trace metal geochemistry and weathering mineralogy in a … · 2013-07-03 · trace metal...
TRANSCRIPT

Trace Metal Geochemistry and Weathering Mineralogy in a
Quaternary Coastal Plain, Bells Creek Catchment, Pumicestone Passage, Southeast Queensland, Australia
Tania Liaghati
Bachelor of Science (University of Urmia, Iran)
Postgraduate Diploma in Applied Science (Queensland University of Technology)
Master of Environmental Science (Griffith University)
School of Natural Resource Sciences
A thesis submitted for the Degree of Doctor of Philosophy of the Queensland University of Technology
2004

ii
STATEMENT OF ORIGINAL AUTHORSHIP
The work contained in this thesis has not been previously submitted for a degree or
diploma at any other higher education institution. To the best of my knowledge and
belief, the thesis contains no material previously published or written by another
person except where due reference is made.
Signed
Date

iii
TABLE OF CONTENTS
ABSTRACT............................................................................................................... vi
ACKNOWLEDGMENTS ........................................................................................ ix
PUBLICATIONS COMPRISING PhD STUDY..................................................... x
CONFERENCE SUBMISSION............................................................................... xi
INTRODUCTION...................................................................................................... 1
LITERATURE REVIEW .......................................................................................... 8
A BACKGROUND TO GEOCHEMISTRY ........................................................... 9
WEATHERING OF THE BEDROCK.................................................................. 11
INTRODUCTION........................................................................................................ 11 FACTORS CONTROLLING CHEMICAL WEATHERING.................................................. 12
Parent material .................................................................................................. 12 Topography ........................................................................................................ 14 Climate ............................................................................................................... 15 Time.................................................................................................................... 16 Vegetation .......................................................................................................... 16
PRODUCTS OF CHEMICAL WEATHERING ................................................................. 16 CLAY MINERALS..................................................................................................... 17 IRON MINERALS......................................................................................................20
METHODOLOGIES FOR ASSESSING WEATHERING PROFILES WIT H REGARD TO TRACE METALS........................................................................... 22
CALCULATION OF CHEMICAL AND MINERALOGICAL INDICES................................. 22 MASS BALANCE CALCULATIONS ............................................................................ 23
SEDIMENTARY ENVIRONMENTS, THEIR PROPERTIES AND GEOCHEMISTRY .................................................................................................. 25
INTRODUCTION........................................................................................................ 25 PROPERTIES OF SEDIMENTARY MATERIAL .............................................................. 26 GEOCHEMISTRY OF SEDIMENTARY SETTINGS......................................................... 27 COASTAL MARINE SEDIMENTARY ENVIRONMENTS ................................................ 28 PYRITIC SEDIMENTS AND TRACE METALS .............................................................. 29
Background and definition................................................................................. 29 formation and Morphology ................................................................................ 30
TRACE METALS.................................................................................................... 31
OCCURRENCE.......................................................................................................... 31 MOBILITY ............................................................................................................... 32 FACTORS INFLUENCING METAL MOBILITY ............................................................. 34
SEDIMENT pH and Eh ...................................................................................... 34 Salinity and Formation of organic and inorganic complexes............................ 35
TRACE METALS AND ENVIRONMENTAL IMPACTS ............. .................... 37
INTRODUCTION........................................................................................................ 37 SURFACE WATER QUALITY AND METALS................................................................. 37
Background ........................................................................................................ 37

iv
inorganic removal in estuaries........................................................................... 39 FACTORS AFFECTING CHEMICAL COMPOSITION OF NATURAL WATERS.................... 40
introduction........................................................................................................ 40 Acidity and redox ............................................................................................... 40 Rock Type........................................................................................................... 41 Relief .................................................................................................................. 42 Time.................................................................................................................... 42 Aluminium .......................................................................................................... 42 Iron and Manganese .......................................................................................... 42
ANALYSIS OF HETEROGENOUS GEOCHEMICAL DATASETS ...... ......... 43
NORMALISATION ..................................................................................................... 43 STATISTICAL ANALYSES ......................................................................................... 45
COMPARABILITY OF ANALYTICAL METHODS................ ......................... 45
COMPARABILITY OF TOTAL DIGESTION METHOD WITH XRF.................................. 45 COMPARABILITY OF AQUA REGIA AND HF-BASED DIGESTION............................... 46
CONCLUSIONS ...................................................................................................... 47
REFERENCES: ....................................................................................................... 49
PAPER 1 - THE INFLUENCE OF MINERALOGY AND GEOLOGICA L SETTING ON TRACE METAL CONCENTRATION WITHIN SUBTROPICAL WEATHERED PROFILES, BELLS CREEK CATCHME NT, QUEENSLAND, AUSTRALIA .............................................................................. 61
PAPER 2 – GEOCHEMICAL METHOD FOR CHARACTERISATION O F SUBTROPICAL WEATHERING AND METAL RELEASE WITHIN SEDIMENTARY BEDROCK: QUEENSLAND, AUSTRALIA......... ................ 94
PAPER 3 - HEAVY METAL DISTRIBUTION AND CONTROLLING FACTORS WITHIN COASTAL PLAIN SEDIMENTS, BELLS CREEK CATCHMENT, SOUTHEAST QUEENSLAND, AUSTRALIA ......... ............. 127
PAPER 4 - DISTRIBUTION OF FE IN WATERS AND BOTTOM SEDIMENTS OF A SMALL TIDAL CATCHMENT, PUMICESTONE REGION, SOUTHEAST QUEENSLAND, AUSTRALIA................................. 163
GENERAL CONCLUSIONS................................................................................ 189
APPENDIX 1 - CHEMICAL WEATHERING PROCESSES IN A SUBTROPICAL COASTAL CATCHMENT AS INDICATED BY SPATI AL VARIATIONS IN TRACE ELEMENTS AND MINERALOGY, SOUTHE AST QUEENSLAND, AUSTRALIA ............................................................................ 195
APPENDIX 2 - DETERMINATION OF QUATERNARY SEDIMENT SOURCES USING MINERALOGY AND GEOCHEMISTRY IN BELLS CREEK CATCHMENT, PUMICESTONE PASSAGE, SOUTHEAST QUEENSLAND...................................................................................................... 199
APPENDIX 3 - SPATIAL VARIATION OF HEAVY METALS WITH IN SURFICIAL SEDIMENTS OF A SUBTROPICAL COASTAL PLAIN, BELLS CREEK CATCHMENT, SOUTHEAST QUEENSLAND, AUSTRALIA ... .... 202

v
APPENDIX 4 - MORPHOLOGICAL VARIATIONS OF FRAMBOIDAL PYRITE IN AN ESTUARINE ENVIRONMENT, PUMICESTONE CATCHMENT, SOUTHEAST QUEENSLAND, AUSTRALIA ......... ............. 210
APPENDIX 5 – SILICATE ROCK ANALYSIS (MAJOR OXIDES, LOSS ON IGNITION AND SULFUR) ..................................................................................215
APPENDIX 6 - TOTAL TRACE METAL ANALYSIS OF SEDIMENT BY HYDROFLUORIC ACID (UNIVERSITY OF QUEENSLAND) ....... .............. 222
APPENDIX 7 - TOTAL ELEMENT ANALYSIS BY X-RAY FLUORESCENCE SPECTROMETRY (XRF) (JAMES COOK UNIVERS ITY).................................................................................................................................. 224
APPENDIX 8 – EXTRACTABLE CATIONS IN SEDIMENTS ...... ................ 226
APPENDIX 9 - ORGANIC CARBON BY W ALKEY-BLACK METHOD .... 229
APPENDIX 10 - X-RAY DIFFRACTION ANALYSIS ........... .......................... 232
APPENDIX 11 - CATIONS IN WATER, INDUCTIVELY COUPLED PLASMA- OPTICAL EMISSION SPECTROSCOPY (ICP-OES).................. 235
APPENDIX 12 - ANIONS IN WATER BY ION CHROMATOGRAPHY (IC).................................................................................................................................. 238
APPENDIX 13 – TITRATION METHOD FOR ALKALINITY...... ................ 241
APPENDIX 14 – ADDITIONAL LABORATORY ANALYSIS DATA.. ......... 245

vi
ABSTRACT The Bells Creek catchment covers an area of 100 km2 in the northern part of the Pumicestone Passage region of southeast Queensland. This catchment is an example of a low-lying sub-tropical coastal plain including both freshwater and estuarine settings. The main creeks drain into Pumicestone Passage, a large shallow estuary, which is a declared marine habitat and a Ramsar listed wading bird location. The Bells Creek catchment has undergone land-use change from bushland to grazing to pine plantations and is now coming under pressure for urban development. Quaternary age unconsolidated sediments are the dominant surface material in this area and formed during the last marine transgression. Of significance for such a setting is that estuarine sediments can retain metals mobilised as a result of natural processes (e.g. weathering) and anthropogenic activities (e.g. land-use disturbance). As trace metals can also occur naturally in rocks and their weathered products, it is of value to clearly distinguish natural and anthropogenic controls over metal source, distribution and mobility. To achieve this aim two approaches were taken: 1) to determine the factors controlling the geochemistry of weathered profiles, unconsolidated sediments, soils and natural waters, and 2) to identify the most effective analytical and numerical methods for evaluating metal concentration in different solid materials. This investigation is structured around four linked papers. The influence of mineralogy, geological setting, location of water table and depth of burial on the geochemistry of weathered profile are assessed in Paper 1. The second paper is an investigation of different analytical approaches for studying weathered sedimentary rocks, as well as the testing of several numerical methods for evaluating geochemical data from weathered profiles. In paper 3, a large heterogeneous geochemical data set including trace metals, total organic carbon and sulfur content, in addition to mineralogy and land use practices are integrated to enable evaluation of geochemical and anthropogenic processes controlling metal distribution. The fourth paper considers the distribution of iron and its transport as well as variations in size and morphology of different forms of framboidal pyrite within a smaller sub-catchment in the southern part of the study area. The labile and heterogeneous nature of the bedrock of the region, the Landsborough Sandstone, along with the sub-tropical climate of the area have resulted in weathering profiles up to 26 m deep. Due to the absence of industrial activity in the Bells Creek catchment, such weathering of the bedrock constitutes the major process governing metal distribution throughout the area. Analysis by X-ray diffraction (XRD) shows that the primary minerals occurring in the weathered profiles are quartz, plagioclase and K-feldspars while kaolinite is the most dominant secondary mineral present. Overall, parent rock silicates have been extensively replaced by clay minerals and Fe oxides. The relative influence of mineralogy, geological setting and groundwater over chemical weathering and geochemical cycling of metals can be summarised as follows:
mineralogy>geological setting>watertable position>depth of profile burial

vii
As the relationship between the total metal composition and the extractable and mobile component has environmental significance, a comparison was made between these forms of metals in weathered material. This comparison shows that metals such as V, Cr and Fe are part of the aluminosilicate matrix and remain largely in primary mineral structures. The retention of these metals may lead to their future release to the environment during on-going weathering. Other elements such as Cu, Zn, Pb, however, are found to be primarily adsorbed to sediment particles and therefore, easily releasable to the environment. As limited information on weathering of sedimentary rocks is reported in the literature, a variety of chemical analysis and numerical assessment methods were used to understand the geochemical processes involved in trace metal mobility in the weathered profiles. Two analytical methods of digestion, hydrofluoric acid and x-ray fluorescence were tested and found to be highly comparable except for refractory elements such as V and Cr. Among the numerical methods applied to the dataset were “chemical and mineralogical indices”, “weight loss factor” and “immobile element approach”. The “immobile element approach” was found to be the most appropriate method to characterise the weathering profiles typical of the catchment. This method considers a weathering system to be open and transforms the absolute values of trace metals enabling a quantitative evaluation of metal mobility. The following sequence of mobility was determined after applying this method to the data generated in this study:
Zn>Pb>Cu>Cr>V
The above sequence of mobility is supported by the comparison between extractable and total metal concentrations where Cr and V were identified as being part of aluminosilicate matrix and less mobile. On the other hand, Zn, Pb and Cu were found to exist in adsorbed form and to be readily released to the environment. Trace elements released through weathering and erosion of the bedrock can accumulate in estuarine and coastal sediments. Therefore, both the lateral and vertical distribution of trace metals within sediments and soils of Bells Creek catchment were investigated. Natural and anthropogenic factors controlling metal distribution were compared and it was concluded that the natural sediment character such as its mineral content is more significant than anthropogenic influences in controlling lateral and vertical metal distribution. Further, due to varying degrees of weathering and the heterogeneous nature of soils and sediments, the data were normalised. After testing several methods, it was concluded that calculation of an enrichment factor was the most appropriate. The enrichment factor revealed that elevated trace metal concentrations at some sites are due to bedrock weathering. Due to the environmental persistence of iron, excess of this common metal has always been of environmental concern in many coastal settings. In the small Halls Creek sub-catchment, for example, iron anomalies were detected in bottom sediments (Fe up to 14%). This finding has significance in the area, as iron has been identified as one of the major contributors in the growth of the toxic cyanobacteria “Lyngbya majuscula” which can negatively impact on aquatic fauna. Iron concentrations were also shown to be high in natural stream waters of this coastal zone (up to 16 mg/L); in the bottom sediments of the creek, iron occurs as hematite

viii
(freshwater section) or pyrite (estuarine section). A variety of pyrite morphologies were identified in both bottom sediments and particulate matter samples including spherical closely packed framboids, and the rare form of euhedra which indicates slow crystallisation. The different components of this investigation have: 1) established the order and extent to which natural factors control weathering, 2) tested a number of analytical and numerical methods in evaluating weathering profiles, 3) assessed natural and anthropogenic factors and established the mobility sequence for trace metals in weathered profiles and, 4) determined the iron mineral speciation and established morphological variations of pyrite. As the area of Bells Creek catchment will be under development pressure in the future, findings of this study represent a baseline of comparison for environmental assessment and are of importance for environmental management.

ix
ACKNOWLEDGMENTS
• The guidance, encouragement and inspiration I have received from my supervisors Dr Malcolm Cox and Dr Micaela Preda have made the completion of this project possible. I would like to thank them sincerely for their willingness to share their knowledge with me. I also wish to thank Micaela for her patience in teaching me technical methods for data interpretations, which made the publication of the findings possible. I would also like to thank Associate Professor Peter Mather whom I always consider as my very first mentor in my professional life in Australia.
• I would like to thank Lensworth Group Pty Ltd for financial assistance; without their support this project would not have been possible.
The practical and professional assistance of many people and institutions have
contributed in the successful completion of this project. Their contribution is gratefully acknowledged:
• My colleague Tim Ezzy assisted with fieldwork, provided samples and mapping information.
• Rob King assisted with fieldwork and provided valuable local knowledge. • Graham Kimber is greatly appreciated for his inputs regarding data quality
control procedures. • Bill Kwiecien, Wathsala Kumar provided practical assistance with chemical
analysis and Tony Raftery assisted with mineralogical analyses. • Dr Alan Craig (the Advanced Centre for Queensland University Isotope
Research Excellence) carried out total digestion analysis of trace elements. • Dr Sharon Ness carried out major and trace element analysis by XRF
(Advanced Analytical Laboratory, James Cook University, Townsville). • Dr Thor Bostrom and Mr Loc Duong assisted the electron microscopy work. • Hayden McDonald from Mipela provided the GIS database. • Queensland Acid Sulfate Soils Investigation Team (QASSIT) provided soil
samples. • Travel grants from QUT Office of Research and the School of Natural
Resource Sciences provided the opportunity to attend conferences in Adelaide, Grenoble-France and Hobart, which has been beneficial to my research and professional development.
• I would also like to thank staff of the School of Natural Resource Sciences, particularly Mark Crase who helped me in many ways.
• Dr Theo Kloprogge for his constructive comments on the thesis is also gratefully acknowledged.
• Finally, I would like to thank my husband Mehdi and daughter Panthea for their patience and support without which I could not have successfully completed this research.

x
PUBLICATIONS COMPRISING PHD STUDY Paper 1:
Liaghati, T., Preda, M. and Cox, M.E. 2003. The influence of mineralogy and
geological setting on trace metal concentration within subtropical weathered
profiles, Bells Creek catchment, Queensland, Australia. Submitted to Pacific
Science
Paper 2:
Liaghati, T., Preda, M. and Cox, M.E. 2003. Geochemical methods for
characterisation of subtropical weathering and metal release within
sedimentary bedrock: Queensland, Australia. Submitted to Journal of
Geochemical Exploration.
Paper 3:
Liaghati, T., Preda, M. and Cox, M.E. 2003. Heavy metal distribution and controlling
factors within coastal plain sediments, Bells Creek catchment, southeast
Queensland, Australia. Environment International, 29: 935-948
Paper 4:
Liaghati, T., Cox, M.E. and Preda, M. 2004. Distribution of Fe in waters and bottom
sediments of a small tidal catchment, Pumicestone Region, southeast
Queensland, Australia. Accepted for publication in The Science of Total
Environment

xi
CONFERENCE SUBMISSIONS
Conference Abstracts
Liaghati T., Preda M. and Cox M. E. 2002. Determination of Quaternary sediment
sources using mineralogy and geochemistry in Bells Creek catchment,
Pumicestone Passage, southeast Queensland. In Preiss V.P. (ed) Proceedings
of the International Conference on Geoscience: Expanding Horizons,
Adelaide, South Australia, July 1-5 2002, Geological Society of Australia
Incorporated, p 457.
Liaghati T., Preda M. and Cox M. E. 2003. Chemical weathering process in a
subtropical coastal catchment as indicated by spatial variations in trace
elements and mineralogy, southeast Queensland, Australia. In The
International Conference on “The Impact of Global Environment Problems
on Continental and Coastal Marine Waters, Geneva, Switzerland 16-18 July
2003”, Centre d’Etudes en Sciences Naturelles de l’Environment and the
Institut F. A. Forel, University of Geneva, p 35-36.
Liaghati T., Cox M. E. and Preda M. 2003. Morphological variations of framboidal
pyrite in an estuarine environment, Pumicestone catchment, southeast
Queensland, Australia. In “The 17th Australian Geological Convention
February 2004, Hobart, Tasmania” Dynamic Earth: Past, Present and Future,
Abstracts 73, Geological Survey of Australia Incorporated, Sydney, p 29.
Refereed Conference Paper
Liaghati T., Preda M. and Cox M. E. 2003. Spatial distribution of heavy metals
within surficial sediments of a subtropical coastal plain, Bells Creek
catchment, southeast Queensland, Australia. In Boutron, C. and Ferrari, C.
(eds.) Journal De Physique IV “XIIth International Conference on Heavy
Metals in the Environment, Grenoble France 26-30 May 2003”, EDP
Sciences, Vol 2: p 773-776.

1
INTRODUCTION The Pumicestone Passage is an environmentally important waterway being a
declared marine habitat and a Ramsar listed wading bird location. During flood
events, a large input of suspended material can be carried by the ten creek systems
that discharge into the Passage providing the potential to transport nutrients and
metals into the water system and affect local water quality. The Bells Creek
catchment is located within the northern part of the Pumicestone Catchment and
drains into a narrow section of Pumicestone Passage.
An important control over the character of sedimentary material deposited in coastal
plains is the variation of sea level during the Quaternary. During the low sea level of
the Pleistocene period (~ 2 Ma), the eastern Australian continental shelf was exposed
and incised by river systems. Subsequent rises in sea level caused the drowning of
river channels and development of estuaries and coastal plains. In southeast
Queensland, the highest Holocene sea level was reached about 6,500 years ago
(Williams et al., 1998); in the Pumicestone region, there is evidence that the sea level
retreated to its present position around 3,000 years BP (Flood, 1981; Lester, 2000).
This region is part of an intracratonic basin, which accommodated a large fluvial
system during the Early to Middle Mesozoic (Cranfield, 1983; Murphy et al., 1987).
A variety of pre-existing, largely volcanic rocks were eroded and supplied the fluvial
system with material that was eventually incorporated in the Landsborough
Sandstone. This formation, which today represents the bedrock of the region,
consists of quartzo-feldspathic sandstone with lithic fragments of volcanic origin and
layers of shale, conglomerate and coal (e.g. Murphy et al., 1987). The heterogenous
character of the bedrock formation and the lability of the sandstone have led to the
development of thick weathering profiles most likely due to exposure during the
Quaternary (Ezzy et al., 2002).
Chemical weathering of rocks is one of the major processes in the geochemical
cycling of elements (e.g. Faure, 1998). An understanding of the processes of rock
weathering and sediment formation requires not only a sound knowledge of the
geochemical behaviour of elements during weathering, but also of sediment

2
redistribution processes. Identification of such processes can provide fundamental
information for environmental management, especially in coastal regions that are
under development pressure.
Furthermore, estuarine and marine sediments are sinks for various metals transported
from the adjacent landmass. Metals may be mobilised as a result of natural processes
(e.g. weathering and erosion of geological formations) as well as by anthropogenic
activities (e.g. land use practices). In the mobilisation process, trace elements may
be adsorbed by clays, can complex with organic compounds or may co-precipitate
with oxides and hydroxides. As many metals occur naturally in weathered materials
and drainage systems due to their presence in local rocks, the relative influence of
natural and anthropogenic sources on the geochemistry of coastal sediments is not
always clear. Therefore, a systematic assessment of metal distributions within such
environment requires discriminiation between metallic elements released by natural
processes and those introduced by human-related activities. The amounts of trace
elements in natural systems can be of environmental significance because where
elevated they may degrade surface water and shallow groundwater. In addition,
marine organisms and vegetation in coastal environments can uptake metals,
increasing the potential for the inclusion of some metals into the food chain.
The Bells Creek catchment and its adjacent estuarine plain (Figures 1&2) form a
typical setting where a range of both natural and human-induced influences interact
to produce the overall characteristics of the drainage system.
It is well established that factors such as geological setting and mineralogy have a
strong influence over the chemistry of stream and marine sediments and on natural
waters, which in turn influence trace metal distribution. However, the extent to
which these factors interact within active sedimentary processes has received only
limited attention. This study employs a variety of analytical and numerical
approaches to establish trace metal occurrence, and the natural and anthropogenic
factors controlling processes of distribution.

3
In order to understand the interaction between the various components of these
drainage systems and test the potential environmental impact, a program of sampling
and analyses was designed to achieve the following objectives:
1. establish the extent to which natural controls (e.g. mineralogy and geological
setting) influence trace metal concentrations within a weathered profile.
2. identify the most appropriate conceptual and analytical methods to assess
weathering and subsequent metal release from sedimentary rocks.
3. understand geochemical and anthropogenic processes that control trace metal
distribution in bedrocks, soils and sediments.
4. investigate iron distribution patterns in natural waters and bottom sediments.
270
Moreton Bay
Brisbane River
PumicestonePassage
Caboolture River
Pine River
0 10 20 30
kilometers
Bribie Island
Moreton Island
North Stradbroke Island
Redcliffe
153000’ 153030’152030’
BrisbaneCity
N
Deception Bay
Bells Creek Catchment
Figure 1: Location of the study area in relation to Moreton Bay and Pumicestone region.
Queensland
270
Moreton Bay
Brisbane River
PumicestonePassage
Caboolture River
Pine River
0 10 20 30
kilometers
Bribie Island
Moreton Island
North Stradbroke Island
Redcliffe
153000’ 153030’152030’
BrisbaneCity
N
Deception Bay
Bells Creek Catchment
Figure 1: Location of the study area in relation to Moreton Bay and Pumicestone region.
Queensland
Moreton Bay
Brisbane River
PumicestonePassage
Caboolture River
Pine River
0 10 20 30
kilometers
Bribie Island
Moreton Island
North Stradbroke Island
Redcliffe
153000’ 153030’152030’ 153000’ 153030’152030’
BrisbaneCity
NN
Deception Bay
Bells Creek Catchment
Figure 1: Location of the study area in relation to Moreton Bay and Pumicestone region.
Queensland

4
Figure 2: Aerial photo of the Bells Creek catchment with its land use practices
Glass House Mountains
Golden Beach
Pine Plantations
Bells Creek
Pumicestone Passage
Golf Course
Native Vegetation

5
This research project consists of four papers, which aim to achieve the above
objectives. Other outcomes of research activities include a number of posters and
conference abstracts (see appendices), which report preliminary results at the time of
presentation.
The influence of mineralogy, geological setting, location of water table and depth of
burial on the geochemistry of weathered profiles is assessed in Paper 1 “The
influence of mineralogy and geological setting on trace metal concentration within
subtropical weathered profiles, Bells Creek catchment, Queensland, Australia”. In
addition, a comparison is made between total and extractable metal contents based on
two methods of digestion namely, aqua regia and hydrofluoric acid; this compares
availability of elements within primary aluminosilicate matrices to metals that are
largely adsorbed to particles. A comparison between the total metal composition and
the extractable and mobile component has environmental significance and is not
commonly reported in the published literature.
Paper 2 “Geochemical methods for characterisation of subtropical weathering and
metal release within sedimentary bedrock: Queensland, Australia.” investigates two
analytical methods for studying weathered sedimentary rocks. In addition, several
numerical methods were tested for evaluating geochemical data from weathered
profiles. Typically, few such studies are reported for sedimentary rocks due to either
their lesser abundance or their limited economic value; this paper is a significant
contribution in evaluating analytical and assessment methods for sedimentary
weathering products.
Paper 3 “Heavy metal distribution and controlling factors within coastal plain
sediments, Bells Creek catchment, southeast Queensland, Australia” gathers a large
heterogeneous geochemical data set including trace metals, total organic carbon and
sulfur content, as well as mineralogy and land use practice, to evaluate geochemical
and anthropogenic processes controlling metal distribution.
Iron is an important metal adsorbent and it is also identified as one of the main
elements supporting the growth of the toxic algae Lyngbya majuscula, in
Pumicestone region; however, a geochemical and mineralogical study focusing on

6
iron species and their source is lacking. Therefore, paper 4 “Distribution of Fe in
waters and bottom sediments of a small tidal catchment, Pumicestone Region,
southeast Queensland, Australia” aims to identify the source and establish the
process of iron transport through bottom sediments, surface, groundwater and by
suspended matter within Halls Creek sub-catchment.
In summary, the study of this subtropical catchment is designed to determine two
aspects: (a) the natural and anthropogenic factors controlling geochemistry of
weathered profiles, sediments, soils and natural waters of the area, and (b) identify
the best analytical and numerical methods for evaluating metal concentrations in
weathered profiles and large heterogeneous geochemical data sets.
References
CRANFIELD L.C. 1983. Shallow stratigraphic drilling in the Brisbane 1:100,000
Sheet area. Record 40, Geological Survey of Queensland.
EZZY T.R., COX M.E. and BROOKE B. 2002. The influence of stratigraphy on the
occurrence and composition of groundwater within a coastal valley-fill:
Meldale, south-eastern Queensland. In: HAIG T., KNAPTON A., GEORGE
D. and TICKELL S. (eds.). Balancing the groundwater budget, CD of
conference proceedings, International Association of Hydrogeologists
groundwater conference, 12-17 May, Darwin, Australia, pp. 6.
FAURE G. 1998. Principles and application of geochemistry, 2nd edition, Prentice
Hall, New Jersey, pp. 600.
FLOOD P.G. 1981. Carbon-14 dates from the coastal plains of Deception Bay,
south-eastern Queensland. Queensland Government Mining Journal, 82: 19-
23.
LESTER J. 2000. Geomorphology, sedimentology and shoreline processes impacting
on the stability of the Bribie Island Spit. Honours thesis, School of Natural
Resource Sciences, QUT.
MURPHY P.R., TREZISE D.L., HUTTON L.J. and CRANFIELD L.C. 1987.
1:100,000 Geological Map Commentary. Caboolture, Sheet 9443,
Queensland Department of Mines, Geological Survey of Queensland.

