universidade estadual de campinas instituto de … · arterial thrombosis is a disease in which...
TRANSCRIPT

UNIVERSIDADE ESTADUAL DE CAMPINAS
INSTITUTO DE BIOLOGIA
ANDRESSA RAYSA DA COSTA E SILVA
EFEITO DE DIFERENTES GLICOSAMINOGLICANOS NO TEMPO DE
TROMBOSE ARTERIAL E RE-ENDOTELIZAÇÃO DA ARTÉRIA CARÓTIDA
DE CAMUNDONGOS APÓS LESÃO INDUZIDA POR CLORETO FERRICO
CAMPINAS
2017

ANDRESSA RAYSA DA COSTA E SILVA
EFEITO DE DIFERENTES GLICOSAMINOGLICANOS NO TEMPO DE
TROMBOSE ARTERIAL E RE-ENDOTELIZAÇÃO DA ARTÉRIA CARÓTIDA
DE CAMUNDONGOS APÓS LESÃO INDUZIDA POR CLORETO FERRICO
Dissertação apresentada ao Programa de Pós-Graduação
em Biologia Celular e Estrutural do Instituto de Biologia
da Universidade Estadual de Campinas como parte dos
requisitos exigidos para a obtenção do título de Mestra
em Biologia Celular e Estrutural, na Área de BIOLOGIA
CELULAR
Orientadora: Profa. Dra. CRISTINA PONTES VICENTE
Este arquivo digital corresponde à versão final da
Dissertação defendida pela candidata Andressa Raysa da
Costa e Silva e orientada pela profa. Dra. Cristina Pontes
Vicente


BANCA EXAMINADORA
Dra. Cristina Pontes Vicente (Orientadora)
Dra. Joyce Maria Annichino Bizzacchi
Dra. Nicola Amanda Conran Zorzetto
Ata da defesa com as respectivas assinaturas dos membros encontra-se no
processo de vida acadêmica do aluno.
Campinas, 30 de março de 2017

Agradecimentos
Agradeço primeiramente ao meu Deus todo poderoso, que sempre está comigo e me dá
forças quando tudo parece contrário e me encoraja a seguir em frente.
Agradeço indiscutivelmente à minha orientadora Cris, por ter me dado uma oportunidade
tão incrível na minha vida, por ter confiado em mim desde a primeira vez que entrei em
sua sala para uma entrevista. Obrigada pelos seus ensinamentos, sua paciência, seus
conselhos e seu cuidado, jamais esquecerei o que você representa para mim, todo meu
crescimento pessoal e profissional durante esses dois anos eu devo muito a você.
Agradeço à minha mãe, pai e irmã por sempre torcerem por mim mesmo de longe com o
coração apertado, por sempre me amar e nunca me deixar desistir, eu amo vocês! A minha
vozinha, que sempre quando me vê se emociona de saudade e sempre me diz palavras de
encorajamento por quê sabe o quão difícil é...., às minhas tias e primas, cada uma com
sua personalidade fazem dessa família “muito unida e muito ouriçada!”, eu amo vocês!
Agradeço ao meu amado Almir Neto por acreditar em mim e me encorajar, mesmo
quando eu dizia que não era capaz, obrigada por me ensinar a ser persistente, hoje eu vejo
que posso chegar onde eu quero com muita luta e garra! Admiro sua confiança, sua garra
e sua paciência e me espelho muito em você, te amo!
Obrigada aos amigos de laboratório Maiara, Carol, Micheli, Luiz, Michel, vou sentir falta
das nossas inúmeras reuniões e das nossas gargalhadas, cada um tem um cantinho
guardado aqui no meu coração! Júlia, por algum motivo bom você entrou na minha vida
e vai ficar para sempre! Agradeço à Deus por essa amizade que me fez tão feliz, amo
você! Toni, obrigada pela excelente pessoa e profissional que és, vou sentir falta das
conversas, das belíssimas fotos e dos conselhos!
Ao professor Claudio, pela ajuda e suporte quando necessário, a Camila, pela pronta
ajuda, conselhos, pelos brigadeiros e bolos que me fizeram extremamente feliz, pelo seu
carinho! Ao neto, pela alegria cotidiana contagiante, pelo suporte durante a realização do
trabalho.

À Giane, professor Edson, Juliano e Júlio meu sincero obrigada pelos conselhos,
avaliações e observações.
À professora Joyce, Nicola e Fernanda Bassora por terem aceito simpaticamente o convite
de participação na banca de defesa.
Aos meus amigos de Manaus, Camila, Duda, Amanda e Fabrício, pelo apoio
incondicional e pela amizade! À Dona Solange pelo suporte, amor, conselhos e alegria.
À professora Ana Frazão, um anjo que a graduação me presenteou, tinha paixão pelo que
fazia, jamais esquecerei seus ensinamentos!
A Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES/Proex pelo
suporte financeiro,
Meu muito obrigada a todos que fizeram parte dessa caminhada árdua, meu coração se
enche de alegria pelas conquistas logradas pois “por mais longa que seja a caminhada, o
mais importante é dar o primeiro passo” (Vinícius de Moraes).

RESUMO
A trombose arterial é uma doença na qual ocorre interrupção do fluxo sanguíneo de uma
artéria, levando a isquemia do vaso. No caso de uma artéria que oxigena o cérebro, pode
levar ao acidente vascular cerebral isquêmico (AVC) e em uma artéria coronariana pode
levar ao infarto do miocárdio. A trombose ativa também o processo inflamatório e, depois
de alguns dias, pode promover a proliferação exacerbada de células musculares lisas da
camada média para a intima do vaso, ocluindo o lúmen do vaso e formando a camada de
neointima que pode obstruir o vaso (1). Agentes como o dermatan sulfato (DS), Heparina
e a Heparina de baixo peso molecular (HBPM) são glicosaminoglicanos (GAGs),
utilizados na clínica médica e que possuem atividade anticoagulante, antiinflamatória e
antitrombótica. No presente estudo, comparamos o efeito destes três agentes na prevenção
inicial de trombose e no processo inflamatório após a lesão vascular e na formação de
neointima quatorze dias após injúria arterial em camundongos C57BL/6 pelo modelo de
lesão arterial por cloreto férrico. O tempo de trombose foi determinado por medição do
fluxo sanguíneo no vaso lesionado. A resposta inflamatória e o remodelamento dos vasos
foram determinados quantificando-se por ensaio de imunofluorescência a presença da
molécula de adesão intercelular I (ICAM) e a molécula marcadora de leucócitos (CD11b).
O marcador CD34 foi analisado para se verificar a presença de progenitores endoteliais e
o CD31 para a presença de endotélio nos vasos. Além disso, analisamos se estes GAGs
interferem na coagulação sanguínea por análise de TTPA, tempo de protrombina e de
sangramento e verificamos se interferem na atividade de metaloproteinases. A formação
de trombo em animais é menor no tratamento com Heparina e o DS é mais eficiente que
a HBPM. O TTPA é prolongado cerca de 5 vezes pela heparina, 1,9 vezes pela HBPM e
1,7 pelo DS em 24 horas após lesão vascular; o tempo de sangramento foi prolongado em
cerca de 5 vezes pela heparina, cerca de 1vez pela HBPM e 3 vezes pelo DS. A presença
de células CD 11b positivas foi cerca de 3 vezes menor no tratamento com DS
comparando-se com Heparina e HBPM e a marcação com ICAM-1 não foi alterada em
animais tratados com DS e cerca de 40% maior que o controle em animais tratados com
Heparina e com HBPM. As artérias lesionadas de animais tratados com DS, Heparina e
HBPM apresentaram cerca de 3 vezes mais CPEs. O tratamento com DS diminuiu a
atividade de gelatinases no tempo inicial em relação a heparina e de forma semelhante
em 14 dias após a lesão e a HBPM foi a menos eficiente neste processo. A formação de

neointima analisada 14 dias após lesão foi menor nos animais tratados com DS que os
com Heparina e HBPM. Desde modo, pudemos observar que o tratamento com os
glicosaminoglicanos melhora de uma forma geral o processo trombótico inicial e o
remodelamento vascular permitindo uma recuperação melhor do endotélio lesionado com
diminuição da formação da neointima. No entanto, observamos que o DS foi o agente
mais eficiente na diminuição da neointima e, embora a heparina seja mais eficiente na
diminuição da formação do trombo inicial e prolongue acentuadamente o tempo para a
formação de trombo, o DS inibe mais eficientemente a migração de células inflamatórias
para o local da lesão e também proporciona grande migração de CPEs. Deste modo
podemos sugerir que o tratamento com DS pode ser adjuvante a lesão arterial, não só
diminuindo restenose vascular, mas também proporcionando a melhor recuperação do
endotélio após a lesão sem os riscos produzidos pelo tratamento com a heparina como
processo hemorrágicos e a trombocitopenia induzida por heparina.

ABSTRACT
Arterial thrombosis is a disease in which arterial blood flow ceases, leading to vessel
ischemia. In the case of an artery that oxygenates the brain, thrombosis can lead to
ischemic stroke (IS)) and in a coronary artery can lead to myocardial infarction.
Thrombosis also activates the inflammatory process and, after a few days, can promote
exacerbated proliferation of smooth muscle cells from the middle layer to the lumen of
the vessel, occluding it and forming the neointima layer that obstruct the vessel. Agents
such as dermatan sulfate (DS), heparin and low molecular weight heparin (LMWH) are
glycosaminoglycans (GAGs), used in medical practice that have anticoagulant, anti-
inflammatory and antithrombotic activities. In the present study, we compared the effects
of these three agents on the initial prevention of thrombosis and inflammatory processes
after vascular injury and in the formation of neointima fourteen days after arterial injury
in C57BL / 6 mice using the ferric chloride-induced thrombosis model. Thrombosis time
was determined by measuring the blood flow in the injured vessel. The inflammatory
response and vessel remodeling were determined by quantifying the presence of
intercellular adhesion molecule I (ICAM) and the leukocyte marker molecule (CD11b).
The CD34 marker was used to analyzed the presence of endothelial progenitors cells and
CD31 for the presence of endothelial cells in the vessels. In addition, we analyzed whether
these GAGs interfere with blood coagulation by APTT analysis, prothrombin time and
bleeding time, and we also verifed whether they interfere with the activity of
metalloproteinases (gelatinases). As a result we observed that thrombus formation in
animals is decreased after heparin treatment and DS is more efficient than LMWH.
Heparin, 1.9 fold by LMWH and 1.7 by DS within 24 hours after vascular injury prolong
about APTT 5 times. Bleeding time was prolonged 5 times by heparin, 1 time by LMWH
and 3 times by DS. The presence of CD 11b positive cells was about 3-fold lower in DS
treatment compared to Heparin and LMWH, and the ICAM-1 labeling was unchanged in
DS treated animals and about 40% higher than the control in animals treated with Heparin
and LMWH. The injured arteries of animals treated with DS, Heparin and LMWH
exhibited about 3 times more CPEs. DS treatment further decreased gelatinases activity
at time zero than heparin and similarly at 14 days post-injury and LMWH was the least
efficient in this process. The formation of neointima analyzed 14 days after injury was
lower in DS treated animals than those with Heparin and LMWH. Thus, it has been

observed that treatment with glycosaminoglycans generally decreases the initial
thrombotic process and vascular remodeling allowing better recovery of the injured
endothelium with decreased neointima formation. However, we observed that DS was the
most efficient agent in the reduction of neointima and that although heparin is more
efficient at decreasing initial thrombus formation and markedly prolongs the time to
thrombus formation. DS more effectively inhibits the migration of inflammatory cells to
the lesion site and also enhances extensive migration of CPEs. Thus we may suggest that
DS may be used as an arterial injury treatment, not only for decreasing vascular
restenosis, but also to provide better recovery of the endothelium after injury without the
risks produced by treatment with heparin, such as hemorrhage and thrombocytopenia
induced by heparin.

LISTA DE FIGURAS
Figura 1. Morte por doenças cardiovasculares ............................................................................ 16
Figura 2. Proporção de óbitos por causa em 2012 no Brasil ....................................................... 17
Figura 3.Corte histológico de artéria de um camundongo .......................................................... 19
Figura 4. Recrutamento de plaquetas .......................................................................................... 20
Figura 6. Novo modelo da cascata de coagulação ....................................................................... 22
Figura 5.Leucócitos no processo inflamatório ............................................................................ 24
Figura 7.Sequência pentassacarídica de ligação entre HNF e AT............................................... 28
Figura 8.Inativação dos fatores de coagulação pelo complexo HNF + antitrombina ................. 29
Figura 9.Estrutura do Dermatan Sulfato...................................................................................... 32
Figura 10.Modelo de indução de lesão por cloreto ferrico .......................................................... 38
Figura 11. Tempo de formação do trombo na artéria carótida .................................................... 45
Figura 12. Análise da formação de trombo por histologia .......................................................... 46
Figura 13.Porcentagem de formação de trombo. ........................................................................ 47
Figura 14.Formação de trombo por microscopia intravital ......................................................... 48
Figura 16.Análise da porcentagem da área de neointima 14 dias após lesão arterial.................. 50
Figura 17.Área da camada média (μm2) ..................................................................................... 51
Figura 18.Tempo Parcial de Tromboplastina ativada (TTPa) ..................................................... 52
Figura 19.Tempo de sangramento ............................................................................................... 53
Figura 20.TP dosado em camundongos C57BL/6 no período de 12 e 24 horas após o
procedimento de lesão arterial..................................................................................................... 54
Figura 21.Extensão da lesão arterial provocada por cloreto férrico ............................................ 56
Figura 22.Contagem de células CD34+ na artéria após lesão provocada por cloreto férrico ..... 57
Figura 23.Contagem de células CD11b+ na artéria .................................................................... 59
Figura 24.Contagem de ICAM-1. ............................................................................................... 60
Figura 25.Atividade de metaloproteinases no vaso no tempo inicial após a lesão ...................... 63
Figura 26.Atividade de gelatinases no vaso 14 dias após a lesão. .............................................. 65
Figura 27.Agregação de plaquetas. ............................................................................................. 67

LISTA DE ABREVIATURAS
AT Antitrombina
CD11b Marcador leucocitário
CD31 Cluster of differentiation 31
CD34 Cluster of differentiation 34
CML Célula muscular lisa
CPE Célula progenitora endotelial
C57BL/6 Camundongos da linhagem black 6
DAPI 4’6 diamino – 2 fenilindole
DS Dermatan sulfato
FITC Isotiocianato de Fluoresceína (Fluorescein isothiocyanate)
FT Fator tecidual
GAG Glicosaminoglicano
GalNAC N-Acetilgalactosamina
Glc Glicoproteínas
HBPM Heparina de baixo peso molecular
HCII Cofator II da heparina
HNF Heparina não fracionada

ICAM-1 Molécula de adesão intercelular
LTA Ácido lipoteicoico
MAC-1 Antígeno de macrófago-1
MEC Matriz extracelular
MMPs Metaloproteinases
MNC Células mononucleares
NAc N-Acetilcisteína
NS Grupo sulfato
PA1 Ativador de Plasminogênio
PAF Fator de ativação plaquetária
PSGL1 Glicoproteína ligante de P-selectina
TFI Inibidor do fator tecidual
TFPI Inibidor da via do fator tecidual
TIMP Inibidor tecidual de metaloproteinase

Sumário
Agradecimentos ............................................................................................................................. 5
RESUMO ...................................................................................................................................... 7
ABSTRACT .................................................................................................................................. 9
LISTA DE FIGURAS ................................................................................................................. 11
I. INTRODUÇÃO .................................................................................................................. 16
1. Mortalidade por doenças cardiovasculares ...................................................................... 16
2. O processo de trombose: ............................................................................................... 18
3. A trombose ..................................................................................................................... 19
4. As Metaloproteinases e o seu papel no remodelamento vascular ............................. 25
5. Formação da camada neoíntima .................................................................................. 26
6. Os Glicosaminoglicanos e suas vias de atuação na trombose .................................... 27
7. A heparina não fracionada ........................................................................................... 27
7.1 Histórico ............................................................................................................................ 27
7.2 Estrutura e mecanismo de ação ......................................................................................... 28
8. Heparina de baixo peso molecular ............................................................................... 30
8.1 Estrutura e mecanismo de ação ......................................................................................... 30
9. O Dermatan Sulfato ...................................................................................................... 31
9.1 Estrutura de mecanismo de ação ....................................................................................... 31
10. Modelo de lesão arterial por cloreto férrico ............................................................... 33
II. OBJETIVO .......................................................................................................................... 35
Objetivo geral ........................................................................................................................ 35
Objetivos específicos ............................................................................................................. 35
III. MATERIAIS E METODOS ........................................................................................... 36
1. Animais ........................................................................................................................... 36
2. Grupos experimentais ................................................................................................... 36
3. Tratamentos com GAGs ............................................................................................... 36
4. Modelo de indução de trombose arterial ..................................................................... 37
5. Tempo de Trombose Arterial ....................................................................................... 38
6. Cálculo da área de trombo ........................................................................................... 39
7. Microscopia Intravital .................................................................................................. 39
8. Determinação do tempo de tromboplastina parcial ativada (TTPA) ....................... 39
9. Determinação do tempo de protrombina (TP) ........................................................... 40
10. Determinação do Tempo de Sangramento (TS) ......................................................... 40
11. Análise Histológica ........................................................................................................ 41

12. Ensaio de Imunofluorescência ...................................................................................... 41
15. Agregação plaquetária .................................................................................................. 42
16. Análise Estatística ......................................................................................................... 43
IV. RESULTADOS ............................................................................................................... 44
1. Peso dos Animais ........................................................................................................... 44
2. Tempo de formação do trombo nos tratamentos com DS, Heparina e Heparina de
baixo peso molecular inicialmente após o procedimento cirúrgico .................................. 44
3. Área do trombo por histologia ..................................................................................... 45
4. Área do trombo por microscopia intravital ................................................................ 47
5. Formação de neoíntima é atenuada no tratamento com DS, Heparina e HBPM .... 49
6. GAGs alteram o TTPA, TP e TS ................................................................................. 51
7. Expressão de marcadores após tratamento com os GAGs ........................................ 54
7.2 Efeito de GAGs na migração de células progenitoras endoteliais ............................ 56
8. Expressão de moléculas marcadores de inflamação: CD11b e ICAM-1 .................. 58
9.1 CD11b .......................................................................................................................... 58
9.2 ICAM-1 ............................................................................................................................ 59
9. Análise da atividade gelatinolítica nas artérias carótidas .......................................... 61
10. Agregação plaquetária em diferentes tratamentos com GAGs ................................. 65
RESUMO DOS DADOS OBTIDOS .......................................................................................... 75
CONCLUSÃO ............................................................................................................................ 76
REFERÊNCIAS BIBLIOGRÁFICAS ........................................................................................ 77
ANEXO ....................................................................................................................................... 87

16
I. INTRODUÇÃO
1. Mortalidade por doenças cardiovasculares
De acordo com os dados da Organização Mundial da Saúde, aconteceram 17,5
milhões de mortes no ano de 2012 devido às doenças cardiovasculares, sendo que 7,4
milhões foram relacionadas a doenças cardíacas isquêmicas e 6,7 milhões ao acidente
vascular cerebral. Apenas em 1999, essas enfermidades representaram um terço da
mortalidade global, no qual os países mais pobres contribuíram com 78% do total
(Mackay et al., 2004). Segundo projeções para as próximas décadas, o número de mortes
por doenças cardiovasculares irá dobrar nos países em desenvolvimento (Boutayeb e
Boutayeb, 2005). O gráfico abaixo mostrado na figura 1 mostra a taxa de mortalidade por
doenças cardiovasculares no mundo no ano de 2012:
Figura 1. Morte por doenças cardiovasculares. Representam as maiores proporções de
morte no grupo de doenças crônicas não transmissíveis (NCDs) no mundo, ocorrendo
principalmente em países de baixa e média renda(Who, 2014).

