tumour-initiating cells: challenges and opportunities for anticancer
TRANSCRIPT

Modern anticancer drug discovery began in the mid-twentieth century with the observation that cytotoxic chemotherapeutic agents could be used to target cancers with high proliferative rates1,2. Since then, the discovery and development of cancer therapies, initially on the basis of empirical observations, has become increasingly dependent on our understanding of human tumour biology. However, despite advances that have led to the development of new therapies, treatment options are still limited for many types of human cancer — particularly those with undifferentiated phenotypes, such as basal subtype breast cancer — and prognosis remains poor. In addition, the ability to manage tumour recurrence and metastasis following successful initial induction of remission continues to be a challenge.
The experimental demonstration of tumour-initiating cells (popularly known as cancer stem cells) in several human tumours in recent years3–10 supports tumour hierarchy as a fundamental concept in tumour biology and promises a new cellular target for anticancer drug discovery. Although the cancer stem cell hypothesis was first proposed decades ago (reviewed in Refs 11,12), many aspects of this hypothesis remain speculative and are still evolving. A minimal operational definition of tumour-initiating cells is: those tumour cells that have the ability to re-grow the tumour from which they were isolated or identified (fIG. 1), which implies that the
tumour-initiating cells can only be defined experimen-tally in vivo12. More generally, tumour-initiating cells are viewed as those cells at the apex of the tumour hierarchy (BOX 1), which highlights the role of aberrant differen-tiation in tumorigenesis. Multipotency of lineage differ-entiation is likely to be a frequent, but not a necessary, property of tumour-initiating cells.
The term ‘cancer stem cell’ does not imply that the cell is derived from a normal stem cell13. Depending on tumour type, the cells originating the tumour can be stem cells, progenitor cells or differentiated cells, and need not match the phenotype of the eventual tumour-initiating cell with respect to self-renewal and differentiation capacity (see the figure in BOX 1). Nevertheless, the origin of the tumour-initiating cells could have implications for the therapeutic window of the strategy that is used to target them. For example, killing normal stem cells along with the tumour-initiating cells could lead to a chronic loss of normal regeneration, whereas destroying the normal progenitor cells may be less of a long-term problem. Another important point is that tumour-initiating cells are not necessarily rare14,15. Moreover, the behaviour and frequency of tumour-initiating cells could also be influ-enced by various environmental factors. The fundamental concept underlying the cancer stem cell hypothesis is not related to the origin, absolute frequency or particular activity level (for example, proliferation rate) of these
*Oncology Discovery, Wyeth Research, 401 North Middletown Road, Pearl River, New York 10965, USA. ‡Cancer Research, Abbott Laboratories, 100 Abbott Park Road, Abbott Park, Illinois 60064, USA. §The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario, M4G 1X8, Canada.Correspondence to B.-B. S. Z. and J. C. G. e-mails: [email protected]; [email protected]:10.1038/nrd2137
Self-renewalThe ability of a cell to reproduce itself without losing developmental potential, characterized by cell divisions in which differentiation is blocked in at least one daughter cell.
Tumour-initiating cells: challenges and opportunities for anticancer drug discoveryBin-Bing S. Zhou*, Haiying Zhang‡, Marc Damelin*, Kenneth G. Geles*, Justin C. Grindley* and Peter B. Dirks§
Abstract | The hypothesis that cancer is driven by tumour-initiating cells (popularly known as cancer stem cells) has recently attracted a great deal of attention, owing to the promise of a novel cellular target for the treatment of haematopoietic and solid malignancies. Furthermore, it seems that tumour-initiating cells might be resistant to many conventional cancer therapies, which might explain the limitations of these agents in curing human malignancies. Although much work is still needed to identify and characterize tumour-initiating cells, efforts are now being directed towards identifying therapeutic strategies that could target these cells. This Review considers recent advances in the cancer stem cell field, focusing on the challenges and opportunities for anticancer drug discovery.
R E V I E W S
806 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
Differentiate ?
?
?
TICs
Target niche
Combinationtherapy
DestroyTICs
Self renewal
Tumour-initiating cell (TIC)NicheTumour progenitor and differentiated tumour cell
Anoikis A form of programmed cell death that is induced in anchorage-dependent cells when they become detached from the surrounding extracellular matrix. NicheCells and/or extracellular matrix components in specific anatomical locations that regulate the participation of the normal stem cells in tissue generation, maintenance and repair. In some cases, the behaviour of tumour-initiating cells might also be influenced by interactions with surrounding cells and matrix.
cells16. Studying cancer cell populations with concepts of stem cell biology in mind is likely to bring further insight into molecular drug targets and clinical strategies.
current failure with cancer treatment is not usually due to a lack of primary response or initial induction of remission, but to relapse or tumour recurrence after therapy, in which tumour-initiating cells are thought to have crucial roles. A major challenge now is to discover agents and strategies that target cancer and tumour relapse at their apparent source. Following an overview of the current status of the cancer stem cell hypothesis, this article therefore focuses on the challenges and opportunities for anticancer drug discovery.
Cancer stem cells: evidence and controversyIn the mid 1990s, John Dick and colleagues demon-strated the existence of tumour-initiating cells in acute myeloid leukaemia (AMl) using the non-obese diabetic–severe combined immunodeficient (NoD–ScID) mouse model3,17,18 (BOX 2). The purified populations of leukaemia-initiating cells contained a chromosomal translocation that was identical to that found in their progeny, the blast cells. As well as supporting the
clonogenic nature of leukaemia, this important finding pointed to the organization of leukaemia as a stem cell hierarchy19.
Similar studies are much more difficult for solid tumours. Solid tumours contain both tumour cells and various stromal cells, and breaking the interactions between cells in such a tumour might induce anoikis or change the properties of the tumour cells. Furthermore, assays that involve injection of cells into a new tissue loca-tion in mice may fail to recapitulate the environment of those tumour cells in the original tumours. Nevertheless, tumour-initiating cells from solid tumours, including human breast, brain, colon, pancreatic, liver and ovar-ian cancer and melanoma, have been successfully iso-lated using appropriate cell surface markers, including cD44, cD24, cD133, epithelial cell adhesion molecule (ePcAM) and ATP-binding cassette sub-family b mem-ber 5 (Abcb5)4–10. These tumour-initiating cells can pro-duce phenocopies of the original primary tumours when transplanted into NoD–ScID mice.
Theoretical and technical questions remain regarding whether the cells isolated are the true and only tumour-initiating cells that function in the solid tumours of patients20. current cell sorting protocols are thought to favour niche-independent tumour-initiating cells (BOX 2), which may resemble cells in activated or mobilized states that are selected when transplantation assays are per-formed on normal stem cells21. certain markers used for prospective sorting may also select for cells that evade the immune system22, which could also be a relevant property for tumour-initiating cells. Nevertheless, a number of markers used in cell sorting are emerging as being predictive of disease progression9,23, indicating that they identify clinically important cell populations.
It was noticed that some experimental mouse models of leukaemia do not follow the cancer stem cell hypoth-esis, suggesting that certain human cancers may not adhere to this model14. However, as with many cell lines that have lost the hierarchical structure of the primary leukaemia from which they originated, some experi-mental mouse models may not accurately reflect the tumour heterogeneity and pathological environment of spontaneously occurring human malignancies16. It was also argued that certain phenotypically distinct popula-tions of human cancers fail to grow when injected into immunodeficient mice because of differences in the mouse and human microenvironment20, so the iden-tification of tumour-initiating cells in mouse models adds credibility to the cancer stem cell hypothesis24–32. Furthermore, transgenic models facilitate lineage-tracing experiments, which can provide evidence for tumour initiation and tumour hierarchies without the limitations and experimental variability of transplanta-tion assays30,31.
Although growing evidence from a wide range of systems favours the existence of tumour-initiating cells, there may nevertheless be diversity of tumour hierarchy in different patients and varying degrees to which differ-ent tumour-initiating cells are stem cell-like. Many can-cers might contain subpopulations of tumour-initiating cells, but some could contain common tumorigenic cells,
Figure 1 | The cancer stem cell hypothesis and therapeutic strategies to target tumour-initiating cells. According to the cancer stem cell hypothesis, tumour cells are heterogeneous and only the tumour-initiating cells have the ability to proliferate extensively, give rise to differentiated cells and form new tumours. There are various therapeutic strategies that could target tumour-initiating cells. Killing these cells could be achieved by inhibiting their survival pathways (for example, with inhibitors of phosphoinositol 3-kinase or interleukin-4-specific monoclonal antibodies), or sensitizing them to chemotherapeutic agents (for example, with a checkpoint kinase inhibitor). Some of the survival pathways that are used by tumour-initiating cells could also be used by the bulk of the tumour, so agents targeting these pathways are expected to kill more than just tumour-initiating cells. Alternatively, differentiating the tumour-initiating cells (for example, with bone morphogenetic proteins or CD44-specific monoclonal antibodies) might be a successful therapeutic strategy, as the bulk of the tumour has limited proliferation potential. Inhibition of developmental signalling that is involved in self-renewal (for example, with inhibitors of Wnt, Hedgehog or Notch pathway signalling) might work by both mechanisms. In addition, anti-angiogenic therapy might work in part by affecting the vascular niche of tumour-initiating cells. However, tumour-initiating cells are probably genetically unstable, and a committed progenitor could regain renewal activity. It might be therapeutically advantageous to combine agents that target tumour-initiating cells with conventional agents that reduce the bulk of the tumour and agents that target the niche.
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 807
© 2009 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
Normal tissue
Differentiated cells
Stem cell
Multipotent progenitor
Progenitor
Initial tumour
Tumour progenitors and differentiated tumour cells
Tumour-initiating cellAdvanced tumour
Target bulk of the tumour cells
Target tumour-initiating cells
Tissue ortumour hierarchy
Clonal evolution
Mutations
Mutations?EMT?
Mutations
with little hierarchical organization15. controversy aside, the cancer stem cell hypothesis provides a novel frame-work to study cellular heterogeneity, aberrant differen-tiation and tumour–host interactions in many cancers.
Framework for understanding cancer propertiesThe cancer stem cell hypothesis does not contradict the established clonal evolution view of cancer, but instead suggests a key role for tumour cell hierarchy in tumour evolution and highlights the importance of an aberrant differentiation programme in tumorigenesis (BOX 1). one popular explanation for tumour heterogeneity is genetic instability, which is a common feature of cancers accord-ing to comprehensive cancer genomic analysis33–35. conversely, a large part of the apparent morphological and phenotypic heterogeneity can be explained by aber-rant differentiation, and epigenetic changes in tumours could also be dynamic and unstable36,37 (see the figure in BOX 1). The behaviour of tumour-initiating cells could be further modulated by tumour–host interactions. In this section, we consider some of the key characteristics of tumour-initiating cells.
Tumour-initiating cells diverge from their cells of origin with increased self-renewal capacity. Although the origins of tumour-initiating cells may vary, they share several properties with normal stem cells, especially the
extensive capability of self-renewal12. If the cancer stem cell hypothesis is correct, the evolution of the tumour is largely the history of changes in self-renewing tumour-initiating cells (BOX 1), and self-renewal pathways might be more conserved than surface markers among tumour-initiating cells.
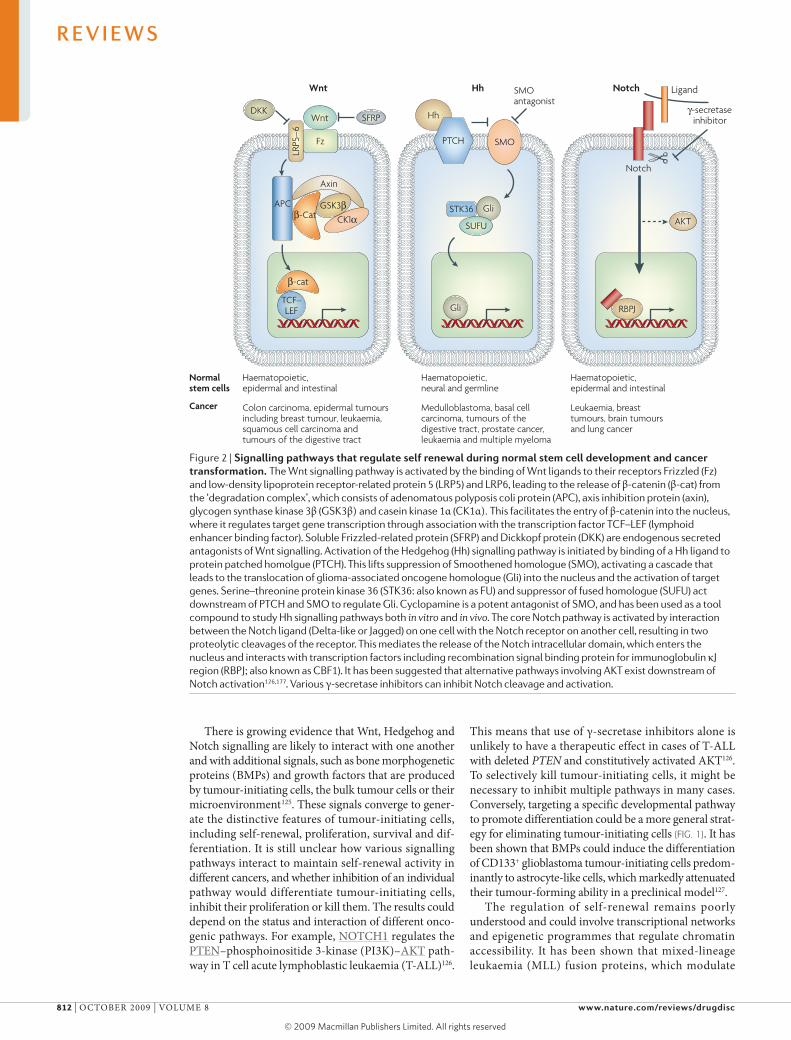
Although pathways that regulate self-renewal are tightly controlled in normal stem cells, in tumour-initiating cells they may be constitutively activated or improperly regulated through genetic and/or epigenetic changes, leading to uncontrolled growth38. Several studies show that bMI1 polycomb ring finger oncogene (BMI1) and the wnt signalling molecule β-catenin regulate the self-renewal of haematopoietic stem cells (HScs) and the proliferation of leukaemia-initiating cells39–41. Actually, many leukaemia-initiating cells have a higher self-renewal capacity than normal HScs3. In addition, it was recently shown that maintenance of cutaneous tumour-initiating cells is dependent on wnt and β-catenin signalling42. Hedgehog signalling regulates tumour-initiating cells from multiple myeloma43 and chronic myeloid leukaemia (cMl)44. Developmental signalling pathways that regulate normal stem cell self-renewal, including wnt, Hedgehog and Notch (fIG. 2), have been shown to be active in numerous human cancers38,45 and may be broadly important for self-renewal in many can-cers. They also have important roles in progenitors and a
Box 1 | Tumour hierarchy and clonal evolution
For decades, tumour initiation and development has been regarded as a multistep process that is reflected by the progressive genetic alterations that drive the transformation of normal human cells into highly malignant derivatives202. As cancers arise only after multiple mutagenic events, long-lived cells are probably the most capable of supporting such cumulative changes. Based on genetic variations already observed in the tumour-initiating cell populations from different tumours76,203 and genetic mutations in developmental pathways in different cancers34,38, it has been proposed that progressive genetic alterations might occur at the level of tumour-initiating cells. The clonal progression to cancer could operate through the ‘stem cell compartment’ (see the figure), as already shown in leukaemia-initiating cells204.
In normal tissues, the heterogeneity of cells reflects a hierarchical programme of differentiation in which multiple mature cell types are derived from a common multipotent stem cell through intermediate progenitors. Heterogeneous populations of cancer cells at various differentiation stages could be the result of both acquired mutations and aberrant but hierarchical differentiation programmes (see the figure). Cancer is both a proliferation and a differentiation disease, and the ‘clonal evolution’ and ‘cancer stem cell’ models might not be mutually exclusive, as initially thought. Owing to genetic instability, the tumour-initiating cells isolated from a clinically detectable tumour would probably have a substantially different genetic profile from the initial transformed cells that originated the tumour (see the figure). Also, as chemotherapeutic agents are often mutagenic, the phenotype and frequency of the tumour-initiating cells in relapsed tumours are expected to be distinct from those of early-stage lesions. In practice, combination treatment involving both traditional therapies and therapies that target tumour-initiating cells will probably be required to ablate all cancer cells, particularly as a genetically unstable tumour cell will present a ‘moving target’. EMT, epithelial–mesenchymal transition.
R E V I E W S
808 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