7
WILLIAMS M., DUNKERLEY D., DE DECKKER P., KERSHAW, P. and
CHAPPELL J. 1998. Quaternary environments. Arnold, Hodder Headline
Group, London.

8
LITERATURE REVIEW

9
A BACKGROUND TO GEOCHEMISTRY
Geochemistry is the application of the principles of chemistry to solve geological
problems. Geochemistry as defined by Goldschmidt and summarised by Mason
(1958, p. 2) is concerned with (1) the determination of the relative and absolute
abundance of elements in the earth, and (2) the study of distribution and migration of
the elements in the various parts of the earth to discover philosophies governing this
distribution and migration. In a more recent definition by Faure (1998), there are
four major goals in geochemistry:
• To understand the distribution of the chemical elements in the Earth and in the
solar system.
• To determine the origin of the observed chemical composition of terrestrial and
extraterrestrial materials.
• To study chemical reactions on the Earth, its interior and in the solar system.
• To congregate these findings into geochemical cycles and to explore their
operational systems in the past and how they may be altered in the future.
Mineral exploration is one of the oldest applications of geochemistry and is defined
as systematic measurement of one or more chemical properties of a naturally
occurring material to locate hidden ores (Hawkes and Webb, 1962). Environmental
geochemistry is a more recent form of geochemistry, which is focused on monitoring
the dispersion of metals and various organic compounds that have anthropogenic
sources. Since the middle of the 20th century, geochemistry has become diversified
into several subdivisions, among them inorganic and organic geochemistry,
cosmochemistry, isotope geochemistry, geochemical prospecting, medical
geochemistry, aqueous geochemistry and trace element geochemistry. This review
will present a summary of relevant literature on aqueous and trace metal
geochemistry.
Environmental geochemistry is of a great importance as it contributes to the
continued well being of human kind and assists in the development and management
of natural resources. The understanding of processes occurring on the Earth will
enable us to monitor the quality of the environment both locally and on a global

10
scale, and to warn humanity against dangerous practices that may threaten the quality
of life in the future (Faure, 1998).
On the basis of variables such as pressure, temperature and the availability of the
most abundant chemical component, it is possible to classify all the natural
environments of the earth into two major groups – primary and secondary (Hawkes
and Webb, 1962). The primary environment is characterised by high temperature
and pressure, restricted circulation of fluids and relatively low free oxygen content;
these conditions occur deep into the earth where most rocks form. The secondary
environment is of low temperatures, nearly constant low pressure, free movement of
solutions containing free oxygen, water and CO2. This latter environment is where
weathering, erosion and sedimentation at the surface of the earth take place.
One of the objectives of this study is to investigate the geochemical and
mineralogical composition of bedrock, soil and sediments of the study area; a brief
review on weathering therefore, is essential to explain the significance of a detailed
chemical and mineralogical analysis of the material throughout the field study area.
As the investigation also focuses on establishing occurrence and distribution of
heavy metals, it is important to understand the geochemical character of the
sedimentary environment. This understanding in turn will help to confirm the source
and distribution, adsorption and mobilisation of trace metals. Identifying and
understanding the geochemistry of post depositional processes associated with metal
occurrence in the Pumicestone Passage is another objective of the study; a brief
background about environmental geochemistry and geochemical cycles is also
presented. Trace elements in the sediments are a potential source of contamination
for surface and shallow groundwater; a summary of the environmental implications
of trace metal presence within such waters is therefore required. As the trace metal
geochemical dataset from coastal sediments produced in the current study is typically
heterogeneous, this review also discusses the approaches to assessment and analysis
of such datasets. There is no consensus about the most appropriate analytical
technique to determine trace metal contents in soils and sediments, therefore, a
variety of procedures have been assessed in the final part of this review.

11
The literature review outlines:
� Chemical weathering and environmental factors influencing it
� Products of chemical weathering
� Methodologies for assessing weathering profiles with regard to trace metals
� Sedimentary environments, their properties and geochemistry
� Trace metal occurrence and mobility
� Trace metals and water quality
� Analysis of heterogeneous geochemical datasets
� Comparability of analytical methods for environmental samples
WEATHERING OF THE BEDROCK
Introduction
Natural processes such as weathering and erosion of the land surface as well as
anthropogenic activities can result in a major input of heavy metals into the coastal
and estuarine environments. Iron, Mn, organic C and S act as metal scavengers in
transportation and deposition of trace elements into the sediments (e.g. Förstner and
Muller, 1973). Many metals occur naturally in weathered materials and drainage
systems due to their presence in local rocks; therefore, in order to understand
distribution and mobility of these elements on the earth surface, it is essential to
identify natural sources of metals and gain knowledge of weathering and associated
processes, which is explained below.
The upper 15 km of the lithosphere is comprised of 95% igneous rocks, 4% shales,
0.75% sandstones, and 0.25% limestones (Carroll, 1970). Only 30% of lithosphere is
dry land of which only the upper surface is affected by some degree of chemical
decomposition or physical weathering (Carroll, 1970). The weathering of rocks
composing the lithosphere occurs through chemical, physical and biological
processes. Details of these processes are not the focus of this review and can be
found in books about weathering and soil formation such as those by (Goldich, 1938;
Colman and Dethier, 1986; Lerman and Maybeck, 1988; Nahon, 1991; Berner and
Berner, 1996; Bland and Rolls, 1998).

12
As it is important to understand variations of major and trace elements related to
chemical and mineralogical changes during intense weathering in a subtropical
environment, products of, and factors controlling chemical weathering will be
discussed in more detail in the following sections.
Factors controlling chemical weathering
The nature of chemical weathering varies widely and is governed by many variables
such as parent rock type, topography, climate, time and vegetation. As the extent of
weathering is a controlling factor on trace metal distribution and mobilisation the
influence of mineralogy and geological setting on trace metal concentration within
weathered profiles has been investigated in the paper “The influence of mineralogy
and geological setting on trace metal concentration within subtropical weathered
profiles, Bells Creek catchment, Queensland, Australia” . The mobilisation and
redistribution of trace elements during weathering is particularly complex because
these elements are affected by various processes such as dissolution of primary
minerals, formation of secondary phases, redox processes, transport of materials,
coprecipitation and ion exchange (e.g. Nesbitt, 1979; Chesworth et al., 1981; Cramer
and Nesbitt, 1983).
PARENT MATERIAL
While the ultimate composition of weathered bedrock formed by near-surface
processes is related to the composition of the source rock, studies have shown that
both chemistry and mineralogy of the weathered profile may differ greatly from
those of the bedrock on which it forms. For example Boggs (1995) demonstrated
that the composition of weathered bedrock is controlled not only by the parent-rock
but also by the nature, intensity and duration of weathering and soil-forming
processes. Furthermore, where there are extreme differences between chemistry and
mineralogy of the weathered profiles and their parent material, in situ chemical
weathering has been accompanied by additional subtractions such as colluvial and /or
alluvial addition that contributed in weathering.
Variations in texture, structure and composition of bedrock can exert a significant
influence on the rate of leaching. Texture affects permeability and therefore, the
degree of penetration of rainwater into the rock. For instance, loose sands are

13
particularly permeable and the soluble constituents can readily leach from areas
above the water table. On the other hand, clayey soils tend to inhibit penetration of
water and increase the loss through surface runoff. In consolidated rocks, fractures
and zones of weakness such as joints, faults, and cleavage offer easy access to water
and accelerate the leaching process as well as providing channels for the subsurface
drainage. In addition, through the development of a mature weathered zone, the rate
of weathering may alter (Loughnan, 1969).
Profiles with different parent material also weather differently. Sedimentary rocks
weather more readily than igneous and metamorphic rocks due to hydrodynamic
processes. For chemical weathering to occur to a significant degree, water must
circulate through the rock. This is more conductive in open structure of sedimentary
rocks compared to most igneous and metamorphic rocks. There are some exceptions
such as the exclusive Carboniferous limestone of England and Wales that is resistant
to weathering (Macias and Chesworth, 1992). When sedimentary rocks are
compared, mudstone has been found to deteriorate to a larger extent compared to
sandstone because the large amount of Fe in both sedimentary rocks behaves
differently. While sandstone is strengthened because of cementation by iron oxides
or hydroxides, mudstone is weakened because it contains a greater amount of clay
size fractions with larger specific surface area than sandstone (Chigira and Sone,
1991; Chigira and Oyama, 1999). Different types of rocks have different mineralogy
and thus chemical character, therefore, influencing the local weathering environment
as they breakdown. For instance, the feldspars in a granite weather to produce a
solution containing K+, Na+, Ca2+, consuming hydrogen ions in the process, thus the
weathering solution becomes more alkaline. Sulfide minerals however, weather by
oxidation, converting the sulfur to sulfuric acid with a marked increase in acidity of
the water (Taylor and Eggleton, 2001).
In areas with homogeneous climates, where sedimentary covering is sparse and
lithological variability pronounced, parent rock has a significant role on weathering.
However, in an edaphological zone (the unconsolidated mineral material on the
immediate surface of the earth that serves as a natural medium for the growth of land
plants) with a relatively homogeneous bedrock type, the parent rock effect can be
masked by the effect of climate and vegetation (Macias and Chesworth, 1992).

14
TOPOGRAPHY
Topography affects the rate of chemical weathering and the nature of the weathered
products by controlling: 1) the rate of surface runoff of rain water and therefore, the
rate of moisture intake by the parent rock, 2) the rate of subsurface drainage and
hence the rate of leaching of the soluble constituents, and 3) the rate of erosion of the
weathered products and thereby the rate of exposure of fresh mineral surfaces.
On very steep slopes most of the rainwater is lost due to surface runoff and at the
same time physical weathering occurs by running water, wind and landslides.
Hence, in such environments, physical disintegration of rocks proceeds at a much
greater rate than chemical breakdown and any accumulation of secondary minerals is
superficial. In low-lying lands such as coastal plains, surface runoff is minimal and
infiltration of rainwater is at a maximum rate. In this type of environment subsurface
drainage is sluggish and soluble products released by hydrolysing reactions are
preserved, thus preventing further breakdown of parent material. The ideal condition
for chemical weathering is attained on gently sloping uplands where surface runoff is
not excessive and the subsurface drainage is unrestrained. Under such condition, the
weathered zone may extend to a depth of 30 m or more (e.g. Jenny, 1941; Loughnan,
1969; Taylor and Eggleton, 2001). Thus, although landscape position is important,
the degree of importance depends on drainage characteristics.
Groundwater movement is another aspect related to weathering. It is the movement
of shallow groundwater that transports solutes from higher parts of the landscape to
lower enabling weathering reactions to continue. In addition, solid particles may be
moved downwards through the weathering material, depositing deeper in the profile.
Fine-grained particles such as clays and Fe-oxides are most easily removed (leached
or eluviated). Landscape shape also has a considerable effect on the groundwater
movement patterns. While under long straight slopes groundwater contributes to the
stream uniformly along the valley, at the valley heads the groundwater flow is
concentrated and tends to intensify solution movement, weathering and erosion by
mass movement. At spurs, groundwater will deliver less solutes and erosion by mass
movement will be less. This means that due to the higher groundwater flows in
valley heads, weathering rates are likely to be higher in such areas compared to

15
anywhere else in the catchment (Taylor and Eggleton, 2001). The role of mineralogy
and that of geological setting on the trace metal concentrations in weathered profiles
has been investigated in the paper “The influence of mineralogy and geological
setting on trace metal concentration within subtropical weathered profiles, Bells
Creek catchment, Queensland, Australia”. A comparison is made between the total
metal composition and the extractable and mobile component which has
environmental significance but is rarely presented in published literature.
CLIMATE
Climate is a paramount factor in chemical weathering as it controls the amount of
rainfall for an environment (Summerfield, 1991). Rainfall in particular, controls the
supply of moisture for chemical reactions and the removal of soluble constituents of
the minerals. Temperature has also considerable influence on the rate and intensity
of chemical weathering. According to the “van’t Hoff’s rule”, for each 10º C rise in
the temperature the velocity of a chemical reaction increases by a factor from 2 to 3
(Taylor and Eggleton, 2001). It is mainly the proportion of the total rainfall which
infiltrates the weathering zone, percolates downward and ultimately finds its way by
subsurface drainage to creeks, rivers, lakes and the ocean, carrying dissolved
constituents that governs the rate of weathering.
In arid regions where evaporation exceeds rainfall, water may penetrate the rocks but
during long dry periods, it is lost through evaporation. Therefore, soluble
constituents of the rocks are not removed and reactions are slowed down. Such areas
are characterised by unaltered or partly altered parent minerals, the presence of salts
such as gypsum and carbonates, alkaline pH values (7.5-9.5), and a general scarcity
of organic matter. The characteristic secondary minerals are montmorillonite, illite,
chlorite and mixed layers of these minerals. In contrast, the rocks of humid areas are
generally well leached due to continual downward movement of the percolating
waters and the soluble products of the hydrolysing reactions ultimately lost through
subsurface drainage. Under these conditions chemical weathering proceeds rapidly
and the most abundant secondary minerals are kaolinite, halloysite and gibbsite as
well as ferric oxide minerals such as hematite and goethite (Crowther, 1930;
Loughnan, 1969). Of note is that a change in temperature does not, however, affect
the processes of weathering. If a rock is so placed that it weathers to bauxite in the

16
tropics, the same rock in the same regolith situation but in Iceland, may also weather
to bauxite, but it will take substantially longer (Taylor and Eggleton, 2001).
TIME
Time can be considered a geological factor. While the effects of weathering can
produce rudimentary soils within times of the order of hundred or several hundred
years, it takes millions of years to produce a ferrasol. Furthermore, in humid climatic
zones, the effect of parent material becomes more difficult to detect as reaction time
increases. On any time-scale, especially on geological ones, there is always a
significant lag between the establishment of a particular set of conditions in the
weathering environment and the adjustment of the mineralogical and physical
properties of the regolith to these conditions. Therefore, weathering profiles are
rarely in full equilibrium with environmental conditions as these conditions
constantly change; in most cases the weathering mantle adjusts to long-term average
conditions rather than to conditions at a specific time (Summerfield, 1991; Macias
and Chesworth, 1992).
VEGETATION
Vegetation directly affects weathering through the release of organic acids and in the
supply of carbon dioxide to soil waters. This occurs by the production of litter which
varies substantially not only between deserts and forest ecosystems but also between
temperate forest with a typical range of 0.1-0.3 × 106 kg/ km2/year and tropical
rainforests which produce 0.4-1.3 × 106 kg/ km2/year (Summerfield, 1991). Organic
activity is closely related to climatic controls, but vegetation type also varies by
topographic factors and soils properties. Therefore, it can indirectly influence
weathering through topography (Summerfield, 1991).
Products of Chemical Weathering
During chemical weathering (dissolution, oxidation, hydrolysis, acidolysis or
alcalinolysis) decomposition of primary minerals leads to the formation of secondary
minerals. Rock-forming minerals are partly dissolved during the weathering process
and hydrolysis and hydration take place. Clay minerals are the most significant type
of secondary minerals due to their complex phyllosilicate properties (e.g. surface
area and internal structure). These characteristics make them metal adsorbents (e.g.

17
Berner, 1971; Chamley, 1989; Summerfield, 1991; Velde, 1992; Hamblin and
Christiansen, 2001) and could be significant for the depositional environment as they
may act as geochemical traps for heavy metals. Furthermore, clay minerals such as
mixed layers of smectite-illite are the species that are likely to be encountered in the
subtropical setting of this project study area (Cox et al., 2000). Therefore, a brief
discussion about clay minerals with special regard to their speciation, distribution
and depositional significance is presented in this review.
Clay Minerals
While clay minerals occur in a variety of forms, only the major groups are discussed
here. Recombination of silica, aluminium and metal cations released during
weathering can form layered phyllosilicate structures. The type of clay minerals
produced in the sedimentary environment depends on the composition of the
circulating pore waters, the mineralogy of primary materials, the intensity of leaching
and the prevailing Eh-pH conditions (Chamley, 1989; Summerfield, 1991).
Under intense leaching conditions, kaolinite is prevalent. Conditions of extreme
leaching can ultimately lead to the formation of iron (goethite) and aluminium-rich
oxides (gibbsite). If leaching is only moderate however, the formation of cation-
bearing clays such as illite and smectite are favoured. Therefore, in a sedimentary
setting the magnitude of the weathering can be predicted from the type of the most
prevalent clay mineral present.
Clay minerals are stable under conditions of normal pressure and temperature; clays
therefore, experience only limited mineralogical transformation during transport and
after deposition in the marine environment. As a result, they are an excellent tracer
for sediment origin, distribution and transport pathways over long distances, as fine-
grained sediments of different origin can often be differentiated by their clay mineral
content (Zollmer and Irion, 1993; Algan et al., 1994). While in this study the clay
speciation is homogeneous throughout the catchment due to the existence of a
relatively uniform bedrock (Landsborough Sandstone), it may have a depositional
significance. Due to limited leaching and physical rework, it is expected that the
fluvial material (upstream) may contain more smectite. In addition, smectite may be
deposited in lower energy sections of the estuarine section and around meanders. In

18
most downstream settings, however, smectite has been weathered to kaolinite (e.g.
Chamley, 1989; Velde, 1992).
While a detailed review of weathering processes is not the focus of this study, a brief
discussion about the mechanism/s involved in silicate weathering and secondary
mineral formation is of significance, as it helps to understand the depositional
significance of clay minerals throughout the study area. When weathering occurs, a
primary mineral (e.g. silicate or carbonate) is attacked by organic acids such as
oxalic acid. However, the overall reaction, as far as groundwater composition is
concerned, can be presented as if the only attacking acid was H2CO3. In other words,
HCO3- and not C2O4
2- is found in most groundwater and river waters (Berner and
Berner, 1996 for detailed reactions between silicates and organic acids). Thus, as
organic acids disappear soon after primary mineral attacking, it is the general
assumption that silicate weathering consists solely of attack by carbonic and sulfuric
acids (e.g. Garrels, 1967). This is a simplification of a series of more complex
chemical weathering reactions which enables the prediction of the origin of ions in
groundwater without concern for the type of organic acid attacking the primary
minerals. As weathering proceeds, aluminium liberated by feldspar dissolution
precipitates to form a secondary clay mineral, except for localised distribution
accompanying chelate transport. Iron, due to its insolubility in the presence of O2,
also accumulates in soils, as ferric oxides.
Precipitation of Al may form gibbsite, smectite or kaolinite under different
conditions. Concentrations of these secondary minerals build up during contact of
the water with primary minerals, however, when the water leaves the rocks, further
build-up will cease. The faster the rock is flushed with water, the shorter will be the
time of contact with the primary mineral, and the higher will be the intensity of the
weathering of the rock. Gibbsite formation represents a high degree of flushing with
removal of both cations and silica. Kaolinite represents less rapid flushing with less
removal of silica, and smectite occurs under stagnant conditions of water flow so that
appreciable build-up of both silica and cations can take place (Berner and Berner,
1996). The following are the chemical reactions occurring in silicate weathering
(Thomas, 1994):

19
The first step is the hydrolysis of albite:
2NaAlSi3O8+3H2O+CO2 → Al2Si2O5(OH)4 + 4SiO2 + 2Na+ + 2HCO3-
albite kaolinite ions in solution
The highly mobile Na+ ion is lost in solution along with some proportion of the silica
which is not recombined to form clay minerals (kaolinite). The silica not combined
as kaolinite goes into solution as silicic acid:
SiO2 + H2O → 4Si(OH)4 (or H4SiO4)
silica silicic acid
Under weakly acid conditions with sufficient water and free drainage, more silica
may be removed, allowing gibbsite to form from kaolinite (incongruent dissolution
of Al and Si):
2Al2Si2O5(OH)4 + 105H2O → 42Al(OH)3 + 42Si(OH)4
kaolinite gibbsite
While gibbsite forms more commonly from the breakdown of kaolinite, it can also
form directly from plagioclase feldspar.
In conditions where water is scarce, the hydrolysis reactions may be retarded and
intermediate clay products will be formed:
8NaAl2Si3O8 +6H+ + 28H2O → 3Na0.66Al2.66Si3.33O10(OH)2 + 14H4SiO4 + 6Na+
albite smectite
or possibly
6KAlSi3O8 + 4H2O+ 4CO2 → 4K+ + K2Al4(Si6Al2O20)(OH4) + 4HCO3- + 12SiO2
illite
These clay minerals are more complex than kaolinite and their physical structure
reflects this complexity (Thomas, 1992).
In general, gibbsite forms in mountainous terrain with high rainfall and good
drainage where there is very rapid flushing. Tropical and subtropical soils tend to
favour formation of kaolinite due to less strong flushing. Finally, smectite is the
characteristic mineral of soils of semiarid regions with low rainfall. The effect of
flushing by water on the weathering of a single rock type is demonstrated by the
studies of Sherman (1952) (correlation between rainfall and clay assemblage) and
Mohr and van Baren (1954) (effect of drainage on clay mineralogy). Mohr and van

20
Baren found that for the same rock type and rainfall, depending on degree of
drainage, soils might have different clay mineralogy. While kaolinite is formed
under a good drainage system, in swampy depressions with poor drainage smectite
was more abundant. Furthermore, differences in water flow path could result in the
formation of different clay assemblages from the same plagioclase-rich rock under
the same climate and relief. In surficial zones where the water residence time was
short due to a small flow path gibbsite was formed. However, at depth both gibbsite
and kaolinite were found where the water travel distance was much greater. In the
slightly weathered and deeply buried underlying rock, the entrapment of water
resulted in the formation of smectite (Velbel, 1984).
Iron Minerals
The common iron minerals forming under sedimentary conditions include hematite,
goethite, siderite, glauconite and pyrite. Hematite and goethite are the oxidation
products of weathering of ferrous minerals and constitute a major source of detrital
iron in sediments. By contrast, glauconite, siderite and the iron sulfides form only
during diagenesis. Fine-grained goethite, FeOOH, is formed by the hydrolysis of
Fe3+ ions released during the oxidation and weathering of Fe-containing phases such
as limonite (e.g. Evans, 1989). While limonite is abundant in modern sediments and
on weathered outcrops, it is rare in buried ancient sedimentary rocks (Fischer, 1963),
and it is an assumption that it is unstable during diagenesis. Hematite however, is a
common mineral of sedimentary rocks and it is believed that if limonite disappears
during diagenesis some of it may be dehydrated to hematite. In order for this to
happen, the original sediment would have to be relatively free of decomposable
organic matter so a high enough Eh can be maintained to stabilise hematite.
Therefore, as organic matter is generally abundant in marine sediments, almost all
hematites are non-marine (Berner, 1971).
Thermodynamical stability of siderite (FeCO3) is severely restricted, as for the stable
form to persist Eh and S2- must be low. This is unlikely to occur in marine
conditions because low Eh is the result of the anaerobic bacterial decomposition of
organic matter; in seawater, which contains abundant dissolved sulfate, anaerobic
decomposition almost always includes the reduction of sulfate to H2S. Thus, if
thermodynamically reversible redox equilibrium between SO4aq2- and HSaq
-, or H2Saq,

21
is only obtained by sulfate reduction, then siderite has no stability field in marine
sediments. By contrast, in both ancient and modern non-marine sediments siderite
occurs commonly in association with coal beds and fresh-water clays (Berner, 1971).
Overall, iron minerals such as hematite and siderite are representative of a non-
marine sedimentary environment, as the marine conditions do not allow for the stable
form of these minerals to persist.
In waterlogged and saline sediments with a significant supply of decomposed organic
matter, bacteria break down this organic matter under anaerobic conditions, reducing
sulfate (SO42-) from seawater to sulfide. The iron source is from detrital ferric (Fe3+)
phases occurring as concretions, coatings or adsorbed by clay minerals oxidising to
Fe2+. The stable end product is pyrite (e.g. Berner, 1971, 1981, 1983; Dent, 1986;
Dent and Pons, 1995). Sea-level changes therefore, can influence pyrite formation;
over the last 6000 years for example, the Holocene sedimentation has kept pace with
sea-level fluctuations and has formed a broad, stable tide-influenced zone. This type
of setting has provided the required conditions for iron sulfide accumulation on many
of the world’s coastal plains (Dent, 1986).
Overall, three environmental systems for accumulation of pyrite have been identified
(Pons et al., 1982). System (1) includes bare tidal flats, marshes with mangrove
swamps in association with tidal creeks. The lower reaches of the system are
inundated most of the time and sediments are reduced; the higher reaches however,
have a predominantly aerated surface soil. In tropical regions, organic carbon
content of the sediments in this system is low (0.15 to 2%) but the system receives a
very high supply of organic matter from mangroves. Tidal flushing kinetically
favours pyrite formation in this system by supplying limited amounts of dissolved
oxygen necessary for complete pyritisation of reduced sulfate. Tidal flushing can
also enhance removal of sedimentary carbonate or bicarbonate from the system,
increasing the potential acidity of the system. This system is the most likely
environment occurring in the southern part of the study area (Halls Creek
catchment), most of which is a tidal-dominated floodplain accumulating large
amounts of pyrite compared with a fluvial-dominated area (e.g. Lin et al., 1995).

