HRSA Health Resources and Services Administration Treasury Department of the Treasury USCFC U.S. Court of Federal Claims VICP National Vaccine Injury Compensation Program
This is a work of the U.S. government and is not subject to copyright protection in the United States. The published product may be reproduced and distributed in its entirety without further permission from GAO. However, because this work may contain copyrighted images or other material, permission from the copyright holder may be necessary if you wish to reproduce this material separately.

Page 1 GAO-15-142 National Vaccine Injury Compensation Program
441 G St. N.W. Washington, DC 20548
November 21, 2014
The Honorable Darrell E. Issa Chairman Committee on Oversight and Government Reform House of Representatives
Dear Mr. Chairman:
Vaccines save lives by preventing disease in the people who receive them. In some instances, however, a vaccine can have severe side effects, including death or an injury requiring lifetime medical care.1
To address this issue, Congress established the National Vaccine Injury Compensation Program (VICP), which is administered by the Department of Health and Human Services (HHS).
In the 1980s, lawsuits stemming from such incidents threatened to affect the availability and cost of vaccines as well as the development of new ones.
2 People who believe they or their child have been injured by vaccines covered by the program may file a petition making a claim with VICP for compensation for medical and other costs and for pain and suffering, as an alternative to civil court.3
1In this report, we use the term injury to refer to an illness, disability, or similar condition.
The program uses a vaccine injury table that lists the injuries and conditions that are presumed to be caused by vaccines covered by the program and thus may entitle a petitioner to compensation. The program may also compensate petitioners for injuries or conditions that are not on the vaccine injury table; however, in those cases causation is not presumed and the petitioner must demonstrate that the vaccine caused the injury or condition. The program pays claims from the Vaccine Injury Compensation Trust Fund, a trust fund supported by an excise tax on each dose of a vaccine covered by the program; since the program
2VICP was established by the National Childhood Vaccine Injury Act of 1986. Pub. L. No. 99-660, title III, §§ 301 et seq., 100 Stat. 3743, 3755 (amending the Public Health Service Act by inserting a new title XXI; codified, as amended, at 42 U.S.C. 300aa-1 et seq.). 3Petitions may also be filed for compensation for deaths from vaccines covered by the program.

Page 2 GAO-15-142 National Vaccine Injury Compensation Program
began in 1988, more than 15,000 claims had been filed and more than $2.8 billion has been awarded to petitioners.
In 1999, we reported that while the program appears to provide an easier process for obtaining compensation than through the civil court system, the claims process was not as quick or easy as expected.4
To address these questions, we used data and information from the federal agencies involved in administering the program and managing the Vaccine Injury Compensation Trust Fund, and from stakeholders. Specifically, to examine how long it has taken to adjudicate VICP claims we examined VICP data and interviewed officials from HHS’s Health Resources and Services Administration (HRSA), which administers VICP; the Department of Justice (DOJ), which represents HHS in VICP proceedings; and the U.S. Court of Federal Claims (USCFC) and the Office of Special Masters within USCFC that adjudicates, or decides, VICP claims.
Recently, concerns have been raised about the timeliness for adjudicating VICP claims and making changes to the vaccine injury table, how the funds in the Vaccine Injury Compensation Trust Fund have been spent, and petitioner experiences and awareness of the program. You asked that we examine these issues. Specifically, this report examines (1) how long it has taken to adjudicate VICP claims and how claims have been adjudicated, (2) the changes to the vaccine injury table and in the types of claims filed, (3) how the balance of and spending from the trust fund have changed, and (4) available information on petitioner experience with VICP and how HHS has informed the public of the availability of VICP.
5 We analyzed USCFC data to examine time frames for the adjudication of claims filed in fiscal years 1999-2014.6
4GAO, Vaccine Injury Compensation Program Challenged to Settle Claims Quickly and Easily,
To examine changes to the vaccine injury table and in the types of claims filed, we reviewed relevant laws, and other documents, including Federal Register notices, studies conducted for HHS by the Institute of Medicine, and HRSA’s proposals for revising the vaccine injury table since fiscal year
GAO/HEHS-00-8 (Washington, D.C.: Dec. 22, 1999). 5For the purposes of this report, we use the term claim to refer to the petition and case filed under VICP. We refer to the individuals filing the claims as petitioners. HRSA administers the program in conjunction with DOJ and USCFC. 6We examined USCFC data showing when VICP petitions were filed and when each claim was adjudicated for petitions filed from fiscal year 1999 through March 31, 2014.

Page 3 GAO-15-142 National Vaccine Injury Compensation Program
1999, and interviewed officials from HRSA and members of its Advisory Commission on Childhood Vaccines.7 We also examined HRSA data on the numbers of claims filed, by vaccine, and USCFC data on the number of claims alleging injuries on the vaccine injury table. To examine the balance of and spending from the Vaccine Injury Compensation Trust Fund, we reviewed reports from the Department of the Treasury (Treasury), which manages the trust fund, to determine the income into and disbursements from the trust fund for fiscal years 2009-2013. We also analyzed HRSA data on the amounts the agency reported for petitioner compensation and attorneys’ fees and costs, and data from HRSA, DOJ, and USCFC on the amounts they obligated for administrative and other expenses related to processing VICP claims for those fiscal years using appropriations from the trust fund.8 Finally, to examine available information on petitioner experience with VICP and how HHS has informed the public of the availability of VICP, we reviewed documents, including the Public Health Service Act, agencies’ congressional budget justifications, agency strategic plans, and studies prepared for HHS. We also reviewed statements and interviewed officials from HRSA, DOJ, and USCFC and stakeholders (from organizations representing providers, petitioners’ attorneys, and parents) regarding available information on petitioner experiences with VICP and steps HHS has taken to inform the public about the availability of the program.9
7The Advisory Commission on Childhood Vaccines (ACCV) consists of nine members appointed by the Secretary of Health and Human Services and advises the Secretary on certain issues relating to VICP. See 42 U.S.C. § 300aa-19. Some individual members of the ACCV provided GAO with their personal views that do not necessarily reflect the official position of the ACCV, which is reflected in consensus recommendations made by the ACCV to the Secretary of HHS.
We determined that the data we used from HRSA, DOJ, and USCFC and from Treasury reports on the trust fund were sufficiently reliable for our purposes by discussing data collection processes and limitations of the data with agency officials, conducting electronic data checks, and comparing the data against other published sources.
8The term obligation refers to a definite commitment by a federal agency that creates a legal liability to make payments immediately or in the future. We examined agency data on obligations for full-time equivalent (FTE) staff, and for other expenses. 9The stakeholders we contacted included the National Vaccine Information Center, Vaccine Injured Petitioners’ Bar Association, American Academy of Family Physicians, and American Academy of Pediatrics. In addition, other individuals and organizations contacted GAO and provided comments, which we considered as we conducted our review.

Page 4 GAO-15-142 National Vaccine Injury Compensation Program
We conducted this performance audit from February 2014 to November 2014 in accordance with generally accepted government auditing standards. Those standards require that we plan and perform the audit to obtain sufficient, appropriate evidence to provide a reasonable basis for our findings and conclusions based on our audit objectives. We believe that the evidence obtained provides a reasonable basis for our findings and conclusions based on our audit objectives.
In general, individuals seeking compensation for a vaccine-related injury or death must first file a petition making a claim for compensation under VICP before suing in civil court.10
Several federal agencies—HHS, DOJ, and USCFC, are involved in administering VICP, and Treasury manages the trust fund which funds compensation for successful claims.
VICP includes a vaccine injury table that lists the vaccines covered by the program and the injuries associated with each those vaccines. (See app. I for the table.) Vaccines are added to the list covered by the program after the Centers for Disease Control and Prevention recommends them for routine administration to children and they are made subject to an excise tax that funds the Vaccine Injury Compensation Trust Fund.11 When individuals submit a claim for an injury listed on the table (called an on-table injury), they do not need to prove that the injury was caused by the vaccine. Instead, if they submit documentation showing that they received a particular vaccine and that they sustained the associated covered injury within the time interval specified on the table, they may receive compensation based on a presumption of causation (unless there is evidence that the injury is due to other factors).12
10Petitioners must first file a VICP claim before suing in civil court unless the civil suit is for $1,000 or less. 42 U.S.C. § 300aa-11(a)(2)(A).
Individuals seeking compensation may submit claims for injuries not listed on the table (called
11The program may provide compensation for individuals who were injured or died after receiving the covered vaccines regardless of their age at the time of vaccination—that is, even though the vaccines are recommended for routine administration for children, adults who are vaccinated with a covered vaccine are also eligible for the program. 1242 U.S.C §§ 300aa-11(c)(1), 300aa-13(a)(1). A petitioner’s claim of an on-table injury must also meet the Qualifications and Aids for Interpretation that define the injuries listed on the vaccine injury table.
Background
Vaccines Covered by the Program and on the Vaccine Injury Table

Page 5 GAO-15-142 National Vaccine Injury Compensation Program
off-table injuries) but they need to demonstrate by the preponderance of the evidence that the vaccine caused the alleged injury.13
HHS has authority to promulgate rules to modify the vaccine injury table when certain criteria are met.
14 HHS is also required to amend the table to include a vaccine within 2 years of the Centers for Disease Control and Prevention’s recommending it for routine administration to children.15 The Advisory Commission on Childhood Vaccines, which was established by the act creating the program, is required to make recommendations concerning changes to the table.16 In 1999, GAO reported that HHS added seven injuries and removed three others from the table in 1995 and 1997, respectively, using findings from Institute of Medicine reviews conducted in 1991 and 1994—in conjunction with public policy considerations provided by the Advisory Commission on Childhood Vaccines, scientific issues raised by HHS’s National Vaccine Advisory Committee, and input from the public.17
Individuals who believe they or their child have been injured by or a death resulted from a vaccine covered by the program may file a petition
13The preponderance of evidence standard is generally interpreted to require that the party with the burden of persuasion—here, that party is the petitioner—must establish that a fact in question is more likely true than not. See 29 Am. Jur. 2d Evid. § 173. This standard is used for VICP claims in a manner similar to its use in civil court but without regard to fault. 14The agency must provide for notice and opportunity for a public hearing and at least 180 days of public comment, as well as provide the Advisory Commission on Childhood Vaccines at least 90 days to provide recommendations and comment on proposed rules to modify the table. 1542 U.S.C. § 300aa-14(e). The effective date for a vaccine added to the vaccine injury table is the effective date of an excise tax on the vaccine. The Omnibus Budget Reconciliation Act of 1993. Pub. L. No. 103-66 § 13632 (a)(3), 107 Stat. 312, 646 (codified at 42 U.S.C. § 300aa-14 note). When a new vaccine or a new injury is added to the vaccine injury table, claims that do not meet the general filing deadlines must be filed within 2 years from the date the vaccine or injury is added to the table for injuries or deaths that occurred up to 8 years before the table change. 16Pub. L. No. 99-660, § 311(a), 100 Stat. 3771 (codified as amended at 42 U.S.C. § 300aa-19). 17GAO/HEHS-00-8.
VICP Claims Process

