trypanosoma cruzi highjacks trkc to enter cardiomyocytes and cardiac fibroblasts while...
TRANSCRIPT

Trypanosoma cruzi highjacks TrkC to entercardiomyocytes and cardiac fibroblasts whileexploiting TrkA for cardioprotection againstoxidative stress
Daniel Aridgides,1,2 Ryan Salvador1,2 andMercio PereiraPerrin1,2*1Graduate Program in Immunology, Sackler School ofGraduate Biomedical Sciences, Tufts University, Boston,MA, USA.2Department of Pathology, Tufts University School ofMedicine, 150 Harrison Avenue, Boston, MA 02111,USA.
Summary
Chronic Chagas cardiomyopathy (CCC), causedby the obligate intracellular protozoan parasiteTrypanosoma cruzi, is a major cause of morbidityand mortality in Latin America. CCC begins whenT. cruzi enters cardiac cells for intracellular multi-plication and differentiation, a process that startswith recognition of host–cell entry receptors.However, the nature of these surface moleculesand corresponding parasite counter-receptor(s)is poorly understood. Here we show that antibodiesagainst neurotrophin (NT) receptor TrkC, but notagainst family members TrkA and TrkB, preventT. cruzi from invading primary cultures of cardio-myocytes and cardiac fibroblasts. Invasion is alsoselectively blocked by the TrkC ligand NT-3, and byantagonists of Trk autophosphorylation and down-stream signalling. Therefore, these results indicatethat T. cruzi gets inside cardiomyocytes andcardiac fibroblasts by activating TrkC preferentiallyover TrkA. Accordingly, short hairpin RNA interfer-ence of TrkC (shTrkC), but not TrkA, selectivelyprevents T. cruzi from entering cardiac cells.Additionally, T. cruzi parasite-derived neurotro-phic factor (PDNF)/trans-sialidase, a TrkC-bindingprotein, but not family member gp85, blocks entrydose-dependently, underscoring the specificityof PDNF as TrkC counter-receptor in cardiac
cell invasion. In contrast to invasion, competitiveand shRNA inhibition studies demonstrate thatT. cruzi–PDNF recognition of TrkA, but not TrkC onprimary cardiomyocytes and the cardiomyocytecell line H9c2 protects the cells against oxidativestress. Thus, this study shows that T. cruzi viaPDNF favours neurotrophin receptor TrkC forcardiac cell entry and TrkA for cardiomyocyte pro-tection against oxidative stress, and suggests anew therapeutic opportunity in PDNF and/or frag-ments thereof for CCC therapy as entry inhibitorsand/or cardioprotection agonists.
Introduction
Chagas disease, caused by infection with the protozoanparasite Trypanosoma cruzi, is a major source of diseaseburden in the Western Hemisphere, including developedcountries (Bern and Montgomery, 2009; Rassi et al.,2010; Machado et al., 2012). It starts with an acute phasewhere robust parasite growth throughout the body coin-cides with non-descript symptoms such as fever, chillsand nausea. Parasitemia typically peaks several weeksafter infection, usually following the bite of reduviidinsects, and is nearly undetectable after eight weeks, atwhich point blood and tissue parasitism wane undercontrol from the immune response, giving rise to asymp-tomatic and pathology-free indeterminate phase that canlast many years or a life-time. For reasons that remainunknown, around 30% of Chagas disease patients sufferor will suffer from chronic Chagas cardiomyopathy (CCC)whose symptoms include arrhythmias, heart failure andsudden death, and for which there are no cure exceptheart transplant (Fiorelli et al., 2011). CCC occurs withouta recrudescence of parasitism, and, in spite of the paucityof parasites in chronically infected heart, progression toCCC appears to be driven by the inflammatory responseto both residual heart parasitism and to cardiac auto-antigens, particularly myosin (Marin-Neto et al., 2007;Machado et al., 2012).
As T. cruzi is an obligate intracellular parasite, it mustgain access into the host cell cytoplasm for multiplication
Received 17 December, 2012; revised 26 January, 2013; accepted31 January, 2013. *For correspondence. E-mail [email protected]; Tel. (+1) 617 636 2933; Fax (+1) 617 636 2990.
Cellular Microbiology (2013) doi:10.1111/cmi.12119
© 2013 Blackwell Publishing Ltd
cellular microbiology

and differentiation and for avoiding killing by the immunesystem. Hence, efficient invasion of cardiac cells is criticalfor survival of T. cruzi and, thus, for the development ofacute heart disease and for parasite persistence inchronic infection, including CCC. Still, mechanisms under-lying T. cruzi recognition of cardiac cell surface receptorsrequired for the intracellular cycle is poorly understood.Although T. cruzi invades both cardiomyocytes andcardiac fibroblasts in vivo (Tafuri, 1970; Wong et al., 1992;Huang et al., 2003), structural and molecular alterationsand immune responses resulting from T. cruzi infection ofthe heart are studied almost exclusively in the context ofcardiomyocytes (Araujo-Jorge et al., 2012; Calvet et al.,2012; Machado et al., 2012), even though cardiac fibrob-lasts are critical for proper cardiomyocyte function eitherby direct cell–cell interaction or paracrine signalling inphysiological conditions and diseases such as myocardialinfarction (Souders et al., 2009; Kakkar and Lee, 2010)and other types of myocarditis (van Nieuwenhoven andTurner, 2012). Therefore, cardiac fibroblasts likely play animportant role in Chagas heart disease and studiesdesigned to understand T. cruzi-cardiac fibroblast interac-tions are sorely needed to better understand Chagasdisease progression.
Early findings suggested that T. cruzi invasion ofprimary cultures of embryonic cardiomyocytes mayinvolve mannosyl residues (Soeiro Mde et al., 1999) andrecent work indicates that cell entry depends on low-density lipoprotein (LDL) receptor in the cardiomyocytecell line H9c2 (Nagajyothi et al., 2012). However, thenature of the T. cruzi ligand that recognizes mannosylresidues and LDL receptor has not been identified. Giventhat the protozoan parasite Leishmania, which infectsa single cell type (macrophage), takes advantage ofat least seven distinct surface receptors for invasion(Cunningham, 2002), and that the relatively simple organ-ism HIV employs three entry receptors (CD4, CCR5 andCXCR4) to invade a single cell type (T-helper cell) (Wilenet al., 2012), then T. cruzi should exploit an array of entryreceptors in the heart and elsewhere as it is able to entera wide range of nucleated host cells (Brener, 1973).