22
System (2), which occurs at the bottom of saline and brackish lagoons, seas and
saline lakes, always contains clastic sediments supplied by rivers. In arctic regions
this system comprises high amounts of organic material. However, where decay of
organic matter is slow, accumulation of sulfate can be considerable.
System (3) consists of poorly drained inland valleys with an influx of sulfate-rich
water. This system is very rare; examples are the pyritic papyrus in a few valleys in
the eastern Netherlands and the sulfidic peat soils of Minnesota, USA.
METHODOLOGIES FOR ASSESSING WEATHERING PROFILES WIT H REGARD TO TRACE METALS A substantial volume of literature is available on methodologies on assessing
weathering of volcanic or igneous rocks (e.g. Chesworth et al., 1981; Middelburg et
al., 1988b; Nesbitt and Wilson, 1992; Hill et al., 2000), however, similar
methodologies for sedimentary rocks are scarce. Therefore, the following are some
methods for evaluating geochemical data from weathered profiles that can be applied
to sedimentary rocks.
Calculation of Chemical and Mineralogical Indices
The chemical behaviour of minor and trace elements for a weathered profile and its
equivalent weathered products as a function of a mineralogical index of alteration
(MIA), rather than depth of sampling is one way of evaluating geochemical data.
Understanding the geochemical behaviour within the weathered profile therefore,
helps to explain the processes involved in mobilisation and deposition of these
metals in unconsolidated sediments throughout the catchment. The degree of
weathering varies for different samples at a similar depth, but in different cores can
be quantitatively measured, using the whole-rock analyses. These values represent
the average weathering index for each sample and can also be used to determine the
weathering index of each separate mineralogical component of the system. The main
assumptions are that the index of alteration of a sample is the same for all its
mineralogical pairs used for the partition of a chemical element between a primary
and its equivalent secondary mineral, and that the system is closed, without mass
transfer (loss or gain) (Voicu et al., 1996, 1997). The first step is to calculate the

23
Chemical Index of Alteration (CIA: Nesbitt and Young, 1982; Fedo et al., 1995) for
each analysed sample using the following equation:
CIA = [Al 2O3 / (Al2O3 + CaO + Na2O + K2O)] × 100 (1)
where oxides are in molecular proportions. While CIA was widely used as a
chemical index to ascertain the degree of source-area weathering (e.g. Bauluz et al.,
2000), according to Voicu and Bardoux (2002), CIA values range between 50 and
100 and cannot be directly applied for the normative calculations. Therefore, they
proposed a second step, the calculation of the mineralogical index of alteration
(MIA), using the following equation (Voicu et al., 1996, 1997):
MIA = 2 × (CIA – 50) (2)
The mineralogical index of alteration indicates the degree of weathering for each
analysed sample, independently of the depth of sampling. The MIA value indicates
incipient (0-20%), weak (20-40%), moderate (40-60%), and intense to extreme (60-
100%) weathering. The value of 100% means complete weathering of a primary
mineral into its equivalent weathered product (Voicu and Bardoux, 2002).
Therefore, the use of a weathering index such as MIA enables the quantification of
the supergene alteration of each individual sample and complements qualitative
estimation of weathering intensity by mineralogical studies such as x-ray diffraction
(XRD). Furthermore, it provides more accurate information about the trends of
major and trace element in weathered material as a comparison to unweathered
parent material.
Mass Balance Calculations
For an accurate assessment of element mobility during weathering, it is necessary to
look at absolute changes in element concentrations. This has been done using two
principal methods. The weight loss factor method (Faure, 1998) is based on the
assumption that during weathering, one of the major-element oxides has remained
constant in amount, although its concentration may appear to have changed. This
procedure is applicable during original transformation of parent rock to weathered
material where original mineral structure is maintained. The constituent chosen most
often for this purpose is Al2O3, which is generally immobile and remains in the
system (e.g. Faure, 1998). Alternatively, Fe2O3 (e.g. Faure, 1998), TiO2 (e.g.

24
Nesbitt, 1979; Eggleton et al., 1987; Hill et al., 2000), or ZrO2 (e.g. Hodson, 2002;
Steyrer and Strum, 2002) may be selected in the cases where Al is not the most
constant oxide.
In more severely weathered profiles however, the original structures and volume are
not preserved and the immobile element approach (Nesbitt, 1979) must be used to
assess element mobility. The percentage increase or decrease of any component (X)
in a weathered rock, relative to the fresh parent rock is calculated according to the
following equation:
Percentage change = [(X / I) weathered / (X / I) parent – 1] × 100 (3)
where I is the concentration of immobile component. In mass balance calculations,
losses are indicated by high (70-100%), average (40-70%) and low (0-40%) and
gains are shown by low / average (40-100%) and high (>100%) (Braun et al., 1993).
The paper “Geochemical methods for characterisation of subtropical weathering
and metal release within sedimentary bedrock: Queensland, Australia” presents the
applicability of the described procedures (chemical and mineralogical indices, weight
loss factor and immobile element) to the sandstones and mudstones present in the
study area.

25
SEDIMENTARY ENVIRONMENTS, THEIR PROPERTIES AND GEOCHEMISTRY
Introduction
Transported aquatic solids comprise a mixture of material inputs from different
sources. These can include eroded rocks and soils, solid waste particles, atmospheric
fallout and autochthonous formations such as inorganic precipitates, biogenic matter,
adsorbents on particles from solution, and other complexed and colloidal matter.
During periods of reduced flow rates, suspended material settles to the bed of the
river, lake or sea and incorporates into the bottom sediments (e.g. Förstner, 1983).
In detecting trace metal pollution sources, it is very important to study and analyse
sedimentary environments. Their significance was highlighted by Förstner (1983)
who stated that sediments with their contaminants have a relationship with the liquid
phases and the organisms; this means that the sediments themselves represent
another environmental contaminant. Sediment analysis has been significant in
identifying sources of trace metals in the aquatic environment for two main reasons:
(1) they exhibit higher accumulation rate (Förstner, 1981) and (2) sediment analysis
allows contaminants that are adsorbed by particulate matter, and thus escaping
detection by water analyses, to be identified (Förstner and Salomons, 1980).
Förstner and Salomons (1980), summarised important problem areas with regard to
the presence of contaminated sediment in the environment as follows:
• contaminants in the sediments are potentially available for aquatic life;
• contaminants in dredged material during and after disposal in dumping area could
cause groundwater pollution;
• vegetation may uptake contaminants from polluted sediments.
To assess the environmental impact of contaminated sediments, vertical sediment
profiles (cores and cuttings) are of importance. This is because the sedimentary
material often preserves the historical sequence of pollution, and at the same time
enables a reasonable estimation of the background levels and the variations in input
of pollutant over an extended period of time. It has been established that vertical

26
sections of the sediment could give a record of level of contamination over time, if
the pollutants are persistent and the sediment stratum has not seriously disturbed by
human activities such as clearing and dredging (Fung, 1993).
Overall, there are two primary aims for environmental studies of sediments: (1) to
identify, monitor and control pollution sources, and (2) to estimate possible effects of
polluted sediments. The results of sediment studies may vary due to sampling
techniques, preparation of samples and analytical procedures. In addition, sediment
metal concentrations are also influenced by sediment properties, for example pH,
redox potential, cation exchange capacity, soil texture and organic content (Ong Che,
1999). Therefore, the above limitations should be considered in making any
conclusions and/or generalisations.
As part of this study is focused on the effect of sediment grain size (mineralogy) on
trace metal chemical behaviour and the resultant geochemistry of estuarine
environments, these will be addressed in the next sections.
Properties of Sedimentary Material
Knowledge of the various characteristics of sediment (e.g. sediment size and
composition with respect to adsorbent material) enables assessment of its character
and evolution. A wide granulometric range, abundant matrix, poor sorting, angular
grains as well as high porosity and permeability characterize immature sediment.
Such sediment is the result of rough hydrodynamic actions, slow or weak, as
encountered in certain fluvial or glacial environments and during marine re-
sedimentation. However, mature sediment is evidence for active and prolonged
hydrodynamic processes in water or air, such as in littoral or desert dunes, beaches,
and other shallow-marine exposed environments (Chamley, 1989).
Sedimentary materials range from the fine dust transported by high-altitude winds to
large erratic blocks moved by glaciers. Sediments transported by and depositing
from waters tend to be within the smaller grain size range. Sedimentary particles
mostly fall in three categories, sand (2-0.063 mm), silt (0.063-0.004 mm), and clay
(below 0.004 mm) (Chamley, 1989). It has been established that the fine fraction of
sediment (<63 µm) has high concentration of heavy metals due to the strong

27
adsorptive surface properties of clay minerals and increased specific surface area
(e.g. Förstner and Salomons 1980; Förstner et al., 1982). This finding has also been
confirmed by other studies (Ellaway et al., 1982; Yocesoy and Ergin, 1992; Irvine
and Birch, 1998; Birch and Taylor, 1999). In a study of the influence of sediment
grain size on the metal concentration, Ellaway et al., (1982) separated samples into
three size fractions (clay < 2 µm, fine silt 2-20 µm, and coarse silt 20-63 µm) slightly
different from categories mentioned earlier, although the influence over metal
adsorption is similar.
Overall, there is some disagreement about the best size fraction to consider as an
indication of trace metal contamination. In the current study, the samples were not
sieved and chemically analysed according to their size, because any sieving may
contaminate the sediments. The effect of the fine-grained fraction on trace metal
content was investigated by determining the mineral composition using x-ray
diffraction analysis, which provides a more precise evaluation of the metal adsorbent
phases. The composition of sediments (see trace metal mobility) with respect to
adsorbent components is also related to the size of sediments because of the potential
influence on mobility of trace metals.
Geochemistry of Sedimentary Settings
An understanding of the geochemical settings of the sedimentary environments
enables the determination of how elements interact and associate with each other.
Consequently, this information can provide an understanding of the distribution of
metals, occurrence and availability to the overall system. Based on the early work of
Krumbien and Garrels (1952), the geochemical environment of sedimentary rocks is
characterised in terms of pH and Eh. However, in more recent studies, the
classification scheme of Berner (1981) is used. In this classification, in order to
determine the relevant environment of sedimentary rocks, the presence or absence of
total dissolved oxygen and sulfide phases (H2S and HS-) are considered. This
scheme is used because aerobic organisms and oxidised minerals cannot tolerate
traces of H2S without death or conversion to sulfide minerals, respectively.
Moreover, sulfide minerals and bacteria that produce H2S in sediments cannot
tolerate traces of oxygen without conversion to oxidised minerals or death. This
simply means H2S and oxygen cannot co-exist in solution (Berner, 1981).

28
Based on the above classification there are two major geochemical categories for
sedimentary environments in this scheme. They are known as oxic and anoxic,
depending on the presence and absence of oxygen, respectively (Berner, 1981). The
implication of anoxic and oxic sedimentary environments with respect to metal
occurrence is that firstly, under reducing conditions (anoxic) trace metals tend to be
less mobile and secondly, pyrite forms under reducing conditions and incorporates
heavy metals if available. Oxidation (oxic) conditions, however, will produce
sulfates, iron, acid and trace metals.
Coastal Marine Sedimentary Environments
There are several types of marine sedimentary environment including deltas and
estuaries, littoral, and shelf settings. As the focus of this project is on an estuarine
setting, they will be explained in more details as follows.
Depending on the approach taken, there are several ways to define estuaries. Based
on Dyer’s (1997) definition estuaries are formed at the mouth of rivers and in the
narrow boundary zone between the sea and the land. Their form and extent is
continuously altered by the erosion and deposition of sediment. A small rising or
lowering of sea level may have drastic effect on estuaries.
A typical estuary represents part of a river under the influence of tides. Although the
salt/freshwater interface is continuously changed, the salinity decreases in the
upstream direction. Therefore, in such environments, marine influences and
especially tidal processes are of significance. For example, deltas and estuaries
represent a continuous series of sedimentary environments at the interface of alluvial
plains and basins where fluvial processes interact with the influences of tides and
waves. An estuary denotes an extreme type of a delta subjected to tidal influences
and if at any stage of its evolution, it is dominated by fluvial supply, it tends to
become a seaward-advancing delta (Chamley, 1989).
While most oceanographers, engineers and natural scientists define estuaries as areas
of interaction between fresh and salt water, there are over 40 different definitions of

29
estuaries (Perillo, 1995). Some consider both tide effects and sediment point of view
(e.g. Chamley, 1989; Dyer, 1997) and some may consider only one of the above.
Pyrite-bearing sediments are typical of estuarine settings and known to be a potential
source of metals to both surface and groundwater. Main metals involved are
aluminium and iron, and therefore, their formation and release to the environment are
discussed in the following section.
Pyritic Sediments and Trace Metals
BACKGROUND AND DEFINITION
The accumulation of pyrite in low-lying coastal sediments is the first step in the
formation of acid sulfate soils (ASS), which is the most common form of acid soils
worldwide. Such soils have been divided into actual acid sulfate soils (AASS), and
potential acid sulfate soils (PASS) (Dent and Pons, 1993). It has been estimated that
AASS cover approximately 12 million ha of land around the world whereas PASS
cover more than 100 million ha (Dent, 1986).
Formation of ASS can be divided into accumulation of pyrite particles and oxidation
of pyrite sediments. The most essential factors involved in pyrite accumulation are:
sufficient amount of Fe3+, decomposed organic matter and sulfate ions under
anaerobic conditions. In different sediments, formation of pyrite is governed by
different factors. For example, in terrigenous marine sediments, mainly organic
matter controls pyrite whereas in non-marine, freshwater sediments accumulation of
pyrite is controlled by concentration of sulfate rather than organic matter (Berner,
1983).
Exposure of pyritic sediments can release acid, which in turn produces iron from
pyrite and aluminium from silicates to the ground and surface waters resulting in
significant acidification (Sammut et al., 1996). Trace metals can also be released,
mobilised and adsorbed to the surface of iron and manganese oxides and hydroxides,
by clays, or complex with organic compounds. The products resulting from
disturbance of pyritic sediments have detrimental environmental impacts in several
ways: (a) sulfuric acid can have an adverse impact on land and aquatic habitats, (b)
elements contained by pyrite are released and mobilised into the environment, and

30
(c) the weathering of pyritic sediments is enhanced by acidic condition, which results
in releasing major and minor metals from silicate structures and trace metals from
minerals (Preda and Cox, 2001). In Australia, major soil acidification has been
reported in Holocene estuarine sediments of coastal flood plains and modification of
these flood plains has accelerated the production and transport of acidified water
(Sammut et al., 1995).
FORMATION AND MORPHOLOGY
Framboids of pyrite are the most common texture of sulfides. They are present in
different environments, from recent sediments (marine and lacustrine) to sedimentary
and metamorphic, to magmatic rocks and hydrothermal deposits. However, their
most typical environment is represented by organic rich marine sediments
(Sawlowicz, 2000). Pyrite framboids are common below the sediment – water
interface (Canfield et al., 1996; Wilkin and Barnes, 1997a, 1997b; Suits and Wilkin,
1998), but are rarely found in the water column (e.g. Skei, 1988; Lyons, 1997).
While the framboids precipitate by gravitational force to form a geopetal fabric
inside the pores of sediment (Kawamura et al., 2002), Ski (1988) demonstrated the
presence and the formation of suspended framboidal pyrite in the highly anoxic
waters of Framvaren Fjord. Other workers have found suspended framboids in water
columns and due to oxic conditions concluded that pyrite was either resuspended or
has been transported from elsewhere. This may be due to the fact that estuarine
water does not contain enough oxygen to rapidly oxidise suspended pyrite framboids,
or alternatively organic inhibitors and surface coatings prevent rapid oxidation (e.g.
Middelburg et al., 1988a).
The morphology of the framboids varies from spherical to euhedral with the latter
more common in large particles. It has been demonstrated that the morphology of
pyrite crystals formed at room temperature is primarily controlled by the degree of
supersaturation in the solution from which pyrite is precipitated (Murowchick and
Barnes, 1987). With the increasing supersaturation, pyrite morphology changes from
cube to (euhedral) octahedron, to spherule (Wang and Morse, 1996). Therefore,
observed pyrite octahedra modified by cubes suggest a decrease in supersaturation
during crystal growth (Bulter and Rickard, 2000). Close spatial association of pyrite
framboids and euhedra in nature may be related to their genetic relationship. The

31
possibility of recrystalisation from framboidal to single grain pyrite has been
suggested (Love and Amstutz, 1966). Sawlowicz (2000) developed this idea and
proposed a continuous growth of microcrystals in the framboids (sometimes towards
euhedra) as long as they are in contact with the initial solution. While pyrite in any
morphological form or size is a major scavenger for chalcophile trace elements in
sedimentary environments, in its framboid form due to its much larger surface area
can accumulate more metals compared to its euhedral form. The spatial
morphological variation of pyrite was investigated in the paper “Distribution of Fe in
waters and bottom sediments of a small tidal catchment, Pumicestone Region,
southeast Queensland, Australia”.
TRACE METALS Occurrence
In sedimentary environments metals may occur as (a) adsorbed on solids, (b)
precipitated and coprecipitated on solids (Fe and Mn as metallic coatings), (c)
incorporated in solid biologic materials, and (d) incorporated in crystal structures
(Förstner and Wittman, 1983). Metals occurrence in rock forming minerals is based
on the type of crystallographic structure, for example, while quartz and alkali-
feldspar have low concentrations of heavy meals, magmatic minerals (biotite,
pyroxene and olivine) contain higher levels of heavy metals.
A major input of various metals to coastal lowlands may occur as a result of
weathering and erosion of geological formations. These trace metals are transported
from their source as either dissolved or particulate forms by streams and rivers and
are deposited on coastal floodplains, and in estuaries and bays. In such aquatic
environments, metals undergo various processes associated with floods, tides, and
waves and can be adsorbed by clays, complex with organic compounds or co-
precipitate as inorganic mineral phases. Human activities are a major source of
introducing trace metals into coastal environments. Hence, in estimating total metal
composition in such settings, both natural regional abundance and local
anthropogenic inputs have to be considered (Preda and Cox, 2001, 2002).
Trace metals therefore, may be introduced to the coastal environment by both natural
processes (e.g. weathering and erosion) and human activities in catchment area or

32
adjacent to the coast (Niencheski et al., 1994; Preda and Cox, 2001). These studies
further explained that in transfer from water to sediment, metal may involve co-
precipitation and adsorption on freshly precipitated Fe/Mn hydroxides and adsorption
on clay minerals and organic matter. Ong Che (1999) showed that metal
concentration in the surface sediments were 4-25% higher than those found in the
deeper sediments, reflecting anthropogenic input to the sediment metal load.
Niencheski et al., (1994) suggested that in assessing metal contamination in coastal
areas, studies of water column concentrations of trace metals are difficult because of
the possibility of sample contamination during collection and handling and
requirements for ultra-trace analysis. Therefore, they concluded that as
measurements of suspended particulate material do not involve such difficulties in
sampling and analysis, they provide a better estimate of anthropogenic contributions
to coastal areas. Ellaway et al., (1982) further explained that sediments have the
capacity to accumulate trace metals and other contaminants over time so that a time-
integrated assessment of contamination in the water body can be obtained. The only
problem associated with this method is standardization of the particle-size range to
be analysed (Förstner and Salomons, 1980). There is disagreement between
scientists in this field, as to which size fraction gives the best indication of trace
metal contamination (see properties of sedimentary material).
Overall, in order to explain trace element distribution in coastal sediments, one has to
distinguish between heavy metal enrichments related to natural sources and
contamination introduced by anthropogenic activities. The paper “Heavy metal
distribution and controlling factors within coastal plain sediments, Bells Creek
catchment, southeast Queensland, Australia” investigates processes influencing trace
metal distribution.
Mobility
In the aquatic environments, trace metals are transported in a variety of ways, such as
soluble chelates and ions, constitutes of particulate matter or by absorption on
suspended organic or inorganic colloids (e.g. De Groot, 1975; Förstner, 1981; Arakel
and Hongjun, 1992). Metal absorption on various (organic and inorganic) colloids
by a variety of processes facilitates their concentration in soils and near-surface

33
sediment. While the organic colloids comprised of various soluble and insoluble
humic substances, the inorganic colloids include a variety of secondary clay
minerals, and Fe-Mn oxides and hydroxides formed due to weathering processes.
Therefore, not only do colloids have the capacity to absorb a great amount of heavy
metals but they may also incorporate the most active phases of the metals (Förstner,
1989).
Overall, some of the most important controls especially on trace metal speciation and
mobility include the pH, Eh, temperature, surface properties of solids, abundance and
speciation of ligands, major cations and anions, presence or absence of dissolved
and/or particulate organic matter, and biological activity (Plant et al., 1996).
Furthermore, there are seasonal variations in the metal levels in floodplain sediments.
During the dry season the metal concentration of interstitial water increases and
metals may form metal-Cl complexes, or be adsorbed by clay and organic particles.
During the wet season however, the sediment pore water becomes slightly acidic as a
result of a rise in watertable. This in turn promotes the desorption and export of
heavy metals (Arakel et al., 1992).
Adsorption of trace metals can result in their precipitation and potential
immobilisation. The mechanism of adsorption comprises the replacement of surface
–OH or –OH+2 groups on variably charged surfaces by the adsorbing ligand. The
specific adsorption of metallic ions occurs in those metals that are readily hydrolysed
in water. The adsorption reaction involves the formation of a complex between the
hydroxo-metal complex and the negatively charged deprotonated surfaces of the
oxides, hydroxides and oxyhydroxides of Al, Mn, and Fe (Evans, 1989). Mantei and
Foster (1991) gave the relative affinities of different heavy metals for the different
metal oxide phases as: hydrous manganese dioxide (Cu2+> Co2+> Mn2+> Zn2+> Ni2+>
Ba2+>); iron oxide (Pb2+> Cu2+> Zn2+> Ni2+> Cd2+>Co2+); and aluminium oxide
(Cu2+> Pb2+> Zn2+> Ni2+> Co2+> Cd2+).
As adsorption of waste products, particularly trace metals, occurs onto particle
surfaces (e.g. Krauskopf, 1956; Krauskopf and Bird, 1995), an understanding of the
physical fate and chemical activity of particulate material is essential to establish the
biogeochemical cycles of pollutant metals. In estuaries, a great percentage of the

34
suspended particulate matter becomes deposited on high intertidal mud flats and salt
marshes. An understanding of post-depositional remobilisation is very important to
provide information on element cycling and potential environmental toxicity. There
are two main factors controlling remobilisation of trace metals from estuarine
material. Mobilisation may take place physically because of natural processes such
as sediment erosion or early diagenetic geochemical processes, or due to artificial
disturbances such as clearing or developing, drainage or dredging (Allen et al.,
1974).
The degradation and dissolution of organic matter is one especially important
diagenetic process governing remobilisation of trace metals. Trace metals are
associated with a variety of organic materials such as living organisms, organic
detritus and organic coatings on mineral grains. As the amount of organic matter
decreases with depth, the proportion of trace metals associated with this fraction
decreases. Moreover, the metals are progressively lost by oxidation from the organic
fraction during early diagenesis and recaptured within the sediment body by a more
stable phase. The phase most likely to recapture a remobilised metal is the Fe-Mn
oxide-hydroxide phase, providing that the grain coatings remain active in the oxic
environment of the buried sediments and the flushing of the remobilised metal is not
too fast (Allen et al., 1974).
Factors Influencing Metal Mobility
Important aspects of trace metals within both aqueous and solid materials are their
distributions, and the controls over their release, transportation and fixation. The
“mobility” of the metals therefore, is of major significance and has been the focus of
several studies (Förstner et al., 1984; Baker, 1990; Davies, 1990; Kiekens, 1990;
McGrath and Smith, 1995; O’Neill, 1990; Alloway, 1990a; 1990b, 1990c). The
following is a summary of the above studies about the factors controlling trace
element mobility in sediments and soils.
SEDIMENT pH and Eh
Two of the most important factors directly controlling metals solubility and mobility
are the pH and Eh of interstitial water (Bell, 1998; Förstner et al., 1984). While the
mobility of an element is controlled significantly by changes in the oxidation state of

35
the environment (Eh), dissolution reactions, including hydrolysis, inorganic
complexation and sorption/desorption are all pH-controlled. For instance, under
high-pH conditions, anions and oxy-anions (e.g. Mo, As, and P) are more mobile
while some cations (Cu, Pb, Hg and Cd) are less mobile.
The relationship between pH and heavy metals mobilisation is significantly affected
by seasonal variations. For example, during the dry season and under oxidised
condition, metals are more mobile; during the wet season and under reducing
conditions metals are less mobile and theoretically their concentration must increase
in soils and sediments. However, this might not occur in situ. Kedron Brook, is an
example of a southeast Queensland estuarine setting where during the dry season
heavy metal concentration correlates positively with the decrease in pH values of
both soils and channel sediments. During the wet season, however, as the pH
increases, the total content of heavy metals in the soils decreases. The explanation
might be that under dry conditions most of the heavy metals in the soils are relatively
more stable than in the aquatic environment (Arakel and Hongjun, 1992). Moreover,
during the wet season, elevated water tables may lead to both dilution and physical
removal of the metals from sediments.
SALINITY AND FORMATION OF ORGANIC AND INORGANIC COMPLEXES
The influence of evaporation on major ion concentration results in a systematic
increase in heavy metal content of many surface waters as salinity increases.
However, since elements such as Cd may form soluble complexes with chlorine at
very low concentrations, hypersaline interstitial waters may also act as activators for
heavy metals variations (Arakel and Hongjun, 1992). In estuarine settings there can
be marked variations in metal concentrations. Förstner et al. (1984) demonstrate the
different behaviour of dissolved Zn and Cd in the salinity gradient of Elbe River
estuary, wherein mixing with relatively clean seawater Zn exhibits the expected
‘dilution’ effect. The general increases of dissolved Cd in the fluvial/estuarine
transect, however, suggest remobilisation of this element from contaminated
particulates.
Due to presence of high amounts of functional groups (-OH, -SH, -OOH and
phenolic), organic material is able to sorb 1-10% dry weight of available amounts of

36
Co, Zn, Cu, Pb, Mo, Ni, Ag, V, Mn and Fe. The larger the molecular mass of humic
substances or fulvic acids, the lower is the capacity of complexing metals. The fact
that many metals are lipophile and accumulate in biota explains the association of
metal-organic compounds to sediments (Saxby, 1973). The organic colloids are
comprised of various soluble and insoluble humic substances whereas clay minerals,
and Fe-Mn oxides and hydroxides may act as inorganic absorbent of heavy metals
(e.g. Arakel and Hongjun, 1992). Where organic matter is present, stable
organometallic complexes are formed increasing the trace metal mobility (Bell,
1998). In an earlier study, Joyce (1984) had explained that nearly all solids are able
to trap soluble ions by sorption (either chemisorption or adsorption) but clay sized
particles especially clays, organic material and colloidal hydrous iron and manganese
oxides are the most significant.
Several investigations have been carried out to characterise the organic compounds
responsible for the mobilisation of the metals. In a study done by DeGroot and
Allersma (1975), freshly deposited sediments from the freshwater tidal regions of the
Rhine and Ems were incubated with distilled water and from the dissolved organic
matter, the fulvic and humic acid fractions were isolated according to Kononova
(1966): fulvic acid soluble in acid and alkali, humic acids insoluble in acid and
soluble in alkali. A calculation based on iron and organic-matter contents of the
fractions revealed that the fulvic acid fraction is mainly responsible for the metal
mobilisation.
Some studies have focused on the mechanism by which organic matter affects metal
mobility. For example, the Kedron Brook floodplain study (Arakel and Hongjun,
1992) showed a positive correlation between total concentration of Cd, organic
matter, and the active phase of Cd; the Fe-Mn oxides form thin coatings on the fine
particulate matter. Due to acidic nature of the soils in the area, the humic acids (the
main absorbent of the heavy metals) occur in the form of solid gels with a very weak
activity. Therefore, metals such as Cd, once they enter the gels, will remain there in
a stable form until the wet season, when the water table rises. During the hot and wet
summer months, favourable Eh conditions for metal oxidation results in subsequent
mobilisation. Furthermore, Gibbs (1993) investigated carbonate free sediments from
the Townsville Harbour and concluded that up to 50% of Cu, Co, Ni, and Cr were