Asymmetrical divisionA form of cellular replication in which a cell renews itself and generates a more differentiated progeny.
Symmetrical divisionA form of cellular replication in which a single cell gives rise to two identical cells.
Epithelial–mesenchymal transitionA cellular program in normal development and in cancer whereby cells of an epithelial origin acquire the properties of mesenchymal cells, typically characterized by loss of cell adhesion, repression of e-cadherin expression, and increased cell motility.
OncomirMicroRNA known to be involved in cancer and tumorigenesis.
wider impact on the lineage, and could have additional roles in the regulation of tumour and stromal cells. In this rapidly expanding field of tumour biology, new routes by which cells may acquire or enhance self-renewal ability are regularly being discovered, and self-renewal roles for classical cancer genes are also emerging. For example, by gaining certain epigenetic programmes or losing certain combinations of tumour suppressor genes, progenitor cells could gain the capacity for self-renewal and become malignant46,47.
Self-renewal can be affected by changes in prolif-eration, differentiation and/or apoptosis. Aberrantly increased self-renewal might therefore be caused by dif-ferent mechanisms, such as an increasing proliferation rate or a shift in the balance of cell division from asymmetrical division to symmetrical division48. Tumour-initiating cells from AMl and cMl are mostly quiescent49,50, whereas tumour-initiating cells from many solid tumours could be proliferative51. The tumorigenic breast and pan-creatic tumour-initiating cells have cell cycle profiles that are similar to those of bulk tumour cells, which do not have tumour-initiating ability, showing it is not a proliferative advantage of these tumour-initiating cells over the non-tumorigenic cells that leads to dif-ferential engraftment4,8. by contrast, slowly proliferat-ing intestinal stem cells from phosphatase and tensin homologue (Pten)-deficient mice initiate intestinal polyposis52. The most important and characteristic feature of tumour-initiating cells therefore seems to be
their increased self-renewal potential, rather than their proliferation rate. Indeed, during the clonal evolution of tumour-initiating cells, there may be stronger selec-tion for increased self-renewal capacity than for higher proliferation53. In addition, a malignant tumorigenic cell could have undifferentiated pathological features associated with increased self-renewal, including meta-static and pro-angiogenic capabilities.
Tumour-initiating cells in metastasis and tumour–host interactions. A corollary of the cancer stem cell hypoth-esis is that macroscopic metastases may arise from migrating or disseminated tumour-initiating cells54. In patients with breast cancer, tumorigenic cD44+cD24–/low cells are readily detectable in metastatic pleural effu-sions, and most early-disseminated cancer cells that are detected in the bone marrow have a putative breast tumour-initiating cell phenotype4,55. Interestingly, cD133+cXcr4+ tumour-initiating cells that are found at the invasive front of pancreatic tumours have been shown to determine the metastatic phenotype of the individual tumour56. Another hypothesis is that meta-static tumours originate when cells in a primary epithelial malignancy undergo an epithelial–mesenchymal transition (eMT)57. like self-renewal, eMT is regulated by develop-mental signalling pathways, such as the wnt and Notch pathways58,59. expression of the oncomir LET7 in a breast tumour-initiating cell model inhibits self-renewal, eMT and xenograft metastasis60. More recently, it was noticed
Box 2 | Mouse models for cancer research and drug discovery
In the mid 1980s, the availability of athymic (nu–nu) mice and the subsequent development of immunodeficient mouse strains with other genetic lesions (including severe combined immunodeficient (SCID) mice, which lack both B and T cells) allowed the widespread use of human tumour xenografts as mouse models for cancer research and drug discovery205. Xenografts can be established either by injecting human tumour cell lines or by direct implantation of patient biopsies into immunodeficient mice. The wide range of tumour cell lines and possibility of ex vivo genetic or pharmacological manipulation before xenotransplantation made human xenograft tumour models popular tools for discovering and developing not only cytotoxic agents but also tumour-targeted agents.
A major drawback of these mouse models is that they have limited utility in measuring tumour self-renewal in vivo, particularly for tumour cells from primary sources. Models that permit efficient engraftment are a prerequisite for assays that directly measure tumour self-renewal by serial transplantation. Animal models with an increased efficiency of engraftment have since been developed. The SCID mouse was crossed with the non-obese diabetic (NOD) mouse, and the NOD–SCID progeny can be engrafted by various tumour types and sustain the serial transplantation that is required for assessing long-term self-renewal potential3–5. However, NOD–SCID assays still have limited efficiency of engraftment and so can lead to underestimations of the frequency of human tumour-initiating cells. Notably, the use of more highly immunocompromised NOD–SCID interleukin-2 receptor γ-chain-null (Il2rg–/–) mice can increase the detection of tumorigenic melanoma cells by several orders of magnitude15. Although engraftment is improved by implantation into NOD–SCID Il2rg–/– mice, it is also clear that immune cells have a role in the progression of many tumours. To properly model certain tumours, disabling the mouse immune system might not be desirable. It may ultimately be necessary to provide a humanized immune repertoire in mice, perhaps by genetic engineering or haematopoietic cell transplantation.
The limitations of xenotransplantation assays are particularly apparent in analysing tumour-initiating cells from solid tumours. The current protocol, even with recent improvements15, is thought to favour niche-independent tumour-initiating cells, which may not be representative of the population of tumour cells that can exhibit stem cell-like properties in their human tumour environment. Orthotopic models might more faithfully maintain the tumour–host interaction, giving tumour-initiating cells a greater chance to interact with and obtain stimulation from a mouse environment. Co-injection with tumour-associated stromal cells might be another approach to improve the clinical relevance of xenotransplantation, and can resolve some of the issues related to the presence of murine stroma in animal models (fIG. 3b). Avoiding the transplant altogether by generating genetic mouse models of tumours may better maintain the tumour–host and tumour-initiating cell–niche interactions30,31. However, owing to the practical difficulty of modelling the multitude of genetic changes in a tumour, this strategy may be more suited to studying tumour-initiating cells that have few defined genetic lesions, perhaps resembling tumour-initiating cells from early-stage human disease.
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 809
© 2009 Macmillan Publishers Limited. All rights reserved

that cells undergoing eMT and tumour-initiating cells share many markers and properties61. These findings reveal the potential plasticity of these tumour cells and suggest that there could be some common regulatory programmes underlying eMT and self-renewal (see the figure in BOX 1).
The behaviour of normal stem cells is tightly regulated by signals that the cells receive from their microenvironmental niches, which are provided by the adjacent cells and/or extracellular matrix compo-nents62–64. A niche, while supporting the self-renewal and maintaining the identity of a stem cell, also con-trols stem cell number and proliferation. This control of cell number and proliferation might be a preventative mechanism against cancer65. Similarly, it is conceivable that the tumour microenvironment could also constrain tumour-initiating cells, as some tumours lie dormant or develop slowly over decades66. one theory of tumour dormancy is that tumour-initiating cells are held in check by a niche–stem cell interaction. cancer could progress if the niche were expanded or altered through genetic or epigenetic means65. It has been shown that an alteration in a HSc niche can lead to myeloproliferative disease67. Tumour-initiating cells could also preferen-tially localize to environments that favour proliferation, such as vascular niches68–70, or even promote the forma-tion of their own niche71. Alternatively, mutations might render tumour-initiating cells independent of niche signals, thereby lifting environmental controls on self-renewal72 and increasing the risk of metastasis.
Why do many therapies fail to eradicate cancers?Many patients with cancer, particularly those with solid tumours, either do not respond to existing cancer ther-apies (including chemotherapeutics, radiotherapy and tumour-targeted agents) or relapse quickly after initial remission. Key possible reasons for this failure include the inherent drug resistance of tumour-initiating cells, the inefficiency of the treatment and/or the genetic instability of cancer cells.
It has been suggested that the more aggressive and refractory cancers contain more tumour-initiating cells73,74. circumstantial evidence in support of this con-nection already exists in medical practice. Following cancer therapy, a patient’s tumour is examined to assess the effects of treatment. If the tumour contains only mature cells, the cancer usually does not recur. However, if a large number of immature cells (probably including a large proportion of tumour-initiating cells) are present in the tumour sample, the cancer is likely to return, and further aggressive treatment is warranted72. It has been shown that a high frequency of stem cells in AMl at diagnosis predicts high minimal residual dis-ease after therapy and poor prognosis75. In melanoma, Abcb5 is a marker for malignant melanoma-initiating cells, and Abcb5+ tumour cells detected in patients with melanoma show a primitive molecular phenotype and correlate positively with clinical melanoma pro-gression9. In breast cancer, cD44+ cell-specific genes included many known stem cell markers and corre-lated with decreased patient survival76. Furthermore,
chemotherapy treatment increased the percentage of cD44+cD24–/low tumour cells in patients with breast cancer77, consistent with the relative chemoresistance of these tumour-initiating cells.
Many tumour-initiating cells are thought to be resistant to chemotherapeutics such as paclitaxel and doxorubicin, for various reasons including their quiescent or slowly proliferating nature78, the high expression level of ATP-binding cassette (Abc) drug pumps9,60,79, the intrinsic high levels of anti-apoptotic molecules11, their relative resistance to oxidative or DNA damage, and their effi-ciency of DNA repair80–82. Although tumour-initiating cells might commonly be more resistant to therapy than the bulk of tumours, there could be variation in sensitivity to therapies among tumour-initiating cells, as has been shown for normal stem cells21,83.
Similar to traditional anticancer drugs, many novel tumour-targeted agents were also designed to target rapidly proliferating cancer cells, so many tumour-initiating cells might also be relatively insensitive to these agents. For example, imatinib (Gleevec; Novartis) targets and inhibits the bcr–Abl kinase, which is the fusion protein product of a chromosomal transloca-tion and is suggested to act as a molecular switch that promotes proliferation and differentiation of multipo-tent progenitors in cMl84. bcr–Abl is required for the survival of proliferating progenitor cells, but not the quiescent cMl stem cells50,85,86. Therefore, although the mutations are thought to accumulate in tumour-initiating cells, their functional effects are mostly manifested fur-ther downstream in the tumour hierarchy, leading to neoplastic proliferation of primitive progenitors. As a result, imatinib eliminates proliferating, committed leu-kaemia progenitors, but not primitive, quiescent tumour-initiating cells, and most patients are still positive for the fusion gene transcripts after treatment87–89. Alterations in Ikaros family zinc finger 1 (IKZF1) are thought to synergize with bcr–Abl to induce lymphoblastic leu-kaemia and contribute to drug resistance and disease progression90. Although it is likely that several factors contribute to the problem, it has been shown that the drug resistance and disease recurrence that are associ-ated with imatinib treatment of cMl might be avoided by targeting an essential stem cell maintenance path-way involving Hedgehog44. As discussed above, there are other cancer types in which tumour-initiating cells could be proliferative, and growth signalling pathways are likely to have important roles in these cells. These pathways are therefore promising therapeutic targets in such cancers.
If a patient has a large number of tumour-initiating cells and only a small number of such cells are required to regenerate a tumour, then therapy has to be highly efficient at killing these cells to avoid relapse91. A ther-apy that kills 95% of cells in a tumour might be consid-ered efficacious based on tumour shrinkage, but may allow for the survival of sufficient tumour-initiating cells to cause eventual relapse. Thus, even if tumour-initiating cells are no more resistant to therapy than bulk tumour cells, they can still be the key to the limitations of treatment.
R E V I E W S
810 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

