universidad de buenos aires - iib-intech - instituto de ... · aparato de golgi ... central o core...
TRANSCRIPT

UNIVERSIDAD DE BUENOS AIRES
FACULTAD DE CIENCIAS EXACTAS Y NATURALES
ESTUDIOS SOBRE EL PROCESAMIENTO Y TRÁFICO INTRACELULAR DE PROTEÍNAS TIPO
MUCINAS EN Trypanosoma cruzi
Andrea C. Mesías
Tesis presentada para optar a la Licenciatura en Ciencias Biológicas
Director: Dr. Carlos A. Buscaglia
Director Asistente: Dr. Gaspar E. Cánepa
Lugar de Trabajo: Laboratorio de Biología Molecular de Protozoarios
Instituto de Investigaciones Biotecnológicas IIB‐INTECH
UNSAM
Ciudad Autónoma de Buenos Aires, diciembre de 2011

2
INDICE
INDICE DE FIGURAS........................................................................................................................................4 INDICE DE TABLAS .........................................................................................................................................4 DEDICATORIAS .............................................................................................................................................5
RESUMEN ....................................................................................................................................... 6
PALABRAS CLAVE ..........................................................................................................................................7 ABREVIATURAS ............................................................................................................................................8
INTRODUCCIÓN............................................................................................................................... 9
1. ENFERMEDAD DE CHAGAS ..........................................................................................................................9 2. TRYPANOSOMA CRUZI..............................................................................................................................10
2.1. Ciclo de vida ...............................................................................................................................10 2.2. Taxonomía..................................................................................................................................11
3. VÍA SECRETORIA EN TRIPANOSOMÁTIDOS ....................................................................................................13 3.1. Entrada de moléculas a la vía secretoria ..................................................................................13 3.2. Retículo endoplásmico ...............................................................................................................14 3.3. Aparato de Golgi ........................................................................................................................16 3.4. Bolsillo flagelar ..........................................................................................................................17 3.5. Reservosomas ............................................................................................................................17 3.6. Vías de secreción alternativas ...................................................................................................18
4. CUBIERTA DE SUPERFICIE (SURFACE COAT) DE T. CRUZI ..................................................................................19 5. MUCINAS EN T. CRUZI .............................................................................................................................20
5.1. Caracterización bioquímica .......................................................................................................20 5.2. Caracterización génica...............................................................................................................22 5.3. Caracterización funcional ..........................................................................................................23
5.3.1. Adhesión ............................................................................................................................................. 24 5.3.2. Protección ........................................................................................................................................... 24 5.3.3. Inmunomodulación............................................................................................................................. 24 5.3.4. Señalización ........................................................................................................................................ 25
ANTECEDENTES Y OBJETIVOS ......................................................................................................... 26
RESULTADOS ................................................................................................................................. 27
PARTE 1: GENERACIÓN DE REACTIVOS ..........................................................................................................27 1.1. Generación de clones de proteínas tipo mucina .......................................................................27
1.1.1. Generación de clones TcSMUG........................................................................................................... 29 1.1.2. Generación de clones TcMUC ............................................................................................................. 32
1.2. Obtención y caracterización de sueros policlonales.................................................................35 1.2.1. Obtención del suero anti‐TcSMUG S .................................................................................................. 35 1.2.2. Obtención del suero anti‐TcMUC ....................................................................................................... 37
PARTE 2: ANÁLISIS DE MOLÉCULAS REPORTERAS ............................................................................................39 2.1. TcMUC ........................................................................................................................................39
2.1.1. Adición de ancla de GPI y localización subcelular.............................................................................. 39 2.1.2. Validación del carácter mucínico........................................................................................................ 42
2.2. TcMUC::GFP y TcSMUG::GFP .....................................................................................................44 2.3. TcMUC∆SP∆GPI::GFP y TcSMUG∆SP∆GPI::GFP ........................................................................45 2.4. TcSMUG∆SP::GFP y TcMUC∆SP::GFP ........................................................................................47 2.5. TcMUC∆GPI::GFP y TcSMUG∆GPI::GFP .....................................................................................49
DISCUSIÓN Y PERSPECTIVAS........................................................................................................... 57

3
1. EL SP ES FUNDAMENTAL PARA EL INGRESO DE LAS MUCINAS A LA VÍA SECRETORIA..............................................57 2. EL PROCESAMIENTO DE LAS SEÑALES DE TRÁFICO INTRACELULAR ES INDEPENDIENTE DEL TIPO DE MUCINA EXPRESADO
...............................................................................................................................................................58 3. LAS PROTEÍNAS TIPO MUCINAS SIN GPI SE ACUMULAN EN VÍA SECRETORIA .......................................................58 4. LAS MUCINAS SIN GPI QUE LOGRAN ESCAPAR DEL MECANISMO DE RETENCIÓN EN LA VÍA SECRETORIA SON
PROCESADAS Y SECRETADAS COMO MOLÉCULAS MADURAS ................................................................................61 5. LA O‐GLICOSILACIÓN DE MUCINAS ESTÁ MODULADA POR DETERMINANTES EN CIS (SECUENCIAS/ESTRUCTURAS DE LAS APO‐MUCINAS) Y EN TRANS (GLICOSIL TRANSFERASAS) ......................................................................................62 6. PERSPECTIVAS ........................................................................................................................................63
MATERIALES Y MÉTODOS .............................................................................................................. 64
SOLUCIONES GENERALES ..............................................................................................................................64 MANIPULACIÓN DE BACTERIAS......................................................................................................................64 TÉCNICAS MOLECULARES..............................................................................................................................66 MANIPULACIÓN DE PARÁSITOS .....................................................................................................................68 TÉCNICAS ANALÍTICAS DE PROTEÍNAS E INMUNOLÓGICAS ...................................................................................72
ANEXO A: ESTRATEGIAS DE CLONADO ............................................................................................ 75
ANEXO B: OLIGONUCLEÓTIDOS Y ANTICUERPOS ............................................................................. 77
ANEXO C: SECUENCIAS NUCLEOTÍDICAS Y AMINOACÍDICAS DE LOS CLONES DE TRABAJO ................. 79
AGRADECIMIENTOS....................................................................................................................... 81
REFERENCIAS ................................................................................................................................ 83

4
Indice de figuras
FIGURA 1: ESQUEMA DEL CICLO DE VIDA DE TRYPANOSOMA CRUZI. .........................................................................11 FIGURA 2: RELACIONES FILOGENÉTICAS DENTRO DEL PHYLUM EUGLENOZOA. ............................................................12 FIGURA 3: ORGANIZACIÓN ESTRUCTURAL INTERNA DE T. CRUZI. .............................................................................14 FIGURA 4: SECRECIÓN DE MICROVESÍCULAS EN TRITRYPS .......................................................................................19 FIGURA 5: SUPERFICIE CELULAR DE TRITRYPS .......................................................................................................20 FIGURA 6: ESTRUCTURA DE PRODUCTOS TCSMUG. .............................................................................................23 FIGURA 7: DIAGRAMA DE LOS PRODUCTOS DEDUCIDOS A PARTIR DE LOS CLONES GENERADOS. .....................................28 FIGURA 8: ANÁLISIS BIOINFORMÁTICO PARA LA SECUENCIA TCSMUG S SELECCIONADA. ............................................30 FIGURA 9: PÉPTIDOS IDENTIFICADOS A PARTIR DE GP35/50 DE EPIMASTIGOTES. ......................................................31 FIGURA 10: MODELO DE PROCESAMIENTO PARA LA PROTEÍNA TCSMUG S. .............................................................32 FIGURA 11: COMPOSICIÓN AMINOACÍDICA RELATIVA Y ALINEAMIENTO DE LOS SP DE LOS PRODUCTOS TCSMUG S Y
TCMUC II USADOS EN ESTE TRABAJO. ..............................................................................................33 FIGURA 12: ANÁLISIS BIOINFORMÁTICO Y MODELO DE PROCESAMIENTO PARA LA PROTEÍNA TCMUC II. .......................34 FIGURA 13: CARACTERIZACIÓN DE LOS SUEROS ANTI‐TCSMUG..............................................................................36 FIGURA 14: ALINEAMIENTO DE PRODUCTOS TCSMUG S PARA DIFERENTES CEPAS.....................................................37 FIGURA 15: CARACTERIZACIÓN DE LOS SUEROS ANTI‐TCMUC II. ............................................................................38 FIGURA 16: ANÁLISIS DEL ANCLA DE GPI DEL PRODUCTO TCMUC. .........................................................................40 FIGURA 17: ENSAYO DE IFI SOBRE LA POBLACIÓN DE EPIMASTIGOTES TCMUC. ........................................................41 FIGURA 18: ENSAYO DE CITOMETRÍA DE FLUJO PARA LA POBLACIÓN TCMUC............................................................42 FIGURA 19: EXTRACCIÓN DE MUCINAS EN PARÁSITOS TRANSFECTADOS CON TCMUC. ................................................43 FIGURA 20: MORFOLOGÍAS OBSERVADAS EN LA POBLACIÓN TCSMUG::GFP. ..........................................................45 FIGURA 21: LOCALIZACIÓN SUB‐CELULAR DE LOS REPORTEROS TCSMUGΔSPΔGPI::GFP Y TCMUCΔSPΔGPI::GFP. ...46 FIGURA 22: ENSAYO DE MICROSCOPÍA DE FLUORESENCIA E IFI SOBRE LOS CLONES TCSMUG∆SP::GFP........................47 FIGURA 23: CARACTERIZACIÓN BIOQUÍMICA DEL REPORTERO TCSMUG∆SP::GFP. ..................................................48 FIGURA 24: RT‐PCR DE LA POBLACIÓN TCMUC∆SP::GFP...................................................................................49 FIGURA 25: ENSAYO DE IFI SOBRE LAS POBLACIONES TCSMUG∆GPI::GFP Y TCMUC∆GPI::GFP. ............................50 FIGURA 26: ENSAYO DE COLOCALIZACIÓN DE TCSMUG∆GPI::GFP Y TCMUC∆GPI::GFP CON MARCADORES DE
DISTINTOS COMPARTIMIENTOS DE LA VÍA SECRETORIA. ........................................................................51 FIGURA 27: CITOMETRÍA DE FLUJO PARA TCSMUG∆GPI::GFP Y TCSMUG∆SP::GFP. ...........................................52 FIGURA 28: ANÁLISIS BIOQUÍMICOS DE TCSMUG∆GPI::GFP Y TCMUC∆GPI::GFP...............................................54 FIGURA 29: ANÁLISIS BIOQUÍMICO DE LAS FORMAS INTRA‐ Y EXTRA‐CELULARES DE TCSMUG∆GPI::GFP Y
TCMUC∆GPI::GFP. ....................................................................................................................55 FIGURA 30: ESQUEMA DEL PROTOCOLO DE FRACCIONAMIENTO SUBCELULAR CON NP‐40. ..........................................69 FIGURA 31: ESQUEMA DEL PROTOCOLO DE PURIFICACIÓN DE MUCINAS MEDIANTE PARTICIÓN EN SOLVENTES ORGÁNICOS.70 FIGURA 32: ESQUEMA DEL PROTOCOLO DE EXTRACCIÓN DE GPI‐PROTEÍNAS EN TRITON X‐114. ................................71 FIGURA 33: DIAGRAMA DE LAS ESTRATEGIAS DE CLONADO UTILIZADAS PARA EL DESARROLLO DE LOS CLONES TCSMUG...75 FIGURA 34: DIAGRAMA DE LAS ESTRATEGIAS DE CLONADO UTILIZADAS PARA EL DESARROLLO DE LOS CLONES TCMUC. ....76
Indice de tablas
TABLA 1: DESCRIPCIÓN DE LAS FAMILIAS GÉNICAS Y PROTEÍNAS TIPO MUCINAS EN T.CRUZI..........................................22 TABLA 2: ROL DEL MOTIVO GPI SOBRE EL TRÁFICO Y PROCESAMIENTO DE PROTEÍNAS EN DIFERENTES SISTEMAS. .............61 TABLA 3: OLIGONUCLEÓTIDOS UTILIZADOS EN EL TRABAJO. ....................................................................................77 TABLA 4: ANTICUERPOS UTILIZADOS EN EL TRABAJO..............................................................................................78

5
Dedicatorias
A Carlos y a Gaspar miles de gracias por el espacio, la ayuda y la paciencia de siempre sobre todo, que ahora se puede visualizar reflejada en este trabajo. A Tito, Tati, el Negro, Facu y el Rulo, mi familia desquiciada e inamovible. A las Pantristes: Be, Da y Solo yo, no tengo palabras para ustedes. A Jancito, Gabi, Pablito, el pastel, Yiyo, Pou, Caro, Mecha, Juan, Edu, Fefo, Tincho, Willy, todos hermanos y primos respectivamente. A Calvin donde sea que esté. A Brukita, Chele, Caro, Ivito por seguir estando. A los más lindos de la FCEN: Lau, Memito, el Pollo, el Pocho, Pable, Mile, Santo, Mati, el mejor de los compañeros de mesada. Bueno, en realidad está muy peleado. A Vero mi hermana mayor, gracias y perdoname. A lxs compañerxs y AMIGXS del Taller, a lxs compas del Bajo, gracias por el aguante! A las Pepas y a la Furia, todas volantes de proyección. Dejenmé también agradecerle a Racing por tantas alegrías (sí), traté de evitarlo pero no pude. A mi familia que me ayudó en esta difícil carrera de Bioquímica, gracias. A mi compañero Martín, muy especialmente. POR ÚLTIMO Y FUNDAMENTAL en todo esto, al 317 que forma así: Ceci, Fabi, Eze, Peter, Fer, Marie, Toti, Nacho y Gasman de nuevo, a quien las gracias exceden a este trabajo, lejos.

6
RESUMEN
El protozoario Trypanosoma cruzi es el agente etiológico de la enfermedad de
Chagas, que afecta a unos 18 millones de personas en Latino América y para la cual no
existe aún una vacuna o tratamiento farmacológico adecuado. Este parásito presenta
un complejo ciclo de vida que involucra un insecto vector y un hospedador mamífero,
pudiéndose distinguir distintos estadios morfológicos dentro de cada uno de ellos. La
transición entre estos estadios incluye, entre otros fenómenos, la remodelación de su
cubierta antigénica. Unos de los componentes principales de esta cubierta son las
mucinas, glicoproteínas de alto contenido de residuos de prolina, treonina y serina, las
cuales son modificados in vivo por la adición de O‐oligosacáridos de composición y
estructura variable. Diferentes líneas experimentales ponen de manifiesto la
importancia de las mucinas en aspectos clave de la biología de T. cruzi, incluyendo la
adhesión e invasión de distintos tipos celulares, la evasión y modulación del sistema
inmunológico y la protección física durante la fase replicativa dentro del insecto
vector.
Estudios genómicos permitieron identificar más de 1.000 genes que codifican
para proteínas tipo mucina en T. cruzi, los cuales pueden agruparse en dos familias
llamadas TcSMUG y TcMUC en base a características estructurales. Interesantemente,
estas familias muestran un perfil de expresión contrapuesto: mientras los productos
TcSMUG constituyen el esqueleto peptídico de las mucinas de los estadios de insecto
(epimastigotes y tripomastigotes metacíclicos), la expresión de los productos TcMUC
está restringida a los estadios del hospedador mamífero (amastigotes y tripomastotes
sanguíneos). Todas estas proteínas presentan una arquitectura común compuesta por
un péptido señal (SP) de unos 20‐30 residuos en el extremo N‐terminal, un dominio
central o core de largo y secuencia variable y un péptido hidrofóbico de 25‐30 residuos
en el extremo C‐terminal que contiene la señal para adición de glicosilfosfatidilinositol
(GPI).
En este trabajo se realizó un análisis del rol de cada uno de estos dominios en el
tráfico intracelular y procesamiento de las mucinas de T. cruzi. Para ello, utilizamos
una estrategia de expresión y seguimiento in vivo de moléculas delecionadas,
generadas a partir de miembros representativos de las familias TcSMUG y TcMUC, las
que fueron etiquetadas y transfectadas en epimastigotes de T. cruzi. La localización
subcelular y el procesamiento de las proteínas reporteras se determinó utilizando
distintas técnicas bioquímicas y de microscopía. Las conclusiones más importantes de
nuestro trabajo son: i) El SP es fundamental para el ingreso de los reporteros a la vía
secretoria; ii) la señal de anclaje por GPI no puede re‐direccionar moléculas a la vía
secretoria per se (en ausencia del SP); iii) moléculas carecientes de GPI se acumulan

7
como intermediarios inmaduros en el retículo endoplásmico (RE); iv) las moléculas sin
GPI que logran escapar a este mecanismo de control, son completamente procesadas y
secretadas al medio extracelular como mucinas maduras; v) el procesamiento de las
señales de tráfico intracelular es independiente del tipo de gen expresado; y vi) los
productos TcMUC son reconocidos como mucinas por el epimastigote aunque sufren
una O‐glicosilación aberrante. Todos estos resultados son discutidos en el marco del
conocimiento actual en el área.
Palabras clave
Trypanosoma cruzi
Mucinas
Tráfico intracelular
Secreción

8
Abreviaturas
• ADN, ácido desoxiribonucleico • ADNc, ADN copia • ADNk, ADN del kinetoplasto • ARN, ácido ribonucleico • ARNm, ARN mensajero • AS, ácido siálico • BiP, Immunoglobulin heavy chain binding protein • CDS, secuencia codificante • dNTPs, mezcla de dATP, dTTP, dGTP y dCTP • Gal, galactosa • Galf, galactofuranosa • GalNAc, N‐acetil Galactosamina • Galp, galactopiranosa • GFP, proteína verde fluorescente • GIPL, glicoinositol fosfolípido • Glc, glucosa • GlcNAc, N‐acetil Glucosamina • GP35/50, mucinas de 35/50 kDa presentes en los estadios de insecto de T. cruzi • GP63, glicoproteína de 63 kDa • GPI, glicosilfosfatidilinositol • GRASP, Golgi reassembly stacking protein • GST, glutatión‐S‐transferasa • HRP, horseraddish peroxidase • IFI, inmunofluorescencia indirecta • Ig, inmunoglobulina • IR, región intergénica • kDa, kilodaltons • Kpb, kilopares de bases • mAb, anticuerpo monoclonal • MASP, mucin‐associated surface protein • MBP, maltosa binding protein • MPT, modificación post‐traduccional • PABP, Poly(A)‐binding protein • PAGE, electroforesis en geles de poliacrilamida • Pb, pares de bases • PCR, reacción en cadena de la polimerasa • PI‐PLC, phosphatidylinositol‐specific phospholipase C • RE, retículo endoplasmático • REt, RE de transición • RT‐PCR, transcripción reversa acoplada a PCR • SP, péptido señal • SRP, Signal Recognition Particle • TcMUC, familia de genes de mucinas expresadas en los estadios de mamífero de T. cruzi • TcSMUG, familia de genes de mucinas expresadas en los estadios de insecto de T. cruzi • tGPI‐mucinas, mucinas de 60/200 kDa presentes en los estadios de mamífero de T. cruzi • Tritryps, denominación operacional que agrupa a T. cruzi, T. brucei y L. major • TS, trans‐sialidasa • VSG, Variant surface glycoprotein

9
INTRODUCCIÓN
1. Enfermedad de Chagas
Hace 102 años en Minas Gerais, el investigador brasileño Carlos Chagas realizaba
la primera descripción de la Tripanosomiasis Americana, hoy más conocida como
enfermedad de Chagas (Chagas C, 1909). Se trata de una enfermedad parasitaria
producida por la infección con el protozoario hemoflagelado Trypanosoma cruzi, que
afecta a unos 10 millones de personas principalmente en Latino América y para la que
se define una población de riesgo en áreas endémicas de más de 25 millones de
personas (Barrett MP, 2003; Lannes‐Vieira J, 1989). Se calcula una incidencia de 40.000
casos/año para la transmisión vectorial y de 14.000 casos/año para la transmisión
congénita. La presencia de más de 130 especies de insectos hematófagos capaces de
transmitir la enfermedad y de diferentes especies de mamíferos silvestres y
domésticos que pueden actuar como reservorios del parásito complica aún más el
cuadro epidemiológico.
Se pueden distinguir 2 fases en la enfermedad de Chagas: aguda y crónica. La
fase aguda, generalmente asintomática, se caracteriza por una alta parasitemia, y
puede durar entre 6 y 8 semanas. En algunos casos, se verifica una inflamación en el
sitio de infección y/o la aparición de edemas conocidos como chagomas,
linfoadenopatías y fiebre. Durante la fase crónica se hace difícil la determinación
directa de parásitos en sangre, recurriéndose a técnicas serológicas y moleculares para
el diagnóstico. Cerca del 40% de los pacientes crónicos desarrollan formas
sintomáticas 20‐30 años post‐infección. Un 30% de ellos desarrolla patologías
cardíacas, como cardiomiopatía dilatada asociada con miocarditis, fibrosis y disfunción
cardíaca (Chiale P, 1989). Cerca de 10% de los individuos infectados desarrollan
patologías en la inervación del tracto gastrointestinal que puede resultar en
megacolon y/o megaesófago (Koberle F, 1968). En algunos casos, ambos tipos de
manifestación clínica se encuentran presentes en el mismo individuo, constituyendo
una forma crónica mixta. La prevalencia de las diferentes formas clínicas de la
enfermedad varía dependiendo de la región geográfica y estaría relacionada con las
diferencias inmunogenéticas de las poblaciones humanas y, sobre todo, con la
variabilidad genética entre las cepas circulantes del parásito (Ver INTRODUCCION 2.2.).
Además de la distribución geográfica de los vectores potenciales (Stevens L,
2011), existen otros factores que contribuyen a explicar la dimensión de la
enfermedad. Entre ellos, el deterioro social, económico y ambiental, que se traduce en
un aumento de los riesgos sanitarios de las poblaciones rurales, de mayor
vulnerabilidad frente a enfermedades emergentes (Boischio A, 2009; Hotez PJ, 2006).
La acción antrópica sobre el medio ambiente también contribuye de forma significativa
para la dispersión de la enfermedad. El correcto manejo de los recursos, la vigilancia

10
epidemiológica sostenida en el tiempo, la educación, un abordaje que contemple
políticas habitacionales y acceso a una mejor calidad de vida por parte de las
comunidades campesinas, podría representar una eficaz forma de control de la
transmisión vectorial (Boischio A, 2009). Resulta también importante la participación
de todos los actores involucrados ‐ el sector público, privado, la comunidad‐ en el
desarrollo e implementación de políticas exitosas contra la enfermedad de Chagas.
2. Trypanosoma cruzi
2.1. Ciclo de vida
T. cruzi es un parásito digenético, en tanto que su ciclo de vida involucra a un
vector invertebrado y un hospedador mamífero (Figura 1). La infección en el insecto
vector comienza cuando éste ingiere la forma tripomastigote del parásito junto a la
sangre de un humano o animal infectado. El parásito ingresa al tracto digestivo donde
se diferencia al estadio epimastigote que se multiplica repetidas veces por fisión
binaria. En la porción final del intestino del insecto, cierta proporción de epimastigotes
se diferencia hacia la forma de tripomastigote metacíclico en un proceso conocido
como metaciclogénesis. Esta forma invasiva y no replicativa es depositada junto a las
heces cuando el insecto se alimenta, ingresando luego al organismo vertebrado a
través de las mucosas o heridas en la piel. El tripomastigote metacíclico es capaz de
invadir diferentes tipos celulares, donde permanece contenido en una vacuola
parasitófora durante algunas horas, para luego escapar al citoplasma celular. Allí se
diferencia al estadio de amastigote, replicativo, y posteriormente a tripomastigote.
Esta forma es liberada al producirse la lisis de la célula y llega a la sangre o fluidos
intersticiales desde donde puede invadir otras células o ser ingerido por el vector para
cerrar el ciclo (Tyler KM, 2001). Existe también un ciclo alternativo establecido por las
formas amastigotes provenientes de la ruptura prematura de la célula hospedadora o
por diferenciación extracelular (Tomlinson S, 1995) en células macrofágicas (Barrett
MP, 2003).