We showed earlier that T. cruzi binds receptor tyrosinekinases TrkA (Chuenkova and PereiraPerrin, 2004) andTrkC (Weinkauf and PereiraPerrin, 2009) and that thebinding leads to invasion of neurons and glial cells (deMelo-Jorge and PereiraPerrin, 2007; Weinkauf et al.,2011). T. cruzi recognition of TrkA and TrkC is throughits surface parasite derived neurotrophic factor (PDNF),a GPI-linked neuraminidase/trans-sialidase; however,PDNF interaction with TrkA and TrkC, and with anotherprotein kinase (Akt) (Chuenkova and PereiraPerrin, 2009)is independent of sialic acid-binding activity (Chuenkovaand PereiraPerrin, 2011). TrkA and TrkC are activatedprimarily by the neurotrophins nerve growth factor (NGF)
and neurotrophin-3 (NT-3), respectively, and the activa-tion is essential for development and maintenance of thenervous system (Huang and Reichardt, 2003). Recently,both TrkC and TrkA have been identified in cardiomyo-cytes (Kawaguchi-Manabe et al., 2007; Meloni et al.,2010), and very recently in our lab, in neonatal and adultcardiac fibroblasts (Aridgides et al., 2013), which led tothe hypothesis that Trks are responsible, at least in part,for T. cruzi homing into the heart.
Here, we report that T. cruzi via PDNF invades bothprimary cardiomyocytes and cardiac fibroblasts preferen-tially through TrkC compared to TrkA, and that TrkA rec-ognition on cardiomyocytes protects the host cells againstoxidative stress. This study is, as far as we know, the firstto identify a molecular mechanism underlying T. cruziinvasion of cardiac fibroblasts and also the first to showthat, prior to homing into the cytosol habitat, direct T. cruzirecognition of TrkA on cardiomyocytes protects the cellsagainst oxidative stress.
Results
Preferential use of TrkC for T. cruzi entryinto cardiomyocytes
To determine whether T. cruzi exploits Trk receptors toinvade cardiomyocytes, we first assessed the ability ofT. cruzi strains Colombian (cardiotropic), CL-Brener (skel-etal muscle tropic) and Tulahuen (reticulotropic) (Brener,1973) to invade primary cultures of cardiomyocytes. Wefind that all three strains readily invade cardiomyocytesdose-dependently, although the Colombian strain isslightly more efficient than the other two (Fig. 1A). We usedthese strains (as indicated) in subsequent experiments.
To determine whether T. cruzi uses Trk receptors toinvade cardiomyocytes, which express all members of theTrk family (TrkA, TrkB and TrkC) (Cai et al., 2006;Kawaguchi-Manabe et al., 2007; Meloni et al., 2010), wepre-incubated primary cultures of cardiomyocytes withneutralizing antibodies against TrkA (a-TrkA), TrkB(a-TrkB), or TrkC (a-TrkC) followed by T. cruzi Colombianstrain. We found that infection is inhibited by TrkC antibod-ies (62% � 2.3 inhibition at 1 mg ml-1) but not by TrkA andTrkB antibodies (Fig. 1B), suggesting that T. cruzi invadescardiomyocytes preferentially via TrkC. To further explorethis point, we pre-incubated cardiomyocytes with NGF,brain-derived neurotrophic factor (BDNF), or NT-3 to blocktheir corresponding primary TrkA, TrkB and TrkC recep-tors, respectively, and then co-cultured the cardiomyocyteswith T. cruzi. Similar to the selective inhibition of infectionby neutralizing TrkC antibody, the TrkC ligand NT-3(10 ng ml-1) potently blocks infection (80% � 3.1),whereas NGF is much less effective (30% � 4.2) andBDNF is without effect (Fig. 1C). Knocking down TrkC in
2 D. Aridgides, R. Salvador and M. PereiraPerrin
© 2013 Blackwell Publishing Ltd, Cellular Microbiology

primary cardiomyocytes (five sets of cardiomyocytesinfected each with distinct lentiviral construct encodingshort hairpin against TrkC (shTrkC) prevents T. cruzi fromefficiently invading cardiomyocytes, whereas knocking
down TrkA does not significantly affect T. cruzi invasion ofcardiomyocytes (Fig. 2A), further establishing TrkC as apreferred invasion receptor for T. cruzi invasion of cardiaccells.
Fig. 1. Cardiomyocytes are preferentially invaded by T. cruzi’s Colombian strain and invasion is specifically inhibited by TrkC antibodies andthe TrkC ligand NT-3.A. Primary cultures of mouse cardiomyocytes were plated in 96-well plates, and infected in triplicate with T. cruzi Colombian, CL-Brener andTulahuen strains at various multiplicity of infection (moi). After 2–3 h, parasites that did not invade were washed away and the ones thatinvaded were allowed to differentiate and multiply for 3 days. Infected cells were counted after Diff-Quik staining (mean � SEM of twoexperiments; *P < 0.05, **P < 0.01 versus Colombian).B. Primary cardiomyocytes were plated in 96-well plates, pretreated with antibodies (1 mg ml-1) against TrkC (a-TrkC), (TrkA (a-TrkA) or TrkB(a-TrkB) followed by T. cruzi Colombian strain as in (A); mean + SEM of two independent experiments are graphed; **P < 0.01 versus vehicle,ns, not significant.C. Primary cardiomyocytes were pretreated with the neurotrophins (NTs) NT-3 (TrkC ligand), NGF (TrkA ligand) and BDNF (TrkB ligand)(10 ng ml-1, 30 min), infected with T. cruzi and the infection quantitated as in (A). Mean + SEM of two independent experiments are graphed;**P < 0.01, *** P< 0.001 versus vehicle, ns, not significant.
Fig. 2. T. cruzi infection of primary cardiomyocytes is inhibited by TrkC knockdown preferentially over TrkA knockdown and by antagonists ofTrk autophosphorylation and Trk downstream signalling.A. Primary cardiomyocytes (in quadruplicate wells per point) were plated on 96-well plates, and after one day, were infected with lentivirusexpressing small hairpin knockdown plasmids for GFP (shGFP), TrkA (shTrkA, four distinct constructs), or TrkC (shTrkC, five distinctconstructs). Six days after lentiviral infection, cells were infected with T. cruzi Tulahuen strain (2 ¥ 105 ml-1) for 3 h, washed to removeunattached parasites, the infection allowed to proceed for two days at 37°C, fixed in 4% paraformaldehyde, and stained with DAPI to visualizehost and parasite nuclei. Infected cells (> two parasites per cell) were counted in > 200 cells per well, and inhibition of infection was calculatedrelative to control infected cells; mean + SEM of two independent experiments; *P < 0.05 relative to control infected cells.B. Primary cardiomyocytes were pretreated (30 min) with AG879 (Trk antagonist), U0126 (Erk signalling antagonist) or IGF1-R (insulin-likegrowth factor-1 receptor antagonist) followed by T. cruzi Tulahuen, as in (A); mean + SEM of two independent experiments; ***P < 0.001versus vehicle.