37
present in the form of a hydrous Fe-Mn oxide coating. As these hydroxides are very
sensitive to pH and Eh changes, a decrease of 1-2 pH units can result in releasing
high amounts of trace metals. If the sedimentation continues, Fe3+ and Mn4+ become
reduced; consequently, trace metals are remobilised and disperse to the upper and
lower parts of the sediments.
While the reduced forms of iron and any pyrite formed are not referred to as metal
scavengers to the same extent as Fe-Mn hydroxides, these sulfides are enriched in
trace metals compared with the bulk sediment. The exposure of the reduced layers of
sediments to aerobic conditions results in organic matter degradation and oxidisation
of sulfides. Thus, the existing metals are remobilised and released to the pore and
bulk water (Volkow and Fomina, 1974).
TRACE METALS AND ENVIRONMENTAL IMPACTS Introduction
Metal pollution has been defined as the presence and / or addition of certain metals to
soils at levels that would have detrimental effects on organisms such as: (1) the
concentration of metals in soils may not influence the growth of vegetation but
would create a health threat to higher organisms in the food chain that consume the
vegetation. (2) metals whose concentration would impair the growth of vegetation
(phytotoxic levels), and (3) since soils serve as a medium through which
groundwater is recharged, therefore, any constituent added to soil may negatively
affect the beneficial use of groundwater (Mattigod and Page, 1983). Furthermore,
anomalies in trace metal concentrations in aquatic sediments can be of environmental
significance because marine organisms and vegetation in coastal environments can
uptake metals, increasing the potential for the entry of some metals into the human
food chain (e.g. Arakel and Hongjun, 1992; Birch and Taylor, 1999).
Surface water quality and metals
BACKGROUND
All natural waters contain small amounts of trace elements at extremely low
concentrations. However, sometimes the concentration of naturally occurring

38
chemical elements can exceed those recommended as the maximum for potable
waters, or domestic use (Edmunds and Smedley, 1996).
The composition of surface waters and shallow groundwater would reflect the local
geology. Groundwater obtains its essential mineral character from reactions between
rainwater and bedrock over a timescale of days or months during percolation,
followed by emergence as springs or as inputs to surface water systems. The
residence times of the water and the primary mineralogy of the aquifer determine the
context of reaction with the host rock. For example, the initial concentration of CO2
in the soil may control the amount of reaction of silicate or carbonate minerals that
occur in the aquifer.
Estuaries are the principal places where the two major types of earth surface water
meet: fresh water and saline ocean water. Besides mixing of fresh and saline water
in estuaries, there are internal processes within the estuary itself that can change the
chemical composition of the water. The sediments on the bottom of estuary and the
overlying water exchange dissolved and particulate matter. Furthermore, biological
activities occur in the estuarine water, in the surrounding marsh tidal areas and in the
bottom sediments (e.g. Berner and Berner, 1996). Therefore, understanding the
dissolved constituents of estuarine waters and processes controlling these
concentrations is significant to understand the processes governing the metal
distribution in an entire catchment area. The dissolved elements of estuarine water
can be divided into two groups (Liss, 1976): 1) those which are more abundant in
seawater than in fresh water (e.g. Ca, Mg, Na, K, Cl and SO4) and 2) those which are
more abundant in fresh water (e.g. Fe, Al, P, N, Si and dissolved organic matter).
Due to its greater salinity, seawater has higher concentration of major dissolved
elements. However, metals such as Fe, Al, Mn, trace metals such as Zn and Cu,
nutrients such as P, N, Si and dissolved organic matter generally have a greater
concentration in fresh water than in seawater (Berner and Berner, 1996).
Dissolved constituents with higher concentration in fresh water must be removed
either in estuaries, where the original mixing occurs, or later, in the ocean
(Mackenzie and Garrels, 1966). Therefore, it is reasonable to consider removal of
elements such as Fe and Al might occur in estuaries. The removal of elements may

39
take place by either inorganic (nonbiogenic) or by biogenic processes. The elements
Fe, Al, and Mn are involved mainly in inorganic phases, while Si, N, P, and organic
matter are mainly biogenic. As the focus of this project is predominantly on fate of
elements such as Fe, Al, and Mn the inorganic process is explained below.
INORGANIC REMOVAL IN ESTUARIES
Laboratory studies of experimental mixing of fresh and saline water (Sholkovitz,
1976; Boyle et al., 1977; Crerar et al., 1981) show that “dissolved” iron - the term
refers arbitrarily to material passing a 0.45-µm filter and may consist of fine colloidal
material and complex organic matter as well as truly dissolved inorganic species
flocculates or precipitates from solution during mixing. The amount of Fe removal
estimated by either of these methods is between 50 and 95%, and the higher the Fe
concentration is in the fresh water; the greater is the total Fe removal (Boyle et al.,
1977). Other laboratory experiment on the mixing of fresh and saline water showed
that in some cases flocculation of Fe, Al, and Mn can be related to their association
with organic matter (Sholkovitz, 1976). For example, Boyle et al., (1977) observed
that river-borne “dissolved” Fe was almost entirely colloidal (Fe oxides particles
coated with an organic film) and not truly dissolved and in the laboratory experiment
more than 50% of it precipitated.
When highly acidic rivers reach the estuaries, the consequent rise in pH can cause
precipitation of some of the dissolved species. The removal of dissolved constituents
occurs by common physical-chemical mechanisms: acid neutralisation combined
with flocculation as it was observed by Crerar et al. (1981). Those workers found
that upon increase of pH during mixing with seawater, the dissolved inorganic Fe
and Al become supersaturated and precipitate as Fe oxyhydroxides floccules along
with the pre-existing Fe colloids and high-molecular-weight humic.
In highly polluted estuaries, such as the Belgian-Dutch Scheldt Estuary (Wollast,
1983) with anoxic bottom waters, reactive Fe3+ hydroxides carried by the river are
reduced to Fe2+ ions. Phosphate, previously adsorbed by the Fe3+ hydroxides, is
released to solution. When the estuarine waters become more oxygenated farther
downstream, the ferrous (Fe2+) iron is oxidised and precipitated as ferric (Fe3+)
hydroxide, which removes dissolved phosphate by reabsorption.

40
The overall removal of Fe in estuaries is controlled by the removal of particulates. In
addition, ferric iron that is precipitated onto the bottom sediments or originally
deposited as Fe coatings on clay minerals can be remobilised as ferrous iron by
reactions in reducing sediments and then released by wave, current, and biological
stirring of bottom sediments or by diffusion into bottom water. Thus, there is
potentially a Fe source in estuarine bottom sediments (Berner and Berner, 1996)
some of which can form pyrite.
Constituents such as Na, K, Mg, Ca and SO4, which are more abundant in seawater
than in fresh water, essentially behave conservatively upon mixing with fresh water
(Liss, 1976). This does not mean that reactions do not occur, but because of their
large concentrations in the seawater, small changes in concentration during estuarine
mixing are difficult to detect.
Factors affecting chemical composition of natural waters
INTRODUCTION
Surface water is the most appropriate environment in which to assess, monitor, and
control metal pollution (Förstner, 1983). However, in river waters there are strong
fluctuations in trace metal concentration due to many variables, such as daily and
seasonal variations in water flow, changing pH and redox conditions, the input of
secondary sewage, detergent levels and temperature. There are also problems related
to inadequate sampling, storage and analysis procedures that can lead to the
conclusion that many of the natural water values quoted in the literature review are
high solely for reason of contamination during sampling and analysis (Förstner,
1983).
ACIDITY AND REDOX
Acidity (pH) and redox potential (Eh) are the most significant factors governing
water quality. Acid groundwater may result either from natural processes such as
flow through non-carbonate rocks (e.g. granite), from pyrite oxidation or from
pollution (acid rain). A pH decrease of one unit may lead to an increase of more than
one order of magnitude in the concentration of certain metals. This consequently
leads to mobilisation and availability of these metals to the natural environment. The

41
mobility of some elements is controlled by the setting conditions whether there are
oxidizing or reducing. At shallow depth, soils or aquifer materials can be high in
organic matter or sulfide minerals and act as the main substrate (electron donor) for
the reduction of oxygen. In organic–deficient sediments, however, oxidizing
conditions may persist for thousands of years (Edmunds and Smedley, 1996).
In measuring metal contamination or pollution in water, not only is the abundance of
a particular metal constituent of importance, but also its availability in the form of
solubilised species. Organic ligands such as fulvic acid, and EDTA
(ethylendiaminetera-acetic acid) can inhibit the uptake of metals and thus many
increase the toxic threshold (Andrew et al., 1976). On the other hand, organic
complexing may enhance toxicity (Förstner, 1983).
The following summary highlights some important factors including environmental
aspects (e.g. rock type, relief and time) and the metal content of the parent rock
affecting freshwater chemistry.
ROCK TYPE
Waters draining igneous and metamorphic rocks are relatively dilute with average
TDS of 100-500 mg/L; HCO3 is the major anion (unless there is an input of
anthropogenic sulfate) and Na and Ca are the major cations (Drever, 1997). Mafic
rocks such as basalt tend to produce waters with higher Ca2+/Na+ and Mg2+/Ca2+
ratios that felsic rocks. In waters draining limestones and dolomites, Ca, Mg and
HCO3 are the major solutes, although sulfate is commonly present from pyrite
oxidation or associated gypsum. Waters draining siliclastic rocks such as sandstones
and shales are more complex than felsic and mafic rocks. These waters contain
chloride and sodium, which are thought to originate from seawater trapped in the
shale at the time of deposition. They usually have sulfate or chloride as major
anions, and lower silica to total cation ratios than waters draining igneous rocks. The
TDS values of sedimentary rocks are highly variable (Drever, 1997).

42
RELIEF
The effect of relief on water chemistry is discussed in chapter “The relationship
between chemical weathering rate and relief” (e.g. Drever, 1997). This relationship
is explained in detail in the chapter “Weathering” (see Topography).
TIME
The time of contact between rock and water is the most important variable in
determining the chemistry of runoff from igneous rocks. However, contact time is
itself a function of other environmental parameters. High rainfall results in rapid
flowing of water and short contact time. Well-drained areas have short contact times
and tend to have kaolinitic soils, whereas poorly drained areas have longer contact
times and tend to contain more smectite.
Through stabilising the soil, vegetation increases the contact time of initial
weathering products with incoming rainwater. Therefore, forested areas tend to have
kaolinite (or gibbsite) in the soil. In the absence of vegetation an initially formed
smectite would probably erode before it could be weathered to kaolinite (Drever,
1997). The effect of plants on water chemistry is explained in Drever (1994).
ALUMINIUM
Aluminium is a common constituent of most rocks because it is a major element in
alumino-silicate minerals. As the solubility of Al is significantly pH dependent, its
significant environmental concentration is found only below pH 5.5 where the
increasing concentration is related to the solubility of microcrystalline gibbsite
(Bache, 1986). The solubility of Al may be increased as a result of the presence of
inorganic ligands, notably F and SO4 (May et al., 1979). At pH greater than five, it is
unlikely that unstable, toxic forms of Al will be present in natural waters, however,
colloidal aluminium and other aluminosilicate colloids may contribute to the total
aluminium in waters (Edmunds and Smedley, 1996).
IRON AND MANGANESE
Concentrations of dissolved Fe and Mn can reach several mg/L under reducing
conditions. Solubility of Fe and Mn also increases at low pH. There is limited
evidence that Mn may be toxic at high concentrations. Iron concentration is

43
relatively high in igneous rock minerals such as the pyroxenes, the amphiboles,
biotite, magnetite and especially, the nesosilicate olivine (note that some olivines do
not contain iron, e.g. forsterite). The iron in these minerals is predominantly in the
form of ferrous, Fe2+ (oxidation state), but ferric Fe3+ may also be present, as
magnetite, Fe3O4. When these minerals are attacked by water, the released iron
precipitates as sedimentary species. Under reducing conditions when sulfur is
available, the ferrous polysulphides such as pyrite may occur. In oxidising
conditions the sedimentary species will be ferric oxides or oxyhydroxides such as
hematite, Fe2O3 and, goethite, FeOOH. Freshly precipitated material may have
poorly developed crystal structure and is commonly designated ferric hydroxide, Fe
(OH)3 (Hem, 1992).
ANALYSIS OF HETEROGENOUS GEOCHEMICAL DATASETS Normalisation
It is well established that trace metals may be introduced to the coastal environment
by both natural processes (e.g. weathering and erosion) and human activities in
catchment areas adjacent to the coast (e.g. Niencheski et al., 1994; Preda and Cox,
2003). Fine grained estuarine and coastal sediments act as sinks for these metals as
they comprise of a mixture of inorganic (detrital) and organic material with a variety
of particle sizes.
The concentration of trace elements in coastal zone sedimentary materials are
dominantly related to inorganic material resulted from physical and chemical
weathering of the continent. For some metals (e.g. Cd and Hg) organic material may
be the metal carrier but due to its low abundance in most sediments (<0.5% by
weight), it is not considered as a predominant contributor to total metal levels
(Loring, 1991). The inorganic detritus is composed mainly of a limited number of
silicate minerals such as quartz, feldspar, micas and clay minerals, and variable
amounts of metal oxides and sulfide phases. Of these minerals, clays tend to adsorb
metals due to their large surface area and negative charge (Windom et al., 1989).
Therefore, the source of natural variation in metal concentration has been placed on
accounting for the “grain size effect” and analyses have been carried out on a
specific size fraction to correct for natural variability (e.g. Förstner and Salomons,

44
1980). This approach requires, however, a separation step and the concentration in
the fine fraction does not necessarily reflect the concentration in the total sediment.
To compensate for this natural variability without sieving, metal concentrations are
normalised. This can be done by calculating the ratio of natural concentrations to
that of a normalising factor whose concentration is not affected by anthropogenic
processes (Daskalakis and O’Connor, 1995). There is no consensus on the
appropriate sediment constituent to be used for normalisation. However, two broad
categories in normalisation of metal concentration have been well established:
granulometric and geochemical. Granulometric techniques rely on normalisation
against the total weight percent of fines (<62.5 µm) or the total clay size particles (<4
µm) present in the sediment (Loring, 1991). Geochemical methods are based on the
comparison of the metal concentration in sediment to the concentration of some other
“reference” elements (Loring, 1992; Trimble and Hoenstine, 1997) such as
aluminium, iron, organic carbon, and lithium (Windom et al., 1989; Loring, 1990;
Loring, 1991; Daskalakis and O’Connor, 1995; Balls et al., 1997; Trimble and
Hoenstine, 1997; Tam and Yao, 1998; Fang and Hong, 1999).
In all the above examples, normalisation of metal concentrations assumes a linear
relationship between either the geochemical or sedimentological characteristics, and
the element of interest. Regardless of the type of normalising method, the
concentration of normalising metal is also used to establish the relationship between
natural trace metal concentrations in sediments from different areas. Overall,
geochemical normalisation is superior to granulometric methods, as it compensates
for both mineralogical and the natural granular variability of trace metal
concentrations in sediments (Loring, 1991).
One of the drawbacks of the geochemical approach is that it generates a ratio instead
of a total concentration. This can be compensated by standardising the contents to a
reference material, and calculating an enrichment factor (EF). For instance, an
enrichment factor for Zn relative to Al = (Zn/Al sample)/(Zn/Al reference material).
The validity of such an enrichment factor differs with values used for the reference
material. Most workers have used metal concentrations in Earth’s crust as reference
to interpret the results. However, concentrations for crustal abundances are not

45
appropriate because they neither represent the regional background level nor the
analytical uncertainties associated with their measurements (Loring, 1991).
Overall, this current study has used a geochemical approach to test correlations
between concentrations of trace metals and three candidate-normalising elements:
iron, aluminium and total organic carbon considering EF for trace metals relative to
both Fe and Al. For organic carbon, however, only a ratio was considered because
the OC values varied extensively between samples (see pages 245-247) and
calculating EF may have been misleading. The issue of normalisation in evaluating
heterogeneous geochemical data is debated in paper “Heavy metal distribution and
controlling factors within coastal plain sediments, Bells Creek catchment, southeast
Queensland, Australia”.
Statistical Analyses
In the current study, correlation indices help to determine the relationship between
different metals. Understanding such relationships not only does elucidate the path
by which these metals are transported and deposited but also explains the processes
involved. Metal oxides such as iron oxyhydroxides and manganese oxides, and
organic carbon commonly act as metal scavengers for heavy metals. Therefore,
correlations between any of these minor metals and the heavy ones may help to
understand the processes involved in metal associations (Rollinson, 1993). As
geochemical data such as those produced in this study are not normally distributed,
Spearman rank correlation was considered more appropriate than the calculation of a
simple linear correlation coefficient such as Pearson. In addition, descriptive data
analysis can provide further information (mean, standard error, standard deviation,
maximum and minimum concentrations).
COMPARABILITY OF ANALYTICAL METHODS Comparability of Total Digestion Method with XRF
There are numerous examples available in the literature confirming that total
digestion based on HF is a common procedure in determining total concentration of
the trace elements (e.g. Windom et al., 1989; Bettinelli et al., 2000; Sastre et al.,
2002). Studies focused on analysing metals within weathering products, however,

46
commonly apply XRF as the primary analytical method (e.g. Bauluz et al., 2000;
Voicu and Bardoux, 2002).
There also are many studies comparing these methods. For instance, Salminen and
Gregorauskiene (2000), compare the methods of total digestion and total analysis by
XRF. They concluded that as there are several analytical methods available for such
geochemical studies, the ultimate choice depends on the purpose of the study. While
total analysis gives absolute element abundance and a true picture of natural relations
of the sample analysed, for many practical purposes methods based on a partial leach
in which only a soluble part of the element is extracted and measured, are preferred.
In a study by Cook et al., (1997), however, it was concluded that HF methods are
recognised internationally as the ‘standard’ technique for total metal determination.
The HF digestion generally fails to recover a substantial proportion of V and Cr
which may be for two reasons. Either the HF attack fails to decompose some
chromium-containing materials (e.g. Cook et al., 1997) or due to presence of lithic
fragments of volcanic material, which are heterogeneously spread throughout the
matrix of sandstone (Cranfield, 1983; Hawkins, 1983), sampling of a representative
sub-sample cannot be achieved. A detailed analytical comparison is presented in
paper “Geochemical methods for characterisation of subtropical weathering and
metal release within sedimentary bedrock: Queensland, Australia”.
Comparability of Aqua Regia and HF-based Digestion
There are many studies in the literature comparing extractable and total digestion
methods for the determination of metals in soils and sediments (e.g. Cook et al.,
1997; Salminen and Gregorauskiene, 2000; Bettinelli et al., 2000; Sastre et al.,
2002). Since the existence of the ISO 11466 method (International Organisation for
Standardisation, ISO 1995), aqua regia extraction is one of the acid leaching methods
more widely used, however, the strong dependence of element recovery on the
applied leaching procedure makes it mandatory in each case to do a comparison with
a total digestion approach, including the use of HF (Sastre et al., 2002). Therefore,
considering the matrix of most environmental samples (soils and sediments), a total
digestion scheme must include the use of hydrofluoric acid to completely release the
trace elements included in the aluminosilicate phase (McGrath, 1998; Sanchez et al.,
1998).

47
Based on the above comparison therefore, in environmental studies where the focus
is on the extractable metals, it is not necessary to apply the total digestion method,
which is both more expensive and time consuming compared to extractable methods.
A comparison between aqua regia and HF-based digestion is presented in paper “The
influence of mineralogy and geological setting on trace metal concentration within
subtropical weathered profiles, Bells Creek catchment, Queensland, Australia”.
CONCLUSIONS From reading the broader literature related to the research topic, and summarised
here several main conclusions can be made:
• Chemical weathering of rocks is one of the major processes that modify the
earth’s surface contributing to the geochemical cycling of elements (Berner and
Berner, 1996). The effect of geological settings (e.g. parent rock type and
topography) climate and position of water table on chemical weathering of rocks is
well documented in literature (e.g. Jenny, 1941; Loughnan, 1969; Summerfield,
1991; Hill et al., 2000; Taylor and Eggleton, 2001). Clay minerals such as kaolinite,
illite and smectite, and iron minerals such as hematite, siderite and pyrite are the
main secondary products. There are several methodologies available on assessing
rock weathering including: calculation of chemical and mineralogical indices, weight
loss factor method and immobile element approach.
• Trace metals are transported from their source as dissolved or particulate
forms by streams and rivers and deposited on coastal floodplains, estuaries and bays.
Human activities (e.g. mining and industrial pollution) as well as natural processes
(e.g. erosion and weathering) are responsible for introducing trace metals into the
coastal environments. Therefore, in understanding metal occurrence in such settings,
both anthropogenic and natural sources have to be considered.
• Detrimental influence of trace metals on water quality has been covered in
this review. Surface water is the most appropriate environment for assessing,
monitoring and controlling metal pollution (Förstner, 1983). Some metals whose

48
concentration may reach toxic levels in surface water are aluminium, iron,
manganese and arsenic and their source can be traced back to adjacent sediments.
• Natural sedimentary metal loads may vary depending on the mineralogy and
grain size distribution. This natural variability can be compensated by normalisation
methods so that anthropogenic metal contribution can be identified and quantified.
Various granulometric and geochemical approaches can be used to normalise heavy
metal data from estuarine and coastal sediments.
• The analytical method chosen in the analyses of the heavy metal content
depends on the purpose of the study. A comparison between HF and XRF confirmed
previous findings that these methods are comparable for most chemical elements
except for refractory elements such as V and Cr. In comparing HF with aqua regia, it
was concluded that while the use of HF ensures a total digestion of the
aluminosilicate matrix, other approaches such as an aqua regia extraction also allows
digesting a high number of samples simultaneously and the extractable metal
amounts are similar to the total content except for V and Cr. In cases where the
focus of study is the extractable cations, using aqua regia is especially important as it
enables the researcher to look at only the extractable form of element, which is
readily available and released to the environment.

49
REFERENCES: ALGAN O., CLAYTON T., TRANTER M. AND COLLINS M.B. 1994. Estuarine
mixing of clay minerals in the Solent region, southern England. Sedimentary
Geology 92: 241-255.
ALLEN A.E., GRIMSHAW H.M., PARKINSON J.A., QUARMBY C. 1974.
Chemical analysis of ecological material. Blackwell Scientific: London.
ALLOWAY B.J. 1990a. Soil processes and the behaviour of metals. Heavy metals in
soils. B. J. Alloway., Blackie, J Wiley and Sons, Inc: New York pp. 7-28.
ALLOWAY B.J. 1990b. The origin of heavy metals in soils. Heavy metals in soils.
B. J. Alloway. Blackie, J Wiley and Sons, Inc: New York pp. 29-40.
ALLOWAY B.J. 1990c. Cadmium. Heavy metals in soils. B. J. Alloway. Blackie, J
Wiley and sons, Inc: New York pp. 100-124.
ANDREW R.W., HODSON P.V., KONASEWICH D.E. 1976. Toxicity to biota of
metal forms in natural water. International Joint Commission: Windsor,
Ontario.
ARAKEL, A.V., HONGJUN T. 1992. Heavy metal geochemistry and dispersion
pattern in coastal sediments, soil and water of Kedron Brook floodplain area,
Brisbane. Environmental Geology and Water Science 20: 219-231.
BACHE B.W. 1986. Aluminium mobilisation in soils and waters. Journal of the
Geological Society 143: 699-706.
BAKER D.E. 1990. Copper. In “Heavy metals in soils” Alloway, B. J. (ed.), Blackie,
John Wiley and Sons, Inc: New York pp. 151-176.
BALLS P.W., HULL S., MILLER B.S., PIRIE J.M, AND PROCTOR W. 1997.
Trace Metal in Scottish estuarine and coastal sediments. Marine Pollution
Bulletin 34(1): 42-50.
BAULUZ B., MAYAYO M.J., FERNANDEZ-NIETO C. AND GONZALEZ
LOPEZ J.M. 2000 Geochemistry of Precambrian and Paleozoic siliciclastic
rocks from the Iberian Range (NE Spain): implications for source-area
weathering, sorting, provenance, and tectonic setting. Chemical Geology 168:
135-150.
BELL F.G. 1998. Environmental geology, principles and practice. Blackwell
Science, University Press Cambridge: London.

50
BERNER R.A. 1971. Principles of chemical sedimentology. McGraw-Hill, Inc:
USA.
BERNER R.A. 1981. A new geochemical classification of sedimentary
environments. Journal of Sedimentary Petrology 51: 359-365.
BERNER R.A. 1983. Sedimentary pyrite formation: an update. Geochimica et
Cosmochimica Acta 48: 605-615.
Berner, E. K. and R. A. Berner 1996. Global environment: water, air and
geochemical cycles. Prentice Hall: New Jersey p 376.
BERNER E.K. AND BERNER R.A. 1996. Global Environment. Prentice Hall: New
Jersey.
BETTINELLI M., BEONE G.M., SPEZIA S. AND BAFFI C. 2000. Determination
of heavy metals in soils and sediments by microwave-assisted digestion and
inductively coupled plasma optical emission spectrometry analysis. Analytica
Chimica Acta 424: 289-296.
BIRCH G.F. AND TAYLOR S. 1999. Source of heavy metals in sediments of the
Port Jackson estuary, Australia. The Science of The total Environment 227:
123-138.
BLAND W. AND ROLLS D. 1998. Weathering: an introduction to the scientific
principles. Arnold: New York.
BOGGS S. 1995 Principles of sedimentology and stratigraphy. Prentice Hall: New
Jersey.
BOYLE E.A., EDMOND, J. M. AND SHOLKOVITZ, E. R. 1977. The mechanism
of iron removal in estuaries. Geochimica et Cosmochimica Acta 41: 1313-
1324.
BRAUN J.J., PAGEL M., HERBILLON A. AND ROSIN C. 1993 Mobilisation and
redistribution of REEs and thorium in a syenitic lateritic profile: a mass
balance study. Geochimica Cosmochimica Acta 57: 4419-4434.
BULTER I.B. AND RICKARD D. 2000. Framboidal pyrite formation via the
oxidation of iron (II) monosulfide by hydrogen sulfide. Geochimica et
Cosmochimica Acta 64(15): 2665-2672.
CARROLL D. 1970. Rock weathering. Plenum Press: New York-London. pp. 203.
CHAMLEY H. 1989. Clay sedimentology. Springer-Verlag: Berlin. Heidelberg.
New York. London. Paris. Tokyo. Hong Kong.

