trinity college dublin qp forum excipient risk …© 2016 mcgee pharma international 1 trinity...
TRANSCRIPT

1© 2016 McGee Pharma International
Trinity College Dublin QP ForumExcipient Risk Assessment Workshop
21April 2016
Presenter: Ms. Fionnuala Burke
Associate Consultant, MPI

2© 2016 McGee Pharma International
your partner in compliance
Definition of Excipient
• Defined in Falsified Medicines Directive 2011/62/EC as
• ‘Any constituent of a medicinal product other than the active substance and the packaging material’

3© 2016 McGee Pharma International
your partner in compliance
FMD
“The holder of the manufacturing authorisation shall ensure that the appropriate good manufacturing practice so ascertained, is applied. The holder of the manufacturing authorisation shall document the measures taken under this paragraph”
• Directive 2011/83/EC, Article 46(f)

4© 2016 McGee Pharma International
your partner in compliance
FMD
‘There is a range of different good manufacturing practices that are suitable for being applied to the manufacturing of excipients. In order to provide for a high level of protection of public health, the manufacturer of the medicinal product should assess the suitability of excipients on the basis of appropriate good manufacturing practices for excipients.’
‘This shall be ascertained on the basis of a formalised risk assessment in accordance with the applicable guidelines referred to in the fifth paragraph of Article 47. ‘

5© 2016 McGee Pharma International
your partner in compliance
Eudralex Volume IV
Chapter 5 ‘Production’ updated and issued 13 Aug 2014
5.29 Excipients
‘Excipients and excipient suppliers should be controlled appropriately based on the
results of a formalised quality risk assessment in accordance with the
European Commission ‘Guidelines on the formalised risk assessment for
ascertaining the appropriate Good Manufacturing Practice for excipients of
medicinal products for human use’.

6© 2016 McGee Pharma International
your partner in compliance
GMP for Excipients – Formalised Risk Assessment
‘A risk assessment as set out in these guidelines should be carried out
for excipients for authorised medicinal product for human use by 21st
March 2016’

7© 2016 McGee Pharma International
your partner in compliance
Excipient Guideline 19 March 2015
‘Guidelines of 19 March 2015 on the formalised risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal
products for human use’ (2015/C 95/02)
• The appropriate GMP for excipients of medicinal products for human use shall be ascertained on the basis of a formalised risk assessment in accordance with these guidelines.
• The excipient risk assessment/risk management procedure should be incorporated in the pharmaceutical quality system of the manufacturing authorisation holder.
States in the Introduction…

8© 2016 McGee Pharma International
your partner in compliance
Challenges and Opportunities for the Industry
Challenges:
• Small customers - difficult to have influence over suppliers regarding GMP & access to audit
• Increased audit focus – costly to implement
• May be complicated to complete the assessment
• May require significant resource – look for opportunity to leverage work previously completed
• Multiple excipients on site with different risks – consider worst case under each risk area
• Maintaining objectivity throughout the process
Opportunities:
• Improved product and patient safety
• Risk assessment expands the knowledge on the role of an excipient in the formulation that might not have been understood previously
• Industry initiatives to manage audit programs – use of 3rd parties

9© 2016 McGee Pharma International
your partner in compliance
Summary
Expectation is that the Manufacturing Authorisation Holders perform a risk assessment to determine the excipient manufacturers risk profile
A number of steps to be taken to achieving this:
1. Classification of excipients risk profile - Low/Medium/High Risk
2. Determine GMP Controls
3. Classification of manufacturers risk profile - Low/Medium/High Risk
4. Risk Control Strategy
Considerations include:
•Source - animal/mineral/vegetable/synthetic
•Past history of supply
•Purpose and function of excipient
•Patient risk (e.g. route of administration, volume consumed)
•Source of excipient and supply chain
•Supplier history etc.

