university of groningen control of translational and
TRANSCRIPT

University of Groningen
Control of translational and rotational movement at nanoscaleStacko, Peter
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2017
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Stacko, P. (2017). Control of translational and rotational movement at nanoscale. University of Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 05-12-2021

Molecular dragsters: Towards controlled translational motion on surfaces
| 41
Chapter II:
Molecular dragsters: Towards controlled
translational motion on surfaces
Molecular dragsters bearing two appending molecular motor units as the
propelling wheels have been synthesized. The two dragsters feature different
sizes of the rotor units. The motors preserve their rotary function in solution and
preliminary experiments regarding movement on a surface under ambient
conditions have been performed.

Molecular dragsters: Towards controlled translational motion on surfaces
42 |
Introduction
Directional motion of molecules offers prospects for development of unique and
unprecedented nanotechnology devices rivaling machinery developed by
Nature1,2 such as motor proteins moving along microtubules3, the ATPase rotary
motion4,5 or myosin V walking on actin filaments6,7. Examples of directional
motion in the literature include DNA walkers8,9, photochemically10,11 and redox-
driven12 motors and catenane, and rotaxane translational systems, although the
systems are predominantly studied in solution.13–17
Regarding surface confined motors, directional movement can be divided to
rotary or translational motion. Achieving rotary motion is the more
straightforward option as the two major conditions to be fulfilled are anchoring
the molecules on the surface18–22 and the surface should not interfere with
process of controlling the motion; such as quenching of the excited state for
photochemically driven molecules, or redox reaction between the molecules and
surface for electrochemically driven processes.
On the other hand, there are many obstacles associated with autonomous
directional translational movement on surfaces. For example, the attractive
forces between the molecules and the surface have to be strong enough to
facilitate physisorption of molecules on the surface, while at the same time, the
forces have to be weak enough to allow for propulsion along the surface upon
application of stimuli. Perhaps due to the delicate balance necessary to achieve
this goal, autonomous directional movement of molecules on surface constitutes
a major challenge in contemporary chemistry.
Though, several examples of directional translational movement of molecules;
such as fullerenes23, porphyrins24, cyclodextrins25 or molecular landers26, on
surfaces can be found in the literature, however, the majority of them is induced
by “pushing” or “pulling” of the molecules with an STM tip (e.g. non-
autonomous).27,28 In addition, most of the observed motion is attributed to
sliding or slipping motion, rather than rolling motion and the exceptions to
these observations will be discussed here.
In a simple example, Grill et. al. have demonstrated that a molecule consisting of
two triptycene units connected with an axle (Figure 1a) can exhibit translational
movement when adsorbed on a Cu (110) surface at 25 K.28 Placing the STM tip

Molecular dragsters: Towards controlled translational motion on surfaces
| 43
on top of the molecule and moving it perpendicular to the axle of the molecules,
4 nm translational movement was observed (Figure 1b-d).
Figure 1. (a) Two triptycene units connected with an axle. (b) Scheme of manipulation with the STM tip. (c) STM image of the molecule on Cu (110) prior to manipulation. d) STM image of the molecule after the manipulation. (reproduced from ref. 24)
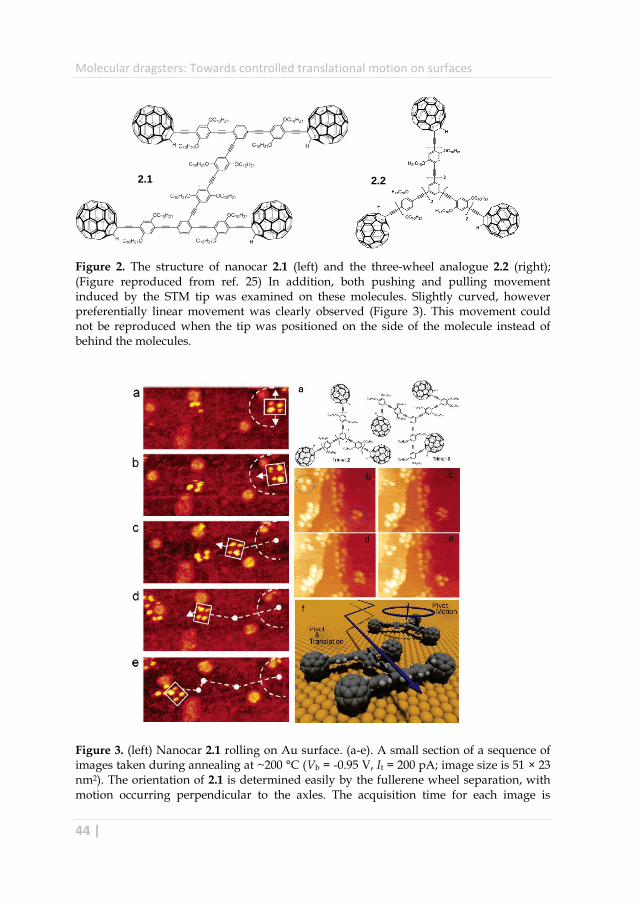
The group of Tour was able to demonstrate the wheel-assisted rolling motion on
gold surface at single molecular level using STM.29–31 For this purpose, the
nanocar 2.1 with four wheels as well as the three-wheel analogue 2.1 has been
synthesized (Figure 2). The comparison of movement for the two molecules
provided the evidence for wheel-assisted rolling motion. Upon heating the Au
surface to 200 °C only the four-wheel nanocar 2.1 showed translational motion
perpendicular to the axes, whereas the analogue 2.1 displayed pivoting motion
(Figure 3). This behavior demonstrates the expected fullerene-facilitated rolling
which is contrast to most examples in the literature where simple pushing is
almost universally accepted as the mechanism for large molecules.32–34
Molecular dragsters: Towards controlled translational motion on surfaces
44 |
Figure 2. The structure of nanocar 2.1 (left) and the three-wheel analogue 2.2 (right); (Figure reproduced from ref. 25) In addition, both pushing and pulling movement induced by the STM tip was examined on these molecules. Slightly curved, however preferentially linear movement was clearly observed (Figure 3). This movement could not be reproduced when the tip was positioned on the side of the molecule instead of behind the molecules.
Figure 3. (left) Nanocar 2.1 rolling on Au surface. (a-e). A small section of a sequence of images taken during annealing at ~200 °C (Vb = -0.95 V, It = 200 pA; image size is 51 × 23 nm2). The orientation of 2.1 is determined easily by the fullerene wheel separation, with motion occurring perpendicular to the axles. The acquisition time for each image is
2.1 2.2

Molecular dragsters: Towards controlled translational motion on surfaces
| 45
approximately 1 min, with a-e selected from a series spanning 10 min, which shows 80° pivot (a) followed by translation interrupted by small-angle pivot perturbations (b-e). (right) Structure and pivot motion of the trimers. (a) Structure of trimers. A sequence of STM images (b-e) acquired approximately 1 min apart during annealing at ~225 °C show the pivoting motion of 2.2 (both circled molecules) and lack of translation in b-e of any molecules. (Vb = -0.7 V, It ) 200 pA; image size is 34 × 27 nm2). Monatomic step edges in these images are lined with clustered molecules. (f) A summary of the two methods of motion for the different structures showing that nanocar 2.1 consecutively pivots and then translates perpendicular to its axles, whereas trimer 2.2 pivots but does not translate on the surface. For clarity, both structures are drawn devoid of the alkoxy units (reproduced from ref. 27).
Another attempt from group of Tour and co-workers to construct a “motorized
nanocar”, rather than a car being pushed or pulled by STM tip contained
p-carborane as a wheel and a light-driven molecular motor as the engine of the
nanocar 2.3.35,36 The molecule was thought to be propelled by the molecular
motor along the substrate surface as it can perform repetitive unidirectional
rotary motion induced by light (Figure 4). The molecule has only been studied
by 1H NMR and UV-vis spectroscopy in the solution and unfortunately, no
reports of movement on surface studied by STM have been made. The function
of the molecular motor was preserved in the presence of p-carborane units
unlike in the case of fullerene-C60 wheels.29
Figure 4. (left) The structure of the nanocar 2.3 with p-carborane units as wheels and molecular motor in the middle. (right) Irradiation (a) of the motor unit leads to rotation of the motor (b) inducing the forward translational motion (c-d). (Reproduced from ref. 25)
Perhaps the most successful demonstration of unidirectional movement on a
surface has been reported by the group of Feringa.37 The molecular nanocar 2.4
features a chassis connected to four molecular motors designated to propel the
molecule along the surface when performing the rotational movement (Figure
2.3