Page 6 GAO-15-142 National Vaccine Injury Compensation Program
making a claim with the USCFC.18 In general, to be eligible for compensation, a petition must be filed (1) for a vaccine-related injury within 3 years of the first symptom of the injury (or significant aggravation of an injury), or (2) for a death within 2 years of the death and within 4 years after the first symptom of the vaccine-related injury (or signification aggravation of an injury) from which the death resulted.19
HHS, as the respondent in the process, receives a copy of the petition, including medical records, and other documentation filed with the USCFC. HRSA sends a report of its medical review including HHS’s recommendation regarding the claim to DOJ. DOJ lawyers, representing HHS in the proceedings, review the HRSA report and the legal aspects of the claim and produce a report that outlines the government’s position as to why compensation should or should not be awarded, provides a summary and medical analysis of the petitioner’s claims, and asserts applicable legal arguments.
After the claim is filed in USCFC, it is assigned to a special master, a judicial officer who examines the evidence and adjudicates the claim.20 The special master reviews the petition and may order the petitioner to provide additional records if they are missing or if they are insufficient.21
18Individuals eligible to file a VICP claim include the person who sustained the vaccine-related injury or that person’s legal representative if that person is a child, disabled, or deceased. 42 U.S.C. § 300aa-11(b)(1)(A).
Additionally, petitioners and DOJ may file expert reports including additional medical evidence from scientific literature or studies. The
19See 42 U.S.C. § 300aa-16. 20The Office of Special Masters is an office within the USCFC, part of the judiciary, comprising up to eight court-appointed special masters who develop expertise in vaccine-injury claims. According to the Office of Special Masters, most claims are assigned to special masters on a random basis, but to the extent that a special master has acquired expertise in a specific injury or vaccine, claims may be directed to a particular special master. 21A special master may suspend activity on a claim while waiting for additional documentation.

Page 7 GAO-15-142 National Vaccine Injury Compensation Program
special master then determines whether a claim should be compensated.22
For claims that are compensated, there are three adjudication categories:
• Concession. In a concession, HHS’s review of medical records, scientific literature, and other documents finds that the petitioner is entitled to compensation, because the evidence meets the criteria of the vaccine injury table or because it is more likely than not that the vaccine caused the injury.
• Negotiated settlement. In a negotiated settlement, the petition is resolved via negotiation between HHS (represented by DOJ) and the petitioner.23
• Contested decision in favor of the petitioner. If HHS does not concede that a petition should be compensated or if both parties do not agree to settle, the special master issues a decision after weighing the evidence presented by both sides, which may involve conducting a hearing.
24
If the petitioner is entitled to compensation as a result of a concession or contested decision in favor of the petitioner, the proceeding then moves to the damages phase, in which the amount of compensation is determined.
25
22Even when HHS concedes a case or a case is settled, the USCFC issues a decision and judgment awarding compensation. A claim is not compensated if the court determines the petitioner is not entitled to compensation or in certain other circumstances, such as where the petitioner elects not to receive compensation or withdraws the petition.
In a negotiated settlement, the amount of compensation is
23According to HRSA, such a settlement is not an admission by HHS that the vaccine caused the petitioner’s alleged injuries, and in settlements, the court does not determine that the vaccine caused the injury. See Health Resources and Services Administration, Statistics Report-August 1, 2014, http://www.hrsa.gov/vaccinecompensation/statisticsreport.pdf, p. 9, accessed Sept. 8, 2014. Once a settlement is reached, it must be formally approved by the decision of the special master, provided the special master determines it is reasonable. 24If the special master rules that a petitioner is not entitled to compensation, the petitioner may request to have the decision reviewed by a judge of the USCFC who may issue a decision in favor of the petitioner. HHS may appeal the decision of the special master if the petitioner is awarded compensation. 25Damages may be agreed to by the parties, or the special master may hold another hearing or rule upon the record without conducting a hearing.

Page 8 GAO-15-142 National Vaccine Injury Compensation Program
included in the settlement presented to the special master. VICP may also pay for attorneys’ fees and costs deemed reasonable even for unsuccessful petitioners, and the amounts of these fees and costs may be part of a settlement between the parties or determined by the special masters.26 After the claim has been adjudicated within VICP, the petitioner may choose to file a suit in civil court. Even if found to be entitled to compensation, the petitioner may elect to reject the compensation awarded and file a suit in civil court.27
The Vaccine Injury Compensation Trust Fund, managed by Treasury, is funded by an excise tax imposed on each dose of vaccine sold in the United States that is routinely recommended for administration to children.
28 Appropriations from the trust fund to HRSA pay compensation awarded under VICP for vaccine-related injury or death to the petitioner and may also pay for petitioner attorneys’ fees and costs. Appropriations from the trust fund to HRSA, DOJ, and USCFC (for the Office of Special Masters) also pay for administrative and other expenses associated with processing VICP claims.29
USCFC has managed large influxes of similar vaccine injury claims through omnibus proceedings or groupings of claims. According to the Office of Special Masters, many of the claims alleging that a particular vaccine caused the same injury will rely on similar evidence, so by using omnibus proceedings or groupings to examine evidence for similar claims, the courts can more efficiently review the evidence.
30
26Attorney fees and costs may be paid if the court determines there is a reasonable basis for the petition and the petition was filed in good faith. These payments generally reflect the actual time and expense devoted to the case.
In omnibus
27See 42 U.S.C. § 300aa-21(a). 28The Vaccine Injury Compensation Trust Fund is funded by a $0.75 excise tax on each dose of vaccine recommended by HHS’s Centers for Disease Control and Prevention for routine administration to children. The excise tax is imposed on each disease that is prevented in a vaccine. Influenza vaccine, for example, is taxed $0.75 because it prevents one disease; measles-mumps-rubella vaccine, which prevents three diseases, is taxed $2.25. The trust fund receives net revenues from the excise tax, plus interest on investments in government securities. 29Within USCFC, only the Office of Special Masters receives appropriations from the trust fund. 30The Chief Special Master is responsible to provide for the efficient, expeditious, and effective handling of petitions under VICP. 41 U.S.C. § 300aa-12(c)(6).

Page 9 GAO-15-142 National Vaccine Injury Compensation Program
proceedings, petitioners select a lead claim in each category of injury, and develop these lead claims while the remaining petitioners choosing to participate in the omnibus elect for their remaining claims to be stayed, or put on hold, until the lead claim reaches a final disposition.
HHS is required to include a statement of the availability of VICP in the vaccine information materials that health care providers are to distribute to the parent or legal representatives of a child or to any other individual to whom the provider intends to administer a covered vaccine.31 These materials—referred to as vaccine information statements by HHS—are intended to explain both the benefits and risks of a vaccine covered by VICP. HHS is also required to undertake reasonable efforts to inform the public of the availability of the program.32
Most of the VICP claims filed since fiscal year 1999 have taken multiple years to adjudicate, but those filed since fiscal year 2009 have taken less time. For many claims, the parties have concluded the proceeding through a negotiated settlement, rather than a contested decision adjudicated by a special master or the courts. Additionally, certain claims were addressed along with similar claims as part of an omnibus proceeding or informal grouping.
VICP claims filed since fiscal year 1999 took an average of about 5 and a half years to adjudicate, according to USCFC data for the nearly 8,800 claims filed since fiscal year 1999 that were adjudicated as of March 31, 2014.33
31See 42 U.S.C. § 300aa-26.
There was wide variation in the amount of time to adjudicate these claims. The claim that took the shortest time to adjudicate was filed in fiscal year 1999 and took 2 days, and the claim that took the longest time to adjudicate was filed in fiscal year 1999 and took 5,276 days (more
3242 U.S.C. § 300aa-10(c). 33USCFC data on processing times for claims runs from the time the petition was filed until judgment was entered.
Requirements for Disseminating Information on the Program
Most Claims Took Multiple Years to Adjudicate and Many Were Adjudicated by Settlement or Addressed by an Omnibus Proceeding
Most Claims Filed Since Fiscal Year 1999 Have Taken Multiple Years to Adjudicate

Page 10 GAO-15-142 National Vaccine Injury Compensation Program
than 14 years). More than 1,000 (11 percent) of the claims filed since fiscal year 1999 were still in process (pending) as of March 31, 2014; most of these had been pending for 2 years or less (see fig. 1.)
Figure 1: Time Taken to Adjudicate National Vaccine Injury Compensation Program Claims Filed Fiscal Years 1999-2014, as of March 31, 2014
Note: This figure shows the time (as of March 31, 2014) to adjudicate claims for which petitions were filed in fiscal year 1999 through March 31, 2014, based on data from the U.S. Court of Federal Claims. Figures do not sum to 100 due to rounding.
For claims filed since fiscal year 2009, a greater percentage of claims were resolved within 1 or 2 years. One possible reason is that the vast majority of claims alleging autism as the injury were filed prior to fiscal year 2009. Autism claims may have taken longer because they were part of an omnibus proceeding, which suspended activity on most autism claims for a period of time. According to USCFC data, for the more than 1,400 claims filed since fiscal year 2009 that were adjudicated as of March 31, 2014, the average amount of time to adjudicate a claim was 587 days (about 1.6 years). More than 900 (40 percent) of the claims filed

Page 11 GAO-15-142 National Vaccine Injury Compensation Program
since fiscal year 1999 were still pending, which could cause this average to increase over time as these pending claims are resolved (see fig. 2).34
Figure 2: Status of National Vaccine Injury Compensation Program Claims Filed Fiscal Years 2009-2014, as of March 31, 2014
Of the pending claims, nearly half had been pending for 1 year or less as of March 31, 2014.
Note: This figure shows the status (as of March 31, 2014) of nearly 2,400 claims for which petitions were filed in fiscal year 2009 through March 31, 2014, based on data from the U.S. Court of Federal Claims.
HHS has reported the program has met its annual target of 1,300 days (about 3.5 years) for the average time to adjudicate non-autism claims in all but 1 year since fiscal year 2009. This target (1,300 days) has been in
34Of the claims filed since fiscal year 2009 that were adjudicated as of March 31, 2014, the claim that took the shortest time to adjudicate took 3 days, and the claim that took the longest time to adjudicate took 1,981 days (nearly 5 and a half years).