10© 2016 McGee Pharma International
your partner in compliance
Requirements of the approach
The RA process should ensure that all elements of the guidance are achieved – holistic risk based management of the excipient manufacturer on the basis of YOUR products
The approach should meet the requirements of the guidance
The approach should be easy to use – this is a complicated process requiring assessment of each excipient across multiple elements (18 minimum metrics)
The approach should not be excessively resource heavy – standardise the approach where possible to make the process more efficient
Where possible, work already completed by the company should be leveraged e.g. development work, toxicological studies etc. – can provide a repository for capturing tacit knowledge on excipients
The process is wider than just the excipient risk profile – it must achieve the end goal of determining the risk profile per excipient, per manufacturer and the control strategy for managing and assessing this on an ongoing basis

11© 2016 McGee Pharma International
TSE
Viral contamination
Micro/endotoxin/pyrogen contamination
Any impurity originating from the raw materials
Any impurity generated as part of the process
Sterility assurance
Impurities carried over from absence of dedicated equipment and/or facilities
Environmental control and storage/transportation conditions including cold chain management
Supply chain complexity
Stability of excipient
Packaging integrity evidence
Risks from Excipient Source and Process

12© 2016 McGee Pharma International
Pharmaceutical form and use of the medicinal product
Function of the excipient in the formulation
Proportion of the excipient in the medicinal product composition;
Daily patient intake of the excipient
Known quality defects/fraudulent adulterations
Whether the excipient is a composite
Known or potential impact on the critical quality attributes of the medicinal product
Other factors as identified or known to be relevant to assuring patient safety
With respect to Excipient Use and Function consider:

13© 2016 McGee Pharma International
your partner in compliance
Where to begin?
Identify all excipients used on site
What approach should be taken where:
• Multiple manufacturers of an excipient are used?
• Excipient is used in different formulations?
• Different functions, and volumes of the excipient in the different formulations?
• Different routes of administration for different formulations
• Product manufactured by CMO?
Identify risks to Quality, Safety and Function from source

14© 2016 McGee Pharma International
your partner in compliance
1. Determining Excipient Risk Profile
What type of RA tool should be used?
• Failure Mode Effect Analysis (FMEA)
• Hazard Analysis Critical Control Point (HACCP)
• Fault Tree Analysis (FTA)
For each excipient how would you categorise risk for each of the elements listed in the excipient RA guideline?
• Qualitative, Quantitative, Semi-quantitative approach?
• How would you ensure consistency in approach across the RA?
How would you determine and categorise the overall Risk Profile for the excipient?
How would you interpret the Risk Profile to determine appropriate level of GMP?

15© 2016 McGee Pharma International
your partner in compliance
2. Determine the Appropriate level of GMP
Document GMP standard considered
appropriate and justify with rationale
• What are the standards that should be considered?
• Rationale for choosing most appropriate standard
• What elements of the standard should be considered?
• Why?

16© 2016 McGee Pharma International
your partner in compliance
3. Determining Manufacturer’s Risk Profile
Determine excipient manufacturers current level of GMP
• How should excipient manufacturers current level of GMP be determined?
Compare excipient manufacturers current level of GMP to required standard and identify gaps
For each excipient how would you categorise risk for each of the GMP elements listed in the excipient RA guideline?
• Qualitative, Quantitative, Semi-quantitative approach
How would you determine and categorise the excipient manufacturer’s overall Risk Profile?

17© 2016 McGee Pharma International
your partner in compliance
4. Risk Control Strategy
Document overall conclusion
• Acceptable
• Acceptable with control
• Unacceptable
What types of controls might
be used to control excipient manufacturer’s
risk?
What Quality Metrics should be established and reviewed?
Established control strategy and the overall assessment process should be
reviewed and revised as appropriate

18© 2016 McGee Pharma International
your partner in compliance
Questions

19© 2016 McGee Pharma International
your partner in compliance
Question 1
• Is a capsule shell considered to be an excipient?o Yes. Refer to EMA ‘Guideline on Excipients in the Dossier for
Application for Marketing Authorisation of a Medicinal Product’ – 19 June 2007.
o This guideline lists capsule shells as an excipient.