Molecular dragsters: Towards controlled translational motion on surfaces
46 |
5). Upon electronic excitation using an STM tip double bond isomerization
occurs followed by a helix inversion resulting in rotary motion pushing the
molecule forward. A correct configuration of the stereogenic centers was
required as only in the meso isomer both of the motor units rotate in the forward
direction from the external point of view. Ultimately, the molecule was shown to
undergo a preferentially linear movement showing that the intrinsic motor
function is capable of converting external energy input into mechanical action
(Figure 6).
Figure 5. (a) The structure of nanocar 2.4 with embedded four molecular motors as wheels. (b) Schematic representation of the concept (Figure reproduced from ref. 33)
Figure 6. (a) STM image (imaging parameters: area 10.2 nm × 39.3 nm, current I = 74 pA, U = 47 mV) of the initial position. The black area was scanned only after the molecule moved into it. (b) Trajectory depicting the individual steps taken. (c) Final position after ten consecutive voltage pulses. (d) The action spectrum for movement shows a voltage threshold at 500 mV. Each data point represents 8 to 40 manipulations performed on various molecules (I = 30–50 pA). Error bars represent the standard deviation from the probability for successful events. (e) STM frames corresponding to individual steps of the trajectory in (b) excluding starting and final position (reproduced from ref. 33).
2.4

Molecular dragsters: Towards controlled translational motion on surfaces
| 47
Design of the dragsters
Some of the initial difficulties in the molecular nanocar project included poor
volatility of the molecule for surface deposition as well as rather high voltage
required for movement of the molecule. Motivated by the success of the
electrically-driven molecular nanocar, we sought to address these issues by
simplifying the design along with enabling modification of the molecule for
future applications such as cargo transport. One could imagine a cargo unit
being attached to the motorized part and once at the desired destination, the
cargo could be released. For this purpose, we envisioned that molecular
dragsters bearing only two rotating motor units (instead of four) could be
sufficient (Figure 7). To first prove that the unidirectionality of the molecular
dragsters on surface is preserved, only a dummy cargo unit, sharing the same
carbazole building block as the other half of the molecule, was attached at this
point. In order to assess the influence of wheel “size” on the unidirectional
movement, we decided to construct two dragsters with different wheel sizes –
one based on simple fluorene units and the second one, larger, based on phenyl
substituted fluorene units.
Figure 7. The design of the molecular dragsters consisting of a motorized part with two motor units and a dummy cargo unit.

Molecular dragsters: Towards controlled translational motion on surfaces
48 |
Synthesis of the dragsters and intermediates
Due to similarities in the design, the synthesis of the molecular dragsters is
based on that of the molecular nanocar 2.4, although with several crucial
modifications in both synthetic and purification procedures.37
The synthesis started with the preparation of 2,7-dibromo-9H-carbazole as it is a
common building block for both parts of the molecule (2.11 and 2.18a-b, vide
infra). For this purpose, 4,4’-dibromobiphenyl was nitrated using fuming nitric
acid in glacial acetic acid (Scheme 1).38,39 The resulting mono-nitrated product
2.7 was then subjected to reductive cyclization using PPh3 in o-DCB at high
temperature to facilitate the formation of 2.8 in a good overall yield.40
Scheme 1. Synthesis of 2.8 as the common intermediate for further synthesis.
From this intermediate, both parts of the dragster have been synthesized. The
carbazole 2.8 was reacted with a large excess (4 eq.) of 1,4-diiodobenzene in
Ullmann coupling catalyzed by CuI and 1,10-phenanthroline as a ligand to
provide the N-substituted carbazole 2.9 (Scheme 2).41 The excess of 1,4-
iodobenzene prevented the formation of the di-substituted product as well as
Ullmann coupling between two molecules of the starting material.

Molecular dragsters: Towards controlled translational motion on surfaces
| 49
Scheme 2. Synthesis of the unpropelled part 2.11 of the dragsters from the carbazole 2.8.
Following a procedure for Sonogashira coupling with TMSA using Pd(PPh3)Cl2
and CuI as the catalysts, a TMS-protected acetylene was installed onto the
molecule to give compound 2.10. The acetylene would then, upon deprotection,
serve as a connection point with the other half of the molecule. It should be
stressed that excess of TMSA was avoided as it led to coupling at the bromo-
substituted positions even at room temperature. Besides that, a complete
selectivity for the iodo-substituted reaction site was observed. Metal-halogen
exchange at -78 °C using n-BuLi, followed by a substitution of the di-lithiated
carbazole with TMSCl and deprotection of the acetylene in situ with KOH
afforded the final building block 2.11 in 84 % yield over the three steps (Scheme
3).
In the next stage, the motorized part 2.18a-b of the dragster was to be
synthesized. Unlike in the case of the nanocar 2.4 where the solubilizing hexyl
chains were introduced via Sonogashira coupling followed by hydrogenation37,
in this synthesis the alkyl moieties were installed in one step using Pd(dppf)Cl2
catalyzed variation of Kumada coupling in 82% yield (Scheme 3, 2.12).42

Molecular dragsters: Towards controlled translational motion on surfaces
50 |
Scheme 3. Synthesis of the diketone 2.14 for double Barton-Kellogg coupling.
Employing the same strategy as for the nanocar, the diketone 2.13 was formed
from the carbazole 2.12 by a Friedel-Crafts-Nazarov cyclization sequence with
methacrylic acid in PPA (Scheme 4).37 Despite the solubilizing chains, the
compound 2.13 is poorly soluble in most organic solvents. Due to this, the
original eluent for flash column chromatography was substituted for
dichloromethane, improving the yield to 39% across multiple steps. Compound
2.13 was isolated as a mixture of the two possible diastereomers (1:1, based on 1H NMR spectroscopy) and used as such throughout the rest of the synthetic
sequence. Both the Kumada coupling and the condensation could be
conveniently carried out on a multigram scale. Reproduction of the procedure
reported for the nanocar 2.4 involving a protection of the carbazole 2.13 with a t-
Boc group followed by a thionation using Lawesson’s reagent was unfortunately
not met with a success due to constant deprotection of the t-Boc group under
various conditions and formation of complex mixtures, presumably due to free
NH present in the molecule. It was therefore decided to circumvent the use of a
protecting group and shorten the synthetic sequence by performing an Ullmann
coupling on the carbazole 2.13 prior to double Barton-Kellogg coupling.
Initially, the Ullmann coupling was planned to be carried out with 1-bromo-4-
iodobenzene to avoid di-substitution on the benzene ring. Various conditions
and catalysts such as Cu43 or CuI44–46 with L-proline47–49, 1,10-phenanthroline50 as
ligands in both toluene and DMF were screened, however, none of the described
methods displayed appreciable selectivity towards the iodine substituent and
either no conversion or a mixture of products (1:2 to 1:4, bromo:iodo-substituted

Molecular dragsters: Towards controlled translational motion on surfaces
| 51
product) was always observed. We have therefore opted for the same approach
as in the case of carbazole 2.8 and used a large excess (6 equivalents) of 1,4-
idodobenzene (Scheme X). Conveniently, the unreacted 1,4-iodobenzene can be
recycled after the reaction by flash column chromatography due to its low
polarity. Finally, this procedure gave the N-substituted carbazole 2.14 in an
excellent yield of 91%.
Scheme 4. Double Barton-Kellogg coupling used to install the two molecular motor units.
At this point, the two motor units needed to be introduced in the molecule via
double Barton-Kellogg coupling (Scheme 5). The sequence started with a
thionation of the ketone 2.14 using Lawesson’s reagent in toluene at 95 °C. The
temperature had to be closely monitored as increase above 100 °C led to a quick
decomposition of the formed thioketone 2.15 presumably due to enolization,
followed by dimerization, trimerization and oligomerization of the thioenol.51,52
This side process could be distinguished by a formation of a yellow spot situated
at the start of the TLC plate. Isolation of the dithioketone by a short column,
followed by a reaction with 9-diazofluorenone 2.16a or 2.16b at 80 °C in toluene
and subsequent desulphurization of the resulting episulfides 2.17a-b with
HMPT afforded the building block 2.18a-b in 49% and 44% yield, respectively,
over three steps.