T. cruzi uses Trk receptors for cardiomyocytes invasion and protection 3
© 2013 Blackwell Publishing Ltd, Cellular Microbiology

In addition, we find that invasion is blocked by the Trkautophosphorylation inhibitor AG879 (Ohmichi et al.,1993) and by U0126, an inhibitor of Erk signalling that isactivated downstream of Trks (Huang and Reichardt,2003), but not by a pharmacological inhibitor of insulin-like growth factor-1 receptor (IGF-1R), which does notmediate T. cruzi invasion of neuronal cells (de Melo-Jorgeand PereiraPerrin, 2007) (Fig. 2B). Thus, T. cruzi invasionof cardiomyocytes via TrkC requires TrkC signalling.
Selective inhibition of cardiomyocyte invasion by theT. cruzi surface protein parasite-derived neurotrophicfactor (PDNF)/trans-sialidase
Given that the T. cruzi surface protein PDNF isthe counter-receptor for TrkA (de Melo-Jorge andPereiraPerrin, 2007) and TrkC (Weinkauf et al., 2011)in invasion of neuronal cells, PDNF may act likewiseduring T. cruzi entry into cardiomyocytes. We find that abacterially expressed short version of PDNF (sPDNF)(Chuenkova et al., 1999) lacking C-terminal tandemrepeat SAPA (Affranchino et al., 1989) effectively blockscardiomyocyte invasion by Colombian, CL-Brener andTulahuen strains (Fig. 3A). PDNF has intrinsic neuramini-dase activity (Pereira, 1983; Scudder et al., 1993) but thisactivity may not be responsible for the ability of sPDNF tocompetitively inhibit cardiomyocyte invasion because pre-treatment of cardiomyocytes with Vibrio cholera neurami-nidase (VCN) (same number of units as sPDNF) doesnot affect invasion (Fig. 3A). In addition, PDNF familymember gp85 (Alves and Colli, 2008) is also inactive inblocking invasion (Fig. 3B), underscoring the specificityof PDNF/TrkC interaction in triggering cardiomyocyteinvasion.
Preferential use of surface receptor TrkC for T. cruzi viaPDNF entry into cardiac fibroblasts
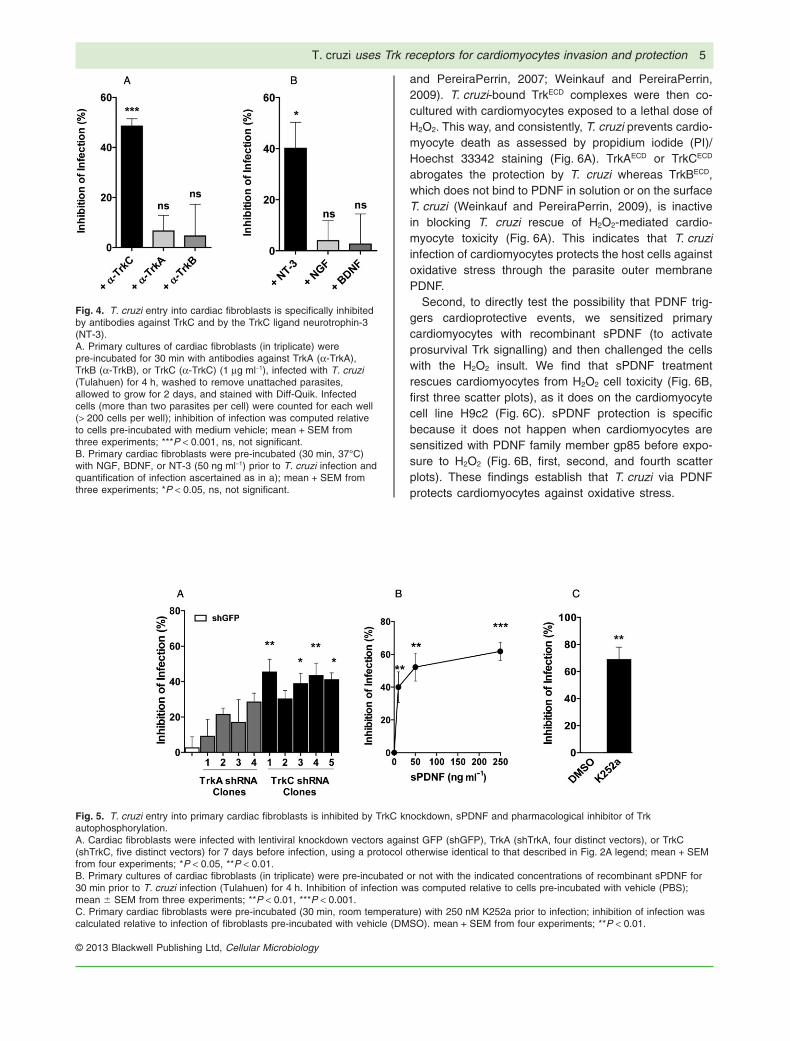
To determine whether T. cruzi invades primary cardiacfibroblasts through Trk receptors (Aridgides et al., 2013)we performed invasion inhibition assays analogous tothose described above for cardiomyocytes. As with car-diomyocytes, we find that antibodies against TrkC specifi-cally inhibit T. cruzi entry (Fig. 4A), as does the TrkCligand NT-3 (Fig. 4B), knockdown of TrkC by shRNA asdetermined with nine distinct lentiviral vectors encodingshTrkC or shTrkA (Fig. 5A), and the Trk autophosphoryla-tion inhibitor K252a (Tapley et al., 1992) (Fig. 5B). Theseresults indicate that T. cruzi uses TrkC preferentially overTrkA to invade cardiac fibroblasts, that TrkC must beactivated to drive cell entry, and that TrkC-mediatedinvasion depends on parasite-bound PDNF as judged bythe effect of soluble recombinant sPDNF in inhibiting inva-sion dose-dependently (Fig. 5C).
Recognition of cardiomyocyte-TrkA by T. cruzi orrecombinant PDNF triggers protection againstoxidative stress
As TrkA activation is a bona fide trigger of cell survival andtrophic events in neural cells (Huang and Reichardt,2003) and cardiomyocytes (Caporali et al., 2008), wesought to determine whether T. cruzi exploits TrkA forcardiomyocyte protection.
First, to test whether T. cruzi infection of cardiomyo-cytes elicits protective events via PDNF, we pre-incubatedT. cruzi with the extracellular domain of TrkA (TrkAECD) or(TrkCECD), which, through their neurotrophin-binding sites,bind to and block T. cruzi-anchored PDNF (de Melo-Jorge
Fig. 3. T. cruzi infection of cardiomyocytes is blocked by PDNF and not by Vibrio cholera neuraminidase or PDNF family member gp85.A. Primary cardiomyocytes (triplicate) were pretreated with sPDNF (250 ng ml-1, 30 min) or Vibrio cholera neuraminidase (VCN, same numberof units as PDNF) followed by Colombian, CL-Brener and Tulahuen strains of T. cruzi at 4, 16 and 32 MOI for 3 h, washed to removeunattached parasites, and allowed infection to proceed for 2 days. Infection was quantified by light microscopy after Diff-Quik staining,mean � SEM of two experiments, ***P < 0.001 versus vehicle.B. Primary cardiomyocytes were pretreated with various doses of sPDNF or gp85 (30 min) followed by T. cruzi Tulahuen, asin (A), mean � SEM of two experiments, ***P < 0.001 versus vehicle.
