tunneling in unimolecular reactions
TRANSCRIPT

8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 1/9
Chem. Rev. 1987. 87. 19-27 19
Tunneling and State Specificity in Unimolecular Reactions
WILLIAM H. MILLER
Deparlment of Chamisfry, Universny of Californle. and the Materials and Molecular Research Division of the Lawrence Berkeley Laba-story,eley, California 94720
Received May28, 1988
(Revised Manuscript Received June27, 1966
1. Introduct ion
In the standard expositions'.' on unimolecular reac-tion rates there is little discussion of the effect ofquantum mechanical tunneling. This is due partly tothe fact that traditional ways of studying such reactions(e.g., thermal excitation, cf. the Lindemann mechanism)tend toobscure subtleties of the reaction dynamics suchas tunneling, state-specific effects, etc. Also, very sel-dom has one had much quantitative information aboutthe potential energy surfaces (i.e., barrier heights and
other properties of the transition state) for these reac-tions, so that these parameters have traditionally beenadjusted to fit the data, and this can also hide specificsof the dynamics.
Both of these situations are beginning to change,however, as molecular beam and a variety of lasermethods make it possible to study unimolecular pro-cesses under near-ideal, collisionless conditions, wherethe molecule of interest is excited t o an extremelywell-defined initial state and then reacts without othercompeting proce~ses . ~ These studies require morerigorous dynamical descriptions in order to understandthe variety of phenomena seen at this new level of de-tail. Also, one is beginning to have usefully reliable ahinitio quantum chemistry calculations for transition-state properties of the reacting molecules.6.'
The purpose of this paper is to review the role oftunneling in unimolecular reaction rates. (Tunnelingin gas-phase species can also manifest itself as splittingsof spectral lines-particularly so for symmetricisomerizations-and though not explicitly discussed inthis review, this phenomena can also be treated by thetheoretical methods described herein. The tunnelingsplitting in malonaldehyde is an excellent example of
Since the state-specific character of unimo-lecular reactions is intimately involved with tunnelingeffects, this feature of unimolecular reactions is alsodiscussed. Though I will try to keep the discussion asgenerally applicable as possible, it will be useful to il-
lustrate a number of points with the specific unimo-lecular reaction for which the significance of tunneling
1)
and state-specificity have been most strongly estab-lished. This is an ideal example because so much de-tailed experimental information is available, becausethe potential energy surface (e.g., barrier heights, vi-brational frequencies, and other transit ion-state prop-erties) is known to an exceptional degree and becausethe dynamics involves hydrogen atom motion whichenhances tunneling effects. The reader interested inthe complete story on the unimolecular dissociation of
HZCO - , + CO
0009-266518710787-0019 06.50/0
WUliamH.Ml l e r was ban, In Kosclusko. MI, in 1941. He receiveda B.S. in Chemistry from Georgia Tech (1963) and a Ph.D. inChemical Physics from HaNard (1967). Postdoctorai years werespent as a NATO Fellow in Freiburg (Germany) and as a JuniorFellow in the Society of Fellows (Haward). He joined the Berkeleyfacuily in 1969 where he has been Professor of Chemistry since1974. His research has dean with various aspects of scatteringtheory and chemical dynamics, most notably w'th the developmentof a general semiclassicalmeOry for such processes. He has been
a Sioan Fellow, a Dreyfus Scholar. a Guggenheim Fellow. an Ov-erseas Fellow of Churchiil College (Cambrklge).and a U.S. SeniwScientist Alexander von Humbolt Awardee and in 1974 receivedthe Annual Prize of the International Academy of Quantum Mo-
lecular Science. In 1985 he was elected to membership n theabove Int em al mi Academy and also received the Ernest OrlandoLawrence Memorial Award in Chemistry.
formaldehyde should see the recent review by Mooreand Weisshaar.
The recent review by Cnm3on unimolecular reactionsfollowing selective (usually laser) excitation is alsorelevant to the topic of this paper, as is the review byDykstra6 on ab initio quantum chemistry calculationsof potential barriers for unimolecular rearrangements.Though not dealing specifically with tunneling, thetopic of intramolecular vibrational energy redistribution(IVR) in highly vibrationally excited states is relevantto state-specific effects in unimolecular reactions, andthere are a number of reviews related to IVR.S.'O Alsoof interest is a series of papers by Kay on unimolecularreactions, though these are not specifically related totunneling effects therein.
Tunneling in unimolecular processes is, of course, nodifferent from tunneling in other phenomena (e.g., bi-molecular rections), and the same theoretical modelsare thus used in these various applications. The in-terested reader should thus take special note of thearticle in this volume by Schatz12which reviews tun-neling effects in bimolecular reactions, which has his-torically received much greater attention. Also relevantis the recent review by Garrett and Truhlar13 which
1987 American Chemical Society

8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 2/9
20 Chemical Reviews, 1987, Vol. 87, No. 1
discusses tunneling corrections to transition-state the-ory. One should also note that there are other theo-retical descriptions of tunneling that employ diabatic(i.e., crossing) potential energy curves, usually with thegolden rule (first-order time-dependent perturbationtheory) used to calculate the rate.14 This approach isoften useful for reactions in condensed phases and whenthe theory is used in a phenomenological framework;it is generally not useful if taken literally and used in
an ab initio fashion for gas phase reactions.Section I1 first discusses tunneling effects within theStatistical approximation to unimolecular reactions, andsection I11 discusses state-specific unimolecular rates.
Miller
If the transition state corresponds to a well-definedreaction barrier (Le., a “tight” transition state), then theenergy levels of the activated complex can also be ap-proximated in a similar fashion
I I . Tunnellng wlthin a Statlsti cal Approximation
The statistical approximation for unimolecular rateconstants, i.e., the well-known RRKM theory,’p2J5 ismicrocanonical transition-state theory. To incorporatetunneling effects in RRKM theory, one thus simplyincorporates tunneling effects into transition-statetheory.16 Because it ismicrocanonical transition-statetheory that is relevant, however, rather than the can-onical version most familiar for bimolecular rate con-stants, the tunneling correction is not a simple multi-plicative factor as i t is in the simplest version of can-onical transition state theory.