Page 12 GAO-15-142 National Vaccine Injury Compensation Program
place since fiscal year 2009, and applies to claims concluded in a given fiscal year, regardless of the year they were filed, excluding claims that alleged autism as the vaccine-related injury.35
Organizations representing petitioners have criticized the program for taking a long time to resolve claims. Petitioners report that long processing times delay receiving compensation to pay medical bills and other expenses related to the alleged injury. A HRSA-contracted survey of petitioners whose claims were adjudicated (regardless of whether or not they received compensation under VICP), found that nearly two-thirds of the 103 respondents indicated they were somewhat or very dissatisfied with the length of the process.
HRSA has reported meeting this goal since fiscal year 2009, except in fiscal year 2012, when VICP claims concluded that year took an average of 1,309 days to complete.
36 Petitioners may withdraw if a VICP decision is not made on their claim within specific time periods but according to the Office of Special Masters, petitioners rarely exercise this option.37
Officials cite certain delays within the process as factors that can increase average claims processing times. Officials at USCFC and DOJ told us
35Performance measures, including a measure for average claims processing time, were developed for VICP during the Program Assessment Rating Tool review conducted by the Office of Management and Budget for the fiscal year 2007 budget. HHS reports the targets and tracks the program’s performance for average claims processing time in HRSA’s annual congressional budget justifications. 36Altarum Institute, Determining the Feasibility of Evaluating the National Vaccine Injury Compensation Program, Final Report, a report prepared for the Health Resources and Services Administration, June 15, 2009. Of 716 petitioners the researchers identified as meeting their inclusion criteria,107 responded to their survey and 103 responded to this question on the length of the process. The results of this study cannot be generalized to the population of all petitioners who completed the VICP process; instead, they reflect only the VICP petitioners who responded to the survey. According to HRSA, petitioners were included in the sample if they (1) had filed a claim that had been compensated or dismissed in fiscal years 2004-2008 and (2) were represented by an attorney. 37If a special master fails to make a decision on a petition within 240 days (excluding any period of suspension), or if USCFC fails to enter a judgment on a petition within 420 days after the date on which the petition was filed (excluding any period of suspension), petitioners can withdraw their claim from VICP. 42 U.S.C. § 300aa-12(g). While USCFC officials report that most claims exceed both the 240-day and the 420-day limits when periods of suspension are not excluded, USCFC does not track suspensions so available data do not show how much time a case was suspended and how many cases met the statutory time frames.

Page 13 GAO-15-142 National Vaccine Injury Compensation Program
that the time petitioners spend gathering supporting documentation or evidence can add significantly to the amount of time required to process a claim. These delays may occur at multiple points in the claims process, from petitioners needing to gather sufficient documentation for the court to begin an initial review, to the court needing documentation to determine the amount of compensation that a successful petitioner will receive.38 According to HRSA, for claims adjudicated as of March 31, 2014, its medical review process averaged over 700 days for claims filed in fiscal year 2010. HRSA attributes the length of time for medical review primarily to time spent waiting for petitioners to submit requested documentation. During the medical review, HRSA may also consult with external experts, who require additional time to review the details of the case; HRSA’s data indicate that over 1,200 outside reviews were conducted from fiscal years 2009 to 2014. Additionally, when special masters are reviewing the claim, a party may request that the special master delay a decision until additional documentation is available.39
Special masters may also request additional information from petitioners—such as a specialist physician’s opinion.
According to HRSA data for claims filed since 2006, most compensated claims were adjudicated through negotiated settlement rather than a concession or a contested decision. HRSA’s data indicated that about 80 percent of the more than 1,500 non-autism VICP claims filed since 2006 for which compensation was awarded were adjudicated through a negotiated settlement between the parties, compared to about 10 percent involving a contested decision in favor of the petitioner and about 10 percent conceded by HHS.40
38Petitioners are required by statute to submit their supporting documentation or evidence at the time they file their petition. See 42 U.S.C. § 300aa-11(c)(2). According to DOJ, petitions are frequently filed without documentation, which delays adjudication.
According to HRSA, claims which HHS does not concede may be resolved via a negotiated settlement for several reasons, including a desire by both parties to resolve a case quickly and efficiently. According to the Office of Special Masters, a special master
39Special masters report allowing these delays in order to accumulate sufficient evidence to adjudicate a claim, but they could not provide a precise number. 40See Health Resources and Services Administration, Statistics Report-October 2, 2014, http://www.hrsa.gov/vaccinecompensation/statisticsreport.pdf, accessed October 8, 2014.
Many Claims Were Adjudicated through Settlement or Grouped in an Omnibus Autism Proceeding

Page 14 GAO-15-142 National Vaccine Injury Compensation Program
may recommend parties settle as an expeditious and efficient method of resolving certain claims.
The Office of Special Masters created an omnibus proceeding in order to address thousands of autism cases systematically and efficiently.41 Beginning in 1999, parents began filing petitions for compensation under VICP alleging that autism or neurodevelopmental disorders similar to autism were caused by the measles-mumps-rubella vaccine or vaccines containing thimerosal, a mercury-containing preservative used in some vaccines, covered by the program, or both. In 2002, the Office of Special Masters held a series of meetings with an informal advisory committee, including attorneys who represented many potential petitioners and legal and medical representatives of HHS, to address the task of dealing with these claims. The Office of Special Masters decided to utilize a two-step procedure: first, looking into whether the vaccinations in question can cause autism and, if so, the circumstances under which this occurs, and second, applying the conclusions from the first step to the individual claims. The omnibus autism proceeding included test cases for two different theories by which vaccines were alleged to cause autism.42 Some petitioners withdrew from the omnibus proceeding and elected to proceed within the vaccine program on other theories of causation. Some petitioners withdrew from the program entirely, as was their statutory right, which enabled them to pursue claims against vaccine manufacturers in civil court.43
41The omnibus autism proceeding was the largest of several instances in which the courts addressed groupings of similar claims. Since 1999, the USCFC has grouped similar claims to manage hepatitis B vaccine injury claims, claims alleging an injury of type 1 diabetes, and claims that certain childhood vaccinations have caused or contributed to autism. According to the Office of Special Masters, the omnibus autism proceeding was the largest of these groupings, involving over 5,000 claims, with the hepatitis B grouping involving several hundred and the type 1 diabetes grouping involving fewer than 50 claims.
42According to the Office of Special Masters, petitioners withdrew a third theory of causation. 43See 42 U.S.C. § 300aa-21(b). By filing a “Short-Form Autism Petition for Vaccine Compensation,” a petitioner could elect to be part of the Omnibus Autism Proceeding and simultaneously choose to stay his or her case-specific proceeding on his own petition until the conclusion of the Omnibus Autism Proceeding. See Office of Special Masters, Autism General Order #1, July 3, 2002 (http://www.uscfc.uscourts.gov/sites/default/files/autism/Autism+General+Order1.pdf), accessed October 14, 2014.

Page 15 GAO-15-142 National Vaccine Injury Compensation Program
The influx of new VICP claims for autism continued in fiscal years 2002-2005, while the number of new non-autism claims remained relatively stable (see fig. 3). HRSA data show that during this period, nearly 90 percent of VICP claims filed alleged autism as the vaccine-related injury. Ultimately, the special masters did not award compensation in any of the test cases,44 and most remaining omnibus autism proceeding claims were dismissed. However, according to the Office of Special Masters, some petitioners who had been part of the omnibus autism proceeding continued with claims separate from the omnibus proceeding.45
44See In Re: Claims for Vaccine Injuries Resulting in Autism Spectrum Disorder or a Similar Neurodevelopmental Disorder, Fed. Cl. Spec. Mstr. Update, p. 3, Jan. 12, 2011 (indicating that proceedings in all test cases had concluded and that decisions in all such cases rejected petitioners’ theories of causation).
45According to the Office of Special Masters, these petitioners proceeded on other theories of causation.

Page 16 GAO-15-142 National Vaccine Injury Compensation Program
Figure 3: Number of National Vaccine Injury Compensation Program Claims Filed Alleging Autism, Fiscal Years 1999-2014, as of March 31, 2014
Note: Data for fiscal year 2014 are as of March 31, 2014.
HHS has added vaccines to the vaccine injury table without adding covered injuries associated with those vaccines. Following their addition to the table, more claims were filed for off-table injuries.
Vaccines Have Been Added to the Vaccine Injury Table since Fiscal Year 1999 without Covered Injuries, Resulting in More Off-Table Claims

Page 17 GAO-15-142 National Vaccine Injury Compensation Program
Since fiscal year 1999, HHS has added six vaccines to the vaccine injury table (but has not added covered injuries associated with these vaccines to the table).46 This means that while individuals may file VICP claims for those vaccines, each petitioner must demonstrate that the vaccine that was administered caused the alleged injury. In general, each of the six vaccines was added within 2 years of the Centers for Disease Control and Prevention’s recommending it for routine administration to children and having an excise tax imposed.47 Since 1999, two vaccines, both of which had covered injuries associated with them, were removed from the vaccine injury table.48 See appendix II for the vaccines and injuries added and removed from the vaccine injury table since 1999. At the end of the fiscal year 2014, 16 vaccines were covered by the program, 8 of which did not have associated covered injuries on the table.49
HHS has been considering adding injuries to the table in association with the eight vaccines that are listed without covered injuries.
50
• In July 2013, HRSA published a proposed rule to add intussusception (obstruction of the bowels) as an injury associated with the rotavirus
Specifically, HRSA officials said they are working on a final rule to add an injury associated with one vaccine and a proposed rule that would add injuries associated with several other vaccines.
46Although the rhesus-based rotavirus vaccine was added to the table with an associated injury in 2002, it was removed in 2008. See 67 Fed. Reg. 48558 (Jul. 25, 2002) (addition to table); 73 Fed. Reg. 59528 (Oct. 9, 2008) (removal from table). 47In October 1999, the Centers for Disease Control and Prevention recommended the hepatitis A vaccine be administered to children in states, counties, and communities with rates of hepatitis A that were twice the 1987-1997 national average or greater. However, the hepatitis A vaccine was not covered by VICP until 2004, when the American Jobs Creation Act of 2004 imposed an excise tax for all hepatitis A vaccines. Pub. L. No. 108-357, § 889(a), 118 Stat. 1418, 1643 (codified at 26 U.S.C. § 4132(a)(I)). 48In addition to the rhesus-based rotavirus vaccine, which was removed in 2008, unconjugated Hemophilus influenza type b (Hib) polysaccharide vaccine was removed from the vaccine injury table in 2002. See 67 Fed. Reg. 48558 (Jul. 25, 2002). 49There were two vaccines on the table prior to fiscal year 1999 that had no covered injuries listed: Hemophilus influenza type b (Hib) vaccine (which prevents meningitis and other serious infections) and varicella vaccine (which prevents chicken pox). 50HHS has also been considering adding injuries in association to covered vaccines that already have associated injuries on the vaccine injury table.
Six Vaccines Have Been Added to the Vaccine Injury Table since Fiscal Year 1999 without Covered Injuries; HHS Is Considering Additional Changes

Page 18 GAO-15-142 National Vaccine Injury Compensation Program
vaccine.51
• HRSA officials said they are developing a proposed rule to add covered injuries for all seven of the remaining vaccines on the table without covered injuries. For example, HRSA is considering proposing to add Guillain-Barré Syndrome (a disorder in which the body’s immune system attacks part of the nervous system and is characterized by muscle weakness and paralysis), as an injury associated with the influenza vaccine. The agency is also considering proposing to add other injuries associated with the influenza, hemophilus influenza type b conjugate, varicella, pneumococcal conjugate, hepatitis A, meningococcal, and human papillomavirus vaccines.
In proposing the addition, HRSA considered reviews and injury claims attributed to another vaccine that protected against rotavirus but had been removed from the table in fiscal year 2009. According to HRSA, the agency is working on a final rule to add this injury.
5178 Fed. Reg. 44512 (Jul. 24, 2013) (to be codified at 42 U.S.C. § 100.3).