17
Ainda de acordo com a WHO, a taxa de mortalidade por doenças crônicas não
transmissíveis no Brasil no ano de 2012 que incluem as doenças cardiovasculares,
doenças respiratórias, câncer, diabetes, entre outras, apresentaram como maior fator de
óbito as doenças cardiovasculares, como exemplificado na figura 2:
Figura 2. Proporção de óbitos por causa em 2012 no Brasil. As doenças
cardiovasculares foram e continuam a ser, apesar de sua diminuição, a principal causa de
morte no Brasil e geram o maior custo referente a internações hospitalares no sistema de
saúde nacional. Gráfico modificado do Atlas global do perfil de doenças crônicas não
transmissíveis por perfil de país, 2014.
(Http://www.who.int/nmh/countries/bra_en.pdf?ua=1).
A OMS destacou que aproximadamente 80% dos óbitos por doenças crônicas não
transmissíveis (doenças cardiovasculares, cânceres, doenças respiratórias crônicas e
diabetes) ocorreram em países de baixa ou média renda com 29% dos óbitos em adultos
com menos de 60 anos, menor renda e escolaridade, por serem exatamente a classe mais
exposta aos fatores de risco e com menor acesso às informações e aos serviços de saúde,
Injúrias; 12%
Condições
comunicável,mater
nal,perinatal e
nutricional ; 13%
Outras NCDs;
15%
Diabetes;
6%
Doenças
respiratórias; 6%
Câncer; 17%
Doenças
cardiovasculares;
31%
Proporção da taxa de mortalidade brasileira em 2012

18
acentuando ainda mais as desigualdades sociais enquanto naqueles de alta renda esse
percentual era de apenas 13% (Alwan et al., 2010).
Em consequência da gravidade das doenças cardiovasculares no atual cenário, essas
enfermidades são responsáveis por um grande impacto econômico nos sistemas de saúde
em todos os países (Roger et al., 2012). De acordo com os custos diretos dos sistemas de
saúde com as doenças cardiovasculares foi de aproximadamente € 106 bilhões em 2009
nos países da Europa; os custos totais levando em conta a perda de produtividade devido
à mortalidade e morbidade e os custos informais, chegam a € 195 bilhões.
2. O processo de trombose:
A Hemostasia é um processo fisiológico que garante o equilíbrio dinâmico da
forma fluida do sangue. Ao mesmo tempo em que previne que o sangue se solidifique
dentro dos vasos sanguíneos, impedindo a formação do trombo (trombose), a hemostasia
protege o organismo da perda sanguínea em um eventual rompimento de vasos
(hemorragia)(Zarbock et al., 2007). As células endoteliais formam o revestimento interno
da parede dos vasos da chamada íntima, esta camada deve estar íntegra pois sua função é
a regulação da homeostase do vaso e a troca de constituintes do sangue circulante para os
tecidos e órgãos, através das junções intercelulares, que, em geral, são impermeáveis às
grandes moléculas. As camadas de células endoteliais devem responder a uma variedade
de estímulos patológicos ajustando suas funções habituais e expressando novas
propriedades adquiridas quando há disfunção no vaso sanguíneo, onde ocorre um
desequilíbrio na produção endotelial de mediadores que regulam o tônus vascular,
agregação plaquetária, coagulação e fibrinólise.
A disfunção endotelial está presente em diversas doenças como obesidade,
diabetes, hipertensão arterial e dislipidemia e em acidentes vasculares ou isquêmicos
como o acidente vascular cerebral (AVC) e a trombose. Estas doenças estão integradas a
fatores de risco herdados e/ou adquiridos, ou seja, algumas condições clínicas como
hipertensão, diabetes, câncer, inflamações, traumas e cirurgias podem aumentar a
incidência destes distúrbios hemostáticos (Lorenzi, 2006).
Nestas doenças as células endoteliais deixam de produzir as substâncias
responsáveis pela homeostase vascular que tornam o endotélio antitrombótico. Elas
passam a liberar então fatores pró-inflamatórios como moléculas de adesão celular e

19
citocinas que estimulam a migração e adesão de monócitos, linfócitos e neutrófilos para
o espaço subendotelial (Previtali et al., 2011) com consequente acontecimento de
processos inflamatórios e trombóticos.
Figura 3.Corte histológico de artéria de um camundongo. O vaso é composto por 3
túnicas (íntima, média e adventícia) sendo a camada íntima que reveste internamente o
vaso contém as células endoteliais. Corte corado com Hematoxilina e Eosina. Foto:
Andressa Costa
3. A trombose
Quando a integridade da parede vascular é interrompida, ocorre a ativação do
sistema hemostático. Este fenômeno envolve diversas interações entre componentes
endoteliais e subendoteliais, plaquetas e outras células sanguíneas periféricas e proteínas
pró-coagulantes circulantes (Hoffbrand et al., 2011) (Figura 4). Após a lesão vascular as
plaquetas encontram constituintes da matriz extracelular, especialmente o colágeno e são
ativadas. Em contato com a matriz, as plaquetas atuam de três formas: 1) aderem-se à ela;
2) secretam conteúdos de seus grânulos; 3) iniciam a agregação a outras plaquetas. As
plaquetas se aderem à matriz extracelular através do fator de von Willebrand (FvW) que
faz a ligação entre o colágeno e o receptor de glicoproteína Ib/IX-V plaquetário. A
secreção do conteúdo granular é promovida pela ligação de agonistas aos receptores de
superfícies nas plaquetas. O difosfato de adenosina (ADP) é um dos elementos secretados
e atua no recrutamento de mais plaquetas (Rondaij et al., 2006). A agregação plaquetária
induzida pelo ADP (contido no grânulo plaquetário), e pelo tromboxano A2 (também
sintetizado com a ativação plaquetária) leva a formação do tampão hemostático. Com a
Lúmen do vaso
Camada íntima do vaso
contendo células
Camada média do vaso
contendo células
musculares lisas
Camada adventícia

20
ativação de outros fatores da cascata de coagulação, a trombina gerada liga-se à superfície
plaquetária, resultando em mais agregação. O fibrinogênio é um outro fator atuante na
agregação plaquetária, que associa-se as plaquetas através de receptores de glicoproteínas
IIb/IIIa (Ruggeri e Mendolicchio, 2007). A Glicoproteína Ib/IX-V liga a plaqueta ao
FvW. (Alevriadou et al., 1993)
Figura 4. Recrutamento de plaquetas. A) A interação entre Gp plaquetária e o colágeno
é estabilizada pelo Fator de von Willebrand. A aderência plaquetária ocorre sobre o
colágeno da superfície subendotelial (endotélio lesado). B) Glicoproteínas da superfície
plaquetária são mediadoras da adesão e ativação plaquetária juntamente com o fator
tecidual (FT). Gp = Glicoproteína; FT = Fator Tecidual; FvW = Fator de von Willebrand.
Imagem: Andressa Costa.
Fibrinogênio Complexo II b-III a
A) Endotélio
Subendotélio
Camada média
GpIb/IX-V FvW
Plaquetas
Lesão vascular
FT FT
Fibrina
Camada média
B)
Adesão e agregação
(Plaquetas ativadas)
Lúmen

21
Simultaneamente a este processo, o sistema de coagulação se ativa através de
clivagens proteolíticas de zimogênios em enzimas. Recentemente foi proposto o novo
modelo de coagulação baseado em superfícies celulares, considerando a interrelação dos
processos físicos, celular e bioquímicos e não em duas vias (intrínseca e extrínseca) como
antes (Hoffman, 2003). O novo modelo propõe a ocorrência das fases de iniciação,
amplificação, propagação e finalização.
A fase de iniciação acontece quando células que expressam o fator tecidual (FT)
em sua superfície são expostas após lesão vascular (Vine, 2009). O FT liga-se ao FVII e
o ativa FVIIa, esse complexo é responsável pela ativação dos fatores FIX e FX. O fator
Xa em associação com o fator Va transforma pequenas quantidades de protrombina em
trombina. Na fase de amplificação pequenas quantidades de trombina produzidas por
células que expressam o FT interagem com as plaquetas e FVIII que está ligado ao fator
de von Willebrand. No processo de lesão as plaquetas interagem com componentes da
matriz extracelular exposta, onde são ativadas, resultando em um tampão plaquetário. A
trombina gerada é responsável pela ativação máxima de plaquetas. Dessa forma, inicia-
se a formação de fibrina estável, que consolida o tampão plaquetário inicial (Monroe e
Hoffman, 2009). As plaquetas também permitem a entrada de íons cálcio e expressão de
quimiocinas que atraem os fatores de coagulação para sua superfície, além de liberar o
FV parcialmente ativado. A fase de propagação é destacada pelo recrutamento de mais
plaquetas para o local lesionado e pela formação do complexo tenase e protrombinase na
superfície das plaquetas ativadas (Boucher e Traub, 2009). Nesse processo, o fator FIXa
liga-se ao fator FVIIIa na superfície de plaquetas (complexo tenase), este por sua vez
ativa o fator X em Xa, o qual associa-se com o fator Va que está ligado à plaqueta,
resultando no complexo protrombinase, que converte a protrombina em trombina, esta
quebra o fibrinogênio em fibrina, que é o principal componente de consolidação do
trombo (Riddel et al., 2007). O trombo de fibrina formado deve limitar-se ao local
lesionado para que não venha a ocorrer oclusão trombótica no vaso. Para isso,
anticoagulantes naturais agem nesse processo, como a antitrombina (AT), cofator II de
heparina, inibidor da via do fator tecidual (TFPI), a proteína C (PC) e proteína S (OS).
As células endoteliais produzem uma variedade de GAGs, que funcionam como sítios de
ligação, de alta afinidade, para a AT, que são importante na inativação da trombina
(Cerinic et al., 2003).

22
A atividade da coagulação sanguínea pode ser feita por análise laboratorial da
coagulação, in vitro, o teste de tromboplastina parcial ativada (TTPa) analisa os níveis de
fatores procoagulante envolvidos na produção de trombina na superfície de plaquetas e o
tempo de protrombina (TP) avalia os níveis de fatores procoagulante envolvidos na fase
de iniciação da coagulação (Spronk et al., 2003).
Figura 5. Novo modelo da cascata de coagulação. Representação do modelo da
coagulação baseado em superfícies celulares compreendendo as fases de iniciação,
amplificação e propagação. Fator tecidual (FT), ativado (a). (Ferreira et al., 2010)
Além da ativação de plaquetas e da cascata de coagulação, uma outra
característica da trombose é a inicialização de uma resposta inflamatória. Nesse processo
ocorre o recrutamento rápido de leucócitos do sangue para o local da lesão, sendo os
neutrófilos as primeiras células a chegarem no sítio (Muller, 2003). Na inflamação tanto
células endoteliais como leucócitos circulantes são ativados por citocinas inflamatórias
circulantes. A interação dos leucócitos ocorre por meio de seus receptores, L-selectinas
(presente em leucócitos e que promove a ligação leucócito e endotélio), com os receptores
P-selectinas (expresso pelo endotélio e plaqueta ativada), onde promovem uma adesão
fraca que produz a fase de rolamento dos leucócitos no endotélio (Mitchell et al., 2006).

23
A adesão firme de leucócitos ao endotélio ocorre em seguida ao rolamento e se dá
através de proteínas transmembranas de superfície celular, as integrinas, as quais irão se
ligar as imunoglobulinas expressas por células endoteliais(Stewart et al., 1995) (figura
5). Cada integrina contém cadeias α e β, definidas em relação a sua cadeia β: β1-integrinas
expressas por leucócitos irão interagir com a molécula de adesão celular vascular
(VCAM-1); β2-integrinas conhecidas são as LFA (também denominadas de CD11a/
CD18 OU αLβ2) e MAC-1 (conhecidas como CD11b/CD18 ou αMβ2). A MAC-1 e LFA-
1 são as moléculas mais estudadas pois atuam na adesão e na transmigração de leucócitos
pelo endotélio (Simon et al., 2000). As imunoglobulinas expressas pelo endotélio como
VCAM-1 (molécula de adesão celular vascular) e ICAM-1 (molécula de adesão
intercelular-1) são os principais receptores para as β2-integrinas dos leucócitos. O ICAM-
1 possui cinco domínios extracelulares, um ligado ao LFA (CD11a/CD18) e outro a
MAC-1 (CD11b/CD18) (Diacovo et al., 1996). Essas moléculas de adesão são
importantes pois fornecem ancoragem firme aos leucócitos (Schneider e Sobel, 2007). A
adesão e transmigração leucocitária são afetadas por mediadores químicos, os quais
podem gerar uma cascata capaz de ampliar e liberar outros fatores estimulantes, esses
mediadores podem ser agentes vasoativos como a histamina, serotonina, prostaglandinas,
leucotrienos e lipoxinas, proteínas plasmáticas, fator de ativação das plaquetas (PAF),
TNF-α, Interleucina-1 e óxido nítrico (Marcos Da Silveira, 2004).

24
Figura 6.Leucócitos no processo inflamatório. Adesão leucocitária é um evento
fundamental para o processo inflamatório, culminando com o carreamento de células de
defesa mediante ação de citocinas e moléculas de adesão. Entre as moléculas de adesão,
três grandes são conhecidas: as integrinas, imunoglobulinas e selectinas. Essas moléculas
coordenam as várias fases da adesão, rolamento e transmigração leucocitária ao endotélio,
contribuindo na resposta inflamatória aguda, segundo (Francischetti, 2010)
Após a formação do trombo e a migração de células inflamatórias, se inicia o
processo de reabsorção do trombo e do remodelamento do vaso, com produção de células
endoteliais, células musculares lisas e matriz extracelular que irão propiciar a regeneração
do tecido vascular. Um dos principais agentes responsáveis pelo remodelamento do vaso
são as enzimas do tipo metaloproteinases. Estas enzimas podem participar do
remodelamento positivo estimulando a modificação da matriz extracelular, crescimento
de células endoteliais e a reconstituição do endotélio funcional ou um processo negativo,
onde ocorre maior proliferação de células musculares lisas e de matriz para o lúmen do
vaso, levando a oclusão deste (Bosman e Stamenkovic, 2003).

25
4. As Metaloproteinases e o seu papel no remodelamento vascular
A angiogênese é a formação de novos vasos a partir de vaso pré-existentes, sendo
um processo estritamente regulado necessário durante a reprodução, desenvolvimento,
inflamação e reparo de lesões (Arnold, 1991). O remodelamento da matriz extracelular é
um evento crucial durante a angiogênese, onde as células endoteliais estimuladas por
fatores angiogênicos produzem enzimas que degradam a matriz, incluindo as
metaloproteinases (MMPs). As MMPs são uma grande família de metaloendopeptidases
que degradam componentes proteicos da matriz extracelular (MEC) e da membrana basal.
Existe um delicado balanço fisiológico entre degradação, reconstrução e remodelamento
dos colágenos na MEC. As MMPs apresentam especificidade por substratos como,
colágeno, colágenos da membrana basal, proteoglicanos, elastinas e fibronectinas.
Algumas MMPs, como MMP-2, MMP-3, MMP-9 e MMP-12 hidrolisam elastina e tem
importância na parede vascular arterial (Busti et al., 2010). As MMPs podem ser
conhecidas como colagenases (MMP-1, 8, 13 e 18), gelatinases (MMP-2 e 9)
estromelisinas (MMP-3, 10 e 11), matrilisinas (MMP-7 e 26) e MMPs de membrana ou
MT-MMPs (MMP-14, 15, 16, 17, 24 e 25). Quanto maior a sinalização e ação das MMPs
na MEC mais robusto será o mecanismo de reparo da síntese de colágeno recém-formado
(Hayden et al., 2005).
O balanço entre os níveis de MMPs e os seus inibidores teciduais (TIMPs) é
conhecido por ter um papel central no remodelamento vascular (Visse e Nagase, 2003).
Sob condições patológicas associadas com um desequilíbrio das atividades das MMPs,
mudanças nos níveis de TIMPs são consideradas importantes porque afetam diretamente
os níveis das atividades das MMPs. O desequilíbrio entre MMPs e TIMPs pode levar a
um excesso do acúmulo ou degradação de MEC (Bode e Maskos, 2003).
A MMP-2 é uma das gelatinases mais expressas por células endoteliais e células
musculares lisas e possui importante papel na angiogênese. MMP-2 está envolvida
também na formação de lesão aterosclerótica e neointima (Kuzuya et al., 2003). A MMP-
9 possui um papel fundamental na neovascularização através do efeito direto no
deslocamento de células progenitoras endoteliais (CPEs) da medula óssea para o tecido
isquêmico. A inibição de MMP-9 promove redução na mobilização CPEs e
vasculogênese, comprovado em animais MMP-9 (-/-) (Huang et al., 2009).