If the cancer stem cell hypothesis is widely applicable, it probably extends to neoplasms that are amenable to cure, such as certain germ cell neoplasms (that is, testicular seminoma) and neuroblastomas. Indeed, chemotherapy treatments for such tumours often eliminate the undif-ferentiated cancer cells and produce residual masses that are benign tumours composed of differentiated cells92. This suggests that their stem cell components might be inherently sensitive to chemotherapeutics and unable to adapt to counteract them. Several recent studies suggested that the survival benefit of trastuzumab (Herceptin; Genentech/roche) — a monoclonal antibody specific for receptor tyrosine protein kinase erbb2 (also known as Her2) — might relate to its ability to target breast tumour-initiating cells93,94. However, most patients with metastatic breast cancer still develop resistance within 1 year of trastuzumab treatment95, which implies the genetic and/or epigenetic plasticity of these tumour-initiating cells permit them to evolve as a function of tumour progression and/or therapeutic challenges. Targeting tumour-initiating cells might not avoid the same problems that have been encountered for decades in treating bulk tumour cell populations: the emergence of drug resistance and the selection of increasingly refractory cell types96. Nevertheless, understanding the molecular basis of tumour-initiating cell behaviour will allow for the design of new strategies, including combi-nation therapies to counter drug resistance.
Therapeutic opportunitiesThe cancer stem cell hypothesis provides a rationale for several therapeutic strategies beyond traditional anti-proliferative agents (fIG. 1). Potential approaches to kill tumour-initiating cells include blocking essential self-renewal signalling, inhibiting the survival mechanisms of these cells, or targeting tumour-initiating cell surface markers through antibody-based cytotoxic approaches. Another strategy is to induce tumour cell differentiation, which can potentially be achieved by inhibiting develop-mental pathways or epigenetic programmes. As many tumour-initiating cells might depend on a niche to main-tain their identity, targeting the niche could be a strategy to indirectly inhibit or differentiate tumour-initiating cells.
Developmental pathways in self-renewal and differen-tiation. As many tumour cells are thought to have an aberrant differentiation programme and deregulated self-renewal could be a key factor in many types of cancer38,45, several developmental signalling pathways have become the recent focus of drug discovery efforts (fIG. 2; TABLe 1). During embryonic development, there is considerable crosstalk between these pathways, with identifiable signalling centres that generate, receive and integrate several pathways97,98. Tumour-initiating cells and their niches might similarly operate as signalling centres, in which multiple developmental pathways are active and converge to control self-renewal.
wnt signalling has been shown to be required for self-renewal of tumour-initiating cells in several can-cers, including cMl and squamous cell carcinomas41,42. extracellular wnt inhibitors, including the secreted
Frizzled-related proteins (SFrPs) and Dickkopf proteins (DKKs), which act at the cell surface to inhibit wnt sig-nalling through its receptors, have been discovered99. Active derivatives of these antagonists, if designed to have desirable pharmacokinetic properties, could be developed into antitumour agents, particularly as they might be able to suppress wnt signalling in cancer even when the genes encoding adenomatous polyposis coli protein (APc) or β-catenin are mutated100. Small-molecule antagonists of the oncogenic transcription factor TcF–β-catenin pro-tein complex have been reported101,102. Antibodies against various wnts103,104, Frizzled proteins and wnt corecep-tor low-density lipoprotein receptor-related protein 5 (lrP5)–lrP6 are also being explored.
Inhibition of the Hedgehog signalling pathway is also a viable therapeutic strategy, and antibodies against Hedgehog and small-molecule inhibitors of the Hedgehog coreceptor Smoothened homologue (SMo) have been identified105,106 (TABLe 1). It has been reported that pharmacological inhibitors of Hedgehog signalling display efficacy in various animal models, including those of basal cell carcinoma, medulloblastoma, small-cell lung cancer and pancreatic cancer106–110. More recently, it was shown that inhibition of Hedgehog signalling kills cMl tumour-initiating cells, impairs the propagation of bcr–Abl-driven cMl and the growth of imatinib-resistant mouse and human cMl44. However, these agents are not very effective and the results vary among cellular models, partly because it is difficult to maintain Hedgehog pathway activity in vitro due to differentia-tion under conventional culture conditions111,112 and/or requirements for the presence of stromal cells63,64,113. Hedgehog ligand expressed by tumour cells can also activate the Hedgehog pathway in the tumour stromal microenvironment, illustrating a paracrine requirement of Hedgehog signalling114. Such challenges complicate in vitro screening assays, as discussed below.
Inhibition of Notch expression by antisense nucleic acid technology or the pharmacological blockade of the protease γ-secretase, which cleaves Notch (fIG. 2), has striking antineoplastic effects in Notch-expressing transformed cells in vitro and in xenograft models115–117. A γ-secretase inhibitor has been shown to induce goblet cell differentiation and regress colon adenomas in mice carrying a mutation of the Apc gene118. Another γ-secretase inhibitor depletes tumour-initiating cells in brain tumours119. However, the therapeutic window of γ-secretase inhibitors is narrow, because of their inhi-bition of multiple Notch pathways and the possible effect on normal stem cells. recently, antibodies that are selective for the Notch ligand Delta-like 4 (Dll4) were shown to inhibit tumour growth by deregulating angiogenesis without much of the toxicity related to γ-secretase inhibition in animal models120,121. Such selective targeting of an individual Notch pathway might provide a viable strategy for impairing niche function. Antagonist antibodies against individual Notch receptors are also being explored122. In addition to developmental signalling pathways, other signalling pathways could be important for the self-renewal of different tumour-initiating cells123,124.
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 811
© 2009 Macmillan Publishers Limited. All rights reserved
Nature Reviews | Drug Discovery
DKK
Normal stem cells
Cancer
Haematopoietic,epidermal and intestinal
Colon carcinoma, epidermal tumoursincluding breast tumour, leukaemia, squamous cell carcinoma andtumours of the digestive tract
Wnt
Wnt Hh Notch
SFRP
SMOantagonist
Notch
γ-secretase inhibitor
Gli
Gli TCF–LEF
Fz
LRP5
–6
Haematopoietic,neural and germline
Medulloblastoma, basal cellcarcinoma, tumours of the digestive tract, prostate cancer, leukaemia and multiple myeloma
SMO
STK36
Haematopoietic,epidermal and intestinal
Leukaemia, breasttumours, brain tumours and lung cancer
Hh
RBPJ
PTCH
SUFU AKTβ-Cat
GSK3β CK1α
Axin
APC
β-cat
Ligand
There is growing evidence that wnt, Hedgehog and Notch signalling are likely to interact with one another and with additional signals, such as bone morpho genetic proteins (bMPs) and growth factors that are produced by tumour-initiating cells, the bulk tumour cells or their microenvironment125. These signals converge to gener-ate the distinctive features of tumour-initiating cells, including self-renewal, proliferation, survival and dif-ferentiation. It is still unclear how various signalling pathways interact to maintain self-renewal activity in different cancers, and whether inhibition of an individual pathway would differentiate tumour-initiating cells, inhibit their proliferation or kill them. The results could depend on the status and interaction of different onco-genic pathways. For example, NoTcH1 regulates the PTeN–phosphoinositide 3-kinase (PI3K)–AKT path-way in T cell acute lymphoblastic leukaemia (T-All)126.
This means that use of γ-secretase inhibitors alone is unlikely to have a therapeutic effect in cases of T-All with deleted PTEN and constitutively activated AKT126. To selectively kill tumour-initiating cells, it might be necessary to inhibit multiple pathways in many cases. conversely, targeting a specific developmental pathway to promote differentiation could be a more general strat-egy for eliminating tumour-initiating cells (fIG. 1). It has been shown that bMPs could induce the differentiation of cD133+ glioblastoma tumour-initiating cells predom-inantly to astrocyte-like cells, which markedly attenuated their tumour-forming ability in a preclinical model127.
The regulation of self-renewal remains poorly understood and could involve transcriptional networks and epigenetic programmes that regulate chromatin accessibility. It has been shown that mixed-lineage leukaemia (Mll) fusion proteins, which modulate
Figure 2 | signalling pathways that regulate self renewal during normal stem cell development and cancer transformation. The Wnt signalling pathway is activated by the binding of Wnt ligands to their receptors Frizzled (Fz) and low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6, leading to the release of β-catenin (β-cat) from the ‘degradation complex’, which consists of adenomatous polyposis coli protein (APC), axis inhibition protein (axin), glycogen synthase kinase 3β (GSK3β) and casein kinase 1α (CK1α). This facilitates the entry of β-catenin into the nucleus, where it regulates target gene transcription through association with the transcription factor TCF–LEF (lymphoid enhancer binding factor). Soluble Frizzled-related protein (SFRP) and Dickkopf protein (DKK) are endogenous secreted antagonists of Wnt signalling. Activation of the Hedgehog (Hh) signalling pathway is initiated by binding of a Hh ligand to protein patched homolgue (PTCH). This lifts suppression of Smoothened homologue (SMO), activating a cascade that leads to the translocation of glioma-associated oncogene homologue (Gli) into the nucleus and the activation of target genes. Serine–threonine protein kinase 36 (STK36: also known as FU) and suppressor of fused homologue (SUFU) act downstream of PTCH and SMO to regulate Gli. Cyclopamine is a potent antagonist of SMO, and has been used as a tool compound to study Hh signalling pathways both in vitro and in vivo. The core Notch pathway is activated by interaction between the Notch ligand (Delta-like or Jagged) on one cell with the Notch receptor on another cell, resulting in two proteolytic cleavages of the receptor. This mediates the release of the Notch intracellular domain, which enters the nucleus and interacts with transcription factors including recombination signal binding protein for immunoglobulin κJ region (RBPJ; also known as CBF1). It has been suggested that alternative pathways involving AKT exist downstream of Notch activation126,177. Various γ-secretase inhibitors can inhibit Notch cleavage and activation.
R E V I E W S
812 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved
chromatin structure through histone modification, can reprogramme differentiated myeloid cells and activate self-renewal in cells with no inherent self-renewal prop-erties46,128. Polycomb group proteins, such as enhancer of zeste homologue 2 (eZH2), are essential components by which stem cells reversibly repress genes that are related to differentiation129. recently, it was reported that hyper-methylation of the gene encoding the bMP receptor 1b in a subset of glioblastoma-initiating cells is linked to the activity of eZH2 (Ref. 130). eZH2 also affects bMI1-mediated suppression of the p16Ink4a–p19Arf locus to avert growth arrest or apoptosis of stem cells131. Invasive basal subtype breast cancers significantly overexpress eZH2, which could lead to downregulation of breast cancer type 1 susceptibility protein (brcA1)132. The possible involvement of these epigenetic programmes in different tumour-initiating cells and their potential as therapeutic targets are areas of intense study.
Survival mechanisms in tumour-initiating cells. Although we know little about survival pathways of tumour-initiating cells in various tumour types, they are potential targets for killing tumour-initiating cells. certain oncogenic path-ways that are distinct from developmental pathways might have a role in the survival of some tumour-initiating cells,
but have no effect on cell differentiation or multipotency. In AMl, nuclear factor κb (NF-κb) was found to be constitutively active in primitive AMl cells (which are considered leukaemia-initiating cells), but not in normal HScs123. MG-132, a proteasome inhibitor with potent NF-κb signalling inhibitory activity, was shown to induce rapid cell death in cD34+cD38– leukaemia-initiating cells, but not normal cD34+cD38– cells. An interleukin-4 (Il-4)-specific antibody reduced the viability of both cD133– and cD133+ colon cancer cells and increased the efficacy of chemotherapy, suggesting that molecular pathways that contribute to bulk tumour growth can also be successfully targeted to sensitize tumour-initiating cells to cytotoxic therapies133. In addition, activation of the PTeN–mammalian target of rapamycin (mTor)–signal transducer and transcription activator 3 (STAT3) pathway in breast tumour-initiating cells is required for their viability and maintenance134, and the PI3K pathway regulates survival of tumour-initiating cells that reside in the perivascular niche of medulloblastoma70. recent studies also suggest an important role for erbb2 in maintaining tumour-initiating cells in breast cancer93,94, in addition to its presumed role in bulk tumour cells. It is unclear how pathways that regulate self-renewal interact with those that regulate the survival of tumour-initiating
Table 1 | Selected agents targeting self-renewal signalling or tumour-initiating cell surface markers
Target or target pathway
Agents cancer indication status references
Hedgehog pathway
5E1, a Sonic Hedgehog neutralizing mAb
Oesophageal and prostate cancer and medulloblastoma
Target validation 105
Cyclopamine, a SMO antagonist Solid tumours Target validation 38
Basal cell carcinoma Phase I 200
GDC-0449, a SMO antagonist Basal cell carcinoma and metastatic colorectal cancer
Phase II Genentech website (www.gene.com)
IPI-926 Advanced and/or metastatic solid tumours
Phase I Infinity Pharmaceuticals website (www.ipi.com)
Wnt pathway A WNT2-specific mAb Melanoma and NSCLC Target validation 103,104
Various TCF–b-catenin inhibitors Colon cancer Target validation 101,102
Notch pathway DBZ, a γ-secretase inhibitor Intestinal adenomas Target validation Preclinical
118
MK-0752, a γ-secretase inhibitor T-ALL and metastatic or advanced breast cancer
Phase I Merck website (www.merck.com)
DLL4-specific mAbs Solid tumours Preclinical 120,121
ABCB5 3C2-1D12, an ABCB5-specific mAb Malignant melanoma Target validation 9
CD44 H90, a CD44-specific mAb Acute myeloid leukaemia Preclinical 148
P245, a CD44-specific mAb Breast cancer Preclinical 201
EPCAM Adecatumumab, an EPCAM-specific human mAb
Metastatic breast cancer Phase II Micromet website (www.micromet.de)
Edrecolomab, an EPCAM-specific mAb
Colon cancer Phase II–III 142
Catumaxomab, a trifunctional antibody against EPCAM and CD3
Malignant ascites and ovarian and gastric cancer
Phase II–III Trion Pharma website (www.trionpharma.de)
MT110, a bispecific single-chain antibody against EPCAM and CD3
Lung and gastrointestinal cancer Phase I Micromet website (www.micromet.de)
ABCB5, ATP-binding cassette sub-family B member 5; DLL4, Delta-like 4; EPCAM, epithelial cell adhesion molecule; mAb, monoclonal antibody; NSCLC, non-small-cell lung cancer; SMO, Smoothened homologue;T-ALL, T cell acute lymphoblastic leukaemia; TCF, T-cell factor.
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 813
© 2009 Macmillan Publishers Limited. All rights reserved

