torquoselectivity of the ring-opening reaction of 3,3
TRANSCRIPT

doi.org/10.26434/chemrxiv.9178121.v1
Torquoselectivity of the Ring-Opening Reaction of 3,3-DihalosubstitutedCyclobutenes: Lone Pair Repulsion and Cyclic Orbital InteractionYuji Naruse, Atsushi Takamori
Submitted date: 31/07/2019 • Posted date: 31/07/2019Licence: CC BY-NC-ND 4.0Citation information: Naruse, Yuji; Takamori, Atsushi (2019): Torquoselectivity of the Ring-Opening Reactionof 3,3-Dihalosubstituted Cyclobutenes: Lone Pair Repulsion and Cyclic Orbital Interaction. ChemRxiv.Preprint.
Three major factors determine torquoselectivity, which is the diastereoselectivity in electrocyclic ring-openingreactions to produce E/Z-double bond(s). One is the interaction between the decomposing sCC bond andlow-lying vacant orbital(s), such as a p*- or s*-orbital on the substituent, which promotes the reaction, resultingin inward rotation of the substituent. Second, for a substituent with a lone pair(s), repulsive interactionbetween the decomposing s-bond and the lone pair(s) hinders inward rotation, so that the products of outwardrotation should be preferred. Finally, a more strongly donating s-electron-donating group (sEDG) rotatesinwardly due to stabilization by phase-continuous cyclic orbital interaction. We compared the latter twointeractions, repulsion between the lone pairs on the substituent and stabilization from phase-continuouscyclic orbital interaction, to determine which has a greater effect on the diastereoselectivity. We considered aseries of model reactions with halogen substituents, and concluded that the diastereoselectivity is mainlycontrolled by cyclic orbital interaction.
File list (1)
download fileview on ChemRxivcycX-YNa9729d.pdf (920.45 KiB)

1
Torquoselectivity of the Ring-Opening Reaction of
3,3-Dihalosubstituted Cyclobutenes: Lone Pair
Repulsion and Cyclic Orbital Interaction
Yuji Naruse1,2* and Atsushi Takamori1
1Department of Materials Chemistry and Processing, Graduate School of Gifu University, 1-1,
Yanagido, Gifu, 501-1193 JAPAN , 2Department of Chemistry and Biomolecular Science, Gifu
University, 1-1, Yanagido, Gifu, 501-1193 JAPAN
KEYWORDS: Torquoselectivity, Electrocyclic Ring-Opening Reaction, Geminal Bond
Participation, Bond Model Analysis.
ABSTRACT. Three major factors determine torquoselectivity, which is the diastereoselectivity
in electrocyclic ring-opening reactions to produce E/Z-double bond(s). One is the interaction
between the decomposing CC bond and low-lying vacant orbital(s), such as a *- or *-orbital
on the substituent, which promotes the reaction, resulting in inward rotation of the substituent.
Second, for a substituent with a lone pair(s), repulsive interaction between the decomposing -
bond and the lone pair(s) hinders inward rotation, so that the products of outward rotation should
be preferred. Finally, a more strongly donating -electron-donating group (EDG) rotates

2
inwardly due to stabilization by phase-continuous cyclic orbital interaction. We compared the
latter two interactions, repulsion between the lone pairs on the substituent and stabilization from
phase-continuous cyclic orbital interaction, to determine which has a greater effect on the
diastereoselectivity. We considered a series of model reactions with halogen substituents, and
concluded that the diastereoselectivity is mainly controlled by cyclic orbital interaction.
1. Introduction
Torquoselectivity is the diastereoselectivity in electrocyclic ring-opening reactions that produces
E-/Z-isomers of the double bond.1-4 Many reports have discussed this diastereoselectivity,
especially with regard to the electrocyclic ring-opening reaction of 3-substituted cyclobutenes.
However, the diastereoselectivity often does not seem to be guided by steric considerations, so
that electronic effects need to be considered. Three major interactions of the electronic effect
have been proposed to explain this torquoselectivity (Figure 1).
Figure 1. Torquoselectivity of 3-substituted cyclobutenes and its proposed electronic effects.

3
One is the interaction between the decomposing CC bond and a low-lying vacant orbital, such
as a *- or *-orbital on the substituent, which promotes the reaction (Figure 1a).3-6 Cyclobutene
with an alkoxycarbonyl group at the 3-position was reported to show torquoselectivity with
inward rotation of the alkoxycarbonyl group. This would be a donor-acceptor interaction, and
hence an attractive interaction, and the TS should be considerably stabilized, resulting in inward
rotation of the substituent. On the other hand, interaction between the decomposing CC bond and
the lone pair(s) on the substituent is a type of donor-donor interaction, which leads to repulsive
destabilization. Substituent(s) with lone pairs should prefer outward rotation (Figure 1b).
In contrast, Inagaki proposed that phase-continuous cyclic orbital interaction8 that includes
geminal bond participation controls the diastereoselectivity (Figures 1c and 1d).7b The cyclic
orbital interactions among *C=C, decomposing CC and C-D of the -donating group (EDG) D
(Figure 1c) and among C=C, decomposing *CC and C-D (Figure 1d) satisfy the phase continuity
requirements, so that electron delocalization among them is enhanced to produce considerable
stabilization at TSs.
We wondered which of these three effects controlled the diastereoselectivity most effectively.
As a first step, we compared the effects of the latter two, i.e., the repulsive interaction between
the decomposing CC bond and the lone pair(s) on the substituent(s) and the cyclic orbital
interaction including geminal bond participation.
2. Expected torquoselectivity
To compare these effects, we should choose a system that is free from bond interaction on the
substituent(s). Thus, we chose 3,3-dihalocyclobutenes 1 as models. Halogens are monovalent, so
that there no other bonds on the substituent(s) and no effects from the * or *-orbital. The

4
atomic radii of halogens are in the order F < Cl < Br and the bond lengths of carbon-halogen
bonds are in the order C-F < C-Cl < C-Br. If repulsion with the decomposing CC bond is the
main contributor to diastereoselectivity, the preference for inward rotation should be in the order
F > Cl > Br, considering the atomic radii. In contrast, if a longer bond length reduces repulsion,
the order of preference should be reversed to F < Cl < Br.