4 D. Aridgides, R. Salvador and M. PereiraPerrin
© 2013 Blackwell Publishing Ltd, Cellular Microbiology
and PereiraPerrin, 2007; Weinkauf and PereiraPerrin,2009). T. cruzi-bound TrkECD complexes were then co-cultured with cardiomyocytes exposed to a lethal dose ofH2O2. This way, and consistently, T. cruzi prevents cardio-myocyte death as assessed by propidium iodide (PI)/Hoechst 33342 staining (Fig. 6A). TrkAECD or TrkCECD
abrogates the protection by T. cruzi whereas TrkBECD,which does not bind to PDNF in solution or on the surfaceT. cruzi (Weinkauf and PereiraPerrin, 2009), is inactivein blocking T. cruzi rescue of H2O2-mediated cardio-myocyte toxicity (Fig. 6A). This indicates that T. cruziinfection of cardiomyocytes protects the host cells againstoxidative stress through the parasite outer membranePDNF.
Second, to directly test the possibility that PDNF trig-gers cardioprotective events, we sensitized primarycardiomyocytes with recombinant sPDNF (to activateprosurvival Trk signalling) and then challenged the cellswith the H2O2 insult. We find that sPDNF treatmentrescues cardiomyocytes from H2O2 cell toxicity (Fig. 6B,first three scatter plots), as it does on the cardiomyocytecell line H9c2 (Fig. 6C). sPDNF protection is specificbecause it does not happen when cardiomyocytes aresensitized with PDNF family member gp85 before expo-sure to H2O2 (Fig. 6B, first, second, and fourth scatterplots). These findings establish that T. cruzi via PDNFprotects cardiomyocytes against oxidative stress.
Fig. 4. T. cruzi entry into cardiac fibroblasts is specifically inhibitedby antibodies against TrkC and by the TrkC ligand neurotrophin-3(NT-3).A. Primary cultures of cardiac fibroblasts (in triplicate) werepre-incubated for 30 min with antibodies against TrkA (a-TrkA),TrkB (a-TrkB), or TrkC (a-TrkC) (1 mg ml-1), infected with T. cruzi(Tulahuen) for 4 h, washed to remove unattached parasites,allowed to grow for 2 days, and stained with Diff-Quik. Infectedcells (more than two parasites per cell) were counted for each well(> 200 cells per well); inhibition of infection was computed relativeto cells pre-incubated with medium vehicle; mean + SEM fromthree experiments; ***P < 0.001, ns, not significant.B. Primary cardiac fibroblasts were pre-incubated (30 min, 37°C)with NGF, BDNF, or NT-3 (50 ng ml-1) prior to T. cruzi infection andquantification of infection ascertained as in a); mean + SEM fromthree experiments; *P < 0.05, ns, not significant.
Fig. 5. T. cruzi entry into primary cardiac fibroblasts is inhibited by TrkC knockdown, sPDNF and pharmacological inhibitor of Trkautophosphorylation.A. Cardiac fibroblasts were infected with lentiviral knockdown vectors against GFP (shGFP), TrkA (shTrkA, four distinct vectors), or TrkC(shTrkC, five distinct vectors) for 7 days before infection, using a protocol otherwise identical to that described in Fig. 2A legend; mean + SEMfrom four experiments; *P < 0.05, **P < 0.01.B. Primary cultures of cardiac fibroblasts (in triplicate) were pre-incubated or not with the indicated concentrations of recombinant sPDNF for30 min prior to T. cruzi infection (Tulahuen) for 4 h. Inhibition of infection was computed relative to cells pre-incubated with vehicle (PBS);mean � SEM from three experiments; **P < 0.01, ***P < 0.001.C. Primary cardiac fibroblasts were pre-incubated (30 min, room temperature) with 250 nM K252a prior to infection; inhibition of infection wascalculated relative to infection of fibroblasts pre-incubated with vehicle (DMSO). mean + SEM from four experiments; **P < 0.01.
T. cruzi uses Trk receptors for cardiomyocytes invasion and protection 5
© 2013 Blackwell Publishing Ltd, Cellular Microbiology

Fig. 6. T. cruzi infection or sPDNF stimulation of cardiomyocytes confers protection against oxidative stress.A. T. cruzi infection protects cardiomyocytes against H2O2-induced cell death and the protection is blocked by T. cruzi interaction with PDNFreceptors TrkAECD and TrkCECD but not with PDNF non-receptor TrkBECD. Primary cultures of cardiomyocytes were exposed to 150 mM H2O2 for4 h following pre-incubation with vehicle media (H2O2) or with T. cruzi (2 ¥ 105 ml-1) pretreated with vehicle (T. cruzi) or with the extracellulardomains (ECDs) of TrkA (TrkAECD), TrkC (TrkCECD) or TrkB (TrkBECD) (200 ng ml-1, 30 min) to block (TrkAECD and TrkCECD) or not (TrkBECD)GPI-anchored PDNF. Cell death was assessed by counting > 200 cells stained with the propidium iodide (PI)/Hoechst 33342 mixture. Scatterplot displays means of five separate experiments, each performed in triplicate. Panel on the right shows representative staining from eachcondition; dead cardiomyocytes are seen a pink dots (the result of the merge of PI and Hoechst staining), while viable cardiomyocytes arestained blue by the vital dye Hoechst 33342 and no PI staining. *P < 0.05.B. sPDNF specifically protects primary cardiomyocytes from H2O2-induced cell death. Primary cardiomyocytes were unexposed (Vehicle) orexposed to 150 mM H2O2 for 4 h without (+ H2O2). Prior to H2O2 exposure, cardiomyocytes were pre-incubated (30 min, room temperature) withsPDNF or with the PDNF/trans-sialidase family member gp85 (150 ng ml-1). Prior to H2O2 exposure and sPDNF treatment, cardiomyocyteswere additionally pre-incubated with antibodies against TrkA (a-TrkA IgG) or TrkC (a-TrkC IgG) (1 mg ml-1). Scatter plot displays means of twoidentical experiments, each performed in triplicate *P < 0.05; ns, not significant statistically.C. T. cruzi sPDNF protects H9c2 cardiomyocytes from H2O2-induced cell death. H9c2 cardiomyocytes were exposed to 150 mM H2O2 for 4 hwith or without pre-incubation with sPDNF (150 ng ml-1). Scatter plot displays means of three identical experiments, each performed intriplicate *P < 0.05, **P < 0.01.D. sPDNF protection of cardiomyocytes is selectively abrogated by TrkA knockdown. Primary cardiomyocytes were lentivirally transfected withshRNA to generate four shTrkA clones and five shTrkC clones, and after 1 week, clones were exposed to 150 ng ml-1 sPDNF and 500 mMH2O2 for four hours in DMEM/0.1% FCS, and cell death quantified by PI/Hoechst 33342. *P < 0.05; mean + SEM from three experiments;*P < 0.05.