cells, although it was recently shown that NoTcH1 regulates the PTeN–PI3K–AKT pathway in T-All126 and erbb2 expression in breast cancer94.
In glioblastomas, DNA damage responses were shown to be preferentially activated in glioma-initiating cells before and after radiation. The radioresistance of cD133+ glioma-initiating cells can be reversed with a specific inhibitor of checkpoint kinase 1 (cHK1) and cHK2 (Ref. 81). In breast cancer, certain tumour-initiating cells have lower levels of reactive oxygen species (roS) than corresponding non-tumorigenic cells. Pharmacological depletion of roS scavengers in tumour-initiating cells markedly increases DNA damage and results in radio-sensitization82. In addition, cell cycle restriction through the expression of cyclin-dependent kinase inhibitor 1A (cDNK1A; also known as p21) limits DNA damage and maintains the self-renewal of leukaemia-initiating cells135. These preliminary studies highlight the potential of inhibiting DNA damage responses136 to overcome the resistance of tumour-initiating cells to therapy.
Cell surface markers and niche interaction. Markers on the surface of tumour-initiating cells are also important targets, particularly for antibody-based therapeutics (TABLe 1). The surface markers cD34, cD44, cD133 and Abcb5 have been used to identify tumour-initiating cells from various tumour types3–9. The surface markers might reflect the cellular origin and history of that particular tumour-initiating cell, and could be highly variable among tumour types, patients and even within the same patient. uniquely among such markers, cD44 could have several roles in tumorigenesis; its expres-sion is induced by oncogenic signals such as β-catenin–TcF4 and ras–raf–extracellular signal-regulated kinase (erK) pathways and negatively regulated by the tumour suppressor p53 (Refs 137,138). Most of these markers are expressed in normal cells, and so finding a therapeutic window could be a challenge. However, there seem to be some phenotypic differences between leukaemia-initiating cells and HScs, including differences in the expression of THY1 membrane glycoprotein, KIT and Il-3 receptor-α (Il3rA)139–141. In particular, Il3rA, which is not present on normal stem cells, is a specific surface marker for AMl-initiating cells141.
In addition to binding to their targets and inhibiting target-dependent signalling, most therapeutic mono-clonal antibodies (mAbs) interact with components of the immune system through antibody-dependent cellular cytotoxicity (ADcc) and/or complement-dependent cytotoxicity, and immune responses might be part of their antitumour mechanisms142. Interestingly, systemic administration of a mAb directed against Abcb5, which induces ADcc in Abcb5+ malignant-melanoma-initiating cells, exerts tumour-inhibitory effects in a melanoma xenograft model9. Some tumour-initiating cells express immune-tolerance markers143, potentially making them resistant to immune attack. However, the antitumour activity of these antibodies can be enhanced by a cytotoxic immunoconjugate or engineered antibody binding to both tumour and immune cells144,145, which might be able to bypass
various immune tolerance mechanisms. Although Abc transporter activity can result in tumour-initiating cells being relatively resistant to many conventional thera-pies, it may make the cells susceptible to alternative strategies that target cells with effective drug efflux146.
Agents that target markers on the surface of tumour-initiating cells could also work by affecting the niche (fIG. 1). endosteal (osteoblastic) niches have been identi-fied for both normal HScs63 and AMl-initiating cells147. A mAb specific for the adhesion molecule cD44 was shown to eradicate human AMl-initiating cells in vivo by blocking the trafficking of leukaemia-initiating cells to supportive microenvironments, and by altering their ‘stem cell’ fate through differentiation148. endothelial niches were also revealed for HScs149, and are a possi-bility for leukaemia-initiating cells. Neural stem cells are thought to localize to vascular niches150–152, and brain tumour-initiating cells reside in a perivascular niche69,70. Moreover, stem cell-like glioma cells have been shown to promote tumour angiogenesis71, sug-gesting that these cells might help to create their own niche. These data indicate that signalling in niche inter-actions can be bidirectional, with tumour-initiating cells as both the source and the target.
It is important to emphasize that much of the niche-interaction data has been obtained from animal models, and the role of the niche in various human tissues and cancers is not yet clear. Nevertheless, the association between tumour-initiating cells and the vasculature does raise the intriguing possibility that anti-angiogenic therapy may work in part by affecting the vascular niche of tumour-initiating cells, and there could be many rea-sons to combine agents that target tumour-initiating cells with anti-angiogenic therapy. Strategies that specifically target other niches of tumour-initiating cells can also be envisioned.
Therapeutic windows and combination strategiesThe potential therapeutic window is always a concern for any anticancer approach, including those that target tumour-initiating cells. However, tumour-initiating cells that exhibit overactive self renewal might be more sensi-tive to agents that inhibit self-renewal pathways than normal stem cells, the self-renewal activity of which depends on the developmental stage and tissue homeo-stasis, among other things. As combined loss of certain tumour suppressor genes in progenitor cells can lead to malignant cells with increased self-renewal activity47, there might be mechanistic differences between tumour-initiating cells and normal stem cells with respect to self renewal. Knockout of the PTEN tumour suppres-sor causes the generation of transplantable leukaemia-initiating cells and the depletion of normal HScs in mice153,154. Interestingly, rapamycin not only depleted leukaemia-initiating cells but also restored normal HSc levels in this model. Although it has not yet been con-firmed in the case of human cancer, this finding high-lights the potential feasibility of achieving a therapeutic window in targeting tumour-initiating cells. recently, wnt–β-catenin signalling was shown to be involved in the maintenance of cutaneous tumour-initiating cells
R E V I E W S
814 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

and malignant human squamous cell carcinomas42, whereas wnt–β-catenin signalling is not essential for normal epidermal homeostasis. As discussed below, the SMo antagonist GDc-0449 was well tolerated in Phase I clinical trials155, providing preliminary clinical evidence of therapeutic windows for strategies that target self-renewal signalling.
Now that markers of tumour-initiating cells, including Il3rA for AMl, are known to exist141, the currently limited repertoire of those that have been identified will grow as tumour-initiating cell populations from various systems are further characterized. As many tumour-initiating cells might originate from progenitor cells or partially differentiated cells46,47, many of them are expected to have surface markers distinct from those of normal stem cells. mAbs that target different epitopes of the same target could have different therapeutic win-dows, owing to differential exposure and signalling, as shown for cD44-specific mAbs148.
Another example relevant to the potential therapeutic window of strategies targeting tumour-initiating cells is the observation that the combination of a proteasome inhibitor (MG-132) with the cytotoxic drug idarubicin induces rapid and extensive apoptosis of the leukaemia-initiating cell population while leaving normal HScs viable both in vitro and in vivo123. The inhibition of Notch signalling by γ-secretase inhibitors is presumed to have a narrow therapeutic window. However, treatment with a γ-secretase inhibitor seemed to be better tolerated in mice when using a pulsed dosing regimen156, suggest-ing that normal stem cells could recover quickly from the treatment. Therefore, based on the pharmacokinetic properties of the agent, the therapeutic window can be improved by optimizing the dosing regimen, similar to the adjustment of dosing schedule of cytotoxic agents.
As discussed above, it is thought that several pathways, including those that regulate self renewal and cell growth, converge to regulate tumour-initiating cells. If there is a sufficiently large therapeutic window, targeting a combi-nation of pathways that are uniquely active in tumour-initiating cells will be more effective than inhibiting a single pathway. It has been reported that simultaneously blocking the Hedgehog and epidermal growth factor receptor (eGFr) pathways using cyclopamine and gefit-inib resulted in growth arrest, apoptosis and a decrease in the invasiveness of prostate cancer cells157. The PI3K–AKT pathway has been reported to regulate Hedgehog signal-ling in part by controlling protein kinase A-mediated glioma-associated oncogene homologue (Gli) activity158, indicating the potential therapeutic value of combining Hedgehog antagonists with PI3K–AKT inhibitors.
Many agents that target tumour-initiating cells dif-ferentiate these cells, and may need to be combined with chemotherapeutics to eliminate cells further down the tumour hierarchy. Inhibition of promyelocytic leukae-mia protein (PMl) by arsenic trioxide disrupted the maintenance of cMl-initiating cells, induced the dif-ferentiation and progression through the cell cycle of these otherwise quiescent tumour cells and sensitized them to pro-apoptotic stimuli159. In addition, the PI3K pathway regulates survival of tumour-initiating cells that
reside in the perivascular niche following radiation in medulloblastoma in vivo, and inhibition of AKT signal-ling sensitizes these cells to radiation-induced apopto-sis70. The PI3K–AKT pathway regulates AbcG2 activity in glioma-initiating cells160, providing another rationale for combining PI3K–AKT inhibitors with chemothera-peutics that are substrates of the Abc drug transporter.
Given the emerging role of Notch and Hedgehog in the tumour stroma and vasculature114,120,121, agents tar-geting these pathways might have an impact on both tumour-initiating cells and tumour vasculature; the effects of these agents on drug delivery should therefore also be considered161. It is conceivable that challenging tumour-initiating cells with both targeted agents and conventional chemotherapy or radiotherapy would not only be more effective in cell killing, but also delay the development of drug resistance compared with either agent alone; however, preclinical evidence for this is not yet available.
It is important to evaluate different agents and combi-nations in the context of the tumour hierarchy and with biomarkers162 (fIG. 3b), to determine their efficacy and to study the emergence of drug resistance in preclinical models. It is also crucial to study their effects on stromal cells and the vasculature, and to follow closely the ther-apeutic window of different combinations. Hopefully, well designed combination strategies based on data from relevant preclinical models can overcome pathway redundancy, counter drug resistance and help to achieve long-term remission in a clinical setting.
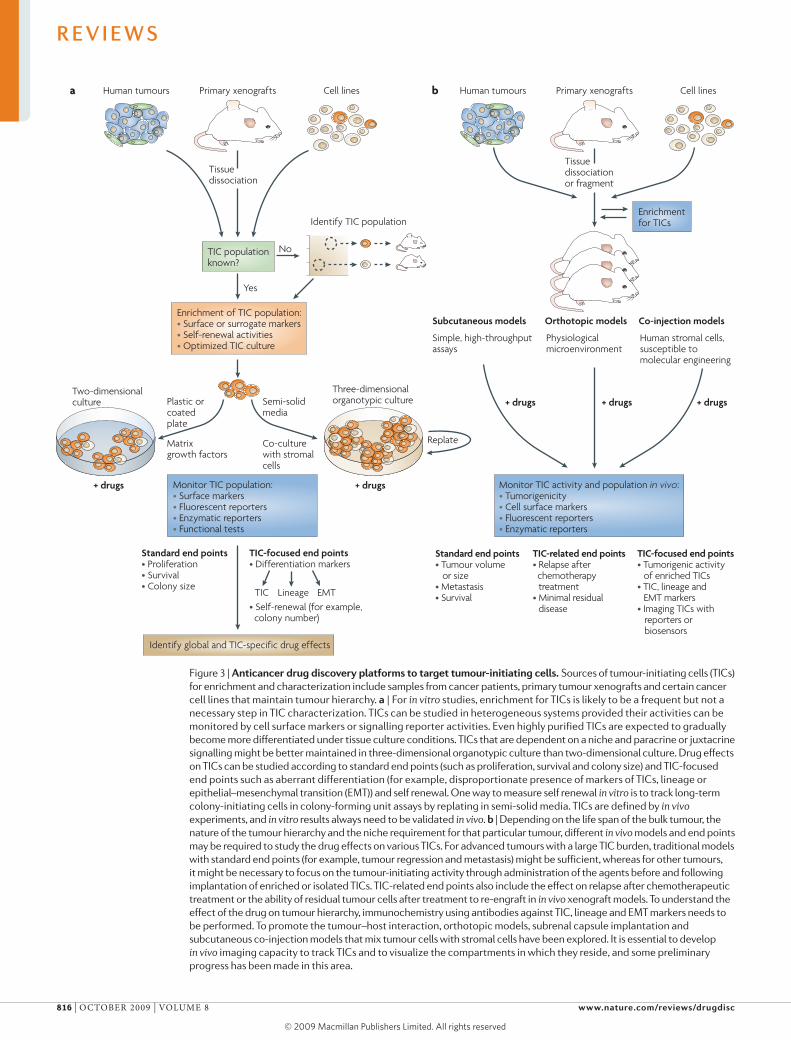
Drug discovery with tumour-initiating cellsThe conventional approach for anticancer drug discovery is to target cell proliferation rather than self renewal and/or differentiation, and so is often biased to select targets with homogeneous expression patterns and potent compounds that kill the bulk tumour cells. In addition, some traditional preclinical models may not reflect clinical complexities such as tumour hierarchy. Tumour-initiating cells that depend on a niche and developmental pathways involving paracrine or juxta-crine signalling may demand more sophisticated drug discovery platforms than the two-dimensional tissue culture or subcutaneous xenograft models that have traditionally been used to characterize autonomous tumour cells and autocrine signalling in cancer. The large body of evidence in support of the cancer stem cell hypothesis and the related therapeutic strategies require adjustments to anticancer drug discovery platforms to make them more clinically relevant (fIG. 3).
Tumour-initiating cell enrichment and in vitro culture con ditions. Sources from which to isolate tumour-initiating cells include samples from patients with primary cancer, primary tumour xenografts and certain cancer cell lines (fIG. 3). Interestingly, it was observed that, as with primary tumour cells, some cancer cell lines cultured under conven-tional conditions have a tumour hierarchy based on estab-lished tumour-initiating cell markers10,60,163–165. Although their relevance to tumour-initiating cells from primary patient samples still requires further characterization,
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 815
© 2009 Macmillan Publishers Limited. All rights reserved
Nature Reviews | Drug Discovery
Tissuedissociation
Primary xenograftsHuman tumours Cell lines
TIC population known?
Enrichment of TIC population:• Surface or surrogate markers• Self-renewal activities• Optimized TIC culture
Monitor TIC population: • Surface markers• Fluorescent reporters• Enzymatic reporters• Functional tests
Monitor TIC activity and population in vivo: • Tumorigenicity• Cell surface markers • Fluorescent reporters• Enzymatic reporters
Enrichmentfor TICs
Standard end points• Proliferation• Survival• Colony size
Standard end points• Tumour volume or size• Metastasis• Survival
TIC-related end points• Relapse after chemotherapy treatment• Minimal residual disease
TIC-focused end points• Tumorigenic activity of enriched TICs• TIC, lineage and EMT markers• Imaging TICs with reporters or biosensors
TIC-focused end points• Differentiation markers
TIC Lineage EMT • Self-renewal (for example, colony number)
Identify global and TIC-specific drug effects
Yes
No
Identify TIC population
Two-dimensionalculture
+ drugs
+ drugs+ drugs + drugsPlastic or coated plate
Matrix growth factors
Three-dimensionalorganotypic culture
+ drugs
Semi-solidmedia
Co-culturewith stromal cells
Replate
a
Tissuedissociationor fragment
Primary xenografts
Co-injection models
Human stromal cells,susceptible tomolecular engineering
Orthotopic models
Physiologicalmicroenvironment
Subcutaneous models
Simple, high-throughputassays
Human tumours Cell linesb
Figure 3 | Anticancer drug discovery platforms to target tumour-initiating cells. Sources of tumour-initiating cells (TICs) for enrichment and characterization include samples from cancer patients, primary tumour xenografts and certain cancer cell lines that maintain tumour hierarchy. a | For in vitro studies, enrichment for TICs is likely to be a frequent but not a necessary step in TIC characterization. TICs can be studied in heterogeneous systems provided their activities can be monitored by cell surface markers or signalling reporter activities. Even highly purified TICs are expected to gradually become more differentiated under tissue culture conditions. TICs that are dependent on a niche and paracrine or juxtacrine signalling might be better maintained in three-dimensional organotypic culture than two-dimensional culture. Drug effects on TICs can be studied according to standard end points (such as proliferation, survival and colony size) and TIC-focused end points such as aberrant differentiation (for example, disproportionate presence of markers of TICs, lineage or epithelial–mesenchymal transition (EMT)) and self renewal. One way to measure self renewal in vitro is to track long-term colony-initiating cells in colony-forming unit assays by replating in semi-solid media. TICs are defined by in vivo experiments, and in vitro results always need to be validated in vivo. b | Depending on the life span of the bulk tumour, the nature of the tumour hierarchy and the niche requirement for that particular tumour, different in vivo models and end points may be required to study the drug effects on various TICs. For advanced tumours with a large TIC burden, traditional models with standard end points (for example, tumour regression and metastasis) might be sufficient, whereas for other tumours, it might be necessary to focus on the tumour-initiating activity through administration of the agents before and following implantation of enriched or isolated TICs. TIC-related end points also include the effect on relapse after chemotherapeutic treatment or the ability of residual tumour cells after treatment to re-engraft in in vivo xenograft models. To understand the effect of the drug on tumour hierarchy, immunochemistry using antibodies against TIC, lineage and EMT markers needs to be performed. To promote the tumour–host interaction, orthotopic models, subrenal capsule implantation and subcutaneous co-injection models that mix tumour cells with stromal cells have been explored. It is essential to develop in vivo imaging capacity to track TICs and to visualize the compartments in which they reside, and some preliminary progress has been made in this area.
R E V I E W S
816 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