Page 19 GAO-15-142 National Vaccine Injury Compensation Program
According to HRSA officials, the factors informing the table changes that they are considering proposing include (1) an Institute of Medicine study of certain vaccines;52 (2) HHS’s independent review of vaccine causation and medical and scientific evidence; (3) the need to clarify injury definitions on the table53
As of September 30, 2014, HRSA had not promulgated regulations to make these changes to the vaccine injury table. According to HRSA officials, the agency plans
; and (4) recent studies related to injuries associated with the influenza vaccine.
• to publish the final rule to add the injury associated with the rotavirus vaccine to the table by July 2015,54
• to publish a proposed rule for the other injuries it is considering adding to the table by August 2015.
and
According to HRSA officials, the process to publish such a proposed rule can take about 9 months to 1.5 years. The officials also said that the process for publishing such a final rule can take about 1.5 years to 2.5 years after the proposed rule is published. In its justification accompanying its fiscal year 2013 budget request, HRSA acknowledged that many stakeholders, including Congress, have voiced interest and concern over keeping the injury table in line with current science. At that time, HRSA reported that other VICP activities, including medical reviews and court deadlines, have taken priority over updating the table.55
52In 2009, HHS commissioned the Institute of Medicine to review specific injuries in association with vaccines that are currently on the table. These vaccines include varicella; influenza, hepatitis B; human papillomavirus; measles, mumps, and rubella; hepatitis A; meningococcal; and vaccines containing tetanus. See Institute of Medicine, Adverse Effects of Vaccines: Evidence and Causality (Washington, D.C.: The National Academies Press: 2012).
53HRSA officials said they are also considering changes in the language defining injuries on the table. 54Public hearings on the proposed rule for adding intussusception as a covered injury associated with the rotavirus vaccine took place on January 13, 2014, and April 28, 2014. 55Health Resources and Services Administration, Justification of Estimates to Appropriations Committees, Fiscal Year 2013, accessed November 4, 2014 http://www.hrsa.gov/about/budget/budgetjustification2013.pdf.

Page 20 GAO-15-142 National Vaccine Injury Compensation Program
While the injuries HRSA is considering have not yet been added to the table, HRSA and DOJ officials report that many claims alleging these injuries that HRSA is considering adding to the table have been conceded or settled. For example, according to DOJ officials, there have been numerous settlements for cases alleging Guillain-Barré Syndrome as an injury associated with the influenza vaccine.
The addition of the six vaccines to the vaccine injury table without associated injuries has contributed to an increase in off-table claims. When we reported on this program in 1999, 2 of the 12 vaccines on the injury table were without associated injuries listed on the table. We reported that about one-quarter (28 percent) of claims filed as of February 1999 were for off-table injuries.56 In contrast, of the 3,007 claims filed since fiscal year 2005 (the year that trivalent influenza vaccine was added to the table) for which the covered vaccine associated with the alleged injury was specified, at least 59 percent were associated with one of five vaccines added to the table without associated table injuries, according to HRSA data.57
Claims alleging injuries to adults also increased as a result of the addition of vaccines that are recommended for administration in adults (as well as children) to the vaccine injury table. Several of the vaccines added to the injury table—in particular, the vaccine to prevent influenza—are
To receive compensation, the petitioners in these claims would not have the presumption of causation associated with an on-table claim. Overall, since 2009, more than 98 percent of the new claims filed alleged off-table injuries that required the petitioner to prove their injury was caused by the vaccine they received, according to the Office of Special Masters.
56GAO/HEHS-00-8. 57The five vaccines included hepatitis A, human pappilomavirus, influenza, meningococcal, and pneumococcal conjugate vaccines. According to HRSA data, 1,245 claims filed since fiscal year 2005, including claims filed as part of the Omnibus Autism Proceeding, did not specify a vaccine and 30 claims specified a nonqualified vaccine—that is, a vaccine not covered under VICP.
Changes in the Vaccine Injury Table Contributed to More Claims for Off-Table Injuries and for Injuries in Adults

Page 21 GAO-15-142 National Vaccine Injury Compensation Program
recommended for routine administration to adults as well as children.58 As a result, although the vaccines were added to the table because they were recommended for children, adults who are vaccinated with them are also eligible for compensation under VICP. More than half (51 percent) of the 4,402 VICP claims filed since fiscal year 1999 (for which the covered vaccine associated with the alleged injury was specified) were for injuries to adults and 1,287 (29 percent) were for adults alleging injuries in association with influenza vaccine, according to HRSA data.59
The Vaccine Injury Compensation Trust Fund balance increased to more than $3 billion in fiscal year 2013 despite increased spending by HRSA, DOJ, and USCFC on petitioner compensation, attorneys’ fees and costs, and other VICP-related expenses.
58Vaccines generally recommended for adults (as well as children) and currently covered by VICP include vaccines to protect against influenza, tetanus, diphtheria, pertussis, varicella, human papillomavirus, pneumococcal disease (pneumococcal conjugate vaccine), measles, mumps, and rubella. See http://www.cdc.gov/vaccines/schedules/downloads/adult/adult-schedule-easy-read-bw.pdf, accessed Oct. 9, 2014. 59According to HRSA data, 5,377 claims filed since fiscal year 1999 (as of March 31, 2014) did not specify which covered vaccine caused an alleged injury and 57 claims specified a vaccine not covered by the program.
Trust Fund Balance Has Increased since 2009, As Has Petitioner and Attorney Compensation and Other VICP-Related Spending

Page 22 GAO-15-142 National Vaccine Injury Compensation Program
The balance in the trust fund increased from $2.9 billion at the end of fiscal year 2009 to nearly $3.3 billion at the end of fiscal year 2013.60 The balance increased because the trust fund’s income outpaced its disbursements to HRSA, DOJ, and USCFC, although disbursements also increased during this period (see fig. 4).61 Treasury reported over $200 million in net revenues from the vaccine excise tax in each of fiscal years 2009-2013. As required by applicable law pertaining to the management of trust funds, Treasury oversees the investment of part of the net revenue from vaccine excise taxes.62
60In this report, investments in U.S. securities issued by the Bureau of Fiscal Service and the fund balance with Treasury comprise the balance of the trust fund at the end of the fiscal year.
Interest from these investments ranged from about $49 million in fiscal year 2012 to about $126 million in fiscal year 2011.
61Net revenue from vaccine excise taxes and interest on investments comprise income for the trust fund. Disbursements to HRSA—for compensation to petitioners, attorneys’ fees and costs, and administrative expenses, and to DOJ and the USCFC for the Office of Special Masters—for expenses associated with processing National Vaccine Injury Compensation Program (VICP) claims, comprise disbursements from the trust fund. 62See 26 U.S.C. § 9602.
From Fiscal Year 2009 to 2013 the Trust Fund Balance Gradually Increased to More than $3 Billion
Page 23 GAO-15-142 National Vaccine Injury Compensation Program
Figure 4: Vaccine Injury Compensation Trust Fund Income, Disbursements, and Balance for Fiscal Years 2009–2013
Note: This figure presents the Vaccine Injury Compensation Trust Fund balance (in terms of investment in U.S. securities issued by the Bureau of Fiscal Service and the fund balance with Treasury) at the end of the fiscal year. It also presents the income to trust fund (in terms of net revenue from excise taxes on vaccines and interest on investments) as well as disbursements to the

Page 24 GAO-15-142 National Vaccine Injury Compensation Program
Health Resources and Services Administration (HRSA)—for compensation to petitioners, attorneys’ fees and costs, and administrative expenses, and to the Department of Justice (DOJ) and the U.S. Court of Federal Claims (USCFC) for the Office of Special Masters—for expenses associated with processing National Vaccine Injury Compensation Program (VICP) claims, as reported in the Treasury reports.
Total compensation to petitioners and the number of claims compensated have both increased since fiscal year 1999. Petitioners’ compensation paid by HRSA using appropriations from the trust fund increased to over $254 million in fiscal year 2013. With the exception of fiscal year 2000, the total amount spent on compensation awarded to petitioners remained under $125 million between fiscal years 1999 and 2009. The total amount spent on compensation to petitioners increased to nearly $180 million in fiscal year 2010 and to more than $250 million in fiscal year 2013 (see fig. 5 and app. III).63
63There is no cap on the amount of an award in a vaccine injury case, but the law does provide certain restrictions, such as a prohibition on punitive damages and limits on pain and suffering and emotional distress. Awards may include compensation for actual and anticipated loss of earnings. In some cases, compensation may be provided in the form of an annuity. In the event of a vaccine-related death, VICP will award $250,000 to the estate of the deceased. See 42 U.S.C.§ 300aa–15. Injury-related damages in addition to the statutory death benefit may be awarded.
Spending on Compensation to Petitioners and Attorneys Has Increased since Fiscal Year 1999

Page 25 GAO-15-142 National Vaccine Injury Compensation Program
Figure 5: Compensation to Petitioners under the National Vaccine Injury Compensation Program
Note: This figure presents the total amounts and numbers of claims paid by the Health Resources and Services Administration to petitioners under the National Vaccine Injury Compensation Program for each of fiscal years 1999-2013. Data are presented by the fiscal year in which the compensation was paid.
According to the Office of Special Masters, the increase in the total amount paid to petitioners in compensation and number of compensated claims is related to the addition of the influenza vaccine to the vaccine injury table. The influenza vaccine, which is administered to millions of people each year, was added to the injury table in fiscal year 2005.
The annual amount the VICP program paid for attorneys’ fees and costs remained relatively steady from fiscal year 1999 to fiscal year 2007, but started to increase in fiscal year 2008, consistent with an increase in the total number of payments to attorneys (see fig. 6 and app. III). In order to help ensure access to the program, VICP may pay for reasonable attorney fees and costs upon a determination that the petition was brought in good faith and there was a reasonable basis for the claim for