6 D. Aridgides, R. Salvador and M. PereiraPerrin
© 2013 Blackwell Publishing Ltd, Cellular Microbiology

Third, to determine whether T. cruzi protects cardio-myocytes against oxidative stress via TrkA, we reactedcardiomyocytes with anti-TrkA IgG or anti-TrkC IgG, andfound that only anti-TrkA IgG effectively reverses theprotective effect of sPDNF (Fig. 6B, first, second, fifthand sixth scatter plots). This underscores the require-ment of cardiomyocyte TrkA for the beneficial effectof sPDNF to take place, the opposite of TrkC usage forcell entry (Fig. 1B). Furthermore, knockdown of TrkAgene expression in primary cardiomyocytes (four setseach infected with lentivirus encoding a distinct shorthairpin TrkA construct) annuls sPDNF-induced neu-tralization of H2O2 cardiotoxicity relative to controlcardiomyocytes bearing lentivirus-encoding shGFP(Fig. 6D). Contrary to TrkA knockdown, interference withTrkC mRNA (five sets of cardiomyocytes transfectedwith different TrkC shRNAs) is much less effective inreversing the protective effect of sPDNF (Fig. 6D).
Discussion
TrkC/NT-3 signalling promotes cardiomyocyte prolifera-tion and ventricular trabeculation in embryogenesis (Linet al., 2000) and TrkC deletion gives rise to defects incardiac septation, valvulogenesis and truncal formation(Tessarollo et al., 1997). However, little is known aboutthe role TrkC signalling plays in cardiomyocyte function inadulthood except that it increases the expression ofcardiac hypertrophic markers (skeletal a-actin and atrialnatriuretic peptide), cardiomyocyte size and phenyla-lanine uptake (Kawaguchi-Manabe et al., 2007). Ourresults suggest a role for TrkC in Chagas myocarditis asthey demonstrate that T. cruzi binding to, and activatingof, TrkC through its PDNF/trans-sialidase, preferentiallyover family members TrkA and TrkB, is a prelude to car-diomyocyte invasion, thereby implicating TrkC in T. cruzihoming into the heart. Earlier studies showed that activa-tion of transforming growth factor-b (TGF-b) receptorenhances T. cruzi entry into cardiomyocyte and other celltypes (Ming et al., 1995; Waghabi et al., 2005; 2007), asdoes binding to kinin receptors (Todorov et al., 2003).Although our results suggest that PDNF family membergp85 does not initiate a cascade of events triggeringTrkC-dependent cardiomyocyte invasion, independentstudies showed that gp85 is important in T. cruzi adhesionto laminin and other ECM components (Alves and Colli,2008; Mattos et al., 2012).
The idea that T. cruzi recognition of TrkC contributes tocardiac homing is further supported by our findings thatT. cruzi entry into cardiac fibroblasts is selectively drivenby TrkC-PDNF interaction. Most studies on Chagas heartdisease focus on T. cruzi recognition of cardiomyocytesand mechanisms underlying T. cruzi-cardiac fibroblastinteraction are rare to find (Tafuri, 1970; Wong et al.,
1992), even though cardiomyocytes require cardiacfibroblasts for proper function either through gap junction-based direct cell–cell interaction or by releasing growthfactors and cytokines such as TGF-b, fibroblast growthfactor-2, and interleukin-6 for paracrine signalling(Souders et al., 2009; Kakkar and Lee, 2010). Cardiacfibroblasts express receptors for growth factors, several incommon with cardiomyocytes and some more abundantin the fibroblasts, like angiotensin receptors, which link therenin–angiotensin–aldosterone system to myocardial andextracellular matrix remodelling and which convey para-crine signals to cardiomyocytes via TGF-b secretion (Grayet al., 1998). Results presented here provide a molecularbasis for T. cruzi entry into cardiac fibroblasts as theyshow, for the first time, that TrkC is an entry receptor forT. cruzi invasion of cardiac fibroblasts, analogous to inva-sion of cardiomyocytes.
Although TrkA is relatively unimportant for T. cruzi entryinto cardiomyocytes and cardiac fibroblasts, our resultsdemonstrate that T. cruzi interaction with TrkA on cardio-myocytes protects the cells against oxidative stress(Fig. 6). This is in concert with cardiomyocyte-TrkA acti-vation by its natural ligand, NGF, which reduces myo-cardial infarction (Meloni et al., 2010) and hypoxia/reoxygenation (Caporali et al., 2008) injury, and with ourrecent findings that T. cruzi–PDNF activation of TrkA incardiac fibroblasts triggers the secretion of NGF thataverts cardiomyocyte damage (Aridgides et al., 2013).
How might T. cruzi-induced cardioprotective events fitin with Chagas heart disease progression? Patients withacute Chagas heart infection present several cardiacdysfunctions including alterations in ventricular repolari-zation and cardiomegaly due to pericardial effusion(Parada et al., 1997; Pinto et al., 2001; Beltrao Hde et al.,2009; Valente et al., 2009). Paradoxically, acute Chagasmyocarditis wanes without sequelae and most patientsremain asymptomatic and pathology-free for many years(indeterminate phase) until CCC emerges. But onlyapproximately 30% chagasic patients progress to CCC(Marin-Neto et al., 2007; Rassi et al., 2010; Machadoet al., 2012). Thus it may be that the interaction of T. cruzi,via PDNF, with cardiac cells in acute infection enhancesmutually beneficial host survival mechanisms, as origi-nally suggested in T. cruzi recognition of neuronal cells(Chuenkova and Pereira, 2000) and in concert with otherbeneficial mammalian host/parasite interactions such assuppression of bacterial infection resulting from herpesvi-rus infection (Barton et al., 2007) and of allergies byworms (Fallon and Mangan, 2007).
Cardiomyocytes are particularly vulnerable to reactiveoxygen species due to their high metabolic rate, andoxidative stress is important in Chagas cardiomyopathy(Machado et al., 2012). A T. cruzi-driven cardioprotectionagainst oxidative stress such as by direct activation of
T. cruzi uses Trk receptors for cardiomyocytes invasion and protection 7
© 2013 Blackwell Publishing Ltd, Cellular Microbiology

TrkA on cardiomyocytes (this study) and by paracrinesignalling though the stimulation of NGF secretion fromcardiac fibroblasts (Aridgides et al., 2013) fits with theconcept of acute heart infection waning without sequelaeand with chronic pathology-free infection (indeterminatephase). Consistent with this idea, earlier studies showedthat T. cruzi can prevent apoptosis in cultured cardiomyo-cytes via activation of the transcription factor NF-kB(Petersen et al., 2006), an activation that must be distinctfrom PDNF signalling because NF-kB is not activated byTrks (Huang and Reichardt, 2003).