11
Figura 1: Esquema del ciclo de vida de Trypanosoma cruzi.
2.2. Taxonomía
T. cruzi, cuyo nombre hace referencia al médico brasileño Oswaldo Cruz (1871‐
1917) por dedicación de Carlos Chagas (Chagas C, 1909), se ubica dentro del reino
parafilético de los Protistas. Su ubicación sistemática (Levine N, 1980), detallada a
continuación, lo emparenta con otros patógenos humanos (Figura 2), incluyendo a
Trypanosoma brucei (agente etiológico de la enfermedad del sueño en África) y
Leishmania major (agente etiológico de la Leishmaniasis a lo largo de los trópicos), con
los que conforma el grupo de los Tritryps (El‐Sayed NM, 2005b).
Reino: Protista.
Phylum: Euglenozoa.
Sub‐phylum: Mastigophora.
Clase: Zoomastigophora.
Orden: Kinetoplastida.
Suborden: Trypanosomatina.
Familia: Trypanosomatidae. Incluye a los géneros Trypanosoma y Leishmania.
Género: Trypanosoma. Se divide en dos grandes secciones según el sitio de producción
de tripomastigotes metacíclicos en el insecto vector (Hoare CA, 1964). Comprende,
además de T. cruzi y T. brucei, a otras especies de patógenos que afectan a animales de

12
ganado y que ocasionan grandes pérdidas económicas, como T. vivax y T. congolense
(Osório AL, 2008; Van den Bossche P, 2011).
Sección: Estercoraria. Incluye tripanosomas cuyo ciclo de desarrollo en el vector
invertebrado se completa en la región del tubo digestivo del mismo. Se transmiten a
través de las heces del insecto.
Especie: Trypanosoma cruzi.
La especie T. cruzi se encuentra representada por un conjunto de poblaciones
discretas con prevalencia diferencial en distintas zonas endémicas y tipos de
hospedadores. Estas poblaciones, que pueden agruparse en 6 linajes evolutivos o
discrete typing units (llamados TcI‐TcVI, (Zingales B, 2009)) presentan una gran
heterogeneidad en cuanto a su comportamiento biológico: virulencia y/o tropismo
tisular en animales experimentales y seres humanos, sensibilidad a drogas, velocidad
de duplicación, etc. (Buscaglia CA, 2003). Estas diferencias fenotípicas guardan una
correlación con la gran variabilidad genética intra‐específica (e incluso intra‐linaje
evolutivo), la cual se atribuye al hecho de que T. cruzi se multiplica mayormente por
división binaria, con raros eventos de intercambio genético (Gaunt MW, 2003; Lewis
MD, 2011). Esta propagación “clonal” favorece la sub‐especiación, ya que el genoma
de cada cepa evoluciona de manera independiente.
Figura 2: Relaciones filogenéticas dentro del phylum Euglenozoa. Dendograma obtenido a partir de secuencias de EF‐1 α (factor de elongación de la traducción), mediante un algoritmo de máxima probabilidad. Se remarca la relación filogenética estrecha entre T. cruzi, T. brucei y L. major. Adaptado de (Gile GH, 2009).

13
3. Vía secretoria en tripanosomátidos
La investigación en protozoos del orden Kinetoplastida llevó a la identificación de
una gran cantidad de características biológicas distintivas, probablemente más que
para cualquier otro grupo comparable de protistas (Donelson JE, 1999; Vickerman K,
1994). Debido a su divergencia evolutiva temprana, algunas de estas características
pudieron haber sido heredadas directamente a partir de los primeros organismos
eucariotas, mientras que muchas otras pueden representar adaptaciones más
recientes hacia el parasitismo. En las siguientes secciones se detallan algunas de las
particularidades relacionadas a las estructuras y procesos biológicos que conforman la
vía secretoria de T. cruzi y Tritryps en general, y que son relevantes para la mejor
interpretación de nuestro trabajo.
3.1. Entrada de moléculas a la vía secretoria
Pueden diferenciarse dos formas alternativas de ingreso de moléculas a la vía
secretoria: dependiente e independiente de la ribonucleoproteína citoplasmática
Signal Recognition Particle (SRP) (Tuteja R, 2007). En organismos eucariotas, los SP
presentes en la mayoría de las proteínas nacientes son reconocidos por SRP, lo que
produce el arresto de la elongación. Luego de la interacción con su receptor en la
membrana del RE, y en un proceso dependiente de la hidrólisis de GTP (Pool MR,
2005), el polipéptido es translocado en forma co‐traduccional al lumen del RE. Los
distintos componentes del SRP pudieron identificarse y caracterizarse en eucariotas
superiores (Walter P, 1981) y levaduras (Hann BC, 1991). Los tripanosomátidos
presentan ciertas diferencias estructurales a nivel de SRP, tales como la co‐existencia
de dos unidades de ARN (7SL RNA y un análogo de ARNt) dentro del complejo (Liu L,
2003). En la vía independiente de SRP, por otro lado, el polipéptido es reconocido por
chaperonas citoplasmáticas capaces de direccionarlo hacia la membrana del RE y éste
es translocado post‐traduccionalmente al lumen del mismo.
Estudios realizados en T. brucei, demostraron la presencia y utilización
simultánea de ambas vías, dependiente e independiente de SRP. Interesantemente, la
interrupción de la vía dependiente de SRP mediante ARN de interferencia
comprometió la expresión en superficie de los reporteros conteniendo segmentos
trans‐membrana pero no de aquellos anclados a la superficie celular por un motivo GPI
(Goldshmidt H, 2008). Este hecho podría correlacionarse con una menor
hidrofobicidad de los SP encontrados en proteínas ancladas por GPI, y sugiere la no
redundancia de las 2 vías en tripanosomátidos.

14
Figura 3: Organización estructural interna de T. cruzi. A; Diagrama esquemático de la distribución de organelas del parásito, adaptado de (Docampo R, 1974). B; Microscopía electrónica mostrando el kinetoplasto (k), el flagelo (f) y el bolsillo flagelar, adaptado de (Rocha GM, 2006). C; Micrografía electrónica de reservosomas (R) observados en la región posterior de epimastigotes, adaptado de (Pereira MG, 2011). D; Imagen de microscopia electrónica de transmisión mostrando una región de la membrana plasmática (PM) asociada a microtúbulos subpeliculares. Adaptado de (Souto‐Padron T, 1984). 3.2. Retículo endoplásmico
El RE en tripanosomátidos se extiende a lo largo de gran parte de la célula,
estableciendo contacto con la membrana plasmática en distintos puntos (Figura 3A).
Diversos estudios revelaron la presencia de subdominios funcionalmente
especializados en el RE de estos organismos, incluyendo un dominio vinculado a la
envoltura nuclear, un dominio asociado a los microtúbulos subpeliculares (Figura 3D) y
B
C
D
A

15
un dominio de transición (REt), desde donde las proteínas y lípidos serían
transportados al Golgi siguiendo distintas vías/cinéticas de acuerdo a sus propiedades
físico‐químicas (Sevova ES, 2009; Silverman JS, 2011). Han sido descriptos varios
marcadores de RE en Tritryps, incluyendo chaperonas como la Immunoglobulin heavy
chain binding protein (BiP) o calreticulina, y enzimas asociadas al ensamblado de
oligosacáridos y precursores de GPI (Labriola CA, 2011).
En el RE tienen lugar 2 procesos clave en la maduración de las proteínas tipo
mucina: el ensamblado y procesamiento de N‐glicanos y la adición del ancla de GPI.
Trabajos realizados fundamentalmente por Parodi y colaboradores establecieron que
los pasos en la biosíntesis de N‐glicanos y los requerimientos estructurales en las
proteínas aceptoras son muy similares a los que ocurren en otros eucariotas (Parodi
AJ, 1993).
Además de la N‐glicosilación, en el RE tiene lugar la adición de anclas de GPI. Al
igual que en las demás ramas eucariotas, las anclas de GPI son ensambladas a partir
de precursores simples y transferidas en bloque por transamidasas a proteínas
conteniendo señales específicas (McConville MJ, 1993). En mamíferos, la adición de
GPI requiere de una secuencia C‐terminal de aproximadamente 17‐31 residuos, que
contiene al menos dos elementos críticos: i) un dominio hidrofóbico y ii) un sitio de
clivaje y conjugación del ancla (residuo ω), ubicado hacia el N‐terminal de esta
secuencia. El residuo ω es en general una serina, aunque alanina, aspartato,
asparagina y glicina pueden también ser conjugados (Ferguson MA, 1999). A pesar de
mostrar requerimientos estructurales muy similares, la decodificación cruzada de
señales de adición de GPI usando sistemas de expresión heteróloga (reporteros
humanos en tripanosomátidos o al revés) es muy ineficiente (Ramirez MI, 1999). De
hecho, las diferencias intrínsecas en esta ruta metabólica la erigen como un de los
blancos estratégicos validados para el control de estos parásitos (Ferguson MA, 2000).
En tripanosomátidos, las señales de GPI más estudiadas han sido las de la familia
de las proteínas variables de superficie (VSGs) de T. brucei. Si bien esta señal se
encuentra extremadamente conservada entre las distintas VSG, estudios mutacionales
determinaron que serían las propiedades estructurales y no el grado de conservación
que guardan estas secuencias los determinantes esenciales para su correcto
procesamiento (Böhme U, 2002).
El anclaje mediante GPI es una alternativa post‐traduccional para la expresión de
proteínas sobre una bicapa lipídica que presenta muchas implicancias a nivel biológico.
Por un lado, la estructura propia del GPI facilita el empaquetamiento de alta densidad
de glicoconjugados sobre la superficie celular, con una perturbación mínima de la
membrana plasmática (Böhme U, 2002). Por otra parte, las proteínas ancladas por GPI
pueden ser liberadas de forma controlada al medio extracelular a través de la actividad
de fosfolipasas de superficie. En tripanosomátidos, este mecanismo además de estar

16
involucrado en la diferenciación celular (Martins V de P, 2010), permitiría la
solubilización de factores de virulencia presentes en la cubierta antigénica, los que
podrían ejercer su efecto a distancia (Mucci et al., 2002; Rifkin MR, 1990). Por último,
la afinidad de los GPIs por dominios ricos en colesterol o esfingolípidos, conlleva la
posibilidad de generar balsas lipídicas discretas (lipid rafts) que podrían involucrarse en
fenómenos de interacción celular y transducción de señales (Paulick MG, 2008).
Existen evidencias que sugieren que la adición de GPI, además de sus roles
estructurales, podría regular el tráfico de proteínas en la vía endocítica‐secretoria. Un
ejemplo de esto es el rol que juegan estas estructuras en el direccionamiento de
moléculas desde el complejo de Golgi hacia la membrana apical en células polarizadas
(Lisanti M, 1990).
3.3. Aparato de Golgi
El aparato de Golgi en tripanosomátidos consiste de un apilamiento de 3 a 10
cisternas y una red trans‐Golgi (TGN) en la porción anterior del parásito, cercano al
kinetoplasto y el bolsillo flagelar (Figura 3A) (McConville MJ, 2002). Como
característica distintiva respecto de lo observado para eucariotas superiores, el
complejo de Golgi de Tritryps no sufre un break down durante el ciclo celular sino que
se fisiona al igual que el cuerpo basal y el kinetoplasto. Existen pocas proteínas que
puedan ser utilizadas como marcadoras de Golgi en tripanosomátidos (Morgado‐Díaz
JA, 2001). Una de ellas es la Golgi reassembly stacking protein (GRASP), que en T.
brucei se localiza a nivel de la matriz (Yelinek JT, 2009).
En el complejo de Golgi tiene lugar la O‐glicosilación, una de las características
más distintivas de las proteínas tipo mucina (Varki A, 1998). Posiblemente el rasgo más
sobresaliente de la O‐glicosilación en T. cruzi, y que por lo tanto constituye un blanco
promisorio para el desarrollo de drogas anti‐parasitarias, sea la conjugación de
aminoácidos S y T a un residuo α‐GlcNAc en vez de α‐GalNAc como en la mayoría de
los sistemas conocidos (McConville MJ, 2002). Una de las causas de esta diferencia
puede residir en la actividad de la enzima UDP‐Glc 4´ epimerasa que en T. cruzi, al
contrario de lo que sucede en vertebrados, no convierte eficientemente UDP‐GlcNAc
en UDP‐GalNAc (Roper and Ferguson, 2003).
La enzima O‐α‐GlcNAc‐transferasa que transfiere el primer residuo de GlcNac a
las proteínas ha sido caracterizada bioquímicamente hace más de 10 años (Previato JO,
1998), aunque el gen correspondiente aún no ha podido ser identificado
inequívocamente (Heise N, 2009). Una vez transferido el core GlcNAc, sobre él se
adicionan principalmente unidades de galactosa (Gal) ‐en sus conformaciones de
piranosa (Galp) o furanosa (Galf) y en distintas configuraciones y tipos de enlace‐
dependiendo del pool de glicosil transferasas presentes en ese estadío/cepa (El‐Sayed
NM, 2005a).

17
A diferencia de la N‐glicosilación, no se ha podido identificar una secuencia
primaria consenso para adición de O‐glicanos. Se presupone que se requiere de ciertos
motivos o estructura tridimensional para su reconocimiento por parte de la
maquinaria enzimática. En general, los residuos P en posición +3 o +1, respecto del
sitio S o T de glicosilación, favorecen este reconocimiento (Yoshida A, 1997).
En términos generales, la presencia de O‐glicanos interviene en procesos de
interacción célula‐célula y de protección/lubricación de epitelios (Tian E, 2009). Datos
recientes obligan a considerar una nueva perspectiva acerca del rol de esta
modificación post‐traduccional. Por un lado, en células de mamífero, se demostró la
intervención de glicosiltransferasas residentes del complejo de Golgi en el proceso de
degradación de proteínas asociado al RE (Pan S, 2011). Por otra parte, la adición de O‐
glicanos (o su consecuente enmascaramiento de dominios ricos en T, P y S) parecería
tener un efecto sobre la secreción de los productos modificados, aunque este efecto
puede ser positivo o negativo, dependiendo del sistema en estudio (Kinlough CL,
2011).
3.4. Bolsillo flagelar
Una región característica de los tripanosomátidos es el bolsillo flagelar, formado
por una invaginación en la membrana plasmática en la región anterior de la célula,
desde donde emerge el flagelo (Figura 3B). Esta región altamente especializada está
posicionada sobre una brecha en el esqueleto de microtúbulos subpeliculares, donde
también se posiciona el Golgi, lo que determina que sea un sitio preferencial de exo y
endocitosis. La membrana que conforma el bolsillo, así mismo, representa un
subdominio de la membrana plasmática en términos de la composición y distribución
de las moléculas presentes (De Souza W, 2002).
Un trabajo reciente en T. brucei relaciona además al bolsillo flagelar con el
fenómeno de remoción o clearance de inmunocomplejos anti‐VSG. Según este
modelo, el propio movimiento del parásito generaría una fuerza hidrodinámica sobre
los inmunocomplejos, tal que éstos son trasladados pasivamente hasta el bolsillo
flagelar para ser luego endocitados (Engstler M, 2007).
3.5. Reservosomas
Un destino alternativo al bolsillo flagelar para las moléculas exportadas desde el
Golgi son los reservosomas, compartimentos grandes (400–600nm) y esféricos
encontrados únicamente en miembros del subgénero Schizotrypanum del género
Trypanosoma (Figura 3C). Los reservosomas comparten características estructurales y
funcionales con los lisosomas, siendo responsables de la digestión celular y reciclaje de
organelas (Sant'Anna C, 2008b). De hecho, en estos compartimentos ácidos se
almacenan proteínas y lípidos de reserva provenientes de la vía endocítica. Análisis

18
proteómicos realizados sobre reservosomas aislados confirmaron la presencia de
varias proteasas, entre ellas la cruzipaína (Cazzulo JJ, 1999), y enzimas asociadas al
metabolismo de lípidos. En T. cruzi, los reservosomas fueron descriptos inicialmente en
el estadio epimastigote, y recientemente encontradas en tripomastigotes y
amastigotes (Sant'Anna C, 2008a).
3.6. Vías de secreción alternativas
Estudios proteómicos en Tritryps indican que i) sólo una minoría de las proteínas
secretadas por estos organismos cuentan con un SP en ausencia de señal de adición de
GPI y ii) las proteínas secretadas no se encuentran en forma soluble sino asociadas a
microvesículas (Geiger A, 2010; Silverman JM, 2008). De hecho, en distintos estadios
de los Tritryps pudieron observarse vesículas tipo secretorias, particularmente en la
zona del bolsillo flagelar (Figura 4). La liberación de vesículas ha sido descripta como
una vía de secreción no clásica o alternativa en distintos tipos celulares y puede
deberse a la exocitosis regulada de compartimentos lisosomales, la expulsión de
polipéptidos por medio de transportadores ABC (Torres C, 2004) o la formación y
liberación de vesículas por evaginación de la membrana plasmática. En
tripanosomátidos, algunas de las posibles funciones de estas vesículas podrían ser la
modulación del sistema inmune del hospedador (Tonelli RR, 2011) o bien alguna forma
de comunicación intra‐específica (Geiger A, 2010). En conjunto, estos hallazgos,
sugieren fuertemente la existencia e importancia de mecanismos no convencionales
en la secreción de moléculas en Tritryps.

19
Figura 4: Secreción de microvesículas en Tritryps A, B; Liberación de pequeñas porciones de la membrana bajo la forma de vesículas en tripomastigotes y
amastigotes de T. cruzi (Extraído de http://www.fiocruz.br/). C; Formas sanguíneas de T. brucei
gambiense, se señala el flagelo (f), mitocondria, (m) y vesículas emergentes (v). Adaptado de (Geiger A,
2010). D; Microvesículas emergiendo desde el bolsillo flagelar de promastigotes de Leishmania sp.,
observadas por microscopía de barrido electrónico. Adaptado de (Silverman JM, 2008).
4. Cubierta de superficie (surface coat) de T. cruzi
La cubierta de superficie de todas las formas de desarrollo de T. cruzi y de otros
patógenos relacionados como T. brucei, Leishmania, Toxoplasma y Plasmodium está
constituida por un arreglo de alta densidad de GPI‐glicoconjugados (McConville MJ,
2002). Esta cubierta actúa como una interfase entre el organismo y su medio externo,
y juega un rol clave en diversos aspectos de su biología: protección, adhesión, invasión,
patogénesis y evasión de la respuesta inmune (Buscaglia CA, 2006; Ferguson MA, 1999;
Gazzinelli RT, 2006).
Además de las mucinas, las cuales se detallarán en las próximas secciones, otros
GPI‐glicoconjugados relevantes de la cubierta del parásito incluyen a las trans‐
sialidasas (TS), que transfieren residuos de ácido siálico (AS) desde gliconjugados del
hospedador hacia Gal terminales de mucinas (Giorgi M, 2011), ortólogos de las GP63,
ya descriptas en Leishmania (Cuevas IC, 2003) y MASPs (mucin‐associated surface
proteins) (El‐Sayed N, 2005). Los glicoinositol fosfolípidos (GIPLs), son también muy
D

20
abundantes, y constituyen un glicocálix por debajo de la cubierta mucínica (Figura 5)
(Lederkremer RM, 2001).
Figura 5: Superficie celular de Tritryps PANEL SUPERIOR: A; Micrografía electrónica de barrido de T. cruzi. B; Micrografía electrónica de barrido
teñida in silico de T. brucei, se observan también glóbulos rojos C; Micrografía electrónica de un
promastigote de L. major. PANEL INFERIOR: D, E, F; Esquema de la cubierta de superficie de cada uno de
estas especies. Se muestra la naturaleza de las modificaciones más comunes: N‐glicanos ( ), O‐glicanos
( ), y fosfoglicanos ( ). La presencia de GIPLs es indicada. Adaptado de (McConville MJ, 2002).
5. Mucinas en T. cruzi
Como ya se mencionó, uno de los componentes principales de la cubierta de T.
cruzi son las proteínas de tipo mucina, cadenas peptídicas ricas en residuos de P, T y S
que proporcionan el andamiaje adecuado para la adición extensiva de O‐glicanos de
composición y estructura variable.
5.1. Caracterización bioquímica
Los estadios que desarrollan dentro del insecto –epimastigote y tripomastigote
metacíclico‐ expresan un grupo de mucinas que pueden visualizarse en SDS‐PAGE
como una doble o triple banda, dependiendo de la cepa estudiada, en el rango de los
35‐50 kDa (llamadas por lo tanto GP35/50). Las principales diferencias entre las
GP35/50 de epimastigotes y tripomastigotes metacíclicos residen en el mayor
contenido aparente de residuos AS en estas últimas, que correlaciona con un aumento
en la actividad TS del parásito (Briones MR, 1995; Chaves LB, 1993), y en la estructura
lipídica del ancla de GPI. La relevancia funcional de este último punto, no ha sido
A B C
D E F

21
demostrada aunque podría la secreción de las GP35/50 de uno y otro estadio
(Lederkremer MR, 2009).
Existe mucha variabilidad en la O‐glicosilación de las GP35/50 entre diferentes
cepas del parásito; y aún dentro de una misma cepa han podido identificarse distintos
tipos de arreglos (Lederkremer RM, 2009). En líneas generales, los residuos GlcNAc
directamente unidos a T o S son conjugados con varias unidades (hasta 5) de Galf y/o
Galp en distintas configuraciones y tipos de enlace. En caso de tener βGalp terminales,
éstos pueden ser decorados extracelularmente con residuos de AS vía acción de TS.
Debe mencionarse también que en todas las cepas estudiadas, una proporción de los
residuos GlcNAc permanecen sin sustituir. En cepas TcI (antiguamente linaje I), Galf se
encuentra sobrerepresentada mientras que en cepas pertenecientes a los grupos TcII‐
TcVI (agrupadas antiguamente dentro del linaje II), la Gal de las mucinas corresponde
únicamente a la forma piranosa (Previato JO, 1994; Todeschini AR, 2001). Este hecho
podría tener consecuencias epidemiológicas, ya que los seres humanos no producen
glicoconjugados conteniendo Galf, y montan una fuerte respuesta inmune contra estos
residuos. (Lederkremer RM, 2009).
Una herramienta para el estudio de la maduración de las GP35/50, además del
uso de lectinas, es el uso de anticuerpos monoclonales (mAbs) dirigidos contra
epítopes sacarídicos presentes en sus O‐oligosacáridos. Entre ellos cabe mencionar el
mAb 10D8 capaz de reconocer epítopes de Galf en cepas TcI y el mAb 2B10, que
reacciona contra epítopes de Galp presentes en todas las cepas (Mortara RA, 1992).
A diferencia de las GP35/50, las mucinas de tripomastigotes sanguíneos (tGPI‐
mucinas) se visualizan en corridas de SDS‐PAGE como un chorreado de masa molecular
comprendida entre los 60‐200 kDa, lo que puede atribuirse a la co‐expresión de
múltiples genes de secuencia y modificación post‐traduccional variable (Buscaglia CA,
2004). Es poco lo que se conoce sobre las estructuras de sus carbohidratos, aunque
difieren de las GP35/50 en que portan epítopes Galp(α1→3)Galp(β1→4)GlcNac muy
antigénicos llamados αGal (Pereira‐Chioccola VL, 2000). El ancla de GPI asociado a las tGPI‐mucinas se encuentra compuesta
exclusivamente de estructuras alquilacilfosfatidil inositol con predominancia de ácidos
grasos insaturados de tipo C18:1 y C18:2 (Buscaglia CA, 2004). Esta última estructura
es la que induce en macrófagos la secreción de citoquinas proinflamatorias como IL‐12,
TNF α y óxido nítrico (Almeida IC, 2000; Previato JO, 2004).
Las mucinas de los amastigotes intracelulares, por último, no fueron
caracterizadas en detalle, aunque ciertos datos inmunológicos sugieren que estarían
más relacionadas con las tGPI‐mucinas que con las GP35/50 (Buscaglia CA, 2004;
Campo VA, 2006).