Molecular dragsters: Towards controlled translational motion on surfaces
52 |
Scheme 5. Connection of the two halves 2.11 and 2.18a-b via Sonogashira coupling
Finally, the unpropelled part 2.11 and the motorized part 2.18a-b were
connected in a classical Sonogashira coupling, using a slight excess of the
alkyne. The final compounds 2.5a-b were isolated as a mixture of two
diastereomers since they were found to be inseparable by flash column
chromatography in all steps of the synthesis. It should be realized that only the
meso diastereomer can be effective for directional movement on a surface. In
case of the (R,R)- and (S,S)-isomers, the motor units rotate in a disrotatory
fashion with respect to each other, thus preventing the molecule from moving in
a linear manner. For this purpose, the diastereomers can be separated using
preparative HPLC or supercritical fluid chromatography (SFC) when required.
Photochemical and thermal isomerization studies in a solution
The photochemical and thermal isomerization behavior of 2.5a-b was examined
in solution using both low temperature UV-vis absorption and 1H NMR
spectroscopy to demonstrate that the rotary function of the two appending
motor units remained uncompromised.
The UV-vis absorption spectra of the dragsters 2.5a and 2.5b in heptane at 293 K
show absorption bands centered at 392 and 434 nm, respectively (Figure 8, black
solid).

Molecular dragsters: Towards controlled translational motion on surfaces
| 53
Figure 8. UV-vis absorption spectra (heptane, 293 K) of stable 2.5a (a) and stable 2.5b (b)
(black solid); irradiation to PSS (365 nm) at 273 K (blue solid); and after heating the samples to 333 K (dashed red).
Irradiation of the sample with UV light (365 nm) at 273 K led to a red-shift of the
absorption bands to 401 and 436 nm, respectively, consistent with a formation of
a higher energy isomer, and hence species containing a more strained central
double bond.53 Throughout the irradiation, a single isosbestic point was
observed in each case, indicating that only a single motor unit undergoes
photochemical EZ isomerization at a time and that the irradiation does not
produce a double unstable form of 2.5a or 2.5b (based on 1H NMR spectroscopy
and presence of a single isosbestic point in UV-vis experiments). Samples were
irradiated until no further changes were observed and the photostationary state
(PSS) was reached (Figure 8, blue solid).
After heating the samples to 333 K, a complete recovery of the original UV-vis
absorption spectra were observed, consistent with the unstable form of 2.5a-b
undergoing thermal helix inversion back to the stable isomers (Figure 8, red
dashed).
The rate constants for the thermal helix inversion were measured at five
different temperatures (heptane) by following the UV-vis absorption spectra
over time. Multivariate analysis was performed on the array of the spectra,
providing the rate constant for each temperature. The Eyring plot was then
constructed and the activation parameters for the thermal process were derived
from the linear fit (Figure 9 and 10).

Molecular dragsters: Towards controlled translational motion on surfaces
54 |
∆‡G°THI 88.4 ± 0.0
kJ.mol-1
∆‡H°THI 65.4 ± 0.2
kJ.mol-1
∆‡S°THI -79.5 ± 0.5
J.K-1.mol-1
k (293 K) (1.10 ± 0.05) × 10-3
s-1
t1/2 (293 K) 10.7 ± 0.0 min
Figure 9. (left) Thermodynamic data for the thermal helix inversion of the unstable 2.5a
to the stable 2.5a (heptane). (right) Eyring plot for the thermal process.
The Gibbs free energy of activation ∆‡G°THI for isomerization of the unstable 2.5a
was determined to be 88.4±0.0 kJ.mol-1 (∆‡H°THI 65.4±0.2 kJ.mol−1, ∆‡S°THI -
79.5±0.5 J.K−1.mol−1), corresponding to a half-life of 10.7±0.0 min at room
temperature (Figure 9, left). These values compare well to those reported for the
nanocar 2.4 which were found to be 88.0 kJ.mol-1 and a half-life of 9.1 min.37
∆‡G°THI 92.6 ± 0.1
kJ.mol-1
∆‡H°THI 67.2 ± 2.3
kJ.mol-1
∆‡S°THI -86.4 ± 7.5
J.K-1.mol-1
k (293 K) (1.95 ± 0.08) × 10-3
s-1
t1/2 (293 K) 59.2 ± 2.5 min
Figure 10. (left) Thermodynamic data for the thermal helix inversion of the unstable 2.5b
to the stable 2.5b (heptane). (right) Eyring plot for the thermal process.
Using the same procedure, the activation Gibbs energy ∆‡G°THI for the thermal
isomerization of the unstable 2.5b to the stable 2.5b was found to be 92.6±0.1
kJ.mol-1 (∆‡H°THI 67.2±2.3 kJ.mol−1, ∆‡S°THI -86.4±7.5 J.K−1.mol−1), corresponding
to a half-life 59.2±2.5 min at room temperature (Figure 10a). The increase of
∆‡G°THI is in accordance with introduction of phenyl substituents in the 2,7-
position of the fluorenyl rotors, resulting in increased steric hindrance in the
fjord region during the thermal helix inversion. One should realize that these
∆‡G°THI values are only relevant for measurements on surfaces at ambient
conditions, as in other instances, such as ultra-high vacuum STM at 7 K, the
barriers are thermally insurmountable and electron tunneling excitation by STM
tip must be used for induction of the thermal step regardless of the activation
barrier.

Molecular dragsters: Towards controlled translational motion on surfaces
| 55
The switching behavior was further probed using 1H NMR spectroscopy. A
sample of 2.5a-b (2 mg) in chloroform-d3 was prepared. 1H NMR spectra were
recorded at 243 K before and after irradiation (at 233 K) with a hand-held UV-
lamp or LED using optical fiber inside the NMR spectrometer (both 365 nm) (3-
16 h). Unfortunately, in neither case a formation of a new set of NMR
absorptions was observed as a consequence of the irradiation, unlike in the case
of nanocar 2.4.37 Lowering the temperature further (223 K) did not improve the
situation. It remains unclear why no change was observed using 1H NMR
spectroscopy. Possible a much higher concentration than in the UV-vis
spectroscopy experiments, low quantum yield or a combination of thereof is
responsible for this.
Examining the photochemical and thermal behavior of 2.5a-b in solution using
UV-vis absorption, it can be concluded that the motor function of the
overcrowded alkenes is preserved in both molecules. The dragsters described
herein are therefore suitable candidate for investigation of a directional
movement on surfaces.
Single molecule STM experiments at ambient conditions
With the knowledge that the molecules 2.5a-b retained their molecular motor
function, we opted to perform preliminary scanning tunneling miscroscopy
(STM) experiments and study their behavior on surfaces under ambient
conditions. For this purpose, HOPG and gold were chosen as the surfaces for
modification with n-pentacontane. N-pentacontane is known to self-assemble on
surfaces and serve as a “ruler” and may exhibit attractive interactions with the
hexyl chains of the dragsters 2.5a-b, facilitating their absorption on the surface.
Upon preparation of the n-pentacontane modified HOPG, the sample was
examined by STM at the liquid-solid interface to ensure that the n-pentacontane
domains are large enough. Subsequently, a drop of the ~7.9× 10-5 M solution of
the dragster 2.5a in n-tetradecane was added. The pentacontane adlayer could
still be resolved after the addition of the dragster, however no dragsters were
observed in these experiments (Figure 13). Measurements on bare HOPG did
not lead to adsorption of the dragsters either.
For the experiments were performed on gold on mica substrates, a 400 nm-thick
layer of gold was deposited (in a home-built thermal deposition system) on a
freshly cleaved mica substrate at a temperature of 375°C and a pressure of 1.8 ×