Figure 2. 3,3-Dihalocyclobutenes considered.
From the perspective of cyclic orbital interaction7b,8, the energy level of C-X is well correlated
with electronegativity, so that the donating character is in the order C-F < C-Cl < C-Br, since
fluorine is the most electronegative (4.0), followed by chlorine (2.8) and bromine (2.7). We
previously confirmed this order by evaluating the energy levels of C-X bond orbitals: C-F (-
1.068 a.u.) < C-Cl (-0.882 a.u.) <C-Br (-0.834 a.u.).7b-d,9 Since the more strongly donating
character of the geminal C-X bond enhances the cyclic orbital interaction among *C=C,
decomposing CC and C-D and among C=C, decomposing *CC and C-D, we can expect that the
preference for inward rotation should be in the order F < Cl < Br.
3. Theoretical calculations
First, we performed theoretical calculations at the M06-2X/6-311++G** level (Figure 3).10
The results are summarized in Table 1.
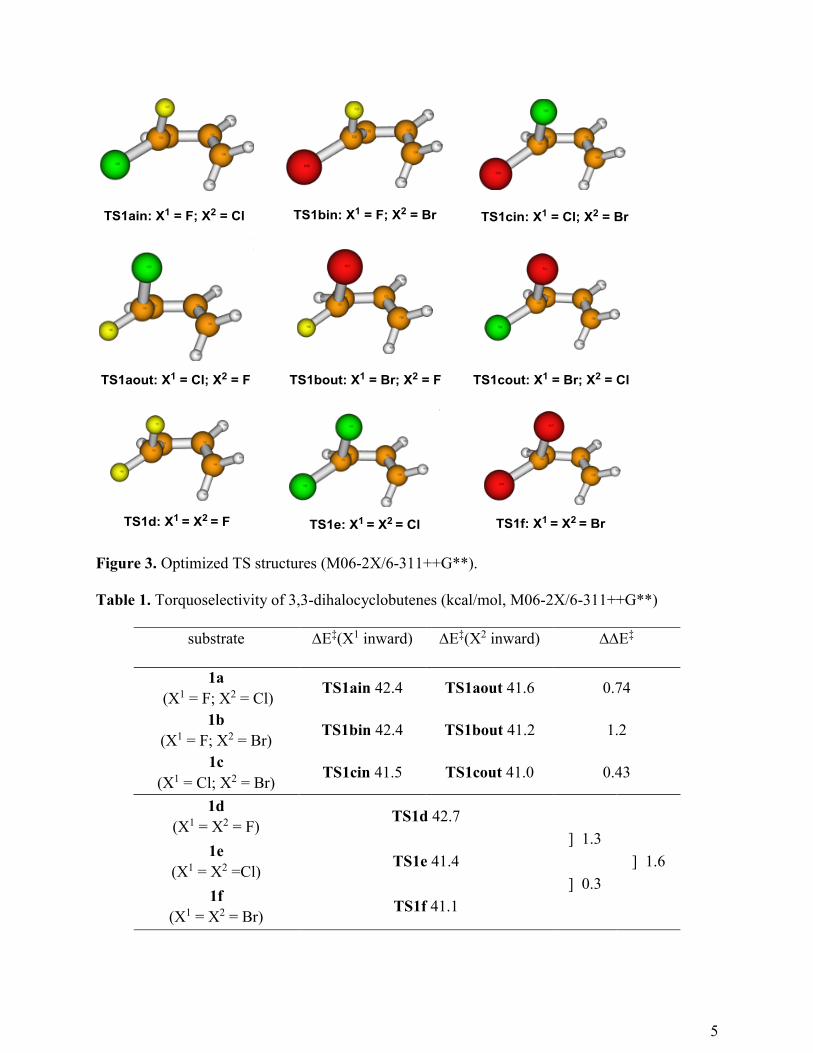
5
Figure 3. Optimized TS structures (M06-2X/6-311++G**).
Table 1. Torquoselectivity of 3,3-dihalocyclobutenes (kcal/mol, M06-2X/6-311++G**)
substrate E‡(X1 inward) E‡(X2 inward) E‡
1a
(X1 = F; X2 = Cl) TS1ain 42.4 TS1aout 41.6 0.74
1b
(X1 = F; X2 = Br) TS1bin 42.4 TS1bout 41.2 1.2
1c
(X1 = Cl; X2 = Br) TS1cin 41.5 TS1cout 41.0 0.43
1d
(X1 = X2 = F) TS1d 42.7
] 1.3
] 1.6 1e
(X1 = X2 =Cl) TS1e 41.4
] 0.3 1f
(X1 = X2 = Br) TS1f 41.1

6
Apparently, the preference for inward rotation is in the order F < Cl < Br. The activation
energies are in the order TS1d (difluoro, 42.7 kcal/mol) > TS1e (dichloro, 41.4 kcal/mol) >
TS1f (41.1 kcal/mol, dibromo), and there is a preference for inward rotation of a chloro group
in TS1a and a bromo group in TS1b and TS1c. There is some regularity in these values. The
activation energies E‡ for fluoro-inward rotation of TS1ain (42.4 kcal/mol) and TS1bin (42.4
kcal/mol) are almost the same as that of difluoro TS1d (42.7 kcal/mol), the activation energies
E‡ for chloro-inward rotation of TS1aout (41.6 kcal/mol) and TS1cin (41.5 kcal/mol) are
almost the same as that of dichloro 1e (41.4 kcal/mol), and finally, the activation energies E‡ for
bromo-inward rotation of TS1bout (41.2 kcal/mol) and TS1cout (41.0 kcal/mol) are almost the
same as that of dibromo TS1e (41.1 kcal/mol). These results clearly indicate that the activation
energy heavily depends on the substituent that rotates inward.
Figure 3. Relationship among the optimized activation energies.