Page 26 GAO-15-142 National Vaccine Injury Compensation Program
which the petition was brought, regardless of whether the petitioner’s claim is compensated or dismissed.64 The majority of VICP payments for attorneys’ fees and costs have been for compensated or dismissed claims; however, since fiscal year 2008, VICP has also paid some interim attorneys’ fees and costs for selected ongoing claims at the special master’s discretion.65
64Attorneys are prohibited from taking a contingent fee on compensation awarded to a petitioner. 42 U.S.C. § 300aa-15(e).
Compensation for attorneys’ fees and costs must be reasonable such that it generally reflects the actual time and expense devoted to the case.
65According to the Office of Special Masters, interim awards are made at the special master’s discretion, depending on a variety of factors, including but not limited to the duration of the litigation (e.g., lengthy proceedings), the stage of the proceedings, the amounts sought (e.g., demonstrating that substantial costs have been incurred), and the documented need for an interim award (e.g., demonstrating undue hardship).
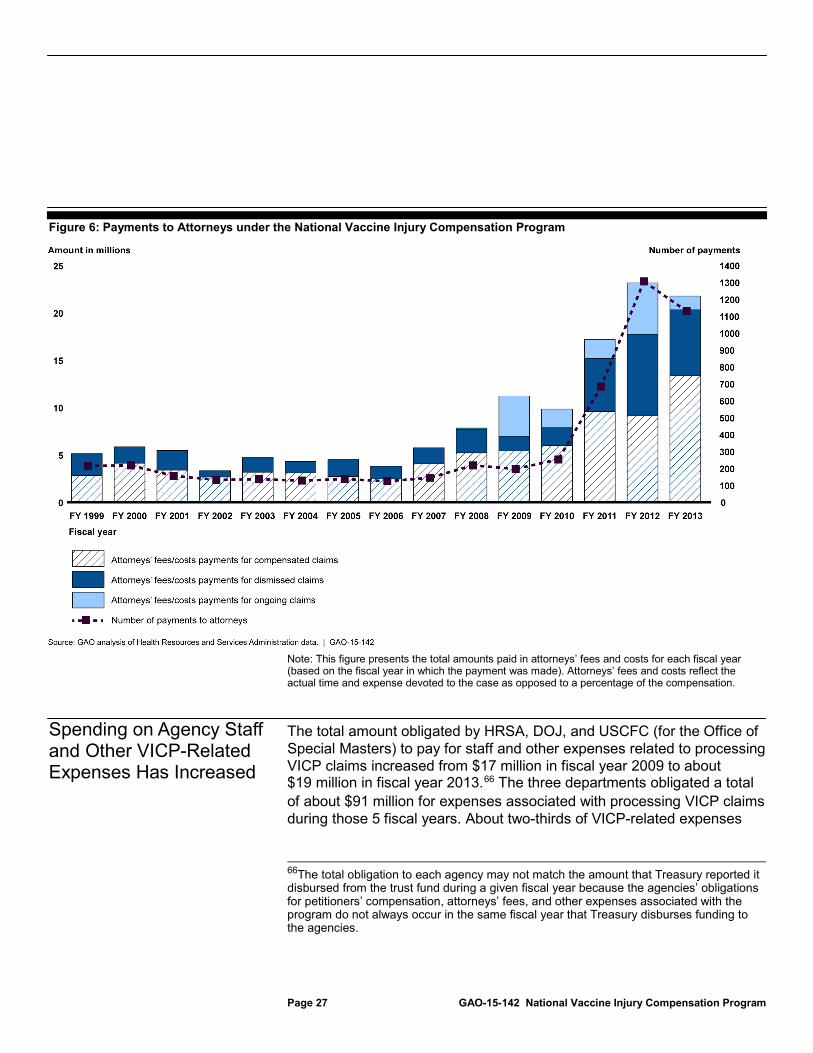
Page 27 GAO-15-142 National Vaccine Injury Compensation Program
Figure 6: Payments to Attorneys under the National Vaccine Injury Compensation Program
Note: This figure presents the total amounts paid in attorneys’ fees and costs for each fiscal year (based on the fiscal year in which the payment was made). Attorneys’ fees and costs reflect the actual time and expense devoted to the case as opposed to a percentage of the compensation.
The total amount obligated by HRSA, DOJ, and USCFC (for the Office of Special Masters) to pay for staff and other expenses related to processing VICP claims increased from $17 million in fiscal year 2009 to about $19 million in fiscal year 2013.66
66The total obligation to each agency may not match the amount that Treasury reported it disbursed from the trust fund during a given fiscal year because the agencies’ obligations for petitioners’ compensation, attorneys’ fees, and other expenses associated with the program do not always occur in the same fiscal year that Treasury disburses funding to the agencies.
The three departments obligated a total of about $91 million for expenses associated with processing VICP claims during those 5 fiscal years. About two-thirds of VICP-related expenses
Spending on Agency Staff and Other VICP-Related Expenses Has Increased

Page 28 GAO-15-142 National Vaccine Injury Compensation Program
($61 million) was obligated to pay the salaries and benefits of full-time equivalent (FTE) agency staff and about one-third ($30 million) was obligated for other VICP-related expenses (see table 1).
Table 1: Amounts Obligated by HRSA, DOJ, and USCFC for the National Vaccine Injury Compensation Program Administrative Expenses
Dollars in millions HRSAa DOJ USCFCb
Fiscal year FTE staff (number)
Other expenses
FTE staff (number)
Other expenses
FTE staff (number)
Other expenses
Total obligations
2009 $2.8 (18) $2.6 $4.5 (34) $3.2 $3.2 (24) $0.8 $17.0 2010 3.4 (22) 3.1 5.0 (34) 2.5 3.3 (25) 1.7 19.0 2011 3.8 (22) 2.7 5.4 (33) 2.4 3.4 (26) 0.8 18.5 2012 4.2 (22) 2.3 5.4 (34) 1.6 3.7 (29) 1.0 18.1 2013 3.8 (20) 2.5 5.5 (34) 2.1 3.7 (29) 1.1 18.7 Total $17.9 $13.2 $25.8 $11.9 $17.3 $5.4 $91.4
Source: GAO summary of HHS, DOJ, and USCFC data. | GAO-15-142
Legend: HRSA = Health Resources and Services Administration; DOJ = Department of Justice; USCFC = U.S. Court of Federal Claims; FTE = Full-time equivalent. Notes: Figures do not sum to totals due to rounding. aAmounts obligated by HRSA do not include compensation to petitioners or attorney costs. bAmounts obligated by USCFC were for the Office of Special Masters.
The amounts obligated for FTE salaries and benefits were for staff of HRSA, DOJ, and USCFC’s Office of Special Masters who process the claims and represent the government’s interest in legal proceedings. For example, USCFC’s Office of Special Masters paid the salaries and benefits for special masters and clerks. The average number of full-time equivalent (FTE) staff supported across the three agencies was 81 per fiscal year.
Each agency also had other VICP-related spending reimbursed by the trust fund. For example, HRSA reported obligating about $9.6 million for medical experts to review petitioner claims and provide expert testimony during adjudication proceedings for fiscal years 2009 through 2013. HRSA also obligated funds to support the Advisory Commission on Childhood Vaccines, including compensation and travel expenses for commission members. The departments reported obligating funds for costs for travel, processing documents, maintaining records, rent, supplies, and equipment. For example, obligations include rental

Page 29 GAO-15-142 National Vaccine Injury Compensation Program
payments to the General Services Administration by DOJ and the cost of court reporters funded by USCFC’s Office of Special Masters.
Information on petitioners’ experience with VICP is limited. HRSA has taken some steps to undertake outreach activities, but the agency has not yet assessed the effect of these efforts.
Other than a study for HRSA on petitioners’ satisfaction with VICP, the agency officials and stakeholders we interviewed and the documents we reviewed did not identify any data or studies regarding the experience of individuals who have filed VICP claims. The study prepared for HRSA, dated 2009, reported the responses from 107 petitioners whose claims were compensated or dismissed in fiscal years 2004-2008. Because this was a voluntary survey with a low response rate, its results cannot be generalized to all petitioners who completed the VICP process; instead, it reflects only the experience of the VICP petitioners who responded to the survey.67
67Altarum Institute, Determining the Feasibility of Evaluating the National Vaccine Injury Compensation Program. HRSA contracted for this study in 2005, which surveyed petitioners regarding their perceptions of access to VICP information, program implementation and processes, financial award decision, and overall satisfaction. According to HRSA, petitioners were included in the sample if they (1) had filed a claim that had been compensated or dismissed in fiscal years 2004-2008 and (2) were represented by an attorney. Of 716 petitioners the researchers identified as meeting their inclusion criteria, 107 responded to their survey.
We also obtained comments from stakeholders, including officials from organizations representing providers, petitioners’ attorneys, and parents. These stakeholder comments, while providing insight into petitioner experiences, are anecdotal and do not represent the experience of all petitioners who have filed VICP claims. Members of the Advisory Commission on Childhood Vaccines we interviewed expressed interest in obtaining additional information on petitioner’s experience with VICP; however, they told us they had not done so, citing concerns about confidentiality and other issues.
Information on VICP Petitioner Experience Is Limited; HHS Is Taking Steps to Address Criticism of Its Outreach Efforts
Information on Petitioners’ Experiences with VICP Is Limited

Page 30 GAO-15-142 National Vaccine Injury Compensation Program
The limited comments on petitioners’ experience from those who responded to the survey prepared for HRSA and from stakeholders included the following:
• Some petitioners responding to the HRSA survey reported being dissatisfied with the claims process and some commented that the process places too great a burden on petitioners and family members, with requests for additional information after the claims were filed. Similarly, one stakeholder said that petitioners view the vaccine injury claims process as confusing, time-consuming, too lengthy, and traumatic. Another stakeholder, on the other hand, commented that while vaccine-related injuries do not happen often, the program handles them efficiently and fairly when they do happen.
• Other comments from petitioners responding to the HRSA survey and stakeholders were related to the payment process and amount of compensation. More than half of the 61 petitioners who responded to a question on the method of payment in the HRSA survey reported being somewhat or very satisfied with the method of award payment; however, 14 respondents suggested more timely and flexible payment mechanisms. About half of the 63 respondents to the question on the amount of compensation reported the award amount was inadequate to cover past and future medical care. Similarly, stakeholders we interviewed reported concerns with the amounts petitioners receive, the method of payment, and a perceived lack of transparency regarding how the money from the Vaccine Injury Compensation Trust Fund has been spent. For example, one stakeholder told us that petitioners felt forced to settle for less than what it will cost them to care for their children or themselves for their lifetimes, and another stakeholder raised concerns about the choice of annuities for petitioners.
• Other comments on the program from stakeholders were related to the perception of an adversarial or unfriendly environment throughout the process, the use of settlements and perceived pressure to settle claims, the use of omnibus proceedings to group claims together, and concerns about confidentiality of the medical information filed by the petitioner. Some petitioners responding to the HRSA survey and some stakeholders also reported difficulties finding an attorney to represent petitioners in the process; however, petitioners responding to the survey were split on this issue—about the same number reported that finding an attorney was difficult as reported that finding an attorney was easy.