Given that Maraviroc and other entry receptor antago-nists of HIV infection of CD4 lymphocytes are currentlyused in the treatment of AIDS (Meanwell and Kadow,2007), and given that sPDNF potently blocks cardiac cellentry of three biologically distinct T. cruzi strains, PDNFand/or PDNF fragments may have an opportunity astherapeutics in CCC for which anti-infective therapy inunavailable (Matta Guedes et al., 2012). Furthermore,TrkA-mediated cardioprotective effects should addanother dimension for PDNF as a therapeutic in CCC.
Experimental procedures
Ethics statement
All mouse work was approved by the Institutional Animal Careand Use Committee of Tufts University School of Medicine andDivision of Laboratory Animal Medicine.
Trypanosoma cruzi culture
Parasites were maintained in monolayers of Vero cells. Trypo-mastigotes, when released from infected cells, were harvestedfrom supernatants by low speed centrifugation (250 g, 5 min) in atable-top centrifuge to remove any contaminating Vero cells, thenat high speed (2500 g, 10 min) to pellet parasites, which werethen resuspended in DMEM/0.1% fetal calf serum (FCS) andcounted in a haematocytometer before use.
Cardiomyocyte and cardiac fibroblast preparation
Neonatal (1–3 days old) C57BL/6 mice (Jackson Laboratories)were sacrificed by decapitation, hearts dissected, and cardiaccells were prepared as described previously (Aridgides et al.,2013) according to a procedure modified from Sreejit et al.(2008). Briefly, hearts were cut into small cubes and digestedwith trypsin, separated by differential adhesion to a gelatin-coated substrate, then plated in 20% FCS and 5% horse serum.Purity of cardiac fibroblasts was assessed by immunofluores-cence microscopy using antibodies to vimentin and of cardio-myocytes using antibodies to myosin heavy chain, and usedwhen purity for each cell type was > 90%; cardiomyocytesshowed a beating phenotype.
In vitro infection assay
Cardiac fibroblasts or cardiomyocytes in 20% FCS/5% horseserum were plated at ~ 2 ¥ 104 ml-1 on 1% gelatin-coated 96-well
plates and, after incubation for 14 h, medium was changed toDMEM/0.1% FCS. For inhibition of infection assays, cells werepretreated for 30 min with a-Trk antibodies (Abcam), neuro-trophins (R&D Systems), Trk chemical inhibitors (Sigma-Aldrich),sPDNF, gp85 (see below), or VCN (same number of units assPDNF). Parasites were added at the concentrations indicated infigure legends of figures, and allowed to infect for 3 h, after whichnon-invaded parasites were washed away twice with PBS andcells were left in DMEM/1% FCS. After 2 days, cells were fixedwith methanol, stained with Diff-Quik, and infected cells werecounted in each well by microscopy. Inhibition of infection wascalculated relative to vehicle-treated cells in each experiment.
Purification of recombinant sPDNF and gp85
sPDNF was cloned into BL21 expression bacteria and purified byNi-chromatography as previously described (Aridgides et al.,2013). gp85 (Giordano et al., 1999; Alves and Colli, 2008) wascloned from CL-Brener T. cruzi DNA using a nested PCRapproach to specifically amplify this single member of a highlyconserved gene family (NCBI Red Seq XM_808586). Flankingprimers (Forward: 5′-GTG GCC TCA CAG TGA TTA AG-3′,Reverse: 5′-ACA GGA AGA GTG CGG AAG AA-3′) generated a3035 base pair product, which was then used as a template forPCR to produce the gp85 cDNA product with additional BamHIand XhoI sites (underlined) (Forward: 5′-GCA GGA TCC ATGCTC TCA CGT GTT G-3′, Reverse: 5′-ATA CTC GAG CGC AGTCGC AAG G-3′). This PCR product was then cloned intothe pET-23b vector (Novagen) and transformed into BL21Escherichia coli for expression. Bacteria were grown in LBmedium to an OD600 of 0.7, then induced for 3 h with 200 mMIPTG (isopropyl b-D-1-thiogalactopyranoside). Bacteria werethen pelleted, lysed by sonication, and gp85 was purified over aNi++ column. Purity and concentration were assessed in SDS-PAGE gels after staining with Coomassie brilliant blue.
Lentiviral transfection and shRNA knockdown
Lentiviral vectors (Open Biosystems) encoding shRNA constructstargeting TrkA, TrkC or GFP mRNA were generated from trans-fected HEK 293 cells according to manufacturer instructions,aliquoted, and frozen at -80°C until use. Lentiviral infection ofprimary cultures was performed after pretreatment with Poly-brene (8 mg ml-1) (Sigma-Aldrich) then adding lentiviral particlesto each well. Optimal concentrations (8 ml vector/100 ml well on96-well plates) were discerned by preliminary dose–responseexperiments on cardiac fibroblasts where cells were infected for7 days, fixed and stained with antibodies specific for TrkA andTrkC (Abcam) and assessed by fluorescence microscopy.
Cardiomyocyte survival assay
Primary cardiomyocytes or H9c2 cardiomyocytes (ATCC) wereplated on 96-well plates, pretreated with indicated proteins for30 min, and hydrogen peroxide or PBS vehicle was added to thecultures for four h. Cells were stained with Hoechst 33342(20 mg ml-1) and PI (10 ml) to visualize live and dead (apoptotic)cells, as we described earlier (Chuenkova and PereiraPerrin,2004). After 5 min, the cells were visualized under ultravioletirradiation at 340–380 nm with an inverted microscope. More
8 D. Aridgides, R. Salvador and M. PereiraPerrin
© 2013 Blackwell Publishing Ltd, Cellular Microbiology

than 200 cells per well were counted and percent viable cells(Hoechst 33342 positive and PI negative) were calculated rela-tive to total cell number.
Statistics
Statistical analyses were performed using GraphPad Prism soft-ware using Student’s t-test or ANOVA with Tukey’s post-test.
Acknowledgements
We thank Dr Ricardo Gazzinelli for the gift of CL-Brener andColombian strains of T. cruzi.
References
Affranchino, J.L., Ibanez, C.F., Luquetti, A.O., Rassi, A.,Reyes, M.B., Macina, R.A., et al. (1989) Identification of aTrypanosoma cruzi antigen that is shed during the acutephase of Chagas’ disease. Mol Biochem Parasitol 34: 221–228.
Alves, M.J., and Colli, W. (2008) Role of the gp85/trans-sialidase superfamily of glycoproteins in the interaction ofTrypanosoma cruzi with host structures. Subcell Biochem47: 58–69.