Furthermore, the activation energy decreases with the outward-rotating substituent in the
order of F > Cl > Br, e.g., 1d (outward F: 42.7 kcal/mol) > 1a (outward Cl: 42.4 kcal/mol) ~ 1b
(outward Br: 42.4 kcal/mol) in the inward-F series, and 1a (outward F: 41.6 kcal/mol) > 1e
(outward Cl:41.4 kcal/mol) > 1c (outward Br: 41.0 kcal/mol) in the inward-Cl series. For the
inward Br series, there are no apparent differences of ca. 0.1 kcal/mol.
We can expect that these relationships can be attributed to the inductive effect. The stronger
-electron-withdrawing group attracts electrons on other atoms in the neighborhood, so that the
-bond energies of the other substituent bonds should be lowered. This inductive effect also
7
affects the charge on the other substituents to reduce steric repulsion while the bond lengths are
almost the same.
Thus, we can conclude that the obtained values are consistent with our expectation. Repulsion
should be reduced with longer bond lengths, and only the -bond of the substituent C-X bond
with inward rotation is involved in the cyclic orbital interaction. Thus, we can expect that one of
these two effects should mainly control the torquoselectivity.
4. Bond model analysis
To determine which effect should contribute more to the torquoselectivity, we performed a
bond model analysis11 to evaluate the bond interactions. We used the interbond energy IBE12 for
this evaluation. Due to the difficulty of separating the lone pairs of the valence electrons and the
core orbitals, we evaluated the sum IBE(CC-nX) of the repulsive interactions between
decomposing CC and the lone pairs on the halogen atom that rotates inward. We summarize our
analysis in Table 2.
Table 2. Charge and bond interactions (IBE in a.u., RHF/6-31G(d)//M06-2X/6-311++G**).
Lone pair repulsion
Cyclic orbital interaction
Among
CC-*C=C-C-D- orbitals
Among
C=C-*CC-C-D- orbitals
Mulliken
charge on
Xa
IBE(C
C-nX)/a.u.
IBE(CC-
*C=C)/a.u.
IBE(C-D-
*C=C)/a.u.
IBE(C=C-
*CC)/a.u.
IBE(C-D-
*CC)/a.u.
1a F-inward 0.0457
(-0.3370) 0.7814 -2.2363 -0.0356 -1.0408 0.0798
1a Cl-inward 0.1492
(0.0126) 0.8466 -2.2637 -0.0601 -1.1634 0.3288
1b F-inward 0.0366
(-0.3898) 0.9466 -2.6783 -0.0406 -1.2335 0.0981
1b Br-inward -0.0232 0.9495 -2.7379 -0.1082 -1.4206 0.4624

8
(-0.1045)
1c Cl-inward 0.1138
(0.0574) 1.1782 -2.6154 -0.0996 -1.5448 0.3961
1c Br-inward -0.0571
(-0.0568) 1.1044 -2.6363 -0.1118 -1.6146 0.3358
1d difluoro -0.0236
(-0.3360) 0.6758 -2.1999 -0.0263 -0.8872 0.0743
1e dichloro 0.1042
(0.0439) 1.0088 -2.2158 -0.0826 -1.1340 0.3229
1e dibromo -0.0343
(-0.0517) 1.2536 -3.0452 -0.1619 -1.8195 0.5669
aM06-2X/6-311++G**; at the level of RHF/6-31G(d)//M06-2X/6-311++G** in parentheses.
First, the evaluated steric repulsion IBE(CC-nX) is in the order F < Cl < Br, which is
opposite the order we expected. From a charge perspective, there is no correlation with the
changes in the activation energies. The steric repulsion should mostly depend on the atomic radii
of the halogens. The only exception is the case of 1c, where less steric repulsion is calculated for
the inward rotation of Br. We suppose that the longer bond length of C-Br (1.957 Å, M06-2X/6-
311++G**) compared to that of C-Cl (1.785 Å) would be the main contributor in this case.
Considering these results, we conclude that steric repulsion does not control the torquoselectivity.
For the cyclic orbital interaction, stabilization due to the bond interactions between CC and
*C=C and between C-D and *C=C in the cyclic orbital interaction among CC-*C=C-C-D-
orbitals, and those between C=C and *CC and between C-D-*CC in the cyclic orbital
interaction among C=C-*CC-C-D- orbitals are all enhanced in the order F < Cl < Br. For inward
rotation of the same atom, repulsive interaction increased in the order F < Cl < Br. In contrast,
the stabilization from the cyclic orbital interactions also increased. They should cancel each other
so that the activation energies remained almost the same regardless of the substituent rotating
outward. For example, in fluoro-inward rotation, repulsion between the decomposing CC and
lone pairs on the halogen IBE(CC-nX) increases according to the outward-rotating substituent

9
in the order F (1d, fluoro-outward: 0.6758 a.u.) < Cl (1a, Cl-outward: 0.7814 a.u.) < Br (1b, Br-
outward: 0.9466 a.u.). On the other hand, stabilization from CC-*C=C in the cyclic orbital
interaction is in the order F (1d, -2.1999 a.u.) < Cl (1a, -2.2363 a.u.) < Br (1b, -2.6783 a.u.).
Note that the geminal interaction between C-D-*CC shows an antibonding character of IBE
values. Inagaki explained this phenomenon in terms of the antibonding nature of geminal
delocalization.13 The geminal bond interaction was erroneously considered to mean that there
was no interaction between the two geminal bonds. However, although the two hybrid orbitals on
the center atom are orthogonal, the geminal bonds still interact mostly via the hybrid orbital at
the terminal positions (Figure 4). The phase between the two geminal bonds is determined by the
two hybrid orbitals on the center atom. Thus, the overlaps between the other three hybrids should
often be out-of-phase combinations for the obtuse bond angle, which results in the antibonding
nature. This characteristic of geminal delocalization leads to the positive values for IBEs. Thus,
we can conclude that a more electron-donating character of EDG prefers inward rotation by
enhancing the cyclic orbital interaction, i.e., the order of electronegativity F > Cl > Br should
result in the donating character of a C-X bond to be in the order C-F < C-Cl < C-Br, so that the
preference for inward rotation is in the order F < Cl < Br due to the stabilization from the cyclic
orbital interaction at the TS. These results are in good agreement with our expectations.