22
5.2. Caracterización génica
Distintos estudios genómicos permitieron identificar más de 1.000 genes ‐
dispuestos en tándem o dispersos en el genoma ‐ que codifican para proteínas tipo
mucina en T. cruzi, los cuales pudieron agruparse en dos familias llamadas TcSMUG y
TcMUC en base a características estructurales (Tabla 1). Todos ellos se caracterizan por
la presencia de una región central variable, codificante para un dominio rico en S, T y
P, flanqueado por secuencias muy conservadas, correspondientes al SP y la señal de
anclaje por GPI. La topología predicha para el producto maduro expuesto en en
superficie, una vez que ha sufrido el correcto procesamiento de todas estas señales, se
presenta en la Figura 6B.
La familia TcMUC es la más variable y extensa (alrededor de 900 miembros),
pudiéndose definir 3 subgrupos de genes en base a la presencia y número de
repeticiones en su dominio central. En el grupo TcMUC I se observa una región
hipervariable corta hacia el extremo N‐terminal y 2‐10 repeticiones de la secuencia
T8KP2, que son un sustrato ideal para la O‐α‐GlcNAc‐transferasa (Previato JO, 1998). En los genes TcMUC II, la región hipervariable se expande a casi la totalidad de la región
central, observándose apenas 1 o 2 repeticiones T8KP2 seguidas de un tracto de T hacia
el extremo C‐terminal. Estudios bioquímicos y proteómicos indicaron que los
productos de estos genes TcMUC II, y en mucha menor medida los de TcMUC I,
constituyen el esqueleto proteico de las tGPI‐mucinas (Buscaglia CA, 2004).
Sugestivamente, un grupo de los genes TcMUC II se encuentra asociado a genes de la
familia TS. El tipo de regulación de la expresión génica en T. cruzi (Vazquez MP, 2005),
indica que esta asociación física favorecería la expresión conjunta de ambas moléculas
y por lo tanto, su asociación funcional (enzima‐aceptor) en la superficie del parásito
(Campo VA, 2004; de Freitas JM, 2006). Existe un tercer grupo denominado TcMUC III o
TSSA (trypomastigote small surface antigen), constituido por un gen multialélico de
copia única cuya expresión también está restringida a los estadios de mamífero
(Bhattacharyya et al., 2009; Di Noia JM, 2002).
Tabla 1: Descripción de las familias génicas y proteínas tipo mucinas en T.cruzi
E/M

23
Los principales estadios se indican como A: Amastigote, T: Tripomastigote, E: Epimastigote, M:
Tripomastigote metacíclico. TSSA es expresado en el estadio T, como así también en estadios
intermedios presentes en el mamífero, indicados como T*. Adaptado de (Buscaglia CA, 2006).
La familia TcSMUG está compuesta por alrededor de 40‐80 genes más pequeños
en promedio que los TcMUC y con mucha menor heterogeneidad entre ellos (Buscaglia
CA, 2006; Di Noia JM, 2000). Dentro de esta familia, pueden distinguirse 2 grupos (S y
L) de acuerdo a la estructura genómica, el tamaño del ARNm y ciertas divergencias en
la zona codificante (Figura 6A). Como rasgo saliente, puede mencionarse la presencia
de secuencias derivadas de elementos desestabilizantes de ARNm en el 3’UTR de los
genes del grupo L (Di Noia JM, 2000).
Recientes estudios proteómicos determinaron que los productos TcSMUG S
constituyen el esqueleto peptídico de las GP35/50 (Nakayasu ES, 2009). Los productos
TcSMUG L, en cambio, son expresados exclusivamente en el estadio epimastigote de
algunas cepas y, si bien son adicionados con O‐glicanos, no constituyen mucinas
funcionales (Urban I, 2011).
Figura 6: Estructura de productos TcSMUG. A; Se indica el porcentaje de similitud entre las regiones conservadas de los grupos S y L. Adaptado de
(Di Noia JM, 2000). B; Se muestra un diagrama esquemático de una mucina madura TcSMUG S,
completamente procesada y expuesta en la superficie del epimastigote.
5.3. Caracterización funcional
Las mucinas juegan un rol clave en cada uno de los diferentes estadios del
parásito. En las secciones siguientes se detallan los distintos procesos biológicos en las
que están involucradas.
A B

24
5.3.1. Adhesión
Los fenómenos de adhesión de T. cruzi a distintos tipos celulares están mediados
principalmente por glicolípidos (Nogueira et al., 2007) y glicoproteínas de las familias
de las TS (particularmente las gp85, gp 82, gp30 y gp90 en los distintos estadios)(Giorgi
M, 2011) y mucinas (Buscaglia CA, 2006). Para las tGPI‐mucinas se ha descripto un
epitope conteniendo AS, denominado Ssp‐3 y reconocido por el mAb 3C9, el cual está
directamente involucrado en el reconocimiento e invasión de la célula blanco
(Schenkman S, 1991). En cuanto a las GP35/50, numerosos estudios han verificado el
binding de estas moléculas a distintos tipos celulares no macrofágicos (Vermelho AB,
1994). Este binding es mayor para las GP35/50 de tripomastigotes metacíclicos, lo que
sugiere un rol del AS en este fenómeno (Ruiz R de C, 1993). Un resultado curioso, sin
embargo, es que este binding parecería funcionar como un regulador negativo de la
internalización, ya que existe una relación inversa entre el contenido de GP35/50 del
tripomastigote metacíclico (o de la cantidad de AS en las mismas) y la capacidad
infectiva de la cepa (Yoshida N, 1997). Datos recientes indican que las GP35/50
estarían también involucradas en el reconocimiento e invasión de células epiteliales
del aparato digestivo, lo que tendría relevancia para el establecimiento de la infección
por vía oral, que se verifica luego de la ingestión de alimentos o bebidas contaminadas
con T. cruzi (Yoshida N, 2008). Los receptores putativos de las tGPI‐mucinas y GP35/50
en la célula de mamífero no han podido ser identificados aún.
5.3.2. Protección
Las sialilación de tGPI‐mucinas contribuye significativamente a la protección del
parásito frente a la respuesta inmune montada por el huésped, escudándolo de
anticuerpos líticos (Pereira‐Chioccola VL, 2000) y posiblemente de factores del
complemento (Norris KA, 1998). Además, ya dentro de la célula blanco, la decoración
de tGPI‐mucinas con AS es clave para la protección del tripomastigote dentro de la
vacuola parasitófora y su posterior escape al citoplasma. Brevemente, la extracción de
estos residuos de la membrana lisosomal del huésped aumenta su sensibilidad frente a
la hemolisina secretada por T.cruzi, al tiempo que protege al propio parásito (Andrews
et al., 1988).
La hiperglicosilación de las GP35/50, por otro lado, resulta muy importante para
la resistencia del epimastigote frente a proteasas del tracto digestivo del insecto
(Mortara RA, 1992).
5.3.3. Inmunomodulación
Se ha propuesto que las tGPI‐mucinas podrían inmunomodular la respuesta
inmune del hospedador mamífero a través de la inducción de un estado de anergia en
las células mononucleares de sangre periférica (Argibay PF, 2002; Kierszenbaum F,

25
1999), lo que se verifica en pacientes durante la infección aguda. La estrategia de
presentación simultánea de múltiples productos TcMUC II conteniendo diferentes
epítopes relacionados podría contribuir en este sentido (Buscaglia CA, 2004), de
manera análoga a lo observado para la familia de las TSs (Kahn SJ, 1999). Estudios
recientes indican que las tGPI‐mucinas utilizan mecanismos complementarios,
posiblemente dependientes de sus O‐glicanos, para alterar el curso de la respuesta
inmune (Erdmann H, 2009; Talvani A, 2008). Por último, la porción lipídica de las tGPI‐
mucinas tiene un efecto inmunomodulador demostrado. La iniciación de esta
respuesta inflamatoria se encuentra mediada por la interacción entre el ancla de GPI y
el toll‐like receptor‐2 sobre la superficie del macrófago (Almeida IC, 2001). En conjunto,
estos datos indican que las tGPI‐mucinas constituyen un arma con múltiples filos
(peptídico, glucosídico, lipídico) capaces de modular el sistema inmune a distintos
niveles.
Algunos estudios dan cuenta de cierta capacidad inmunomoduladora de las
GP35/50 (Alcaide P, 2004). La relevancia de estos resultados, sin embargo, es
discutible ya que el tiempo de interacción de estas moléculas con el sistema inmune se
limita a la infección inicial del tripomastigote metacíclico (Figura 1).
5.3.4. Señalización
Las GP35/50 son capaces de inducir distintas clases de señalización,
particularmente cascadas de Ca2+, en la célula blanco. Esta capacidad es mayor en las
cepas del linaje evolutivo TcI y, al igual que en el caso del binding, es exacerbada por la
remoción de los residuos de AS. En este sentido, una posibilidad es que la señalización
esté relacionada con la presencia de residuos de Galf (Yoshida N, 1989).
Más allá del efecto ya comentado de las tGPI‐mucinas sobre células inmunes,
trabajos recientes detallan el gatillado de una compleja cascada de señalización
mediada por un factor no definido, aunque estructuralmente relacionado a las tGPI‐
mucinas, en fibroblastos infectados por T. cruzi (Mott GA, 2011). Esta señalización
termina alterando el perfil de expresión genética del fibroblasto en beneficio del
parásito.

26
ANTECEDENTES Y OBJETIVOS
Las mucinas son componentes mayoritarios y distintivos de la cubierta de todos
los estadios de T. cruzi, por lo que constituyen un interesante blanco de intervención
potencial contra este organismo. La falta de herramientas genéticas apropiadas
sumado a la presencia de cientos de genes relacionados que se expresan en forma
simultánea dificulta la realización de estudios definidos sobre estas moléculas.
En el laboratorio se demostró recientemente que genes de la familia TcSMUG
marcados mediante la etiqueta molecular FLAG y expresados en poblaciones de
epimastigotes de T. cruzi son presentados en la superficie celular como mucinas
maduras, estructural y funcionalmente indistinguibles de las GP35/50 endógenas
(Urban I, 2011). En vista de este antecedente, decidimos usar un modelo experimental
similar para el estudio del tráfico y procesamiento intracelular de las mucinas de T.
cruzi. Para ello se construyeron distintas mutantes de deleción a partir de miembros
representativos de las familias TcSMUG y TcMUC, las cuales fueron fusionadas al
epítope FLAG y/o a GFP, luego se transfectaron en epimastigotes de T. cruzi. La
localización subcelular y el procesamiento de las proteínas reporteras quiméricas se
realizó utilizando técnicas bioquímicas y de microscopía.

27
RESULTADOS
PARTE 1: Generación de reactivos
1.1. Generación de clones de proteínas tipo mucina
La estructura modular característica y conservada, propia de las diferentes
familias mucínicas de T. cruzi (Buscaglia CA, 2006), lleva a suponer que cada uno de sus
dominios (SP, región central o core y señal de adición de GPI) contribuye a asegurar el
correcto procesamiento y expresión de estas glicoproteínas en superficie. Con el
objetivo de caracterizar la funcionalidad de cada uno de ellos en el marco de la vía
secretoria del parásito, se diseñaron diferentes variantes de deleción a partir del clon
TcSMUG S. Para hacerlo más abarcativo, se generaron y analizaron en paralelo
construcciones equivalentes derivadas de un clon TcMUC II, representativo de la
familia TcMUC. En líneas generales, los clones generados presentan la deleción del SP
(variantes ∆SP), la deleción de la señal de anclaje por GPI (∆GPI) o la deleción conjunta de ambos motivos (variantes ∆SP∆GPI). Los clones TcSMUG S y TcMUC II completos
(variantes full length), etiquetados y transfectados en las mismas condiciones que las
distintas variantes de deleción, fueron analizados como controles en uno y otro caso.
Como se mencionó, todos los clones están etiquetados con el epítope FLAG
(DYKDDDD) en posición análoga a la del clon TcSMUG S original (Urban I, 2011) (Figura
7). La presencia de este epítope en esa posición nos asegura en principio su asociación
con la proteína madura, considerándose los procesamientos post‐traduccionales
predichos (Figuras 10 y 12C) y nos permite seguir las moléculas reporteras usando
anticuerpos comerciales altamente específicos. De cada uno de estos clones, a su vez,
se generó una variante alternativa que, además del epítope FLAG, se encuentra
fusionada al reportero eGFP (variantes ::eGFP). La presencia de eGFP, una mutante
puntual de la GFP original que le confiere una mayor eficiencia en el plegado sin
afectar su espectro de excitación y emisión (488/507 nm) (Zhang G, 1996), nos brinda
la posibilidad de visualizar las moléculas reporteras de una forma más directa, por
simple microscopía de epifluorescencia. Como contrapartida, existe una probabilidad
mayor de que la fusión a una molécula grande como eGFP altere en cierta medida el
tráfico intracelular y/o procesamiento de los reporteros. En la Figura 7, se presenta un
diagrama de todos los clones generados durante este trabajo. Algunos de estos clones
están todavía en proceso de selección, o bien los resultados obtenidos con ellos son
todavía preliminares por lo que no se presentan aquí.

28
Figura 7: Diagrama de los productos deducidos a partir de los clones generados. Cada conjunto de clones (identificado con naranja para TcSMUG y azul para TcMUC) se encuentran fusionados al epítope antigénico FLAG (F) entre el dominio central (core) y la señal de adición de GPI o el reportero eGFP, según corresponda en cada caso.
Para la generación de toda esta batería de clones se utilizó una estrategia de PCR
(Ver M&M), para lo que se diseñaron oligonucleótidos específicos que, de acuerdo a
las necesidades, pueden contener sitios de restricción, codones de iniciación o
terminación de la traducción u otro tipo de secuencias (Ver M&M, ANEXO A y ANEXO
B, Tabla 4). El vector de expresión elegido fue, en todos los casos, una versión
modificada del vector pTREX (Vazquez MP, 1999) denominada pTREX omni (Bouvier L,
resultados sin publicar). Dicho vector posee múltiples sitios de clonado, secuencias
codificantes para distintas etiquetas moleculares (FLAG, HA, eGFP) y sitios blanco de
proteasas, constituyendo una herramienta de utilidad para la investigación en T. cruzi.
Todas las construcciones generadas se transfectaron en epimastigotes de la cepa
Adriana (perteneciente al linaje evolutivo TcI) y se analizaron como poblaciones (sin
proceso de clonado o enriquecimiento previo) al menos 45 días después de iniciado el
proceso de selección.

29
1.1.1. Generación de clones TcSMUG
Para la generación de los clones TcSMUG, se partió de un gen TcSMUG S
previamente aislado de la cepa SC43 cl92 (perteneciente al linaje evolutivo TcV)
(ANEXO C, GenBank XM_815949.1). La proteína deducida cuenta con 122 aminoácidos
considerando al epítope FLAG. Si bien la expresión de una versión etiquetada con FLAG
del clon TcSMUG S full length permitió validar el correcto procesamiento de las
distintas señales de tráfico intracelular y modificaciones post‐traduccionales (MPTs)
predichas (SP, anclaje en superficie a través de un motivo GPI, adición de N‐ y O‐
glicanos) (Urban I, 2011), la fijación de los límites exactos de cada módulo para la
generación de las variantes de deleción se estableció de forma arbitraria sobre la
combinación de datos de base experimental y computacional que se detallan a
continuación.
Para identificar el sitio de clivaje del SP más probable recurrimos al algoritmo
Signal‐P (Bendtsen JD, 2004). Para cada análisis bajo este utilitario, son utilizadas dos
redes neurales diferentes, una para la predicción del SP y otra para la predicción de la
posición del sitio de corte por parte de la peptidasa de la señal presente en RE (Paetzel
M, 2002). El S‐score, por un lado, asigna un valor a cada residuo indicando su
probabilidad de pertenecer a una secuencia señal. Valores bajos del S‐score indican
que dicho residuo forma parte de la proteína madura. El C‐score, por otro lado,
correlaciona con la probabilidad de clivaje para ese residuo (Cleavage site). El Y‐score
no es más que una combinación de los estadísticos del C y S‐score, proporcionando
una mejor visualización de la predicción. Los resultados arrojados predicen un sitio de
corte con una probabilidad máxima (0.982) entre las posiciones A23 y Q24.

30
Figura 8: Análisis bioinformático para la secuencia TcSMUG S seleccionada. A; Resultado arrojado por el algoritmo Signal‐P, se observan para los primeros 70 residuos de la
secuencia primaria el valor de los estadísticos C, S e Y. B; Análisis obtenido a partir del utilitario
NetOGlyc.
Un análisis directo de péptidos a partir de fracciones purificadas de GP35/50 de
epimastigotes de la cepa Y (TcII), permitió identificar secuencias N‐terminales
correspondientes a productos TcSMUG S (Dr. Igor Almeida, comunicación personal)
(Figura 9). Estos datos son compatibles con la predicción in silico, aunque sugieren que
el sitio de corte del péptido señal de TcSMUG S podría encontrarse en residuos
ubicados más hacia el dominio central del polipéptido o, más posiblemente, que
ocurre cierto procesamiento proteolítico de las regiones N‐terminales de estos
productos in vivo.
B
A

31
Figura 9: Péptidos identificados a partir de GP35/50 de epimastigotes. Se muestra un alineamiento de secuencias entre los péptidos identificados sobre mucinas GP35/50
maduras (Dr. Igor C. Almeida, comunicación personal) y los productos TcSMUG S deducidos (línea
superior). Se indican las variantes encontradas de TcSMUG S en cepas pertenecientes a distintos linajes
evolutivos (Urban et al., 2011) y el sitio de clivaje de SP predicho por el análisis bioinformático (*).
En el extremo C‐terminal de TcSMUG S se predice una secuencia aceptora para la
adición de GPI, la cual es procesada in vivo (Urban I, 2011). De acuerdo al análisis con
el algoritmo GPI‐SOM (http://www.expasy.org/proteomics), la posición D89 presenta
el mayor puntaje entre los posibles sitios ω chequeados, con un valor de P=1.928343x10‐2. Recientes análisis proteómicos sobre péptidos unidos a GPI,
purificados a partir de GP35/50 de epimastigotes de la cepa Y, demuestran que los
genes que proveen el esqueleto para estas mucinas son los TcSMUG S y, lo que es más
importante, que este residuo de aspartato (D) es el que funciona como aceptor del
ancla de GPI in vivo (Nakayasu ES, 2009). Por lo tanto, si bien el residuo aceptor para el
producto de nuestro gen en particular no ha sido demostrado, el conjunto de estas
evidencias nos permiten asumir con razonable certeza que se trata del residuo D89 y,
por lo tanto fue determinado como límite entre el core y el motivo GPI.
El dominio central del gen TcSMUG S seleccionado es aceptor de N y O‐
glicosilación (Urban I, 2011). En el primer caso, el análisis in silico realizado a través del
algoritmo NetNGlyc (http://www.expasy.org/proteomics), predice un único sitio
consenso de N‐glicosilación N66TT. Hemos generado clones en donde se mutó esta
señal de N‐glicosilación, tal que nos permita estudiar de la misma en el tráfico y
procesamiento de TcSMUG S. El análisis de estas construcciones no se incluye en el
presente trabajo debido al tiempo que demanda el proceso de selección de
transfectantes y el análisis bioquímico de las proteínas reporteras.
Como se mencionó, la identificación in silico de residuos S o T aceptores de O‐
glicanos resulta poco confiable (Julenius K, 2005). Esta limitación es aún mayor para
proteínas de T. cruzi, por las particularidades propias de su sistema de O‐glicosilación,
y porque los distintos sets de datos con los que fueron entrenados los algoritmos son
obtenidos, en general, de células de mamífero. De todas formas, el análisis
bioinformático usando el programa NetOGlyc (http://www.expasy.org/proteomics)

32
determinó la presencia de 27 sitios aceptores putativos (Figura 8B), de los cuales 23
tienen un potencial considerado como significativo.
En definitiva, integrando todo lo anterior, se propone el siguiente procesamiento
para el producto del gen TcSMUG (Figura 10), y en base a este modelo se realizaron las
distintas construcciones.
Figura 10: Modelo de procesamiento para la proteína TcSMUG S. AE, aminoetil fosfonato; EP, etanolamina fosfato; Gn, GlcN/GlcNAc; M, manosa. (*) Se muestra la
estructura detallada de un arreglo de GPI típico del estadio de epimastigote (McConville MJ, 1993).
1.1.2. Generación de clones TcMUC
Para la generación de los clones TcMUC, se partió de un gen TcMUC II
previamente identificado en la cepa RA (perteneciente al linaje evolutivo TcVI) (ANEXO
C, GenBank U32448.1). La proteína deducida cuenta con 217 aminoácidos y un peso
molecular de alrededor de 23 kDa, sin considerarse las posibles MPTs (ANEXO C). Para
determinar los límites exactos de cada módulo, se procedió de manera análoga a lo
descrito en la sección anterior.
En la región N‐terminal de la proteína deducida, pueden observarse algunas
características estructurales típicas de un SP (von Heijne, 1985): (i) una región N‐
terminal con carga positiva neta centrada en los primeros 5 o 10 aminoácidos, (ii) una
región central más hidrofóbica de 10 aminoácidos y por último (iii) una región más
polar de 5 aminoácidos, en este caso conformada por un triplete de cisteínas (Figura
11) hacia el extremo C‐terminal de la secuencia. Su análisis mediante el servicio de
Signal‐P 3.0, reveló la presencia de un SP con una probabilidad de 0.994 y valores de
los estadísticos mayores al umbral de 0.5 establecido por el algoritmo. Como se

33
muestra en la Figura 12A, el sitio de corte del SP de TcMUC más probable estaría dado
entre los aminoácidos C23 y I24.
Figura 11: Composición aminoacídica relativa y alineamiento de los SP de los productos TcSMUG S y TcMUC II usados en este trabajo. Aminoácidos idénticos se sombrearon en amarillo y aquellos estructuralmente relacionados, en verde.
El análisis proteómico de tGPI‐mucinas de la cepa Y permitió la identificación de
algunos péptidos no glicosilados (Buscaglia CA, 2004). Uno de estos péptidos (de
secuencia ANGVDEQVAPDMTK) es idéntico al fragmento A26‐K39 del producto del gen
TCMUC II utilizado aquí (ANEXO C). La presencia de este péptido en las tGPI‐mucinas
maduras es perfectamente compatible con la utilización del sitio de clivaje predicho in
silico.
Por otro lado, no se encontraron motivos consenso de N‐glicosilación, pero sí 51
sitios putativos de O‐glicosilación en el producto deducido de TcMUC II (Figura 12B). 38
de estos sitios presentan un potencial superior al umbral y 36 de ellos (25 residuos T y
11 S) se encuentran comprendidos en el dominio central, y por lo tanto estarían
presentes en la molecula madura. Finalmente, la proteína deducida cuenta con una
secuencia C‐terminal compatible con una señal de adición de GPI, el cual fue analizado
con el algoritmo GPI‐SOM. De todas las posiciones chequeadas, la posición D182
presentó el mayor puntaje con un valor de P=2.098945x10‐2. Este aminoácido predicho
como residuo ω, nuevamente resultó coincidente con el sitio identificado
experimentalmente para tGPI‐mucinas (Buscaglia CA, 2004).

34
A partir de la integración de todos estos análisis se generó el modelo más
probable de proteína TcMUC madura (Figura 12C).
Figura 12: Análisis bioinformático y modelo de procesamiento para la proteína TcMUC II. A; Resultado arrojado por el algoritmo Signal‐P, se observan para los primeros 70 residuos de la
secuencia primaria el valor de los estadísticos C, S e Y. B; Análisis obtenido a partir del utilitario
NetOGlyc. C; Modelo de procesamiento de la proteína TcMUC (ver leyenda Figura 10).
A
C
B

35
1.2. Obtención y caracterización de sueros policlonales
Paralelamente a la generación de los clones, se avanzó en la inmunización de
ratones con proteínas recombinantes con el objetivo de generar herramientas
adicionales que nos facilitaran el análisis de las moléculas reporteras. El vector de
expresión utilizado en todos los casos fue pGEX, que permite la expresión de proteínas
de fusión al C‐terminal de la Glutation‐S‐Transferasa (GST). Desde el punto de vista
inmunológico, GST provee epítopes B y T co‐estimulatorios para el desarrollo de una
respuesta inmune eficiente. Desde un punto de vista operativo, la presencia de GST
favorece la expresión en bacterias de proteínas de fusión en forma soluble y,
fundamentalmente, facilita la purificación de las mismas mediante columnas de
afinidad (Kaelin WG, 1992).
Para el testeo de los antisueros obtenidos, también resultaba de utilidad contar
con una proteína conteniendo el péptido o construcción usada para inmunizar
fusionado a un carrier diferente. En este caso se utilizó el vector pMAL que permite la
expresión de proteinas recombinantes como fusión al C‐terminal de la Maltosa Binding
Proteín (MBP) y que posibilita la subsecuente purificación de las mismas usando
cromatografía de afinidad (Guan C, 1987).
1.2.1. Obtención del suero anti‐TcSMUG S
Para la obtención de un antisuero específico para TcSMUG S, se inmunizaron
animales con un péptido correspondiente al extremo N‐terminal de la proteína madura
el cual fue elegido de acuerdo a los siguientes criterios:
• Datos proteómicos indican que este péptido, carente de modificaciones post‐
traduccionales, está presente en las GP35/50 maduras, purificadas de
superficie de epimastigotes (Figura 9).
• Esta secuencia es una de las pocas diferenciales con respecto a los productos
TcSMUG L, también presentes en el estadio epimastigote (Urban I, 2011).
• Los epítopes peptídicos presentes en el resto de la región central se encuentran
enmascarados casi en su totalidad por la presencia de O‐glicanos y, por lo
tanto, no son accesibles al reconocimiento por anticuerpos.
La estrategia de clonado seguida (Ver M&M), se basó en el apareamiento de
primers complementarios. El clonado de este dúplex en un vector pGEX modificado
(Urban I, 2011), permitió la expresión de una proteína con la siguiente estructura: GST‐
(V2EAGEGQDQT2ST). Finalmente, luego de la purificación por cromatografía de afinidad
(Figura 13A) se procedió con el protocolo de inmunización.
La especificidad de los 3 sueros policlonales obtenidos se evaluó enfrentándolos
a las proteínas MBP‐SMUG S y MBP. De acuerdo a los resultados del ensayo de Dot
blot (Figura 13B), se seleccionó el suero #1 para continuar con los análisis, ya que es el

36
que presenta una mejor relación señal específica (MBP‐SMUG S)/señal no específica
(MBP).
Figura 13: Caracterización de los sueros anti‐TcSMUG. A; SDS‐PAGE teñido con Coomasie blue de los productos de inducción (Ind) y purificación (Pur) del
producto de fusión GST‐TcSMUG (27.7 kDa). B; Dot blot realizado a partir de enfrentar los 3 sueros
obtenidos en una dilución 1:500 frente a diluciones seriadas de los antigenos MBP o MBP‐TcSMUG S. C;
Tinción con Ponceau S y Western blot revelado con αGST‐SmugS #1 de extractos totales de
epimastigotes de las cepas Adriana, Y, Cl‐Brener y RA de T. cruzi. D; Ensayo de desplazamiento
competitivo con MBP o MBP‐Smug S, sobre extractos totales de las cepas Adriana y CL.
Como parte de la caracterización de este antisuero, se evaluó si era capaz de
detectar las GP35/50 nativos. Para ello, se realizaron ensayos de Western blot sobre
lisados totales de epimastigotes de distintas cepas cultivadas en el Instituto. En la
Figura 13C se observa la presencia de una banda de aproximadamente 45 kDa en
epimastigotes de las cepas CL‐Brener y RA. El tamaño, y el hecho de que la reactividad
pudo ser competida por la adición de proteína MBP‐SMUG pero no MBP (Figura 13D),
sugieren que esta banda correspondería a las GP35/50 (o a una fracción de ellas).
Interesantemente, esta banda no se observa en las calles correspondientes a las cepas
Adriana e Y, a pesar de que la carga total de proteínas evaluadas por tinción de
Ponceau es similar (Figura 13C, panel superior). Si bien el análisis de las bases
moleculares que promueven este reconocimiento diferencial entre cepas excede el
presente trabajo, se proponen las siguientes hipótesis:

37
a) El procesamiento proteolítico de las GP35/50 (y posiblemente de otros
componentes de la superficie) es diferencial en las distintas cepas, tal que el
epítope reconocido por nuestro antisuero no se encuentra presente en las
mucinas maduras de las cepas Adriana e Y.
b) Estas diferencias de reconocimiento responden a variaciones en la secuencia
del péptido usado como inmunógeno para los productos TcSMUG S
correspondientes a los distintos grupos evolutivos presentes en T. cruzi (Urban
I, 2011) (Figura 14).
Estos resultados fueron muy interesantes y merecen por supuesto un análisis
más profundo. A los fines de nuestro trabajo, el anticuerpo obtenido ofrece la ventaja
adicional de no “dar fondo” en la cepa Adriana utilizada, permitiendo detectar
específicamente a las proteínas recombinantes TcSMUG.
Figura 14: Alineamiento de productos TcSMUG S para diferentes cepas. Se marcan en rojo aquellas posiciones variables respecto del inmunógeno utilizado (cepa SC43 cl92,
TcV). X, D o E; Z, G o D; B, E o K.
1.2.2. Obtención del suero anti‐TcMUC
Resultados previos indicaban que las regiones centrales completas de los
productos TcMUC II podían ser expresadas en forma recombinante sin mayores
problemas y resultaban muy antigénicas (Buscaglia CA, 2004). La utilidad de estos
anticuerpos para el reconocimiento de tGPI‐mucinas in vivo es, sin embargo, relativa,
ya que los epítopes peptídicos presentes en esta región central se encuentran
enmascarados casi en su totalidad por la presencia de O‐glicanos. La única excepción
parece ser un pequeño segmento C‐terminal, ubicado entre la zona rica en treonina y
el residuo aceptor de GPI, que se encuentra conservado entre muchos de los
productos TcMUC II (Buscaglia CA, 2004).