51
CANFIELD D.E., LYONS T.W., RAISWELL R. 1996. A model for iron deposition
to euxinic Black Sea sediments. American Journal of Science. 296:818-834.
CHESWORTH W., DEJOU J. AND LARROQUE P. 1981. The weathering of basalt
and relative motilities of the major elements at Belbex, France. Geochimica
Cosmochimica Acta 45: 1235-1243.
CHIGIRA M., SONE K. 1991. Chemical weathering mechanisms and their effects
on engineering properties of soft sandstone and conglomerate cemented by
zeolite in a mountainous area. Engineering Geology 30: 195-219.
CHIGIRA M., OYAMA T. 1999. Mechanism and effect of chemical weathering of
sedimentary rocks. Engineering Geology 55: 3-14.
COLMAN S.M. AND DETHIER D.P. 1986. Rates of chemical weathering of rocks
and minerals. Academic Press: Orlando. pp. 603.
COOK J.M., GARDNER M.J., GRIFFITHS A.H., JESSEP M.A., RAVENSCROFT
J.E. AND YATES R. 1997. The comparability of sample digestion
techniques for the determination of metals in sediments. Marine Pollution
Bulletin 34 (8): 637-644.
COX M.E., PREDA M. AND BROOKE B. 2000. General features of the geo-setting
of the Pumicestone Region. PASSCON 2000, Brisbane, School of Natural
Resource Sciences, Queensland University of Technology. pp 7-12.
CRAMER J.J. AND NESBITT H.W. 1983. Mass-balance relations and trace element
mobility during continental weathering of various igneous rocks.
Symp.Petrol. Weather Soils. Sci.Geol.Mem 73: 63-73.
CRANFIELD L.C. 1983 Shallow stratigraphic drilling in the Brisbane 1:100,000
sheet area, Record 40. Geological Survey of Queensland.
CRERAR D.A., MEANS J.L., YURETICH R.F, BORCSIK M.P., AMSTER J.L.,
HASTINGS D.W., KNOX G.W., LYON K.E. AND QUIETT R.F. 1981.
Hydrogeochemistry of the New Jersey coastal plain, transport and deposition
of iron, aluminum, dissolved organic matter, and selected trace elements in
stream, ground and estuary water. Chemical Geology 33: 23-44.
CROWTHER M.E. 1930 The relationship of climate and geological factors to the
composition of the clay and the distribution of soil types. Proceedings of
Royal Society 107: 10-30.

52
DASKALAKIS, K.D., AND O'CONNOR T.P. 1995. Normalization and Elemental
Sediment Contamination in the Coastal United States. Environmental Science
and Technology 29(2): 470-477.
DAVIES B.E. 1990. Lead. In “Heavy metals in soils” Alloway, B. J. (ed.), Blackie,
John Wiley and Sons, Inc: New York pp. 177-196.
DEGROOT A.J. AND ALLERSMA E. 1975. Field observations on the transport of
heavy metals in sediments. Heavy Metals in the Aquatic Environment,
Pergmon Press: Tennessee, USA.
DENT D. 1986. Acid sulfate soils: a base line for research and development. ILRI:
Wagenington, The Netherlands.
DENT D. AND PONS L. 1993. Acid and muddy thoughts. National Conference on
Acid Sulfate Soils, NSW Agriculture: Orange, NSW.
DENT D. AND PONS L. 1995. A world prospective on acid sulfate soils.
Geoderma, 67: 263-176.
DREVER J.I. 1994. The effect of land plants on weathering rates of silicate mineral.
Geochimica et Cosmochimica Acta 58: 2325-2332.
DREVER J.I. 1997. The geochemistry of natural waters: surface and groundwater
environments. Upper Saddle River: New Jersey.
DYER K.R. 1997. Estuaries: a physical introduction. John Wiley and Sons:
Chichester.
EDMUNDS W.M. AND SMEDLEY P.L. 1996. Groundwater geochemistry and
health: an overview. In “Environmental geochemistry and health”. Appleton,
J. D., Fuge, R. and McCall, G. J. H. (eds.), Geological Society. 113: 91-105.
EGGLETON R.A., FOUDOULIS C. AND VARKEVISSER D. 1987 Weathering of
basalt: changes in rock chemistry and mineralogy. Clays and Clay Minerals
35(3): 161-169.
ELLAWAY M., HART B.T., BECKETT R. 1982. Trace metals in sediments from
the Yarra River. Australian Journal of Marine and Freshwater Research 33:
761-778.
EVANS L.J. 1989. Chemistry of metal retention by soils. Environmental Science and
Technology 23: 1046-1056.
FANG T., AND HONG E. 1999. Mechanisms Influencing the Spatial Distribution of
Trace Metals in Surficial Sediments off the South-Western Taiwan. Marine
Pollution Bulletin 38(11): 1026-1037.

53
FAURE, G. 1998. Principles and application of geochemistry. Prentice Hall: New
Jersey.
FEDO C.M., NESBITT H.W. AND YOUNG G.M. 1995 Unraveling the effects of
potassium metasomatism in sedimentary rocks and paleosols, with
implications for paleoweathering conditions and provenance. Geology 23:
921-924.
FISCHER A.G. 1963. Essay review of descriptive paleoclimatology. American
Journal of Science 261: 281-293.
FÖRSTNER U., AND MULLER G. 1973. Heavy metal accumulation in river
sediments, a response to environmental pollution. Geoforum 14: 53-62.
FÖRSTNER U. AND SALOMONS W. 1980. Trace metal analysis on polluted
sediments. Part I. Assessment of sources and intensities. Environmental
Technology Letters 1: 494-506.
FÖRSTNER U. 1981. Metal pollution assessment from sediment analysis. Metal
pollution in the aquatic environment. U. Forstner and G. T. W. Wittmann.
Springer-Verlag: Berlin pp. 110-196.
FÖRSTNER U., CALMANO, W. AND SCHOER, J. 1982. Metals in sediments
from the Elbe, Weser and Ems estuaries and from the German Bight: grain
size effects and chemical forms. Thalassia Jugostavica 18(1-4): 97-122.
FÖRSTNER U. 1983. Assessment of metal pollution in rivers and estuaries. Applied
environmental geochemistry. I. Thornton. Academic Press: New York.
FÖRSTNER U., AND WITTMANN, G.T.W. 1983. Metal pollution in the aquatic
environment. Springer Verlag: Berlin.
FÖRSTNER U., WOLFGANG A, WOLFGANG C., KERSTEN M., AND
SALOMONS W. 1984. Mobility of heavy metals in dredged harbour
sediments. Sediment and water interaction: proceedings of the third
international symposium on interactions between sediments and water,
Geneva, Switzerland, Springer-Verlag: New York.
FÖRSTNER U. 1989. Contaminated sediments. Springer-Verlag: Berlin.
FUNG Y.S. 1993. Analysis and assessment of heavy metal pollution in Hong Kong's
marine environment. In “The marine biology of South China Sea. Proceeding
of the first international conference on the marine biology of Hong Kong and
the South China Sea, Hong Kong, 28 October-3 November 1990 (B. Morton,
ed), pp 261-272, Hong Kong University Press, Hong Kong.

54
GARRELS R.M. 1967. Genesis of some groundwaters from igneous rocks. In
Abelson, P. H. (ed.) Researches in geochemistry, John Wiley: New York, pp
405-420.
GIBBS R.J. 1993. Metals of the bottom muds in Townsville Harbor, Australia.
Environmental Pollution 81: 297-300.
GOLDLICH S.S. 1938. A study in rock weathering. Journal of geology 46: 17-58.
HAMBLIN W.K. AND CHRISTIANSEN E.H. 2001 Earth's Dynamic Systems.
Brigham Young University Provo: Utah.
HAWKES H.E. AND WEBB J.S. 1962. Geochemistry in Mineral Exploration,
Happer and Row and Weatherhill, Inc.: Tokyo, p. 415.
HAWKINS P.J. 1983. Petrology of sandstone samples from road cuttings, southeast
Queensland, Record 22. Geological Survey of Queensland.
HILL I.G., WORDEN R.H. AND MEIGHAN I.G. 2000. Geochemical evolution of a
palaeolaterite: the interbasaltic formation, Northern Ireland. Chemical
Geology 166: 65-84.
HEM J.D. 1992. Study and interpretation of the chemical characteristics. 4th ed. U.S.
Geological Survey Water Supply Paper 2254.
HODSON M. 2002 Experimental evidence for mobility of Zr and other trace
elements in soils, Geochimica et Cosmochimica Acta, 66(5): 819-828.
IRVINE I. AND G.F. BIRCH 1998. Distribution of heavy metals in surficial
sediments of Port Jackson, Sydney, NSW. Australian Journal of Earth
Sciences 45: 297-304.
ISO 11466 1995. Soil quality: Extraction of trace metals soluble in aqua regia.
International Organisation for Standardisation.
JENNY H. 1941 Factors in soil formation. McGraw-Hill: New York.
JOYCE A.S. 1984. Geochemical exploration. Glenside, Australian Mineral
Foundation.
KAWAMURA K., OGAWA Y., IKEHARA K. AND TANIGUCHI H. 2002.
Growth process of framboidal pyrite in deep-sea sediment. Geological
Society of America Abstracts with Program, 34 (6): 207.
KIEKENS L. 1990. Zinc. In “Trace metals in soils”. Alloway, B. J. (ed.) Blackie,
John Wiley and Sons, Inc: New York, pp. 261-279.
KONONOVA M.M. 1966. Soil organic matter. Pergamon Press: New York.

55
KRAUSKOPF K.B. 1956. Factors controlling the concentration of thirteen rare
metals in seawater. Geochimica et Cosmochimica Acta 9: 1-32.
KRAUSKOPF K.B. AND BIRD D.K. 1995. Introduction to Geochemistry.
McGraw-Hill, Inc: New York.
KRUMBIEN W.C. AND GARRELS R.M. 1952. Origin and classification of
chemical sediments in terms of pH and oxidation-reduction potentials.
Journal of Geology 60: 1-33.
LERMAN A. AND MEYBECK M. 1988. Physical and chemical weathering in
geochemical cycles. Kulwar Academic: Dordrecht, pp. 375.
LIN C., MELVILLE M.D. AND HAFER S. 1995. Acid sulfate soil – landscape
relationship in an undrained , tide-dominated estuarine floodplain, eastern
Australia, Catena 24: 177-194.
LISS P.S. 1976. Conservative and non-conservative behaviour of dissolved
constituents during estuarine mixing. In “Estuarine Chemistry”. Burton, J.D.
and Liss, P.S. (eds.), Academic Press: New York, pp. 93-130.
LORING D.H. 1990. Lithium - a new approach for the granulometric normalization
of trace metal data. Marine Chemistry 29: 155-168.
LORING D.H. 1991. Normalization of heavy-metal data from estuarine and coastal
sediments. Journal of Marine Science 48: 101-115.
LOUGHNAN F.C. 1969 Chemical weathering of silicate minerals. American
Elsevier Publishing Company, Inc.: New York.
LOVE L.G., AMSTUTZ G.C. 1966. Review of microscopic pyrite from the
Devonian Chattanooga shale and Rammelsberg Banderz. Fortschr. Mineral.
43: 273-309.
LYONS T.W. 1997. Sulfur isotopic trends and pathways of iron sulfide formation in
upper Holocene sediments of the anoxic Black Sea. Geochimica et
Cosmochimica Acta. 61: 3367-3382.
MACIAS F. AND CHESWORTH W. 1992 Weathering in humid regions, with
emphasis on igneous rocks and their metamorphic equivalents. In:
Weathering, soils and paleosols, Martini IP and Chesworth W (eds.). Elsevier
Science Publishers B.V.: Amsterdam, The Netherlands.
MACKENZIE F.T. AND GARRELS R.M. 1966. Chemical mass balance between
rivers and oceans. American Journal of Science 264: 507-525.

56
MANTEI E.J., AND FOSTER M.V. 1991. Heavy metals in stream sediments: effects
of human activities” Environmental Geology and Water Sciences. 18(2): 95-
104.
MASON B. 1958. Principles of Geochemistry. Wiley: New York, 2nd edition, p. 310.
MATTIGOD S.V. AND PAGE A.L. 1983. Assessment of metal pollution in soils. In
“Applied environmental geochemistry”. Thornton, I. (ed.), Academic Press:
New York.
MAY H.M., HELMKE P.A., JACKSON M.L. 1979. Gibbsite solubility and
thermodynamic properties of hydroxy-aluminium ions in aqueous solution at
25°C. Geochimica et Cosmochimica Acta 43: 861-868.
MCGRATH S.P., AND SMITH S. 1995. Chromium and nickel. In “Heavy metals in
soils”. Alloway, B. J. (ed.), Blackie, John Wiley and Sons, Inc: New York,
pp. 125-150.
MCGRATH D. 1998. Use of microwave digestion for estimation of heavy metal
content of soils in a geochemical survey. Talanta 46: 439-448.
MIDDELBURG J.J., DE LANGE G.J., VAN DER SLOOT H.A., VAN EMBURG
P.R. AND SOPHIAH S. 1988a. Particulate manganese and iron framboids in
Kau Bay, Halmahera (Eastern Indonesia). Marine Chemistry. 23: 353-364.
MIDDELBURG J.J., CORNELIS H., WEIJDEN V.D. AND WOITTIEZ J.R.W.
1988b. Chemical processes affecting the mobility of major, minor and trace
elements during weathering of granitic rocks. Chemical Geology 68: 253-
273.
MOHR E.J.C. AND VAN BAREN F.A. 1954. Tropical soils. Interscience: New
York.
MUROWCHICK J.B. AND BARNES H.L. 1987. Effects of temperature and degree
of supersaturation on pyrite morphology. American Mineralogy 72: 1241-
1250.
NAHON D.B. 1991. Introduction to the petrology of soils and chemical weathering.
John Wiley and Sons: New York, pp. 313.
NESBITT H.W. 1979. Mobility and fractionation of rare earth elements during
weathering of a granodiorite. Nature 279: 206-210.
NESBITT H.W. AND YOUNG G.M. 1982. Early Proterozoic climate and plate
motions inferred from major element chemistry of lutites. Nature 299: 715-
717.

57
NESBITT H.W. AND WILSON R.E. 1992 Recent chemical weathering of basalts.
American Journal of Science 292: 740-777.
NIENCHESKI L.F., WINDOM H.L. AND SMITH R. 1994. Distribution of
Particulate Trace Metal in Patos Lagoon Estuary (Brazil). Marine Pollution
Bulletin 28(2): 96-102.
O'NEILL P. 1990. Arsenic. In “Heavy metals in soils”. Alloway, B. J. (ed.) Blackie,
John Wiley and Sons, Inc: New York, pp. 83-99.
ONG CHE R.G. 1999. Concentration of seven heavy metals in sediment and
mangrove root samples from Mai Po, Hong Kong. Marine Pollution Bulletin.
Marine Pollution Bulletin 39: 169-279.
PERILLO G. 1995. Geomorphology and sedimentology of estuaries. Elsevier:
Amsterdam.
PLANT J.A., BALDOCK J.W., SMITH B. 1996. The role of geochemistry in
environmental and epidemiological studies in developing countries: a review.
In “Environmental geochemistry and health with special reference to
developing countries”. Appleton, J. D., Fuge, R. and McCall, G. J. H. (eds.),
Geological Society: London. No. 113: 7-22.
PONS L.J., VAN BREEMAN N., AND DRIESSEN P.M. 1982. Physiography of
coastal sediments and development of potential acidity. In “ASA special
publication”. Soil Science Society of America: Madison, Wisconsin, pp. 1-
18.
PREDA M. AND. COX M.E 2001. Trace metals in acid sediments and waters,
Pimpama River, Southeast Queensland. Environmental Geology 40(6): 755-
768.
PREDA M. AND COX M.E. 2002. Trace metals occurrence and distribution in
sediments and mangroves, Pumicestone Region, southeast Queensland,
Australia, Environment International 28: 433-449.
ROLLINSON H.R. 1993. Using geochemical data: evaluation, presentation,
interpretation. John Wiley and Sons: New York, pp. 352.
SALMINEN R. AND GREGORAUSKIENE V. 2000. Considerations regarding the
definition of a geochemical baseline of elements in the surficial materials in
areas differing in basic geology. Applied Geochemistry 15: 647-653.

58
SAMMUT J., MELVILLE M.D., CALLINAN R.B., FRASER G.C. 1995. Estuarine
acidification: Impacts on aquatic biota of draining acid sulfate soils.
Australian Geographical Studies 33(1): 89-100.
SAMMUT J., CALLINAN R.B., FRASER G.C. 1996. An overview of the ecological
impacts of acid sulfate soils in Australia. Second National Conference on
Acid Sulfate Soils, NSW, Alstonville and ASSMAC: NSW.
SANCHEZ J., MARINO N., VAQUERO M.C., ANSORENA J. AND
LEGORBURU I. 1998. Metal pollution by old lead-zinc mines in Urumea
River Valley (Basque country, Spain). soil, biota and sediment. Water, Air,
and Soil Pollution 107: 303-319.
SASTRE J., SAHUQUILLO A., VIDAL M. AND RAURET G. 2002. Determination
of Cd, Cu, Pb and Zn in environmental samples: microwave assisted total
digestion versus aqua regia and nitric acid extraction. Analytica Chimica Acta
462: 59-72.
SAWLOWICZ Z. 2000. Framboids: from their origin to application, Prace
Mineralog. Pan 88: 1-80.
SAXBY J.D. 1973. Diagenesis of metal-organic complexes in sediments: formation
of metal sulfides from crystalline complexes. Chemical Geology 12: 241-288.
SHERMAN G.D. 1952. Problems in clay and laterite genesis. American Institute of
Mining and Metallurgical Engineers: New York.
SHOLKOVITZ E.R. 1976. Flocculation of dissolved organic and inorganic matter
during the mixing of river and seawater. Geochimica et Cosmochimica Acta
40: 831-845.
SKEI J.M. 1988. Formation of framboidal iron sulfide in the water of a permanently
anoxic fjord, Framvaren, South Norway. Marine Chemistry. 23: 345-352.
STEYRER H.P. AND STRUM R. 2002 Stability of zircon in a low-grade
ultramylonite and its utility for chemical mass balancing: the shear zone at
Miéville, Switzerland, Chemical Geology 187: 1-19.
SUITS N.S., WILKIN R.T. 1998. Pyrite formation in the water column and
sediments of a meromictic lake. Geology. 26: 1099-1102.
SUMMERFIELD M.A. 1991. Global Geomorphology. John Wiley and Sons, Inc:
New York.

59
TAM N.F.Y., AND YAO M.W.Y. 1998. Normalization and heavy metal
contamination in mangrove sediments. The Science of The Total Environment
216: 33-39.
TAYLOR G. AND EGGLETON R.G. 2001. Regolith geology and geomorphology.
John Wiley and Sons: England.
THOMAS M.F 1994 Geomorphology in the tropics: A study of weathering and
denudation in low latitude. John Wiley and sons: Chichester, England.
TRIMBLE C.A., AND HOENSTINE R.W. 1997. Baseline investigation of estuarine
sediment metals for the Steinhatchee River area of the Florida Big Bend.
Environmental Geosciences 4(2): 95-103.
VELBEL M.A. 1984 Mineral transformations during rock weathering and
geochemical mass-balances in forested watersheds of the southern
Appalachians. PhD dissertation, Yale university: New Haven.
VELDE B. 1992. Introduction to clay minerals, chemistry, origin, uses and
environmental significance. Chapman and Hall: London. New York. Tokyo.
Melbourne.
VOICU G., BARDOUX M., JEBRAK M. AND VOICU D. 1996. Normative
mineralogical calculations for tropical weathering profiles, Winnipeg '96,
Annual meeting: Geological Association of Canada and Mineral Association
of Canadian Program with Abstract 21 (A-69).
VOICU G., BARDOUX M., HARNOIS L. AND CREPEAU R. 1997. Lithological
and geochemical environment of igneous and sedimentary rocks at the Omai
gold mine, Guyana, South America. Exploration and Mining Geology 6: 153-
170.
VOICU G. AND BARDOUX M. 2002. Geochemical behaviour under tropical
weathering of the Barama-Mazaruni greenstone belt at Omai gold mine,
Guiana Shield. Applied Geochemistry 17: 321-336.
VOLKOV I.I. AND FOMINA L.S. 1974. Influence of organic material and
processes of sulfide formation on distribution of some trace elements in
deepwater sediment of the Black Sea. The Black Sea, Geology, Chemistry
and Biology. E. T. Degens, and Ross, D.A. Tulsa, Okla, American
Association of Petrology and Geology: 456-476.

60
WANG Q.W., MORSE J.W. 1996. Pyrite formation under conditions approximating
those in anoxic sediments. 1. Pathway and morphology. Marine Chemistry.
52: 99-121.
WILKIN R.T., BARNES H.L. 1997a. Formation processes of framboidal pyrite.
Geochimica et Cosmochimica Acta. 61: 323-339.
WILKIN R.T., BARNES H.L. 1997b. Pyrite formation in an anoxic estuarine basin.
American Journal of Science. 297: 620-650.
WINDOM H.L., SCHROPP S.J., CALDER F.D., RYAN J.D., SMITH R.G.,
BURNEY L.C., LEWIS F.G., AND RAWLINSON C.H. 1989. Natural trace
metal concentrations in estuarine and coastal marine sediments of the
southern United States.” Environmental Science and Technology 23(3): 314-
320.
WOLLAST R. 1983. Interactions in estuaries and coastal waters. In “The major
biogeochemical cycles and their interactions”. Bolin, B. and Cook, R.B.
(eds.), John Wiley: England.
YOCESOY F. AND ERGIN M. 1992. Heavy-metal geochemistry of surface
sediments from the southern Black Sea shelf and upper slope. Chemical
Geology 99: 265-287.
ZOLLMERV, AND IRION G. 1993. Clay mineral and heavy metal distributions in
the northeastern North Sea. Marine Geology 111: 223-230.

61
PAPER 1 - THE INFLUENCE OF MINERALOGY AND GEOLOGICA L SETTING ON TRACE METAL CONCENTRATION WITHIN
SUBTROPICAL WEATHERED PROFILES, BELLS CREEK CATCHME NT, QUEENSLAND, AUSTRALIA
Tania Liaghati, Micaela Preda and Malcolm Cox
School of Natural Resource Sciences
Queensland University of Technology (QUT)
Journal of Pacific Science (in press)

94
PAPER 2 – GEOCHEMICAL METHOD FOR CHARACTERISATION O F SUBTROPICAL WEATHERING AND METAL RELEASE WITHIN
SEDIMENTARY BEDROCK: QUEENSLAND, AUSTRALIA
Tania Liaghati, Micaela Preda and Malcolm Cox
School of Natural Resource Sciences
Queensland University of Technology (QUT)
Journal of Geochemical Exploration (submitted)

127
PAPER 3 - HEAVY METAL DISTRIBUTION AND CONTROLLING FACTORS WITHIN COASTAL PLAIN SEDIMENTS, BELLS CREEK
CATCHMENT, SOUTHEAST QUEENSLAND, AUSTRALIA
Tania Liaghati, Micaela Preda and Malcolm Cox
School of Natural Resource Sciences
Queensland University of Technology (QUT)
Environment International (published)

163
PAPER 4 - DISTRIBUTION OF Fe IN WATERS AND BOTTOM SEDIMENTS OF A SMALL TIDAL CATCHMENT, PUMICESTONE
REGION, SOUTHEAST QUEENSLAND, AUSTRALIA
Tania Liaghati, Malcolm E. Cox, Micaela Preda
School of Natural Resource Sciences
Queensland University of Technology (QUT)
The Science of Total Environment (published)

189
GENERAL CONCLUSIONS This research project had two main objectives (a) to determine the natural factors
controlling the geochemistry of weathered profiles, unconsolidated sediments, soils
and natural waters, and (b) to identify the most effective analytical and numerical
methods for evaluating metal concentrations, in particular for large heterogeneous
geochemical data sets. A variety of approaches, including physical and chemical
analytical techniques as well as numerical methods have been applied to understand
the relationship and interaction between various system components in a small
coastal catchment. The location of the study, Bells Creek drainage system in
northern Moreton Bay provides a good example of a low-lying sub-tropical
catchment incorporating both freshwater and estuarine settings which formed during
the last marine transgression.
From a geochemical point of view, estuarine sediments can act as sinks for metals
mobilised as a result of natural processes such as weathering, as well as
anthropogenic activities. Many metals occur naturally in weathered materials and in
drainage system sediments due to their presence in local rocks; it is therefore, of
value to clearly differentiate natural and anthropogenic controls over the
geochemistry of a broad suite of geological materials within a coastal setting.
The Landsborough Sandstone, the bedrock of the region, is both labile and
heterogeneous; these characteristics along with the sub-tropical climate of the area
have resulted in development of thick weathering profiles. Bells Creek catchment
remains largely unpolluted, and therefore, weathering of the bedrock is considered to
be the major natural process controlling the geochemical cycling of the metals within
it. Paper 1 “The influence of mineralogy and geological setting on trace metal
concentration within subtropical weathered profiles, Bells Creek catchment,
Queensland, Australia” investigates chemical weathering, the natural factors
affecting it, and the relative influence of this weathering on trace metal distribution
and mobility. The outcomes of this paper were:
• Lithological variations (e.g. mineralogy) and geological setting along with
topography, water table position and depth of profile burial are shown to control the
nature of the weathered profile and the intensity of weathering.