the finding that certain cancer cell lines may contain a subpopulation of tumour-initiating cells is important: cell lines can provide sufficient material for extensive molecular and signalling profiling of these cells.
There are several ways to enrich for tumour-initiating cells: using cell sorting techniques to select for combi-nations of cell-surface markers, sorting to select for a subpopulation of cells that efflux dyes166,167, or various serum-free stem cell culture conditions111,168,169 (fIG. 3a). Although cell sorting for cell surface markers is use-ful for isolation of tumour-initiating cells that express established markers81,123, it could be challenging to use this technique in many other tumours owing to a lack of markers and/or the requirement for multiple markers15.
As the expression of the genes encoding Abc drug transporters such as AbcG2 is a conserved feature of several stem cell populations from a range of sources, the efflux of the DNA-binding dye Hoechst 33342 has been used to identify and enrich for certain types of tumour-initiating cells160,166,167. Theoretically, this method may be more suitable to enrich for potential tumour-initiating cells as it is less constrained by tissue specificity than the cell surface makers discussed above. However, the kinetics of dye exclusion, a potentially small window for detecting stem cells that exclude the dye and the toxicity of various dyes might limit functional analysis of the enriched cell populations. It is also possible to enrich for tumour-initiating cells by selecting other surrogate func-tional properties of stem cells, such as aldehyde dehydro-genase activity165,170 and chemotherapeutic resistance60.
Serum-free non-adherent culture has been shown to enrich for and propagate several types of tumour-initiating cells, including those from brain, breast and colon cancer7,73,111,168,171. The serum-free ‘sphere’ culture was developed when it was observed that central nerv-ous system (cNS) cells grown on non-adherent surfaces form spheroid colonies (neurospheres) that have the capacity for self renewal and can generate all of the prin-cipal cell types of the brain (that is, neurons, astrocytes and oligodendrocytes)172. conversely, cells in serum-free adherent culture can also maintain stem cell-like proper-ties if the plates used have a unique surface (for example, incorporating various nitrogen-containing functional groups), or are coated with an appropriate matrix and/or ligand111,169,173.
It is crucial to develop culture methods and con-ditions that simulate the growth and differentiation-inhibitory signalling that is provided by the niche, particularly for culturing cells from primary tumours. Serum-free culture conditions that have been estab-lished for normal stem cells154,172,174 provide useful starting points. A new generation of high-throughput platforms, such as microfabricated arrays of extracell-ular matrix or other molecules that are involved in paracrine and juxtacrine signalling, can be used to identify relevant microenvironmental signals for differ-ent tumour-initiating cells175. Successful approaches to sustaining and expanding normal stem cells in serum-free culture have included stimulation of the wnt, Hedgehog and/or Notch pathways and inhibition of
the bMP pathway174,176,177. Appropriate growth factors must also be provided111,154,174 and favourable micro-environments, such as laminin-coated plates or lam-inin-rich matrigel, could be necessary or helpful111,169,174. Metabolic activity and oxygen tension are other varia-bles to consider in the culture of stem cells and tumour-initiating cells178,179.
As well as considering suitable screening end points, efforts to adapt these stem cell culture systems to tumour-initiating cells must take into account the distinct origins and characteristics of these cells. Developmental pathways that regulate self-renewal in culture may also provide therapeutic targets, so the balance of exogenous factors may be crucial for certain screens. unlike nor-mal stem cells, tumour-initiating cells commonly carry mutations that alter their growth factor dependence or responses. Further differences in culture requirements may be expected when the origin or characteristics of a tumour-initiating cell is progenitor-like. Finally, the mutational and pathway profiles of tumour-initiating cells will vary with tumour subtype and grade, and so culture conditions might have to be optimized accordingly.
In vitro assays and screening methods. In many cases, including under sphere conditions, in vitro culture of tumour-initiating cells is expected to produce mixed populations of tumour-initiating cells and more dif-ferentiated progeny. This presents both a challenge (isolating the effect of experimental intervention on tumour-initiating cells) and an opportunity (the ability to use differentiation as an end point). Monitoring stem cell markers with immunofluorescence or fluorescent reporter gene expression is amenable to high-through-put analysis, and both readouts have been successfully used to screen for novel regulators of self-renewal in embryonic stem (eS) cells180,181. Differentiation mark-ers have also been used as reporters to screen for small molecules or genes that drive or inhibit stem cell dif-ferentiation182,183. based on such successes, imaging platforms and other marker-based screens for modula-tors of tumour-initiating cell behaviour can be readily envisioned (fIG. 3a). Nevertheless, efficiently and reliably obtaining quantitative data from images presents design challenges in terms of the data collection, data handling and image processing184. The other challenge is that few markers and reporter genes have been established for various tumour-initiating cells and their differentiated progeny.
ultimately, the complexity of tumour–stroma inter-actions and tumour–matrix interactions in vivo might be more accurately reproduced by cultivating mixed cell populations in three-dimensional organotypic cultures that can maintain various aspects of in vivo tumour–host interactions and might enrich for tumour-initiating cells. A more successful in vitro screening strategy might be to use mixed three-dimensional organotypic cultures and quantify and track tumour-initiating cells within them using various markers or built-in quantitative fluores-cent or enzymatic reporters. one challenge in screen-ing three-dimensional cultures is the production of a
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 817
© 2009 Macmillan Publishers Limited. All rights reserved

consistent organotypic structure in a high-throughput fashion, but aggregation methods that were developed for reliable production of embryoid bodies from eS cells185 can perhaps be adapted. To quantify specific cell populations in three-dimensional culture formats, high-speed imaging systems and automated image analysis methods can be combined186.
Progress has been made with leukaemia-initiating cells and cNS tumour-initiating cells in terms of in vitro assay conditions123,169,171, and it has become feasible to conduct high-throughput in vitro analyses to search for compounds that differentiate or kill these cells124,169. In addition, in-depth knowledge of the stem cells and progenitor cells from haematopoietic and cNS systems is beginning to allow direct comparison of normal stem cells and tumour-initiating cells from these tissues123,124,169. eventually, similar assays for other tumour-initiating cells will be established and optimized.
Ideal culture conditions should support cancer cell proliferation in vitro without genotypic alterations and with the retention of phenotypic behaviour — most importantly in vivo tumorigenic ability over passages111. However, even short-term culture can cause differences in the in vivo repopulation ability of HScs that have identi-cal cell surface markers, emphasizing the caution that is needed in drawing conclusions from in vitro results21,187. Alternatively, using genetic manipulations to achieve a stabilized mesenchymal-like state that captures many tumour-initiating cell properties can allow high-through-put screening in vitro, which has recently generated novel leads against tumour-initiating cells188.
In vivo tumour models. To evaluate agents that specifically target tumour-initiating cells (fIG. 3b), it may be necessary to focus on the tumour-initiating activity through admin-istration of the agents before and following implantation of tumorigenic cells; the effect on established tumours might not be as dramatic as in nascent tumours and could take longer9. Alternative end points include the effect on relapse after chemotherapeutic treatment, the effect on metastasis and the ability of residual tumour cells after treatment to re-engraft in in vivo xenograft models. However, the frequency of tumour-initiating cells in solid tumours seems to be substantially higher than that of leukaemia-initiating cells in leukaemia3,5, and recent mathematical analyses have further indicated that tumour-initiating cells in advanced tumours may not occur as a small fraction189. Mathematical model-ling predicts that if progenitors acquire self-renewal ability then self-renewing cells can come to dominate a tumour53. Agents that target tumour-initiating cells might therefore show dramatic activity in certain tra-ditional models of advanced tumours that have a large tumour-initiating cell burden.
The function of a tumour-initiating cell may be more effectively assessed when the cell is orthotopically engrafted4,5,190,191, and tumour metastasis to specific organs can often be reproduced in an orthotopic model (BOX 2). In many epithelial tumours, an eMT or loss of differentiation is frequently evident at the invading edge of the tumour and is likely to mediate cellular detachment and eventual
metastasis57. cells that are undergoing an eMT could conceivably be the precursors to metastatic tumour-initiating cells, and so eMT markers could be used as biomarkers for evaluating agents that target tumour-initiating cells in metastasis models. Despite an increasing awareness of orthotopic models, for some cancer types (such as colon cancer), performance of an orthotopic injection can be technically difficult. In these cases, alternative approaches, including subcutaneous models with cells suspended in matrigel (or mixed with stromal cells) and xenograft models featuring subrenal capsule implantation, have been explored6,7.
In vivo drug discovery screening demands a repro-ducible, cost-effective system. Tumour-initiating cell models involving xenotransplantation of primary tumour cells4,5,17 have limitations for medium- or high-through-put assays due to the intrinsic variation between tumours and practical challenges of using freshly resected mate-rial, but could be a choice for testing candidate agents. An alternative approach is to generate cancer cell lines that are enriched with tumour-initiating cells. A highly malignant breast cancer cell line (SK-3rd) was generated by sequential in vivo passage in epirubicin-treated NoD–ScID mice, taking advantage of the chemotherapy resist-ance of the tumour-initiating cells60. The SK-3rd cell line is enriched for cells that display all the putative properties of breast tumour-initiating cells13; moreover, these cells metastasize and are capable of serial transplantation60. More recently, glioma-initiating cell lines that are derived directly from primary malignant gliomas were success-fully cultured and expanded under serum-free adherent culture conditions169. Genetic manipulation of differ-entiation status can also be used to produce an undif-ferentiated, tumorigenic character in cancer cell lines188. These cell lines could be more clinically relevant than conventional cancer cell lines, although the relevance of any results obtained in cell lines needs to be confirmed in primary cancer cells.
To address the limitations of cell lines and primary tumour cells, primary tumour xenografts that have been passaged in vivo offer a unique system for the study of tumour heterogeneity and hierarchy in preclinical models. Fragments of surgically resected tumour are implanted directly into immunocompromised mice (orthotopi-cally or subcutaneously). The resulting xenografts are passaged to new animals and are therefore maintained exclusively in vivo192. The cellular architecture and heterogeneity of a primary tumour xenograft closely resemble those of the original patient tumour and are more complex than the corresponding features of tra-ditional cell line xenografts. Therefore, primary tumour xenografts coupled with appropriate experimental analysis tools constitute a tractable preclinical model for effectively evaluating lead compounds and developing drug combination and biomarker strategies162.
In vivo biomarker and imaging studies. Given that tumour-initiating cells represent only a subpopulation of the cells in a tumour and their existence might depend on a niche, it is desirable to track them and visualize the com-partments in which they reside before, during and after
Orthotopic modelA system in which tumour cells are implanted at the site of the organ of origin.
R E V I E W S
818 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