A. Separable Reactlo n Coordinate
The simplest way to incorporate tunneling intotransition-state theory is to assume that motion alongthe reaction coordinate is separable from the otherdegrees of freedom. The unimolecular rate constant fora molecule with total energy E and total angular mo-mentum J is then given by1’J8
k(E,J) = [27hp(E,J)]-l P ( E c n J K * ) 2.1)nJc
where { E ~ ~ * }re the vibrational and rotational energylevels of the “activated complex” in terms of the F1 vibrational quantum numbers n = n1,n2, ..., ~ F - J
and angular momentum quantum numbers ( J ,K ) . ( Jis the conserved total angular momentum quantumnumber, and K is the nonconserved projection of totalangular momentum onto a body-fixed axis.) P(EF) isthe one-dimensional unneling probability as a functionof the energy EF along the reactant coordinate a t thetransition state. p is the density of reactant states perunit energy
(2.2)
where en J K) are the energy levels of the reactant mol-ecule in terms of the F vibrational quantum numbersn = (nl, .., F ) and angular momentum quantum num-bers J , K ) .
In practice the energy levels tnJK are usually ap-
proximated by those of a harmonic oscillator rigid rotor
p(E,J) = c 6(E EnJK)n,K
F
1=1
en j K hwi(nl+ 72) + A J J + 1 + C - A ) P
(2.3)
where w,)are the vibrational frequencies of the reactantmolecule and A is the average of the two most nearlyequal of the three rotation constants and C is the thirdone.
F-1
V o+ 1 wis(ni+ Y2 A * J J 1) C’ - A ’ ) Pi= l
where Vo s the potential energy and {wi*)and A*, * )are vibrational frequencies and rotational constants, atthe transition-state geometry. If the transition statedoes not correspond to a well-defined potential energybarrier, e.g., a “loose” transition state, then one mustdetermine Vo, wi*),* ,and C* s functions of a reactioncoordinate and choose the transition state as the loca-tion where the rate constant is a minimum.lnl It is alsousually necessary in this case to recognize that some ofthe vibrational modes of the transition state becomerotational degrees of freedom of the fragment molecules;it is thus necessary to take anharmonicity of these de-grees of freedom into account, by using either hinderedrotor energy levels or simply an empirical interpolationformula between the “tight” harmonic energy levels andthe “loose” rotational energy levels.22
It is easy to see that the classical RRKM expres-sion1*2J5or the rate constant is regained if one ignorestunneling, i.e., replaces the tunneling probability P (E F )by a step function
(2 .5 )E > O
P(EF)- EF) = l o : < o
B. What Is k ( E , J )
Before discussing specific examples, it is useful t oemphasize the physical meaning of k E , J ) , he unimo-lecular rate constant for a molecule with total energy
E and total angular momentum J . (For the presentdiscussion J will not be indicated explicitly.) Specifi-cally, K E ) is the average unimolecular rate constant fora molecule with energy E whether the molecule is either(a) intrinsically statistical, i.e., all states with essentiallythe same energy have essentially the same rate constant,or (b) state specific, i.e., different states with essentiallythe same total energy have significantly different rateconstants. A statistical approximation to the rate willbe adequate if either the molecule itself is statistical,case a above, or the exper im en t is statistical, i.e., av-erages over many states. To distinguish between casesa and b obviously requires experiments (or theories)that measure (or calculate) rate constants for individual
quantum states of the molecule. In either case, though,the microcanonical transition-state theory rate constant
of section A is an approximation to the average rate atenergy E .
To illustrate the fact that statistical theory yields thecorrect average rate constant k E) ven if the moleculeis intrinsically state-specific, it is useful to consider acompletely separable Hamiltonian for a system of twodegrees of freedom23
Px2 Py2H@,,x,p,,y) =- - u , (x ) + ‘ / m w y 2 2 (2.6)
2 m 2 m
Mode y is harmonic, but the potential u , ( x ) is assketched in Figure 1;x is thus the reaction coordinate
8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 3/9
Tunneling and State Specificity in Unimoiecular Reactions Chemical Reviews, 1987, Vol. 87, No. 1 2
Figure 1. Sketch of the potential u, T) for the diss ociative degreeof freedom for the Hamiltonian of eq 2.6.
along which the particle can dissociate by tunneling
through the barrier. Because the x and y motions areseparable, both n, and nyare good quantum numbers.The energy of the state characterized by quantumnumbers n x , n y )s
where tnz is the energy level of potential u x , and theunimolecular decay (Le., tunneling) rate constant is
Here P is the tunneling probability for the x motion,
and d t n Z . / d n , / 2 a h )s the frequency of motion in thex potential. It is clear that eq 2.7 and 2.8 describe anextremelystate-specific situation; i.e., the unimoleculardecay rate of eq 2.8 depends only on the energy tnx nthe mode. Thus states with essentially the same totalenergy EnZnyan have vastly different rate constants,depending on how much of the total energy is in thereaction coordinate (modex). The average rate at totalenergy E is defined by
n,,nY
where in practice the 6 functions should be broadenedsomewhat so tha t a t least several states contribute tothe sums. Replacing the sum over n, by an integral andusing eq 2.7 and 2.8 and the definition of the densityp of reactant states
2.10)
gives
k E ) =
which is recognized as the standard microanonicaltransition state rate (i,e., eq 2 . 1 ) for this example. Thuseven for this extreme limit of state specificity, the au-erage rate constant for energyE is seen to be given bystatistical theory.
C . Example: H CO - H i O
Figure 2 shows a sketch of the potential energy sur-face for the unimolecular decomposition of form-aldehyde in its ground electronic state So), H2C0-H2 CO, and Figure 3 shows the unimolecular rate
V s 1
I
5
Figure 2. Sketc h of the potential energy surface for the form-aldehyde dissociation HzC O- z + C O . Th e upper f igure is acontour plot of the potential as a function of the reaction coor-dinate s and th e out-of-plane bend coordinate Q. he lower figureis the poten tial along the reaction pa th, i .e., along the line Q =0 of the upper figure. Th e barrier height for this reaction is-80-90 kcal/mol.