20© 2016 McGee Pharma International
your partner in compliance
Useful Reference Document for Excipients

21© 2016 McGee Pharma International
your partner in compliance
Question 2
• If you audit all your excipient suppliers do you need to do the risk assessment?
o Yes- audit is not sufficient to meet the requirements of the Risk Assessment guideline or of Chapter 5 of the EU GMP Guide, which states:.
“Excipients and excipient suppliers should be controlled appropriately based on the results of a formalised quality risk assessment in accordance with the European Commission ‘Guidelines on the formalised risk assessment for ascertaining the appropriate Good Manufacturing Practice for excipients of medicinal products for human use”.

22© 2016 McGee Pharma International
your partner in compliance
Question 3
• What, if any, additional challenges could be foreseen in addressing the requirements of the EU guidelines of 19 March 2015 where contract manufacturing organisations are used?
• Additional challenges include but are not limited to:o The Contract Giver may be a small customer of the CMO and
therefore may have difficulty gaining information, support and time from the CMO in performing the risk assessment.
o The CMO may be located in a third country and does not have to comply with the guideline.
o There may be cultural and communication barriers.
o The CMO may not operate to GMP standards.

23© 2016 McGee Pharma International
your partner in compliance
Question 4
• Which elements of the Quality System should be higher priority if some elements may not be required at the excipient manufacturing site based on the excipient risk profile?o By performing the excipient risk assessment in accordance
with the guideline and identifying, as a first step, areas of most risk for the excipient itself, the appropriate GMP standard for the excipient based on its type, use and function in the medicinal product may be determined.
o The elements of the Quality System to be prioritised during audit or evaluation of the excipient supplier should be determined based on the outcome of the risk assessment for the excipient itself.

24© 2016 McGee Pharma International
your partner in compliance
Question 5
• Is bracketing of excipient/formulations allowed?
o The guideline does not exclude the use of bracketing in performing the excipient risk assessment.
o If a bracketing approach is used, document the scientific rationale to support the approach used.

25© 2016 McGee Pharma International
your partner in compliance
Question 6
• Could an example of a Risk Assessment of a critical excipient be shown?
o See example of ‘Excipient Risk Assessment – Worked Example’ below. This example demonstrates a semi-quantitative approach that can be used to address all the requirements of the guideline in a formalised structured manner.
Please contact MPI for further information if required

26© 2016 McGee Pharma International
your partner in compliance
Question 7
• What are the challenges/expectations relating to brokers and visibility to establish pedigree?o Brokers are involved in activities in relation to the sale or
purchase of medicinal products that consists of negotiating independently and on behalf of another legal or natural person. They do not physically handle or store materials or medicinal products.
o Specific provisions for brokers are detailed in chapter 10 of GDP guideline 2013/C 343/01.
o Brokers are subject to a registration requirement. They must have a permanent address and contact details in the Member State where they are registered.
o The quality system of a broker should be defined in writing, approved and kept up-to-date. It should set out responsibilities, processes and risk management in relation to their activities.

27© 2016 McGee Pharma International
your partner in compliance
Question 8
• How do you define the start of an excipient?
(Boundary as it relates to your product ie how far back in the supply chain should the risk assessment go?)
o An excipient is defined as: “Any constituent of a medicinal product other than the active substance and the packaging material”.
o The specifications for the quality attributes of excipients are defined in the marketing authorisation dossier.
o The risk assessment should be performed on the excipients as defined in the marketing authorisation.
o Additionally (e.g. for biological products), MPI would recommend that you document the materials used in manufacture that you consider to be and not to be excipients & your rationale for those excluded; refer to existing guidelines for this exercise.

28© 2016 McGee Pharma International
your partner in compliance
Question 9
• How can you define what an excipient is within your process?o The excipients used within the process are defined in the
Chemical Manufacturing Control (CMC) section/Module 3 (Quality) of the Common Technical Document (CTD).
o The CTD is the dossier submitted for marketing authorisation approval to the regulator.
o Additionally, refer to current guidance on the definition of excipients. Refer to question 8.