treatment in vivo. A preliminary understanding of the effect of experimental intervention on tumour hierarchy and tumour–host interaction in vivo can be achieved by immunohistochemistry using antibodies against markers of tumour-initiating cells, eMT, lineage differentiation and different stromal cells (fIG. 3b). For tumour models that have a large tumour-initiating cell burden, gene signature and microarray analysis can also provide biomarkers of tumour-initiating cells. Through in vivo lineage tracking of cultured melanoma cells that are differentially fluorochome-conjugated, co-xenografted Abcb5+ melanoma-initiating cells and Abc5– subpopu-lations have been assayed for their relative contribution to tumour growth, self renewal and differentiation9, and the effect of treatment can in principle be similarly studied. currently, there are no obvious paths by which to pursue direct imaging, although the use of labelled antibodies against specific tumour-initiating cell markers might offer an entry point. To evaluate metastasis mediated by tumour-initiating cells, non-invasive magnetic resonance imaging of magnetically labelled tumour-initiating cells, as has been used for normal stem cells193, could be useful. It would also be desirable to develop surrogate reporter genes or other biosensors that will allow for in vivo mon-itoring of the activities of self-renewal signalling and tumour-initiating cells194. Targeted delivery of a reporter gene has already allowed the locations of normal HScs to be identified and monitored by whole-animal live imag-ing195. More markers and reporter genes of tumour stem cell-like properties need to be established.
Hopefully, the improvement and modification of anticancer drug discovery platforms in light of the cancer stem cell hypothesis will improve the clinical relevance of preclinical assays and models. These models will not only help us to understand how current chemotherapeutic and tumour-targeted agents affect different levels of the tumour hierarchy, but also reveal novel agents that target tumour-initiating cells. In addition, preclinical studies using these models might provide data that support unique clinical combinations and biomarker strategies for agents that target tumour-initiating cells.
Clinical strategies and outlookMany aspects of the aberrant differentiation that is associ-ated with poor prognosis in cancer can be best explained by the cancer stem cell hypothesis. This is underscored by the fact that, in clinical trials for advanced cancers, tumour regression often does not translate into clinically significant increases in patient survival. efficacy against minimal residual disease, metastasis, delayed relapse and tumour-free survival are expected to correlate with the mechanism-based activity of agents that target tumour-initiating cells.
Given that tumour regression might not be the most relevant early end point, biomarkers for tumour-initiating cells in patients who are receiving cancer therapy need to be developed. However, translating the markers that are used to enrich for tumour-initiating cells into clinical biomarkers is not necessarily straight-forward. For example, the cD44+ cD24–/low expres-sion in patients with breast cancer that is detected by
immunohistochemistry does not correlate with event-free or overall survival196, suggesting that not all cD44+ cD24–/low
cancer cells are tumorigenic and surface markers might not be as conserved as was originally thought. However, gene signatures derived by comparing cD44+ cD24–/low breast cancer cells with normal breast epithelial cells, which might reflect the epigenetic state of the tumour cells, are correlated with decreased patient survival76,197. circulating tumour cells, although extremely rare, are a potential alternative to invasive biopsies as a source of tumour tissue for the detection, characterization and monitoring of tumour-initiating cells from solid tumours. A microchip technology that is based on microfluidics was shown to be sensitive and able to detect circulating tumour cells in almost all of the examined patients with recurrent carcinomas198.
It is also important to study the relevance to tumour-initiating cells of current biomarkers, such as prostate-specific antigen (PSA) for prostate cancer and mucin 16 (Muc16; also known as cA125) for ovarian cancer, which are beginning to guide clinical trials and therapy. both PSA and Muc16 are expressed in differentiated tumour cells; it is still unclear whether they are surrogate only to a bulk tumour cell population or to a tumour-initiating cell population as well. If genetic (for example, gene amplification) or epigenetic (for example, promoter methylation) changes or multiple changes of related pathways could be used to predict the dependence of a tumour-initiating cell on certain oncogenic pathways, some of the self-renewal signalling molecules could represent an Achilles’ Heel of cancer. Inhibitors of these pathways would have considerable antitumour activity alone199. In this regard, a diagnostic procedure to pre-screen patients with such genetic or epigenetic changes might be essential for drug development.
Among various agents that target self-renewal path-ways, small molecules that target the Hedgehog pathway are in early clinical studies, and have shown promising results (TABLe 1). The SMo antagonist cyclopamine was shown to lead to rapid regression of basal cell carcinoma in all four patients in which it was tested200. In addition, an orally-administered small-molecule antagonist of SMo, GDc-0449, has shown limited toxicity and partial responses in advanced basal cell carcinoma tumours in a Phase I clinical trial. It is advancing to Phase II trials for metastatic colorectal cancer and other advanced epithe-lial tumours155. The majority of patients with basal cell carcinoma have genetic mutations in Hedgehog path-way mediators38; it is unclear whether GDc-0449 will be as effective in other tumour types that do not have such mutations or whether it must be combined with other agents to show clinical activity. This combination approach could be further complicated by the emerging role of Hedgehog signalling in tumour stromal cells114,161. GDc-0449 and other SMo antagonists (TABLe 1) will therefore provide an interesting test of clinical strategies in targeting renewal signalling.
There are several antibodies against cell surface markers of tumour-initiating cells in clinical studies (TABLe 1). ePcAM is highly expressed in numerous solid tumours, and was recently shown to be expressed
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 819
© 2009 Macmillan Publishers Limited. All rights reserved

on tumour-initiating cells from breast, prostate, colon and pancreatic cancer4,7,8. ePcAM-specific mAbs have shown a limited efficacy in clinical trials142, suggesting that immune tolerance or ADcc stimulated by these mAbs by itself might not be effective in killing ePcAM-overexpressing tumour cells in clinical settings. To overcome the limitations of the naked antibodies, catu-maxomab was designed to bind to both human ePcAM (the target on the tumour) and human cD3 (the target on T cells), bringing cancer cells into proximity with the immune-system cells that can destroy them. In addi-tion, catumaxomab induces ADcc and is undergoing advanced study in patients with malignant ascites145. conversely, mAbs against cD44 can differentiate tumour-initiating cells and have single-agent activity in certain preclinical models148,201 (TABLe 1). It remains to be seen whether these mAbs will show single-agent activity in clinical settings or whether they will also need to be coupled with cytotoxic approaches.
Various drug screening platforms that were specifically designed to target tumour-initiating cells have begun to identify novel drug leads169,188. whether tumour-initiating cell-targeted therapies are effective as single agents, in terms of short- and long-term clinical end points, will probably depend on the lifespan of the bulk tumour cells and the symptoms they cause. For tumours in which most tumour cells are short-lived or the tumour-initiating cell burden is large, continued tumour growth and maintenance may be highly dependent on the activity of tumour-initiating cells. Tumours with populations of proliferating progenitor-like cells (BOX 1) may take a long time to regress, even if tumour-initiating cells are destroyed. For some cancers, continued although lim-ited proliferation of bulk tumour cells might be sufficient to cause irreversible pathological damage. It is necessary to
kill all cancer cells that have the potential to contribute to disease, particularly for cancers with little evidence of hierarchical organizations15. Genetic and epigenetic instability could also confound the effectiveness of agents that otherwise efficiently target tumour-initiating cells. It may be important to combine agents that target tumour-initiating cells with conventional agents that reduce the bulk of the tumour, and the optimal manner of combination could depend on the therapeutic win-dow of the agents, the life span of the bulk tumour cells, and the nature and stability of the tumour hierarchy in that particular tumour type.
More work is required to understand how current, partially effective, chemotherapeutic and tumour-targeted agents affect different levels of the tumour hierarchy (see the figure in BOX 1). Some combination treatment strate-gies have already emerged from preclinical studies, as highlighted in previous sections. In the future, combi-nation therapy, including cytotoxic, tumour-targeted drugs and agents that target tumour-initiating cells, may be aimed at tumour-initiating cells, rapidly proliferat-ing tumour cells and their niches — simultaneously or sequentially (fIG. 1). It is hoped that this new strategy will result in the rapid removal of all tumour cell subpopula-tions and avoid the possible repopulation of the tumour mass by tumour-initiating cells or by originally differen-tiated tumour cells that have regained renewal activity (see the figure in BOX 1). Although the paths for devel-oping agents that target tumour-initiating cells are not straightforward, the cancer stem cell hypothesis provides an important framework for drug discovery and cancer treatment, with the potential to find novel antitumour activities, to have an impact on cancers with undiffer-entiated phenotypes and to yield long-term benefits for many patients with cancer.
1. Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 105, 574–578 (1963).
2. Kohn, K. W. Beyond DNA cross-linking: history and prospects of DNA-targeted cancer treatment — fifteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 56, 5533–5546 (1996).
3. Bonnet, D. & Dick, J. E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Med. 3, 730–737 (1997).
4. Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J. & Clarke, M. F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl Acad. Sci. USA 100, 3983–3988 (2003).
5. Singh, S. K. et al. Identification of human brain tumour-initiating cells. Nature 432, 396–401 (2004).References 4 and 5 provide an early description of the purification of tumour-initiating cells that give rise to solid malignancies.
6. O’Brien, C. A., Pollett, A., Gallinger, S. & Dick, J. E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, 106–110 (2007).
7. Ricci-Vitiani, L. et al. Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111–115 (2007).
8. Li, C. et al. Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037 (2007).
9. Schatton, T. et al. Identification of cells initiating human melanomas. Nature 451, 345–349 (2008).
10. Yang, Z. F. et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 13, 153–166 (2008).
11. Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001).
12. Pardal, R., Clarke, M. F. & Morrison, S. J. Applying the principles of stem-cell biology to cancer. Nature Rev. Cancer 3, 895–902 (2003).
13. Clarke, M. F. et al. Cancer stem cells — perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344 (2006b).
14. Kelly, P. N., Dakic, A., Adams, J. M., Nutt, S. L. & Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 317, 337 (2007).This paper described the observation that three mouse models of leukaemia and lymphoma are maintained by a dominant tumour cell population. The authors posit that xenotransplantation may select for tumour cells that are capable of surviving in a foreign environment.
15. Quintana, E. et al. Efficient tumour formation by single human melanoma cells. Nature 456, 593–598 (2008).
16. Kennedy, J. A., Barabe, F., Poeppl, A. G., Wang, J. C. & Dick, J. E. Comment on “Tumor growth need not be driven by rare cancer stem cells”. Science 318, 1722 (2007); author reply 318, 1722 (2007).
17. Lapidot, T. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648 (1994).The original report that showed the existence of stem cells in leukaemia.
18. Wang, J. C. et al. High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase. Blood 91, 2406–2414 (1998).
19. Miyamoto, T., Weissman, I. L. & Akashi, K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc. Natl Acad. Sci. USA 97, 7521–7526 (2000).This study showed that purified populations of leukaemia stem cells contained the identical translocation as that found in their progeny, the blast cells, suggesting that the clonal progression to cancer could operate through the ‘stem cell compartment’.
20. Hill, R. P. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 66, 1891–1895; discussion 1890 (2006).
21. Haug, J. S. et al. N-cadherin expression level distinguishes reserved versus primed states of hematopoietic stem cells. Cell Stem Cell 2, 367–379 (2008).
22. Chen, G. Y., Tang, J., Zheng, P. & Liu, Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 323, 1722–1725 (2009).
23. Zeppernick, F. et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 14, 123–129 (2008).
24. So, C. W. et al. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 3, 161–171 (2003).
R E V I E W S
820 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