190
• The influence of mineralogy, geological setting and groundwater occurrence
on chemical weathering and trace metal distribution can be summarised as follows:
mineralogy>geological setting>water table position>depth of profile burial
Based on data analysed (Paper 1, Figures 3 and 4), it is also concluded that:
• Flat areas with a shallow water table and fine-grained sediments such as
mudstone as parent rock are more prone to leach metals.
• Further, applying both extractable and total methods of digestion for selected
samples enabled a comparison between the amount of trace metals contained in
silicate mineral structures with that weakly adsorbed and readily extracted.
• Metals such as V, Cr and Fe were found to be part of the aluminosilicate
matrix and therefore, they are not readily releasable to the environment; these
elements therefore, remain in primary mineral structure and can undergo further
release during weathering.
• Other metals such as Cu, Zn and Pb, however, are predominantly adsorbed to
sediment grains and easily available to the environment.
Paper 2 “Geochemical methods for characterization of subtropical weathering and
metal release within sedimentary bedrock: Queensland, Australia” compared a
variety of analytical and numerical methods to understand the processes involved in
trace metal mobility and to establish their sequence of mobility in weathered profiles
of sedimentary rocks.
While total digestion based on HF is commonly used in determining total
concentration of trace elements, environmental studies of metal concentrations
within weathered products apply XRF as the primary analytical method. Both
methods of digestion are compared and it is concluded that:
• For most chemical elements, especially for major oxides (Table 1 Appendix
14), HF and XRF methods are comparable except for elements such as V and Cr
(Table 2, Appendix 14) where the HF digestion produces relatively lower recovery
levels (up to 35% for V, and 77% for Cr) largely due to the refractory nature of these
metals. These findings are in agreement with previous reported studies in which

191
total digestion by HF and analysis by XRF were compared. The general conclusion
is that as there are several analytical methods available for geochemical studies, the
ultimate choice depends on the purpose of the study; HF digestion however, is a
fundamental and internationally recognised technique in monitoring programs.
It was also established that the local sandstone-dominated bedrock is the major
source of metals in unconsolidated material of this area (Appendix 2). Therefore, a
further objective of Paper 2 was to understand the geochemical processes during
mobilisation and deposition of these elements throughout the catchment. In order to
achieve that, it was essential to assess the trace metal geochemistry within the
weathered profiles (Paper 2). The numerical tools applied to the dataset were
“chemical and mineralogical indices” such as chemical and mineralogical indices of
alteration, “weight loss” and “immobile element approach”. The first two methods
were not considered to be appropriate for this current application as they are both
based on the assumption that the weathering profile is a closed system without mass
transfers. Moreover, as quantitative differences between the weathering indices were
too small to enable comparisons, MIA did not provide a clear evaluation of the metal
distribution throughout weathered profiles. The findings of this part of the study can
be summarised as:
• The “immobile element approach” was identified as the most appropriate
method, as it considers the weathering system to be open; the approach transforms
the absolute values of trace metals to quantitatively evaluate their mobility and
enables a calculation of loss and gains.
• Applying the method, led to predicting a sequence of mobility for metals in
the study area, which, however, could not be readily generalised in other areas as it is
greatly influenced by bedrock mineralogy. The sequence of mobility for trace metals
in the study area can be summarised as:
Zn>Pb>Cu>Cr>V
While based on general literature, Pb is considered an immobile element, in the
sequence of mobility in this study, Pb appears to be among the mobile elements.
This finding is in agreement with Paper 1 where two methods of extractable and total
digestion were compared and it was concluded that Cu, Zn and Pb are adsorbed to
the sediments and easily available to the environment. Whereas, V and Cr were

192
identified to be part of aluminosilicate matrix which is not easily available to the
environment resulting in immobility of these elements. Therefore, it can be
concluded that there is a relationship between the sequence of mobility and the
source of metals and one can predict the other. Further, whether applying the
analytical digestion methods or taking the numerical approach both sequence of
mobility and source of metals can be established.
As noted above, trace metals transported from natural and anthropogenic sources can
accumulate in estuarine and coastal sediments. As investigating the processes
controlling adsorption and mobilisation of metals in soils and sediments of this
coastal plain was an objective of this study, Paper 3 “Heavy metal distribution and
controlling factors within coastal plain sediments, Bells Creek catchment, southeast
Queensland, Australia” examined natural and anthropogenic controls governing
trace metal distribution in soil and sediments of the coastal plain. Factors considered
were sediment source (fluvial/estuarine), organic matter as organic carbon,
mineralogy (in particular clay speciation) and local land use practices (e.g. pine
plantation versus native mangrove and Melaleuca forests). This paper is an
investigation into the lateral and vertical distribution of trace metals within a non-
industrialised coastal plain. As varying degrees of weathering have resulted in the
heterogeneous nature of the soils and sediments, a normalisation procedure was
applied to the data set and an enrichment factor was calculated to describe patterns of
metal distribution. A comparison between absolute and normalised metal contents in
soil cores with respect to Al content was made and it was concluded that:
• Elevated trace metal concentrations at some sites could be due to natural
enrichment resulted from bedrock weathering.
• In a highly heterogeneous setting, interpretations based on absolute
concentration of different elements alone are not sufficient.
• In Bells Creek catchment, natural sediment characteristics such as mineralogy
are dominant over anthropogenic inputs in controlling lateral / vertical metal
concentrations.
While iron is a biologically and geochemicaly important trace element in marine
systems, due to its environmental persistence and ecological risks, iron excess has
always been of environmental concern in such settings. In particular, in Halls Creek

193
sub-catchment, an area with anomalously high iron was detected earlier in this study
(Paper 3, Fe up to 14%). Of note, recent studies in the Pumicestone region have
identified iron as one of the major elements supporting the growth of Lyngbya
majuscula, which negatively affects the aquatic fauna.
The final part of the study, Paper 4 “Distribution of Fe in waters and bottom
sediments of a small tidal catchment, Pumicestone Region, southeast Queensland,
Australia” therefore, concentrated on identifying iron species and elucidating its path
from the source to sediments, surface and groundwater, and suspended matter.
Moreover, previously found pyrite in the area provided the opportunity to consider
the morphological variations that occur in framboidal pyrite. Major findings are:
• Iron concentration is generally high in natural waters of this
catchment with highest amount occurring at the boundary between the fresh and
saline sections of the catchment (Fe = 15.7 mg/L).
• In bottom sediments iron predominantly occurs as hematite or pyrite,
which demonstrate depositional significance.
• Iron released from adjacent land during weathering precipitates as
hematite in the fresher water section, however, anaerobic degradation of organic
matter in aquatic sediments results in reduction of hematite; the reduced Fe2+ along
with sulfate from seawater and high organic matter therefore, forms pyrite in the
estuarine sediments.
• Pyrite was identified in both bottom sediment and particulate matter
samples. While perfectly spherical closely packed framboids were only found in
suspended matter, the rare form of euhedra was present only in bottom sediments
indicating slow crystallisation and minimum degree of disturbance for this setting.
• Further, based on the elevated oxygen content of water it was
concluded that the framboids of pyrite found floating with suspended material had
not formed in water and they were a result of re-suspension and mobilisation from
bottom sediments.
Overall, this study has scientific significance as well as environmental management
application on a regional scale. As the study area will be under development

194
pressure in the future, this study will represent a baseline of comparison for future
environmental assessment projects as well as provide a scientific framework to gauge
suitable development strategies.
The broad findings of the study have:
(1) Established the order and degree in which natural factors control weathering.
(2) Tested and proposed a number of analytical and assessment methods in
evaluating weathering and subsequent metal release from sedimentary rocks.
(3) Assessed natural versus anthropogenic factors and determined the order of
mobility of trace metals in weathered profiles.
(4) Determined the iron mineral speciation for an anomalous area and established
pyrite morphology.

195
APPENDIX 1 - CHEMICAL WEATHERING PROCESSES IN A SUBTROPICAL COASTAL CATCHMENT AS INDICATED BY SPATI AL
VARIATIONS IN TRACE ELEMENTS AND MINERALOGY, SOUTHE AST QUEENSLAND, AUSTRALIA

199
APPENDIX 2 - DETERMINATION OF QUATERNARY SEDIMENT SOURCES USING MINERALOGY AND GEOCHEMISTRY IN BELLS
CREEK CATCHMENT, PUMICESTONE PASSAGE, SOUTHEAST QUEENSLAND

200
16th Australian Geological Convention, 1-5 July 2002, Adelaide, South Australia
Determination of Quaternary sediment sources using mineralogy and
geochemistry in Bells Creek catchment, Pumicestone Passage, southeast
Queensland
Tania Liaghati, Micaela Preda and Malcolm Cox
School of Natural Resource Sciences, Queensland University of Technology,
Brisbane, QLD 4001

202
APPENDIX 3 - SPATIAL VARIATION OF HEAVY METALS WITH IN SURFICIAL SEDIMENTS OF A SUBTROPICAL COASTAL PLAIN, BELLS
CREEK CATCHMENT, SOUTHEAST QUEENSLAND, AUSTRALIA
Tania Liaghati, Micaela Preda and Malcolm Cox
School of Natural Resource Sciences Queensland University of Technology, Brisbane, QLD, Australia
In Proceedings of the XIIth International Conference on Heavy Metals in the Environment 26-30 May 2003, Grenoble France

210
APPENDIX 4 - MORPHOLOGICAL VARIATIONS OF FRAMBOIDAL PYRITE IN AN ESTUARINE ENVIRONMENT, PUMICESTONE
CATCHMENT, SOUTHEAST QUEENSLAND, AUSTRALIA
Tania Liaghati, Malcolm Cox and Micaela Preda School of Natural Resource Sciences
Queensland University of Technology, Brisbane, QLD 4001
In Proceedings of 17th Australian Geological Convention 8-13th February 2004, Hobart Tasmania

215
APPENDIX 5 – SILICATE ROCK ANALYSIS (MAJOR OXIDES, LOSS ON IGNITION AND SULFUR)

216
MAJOR OXIDE IN SILICATES
SILICATE ROCK ANALYSIS BY ICP
This method allows for the determination of all 10 major oxides from the one sample solution using standard silicate reference materials. Iron is determined as total ferric iron and expressed as Fe2O3T. APPARATUS: Digestion bottles: Nalgene 125ml polypropylene or low-density polyethylene (code number 2003-0004) with a machined gas tight tapered plastic screw cap, Storage bottles: Nalgene 250ml low density polyethylene storage bottles, 200ml borosilicate volumetric flask. 50mm polyethylene funnel. 10ml polyethylene pipette. 250ml polyethylene beaker. 25ml polyethylene measuring cylinder. 50ml polyethylene measuring cylinder. Pipette sucker. Wash bottle. Water bath.
REAGENTS: Hydrofluoric Acid (AR grade or better) 50%. 4mls per sample. Aqua regia (1:3 Nitric / Hydrochloric Acid - AR grade). 1ml per sample. Boric Acid (AR grade or better) 50g/litre. 50mls per sample. (It will be necessary to heat to 70°C and stir on magnetic stirrer to dissolve). Deionised water in preference to distilled water (check Si content of distilled water). Calibrating Standards: USGS reference standards or other silicate rocks covering the expected compositional range of the rocks to be analysed. APPARATUS PREPARATION: (a) Preparation of storage bottles. Transfer approximately 20mls 1:3 nitric acid to a bottle, shake vigorously for about 30 seconds, and transfer to the next bottle. Rinse the bottle thoroughly at least 3 times removing all traces of nitric acid and oven dry at 50°C. (b) Volumetric Flasks. Treat with nitric acid similar to the above procedure.

217
PROCEDURE: 1. Dry the powdered* rock sample in an oven at 105-110°C for 1 hour and allow to cool in a desiccator. *(This procedure relies on all of the constituents being ground extremely fine for complete dissolution to occur) 2. Weigh exactly 0.2000g (± 0.1mg) of dry - 200* mesh rock powder into a glass weighing bottle and quantitatively transfer to a pre-cleaned, labelled digestion bottle, and recap. Re-weigh the weighing bottle and calculate the exact weight of sample used. Write this weight on the digestion bottle if the weight is not 0.2000g. After all samples have been weighed, weigh out at least one control standard in the same manner as your samples were weighed. (Ask staff for a standard). Each student is required to include at least one control standard in each batch of analysis in order to monitor analytical technique, instrument calibration and drift. (A control standard is not to be confused with a blank). Leave one digestion bottle spare in each batch for a blank. The next two steps will be done by staff. Please ensure you have notified and allowed enough time for staff to do this step. 3. Premix the required volume of digestion acid in a polyethylene beaker allowing extra acid for the blank preparation. Prepare one blank for each batch of analysis. The ratio for 20mls of digestion acid (4 samples) is as follows; 1ml Nitric acid, 3mls hydrochloric acid and 16mls hydrofluoric acid. 4. Remove cap from the digestion bottle and lightly tap the corner of the bottle to consolidate the rock powder to one side. (as per diagram). Slowly rotate the bottle 180° and pipette in (plastic pipette) 5mls of acid mixture into the side of the bottle not allowing it to come in contact with the silicate sample. Recap the bottle tightly and swirl the contents. Allow the sample to stand overnight. Check for complete dissolution and if necessary the bottles may be heated in a water bath at 50°-60°C until the sample is dissolved NOTE Do not confuse incomplete digestion of the sample with white precipitates of Ca and Mg fluorides. The absence of any black mafic material is usually a dependable criterion for complete dissolution. 5. After digestion cool the bottles in a deep freeze (-15°C) for 15 minutes. Remove the bottles one at a time to the fume cupboard and rapidly add 50mls of boric acid solution by pipette (50g/litre)(The boric acid must be at room temperature). Alternatively and preferably, the digestion bottles may be transferred to the fume cupboard half submerged in a tray of ice during the addition of the boric acid solution. Recap the bottle and shake vigorously. Allow to stand for about 10 minutes and re-shake. If the solution is not completely clear heat the bottles to 60°C in a water bath for 30 minutes to dissolve all precipitated fluorides.

218
6. Remove cap from digestion bottle and add approximately 30mls deionised water. Transfer the contents to a 200ml borosilicate volumetric flask via a 50mm polyethylene funnel. Rinse the bottle at least three times transferring the contents into the funnel. Make up to the mark, shake vigorously and rapidly transfer to a pre-cleaned 250ml polyethylene storage bottle. The final concentration of silicate rock is 0.2000g/200ml. Determine all 10 major oxides directly from this solution using silicate reference materials (calibrating standards) prepared in the same manner. For each batch of samples, a solution blank must be prepared. (Carry out the above procedure without using any rock powder). For a complete silicate analysis 'loss on ignition' must also be performed see LOI procedure for more information. ICP DETERMINATION The Silicate Rock Analysis method is stored on disk in the ICP. This program has been optimised for the determination of all 10 major oxides. Three separate runs are carried out on each batch of analysis, for specific groups of elements have specific parameters for optimum precision, which is required for silicate analysis. Appropriate calibration standards and a blank are run first in order to obtain a calibration graph. The samples are run in batches of 10 and the ICP is then recalibrated to minimise drift. One or 2 control standards are run with each batch of 10 samples to monitor calibration and drift. Should the calibration of any of the elements fail, an error message will appear informing the operator immediately. All data is to be stored to disk and at the end of the run, if necessary, the calibration may be adjusted and the results reprocessed. All the major oxides together with barium and strontium are run through the ICP at a concentration of 0.1g/100mls (1000 dilution).
After summation of Oxide and Loss on Ignition values samples may be rerun for SiO2 and Al2O3 to obtain improved results. Acceptable results are 100.00 ± 1.00% ie between 99.00% and 101.00%
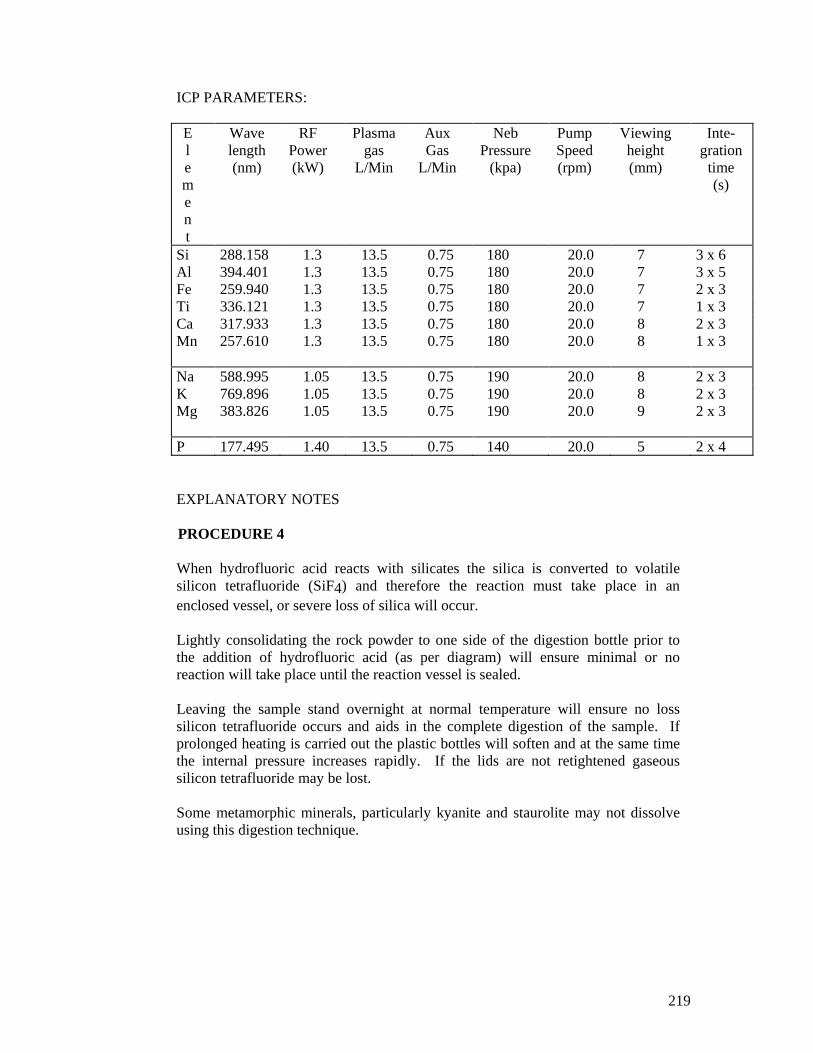
219
ICP PARAMETERS: E l e m e n t
Wave length (nm)
RF Power (kW)
Plasma gas
L/Min
Aux Gas
L/Min
Neb Pressure
(kpa)
Pump Speed (rpm)
Viewing height (mm)
Inte-gration time (s)
Si 288.158 1.3 13.5 0.75 180 20.0 7 3 x 6 Al 394.401 1.3 13.5 0.75 180 20.0 7 3 x 5 Fe 259.940 1.3 13.5 0.75 180 20.0 7 2 x 3 Ti 336.121 1.3 13.5 0.75 180 20.0 7 1 x 3 Ca 317.933 1.3 13.5 0.75 180 20.0 8 2 x 3 Mn
257.610 1.3 13.5 0.75 180 20.0 8 1 x 3
Na 588.995 1.05 13.5 0.75 190 20.0 8 2 x 3 K 769.896 1.05 13.5 0.75 190 20.0 8 2 x 3 Mg 383.826 1.05 13.5 0.75 190 20.0 9 2 x 3
P 177.495 1.40 13.5 0.75 140 20.0 5 2 x 4 EXPLANATORY NOTES
PROCEDURE 4 When hydrofluoric acid reacts with silicates the silica is converted to volatile silicon tetrafluoride (SiF4) and therefore the reaction must take place in an enclosed vessel, or severe loss of silica will occur. Lightly consolidating the rock powder to one side of the digestion bottle prior to the addition of hydrofluoric acid (as per diagram) will ensure minimal or no reaction will take place until the reaction vessel is sealed. Leaving the sample stand overnight at normal temperature will ensure no loss silicon tetrafluoride occurs and aids in the complete digestion of the sample. If prolonged heating is carried out the plastic bottles will soften and at the same time the internal pressure increases rapidly. If the lids are not retightened gaseous silicon tetrafluoride may be lost. Some metamorphic minerals, particularly kyanite and staurolite may not dissolve using this digestion technique.

220
PROCEDURE 5
The digestion bottles are cooled in a deep freeze to -15°C prior to the addition of boric acid to create a negative pressure in the bottle when the cap is removed. This reduces the possibility of any silicon tetrafluoride loss. The addition of boric acid is a critical component of the analysis. It performs the following functions; (i) It reacts with residual hydrofluoric acid to form fluoroboric acid which does not immediately react with glass. This enables the final volume of solution (200ml) to be made up in a volumetric flask. (ii) It reacts with and stabilises the volatile silicon tetrafloride and prevents any further volatilisation losses of silica. (iii) It reacts with and dissolves the precipitated calcium and magnesium fluorides formed during the digestion. (iv) Does not introduce any analyte elements into the digestion. LOSS ON IGNITION
Volatiles determined by loss on ignition will be most accurate for samples containing low concentrations of ferrous iron. The oxidation of ferrous iron during ignition increases the sample weight and if this is not subtracted from the LOI value, an incorrect result will be obtained. For samples containing low concentrations of volatiles and high concentrations of ferrous iron, the sample may actually gain weight rather than lose it after it has been ignited.
SULFUR
Sulfur is determined on the Leco R432 Sulfur analyser, by oxidation of the powdered sample in an atmosphere of pure oxygen at 1050°C, and quantitatively measuring the evolved sulfur dioxide by infra-red spectroscopy using appropriate calibrating standards.
To obtain significant results it is important the sample be ground extremely fine to ensure homogeneity of low concentration sulphides throughout the sample.
SAMPLE WEIGHT Approximately 0.250-0.300g of dried (110°C) sample is required for the Leco R432 Sulfur analyser.

221
ANALYSIS TIME Approximately 3 minutes per sample plus 3 minutes per calibrating standard and blank. REFERENCES ABBEY S. 1978. Calibration Standards. X-Ray Spectrometry, 7(2), 99-121. BERNAS B. 1968. A new method for decomposition and comprehensive analysis of silicates by atomic absorption spectrometry. Anal. Chem. 40(11), 1682. BUCKLEY D. E. & CRANSTON R. E. 1971. Atomic absorption analysis of 18 elements from a single decomposition of an aluminosilicate. Chem. Geol., 7, 273-284. EASTON A. J. 1972. Chemical analysis of silicate rocks. Elsevier Publishing Company, New York. FITTON J. G. & GILL R. C. O. 1970. The oxidation of ferrous iron in rocks during mechanical grinding. Geochim. Cosmochim. Acta 34, 518-524. FRENCH W. J. & ADAMS S. J. 1973. Polypropylene bottles in the decomposition of silicate rocks. Analytica Chimica Acta 62, 324-328. KWIECIEN W. 1990. Silicate rock analysis by AAS. School of Geology, Queensland University of Technology, Australia. LORING D. H. & RANTALA R. T. T. 1992. Manual for geochemical analysis of marine sediments and suspended particulate matter. Earth Science Reviews 32, 235-283. MAGILL W. A. & SUEHLA G. I. 1974. The study on the elimination of interferences in the determination of calcium by atomic absorption spectrophotometry. Anal. Chem. 268, 177-180. POTTS P. J. 1987. A handbook of silicate rock analysis. Chapman and Hall, New York. RANTALA R. T. T. & LORING D. H. 1989. Teflon bomb decomposition of silicate materials in a microwave oven. Anal. Chim. Acta, 220, 263-267. SAMCHUK A. I. & PILIPENKO A.T. 1987. Analytical chemistry of minerals. VNU Sciences Press, Utrecht, Netherlands.