26
Uma alteração na atividade das MMPs pode estar diretamente relacionada com o
remodelamento positivo ou negativo do vaso lesionado (Giagtzidis et al., 2015). Após a
ativação da trombose, do processo inflamatório e da ativação das enzimas de matriz
extracelular o vaso começa o processo de recuperação, no qual poderá ou não resultar na
formação de neointima como será descrito a seguir.
5. Formação da camada neoíntima
O procedimento cirúrgico mais utilizado na restauração do fluxo sanguíneo
vascular após obstrução das artérias coronarianas é a angioplastia por implante de
próteses medicamentadas (stents), com taxa de lesões residuais menores que 50% e
ausência de complicações maiores que 90% (Bauters et al., 1996). A reestenose é a
principal limitação desses procedimentos, ocorre em cerca de 20 a 40% das desobstruções
feitas com sucesso. A reestenose desenvolve-se, fundamentalmente, nos primeiros meses
após a angioplastia, tendo máxima incidência entre o 3º e o 6º mês após o procedimento
(Marquez-Gonzalez et al., 2017).
Inicialmente após o procedimento cirúrgico, plaquetas e fibrina são depositadas
no vaso lesionado e as plaquetas ativadas expressam moléculas de adesão como as p-
selectinas, que interagem com prostanglandinas-1 (PSGL-1) na superfície dos leucócitos,
ocasionando a adesão e rolamento dos mesmos na superfície do vaso. A migração dos
leucócitos pela camada de plaquetas ocorre também por agentes quimiotáticos que são
liberados por células musculares lisas e macrófagos que estão na parede do vaso (Gawaz
et al., 1996).
A próxima etapa é a de proliferação celular que ocorre quando plaquetas,
leucócitos e células musculares lisas liberam fatores de crescimento e estimulam a
migração de mais células musculares lisas da camada média para a camada íntima,
formando a neoíntima. Esta consiste na proliferação exacerbada de células musculares
lisas, matriz extracelular e macrófagos durante o período de várias semanas (Schwartz Rs
Fau - Holmes et al., 1992). Dependendo do estímulo que recebe, as CMLs podem assumir
dois diferentes fenótipos. O fenótipo contrátil, ou quiescente, é o aspecto normal da CML
da camada média arterial ou fenótipo ativado ou proliferativo, que é a forma
predominante em vasos lesionados (Kocher et al., 1991). Após o período de meses é
observado o remodelamento vascular com a degradação de matriz extracelular e sua re-

27
síntese no lúmen do vaso juntamente com as CMLs proliferativas, reduzindo o diâmetro
do vaso e bloqueando a passagem de fluxo sanguíneo, sendo o principal mecanismo da
formação de neoíntima em artérias tratadas com implante de stent (Carter et al., 1994). A
necessidade de procedimentos diagnósticos e terapêuticos repetitivos determina um
marcante prejuízo à evolução dos pacientes que desenvolvem reestenose e tornam altos
os custos do tratamento.
Uma variedade de agentes terapêuticos tem sido empregada para tentar modular a
função vascular e regenerar as células endoteliais nos estágios pós-operatórios de
pacientes submetidos à angioplastia, dentre estes agentes, pode-se destacar a utilização
de glicosaminoglicanos, que estão diretamente envolvidos em processos anti-
inflamatórios, antitrombóticos e anticoagulantes.
6. Os Glicosaminoglicanos e suas vias de atuação na trombose
Os glicosaminoglicanos são heteropolissacarídeos de caráter ácido, compostos de
cadeias lineares de unidades dissacarídicas repetitivas, formadas por unidades alternadas
de hexosamina e um ácido urônico. Dentre os GAGs presentes em tecidos animais estão
a heparina, heparan sulfato, dermatan sulfato, condroitin 4 e 6 sulfato, queratam sulfato e
o ácido hialurônico (Taylor e Gallo, 2006). Muitos glicosaminoglicanos são utilizados
como drogas antitrombóticas e anticoagulantes no tratamento de doenças
cardiovasculares, portanto o estudo comparativo dos efeitos destas moléculas na
trombose e na revascularização do endotélio são de grande interesse atualmente.
7. A heparina não fracionada
7.1 Histórico
A heparina não fracionada (HNF) foi descoberta por McLean em 1916 que a
caracterizou como um eficiente anticoagulante, (Mc, 1959) utilizado mundialmente em
muitos distúrbios trombóticos arteriais e venosos, até os dias atuais. Conhecida por ser
um dos anticoagulantes mais antigos, ela é amplamente utilizada em medicina desde 1930
(Jaques, 1979). O seu principal mecanismo de ação consiste em acelerar em cerca de 1000
vezes, a velocidade com que a antitrombina (AT), uma proteína anticoagulante circulante
no plasma atua sobre as serino-proteases da cascata de coagulação sanguínea, dentre elas
a trombina e o fator X ativo (Rosenberg e Damus, 1973).

28
7.2 Estrutura e mecanismo de ação
A HNF é constituída por uma cadeia linear de tamanho variável entre 10 e 200
unidades dissacarídicas de glucosamina e ácido urônico, ligados através de ligações
glicosídicas α ou β (1 → 4). O ácido urônico pode ser ácido β-D-glicurônico (GlcA) ou
α-L-idurônico (IdoA), enquanto que a glucosamina pode ser apenas α-D-glucosamina
(GlcN) (Silva, 1975). A presença de vários grupos sulfatos ligados a HNF, conferem a
ela a maior densidade de carga aniônica que qualquer outra molécula biológica conhecida
(figura 7). Ela apresenta em média 3,5 cargas negativas por unidade dissacarídica (Nader
e Dietrich, 1994). A HNF é encontrada exclusivamente em grânulos secretores no interior
de mastócitos, enquanto o heparan sulfato é abundante em superfícies celulares, ligado a
receptores de diversos tecidos (Sasisekharan e Venkataraman, 2000).
Ela atua aumentando a ação inibitória exercida pelas serpinas, como a
antitrombina (AT) e o cofator II da heparina, uma outra proteína anticoagulante presente
no plasma. (Rosenberg e Lam, 1979) demonstraram a presença de uma sequência
pentassacarídica, na molécula de heparina, importante para a ligação com a AT. Essa
sequência contém uma unidade glucosamina, com um único grupo O-sulfato na posição
3, que é crítico para o papel anticoagulante da heparina (Choay et al., 1983) (Figura 6).
Figura 7.Sequência pentassacarídica de ligação entre HNF e AT. Seta vermelha: pode
ser GlcNAc6SO3 ou GlcNSO3,6SO3; seta verde: GlcNSO3-3, 6SO3; seta laranja: Glc;
seta preta: IdoA2SO3. O asterisco na seta laranja indica o resíduo de glucosamina
supersulfatado característico dessa unidade. Círculo amarelo: grupo sulfato essencial para
a interação com AT. (Casu et al., 2007).
A ligação HNF/AT causa uma mudança conformacional neste cofator, resultando
em uma inativação acelerada das serino-proteases como os fatores IXa, Xa, XIa, XIIa e
IIa (trombina)(Linhardt, 2003). Neste processo é formado um complexo covalente
AT/trombina/HNF que inativa a enzima ligada permanentemente. Aproximadamente,
*

29
apenas um terço das moléculas de HNF se liga à AT as quais contêm a sequência
pentassacarídica.
Figura 8.Inativação dos fatores de coagulação pelo complexo HNF + antitrombina. A trombina e o fator Xa são as proteínas mais suscetíveis, sendo a trombina 10 vezes mais
sensível à inibição que o fator Xa. Adaptado de (Hirsh et al., 1995)
A HNF atua também através da interação com o cofator II da heparina, inibindo
apenas a ação da trombina e não outras enzimas da cascata de coagulação (Young, 2008).
No mecanismo antitrombótico da HNF também se observa redução da atividade do
complexo fator tecidual/fator VIIa, pois há aumento do inibidor da via do fator tecidual
(TFPI) pelo endotélio e, também, há redistribuição do TFPI, com sua diminuição por ser
direcionado a superfície celular. O TFPI forma um complexo com fator Xa, inativando-
o, e este complexo inativa o complexo fator VIIa + fator tecidual (Lupu et al., 1999).
(Nader et al., 1989) demonstraram outro mecanismo de inibição antitrombótica e
observaram que ela aumentava, em cerca de 2 a 3 vezes, a síntese de heparan sulfato pelas
células endoteliais da aorta de coelhos, em meio de cultura, e que esse aumento ocorria
imediatamente após a exposição das células endoteliais à HNF. Demonstraram também,
que ocorria aumento do grau de sulfatação nos resíduos de ácido urônico do heparan
sulfato proveniente destas células.
A HNF é efetiva para prevenção e tratamento das tromboses arteriais, venosas e
embolia pulmonar. É também empregada no tratamento de pacientes com angina e infarto
agudo do miocárdio e em alguns casos iniciais de coagulação intravascular disseminada.
É importante durante cirurgia cardíaca com circulação extracorpórea, hemodiálise,

30
cirurgia vascular, angioplastia e colocação de stents e endopróteses vasculares (Hirsh et
al., 2001).
Apesar de ser efetiva como droga e amplamente utilizada, seus efeitos adversos
levam a procura de outros substitutos. Dentre esses efeitos adversos pode-se citar a
hemorragia e a trombocitopenia induzida por heparina (TIH) (Gupta et al., 2015). A TIH
é uma resposta imunológica devido ao uso da HNF. Essa reposta é atribuída a anticorpos
que identificam a formação do complexo heparina mais fator plaquetário 4, os quais
ligam-se a esse complexo levando à formação de lesões no vaso, trombose e coagulação
disseminada podendo se expressar depois de cinco a sete dias ou até mesmo no primeiro
contato com o fármaco (Warkentin et al., 2008). A HNF também liga-se a proteínas
plasmáticas durante sua administração e células endoteliais, resultando em uma resposta
anticoagulante variável (Barzu et al., 1986). Além disso, o tratamento anticoagulante
com demanda monitoramento diário de sua atividade que, em geral, demanda
internação para a observação do paciente.
8. Heparina de baixo peso molecular
8.1 Estrutura e mecanismo de ação
Em 1980 teve início a introdução das heparinas de baixo peso molecular. As
HBPM são porções da heparina não fracionada produzidas pela despolimerização de
cadeias de polissacarídeos, seja quimicamente ou por uma reação enzímica, processo que
reduz seu peso molecular cerca de 4.000 a 6.000 Da. As HBPM são extraídas de mucosa
intestinal porcina (Fareed et al., 2004) e também do tecido pulmonar bovino (Andréo
Filho, 2009).
A interação da HBPM ocorre pela antitrombina, sendo mediada por uma
sequência de pentassacarídeo, a qual ocorre em menos de um terço das moléculas das
HBPM que está distribuída de forma aleatória ao longo de sua cadeia. Possuindo peso
molecular menor, a capacidade para inibir a trombina diminui, pois moléculas com menos
de 18 sacarídeos não são capazes de formar o complexo ternário heparina-AT-trombina
(Harenberg, 1990). Esta interação entre pentassacarídeo e antitrombina é também
responsável pelo efeito anticoagulante das HBPM, via inibição do fator Xa.

31
A redução do peso molecular reflete no mecanismo de inibição sobre a trombina
e o fator Xa. A trombina, além de converter o fibrinogênio, promove uma
retroalimentação positiva ativando os fatores V e VIII. Como as HBPM possuem baixa
atividade inibitória de trombina devido ao seu tamanho reduzido, não são capazes de
impedir a formação dos fatores V e VIII, importantes complexos procoagulante (Holmer
et al., 1986). Há, portanto, diferença entre a potência das HBPM e HNF, cuja atividade
anticoagulante permanece maior.
As HBPM possuem menor interação com as plaquetas, fator de von Willebrand,
endotélio e atuam menos sobre a permeabilidade vascular, o que proporcionaria menor
ocorrência de processos hemorrágicos em relação as heparinas não fracionadas
(Horlocker e Heit, 1997). Possuem uma meia-vida plasmática duas a quatro vezes maior
do que as heparinas não fracionadas, além de maior absorção subcutânea, o que permite
a aplicação de doses mais espaçadas. O monitoramento laboratorial é dispensável na
maioria dos pacientes, consequência da sua relação dose-efeito (Hull et al., 1992). Este
melhor perfil de desempenho das HBPM reflete-se em seu custo que é cerca de 10 a 20
vezes maior. A meia-vida da atividade de inibição do fator Xa das HBPM varia entre 3 e
10 minutos após injeção intravenosa e entre 3 e 5 horas após injeção subcutânea
apresentando variação também entre uma e outra HBPM (Andrassy e Eschenfelder, 1996)
9. O Dermatan Sulfato
9.1 Estrutura de mecanismo de ação
O dermatan sulfato é um glicosaminoglicano composto de cadeias lineares
polissacarídicas formadas por unidades dissacarídicas contendo uma hexosamina, N-
acetil galactosamina (GalNAC) ou ácido idurônico (GlcA) unidos por ligações β 1→4 ou
β 1→3 respectivamente.
O dermatan Sulfato possui um mecanismo de ação na coagulação como inibidor
seletivo da trombina através da catálise do cofator II da heparina (Tovar, M. F. et al.,
2005). Diferente das heparinas não fracionadas e de baixo peso molecular, o dermatan
sulfato não influencia na antitrombina III e, portanto, não inibe o Fator Xa. Ele é efetivo
não somente na trombina livre, mas também na trombina ligada à fibrina. O dermatan
sulfato não interfere com as plaquetas ou na função plaquetária. Assim, quando complexo

32
cofator II de heparina e DS é formado, o efeito anticoagulante é aumentado em até 1000
vezes (Tollefsen, 2007).
Figura 9.Estrutura do Dermatan Sulfato. Possui alta afinidade para o cofator II de
heparina. Um pequeno fragmento de DS que se liga ao HCII com alta afinidade é um
hexassacarídeo contendo 3 unidades dissacarídicas de IdoA2SO3 → GalNAc4SO3
(Maimone e Tollefsen, 1990).
Foi observado que o DS é o principal polissacarídeo antitrombótico e
anticoagulante nas paredes dos vasos sanguíneos de artéria aorta humana e veia safena
em relação aos outros GAGs, em tecidos afetados pela aterosclerose. Foi observado haver
uma mudança entre a proporção de heparan sulfato/dermatan ao longo do
desenvolvimento da doença no vaso afetado (Tovar, A. M. et al., 2005). Foi demostrando
por (He et al., 2008) que o DS está localizado principalmente na camada adventícia da
artéria carótida e que após a injúria arterial o HCII é capaz de se difundir do plasma para
esta camada onde é ativado.
Um estudo de nosso laboratório demonstrou que o DS foi capaz de diminuir a
presença de P-selectina (moléculas de adesão presente no endotélio que promove a adesão
de leucócitos às células endoteliais ativadas durante a resposta inflamatória) no local da
lesão vascular, indicando uma possível função antiinflamatória desse GAG (Godoy et al.,
2015). (Vicente et al., 2007) demonstraram que a administração de dermatan sulfato 48
horas após a injúria vascular inibiu a formação da neointima em camundongos normais,
mas não em camundongos deficientes de HCII. Em outro trabalho de nosso laboratório
observamos que a administração de células mononucleares da medula óssea de
camundongos C57BL6 juntamente com o DS ajudou a prevenir formação da neoíntima
após lesão arterial e aumentou a migração destas células para o local da lesão arterial
(Godoy et al., 2011).

33
10. Modelo de lesão arterial por cloreto férrico
Estudo in vivo em modelos de animais são críticos para melhor entendimento dos
mecanismos envolvidos em desordens trombóticas (Bayes-Genis et al., 2000). Dentre os
diversos métodos utilizados para induzir a formação do trombo em modelos de animais,
encontra-se o modelo químico utilizando o cloreto férrico (FeCl3).
Em 1994, o FeCl3 foi inicialmente utilizado na veia cava inferior de coelhos a fim
de induzir a trombose (Reimann-Hunziker, 1944), este método foi adaptado para carótida
de ratos em 1990 por Kurz e colaboradores mostrando que uma solução de FeCl3 aplicada
na artéria carótida exposta de ratos, induzia trombose oclusiva. Em 1998, o modelo de
trombose arterial causada pelo FeCl3 foi adaptado para camundongos no estudo da
deficiência do inibidor do ativador do plasminogênio (Konstantinides et al., 2001).
Um dos benefícios desse modelo é que diferentes concentrações de cloreto férrico
geram lesões de níveis diferentes e consequentemente, tempo de oclusão do vaso
sanguíneo variado. Dessa forma, com estabelecimento da concentração de FeCl3 e tempo
de aplicação, a técnica tem se mostrado reprodutível (Ciciliano et al., 2015).
Este modelo é simples e sensitivo, tanto a drogas anticoagulantes como
antiplaquetárias, motivo pelo qual foi escolhido como modelo para estudo neste trabalho.
Tem sido realizado em artérias carótida e femoral, veia jugular, arteríolas e vênulas
mesentérica e cremastérica em ratos, camundongos, mini pigs e coelhos (Wiedermann et
al., 1994; Hehrlein et al., 1996; Day et al., 2004)
A técnica consiste na colocação de um papel filtro com medida de um mm²
embebido em FeCl3 sobre o vaso e deixado por alguns minutos. A lesão é devida à
capacidade do cloreto férrico de penetrar a parede vascular, causando desnudação de
células endoteliais e expondo o subendotélio juntamente com fibras de colágeno. A
exposição ativa a cascata de coagulação, culminando com formação de trombo e posterior
crescimento da camada neointimal (Eckly et al., 2011)
Neste modelo o tempo de oclusão é considerado como o tempo ocorrido após o
início da lesão vascular até a completa oclusão do vaso através da interrupção do fluxo
sanguíneo, que pode ser medido por um equipamento de aferição de fluxo de Doppler ou
análise com microscopia intravital. Trabalhos anteriores utilizando camundongos
C57BL6 observaram um tempo de oclusão variando de 5 a 30 minutos, devido as

34
diferentes concentrações de FeCl3 que são utilizadas, tempo de exposição, tipos de
anestesia, técnicas cirúrgicas, a idade e o background genético dos animais, o método de
medição do fluxo sanguíneo, e outras variáveis ambientais surtirão efeito significativo
neste modelo (Robertson et al., 2009; Owens e Mackman, 2010).

35
II. OBJETIVO
Objetivo geral
Analisar o efeito do dermatan sulfato, heparina e heparina de baixo peso molecular
na formação e dissolução do trombo na artéria carótida, remodelamento e reendotelização
do vaso após lesão arterial promovida por cloreto ferrico in vivo.
Objetivos específicos
1. Determinar o tempo de trombose e o tamanho de trombo arterial no tempo
imediatamente após a lesão química com cloreto férrico nos diferentes grupos
tratados com GAGs (tempo zero);
2. Avaliar a influência dos tratamentos dos diferentes GAGs no tempo de
sangramento, tempo de trombina e tempo de tromboplastina parcial ativada no
plasma obtido dos animais tratados;
3. Observar o processo de recuperação do vaso após lesão arterial através da
quantificação de células progenitoras endoteliais para o local da lesão (células
CD34) e de células endoteliais (CD31) nos diferentes grupos tratados com GAGs;
4. Avaliar a influência dos GAGs na inflamação inicial pela presença células
inflamatórias no vaso (CD11b e ICAM-1);
5. Analisar a influência dos diferentes GAGs na formação da camada neointima
quatorze dias após a lesão química;
6. Quantificar a atividade das metaloproteinases (gelatinases) no remodelamento
vascular, observando a atividade destas nos vasos em diferentes tratamentos com
GAGs.