29© 2016 McGee Pharma International
your partner in compliance
Question 10
• Does the guideline apply to clinical supply?o The guideline states: “A risk assessment as set out in these
guidelines should be carried out for excipients for authorised medicinal products for human use by 21 March 2016”.
o Therefore, there is no stated requirement to apply the guideline to clinical supply.
o However, Investigational Medicinal Products are high risk products and their manufacture is required to be in accordance with GMP. It is therefore considered appropriate that the principles of the excipient risk assessment guideline be applied to clinical product.

30© 2016 McGee Pharma International
your partner in compliance
Question 11
• How can CMO’s incorporate requirements/details of the final formulation as part of the consideration of risk?
o The Contract Giver must work closely with its CMOs to ensure that the excipient risk assessments are carried out in accordance with the guideline.

31© 2016 McGee Pharma International
your partner in compliance
HPRA Question 1
• CMO’s deal with multiple Contract Givers that have procedural requirements/ expectations of their own. The excipient risk assessment guideline requires the MIA holders to conduct risk assessments. What is the Regulators’ perspective and expectation for:
1. MAH’s to ensure CMO’s comply with the guideline?
2. QP’s knowledge and oversight of this area and link to batch certification?
3. Engaging with CMO’s that are not knowledgeable of the requirements and not willing to engage?
Note: Answer will be posted on the QP Forum website when received

32© 2016 McGee Pharma International
your partner in compliance
HPRA Question 2
• How does HPRA plan to inspect for implementation of this guideline?
e.g. expectation for status of implementation, timelines for completion of the initial risk assessments? (Companies may have hundreds of excipients and significant numbers of formulations)
Note: Answer will be posted on the QP Forum website when received

33© 2016 McGee Pharma International
your partner in compliance
HPRA Question 3
• Can the HPRA (or EMA) provide guidance on the definition of an excipient - particularly in relation to Biotech products?
(Old EMA guidance document exists relating to excipients and the regulatory filing. It doesn’t cover biotech products or advances in formulation/product innovation)
Note: Answer will be posted on the QP Forum website when received