38
Los antisueros obtenidos se evaluaron a través de un ensayo de Dot blot similar
al descripto (Figura 15B), utilizando en este caso como antígenos a MBP, MBP‐MUC II,
GST y GST‐MUCII y variando la dilución del antisuero. Nuevamente, este análisis
permitió identificar aquellos sueros más reactivos frente al péptido de interés. Se
seleccionó en este caso al suero #2 para análisis posteriores.
Tal cual lo esperado, estos sueros no presentaron reactividad clara en ensayos de
Western blot sobre tGPI‐mucinas presentes en extractos totales de las formas
amastigote y tripomastigote de la cepa CL Brener (datos no mostrados).
Figura 15: Caracterización de los sueros anti‐TcMUC II. A; Esquema de clonado de la región amplificada para su introducción en el plásmido pGEX 2T (Ver
M&M). B; Ensayo de Dot blot de los 3 sueros obtenidos, probados en diluciones de 1:1.500 (paneles de
la izquierda) y 1:3.000 (paneles de la derecha), respectivamente.

39
PARTE 2: Análisis de moléculas reporteras
2.1. TcMUC 2.1.1. Adición de ancla de GPI y localización subcelular
Para evaluar el procesamiento de la señal de anclaje de GPI en el producto
TcMUC, se utilizó el detergente neutro Tritón X‐114 (Tx114), que presenta afinidad por
las anclas de GPI (Cordero EM, 2009). Lisados totales de epimastigotes transfectados
fueron sometidos a extracciones sucesivas en este detergente y las distintas fracciones
(Ver M&M, Figura 32) fueron precipitadas con acetona y analizadas por Western blot
usando el mAb anti‐FLAG. La Figura 16A muestra una única banda reactiva de ~25 kDa en la fracción S4. La misma contiene la fracción del detergente obtenida después de
numerosos lavados y es donde particionan también el reportero TcSMUG S (Urban I,
2011) y las GP35/50 nativas (datos no mostrados, Urban I, 2011). Este perfil de
partición en Tx114 sugiere que, al igual que lo que ocurre en estas moléculas, la señal
de adición de GPI en el producto TcMUC es procesada correctamente en nuestro
modelo.
Un método alternativo para evaluar la presencia del ancla de GPI se basó en el
tratamiento del producto TcMUC parcialmente purificado (fracción S4, Figura 16A) con
la enzima fosfolipasa C específica para fosfatidil inositol (PI‐PLC) de Bacillus cereus, que
cliva la mayoría de las anclas de GPI de tripanosomátidos (Stanton et al., 2002). Luego
del tratamiento con PI‐PLC, se indujo la separación de fases a 37ºC y la fase acuosa
(Sec) y detergente (Tx) resultantes fueron analizadas por Western blot con el mAb anti‐
FLAG. En la Figura 16B, se observa que TcMUC desaparece de la fase detergente (Tx)
luego del tratamiento con PI‐PLC, lo que podría atribuirse a la pérdida del motivo
lipídico de naturaleza fuertemente hidrofóbica del ancla. Interesantemente, la
proteína TcMUC tratada con PI‐PLC no pudo ser recuperada tampoco en la fracción
acuosa (Sec). Este comportamiento ya había sido previamente observado para otras
proteínas tipo mucinas de T. cruzi (Di Noia JM, 2002; Urban I, 2011) y creemos que
podría deberse a la incapacidad de la proteína sin GPI, altamente hidrofílica, de ser
inmovilizada en membranas de PVDF o nitrocelulosa. A modo de ejemplo, en el panel
inferior de la Figura 16B se presenta un Western blot de las mismas fracciones del
panel superior pero revelado con el mAb 10D8, específico para las GP35/50. De
manera análoga a lo observado para TcMUC, y tal cual lo mostrado (Urban I, 2011),
estas moléculas son solubilizadas luego del tratamiento con PI‐PLC (desaparecen de la
fase detergente) pero no pueden ser detectadas de manera concomitante en la fase
acuosa.

40
Figura 16: Análisis del ancla de GPI del producto TcMUC. A; Western blot revelado con mAb anti‐FLAG sobre muestras de extracción con Tx114 (ver M&M, Fig 32). B; El producto TcMUC parcialmente purificado (fracción S4 del panel A) se trató con la enzima PI‐PLC y se volvió a inducir separación de fases a 37ºC. Las fases acuosas (Aq) y detergente (Tx) obtenidas antes y después del tratamiento con PI‐PLC fueron precipitadas en acetona y analizadas por Western blot con los mAb indicados debajo de cada panel.
En conjunto, estos resultados sugieren que la señal de adición de GPI presente
en TcMUC es funcional y correctamente procesada por la maquinaria del epimastigote.
Para determinar de manera directa la localización subcelular del producto TcMUC
se utilizó, como un primer abordaje, la técnica de inmunofluorescencia indirecta (IFI).
Para esto, los parásitos transfectantes, sin permeabilizar, fueron fijados y tratados con
el mAb anti‐FLAG seguido por un anticuerpo secundario conjugado al fluorocromo
Alexa 568, que al exitarse emite señal en una longitud de onda correspondiente al
color rojo. Tal como se ve en la Figura 17, TcMUC presenta distribución en focos
discretos o dots de localización perimetral al parásito, típico de moléculas de
superficie. En el mismo preparado pueden observarse algunos parásitos no
transfectantes (flecha verde), que subsisten en cierta proporción aún en presencia del
agente de selección. La presencia de estos parásitos actúa a modo de control interno
del experimento, evidenciando la especificidad de la señal anti‐FLAG obtenida. Una
observación interesante, que desde ya merece un desarrollo posterior, es que puede
detectarse la presencia de puntos reactivos para FLAG no asociados al parásito (flecha
amarilla), lo que sugeriría cierto nivel de liberación o secreción de TcMUC hacia el
medio extracelular. Este fenómeno, que podría atribuirse a la actividad de la enzima
PI‐PLC endógena u otra vía de secreción no clásica, fue previamente observado para el
reportero TcSMUG S (Urban I, 2011).

41
Figura 17: Ensayo de IFI sobre la población de epimastigotes TcMUC. Se puede observar la presencia de puntos reactivos por fuera de la superficie del parásito, sobre todo en la zona cercana al bolsillo flagelar (flecha amarilla). En la imagen de contraste de fase se señala un parásito no transfectante (flecha verde).
Para verificar la localización superficial de TcMUC, decidimos implementar un
ensayo de citometría de flujo sobre parásitos sin permeabilizar. La técnica fue puesta a
punto utilizando el mAb anti‐FLAG y, como anticuerpo secundario, uno acoplado al
fluorocromo Alexa 488, que emite señal en el rango del verde. A su vez, se realizó la
siguiente serie de controles:
Negativos: i) de fluorescencia endógena y no internalización del anticuerpo
primario, usando una población de parásitos transfectados con una construcción que
codifica para la citosina deaminasa ADAT2 de T. cruzi, localizada en citoplasma y
núcleo, fusionada al epítope FLAG (Bertotti et al., resultados sin publicar); y ii) de
inespecifidad del anticuerpo secundario, incubándolo con los parásitos en ausencia del
mAb anti‐FLAG.
Positivo: i) Parásitos transfectados con el clon TcSMUG fusionado al epítope FLAG,
como control positivo de localización superficial (Urban I, 2011).
En la Figura 18 se muestran los Dot blots e histogramas correspondientes a la
lectura de 10.000 eventos por corrida para cada muestra. Puede observarse que en los
gráficos obtenidos para las muestras TcSMUG y TcMUC, pero no para TcADAT2, la
mayor parte de los eventos poseen señal en FL1 (verde), indicando la presencia de
reactividad anti‐FLAG en superficie de los parásitos.
Tomados en conjunto, los experimentos de partición en Tx114, sensibilidad a PI‐
PLC, IFI y citometría mostrados en esta sección, sugieren fuertemente que, similar a lo
demostrado para TcSMUG S (Urban I, 2011), el reportero TcMUC se encuentra
expuesto en la superficie de los epimastigotes transfectados, muy posiblemente
anclado por un motivo GPI.
FASE TcMUC

42
Figura 18: Ensayo de citometría de flujo para la población TcMUC. Las poblaciones TcMUC (paneles A), TcSMUG S (paneles B) y TcADAT2 (paneles C) fueron analizadas por citometría de flujo de acuerdo a lo descripto en texto. Los gráficos de SSC vs FSC permiten ver la integridad de los parásitos. Los histogramas de la izquierda, corresponden a la marca observada en ausencia del anticuerpo primario y los histogramas de la derecha fueron obtenidos a través del sistema completo de anticuerpos. Se indica en cada caso los porcentajes de señal positiva en FL1 en el cuadrante 2 (C2), que contiene a los parásitos integros.
2.1.2. Validación del carácter mucínico
Como una primera aproximación para evaluar la glicosilación de TcMUC, las
proteínas tipo mucinas totales de parásitos transfectados con esta construcción y
delipidados en solventes orgánicos fueron purificadas siguiendo un protocolo estándar
de extracción con butanol (Almeida IC, 2001)(Ver M&M, Figura 31). Como muestra la
Figura 19, el análisis por Western blot procesado con el mAb anti‐FLAG reveló especies
reactivas del tamaño de TcMUC (~25 kDa) exclusivamente en las fracciones F2 y F3. En
la fracción F2 puede observarse una banda reactiva adicional de mayor tamaño (~70
kDa) que podría corresponder a agregados de TcMUC o a formas procesadas
diferencialmente. Análisis similares revelaron el mismo perfil de partición para las
GP35/50 endógenas y para el reportero TcSMUG S (Urban I, 2011), lo que sugiere que,
al igual que estas moléculas, el producto recombinante TcMUC es reconocido como
mucina y glicosilado como tal por la maquinaria del epimastigote. El tamaño de
TcMUC, sin embargo, es significativamente menor del verificado para las tGPI‐mucinas

43
de tripomastigote de las cuales forma parte (60‐200 kDa), sugiriendo diferencias en su
procesamiento en uno y otro estadio.
Figura 19: Extracción de mucinas en parásitos transfectados con TcMUC. Western blot revelado con mAb anti‐FLAG sobre fracciones de extracción butanólica de parásitos delipidados (ver M&M, Figura 31).
Tal como se comentó en secciones anteriores, existen ciertos criterios que
pueden usarse como indicadores de maduración de los O‐glicanos de las mucinas de
los distintos estadios. Uno de ellos es la adquisición de epítopes glicosídicos definidos
que pueden ser reconocidos mediante lectinas o anticuerpos. Para iniciar la
caracterización de los O‐oligosacáridos de TcMUC, se enfrentaron extractos totales de
parásitos transfectantes con los mAb 3C9 (Schenkman S, 1991), que reconoce epítopes
sialilados en tGPI‐mucinas, y 10D8 y 2B10, que reconocen, respectivamente, epitopes
de Galf y Galp terminales en las GP35/50 kDa (Yoshida N, 1989). En ninguno de los 3
casos se observó una banda diferencial del tamaño de TcMUC con respecto al lisado de
epimastigotes de la cepa Adriana sin transfectar (datos no mostrados).
Adicionalmente, se evaluó la capacidad del producto TcMUC de funcionar como
aceptor de AS. Para ello se realizó una marcación de superficie de parásitos TcMUC
intactos utilizando un dador de AS conteniendo un grupo azida y enzima TS
recombinante de T. cruzi, expresada en bacterias (Urban I, 2011). Luego de la reacción,
los parásitos fueron lavados e incubados en presencia de fosfino‐biotina, éste
reacciona de manera covalente con el grupo azida libre generando un neo‐epitope AS‐
biotina (Yu H, 2005). La especificidad y selectividad de esta marcación ya había sido
validada para distintos estadios de T. cruzi (Muiá RP, 2010; Urban I, 2011).
Parásitos TcMUC marcados según este protocolo fueron lisados y los extractos
sometidos a cromatografía de afinidad en una resina de estreptoavidina. Análisis de
Western blot revelados con mAb anti‐FLAG no detectaron ninguna especie reactiva en
las muestras retenidas en la columna (datos no mostrados), sugiriendo que la proteína

44
TcMUC reportera, a diferencia del reportero TcSMUG S (Urban I, 2011), no contiene
residuos βGalp terminales en la configuración apropiada como para actuar de
aceptores de AS en la reacción catalizada por TS.
Estos ensayos indican que el tipo de glicosilación que sufre TcMUC es distinto al
de las tGPI‐mucinas y también al que el que sufren el reportero TcSMUG y las GP35/50
endógenas del estadio epimastigote usado como modelo de análisis. Para intentar
esclarecer la estructura de los oligosacáridos unidos a TcMUC, el producto se
inmunoprecipitó a partir de extractos totales de parásitos transfectados usando
cromatografía de afinidad por el epítope FLAG y será analizado en colaboración con el
grupo de la Dra. Rosa M. Lederkremer, del departamento de Química Orgánica de la
FCEyN‐UBA.
2.2. TcMUC::GFP y TcSMUG::GFP
Si bien para algunos de los clones fue necesario transfectar reiteradas veces
hasta obtener una población recombinante estable, esto no pudo ser posible para el
caso de la construcciones completas o full lenght fusionadas al reportero eGFP
(TcMUC::GFP y TcSMUG::GFP). Resultados (negativos) semejantes habían sido
obtenidos por otros laboratorios al tratar de expresar TcSMUG S fusionada a eGFP
utilizando diferentes vectores de expresión y distintas cepas de T. cruzi (Dra. Rosa
Maldonado, comunicación personal). En la Figura 20 se muestran las diferentes
morfologías encontradas en la población TcSMUG::GFP seleccionada. Más allá de la
baja densidad y poca viabilidad de estos cultivos, se intentó detectar la expresión de
ambas construcciones por Western blot y RT‐PCR, sin ningún resultado positivo (datos
no mostrados).
Durante los últimos años, GFP pudo ser usado exitosamente como un marcador
de diferentes compartimentos celulares en tripanosomátidos (Ha DS, 1996). Mediante
la adición de señales apropiadas de palmitoilación y miristoilación, esta proteína pudo
incluso ser dirigida a la superficie de epimastigotes de T. cruzi (Martins V de P, 2010).
Sin embargo, no existen reportes en donde GFP, ni otros reporteros comúnmente
usados en biología celular, hayan podido ser anclados en la superficie de
tripanosomátidos a través de un motivo GPI (Böhme U, 2002). Las bases moleculares
de este fenómeno son inciertas, pero podría hipotetizarse que las propiedades físico‐
químicas de estas moléculas altera el empaquetamiento de alta densidad de
glicoconjugados en la cubierta de superficie de estos parásitos y, por lo tanto,
compromete su viabilidad.

45
Figura 20: Morfologías observadas en la población TcSMUG::GFP.
2.3. TcMUC∆SP∆GPI::GFP y TcSMUG∆SP∆GPI::GFP Ensayos de microscopía de fluorescencia e IFI mostraron que los epimastigotes
transfectados con los constructos TcMUC∆SP∆GPI::GFP y TcSMUG∆SP∆GPI::GFP presentan una señal intensa (de FLAG y de eGFP), de distribución homogénea a lo
largo de las células, lo que sugiere en principio una localización citoplasmática para
estos reporteros (Figura 21A). Ensayos de co‐localización con la proteína Poly(A)‐
binding protein (TcPABP), de localización citoplasmática en epimastigotes no
sometidos a estrés nutricional (D'Orso I, 2002; Gingras AC, 1999), refuerzan esta idea
(Figura 21B).
Para obtener una confirmación de bases bioquímicas, se llevó adelante un
fraccionamiento subcelular basado en centrifugaciones secuenciales en
concentraciones crecientes del detergente NP‐40 (ver M&M, Figura 30) (Labriola CA,
2010). Este protocolo permite obtener una fracción citoplasmática (C), una fracción
enriquecida en microsomas (R) y un pellet final conteniendo los núcleos celulares y
componentes del citoesqueleto, el cual no fue analizado. La resolución de nuestro
fraccionamiento fue evaluada a través de la técnica de Western blot, usando moléculas
marcadoras de la fracción citoplasmática (TcPABP) y microsomal (BiP),
respectivamente. La Figura 21C muestra reactividad para TcPABP restringida a la
fracción citoplasmática mientras que la reactividad para BiP se observa en ambas
fracciones, aunque enriquecida en la fracción microsomal. Esto indica que existe cierta
contaminación desde la fracción microsomal hacia el sobrenadante citoplasmático
pero no en el sentido opuesto. Fraccionamientos de parásitos transfectantes seguidos
de ensayos de Western blot con el mAb anti‐FLAG indicaron que tanto
TcMUC∆SP∆GPI::GFP como TcSMUG∆SP∆GPI::GFP localizan exclusivamente en la
fracción citoplasmática (Figura 21C). Ambos reporteros migran en SDS‐PAGE como
bandas discretas de ~55 y ~45 kDa, respectivamente, lo cual es compatible con el
tamaño predicho in silico sin MPT (Figura 7). En conjunto, los resultados de
microscopía y fraccionamiento subcelular con NP‐40 indican que en ausencia del SP y
de la señal de anclaje por GPI, las proteínas recombinantes quedan excluidas de la vía
secretoria.

46
Figura 21: Localización sub‐celular de los reporteros TcSMUGΔSPΔGPI::GFP y TcMUCΔSPΔGPI::GFP. A; Imágenes de microscopía de fluorescencia y fase de las poblaciones transfectantes TcSMUG
ΔSPΔGPI::GFP a la izquierda y TcMUC ΔSPΔGPI::GFP en los paneles de la derecha. B; Ensayo de co‐
localización con el marcador citoplasmático TcPABP para la variante TcSMUG∆SP∆GPI::GFP. C; Análisis
por Western blot de las fracciones citoplasmáticas (C) y microsomal (R) obtenidas de los parásitos
indicados arriba de cada panel y revelados con los anticuerpos indicados debajo.
C
B TcSMUGΔSPΔGPI::GFP PABP MERGE FASE
A TcSMUGΔSPΔGPI::GFP TcMUCΔSPΔGPI::GFP FASE FASE

47
2.4. TcSMUG∆SP::GFP y TcMUC∆SP::GFP La localización subcelular del reportero TcSMUG∆SP::GFP se investigó
primeramente mediante la técnica de IFI (Figura 22). Las imágenes reflejan un patrón
de puntos discretos o focos de agregación de variados tamaños distribuidos por toda la
célula. Careciendo de un SP y de acuerdo a lo observado para las construcciones de
tipo ΔSPΔGPI (ver sección anterior), puede suponerse que esta proteína también se
encuentra excluída de la vía secretoria.
Para analizar mejor la localización de este reportero, utilizamos nuevamente el
método de fraccionamiento subcelular basado en NP‐40. Como muestra la Figura 23A,
la proteína TcSMUG∆SP::GFP migra como una banda citoplasmática (C) discreta de ~55 kDa, compatible con la predicción in silico. Análisis de extracción con Tx‐114 indican
que este reportero particiona en distintas fracciones acuosas (S1 y S2) y en el pellet 1
(P1), pero no en la fracción S4. Estos datos indican que la señal de anclaje por GPI
presente en esta molécula no es procesada (Fig 23B), lo cual es compatible con su
exclusión aparente de la vía secretoria. Las bandas de menor tamaño reactivas con el
mAb anti‐FLAG en la Fig 23B, podrían atribuirse a proteólisis parcial del reportero
durante el experimento de extracción. En cuanto a la presencia de especies reactivas
en la fracción P1, podría proponerse que corresponden a los focos discretos
observados en los ensayos de IFI, y podrían deberse a agregados de localización
citoplasmática del reportero debidas a la presencia de la señal de GPI, altamente
hidrofóbica. Ensayos adicionales serían necesarios para evaluar esta posibilidad.
Figura 22: Ensayo de microscopía de fluoresencia e IFI sobre los clones TcSMUG∆SP::GFP. Se observa la co‐localización de señales de eGFP y FLAG como puntos discretos en los parásitos.
Estos resultados, en conjunto, indican que la proteína TcSMUGΔSP::GFP es
fundamentalmente citoplasmática. Esta observación permite concluir, además, que la
señal de anclaje por GPI es incapaz de redireccionar al reportero hacia la vía secretoria
per se (en ausencia de SP).
GFP MERGE FASE TcSMUGΔSP::GFP

48
Figura 23: Caracterización bioquímica del reportero TcSMUG∆SP::GFP. A; Fraccionamiento subcelular con NP‐40, se presentan las fracciones citoplasmática (C) y microsomal
(R). B; Extracción en Tx114 (TX‐114). A y B fueron revelados con mAb anti‐FLAG.
En cuanto al reportero TcMUC∆SP::GFP, la expresión del mismo no pudo ser
detectada a través de las técnicas de Western blot, IFI o microscopía de fluorescencia
ya descriptas. Los epimastigotes transfectados, sin embargo, lograron constituirse
como una población estable en presencia del agente de selección G418.
Para verificar la transcripción del constructo, se realizó una reacción de RT‐PCR a
partir de ARN total de parásitos transfectados (Figura 24). Brevemente, se realizó
primero una reacción de retrotranscripción sobre el molde de ARN total utilizando un
primer Oligo dT y la enzima transcriptasa reversa y luego, sobre ese templado, una
amplificación por PCR utilizando un oligonucleótido complementario a la secuencia del
mini‐exon o spliced leader en combinación con un primer específico del clon
transfectado (RvFLAG Hind, ver ANEXO B). Como resultado se obtuvo una banda única
de amplificación de tamaño esperado (~450 bp) en la calle experimental, la cual está
ausente en la calle correspondiente a la reacción control, en donde no se adicionó el
templado. Este resultado es compatible con la presencia del ARNm específico para el
constructo TcMUC∆SP::GFP en los parásitos transfectantes y sugiere que la falta de expresión del reportero se debe a fenómenos post‐transcripcionales.