36
III. MATERIAIS E METODOS
1. Animais
Foram utilizados camundongos machos selvagens de linhagem C57BL/6
adquiridos no CEMIB/UNICAMP, mantidos em biotério do Departamento de Biologia
Celular e Estrutural da UNICAMP sob responsabilidade da Profa. Dra. Cristina Pontes
Vicente. Este trabalho teve a aprovação do comitê de ética da UNICAMP (CEUA n°
3892-1).
2. Grupos experimentais
Neste estudo foram testadas quatro condições de tratamento em camundongos
C57BL/6: i) animais lesionados e não tratados ii) animais lesionados e tratados com
dermatan sulfato iii) animais lesionados e tratados com heparina não fracionada iv)
animais lesionados e tratados com heparina de baixo peso molecular.
Os quatro grupos de animais foram analisados sob as circunstâncias:
3. Tratamentos com GAGs
Nos grupos de animais com lesão arterial (LA) e analisados imediatamente após
a lesão (tempo 0) a administração foi realizada da seguinte forma:
HNF – administração intravenosa (Cristália, SP) (1 mg/kg animal), 10 minutos
antes do procedimento de lesão arterial ocorrer;

37
HBPM – administração intravenosa (Clexane) (1 mg/kg animal), 10 minutos antes
do procedimento de lesão arterial ocorrer;
DS – administração intravenosa (Sigma, Saint Louis, MO) (10 mg/kg animal), 10
minutos antes do procedimento de lesão arterial ocorrer;
Nos grupos de animais com lesão arterial e analisados 14 dias após a
administração foi realizada da seguinte forma:
HNF – administração intravenosa (1 mg/kg animal), 10 minutos antes do
procedimento de lesão arterial ocorrer; 12 horas, 24 horas e 48 horas após a lesão
arterial ocorrer;
HBPM – administração intravenosa (1 mg/kg animal), 10 minutos antes do
procedimento de lesão arterial ocorrer; 12 horas, 24 horas e 48 horas após a lesão
arterial ocorrer;
DS – administração intravenosa (10 mg/kg animal), 10 minutos antes do
procedimento de lesão arterial ocorrer; 12 horas, 24 horas e 48 horas após a lesão
arterial ocorrer.
4. Modelo de indução de trombose arterial
Camundongos pesando 25g entre 6-7 semanas de idade foram anestesiados com
16 mg/kg de cloridrato de xilazina 2% e 100 mg/kg de ketamina. Após uma incisão na
cervical média e após isolar a artéria carótida comum direita (figura 10A e 10B), foi
provocada a lesão química (figura 10C). Colocou-se sobre a artéria um papel de 1mm²
(Whatmann 3MM) previamente preparado com 15% de concentração de cloreto férrico,
por 2 minutos (figura 10C), após este tempo o papel foi removido e o vaso lavado com
PBS para evitar lesões em locais indesejados. Depois desse procedimento, foi posicionada
uma sonda de ultrassom abaixo do vaso para medição do fluxo sanguíneo. A mensuração
do fluxo foi registrada usando um modelo de ultrassom (Transonic System TS 420)
(figura 10D). Foram utilizados 6 camundongos por grupo.
Para a preparação do papel com cloreto férrico, cortou-se papeis filtro no tamanho
de 1 mm², sendo estes embebidos em uma solução de 15% de cloreto férrico por 24h.
Após este tempo estes papeis foram secos em estufa por um período de 1h, e utilizados
então, para o procedimento de lesão arterial.

38
Figura 10.Modelo de indução de lesão por cloreto ferrico. (A e B) Isolamento da
artéria carótida comum, (C) indução da trombose arterial com papel saturado com cloreto
férrico, (D) posicionamento da sonda de ultrassom para medição do fluxo sanguíneo.
5. Tempo de Trombose Arterial
Após a denudação arterial por modelo de cloreto férrico mencionada
anteriormente, foi posicionada uma sonda de ultrasson (Transonic Flowprobe MAO 5
PBS) no vaso lesionado, e o fluxo sanguíneo registrado pelo programa DATAQ. O tempo
entre a retirada do papel contendo a solução de cloreto férrico e o posicionamento da
sonda de ultrasson na artéria carótida não ultrapassou o tempo de 1 minuto nos animais
analisados.
Para o tempo de oclusão foi utilizada uma medida do tempo entre o fluxo inicial
e o tempo onde o fluxo parou por oclusão do vaso pela formação do trombo. Foi medido
o tempo de oclusão nos animais lesionados sem tratamento, lesionado e tratados com
heparina, lesionados e tratados com HBPM, lesionados e tratados com DS.

39
6. Cálculo da área de trombo
A área de trombo analisada no tempo inicial após lesão arterial foi calculada pela
subtração da área oclusa de lúmen da área total encoberta pela lâmina elástica interna. A
área média foi calculada, em μm², pela subtração da área encoberta pela lâmina elástica
interna da lâmina elástica externa.
7. Microscopia Intravital
A análise da formação de trombo nas artérias carótidas por meio da microscopia
intravital foi realizada nos 4 grupos: 1) Lesão arterial e não tratado, 2) Lesão arterial e
tratado com DS, 3) Lesão arterial e tratado Heparina e 4) Lesão arterial e tratado com
HBPM.
A administração dos fármacos nos respectivos grupos foi feita como descrito no
item 3, 10 minutos antes do procedimento de lesão ocorrer por cloreto férrico acontecer.
Após esse período, foi aplicada na veia da cauda desses animais uma solução de
Rodamina 6G na concentração de 0,1% em água (Sigma Aldrich, St Louis-USA). A
Rodamina 6G é um corante fluorescente que tem afinidade pela membrana mitocondrial,
que cora plaquetas e glóbulos brancos, excluindo os eritrócitos. Injetada a solução, os
animais foram lesionados na artéria carótida direita com o papel embebido em solução de
cloreto ferrico 15% por 2 minutos. As imagens foram captadas com auxílio de câmera
Axiocam HSm e o experimento foi realizado no microscópio modelo Carl Zeiss Imager.
A.2 gentilmente cedido pelo prof. Fábio Maranhão Costa. A filmagem foi feita por 12
minutos e as fotos obtidas nos tempos 0, 5 e 10 min foram analisadas para a verificação
do tamanho do trombo. Após a captura de imagens, o software ImageJ foi utilizado para
medição da fração do trombo formado no vaso nos três tempos analisados (0, 5 e 10
minutos).
8. Determinação do tempo de tromboplastina parcial ativada (TTPA)
O TTPa é o tempo medido entre a adição de cálcio, na presença de uma cefalina
(fator de contato) e a coagulação do plasma. A presença de um fator de contato (cefalina)
ativa as reações da via intrínseca da coagulação. O TTPa corresponde ao tempo gasto para
ocorrer a coagulação do plasma recalcificado em presença de cefalina. Esta substitui o
fosfolipídio da membrana plaquetária. O TTPa foi medido no plasma com citrato de sódio
3,8% de camundongos selvagens nos grupos controle, lesão tratados com DS, lesão

40
tratados com Heparina e lesão tratados com HBPM utilizando o kit TTPA Clot (Bios
Diagnostica, SP). Em cada grupo foi feita a análise 12 e 24 após a lesão arterial, avaliada
em triplicata. Para cada grupo foi utilizado n=3 animais.
O teste é usado na avaliação da via intrínseca de coagulação e monitoramento de
terapias com agentes anticoagulantes. O ensaio foi realizado da seguinte maneira: 50 uL
de amostras foram pré-aquecidas à 37°C durante 2 minutos em um bloco térmico; foram
adicionados 50uL de reagente de TTPA clot ao tubo e incubou-se por 2 minutos à 37°C.
Em seguida, adicionou-se 50 uL de cloreto de cálcio (CaCl2 pré-aquecido à 37°C) e
iniciou-se a medição do tempo no coagulômetro CLOTimer (Clot, S.P, Brasil).
9. Determinação do tempo de protrombina (TP)
O TP avalia a atividade extrínseca do sistema de coagulação. O reagente para
análise contém fator tecidual, fosfolipídios e cálcio. A determinação do tempo de
protrombina é adequada para acompanhamento de terapias com drogas antitrombóticas.
O tempo de protrombina foi dosado em plasma citratado (1 parte de citrato de sódio 3,8%:
9 partes de sangue venoso) de camundongos nos 4 grupos citados anteriormente neste
estudo. A análise foi feita utilizando-se o kit TP Clot (Bios Diagnostica, SP). O ensaio
foi realizado da seguinte maneira: pipetou-se em uma cubeta pré-aquecida à 37°C 50uL
de plasma e incubou-se por 1 minuto à 37°C; adicionou-se 100uL de reagente para teste
de protrombina previamente preparado e também pré-aquecido à 37°C. Com a adição do
reagente, iniciou-se a medição no coagulômetro (tempo medido em segundos). As
amostras foram feitas em triplicata para cada grupo de animal, onde foi utilizado n=3
animais.
10. Determinação do Tempo de Sangramento (TS)
Realizou-se uma incisão longitudinal de 3 mm de comprimento por 2 mm de
profundidade, na face ventral da cauda do animal, entre 1,5 cm do final desta, evitando-
se grandes veias. A cauda foi mergulhada em uma proveta com solução salina a 37ºC
(Dejana et al., 1979). O tempo de sangramento foi mensurado desde o momento da incisão
até o final do sangramento (Liu et al.; Broze et al., 2001).

41
11. Análise Histológica
As artérias carótidas obtidas nos diferentes tempos foram coletadas e embebidas
em OCT (Tissue Tek Optimal Cutting temperature compound, Torrance – USA), e
armazenadas no biofreezer a -80°C. Em seguida foram feitos cortes transversais de 8 µm
de espessura em criostato, com 20 lâminas de cada vaso com 5 cortes por lâmina, também
armazenados em biofreezer a -80°C. Os cortes foram descongelados e as lâminas 1, 5, 10,
15 e a 20 foram coradas com hematoxilina-eosina. As lâminas foram montadas com
Cytoseal 60 (Richard Allan Scientific) e examinadas em microscópio Olympus BX600.
As imagens foram feitas com câmera Olympus Optical U-ULH e analisadas com o
programa ImageJ. Para a área de trombo foi escolhido o corte com maior obstrução ao
longo do vaso, e a área do trombo foi calculada pela subtração da área ocluída do vaso
pela área total do vaso delimitada pela lâmina elástica.
12. Ensaio de Imunofluorescência
Realizou-se imunohistoquímica para a análise de marcadores expressos pelas
células no vaso. Para isso, foram utilizados os anticorpos rat anti-mouse CD 31
(Invitrogen), para a visualização das células endoteliais maduras, para que houvesse uma
análise da capacidade dos glicosaminoglicanos na proteção dessas células após a
denudação endotelial, rat anti mouse CD 34 (eBioscience), um marcador para células
progenitoras endoteliais para que se identificasse a mobilidade destas para o vaso após
lesão, o rat anti-mouse CD11b, característicos de células leucocitárias envolvidas na
inflamação e anti-ICAM-1 (eBioscience, San Diego, CA), molécula de adesão
intercelular envolvida na transmigração de leucócitos. Os anticorpos foram diluídos
conforme instruções do fabricante, e depois de incubados por pelo menos 2 horas, lavou-
se para retirar o excesso de anticorpo e incubou-se com o anticorpo secundário anti-rat
FITC (eBiocience) por 45 minutos. Em seguidas as amostras foram lavadas e os núcleos
corados com DAPI por 15 minutos. As lâminas foram montadas e examinou-se em
microscópio Olympus BX600. As imagens foram feitas com câmera Olympus Optical U-
ULH. Após revelação das imagens, utilizou-se o programa ImageJ para quantificação
das marcações fluorescentes dos cortes de artérias carótidas. Uma lâmina sem o anticorpo
primário (controle negativo) foi feita para subtração da autofluorescência das artérias. Em
cada lâmina contendo cinco cortes, foi escolhido o corte com mais marcações. Para cada
grupo analisado, foram utilizadas três lâminas diferentes de animais.

42
13. Cálculo área da neoíntima
A área de neoíntima analisada 14 dias após lesão arterial foi calculada pela
subtração da área de neoíntima formada no lúmen da área total encoberta pela lâmina
elástica interna. A área média foi calculada, em μm², pela subtração da área encoberta
pela lâmina elástica interna da lâmina elástica externa.
14. Zimografia ‘in situ’
Metaloproteinases (MMPs), como as colagenases, gelatinases degradam
colágeno, gelatina e outros componentes da matriz extracelular, contribuindo no processo
inflamatório e mediando o remodelamento vascular. As principais gelatinases envolvidas
no processo de remodelamento tecidual são MMP-2 e MMP-9. A atividade destas
enzimas foi analisada artérias carótidas obtidas dos diferentes grupos de tratamento.
Considerou-se o aumento da fluorescência como proporcional a atividade proteolítica das
gelatinases. As artérias carótidas foram obtidas como descrito anteriormente; os cortes
foram incubados em câmara úmida escura por 2 horas com 100 μg/mL de DQ gelatina
em solução tampão de acordo com as instruções do fabricante. Depois os cortes foram
analisados com microscópio de fluorescência (Observer Z1, Zeiss) usando lentes
objetivas de 20x e câmera AxioCam MRm (Zeiss) e as imagens capturadas usando o
programa AxioVision Release 4.8.2.0. A atividade proteolítica foi detectada por um
aumento da intensidade de fluorescência devido à atividade gelatinolítica, e a intensidade
de fluorescência nos diferentes cortes foi quantificada no programa Image J (NIH). O
corte apresentando maior fluorescência foi selecionado. Foi feita lâmina sem a solução
de DQ-gelatina como controle negativo. Foram utilizados n=3 animais em cada grupo. A
análise de zimografia ‘in situ’ foi feita no tempo inicial após lesão arterial e 14 dias após
lesão arterial.
15. Agregação plaquetária
Realizou-se a coleta de sangue venoso periférico humano de doador em jejum
(período de 12 horas) em tubos contendo citrato de sódio a 3,2%. Após a centrifugação
do sangue por 15 minutos a 100xg foi obtido o plasma rico em plaqueta (PRP) e por 20
minutos a 2400xg para obtenção do plasma pobre em plaqueta (PPP). Para medição da
agregação foi utilizado um agregômetro de dois canais (490-2D, Chrono-log Corporation,
Havertown, PA, USA). A calibração inicial do aparelho foi feita com concentração de

43
500 μL de PPP. Em cada amostra foi utilizado 500 μL de PRP, o qual foi mantido em
37°C antes de iniciar o ensaio. Após isso, adicionou-se o agente agonista trombina na
concentração de 0,25 U/ml no plasma e mediu-se a agregação plaquetária por 14 minutos.
Os grupos analisados foram controle: PRP+ trombina; DS: PRP + dermatan sulfato +
trombina; Heparina: PRP + Heparina+ trombina e HBPM: PRP + HBPM + trombina.
16. Análise Estatística
A análise de cada grupo individual foi expressa pela média ± desvio-padrão. A
avaliação da média entre os grupos foi realizada através do teste Mann Whitney, com
critério de significância estatística p≤ 0,05.

44
IV. RESULTADOS
1. Peso dos Animais
Não houve diferença estatisticamente significativa no peso dos animais, entre os grupos
experimentais (p < 0,05).
2. Tempo de formação do trombo nos tratamentos com DS, Heparina e
Heparina de baixo peso molecular inicialmente após o procedimento cirúrgico
A trombose foi induzida no vaso conforme descrito anteriormente, através da
lesão por cloreto férrico a 15% de concentração na artéria carótida comum direita. Após
a lesão, posicionou-se uma sonda de ultrassom no local para medição do tempo de oclusão
do vaso (figura 11). A formação de trombo no grupo lesionado e não tratado com GAG
foi de 5,5 minutos ( ± 0,5); nos animais lesionados e tratados com a heparina de baixo
peso molecular 10,3 minutos ( ± 0,51 com p=0,02); em animais tratados com dermatan
sulfato ocorreu no tempo de 14,6 (± 0,51 com p=0,02) e nos animais lesionados e tratados
com heparina (30,8 ± 2,0 com p<0,01). Todos os grupos foram significativamente
maiores que o controle com p<0.05. (Figura 10).
O DS aumentou o tempo de oclusão em 2,8x em relação ao controle e 1,4x mais
que a HBPM. A Heparina aumentou o tempo de oclusão 6x em relação ao controle, cerca
de 2x mais que o DS e em 3x mais que HBPM. A HBPM prolongou em 2x o tempo de
oclusão em relação ao controle.