Molecular dragsters: Towards controlled translational motion on surfaces
56 |
10-6M. After flame annealing the substrate, a ~9.6 × 10-4M solution of n-
pentacontane in n-tetradecane was drop-casted on the gold/mica substrate. One
hour later, a ~7.9× 10-5M solution of the dragster 2.5a in n-tetradecane was drop-
casted on top of the pentacontane solution. In this system, the STM
measurements did not reveal adsorped dragster molecules, neither the
pentacontane adlayer. However, it is necessary to carry out more experiments in
order to conclude whether the dragsters are suitable for use on surfaces under
ambient conditions.
Figure 13. STM images of the n-pentacontane adlayer after deposition of the
dragster 2.5a.
Conclusions
Two molecular dragsters with different wheel size have been synthesized.
As anticipated, the motors units in the dragsters have been proven to undergo
photochemically driven rotation in a solution using UV-vis spectroscopy.
Kinetic experiments were performed and thermodynamic data for the thermal
helix inversion step of the rotary cycle have been determined by Eyring analysis.
The barriers of THI were found to be 88.4±0.0 kJ.mol-1 and 92.6±0.1 kJ.mol-1 for
2.5a and 2.5b respectively, which corresponds to a half-life of 10.7±0.1 and
59.2±2.5 min, respectively, at room temperature.
No movement of the molecules on the pentacontane modified surface has been
observed under UV irradiation (365 nm) in the preliminary STM measurements
5.0nm 10nm

Molecular dragsters: Towards controlled translational motion on surfaces
| 57
so far, presumably due to strong attractive forces between the dragsters and the
surface.
Acknowledgment
The single molecule STM experiments on the surfaces described herein have
been performed by G. H. Heideman, who is gratefully acknowledged for this
contribution.
Experimental section
General remarks
Reagents were purchased from Aldrich, Merck or Fluka and were used as
provided unless otherwise stated. The solvents were distilled and dried, if
necessary, by standard methods. Column chromatography was performed on
silica gel (Merck type 9385 230-400 mesh) unless stated otherwise using positive
pressure, TLC: silica gel 60, Merck, 0.25 mm. High Resolution Mass spectra
(HRMS) were recorded on an LTQ Orbitrap XL. NMR spectra were obtained
using a a Varian Mercury Plus (1H: 400 MHz, 13C: 100 MHz), a Varian Unity Plus
(1H: 500 MHz, 13C: 125 MHz) or a Varian Innova (1H: 600 MHz,) in CDCl3, d8-
toluene or CD2Cl2. Chemical shifts are reported in δ units (ppm) relative to the
residual deuterated solvent signal of CDCl3 (1H NMR, δ 7.26 ppm; 13C NMR, δ
77.23 ppm), d8-toluene (1H NMR, δ 2.09 ppm), or CD2Cl2 (1H NMR, δ 5.32 ppm; 13C NMR, δ 54.0 ppm). The splitting patterns are designated as follows: s
(singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), td (triplet
of doublets), qt (quartet of triplets), m (multiplet) and br (broad). SFC was
performed on a Thar SFC system consisting of a fluid delivery module (FDM10-
1), an autosampler (a modified Alias 840), a semi-prep column oven, PDA
detector, a back-pressure regulator (ABPR20), heat-exchanger, and a fraction
collector (modified Thar SFC-FC). UV-vis absorption spectra were measured on
a Jasco V-630 or a Hewlett-Packard 8453 spectrometer. CD spectra were
measured on a Jasco J-815 CD spectrometer. Solvents used for spectroscopic
studies was of spectroscopic grade (UVASOL Merck). Irradiations were
performed using a spectroline ENB-280C/FE lamp (λmax = 365 nm), or an LED
(5 W, 365 nm, 10 nm width at half-height), mounted in a modified Nalorac Z-
Spec probe in the Varian Innova-600 NMR. Samples irradiated for 1H NMR
spectroscopy were placed 3-5 cm from the lamp. Irradiations at low

Molecular dragsters: Towards controlled translational motion on surfaces
58 |
temperatures were performed in standard EtOH/N2 bath. Photostationary states
were determined by monitoring changes in UV-vis spectra or 1H NMR spectra
until no further changes were observed. Kinetic analysis of the thermal
isomerization steps was performed by UV-vis spectroscopy. Changes in UV-vis
absorptions were monitored at different temperatures. The array of the UV-vis
spectra was processed using multivariate analysis (from 200 to 800 nm) to obtain
the corresponding rate constants from which an Eyring plot was constructed.
∆‡G°, ∆‡H°, ∆‡S° and t1/2 (20 °C) were extracted from this plot.
STM visualization of 2.5a on HOPG
Prior to imaging, the n-pentacontane (TCI Europe) molecules were dissolved in
n-tetradecane (Sigma-Aldrich) by heating (40°C) and sonication for at least 2
hours. The non-volatile solvent n-tetradecane allows us to perform STM
measurements at the solid-liquid interface. A ~9.6 × 10-4 M solution of n-
pentacontane in n-tetradecane was drop-casted on a freshly cleaved highly
oriented pyrolytic graphite (HOPG) crystal (SPI supplies, SPI-3 grade). The STM
tips were prepared by mechanical cutting from Pt/Ir wire (90:10, diameter 0.25
mm, Goodfellow). All experiments were performed at room temperature using a
PicoSPM instrument (Molecular Imaging, Scientec) and PicoScan imaging
software.
Synthesis of the dragsters and intermediates
4,4'-Dibromo-2-nitro-1,1'-biphenyl (2.7).
Fuming nitric acid (92.5 %, 120 mL, 2.5 mol) was added into a
solution of 4,4'-dibromo-1,1'-biphenyl (29.0 g, 93 mmol) in
acetic acid (300 mL) over 10 min. The resulting suspension
was heated to 100 °C for 30 min. The solution was cooled down to 0 °C, and the
pale yellow solid was filtered through a glass filter. The solid was dried on air
and then triturated with ethanol (350 mL) to give the pure product 2.7. Yield:
27.53 g (83%). Pale yellow solid. Mp 124.3127.5 °C. 1H NMR (400 MHz, CDCl3):
δ (ppm) 8.03 (dd, 1H, J1 = 1.7 Hz, J2 = 1.7 Hz), 7.76 (ddd, 1H, J1 = 8.2 Hz, J2 = 1.8
Hz, J3 = 1.8 Hz), 7.57 (dd, 2H, J1 = 8.5 Hz, J2 = 1.6 Hz), 7.29 (dd, 1H, J1 = 8.2 Hz, J2
= 1.5 Hz), 7.16 (dd, 2H, J1 = 8.5 Hz, J2 = 1.6 Hz). 13C NMR (100 MHz, CDCl3): δ
(ppm) 136.8, 135.8, 135.5, 134.3, 133.2, 132.2, 129.6, 127.5, 123.3, 122.0.