49
Figura 24: RT‐PCR de la población TcMUC∆SP::GFP. Gel de agarosa conteniendo las muestras de RT‐PCR obtenidas según se indica en texto. Se indica la
banda de 400 bp del marcador de peso molecular .
2.5. TcMUC∆GPI::GFP y TcSMUG∆GPI::GFP
En ensayos de IFI, los reporteros TcMUC∆GPI::GFP y TcSMUG∆GPI::GFP presentan un patrón común, dado por múltiples focos discretos a lo largo del toda la
célula, aunque concentrados en la región perinuclear (Figura 25). De acuerdo a lo
observado en otros trabajos (Wilkinson SR, 2002), este patrón es compatible con una
localización en el RE.
Para evaluar esta posibilidad se realizó un análisis de co‐localización con la
chaperona BiP de acuerdo a lo descripto (Figura 26). En este caso, se observa en verde
la marcación de FLAG y en rojo la señal generada para BiP. De esta manera, la señal
que pudiera generar el dominio eGFP presente en los reporteros se ve solapada con la
producida por la marcación de FLAG. Cabe aclarar, de todas maneras, que la señal
emitida por la proteína eGFP para estos reporteros resultó ser muy débil
posiblemente debido a su localización en RE. Se conoce que, en otros sistemas, la
traslocación de GFP al RE conlleva problemas de plegamiento y/o agregación que
comprometen su emisión de fluorescencia (Aronson DE, 2011).
400 pb

50
Figura 25: Ensayo de IFI sobre las poblaciones TcSMUG∆GPI::GFP y TcMUC∆GPI::GFP. Epimastigotes TcSMUG∆GPI::GFP (paneles A) o TcMUC∆GPI::GFP (paneles B) fueron tratados con al mAb
anti‐FLAG y un anticuerpo secundario conjugado al fluorocromo Alexa 568. El núcleo y el kinetoplasto se
tiñeron con DAPI.
Como se muestra en las Figuras 26A y B, existe una co‐localización casi total entre
las moléculas recombinantes –TcMUC∆GPI::GFP y TcSMUG∆GPI::GFP ‐ y BiP. Es interesante observar que en la población seleccionada permanecen aún parásitos no
transfectantes sobre los cuales podemos observar, a modo de control interno, el
mismo patrón de localización de BiP. Ensayos de co‐localización con calreticulina, otro
marcador del RE (Labriola CA, 2010), arrojaron similares resultados (datos no
mostrados). Ensayos de co‐localización con marcadores de otros compartimientos de
la vía secretoria como GRASP (de Golgi, (Yelinek JT, 2009)) y cruzipaína (de
reservosomas, (Cazzulo JJ, 1999), en cambio, mostraron un solapamiento mínimo con
la señal de los reporteros (Figura 26C).
B
A TcSMUG∆GPI::GFP
TcMUC∆GPI::GFP FASE
FASE
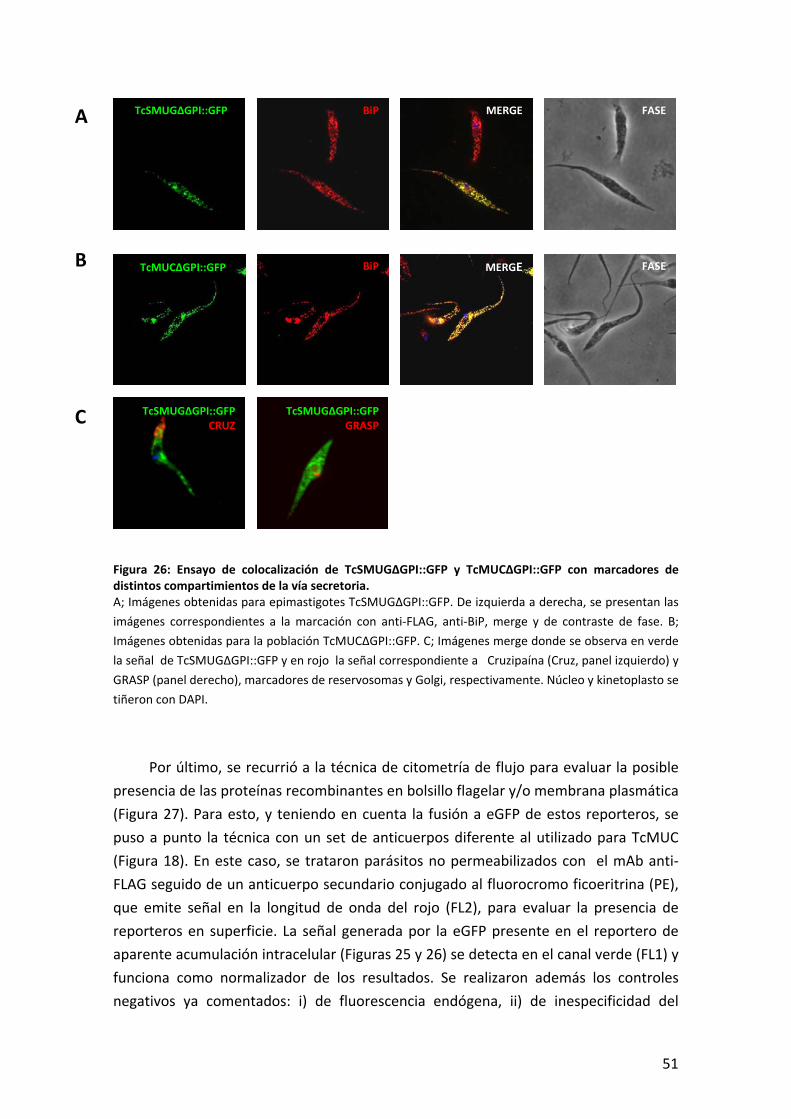
51
Figura 26: Ensayo de colocalización de TcSMUG∆GPI::GFP y TcMUC∆GPI::GFP con marcadores de distintos compartimientos de la vía secretoria. A; Imágenes obtenidas para epimastigotes TcSMUG∆GPI::GFP. De izquierda a derecha, se presentan las
imágenes correspondientes a la marcación con anti‐FLAG, anti‐BiP, merge y de contraste de fase. B;
Imágenes obtenidas para la población TcMUC∆GPI::GFP. C; Imágenes merge donde se observa en verde
la señal de TcSMUG∆GPI::GFP y en rojo la señal correspondiente a Cruzipaína (Cruz, panel izquierdo) y
GRASP (panel derecho), marcadores de reservosomas y Golgi, respectivamente. Núcleo y kinetoplasto se
tiñeron con DAPI.
Por último, se recurrió a la técnica de citometría de flujo para evaluar la posible
presencia de las proteínas recombinantes en bolsillo flagelar y/o membrana plasmática
(Figura 27). Para esto, y teniendo en cuenta la fusión a eGFP de estos reporteros, se
puso a punto la técnica con un set de anticuerpos diferente al utilizado para TcMUC
(Figura 18). En este caso, se trataron parásitos no permeabilizados con el mAb anti‐
FLAG seguido de un anticuerpo secundario conjugado al fluorocromo ficoeritrina (PE),
que emite señal en la longitud de onda del rojo (FL2), para evaluar la presencia de
reporteros en superficie. La señal generada por la eGFP presente en el reportero de
aparente acumulación intracelular (Figuras 25 y 26) se detecta en el canal verde (FL1) y
funciona como normalizador de los resultados. Se realizaron además los controles
negativos ya comentados: i) de fluorescencia endógena, ii) de inespecificidad del
TcSMUGΔGPI::GFP BiP MERGE FASE
TcMUCΔGPI::GFP BiP MERGE FASE
A
B
C TcSMUGΔGPI::GFP CRUZ
TcSMUGΔGPI::GFP GRASP

52
anticuerpo primario, sobre un cultivo de parásitos sin transfectar, y iii) de
inespecificidad del anticuerpo secundario en ausencia del primario. A partir de estos
controles se fijaron los cuadrantes, tal que estos eventos sean visualizados como
negativos para la señal de GFP (FL1) y de PE (FL2), en un 99%.
En la Figura 27 se presentan los Dot blots obtenidos en estas condiciones para las
poblaciones TcSMUG∆GPI::GFP, TcSMUG∆SP::GFP y TcSMUG, de nuevo usado a modo
de control positivo de localización superficial. Como puede observarse, ninguna de las
poblaciones de parásitos transfectados con reporteros carecientes de señal de SP o de
anclaje por GPI, presentó reactividad significativa para FLAG, indicando que los mismos
no se encuentran expuestos en superficie. La expresión de todos estos reporteros en
las células analizadas (salvo TcSMUG), sin embargo, queda demostrada por la
presencia de alrededor de un 23‐26% de eventos GFP positivos.
TcSMUG
10 0 10 1 10 2 10 3 10 0 10 1 10 2 10 310 0 10 1 10 2 10 3
TcSMUG∆GPI::GFPTcSMUG∆SP::GFP
FL1
FL2
Figura 27: Citometría de flujo para TcSMUG∆GPI::GFP y TcSMUG∆SP::GFP. Para cada Dot blot el eje FL1 correponde a la señal de GFP, mientras que en el eje FL2 se observan
parásitos marcados superficialmente con mAb anti‐FLAG seguido por un anticuerpo secundario
conjugado a PE. Se señala dentro de cada gráfico el porcentaje de eventos correspondiente a cada
cuadrante.
Ensayos de fraccionamiento subcelular con NP‐40 revelaron la presencia de los
reporteros TcSMUG∆GPI::GFP y TcMUC∆GPI::GFP en la fracción microsomal (Figura
28A), lo que es coincidente con los datos de microscopía. Las productos
TcSMUG∆GPI::GFP y TcMUC∆GPI::GFP migran como bandas discretas de ~55 y ~70 kDa, respectivamente. Puede verse, además, una serie de bandas reactivas de menor
PM aparente en las fracciones de TcMUC∆GPI::GFP, que podrían corresponder a productos de degradación.
Otra evidencia en favor de una localización intra‐RE sería la presencia de N‐
glicanos sobre la molécula reportera TcSMUG∆GPI::GFP. Para evaluar esta posibilidad, se sometió a un lisado completo de parásitos transfectados con esta construcción a

53
cromatografía de afinidad en columnas de Concanavalina A (Urban I, 2011). Como se
muestra en la Figura 28B, TcSMUG∆GPI::GFP interacciona con esta lectina (fracción unida o B) mientras que TcMUC∆GPI::GFP, que no tiene sitios predichos de N‐glicosilación fue detectada principalmente en la fracción no unida (U). Como control
adicional, se realizó el mismo experimento sobre lisados de parásitos transfectados
con TcSMUG∆SP::GFP. La secuencia del core central de este reportero, incluyendo el sitio consenso para N‐glicosilación, es idéntica a la de TcSMUG∆GPI::GFP pero sin embargo no presenta afinidad por Concanavalina A (Figura 28B, panel derecho). Este
resultado refuerza aún más la exclusión de la vía secretoria del reportero
TcSMUG∆SP::GFP (ver sección 2.4), lo que evitaría el procesamiento de la señal de
adición de N‐glicanos.
El tratamiento del reportero TcSMUG∆GPI::GFP con la glicosidasa PNGasa F genera un pequeño cambio en su movilidad aparente, lo que confirmaría la presencia
del N‐glicano (Figura 28C). En esta misma figura, se presentan los resultados para los
controles positivos serincarboxipeptidasa de T. cruzi (TcSCP)(Urban I, 2011)) y
negativos (TcMUC∆GPI::GFP) incluidos para esta reacción. Estos resultados, en conjunto, indican que el reportero TcSMUG∆GPI::GFP –y de
manera análoga TcMUC∆GPI::GFP‐ se acumula en algún punto de la vía secretoria,
muy probablemente en el RE.
Para evaluar si una fracción de los reporteros ΔGPI pueden eventualmente
escapar a este mecanismo de retención en el RE, se incubaron parásitos
transfectantes en medio BHT sin suero por un lapso de 2 horas y, luego de
centrifugaciónes y lavados, se separó el medio condicionado o sobrenadante (S) del
pellet de epimastigotes, los que fueron lisados en loading buffer para generar los
extractos totales (ET). Estas muestras fueron analizadas por Western blot revelado con
mAb anti‐FLAG. Como controles, se sembraron muestras equivalentes de parásitos
transfectados con el vector pTREX omni vacío, que genera la proteína quimérica FLAG‐
eGFP, también de localización citoplasmática. En este último caso, no se detectaron
especies conteniendo FLAG en el sobrenadante (Figura 29A), lo que indica una tasa
despreciable de lisis celular durante el ensayo. En el caso de los productos
TcMUC∆GPI::GFP y TcSMUG∆GPI::GFP, en cambio, pudieron detectarse bandas tanto
en el extracto total de parásitos como en el medio condicionado, indicando que una
fracción de estos reporteros es secretada activamente. En ambos casos se observó una
serie de bandas de menor tamaño, que en principio podrían atribuirse a productos de
degradación parcial.

54
Figura 28: Análisis bioquímicos de TcSMUG∆GPI::GFP y TcMUC∆GPI::GFP. A; Fraccionamiento subcelular con NP‐40. Se muestran ensayos de Western blot correspondientes a las fracciones citoplasmática (C) y microsomal (R) de los parásitos transfectados con los clones indicados arriba de cada panel y revelados con el anticuerpo indicado debajo de cada panel. B; Fraccionamiento por afinidad a Concanavalina A, se muestran las fracciones de unión (B) y la no retenida por la resina (U). C; Western blot de las muestras de los parásitos transfectados con los clones indicados arriba de cada panel tratadas (+) o no (‐) con la enzima PNGasa F. Los anticuerpos de revelado se indican debajo de cada panel
Un detalle interesante de estos resultados es que, tanto para TcMUC∆GPI::GFP como para TcSMUG∆GPI::GFP, el PM aparente de las formas secretadas es
significativamente mayor que el de las especies presentes en los extractos totales
(Figura 29A). Esta diferencia sugiere que las moléculas que escapan al mecanismo de
retención en RE, antes de ser secretadas, sufren un procesamiento adicional durante
su tránsito por la vía secretoria, posiblemente la adición de O‐glicanos en Golgi.
A fin de evaluar esta posibilidad, se sometió a extractos totales y a
sobrenadantes de ambas poblaciones transfectantes a una inmunoprecipitación
mediante mAb anti‐FLAG y se analizaron las fracciones retenidas por Western blot. Se
utilizó un anticuerpo policlonal anti‐FLAG hecho en conejo, como control positivo y
como marcador de adquisición de O‐glicanos, el mAb 10D8. Como se observa en la
Figura 29B, la forma secretada del reportero TcSMUG∆GPI::GFP, pero no la retenida en RE, presentó reactividad frente al mAb 10D8, indicando la presencia de epitopes
sacarídicos marcadores de mucinas GP35/50 maduras. Resultados muy similares se

55
obtuvieron al revelar estas fracciones con el mAb 2B10 (datos no mostrados). En el
caso de TcMUC∆GPI::GFP, ni la forma retenida en RE ni la secretada presentaron
reactividad frente al mAb 10D8 o 2B10, lo cual es compatible con lo verificado para la
construcción TcMUC full length (ver sección 2.1).
Figura 29: Análisis bioquímico de las formas intra‐ y extra‐celulares de TcSMUG∆GPI::GFP y TcMUC∆GPI::GFP. A; Ensayo de secreción. Se presentan calles correspondientes a un extracto total (ET) de cada muestra y producto secretado (S), precipitado con acetona. pTREXo: Epimastigotes transfectados con el vector pTREX omni vacío. B; Ensayo de inmunoprecipitación (IP) con anti‐FLAG monoclonal. Izq.: Los productos de IP fueron revelados con un antisuero policlonal anti‐FLAG hecho en conejo, como control. Centro: productos de inmunoprecipitación se revelaron con el anticuerpo mAb 10D8. Der.: Se revelaron el extracto total (ET) y producto de secreción (S) para la muestra TcMUC∆GPI::GFP, con anti‐GST‐TcMUC II.

56
Por último, y dado que no disponíamos de un marcador específico de
maduración de oligosacáridos para el producto TcMUC recombinante, evaluamos la
reactividad de las formas intracelulares y secretadas del reportero TcMUC∆GPI::GFP usando un antisuero policlonal anti‐TcMUC generado en el laboratorio (ver sección
1.2.2). La idea era que la maduración del reportero por adición de O‐oligosacáridos
podría correlacionarse con un enmascaramiento de epítopes de base peptídica y, por
lo tanto, comprometiera su reactividad frente a este antisuero. Los resultados
presentados en el Figura 29B, si bien no son cuantitativos ni concluyentes, sugieren
efectivamente una menor reactividad (y por ende un mayor “procesamiento”) de la
forma secretada de TcMUC∆GPI::GFP. En conjunto, estos resultados sugieren fuertemente que los reporteros sin GPI
que escapan el mecanismo de retención putativo en el RE son procesados y secretados
como mucinas maduras. Datos preliminares indican que la forma secretada del
reportero TcSMUG∆GPI::GFP, pero no la forma retenida en RE, pueden incorporar
residuos de AS, lo cual refuerza aún más esta hipótesis (datos no mostrados).

57
DISCUSIÓN Y PERSPECTIVAS
T. cruzi posee un ciclo de vida complejo, que involucra una variedad de
ambientes muy diferentes como el tracto digestivo del insecto vector, la sangre y el
citoplasma celular del hospedador mamífero. Esto implica la necesidad de desarrollar
estrategias de adaptación rápida a condiciones ambientales o nichos diversos y, en
principio, desfavorables. Entre estas estrategias resulta fundamental la remodelación
de su cubierta antigénica y, más precisamente, el recambio estadio‐específico de sus
glicoconjugados de superficie. En este contexto, y considerando que ninguno de sus
muchos puntos diferenciales con respecto a la fisiología del hospedador ha podido ser
capitalizado como blanco terapéutico efectivo para el tratamiento de esta parasitosis,
el estudio de los mecanismos subyacentes a la expresión, procesamiento y secreción
de uno de sus glicoconjugados mayoritarios – las mucinas – resulta relevante.
La existencia de numerosos genes de expresión simultánea, sumado al poco
desarrollo de herramientas para la manipulación genética en este organismo,
dificultan el análisis de las proteínas tipo mucinas en T. cruzi. Para este trabajo, resultó
fundamental contar con una validación previa del sistema experimental elegido, en el
que se había demostrado el correcto procesamiento de todas las señales de tráfico y
MPT presentes en el reportero TcSMUG (SP, GPI, N‐glicosilación, O‐glicosilación)
(Urban I, 2011). En base a estos antecedentes, se planteó como estrategia de estudio
la generación de versiones truncadas en cada una de estas señales de manera
individual, para evaluar el impacto de cada una de ellas en la maduración global del
reportero. Para hacerlo más abarcativo, se planteó el mismo esquema de trabajo para
un miembro representativo de la familia TcMUC, para el cual debimos realizar una
validación previa del procesamiento del reportero full length (ver sección 2.1). Cabe
aclarar que esfuerzos previos por expresar reporteros TcMUC II en superficie de
epimastigotes de T. cruzi resultaron infructuosos, posiblemente debido a la utilización
de sistemas inapropiados de etiquetado y/o expresión (Pollevick GD, 2000).
Del análisis integrado de estos reporteros (sumado a numerosos datos no
mostrados obtenidos a partir de reporteros análogos sin eGFP) pueden desprenderse
algunas conclusiones generales relacionadas al tráfico, procesamiento y maduración
de las proteínas tipo mucina de T. cruzi, las cuales son discutidas a continuación. Más
importante aún, este análisis permite, a su vez, plantear nuevos interrogantes sobre
los que focalizar futuras investigaciones en el área.
1. El SP es fundamental para el ingreso de las mucinas a la vía secretoria
La deleción del SP tanto en las series ΔSP como ΔSPΔGPI llevó a la localización
invariablemente citoplasmática de los reporteros, de acuerdo a lo observado en los

58
análisis bioquímicos y de microscopía. Si bien en este trabajo se analizaron variantes
basadas en un único miembro de las familias TcMUC y TcSMUG, el grado de
conservación de los motivos SP entre los miembros de cada una de las familias
(Buscaglia CA, 2006), permite en principio generalizar esta proposición para el
conjunto de las mucinas de T. cruzi. Este hallazgo, si bien intuitivo y aparentemente
menor, debe ser revalorizado en el contexto de un organismo donde predominan los
mecanismos no convencionales de secreción de moléculas.
2. El procesamiento de las señales de tráfico intracelular es independiente del tipo de
mucina expresado
Nuestros resultados indican que el reportero TcMUC II full length, al igual que un
reportero análogo derivado de TcMUC I (Pollevick GD, 2000), es reconocido como una
apo‐mucina y expuesto correctamente en membrana plasmática a través de un ancla
de GPI. Este hecho sugiere que, más allá de las glicosil transferasas que se discuten en
otra sección, aquellas enzimas involucradas en las rutas de MPT de mucinas del estadio
epimastigote como chaperonas citoplasmáticas y de RE, peptidasas y transamidasas,
son en principio capaces de procesar indistintamente sustratos TcMUC y TcSMUG.
Estos resultados son compatibles con el alto grado de conservación que hay entre las
secuencias N‐ y C‐terminales de estas proteínas (Figura 11). Más importante, estos
resultados sugieren que la expresión marcadamente estadio‐específica de mucinas en
T. cruzi (genes TcSMUG –> GP35/50 en estadios de insecto y genes TcMUC –> tGPI‐
mucinas en estadios de mamífero) opera a nivel post‐transcripcional, con una mínima
o nula contribución de mecanismos de regulación post‐traduccional.
3. Las proteínas tipo mucinas sin GPI se acumulan en vía secretoria
El anclaje de proteínas a través de GPI es una modificación post‐traduccional
ubicua en eucariotas, particularmente prevalente en tripanosomátidos y otros
protozoarios de relevancia médica y veterinaria (McConville MJ, 1993). Más allá de sus
roles estructurales, distintas evidencias indican que el GPI también podría jugar un rol
en el tráfico intracelular de moléculas en distintos sistemas (Maeda Y, 2011). En
tripanosomátidos, diversos abordajes experimentales (deleción o mutación de la señal
de anclaje, bloqueo de la maquinaria de síntesis y/o transferencia de anclas de GPI,
etc.) llevaron, en líneas generales, a la conclusión de que el ancla de GPI actúa como
una señal positiva para la progresión de moléculas dentro de la vía secretoria
(McConville MJ, 1993). Brevemente, los resultados obtenidos, si bien presentan gran
variabilidad de acuerdo al sistema de estudio, indican que los reporteros deficientes en
GPI sufren i) una acumulación en vía secretoria y/o ii) un incremento en su
degradación por diferentes vías metabólicas y/o iii) una disminución parcial o total en

59
sus niveles de secreción. La Tabla 2 muestra un resumen comparativo de todos estos
trabajos.
En el presente trabajo verificamos un fenómeno similar de retención activa en la
vía secretoria de T. cruzi. Nuestros resultados ‐si bien no son concluyentes‐ sugieren
que los reporteros ΔGPI de ambas familias de mucinas se acumulan a nivel del RE y
que, en una pequeña proporción, terminan siendo secretados al medio extracelular
como moléculas “maduras”. La identificación de las bases moleculares subyacentes a
este mecanismo putativo de retención exceden los objetivos del trabajo, pero sería
esperable que presenten sustanciales analogías con las verificadas en estadios
procíclicos de T. brucei, que es el modelo experimental con el que presenta más
paralelismos (Tabla 2). Brevemente, en T. brucei se observa que las reporteros
VSGΔGPI son acumulados en vía secretoria a través de mecanismo de retención que
opera en la interfase entre el RE y el Golgi (REt) y que parece involucrar la segregación
de anclas de GPI hacia vesículas ricas en lipid rafts, de las que son excluídas las
moléculas solubles o conteniendo dominios trans‐membrana. Las vesículas
conteniendo GPI‐glicoconjugados son luego transportadas a membrana plasmática
siguiendo una cinética particular (Silverman JS, 2011). En las formas procíclicas de T.
brucei y en forma análoga a los experimentos presentados en este trabajo, parte de las
VSGΔGPI acumuladas en REt terminan siendo secretadas al medio extracelular
mientras que en las formas sanguíneas del parásito, por el contrario, éstas son
totalmente degradadas en lisosoma (Triggs VP, 2003).
Un detalle relevante es que el hecho de que los reporteros TcSMUG y TcMUC sin
GPI se comporten de manera similar sugiere que, a diferencia de lo que se verifica para
los reporteros VSGΔGPI en procíclicos de T. brucei, la N‐glicosilación cumple un rol
menor o despreciable en el tráfico de las moléculas sin GPI en epimastigotes de T.
cruzi. De todas formas, este punto debe ser analizado más en profundidad mediante el
uso de drogas específicas para esta maquinaria y/o la evaluación de reporteros
TcSMUG delecionados en su sitio aceptor de N‐glicosilación, los que están siendo
desarrollados al momento de presentar este trabajo.