45
Figura 11. Tempo de formação do trombo na artéria carótida. O tempo de formação
do trombo foi medido através da colocação da sonda de ultrassom na artéria carótida
direita dos animais e retirada após completa oclusão do vaso pela lesão por cloreto férrico.
Média do tempo de oclusão da artéria em animais controle, tratados com HBPM, tratados
com DS e tratados com Heparina, sendo n= 6 em cada grupo. Diferença significativa com
p<0,05 em relação ao controle representada por (*).
3. Área do trombo por histologia
Para medição da formação de trombo no vaso, as artérias carótidas foram
coletadas no tempo inicialmente após a lesão com cloreto férrico e mantidas em OCT.
Depois, foram realizados cortes histológicos e coloração HE (Hematoxilina e Eosina)
para posterior análise da área do trombo através do programa Image J (figura 12).
O grupo de animais lesionados e não tratados obtiveram média de porcentagem
de trombo arterial imediatamente após a lesão de 86,5% ± 4,72 (figura 12A). No grupo
de animais lesionados e tratados com a HBPM a média observada foi de 80,4% ± 5,0
(figura 12B). Em animais lesionados e tratados com o dermatan sulfato, essa média foi
de 50,8% ± 4,33 (figura12C). A média nos grupos lesionados e tratados com a heparina
foi de 30% ± 3,47 (figura 12D).
O tratamento no grupo de animais lesionados e injetados com HBPM foi capaz de
diminuir a formação de trombo em aproximadamente 20% (p=0,03 16433,48 vs.
132125,17); no grupo lesionado e tratado com o DS houve diferença significativa (p=
0
5
10
15
20
25
30
35
Lesão sem
tratamento
Lesão + DS Lesão+ Heparina Lesão + HBPM
Min
uto
s
Tempo de oclusão arterial
*
*
*

46
0,02), onde o tratamento conseguiu a diminuição na formação de trombo em
aproximadamente 45% em relação à formação de trombo no grupo controle (164334,8
vs. 90055,47); no grupo lesionado e tratado com heparina houve diferença significativa
em relação ao grupo controle sendo a formação de trombo aproximadamente 67% menor
(p=0,01 16433,48 vs. 53737,47). A heparina, portanto, mostrou maior eficiência em
diminuir o trombo arterial e o tratamento com HBPM mostrou menor capacidade neste
processo. O tratamento com DS obteve formação de trombo nos vasos em cerca de 1,4x
menor (43% p=0,02) em relação a HBPM.
Figura 12. Análise da formação de trombo por histologia. Corte transversais de vasos
arteriais de camundongos C57BL/6 foram analisados pela técnica de coloração
Hematoxilina e Eosina. (A) artéria lesionada e não tratada, (B) artéria lesionada e tratada
com 1 mg/kg de HBPM, (C) artéria lesionada e tratada com DS em concentração de 10
mg/kg, (D) artéria lesionada e tratada com heparina com concentração de 1 mg/kg. Seta
azul indica deposição de Fe3+ decorrente do processo de lesão. Barra= 50 μm
A B
A
A
C
A
D
A
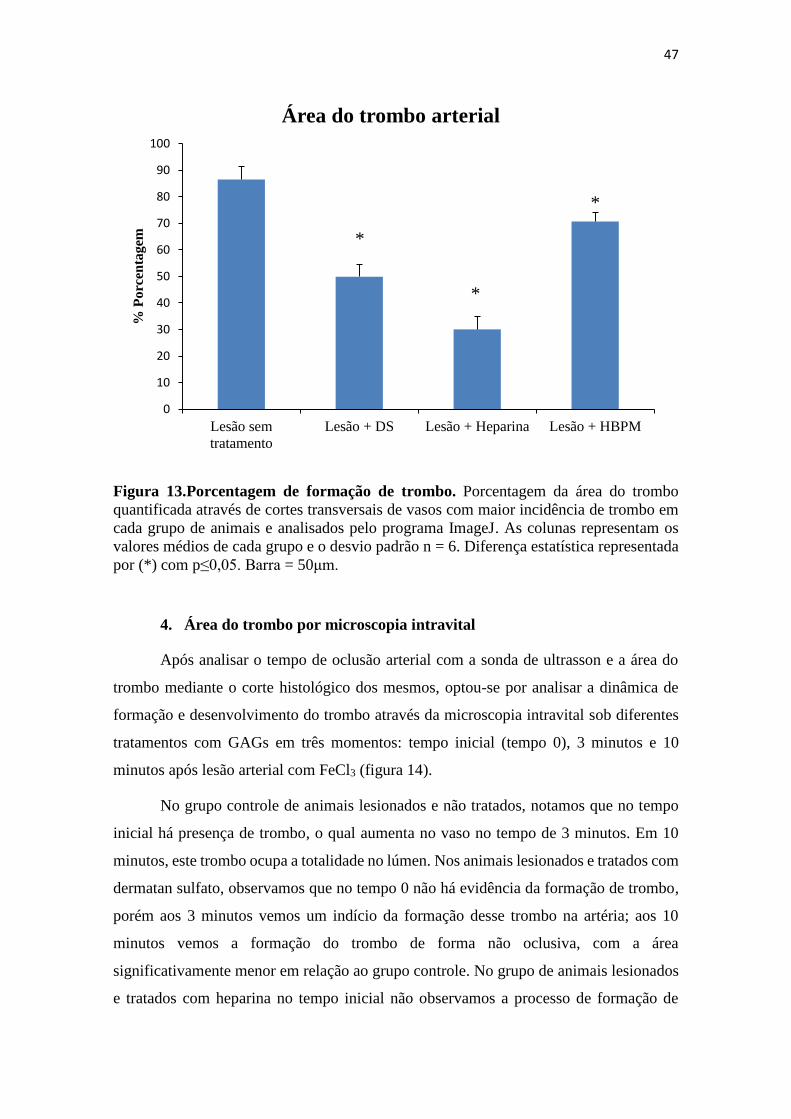
47
Figura 13.Porcentagem de formação de trombo. Porcentagem da área do trombo
quantificada através de cortes transversais de vasos com maior incidência de trombo em
cada grupo de animais e analisados pelo programa ImageJ. As colunas representam os
valores médios de cada grupo e o desvio padrão n = 6. Diferença estatística representada
por (*) com p≤0,05. Barra = 50μm.
4. Área do trombo por microscopia intravital
Após analisar o tempo de oclusão arterial com a sonda de ultrasson e a área do
trombo mediante o corte histológico dos mesmos, optou-se por analisar a dinâmica de
formação e desenvolvimento do trombo através da microscopia intravital sob diferentes
tratamentos com GAGs em três momentos: tempo inicial (tempo 0), 3 minutos e 10
minutos após lesão arterial com FeCl3 (figura 14).
No grupo controle de animais lesionados e não tratados, notamos que no tempo
inicial há presença de trombo, o qual aumenta no vaso no tempo de 3 minutos. Em 10
minutos, este trombo ocupa a totalidade no lúmen. Nos animais lesionados e tratados com
dermatan sulfato, observamos que no tempo 0 não há evidência da formação de trombo,
porém aos 3 minutos vemos um indício da formação desse trombo na artéria; aos 10
minutos vemos a formação do trombo de forma não oclusiva, com a área
significativamente menor em relação ao grupo controle. No grupo de animais lesionados
e tratados com heparina no tempo inicial não observamos a processo de formação de
0
10
20
30
40
50
60
70
80
90
100
Lesão sem
tratamento
Lesão + DS Lesão + Heparina Lesão + HBPM
% P
orc
enta
gem
Área do trombo arterial
*
*
*

48
trombo, aos 3 minutos não há evidência da presença do trombo. Aos 10 minutos vemos o
início do trombo ser formado, com a área deste significativamente menor em relação ao
grupo controle e ao grupo lesionados + DS. Em animais lesionados e tratados com HBPM
notamos que no tempo 0 há presença de trombo. Este aumenta ao longo do tempo, aos 3
minutos ocupa grande parte do lúmen e aos 10 minutos o trombo apresenta indícios de
embolização como representado na seta abaixo:
Figura 14.Formação de trombo por microscopia intravital. Após a lesão química com
cloreto férrico, a formação de trombo nas artérias carótidas foi observada por 12 minutos
nos grupos lesionados sem tratamentos, lesionados tratados com DS, lesionados tratados
Lesão sem
tratamento
Lesão + DS
Lesão +Heparina
Lesão + HBPM
0 minutos 3 minutos 10 minutos
10x
5x
5x
5x

49
com heparina e lesionados tratados com HBPM. Aumentos de 5x e 10x. Microscópio
Intravital Zeiss – Imager A2.
5. Formação de neoíntima é atenuada no tratamento com DS, Heparina e
HBPM
Dados anteriores observados em nosso laboratório (Sielski, 2015) demonstraram
que 14 dias após a lesão arterial, a proliferação de células musculares lisas já está
estabelecida, ocupando cerca 90% do lúmen do vaso, com células musculares lisas. Por
isso optamos por utilizar este tempo em nossos experimentos. Por análise histológica
escolhemos o corte com a maior formação de neointima ao longo do vaso e a partir destes
foi calculada a área de neointima. (figura 16.I). Em nossos resultados observamos que a
área média de neoíntima ocupada no lúmen do vaso nos animais controle foi de 88%
(figura 16B), em animais tratados com DS 12,79% (figura 16C); com heparina 22,35%
(figura 16D) e com HBPM 71,4%. (Figura 16E). O DS obteve média de inibição de
neoíntima 13,7% maior que Heparina (p=0,04) e 58,8% maior que o HBPM (p=0,02).
Apesar de observar intensa proliferação de células musculares lisas no lúmen do
vaso, representada pelas setas pretas nas imagens abaixo, em nenhum dos tratamentos
houve aumento significativo da camada média do vaso, indicando que o método de lesão
arterial que utilizamos não lesiona a lâmina elástica o que em geral pode promover um
aumento significativo da camada media. (Figura 17).
A B
I)

50
Figura 15.Análise da porcentagem da área de neointima 14 dias após lesão arterial.
Figura I) Cortes transversais de artérias carótidas esquerdas coradas com Hematoxilina
e Eosina. Camundongos C57BL/6 tratados e não tratados com os glicosaminoglicanos 14
dias após lesão arterial com cloreto férrico. (A) artéria não lesionada sem presença de
trombo, (B) artéria lesionada e não tratada, (C) artéria lesionada e tratada com DS em
concentração de 10 mg/kg, (D) artéria lesionada tratada com Heparina com concentração
de 1 mg/kg e (E) artéria lesionada tratada com HBPM com concentração de 1 mg/kg.
Barra= 50 μm. Figura II) Porcentagem da área de neointima quantificada como descrita
em materiais e métodos. As colunas representam os valores médios de cada grupo e o
desvio padrão. Utilizado n=6 animais em cada grupo. Diferença estatística representada
por (*) p≤0,05. Barra = 50μm
0
10
20
30
40
50
60
70
80
90
100
Lesão sem
tratamento
Lesão + DS Lesão + Heparina Lesão + HBPM
% P
orc
enta
gem
Área da neoíntima
* *
C D E
II)

51
Figura 16.Área da camada média (μm2). Quantificada através da subtração da medida
da lâmina elástica interna pela lâmina elástica externa do vaso. As colunas representam
os valores médios para cada grupo ± desvio padrão; n=6 para cada grupo.
6. GAGs alteram o TTPA, TP e TS
Como o tratamento com os três GAGs aumentam o tempo de formação de trombo
nas artérias, optou-se por realizar testes de coagulação in vitro para o acompanhamento
da capacidade anticoagulante e antitrombótica dessas drogas. Realizamos três testes,
TTPA, TP e TS.
Observou-se um prolongamento do TTPa em todos os grupos analisados no
período de 12 horas após a lesão arterial quando comparados aos animais do grupo
controle: animais tratados com heparina prolongaram o TTPA em 5 vezes (503%) (240
vs. 39,8 segundos p <0,05); animais tratados com DS prolongaram o TTPA em 1,7 vezes
(73,4%) (69,03 vs. 39,8 segundos p <0,05) e animais tratados HBPM prolongaram o
TTPA em 1,9 vezes (90%) (75,68 vs. 39,8 segundos p<0,05). Em 24 horas, os animais
injetados com heparina apresentaram aumento do TTPa em 1,4 vezes (146%) em relação
ao grupo de animais lesionados e não tratados (89,6 vs. 36,3 segundos p<0,05); em
animais injetados com DS houve aumento do TTPA em 1 vez aproximadamente (8,8%)
em relação ao grupo de animais sem tratamento (39,5 vs. 36,3 p>0,05) considerado não
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
Lesão sem
tratamento
Lesão + DS Lesão + Heparina Lesão + HBPM
Esc
ala
-μ
m2
Área da camada média

52
significativo; animais tratados com HBPM diminuíram o TTPa em 10,8%. (32,4 vs. 36,3
p > 0,05) e não obtiveram diferença significativa (figura 18).
Figura 17.Tempo Parcial de Tromboplastina ativada (TTPa). O TTPa foi dosado em
plasma de camundongos C57BL/6 12 e 24 após lesão química provocada por cloreto
ferrico na artéria carótida comum. Os grupos analisados foram: Lesão (sem tratamento),
Lesão tratados com Heparina, Lesão tratados com DS e Lesão tratados com HBPM. (*).
Representa significância estatística entre os grupos. Experimento feito para n=3 animais
em cada grupo feito em triplicata. (*) Representa significância em relação ao grupo
controle (p≤0,05).
Em relação ao tempo de sangramento (TS), no tempo de 40 minutos após o
procedimento de lesão arterial o aumento foi de 370% no grupo de animais injetados com
heparina (p≤0,01 400 vs. 85 segundos), nos animais tratados com DS o aumento foi de
48% (p =0,02 126,3 vs. 85 segundos), animais tratados com HBPM o aumento foi de
22,3% (p=0,07 104 vs. 85 segundos) considerado não significativo. No período de 12
horas observou-se um aumento em todos os grupos analisados: em animais tratados com
heparina o TS prolongou em 305% em relação ao grupo de animais lesionados sem
tratamento (p=0,01 381,3 vs. 94 segundos); animais tratados com DS o TS prolongou-se
em 39% considerado não significativo (p >0,05 131,4 vs. 94 segundos) e em animais
tratados com HBPM o TS prolongou-se em 20%, considerado não significativo (p>0,05
113,3 vs. 94 segundos). No período de 24 horas houve um aumento do TS em 269% em
animais tratados com heparina (p<0,05 339,5 vs. 92 segundos), considerado significativo;
0
50
100
150
200
250
300
12 horas 24 horas
Tem
po
-se
gu
nd
os
Tempo de Tromboplastina parcial ativada
(TTPA)
Lesão sem tratamento
Lesão + DS
Lesão + Heparina
Lesão + HBPM
*
* * *

53
animais tratados com DS o aumento foi de 46% (p <0,05 135 vs. 92 segundos) houve
diferença significativa e em animais tratados com HBPM o aumento foi de 30,4% (p >
0,05 120 vs. 92 segundos) considerado não significativo (figura 19.II).
Figura 18.Tempo de sangramento. Figura I) Imagem dos tubos obtidos durante a
medida do tempo de sangramento nos grupos tratados e não tratado. (A) grupo lesionado
e não tratado, (B) grupo lesionado e tratado com DS, (C) grupo lesionado tratado com
heparina e (D) grupo lesionado tratado com HBPM. Figura II) O tempo entre a incisão
e a interrupção do sangramento foi cronometrado e considerado como o TS. O TS foi
calculado em camundongos C57BL/6 40 minutos, 12 e 24 horas após lesão química
promovida por cloreto férrico. Experimento feito para n=3 animais em cada grupo. (*).
Representa significância estatística em relação ao grupo controle (p≤0,05)
0
50
100
150
200
250
300
350
400
450
40 minutos 12 horas 24 horas
Tem
po
-se
gun
do
s
Tempo
Tempo de sangramentoLesão sem tratamento
Lesão + DS
Lesão + Heparina
Lesão + HBPM
* *
*
*
C
A
D
A
A
A
B
A
I)
II)
*

54
O tempo de protrombina (TP) foi dosado e apenas nos grupos de animais tratados
com heparina foram observados aumentos do TP (22,8%), porém esse aumento não foi
considerado significativo comparado ao grupo controle (figura 20).
Figura 19.TP dosado em camundongos C57BL/6 no período de 12 e 24 horas após o
procedimento de lesão arterial. Experimento feito para n=3 animais em cada grupo feito
em triplicata.
7. Expressão de marcadores após tratamento com os GAGs
Após o tempo imediato do procedimento de lesão arterial, as artérias carótidas dos
diferentes grupos foram coletadas, emblocadas e feito cortes histológicos nos grupos.
Realizou-se a técnica de imunofluorescência nos cortes usando os seguintes marcadores
CD31 conjugado com anti rat FITC (Figura 19), CD34 conjugado com anti rat FITC
(Figura 21), CD11b conjugada com anti rat FITC (figura 22) e ICAM-1 conjugada com
anti rat FITC (Figura 23).
7.1 Extensão da lesão arterial por lesão com cloreto férrico
Utilizamos a marcação com o anticorpo anti CD31 para identificar presença de
células endoteliais após os tratamentos com os GAGs, deste modo podemos verificar a
extensão da lesão arterial em cada grupo e se o tratamento prévio com os GAGS altera a
0
5
10
15
20
25
12 horas 24 horas
Tem
po
-se
gun
do
s
Tempo
Tempo de protrombina
Lesão sem tratamento
Lesão + DS
Lesão + Heparina
Lesão + HBPM

55
extensão da lesão. De acordo com os resultados, o processo de injúria nos vasos
demonstrou a perda de 64,7% de células endoteliais em animais lesionados e não tratados
(figura 21B), em animais tratados com DS a perda foi de 51,2% (figura 21C), com HBPM
23,4% (figura 21D) e com Heparina 31,4% (figura 21E). Assim, a média de desnudação
endotelial do grupo DS não foi significativamente diferente do controle. Entretanto,
apresentou vasos com maiores perdas de endotélio em comparação aos tratados com HNF
e HBPM (p= 0.008). Embora apresente maior lesão arterial o DS conseguiu diminuir a
velocidade de formação de trombo em comparação com o grupo controle (ver Figura 12).
0
10
20
30
40
50
60
70
80
90
100
Sem lesão Lesão sem
tratamento
Lesão + DS Lesão +
Heparina
Lesão + HBPM
Inte
nsi
dad
e d
e F
luore
scên
cia
Expressão de células CD31+ após lesão
* *
E
A
A
D
D
A
A
F
A
A
A C
A
A
B I)
I)
II)

56
Figura 20.Extensão da lesão arterial provocada por cloreto férrico. Figura I) Imunofluorescência da artéria carótida usando anticorpo anti-CD31 conjugado com FITC
no tempo imediatamente após a lesão. (A) Artéria não lesionada. (B) Artéria lesionada de
animais sem tratamento; (C) Artéria lesionada de animais tratados com DS; (D) Artéria
lesionada de animais tratados com HBPM; (E) Artéria de animais lesionada e tratados
com Heparina; (F) Artéria sem adição de anticorpo primário (F). Figura II) A quantidade
de células CD31 foi determinada através da contagem das mesmas. Resultados de
expressão com valores médios ± desvio padrão. Barra = 50μm. Contagem para n = 4
animais em cada grupo. (*). Representa em relação ao grupo controle (p<0,05)
7.2 Efeito de GAGs na migração de células progenitoras endoteliais
Após quantificar a perda de células endoteliais nas artérias carótidas após lesão
química, nós analisamos a migração de células progenitoras endoteliais (CPEs) para o
local de lesão. Utilizou-se corte das artérias carótidas inicialmente após o procedimento
de lesão química de animais tratados com dermatan (figura C), tratados com heparina
(figura D), animais tratados com HBPM (figura E) e compararam-se os resultados com
cortes de artérias carótidas de animais lesionados e não tratados (Figura B).
De acordo com os dados, os vasos arteriais de animais lesionados e tratados com
DS acumularam 3 vezes mais células progenitoras marcadas para o local de injúria em
comparação ao grupo de animais lesionados e não tratados ( p <0,005 n=4); o grupo de
animais lesionados e tratados com HBPM acumularam cerca de 2,5 vezes mais células
progenitoras na parede arterial (p<0,005 n=4); animais lesionados e tratados com HNF
acumularam cerca de 3,5 vezes mais células progenitoras no local da lesão (p<0,005 n=4)
arterial em relação ao animais não tratados (figura 22.II). Em relação aos três GAGs, não
houve diferença significativa nos resultados (p=0,08).