E - V,Zp kcal lmole)
Figure 3. The microcanonical unimolecular rate constant for th eform aldehyd e dissociation J = 0) as a fu nction of energy relativeto th e classical threshold. Th e solid line includes the effects oftunneling, and t he broken line is the classical result th at neglectstunneling. [If E is the total energy in the molecule ( E =I E,, +E,, where E,, is the zeroth point energy of H 2C 0 an d E,, t heexcitation energy), then Vow= Vo+ E,,*, where Vo is the “bare”barrier height and E,,* the zero-point energy of the transitions t a t e . Thus E VozP = E,, - (Vo + Ezp* E,,).]
constant k E , J ) for J = 0 as a function of total energyE (relative to the classical threshold).la The dashed linein Figure 3 shows, for comparison, the classical RRKMrate constant. One sees that the rate is somewhatgreater than lo9 s-l at the classical threshold and is- o6s-l at an energy - 0 kcal/mol below the classicalthreshold. Similar calculations have been carried outfor J > O; 8924 the rate in general decreases with in-creasingJ,which is understandable since increasing J
takes energy out of the reaction coordinate.The most dramatic aspect of tunneling will thus bemanifested if one excites the isolated molecule to anenergy below the classical threshold. Without inclusionof tunneling the unimolecular decay rate is zero, whiletunneling allows a finite rate, which this example showscan be physically significant for energies as much as 10kcal/mol below the classical threshold.
Experiments on formaldehyde are actually accom-plishing this, though the interpretation is far fromstraightforward. The most relevant experiments exciteH2C0 o individual rotational-vibrational states of SI,the first electronically excited singlet state; this is fol-lowed by a radiationless transition Si - So* (highlyvibrationally excited), and it is So* which undergoes

8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 4/9
22 Chemical Reviews, 1987, ol. 87,No. Miller
so s
F i g u r e 4. Schematic p icture of th e photochem ical dissociationof formaldeh yde; cf. eq 2.13 a nd 2.14). E , is the energy of th e
state Is) of SI ha t is excited by th e laser, Vl are the nonadiabaticcouplings between Is) and t he various states I I of So, a nd IF,)arethe “widths” of these stat es with respect to unimolecular de-composition. AEl is the spacing of the levels E l .
Appl ied Vol ts k V )20
1-0.20
S, Energy an-‘)
F i g u r e 5. Lifetime of th e 111 M = 1 otational state of the 4lvibrational s tate of S1 D,CO, as a function of the energy of thislevel as tune d by a n applied electric (Stark ) field, from ref 30.T he zero of energy here is an ex citation energy of 28375 cm-’ =81.1 kcal/mol.
unimolecular decomposition. The reader should see ref4 or a more complete discussion of these experiments.
Figure 4 shows a schematic picture of this mecha-nism. The widths (r,)f the individual highly excitedvibrational states of Sogive the state-specific unimo-
lecular decay rates klk2= r l / h (2.12)
and they are the objects of interest with regard todiscussing the unimolecular reaction dynamics in theground electronic state. The quantity directly measuredin these experiments, however, is the decay rate (i.e.,inverse lifetimes) of the initially excited state Is) of SI,and within a perturbative approximation to the -Socoupling it is given byz5
k, = h-l C Iv,,i2r,/[(E,E,)2 + (r1/2l2] (2.13)
It is thus not trivial to obtain the individual widths r,]
from such measurements. In fact, if many terms con-tribute to the sum in eq 2.13, then it is well-approxi-mated by
(2 .14)
so that no information about (I?,}is obtainable. Non-perturbative t reatments of the decay can lead to non-exponential decay (i.e., multiple exponential decay withquantum beats), and if these features are observed, itis possible to extract not only the energies (El)andwidths {I’l]but also the nonadiabatic coupling matrixelements V,,.26
A major breakthrough occurred when Weisshaar andM ~ o r e ~ ’ - ~ ~irst carried out these experiments in a
I 1 I
0 2 4 6 8 1 0 1 2
E - V i p k c a l / m o l e )
F i g u r e 6. Average width-to-spacing ratio for levels in SoHzCD( J = 0), as a fu nction of energy relative to th e classical threshold.(See Figure 3 for a discussion of the abscissa.)
T A B L E I. Expe r ime n ta l ly D e termine d Ene rg ie s a n dU n imo lec u lar D i ssoci a ti on Ra te s fo r Ind iv idua l Q u a n tumStates of So Formaldehyde
rateref J kca l/m ol (101 s-’Y
27272727
2929313131
510114
22111
81.4082.7082.7981.37
80.9481.0380.9880.98
80.98
5.65.05.0
7.12.8
5.61.3
530
17
“T hi s is the excitation energy above the ground sta te of form-aldehyde. bT he analysis is still tentative in some cases, so t ha tthese values should be considered preliminary.
variable electric (Le., Stark) field, which modulates thelevel E , relative to the So levels (E,} .Figure 5 shows anexample30 of these experimental results, the decay ratek, as a function of the electric field strength, for aparticular ro-vibrational state of SI of D2C0. The ob-served structure is assumed4 o be due to the fact tha tthe levels of So*constitute a “lumpy continuum”; i.e.,the broadened levels (E ,}are sufficiently sparse (cf.Figure 4) hat they do not overlap and form a “smoothcontinuum” into which the s tate Is) decays.