34© 2016 McGee Pharma International
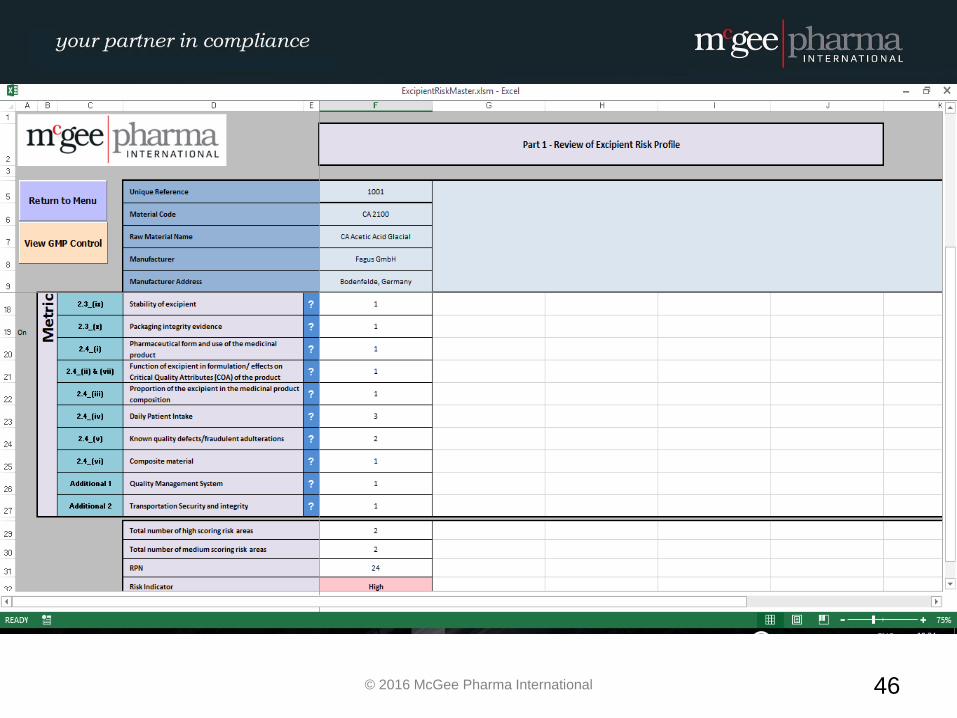
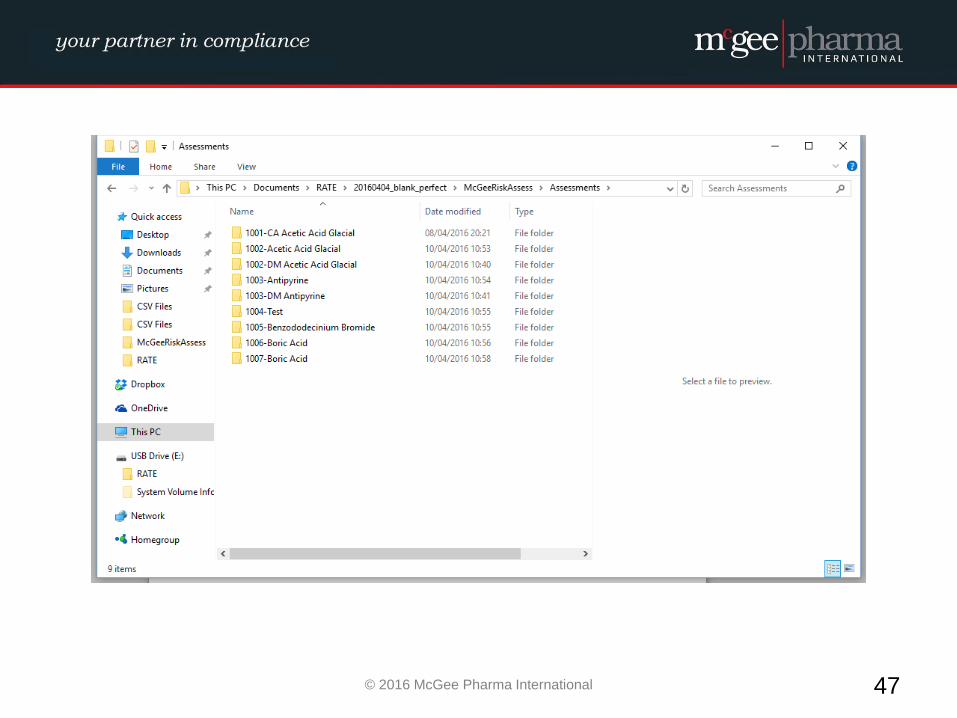