Page 31 GAO-15-142 National Vaccine Injury Compensation Program
HRSA has acknowledged being criticized for years for not adequately promoting public awareness of VICP, and has recently taken some steps, such as developing and starting to implement an outreach plan for fiscal year 2014 and developing an outreach plan for fiscal year 2015, to improve its efforts to reach out to providers and the public.68 In its 2006 VICP strategic plan, HRSA noted that one of the critical issues facing the program from 2005 to 2010 was that many parents, the general public, attorneys, and health care professionals were not aware VICP existed.69 In 2009, HRSA contracted for the development of a comprehensive national marketing and outreach communication plan; the contactor presented the plan to HRSA in November 2010.70 According to HRSA, the agency used this plan to guide outreach efforts.71 Prior to the fiscal year 2014 plan, HRSA also reported exhibiting at professional conferences; updating the VICP website and the VICP booklet that is available from the website (including translating the booklet into Spanish); facilitating the review of vaccine information statements (which include a statement on VICP) by the Advisory Commission on Childhood Vaccines; and responding to media inquiries and inquiries received via e-mail, letters, and the program’s toll-free number.72
68In each of HRSA’s annual justification of estimates for appropriations committees for fiscal years 2011-2014, HRSA noted that the agency has been criticized for not adequately promoting public awareness of the VICP.
HRSA officials also noted the need to carefully balance messages that increase awareness of VICP with public health messages that encourage and promote immunizations.
69Division of Vaccine Injury Compensation, National Vaccine Injury Compensation Program Strategic Plan (April 2006). 70Banyan Communications, Inc., The Comprehensive National Vaccine Injury Compensation Program Marketing and Outreach Communication Plan, a report prepared for the Health Resources and Services Administration, (Nov. 2010). 71According to HRSA officials, the agency has been implementing some of the activities recommended in the November 2010 plans since that plan was issued, but the agency did not formally document its outreach efforts until the fiscal year 2014 VICP outreach plan. Agency officials report that the 2014 plan was developed because HRSA is taking a more strategic approach to its outreach and wanted to formally document these efforts. 72As part of its fiscal year 2011 budget justification, HRSA reported that exhibiting at conferences had proven beneficial in increasing knowledge of the availability of the program and that it planned exhibiting at medical and legal conferences in fiscal year 2011.
HHS Is Taking Steps to Address Criticism of Its Efforts to Inform the Public of the Program

Page 32 GAO-15-142 National Vaccine Injury Compensation Program
HRSA shared an overview of its outreach plans with its Advisory Commission on Childhood Vaccines in September 2014. HRSA reported that many of the activities in the agency’s 2014 outreach plan were in process at the end of the fiscal year. These activities included reviewing the VICP booklets to use plain language and make them more user-friendly, reviewing and upgrading the VICP website to improve navigation, developing VICP message points for target audiences and slides about the program to be used in speeches and other presentations by HRSA staff, and requesting federal websites to provide information on the program and to link to the VICP website. In its outreach plan for fiscal year 2015, the agency is targeting health care providers, parents and expectant parents, adults aged 50 years and older (including Spanish-speaking older adults), and civil litigation and health attorneys, with the goal of informing target audiences of the availability of the program. According to HRSA, these target audiences were selected because they include individuals who administer vaccines and individuals (or their caregivers) who receive vaccinations. HRSA has identified a number of measures to assist in tracking performance of its fiscal year 2015 plan, including website metrics, the number of “retweets” and “shares” from social media initiatives, the number of media inquiries, and the number of attendees or participants at outreach events. Because the agency has not completed many of its planned efforts to improve how it informs the public of the availability of the program, it is too early to determine the effect of HRSA’s current and planned outreach efforts.
Without awareness of the program, individuals who might otherwise receive compensation for a vaccine-related injury or death could be denied compensation because of a failure to file their claim within the statutory deadlines. One stakeholder commented that the public is largely unaware of the program, and this lack of awareness contributes to missing filing deadlines and individuals being denied the opportunity for compensation. Members of the Advisory Commission on Childhood Vaccines also told us that many individuals may not know there is a statute of limitations on filing a claim and many miss the opportunity to file

Page 33 GAO-15-142 National Vaccine Injury Compensation Program
a claim because of the statute of limitations.73 In December 2013, the commission recommended extending the statute of limitations for vaccine-related injuries and deaths. Extending the statute of limitations would require amending the applicable statutory provision.74
It has been more than 25 years since VICP went into effect in 1988. In that time, the program has awarded more than $2.8 billion to thousands of petitioners. Several aspects of the program have changed over the years. First, while claims alleging injuries on the vaccine injury table made up the majority of claims filed in the first decade of the program, today—after the addition of new vaccines, particularly influenza vaccine, to the table without associated injuries—the majority of the claims filed involve off-table injuries. Stakeholders report that the program has an adversarial environment, as petitioners are required to demonstrate a covered vaccine caused the injury on their VICP claim when there are no associated injuries on the table. The extent to which this will change if HHS updates the vaccine injury table to include more injuries, as expected, is yet to be seen. Second, most of the compensated cases are now adjudicated through negotiated settlement, rather than contested decisions before the special master. And while the vaccines covered by the program are included because they are recommended for children, many of the program’s petitioners in recent years are adults who received covered vaccines.
Addressing one criticism of the program by stakeholders—specifically the need to increase the statute of limitations—would require a statutory change in the program. Regardless of whether the statute of limitations is
73The current statute of limitations, in general, requires that claims be filed within 3 years of the date of the first symptom or manifestation of the onset or significant aggravation of an injury and within 2 years of a death (and within 4 years of the date of the first symptom or manifestation of the onset or significant aggravation of the injury from which a death resulted). Generally, when a vaccine or injury is added to the vaccine injury table, petitioners are required to file claims within 2 years from the date the vaccine or injury is added to the table for injuries or deaths that occurred up to 8 years before the table change. See 42 U.S.C. § 300aa-16. 74HHS has proposed extending the statutory time limits for filing a VICP claim in the past. For example, in commenting on a draft of our 1999 report, the department stated that the legislative proposals it had sent to the Congress included doubling the statutory time limit for filing a claim. GAO/HEHS-00-8, 39. DOJ officials also reported they believed their department had supported previous proposals to extend the program’s statute of limitations.
Concluding Observations

Page 34 GAO-15-142 National Vaccine Injury Compensation Program
increased, HHS’s efforts to increase awareness of the availability of the program will be important to help ensure that potential petitioners are aware of the program and can file claims in time. While HHS has recently taken or planned steps to improve its outreach activities, what effect, if any, these efforts will have remains to be seen. As the agency moves forward, it will be important for HRSA to identify which activities are reaching its target audiences.
We provided a draft of this report to HHS, DOJ, USCFC, and Treasury. HHS and USCFC agreed with our findings and provided written comments, which are reprinted in appendixes IV and V, respectively. In its comments, HHS emphasized that it administers VICP jointly with DOJ and USCFC, with HHS responsible for reviewing petitioners’ claims, providing recommendations for entitlement to compensation, and making payments to petitioners and attorneys. In commenting on our identification of its efforts on VICP outreach, HHS noted that it is strengthening its outreach efforts by implementing its fiscal year 2015 VICP outreach plan, which increases outreach to target populations and includes performance measures. In its written comments, USCFC noted that the Administrative Office of the United States Courts and USCFC both supply administrative support to the Office of Special Masters and VICP without reimbursement from the trust fund. The court also commented that, while considerable strides have been made in reducing the average processing time for claims in recent years, the climbing and changing nature of the caseload, coupled with the statutory cap on the number of special masters, present a continuing challenge to the Office of Special Masters’ ability to continue to reduce average processing times. USCFC also commented that following omnibus proceedings, claims have been resolved more expeditiously in recent years, and often through settlements. USCFC also commented that the Office of Special Masters strives to resolve all cases fairly and expeditiously. HHS, USCFC, DOJ, and Treasury also provided technical comments that were incorporated, as appropriate.
We are sending copies of this report to the Secretary of Health and Human Services, the Attorney General of the United States, the Chief Judge of the United States Court of Federal Claims, and the Secretary of the Treasury. In addition, the report is available at no charge on the GAO website at http://www.gao.gov.
Agency Comments

Page 35 GAO-15-142 National Vaccine Injury Compensation Program
If you or your staff have any questions about this report, please contact me at (202) 512-7114 or [email protected]. Contact points for our Offices of Congressional Relations and Public Affairs may be found on the last page of this report. GAO staff who made key contributions to this report are listed in appendix VI.
Sincerely yours,
Marcia Crosse Director, Health Care

Appendix I: Vaccine Injury Table, September 2014
Page 36 GAO-15-142 National Vaccine Injury Compensation Program
Vaccine Injurya Time periodb Vaccines against tetanus (e.g., DTaP, DTP, DT, Td, or TT) Anaphylaxis or anaphylactic shock 4 hours Brachial neuritis 2-28 days Vaccines against pertussis (e.g., DTP, DTaP, P, DTP-Hib) Anaphylaxis or anaphylactic shock 4 hours Encephalopathy (or encephalitis) 72 hours Vaccines against measles, mumps, rubella in any combination (e.g., MMR, MR, M, R)
Anaphylaxis or anaphylactic shock 4 hours Encephalopathy (or encephalitis) 5-15 days
Vaccines against measles (e.g., MMR, MR, M) Thrombocytopenic purpura 7-30 days Vaccine-strain measles viral infection in an
immunodeficient recipient 6 months
Vaccines against rubella (e.g., MMR, MR, R) Chronic arthritis 7-42 days Vaccines against polio (polio live virus-containing (OPV)) Paralytic Polio —In a nonimmunodeficient recipient 30 days —In an immunodeficient recipient 6 months —In a vaccine-associated community case Not applicable Vaccine-strain polio viral infection —In a nonimmunodeficient recipient 30 days —In an immunodeficient recipient 6 months —In a vaccine-associated community case Not applicable Vaccines against polio (polio inactivated virus-containing (IPV)) Anaphylaxis or anaphylactic shock 4 hours Vaccines against hepatitis B Anaphylaxis or anaphylactic shock 4 hours Vaccines against hemophilus influenzae type b (Hib conjugate vaccine)
No condition specified Not applicable
Vaccines against varicella No condition specified Not applicable Vaccines against rotavirusa No condition specified Not applicable Vaccines against pneumococcal disease (pneumococcal conjugate vaccine)a
No condition specified Not applicable
Vaccines against hepatitis Aa No condition specified Not applicable Vaccines against influenza (trivalent vaccine)a,c No condition specified Not applicable Vaccines against meningococcal diseasea No condition specified Not applicable Vaccines against human papillomavirus (HPV) No condition specified Not applicable
Source: GAO summary of information from the Health Resources and Services Administration. | GAO-15-142
Notes: In addition to the specific vaccines currently listed on the table, the National Vaccine Injury Compensation Program (VICP) also covers new vaccines recommended by the Centers for Disease Control and Prevention for routine administration to children after publication by the Secretary of a notice of coverage, effective on the date of an applicable excise tax. aThis column includes the illness, disability, injury, or condition covered by the program. In addition, covered injuries include any acute complication or sequela (including death) of the listed injuries (for all but the Hib, varicella, rotavirus, pneumococcal conjugate, hepatitis A, trivalent influenza, meningococcal, and HPV vaccines).
Appendix I: Vaccine Injury Table, September 2014

Appendix I: Vaccine Injury Table, September 2014
Page 37 GAO-15-142 National Vaccine Injury Compensation Program
bFor first symptom, onset, or aggravation of injury after vaccination. cTrivalent influenza vaccine was added to the table effective July 1, 2005. All additional seasonal influenza vaccines, including quadrivalent vaccine, are covered by VICP, effective November 12, 2013.