Molecular dragsters: Towards controlled translational motion on surfaces
| 59
2,7-Dibromo-9H-carbazole (2.8).
A mixture of the biphenyl 2.7 (28.5 g, 80 mmol) and
triphenylphosphine (52.3 g, 200 mmol) in 1,2-dichlorobenzene
(160 mL) was heated at 190 °C for 6 h. The solvent was then
distilled out at reduced pressure and the residue was purified by column
chromatography on silica gel (pentane : dichloromethane – 5 : 1 to 3 : 1) give the
pure carbazole 2.8. Yield: 20.3 g (78%). Tan solid. Mp 230.1231.8 °C. 1H NMR
(400 MHz, CDCl3): δ (ppm) 8.07 (s, 1H), 7.88 (d, 2H, J = 8.5 Hz), 7.58 (d, 2H, J =
1.6 Hz), 7.36 (dd, 2H, J1 = 8.5 Hz, J2 = 1.6 Hz). 13C NMR (100 MHz, CDCl3): δ
(ppm) 138.5, 129.4, 124.2, 123.3, 112.7, 112.2.
2,7-Dibromo-9-(4-iodophenyl)-9H-carbazole (2.9).
A mixture of the carbazole 2.8 (1.95 g, 6.0 mmol),
1,4-diiodobenzene (7.92 g, 24.0 mmol), 1,10-phenanthroline
(216 mg, 1.2 mmol), Cs2CO3 (3.91 g, 12.0 mmol) and CuI
(229 mg, 1.2 mmol) in toluene (30 mL) was heated at 110 °C.
When the starting material was completely consumed
(67 h, TLC), the solvents were evaporated at reduced pressure and the residue
was purified by column chromatography on silica gel (pentane to pentane :
dichloromethane – 5 : 1) to give the pure product 2.9. Yield: 2.43 g (77%). White
solid. Mp 188.1189.3 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.97 (d, 2H, J = 8.1
Hz), 7.93 (d, 2H, J = 8.3 Hz), 7.47 (s, 2H), 7.41 (d, 2H, J = 8.3 Hz), 7.25 (d, 2H, J =
8.1 Hz). 13C NMR (100 MHz, CDCl3): δ (ppm) 141.6, 139.7, 136.3, 129.0, 124.1,
121.9, 121.7, 120.3, 113.0, 93.5.
2,7-Dibromo-9-(4-iodophenyl)-9H-carbazole (2.10).
A mixture of the carbazole 2.9 (2.30 g, 4.36 mmol),
ethynyltrimethylsilane (620 l, 4.36 mmol), Pd(PPh3)2Cl2
(153 mg, 0.22 mmol) and CuI (42 mg, 0.22 mmol) in dry THF
(50 mL) and Et3N (25 mL) was degassed by four freeze-
pump-thaw cycles. The resulting solution was stirred at
room temperature under N2 atmosphere overnight. The volatiles were
evaporated at reduced pressure and the residue was purified by column
chromatography on silica gel (pentane to pentane : dichloromethane – 5 : 1) to
give the pure product 2.10. Yield: 1.78 g (82%). White solid. Mp 239.1240.3 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.93 (d, 2H, J = 8.3 Hz), 7.74 (d, 2H, J = 8.5
Hz), 7.49 (d, 2H, J = 1.2 Hz), 7.46 (d, 2H, J = 8.5 Hz), 7.41 (dd, 2H, J1 = 8.3 Hz, J2 =

Molecular dragsters: Towards controlled translational motion on surfaces
60 |
1.5 Hz), 0.32 (s, 9H). 13C NMR (100 MHz, CDCl3): δ (ppm) 141.7, 136.4, 134.0,
126.9, 124.0, 123.4, 122.0, 121.7, 120.2, 113.2, 103.9, 96.3, 0.1.
9-(4-Ethynylphenyl)-2,7-bis(trimethylsilyl)-9H-carbazole (2.11).
The solution of carbazole 2.10 (710 mg, 1.4 mmol) in dry
THF (30 mL) was cooled down to -78 °C under nitrogen
atmosphere. Solution of n-BuLi (3.1 mL, 5.0 mmol) was
added dropwise over 10 minutes. After stirring for 30 min,
TMSCl (730 l, 5.7 mmol) was added at once and the
reaction was left to warm to room temperature. Aq. KOH (3 ml, 2 M) was added
and the mixture was stirred for 6 h. The reaction mixture was partitioned
between water (60 mL) and ethyl acetate (50 mL). The water layer was washed
with ethyl acetate (2 × 50 mL) and the combined organic extracts were dried
with MgSO4. The volatiles were evaporated at reduced pressure and the residue
was purified by column chromatography on silica gel (pentane : ethyl acetate –
50 : 1) to give the pure product 2.11. Yield: 505 mg (86%). White solid. Mp 221.6-
222.3 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.17 (d, 2H, J = 7.7 Hz), 7.81 (d,
2H, J = 8.3 Hz), 7.617.63 (m, 4H), 7.49 (d, 2H, J = 7.7 Hz), 3.24 (s, 1H), 0.36 (s,
18H). 13C NMR (100 MHz, CDCl3): δ (ppm) 140.4, 138.7, 138.4, 133.9, 127.1, 125.1,
124.2, 121.1, 120.0, 114.4, 83.2, 78.3, -0.5. HRMS (ESI+): calcd for C26H30NSi2+ (M +
H+) 412.1911 found 412.1909.
2,7-Dihexyl-9H-carbazole (2.12).
A solution of hexylmagnesium bromide (38.5 mL, 2.0 M in
Et2O) was added slowly to a degassed solution of 2.8 (5.0 g,
15.4 mmol) and Pd(dppf)Cl2 (450 mg, 0.62 mmol) in dry
THF (100 mL) at room temperature under N2 atmosphere. The resulting mixture
was then heated at reflux for 4 h. After no more starting material could be
observed by TLC, the reaction mixture was cooled down and the reaction was
quenched by slow addition of methanol (5 mL). The solvents were evaporated at
reduced pressure and the residue purified by column chromatography on silica
gel (pentane : dichloromethane – 5 : 1) to give the pure product 2.12. Yield: 4.21
g (81%). White flaky solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.93 (d, 2H, J =
7.9 Hz), 7.76 (brs, 1H), 7.17 (s, 2H), 7.07 (dd, 2H, J1 = 8.0 Hz, J2 = 1.2 Hz), 2.78 (t,
4H, J = 7.7 Hz), 1.72 (m, 4H), 1.331.39 (m, 12H), 0.92 (t, 6H, J = 7.0 Hz). 13C NMR
(100 MHz, CDCl3): δ (ppm) 140.8, 140.1, 121.5, 120.4, 119.8, 110.2, 36.7, 32.1, 32.0,
29.3, 22.8, 14.3.

Molecular dragsters: Towards controlled translational motion on surfaces
| 61
4,8-Dihexyl-2,10-dimethyl-10,11-dihydro-1H-dicyclopenta[c,g]carbazole-
3,9(2H,6H)-dione (2.13).
Methacrylic acid (10.4 mL, 122 mmol) was added to a
mechanically stirred mixture of the carbazole 2.12 (4.10 g,
12.2 mmoL) and in PPA (115 %, 100 mL) and heated at
100 °C overnight. The reaction was quenched by addition
of ice (300 mL) and the resulting mixture was extracted
with dichloromethane (3 x 100 mL). The combined organic extracts were washed
with sat. aq. K2CO3 (100 mL) and dried with MgSO4. The solvents were
evaporated at reduced pressure and the residue purified by column
chromatography on silica gel (dichloromethane) to give the pure product 2.13.
Yield: 1.94 g (34%). Pale yellow solid. Mp 197.5198.3 °C. 1H NMR (400 MHz,
CDCl3): δ (ppm) 8.91 (brs, 1H), 7.22 (s, 2H), 4.06 (dd, 2H, J1 = 17.0 Hz, J2 = 7.9
Hz), 3.143.36 (m, 6H), 2.802.91 (m, 2H), 1.641.72 (m, 4H), 1.401.46 (m, 10H),
1.281.38 (m, 8H) 0.88 (t, 6H, J = 7.0 Hz). 13C NMR (100 MHz, CDCl3): δ (ppm)
209.0, 208.9, 150.07, 150.04, 143.8, 143.0, 127.58, 127.55, 118.65, 118.63, 111.5, 42.3,
42.2, 36.9, 36.8, 32.5, 32.0, 31.2, 29.6, 22.9, 17.2, 17.0, 14.3.
4,8-Dihexyl-6-(4-iodophenyl)-2,10-dimethyl-10,11-dihydro-1H-
dicyclopenta[c,g]carbazole-3,9(2H,6H)-dione (2.14).
A mixture of the carbazole 2.13 (250 mg, 0.53 mmol),
1,4-diiodobenzene (1.05 g, 3.2 mmol), 1,10-phenanthroline
(38.2 mg, 0.21 mmol), Cs2CO3 (345 mg, 1.1 mmol) and CuI
(40.4 mg, 0.21 mmol) in toluene (2 mL) was heated to
110 °C. When the starting material was completely
consumed (67 h, TLC), the solvents were evaporated at
reduced pressure and the residue purified by column chromatography on silica
gel (dichloromethane to dichloromethane : ethyl acetate – 40 : 1) to give the pure
product 2.13. Yield: 321 mg (90 %). Pale yellow solid. Mp 79.781.6 °C. 1H NMR
(400 MHz, CDCl3): δ (ppm) 8.02 (d, 2H, J = 8.5 Hz), 7.23 (d, 2H, J = 8.4 Hz), 6.96
(s, 2H), 4.09 (dd, 2H, J1 = 16.8 Hz, J2 = 7.9 Hz), 3.303.37 (m, 2H), 3.043.23 (m,
4H), 2.792.89 (m, 2H), 1.58 (m, 4H), 1.261.44 (m, 18H), 0.87 (t, 6H, J = 7.0 Hz). 13C NMR (100 MHz, CDCl3): δ (ppm) 208.7, 208.6, 149.89, 149.85, 145.2, 143.3,
139.8, 136.3, 130.0, 128.1, 128.0, 118.69, 118.67, 110.6, 94.6, 42.33, 42.30, 37.1, 37.0,
32.7, 31.9, 31.5, 29.6, 22.9, 17.2, 17.0, 14.3. HRMS (ESI+): calcd for C38H45INO2+ (M
+ H+) 674.2489 found 674.2484.