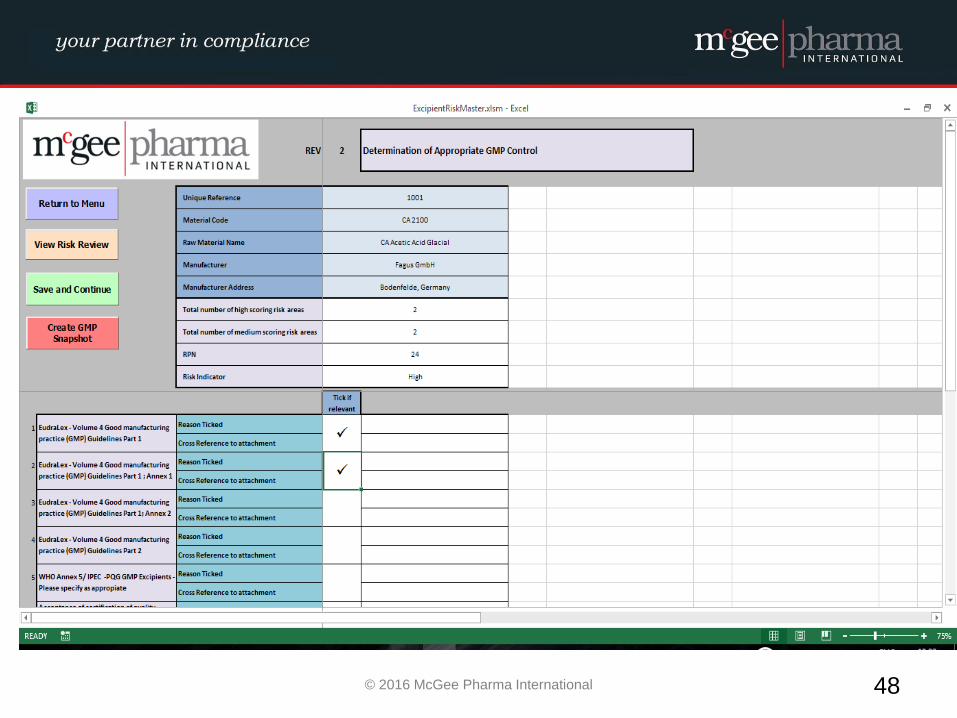
EXCIPIENT RISK ASSESSMENT
WORKED EXAMPLE