Appendix II: Covered Vaccines and Injuries on the Vaccine Injury Table, Fiscal Years 1999-2014
Page 38 GAO-15-142 National Vaccine Injury Compensation Program
Fiscal year Vaccine 1999 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013 2014a Vaccines and Injuries Added to the Table before Fiscal Year 1999 Tetanus-containing
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Pertussis-containing
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Measles, mumps, rubella in any combination
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Measles-containing
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Rubella-containing
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Polio live virus-containing
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Polio inactivated virus-containing
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Hepatitis B ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● Hemophilus influenza type b (Hib) conjugateb
○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○
Varicellab ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ Vaccines and Injuries Added and Proposed to be Added to the Table During and After Fiscal Year 1999 Rotavirusc ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ◐c ◐c Pneumococcal conjugate
— ○d ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○
Hepatitis A — — — — — — ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ Trivalent influenzae
— — — — — — ○ ○ ○ ○ ○ ○ ○ ○ ○ ○
Meningococcal — — — — — — — — ○ ○ ○ ○ ○ ○ ○ ○ Human Papillomavirus
— — — — — — — — ○ ○ ○ ○ ○ ○ ○ ○
Vaccines and Injuries Added and Removed from the Table since Fiscal Year 1999 Hib polysaccharide (unconjugated)
● ● ● ● — — — — — — — — — — — —
Rotavirus rhesus-based
— — — ● ● ● ● ● ● ● ● — — — — —
Source: GAO analysis. | GAO-15-142
Appendix II: Covered Vaccines and Injuries on the Vaccine Injury Table, Fiscal Years 1999-2014

Appendix II: Covered Vaccines and Injuries on the Vaccine Injury Table, Fiscal Years 1999-2014
Page 39 GAO-15-142 National Vaccine Injury Compensation Program
Legend: ● = at least one injury specified for this vaccine on the table at any point during the fiscal year ◐ = HHS issued a proposed rule to add an injury to the table during the fiscal year ○ = there are no table injuries specified for this vaccine — = the vaccine is not on the table Notes: When no injury is identified or specified for the vaccine, the petitioner must prove that the injury was caused by the vaccine. In addition to the vaccines listed on the table, the National Vaccine Injury Compensation Program (VICP) may also cover any new vaccines that are recommended by the Centers for Disease Control and Prevention’s for routine administration to children and which are subject to an excise tax, but are not yet specifically listed on the vaccine injury table. aThe information in this column is up to date as of September 17, 2014. bHemophilus influenza type b conjugate and varicella vaccines were added to the table effective August 6, 1997. cRotavirus was added to the table effective October 22, 1998. HHS issued a proposed rule on July 24, 2013, to add an injury to the table associated with rotavirus vaccine. However, the rule was not finalized before the end of fiscal year 2014. dPneumococcal conjugate vaccine was added to the table effective December 18, 1999. eTrivalent influenza vaccine was added to the table effective July 1, 2005. All additional seasonal influenza vaccines, including quadrivalent vaccine, are covered by VICP, effective November 12, 2013.

Appendix III: Compensation to Petitioners and Attorneys’ Fees and Costs, Fiscal Years 1999-2013
Page 40 GAO-15-142 National Vaccine Injury Compensation Program
This appendix shows the total amount paid in compensation to petitioners under the National Vaccine Injury Compensation Program and the number of compensated claims in fiscal years 1999-2013 (see table 2). It also shows the amounts the program paid in attorneys’ fees and costs for those same fiscal years (see table 3).
Table 2: Compensation to Petitioners under the National Vaccine Injury Compensation Program, Fiscal Years 1999-2013
Fiscal year Petitioners’ award amount Number of compensated claims 1999 $95,917,681 96 2000 $125,945,196 136 2001 $105,878,633 97 2002 $59,799,604 80 2003 $82,816,240 65 2004 $61,933,764 57 2005 $55,065,797 64 2006 $48,746,163 68 2007 $91,449,434 82 2008 $75,716,552 141 2009 $74,142,491 131 2010 $179,387,341 173 2011 $216,319,428 251 2012 $163,511,999 250 2013 $254,666,327 375 Total $1,691,296,649 2,066
Source: HRSA Data and Statistics Report, August 1, 2014. | GAO-15-142
Appendix III: Compensation to Petitioners and Attorneys’ Fees and Costs, Fiscal Years 1999-2013

Appendix III: Compensation to Petitioners and Attorneys’ Fees and Costs, Fiscal Years 1999-2013
Page 41 GAO-15-142 National Vaccine Injury Compensation Program
Table 3: Payments to Attorneys under the National Vaccine Injury Compensation Program, Fiscal Years 1999-2013
Fiscal year
Compensated attorneys’ fees/cost
payments
Dismissed attorneys’ fees/cost
payments
Interim attorneys’ fees/cost
payments
Number of payments to
attorneys 1999 $2,799,911 $2,306,957 $0.00 213 2000 $4,112,369 $1,724,451 $0.00 216 2001 $3,373,866 $2,066,225 $0.00 154 2002 $2,653,599 $656,245 $0.00 130 2003 $3,147,755 $1,545,655 $0.00 134 2004 $3,079,329 $1,198,616 $0.00 126 2005 $2,694,664 $1,790,587 $0.00 135 2006 $2,441,199 $1,353,633 $0.00 122 2007 $4,034,154 $1,692,020 $0.00 143 2008 $5,191,771 $2,511,313 $117,265 216 2009 $5,404,712 $1,557,140 $4,241,363 195 2010 $5,961,744 $1,886,240 $1,978,804 251 2011 $9,572,043 $5,589,417 $2,001,771 682 2012 $9,104,489 $8,621,182 $5,420,258 1,304 2013 $13,333,180 $6,970,279 $1,454,852 1,128 Total $76,904,784 $41,469,960 $15,214,312 5,149
Source: HRSA Data and Statistics Report, August 1, 2014. | GAO-15-142

Appendix IV: Comments from the Department of Health and Human Services
Page 42 GAO-15-142 National Vaccine Injury Compensation Program
Appendix IV: Comments from the Department of Health and Human Services

Appendix IV: Comments from the Department of Health and Human Services
Page 43 GAO-15-142 National Vaccine Injury Compensation Program

Appendix V: Comments from the United States Court of Federal Claims
Page 44 GAO-15-142 National Vaccine Injury Compensation Program
Appendix V: Comments from the United States Court of Federal Claims

Appendix V: Comments from the United States Court of Federal Claims
Page 45 GAO-15-142 National Vaccine Injury Compensation Program

Appendix VI: GAO Contact and Staff Acknowledgments
Page 46 GAO-15-142 National Vaccine Injury Compensation Program
Marcia Crosse, (202) 512-7114 or [email protected]
In addition to the contact named above, Kim Yamane, Assistant Director; George Bogart; Carolyn Garvey; Cathleen Hamann; Katherine Perry; Fatima Sharif; and Eric Wedum made key contributions to this report.
Appendix VI: GAO Contact and Staff Acknowledgments
GAO Contact
Staff Acknowledgments
(100007)

The Government Accountability Office, the audit, evaluation, and investigative arm of Congress, exists to support Congress in meeting its constitutional responsibilities and to help improve the performance and accountability of the federal government for the American people. GAO examines the use of public funds; evaluates federal programs and policies; and provides analyses, recommendations, and other assistance to help Congress make informed oversight, policy, and funding decisions. GAO’s commitment to good government is reflected in its core values of accountability, integrity, and reliability.
The fastest and easiest way to obtain copies of GAO documents at no cost is through GAO’s website (http://www.gao.gov). Each weekday afternoon, GAO posts on its website newly released reports, testimony, and correspondence. To have GAO e-mail you a list of newly posted products, go to http://www.gao.gov and select “E-mail Updates.”
The price of each GAO publication reflects GAO’s actual cost of production and distribution and depends on the number of pages in the publication and whether the publication is printed in color or black and white. Pricing and ordering information is posted on GAO’s website, http://www.gao.gov/ordering.htm.
Place orders by calling (202) 512-6000, toll free (866) 801-7077, or TDD (202) 512-2537.
Orders may be paid for using American Express, Discover Card, MasterCard, Visa, check, or money order. Call for additional information.
Connect with GAO on Facebook, Flickr, Twitter, and YouTube. Subscribe to our RSS Feeds or E-mail Updates. Listen to our Podcasts. Visit GAO on the web at www.gao.gov.
Contact:
Website: http://www.gao.gov/fraudnet/fraudnet.htm E-mail: [email protected] Automated answering system: (800) 424-5454 or (202) 512-7470
Katherine Siggerud, Managing Director, [email protected], (202) 512-4400, U.S. Government Accountability Office, 441 G Street NW, Room 7125, Washington, DC 20548
Chuck Young, Managing Director, [email protected], (202) 512-4800 U.S. Government Accountability Office, 441 G Street NW, Room 7149 Washington, DC 20548
GAO’s Mission
Obtaining Copies of GAO Reports and Testimony
Order by Phone
Connect with GAO
To Report Fraud, Waste, and Abuse in Federal Programs
Congressional Relations
Public Affairs
Please Print on Recycled Paper.