Molecular dragsters: Towards controlled translational motion on surfaces
62 |
General procedure for Barton-Kellogg coupling (2.18a-b).
A mixture of the diketone 2.14 (200 mg, 0.30 mmol)
and Lawesson’s reagent (480 mg, 1.2 mmol) in dry
toluene (20 mL) was heated at 90 °C for 3 h. The
mixture was left to cool down to room temperature
and passed through a short column of silica gel. The
dithioketone was eluted (pentane : ethyl acetate –
10 : 1) as the first red band. The solvents were
evaporated at reduced pressure and the residue was
redissolved in dry toluene (20 mL). The solution was heated to 80 °C and
diazofluorenone54 or 2,7-biphenyldiazofluorenone (1.05 mmol) was added at
once. The resulting solution was heated at 80°C overnight. HMPT (160 l, 0.89
mmol) was added and the mixture was heated for additional 3 h. The solvents
were evaporated at reduced pressure and the residue was purified by column
chromatography.
3,9-Di(9H-fluoren-9-ylidene)-4,8-dihexyl-6-(4-iodophenyl)-2,10-dimethyl-
2,3,6,9,10,11-hexahydro-1H-dicyclopenta[c,g]carbazole (2.18a).
Prepared according to the general procedure from the ketone 2.14 (200 mg, 0.30
mmol) and diazafluorenone (228 mg, 1.2 mmol). The product was purified by
column chromatography on silica gel (pentane : dichloromethane - 7:1 to 5:1) to
give 2.18a as a mixture of diastereomers (~ 1:1). Yield: 132 mg (46 %). Orange
solid. Mp >250 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.04 (dd, 2H,
J1 = 8.3 Hz, J2 = 2.4 Hz), 7.93 (dd, 2H, J1 = 7.2 Hz, J2 = 7.2 Hz), 7.81 (dd, 2H, J1 =
7.6 Hz, J2 = 7.6 Hz), 7.75 (d, 1H, J = 7.6 Hz), 7.71 (d, 1H, J = 7.5 Hz), 7.61 (d, 1H, J
= 7.9 Hz), 7.49 (dd, 2H, J1 = 8.2 Hz, J2 = 8.2 Hz), 7.45 (d, 1H, J = 8.3 Hz), 7.307.40
(m, 4H), 7.26 (dd, 2H, J1 = 7.7 Hz, J2 = 7.4 Hz), 7.20 (d, 1H, J = 7.9 Hz), 7.14 (s,
1H), 7.12 (s, 1H), 7.08 (dd, 1H, J1 = 7.5 Hz, J2 = 7.5 Hz), 7.02 (dd, 1H, J1 = 7.6 Hz, J2
= 7.6 Hz), 4.30 (m, 2H), 3.87 (dd, 1H, J1 = 14.9 Hz, J2 = 5.6 Hz), 3.75 (dd, 1H, J1 =
15.2 Hz, J2 = 5.6 Hz), 3.41 (d, 1H, J = 15.0 Hz), 3.20 (d, 1H, J = 15.0 Hz), 3.07 (m,
2H), 2.64 (m, 2H), 1.50 (d, 3H, J = 6.5 Hz), 1.331.43 (m, 7H), 0.881.11 (m, 12H),
0.68 (t, 3H, J = 7.4 Hz), 0.64 (t, 3H, J = 7.4 Hz). 13C NMR (100 MHz, CDCl3): δ
(ppm) 153.0, 152.9, 143.4, 143.2, 141.5, 141.4, 140.5, 140.2, 139.99, 139.95, 139.8,
139.6, 139.5, 139.15, 139.11, 138.3, 138.1, 137.5, 137.4, 133.6, 133.5, 130.0, 129.7,
128.6, 128.5, 127.05, 127.01, 126.99, 126.95, 126.6, 126.59, 126.54, 123.8, 122.8,
119.82, 119.80, 119.4, 119.33, 119.30, 119.2, 108.9, 108.8, 93.4, 93.1, 44.9, 44.3, 43.3,
43.1, 35.1, 35.0, 32.8, 32.5, 31.8, 31.7, 28.76, 28.75, 22.66, 22.61, 20.0, 19.6, 14.19,
14.15. HRMS (ESI+): calcd for C64H61IN+ (M + H+) 970.3843 found 970.3823.

Molecular dragsters: Towards controlled translational motion on surfaces
| 63
3,9-Bis(2,7-diphenyl-9H-fluoren-9-ylidene)-4,8-dihexyl-6-(4-iodophenyl)-2,10-
dimethyl-2,3,6,9,10,11-hexahydro-1H-dicyclopenta[c,g]carbazole (2.18b).
Prepared according to the general procedure from the ketone 2.14 (200 mg, 0.30
mmol) and 2,7-diphenyldiazafluorenone (360 mg, 1.08 mmol). The product was
purified by column chromatography on silica gel (pentane : dichloromethane -
5:1 to 3:1) to give 2.18b a mixture of diastereomers (~1:1). Yield: 195 mg (52 %).
Orange solid. Mp >250 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.19
(d, 2H, J = 8.4 Hz), 8.10 (d, 2H, J = 8.2 Hz), 7.96 (s, 1H), 7.88 (dd, 2H, J1 = 7.2 Hz,
J2 = 7.2 Hz), 7.82 (m, 3H), 7.73 (dd, 4H, J1 = 7.8 Hz, J2 = 7.8 Hz ), 7.607.64 (m, 4
H), 7.287.52 (m, 18H), 7.22 (s, 1H), 7.17 (s, 1H), 4.40 (m, 2H), 3.89 (dd, 1H, J1 =
15.0 Hz, J2 = 5.7 Hz), 3.80 (dd, 1H, J1 = 15.1 Hz, J2 = 5.7 Hz), 3.45 (d, 1H, J = 15.2
Hz), 3.22 (m, 3H), 2.712.76 (m, 2H), ), 1.59 (d, 3H, J = 6.5 Hz), 1.381.45 (m, 7H),
0.891.12 (m, 12H), 0.69 (t, 3H, J = 6.9 Hz), 0.63 (t, 3H, J = 6.8 Hz). 13C NMR (100
MHz, CDCl3): δ (ppm) 153.56, 153.42, 143.66, 143.37, 142.37, 142.23, 141.87,
141.76, 141.74, 141.68, 140.92, 140.87, 140.53, 140.42, 140.11, 140.06, 139.69, 139.62,
139.36, 139.31, 139.24, 139.09, 138.71, 138.00, 137.95, 137.62, 133.65, 133.50, 129.89,
129.48, 129.10, 128.75, 128.71, 128.52, 128.46, 127.42, 127.36, 127.29, 127.27, 127.12,
127.02, 126.97, 126.1, 125.95, 125.92, 122.82, 122.78, 121.50, 121.47, 120.3, 120.2,
119.86, 119.76, 119.52, 119.3, 108.94, 108.89, 93.6, 93.1, 44.7, 44.3, 43.3, 43.2, 35.4,
35.3, 33.0, 32.8, 31.84, 31.77, 28.82, 28.81, 22.67, 22.60, 20.12, 19.7, 14.19, 14.14.
HRMS (APCI+): calcd for C88H76IN+ (M+) 1273.5011 found 1273.5017.