From the transition-state theory rate expression onecan actually obtain a useful quantitative measure of the“lumpy continuum” character of the So*states. Theaverage width F l is given in terms of the average ratek of eq 2.1
F l Z h k E , J ) (2.15)
of So* states is the
= l /p(E,J) (2.16)
Thus the average width-to-spacing ratio is given by
iand the average level spacingreciprocal of the density of states p
This quantity is shown in Figure 6 , as a function ofenergy relative to the classical threshold, for J = 0. Ifone calls the continuum “lumpy” if
(2.18)
then one sees from Figure 6 that the So* continuum isstill lumpy (for J = 0) for energies up to -3 kcal/mol
rla 5 2

8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 5/9
Tunneling and State Speclflclty in Unlmolecular Reactions Chemical Reviews, 1987, Vol. 87, No. 1 23
there is no more than a factor of 2-there are somecases where the error this causes can be an order ofmagnitude or greater. For a standard bimolecular testreaction
H H2 + Hz H
the error is a factor of -50-100 in the importantthreshold region.34
There are a variety of still relatively simple models
for including, at least approximately, the effect ofcoupling of the reaction coordinate to other degrees offreedom of the molecular system, i.e., the effect ofnonseparability of the reaction coordinate. Most ofthese are derived from a reaction path description ofthe p ~ o c e s s , ~ ~ - ~ ~hereby the reaction coordinate s isdefined as the distance along the steepest descent pathin mass-weighted Cartesian coordinates-the reactionpath-that passes through the transition state fromreactants to products. The other internal (Le., vibra-tional) coordinates of the system are chosen as localharmonic normal modes tha t are orthogonal to the re-action path. The Hamiltonian of the system in thesecoordinates is of the form, for J = 036
F- 1
Y 2 b S C QJ'ktB~k,ds)l~
[1 + C kBk,ds)l2
k,k '=l
Hb,,s,P,Q) = F- 1
k = lF-1
vo S)+ ('/ -t /,(Jk(s)'QE)2.22)
where (Qk,Pk)(k = 1,...,F - 1 are the coordinates andmomenta for the local harmonic modes perpendicularto the reaction path, Vo(s) is the potential energy alongthe reaction path, and the functions BL F s ) )k = 1,...,F 1 describe coupling of the vibrational modes to thereaction coordinate (labeled as mode F);he functions
B k , k t ~ ) )k,k = 1, ...,F 1 describe coupling of themodes among themselves. This reaction path Hamil-t ~ n i a n ~ ~nd its applications have been reviewed severaltimes34b~38~39ecently, and the reader should see thesefor more information.
The primary effect which couples the reaction coor-dinate s to the other degrees of freedom is the curvatureof the reac t ion path, which is characterized by thecurvature coupling functions B k p s ) ) .One can showthat the total curvature K ( S ) of the reaction path in theF-dimensional space at distance s along it is
k = l
F-1
4 s = [C Bkp s)211/2 (2.23)
The individual coupling functions &+&) are a measureof how the total curvature of the reaction path projectslocally onto the various modes k orthogonal to it. It isthis curvature of the reaction path which causes thecoupling. Synonymous with the effect of nonsepara-bility of the reaction coordinate on tunneling, therefore,is the effect of reaction path curvature on tunneling.
The simplest approximate expression which takesaccount of reaction path curvature on the tunnelingprobability was first suggested by Marcus and Coltrina
P, E) = e -2W)n e20k E) (2.24)
and also results from other theoretical a p p r o a ~ h e s . ~ ~ *
k = l
F-1
k = l
l o g ,o mol / c m 3
Figure 7. Activation energy for the effective unimolecular rateconstant (see eq 2.19 and 2.21) as a function of density (n/V)(which is proportion al to pressure via p V = nRT). Th e solid curveincludes the effect of tunneling in K E),nd the dashed curveneglects i t .
above the classical threshold for the reaction.Table I lists some energies E l ] nd decay rates (rl /h)
for individual ro-vibrational states of Sostates that havebeen determined27v29~31y using the above Stark shiftmeasurements. The agreement with the values com-puted via the statistical approximation (Figure 3) is ingeneral quite good, demonstrating that at least theorder of magnitude given by the tunneling mechanismis correct. Perhaps even more interesting, however, isthat the experiments appear to demonstrate statespecificity; i.e., the rate is not a smooth, monotonicallyincreasing function only of the total energy. Sta tespecificity will be discussed more fully in section 111.
Finally, to conclude this discussion of the form-aldehyde dissociation within the statistical approxi-mation, i t should be noted that F o r ~ t ~ ~as consideredthe effect of tunneling on the tradi tional thermal de-composition of formaldehyde. Here the effective,pressure-dependent unimolecular rate constant is'J
1 mkUi = QL E p E ) e- E /k T k E ) / [ w+ k E)] (2.19)
where Q is the partition function of reactants
Q = LmdEE)e -E/kT (2.20)
and J is the collision frequency (proportional to pres-sure). k E) s the microcanonical unimolecular rateconstant (eq 2.1), for J = 0. Forst finds that tunnelingcauses the effective activation energy
(2.21)
to fall off forever with decreasing pressure; cf. Figure7. He concludes that the thermal decomposition ex-perimental data are consistent with the photochemicalexperiments discussed above and in particular with thenotion that the decomposition into Hz + CO proceedsby tunneling.
D. Effect of React ion Path Curvature
The treatment of tunneling described in section IIAignores any coupling of reaction coordinate motion toother degrees of freedom in the molecule. Though thismay often be reasonable-more detailed calculationsfor the formaldehyde di ss ~c ia ti on ~~iscussed in section
IIC, for example, suggests that the error introduced
8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 6/9
24 Chemical Reviews, 1987, Vol. 87, No. 1
Here 80 is the vibrationally adiabatic action integral0 -
2
- 4
-6
- 8
x -10-0
9 - 1 2 -
- 14-
- 16-
Miller
I I I I ~-. .-e .
b a -
- -e . . .
* * a .
. e . . .
-
where
0 -
2
-4
- 6
- 8
-10-
E - 1 2 -
- 14
- 16
- 18 -
-20
F-1
I I I I .a -
- * . a -
- a .
.a
0 -.w
e .
-
8.-
0.- -
I I I I
k = l
Le., the factor in eq 2.24 is the zeroth-order tun-neling probability. The action integrals (8,) contain theinfluence of reaction path curvature
(2.26)
and one sees that the factors { ezob ]n eq 2.24 in generalincrease the tunneling probability beyond what it wouldbe if reaction path curvature were ignored.