Contains Nonbinding Recommendations
Use of the Term “Healthy” in the Labeling of Human Food Products:
Guidance for Industry
Additional copies are available from: Office of Nutrition and Food Labeling
Nutrition Program Staff, HFS-830 Center for Food Safety and Applied Nutrition
Food and Drug Administration 5001 Campus Drive
College Park, MD 20740 (Tel) 240-402 -1450
http://www.fda.gov/FoodGuidances
U.S. Department of Health and Human Services Food and Drug Administration
Center for Food Safety and Applied Nutrition
September 2016

Contains Nonbinding Recommendations
2
Table of Contents
I. Introduction II. Background III. Discussion
A. Low Fat B. Beneficial Nutrients
IV. References

Contains Nonbinding Recommendations
3
Use of the Term “Healthy” in the Labeling of Human Food Products:
Guidance for Industry1
This guidance represents the current thinking of the Food and Drug Administration's (FDA or we) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the FDA staff responsible for this guidance as listed on the title page.
I. Introduction
The purpose of this guidance is to advise manufacturers who wish to use the implied nutrient content claim “healthy” to label their food products as provided by our regulations. More specifically, this guidance is intended to advise food manufacturers of our intent to exercise enforcement discretion relative to foods that use the implied nutrient content claim “healthy” on their labels which: (1) Are not low in total fat, but have a fat profile makeup of predominantly mono and polyunsaturated fats; or (2) contain at least ten percent of the Daily Value (DV) per reference amount customarily consumed (RACC) of potassium or vitamin D.
This guidance is immediately effective because the agency has determined that prior public participation is not feasible or appropriate (21 CFR 10.115(g)(2)).
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe our current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in FDA guidances means that something is suggested or recommended, but not required.
II. Background
Under section 403(r)(1)(A) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act) (21 U.S.C. 343(r)(1)(A)), a food2 is misbranded if it bears claims, either express or implied, that characterize the level of a nutrient which is of a type required to be declared in nutrition
1 This guidance has been prepared by the Office of Nutrition and Food Labeling, Nutrition Program Staff, in the Center for Food Safety and Applied Nutrition at the U.S. Food and Drug Administration. 2 Section 201(f) of the FD&C Act (21 U.S.C. 321(f)) defines the term “food” to mean articles used for food or drink for man or other animals, chewing gum, and articles used for components of any such article.

Contains Nonbinding Recommendations
4
labeling3 unless the claim is made in accordance with a regulatory definition established by FDA (see section 403(r)(2) of the FD&C Act, 21 U.S.C. 343(r)(2)). 21 CFR 101.65(d) provides such regulatory definition for use of the term “healthy” or related terms (such as “health,” “healthful,” “healthfully,” “healthfulness,” “healthier,” “healthiest,” “healthily,” and “healthiness”) as an implied nutrient content claim on the label or in labeling of a food. The “healthy” nutrient content claim can be used if the food meets certain nutrient conditions; and, when used with an explicit or implicit claim or statement about a nutrient (e.g. “healthy, contains 3 grams of fat”), suggests that a food, because of its nutrient content, may be useful in creating a diet that is consistent with dietary recommendations. The nutrient conditions for bearing a “healthy” nutrient content claim include specific criteria for nutrients to limit in the diet, such as total fat, saturated fat, cholesterol, sodium, as well as requirements for nutrients to encourage in the diet, including vitamin A, vitamin C, calcium, iron, protein, and fiber. The criteria are linked to elements in the Nutrition Facts label and serving size regulations (see 21 CFR §§ 101.9 and 101.12). The nutrient criteria to use the claim can vary for different food categories (e.g., fruits and vegetables, or seafood and game meat) (see 21 CFR 101.65(d)(2)).
In addition, it is also important to note that under section 403(a)(1) of the FD&C Act (21 U.S.C. 343(a)(1)), a food is misbranded if its labeling is false or misleading in any particular. Section 201(n) of the FD&C Act (21 U.S.C. 321(n)) provides that labeling is misleading if, among other things, it fails to reveal facts that are material in light of representations made or suggested in the labeling, or material with respect to consequences that may result from the use of the food to which the labeling relates under the conditions of use prescribed in the labeling, or under such conditions of use as are customary or usual.
III. Discussion In the Federal Register of May 27, 2016, we issued final rules updating the Nutrition Facts label and serving size information for packaged foods to reflect new scientific information, including the link between diet and chronic diseases such as obesity and heart disease (see 81 FR 33742, “Food Labeling: Revision of the Nutrition and Supplement Facts Labels”; 81 FR 34000, “Food Labeling: Serving Sizes of Foods That Can Reasonably Be Consumed At One Eating Occasion; Dual-Column Labeling; Updating, Modifying, and Establishing Certain Reference Amounts Customarily Consumed; Serving Size for Breath Mints; and Technical Amendments”). Updates to the Nutrition Facts label include changes in the individual nutrients that must be declared and also changes to the Daily Value (DV) of other individual nutrients, reflecting changes in recommended intake levels, based on current science. The individual nutrients included in Nutrition Facts and their DVs significantly influence the regulations on nutrient content and health claims. Because the framework for many of FDA’s other nutrition labeling regulations is linked to elements in the Nutrition Facts label and serving size regulations, we plan to update those other regulations, such as those for health claims and nutrient content claims (including the implied nutrient content claim “healthy”), to align with these most recent updates.
3 Section 201(m) of the FD&C Act (21 U.S.C. 321(m)) defines “labeling” as all labels and other written, printed, or graphic matter upon any article or any of its containers or wrappers or accompanying such article.

Contains Nonbinding Recommendations
5
The science underlying the final rules, referenced above, is also reflected in the 2015-2020 Dietary Guidelines for Americans (2015-2020 Dietary Guidelines) (Ref. 1). The Dietary Guidelines is designed for professionals to help all individuals ages 2 years and older and their families consume a healthy, nutritionally adequate diet. The Dietary Guidelines is the foundation of federal nutrition guidance and is fundamental in shaping federal policies and programs related to food, nutrition, and health. The Dietary Guidelines provides information and perspectives on healthy eating patterns and consumption of foods from various food groups, as well as the intake of specific macronutrients such as fats, sugars, and micronutrients such as vitamins and minerals. Specific recommendations in the Dietary Guidelines have evolved over time, as nutrition science has advanced. For example, scientific understanding and nutrition guidance has shifted from recommending diets low in total fat (2005 Dietary Guidelines) (Ref. 2) to no longer recommending limiting overall fat intake, and instead prioritizing increasing intakes of polyunsaturated and monounsaturated fats and decreasing intakes of saturated fat and trans fat (2015-2020 Dietary Guidelines). The 2015-2020 Dietary Guidelines, also emphasizes the importance of dietary patterns as a whole, the combination of foods and drinks that people consume over time. The body of scientific evidence and the recommendations based on that science in the Dietary Guidelines will help FDA shape additional updates to regulations on nutrition related claims and information that are permitted on the food label. We are re-evaluating the regulatory criteria for use of the implied nutrient content claim “healthy” in light of the latest nutrition science and the current dietary recommendations and seek input on possible future rulemaking to update the existing regulations for this claim. Because the rulemaking process can sometimes be lengthy, we intend to exercise enforcement discretion in the interim with respect to some of the existing criteria for the nutrient content claim “healthy” if the alternative nutrient criteria described below are met. We intend to exercise enforcement discretion until we amend 21 CFR 101.65(d)(2). A. Low Fat As stated above, since we published the final rule defining “healthy” (58 FR 2302, January 6, 1993), the science related to public health recommendations for intake of dietary fats has evolved. The focus of the most recent dietary fat recommendations has shifted away from limiting total fat intake to encouraging intakes of mono and polyunsaturated fats. Foods that use the term “healthy” on their labels that are not low in total fat should have a fat profile makeup of predominantly mono and polyunsaturated fats (i.e., sum of monounsaturated fats and polyunsaturated fats are greater than the total saturated fat content of food). Consistent with consensus science and public health recommendations for dietary fats which no longer recommend low total fat intake, we intend to exercise enforcement discretion with respect to the current requirement that any food bearing the nutrient content claim “healthy” meet the low fat requirement (§101.65(d)(2)(i)), provided that: (1) The amounts of mono and polyunsaturated fats are declared on the label and (2) the amounts declared constitute the majority of the fat content. These conditions are necessary so that consumers are made aware that the total fat is mostly made up of fats that are encouraged by current dietary recommendations.

Contains Nonbinding Recommendations
6
B. Beneficial Nutrients The definition for “healthy” also includes a nutrient contribution criterion. Healthy dietary patterns not only restrict nutrients that increase risk of chronic disease, but also help assure nutrient adequacy to ensure sufficient intake of nutrients that are important in sustaining body function and reducing the risk of disease. The current definition of “healthy” has focused on foods providing a good or excellent source of nutrients for which there had been public health concern. Historically, these nutrients have been vitamin A, vitamin C, iron, calcium, and dietary fiber. Nutrient intakes have shifted over time, however, and vitamins A and C are no longer nutrients of public health concern. The nutrients of public health concern now include potassium and vitamin D, in addition to iron and calcium (2015-2020 Dietary Guidelines). This change in nutrients of public health concern has been reflected in the mandatory nutrients to be labeled in the Nutrition Facts Label (see 81 FR 33742, May 27, 2016). In recognition of this change, we intend to exercise enforcement discretion with respect to the current requirement that any food bearing the nutrient content claim “healthy” contain at least ten percent of the Daily Value (DV) per reference amount customarily consumed (RACC) of vitamin A, vitamin C, calcium, iron, protein, or fiber, if the food instead contains at least ten percent of the DV per RACC of potassium or vitamin D. The regulations updating the Nutrition Facts label have provided for new DVs for potassium and vitamin D and manufacturers have been given some time to come into compliance with these regulations. If a manufacturer has not yet implemented the updated Nutrition Facts label, they may use the old DVs for potassium and vitamin D. If a manufacturer has already implemented the updated Nutrition Facts label, they must use the new, updated DVs for potassium and vitamin D. In either case, if a food is basing its eligibility for bearing a “healthy” claim on potassium or vitamin D, whichever nutrient is being used as the basis for eligibility should be declared in the Nutrition Facts label.
IV. References
We have placed the following references on display in the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane, rm. 1061, Rockville, MD 20852. You may see them at that location between 9 a.m. and 4 p.m., Monday through Friday. As of June 1, 2016, FDA had verified the Web site address for the references we make available as hyperlinks from the Internet copy of this guidance, but we are not responsible for any subsequent changes to Non-FDA Web site references after June 1, 2016. 1. U.S. Department of Health and Human Services and U.S. Department of Agriculture. 2015 –
2020 Dietary Guidelines for Americans. 8th Edition. December 2015. Available at http://health.gov/dietaryguidelines/2015/guidelines/.
2. U.S. Department of Health and Human Services and U.S. Department of Agriculture. Dietary Guidelines for Americans, 2005. 6th Edition, Washington, DC: U.S. Government Printing Office, January 2005. Available at http://health.gov/dietaryguidelines/dga2005/.