Molecular dragsters: Towards controlled translational motion on surfaces
64 |
General procedure for Sonogashira coupling of 2.18a-b and 2.11.
A mixture of aryl iodide 2.18a-b (1 eq.), ethyne 2.11 (1.6-1.8 eq), Pd(PPh3)2Cl2 (5
% mmol) and CuI (5 % mmol) in dry THF (5 mL) and Et3N (5 mL) was degassed
by four freeze-pump-thaw cycles. The resulting solution was stirred at room
temperature under N2 atmosphere overnight. The volatiles were evaporated at
reduced pressure and the residue was purified by column chromatography.
6-(4-((4-(2,7-Bis(trimethylsilyl)-9H-carbazol-9-yl)phenyl)ethynyl)phenyl)-3,9-
di(9H-fluoren-9-ylidene)-4,8-dihexyl-2,10-dimethyl-2,3,6,9,10,11-hexahydro-
1H-dicyclopenta[c,g]carbazole (2.5a).
Prepared according to the general procedure from 2.18a (194 mg, 0.20 mmol)
and 2.11 (154 mg, 0.37 mmol). The product was purified by column
chromatography on silica gel (pentane : dichloromethane - 7:1 to 5:1) to give
2.18a as a mixture of diastereomers (~ 1:1). Yield: 236 mg (94 %). Orange wax. 1H
NMR (400 MHz, CDCl3): δ (ppm) 8.11 (d, 2H , J = 7.6 Hz), 7.687.95 (m, 12H),
7.597.65 (m, 3H), 7.407.51 (m, 3H), 7.167.39 (m,10H), 7.08 (dd, 1H, J1 = 7.6 Hz,
J2 = 7.6 Hz), 7.02 (dd, 1H, J1 = 7.7 Hz, J2 = 7.7 Hz), 4.30 (m, 2H), 3.87 (dd, 1H, J1 =
15.3 Hz, J2 = 5.5 Hz), 3.77 (dd, 1H, J1 = 15.1 Hz, J2 = 5.6 Hz), 3.42 (d, 1H, J = 14.9
Hz), 3.20 (d, 1H, J = 15.0 Hz), 3.06 (m, 2H), 2.63 (m, 2H), 1.50 (d, 3H, J = 6.5 Hz),
1.331.43 (m, 8H), 0.881.11 (m, 12H), 0.67 (t, 3H, J = 7.4 Hz), 0.63 (t, 3H, J = 7.4
Hz), 0.30 (s, 18H). 13C NMR (100 MHz, CDCl3): δ (ppm) 153.12, 152.97, 143.50,
143.31, 141.52, 141.51, 140.61, 140.50, 140.29, 140.02, 139.98, 139.85, 139.17, 139.12,
138.79, 138.35, 138.29, 138.27, 138.20, 137.80, 137.74, 134.89, 133.73, 133.66, 133.61,

Molecular dragsters: Towards controlled translational motion on surfaces
| 65
133.53, 129.29, 128.62, 128.56, 128.15, 127.84, 127.24, 127.06, 127.01, 126.96, 126.68,
126.61, 126.55, 125.19, 124.54, 124.31, 123.90, 123.21, 122.92, 122.87, 121.96, 121.94,
120.51, 120.10, 119.83, 119.56, 119.40, 119.37, 119.33, 114.48, 109.11, 109.06, 90.36,
90.27, 89.76, 89.70, 44.97, 44.37, 43.38, 43.22, 35.13, 35.04, 32.82, 32.56, 31.83, 31.75,
29.92, 28.79, 28.77, 27.14, 22.67, 22.62, 20.08, 19.64, 14.20, 14.15, -0.57. HRMS
(ESI+): calcd for C90H88N2Si2+ (M+) 1253.6481 found 1252.6472.
6-(4-((4-(2,7-Bis(trimethylsilyl)-9H-carbazol-9-yl)phenyl)ethynyl)phenyl)-3,9-
bis(2,7-diphenyl-9H-fluoren-9-ylidene)-4,8-dihexyl-2,10-dimethyl-2,3,6,9,10,11-
hexahydro-1H-dicyclopenta[c,g]carbazole (2.5b).
Prepared according to the general procedure from 2.18b (190 mg, 0.15 mmol)
and 2.11 (98 mg, 0.24 mmol). The product was purified by column
chromatography on silica gel (pentane : dichloromethane - 5:1 to 3:1) to give
2.18b as a mixture of diastereomers (~ 1:1). Yield: 195 mg (84 %). Orange wax. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.19 (d, 2H, J = 9.9 Hz), 8.14 (d, 2H, J = 7.7
Hz), 7.978.01 (m, 3H), 7.777.93 (m, 6 H), 7.217.75 (m, 35H), 4.41 (m, 2H), 3.92
(dd, 1H, J1 = 15.0 Hz, J2 = 5.6 Hz), 3.83 (dd, 1H, J1 = 15.0 Hz, J2 = 5.5 Hz), 3.48 (d,
1H, J = 15.2 Hz), 3.22 (m, 3H), 2.74 (m, 2H), ), 1.59 (d, 3H, J = 6.6 Hz), 1.361.47
(m, 7H), 0.821.12 (m, 12H), 0.69 (t, 3H, J = 6.9 Hz), 0.63 (t, 3H, J = 6.8 Hz), 0.33
(s, 18H). 13C NMR (100 MHz, CDCl3): δ (ppm) 153.63, 153.49, 143.73, 143.43,
142.44, 142.39, 142.26, 141.89, 141.77, 141.73, 141.68, 140.94, 140.89, 140.53, 140.50,
140.42, 140.14, 140.08, 139.35, 139.30, 139.27, 139.11, 138.80, 138.72, 138.35, 138.32,
138.01, 137.95, 137.84, 137.80, 133.71, 133.68, 133.53, 129.16, 129.11, 128.95, 128.79,
128.74, 128.49, 128.45, 128.14, 128.03, 127.57, 127.44, 127.39, 127.31, 127.27, 127.16,
127.05, 126.99, 126.04, 125.95, 125.21, 124.32, 123.26, 122.98, 122.84, 122.80, 121.94,
121.52, 120.97, 120.25, 120.19, 120.11, 119.86, 119.76, 119.59, 119.38, 114.49, 109.15,
109.07, 90.54, 90.45, 89.70, 89.67, 44.63, 44.27, 43.37, 43.23, 35.39, 35.28, 33.02,
32.82, 31.87, 31.79, 29.92, 28.85, 27.14, 22.69, 22.61, 20.15, 19.73, 14.15, -0.56.
HRMS (ESI+): calcd for C114H105N2Si2+ (M + H+) 1557.7811 found 1557.7751.