For the well-studied test case H Hz- 2+ H, thissimple model (eq 2.24-2.26) corrects the tunnelingprobability from being a factor of 50-100 too small if
reaction path curvature is ignored to within a factor of2 of the correct value.34bp40f the curvature correctionof the tunneling probability is smaller than this, thenone has some confidence that this model will be ableto described it at least semiquantitatively. As notedabove, the curvature correction to the tunneling prob-ability given by eq 2.24-2.26 for the formaldehydedissociation is less than a factor of 2.
Other, more sophisticated and accurate m ~ d e l s ~ ~ ? ~ordescribing the effects of reaction path curvature aredescribed in Schatz’ paper12 n this volume on tunnelingin bimolecular reactions. They can also be applied tothe present unimolecular case.
I I I . State-Specif ic Unlmolecular Reactions
As discussed in section IB, he “ultimate” descriptionof a unimolecular reaction, theoretically and experi-mentally, is the determination of the unimolecular rateconstants for individual quantum states of the reactantmolecule, i.e., the determination of the energies andlifetimes of the “metastable eigenstates”. (These aresolutions of the Schrodinger equation with outgoingwave boundary conditions and thus have complex ei-genvalues E l - iFl/2.) Only then can one answer thequestion of whether a particular unimolecular reactionis in t r ins ica l ly s ta t i s t ica l , case a of section IIB, ors ta t e - spec i f i c ,case b of section IIB.
[It is interesting to point out that it is in the tunnelingregion where it should be easiest to determine state-specific unimolecular rates. This follows from thediscussion related to eq 2.15-2.18 of section IIC, whichshowed that it is the tunneling region where the averagewidth f lof the eigenmetastable states is less than theiraverage spacinga l ith a sufficiently narrow laser,therefore, it is straightforward (in principle) to exciteindividual eigenmetastable states since they do notoverlap.]To illustrate state specificity, particularly for a tun-
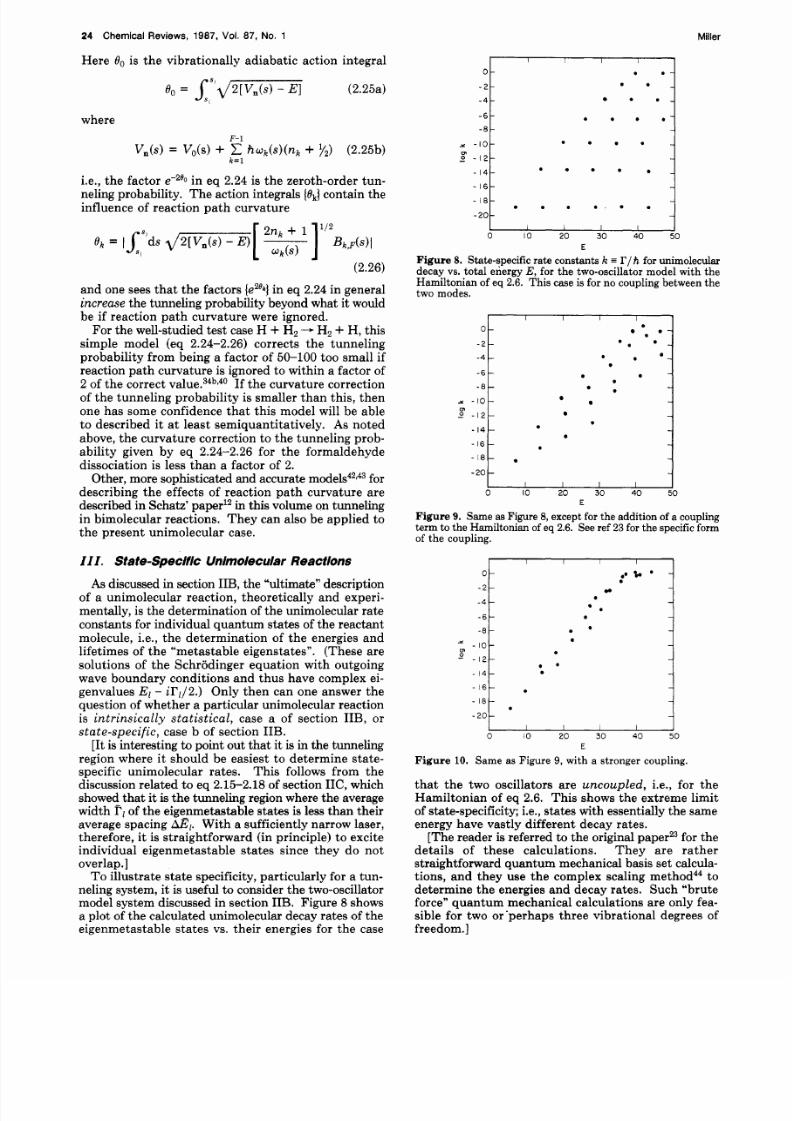
neling system, it is useful to consider the two-oscillatormodel system discussed in section IIB. Figure 8 showsa plot of the calculated unimolecular decay rates of theeigenmetastable states vs. their energies for the case
1 I 1 I I0 I O 20 3 40 50
E
Figure 8. State-specific rate con stants k = I’/h for unimoleculardecay vs. tota l energy E, for the two-oscillator m odel with theHamiltonian of eq 2.6. Th is case is for no coupling between th etwo modes.
- 1 4 1 .208 1 .
I I J
F i g u r e 10. S a m e as Figure 9, with a stronger coupling.
that the two oscillators are u n c o u p k d , i.e., for theHamiltonian of eq 2.6. This shows the extreme limitof state-specificity; i.e., states with essentially the sameenergy have vastly different decay rates.
[The reader is referred to the original paperz3 or thedetail s of these calculations. They are ratherstraightforward quantum mechanical basis set calcula-tions, and they use the complex scaling method44 odetermine the energies and decay rates. Such “bruteforce” quantum mechanical calculations are only fea-sible for two or‘perhaps three vibrational degrees offreedom.]