57
Figura 21.Contagem de células CD34+ na artéria após lesão provocada por cloreto
férrico. Figura I) Imunofluorescência da artéria carótida usando anticorpo anti-CD34
conjugado com FITC no tempo imediatamente após a lesão. (A) Artéria sem adição de
anticorpo primário; (B) Artéria lesionada de animais sem tratamento; (C) lesionados e
tratados com DS; (D) lesionados e tratados com Heparina e (E) Artéria lesionada de
animais tratados com HBPM. Figura II) A quantidade de células CD34 foi determinada
através da contagem por marcações de intensidade de fluorescência no programa Imagej.
Resultados expressão como valores médios ± desvio padrão. Barra = 50μm. Contagem
para n = 4 animais em cada grupo. (*). Representa significância estatística em relação ao
grupo controle (p<0,05).
0
10
20
30
40
50
60
70
Lesão sem tratamento Lesão + DS Lesão + Heparina Lesão + HBPM
Inte
nsi
dad
e d
e Fl
uo
resn
cên
cia
Expressão de células CD34+ após lesão arterial
E
*
*
*
A C B
D
II)

58
8. Expressão de moléculas marcadores de inflamação: CD11b e ICAM-1
9.1 CD11b
O recrutamento de células CD11b+ foi determinada contando-se a quantidade de
células presente no local da lesão no tempo inicial após o procedimento de injúria arterial
na presença ou ausência de glicosaminoglicanos. O anticorpo anti-CD11b usado nos
ensaios imunohistoquímicos detecta leucócitos em diferentes estágios de maturação.
(figura 23 A,B,C,D).
O tratamento com DS diminuiu o recrutamento de células CD11b+ na parede
vascular após a injúria arterial em aproximadamente 39,6% (figura 23C) (p=0,03).
Animais que foram lesionados e receberam injeções de HBPM obtiveram redução das
células leucocitárias em aproximadamente 14,11% (figura 23E) (p > 0,005); animais que
foram tratados com HNF obtiveram redução de células leucocitárias em 10,89% (23D) (p
> 0,005 n=3 animais). Esses dados indicam que o DS mostrou grande capacidade em
inibir células inflamatórias até o local da lesão arterial, no tratamento com HBPM a
inibição ocorreu de modo inferior ao DS e interessantemente, no tratamento com heparina
essa inibição não aconteceu.
I)
A B
A
C
D
A
E
A

59
Figura 22.Contagem de células CD11b+ na artéria. Resultados de expressão como
valores médios ± desvio padrão. Contagem para n = 4 animais em cada grupo. Figura
I) Imunofluorescência da artéria carótida usando anticorpo anti-CD11b conjugado com
FITC no tempo imediatamente após a lesão. (A) Artéria sem anticorpo primário (controle
negativo); (B) Artéria lesionada de animais sem tratamento. (C) Artéria lesionada de
animais tratados com DS; (D) artéria de animais tratados com Heparina; (E) artéria de
animais tratados com HBPM e (E) artéria de animais sem adição de anticorpo primário
(controle negativo). Figura II) A quantidade de células CD11b foi determinada através da
contagem por marcações de intensidade de fluorescência no programa Imagej. Setas
vermelhas indicam leucócitos ao redor, lúmen e fora do vaso. Resultados de expressão
como valores médios ± desvio padrão. Barra = 50 μm. (*). Representa significância
estatística em relação ao grupo controle (p≤0,05)
9.2 ICAM-1
A presença das moléculas de adesão intercelular-1 (ICAM-1) foi determinada
contando-se a quantidade de células presentes no vaso no tempo inicial após o
procedimento de injúria arterial na presença ou ausência de glicosaminoglicanos. Este
tempo foi escolhido pois a expressão dessa molécula ocorre nas primeiras horas após
lesão e promove o recrutamento das células inflamatórias neste local. O anticorpo anti-
ICAM usado nos ensaios imunohistoquímicos identifica a molécula de ICAM que é
expressa na superfície de células endoteliais, epiteliais, macrófagos e fibroblastos em
condições inflamatórias aguda. As alterações dos níveis destas moléculas podem indicar
0
20
40
60
80
100
120
Lesão sem
tratamento
Lesão + DS Lesão + Heparina Lesão + HBPM
Inte
nsi
da
de
de
flu
ore
scên
cia
Expressão de células CD11b+ após lesão arterial
E
II)
*

60
um aumento ou diminuição do processo inflamatório na parede dos vasos lesionados. Em
relação ao grupo de animais controle, o tratamento com DS foi capaz de diminuir a
marcação de ICAM-1 nas artérias em 37,5%, porém essa diminuição não foi significativa
(p=0,08) ; animais tratados com HNF diminuíram 15,6% e animais tratados com HBPM
diminuíram a expressão de ICAM-1 em 25,1%, ambos não significativo (p= 0,1).
0
20
40
60
80
100
120
140
Lesão sem
tratamento
Lesão + DS Lesão + Heparina Lesão + HBPM
Inte
nsi
dad
e d
e fl
uore
scên
cia
Expressão de ICAM-1
A E
I)

61
Figura 23.Contagem de ICAM-1. Resultados de expressão como valores médios ±
desvio padrão. Contagem para n = 4 animais em cada grupo. Figura I)
Imunofluorescência da artéria carótida usando anticorpo anti-CD11b conjugado com
FITC no tempo imediatamente após a lesão. (A) Artéria sem adição de anticorpo primário
(controle negativo); (B) Artéria lesionada de animais sem tratamento. (C) Artéria
lesionada de animais tratados com DS; (D) artéria de animais tratados com Heparina; (E)
artéria de animais tratados com HBPM. Resultados de expressão como valores médios ±
desvio padrão. Barra = 50 μm. Figura II) A quantidade de moléculas de ICAM-1 foi
determinada através da contagem por marcação de intensidade de fluorescência no
programa Imagej.
9. Análise da atividade gelatinolítica nas artérias carótidas
A análise da atividade de gelatinases ‘in situ’ foi feita após o procedimento
cirúrgico e após 14 dias. Para tal utilizamos a DQ gelatina, que é um substrato
fluorogênico usado para a identificação da atividade de proteases com alta sensibilidade.
Esse substrato é composto por gelatina conjugada com fluoresceína (FITC); caso ocorra
a digestão proteolítica, há revelação de uma fluorescência verde, a qual pode ser usada
para medir a atividade enzimática. Para medição de tal atividade, analisamos os cortes
corados e determinamos a intensidade de fluorescência com o programa ImageJ. Foi
utilizado n=3 animais em cada grupo. Para a determinação da fluorescência específica,
subtraímos a fluorescência natural da lâmina elástica, pois estas apresentam uma
autofluorescência característica, do valor encontrado nas artérias incubadas com DQ
gelatina, figura 24.I (M). A administração de DS promoveu uma diminuição de 41,5% na
atividade gelatinolítica nas artérias carótidas (figura D-F) quando comparado ao controle
(A-C) (30824,6 vs. 52675,5 p=0,04). Nos grupos com heparina houve diminuição em
11,8% dessas gelatinases (figura G-I) (465053,3 vs. 526755 p=0,2) e com HBPM houve
diminuição em 12,1% (figura J-L)(463300,3 vs. 526755 p=0,2). Em relação aos três
GAGs, o DS diminuiu a expressão de MMPs 29,7% a mais que a HNF (p=0,05) e 29,3%
a mais que a HBPM (p=0,05).

62
M
B
A
Lesão + sem
tratamento
Lesão + DS
Lesão + Heparina
A B C
G
F
Lesão + HBPM
E D
H
K L
Atividade de gelatinases – tempo inicial
I
J
I) DAPI DQ-Gelatina DAPI + DQ-Gelatina
Sem adição de
DQ-Gelatina
L N
L
M

63
Figura 24.Atividade de metaloproteinases no vaso no tempo inicial após a lesão.
Figura I) Zimografia realizada com o uso de DQ-gelatina conjugada com fluoresceína
(FITC) (Invitrogen); a expressão foi analisada subtraindo-se a auto-fluorescência do
tecido da fluorescência presente nas lâminas submetidas à técnica. 1a coluna: núcleos
corados com DAPI (A, D, G); 2a coluna: Artérias com DQ-gelatina; (B, E, F); 3a coluna:
sobreposição de imagens (C, F, I) e artéria sem adição de DQ-gelatina para análise da
subtração fluorescência natural da lâmina elástica (J, K, L). Barra = 50μm. Figura II)
Análise de densidade integrada da atividade proteolítica de gelatinases quantificada no
software ImageJ. (*). Representa significância estatística em relação ao grupo controle
(p≤0,05).
No período de 14 dias após lesão química nas artérias carótidas, observou-se que
a atividade de gelatinases diminuiu 71,44% (p=0,02) no grupo administrado DS; nos
grupos administrado HNF houve diminuição da atividade de gelatinases em que alcançou
78,44% (p=0,02) e nos grupos de animais em que foi administrado HBPM não houve
diferença significativa (figura 25.II). O DS e a Heparina não apresentaram diferença
estatística entre eles quanto a atividade de gelatinases (p=0,09). Em animais tratados com
DS houve diminuição significativa da atividade de MMPs em 29,4% em comparação a
animais tratados com HBPM (p=0,03).
0
100
200
300
400
500
600
700
800
Lesão sem
tratamento
Lesão + DS Lesão + Heparina Lesão + HBPM
Den
sid
ad
e In
teg
rad
a (
DI)
II)
*

64
Atividade de gelatinases -14 dias
M N O
Sem adição de
DQ-gelatina
Lesão + HBPM
Lesão + sem
tratamento
Lesão + DS
Lesão + Heparina
A B C
DAPI
I) DQ-Gelatina
F
DAPI + DQ-Gelatina
E D
G H I
J K L

65
Figura 25.Atividade de gelatinases no vaso 14 dias após a lesão. Figura I) Zimografia
in situ realizada com o uso de DQ-gelatina conjugada com fluoresceína (FITC), os vasos
foram contracorados com DAPI para a visualização dos núcleos. Figura II) Análise de
densidade integrada da atividade proteolítica de gelatinases quantificada no software
ImageJ. (*) Representa significância em relação ao grupo controle (p≤0,05). N=3 animais
em cada grupo.
10. Agregação plaquetária em diferentes tratamentos com GAGs
Com a finalidade de monitorar o efeito da terapia com os GAGs, analisou-se a
atividade agregante das plaquetas na presença de dermatan sulfato, heparina e heparina
de baixo peso molecular. O ensaio foi realizado in vitro utilizando-se a trombina como
agonista do processo de agregação na concentração de 0,25 U/ml. A trombina é uma
serina protease conhecida por ser o mais potente indutor da agregação e ativação
plaquetária, sendo responsável pela conversão do fibrinogênio em fibrina na coagulação.
O perfil de agregação das plaquetas foi analisado ao longo de 14 minutos e pode
ser observado nas imagens abaixo: o gráfico com curva de agregação destacado em cor
preta representa o grupo controle, onde há presença do plasma rico em plaqueta (PRP) e
trombina. A média dos controles de todos os grupos obteve agregação em 97,4%. O
tratamento com DS apresentou valor de agregação em 46,7% durante o tempo estipulado
(figura 15A) com diferença estatisticamente significativa (p<0,05). Em contato com a
0
50
100
150
200
250
Lesão sem
tratamento
Lesão + DS Lesão + Heparina Lesão + HBPM
Den
sid
ad
e I
nte
gra
da
(D
I)Atividade de gelatinases - 14 dias
*
*
II)

66
heparina, a agregação plaquetária mostrou-se evidentemente menor chegando ao valor de
2% com diferença significativa em relação ao grupo controle (p<0,05) (figura 15B).
Ensaios com a HBPM mostraram uma taxa de agregação plaquetária em 79%, sendo que
esta taxa não apresentou diferença estatística em relação ao grupo controle (p>0,05)
(figura 15C).
0
20
40
60
80
100
120
Controle DS Heparina HBPM
% P
orc
enta
gem
de
agre
gaç
ão
Agregação plaquetária
a) Dermatan sulfato c) HBPM
*
*
b) Heparina

67
Figura 26.Agregação de plaquetas. I) Representação da curva de agregação das
plaquetas sob tratamento com DS, Heparina e HBPM em 14 minutos na presença de
trombina. A linha azul do gráfico indica o plasma rico em plaquetas + drogas analisadas
+ trombina e a linha preta do gráfico indica o grupo controle (plasma rico em plaqueta +
trombina). II) Porcentagem em gráfico da agregação plaquetária, a imagem abaixo do
gráfico indica plaquetas agregadas após os ensaios. a) plasma + trombina; b) plasma+
DS+ trombina; c) plasma + heparina trombina; d) plasma + HBPM + trombina. (*) indica
diferença estatística em relação ao grupo controle (p≤0,05). N=1 em triplicata.
A
A B
A
C
B
A
A
D
B
A
A

68
DISCUSSÃO
Uma variedade de agentes terapêuticos tem sido empregada para tentar modular a
disfunção vascular e regenerar o endotélio após a lesão vascular. Neste trabalho, nós
utilizamos a terapia com três diferentes glicosaminoglicanos heparina não fracionada,
heparina de baixo peso molecular e dermatan sulfato em camundongos C57Bl6/J como
forma de modulação na trombose arterial, com possível atuação na diminuição do trombo
e da formação de neoíntima com restenose do vaso.
Para isso, nós usamos o modelo físico e químico para a perda do endotélio por
cloreto férrico, devido à ampla aplicabilidade do mesmo em diversos modelos animais, a
facilidade de reprodução da técnica, uma vez estabelecida a concentração do cloreto
férrico, além de não exigir equipamentos específicos ou caros (Tseng et al., 2006) e ainda
segundo (Farrehi et al., 1998) trombos induzidos por esta técnica em camundongos são
ricos em plaquetas e são muito semelhantes aos trombos arteriais humanos. A
concentração de FeCl3 utilizada para denudação endotelial varia muito, podendo ser de
2,5 % a 80%. Em nosso laboratório utilizamos o modelo de denudação endotelial com
uma concentração de 15% (Sielski, 2015).
Este trabalho estabelece pela primeira vez em nosso laboratório o uso da técnica
de denudação endotelial para indução da trombose por cloreto férrico em artérias
carótidas em grupos de animais C57Bl6 que foram tratados com Heparina e Heparina de
baixo peso molecular, visto que não há dados descritos em literatura com este modelo.
Em nosso trabalho nós analisamos o tempo de formação completa do trombo
inicialmente após a lesão por cloreto férrico em cada grupo de animais tratados com
glicosaminoglicanos e observamos que no grupo de animais em que foram aplicadas
injeções de heparina na concentração de 1 mg/kg, o tempo de formação do trombo ocorreu
em trinta minutos e no grupo tratado com injeções de HBPM na concentração de 1 mg/kg
o tempo de formação do trombo ocorreu em dez minutos. Quando comparamos as 2
heparinas com o DS vimos que este prolonga o tempo de trombose para 14,6 min,
apresentando um valor intermediário entre a heparina e a HBPM. Nós também
observamos que a área do trombo formado no lúmen dos vasos sanguíneos em todos os
grupos, foi estatisticamente menor no grupo de animais tratados com heparina; no grupo
de animais tratados com HBPM a área do trombo observada foi maior em relação ao

69
tratamento com os demais GAGs e o tamanho do trombo com DS foi intermediário
também, assim como o tempo de trombose. O dermatan sulfato atua como agente
antitrombótico através da interação com o cofator II de heparina, uma molécula da família
das serpinas que inibe a trombina e que ao ligar-se com o DS, inibe a trombina pelo menos
mil vezes mais rápido(He et al., 2008). Estudos anteriores demonstraram que a injeção
de DS é capaz de inibir a atividade de trombina presente na parede vascular após injúria
arterial (Vicente et al., 2004) e que o HCII migra do sangue circulante para a camada
adventícia após lesão arterial, onde ele se co-localiza com o DS na parede arterial (Vicente
et al., 2007). Outro estudo em coelhos evidenciou que o DS previne trombose venosa
com um menor efeito hemorrágico que a heparina: observou-se que o máximo de efeito
de antitrombótico se obtinha com 70 μg de DS e 500 μg de Heparina e que um aumento
de 20 vezes na dose de heparina causava sangramento. Em contraste, um aumento de 40
vezes na dose de DS não aumentou o sangramento(Bendayan et al., 1994). Utilizamos
concentração de dermatan sulfato de 10 mg/kg peso do animal e observamos que o tempo
de formação do trombo ocorreu em aproximadamente quinze minutos, a área do trombo
observada estatisticamente mostrou-se menor quando comparada aos animais tratados do
grupo controle e aos animais tratados com HBPM e não foi observado efeito hemorrágico.
Como observamos um prolongamento dos tempos de trombose nos animais
tratados com os GAGs, verificamos o efeito destes no tempo de sangramento, TTPA e
TT. O TTPA foi prolongado no tratamento com heparina, HBPM e DS nas primeiras 12
horas após lesão arterial. Esses dados demonstram que a heparina e a hbpm exerceram
seus efeitos através da ligação com a antitrombina. No caso da heparina sabe-se que seu
efeito culmina com a inibição de fatores de coagulação como IXa, Xa, XIa, XIIa e da
trombina através da formação do complexo antitrombina-heparina que se liga as serina
proteases da cascata de coagulação deste modo produzindo seu efeito (Danielsson et al.,
1986). No caso da HBMP seu efeito está diretamente relacionado a inibição do fator Xa,
por isso seu efeito é mais controlado que o observado com a heparina. O DS também
mostrou efeito anticoagulante ao prolongar o TTPa nas doses administradas. No período
de 24 horas, o TTPa apresentou uma diminuição em todos os grupos, voltando aos níveis
controle no DS e HBPM pois os níveis desses GAGs no organismo diminuem, logo a
capacidade anticoagulante diminui. Como a dose de heparina utilizada é considerada alta,
espera-se que seu efeito anticoagulante no teste de TTPa ainda se mantivesse alto.