222
APPENDIX 6 - TOTAL TRACE METAL ANALYSIS OF SEDIMENT BY HYDROFLUORIC ACID (UNIVERSITY OF QUEENSLAND)

223
Hot Plate Digestion Method applied in UQ
All acids used are concentrated and are distilled in-house and 18.2 megaohm Milli-Q water is used. All sample preparation and analytical procedures are carried out in HEPA filtered laboratories. � 100 mg of powdered sample is weighed into a PFA teflon beaker with a screw top lid and 2.5 ml of Hydrofluoric and 1 ml of Nitric acid is added. The lid is screwed on and the beaker left on a hot plate at 130C overnight. � The lid is removed and the solution dried down. � 1ml of Nitric acid is then added and dried down. � Another 1 ml of Nitric acid is added and dried down. � 2 ml of Nitric acid and 5ml of Milli-Q water is added and the lid screwed on and the beaker is left on a hotplate overnight at 100C. The solution is checked to ensure no insoluble fluorides remain. � 2 g of a 500ppb solution of internal standards is added to a cleaned 125ml LDPE bottle, the sample solution is added and then Milli-Q is added to give a total solution weight of 100g. The internal standards used are Rh, In, Re, Bi and artificially enriched isotopes of Li (6), Sr (84), Sm (147) and U (235). Samples were analysed on a Fisons PQ2+ Plasmaquad ICP-MS. Instrument operating parameters and data reduction procedures are as described in Eggins et al. (1997), except Tm is not used as one of the internal standards and a dolerite standard, W-2, was used for external calibration. Concentrations used for W-2 were derived partly by analysing it relative to synthetic standards (Li, Cr, Ni, Ga, Rb, Sr, Y, Zr, Nb, Cs, Ba, Hf, Ta, Pb, Th, U) or are based on an assessment of published, mainly ID-TIMS, data. Long-term reproducibility for the elements Li and Ga to U is about 1% and for Be to Zn is 2% to 4% as determined by repeated analyses of multiple digestions of basalt standards. REFERENCE EGGINS, S.M., WOODHEAD, J.D., KINSLEY, L.P.J., MORTIMER, G.E., SYLVESTER, P., MCCULLOCH, M.T., HERGT, J.M. & HANDLER, M.R. 1997. A simple method for the precise determination of >40 trace elements in geological samples by ICPMS using enriched isotope internal standardisation. Chemical Geology 134: 311-326,

224
APPENDIX 7 - TOTAL ELEMENT ANALYSIS BY X-RAY FLUORESCENCE SPECTROMETRY (XRF) (JAMES COOK UNIVERS ITY)

225
METHOD X-ray fluorescence (XRF) analysis provides analysis of the bulk sample in the solid form. Samples are usually prepared as fused discs (usually for major element analysis) and pressed powder pellets (for minor and trace element analysis). Both preparation methods give whole rock analysis, with none of the problems associated with the dissolution techniques cited above. XRF does have its problems with the analysis of the lighter elements and those elements tending to be volatile. For these analyses, slightly different preparation techniques are employed to keep these in the sample. Major elements (Na, Mg, Al, Si, P, S, K, Ca, Ti, Mn and Fe) are analysed with the fused disc preparation. This method minimises the mineralogical effects of the samples and produces a more accurate and precise analysis especially for the lighter elements. The glass bead from the fusion process effectively homogenises the sample so that the analysis at the surface of the bead is the same as from the bulk of the bead. This is especially important for the sodium and magnesium. However, fused beads are usually not suitable for low level analysis of trace elements: firstly because the sample is diluted, very long count times are necessary to achieve the lower limit of detection and secondly, there is usually significant contamination from the platinum crucibles that interferes with low level analysis of some elements. Therefore, trace elements are analysed from pressed powder pellets. Because most trace elements are heavy elements and thus have shorter wavelengths they are much less affected by the mineralogy of the sample and the analysis is typical of the bulk sample. JCU PREPARATION METHOD: Samples are accepted as ground powders. FUSED GLASS DISC Approximately 0.600 gms of sample is mixed with approximately 3.143 g of Norrish Hutton flux (contains La2O3) in a platinum crucible and heated at 1100oC for 15 minutes in a fusion furnace fitted with rocking device. The molten sample is quench pressed onto a graphite die and then annealed at 200oC for 30 minutes. The size of the glass disc is 28mm diameter. Weights of the sample and flux are recorded electronically for correction of the analysis for non-standard dilution. PRESSED POWDER PELLET A 5.5 gm aliquot of the ground sample is mixed with 5 drops of PVA and hydraulically pressed into an Aluminium cap. This forms a pellet of 28mm diameter. INSTRUMENTATION Both major and trace element analyses are obtained from a Bruker SRS3000 Sequential XRF Spectrometer. The Spectrometer is fitted with an Rh end-window 3kW tube, which operates under conditions suitable for each element and also a flow counter and a scintillation counter, which can be used simultaneously where possible. Analysing crystals are also chosen specific to the element. Count times and other analysing conditions are given in the results sheet. REFERENCE POTTS, P.J. 1996. A Handbook of Silicate Rock Analysis, Chapman and Hall, UK

226
APPENDIX 8 – EXTRACTABLE CATIONS IN SEDIMENTS

227
EXTRACTABLE CATIONS
IN SEDIMENT The determination of extractable cations in sediment by aqua-regia should not be confused with a total cation content. Aqua-regia (1 HNO3:3 HCl) used in this determination is unable to attack the silicate lattice therefore only adsorbed cations, oxides, hydroxides, carbonates and sulfides are digested. The following elements are routinely analysed: Cu, Pb, Co, Ni, Cr, Mo, Mn, V, Fe and Zn. Other extractable cation species may also be determined. SAFETY EQUIPMENT: Laboratory coat, safety glasses. Risk Assessments: Hydrochloric Acid (32%)/ Nitric Acid (69%)/Aqua Regia – Student usage APPARATUS:
50 mL test tubes with a 25mL calibration mark, 50 mL test tube uncalibrated, Whatman glass microfibre filters (GF/A), water bath,
REAGENTS:
SAFETY: Always use concentrated acids in an operational fume cupboard. Avoid inhalation of acid fumes. Hydrochloric Acid: Concentrated HCl (32%). Nitric Acid: Concentrated HNO3 (69%)
PROCEDURE: 1. Oven dry samples at 100-110oC until moisture free: usually overnight
depending upon volume and nature of sediment. 2. Accurately weigh 1.000 g of -80 mesh dried sediment onto a plastic weigh dish
and quantitatively transfer the sample into a 50 mL digestion tube graduated to 25 mLs. Transfer test tubes to a stainless steel test tube rack for digestion.
3. Add 1 mL conc HNO3 from an automatic acid dispenser and allow any vigorous effervescence to cease before continuing.
Add 3mLs conc HCl from automatic acid dispenser and allow any vigorous effervescence to cease before continuing.
4. SAFETY: Digestions must be carried out in an operational fume cupboard, ie set up water bath in a fume cupboard. Immerse racks into a water bath preset at about 50oC and observe reaction for at least 10 minutes. If reaction mixture effervesces vigorously, and rises rapidly above the 25 mL graduation mark, remove rack from water bath immediately and allow reaction to subside before re-entering into bath. if only one or two tubes react in this manner the reaction may be slowed down by the addition of a few mLs of distilled water from a wash bottle.

228
Increase the temperature of the water bath to 95oC and digest samples for 4 hours.
5 Allow samples to cool, add distilled water and make up to the 25 mL calibration mark. Swirl each test tube vigorously to intermix contents.
6. Re-label a series of 50 mL test-tubes in exactly the same sequence as the digested samples.
7. Filter the mixture through Whatman 41 papers or Whatman glass micro fibre filters GF/A.
8 Transfer filtered samples to plastic coated test-tube rack and seal test tubes with plastic inset caps or plastic wrap.
9. Determine trace element content in samples by ICP-OES analysis using an aqua regia blank and calibrating standards containing approximately the same matrix materials as the soil samples.
REFERENCES
LORING D. H. & RANTALA R. T. T. 1992. Manual for geochemical analysis of
marine sediments and suspended particulate matter. Earth Science Reviews 32, 235-283.

229
APPENDIX 9 - ORGANIC CARBON BY W ALKEY-BLACK METHOD

230
APPARATUS: 1 x 50 mL burette, 10mL pipette, 2 x 100mL volumetric flask, 500 mL conical
flasks SAFETY EQUIPMENT: Laboratory Coat, safety glasses, rubber gloves and fume cupboard. WARNING:
This procedure involves the use of strong acid and oxidising agents. Rubber gloves must be worn for all parts of this procedure.
Risk Assessment/s: Potassium Dichromate – Student usage Potassium Dichromate Solution – Student usage Orthophosphoric Acid – Student usage Sulfuric Acid – Student usage Sodium Fluoride – Student usage Barium Diphenylamine-4-sulphonate – Student usage REAGENTS: Orthophosphoric Acid: 85% H3PO4 Sodium Fluoride: Solid NaF Silver Sulfate and Sulfuric Acid: Weigh 1.25g Ag2SO4 into a 500 mL glass
bottle. Add 200 mL of conc. H2SO4. Shake to dissolve Ag2SO4. Add another 800 mLs of conc. to give a total of 1000 mLs H2SO4.
Standard 1 N Potassium Dichromate Solution: Weigh exactly 49.04g of
K2Cr2O7 and dilute to 1000 mL in a volumetric flask. 0.5 N Ferrous Solution: Weigh exactly 196.1 g of Fe(NH4)2SO4.6H2O.
Dissolve in 20 mL H2SO4. Transfer to a 1000 mL volumetric flask and dilute to the mark.
Diphenylamine Indicator: Weigh 0.5 g of Diphenylamine or BDS and
dissolving 20 mL of water and 100 mL of conc. H2SO4. PROCEDURE: 1) Weigh 0.5 g of dried dextrose into a 500 mL conical flask. 2) Add exactly 10 mL of 1 N K2Cr2O7 by pipette. 3) Add 20 mL of the H2SO4/Ag2SO4 mix. SAFETY: This must be
carried out in an operating fume cupboard. Mix by gently rotation flask to ensure complete mixing of reagents and sample while avoiding splashing sample onto sides of the flask out of contact with reagents.
4) Let mixture stand for 10 mins. 5) Add 200 mLs of distilled water, 10 mLs of 85% H3PO4 and 0.2g of

231
NaF. 6) Add 15 drops of BDS indicator. 7) Titrate the solution with 0.5 N Ferrous solutions to a brilliant green
end point. During the titration the solution will change from an opaque green-brown to bluish-black-grey and finally to a one drop end point of brilliant green.
8) Prepare a blank by adding all reagents except the sediment sample to the conical flask and treat using the same procedure as above.
DISPOSAL: All solutions containing chromium must be disposed of into the heavy metal
residues collection bottle located in the fume cupboards. CALCULATION:
% C = 10 (1 - T/B) x F
Where: B = standardisation blank titration (mL of ferrous
solution) T = Sample titration (mL of ferrous solution) F = (1.0 N) x 12/4000 x 1.72 x 100 / sample weight Standardisation of this procedure may be performed by following the procedure
using 0.01g dextrose instead if sediment sample. (Dextrose should contain 39.99% C.) The calculation is as above except:
F = (1.0 N) x 12/4000 x 100 / sample weight REFERENCE: LORING, D.H. and RANTALA, R.T.T. 1992 Manual for geochemical analyses of marine sediments and suspended particulate matter, Earth Science Reviews 32: 235 - 283.

232
APPENDIX 10 - X-RAY DIFFRACTION ANALYSIS

233
X-RAY DIFFRACTION ANALYSIS GENERAL XRD is a widely used technique for mineral identification, particularly for fine-grained materials where the grain size is too small to be usefully studied with the optical microscope. In addition, the XRD analysis can provide information on the degree of structural disorder, particle size, and the nature of isomorphous substitutions. The method is based on the fact that X-rays are scattered by the electrons around atoms, which form the atomic layers in crystals (lattice spacings). A particular crystalline material has a particular structure or lattice. The scattered x-rays reinforce each other in directions that depend on the lattice repeat distances and the wavelength of the x-rays. The angles of diffraction give an indirect indication of the spacings (d spacings) between atomic layers and therefore can be used for mineral identification. The advantages of the method include the fact that: 1) It is non-destructive, 2) The samples are reasonably easy to prepare, 3) The material can be processed even in very small quantities, 4) Modern computer-linked instruments are quite straightforward to operate and
maintain. The limitations of the XRD analysis include: 1) The method is capable of identifying only crystalline materials, 2) Components of the same mineral series (i.e. micas, feldspars, amphiboles) which
have very similar crystallographic structures are difficult to separate due to their very similar XRD patterns.
MICRONIZING The micronizing vessel consists of a plastic cylinder filled with 48 stacked small agate or corundum cylinders. The particle size of the sample material to be crushed in this type of mill is to be no larger than 100 microns (i.e. what is obtainable from a swing mill). Approximately 3 g of sample and 10-12 ml of alcohol are placed into the micronization vessel and then into the arm of the mill. The timer on the mill is typically set to 0.2 (hr) (i.e. 12 minutes). Other settings of the timer can be made. The slurry obtained is homogenous and the particle size is ideally in the range of 1 to 5 microns. The mixture of sample/alcohol is placed in a pre-labelled beaker and left to dry overnight in an oven at 50-600 C. The sample will require remixing prior its use to counteract any segregation of phases during the drying step. The micronized powder is used to identify all the mineral phases of the sample providing that the phases are present in sufficient abundance. RANDOMLY ORIENTATED POWDER SAMPLES About 1.5-2 g of powder is lightly packed (to avoid as much as practical pressure orientation), into the backside of a circular cavity of an aluminium plate. The front

234
face of the sample holder rests on a polished metal block. The pressing is done using a small plastic cylinder and a metal ring for guidance. After the powder is packed, the plastic cylinder and metal ring are removed and the second half of the holder is carefully clicked on. The entire holder is then lifted, inverted and placed face upward into the auto sample changer carousel. When the entire batch is ready, the carousel is placed into the auto sample changer and the data acquisition task begun. ORIENTATED SPECIMENS Preparation of orientated samples is suitable and sometimes absolutely necessary for identification of clay minerals. Clay silicates have a particular structure developed along the (001) crystallographic plane, which makes them difficult to identify in a non-orientated (random) sample preparation, especially when the clay phases are in small quantities. The mechanism of orientating clay crystals exploits their sheet structure and to make the sheets lay one atop of the other in the same plane. This produces a pseudo-macrocrystal and creates a more intense diffraction pattern of the (001) basal spacing series. The first step is to disperse the sample in about ten times its volume of distilled water. Light grinding and shaking of the mixture (or ultrasonic dispersion) leads to a dissociation of the clay particles and separation of clay size material from the coarser (mineral) fractions. The material left in suspension is generally finer than 2 microns and can be taken with a pipette, spread over a glass slide and the slide then placed on top of a warm surface to dry. In about an hour at 500 C, the water evaporates leaving behind a gravimetrically deposited clay fraction. The glass slide is placed on a plastic holder, which fits on the back of the Co-machine holders. REFERENCES JENKINS R. & SNYDER R. L. 1996. Introduction to X-ray Powder Diffractometry, Chapter 9 (Specimen preparation), Chemical Analysis, Vol. 138, pp. 231-259, Wiley. BISH D. L. & POST J. E. 1989. Sample Preparation for X-ray Diffraction, Chapter 4 (Modern powder X-ray diffraction), Rev. Mineral, Vol 20, pp. 72-99. Mineral Soc. Am., Washington DC. NOTE The identification and quantification of sediment mineral phases presented in this thesis was assisted by several computer programs: 1) TRACES (plot of XRD traces, locate peaks and export data), 2) JADE (search-match program) and 3) SIROQUANT (quantification program which expresses the composition of the sample in percentages of dry weight). The error of the quantification was calculated as Chi Squared (goodness of fit between the experimental XRD trace and the calculated one) and ranged between 3 and 7.

235
APPENDIX 11 - CATIONS IN WATER, INDUCTIVELY COUPLED PLASMA- OPTICAL EMISSION SPECTROSCOPY (ICP-OES)

236
Cations in Water by Inductively Coupled Plasma – Optical Emission
Spectroscopy (ICP – OES) Cations were analysed by a Varian Liberty 200 inductively coupled plasma optical emission spectrometer (ICP – OES). The instrument was calibrated using synthetic standards. Major cations analysed were Na, K, Ca and Mg; minor and trace cations were Fe, Al, Zn, Cu, Mn, Sr, Ba, Ti, Li and V. Silica, although not in ionic form in natural waters, was also analysed with this suite of cations. The detection limits used are shown in Table 1. Major Cations: Na, K, Mg and Ca Minor Cations: Al, Si, Sr, Mn, Fe, Zn and Cu Table 1. Detection limits. Element
Working Detection Limits
Na 0.015 – 1500 mg/L
K 0.20 – 150 mg/L
Mg 0.001 – 150 mg/L
Ca 0.0003 – 250 mg/L
Al 0.015 – 75 mg/L
Si 0.011 – 75 mg/L
Sr 0.0006 – 75 mg/L
Mn 0.003 – 7.5 mg/L
Fe 0.015 – 7.5 mg/L
Zn 0.009 – 7.5 mg/L
Cu 0.02 – 0.75 mg/L
THEORY OF OPERATION: The cation and sulfur concentrations are measured using inductively coupled plasma - optical emission spectroscopy (ICP-OES). This technique involves the water sample being aspirated into a plasma. The intensity of characteristic wavelengths emitted by the excited analyte ions in the plasma are measured by a spectrophotometer. The measured intensity is proportional to concentration, thus concentration of ions in the sample can be determined. SAMPLE PREPARATION: Little or no sample preparation is required for analysis of aqueous samples by ICP-OES except for highly turbid samples, which must be filtered and samples of high

237
conductivity, which must be diluted to <4000 µS before analysis. Also, concentration of elements determined must be within the detection limits of the ICP-OES for the results to have analytical meaning. Filter turbid samples through a 0.45 or 0.8 µm membrane filter, collect and analyse the filtrate, diluting if necessary. It is possible for cations other than those listed above to be analysed, however it may not be feasible if the selected analyte ions are present only in trace amounts i.e. at levels below the limits of ICP-OES detection. ANALYTICAL ERROR: Approximate error (based on repeat analyses) of approximately 5%.

238
APPENDIX 12 - ANIONS IN WATER BY ION CHROMATOGRAPHY (IC)

239
Anions in Water by Ion Chromatography (IC)
The following anions, chloride, sulfate, nitrate, phosphate and bromide, have been determined by ion chromatography (IC) using a Dionex DX 300 ion chromatograph with suppressed conductivity detection. The system utilises Dionex AS14 analytical column and AG14 guard column. Conductivity suppression was by a micromembrance suppressor, Dionex CMMS-II. Cl-, SO4
2-, Fl-, Br-, NO3- and PO4
3- (NO2
- and SO32- can also be analysed but are not included in the routine analysis.)
DECTECTION LIMITS: A working range has been given below. This range is based on a combination of standard concentration range and instrument working range. Fl- 0.05 to 12 ppm Cl- 0.5 to 150 ppm SO4
2- 0.5 to 100 ppm Br- 0.05 to 12 ppm NO3
- 0.05 to 12 ppm PO4
3- 0.05 to 12 ppm THEORY OF OPERATION: THE ION CHROMATOGRAPHIC PROCESS: The sample is introduced in the flowing stream and carried into the anion exchange column. Ions interact with the ion exchange sites on the stationary phase in the column. Mobile phase ions (or eluent ions) compete with the sample ions for ion exchange sites on the column. Separation depends upon the different ions having different affinities for both phases. In the case of anion separations the differing affinities for stationary and mobile phases are due to the ionic charge and ion size (ionic radius) of each anion species. Once anions are separated the concentration of each species present in the sample is measured using a conductivity detector. A chromatogram displays peaks in conductivity at various retention times. Each anionic species is identified by its retention time, which remains constant throughout successive runs. STATIONARY PHASE: the column packing material containing functionalised active sites. For anion determinations the Dionex AS14 anion exchange column is used. MOBILE PHASE (OR ELUENT): The liquid flowing though the column that contains competing ion for the active sites. SAMPLE PREPARATION: Little of no sample preparation is required of analysis of aqueous sample by ion chromatography. However highly turbid samples must be filtered before analysis

240
and sample of high conductivity require diluting before analysis. Samples analysed must have a conductivity of less than 700 µS, if not dilution is required. Filter turbid samples through a 0.45 of 0.8 um membrane filter, collect and analyse the filtrate, diluting if necessary. REAGENTS: Eluent: 3.5mM Na2CO3/1.0 mM NaHCO3. Prepare diluting the 100x concentrate 100 fold. I.e. pipette 10 mL of 100x concentrate into a 1000 mL volumetric flask and dilute to the mark with ultra pure water. (Obtain ultra pure water from the purification unit located on the back island bench located in the Geochem lab R431.) Fill eluent bottle with this solution and sparge with argon for at least ten minutes before starting eluent pump. Regenerant solution: Add 2.4 mL of conc. H2SO4 to 1000 mL of ultra pure water and dilute further to 2000mLs. Fill regent bottle with this solution recap and allow pressurising. After several minutes ensure regent solution is flowing through suppressor. RESULTS: Ion chromatography is an excellent method of anion species determination in water samples. It has an extremely good precision with a %RSD of <2%. However it is important that results obtained are not taken on face value but are checked to assure data is reasonable. This is particularly important as peaks can be misnamed due to small shifts in retention time. The retention time can change due to a variety of reasons most commonly due to problems with the eluent pump, blockages and inaccurate preparation of eluent. Always check with previous days data to determine if retention times have not changed (refer to daily log, located next to instrument for this information). Also data should be with the working range of each species listed above, if not a dilution may be requiring before rerunning samples or an alternative method of analysis may be required. In particular, high chloride data should be checked by titration as concentrations over 150-200 ppm may not be linear, giving inaccurate results.

241
APPENDIX 13 – TITRATION METHOD FOR ALKALINITY

242
Alkalinity and Acid Titration Methods
DETECTION LIMIT: 0.25 ppm CaCO3 (mg/L water) APPARATUS: 250 mL conical flask, calibrated pH meter and 25 mL burette SAFETY EQUIPMENT: Laboratory coat, safety glasses. Refer to Risk Assessment/s: Hydrochloric acid (32%) - Student usage REAGENTS: 0.1N Standard HCl: SAFETY: This dilution must be carried out in a fume
cupboard. Pipette 10 mLs of conc HCl (10 M) into a 1000 mL volumetric flask and dilute to mark.
Standardisation of 0.1 N HCl: Weigh 0.7 - 0.8 g of pure sodium tetraborate by difference into a 150 mL
conical flask, dissolve in about 50 mLs of distilled water and add a few drops of methyl red indicator. Titrate the sodium tetraborate solution with the 0.1N HCl as the titrant until the colour changes to pink. Record the volume of HCl used. Carry out this procedure in triplicate. Use the following equation to calculated the normality of the acid solution.
N HCl = Weight of Na2B4O7 / 190.72 x Vol of Titrant (HCl)
0.02 N Standard HCl: Pipette 200 mLs of standard 0.1N HCl into a 1000 mL volumetric flask and dilute to the mark.
PROCEDURE: The alkalinity of a sample is due to the presence of hydroxide, carbonate or
bicarbonate ions. The concentration of each of these ions in a sample can be calculated once the phenolphthalein and total alkalinity have been determined.
1) Determination of phenolphthalein alkalinity or P
a) Pipette 100 mLs of sample into a 250 mL beaker. Measure the pH of the sample. If pH is less than 8.3 go on to step 2) as P=0.
b) If pH is greater than 8.3 then titrate the sample with 0.1N HCl to pH
8.3. Use a magnetic stirrer and leave pH probe in sample while titrating.

243
Record volume of HCl used. Calculate alkalinity due to hydroxide, P, by
using Calculation (a). Go on to step 2).
2) Determination of total alkalinity or T
a) Titrate the sample to the pH 4.7 if the sample alkalinity is unknown. If known choose the appropriate total alkalinity equivalence point from the following table.
These pH values are suggested equivalence points for the corresponding
alkalinity concentrations.
Alkalinity (mg/L CaCO3)
End Point pH: Total
30 4.9 150 4.6 500 4.3
Silicates, phosphates known or suspected
4.5
Industrial waste or complex system
4.5
b) Record total volume of HCl titrated ie. include volume of titrant used in
step 1 if appropriate. Calculate the Total Alkalinity, T, using calculation (b). If Total Alkalinity, T, is less than 20 mg/L CaCO3 go to step 3). If Total Alkalinity, T is greater than 20 mg/L CaCO3 go to step 4.
3) Determination of Total Alkalinity less the 20mg/L CaCO3 a) Pipette 100 mLs of sample into a 250 mL beaker and titrate using
0.01M HCl to an end point in the range of 4.3 to 4.7. Record the volume and the exact pH.
b) Titrate the solution further to reduce the pH exactly 0.30 pH units and record volume. Use Calculation (c) to determine Total Alkalinity, T.
4) Determine the relationship between Hydroxide, Carbonate and Bicarbonate
Alkalinity using Table 2. NOTE: As the end point is approached make smaller additions of acid and be sure
that pH equilibrium is reached before adding more titrant. CALCULATIONS: a) P (Phenolphthalein Alkalinity) P mg/L CaCO3 = A x N x 50 000 / volume of sample where A = mL standard acid used N = normality of standard acid b) T (Total Alkalinity)

244
T mg/L CaCO3 = A x N x 50 000 / volume of sample where A = mL standard acid used N = normality of standard acid c) Potentiometric titration of low alkalinity (<20mg/L CaCO3): T (Total alkalinity), T mg/L CaCO3 = (2B - C) x N 50 000 / volume of sample where B = mL of titrant to first recorded pH C = total mL of titrant of reach pH 0.3 unit lower N = normality of acid Table 2 Calculation of alkalinity relationships
Result of titration Hydroxide Alkalinity as CaCO3
Carbonate Alkalinity as CaCO3
Bicarbonate Alkalinity as CaCO3
P = 0 0 0 T P < 1/2T 0 2P T - 2P P >=1/2T 0 2P 0 P > 1/2T 2P-T 2(T-P) 0
P=T T 0 0 Where P = phenolphthalein alkalinity T = total alkalinity Report total alkalinity as: "The alkalinity to pH ____ = ____ mg CaCO3/L" To convert hydroxide, carbonate and bicarbonate expressed as alkalinity to concentration of their own species to be used in a mass balance multiply by the following factors.
Hydroxide mg/L OH- = mg/l CaCO3 x 0.34
Carbonate mg/L CO32- = mg/L CaCO3 x 0.60
Bicarbonate mg/L HCO3- = mg/L CaCO3 x 1.22 REFERENCE
GREENBERG A. E., CLESCERI L. S. & EASTON A. D. 1992. Standard methods
for the examination of water and wastewater. 18th Ed. APHA. AWWA. WEF.

245
APPENDIX 14 – ADDITIONAL LABORATORY ANALYSIS DATA

246
These additional data complement Paper 2. The samples were analysed following
the procedures described in the paper and Appendices 5 (HF) & 7 (XRF).
0 1.5 3
kilometres
Weathered bedrock
Alluvium
weathered profilesSand
Clay rich in organic matter
Brib
ie Is
land
Lamerough Ck
Bells North Arm Ck
Bel ls South Arm Ck
Halls Ck
N
Coral Sea
S1
B3
GB1
Caloundra
Pum
ices
tone
Pas
sage
Queensland
0 1.5 3
kilometres
Weathered bedrock
Alluvium
weathered profilesSand
Clay rich in organic matter
Brib
ie Is
land
Lamerough Ck
Bells North Arm Ck
Bel ls South Arm Ck
Halls Ck
NN
Coral Sea
S1
B3
GB1
Caloundra
Pum
ices
tone
Pas
sage
Queensland
Figure 1. Location of sample sites studied in paper 2.