0 10 20 30 40 5
E

8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 7/9
Tunneling and State Speclficity in Unlmolecular Reactio ns hemical Reviews, 1987, Vol. 87, No. 1 25
1 I7 8 90 100
E k c a l / m o l e )
Figure 11. State-specific unimolecular rate constants for atwo-mode model of the formaldehyde d issociateH2 CO. Seeref 26. The solid squares and circles are states of AI and B1symmetry, respectively, and open squares and circles are A2 andB2 states.
Figure 9 shows how the rate vs. energy plot changesif a nonseparable term is added to the Hamiltonianwhich couples the x and y degrees of freedom, andFigure 10 shows the most statistical-like result that wasobtained when the coupling term and relative and y
frequencies were chosen to produce the most effectivemixing. This latter case is beginning to approach theideal limit of intrinsic statisticallly behavior, when theindividual rates (kl}re a smooth function only of their
ki = ME,) (3.1)
Here k E ) is (by definition) the statistical rate, theaverage rate a t energy E . Even in Figure 10, though,one sees that there remains a significant degree of statespecificity; rates for states of essentially the same energyvary approximately 1 order of magnitude.
Similar calculations have been carried out for atwo-mode model of the formaldehyde decomposition,26the two modes being the reaction coordinate and theout-of-plane bend; cf. the potential surface in the toppart of Figure 2. The out-of-plane bend was chosenbecause it is the mode least strongly coupled to thereaction coordinate (via the reaction path Hamiltonianasdescribed in section IID) and the one thus most likelyto show significant state specificity. Figure 11 showsthe results of this calculation. The overall trend followsthe statistical rate very well, but one sees that there isa significant degree of s tate specificity: rates vary byapproximately 2 orders of magnitude for various stateswith essentially the same total energy. Interestingly,this is roughly the same degree of variation seen in thestate-specific unimolecular decay rates that have beendetermined experimentally; cf. Table I.
How should one interpret the various degrees of statespecificity illustrated by Figures 8-11? One notion thatoccurred to usz3 arly-which actually turns out not tobe very useful in this case-was to see how state spe-cificity in the eigenmetastable states correlates withchaotic dynamicsloblc n the (classical) intramoleculardynamics. The idea was that chaotic dynamics shouldcorrelate with statistical behavior of the rates, whilequasiperiodic dynamics should correlate with s tatespecificity. If this were true, then one should see statespecificity at low energies and then a more-or-less ab-rupt transition energy above which the rates would bemore statistical-like. This behavior was never observed,
energy lEll
IO”, , , I l I , l y
-I-c
c
0
Figure 12. Microcanonical rate constant for the reaction H2C0- 2 CO (J = 0) for the symmetry A’ and A” as a functionof totalenergy relative to the zero-point energy adjusted barrierheight.
even though the classical dynamics of some of thesesystems do show transitions from quasiperiodic dy-namics to chaotic dynamics. (It should be noted,
though, tha t in other kinds of studies& there have beenstrong correlations seen between predissciation ratesand the underlying quasiperiodic-chaotic character ofthe classical mechanics.)
Another way to think about state specificity in theunimolecular rate constants is to suppose that there aresome approximate ly conserved quantum numberswhich are operative. Total angular momentum, forexample, is a rigorously conserved quantum number (infree space, i.e., no electronic or magnetic fields), and aswas done in section I1 one applies the statistical ap-proximation sepa rately for each value of J when cal-culating the average rate constant a t energyE . Thereis thus a family of k vs.E curves, a separate one for each
value ofJ . If
there are other good (or approximatelygood) quantum numbers A by which the energy levelsof the reactant molecule and the transition state canbe labeled, then here too one should apply the statisticalapproximation separately for each value of A i.e., eq 2.1should be modified to read
k, E,J) = [2ahpx E ,J)- l C P E ~ ~ J K * I (3.2a)nJOh
pA(E,J) = C & ( E EnJK) (3.2b)nJOr
where the notation nmx eans that only vibration-al-rotational states of type X are included in the sum.
The calculated results of the two-dimensionalmodelproblems in Figures 8-11 do suggest this behavior.Certainly the separable limit, Figure 8, shows it sincehere the quantum numbers n , and nr are separatelyconserved. But even in the nonseparable cases theserates seem to fall on several separate k vs. E curveswhich could be labeled by a discrete index A.
Another example of conserved quantum numbers isseen in the formaldehyde dissociation for J = 0. Be-cause the reaction path% s planar, the A’/”’ symmetry(even/odd on reflection in the plane) is conservedduring the dissociation dynamics, so the statistical ap-proximation should be applied separately for eachsymmetry.& Figure 12 shows these two rates, and onesees that in the tunneling region this symmetry-inducedstate specificity accounts for about 1order ofmagnitude

8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 8/9
26 Chemical Reviews, 1987, Vol. 87, No.
I O 0 I I
--
Miller
1
d
o-2Oo005 10 0.15 0 2 0
E
Figure 13. State-specific rate constants for unimolecular decayin th e H enon-Heiles potent ia l V x , y )= 1/2x2 1/3x3t ‘/y2.x y 2 ,as a function of energy; see ref 47 for complete details. Solidpoints, squares, and circles denote sta tes of symmetry AI,A,, F dE, respectively. The solid curve is the result of the microcanonicalt ransi t ion sta te theory (eq 2.1).
variation in the rate constants for states with the same
energy.A final example of state specificity is seen in calcu-lations carried out by Waite e t al.47and Taylor et al.48on another two-dimensional model potential, thewell-studied Henon-Heiles potential. Figure 13 showsan example of these results; this potential has CSv ym-metry, and the AI, Az, and E states are indicated sep-arately. One sees a large symmetry-induced effect; Le.,the Az rates are much less than the AI and E rates (forthe same energy). Beyond this, one also sees about anorder of magnitude of state specificity among the Estates themselves (the open circles) and a hint a t somestate specificity among the highest energy A, states.Taylor et al. are able to explain some of this state
specificity, at least qualitatively, by using ideas of Hoseand Taylor49 o classify various states. The basic ideais tha t many of the states correspond to quasiperiodicmotion, i.e., have approximately conserved quantumnumbers and thus do not explore all the energeticallyaccessible space. Some thus correspond to motion thatavoids the transition-state region, while others corre-spond to motion that is localized in its vicinity, andthese states decay slower and faster, respectively, thanthe average.