Araujo-Jorge, T.C., Waghabi, M.C., Bailly, S., and Feige, J.J.(2012) The TGF-beta pathway as an emerging target forChagas disease therapy. Clin Pharmacol Ther 92: 613–621.
Aridgides, D., Salvador, R., and PereiraPerrin, M. (2013)Trypanosoma cruzi coaxes cardiac fibroblasts into pre-venting cardiomyocyte death by activating nerve growthfactor receptor TrkA. PLoS ONE 8: e57450.
Barton, E.S., White, D.W., Cathelyn, J.S., Brett-McClellan,K.A., Engle, M., Diamond, M.S., et al. (2007) Herpesviruslatency confers symbiotic protection from bacterial infec-tion. Nature 447: 326–329.
Beltrao Hde, B., Cerroni Mde, P., Freitas, D.R., Pinto, A.Y.,Valente Vda, C., Valente, S.A., et al. (2009) Investigationof two outbreaks of suspected oral transmission of acuteChagas disease in the Amazon region, Para State, Brazil,in 2007. Trop Doct 39: 231–232.
Bern, C., and Montgomery, S.P. (2009) An estimate of theburden of Chagas disease in the United States. Clin InfectDis 49: e52–e54.
Brener, Z. (1973) Biology of Trypanosoma cruzi. Annu RevMicrobiol 27: 347–382.
Cai, D., Holm, J.M., Duignan, I.J., Zheng, J., Xaymardan, M.,Chin, A., et al. (2006) BDNF-mediated enhancement ofinflammation and injury in the aging heart. Physiol Genom-ics 24: 191–197.
Calvet, C.M., Melo, T.G., Garzoni, L.R., Oliveira, F.O., Jr,Neto, D.T., Meirelles, M.N.S.L., et al. (2012) Current under-standing of the Trypanosoma cruzi-cardiomyocyte interac-tion. Front Immunol 3: 327.
Caporali, A., Sala-Newby, G.B., Meloni, M., Graiani, G., Pani,E., Cristofaro, B., et al. (2008) Identification of the prosur-vival activity of nerve growth factor on cardiac myocytes.Cell Death Differ 15: 299–311.
Chuenkova, M., Pereira, M., and Taylor, G. (1999) trans-sialidase of Trypanosoma cruzi: location of galactose-binding site(s). Biochem Biophys Res Commun 262:549–556.
Chuenkova, M.V., and Pereira, M.A. (2000) A trypanosomalprotein synergizes with the cytokines ciliary neurotrophicfactor and leukemia inhibitory factor to prevent apoptosis ofneuronal cells. Mol Biol Cell 11: 1487–1498.
Chuenkova, M.V., and PereiraPerrin, M. (2004) Chagas’disease parasite promotes neuron survival and differentia-tion through TrkA nerve growth factor receptor. J Neuro-chem 91: 385–394.
Chuenkova, M.V., and PereiraPerrin, M. (2009) Trypanosomacruzi targets Akt in host cells as an intracellular antiapop-totic strategy. Sci Signal 2: ra74.
Chuenkova, M.V., and PereiraPerrin, M. (2011) Neurodegen-eration and neuroregeneration in Chagas disease. AdvParasitol 76: 195–233.
Cunningham, A.C. (2002) Parasitic adaptive mecha-nisms in infection by leishmania. Exp Mol Pathol 72: 132–141.
Fallon, P.G., and Mangan, N.E. (2007) Suppression of TH2-type allergic reactions by helminth infection. Nat RevImmunol 7: 220–230.
Fiorelli, A.I., Santos, R.H., Oliveira, J.L., Jr, Lourenco-Filho,D.D., Dias, R.R., Oliveira, A.S., et al. (2011) Heart trans-plantation in 107 cases of Chagas’ disease. TransplantProc 43: 220–224.
Giordano, R., Fouts, D.L., Tewari, D., Colli, W., Manning, J.E.,and Alves, M.J. (1999) Cloning of a surface membraneglycoprotein specific for the infective form of Trypanosomacruzi having adhesive properties to laminin. J Biol Chem274: 3461–3468.
Gray, M.O., Long, C.S., Kalinyak, J.E., Li, H.T., and Karliner,J.S. (1998) Angiotensin II stimulates cardiac myocytehypertrophy via paracrine release of TGF-beta 1 andendothelin-1 from fibroblasts. Cardiovasc Res 40: 352–363.
Huang, E.J., and Reichardt, L.F. (2003) Trk receptors: roles inneuronal signal transduction. Annu Rev Biochem 72: 609–642.
Huang, H., Petkova, S.B., Cohen, A.W., Bouzahzah, B.,Chan, J., Zhou, J.N., et al. (2003) Activation of transcriptionfactors AP-1 and NF-kappa B in murine Chagasic myocar-ditis. Infect Immun 71: 2859–2867.
Kakkar, R., and Lee, R.T. (2010) Intramyocardial fibroblastmyocyte communication. Circ Res 106: 47–57.
Kawaguchi-Manabe, H., Ieda, M., Kimura, K., Manabe, T.,Miyatake, S., Kanazawa, H., et al. (2007) A novel cardiachypertrophic factor, neurotrophin-3, is paradoxicallydownregulated in cardiac hypertrophy. Life Sci 81: 385–392.
Lin, M.I., Das, I., Schwartz, G.M., Tsoulfas, P., Mikawa, T.,and Hempstead, B.L. (2000) Trk C receptor signalingregulates cardiac myocyte proliferation during early heartdevelopment in vivo. Dev Biol 226: 180–191.
Machado, F.S., Dutra, W.O., Esper, L., Gollob, K.J., Teix-eira, M.M., Factor, S.M., et al. (2012) Current understand-ing of immunity to Trypanosoma cruzi infection andpathogenesis of Chagas disease. Semin Immunopathol34: 753–770.
T. cruzi uses Trk receptors for cardiomyocytes invasion and protection 9
© 2013 Blackwell Publishing Ltd, Cellular Microbiology

Marin-Neto, J.A., Cunha-Neto, E., Maciel, B.C., and Simoes,M.V. (2007) Pathogenesis of chronic Chagas heartdisease. Circulation 115: 1109–1123.
Matta Guedes, P.M., Gutierrez, F.R.S., Nascimento, M.S.L.,Do-Valle-Matta, M.A., and Silva, J.S. (2012) Antiparasiticalchemotherapy in Chagas’ disease cardiomyopathy: currentevidence. Trop Med Int Health 17: 1057–1065.
Mattos, E.C., Schumacher, R.I., Colli, W., and Alves, M.J.(2012) Adhesion of Trypanosoma cruzi trypomastigotes tofibronectin or laminin modifies tubulin and paraflagellar rodprotein phosphorylation. PLoS ONE 7: e46767.