Molecular dragsters: Towards controlled translational motion on surfaces
66 |
References
(1) Molecular Motors; Schliwa, M., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, FRG, 2002.
(2) Schliwa, M.; Woehlke, G. Nature 2003, 422 (6933), 759. (3) Finer, J. T.; Simmons, R. M.; Spudich, J. A. Nature 1994, 368 (6467), 113. (4) Boyer, P. D. Nature 1999, 402 (6759), 247. (5) Yoshida, M.; Muneyuki, E.; Hisabori, T. Nat. Rev. Mol. Cell Biol. 2001, 2
(9), 669. (6) van den Heuvel, M. G. L.; Dekker, C. Science 2007, 317 (5836), 333. (7) Kodera, N.; Yamamoto, D.; Ishikawa, R.; Ando, T. Nature 2010, 468
(7320), 72. (8) Lund, K.; Manzo, A. J.; Dabby, N.; Michelotti, N.; Johnson-Buck, A.;
Nangreave, J.; Taylor, S.; Pei, R.; Stojanovic, M. N.; Walter, N. G.; Winfree, E.; Yan, H. Nature 2010, 465 (7295), 206.
(9) Gu, H.; Chao, J.; Xiao, S.-J.; Seeman, N. C. Nature 2010, 465 (7295), 202. (10) Koumura, N.; Geertsema, E. M.; Meetsma, A.; Feringa, B. L. Journal of the
American Chemical Society. American Chemical Society December 2000, pp 12005–12006.
(11) Feringa, B. L.; Koumura, N.; Zijlstra, R. W. J.; van Delden, R. A.; Harada, N. Nature 1999, 401 (6749), 152.
(12) Huang, T. J.; Brough, B.; Ho, C.-M.; Liu, Y.; Flood, A. H.; Bonvallet, P. A.; Tseng, H.-R.; Stoddart, J. F.; Baller, M.; Magonov, S. Appl. Phys. Lett. 2004, 85 (22), 5391.
(13) Erbas-Cakmak, S.; Leigh, D. A.; McTernan, C. T.; Nussbaumer, A. L. Chem. Rev. 2015, 115 (18), 10081.
(14) Coskun, A.; Banaszak, M.; Astumian, R. D.; Stoddart, J. F.; Grzybowski, B. A. Chem. Soc. Rev. 2012, 41 (1), 19.
(15) Browne, W. R.; Feringa, B. L. Nat. Nanotechnol. 2006, 1 (1), 25. (16) Stoddart, J. F. Acc. Chem. Res. 2001, 34 (6), 410. (17) Balzani, V.; Gómez-López, M.; Stoddart, J. F. Acc. Chem. Res. 1998, 31 (7),
405. (18) Chen, J.; Chen, K.-Y.; Carroll, G. T.; Feringa, B. L. Chem. Commun. 2014, 50
(84), 12641. (19) Carroll, G. T.; London, G.; Landaluce, T. F.; Rudolf, P.; Feringa, B. L. ACS
Nano 2011, 5 (1), 622. (20) Chen, K.-Y.; Ivashenko, O.; Carroll, G. T.; Robertus, J.; Kistemaker, J. C.
M.; London, G.; Browne, W. R.; Rudolf, P.; Feringa, B. L. J. Am. Chem. Soc. 2014, 136 (8), 3219.
(21) Chen, K.-Y.; Wezenberg, S. J.; Carroll, G. T.; London, G.; Kistemaker, J. C. M.; Pijper, T. C.; Feringa, B. L. J. Org. Chem. 2014, 79 (15), 7032.
(22) Zheng, X.; Mulcahy, M. E.; Horinek, D.; Galeotti, F.; Magnera, T. F.;

Molecular dragsters: Towards controlled translational motion on surfaces
| 67
Michl, J. J. Am. Chem. Soc. 2004, 126 (14), 4540. (23) Griessl, S. J. H.; Lackinger, M.; Jamitzky, F.; Markert, T.; Hietschold, M.;
Heckl, W. M. J. Phys. Chem. B 2004, 108 (31), 11556. (24) Moresco, F.; Meyer, G.; Rieder, K.-H.; Tang, H.; Gourdon, A.; Joachim, C.
Appl. Phys. Lett. 2001, 78 (3), 306. (25) Shigekawa, H.; Miyake, K.; Sumaoka, J.; Harada, A.; Komiyama, M. J.
Am. Chem. Soc. 2000, 122 (22), 5411. (26) Rosei, F.; Schunack, M.; Jiang, P.; Gourdon, A.; Laegsgaard, E.;
Stensgaard, I.; Joachim, C.; Besenbacher, F. Science 2002, 296 (5566), 328. (27) Rosei, F.; Schunack, M.; Naitoh, Y.; Jiang, P.; Gourdon, A.; Laegsgaard,
E.; Stensgaard, I.; Joachim, C.; Besenbacher, F. Prog. Surf. Sci. 2003, 71 (5), 95.
(28) Grill, L.; Rieder, K.-H.; Moresco, F.; Rapenne, G.; Stojkovic, S.; Bouju, X.; Joachim, C. Nat. Nanotechnol. 2007, 2 (2), 95.
(29) Shirai, Y.; Morin, J.-F.; Sasaki, T.; Guerrero, J. M.; Tour, J. M. Chem. Soc. Rev. 2006, 35 (11), 1043.
(30) Shirai, Y.; Osgood, A. J.; Zhao, Y.; Kelly, K. F.; Tour, J. M. Nano Lett. 2005, 5 (11), 2330.
(31) Shirai, Y.; Osgood, A. J.; Zhao, Y.; Yao, Y.; Saudan, L.; Yang, H.; Yu-Hung, C.; Alemany, L. B.; Sasaki, T.; Morin, J.-F.; Guerrero, J. M.; Kelly, K. F.; Tour, J. M. J. Am. Chem. Soc. 2006, 128 (14), 4854.
(32) Crommie, M. F.; Lutz, C. P.; Eigler, D. M.; Tang, H.; Joachim, C. Science 1993, 262 (5131), 218.
(33) Gimzewski, J. K.; Joachim, C. Science 1999, 283 (5408), 1683. (34) Grill, L.; Rieder, K.-H.; Moresco, F.; Stojkovic, S.; Gourdon, A.; Joachim,
C. Nano Lett. 2006, 6 (12), 2685. (35) Chiang, P.-T.; Mielke, J.; Godoy, J.; Guerrero, J. M.; Alemany, L. B.;
Villagómez, C. J.; Saywell, A.; Grill, L.; Tour, J. M. ACS Nano 2012, 6 (1), 592.
(36) Morin, J.-F.; Shirai, Y.; Tour, J. M. Org. Lett. 2006, 8 (8), 1713. (37) Kudernac, T.; Ruangsupapichat, N.; Parschau, M.; Maciá, B.; Katsonis, N.;
Harutyunyan, S. R.; Ernst, K.-H.; Feringa, B. L. Nature 2011, 479 (7372), 208.
(38) Dierschke, F.; Grimsdale, A. C.; Müllen, K. Synthesis (Stuttg). 2003, 2003 (16), 2470.
(39) Liu, X.; Sun, Y.; Zhang, Y.; Miao, F.; Wang, G.; Zhao, H.; Yu, X.; Liu, H.; Wong, W.-Y. Org. Biomol. Chem. 2011, 9 (10), 3615.
(40) Freeman, A. W.; Urvoy, M.; Criswell, M. E. J. Org. Chem. 2005, 70 (13), 5014.
(41) Jiang, W.; Duan, L.; Qiao, J.; Zhang, D.; Dong, G.; Wang, L.; Qiu, Y. J. Mater. Chem. 2010, 20 (29), 6131.
(42) Park, M.; Buck, J. R.; Rizzo, C. J. Tetrahedron 1998, 54 (42), 12707. (43) Xiong, M. J.; Li, Z. H.; Wong, M. S. Australian Journal of Chemistry. 2007,

Molecular dragsters: Towards controlled translational motion on surfaces
68 |
pp 603–607. (44) Cho, J.-H.; Ryu, Y.-S.; Oh, S.-H.; Kwon, J.-K.; Yum, E.-K. Bull. Korean
Chem. Soc. 2011, 32 (7), 2461. (45) Lv, H.; Ma, R.; Zhang, X.; Li, M.; Wang, Y.; Wang, S.; Xing, G. Tetrahedron
2016, 72 (35), 5495. (46) Sun, M.; Zhu, L.; Kan, W.; Wei, Y.; Ma, D.; Fan, X.; Huang, W.; Xu, H. J.
Mater. Chem. C 2015, 3 (36), 9469. (47) Ma, D.; Cai, Q. Acc. Chem. Res. 2008, 41 (11), 1450. (48) Zhang, H.; Cai, Q.; Ma, D. J. Org. Chem. 2005, 70 (13), 5164. (49) Ma, D.; Cai, Q.; Zhang, H. Org. Lett. 2003, 5 (14), 2453. (50) Zheng, Z.; Dong, Q.; Gou, L.; Su, J.-H.; Huang, J. J. Mater. Chem. C 2014, 2
(46), 9858. (51) Greidanus, J. W. Can. J. Chem. 1970, 48 (22), 3530. (52) Ishii, A.; Nakayama, J.; Ding, M. X.; Kotaka, N.; Hoshino, M. J. Org. Chem.
1990, 55 (8), 2421. (53) Koumura, N.; Geertsema, E. M.; van Gelder, M. B.; Meetsma, A.; Feringa,
B. L. J. Am. Chem. Soc. 2002, 124 (18), 5037. (54) Bauer, J.; Hou, L.; Kistemaker, J. C. M.; Feringa, B. L. J. Org. Chem. 2014,
79 (10), 4446.