To summarize, state specificity in unimolecular re-action rates has been seen in a variety of rigorousquantum mechanical calculations for model systemswith two degrees of freedom. In fact, for none of these
model problems has one seen any less than approxi-mately 1 order of magnitude in variation of the uni-molecular rates about their average rate a t a given en-ergy. The formaldehydedissociation is the best studiedsystem experimentally, and though the analysis is notcompletely settled, i t appears tha t states of essentiallythe same energy (and symmetry) have rates tha t varyby 1-2 orders of magnitude. It may turn out-and thisis in fact my belief-that essentially all unimolecularreactions will show a significant degree of state spe-cificity at this most microscopic level of individualquantum states.
To model state-specific unimolecular rate constantsrequires theoretical treatments that go beyond thestatistical approximations described in section 11,and
this is very challenging. Such calculations have so farbeen carried out only for two-dimensional IT delproblems. The reason that statistical approximationshave been and continue to be so useful, of course, is thatmost experiments do not determine the rates for in-dividual quantum states but rather an average overmany of them, and for these average rates statisticalapproximations are quite adequate. In any event theyare adequate for determining the average rate even if
one has available state-specific rates.
I V. Concluding Remarks
Statistical approximations do a very good job of de-scribing the aueruge unimolecular reaction rate for amolecule with energy E (and angular momentum J),even in cases where the reaction proceeds by tunneling.Sometimes i t is necessary to take account of nonse-parability of the reaction coordinaj : from the otherJegrees of freedom of the molecule (*.e., reaction pathcurvature), but there are a variety of relatively simplemodels that describe this reasonably well.
In the tunneling region, where the widths {r,jf in-dividual eigenmetastable states are less than their av-erage spacing A it is meaningful to consider theunimolecular reaction rates k , I’l/ h of these individualquantum states. A t this most fine-grained level, theo-retical calculations on various model problems show asignificant degree of state specificity; i.e., rates fluctuateapproximately1order of magnitude or more about theaverage rate at given energy. (The average rate iswell-approximated by statistical theory.) Experimentalresults on the unimolecular decomposition of form-aldehyde appear to show state specificity; i.e., rates forindividual quantum states of essentially the same en-ergy (and angular momentum) vary 1-2 orders ofmagnitude.
As high resolution laser studies of molecular spec-troscopy and dynamics continue to develop, one expectsto see more experimental examples of s tate specificityin various aspects of reaction dynamics. This willprovide a rigorous challenge for theoretical models andmethods.
Acknowledgments. This work has been supported bythe Director, Office of Energy Research, Office of BasicEnergy Sciences, Chemical Sciences Division of the U.S.Department of Energy, under Contract DE-AC03-76SF00098 and also in part by the National ScienceFoundation Grant CHE84-16345. I wish to thank Drs.H. L. Dai, C. B. Moore, W. Polik, and R. W. Field forhelpful discussions regarding their experimental results
on photodissociation of formaldehyde. Special thanksare due H. L. Dai for making available some of the
values in Table I (ref 31) and W. Polik for providingFigure 5 (ref 30), both prior to publication.
References
1) P. J. Robinson and K. A. Holbrook, “Unimolecular Reactions”,Wiley, New York, 1972.
2) W. Forst, “Theorv of Unimolecular Reactions”, AcademicPress, New York, 3973.
(3) F. F. Crim, Annu. Reu. Phys. Chem., 35, 657 (1984).(4) C. B. Moore and J. C. Weisshaar. Annu. Reu. Phvs. Chem.. 34.. . .
525 (1983).( 5 ) Note. for examd e. th e International CECAM WorkshoD on
Intramolecular’ Vibrational R edistribution and Quan’tumChaos, University of R ochester, October 3-5, 1985.
(6) C. E. Dykstra, Annu. R e v . Phys. Chem., 32, 25 (1981).

8/12/2019 Tunneling in Unimolecular Reactions
http://slidepdf.com/reader/full/tunneling-in-unimolecular-reactions 9/9
Tunneling and State Specificity in Unimolecular Reactions
(7) See, for example, th e pa ers from the Fifth American Con-ference on Theoretical 8hemistry, June 15-20, 1984, n J .Ph s. Chem.,89,2121-2234 (1985).
(8) S. l. a hcum, Z. Smith, E. B. Wilson, Jr., an d R. W. Duerst,J . Am. em. SOC.,106, 2265 (1984).
(9) T. Carrington, Jr., and W. H. Miller, J.Chem. Phys., 84, 4364(1986).
(10) See, for exam ple, (a) A. H. Zewail, Ed., Advances in LaserChemistry , Springer, New York, 1978. (b) D. W. Noid, M. L.Koszykowski, and R. A. Marcus, Annu. Rev. Phys. Chem.,32,267 (1981); c) E. B. Stechel and E. J. Heller, Annu. Reu. Phys.Chem.,35, 563 (1984); d) V. E. Bondybey, Annu. Reu. Phys.Chem.,35,591 (1984).
11) K. G. Kay, J.Chem. Phys.,64,2112 1976);65,3813 1976);68,434 (1978): R. W. Numrich and K. G. Kav. ibid.. 70. 4343
Chemical Revlews, 1987, Vol. 87, No. 1 27
(31) A. Ritter , W. Merry, D. Frye, and H . L. Dai, private commu-
32) W. Forst, J.Phys. Chem., 87,4489 (1983).33) S. K. Gray, W. H. Miller, Y. Yamaguchi, and H. F. Schaefer,
J.Am. Chem.SOC.,03, 1900 (1981).(34) (a) R. T. Skodje, D. G. Truhlar, and B. C. Gar rett, J . Chem.