Meanwell, N.A., and Kadow, J.F. (2007) Maraviroc, a chem-okine CCR5 receptor antagonist for the treatment of HIVinfection and AIDS. Curr Opin Investig Drugs 8: 669–681.
de Melo-Jorge, M., and PereiraPerrin, M. (2007) The Chagas’disease parasite Trypanosoma cruzi exploits nerve growthfactor receptor TrkA to infect mammalian hosts. Cell HostMicrobe 1: 251–261.
Meloni, M., Caporali, A., Graiani, G., Lagrasta, C., Katare, R.,Van Linthout, S., et al. (2010) Nerve growth factor pro-motes cardiac repair following myocardial infarction. CircRes 106: 1275–1284.
Ming, M., Ewen, M.E., and Pereira, M.E. (1995) Trypano-some invasion of mammalian cells requires activation ofthe TGF beta signaling pathway. Cell 82: 287–296.
Nagajyothi, F., Weiss, L.M., Silver, D.L., Desruisseaux, M.S.,Scherer, P.E., Herz, J., and Tanowitz, H.B. (2012) Trypano-soma cruzi utilizes the host low density lipoprotein receptorin invasion. PLoS Negl Trop Dis 5: e953.
van Nieuwenhoven, F.A., and Turner, N.A. (2012) The role ofcardiac fibroblasts in the transition from inflammation tofibrosis following myocardial infarction. Vascul Pharmacolhttp://dx.doi.org/10.1016/j.vph.2012.07.003.
Ohmichi, M., Pang, L., Ribon, V., Gazit, A., Levitzki, A., andSaltiel, A.R. (1993) The tyrosine kinase inhibitor tyrphostinblocks the cellular actions of nerve growth factor. Biochem-istry 32: 4650–4658.
Parada, H., Carrasco, H.A., Anez, N., Fuenmayor, C.,and Inglessis, I. (1997) Cardiac involvement is a con-stant finding in acute Chagas’ disease: a clinical, parasi-tological and histopathological study. Int J Cardiol 60:49–54.
Pereira, M.E. (1983) A developmentally regulated neuramini-dase activity in Trypanosoma cruzi. Science 219: 1444–1446.
Petersen, C.A., Krumholz, K.A., Carmen, J., Sinai, A.P., andBurleigh, B.A. (2006) Trypanosoma cruzi infection andnuclear factor kappa B activation prevent apoptosis incardiac cells. Infect Immun 74: 1580–1587.
Pinto, A.Y., Harada, G.S., Valente, V., Abud, J.E., Gomes, F.,Souza, G.C., and Valente, S.A. (2001) Cardiac attacks inpatients with acute Chagas disease in a family micro-outbreak, in Abaetetuba, Brazilian Amazon. Rev Soc BrasMed Trop 34: 413–419.
Rassi, A., Jr, Rassi, A., and Marin-Neto, J.A. (2010) Chagasdisease. Lancet 375: 1388–1402.
Scudder, P., Doom, J.P., Chuenkova, M., Manger, I.D., andPereira, M.E. (1993) Enzymatic characterization of beta-D-galactoside alpha 2,3-trans-sialidase from Trypanosomacruzi. J Biol Chem 268: 9886–9891.
Soeiro Mde, N., Paiva, M.M., Barbosa, H.S., MeirellesMde, N., and Araujo-Jorge, T.C. (1999) A cardiomyocytemannose receptor system is involved in Trypanosoma cruziinvasion and is down-modulated after infection. Cell StructFunct 24: 139–149.
Souders, C.A., Bowers, S.L., and Baudino, T.A. (2009)Cardiac fibroblast: the renaissance cell. Circ Res 105:1164–1176.
Sreejit, P., Kumar, S., and Verma, R.S. (2008) An improvedprotocol for primary culture of cardiomyocyte from neonatalmice. In Vitro Cell Dev Biol Anim 44: 45–50.
Tafuri, W.L. (1970) Pathogenesis of lesions of the autonomicnervous system of the mouse in experimental acuteChagas’ disease. Light and electron microscope studies.Am J Trop Med Hyg 19: 405–417.
Tapley, P., Lamballe, F., and Barbacid, M. (1992) K252a is aselective inhibitor of the tyrosine protein kinase activity ofthe trk family of oncogenes and neurotrophin receptors.Oncogene 7: 371–381.
Tessarollo, L., Tsoulfas, P., Donovan, M.J., Palko, M.E., Blair-Flynn, J., Hempstead, B.L., and Parada, L.F. (1997) Tar-geted deletion of all isoforms of the trkC gene suggests theuse of alternate receptors by its ligand neurotrophin-3 inneuronal development and implicates trkC in normal car-diogenesis. Proc Natl Acad Sci USA 94: 14776–14781.
Todorov, A.G., Andrade, D., Pesquero, J.B., Araujo Rde, C.,Bader, M., Stewart, J., et al. (2003) Trypanosoma cruziinduces edematogenic responses in mice and invades car-diomyocytes and endothelial cells in vitro by activatingdistinct kinin receptor (B1/B2) subtypes. FASEB J 17:73–75.
Valente, S.A., da Costa Valente, V., das Neves Pinto, A.Y.,de Jesus Barbosa Cesar, M., dos Santos, M.P., Miranda,C.O., et al. (2009) Analysis of an acute Chagas diseaseoutbreak in the Brazilian Amazon: human cases, triatom-ines, reservoir mammals and parasites. Trans R Soc TropMed Hyg 103: 291–297.
Waghabi, M.C., Keramidas, M., Feige, J.J., Araujo-Jorge,T.C., and Bailly, S. (2005) Activation of transforming growthfactor beta by Trypanosoma cruzi. Cell Microbiol 7: 511–517.
Waghabi, M.C., Keramidas, M., Calvet, C.M., Meuser, M.,de Nazare, C.S.M., Mendonca-Lima, L., et al. (2007)SB-431542, a transforming growth factor beta inhibitor,impairs Trypanosoma cruzi infection in cardiomyocytes andparasite cycle completion. Antimicrob Agents Chemother51: 2905–2910.
Weinkauf, C., and PereiraPerrin, M. (2009) Trypanosomacruzi promotes neuronal and glial cell survival through theneurotrophic receptor TrkC. Infect Immun 77: 1368–1375.
Weinkauf, C., Salvador, R., and PereiraPerrin, M. (2011)Neurotrophin receptor TrkC is an entry receptor forTrypanosoma cruzi in neural, glial, and epithelial cells.Infect Immun 79: 4081–4087.
Wilen, C.B., Tilton, J.C., and Doms, R.W. (2012) Molecularmechanisms of HIV entry. Adv Exp Med Biol 726: 223–242.
Wong, W.C., Tan, C.K., Singh, M., and Yick, T.Y. (1992)Ultrastructure of murine cardiac ganglia in experimentalChagas’ disease. Histol Histopathol 7: 371–378.
10 D. Aridgides, R. Salvador and M. PereiraPerrin
© 2013 Blackwell Publishing Ltd, Cellular Microbiology