70
O TP mostrou-se prolongado no grupo de animais lesionados e tratados com
heparina. A ação anticoagulante da heparina baseia-se fundamentalmente no efeito
inibidor sobre a trombina e o fator X ativado. A heparina de baixo peso molecular que
não apresenta longevidade suficiente para catalisar a inibição da trombina, produz efeito
anticoagulante por inibição do fator Xa pela antitrombina. Em concordância com alguns
estudos anteriores, não foram evidenciadas alterações do TP nos grupos tratados com
HBPM (Palareti et al., 1989; Monreal et al., 1993; Decarolis et al., 2012). O tratamento
com DS não prolongou o TP, evidenciando que a ação inibitória de DS ocorre pela
interação com o cofator II de heparina presente.(Sie et al., 1993)
O tempo de sangramento (TS) foi avaliado aos 40 minutos com o intuito de se
observar os valores dos mesmos no momento inicial após a trombose, 12 e 24 horas após
lesão endotelial. Observou-se aumento em todos tempos analisados nos grupos de animais
tratados com heparina, sugerindo a atuação da heparina na diminuição plaquetária,
consequente à inibição da trombina (Colman et al., 1994). Não houve alteração
significativa do TS nos animais tratados com HBPM em relação ao grupo controle.
Resultados similares com HBPM foram encontrados nos trabalhos de (Carter et al., 1982)
analisando o TS em coelhos, (Andriuoli et al., 1985) observou em ratos,(Nimmerfall e
Mischke, 1999) analisaram o TS em cachorros tratados com HBPM e (Lim et al., 2004)
em pacientes humanos com insuficiência renal que estavam sob o procedimento de
hemodiálise. Animais tratados com DS obtiveram prolongamento no tempo de
sangramento 40 minutos e 24 horas após lesão, porém esse efeito mostrou-se menor em
relação a heparina. Iacoviello et al. (1996) mostraram efeito similar do DS em modelo de
lesão arterial abdominal em ratos, onde o TS foi significativamente menor em relação aos
animais tratados heparina em dosagens iguais de 8 mg/kg.
Os testes de coagulação dessa forma, demonstram dados interessantes que
podemos associar aos experimentos do tempo de trombose arterial: os animais tratados
com HNF obtiveram maior tempo de formação e menor área de trombo como já citado
anteriormente, em contrapartida apresentaram de forma drástica um aumento nos testes
de TTPA e TS considerado fora do padrão médio para os valores apresentados. Animais
tratados com DS aumentaram o tempo de formação e diminuíram a área de trombo
significativamente em relação ao grupo controle e a HBPM apresentaram um aumento
significativo nos testes de coagulação, porém, em comparação com a HNF mostrou-se
significativamente baixo. E em animais tratados com HBPM o tempo de formação do

71
trombo foi maior e a área mostrou-se menor e nos testes de coagulação observamos que
apenas no período de 12 horas após lesão arterial houve um prolongamento do TTPA.
Os dados dos testes de coagulação e do tempo de trombose arterial podem ser
também correlacionados com os resultados de agregação plaquetária, onde os animais
tratados com a HNF apresentaram significativamente menor agregação de plaquetas em
relação ao grupo controle e em relação aos demais grupos de animais. O grupo tratado
com DS demonstrou menor agregação de plaquetas e diferentemente do aumento do
tempo de trombose em animais tratados com HBPM, não foi observada diferença
significativa na agregação plaquetária neste grupo em relação ao grupo controle.
A reação vascular à lesão endotelial ocorre após a denudação endotelial e está
associada a deposição de plaquetas e leucócitos e outras moléculas inflamatórias. Após
algumas semanas, células musculares lisas iniciam a migrar e a proliferar para a camada
íntima com deposição de matriz extracelular no lúmen do vaso. A trombose pode servir
de base para a formação de neoíntima(Waller et al., 1991). Em nosso trabalho observamos
que em animais C57BL6 lesionados na artéria carótida, a formação de neoíntima foi
maior nos grupos que receberam injeções de HBPM. Por outro lado, animais que
receberam injeções de DS apresentaram menor formação de neoíntima. Vicente et. al
(2007) observaram que a administração de DS no período de 48 horas foi capaz de inibir
o crescimento da neoíntima em artérias de camundongos positivos para HCII mas não em
artérias de animais deficientes no cofator II de heparina.
Uma explicação possível para neoíntima acentuada nos grupos injetados com
HBPM seria a grande expressão de metaloproteinases nos vasos arteriais destes grupos
de animais comparado com os outros tratamentos de GAGs nos dois tempos analisados
(tempo inicial e 14 dias após a lesão). A degradação e o remodelamento da matriz
extracelular contribuem na alteração vascular e no comportamento funcional das células
musculares lisas através da atuação de MMPs (Cho e Reidy, 2002). Jhonson et al.,
observou que a deficiência da MMP-2 resultou na diminuição de CMLs e atenuação da
neointima em artérias carótidas in vivo. Em outro trabalho resultados similares foram
vistos em CMLs da veia safena, onde a replicação de CMLs é significativamente
diminuída em camundongos MMP-9 (-/-)(Turner et al., 2007). Assim, a menor proporção
de neointima observada nos grupos de animais tratados com DS pode ser explicada pela
menor expressão de MMPs nos vasos desses animais nas duas situações analisadas
(tempo inicial e 14 dias após a lesão).

72
Mensuramos a perda de células endoteliais nos vasos arteriais após lesão química
de animais C57BL6 através do ensaio de imunofluorescência com o anticorpo anti-CD31
no tempo inicial após a injúria e observamos que essa perda se mostrou maior no grupo
de animais tratados com DS. Grupos de animais tratados com heparina apresentaram
menor perda endotelial no vaso. O intuito desse experimento foi o de avaliar a capacidade
dos GAGs na proteção das células endoteliais no momento da lesão, provavelmente
inibindo a apoptose após a lesão ou diminuindo o processo inflamatório, que pode
estimular esta perda e alterar o recrutamento e regeneração endotelial no vaso após o
processo inflamatório inicial. Após observarmos a porcentagem de perda endotelial nos
grupos, realizamos o ensaio de imunofluorescência com o anticorpo anti-CD34, um
marcador de células progenitoras endoteliais (CPEs). Essas células são capazes de
proliferar, migrar e diferenciar-se em células endoteliais (Kirton e Xu, 2010). Notamos
que os animais tratados com heparina acumularam maior número de CPEs no vaso,
seguido de animais tratados com DS e por último no grupo de animais tratados com
HBPM. O recrutamento de CPEs para o local da lesão pode contribuir para atenuar a
resposta trombótica e promover a reendotelização e reparo vascular (Napoli et al., 2011),
o que poderia explicar a menor formação de trombo nos vasos de animais tratados
heparina já que obtiveram maior número de CPEs. Por outro lado, os animais tratados
com DS obtiveram dentre os demais tratamentos, maior perda de células endoteliais no
tempo inicial após lesão, porém, apresentaram grande expressão de CPEs indicando,
portanto, maior capacidade no reparo vascular. A presença dos glicosaminoglicanos no
local da lesão pode criar um ambiente atrativo para a migração das CPEs alterando
positivamente a recuperação do endotélio.
Investigamos também o efeito anti-inflamatório dos glicosaminoglicanos após
lesionar as artérias carótidas desses animais. Para isso, foi realizada a técnica de
imunofluorescência nos vasos para identificar presença do marcador relacionado à
inflamação, que neste caso foi o CD11b, uma glicoproteína encontrada na membrana de
leucócitos incluindo monócitos, neutrófilos, granulócitos e macrófagos. O recrutamento
de leucócitos é um evento de extrema relevância para uma resposta adequada do
organismo à invasão de patógenos ou danos teciduais, uma vez que a participação dessas
células tem papel fundamental nos mecanismos de reparo tecidual(Mitchell et al., 2006).
Em nosso modelo de lesão arterial, foi observada grande quantidade de marcação
de células CD11b positivas na parede do vaso no grupo de animais que receberam

73
injeções de HNF e Heparina de baixo peso molecular nas concentrações de 1 mg/kg. Uma
das limitações do uso de heparina é causada pela ligação desta com proteínas plasmáticas,
proteínas liberadas pelas plaquetas e por células endoteliais, levando a uma resposta
anticoagulante variável, além disso a heparina não consegue ativar o fator X do complexo
protrombinase (fator X/fator V), a trombina ligada à fibrina ou a superfícies
subendoteliais(Glimelius et al., 1978). Assim, a heparina nas dosagens utilizadas neste
trabalho possivelmente pode ter estimulado o recrutamento de moléculas pró
inflamatórias como CD11b, devido a sua propriedade de se ligar às células citadas, no
qual a maior expressão de leucócitos foi identificada em animais tratados com heparina.
Em relação aos animais tratados com DS, a expressão de CD11b é significativamente
diminuída em relação aos outros GAGs. O DS também causa a diminuição da geração de
trombina através de sua ligação com o cofator II da heparina (HCII)(Tollefsen, 1995).
Como o complexo HCII-DS pode inibir trombina livre e ligada ao tecido, isto pode
explicar sua maior eficiência em inibir a trombina tecidual e a inibição do crescimento da
neointima. A trombina atua em uma série de processos que abrangem reparo tecidual, na
permeabilidade vascular, tônus vascular, inflamação e processos que afetam a migração
leucocitária (Schuepbach et al., 2009). Assim, por diminuir a geração de trombina, o DS
pode estar afetando o recrutamento de células inflamatórias de forma independente das
moléculas de adesão.
A ativação de leucócitos é capaz de promover a expressão da molécula de adesão
intercelular-1 (ICAM-1), a qual provoca a ligação de leucócitos as células endoteliais
através da interação com o complexo MAC-1 (CD11b/CD18) e ajuda na transmigração
desses leucócitos através dos vasos ao local lesionado. O ICAM-1 ativo também pode
recrutar monócitos do sangue até a parede vascular, aumentando a expressão de citocinas
pró inflamatórias (Pacurari et al., 2014). Em nosso trabalho, observamos que em animais
tratados com DS houve menor expressão de ICAM-1 em relação aos animais tratados
com heparina e a HBPM. Esses dados demonstram que o DS ao inibir o recrutamento de
leucócitos ao local da lesão no tempo inicial, provavelmente interferiu na expressão de
ICAM-1 no vaso. Godoy et. al (2015) observaram a diminuição da expressão de ICAM-
1 no período de 3 dias após lesão arterial em animais C57BL6J, porém essa diminuição
não foi observada em 24 horas. O tratamento com HBPM obteve maior expressão de
ICAM-1 dentre os GAGs, possivelmente devido à grande expressão de leucócitos nas

74
artérias desses animais no tempo inicial após lesão, o que induziu a expressão de ICAM-
1 no processo inflamatório inicial.

75
RESUMO DOS DADOS OBTIDOS
Dermatan sulfato
(10 mg/kg)
Heparina
(1 mg/kg)
HBPM
(1 mg/kg)
Tempo de
oclusão arterial
Área do trombo
Células CD31+
no vaso após
lesão
Células CD34+
após lesão
TTPA
12 horas após
lesão
12 e 24 horas após
lesão
12 horas após lesão
TT
TS
40 min e 24 horas
após lesão
40 min, 12 e 24 horas
após lesão
40 min e 24 horas
após lesão
Neoíntima
Atividade de
gelatinases tempo
inicial
Atividade de
gelatinases 14
dias após lesão
CD11b
ICAM-1
= Aumenta = Diminui = Não houve diferença significativa

76
CONCLUSÃO
Neste estudo, o dermatan sulfato foi capaz de proporcionar menor formação de
trombo nos vasos sanguíneos em comparação com a HBPM e diminuir o recrutamento
leucocitário, obteve maior resposta na prevenção da formação de neointima comparado
ao tratamento com Heparina e HBPM e além disso, pelo modelo físico e químico de
denudação endotelial foi o GAG que obteve maior capacidade de perda endotelial e por
sua vez, mobilizou mais células progenitoras para o vaso lesionado, o que significa que o
tratamento com este GAG possivelmente as mobilizou para os locais lesionados afim de
promover o remodelamento vascular positivo, de forma a atuar no processo de reparo dos
vasos. Os dados de TTPA e TS mostraram que houve aumento desses tempos após 24
horas da lesão arterial em animais tratados com DS, porém este foi menor em relação aos
animais tratados com heparina (3,5 vezes menor).
A heparina em contraste, obteve melhor ação antitrombótica após a indução de
denudação arterial inicial por cloreto ferrico pela menor formação de trombo nas artérias
carótidas e ainda, obteve maior expressão de células progenitoras endoteliais no vaso.
Apesar da heparina ter promovido evidências anticoagulantes satisfatórias, uma de suas
principais limitações durante o uso são a probabilidade de processos hemorrágicos, o qual
tem sido frequentemente relacionado ao seu efeito anticoagulante, podendo estar
relacionado a dose, com a resposta anticoagulante do indivíduo, doenças, entre outros
fatores. Além disso, há necessidade de monitorização laboratorial durante sua utilização
e seu uso constante pode provocar a trombocitopenia induzida pela heparina. Os dados
de teste de coagulação de TTPA e TS em animais tratados com heparina neste projeto
mostraram um amplo prolongamento (5 vezes) nos tempos de 12 e 24 horas após lesão
arterial, considerado um tempo de prolongamento anormal nesses testes.
O DS dessa forma, pode ser considerado como um possível agente na atuação de
injúrias vasculares devido a sua propriedade antihemorrágica, antitrombótica na inibição
de trombo inicial nos vasos e antiinflamatória na formação de neointima, promovendo
maior remodelamento positivo através do recrutamento de CPEs para o vaso lesionado,
podendo ser considerado como uma droga alternativa ao tratamento com HNF e HBPM.

77
REFERÊNCIAS BIBLIOGRÁFICAS
ALEVRIADOU, B. R. et al. Real-time analysis of shear-dependent thrombus formation
and its blockade by inhibitors of von Willebrand factor binding to platelets. Blood, v. 81,
n. 5, p. 1263-1276, 1993.
ALWAN, A. et al. Monitoring and surveillance of chronic non-communicable diseases:
progress and capacity in high-burden countries. Lancet, v. 376, n. 9755, p. 1861-8, Nov
27 2010.
ANDRASSY, K.; ESCHENFELDER, V. Are the pharmacokinetic parameters of low
molecular weight heparins predictive of their clinical efficacy? Thrombosis research, v.
81, n. 2, p. S29-S38, 1996.
ANDRÉO FILHO, N. Challenges in the quality of heparin. Revista Brasileira de
Hematologia e Hemoterapia, v. 31, n. 5, p. 306-307, 2009. ISSN 1516-8484.
ANDRIUOLI, G. et al. Comparison of the antithrombotic and haemorrhagic effects of
heparin and a new low molecular weight heparin in rats. Pathophysiology of
Haemostasis and Thrombosis, v. 15, n. 5, p. 324-330, 1985.
ARNOLD, F. W., D. C. Angiogenesis in wound healing. Pharmacol Ther, v. 52, p. 407-
422, 1991.
BARZU, T. et al. Endothelial binding sites for heparin. Specificity and role in heparin
neutralization. Biochemical Journal, v. 238, n. 3, p. 847-854, 1986.
BAUTERS, C. et al. Restenosis after coronary angioplasty for rapidly progressive
coronary stenosis. Eur Heart J, v. 17, n. 11, p. 1671-1677, 1996.
BAYES-GENIS, A. et al. Restenosis and Hyperplasia: Animal Models. Current
interventional cardiology reports, v. 2, n. 4, p. 303-308, 2000.
BENDAYAN, P. et al. Dermatan sulfate is a more potent inhibitor of clot-bound
thrombin than unfractionated and low molecular weight heparins. Thromb Haemost, v.
71, n. 5, p. 576-80, May 1994.
BODE, W.; MASKOS, K. Structural basis of the matrix metalloproteinases and their
physiological inhibitors, the tissue inhibitors of metalloproteinases. Biochem J, v. 6, n.
384, p. 863-872, 2003.

78
BOSMAN, F. T.; STAMENKOVIC, I. Functional structure and composition of the
extracellular matrix. J. Pathol, v. 200, , n. 4, p. 423-428, 2003.
BOUCHER, B. A.; TRAUB, O. Achieving hemostasis in the surgical field.
Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, v. 29,
n. 7P2, 2009. ISSN 1875-9114.
BOUTAYEB, A.; BOUTAYEB, S. The burden of non communicable diseases in
developing countries. Int J Equity Health, v. 4, n. 1, p. 2, Jan 14 2005.
BROZE, G. J., JR.; YIN, Z. F.; LASKY, N. A tail vein bleeding time model and delayed
bleeding in hemophiliac mice. Thromb Haemost, v. 85, n. 4, p. 747-8, Apr 2001.
BUSTI, C. et al. Matrix metalloproteinases and peripheral arterial disease. Intern Emerg
Med, v. 5, n.1, p. 13-25, 2010.
CARTER, A. J. et al. Morphologic characteristics of lesion formation and time course of
smooth muscle cell proliferation in a porcine proliferative restenosis model. J Am Coll
Cardiol, v. 24, n. 5, p. 1398-1405, 1994.
CARTER, C. J. et al. The relationship between the hemorrhagic and antithrombotic
properties of low molecular weight heparin in rabbits. Blood, v. 59, n. 6, p. 1239-45, Jun
1982. ISSN 0006-4971 (Print)
0006-4971 (Linking).
CASU, B.; VLODAVSKY, I.; SANDERSON, R. D. Non-anticoagulant heparins and
inhibition of cancer. Pathophysiology of haemostasis and thrombosis, v. 36, n. 3-4, p.
195-203, 2007.
CERINIC, M. M. et al. Blood coagulation, fibrinolysis, and markers of endothelial
dysfunction in systemic sclerosis. Seminars in arthritis and rheumatism, 2003, Elsevier.
p.285-295.
CHO, A.; REIDY, M. A. Matrix metalloproteinase-9 is necessary for the regulation of
smooth muscle cell replication and migration after arterial injury. Circ Res, v. 91, n. 9,
p. 845-51, Nov 01 2002.
CHOAY, M. et al. Structure-activity relationship in heparin: a synthetic pentasaccharide
with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochem
Biophys Res Commun, v. 116, n. 2, p. 492-499, 1983.