Figure 4. Phase relationship and overlaps in geminal delocalization
Conclusion

10
We evaluated the electronic effects that affect torquoselectivity in the electrocyclic ring-
opening reaction of 3,3-dihalocyclobutenes. According to theoretical calculations, the change in
activation energies is in the order F > Cl > Br. From the perspective of repulsion between the
decomposing CC and the lone pair(s) on the halogens, the order was assumed to result from
longer bond lengths. On the other hand, from the perspective of cyclic orbital interaction, the
order was believed to be due to phase-continuous cyclic orbital interactions, where the electron-
donating ability of the C-X bond is essential. Electronegativity is in the order F > Cl > Br, and
thus, the -bond orbital energy is in the orderC-F < C-Cl < C-Br. The cyclic orbital interaction
is enhanced in the order F < Cl < Br. According to our bond model analysis, the change in cyclic
orbital interaction is consistent with our expectation, while repulsive interaction between the
decomposing CC and the lone pair(s) on the halogens does not follow the change in the
activation energies. Thus, we conclude that the cyclic orbital interaction controls
torquoselectivity in the electrocyclic ring-opening reaction of 3,3-dihalocyclobutenes.
ASSOCIATED CONTENT
Supporting Information. Summary of the theoretical calculations. This material is available
free of charge via the Internet.
Author Contributions
Both authors contributed to writing the manuscript and approved the final version of the
manuscript.
REFERENCES
1. Some early reports: (a) Frey, H. M.; Marshall, D. C. Trans. Faraday Soc. 1965, 61, 1715-
1721. (b) Rudolf, K.; Spellmeyer, D. C.; Houk, K. N. J. Org. Chem. 1987, 52, 3708-3710. (c)

11
Houk, K. N.; Spellmeyer, D. C.; Jefford, C. W.; Rimbault, C. G.; Wang, Y.; Miller, R. D. J. Org.
Chem. 1988, 53, 2125-2127. (d) Dolbier, W. R.; Grat, T. A.; Keaffaber, J. J.; Calewicz, L.;
Koroniak, H. J. Am. Chem. Soc. 1990, 112, 363-367. (e) Niwayama, S.; Houk, K. N.
Tetrahedron Lett. 1993, 34, 1251-1254.
2. (a) Kirmse, W.; Rondan, N. G.; Houk, K. N. J. Am. Chem. Soc. 1984, 106, 7989-7991. (b)
Rondan, N. G.; Houk, K. N. J. Am. Chem. Soc. 1985, 107, 2099-2111. (c) Nakamura, K.; Houk,
K. N. J. Org. Chem. 1995, 60, 686-691.
3. Jefford, C. W.; Bernardinelli, G.; Wang, Y.; Spellmeyer, D. C.; Buda, A.; Houk, K. N. J. Am.
Chem. Soc. 1992, 114, 1157-1165.
4. (a) Murakami, M.; Miyamoto, Y.; Ito, Y. Angew. Chem., Int. Ed. 2001, 40, 189-190. (b)
Murakami, M.; Miyamoto, Y.; Ito, Y. J. Am. Chem. Soc. 2001, 123, 6441-6442. See also: (c)
Murakami, M.; Miyamoto, Y.; Ito, Y. Yuki Gosei Kagaku Kyokaishi 2002, 60, 1049-1054.
5. (a) Shindo, M.; Matsumoto K.; Mori, S.; Shishido, K. J. Am. Chem. Soc. 2002, 124, 6840-
6841. (b) Shindo, M.; Sato, Y.; Yoshikawa, T.; Koretsune, R.; Shishido K. J. Org. Chem. 2004,
69, 3912-3916.
6. Lee, P. S.; Zhang, X.; Houk, K. N. J. Am. Chem. Soc. 2003, 125, 5072-5079.
7. (a) Ikeda, H.; Kato, T.; Inagaki, S. Chem. Lett. 2001, 270-271. (b) Yasui, M.; Naruse, Y.;
Inagaki, S. J. Org. Chem. 2004, 69, 7246-7249. (c) Naruse, Y.; Ichihashi, Y.; Shimizu, T.;
Inagaki, S. Org. Lett. 2012, 14, 3728-3731. (d) Naruse, Y.; Tokunaga, M. Tetrahedron Lett. 2015,
56, 3813-3815.
8. Inagaki, S. Top. Curr. Chem. 2009, 289, 83-127.
9. (a) Ma, J.; Ding, Y.; Hattori, K.; Inagaki, S. J. Org. Chem. 2004, 69, 4345-4255.
10. Gaussian 09, Rev. D.01. Gaussian, Inc., Wallingford CT, 2013. Full citation is in the
Supporting Information.

12
11. (a) Bond Model Analysis 1.1 Inagaki, S.; Ikeda, H.; Ohwada, T.; Takahama, T. 1996-2013.
See: (b) Inagaki, S.; Ikeda, H. J. Org. Chem. 1999, 63, 7820-7824; (c) Iwase, K.; Inagaki, S. Bull.
Chem. Soc. Jpn. 1996, 69, 2781-2789; (d) Inagaki, S.; Ohashi, S.; Yamamoto, T. Chem. Lett.
1997, 26, 977-978; (e) Ikeda, H.; Inagaki, S. J. Chem. Phys. A 2001, 47, 10711-10718.
12. Interbond energy IBE is derived using the following equation:
IBEij = Pij(Hij + Fij)
where Pij, Hij, and Fij are the elements of the density, Fock and core Hamiltonian matrices of the
bond orbitals, i and j, respectively.