60
Organismo Estadio/
Línea celular
Reportero Tipo de ensayo Localización subcelular/
Procesamiento
Citas
Ssp‐4 y p75 Generación de parásitos
deficientes en GPI por
expresión heteróloga de
GPI‐PLC de T. brucei
Secreción Garg N et al.,
1997
T. cruzi Epimastigote
gp50/55 y
p60
Generación de parásitos
deficientes en GPI por
expresión heteróloga de
GPI‐PLC de T. brucei
Degradación intracelular Garg N et al.,
1997
VSG Deleción de la señal de
GPI
Retículo endoplasmático y
secreción
Triggs VP y
Bang JD,
2003, Bangs
et al., 1997
Procíclico
Prociclina Defecto en la biosíntesis
de GPI
Lisosomas Sampaio
Guther ML,
2009
Wild type Superficie celular Triggs VP y
Bang JD, 2003
Deleción de la señal de
GPI
Retículo endoplasmático y
degradación via lisosoma
Triggs VP y
Bang JD, 2003
VSG
Generación de mutantes
puntuales y de deleción
Retículo endoplasmático y
degradación vía lisosoma
Böhme U
y Cross GA,
2002
Wild type Bolsillo Flagelar Steverding et
al., 1995,
Schwartz KJ et
al., 2005
TfR
Deleción del dominio GPI Lisosomas Biebinger et
al., 2003
Wild type Superficie celular, bolsillo
Flagelar y secreción con
ancla de GPI intacta (50%)
y degradación en lisosoma
(50%)
Schwartz KJ et
al., 2005
Prociclina
Deleción del dominio GPI Lisosomas Schwartz KJ et
al., 2005
p67HP Generación de variante
del marcador de lisosoma
p67 con señal de GPI
Degradación en lisosoma
(más del 50%) y secreción
(15%)
Schwartz KJ et
al., 2005
Wild type Superficie celular y bolsillo
flagelar, secreción con
ancla de GPI intacta
Schwartz KJ et
al., 2005
EPMH
Deleción del dominio GPI Lisosomas Schwartz KJ et
al., 2005
T. brucei
Sanguíneo
BipNHP Generación de variante
del marcador de RE BiP
con señal de GPI
Secreción Schwartz KJ et
al., 2005
Wild type Superficie celular,
independiente de la
glicosilación
Ellis M et al.
2002
Leishmania
mexicana
Promastigote GP63
Defecto en la actividad
GPI‐transamidasa, gp63
delecionada para la señal
de GPI
Secreción dependiente de
la glicosilación
Ellis M et al.
2002

61
Organismo Estadio/
Línea celular
Reportero Tipo de ensayo Localización subcelular/
Procesamiento
Citas
Wild type Superficie celular Delahunty
MD, et al.
1993
Defecto en la biosíntesis
de GPI
Retículo endoplasmático y
degradación via
proteasoma, secreción de
una minoría procesada
proteolíticamente
Delahunty
MD, et al.
1993
CHO Q7
Mutación del sitio ω Retículo endoplasmático y
secreción de una minoría
procesada
proteolíticamente
Delahunty MD
et al., 1993
Wild type Superficie celular en
membrana apical
Brown DA et
al., 1989
Mamífero
MDCK VSV G
Variante asociada a un
dominio transmembrana
Superfice celular en
membrana basolateral
Brown DA et
al., 1989
Saccharomyces
cerevisiae
Gas1p Complementación de
mutantes en la biosíntesis
de GPI por expresión
heteróloga de enzima de T.
brucei
Restauración del transporte forward de
RE‐Golgi
Zhu Y et al.,
2006
Tabla 2: Rol del motivo GPI sobre el tráfico y procesamiento de proteínas en diferentes sistemas.
Los datos genómicos de T. cruzi indican que una significativa proporción (~25%)
de genes TcMUC II presentan codones terminadores de la traducción en su dominio
central, las que han sido arbitrariamente clasificadas como pseudogenes (Di Noia JM,
1995; El‐Sayed NM, 2005a). De manera análoga a lo verificado en las VSG de T. brucei y
en familias de proteínas de superficie de Anaplasma marginale (Brayton et al., 2002),
se ha especulado que estos “pseudogenes” podrían proveer un pool de variabilidad
genética adicional para la generación de nuevas variantes de mucinas a través de
mecanismos de recombinación o conversión génica, y que por eso habrían sido
retenidas en el genoma de T. cruzi (El‐Sayed NM, 2005a). Sin embargo, la expresión de
algunos de ellos a nivel de ARNm ha sido demostrada, por lo que podrían generar in
vivo productos estructuralmente relacionados a los reporteros ΔGPI presentados aquí
(Di Noia JM, 1998). La acumulación en RE y/o la secreción constitutiva de estas
moléculas podría cumplir algún rol particular en la fisiología del parásito o en su
interacción con el hospedador.
4. Las mucinas sin GPI que logran escapar del mecanismo de retención en la vía
secretoria son procesadas y secretadas como moléculas maduras
Como se mostró, una cierta proporción de los reporteros ΔGPI logra escapar al
mecanismo de retención activa propuesto, y terminan siendo secretados como
moléculas “maduras”, entendiendo por esto la adquisición de epitopes sacarídicos

62
distintivos del reportero full length correspondiente o de las mucinas endógenas del
estadio correspondiente del parásito. En el caso de la serie TcMUC, dado que el
reportero full length no expone marcadores sacarídicos distintivos de las tGPI‐mucinas
ni tampoco de las GP35/50, se tomó como criterio de “maduración” el incremento de
su tamaño aparente y la desaparición parcial de epítopes peptídicos accesibles en la
molécula secretada.
La secreción de moléculas “maduras” sugiere que la topología (soluble y no
anclado a membrana) o directamente la ausencia del GPI en los reporteros ΔGPI no constituye un impedimento para su correcto reconocimiento y procesamiento por las
glicosiltransferasas del Golgi o para su progresión ulterior en vía secretoria. Una
posibilidad interesante, no analizada aquí, es que la propia adición de O‐glicanos
constituya una señal positiva o negativa de localización sub‐celular o secreción de los
reporteros, de manera análoga a lo observado en otros sistemas (Boisrame A, 2011;
Ellis M, 2002a; Ellis M, 2002b; Kinlough CL, 2011). De vuelta, el uso de ciertas drogas
y/o la generación de mucinas modificadas en su región central serían fundamentales
para explorar esta hipótesis.
5. La O‐glicosilación de mucinas está modulada por determinantes en cis
(secuencias/estructuras de las apo‐mucinas) y en trans (glicosil transferasas)
De acuerdo a nuestros resultados y otros reportes previos (Pollevick GD, 2000;
Urban I, 2011), la maquinaria del estadio epimastigote de T. cruzi es capaz de
reconocer como apo‐mucinas y procesar en consecuencia a reporteros de distintas
familias, independientemente del perfil de expresión de estas moléculas in vivo. El tipo
de O‐glicosilación de estos reporteros, sin embargo, es diferencial, lo que sugiere la
acción de pooles distintos (al menos parcialmente) de glicosil transferasas sobre cada
uno de ellos. Teniendo en cuenta que los niveles de expresión de estos reporteros no
presentan diferencias significativas, estas variaciones parecen ser determinadas por
motivos estructurales (relación T/S, presencia de repeticiones o tractos de T, contenido
de P, etc.) presentes en la región central de los mismos (elementos en cis). El nivel de
discriminación del sistema es bastante exquisito, ya que variaciones estructurales
mínimas, tales como las verificadas entre productos TcSMUG S y TcSMUG L gatillan
distintos arreglos de oligosacáridos (Urban I, 2011).
El hecho de que la glicosilación de TcMUC presente diferencias con las que se
verifican in vivo para las tGPI‐mucinas del tripomastigote de las cuales forma parte
(Buscaglia CA, 2004), además, sugiere la expresión de pooles distintos (al menos
parcialmente) de glicosil transferasas en los diferentes estadios morfológicos del
parásito. En este sentido, cabe mencionar que el genoma de T. cruzi contiene más de
50 glicosil transferasas putativas, de las que al menos 12 están anotadas como Gal‐
transferasas (El‐Sayed NM, 2005a).

63
En base a todo lo anterior podría concluirse que el perfil de glicosilación de
proteínas tipo mucina en T. cruzi está determinado por la estructura
primaria/secundaria de los motivos ricos en T/S/P de las apo‐mucinas y por el pool
estadio‐específico de glicosil transferasas disponible. En general, el procesamiento de
glicoproteínas en eucariotas está gobernado por el fenotipo de la célula o factores en
trans (es decir el nivel de expresión de exoglicosidasas, glicosiltransferasas, sustratos,
etc.) y la estructura de la propia proteína alrededor del sitio de glicosilación, como
factor de regulación en cis (Rudd PM, 1997; Varki A, 1998).
6. Perspectivas
Frente a las propiedades de T. cruzi como patógeno y los más de 100 años de
investigación en el tema, se prefigura aún como difícil el desarrollo de una vacuna de
acción profiláctica. En este contexto, sigue siendo importante avanzar en nuestro
conocimiento sobre los procesos y particularidades biológicas de este parásito, tal
como las abordadas en este trabajo.
Nuestros estudios serán completados en el corto plazo por la inclusión de clones
complementarios. Tal es el caso de las variantes quiméricas sin el marcador GFP
(Figura 7), para los cuales ya tenemos algunos datos preliminares que son en líneas
generales coincidentes con los presentados aquí, o los mutantes de N‐glicosilación de
TcSMUG. En función de su análisis, podremos evaluar con mayor precisión,
respectivamente, el impacto de la GFP sobre el procesamiento, las propiedades físicas
o la estabilidad de los reporteros y el rol de la N‐glicosilación en el tráfico de proteínas
tipo mucinas. En el mediano plazo, esperamos utilizar los diversos reactivos generados
en este trabajo (variantes de mucinas, anticuerpos, parásitos recombinantes) como
herramientas de análisis para el desarrollo de estudios funcionales. Entre los posibles
caminos a seguir, uno de los más interesantes podría ser el estudio de la capacidad
infectiva de distintos estadios del parásito sobre‐expresando mucinas de diferentes
familias. En la misma línea, aunque sin requerimientos de diferenciación previa de los
epimastigotes transfectantes, podría estudiarse la capacidad y naturaleza de la
adhesión de los transfectantes a células del tracto digestivo de distintos insectos
vectores. Por último, la obtención de moléculas secretadas, aparentemente maduras y
portando etiquetas moleculares, podría facilitar la identificación de ligandos de
superficie y/o el estudio detallado de la señalización intracelular gatillado por mucinas
de T. cruzi.

64
MATERIALES Y MÉTODOS
Soluciones generales • Tris‐Glicina‐SDS 10X: (1 litro) 30,3 g Tris Base, 144 g glicina, 1% (v/v) SDS. • Solución de siembra para muestras de proteínas (CB 5X): 50% (v/v) glicerol, 7,7 %
(p/v) DTT, 10% (p/v) SDS, 0,4 M Tris‐HCl pH 6,8, 0,002% (p/v) azul de bromofenol. • Azul de Coomassie: 0,25% (v/v) Coomassie R250, 30% metanol, 10% ácido acético. • Solución de transferencia: Tris‐Glicina‐SDS 1X, 20% metanol. • TBS: 150 mM NaCl, 50 mM Tris‐HCl (pH 7). • T‐TBS: TBS, 0,05% Tween 20. • PI: 25 mM Tris‐HCl (pH 8), 10 mM EDTA (pH 8), 50 mM glucosa. • PII: 0,2 N NaOH, 1% SDS. • PIII: 3M acetato de potasio pH 4,8. • TBE 10X: 0.45 M Tris‐Borato, 0.01 M EDTA pH 8.3. • TAE: 40 mM Tris‐Acetato, 1mM EDTA. • Solución de siembra para muestras de ADN (LB‐6X): 0,25% (p/v) Azul de
Bromofenol, 0,25 % (p/v) Xilen Cianol, 30% glicerol. • Solución de lisis para parásitos: TBS, 1% TRITON X‐100, 1 mM fenilmetilsulfonil
fluoruro (PMSF), 1 µM trans‐Epoxisuccinil‐L‐leucilamido (4‐guanidino) butano (E‐64).
• Solución Inoue: (1 litro) 10,88 g MnCl2‐4H2O, 2,2 g CaCl2, 18,65 g KCl, 10 ml PIPES (0,5 M pH 6,7), H2O a un litro. Esterilizar por filtrado.
• PBS: 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7. • Buffer GPI: 10 mM Tris‐HCl pH 7.4, 150 mM NaCl, 2% TX‐114, 1 µM E64. • PIC: Fenol:cloroformo:alcohol isoamílico, 25:24:1, equilibrado con Tris. • TE: 10 mM Tris‐HCl pH 8, 1 mM EDTA. Manipulación de bacterias Cepas de Escherichia coli utilizadas en el trabajo
DH5α: F‐ φ80lacZΔM15 Δ(lacZYA‐argF)U169 recA1 endA1 hsdR17(rk‐, mk+) phoA supE44 thi‐1 gyrA96 relA1 λ‐. Cepa de bacterias empleada en la manipulación de ácidos nucleicos. BL21(DE3): F‐ ompT hsdSB (rB‐mB‐) gal dcm (DE3). Cepa de bacterias empleada para la expresión de proteínas recombinantes. Es nula para proteasas y lisógena para el derivado del bacteriófago lambda DE3. Este último codifica para la ARN polimerasa del bacteriófago T7, cuya transcripción es regulada por el represor lac. Se empleó una cepa previamente transformada con el siguiente plásmido: pRIL: Este plásmido confiere resistencia a cloranfenicol y contienen el origen P15a. Codifican numerosos ARNs de transferencia para codones de baja frecuencia en E. coli. Asisten en la traducción de proteínas codificadas en vectores de expresión cuando el uso de codones se aparta significativamente.

65
Medios de cultivo para bacterias ‐ Luria Bertani (LB): Se disuelven: 10 g Triptona de caseína, 5 g extracto de levadura y 5 g NaCl en 1 litro de H2O mQ. Se autoclava a 121ºC por 20 minutos. ‐SOB (Para obtener bacterias competentes): Se disuelve: 20 g Triptona de caseína, 5 g extracto de levadura, 0,5 NaCl g, 0,186 g KCl en 950 ml de H2O mQ. Se ajusta el pH a 7 con una solución de 5 N NaHO. Se lleva a 1000 ml. Se autoclava a 121ºC por 20 min. Antes de utilizar se adicionan 5 ml de una solución de 2 M MgCl2 estéril. ‐LB‐Agar: Se prepara LB y se adiciona agar 20 g/l antes de autoclavar. Posteriormente se deja enfriar a 50ºC y se adicionan los antibióticos (opcional). Se deja solidificar en placas de Petri. ‐Antibióticos y concentraciones de utilización: Los antibióticos fueron suplementados a las siguientes concentraciones finales: Ampicilina: 100 mg/ml. Cloranfenicol: 40 mg/ml. Preparación de bacterias competentes DH5α o BL21(DE3)
A partir de una placa fresca de DH5α o BL21 se picó una colonia aislada y se inoculó a 5 ml de LB en erlenmeyer de 125 ml. Se creció con agitación a 37ºC hasta una DO600nm= 0.3. Luego, se pasaron 5 ml de este cultivo a 100 ml de SOB en un erlenmeyer de 1.000 ml. Se creció con agitación a 37ºC hasta una DO600nm = 0.44 y luego se colocó en hielo por 10 min. Se centrifugó a 3.000 rpm por 5’ a 4ºC. Se descartó el sobrenadante. El pellet se resuspendió cuidadosamente en 40 ml de solución Inoue fría y se dejó en hielo 5’. Luego se centrifugó a 3.000 rpm por 5’ a 4ºC y se descartó el sobrenadante. El pellet se resuspendió cuidadosamente y siempre en hielo con 4 ml de Inoue preenfriado. Se incubó en hielo por 15’. Las células se alicuotaron en tubos eppendorfs estériles en volúmenes de 50 a 200 μl, se congelaron inmediatamente en nitrógeno líquido y se guardaron a – 70ºC hasta el momento de usar. Por transformación se utilizaron 50 μl. Adaptado de (Sambrook J, 2001). Cultivo y transformación de bacterias
Para la amplificación de las diferentes construcciones se transformaron bacterias competentes de E. coli, de la cepa DH5α. Las bacterias se transformaron con la mitad de la reacción de ligación, mediante la técnica de shock térmico, manteniendo al cultivo en hielo por 30 min. para luego llevarlo a 42ºC por 90 seg. y hielo nuevamente. Se dejaron recuperar finalmente en medio LB por 1 hr a 37ºC. Del total del volumen de recuperación se sembraron 150 µl en placas de LB con el antibiótico correspondiente para ser incubadas en cuarto de cultivo a 37ºC. Para el caso de expresión de proteínas recombinantes, se transformaron bacterias de la cepa BL21, utilizando el mismo protocolo.

66
Técnicas moleculares Técnicas analíticas de ácidos nucleicos Reacción en cadena de la polimerasa (PCR)
Se realizaron reacciones de PCR utilizando tanto las enzimas Taq DNA polimerasa (Invitrogen®) o Pfu DNA polimerasa (Fermentas®). Se llevaron a cabo con una concentración de alrededor de 1.5 mM de MgCl2, 20 ng por reacción de los desoxinucleótidos dATP, dTTP, dCTP y dGTP, y 500 ng de ADN total de epimastigotes en un volumen final de 25 μl. La temperatura de elongación fue la adecuada para cada polimerasa (72ºC Taq y 68ºC Pfu). La temperatura de anidado y el tiempo de elongación utilizada para cada reacción fue la sugerida por el programa Vector NTI v10 de Invitrogen®, teniendo en cuenta las velocidades de incorporación de nucleotidos por minuto de las enzimas utilizadas (1000 y 500 nucleótidos por minuto para la enzima Taq y Pfu, respectivamente). Los oligonucleótidos empleados para cada construcción realizada, se detallan en la Tabla 1 del ANEXO B. En el caso de los ensayos de Colony PCR, se procedió de igual forma, repicando las colonias evaluadas en medio LB Agar, suplementado con el antibiótico correspondiente. Las estrategias de clonado de las construcciones realizadas en el siguiente trabajo se encuentran en el ANEXO A. Digestión con enzimas de restricción
Para todas las estrategias de clonado empleadas, se utilizó alrededor de 1U de enzima por μg de ADN a tratar, utilizando el buffer óptimo para cada una de acuerdo a las condiciones informadas por el fabricante (New England Biolabs® o Fermentas®), incubando por períodos de 2‐3 hr. Para el caso de digestiones dobles con enzimas no compatibles, se llevó la primer digestión a un volumen de 100 µl, agregando luego 0.1 volumen de solución PIII y 2.5 volúmenes de 100% etanol, manteniendo las muestras en en frio durante 1 hr. Para precipitar el ADN, se lo centrifugó a 13.000 rpm durante 20 minutos. Luego de lavar el pellet en 70% etanol, se lo resuspendió en la mezcla de reacción de la segunda endonucleasa. Ligación de fragmentos de ADN
Para todas las ligaciones de fragmentos se utilizó la enzima DNA ligasa, del bacteriófago T4 (New England Biolabs®), 0,4 U por reacción, y su buffer correspondiente, realizando las incubaciones overnight a 4ºC o bien a temperatura ambiente y una incubación de 3‐4 hrs. Las reacciones se realizaron en un volumen final de 10‐30 μl, utilizando 50 ng de vector y la cantidad necesaria de inserto en una relación 1:3 (vector:inserto). Los vectores digeridos fueron previamente tratados con fosfatasa alcalina intestinal (Promega®), utilizando 1U de enzima por restricción. Electroforesis en geles de agarosa
Los fragmentos de ADN generados a través de las diferentes restricciones fueron sembrados con LB‐6X y separados en geles de agarosa típicamente al 1% para geles de rutina, conteniendo 0,5 μg/ml bromuro de etidio para su revelado por exposición a luz ultravioleta (Sambrook J, 2001). Las corridas se llevaron a cabo bajo voltajes constantes de 5 V/cm durante aproximadamente 1h. Se utilizó buffer TBE 0.5x o bien

67
TAE en aquellos casos donde se requirió una purificación de banda posterior. El ADN fue visualizado por la fluorescencia inducida al exponer el gel teñido con bromuro de etidio a luz ultravioleta. Purificación de fragmentos de ADN de geles de agarosa
Para la purificación de ADN a partir de un taco de gel se recurrió al kit de extracción HiYield Gel/PCR DNA (Real Genomics®), basado en la retención por afinidad del ADN en columnas de silica en altas concentraciones de sal, según indicaciones del fabricante. Aislamiento de ADN plasmídico
Todas las purificaciones se realizaron por el método de lisis alcalina (Sambrook J, 2001). Se partió rutinariamente de 3‐5 ml de cultivo, el cual se cosechó mediante centrifugación a 4.000 rpm. El pellet obtenido se resuspendió 100 μl en la solución PI conteniendo 1 µl de RNAsa A, se agregaron 200 µl de la solución de lisis alcalina PII y se agregaron luego de agitar por inversión 150 µl de la solución de renaturalización PIII. Luego, se centrifugó durante 20 min. a 13.000 rpm. Al sobrenadante obtenido se le adicionó una solución de Cloroformo‐Alcohol Isoamílico en una relación 24:1. Después de una agitación por inversión se centrifugó, desechando la fase orgánica. Sobre la fase acuosa se realizó la precipitación del ADN plasmídico mediante la adición 1 volumen de isopropanol. El pellet obtenido en la centrifugación se limpió de sales remanentes con 70% etanol y se resuspendió finalmente en agua deionizada. El ADN obtenido se purificó para su secuenciación automática, a través de la incubación en frío con 4 M NaCl y 4% Polietilenglicol 8.000. Luego de una centrifugación a 12.000 rpm se procedió a lavar el ADN precipitado con 70% etanol y posterior resuspensión en agua. En todos los casos se cuantificó y se evaluó la pureza del producto obtenido mediante espectrofotometría, por absorbancia a 260/280 nm. El ADN utilizado para las transfecciones se obtuvo a partir del kit Gene Jet Plasmid Miniprep (Fermentas®) para una mayor pureza de los plásmidos extraídos. Preparación de ADN genómico de tripanosomas
Aproximadamente 108 parásitos en fase logarítmica tardía se centrifugaron a 3.000 rpm durante 5 min. Al precipitado se lo lavo con PBS y se resuspendieron los parásitos en 1 ml de solución de lisis para ADN y se les agregó 0,5 ml de solución PIC. Se mezclo por inversión y se centrifugo a 13.200 rpm por 1 minuto. Se repitió nuevamente el tratamiento con solución PIC. Luego se extrajo con 0,5 ml de cloroformo:alcohol isoamílico (24:1) y se precipito el ADN con el agregado de un volumen de isopropanol centrifugando durante 20 minutos a 13.200 rpm. El precipitado se lavo con 70% etanol y se resuspendieron en 100‐200 μl de solución TE 1X. Purificación de ARN total de tripanosomas
Se utilizó el reactivo comercial TRIzol (Life Technologies®), basado en el método de isotiocianato de guanidina y fenol/cloroformo, según las especificaciones del fabricante. Se utilizo 1 ml de TRIzol por cada 2x108 parásitos. El ARN extraído se cuantificó por espectrofotometría, de acuerdo a su absorbancia a 260 nm.

68
RT‐PCR Para la síntesis de la primera cadena de ADN, se llevaron adelante reacciones con
3 µg de ARN total, con 0.5 mM desoxiribonucleótidos y 25 ng de oligonucleótidos Oligo dT12‐18. Estos oligonucleótidos, contienen a su vez un ancla T7 hacia el extremo 3’ terminal, como sitio de pegado para el primer en una PCR posterior. La reacción se realizó en el buffer provisto por el fabricante, suplementado con 10 mM DTT. Al cabo de una desnaturalización de las estructuras secundarias del templado a 65ºC durante 5 min, se lleva el ARN rápidamente a hielo para luego llevar adelante la reacción adicionando 200 U de Transcriptasa Reversa (SuperScript II, Invitrogen®), con una incubación a 42ºC por 50 min. La enzima se inactiva luego llevando la temperatura a 70°C por 15 min. La PCR se realizó en forma estándar con un 10% del volumen de reacción representado por la mezcla de síntesis de la primera cadena, luego de su tratamiento con RNAsa A por 20 min. a 37ºC. Manipulación de parásitos Cultivo de parásitos
Para todos los ensayos se utilizó una población de epimastigotes de la cepa Adriana de T.cruzi (TcI) (Urban I, 2011), obtenida a partir de un cultivo axénico. Los parásitos fueron mantenidos mediante pases sucesivos cada 3‐4 días, en medio de cultivo monofásico BHT (infusión cerebro‐corazón, 0.3% triptosa, 0.002% hemina bovina 100 U/ml de penicilina y 100 ug/ml de estreptomicina), suplementado con suero fetal bovino inactivado al 10% y/o 20% durante la selección luego de su transfección. Fueron mantenidos a una temperatura de 28ºC sin agitación, los parásitos transfectados se cultivaron en presencia del agente de selección G418 en una concentración de 200 ng/ml. Transfección de parásitos
Para las transfecciones, se utilizaron 150x106 parásitos por muestra. Brevemente, los parásitos en fase exponencial se cosecharon mediante una centrifugación a 3.000 rpm durante 10 min., luego de lo cual se realizaron dos lavados en medio BHT sin suero. Para la electroporación se utilizaron de 20 a 30 µg de ADN, preincubándolo durante 10 min. en hielo con la suspensión de epimastigotes, se transvasó luego todo el volumen a las cubetas de electroporación previamente enfriadas. Las condiciones del electroporador (BTX 630 A) para el pulso fueron 335 V, 24 ohm y 1500 µF, obteniéndose valores de constantes de tiempo entre los 10 y 12 s‐1. Se dejaron recuperar las muestras en hielo durante 5 min., luego de lo cual se inocularon los parásitos en botellas con 3 ml de medio BHT, suplementado con 20% de suero fetal bovino. Para su posterior selección, se adicionó el agente G418, en una concentración de 200 ng/ml a las 24 hr de este tratamiento. Los análisis bioquímicos y de microscopía se realizaron sobre la población total de parásitos (sin previo clonado) al menos 45 días después de iniciada la selección.