Phys., 77, 5955 (1982); b) C. J. Cerjan, S.-h. Shi, and W. H.Miller, J. Phys. Chem., 86,2244 (1982).
(35) R. A. Marcus, J. Chem. Phys., 49,2610 (1968).(36) W. H. Miller, N. C. Handy , and J. E. Adams, J.Chem. Phys.,
72,99 (1980).(37) For other papers on reaction path approaches, see (a) G. L.
Hofacker, Z.Naturforsch.,A: Astrophys., Phys. Phys. Chem.,MA, 607 (1963); (b) S. F. Fischer, G. L. Hofacker, and R.Seiler, J. Chem. Phys., 51,3941 (1969); c) S. F. Fischer andM. A. Ratner, J.Chem. Phys.,57,2769 1972); d) P. Russeggerand J. Brickman, ibid., 62,1086 (1976);60, (1977); e) M. V.Basilevsky, Chem. Ph s., 24,81 (1977);67,337 (1982);M. V.Basilevsky and A. G. ghamov, ibid., 60,347 1981); f) K. Fu-kui, S.Kato, and H. Fujimoto, J.Am. Chem.SOC., 7, l 1975);K. Yamashita, T. Yamabe, and K. F ukui, Chem. Phys. Lett .84,123 (1981);K. Fukui, Acc. Chem. Res., 14,363 (1981); 9)K. Ishida, K. M orokuma, and A. Komornicki, J.Chem. Phys.,66,2153 1977); h)A. Nauts an d X . Chapuisat, Chem. Phys.Lett., 85,212 1982);X. Chapuisat, A. Nauts, and G. Durand,Chem. Phys., 56,91 1981); i) J Pancir, Collect. Czech. Chem.Commun.,40, 1112 (1975); 42, 16(1977); j) . A. Natanson,Mol. Phys., 46,481 (1982).
(38) W. H. Miller in Potential Energy Surfaces and DynamicalCalculations, D. G. Truhlar, Ed.; Plenum Press, New York,1981, 265.
(39) W. H. Miller, J. Phys. Chem., 87,3811 (1983).(40) R. A. Marcus and M. E. Coltrin, J.Chem. Phys., 67, 2609
(1977).(41) R. T. Skodje and D. G. Truhlar, J. Chem. Phys., 79, 4882
(1983).(42) B. C. Garrett and D. G. Truhlar, J. Chem. Phys., 79, 4931
(1983).(43) J. M. Bowman, G.-Z. Ju, and K. T. Lee, J. Chem. Phys., 75,
5199 (1981);Chem. Phys. Lett ., 94,363 (1983).(44) See, for example, Znt. J. Quantum Chem., 14,No. 4 (1978), he
entire volume of which is devoted to th e method of complex
nication.
I
(i 979);71; 5352 1979).12) G. C. Schatz, this volume.13) D. G. Truhlar and B. C. Garrett, Annu. Reu. Phys. Chem., 35,
159 (1984).(14) W. Siebrand,T.A. Wildman, and M. Z. Zgierski,J.Am. Chem.
SOC.,106,4083 (1984).5 ) R. A. Marcus and 0. K. Rice, J.Phys. Colloid Chem.,55,894
(1951); . A. Marcus, J.Chem. Phys., 20, 359 (1952); 43,2658(1965).
(16) (a)W . Miller, Acc. Chem. Res., 9,306 (1976); (b) P. Pe-chukas, Annu. Reu. Phys. Chem., 32, 159 (1981); c) D. G.Tru hlar, W. L. Hase, and J. T.Hynes, J.Phys. Chem.,87, 2664(1983).
(17) R. A. Marcus, J. Chem. Phys., 45,2138 (1966).(18) W. H. Miller, J.Am. Chem.SOC.,101, 6810 (1979).(19) E. Wigner, Trans. Faraday SOC.,4,29(1938).(20) J. C. Keck, Adu. Chem. Phys., 13, 85 (1967);Adu. At. Mol.
Phys., 8, 39 (1972).(21) W. H. Wang and R. A. Marcus, J. Chem. Phys., 55, 5625
(1971).22) M. Quack and J. Troe, Ber. Bunsenges. Phys. Chem., 78, 240
(1974).(23) B. A. Waite and W. H. Miller,J.Chem. Phys., 73,3713(1980).(24) J. Troe, J.Phys. Chem., 88,4375 (1984).(25) See, for example, W. M. Gelba rt, K. C. Freed, and S.A. Rice,
J . Chem. Phys., 52,2460 (1970);P. Avouris, W. M. Gelbart,and M. A. El-Sayed, Chem. Rev., 77,793 (1977).
(26) B. A. Waite, S. K. Gray, and W. H. Miller, J.Chem. Phys., 78,259 (1983).
(27) J C. Weisshaar and C. B. Moore, J. Chem. Phys., 72,2875,5415 (1980).
(28)W .'Henke, H. L. Selzle, T. R. Hays, E. W. Schlag, and S.H. Lin, J. Chem. Phys., 76, 1335 (1982).
(29) H. L. Dai, R. W. Field, and J. L. Kinsey, J.Chem. Phys., 82,1606 (1985).
(30) D. R. Guyer, W. F. Polik, and C. B. Moore, J.Chem. Phys. 84,
6519 (1986).
scaling.(45) R. L. Sundberg and E. J. Heller, J . Chem. Phys., 80, 3680
(1984).(46) W. H. Miller, J. Am. Chem. SOC.,105, 216 (1983); . Chem.
Phys., 78,6640 (1983).(47) B. A. Waite and W. H. M iller,J.Chem. Phys.,74,3910 (1981).(48) Y. Y. Bai, G. Hose, C. W. McCurdy, and H. S.Taylor, Chem.
Phys. Lett., 99,342 (1983).
(49) G. Hose and H. S. Taylor, J. Chem. Phys., 76,5356 (1982).