79
CICILIANO, J. C. et al. Resolving the multifaceted mechanisms of the ferric chloride
thrombosis model using an interdisciplinary microfluidic approach. Blood, v. 126, n. 6,
p. 817-824, 2015.
COLMAN, R. et al. Hemostasis and Thrombosis [’n]: Philadelphia, PA: JB Lippincott
Co 1994.
DANIELSSON, A. et al. Role of ternary complexes, in which heparin binds both
antithrombin and proteinase, in the acceleration of the reactions between antithrombin
and thrombin or factor Xa. J Biol Chem, v. 261, n. 33, p. 15467-73, Nov 25 1986.
DAY, S. M. et al. Murine thrombosis models. Thromb Haemost, v. 92, n. 3, p. 486-94,
Sep 2004.
DECAROLIS, D. D. et al. Enoxaparin outcomes in patients with moderate renal
impairment. Archives of Internal Medicine, v. 172, n. 22, p. 1713-1718, 2012.
DEJANA, E. et al. Bleeding time in laboratory animals. II - A comparison of different
assay conditions in rats. Thromb Res, v. 15, n. 1-2, p. 191-7, 1979.
DIACOVO, T. G. et al. Neutrophil rolling, arrest, and transmigration across activated,
surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin
CD11b/CD18. Blood, v. 88, n. 1, p. 146-57, Jul 01 1996.
ECKLY, A. et al. Mechanisms underlying FeCl3‐induced arterial thrombosis. Journal
of Thrombosis and Haemostasis, v. 9, n. 4, p. 779-789, 2011.
FAREED, J. et al. Biochemical and pharmacologic heterogeneity in low molecular
weight heparins. Impact on the therapeutic profile. Curr Pharm Des, v. 10, n. 9, p. 983-
99, 2004.
FARREHI, P. M. et al. Regulation of arterial thrombolysis by plasminogen activator
inhibitor-1 in mice. Circulation, v. 97, n. 10, p. 1002-8, 1998.
FERREIRA, C. N. et al. O novo modelo da cascata de coagulação baseado nas superfícies
celulares e suas implicações. Revista Brasileira de Hematologia e Hemoterapia, v. 32,
p. 416-421, 2010.
GAWAZ, M. et al. Platelet Function in Acute Myocardial Infarction Treated With Direct
Angioplasty. Circulation, v. 93, n. 2, p. 229, 1996.

80
GIAGTZIDIS, I. et al. Matrix metalloproteinases and peripheral arterial disease. Int
Angiol, v. 3, n. 34, p. 195-201, 2015.
GLIMELIUS, B.; BUSCH, C.; HÖÖK, M. Binding of heparin on the surface of cultured
human endothelial cells. Thrombosis research, v. 12, n. 5, p. 773-782, 1978.
GODOY, J. A. et al. Dermatan sulfate and bone marrow mononuclear cells used as a new
therapeutic strategy after arterial injury in mice. Cytotherapy, v. 13, n. 6, p. 695-704, Jul
2011.
GODOY, J. A. et al. Combined dermatan sulfate and endothelial progenitor cell
treatment: action on the initial inflammatory response after arterial injury in C57BL/6
mice. Cytotherapy, v. 17, n. 10, p. 1447-1464, 2015.
GUPTA, S. et al. Heparin induced thrombocytopenia in critically ill: Diagnostic
dilemmas and management conundrums. World J Crit Care Med, v. 4, n. 3, p. 202-212,
2015.
HARENBERG, J. Pharmacology of low molecular weight heparins. Semin Thromb
Hemost, v. 16 Suppl, p. 12-8, Oct 1990.
HAYDEN, M. R.; SOWERS JR FAU - TYAGI, S. C.; TYAGI, S. C. The central role of
vascular extracellular matrix and basement membrane remodeling in metabolic syndrome
and type 2 diabetes: the matrix preloaded. Cardiovasc Diabetol, v. 4, p. 14-19, 2005.
HE, L. et al. Vascular dermatan sulfate regulates the antithrombotic activity of heparin
cofactor II. Blood, v. 111, n. 8, p. 4118-4125, 2008.
HEHRLEIN, C. et al. Pure β-particleemitting stents inhibit neointima formation in
rabbits. Circulation, v. 93, n. 4, p. 641-645, 1996.
HIRSH, J. et al. Mechanism of action and pharmacology of unfractionated heparin.
Arterioscler Thromb Vasc Biol, v. 21, n. 7, p. 1094-6, Jul 2001.
HIRSH, J. et al. Heparin: mechanism of action, pharmacokinetics, dosing considerations,
monitoring, efficacy, and safety. Chest, v. 108, n. 4 Suppl, p. 258S-275S, Oct 1995.
HOFFBRAND, A. V.; MOSS, P. A. H.; PETTIT, J. E. Essential haematology. 6th ed.
Wiley-Blackwell, 2011.
HOFFMAN, M. A cell-based model of coagulation and the role of factor VIIa. Blood
reviews, v. 17, p. S1-S5, 2003. ISSN 0268-960X.

81
HOLMER, E. et al. Heparin and its low molecular weight derivatives: anticoagulant and
antithrombotic properties. Haemostasis, v. 16 Suppl 2, p. 1-7, 1986.
HORLOCKER, T. T.; HEIT, J. A. Low molecular weight heparin: biochemistry,
pharmacology, perioperative prophylaxis regimens, and guidelines for regional anesthetic
management. Anesth Analg, v. 85, n. 4, p. 874-85, 1997.
HUANG, P.-H. et al. Matrix Metalloproteinase-9 Is Essential for Ischemia-Induced
Neovascularization by Modulating Bone Marrow–Derived Endothelial Progenitor Cells.
Arteriosclerosis, Thrombosis, and Vascular Biology, v. 29, n. 8, p. 1179-1184, 2009.
HULL, R. D. et al. Subcutaneous low-molecular-weight heparin compared with
continuous intravenous heparin in the treatment of proximal-vein thrombosis. N Engl J
Med, v. 326, n. 15, p. 975-82, Apr 09 1992.
JAQUES, L. B. Heparins--anionic polyelectrolyte drugs. Pharmacol Rev, v. 31, n. 2, p.
99-166, Jun 1979.
KIRTON, J. P.; XU, Q. Endothelial precursors in vascular repair. Microvascular
research, v. 79, n. 3, p. 193-199, 2010.
KOCHER, O. et al. Phenotypic features of smooth muscle cells during the evolution of
experimental carotid artery intimal thickening. Biochemical and morphologic studies.
Lab Invest, v. 65, n. 4, p. 459-470, 1991.
KONSTANTINIDES, S. et al. Plasminogen activator inhibitor-1 and its cofactor
vitronectin stabilize arterial thrombi after vascular injury in mice. Circulation, v. 103, n.
4, p. 576-583, 2001.
KUZUYA, M. et al. Deficiency of Gelatinase A Suppresses Smooth Muscle Cell
Invasion and Development of Experimental Intimal Hyperplasia. Circulation, v. 108, n.
11, p. 1375, 2003.
LIM, W.; COOK, D. J.; CROWTHER, M. A. Safety and efficacy of low molecular weight
heparins for hemodialysis in patients with end-stage renal failure: a meta-analysis of
randomized trials. J Am Soc Nephrol, v. 15, n. 12, p. 3192-206, Dec 2004. ISSN 1046-
6673 (Print)
1046-6673 (Linking).
LINHARDT, R. J. Award address in carbohydrate chemistry. Heparin: structure and
activity. J Med Chem, v. 46, n. 13, p. 2551-2564, 2003.

82
LIU, J. Y. et al. Metabolic profiling of murine plasma reveals an unexpected biomarker
in rofecoxib-mediated cardiovascular events.
LORENZI, T. F. Manual de Hematologia: propedêutica e clínica. 4 Rio de janeiro:
2006. 711.
LUPU, C. et al. Cellular effects of heparin on the production and release of tissue factor
pathway inhibitor in human endothelial cells in culture. Arterioscler Thromb Vasc Biol,
v. 19, n. 9, p. 2251-62, Sep 1999.
MACKAY, J. et al. The atlas of heart disease and stroke. Geneva: World Health
Organization, 2004. 112 p.
MAIMONE, M. M.; TOLLEFSEN, D. M. Structure of a dermatan sulfate hexasaccharide
that binds to heparin cofactor II with high affinity. J Biol Chem, v. 265, n. 30, p. 18263-
71, Oct 25 1990.
MARCOS DA SILVEIRA, W. B. Y. Isquemia e reperfusão em músculo esquelético:
mecanismos de lesão e perspectivas de tratamento. J. Vasc. Bras, Faculdade de Medicina
de Botucatu – UNESP, v. 3, p. 367-378, 2004.
MARQUEZ-GONZALEZ, H. et al. Reintervention with percutaneous balloon
angioplasty in patients with congenital heart disease with left-sided obstructions. Rev
Med Ins Mex Seguro Soc, v. 55, n. 7, p. 86-91, 2017.
MC, L. J. The discovery of heparin. Circulation, v. 19, n. 1, p. 75-8, Jan 1959.
MITCHELL, R. et al. Inflamação aguda e crônica. Robbins & Cotran: fundamentos
de patologia. 7ª ed. Rio de Janeiro: Elsevier, p. 29-54, 2006.
MONREAL, M. et al. Comparative study on the antithrombotic efficacy of hirudin,
heparin and a low-molecular weight heparin in preventing experimentally induced venous
thrombosis. Haemostasis, v. 23, n. 3, p. 179-83, May-Jun 1993.
MONROE, D. M.; HOFFMAN, M. The coagulation cascade in cirrhosis. Clinics in liver
disease, v. 13, n. 1, p. 1-9, 2009. ISSN 1089-3261.
MULLER, W. A. Leukocyte-endothelial-cell interactions in leukocyte transmigration and
the inflammatory response. Trends Immunol, v. 24, n. 6, p. 327-34, Jun 2003.

83
NADER, H. B. et al. Heparin stimulates the synthesis and modifies the sulfation pattern
of heparan sulfate proteoglycan from endothelial cells. J Cell Physiol, v. 140, n. 2, p.
305-10, Aug 1989.
NADER, H. B.; DIETRICH, C. P. Anticoagulant, antithrombotic and antihemostatic
activities of heparin: structural requeriments, mechanism of action and clinical
applications. Ciênc. cult.(Säo Paulo), v. 46, n. 4, p. 297-302, 1994.
NAPOLI, C. et al. Endothelial progenitor cells as therapeutic agents in the
microcirculation: an update. Atherosclerosis, v. 215, n. 1, p. 9-22, 2011.
NIMMERFALL, K.; MISCHKE, R. [Effect of unfractionated and low-molecular-weight
heparin on platelet aggregation and in vitro bleeding time in dogs]. Dtsch Tierarztl
Wochenschr, v. 106, n. 10, p. 439-44, Oct 1999.
OWENS, A. P., 3RD; MACKMAN, N. Tissue factor and thrombosis: The clot starts here.
Thromb Haemost, v. 104, n. 3, p. 432-9, Sep 2010.
PACURARI, M. et al. The Renin-Angiotensin-aldosterone system in vascular
inflammation and remodeling. International journal of inflammation, v. 2014, 2014.
PALARETI, G. et al. Pharmacodynamic effects on blood coagulation of a new low
molecular weight heparin (alfa-LMWH) in healthy volunteers and gynecological surgery
patients. Int Angiol, v. 8, n. 1, p. 47-52, Jan-Mar 1989.
PREVITALI, E. et al. Risk factors for venous and arterial thrombosis. Blood Transfus,
v. 9, n. 2, p. 120-38, Apr 2011.
REIMANN-HUNZIKER, G. Uber experimentelle Thrombose und ihre Behandlung mit
Heparin. Schweizerische Medizinische Wochenschrift, v. 74, p. 66-69, 1944.
RIDDEL, J. P., JR. et al. Theories of blood coagulation. Journal of Pediatric Oncology
Nursing, v. 24, n. 3, p. 123-131, 2007.
ROBERTSON, J. O. et al. Deficiency of LRP8 in mice is associated with altered platelet
function and prolonged time for in vivo thrombosis. Thromb Res, v. 123, n. 4, p. 644-
52, Feb 2009.
ROGER, V. L. et al. Executive summary: heart disease and stroke statistics--2012 update:
a report from the American Heart Association. Circulation, v. 125, n. 1, p. 188-97, Jan
03 2012.

84
RONDAIJ, M. G. et al. Dynamics and plasticity of Weibel-Palade bodies in endothelial
cells. Arteriosclerosis, thrombosis, and vascular biology v. 25, n. 6, p. 1002-1007,
2006 2006.
ROSENBERG, R. D.; DAMUS, P. S. The purification and mechanism of action of human
antithrombin-heparin cofactor. J Biol Chem, v. 248, n. 18, p. 6490-505, Sep 25 1973.
ROSENBERG, R. D.; LAM, L. Correlation between structure and function of heparin.
Proc Natl Acad Sci U S A, v. 76, n. 3, p. 1218-22, Mar 1979.
RUGGERI, Z. M.; MENDOLICCHIO, G. L. Adhesion mechanisms in platelet function.
Circ Res, v. 100, n. 12, p. 1673-85, Jun 22 2007.
SASISEKHARAN, R.; VENKATARAMAN, G. Heparin and heparan sulfate:
biosynthesis, structure and function. Curr Opin Cell Biol, v. 4, n. 6, p. 626-631, 2000.
SCHNEIDER, D. J.; SOBEL, B. E. Conundrums in the Combined Use of Anticoagulants
and Antiplatelet Drugs. Circulation, v. 116, n. 3, p. 305, 2007.
SCHUEPBACH, R. A. et al. Protection of vascular barrier integrity by activated protein
C in murine models depends on protease-activated receptor-1. Thromb Haemost, v. 101,
n. 4, p. 724-33, Apr 2009.
SCHWARTZ RS FAU - HOLMES, D. R., JR.; HOLMES DR JR FAU - TOPOL, E. J.;
TOPOL, E. J. The restenosis paradigm revisited: an alternative proposal for cellular
mechanisms. J Am Coll Cardiol, v. 20, n. 8, p. 1282-1293, 1992.
SIE, P. et al. Antithrombotic properties of a dermatan sulfate hexadecasaccharide
fractionated by affinity for heparin cofactor II. Blood, v. 81, n. 7, p. 1771-1777, 1993.
SIELSKI, M. S. Tese de mestrado. 2015. 83 Dissertação (Mestrado). Biologia Estrutural
e Funcional, Universidade Estadual de Campinas, Campinas.
SILVA, M. D., C. P. Structure of heparin. Characterization of the products formed from
heparin by the action of a heparinase and a heparitinase from Flavobacterium heparinum.
J Biol Chem, v. 250, n. 17, p. 6841-1846, 1975.
SIMON, D. I. et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte
integrin Mac-1 (CD11b/CD18). J Exp Med, v. 192, n. 2, p. 193-204, Jul 17 2000.

85
SPRONK, H. M.; GOVERS-RIEMSLAG JW FAU - TEN CATE, H.; TEN CATE, H.
The blood coagulation system as a molecular machine. Bioessays, v. 12, p. 1220-1228,
2003.
STEWART, M.; THIEL, M.; HOGG, N. Leukocyte integrins. Curr Opin Cell Biol, v.
7, n. 5, p. 690-6, Oct 1995.
TAYLOR, K. R.; GALLO, R. L. Glycosaminoglycans and their proteoglycans: host-
associated molecular patterns for initiation and modulation of inflammation. FASEB J,
v. 20, n. 1, p. 9-22, 2006.
TOLLEFSEN, D. Insight into the mechanism of action of heparin cofactor II.
Thrombosis and haemostasis, v. 74, n. 5, p. 1209-1214, 1995.
TOLLEFSEN, D. M. Heparin Cofactor II Modulates the Response to Vascular Injury.
Arteriosclerosis, Thrombosis, and Vascular Biology, v. 27, n. 3, p. 454-460, 2007.
TOVAR, A. M. et al. Dermatan sulfate is the predominant antithrombotic
glycosaminoglycan in vessel walls: implications for a possible physiological function of
heparin cofactor II. Biochim Biophys Acta, v. 1740, n. 1, p. 45-53, 2005.
TOVAR, M. F. et al. Dermatan sulfate is the predominant antithrombotic
glycosaminoglycan in vessel walls: implications for a possible physiological function of
heparin cofactor II. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease,
v. 1740, n. 1, p. 45-53, 2005.
TSENG, M. T. et al. Transendothelial migration of ferric ion in FeCl3 injured murine
common carotid artery. Thromb Res, v. 118, n. 2, p. 275-80, 2006.
TURNER, N. A. et al. Selective gene silencing of either MMP-2 or MMP-9 inhibits
invasion of human saphenous vein smooth muscle cells. Atherosclerosis, v. 193, n. 1, p.
36-43, Jul 2007.
VICENTE, C. P. et al. Antithrombotic activity of dermatan sulfate in heparin cofactor
II-deficient mice. Blood, v. 104, n. 13, p. 3965-70, Dec 15 2004.
VICENTE, C. P.; HE, L.; TOLLEFSEN, D. M. Accelerated atherogenesis and neointima
formation in heparin cofactor II deficient mice. Blood, v. 110, n. 13, p. 4261-7, 2007.
VINE, A. K. Recent advances in haemostasis and thrombosis. Retina, v. 29, n. 1, p. 1-7,
2009. ISSN 0275-004X.

86
VISSE, R.; NAGASE, H. Matrix Metalloproteinases and Tissue Inhibitors of
Metalloproteinases. Structure, Function, and Biochemistry, v. 92, n. 8, p. 827-839,
2003.
WALLER, B. F. et al. Morphological observations late (greater than 30 days) after
clinically successful coronary balloon angioplasty. Circulation, v. 83, n. 2 Suppl, p. I28-
41, 1991.
WARKENTIN, T. E. et al. Treatment and prevention of heparin-induced
thrombocytopenia: American College of Chest Physicians Evidence-Based Clinical
Practice Guidelines (8th Edition). Chest, v. 133, n. 6, p. 340-380, 20080624 DCOM-
20080826 2008.
WHO. The Atlas of Heart Disease and Stroke. 2014. Disponível em: <
http://gamapserver.who.int/mapLibrary/Files/Maps/Global_NCD_mortality_CVD_2012
.png >.
WIEDERMANN, J. G. et al. Intracoronary irradiation markedly reduces restenosis after
balloon angioplasty in a porcine model. J Am Coll Cardiol, v. 23, n. 6, p. 1491-8, 1994.
YOUNG, E. The anti-inflammatory effects of heparin and related compounds. Thromb
Res, v. 122, n. 6, p. 743-52, 2008.
ZARBOCK, A.; POLANOWSKA-GRABOWSKA RK FAU - LEY, K.; LEY, K.
Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood, v. 21, n. 2,
p. 99-111, 2007 2007.

87
ANEXO

88