247
Table 1: A comparison between XRF and HF methods for major element
concentrations (%)
Sample Na2O K2O MgO CaO TiO2 MnO Fe2O3 Al2O3 SiO2
XRF HF XRF HF XRF HF XRF HF XRF HF XRF HF XRF HF XRF HF XRF HF
S1-2 bd 0.2 1.2 1.5 0.4 0.5 0.1 0.02 0.8 0.7 bd 0.01 6.2 7.1 16.4 19 67.1 66
S1-5 0.8 0.8 2.0 2.4 0.7 0.9 0.2 0.3 0.5 0.6 bd 0.1 3.0 3.6 11.2 13 75.9 76
B3-1 2.2 2.4 1.9 2.1 0.4 0.4 0.3 0.3 0.5 0.4 0.1 0.1 2.3 2.4 10.2 11 78.9 80
B3-4 2.1 2.2 1.5 1.5 0.6 0.6 1.1 1.2 0.5 0.4 0.2 0.2 4.1 4.1 9.0 9.1 75.9 76
GB1-3 0.5 0.5 0.2 0.1 0.8 0.2 0.2 0.2 2.9 3.4 bd 0.02 22.7 24 18.1 18 38.9 39
GB3-5 1.9 2.1 1.4 1.6 0.5 0.2 0.2 0.2 0.8 0.9 bd 0.02 2.7 3.2 10.3 12 78.9 79
Table 2: A comparison between XRF (%) and HF (mg/kg) methods for trace metal
concentrations
Sample VXRF VHF CrXRF CrHF CoXRF CoHF NiXRF NiHF CuXRF CuHF ZnXRF ZnHF PbXRF PbHF
S1-2 78 75 76 42 1 1 5 10 bd 1 24 11 5 12
S1-5 67 53 85 29 18 16 15 23 14 15 105 117 10 17
B3-1 67 43 75 37 12 10 8 15 6 5 97 103 4 13
B3-4 59 43 78 20 12 12 10 18 6 4 49 49 4 11
GB1-3 259 290 1056 842 47 35 227 196 51 60 97 93 5 7
GB3-5 95 63 157 75 30 23 67 66 15 16 90 86 3 12

248
The following data are the analyses of soil and sediment samples (locations in the attached figure) following the procedures in Appendices 5 (Sulfur), 8 (extractable method) & 9 (OC) and complement Paper 3.
Soil developed on sandstone bedrock from QASSIT Sample V Cr Mo Ni Cu Zn Pb Fe Mn Al S OC 570-0* 5.4 5.7 1.8 1.4 0.7 30 5 3336 3 5767 0.05 2.2 570-140 3.2 16.2 2.6 4.1 1.3 35 1 995 3 1903 0.04 0.5 570-220 2.6 6.3 1.8 4.7 bd 38 1 1492 2 2677 0.10 0.2 566-0 1.8 6.1 1.1 0.5 0.6 24 2 831 1 537 0.02 0.8 566-50 13.4 6.6 0.8 bd bd 44 4 7523 2 6263 0.02 0.3 566-180 7.5 4.6 3.9 0.0 0.6 48 5 2040 1 4939 0.03 0.1 566-380 9.2 5.6 0.8 0.8 bd 52 3 8596 1 5855 0.02 0.0 565-0 14.0 15.8 6.2 3.2 12.5 35 19 4553 6 19342 0.07 7.2 565-80 7.1 4.8 5.6 0.2 0.8 40 7 2099 2 4726 0.04 0.6 565-380 2.5 3.0 1.9 1.5 bd 43 5 2459 18 3634 0.02 0.1 561-0 3.6 5.6 2.2 0.9 0.6 41 2 2604 3 2135 0.00 1.3 561-80 23.9 13.0 4.6 1.7 bd 43 2 9350 3 10071 0.02 0.1 561-180 4.4 4.3 4.8 0.0 0.5 35 7 1128 1 5585 0.04 0.0 561-280 18.1 5.5 2.5 bd bd 39 4 7198 5 7780 0.03 0.5 560-0 2.0 1.7 0.6 0.5 bd 21 0 1255 2 960 0.01 1.7 560-80 2.6 3.8 bd 1.1 bd 22 0 1300 1 1239 0.01 2.7 560-180 9.1 10.4 0.4 3.4 1.1 30 5 2200 4 4697 0.02 0.8 660-0 11.7 5.7 0.3 1.3 bd 36 8 8288 6 8828 0.05 3.9
660-130 5.0 5.9 1.7 1.1 1.0 32 0 1094 3 3223 0.04 0.6 663-0 8.7 4.6 0.0 1.1 0.8 54 6 3755 7 6819 0.08 8.6 663-50 4.0 2.8 1.0 0.5 bd 39 2 1092 2 3737 0.03 2.4 663-180 5.6 4.7 0.5 0.0 bd 28 2 1530 3 4854 0.03 0.6 1120-0 40.8 14.6 14.4 2.4 14.5 30 6 27001 21 11343 0.05 4.1 1120-50 19.7 9.8 1.7 1.6 bd 25 3 4121 5 8468 0.02 0.7 1120-180 25.1 8.7 1.5 0.7 bd 23 3 755 2 4576 0.03 0.8
* Indicates depth of sampling bd=below detection limit all in mg/kg except for S in %

249
Soils with estuarine origin from QASSIT Sample V Cr Mo Ni Cu Zn Pb Fe Mn Al S OC 573-0* 10.5 10.4 6.4 4.2 2.4 54 17 3848 16 22592 0.13 7.8 573-80 10.2 12.6 4.0 5.9 1.7 61 16 3785 10 28112 0.13 5.3 573-140 6.8 7.0 1.8 2.5 0.7 44 bd 2964 9 3567 0.54 3.6 573-170 7.9 8.8 1.5 4.3 1.1 62 4 8585 32 5692 1.01 4.2 573-210 3.6 6.5 2.4 2.9 0.5 32 bd 3485 12 2099 0.48 1.0 573-250 4.6 10.7 2.3 4.1 0.7 28 3 3866 14 3296 0.44 2.1 542-0 4.5 25.0 3.3 8.3 0.6 47 bd 3522 5 2825 0.03 2.5 542-80 7.0 30.3 1.5 8.3 0.5 36 bd 1114 5 5055 0.02 0.4 542-230 2.1 43.8 2.9 13.4 0.5 40 4 774 4 5757 0.03 0.0 563-0 21.7 13.8 2.7 3.0 1.3 46 11 5214 14 21946 0.14 10.1 563-50 13.1 14.2 5.0 2.1 0.7 25 9 1523 4 17687 0.09 5.1 563-130 2.5 5.5 2.6 0.0 0.5 40 10 670 3 8836 0.03 5.7 545-0 2.3 95.0 2.1 36.0 1.0 27 bd 2431 8 1016 0.02 2.6 545-80 0.6 5.0 1.0 bd 0.8 41 bd 624 1 260 0.01 0.1 545-160 1.2 5.0 1.0 bd 1.4 38 bd 660 1 1143 0.06 2.7 562-0 33.3 23.6 4.9 4.3 2.9 45 9 7165 12 12716 0.10 2.0 562-80 42.6 22.5 2.9 3.3 3.0 29 4 11534 15 10179 0.12 1.5 562-130 29.1 20.1 7.8 2.3 3.0 35 10 4065 11 8629 0.14 1.0 562-230 47.7 21.2 1.0 2.5 0.7 35 11 21329 117 12264 0.07 0.8 651-0 4.9 2.4 1.0 bd 0.8 31 bd 491 2 1058 0.07 1.3
651-130 17.0 11.3 1.1 bd 1.1 28 bd 15742 5 3473 0.03 0.4 651-330 34.6 32.6 2.3 3.4 3.4 41 10 2312 14 12293 0.15 1.7 509-0 0.7 35.4 1.0 10.7 0.8 23 bd 471 4 286 0.06 4.1
509-130 2.5 38.7 1.0 14.6 1.8 34 bd 578 5 3564 0.08 1.3 509-280 1.6 14.1 1.0 5.0 0.6 22 bd 543 2 799 0.05 0.3 652-0 0.7 1.4 1.0 bd 0.5 23 bd 278 2 228 0.02 3.0
652-230 1.4 1.8 1.0 bd 0.5 29 bd 231 0 1176 0.06 1.8 652-480 31.4 28.0 1.5 9.4 3.7 45 11 18435 35 15043 1.54 4.4 569-0 19.6 17.1 2.7 4.9 3.6 33 4 8086 7 7638 0.11 7.1
569-130 5.9 6.6 1.0 1.8 1.5 38 bd 2140 6 2583 0.07 0.7 569-280 14.0 11.6 1.0 5.0 2.6 45 bd 9973 36 5954 0.38 1.2 569-370 16.2 10.2 1.8 5.0 2.0 31 3 10924 26 5714 0.60 2.2 661-0 29.0 19.2 3.7 5.0 0.5 58 5 9426 11 15404 0.11 9.2 661-80 45.0 46.6 4.8 7.3 5.8 63 12 35105 22 23478 0.06 2.2 661-230 6.9 5.3 1.0 4.6 1.0 27 bd 5799 23 3765 0.44 1.4 659-100 8.0 5.6 1.0 1.3 0.8 42 7 2182 5 9066 0.09 7.8 659-230 11.5 8.2 1.0 2.5 1.3 34 9 2633 6 10138 0.09 6.8 659-330 12.3 6.8 1.0 2.8 2.0 34 8 2916 7 7507 0.44 10.2 662-0 34.6 14.0 15.3 6.8 0.9 35 12 34064 13 16322 0.20 10.3 662-70 15.4 8.6 1.0 8.6 1.5 36 5 14619 15 4750 1.52 3.6 662-180 4.7 4.2 1.0 bd 0.5 44 bd 392 2 3198 0.04 0.9 655-0 0.4 1.0 1.0 bd 0.5 22 bd 179 1 210 0.02 1.9 655-80 1.1 4.6 1.1 1.5 0.5 20 bd 261 1 1589 0.02 2.2 655-330 2.7 2.8 1.8 3.8 1.0 29 bd 2503 5 1633 0.21 0.6 655-380 23.3 11.8 3.2 3.7 0.6 31 16 4080 8 18405 0.12 0.8
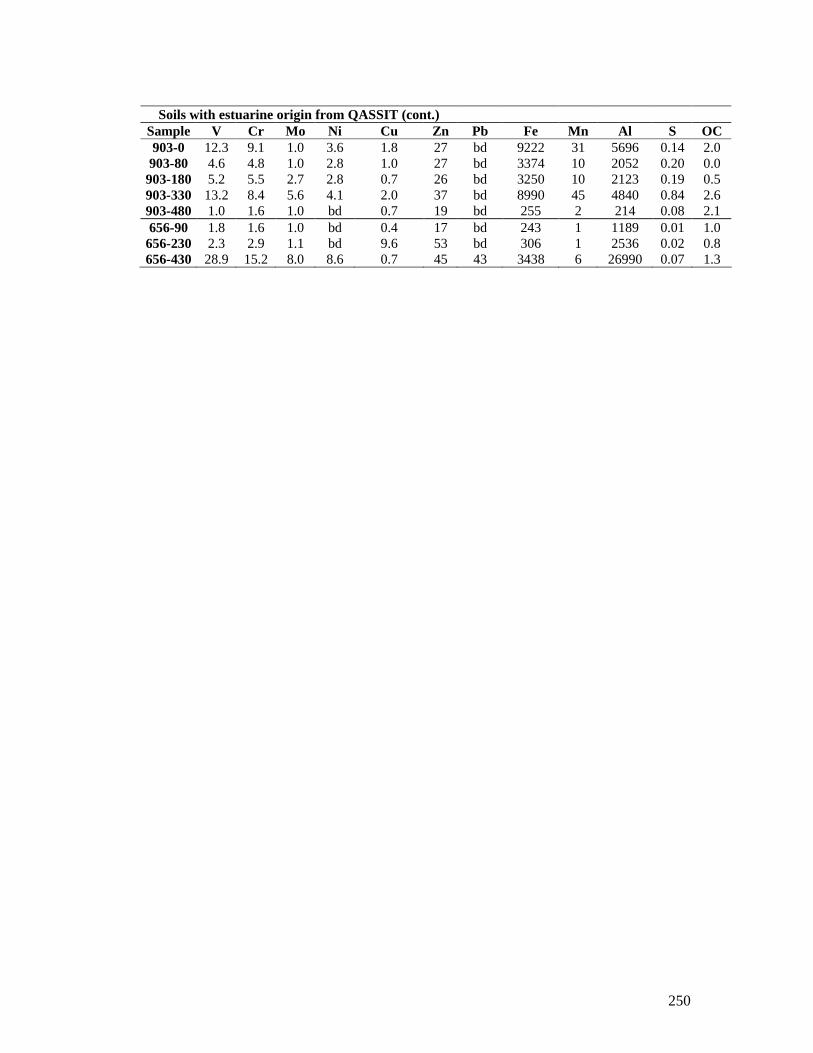
250
Soils with estuarine origin from QASSIT (cont.)
Sample V Cr Mo Ni Cu Zn Pb Fe Mn Al S OC 903-0 12.3 9.1 1.0 3.6 1.8 27 bd 9222 31 5696 0.14 2.0 903-80 4.6 4.8 1.0 2.8 1.0 27 bd 3374 10 2052 0.20 0.0 903-180 5.2 5.5 2.7 2.8 0.7 26 bd 3250 10 2123 0.19 0.5 903-330 13.2 8.4 5.6 4.1 2.0 37 bd 8990 45 4840 0.84 2.6 903-480 1.0 1.6 1.0 bd 0.7 19 bd 255 2 214 0.08 2.1 656-90 1.8 1.6 1.0 bd 0.4 17 bd 243 1 1189 0.01 1.0 656-230 2.3 2.9 1.1 bd 9.6 53 bd 306 1 2536 0.02 0.8 656-430 28.9 15.2 8.0 8.6 0.7 45 43 3438 6 26990 0.07 1.3
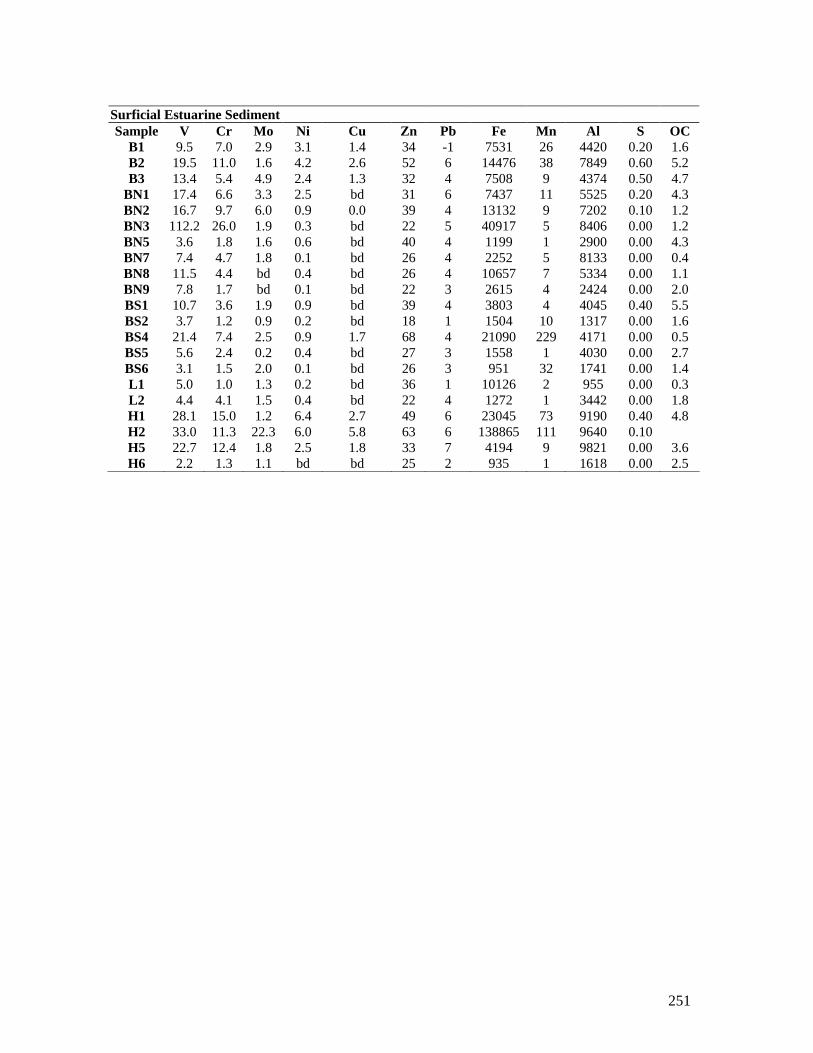
251
Surficial Estuarine Sediment Sample V Cr Mo Ni Cu Zn Pb Fe Mn Al S OC
B1 9.5 7.0 2.9 3.1 1.4 34 -1 7531 26 4420 0.20 1.6 B2 19.5 11.0 1.6 4.2 2.6 52 6 14476 38 7849 0.60 5.2 B3 13.4 5.4 4.9 2.4 1.3 32 4 7508 9 4374 0.50 4.7
BN1 17.4 6.6 3.3 2.5 bd 31 6 7437 11 5525 0.20 4.3 BN2 16.7 9.7 6.0 0.9 0.0 39 4 13132 9 7202 0.10 1.2 BN3 112.2 26.0 1.9 0.3 bd 22 5 40917 5 8406 0.00 1.2 BN5 3.6 1.8 1.6 0.6 bd 40 4 1199 1 2900 0.00 4.3 BN7 7.4 4.7 1.8 0.1 bd 26 4 2252 5 8133 0.00 0.4 BN8 11.5 4.4 bd 0.4 bd 26 4 10657 7 5334 0.00 1.1 BN9 7.8 1.7 bd 0.1 bd 22 3 2615 4 2424 0.00 2.0 BS1 10.7 3.6 1.9 0.9 bd 39 4 3803 4 4045 0.40 5.5 BS2 3.7 1.2 0.9 0.2 bd 18 1 1504 10 1317 0.00 1.6 BS4 21.4 7.4 2.5 0.9 1.7 68 4 21090 229 4171 0.00 0.5 BS5 5.6 2.4 0.2 0.4 bd 27 3 1558 1 4030 0.00 2.7 BS6 3.1 1.5 2.0 0.1 bd 26 3 951 32 1741 0.00 1.4 L1 5.0 1.0 1.3 0.2 bd 36 1 10126 2 955 0.00 0.3 L2 4.4 4.1 1.5 0.4 bd 22 4 1272 1 3442 0.00 1.8 H1 28.1 15.0 1.2 6.4 2.7 49 6 23045 73 9190 0.40 4.8 H2 33.0 11.3 22.3 6.0 5.8 63 6 138865 111 9640 0.10 H5 22.7 12.4 1.8 2.5 1.8 33 7 4194 9 9821 0.00 3.6 H6 2.2 1.3 1.1 bd bd 25 2 935 1 1618 0.00 2.5

252
Mineralogical data for selected samples complementing Paper 3. Samples (locations
in the attached figure) were analysed following a procedure in Appendix 10.
Soil developed on sandstone bedrock Sample Quartz Feldspars Kaolinite Mixed
layer Illite Pyrite Hematite NaCl Goethite Jarosite Anatase
1120-0 77.4 2.8 17.1 2.0 0.0 0.0 0.7 0.0 0.0 0.0 traces 1120-50 81.1 1.1 17.8 0.0 0.0 0.0 0.0 0.0 0.0 0.0 traces 1120-180 78.3 1.1 20.6 0.0 0.0 0.0 0.0 0.0 0.0 0.0 traces
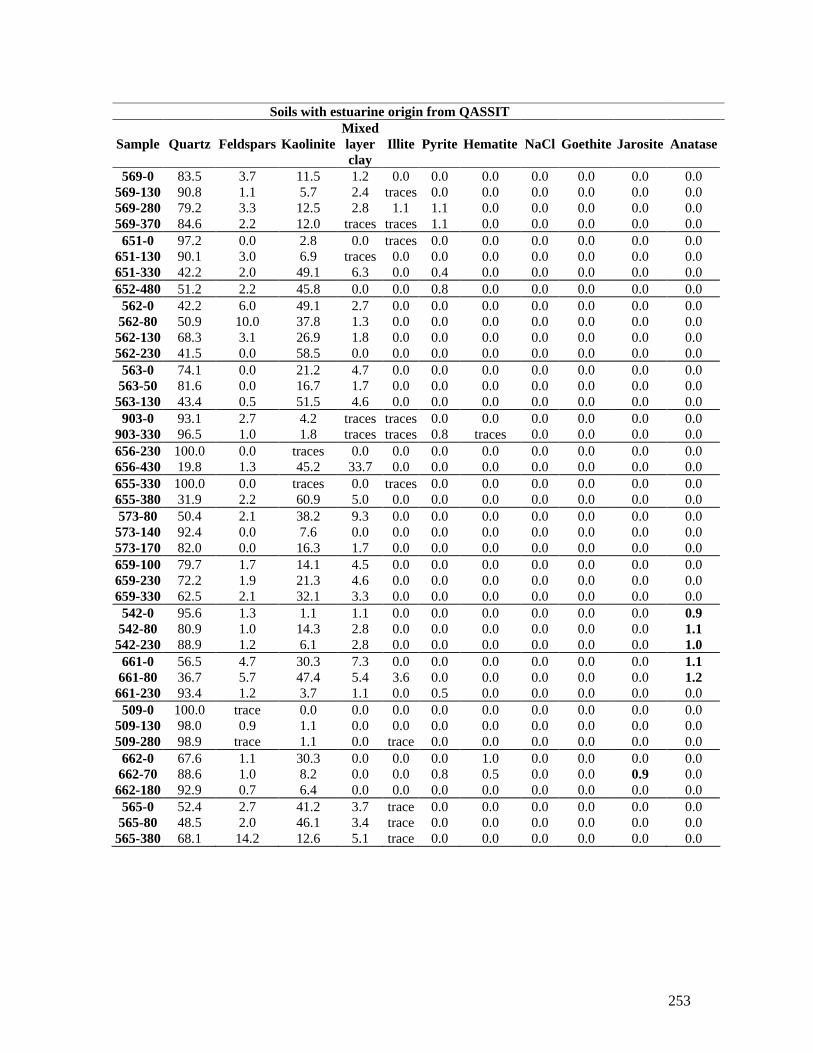
253
Soils with estuarine origin from QASSIT
Sample Quartz Feldspars Kaolinite Mixed layer clay
Illite Pyrite Hematite NaCl Goethite Jarosite Anatase
569-0 83.5 3.7 11.5 1.2 0.0 0.0 0.0 0.0 0.0 0.0 0.0 569-130 90.8 1.1 5.7 2.4 traces 0.0 0.0 0.0 0.0 0.0 0.0 569-280 79.2 3.3 12.5 2.8 1.1 1.1 0.0 0.0 0.0 0.0 0.0 569-370 84.6 2.2 12.0 traces traces 1.1 0.0 0.0 0.0 0.0 0.0 651-0 97.2 0.0 2.8 0.0 traces 0.0 0.0 0.0 0.0 0.0 0.0
651-130 90.1 3.0 6.9 traces 0.0 0.0 0.0 0.0 0.0 0.0 0.0 651-330 42.2 2.0 49.1 6.3 0.0 0.4 0.0 0.0 0.0 0.0 0.0 652-480 51.2 2.2 45.8 0.0 0.0 0.8 0.0 0.0 0.0 0.0 0.0 562-0 42.2 6.0 49.1 2.7 0.0 0.0 0.0 0.0 0.0 0.0 0.0 562-80 50.9 10.0 37.8 1.3 0.0 0.0 0.0 0.0 0.0 0.0 0.0 562-130 68.3 3.1 26.9 1.8 0.0 0.0 0.0 0.0 0.0 0.0 0.0 562-230 41.5 0.0 58.5 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 563-0 74.1 0.0 21.2 4.7 0.0 0.0 0.0 0.0 0.0 0.0 0.0 563-50 81.6 0.0 16.7 1.7 0.0 0.0 0.0 0.0 0.0 0.0 0.0 563-130 43.4 0.5 51.5 4.6 0.0 0.0 0.0 0.0 0.0 0.0 0.0 903-0 93.1 2.7 4.2 traces traces 0.0 0.0 0.0 0.0 0.0 0.0
903-330 96.5 1.0 1.8 traces traces 0.8 traces 0.0 0.0 0.0 0.0 656-230 100.0 0.0 traces 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 656-430 19.8 1.3 45.2 33.7 0.0 0.0 0.0 0.0 0.0 0.0 0.0 655-330 100.0 0.0 traces 0.0 traces 0.0 0.0 0.0 0.0 0.0 0.0 655-380 31.9 2.2 60.9 5.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 573-80 50.4 2.1 38.2 9.3 0.0 0.0 0.0 0.0 0.0 0.0 0.0 573-140 92.4 0.0 7.6 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 573-170 82.0 0.0 16.3 1.7 0.0 0.0 0.0 0.0 0.0 0.0 0.0 659-100 79.7 1.7 14.1 4.5 0.0 0.0 0.0 0.0 0.0 0.0 0.0 659-230 72.2 1.9 21.3 4.6 0.0 0.0 0.0 0.0 0.0 0.0 0.0 659-330 62.5 2.1 32.1 3.3 0.0 0.0 0.0 0.0 0.0 0.0 0.0 542-0 95.6 1.3 1.1 1.1 0.0 0.0 0.0 0.0 0.0 0.0 0.9 542-80 80.9 1.0 14.3 2.8 0.0 0.0 0.0 0.0 0.0 0.0 1.1 542-230 88.9 1.2 6.1 2.8 0.0 0.0 0.0 0.0 0.0 0.0 1.0 661-0 56.5 4.7 30.3 7.3 0.0 0.0 0.0 0.0 0.0 0.0 1.1 661-80 36.7 5.7 47.4 5.4 3.6 0.0 0.0 0.0 0.0 0.0 1.2 661-230 93.4 1.2 3.7 1.1 0.0 0.5 0.0 0.0 0.0 0.0 0.0 509-0 100.0 trace 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0
509-130 98.0 0.9 1.1 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 509-280 98.9 trace 1.1 0.0 trace 0.0 0.0 0.0 0.0 0.0 0.0 662-0 67.6 1.1 30.3 0.0 0.0 0.0 1.0 0.0 0.0 0.0 0.0 662-70 88.6 1.0 8.2 0.0 0.0 0.8 0.5 0.0 0.0 0.9 0.0 662-180 92.9 0.7 6.4 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 565-0 52.4 2.7 41.2 3.7 trace 0.0 0.0 0.0 0.0 0.0 0.0 565-80 48.5 2.0 46.1 3.4 trace 0.0 0.0 0.0 0.0 0.0 0.0 565-380 68.1 14.2 12.6 5.1 trace 0.0 0.0 0.0 0.0 0.0 0.0

254
Surficial Estuarine Sediment
Sample Quartz Feldspars Kaolinite Mixed layer clay
Illite Pyrite Hematite NaCl Goethite Jarosite Anatase
B2 78.9 3.1 16.3 trace trace 0.8 0.4 0.4 0.0 0.0 0.0 B3 88.8 1.7 8.3 trace trace 0.7 0.3 0.2 0.0 0.0 0.0
BN3 89.9 1.0 8.1 trace trace 0.3 0.6 0.0 0.0 0.0 0.0 BN5 93.5 0.0 6.5 traces 0.0 0.0 0.0 0.0 0.0 0.0 0.0 BN7 69.4 0.0 30.0 0.6 0.0 0.0 0.0 0.0 0.0 0.0 0.0 BN8 76.5 1.8 15.9 5.5 trace trace 0.3 0.0 0.0 0.0 0.0 BN9 81.3 3.8 13.5 1.4 0.0 0.0 0.0 0.0 0.0 0.0 0.0 BS1 82.9 0.0 14.3 0.0 0.0 0.8 0.0 2.0 0.0 0.0 0.0 BS4 58.6 24.5 7.3 1.8 7.2 trace 0.6 0.0 0.0 0.0 0.0 BS6 88.5 0.0 10.4 1.1 0.0 0.0 0.0 0.0 0.0 0.0 0.0 H1 64.1 7.7 18.7 3.0 2.7 0.8 0.0 2.7 0.3 0.0 0.0 H5 76.1 3.0 20.4 trace trace 0.1 0.3 0.0 0.0 0.0 0.0
Figure 2. Samples of unconsolidated material studied in Paper 3.
0 1.5 3
kilometres
Weathered bedrock
Alluvial material
Creek bank sedimentsSoil samples
Sand
Organic clay
H1H2
560
569
561
565
H6
563
509652
562
H5
651566
B2
B1
656903
655
661
660BN3
573
BN5BN7
BN8BN9
BN2
BS1
BN1B3
1120
542
662
570BS5
BS6
BS2
663
659
BS4
545
Brib
ie Is
land
Lamerough Ck
Bells North Arm Ck
Bells South Arm Ck
Halls Ck
NN
Coral Sea

255