25. Jaiswal, S. et al. Expression of BCR/ABL and BCL-2 in myeloid progenitors leads to myeloid leukemias. Proc. Natl Acad. Sci. USA 100, 10002–10007 (2003).
26. Huntly, B. J. et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6, 587–596 (2004).
27. Liu, J. C., Deng, T., Lehal, R. S., Kim, J. & Zacksenhaus, E. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res. 67, 8671–8681 (2007a).
28. Cho, R. W. et al. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells 26, 364–371 (2008).
29. Read, T. A. et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell 15, 135–147 (2009).
30. Barker, N. et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 (2009).
31. Zhu, L. et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 457, 603–607 (2009).References 30 and 31 described lineage-tracing experiments using transgenic models that can bypass the limitations and experimental variability of the transplantation assay.
32. Ward, R. J. et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res. 69, 4682–4690 (2009).
33. The Cancer Genome Atlas Research network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
34. Jones, S. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806 (2008).
35. Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008).
36. Warner, J. K., Wang, J. C., Hope, K. J., Jin, L. & Dick, J. E. Concepts of human leukemic development. Oncogene 23, 7164–7177 (2004).
37. Hess, A. R., Margaryan, N. V., Seftor, E. A. & Hendrix, M. J. Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: role of the Eph receptors. Dev. Dyn. 236, 3283–3296 (2007).
38. Beachy, P. A., Karhadkar, S. S. & Berman, D. M. Tissue repair and stem cell renewal in carcinogenesis. Nature 432, 324–331 (2004).
39. Park, I. K. et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423, 302–305 (2003).
40. Lessard, J. & Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423, 255–260 (2003).
41. Zhao, C. et al. Loss of β-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 12, 528–541 (2007).
42. Malanchi, I. et al. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 452, 650–653 (2008).This paper described different requirements for Wnt signalling in cutaneous tumour-initiating cells and normal stem cells.
43. Peacock, C. D. et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl Acad. Sci. USA 104, 4048–4053 (2007).
44. Zhao, C. et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779 (2009).
45. Reya, T. & Clevers, H. Wnt signalling in stem cells and cancer. Nature 434, 843–850 (2005).
46. Krivtsov, A. V. et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822 (2006).
47. Akala, O. O. et al. Long-term haematopoietic reconstitution by Trp53–/–p16Ink4a–/–p19Arf–/– multipotent progenitors. Nature 453, 228–232 (2008).
48. Wu, M. et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell 1, 541–554 (2007).
49. Guan, Y., Gerhard, B. & Hogge, D. E. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML). Blood 101, 3142–3149 (2003).
50. Holyoake, T., Jiang, X., Eaves, C. & Eaves, A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 94, 2056–2064 (1999).
51. Yang, Z. J. et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14, 135–145 (2008).
52. He, X. C. et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nature Genet. 39, 189–198 (2007).
53. Ashkenazi, R., Gentry, S. N. & Jackson, T. L. Pathways to tumorigenesis — modeling mutation acquisition in stem cells and their progeny. Neoplasia 10, 1170–1182 (2008).
54. Brabletz, T., Jung, A., Spaderna, S., Hlubek, F. & Kirchner, T. Opinion: migrating cancer stem cells — an integrated concept of malignant tumour progression. Nature Rev. Cancer 5, 744–749 (2005).
55. Balic, M. et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 12, 5615–5621 (2006).
56. Hermann, P. C. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1, 313–323 (2007).
57. Thiery, J. P. Epithelial–mesenchymal transitions in tumour progression. Nature Rev. Cancer 2, 442–454 (2002).
58. Eger, A. et al. β-Catenin and TGFβ signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene 23, 2672–2680 (2004).
59. Timmerman, L. A. et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 18, 99–115 (2004).
60. Yu, F. et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131, 1109–1123 (2007).
61. Mani, S. A. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
62. Moore, K. A. & Lemischka, I. R. Stem cells and their niches. Science 311, 1880–1885 (2006).
63. Zhang, J. et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841 (2003).
64. Calvi, L. M. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003).
65. Li, L. & Neaves, W. B. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 66, 4553–4557 (2006).
66. Bissell, M. J. & Labarge, M. A. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell 7, 17–23 (2005).
67. Walkley, C. R. et al. A microenvironment-induced myeloproliferative syndrome caused by RARγ deficiency. Cell 129, 1097–1110 (2007).
68. Sipkins, D. A. et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435, 969–973 (2005).
69. Calabrese, C. et al. A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82 (2007).
70. Hambardzumyan, D. et al. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 22, 436–448 (2008).
71. Bao, S. et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 66, 7843–7848 (2006).
72. Clarke, M. F. & Becker, M. W. Stem cells: the real culprits in cancer? Sci. Am. 295, 52–59 (2006).
73. Singh, S. K., Clarke, I. D., Hide, T. & Dirks, P. B. Cancer stem cells in nervous system tumors. Oncogene 23, 7267–7273 (2004).
74. Al-Hajj, M., Becker, M. W., Wicha, M., Weissman, I. & Clarke, M. F. Therapeutic implications of cancer stem cells. Curr. Opin. Genet. Dev. 14, 43–47 (2004).
75. van Rhenen, A. et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin. Cancer Res. 11, 6520–6527 (2005).
76. Shipitsin, M. et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 11, 259–273 (2007).
77. Li, X. et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl Cancer Inst. 100, 672–679 (2008).
78. Gottesman, M. M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 53, 615–627 (2002).
79. Zhou, S. et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nature Med. 7, 1028–1034 (2001).
80. Ito, K. et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002 (2004).
81. Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006).
82. Diehn, M. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783 (2009).
83. Potten, C. S., Wilson, J. W. & Booth, C. Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells 15, 82–93 (1997).
84. Era, T. Bcr-Abl is a “molecular switch” for the decision for growth and differentiation in hematopoietic stem cells. Int. J. Hematol. 76, 35–43 (2002).
85. Bedi, A. et al. BCR-ABL gene rearrangement and expression of primitive hematopoietic progenitors in chronic myeloid leukemia. Blood 81, 2898–2902 (1993).
86. Graham, S. M. et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 99, 319–325 (2002).
87. Hughes, T. P. et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 349, 1423–1432 (2003).
88. Bhatia, R. et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 101, 4701–4707 (2003).
89. Hamilton, A. et al. BCR-ABL activity and its response to drugs can be determined in CD34+ CML stem cells by CrkL phosphorylation status using flow cytometry. Leukemia 20, 1035–1039 (2006).
90. Mullighan, C. G. et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453, 110–114 (2008).
91. Huff, C. A., Matsui, W., Smith, B. D. & Jones, R. J. The paradox of response and survival in cancer therapeutics. Blood 107, 431–434 (2006).
92. Wicha, M. S., Liu, S. & Dontu, G. Cancer stem cells: an old idea — a paradigm shift. Cancer Res. 66, 1883–1890; discussion 1895–1986 (2006).
93. Korkaya, H., Paulson, A., Iovino, F. & Wicha, M. S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 27, 6120–6130 (2008).
94. Magnifico, A. et al. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin. Cancer Res. 15, 2010–2021 (2009).
95. Nahta, R., Yu, D., Hung., M. C., Hortobagyi, G. N. & Esteva, F. J. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nature Clin. Pract. Oncol. 3, 269–280 (2006).
96. Jordan, C. T. Cancer stem cells: controversial or just misunderstood? Cell Stem Cell 4, 203–205 (2009).An excellent review discussing the issues and misconceptions in the cancer stem cell field.
97. Hogan, B. L. et al. Branching morphogenesis of the lung: new models for a classical problem. Cold Spring Harb. Symp. Quant. Biol. 62, 249–256 (1997).
98. Bitgood, M. J. & McMahon, A. P. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 172, 126–138 (1995).
99. Kawano, Y. & Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 116, 2627–2634 (2003).
100. Suzuki, H. et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nature Genet. 36, 417–422 (2004).
101. Lepourcelet, M. et al. Small-molecule antagonists of the oncogenic Tcf/β-catenin protein complex. Cancer Cell 5, 91–102 (2004).
102. Emami, K. H. et al. A small molecule inhibitor of β-catenin/CREB-binding protein transcription [corrected]. Proc. Natl Acad. Sci. USA 101, 12682–12687 (2004).
103. You, L. et al. An anti-Wnt-2 monoclonal antibody induces apoptosis in malignant melanoma cells and inhibits tumor growth. Cancer Res. 64, 5385–5389 (2004).
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 821
© 2009 Macmillan Publishers Limited. All rights reserved

104. You, L. et al. Inhibition of Wnt-2-mediated signaling induces programmed cell death in non-small-cell lung cancer cells. Oncogene 23, 6170–6174 (2004).
105. Sanchez, P. et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl Acad. Sci. USA 101, 12561–12566 (2004).
106. Romer, J. T. et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1+/– p53–/– mice. Cancer Cell 6, 229–240 (2004).
107. Berman, D. M. et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425, 846–851 (2003).
108. Thayer, S. P. et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856 (2003).
109. Watkins, D. N. et al. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 422, 313–317 (2003).
110. Athar, M. et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 64, 7545–7552 (2004).
111. Lee, J. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9, 391–403 (2006).
112. Sasai, K. et al. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 66, 4215–4222 (2006).
113. Fan, L. et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 145, 3961–3970 (2004).
114. Yauch, R. L. et al. A paracrine requirement for hedgehog signalling in cancer. Nature 455, 406–410 (2008).
115. Weijzen, S. et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nature Med. 8, 979–986 (2002).
116. Bocchetta, M., Miele, L., Pass, H. I. & Carbone, M. Notch-1 induction, a novel activity of SV40 required for growth of SV40-transformed human mesothelial cells. Oncogene 22, 81–89 (2003).
117. Weng, A. P. et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004).
118. van Es, J. H. et al. Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435, 959–963 (2005).
119. Fan, X. et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 66, 7445–7452 (2006).
120. Ridgway, J. et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444, 1083–1087 (2006).
121. Noguera-Troise, I. et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444, 1032–1037 (2006).
122. Li, K. et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J. Biol. Chem. 283, 8046–8054 (2008).
123. Guzman, M. L. et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc. Natl Acad. Sci. USA 99, 16220–16225 (2002).
124. Diamandis, P. et al. Chemical genetics reveals a complex functional ground state of neural stem cells. Nature Chem. Biol. 3, 268–273 (2007).
125. Ayyanan, A. et al. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proc. Natl Acad. Sci. USA 103, 3799–3804 (2006).
126. Palomero, T. et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nature Med. 13, 1203–1210 (2007).
127. Piccirillo, S. G. et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444, 761–765 (2006).References 118 and 127 describe differentiation as a strategy to combat tumour-initiating cells.
128. Cozzio, A. et al. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 17, 3029–3035 (2003).
129. Sparmann, A. & van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nature Rev. Cancer 6, 846–856 (2006).
130. Lee, J. et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 13, 69–80 (2008).
131. Bracken, A. P. et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525–530 (2007).
132. Gonzalez, M. E. et al. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 28, 843–853 (2009).
133. Todaro, M. et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 1, 389–402 (2007).
134. Zhou, J. et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl Acad. Sci. USA 104, 16158–16163 (2007).
135. Viale, A. et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 457, 51–56 (2009).
136. Zhou, B. B. & Elledge, S. J. The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439 (2000).
137. Evan, G. I. The ever-lengthening arm of p53. Cancer Cell 14, 108–110 (2008).
138. Godar, S. et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 134, 62–73 (2008).
139. Blair, A., Hogge, D. E., Ailles, L. E., Lansdorp, P. M. & Sutherland, H. J. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood 89, 3104–3112 (1997).
140. Blair, A. & Sutherland, H. J. Primitive acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo lack surface expression of c-kit (CD117). Exp. Hematol. 28, 660–671 (2000).
141. Jordan, C. T. et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 14, 1777–1784 (2000).
142. Adams, G. P. & Weiner, L. M. Monoclonal antibody therapy of cancer. Nature Biotech. 23, 1147–1157 (2005).
143. Kawasaki, B. T., Mistree, T., Hurt, E. M., Kalathur, M. & Farrar, W. L. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem. Biophys. Res. Commun. 364, 778–782 (2007).
144. Wu, A. M. & Senter, P. D. Arming antibodies: prospects and challenges for immunoconjugates. Nature Biotech. 23, 1137–1146 (2005).
145. Burges, A. et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clin. Cancer Res. 13, 3899–3905 (2007).
146. Szakacs, G., Paterson, J. K., Ludwig, J. A., Booth-Genthe, C. & Gottesman, M. M. Targeting multidrug resistance in cancer. Nature Rev. Drug Discov. 5, 219–234 (2006).
147. Ishikawa, F. et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nature Biotech. 25, 1315–1321 (2007).
148. Jin, L., Hope, K. J., Zhai, Q., Smadja-Joffe, F. & Dick, J. E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nature Med. 12, 1167–1174 (2006).
149. Kiel, M. J. et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121 (2005).
150. Palmer, T. D., Willhoite, A. R. & Gage, F. H. Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 425, 479–494 (2000).
151. Capela, A. & Temple, S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron 35, 865–875 (2002).
152. Louissaint, A. Jr, Rao, S., Leventhal, C. & Goldman, S. A. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron 34, 945–960 (2002).
153. Yilmaz, O. H. et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482 (2006).
154. Zhang, J. et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441, 518–522 (2006).
155. Molckovsky, A. & Siu, L. L. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American Society of Clinical Oncology meeting. J. Hematol. Oncol. 1, 20 (2008).
156. Cullion, K. et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 24, 6172–6181 (2009).
157. Mimeault, M. et al. Cytotoxic effects induced by a combination of cyclopamine and gefitinib, the selective hedgehog and epidermal growth factor receptor signaling inhibitors, in prostate cancer cells. Int. J. Cancer 118, 1022–1031 (2006).
158. Riobo, N. A., Lu, K., Ai, X., Haines, G. M. & Emerson, C. P. Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl Acad. Sci. USA 103, 4505–4510 (2006).
159. Ito, K. et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453, 1072–1078 (2008).
160. Bleau, A. M. et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 4, 226–235 (2009).
161. Olive, K. P. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009).
162. Jimeno, A. et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol. Cancer Ther. 8, 310–314 (2009).
163. Kondo, T., Setoguchi, T. & Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl Acad. Sci. USA 101, 781–786 (2004).
164. Fillmore, C. M. & Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 10, R25 (2008).
165. Charafe-Jauffret, E. et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 69, 1302–1313 (2009).
166. Hirschmann-Jax, C. et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl Acad. Sci. USA 101, 14228–14233 (2004).
167. Patrawala, L. et al. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2– cancer cells are similarly tumorigenic. Cancer Res. 65, 6207–6219 (2005).
168. Ponti, D. et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 65, 5506–5511 (2005).
169. Pollard, S. M. et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4, 568–580 (2009).
170. Ginestier, C. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007).
171. Singh, S. K. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828 (2003).
172. Reynolds, B. A. & Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710 (1992).
173. Ince, T. A. et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 12, 160–170 (2007).
174. Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
175. Underhill, G. H. & Bhatia, S. N. High-throughput analysis of signals regulating stem cell fate and function. Curr. Opin. Chem. Biol. 11, 357–366 (2007).
176. Bhardwaj, G. et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nature Immunol. 2, 172–180 (2001).
177. Androutsellis-Theotokis, A. et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442, 823–826 (2006).
178. Ezashi, T., Das, P. & Roberts, R. M. Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl Acad. Sci. USA 102, 4783–4788 (2005).
179. Olivotto, M. & Dello Sbarba, P. Environmental restrictions within tumor ecosystems select for a convergent, hypoxia-resistant phenotype of cancer stem cells. Cell Cycle 7, 176–187 (2008).
R E V I E W S
822 | ocTober 2009 | VoluMe 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