13. Inagaki, S.; Goto, N.; Yoshikawa, K. J. Am. Chem. Soc. 1991, 113, 7144-7146.
Insert Table of Contents Graphic and Synopsis Here
13
Supporting Information
Gaussian 09, Revision D.01,
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,
M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci,
G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian,
A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada,
M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima,
Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr.,
J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers,
K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand,
K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi,
M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross,
V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann,
O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski,
R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth,
P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels,
O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski,
and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
M06-2X/6-311++G**
1a F-Cl reactant
Frequencies -- 165.2802 281.2907 359.8175
Sum of electronic and zero-point Energies= -714.712300
Sum of electronic and thermal Energies= -714.706882
Sum of electronic and thermal Enthalpies= -714.705938
Sum of electronic and thermal Free Energies= -714.741704
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.333858
3 6 0 1.498834 0.000000 -0.065359
4 6 0 1.513169 -0.011645 1.483776
5 1 0 -0.750279 0.013383 -0.778079
6 1 0 -0.791779 0.017608 2.071658
7 9 0 2.066264 -1.096703 -0.640447
8 17 0 2.237218 1.438952 -0.830967
9 1 0 1.962980 0.867000 1.945668
10 1 0 1.943919 -0.925880 1.894438
TS1ain F-Cl-ts F-inward
Frequencies -- -673.6097 190.1844 249.6412
Sum of electronic and zero-point Energies= -714.644809
Sum of electronic and thermal Energies= -714.639348
Sum of electronic and thermal Enthalpies= -714.638404
Sum of electronic and thermal Free Energies= -714.674311
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.384206
3 6 0 1.376690 0.000000 -0.287797
4 6 0 1.221007 0.578411 1.777391
5 1 0 -0.799636 0.097911 -0.723700
6 1 0 -0.733195 -0.444502 2.050433
7 9 0 2.167777 -1.061690 -0.041259
8 17 0 1.990886 0.820251 -1.704952
9 1 0 1.484848 1.550774 1.386172
10 1 0 1.743803 0.282108 2.684956
.delta.E++ = 0.067491 a.u. = 42.4 kcal/mol
.delta.G++ = 0.067393 a.u. = 42.3 kcal/mol
14
.delta..delta.E++ = 0.001173 a.u. = 0.74 kcal/mol
.delta..delta.G++ = 0.001105 a.u. = 0.69 kcal/mol
IRC final strucures
stru. 1
1 6 0 -0.983558 0.414002 0.321754
2 6 0 -1.375566 0.131515 1.704705
3 6 0 0.129625 0.012088 -0.280396
4 6 0 -0.654817 -0.462413 2.658955
5 1 0 -1.660857 0.997818 -0.287271
6 1 0 -2.379485 0.460910 1.953801
7 9 0 1.057699 -0.721609 0.320084
8 17 0 0.552529 0.358154 -1.910693
9 1 0 0.355295 -0.815280 2.501780
10 1 0 -1.081773 -0.600912 3.644002
stru. 2
1 6 0 -1.406354 -0.462921 0.229273
2 6 0 -1.449988 0.167620 1.403871
3 6 0 0.072893 -0.252027 0.096322
4 6 0 0.038035 0.474464 1.464592
5 1 0 -2.122061 -0.942750 -0.423151
6 1 0 -2.257205 0.400866 2.086047
7 9 0 0.832221 -1.382919 0.080858
8 17 0 0.589654 0.749443 -1.293704
9 1 0 0.319602 1.527112 1.433497
10 1 0 0.601297 -0.052060 2.236347
TS1aout Cl-F-ts Cl-inward
Frequencies -- -608.2079 213.0030 244.4735
Sum of electronic and zero-point Energies= -714.645982
Sum of electronic and thermal Energies= -714.640578
Sum of electronic and thermal Enthalpies= -714.639634
Sum of electronic and thermal Free Energies= -714.675416
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.381666
3 6 0 1.368927 0.000000 -0.321843
4 6 0 1.234819 0.542313 1.779298
5 1 0 -0.802243 0.088057 -0.723274
6 1 0 -0.740624 -0.434921 2.044524
7 17 0 2.435739 -1.387228 -0.145812
8 9 0 1.765337 0.670406 -1.413049
9 1 0 1.528559 1.509146 1.396549
10 1 0 1.751125 0.223507 2.683434
.delta.E++ = 0.066318 a.u. = 41.6 kcal/mol
.delta.G++ = 0.066288 a.u. = 41.6 kcal/mol
IRC final structures
stru. 1
1 6 0 -1.496455 -0.037433 -0.009824
2 6 0 -1.542503 0.509225 1.204500
3 6 0 -0.028485 0.238433 -0.150544
4 6 0 -0.072133 0.893265 1.252605
5 1 0 -2.198849 -0.527153 -0.669971
6 1 0 -2.338023 0.635678 1.928363
7 17 0 1.012234 -1.210570 -0.288519
15
8 9 0 0.324494 1.084631 -1.158077
9 1 0 0.144570 1.962235 1.228023
10 1 0 0.535834 0.394044 2.008384
stru. 2
1 6 0 -1.036137 0.753564 -0.024922
2 6 0 -1.401698 0.550191 1.379371
3 6 0 -0.045078 0.195836 -0.708016
4 6 0 -0.578456 0.307570 2.398302