69
Fraccionamiento subcelular con NP‐40 Para el ensayo de partición subcelular, se procedió de acuerdo a Labriola et al.
(Labriola CA, 2010). Básicamente, cultivos de T. cruzi en fase exponencial fueron lavados en 0.25 M Sacarosa, 5 mM KCl y los pellets congelados por 24‐48 hrs a ‐20ºC. Luego se procedió al descongelado a 4ºC y resuspención en TBS, 1 uM E‐64. Se centrifugó a 15.000 g durante 10 min. obteniéndose el sobrenadante 1 o fracción citoplasmática y el pellet se procesa luego con el mismo buffer, suplementado con el detergente NP‐40 al 1%. Repitiéndose la centrifugación, se obtiene el sobrenadante 2 o fracción microsomal y el pellet 2 o fracción nuclear, como se detalla en el siguiente esquema:
Figura 30: Esquema del protocolo de fraccionamiento subcelular con NP‐40. Purificación de proteínas tipo mucinas
Se delipidaron 3x109 parásitos con un tratamiento con agua/cloroformo/metanol de acuerdo a lo descrito por Almeida et al. (Almeida IC, 2001). Brevemente, las fracciones soluble e insoluble se evaporaron mediante una corriente de N2, el sobrenadante obtenido fue re‐extraído con butanol 66% en agua a 4°C. La fase de butanol (F1) contiene principalmente lípidos, fosfolípidos y GIPLs, la fase acuosa (F2) se encuentra enriquecida en mucinas. Los pellets delipidados también fueron extraídos en butanol 9%. El sobrenadante se extrajo con butanol 91% obteniéndose una fase acuosa (F3), también enriquecida en mucinas, y una fase butanolica (F4). Los pellets finales obtenidos se resuspendieron en buffer de carga desnaturalizante conteniendo 6M urea. Como se detalla en el siguiente esquema:

70
Figura 31: Esquema del protocolo de purificación de mucinas mediante partición en solventes orgánicos. Purificación de proteínas ancladas por GPI y tratamiento con PI‐PLC
Pellets conteniendo 1.5 x 108 parásitos fueron homogeneizados en 2 ml de buffer GPI e incubados en hielo durante 1 hr, los homogenatos se procesaron de acuerdo a lo descrito en por (Cordero EM, 2009); como se demuestra en el esquema de abajo. Para los tratamientos con PI‐PLC, se tomaron alícuotas de cada fracción parcialmente purificada para precipitarlas con acetona fría y resuspenderlas en 200 μl de Tris‐HCl 10 mM pH 7.2, deoxicolato de sodio 0.1%. Luego fueron tratados con 0,3 U de PI‐PLC de Bacillus cereus (Invitrogen®) por 4 hr a 37°C. A continuación, se precipitaron y resuspendieron las muestras en 200 μl de buffer GPI para someterlas a una nueva separación de fases. Tanto la fase acuosa como la enriquecida en detergente fueron precipitadas con acetona fría y resuspendidas en buffer de carga desnaturalizante conteniendo 6M urea.

71
Figura 32: Esquema del protocolo de extracción de GPI‐proteínas en TRITON X‐114. Ensayo de secreción
Para evaluar la presencia de proteínas recombinantes secretadas, se realizó un lavado en BHT sin suero de 1,5x107 epimastigotes, incubándolos luego a 28ºC, durante 3 hr. en 200 μl del mismo medio. Finalmente se cosecharon los parásitos y el sobrenandante extraído se paso por un filtro de 0.2 μm y se precipito con 5 volúmenes de acetona por 24 hr. a ‐70ºC. Tratamiento con N‐Glicosidasa F
Para deglicosidar 200 μg de glicoproteína, las muestras se llevaron a un volumen de 35 μl con agua, se agregaron 10μl de 250 mM buffer fosfato pH 7.5 y 2.5 μl de SDS 2% con 1M 2‐mercaptoetanol. Se calentaron a 100ºC durante 5 minutos. Luego de enfriarse, se agregaron 2.5 μl de 15% v/v TRITON X‐100. Por último, se agregó 1 μl de PNGasa F de Elizabethkingia meningoseptica (Sigma®) y se incubó por 3 hrs a 37ºC. Marcación de superficie de Epimastigotes con Azido‐Acido Sialico
Epimastigotes (50‐100x106) fueron lavados exhaustivamente con PBS y marcados en presencia de 30 ng de TS recombinante (Risso MG, 2004), 10 mM 2‐deoxiglucosa y 1 mM del análogo de azido‐sialillactosa Neu5Azα2‐3LacßOMe (Urban I, 2011) por 30 min a temperatura ambiente. La reacción fue incubada a 65ºC para inactivar la TS recombinante y marcada luego con 250 μM Fosfino‐Biotina (Sigma®) por 2 hr. a temperatura ambiente. Los parásitos fueron luego procesados según los tratamientos indicados arriba. Paralelamente para la marcación de extractos de secreción se utilizo la misma metodología pero el producto final fue precipitado con 5 volúmenes de acetona por 24 hr. a ‐70ºC.

72
Técnicas analíticas de proteínas e inmunológicas SDS‐PAGE
Las muestras de proteínas fueron corridas en una matriz de acrilamida‐bisacrilamida 29:1 de 10 al 15%. Se sembraron en geles desnaturalizantes mezclándolas en una relación 5:1 con Laemmli Buffer, las corridas se realizaron bajo un voltaje constante de 100 V. Western blot
La transferencia a membrana de PVDF activada con metanol se realizó por medio de un sistema semi‐dry, bajo un voltaje de 15V durante 40 min. en solución de transferencia. Las membranas se bloquearon luego durante una hr. a temperatura ambiente en T‐TBS 5% Leche. A continuación se incubaron las membranas con el anticuerpo específico overnight a 4ºC, en solución de T‐TBS Leche 5%. Al cabo de este tiempo, se llevaron a cabo una serie de 3 lavados de 10 min. en T‐TBS, para incubar luego con un anticuerpo secundario conjugado con peroxidasa (HRP) durante 1 hr. Finalmente, luego de una nueva ronda de lavados, se incubaron las membranas con un sustrato quimioluminiscente (SuperSignal West Pico y Phemto Chemiluminescent Substrate, PIERCE®). La señal quimioluminiscente producida se registró mediante autoradiografía. Las concentraciones de cada anticuerpo utilizado se encuentran en la Tabla 4 del Anexo B. En el caso del ensayo de competencia, se pre‐incubó el antisuero con el antígeno recombinante purificado o con la proteína carrier sin conjugar al péptido, luego se procedió de acuerdo a lo detallado previamente. Dot blot
Para los ensayos de Dot blot se sembraron de 0.05 ng a 1 µg de proteína recombinante en membranas de nitrocelulosa. Luego adsorbidas las muestras, se prosiguió con la misma metodología que se utilizo en los ensayos de Western blot. Inmunoprecipitación
Para los ensayo de inmunoprecipitación se partió de 50‐100x106 parásitos los cuales fueron tratados con solución de lisis de parásitos. Los extractos totales fueron precipitados con una resina acoplada al anticuerpo monoclonal anti‐FLAG (Sigma A2220) según las especificaciones del fabricante. Expresión y purificación de proteínas recombinantes
Las proteínas recombinantes se expresaron como fusión al C‐terminal de la glutatión‐S‐transferasa (GST) de Schistosoma japonicum en el vector pGEX (GE®) modificado según Buscaglia et al. (Buscaglia CA, 2004). Para el clonado del péptido correspondiente a un fragmento de la región N‐terminal de TcSMUG S (VV33VEAGEGQDQ), se realizó la hibridación de los oligonucleótidos TcSMUGLHVs y antisense TcSMUGLHVa diseñados para tal fin, dicha hibridación posee como extremos libres los sitios de union para las enzimas de restricción BamH I y Spe I por lo que se digirió el vector con las mismas endonucleasas. Para el clonado correspondiente un fragmento de la región N‐terminal de TcMUC II se realizo una PCR convencional con los oligonucleótidos muciiab f y muciiab rv (ANEXO B). Los clones resultantes se

73
verificaron mediante su secuenciación utilizando los oligonucleótidos de secuenciación pGEX5´ y pGEX3´ y transformados en la cepa bacteriana BL21 como se indica arriba.
La sobre‐expresión de las proteínas de fusión a GST se realizó a partir de un cultivo bacteriano starter crecido durante una noche en medio selectivo de Ampicilina y Cloranfenicol. Este fue luego diluido 25 veces en 250 ml de LB y crecido a 37ºC con agitación hasta una DO600nm de 0,5. El cultivo fue inducido con el agregado de 250 µM Isopropiltio‐D‐galactósido (IPGT) (Sigma®) e incubado por 12 hr. a 28°C en agitación. El pellet de bacterias cosechado se resuspendió en 10 ml en PBS suplementado con inhibidor de proteasas TLCK, 1 mM PMSF y 5 mM EDTA. Las bacterias se lisaron por sonicación (4 pulsos de 20 segundos en hielo). Se centrifugó a 10.000 rpm durante 10 min. y el sobrenadante se llevo a 1% TRITON X‐100. Para su purificación, se recurrió a una columna de afinidad de 0.5 ml de glutation acoplado a sefarosa (GE®). La proteína de fusión se eluyó con 25 mM glutatión reducido en 50 mM Tris‐HCl pH 7,6 y se dializó contra una solución de PBS. Las fracciones fueron visualizadas por electroforesis en geles desnaturalizantes de poliacrilamida (SDS‐PAGE), junto con el extracto total para evaluar la sobre‐expresión de la proteína recombinante. Obtención de sueros de inmunización
Las proteínas de fusión purificadas (25 μg) se inyectaron en ratones BALB/c machos (de 60‐90 días de vida) en forma intraperitoneal en 1ml de PBS con 1ml de adyuvante completo de Freund (Sigma®), con dos refuerzos de 12,5 μg de proteína en adyuvante incompleto con intervalos de 14 días. Los ratones se sangraron a blanco y los sueros se obtuvieron por centrifugación después de dejar coagular la sangre por 1hr. a 37°C. Inmunofluorescencia indirecta
Se emplearon alrededor de 5x106 epimastigotes por muestra que se lavaron tres veces en PBS y se inmovilizaron sobe cubreobjetos tratados previamente con polilisina 1:10, durante 30 minutos a temperatura ambiente. Se fijaron luego con solución de PBS y Parafolmaldehído al 4% por 20 min. Posteriormente se realizó un lavado adicional en 25mM NH4Cl en el mismo buffer para la disminución de la autofluorescencia de las células. Para el bloqueo se utilizó una solución de BSA 2% con saponina 0.2% o bien sin detergente para observar marcación de membrana plasmática, con una incubación de 1 hr. a temperatura ambiente. Los anticuerpos primarios y secundarios utilizados (ANEXO B, Tabla 2) fueron incubados en solución de bloqueo durante una hr. con 3 lavados con PBS entre cada tratamiento. Luego de tratar con PBS‐DAPI por 10 minutos y una serie de 3 lavados en PBS se montaron sobre un portaobjetos con FluorSave Reagent. Los preparados obtenidos fueron estudiados en microscopio de epifluorescencia Nikon Eclipse E600 y el software Spot Advanced. Citometría de flujo
Los parásitos fueron lavados con PBS y resuspendidos en 50 µl de PBS‐FCS 1% durante 30 minutos para el bloqueo, en baño agua‐hielo. Los anticuerpos primarios y secundarios utilizados (ANEXO B, Tabla 2) fueron incubados en PBS‐FCS 1% por 30 min., con 3 lavados de PBS‐FCS 1% entre cada tratamiento. Luego se realizó un último lavado en PBS‐FCS 1% y se resuspendieron los parásitos en PBS‐Parafolmaldehido 4%. Para la lectura en el citómetro (Partec), las muestras se diluyeron en PBS y se

74
registraron 100.000 eventos para cada una. Los resultados se analizaron con el software WinMDi 2.8 o FlowJo (TreeStar, San Carlos, CA). Análisis bioinformático de secuencias
Las secuencias de genes de mucinas son actualmente de dominio público y se pueden obtener de la baso de datos TrytripDB v2.4 (http://tritrypdb.org/tritrypdb). Para el análisis y edición de estas secuencias se utilizó el software Vector NTI v10 de Invitrogen®. Se utilizó para la identificación de péptido señal el software SignalP 3.0 (CBS, University of Denmark). Asimismo se utilizó la herramienta GPI‐SOM del mismo servidor para la determinación de señales aceptoras de GPI. Para la predicción de los sitios potenciales de glicosilación se recurrió a las herramientas NetOGlyc y NetNGlyc. Todos disponibles entre los utilitarios online en http://www.expasy.ch.

75
ANEXO A: Estrategias de Clonado
Figura 33: Diagrama de las estrategias de clonado utilizadas para el desarrollo de los clones TcSMUG.
XbaI
XbaI
XbaI
pTR
EX
-Om
ni e
G7
141
bp
Neo
gapd
h
AmpR
FLG
FLAG tri
FLAG
EK TEV
HA EEF
eGFP
gapd
h
gapd
h
HX1
FLAG
Rib.
pro
m.
P Bl
a
FwSP
Rv
Cor
e
XbaI
+ Ec
oRI
FwC
ore
FwG
PI
Rv
GP
I
XbaI
+ C
laI
XbaI
+ H
indI
II
XbaI
TcSM
UG
S
TcSM
UG∆G
PI::G
FP
EcoRI
XbaI
XbaI
+ Cl
aI
HindIII
SpeI
GFP
::GPI
(*)
Eco
RI
Hin
dIII
+ Sp
eI
XbaI
Cla
IXb
aI
HindIII
HindIII
XbaI
+ Ec
oRI
TcSM
UG∆S
P∆G
PI::G
FP
TcSM
UG∆S
P::G
FP
TcSM
UG::G
FP
TcSM
UG∆G
PI
TcSM
UG∆S
P∆G
PI
TcSM
UG
XbaI
+ H
indI
II

76
Figura 34: Diagrama de las estrategias de clonado utilizadas para el desarrollo de los clones TcMUC.
XbaI
Hind
III
FwSP
Rv
Cor
e
XbaI
+ Cl
aI
FwC
ore
FwG
PI
Rv
GP
I
XhaI
+ Xh
oI
FwSP
Rv
Flag
Xba
I+ H
indI
II
XbaI + ClaI
Bam
HI
Bam
HI
XbaI
BglI
I
Bam
HI
Bam
HI
Bam
HI
Bam
HI
BamHI
Xba
I + B
glII
pTREX-O
mnieG
7141
bp
Neo
AmpR
FLAG triF
LAG
EK TEV
HA EE
F
eGFP
gapd
hga
pdh
HX1
FLAG
Rib
. pro
m.
P B
la
Hind
III
(18
62)
Xba
I (1
038
)
Xho
I (1
962)
Ba
mHI
(818
)
Ba
mHI
(113
4)
ClaI
(10
57)
ClaI
(10
99)
ClaI
(35
36)
pTREX-O
mnieG
7141
bp
Neo
AmpR
FLAG triF
LAG
EK TEV
HA EE
F
eGFP
gapd
hga
pdh
HX1
FLAG
Rib
. pro
m.
P B
la
Hind
III
(18
62)
Xba
I (1
038
)
Xho
I (1
962)
Ba
mHI
(818
)
Ba
mHI
(113
4)
ClaI
(10
57)
ClaI
(10
99)
ClaI
(35
36)
XbaI + ClaI
TcM
UC∆G
PI::G
FP
GFP
::GPI
(*)
TcM
UC∆S
P∆G
PI::G
FP
TcM
UC∆S
P::G
FP
TcM
UC
::G
FP
TcM
UC∆G
PI
TcM
UC∆S
P∆G
PI
TcSM
UG
TcM
UC∆S
P
TcM
UC
II
Bam
HI

77
ANEXO B: Oligonucleótidos y anticuerpos
Tabla 3: Oligonucleótidos utilizados en el trabajo.

78
Tabla 4: Anticuerpos utilizados en el trabajo.

79
ANEXO C: Secuencias nucleotídicas y aminoacídicas de los clones de trabajo
TcMuc II Cepa RA‐1 (GenBank: U32448.1)
Secuencia nucleotídica
ccactgaacgaccacactcacgATGATGACGACGTGCCGTCTGCTGTGCGCCCTGTTGGTTCTCGTCC
TGTGCTGCTGCCCGTCCGTGTGCATAAAAGCCAATGGTGTCGACGAACAGGTGGCTCCCGATA
TGACAAAGGTACCAGGGGCACAGCTGCCAGTTAAAAAAGAAGTGGAACAAGGCAGAGGAGT
AGAGAAACCACTTATTGTGACCGGTAAGGAAGAAGCACAAGGTACGGTATCCGGTGGGGCG
TCAAACAATCAAGGTGCATCAGGTGCGTCCGGTCAGTCACAGCTACAAAGTCCTAGCGTTCCG
CTACAGCCACCTACTGCACCAACCGTCACGTCATGGGCACAAAGTGGGCCGATTTTACCCGAA
AAAGAACAGGAAAAGGAGACGACTGCATCAACGACCATAACGACCAAAACCACCAAGGCACC
AACCACAACTACGACTACGACCACGACCACCGCCGCGCCAGAGGCACCAAGTGATGCGGTCA
ACAGTGCAGAGGCACCAACCACGACGACCACCCGCGCACCGTCACGTCTTCGCGAAGTTGAC
GGCAGCCTTAGCAGCTCTGCGTGGGTGTGTGCCCCGCTGCTGCTCGCCGCATCCGCGCTGGCG
TACACCGCTGTGGGCTGAaggggagcgtgggctgtgcgtgccagcacttagccacggg
Secuencia aminoacídica predicha
MMTTCRLLCALLVLVLCCCPSVCIKANGVDEQVAPDMTKVPGAQLPVKKEVEQGRGVEKPLIVTG
KEEAQGTVSGGASNNQGASGASGQSQLQSPSVPLQPPTAPTVTSWAQSGPILPEKEQEKETTAST
TITTKTTKAPTTTTTTTTTTAAPEAPSDAVNSAEAPTTTTTRAPSRLREVDGSLSSSAWVCAPLLLAA
SALAYTAVG

80
TcSMUG S – FLAG (GenBank: AY298907.1)
Secuencia nucleotídica
ATGATGCTGCGCCGCGTCCTCTGCGTGCTTTTGTTTGCCCTCTGCTGTGCGTGCGTGTGCGCCA
CGGCTCAGGAGGAGGGGCAGTACGATGCTGCTGTTGTCGAGGCTGGGGAGGGCCAGGATCA
AACTACAACCACCACCACCACAACAACTACTACTACGACGACCACCACCACCACCAATGCACCC
GCCAAGAACACAACCACCTccaaggattacaaggatgacgacgacaaggcaccgactcctgGCGACGGGAG
CGGCCTCGGCGGCCCTTTGTGGGTTCAGGTGCCGCTGCTGCTGCTTGTCAGTGCCGCTGTGGC
GACCGCCGGTGCTGCGTGCTgAATCGAATTC
Secuencia aminoacídica predicha
MMLRRVLCVLLFALCCACVCATAQEEGQYDAAVVEAGEGQDQTTTTTTTTTTTTTTTTTTNAPAK
NTTTSKDYKDDDDKAPTPGDGSGLGGPLWVQVPLLLLVSAAVATAGAAC

81
AGRADECIMIENTOS
• Agustina Chidichimo, Liliana Sferco y Berta Franke de Cazzulo, por millones de
parásitos
• Alejandro Cassola (IIB‐INTECH), por el antisuero anti‐TcPAbP
• Carlos Labriola (FIL), por el antisuero anti‐TcCalreticulin
• Cynthia He (Department of Biological Science, National University of Singapore),
por el antisuero anti‐TbGRASP
• Fabio Fraga, por la manipulación de los ratones inmunizados
• Gastón Ortiz (IIB‐INTECH), por los ensayos de citometría
• Igor C. Almeida y Rosa Maldonado (UTEP, El Paso TX, USA), por los resultados sin
publicar
• Jay Bangs (Department of Medical Microbiology and Immunology, University of
Wisconsin‐Madison, USA), por el antisuero anti‐TbBiP
• León Bouvier y Claudio Pereira (Instituto de Investigaciones Médicas Alfredo
Lanari), por el vector pTREX omni
• Nobuko Yoshida (Universidade de Sao Paulo, Brazil), por los mAbs 10D8 y 2B10
• Rosa Lederkremer y Rosalía Agusti, por el análisis de carbohidratos de TcMUC
• Santiago Bertotti (IIB‐INTECH), por los parásitos transfectantes TcADAT2
• Sergio Schenkman (Universidade de Sao Paulo, Brazil), por el mAb 3C9
• Vanina Alvarez y Juan José Cazzulo (IIB‐INTECH), por los antisueros anti‐
TcCruzipaina y TcSCP
• Xi Chen (Department of Chemistry, University of California‐Davis, USA), por la
azido‐sialillactosa Neu5Azα2‐3LacßOMe
• UNICEF / UNDP / World Bank / WHO Special Programme for Research and Training
in Tropical Diseases (TDR), Agencia Nacional de Promoción Científica y Tecnológica
(ANPCyT), Fundación Florencio Fiorini, CONICET y UNSAM, por el soporte
económico

82
DIFUSION DE RESULTADOS Trabajos presentados en congresos: Sorting of Trypanosoma cruzi mucins and their role in invasión Canepa GE, Mesias AC, Buscaglia CA. Biology of Pathogens Meeting. Guaruja, Brasil. Agosto‐2011

83
REFERENCIAS
Alcaide P, F. M. (2004). The Trypanosoma cruzi membrane mucin AgC10 inhibits T cell activation and IL‐2 transcription through L‐selectin. Int Immunol 16, 1365‐1375.
Almeida IC, C. M., Procópio DO, Silva LS, Mehlert A, Travassos LR, Gazzinelli RT, Ferguson MA (2000). Highly purified glycosylphosphatidylinositols from Trypanosoma cruzi are potent proinflammatory agents. EMBO J 19(7):1476‐85. Almeida IC, G. R. (2001). Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J Leukoc Biol 70(4):467‐77 .
Andrews, N. W., Schenkman, S., Ley, V., Whitlow, M. B., Robbins, E. S., and Nussenzweig, V. (1988). Trypanosoma cruzi: mechanisms of cell‐invasion and intracellular survival. Mem Inst Oswaldo Cruz 83 Suppl 1, 452‐455.
Argibay PF, D. N. J., Hidalgo A, Mocetti E, Barbich M, Lorenti AS, Bustos D, Tambutti M, Hyon SH, Frasch AC, Sánchez DO (2002). Trypanosoma cruzi surface mucin TcMuc‐e2 expressed on higher eukaryotic cells induces human T cell anergy, which is reversible. Glycobiology 12(1):25‐32.
Aronson DE, C. L., Snapp EL (2011). Superfolder GFP is fluorescent in oxidizing environments when targeted via the Sec translocon. Traffic 12(5):543‐8.
Barrett MP, B. R., Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S (2003). The trypanosomiases. Lancet 362(9394):1469‐80.
Bendtsen JD, N. H., von Heijne G, Brunak S (2004). Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340(4):783‐95.
Bhattacharyya, T., Brooks, J., Yeo, M., Carrasco, H. J., Lewis, M. D., Llewellyn, M. S., and Miles, M. A. (2009). Analysis of molecular diversity of the Trypanosoma cruzi trypomastigote small surface antigen reveals novel epitopes, evidence of positive selection and potential implications for lineage‐specific serology. Int J Parasitol 40, 921‐928.
Böhme U, C. G. (2002). Mutational analysis of the variant surface glycoprotein GPI‐anchor signal sequence in Trypanosoma brucei. J Cell Sci 115(Pt 4):805‐16.
Boischio A, S. A., Orosz Z, Charron D (2009). Health and sustainable development: challenges and opportunities of ecosystem approaches in the prevention and control of dengue and Chagas disease. Cad Saude Publica, 25 Suppl 1:S149‐54. Boisrame A, C. A., Da Costa G, Richard ML (2011). Unexpected role for a serine/threonine‐rich domain in the Candida albicans Iff protein family. Eukaryot Cell 10, 1317‐1330.
Briones MR, E. C., Schenkman S (1995). Trypanosoma cruzi trans‐sialidase gene lacking C‐terminal repeats and expressed in epimastigote forms. Mol Biochem Parasitol 70(1‐2):9‐17.
Buscaglia CA, C. V., Di Noia JM, Torrecilhas AC, De Marchi CR, Ferguson MA, Frasch AC, Almeida IC (2004). The surface coat of the mammal‐dwelling infective trypomastigote stage of Trypanosoma cruzi is formed by highly diverse immunogenic mucins. J Biol Chem 279(16):15860‐9 .