180. Chen, S. et al. Self-renewal of embryonic stem cells by a small molecule. Proc. Natl Acad. Sci. USA 103, 17266–17271 (2006).
181. Desbordes, S. C. et al. High-throughput screening assay for the identification of compounds regulating self-renewal and differentiation in human embryonic stem cells. Cell Stem Cell 2, 602–612 (2008).
182. Falk, A., Karlsson, T. E., Kurdija, S., Frisen, J. & Zupicich, J. High-throughput identification of genes promoting neuron formation and lineage choice in mouse embryonic stem cells. Stem Cells 25, 1539–1545 (2007).
183. Saxe, J. P. et al. A phenotypic small-molecule screen identifies an orphan ligand-receptor pair that regulates neural stem cell differentiation. Chem. Biol. 14, 1019–1030 (2007).
184. Bushway, P. J. & Mercola, M. High-throughput screening for modulators of stem cell differentiation. Methods Enzymol. 414, 300–316 (2006).
185. Ungrin, M. D., Joshi, C., Nica, A., Bauwens, C. & Zandstra, P. W. Reproducible, ultra high-throughput formation of multicellular organization from single cell suspension-derived human embryonic stem cell aggregates. PLoS ONE 3, e1565 (2008).
186. Kim, K. H. et al. Three-dimensional tissue cytometer based on high-speed multiphoton microscopy. Cytometry A 71, 991–1002 (2007).
187. Mazurier, F., Gan, O. I., McKenzie, J. L., Doedens, M. & Dick, J. E. Lentivector-mediated clonal tracking reveals intrinsic heterogeneity in the human hematopoietic stem cell compartment and culture-induced stem cell impairment. Blood 103, 545–552 (2004).
188. Gupta, P. B. et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659 (2009).
189. Kern, S. E. & Shibata, D. The fuzzy math of solid tumor stem cells: a perspective. Cancer Res. 67, 8985–8988 (2007).
190. Galli, R. et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 64, 7011–7021 (2004).
191. McKenzie, J. L., Gan, O. I., Doedens, M. & Dick, J. E. Human short-term repopulating stem cells are efficiently detected following intrafemoral transplantation into NOD/SCID recipients depleted of CD122+ cells. Blood 106, 1259–1261 (2005).
192. Shimosato, Y. et al. Transplantation of human tumors in nude mice. J. Natl Cancer Inst. 56, 1251–1260 (1976).
193. Anderson, S. A. et al. Noninvasive MR imaging of magnetically labeled stem cells to directly identify neovasculature in a glioma model. Blood 105, 420–425 (2005).
194. Vlashi, E. et al. In vivo imaging, tracking, and targeting of cancer stem cells. J. Natl Cancer Inst. 101, 350–359 (2009).
195. Xie, Y. et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 457, 97–101 (2009).
196. Abraham, B. K. et al. Prevalence of CD44+/CD24-/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin. Cancer Res. 11, 1154–1159 (2005).
197. Liu, R. et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 356, 217–226 (2007).
198. Nagrath, S. et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239 (2007).
199. Weinstein, I. B. Cancer. Addiction to oncogenes — the Achilles heal of cancer. Science 297, 63–64 (2002).
200. Tabs, S. & Avci, O. Induction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivity. Eur. J. Dermatol. 14, 96–102 (2004).
201. Marangoni, E. et al. CD44 targeting reduces tumour growth and prevents post-chemotherapy relapse of human breast cancers xenografts. Br. J. Cancer 100, 918–922 (2009).
202. Nowell, P. C. The clonal evolution of tumor cell populations. Science 194, 23–28 (1976).
203. Beier, D. et al. CD133+ and CD133– glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 67, 4010–4015 (2007).
204. Barabe, F., Kennedy, J. A., Hope, K. J. & Dick, J. E. Modeling the initiation and progression of human acute leukemia in mice. Science 316, 600–604 (2007).
205. Sausville, E. A. & Burger, A. M. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 66, 3351–3354, (2006).
AcknowledgementsWe thank J. Dick, L. Li, K. Arndt, R. Abraham, J. Rosen and F. Behbod for discussions and comments on the manuscript.
Competing interests statementThe authors declare competing financial interests: see web version for details.
DATABASESEntrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=geneBMI1 | PtenUniProtKB: http://www.uniprot.orgABCB5 | AKT | APC | BRCA1 | b-catenin | CD24 | CD44 | CD133 | CDNK1A | DLL4 | EGFR | EPCAM | ERBB2 | EZH2 | IL3RA | LRP5 | LRP6 | mTOR | MUC16 | NOTCH1 | PSA | PTEN | SMO | STAT3
FURTHER INFORMATIONGenentech website: http://www.gene.comInfinity Pharmaceuticals website: http://www.ipi.comMicromet website: http://www.micromet.deTrion Pharma website: http://www.trionpharma.de
All links Are AcTive in The online pDf
R E V I E W S
NATure reVIewS | Drug Discovery VoluMe 8 | ocTober 2009 | 823
© 2009 Macmillan Publishers Limited. All rights reserved

Author biographies
bin-bing S. Zhou is Senior Director of oncology Discovery at wyeth research and adjunct Associate Professor of Pharmacology at the robert wood Johnson Medical School, university of Medicine and Dentistry of New Jersey, uSA. He has been interested in cancer drug discovery and drug resistance mechanisms, and has contributed to one approved and two Phase II clinical trial anticancer agents. His research interests range from DNA damage response, erbb signalling and Notch signalling to tumour-initiating cells. He received his Ph.D. from the university of california, berkeley, uSA, and was a postdoc-toral fellow at Harvard Medical School, Massachusetts, uSA.
Haiying Zhang is Senior Group leader of Abbott laboratories and Associate research Fellow of Abbott Volwiler Society. At Abbott, she led several biology teams, including those investigating farnesyl trans-ferase inhibitors, erythropoietin mimetics and checkpoint kinase 1 (cHK1) inhibitors. Her research in cHK1 uncovered a novel mecha-nism that could affect the biomarker and clinical strategy of cHK1 inhibitors. She obtained her Ph.D. in medical microbiology and immunology from ohio State university, uSA, and was a postdoc-toral fellow in neuroscience.
Marc Damelin is Principal research Scientist in oncology Discovery at wyeth research. He helped instigate research on tumour-initiat-ing cells at wyeth and is developing biotherapeutics that target these cells. He received his Ph.D. in biophysics from Harvard university, Massachusetts, uSA, and was a postdoctoral fellow of the Damon runyon cancer research Foundation at columbia university, New York, uSA.
Kenneth G. Geles is Principal research Scientist in oncology Discovery at wyeth research and the project leader of an antibody-based drug discovery programme. He helped pioneer the use of tumor-initiating cells in drug development platforms at wyeth. His current research activities are focused on the isolation, characterization and biotherapeutic targeting of tumour-initiating cells. He received his Ph.D. in cancer biology from Northwestern university, Illinois, uSA, and completed an American cancer Society Postdoctoral Fellowship at the university of california, berkeley.
Justin c. Grindley recently joined wyeth research, where he is expanding the research into and targeting of tumour-initiating cells as a Principal research Scientist in oncology Discovery. His background is in developmental genetics and stem cell biology. He received his Ph.D. in molecular biology from the university of edinburgh, uK, and carried out postdoctoral work in cell biology and paediatrics at Vanderbilt university, Tennessee, uSA. Prior to joining wyeth, he was a Senior research Associate at the Stowers Institute for Medical research, Missouri, uSA, where he studied the control of intestinal and haematopoietic stem cell behaviour in homeostasis and cancer.
Peter b. Dirks is Professor of Neurosurgery at the university of Toronto, ontario, canada, and a staff surgeon and scientist at the Hospital for Sick children, ontario, canada. His laboratory was among the first to show that cells in human brain tumours are het-erogeneous for tumour-initiating ability, with initiation and main-tenance of brain cancers dependent on subpopulations of cells with stem cell properties. Dirks has an M.D. from Queen’s university, ontario, canada, and a Ph.D. in molecular and cellular pathology from the university of Toronto.
xxx Tumour-initiating cells: challenges and opportunities for anticancer drug discoveryBin-Bing S. Zhou, Haiying Zhang, Marc Damelin, Kenneth G. Geles, Justin C. Grindley and Peter B. DirksThe hypothesis that cancer is driven by tumour-initiating cells — popularly known as cancer stem cells — has recently attracted considerable attention, owing to the promise of a novel cellular target for the treatment of haematopoietic and solid malignancies. This Review considers recent advances in the cancer stem cell field, focusing on the challenges and opportunities for anticancer drug discovery.
Competing financial interests statementb.-b.S.Z., M.D., K.G.G. and J.c.G. are employees of wyeth Pharmaceuticals. H.Z. is an employee of Abbott laboratories. P.b.D declares no conflict of interest.
Online summary
• The experimental demonstration of tumour-initiating cells (popu-larly known as cancer stem cells) in several human tumours in recent years supports tumour hierarchy as a fundamental concept in tumour biology.
• Many patients with cancer, particularly those with solid tumours, either do not respond to existing cancer therapies or relapse quickly after initial remission. Key possible reasons for this failure include the inherent drug resistance of tumour-initiating cells, the inef-ficiency of the treatment and/or the genetic instability of cancer cells.
• The cancer stem cell hypothesis provides a rationale for several therapeutic strategies beyond traditional antiproliferative agents. Potential approaches to kill tumour-initiating cells include inhib-iting the survival mechanisms of these cells, blocking essential self-renewal signalling, or targeting tumour-initiating cell surface markers through antibody-based cytotoxic approaches.
• Another strategy is to induce tumour cell differentiation, which can potentially be achieved by inhibiting developmental pathways or epigenetic programmes. As many tumour-initiating cells might be dependent on a niche for their identities, targeting the niche could be a strategy to indirectly inhibit or differentiate tumour-initiating cells.
• The conventional approach for anticancer drug discovery is to target cell proliferation rather than self-renewal and/or differen-tiation, and so is often biased to select targets with homogeneous expression patterns and potent compounds that kill the cells of the bulk tumour. In addition, some traditional preclinical models may not reflect clinical complexities such as tumour hierarchy.
• The large body of evidence in support of the cancer stem cell hypothesis and the related therapeutic strategies suggest that adjustments to anticancer drug discovery platforms are required to make them more clinically relevant, which are discussed in this article.
• Although the paths for developing agents that target tumour-initi-ating cells are not straightforward, the cancer stem cell hypothesis provides an important framework for drug discovery and cancer treatment, with the potential to find novel antitumour activities, to
o n l I n E o n ly
© 2009 Macmillan Publishers Limited. All rights reserved

have an impact on cancers with undifferentiated phenotypes and to yield long-term benefits for many patients with cancer.
o n l I n E o n ly
© 2009 Macmillan Publishers Limited. All rights reserved