5 1 0 -1.666682 1.407025 -0.619606
6 1 0 -2.464897 0.642391 1.580954
7 17 0 1.106429 -0.949303 -0.150959
8 9 0 0.141654 0.474584 -1.997383
9 1 0 0.495702 0.245021 2.277389
10 1 0 -0.973376 0.179960 3.398465
1b F-Br
Frequencies -- 149.1768 255.9505 318.4960
Sum of electronic and zero-point Energies= -2828.683950
Sum of electronic and thermal Energies= -2828.678306
Sum of electronic and thermal Enthalpies= -2828.677362
Sum of electronic and thermal Free Energies= -2828.714491
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.334380
3 6 0 1.496179 0.000000 -0.064296
4 6 0 1.512916 0.002405 1.484952
5 1 0 -0.749827 0.007329 -0.778374
6 1 0 -0.792194 0.015501 2.072087
7 9 0 2.065859 -1.098102 -0.631525
8 35 0 2.298178 1.566305 -0.922290
9 1 0 1.953170 0.887789 1.942994
10 1 0 1.953143 -0.905204 1.900490
TS1bin F-Br-ts F-inward
Frequencies -- -675.0412 171.1218 229.1909
Sum of electronic and zero-point Energies= -2828.616305
Sum of electronic and thermal Energies= -2828.610620
Sum of electronic and thermal Enthalpies= -2828.609676
Sum of electronic and thermal Free Energies= -2828.646931
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.384611
3 6 0 1.377343 0.000000 -0.283261
4 6 0 1.218415 0.582491 1.778838
5 1 0 -0.800536 0.094433 -0.722791
6 1 0 -0.730532 -0.449396 2.050816
7 9 0 2.168723 -1.061174 -0.035794
8 35 0 2.041547 0.887188 -1.844619
9 1 0 1.482419 1.553663 1.384952
10 1 0 1.740983 0.287637 2.686981
.delta.E++ = 0.067645 a.u. = 42.4 kcal/mol
.delta.G++ = 0.067560 a.u. = 42.4 kcal/mol
.delta..delta.E++ = 0.001960 a.u. = 1.2 kcal/mol
.delta..delta.G++ = 0.001870 a.u. = 1.2 kcal/mol
IRC final structures
stru. 1
16
1 6 0 -1.190872 0.249613 0.937540
2 6 0 -1.623618 -0.088020 2.296814
3 6 0 -0.050515 -0.109529 0.359075
4 6 0 -1.021283 -0.890708 3.177203
5 1 0 -1.854032 0.855994 0.335316
6 1 0 -2.556442 0.384707 2.589427
7 9 0 0.873068 -0.843866 0.968571
8 35 0 0.457051 0.346192 -1.390935
9 1 0 -0.094903 -1.407640 2.967544
10 1 0 -1.469054 -1.048626 4.150532
stru. 2
1 6 0 -1.590447 -0.702194 0.720900
2 6 0 -1.634163 -0.005357 1.858166
3 6 0 -0.114443 -0.495828 0.574981
4 6 0 -0.146979 0.304792 1.901565
5 1 0 -2.305686 -1.219396 0.097345
6 1 0 -2.442392 0.269915 2.524193
7 9 0 0.653102 -1.617964 0.609029
8 35 0 0.426421 0.523344 -1.006522
9 1 0 0.136487 1.353463 1.820697
10 1 0 0.416472 -0.185356 2.696989
TS1bout Br-F-ts Br-inward
Frequencies -- -595.7911 187.9116 223.4353
Sum of electronic and zero-point Energies= -2828.618265
Sum of electronic and thermal Energies= -2828.612648
Sum of electronic and thermal Enthalpies= -2828.611704
Sum of electronic and thermal Free Energies= -2828.648801
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.381955
3 6 0 1.366647 0.000000 -0.326626
4 6 0 1.238521 0.533396 1.779041
5 1 0 -0.803500 0.083906 -0.722638
6 1 0 -0.739594 -0.436473 2.044735
7 35 0 2.533385 -1.518755 -0.150251
8 9 0 1.756848 0.666226 -1.423594
9 1 0 1.539952 1.498357 1.397611
10 1 0 1.753671 0.209429 2.682168
.delta.E++ = 0.065685 a.u. = 41.2 kcal/mol
.delta.G++ = 0.065690 a.u. = 41.2 kcal/mol
IRC fibnal structures
stru. 1
1 6 0 -1.848625 0.353115 0.062669
2 6 0 -1.914695 0.928495 1.265012
3 6 0 -0.389358 0.658168 -0.075323
4 6 0 -0.451307 1.338718 1.315509
5 1 0 -2.536424 -0.161975 -0.592176
6 1 0 -2.716836 1.054727 1.980214
7 35 0 0.779053 -0.910050 -0.187183
8 9 0 -0.044250 1.487944 -1.096046
9 1 0 -0.254403 2.410633 1.265309
10 1 0 0.156495 0.873040 2.090390
stru. 2
1 6 0 -1.315578 1.196492 0.060086
17
2 6 0 -1.705458 0.951984 1.451772
3 6 0 -0.372262 0.597066 -0.653790
4 6 0 -0.889044 0.692367 2.470577
5 1 0 -1.885190 1.934788 -0.498155
6 1 0 -2.771805 1.035291 1.641699
7 35 0 0.762541 -0.802297 -0.131982
8 9 0 -0.157249 0.941219 -1.923494
9 1 0 0.186045 0.640510 2.351697
10 1 0 -1.286856 0.536372 3.465678
1c Cl-Br
Frequencies -- 148.9661 221.1986 250.9802
Sum of electronic and zero-point Energies= -3189.041367
Sum of electronic and thermal Energies= -3189.035344
Sum of electronic and thermal Enthalpies= -3189.034400
Sum of electronic and thermal Free Energies= -3189.072793
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.333598
3 6 0 1.495007 0.000000 -0.082448
4 6 0 1.510681 0.002744 1.475640
5 1 0 -0.745972 -0.000127 -0.782311
6 1 0 -0.790454 0.003027 2.072935
7 17 0 2.213747 -1.448128 -0.838651
8 35 0 2.251288 1.608283 -0.901148
9 1 0 1.950986 0.897293 1.916322
10 1 0 1.954136 -0.894521 1.908216
TS1cin Cl-Br-ts Cl-inward
Frequencies -- -632.6326 171.9890 222.1517
Sum of electronic and zero-point Energies= -3188.975235