84
Buscaglia CA, C. V., Frasch AC, Di Noia JM (2006). Trypanosoma cruzi surface mucins: host‐dependent coat diversity. Nat Rev Microbiol 4(3):229‐36.
Buscaglia CA, D. N. J. (2003). Trypanosoma cruzi clonal diversity and the epidemiology of Chagas' disease. Microbes Infect (5):419‐27.
Campo VA, B. C., Di Noia JM, Frasch AC (2006). Immunocharacterization of the mucin‐type proteins from the intracellular stage of Trypanosoma cruzi Microbes Infect 8(2):401‐9.
Campo VA, D. N. J., Buscaglia CA, Agüero F, Sánchez DO, Frasch AC (2004). Differential accumulation of mutations localized in particular domains of the mucin genes expressed in the vertebrate host stage ofTrypanosoma cruzi. Mol Biochem Parasitol 133(1):81‐91. Cazzulo JJ (1999). Cruzipain, major cysteine proteinase of Trypanosoma cruzi: sequence and genomic organization of the codifying genes. Medicina (B Aires) 59 Suppl 2:7‐10.
Cordero EM, N. E., Gentil LG, Yoshida N, Almeida IC, da Silveira JF (2009). Proteomic analysis of detergent‐solubilized membrane proteins frominsect‐developmental forms of Trypanosoma cruzi. J Proteome Res 8(7):3642‐52.
Cuevas IC, C. J., Sánchez DO (2003). gp63 homologues in Trypanosoma cruzi: surface antigens with metalloprotease activity and a possible role in host cell infection. Infect Immun 71(10):5739‐49.
Chagas C (1909). Nova tripanozomiase humana. Estudos sobre a morfolojía e o ciclo evolutivo de Schizotrypanum cruzi n. gen., n. sp., ajente etiolójico de nova entidade morbida do homen. Mem Inst Oswaldo Cruz 1: 159‐218 Chaves LB, B. M., Schenkman S (1993). Trans‐sialidase from Trypanosoma cruzi epimastigotes is expressed at the stationary phase and is different from the enzyme expressed in trypomastigotes. Mol Biochem Parasitol 61(1):97‐106.
Chiale P, R. M. (1989). Clinical and pharmacological characterization and treatment of potentially malignant arrythmias of chronic chagasic cardiomyopathy. Springer‐Verlag KG, Berlin, 1989, 601‐620.
D'Orso I, F. A. (2002). TcUBP‐1, an mRNA destabilizing factor from trypanosomes, homodimerizes and interacts with novel AU‐rich element‐ and Poly(A)‐binding proteins forming a ribonucleoprotein complex. J Biol Chem 277(52):50520‐8.
de Freitas JM, A.‐P. L., Pimenta JR, Bastos‐Rodrigues L, Gonçalves VF, Teixeira SM, Chiari E, Junqueira AC, Fernandes O, Macedo AM, Machado CR, Pena SD (2006). Ancestral genomes, sex, and the population structure of Trypanosoma cruzi. PLoS Pathog 2(3):e24
De Souza W (2002). Basic cell biology of Trypanosoma cruzi. Curr Pharm Des 8, 269‐285.
Di Noia JM, B. C., De Marchi CR, Almeida IC, Frasch AC (2002). A Trypanosoma cruzi small surface molecule provides the first immunological evidence that Chagas' disease is due to a single parasite lineage. J Exp Med 195(4):401‐13.
Di Noia JM, D. O. I., Sánchez DO, Frasch AC (2000). AU‐rich elements in the 3'‐untranslated region of a new mucin‐type gene family of Trypanosoma cruzi confers mRNA instability and modulates translation efficiency. J Biol Chem 275(14):10218‐27

85
Di Noia JM, S. D., Frasch AC (1995). The protozoan Trypanosoma cruzi has a family of genes resembling the mucin genes of mammalian cells. J Biol Chem 270(41):24146‐9.
Docampo R, d. B. J., Stoppani AO (1974). Metabolic differences in Trypanosoma cruzi depending of the culturing conditions. Rev Asoc Argent Microbiol 6(1):7‐14.
Donelson JE, G. M., El‐Sayed NM (1999). More surprises from Kinetoplastida. Proc Natl Acad Sci USA 96(6):2579‐81.
El‐Sayed NM, M. P., Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B (2005a). The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309(5733):409‐15.
El‐Sayed NM, M. P., Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz‐Fowler C, Ghedin E, Peacock, C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark UC, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM, Hauser C, Ivens AC, Kummerfeld SK, Pereira‐Leal JB, Nilsson D, Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall N (2005b). Comparative genomics of trypanosomatid parasitic protozoa. Science 309, 404‐409.
Ellis M, S. D., Hilley JD, Coombs GH, Mottram JC (2002a). Analysis using GP18 mutants deficient in glycosylphosphatidylinositol protein anchoring. J Biol Chem 277(31):27968‐74.
Ellis M, S. D., Hilley JD, Coombs GH, Mottram JC (2002b). Processing and trafficking of Leishmania mexicana GP63. Analysis using GP18 mutants deficient in glycosylphosphatidylinositol protein anchoring. J Biol Chem 277(31):27968‐74.
Engstler M, P. T., Herminghaus S, Boshart M, Wiegertjes G, Heddergott N, Overath P (2007). Hydrodynamic flow‐mediated protein sorting on the cell surface of trypanosomes. Cell 131(3):505‐15.
Erdmann H, S. C., Koch‐Nolte F, Fleischer B, Jacobs T (2009). Sialylated ligands on pathogenic Trypanosoma cruzi interact with Siglec‐E (sialic acid‐binding Ig‐like lectin‐E). Cell Microbiol 11(11):1600‐11.
Ferguson MA (1999). The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J Cell Sci 112 ( Pt 17):2799‐809.
Ferguson MA (2000). Glycosylphosphatidylinositol biosynthesis validated as a drug target for African sleeping sickness. Proc Natl Acad Sci U S A 97(20):10673‐5.
Gaunt MW, Y. M., Frame IA, Stothard JR, Carrasco HJ, Taylor MC, Mena SS, Veazey P, Miles GA, Acosta N, de Arias AR, Miles MA (2003). Mechanism of genetic exchange in American trypanosomes. Nature 421(6926):936‐9.

86
Gazzinelli RT, D. E. (2006). Protozoan encounters with Toll‐like receptor signalling pathways: implications for host parasitism. . Nat Rev Immunol 6(12):895‐906.
Geiger A, H. C., Bécue T, Bellard E, Centeno D, Gargani D, Rossignol M, Cuny G, Peltier JB. (2010). Exocytosis and protein secretion in Trypanosoma. BMC Microbiol 10:20.
Gile GH, F. D., Castlejohn CA, Burger G, Lang BF, Farmer MA, Lukes J, Keeling PJ (2009). Distribution and phylogeny of EFL and EF‐1alpha in Euglenozoa suggest ancestral co‐occurrence followed by differential loss. PLoS One 4(4):e5162.
Gingras AC, R. B., Sonenberg N ( 1999). eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68:913‐63.
Giorgi M, d. L. R. (2011). Trans‐sialidase and mucins of Trypanosoma cruzi: an important interplay for the parasite. Carbohydr Res 346(12):1389‐93.
Goldshmidt H, S. L., Bütikofer P, Roditi I, Uliel S, Günzel M, Engstler M, Michaeli S (2008). Role of protein translocation pathways across the endoplasmic reticulum in Trypanosoma brucei. J Biol Chem 283(46):32085‐98.
Guan C, L. P., Riggs PD, Inouye H (1987). Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose‐binding protein. Gene 67:21‐30.
Ha DS, S. J., Turco SJ, Beverley SM (1996). Use of the green fluorescent protein as a marker in transfected Leishmania. Mol Biochem Parasitol 77(1):57‐64.
Hann BC, W. P. (1991). The signal recognition particle in S. cerevisiae. Cell 67(1):131‐44.
Heise N, S. D., van der Wel H, Sassi SO, Johnson JM, Feasley CL, Koeller CM, Previato JO, Mendonça‐Previato L, West CM (2009). Molecular analysis of a UDP‐GlcNAc:polypeptide alpha‐N‐acetylglucosaminyltransferase implicated in the initiation of mucin‐type O‐glycosylation in Trypanosoma cruzi. Glycobiology 19(8):918‐33.
Hoare CA (1964). Morphological and taxonomic studies on mammalian trypanosomes. Revision of the systematics. J Protozool 11:200‐7
Hotez PJ, F. M. (2006). The antipoverty vaccines. Vaccine 24(31‐32):5787‐99.
Julenius K, M. A., Gupta R, Brunak S (2005). Prediction, conservation analysis, and structural characterization of mammalian mucin‐type O‐glycosylation sites. Glycobiology 15(2):153‐64.
Kaelin WG, K. W., Sellers WR, DeCaprio JA, Ajchenbaum F, Fuchs CS, Chittenden T, Li Y, Farnham PJ, Blanar MA (1992). Expression cloning of a cDNA encoding a retinoblastoma‐binding protein with E2F‐like properties. Cell 70, 351‐364.
Kahn SJ, N. D., Norsen J, Wleklinski M, Granston T, Kahn M (1999). Trypanosoma cruzi: monoclonal antibodies to the surface glycoprotein superfamily differentiate subsets of the 85‐kDa surface

87
glycoproteins and confirm simultaneous expression of variant 85‐kDa surface glycoproteins. Exp Parasitol 92(1):48‐56.
Kierszenbaum F, d. D. J., Fresno M, Sztein MB (1999). Inhibitory effects of the Trypanosoma cruzi membrane glycoprotein AGC10 on the expression of IL‐2 receptor chains and secretion of cytokines by subpopulations of activated human T lymphocytes. Eur J Immunol 29(5):1684‐91.
Kinlough CL, P. P., Gendler SJ, Mattila PE, Mo D, Weisz OA, Hughey RP (2011). Core‐glycosylated Mucin‐like Repeats from MUC1 Are an Apical Targeting Signal. J Biol Chem 286(45):39072‐81.
Koberle F (1968). Chagas' disease and Chagas' syndromes: the pathology of American trypanosomiasis. Adv Parasitol 6:63‐116
Labriola CA, C. I., López Medus M, Parodi AJ, Caramelo JJ (2010). Endoplasmic reticulum calcium regulates the retrotranslocation of Trypanosoma cruzicalreticulin to the cytosol. PLoS One 5(10) pii: e13141.
Labriola CA, G. A., Parodi AJ, Caramelo JJ (2011). Functional cooperation between BiP and calreticulin in the folding maturation of a glycoprotein in Trypanosoma cruzi. Mol Biochem Parasitol 175(2):112‐7.
Lannes‐Vieira J, S. J., Pereira IR, Vinagre NF, Carvalho CM, Paiva CN, Silva da AA, Levin MJ, Mesri E, Benarous R, Levitus G, Schijman A, Levy‐Yeyati P, Chiale PA, Ruiz AM, Kahn A, Rosenbaum MB (1989). Identification of major Trypanosoma cruzi antigenic determinants in chronic Chagas' heart disease. Am J Trop Med Hyg 41(5):530‐8
Lederkremer RM, A. R. (2009). Glycobiology of Trypanosoma cruzi. Elsevier Inc, Advances in carbohydrate chemistry and biochemistry, VOL 62.
Lederkremer RM, B. L. (2001). Glycoinositolphospholipids, free and as anchors of proteins, in Trypanosoma cruzi. Curr Pharm Des 7(12):1165‐79.
Levine N, C. J., Cox F, Devoux G, Grain J, Honigberg B, Leedale G, Polzansky G, Spragne V, Vassa J, Wallace F (1980). A newly revised classification of the protozoa. Journal of Protozoology 27:37‐54.
Lewis MD, L. M., Yeo M, Acosta N, Gaunt MW, Miles MA (2011). Recent, Independent and Anthropogenic Origins of Trypanosoma cruzi Hybrids. PLoS Negl Trop Dis (10):e1363.
Lisanti M, R.‐B. E. (1990). Glycolipid membrane anchoring provides clues to the mechanism of protein sorting in polarized epithelial cells. Trends Biochem Sci 15:113‐118.
Liu L, B.‐S. H., Xu YX, Stern MZ, Goncharov I, Zhang Y, Michaeli S (2003). The trypanosomatid signal recognition particle consists of two RNA molecules, a 7SL RNA homologue and a novel tRNA‐like molecule. J Biol Chem 278(20):18271‐80.
Maeda Y, K. T. (2011). Structural remodeling, trafficking and functions of glycosylphosphatidylinositol‐anchored proteins. Prog Lipid Res 50, 411‐424.

88
Martins V de P, G. M., Salto ML, Docampo R, Moreno SN (2010). Developmental expression of a Trypanosoma cruzi phosphoinositide‐specific phospholipase C in amastigotes and stimulation of host phosphoinositide hydrolysis. Infect Immun 78(10):4206‐12.
McConville MJ, F. M. (1993). The structure, biosynthesis and function of glycosylated phosphatidylinositols in the parasitic protozoa and higher eukaryotes. Biochem J 294 ( Pt 2):305‐24.
McConville MJ, M. K., Ilgoutz SC, Teasdale RD (2002). Secretory pathway of trypanosomatid parasites. Microbiol Mol Biol Rev 66(1):122‐54.
Morgado‐Díaz JA, N. C., Agrellos OA, Dias WB, Previato JO, Mendonça‐Previato L, De Souza W (2001). Isolation and characterization of the Golgi complex of the protozoan Trypanosoma cruzi. Parasitology 123(Pt 1):33‐43.
Mortara RA, d. S. S., Araguth MF, Blanco SA, Yoshida N (1992). Polymorphism of the 35‐ and 50‐kilodalton surface glycoconjugates of Trypanosoma cruzimetacyclic trypomastigotes. Infect Immun 60(11):4673‐8.
Mott GA, C. J., Burleigh BA (2011). A soluble factor from Trypanosoma cruzi inhibits transforming growth factor‐ss‐induced MAP kinase activation and gene expression in dermal fibroblasts. PLoS One 6, e23482.
Mucci, J., Hidalgo, A., Mocetti, E., Argibay, P. F., Leguizamon, M. S., and Campetella, O. (2002). Thymocyte depletion in Trypanosoma cruzi infection is mediated by trans‐sialidase‐induced apoptosis on nurse cells complex. Proc Natl Acad Sci U S A 99, 3896‐3901.
Muiá RP, Y. H., Prescher JA, Hellman U, Chen X, Bertozzi CR, Campetella O (2010). Identification of glycoproteins targeted by Trypanosoma cruzi trans‐sialidase, a virulence factor that disturbs lymphocyte glycosylation. Glycobiology 20(7):833‐42.
Nakayasu ES, Yashunsky DV, Nohara LL, Torrecilhas AC, Nikolaev AV, Almeida IC (2009). GPIomics: global analysis of glycosylphosphatidylinositol‐anchored molecules of Trypanosoma cruzi. Mol Syst Biol. 5:261‐271. Nogueira, N. F., Gonzalez, M. S., Gomes, J. E., de Souza, W., Garcia, E. S., Azambuja, P., Nohara, L. L., Almeida, I. C., Zingales, B., and Colli, W. (2007). Trypanosoma cruzi: involvement of glycoinositolphospholipids in the attachment to the luminal midgut surface of Rhodnius prolixus. Exp Parasitol 116, 120‐128.
Norris KA (1998). Stable transfection of Trypanosoma cruzi epimastigotes with the trypomastigote‐specific complement regulatory protein cDNA confers complement resistance. Infect Immun 66(6):2460‐5.
Osório AL, M. C., Desquesnes M, Soares CO, Ribeiro LR, Costa SC (2008). Trypanosoma (Duttonella) vivax: its biology, epidemiology, pathogenesis, and introduction in the New World‐‐a review. Mem Inst Oswaldo Cruz 103(1):1‐13.
Paetzel M, K. A., Strynadka NC, Dalbey RE (2002). Signal peptidases. Chem Rev 102(12):4549‐80.
Pan S, W. S., Utama B, Huang L, Blok N, Estes MK, Moremen KW, Sifers RN (2011). Golgi localization of ERManI defines spatial separation of the mammalian glycoprotein quality control system. Mol Biol Cell 22(16):2810‐22.

89
Parodi AJ (1993). N‐glycosylation in trypanosomatid protozoa. Glycobiology 3:193‐199.
Paulick MG, B. C. (2008). The glycosylphosphatidylinositol anchor: a complex membrane‐anchoring structure for proteins. Biochemistry 47(27):6991‐7000
Pereira‐Chioccola VL, A.‐S. A., Correia de Almeida I, Ferguson MA, Souto‐Padron T, Rodrigues MM, Travassos LR, Schenkman S (2000). Mucin‐like molecules form a negatively charged coat that protects Trypanosoma cruzi trypomastigotes from killing by human anti‐alpha‐galactosyl antibodies. J Cell Sci 113 ( Pt 7):1299‐307.
Pereira MG, N. E., Sant'Anna C, De Cicco NN, Atella GC, de Souza W, Almeida IC, Cunha‐e‐Silva N (2011). Trypanosoma cruzi epimastigotes are able to store and mobilize high amounts of cholesterol in reservosome lipid inclusions. PLoS One 6(7):e22359.
Pollevick GD, D. N. J., Salto ML, Lima C, Leguizamón MS, de Lederkremer RM, Frasch AC (2000). Trypanosoma cruzi surface mucins with exposed variant epitopes. J Biol Chem 275(36):27671‐80.
Pool MR (2005). Signal recognition particles in chloroplasts, bacteria, yeast and mammals. Mol Membr Biol 22(1‐2):3‐15.
Previato JO, J. C., Gonçalves LP, Wait R, Travassos LR, Mendonça‐Previato L (1994). O‐glycosidically linked N‐acetylglucosamine‐bound oligosaccharides from glycoproteins of Trypanosoma cruzi. Biochem J 301:151‐9.
Previato JO, S.‐P. M., Agrellos OA, Jones C, Oeltmann T, Travassos LR, Mendonça‐Previato L (1998). Biosynthesis of O‐N‐acetylglucosamine‐linked glycans in Trypanosoma cruzi. Characterization of the novel uridine diphospho‐N‐acetylglucosamine:polypeptide N‐acetylglucosaminyltransferase‐catalyzing formation of N‐acetylglucosamine alpha1‐‐>O‐threonine. J Biol Chem 273(24):14982‐8.
Previato JO, W. R., Jones C, DosReis GA, Todeschini AR, Heise N, Previato LM (2004). Glycoinositolphospholipid from Trypanosoma cruzi: structure, biosynthesis and immunobiology. Adv Parasitol 56:1‐41.
Ramirez MI, B. S., Ruiz RC, Han SW, Paranhos‐Baccala GS, Yoshida N, Mortara RA, Silveira JF (1999). Heterologous expression of a trypanosoma cruzi surface glycoprotein (gp82) indicates that requirements for glycosylphosphatidylinositol anchoring are different in mammalian cells and this trypanosome. Mem Inst Oswaldo Cruz 94(4):527‐30.
Rifkin MR, L. F. (1990). Trypanosome variant surface glycoprotein transfer to target membranes: a model for the pathogenesis of trypanosomiasis. Proc Natl Acad Sci U S A 87(2):801‐5
Risso MG, G. G., Mocetti E, Campetella O, Gonzalez Cappa SM, Buscaglia CA, Leguizamon MS (2004). Differential expression of a virulence factor, the trans‐sialidase, by the main Trypanosoma cruzi phylogenetic lineages. J Infect Dis 189(12):2250‐9.
Rocha GM, B. B., Mortara RA, Attias M, De Souza W, Carvalho TM (2006). The flagellar attachment zone of Trypanosoma cruzi epimastigote forms. . J Struct Biol 154:89‐99.

90
Roper, J. R., and Ferguson, M. A. (2003). Cloning and characterisation of the UDP‐glucose 4'‐epimerase of Trypanosoma cruzi. Mol Biochem Parasitol 132, 47‐53.
Rudd PM, D. R. (1997). Rapid, sensitive sequencing of oligosaccharides from glycoproteins. Curr Opin Biotechnol 8(4):488‐97.
Ruiz Rde C, R. V., Gonzalez J, Yoshida N (1993). The 35/50 kDa surface antigen of Trypanosoma cruzi metacyclic trypomastigotes, anadhesion molecule involved in host cell invasion. Parasite Immunol 15(2):121‐5
Sambrook J, R. D. (2001). Molecular Cloning: A Laboratory Manual. New York:Cold Spring Harbor Laboratory Press.
Sant'Anna C, P. F., Lourenço D, De Souza W, Cazzulo JJ (2008a). All Trypanosoma cruzi developmental forms present lysosome‐related organelles. Histochem Cell Biol 130:1187‐1198.
Sant'Anna C, P. M., Lemgruber L, de Souza W, Cunha e Silva NL (2008b). New insights into the morphology of Trypanosoma cruzi reservosome. Microsc Res Tech 71(8):599‐605.
Schenkman S, J. M., Hart GW, Nussenzweig V (1991). A novel cell surface trans‐sialidase of Trypanosoma cruzi generates a stage‐specific epitope required for invasion of mammalian cells. Cell 65(7):1117‐25.
Sevova ES, B. J. (2009). Streamlined architecture and glycosylphosphatidylinositol‐dependent trafficking in the early secretory pathway of African trypanosomes. Mol Biol Cell 20(22):4739‐50.
Silverman JM, C. S., Robinson DP, Dwyer DM, Nandan D, Foster LJ, Reiner NE (2008). Proteomic analysis of the secretome of Leishmania donovani. Genome Biol 9(2):R35.
Silverman JS, S. K., Hajduk SL, Bangs JD (2011). Late endosomal Rab7 regulates lysosomal trafficking of endocytic but not biosynthetic cargo in Trypanosoma brucei. Mol Microbiol 82(3):664‐78.
Souto‐Padron T, D. S. W., Heuser JE (1984). Quick‐freeze, deep‐etch rotary replication of Trypanosoma cruzi and Herpetomonas megaseliae. J Cell Sci 69: 167‐168.
Stevens L, D. P., Schmidt JO, Klotz JH, Lucero D, Klotz SA (2011). Kissing bugs. The vectors of chagas Adv Parasitol 75:169‐92.
Talvani A, C. S., Barcelos Lda S, Teixeira MM (2008). Cyclic AMP decreases the production of NO and CCL2 by macrophages stimulated with Trypanosoma cruzi GPI‐mucins. Parasitol Res 104(5):1141‐8.
Tian E, T. H. K. (2009). Recent insights into the biological roles of mucin‐type O‐glycosylation. Glycoconj J 26(3):325‐34.
Todeschini AR, d. S. E., Jones C, Wait R, Previato JO, Mendonça‐Previato L (2001). Structure of O‐glycosidically linked oligosaccharides from glycoproteins of Trypanosoma cruzi CL‐Brener strain: evidence for the presence of O‐linked sialyl‐oligosaccharides. Glycobiology 11(1):47‐55.
Tomlinson S, V. F., Frevert U, Nussenzweig V (1995). The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology 110 ( Pt 5):547‐54.

91
Tonelli RR, T. A., Jacysyn JF, Juliano MA, Colli W, Alves MJ (2011). In vivo infection by Trypanosoma cruzi: the conserved FLY domain of the gp85/trans‐sialidase family potentiates host infection. Parasitology 138, 481‐492.
Torres C, P.‐V. F., Parodi‐Talice A, Castanys S, Gamarro F (2004). Characterization of an ABCA‐like transporter involved in vesicular trafficking in the protozoan parasite Trypanosoma cruzi. Mol Microbiol 54, 632‐646.
Triggs VP, B. J. (2003). Glycosylphosphatidylinositol‐dependent protein trafficking in bloodstream stage Trypanosoma brucei. American Society for Microbiology 2(1):76‐83
Tuteja R (2007). Unraveling the components of protein translocation pathway in human malaria parasite Plasmodium falciparum. Arch Biochem Biophys 467(2):249‐60
Tyler KM, E. D. (2001). The life cycle of Trypanosoma cruzi revisited. Int J Parasitol 31(5‐6):472‐81
Urban I, B. S. L., Chidichimo A, Yu H, Chen X, Mucci J, Agüero F, Buscaglia CA (2011). Molecular diversity of the Trypanosoma cruzi TcSMUG family of mucin genes and proteins. Biochem J 438(2):303‐13
Van den Bossche P, C. S., Masumu J, Marcotty T, Delespaux V (2011). Virulence in Trypanosoma congolense Savannah subgroup. A comparison between strains and transmission cycles. Parasite Immunol 33(8):456‐60
Varki A (1998). Factors controlling the glycosylation potential of the Golgi apparatus. Trends Cell Biol 8(1):34‐40.
Vazquez MP, L. M. (1999). Functional analysis of the intergenic regions of TcP2beta gene loci allowed the construction of an improved Trypanosoma cruzi expression vector. Gene 239(2):217‐25.
Vermelho AB, M. M. (1994). Sialoglycoconjugates in Trypanosoma cruzi‐host cell interaction: possible biological models. Mem Inst Oswaldo Cruz 89(1):69‐79, Erratum in: Mem Inst Oswaldo Cruz 1994 89(2):298.
Vickerman K (1994). The evolutionary expansion of the trypanosomatid flagellates. Int J Parasitol 24(8):1317‐31
von Heijne, G. (1985). Signal sequences. The limits of variation. J Mol Biol 184(1):99‐105.
Walter P, B. G. (1981). Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence‐dependent and site‐specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol 91(2 Pt 1):557‐61.
Wilkinson SR, T. M., Touitha S, Mauricio IL, Meyer DJ, Kelly JM (2002). TcGPXII, a glutathione‐dependent Trypanosoma cruzi peroxidase with substrate specificity restricted to fatty acid and phospholipid hydroperoxides, is localized to the endoplasmic reticulum. Biochem J 15;364(Pt 3):787‐94.
Yelinek JT, H. C., Warren G (2009). Ultrastructural study of Golgi duplication in Trypanosoma brucei. Traffic10(3):300‐6.

92
Yoshida A, S. M., Ikenaga H, Takeuchi M (1997). Discovery of the shortest sequence motif for high level mucin‐type O‐glycosylation. J Biol Chem 272(27):16884‐8.
Yoshida N (2008). Trypanosoma cruzi infection by oral route: how the interplay between parasite and host components modulates infectivity. Parasitol Int 57(2):105‐9.
Yoshida N, D. M., Ferreira AT, Oshiro ME, Mortara RA, Acosta‐Serrano A, Favoreto Júnior S (1997). Removal of sialic acid from mucin‐like surface molecules of Trypanosoma cruzi metacyclic trypomastigotes enhances parasite‐host cell interaction. Mol Biochem Parasitol 84(1):57‐67.
Yoshida N, M. R., Araguth MF, Gonzalez JC, Russo M (1989). Metacyclic neutralizing effect of monoclonal antibody 10D8 directed to the 35‐ and 50‐kilodalton surface glycoconjugates of Trypanosoma cruzi. Infect Immun 57, 1663‐7.
Yu H, C. H., Karpel R, Wu B, Zhang J, Zhang Y, Jia Q, Chen X (2005). A multifunctional Pasteurella multocida sialyltransferase: a powerful tool for the synthesis of sialoside libraries. J Am Chem Soc 127:17618–17619.
Zhang G, G. V., Kain SR (1996). An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem Biophys Res Commun 227(3):707‐11.
Zingales B, A. S., Briones MR, Campbell DA, Chiari E, Fernandes O, Guhl F, Lages‐Silva E, Macedo AM, Machado CR, Miles MA, Romanha AJ, Sturm NR, Tibayrenc M, Schijman AG (2009). A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz 104(7):1051‐4.