35© 2016 McGee Pharma International
your partner in compliance
Section 2.1 of 2015/C 95/02 states that ICH Q9 ‘Quality Risk Management’ can be used
Part 5 of Q9 states:
‘The degree of rigor and formality of quality risk management shouldreflect available knowledge and be commensurate with thecomplexity and/ or criticality of the issue to be addressed’
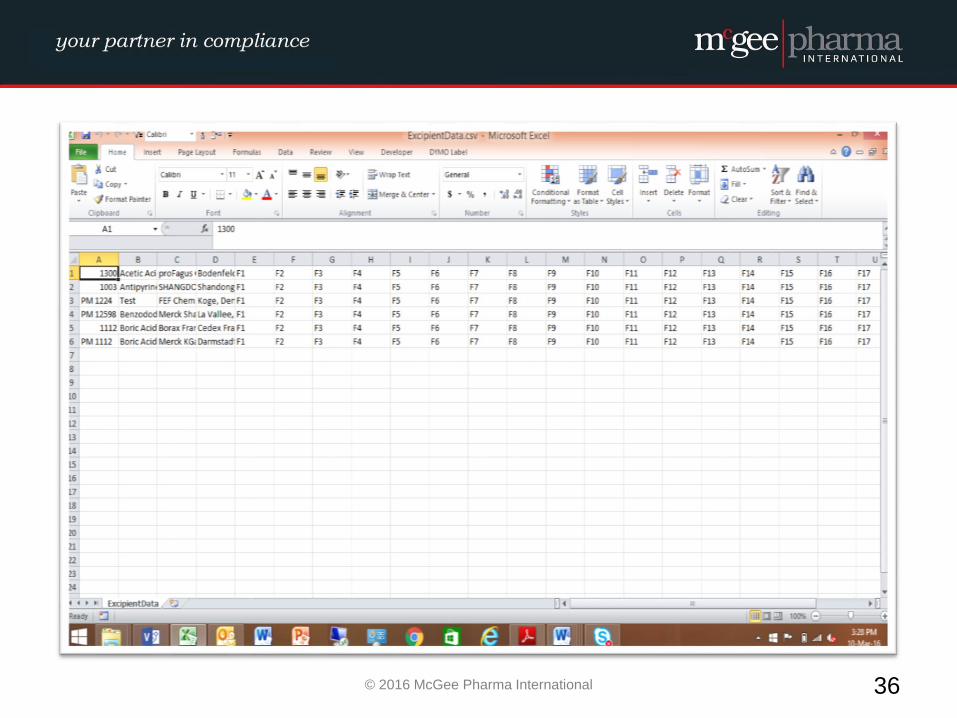
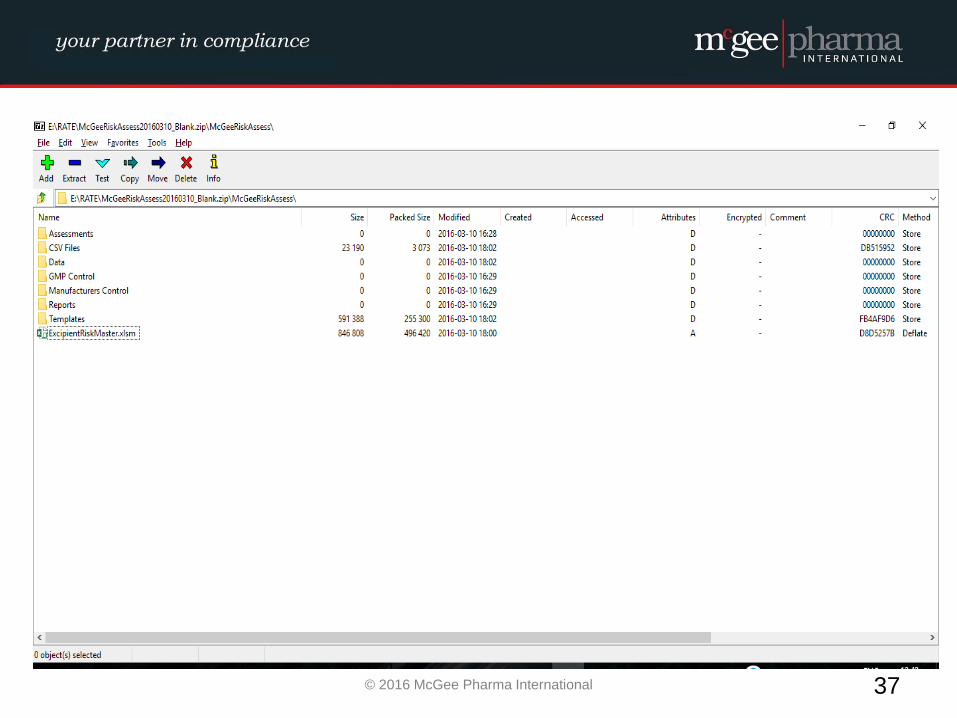
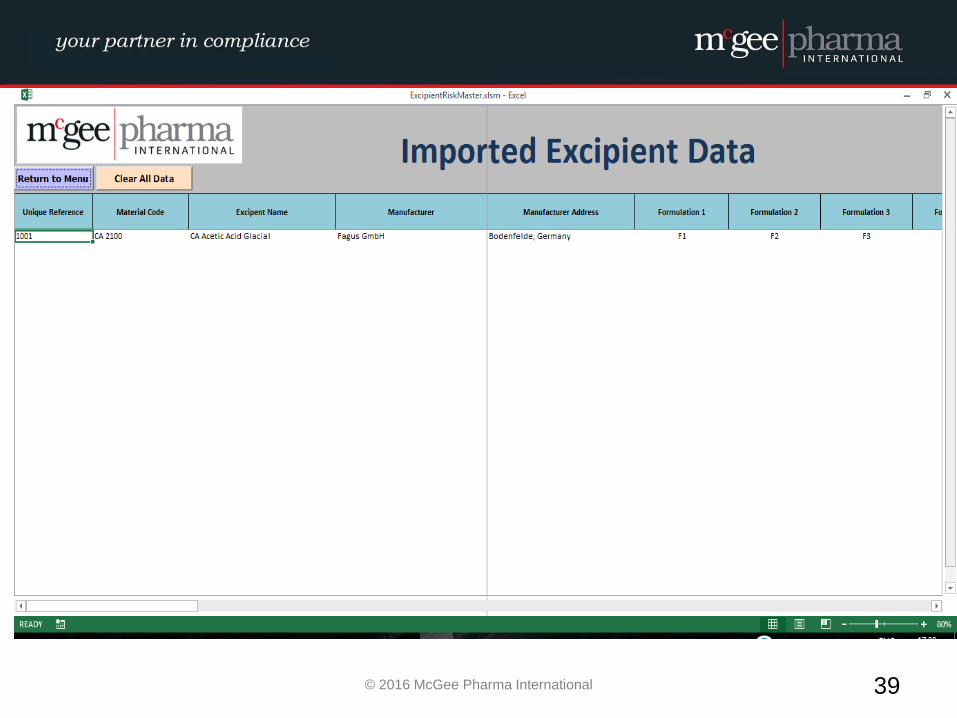
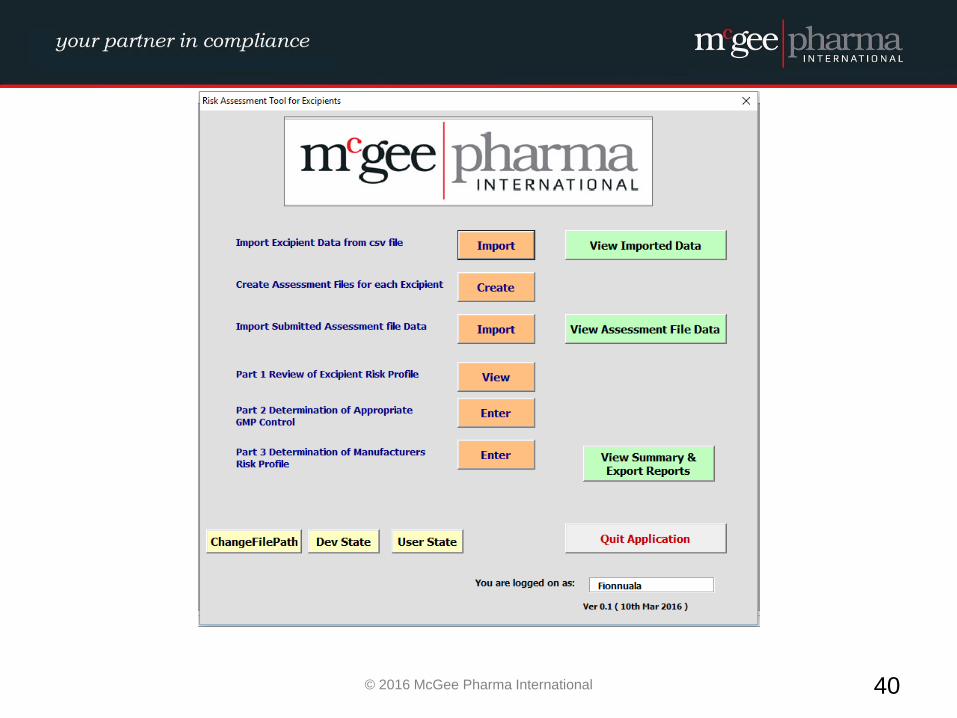
Example of a formalised Excipient Risk Assessment
MPI therefore have developed a management tool to be used by
companies to facilitate the conducting of these risk assessments in a
formalised manner which will be incorporated into their quality
system. This tool is a hybrid electronic system which will comply with
EU GMP Guidelines and 21 CFR Part 11 where relevant.
36© 2016 McGee Pharma International
37© 2016 McGee Pharma International

38© 2016 McGee Pharma International
39© 2016 McGee Pharma International
40© 2016 McGee Pharma International

41© 2016 McGee Pharma International

42© 2016 McGee Pharma International

43© 2016 McGee Pharma International

44© 2016 McGee Pharma International

45© 2016 McGee Pharma International
46© 2016 McGee Pharma International
47© 2016 McGee Pharma International
48© 2016 McGee Pharma International

49© 2016 McGee Pharma International

50© 2016 McGee Pharma International

51© 2016 McGee Pharma International

52© 2016 McGee Pharma International
Contact
A | Suite 2, Stafford House, Strand Road, Portmarnock, Co. Dublin, Ireland
P | +353 (0)1 846 47 42 E | [email protected]
F | +353 (0)1 846 4898 W| www.mcgeepharma.com