Sum of electronic and thermal Energies= -3188.969236
Sum of electronic and thermal Enthalpies= -3188.968291
Sum of electronic and thermal Free Energies= -3189.006706
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.380170
3 6 0 1.372040 0.000000 -0.332864
4 6 0 1.238948 0.539550 1.772238
5 1 0 -0.810392 0.093442 -0.713225
6 1 0 -0.742491 -0.423632 2.048892
7 17 0 2.411141 -1.409619 -0.132124
8 35 0 1.904940 1.016289 -1.869264
9 1 0 1.545980 1.496492 1.374380
10 1 0 1.757706 0.220802 2.675168
.delta.E++ = 0.066132 a.u. = 41.5 kcal/mol
.delta.G++ = 0.066087 a.u. = 41.5 kcal/mol
.delta..delta.E++ = 0.000681 a.u. = 0.43 kcal/mol
.delta..delta.G++ = 0.000686 a.u. = 0.43 kcal/mol
IRC final structures
stru. 1
1 6 0 -1.653772 -0.523656 0.692222
2 6 0 -1.681432 0.134750 1.851394
3 6 0 -0.187061 -0.293441 0.501217
4 6 0 -0.203669 0.479295 1.854428
5 1 0 -2.368566 -1.046128 0.072486
6 1 0 -2.472016 0.356660 2.556448
18
7 17 0 0.816522 -1.770463 0.494370
8 35 0 0.260915 0.804450 -1.053797
9 1 0 0.045492 1.537047 1.768468
10 1 0 0.396878 0.007550 2.632808
stru. 2
1 6 0 -1.116316 0.345733 1.022186
2 6 0 -1.536433 -0.170747 2.329719
3 6 0 -0.131558 -0.087658 0.235799
4 6 0 -0.756819 -0.672214 3.286420
5 1 0 -1.708487 1.160706 0.619385
6 1 0 -2.602395 -0.079825 2.519172
7 17 0 0.906363 -1.420285 0.586876
8 35 0 0.203005 0.726874 -1.439912
9 1 0 0.317456 -0.748797 3.180270
10 1 0 -1.187804 -1.006129 4.222287
TS1cout Br-Cl-ts Br-inward
Frequencies -- -618.3863 174.8776 208.9335
Sum of electronic and zero-point Energies= -3188.975916
Sum of electronic and thermal Energies= -3188.969898
Sum of electronic and thermal Enthalpies= -3188.968954
Sum of electronic and thermal Free Energies= -3189.007392
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.380282
3 6 0 1.369832 0.000000 -0.338722
4 6 0 1.243387 0.528790 1.771782
5 1 0 -0.809598 0.090385 -0.715199
6 1 0 -0.742839 -0.422881 2.048569
7 35 0 2.499957 -1.550374 -0.117031
8 17 0 1.872016 0.914565 -1.745465
9 1 0 1.557013 1.485287 1.378084
10 1 0 1.761019 0.203878 2.673344
.delta.E++ = 0.065451 a.u. = 41.0 kcal/mol
.delta.G++ = 0.065401 a.u. = 41.0 kcal/mol
IRC final structures
stru. 1
1 6 0 -1.843056 0.189414 0.234852
2 6 0 -1.900701 0.811767 1.412885
3 6 0 -0.382111 0.470583 0.064364
4 6 0 -0.434530 1.200313 1.440185
5 1 0 -2.535838 -0.337024 -0.406314
6 1 0 -2.701976 0.984491 2.119714
7 35 0 0.747027 -1.125134 0.038547
8 17 0 0.031339 1.517104 -1.321647
9 1 0 -0.218755 2.267330 1.376155
10 1 0 0.172660 0.734047 2.215927
stru. 2
1 6 0 -1.286954 1.073340 0.384872
2 6 0 -1.732020 0.673071 1.724372
3 6 0 -0.332088 0.540839 -0.375309
4 6 0 -0.959274 0.290560 2.738710
5 1 0 -1.833289 1.892000 -0.075577
6 1 0 -2.803605 0.757480 1.883520
7 35 0 0.725116 -0.952877 0.079897
19
8 17 0 0.013182 1.181682 -1.950311
9 1 0 0.118704 0.234203 2.655234
10 1 0 -1.397391 0.037182 3.696039
1d F-F
Frequencies -- 181.9022 332.0805 463.5757
Sum of electronic and zero-point Energies= -354.359699
Sum of electronic and thermal Energies= -354.354657
Sum of electronic and thermal Enthalpies= -354.353713
Sum of electronic and thermal Free Energies= -354.388125
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.334270
3 6 0 1.501968 0.000000 -0.052370
4 6 0 1.515273 -0.000165 1.491390
5 1 0 -0.756108 0.000407 -0.772637
6 1 0 -0.793129 0.000126 2.070711
7 9 0 2.078687 -1.084856 -0.631276
8 9 0 2.078564 1.085116 -0.630949
9 1 0 1.952968 0.897454 1.929566
10 1 0 1.952828 -0.897846 1.929597
TS1d F-F-ts
Frequencies -- -651.5445 246.0325 276.5491
Sum of electronic and zero-point Energies= -354.291584
Sum of electronic and thermal Energies= -354.286530
Sum of electronic and thermal Enthalpies= -354.285586
Sum of electronic and thermal Free Energies= -354.320066
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.385278
3 6 0 1.373756 0.000000 -0.285239
4 6 0 1.221186 0.569654 1.786696
5 1 0 -0.796031 0.104192 -0.726889
6 1 0 -0.739314 -0.443708 2.044663
7 9 0 2.181062 -1.043801 -0.070149
8 9 0 1.838978 0.617629 -1.369563
9 1 0 1.483165 1.547377 1.407980
10 1 0 1.735945 0.269472 2.697347
.delta.E++ = 0.068115 a.u. = 42.7 kcal/mol
.delta.G++ = 0.068059 a.u. = 42.7 kcal/mol
IRC final structures
stru.1
1 6 0 -1.278797 -0.297494 -0.086369
2 6 0 -1.321587 0.255698 1.127000
3 6 0 0.199841 -0.054513 -0.202882
4 6 0 0.164179 0.583754 1.202856
5 1 0 -1.997943 -0.749125 -0.755403
6 1 0 -2.124954 0.423146 1.832467
7 9 0 0.980684 -1.160169 -0.312987
8 9 0 0.588932 0.780487 -1.201020
9 1 0 0.424036 1.643012 1.214625
10 1 0 0.738179 0.036397 1.951555
stru.2
1 6 0 -0.913165 0.398558 -0.098420
2 6 0 -1.175673 0.345603 1.341538
3 6 0 0.192876 0.004303 -0.707436
20
4 6 0 -0.381173 -0.128040 2.303182
5 1 0 -1.677479 0.792605 -0.754378
6 1 0 -2.143845 0.744513 1.627177
7 9 0 1.255334 -0.502804 -0.119514
8 9 0 0.391714 0.063809 -2.008485
9 1 0 0.598148 -0.544027 2.106129
10 1 0 -0.711710 -0.104933 3.333881
1e Cl-Cl
Frequencies -- 160.1446 266.0103 267.7204
Sum of electronic and zero-point Energies= -1075.068917
Sum of electronic and thermal Energies= -1075.063124
Sum of electronic and thermal Enthalpies= -1075.062180
Sum of electronic and thermal Free Energies= -1075.099242
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.333130
3 6 0 1.497399 0.000000 -0.081701
4 6 0 1.510951 0.001042 1.475573
5 1 0 -0.745874 -0.000554 -0.782546
6 1 0 -0.790450 -0.000509 2.072314
7 17 0 2.208413 -1.459594 -0.832344
8 17 0 2.210712 1.456446 -0.835587
9 1 0 1.952342 0.897942 1.910626
10 1 0 1.953814 -0.894388 1.912144
TS1e Cl-Cl-ts
Frequencies -- -630.6632 186.9043 235.0402
Sum of electronic and zero-point Energies= -1075.002871
Sum of electronic and thermal Energies= -1074.997085
Sum of electronic and thermal Enthalpies= -1074.996141
Sum of electronic and thermal Free Energies= -1075.033258
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.380134
3 6 0 1.372497 0.000000 -0.331687
4 6 0 1.238017 0.541945 1.771819
5 1 0 -0.807978 0.091767 -0.716520
6 1 0 -0.741948 -0.424379 2.048688
7 17 0 2.408671 -1.413333 -0.117193
8 17 0 1.884666 0.912571 -1.737057
9 1 0 1.540993 1.501084 1.376276
10 1 0 1.758155 0.224129 2.674310
.delta.E++ = 0.066046 a.u. = 41.4 kcal/mol
.delta.G++ = 0.065984 a.u. = 41.4 kcal/mol
IRC final structures
stru. 1
1 6 0 -1.553392 -0.229938 0.256018
2 6 0 -1.594611 0.387883 1.436746
3 6 0 -0.085692 0.022672 0.082098
4 6 0 -0.119772 0.745099 1.461841
5 1 0 -2.258738 -0.737966 -0.386229
6 1 0 -2.390678 0.575482 2.145211
7 17 0 0.931542 -1.447831 0.034596
8 17 0 0.335955 1.075414 -1.300885
9 1 0 0.120364 1.807426 1.409524
10 1 0 0.479418 0.254616 2.229514
21
stru. 2
1 6 0 -1.034566 0.681189 0.389812
2 6 0 -1.474844 0.322376 1.740999
3 6 0 -0.044197 0.167677 -0.339391
4 6 0 -0.752857 -0.193566 2.737265
5 1 0 -1.616940 1.445596 -0.111417
6 1 0 -2.519481 0.547638 1.929167
7 17 0 1.011326 -1.100182 0.161449
8 17 0 0.269056 0.730524 -1.950694
9 1 0 0.302177 -0.413582 2.649655
10 1 0 -1.222147 -0.394515 3.692370
1f Br-Br
Frequencies -- 139.5995 170.0888 240.6337
Sum of electronic and zero-point Energies= -5303.013911
Sum of electronic and thermal Energies= -5303.007644
Sum of electronic and thermal Enthalpies= -5303.006700
Sum of electronic and thermal Free Energies= -5303.046422
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.334053
3 6 0 1.492783 0.000000 -0.083299
4 6 0 1.510275 -0.000875 1.475802
5 1 0 -0.746196 0.000421 -0.781993
6 1 0 -0.790470 0.000341 2.073528
7 35 0 2.256892 -1.596414 -0.906713
8 35 0 2.255492 1.598966 -0.903767
9 1 0 1.952465 0.894273 1.913752
10 1 0 1.951199 -0.897244 1.912553
TS1f Br-Br-ts
Frequencies -- -620.4469 161.4098 181.9885
Sum of electronic and zero-point Energies= -5302.948412
Sum of electronic and thermal Energies= -5302.942169
Sum of electronic and thermal Enthalpies= -5302.941225
Sum of electronic and thermal Free Energies= -5302.980953
1 6 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 1.380348
3 6 0 1.369427 0.000000 -0.339810
4 6 0 1.243946 0.527271 1.772062
5 1 0 -0.812227 0.091121 -0.711766
6 1 0 -0.743053 -0.422576 2.048860
7 35 0 2.501783 -1.546296 -0.130623
8 35 0 1.890961 1.018226 -1.877984
9 1 0 1.561121 1.481573 1.375726
10 1 0 1.760688 0.201756 2.673907
.delta.E++ = 0.065499 a.u. = 41.1 kcal/mol
.delta.G++ = 0.067469 a.u. = 42.3 kcal/mol
IRC final structures
stru. 1
1 6 0 -1.854231 -0.133776 0.579138
2 6 0 -1.902326 0.538028 1.730566
3 6 0 -0.394667 0.129558 0.389986
4 6 0 -0.433879 0.917469 1.734942
5 1 0 -2.553403 -0.682269 -0.035989

22
6 1 0 -2.700448 0.747481 2.431494
7 35 0 0.726280 -1.467615 0.415776
8 35 0 0.038470 1.214767 -1.173756
9 1 0 -0.210355 1.979478 1.632264
10 1 0 0.174059 0.474570 2.523887
stru. 2
1 6 0 -1.344525 0.711695 0.847152
2 6 0 -1.812611 0.217046 2.147835
3 6 0 -0.353050 0.250588 0.085875
4 6 0 -1.048910 -0.181287 3.162550
5 1 0 -1.900168 1.546721 0.429625
6 1 0 -2.891151 0.244744 2.277627
7 35 0 0.714161 -1.251676 0.486102
8 35 0 0.058974 1.085836 -1.561957
9 1 0 0.032095 -0.185473 3.104315
10 1 0 -1.497173 -0.503269 4.094658

download fileview on ChemRxivcycX-YNa9729d.pdf (920.45 KiB)