using large-scale genomic data sets to understand the ... · using large-scale genomic data sets to...
TRANSCRIPT

Usinglarge-scalegenomicdatasetstounderstandtheimpactofhuman
geneticvariation
DanielMacArthurMassachusettsGeneralHospital
BroadInstituteofMITandHarvardHarvardMedicalSchoolTwitter:@dgmacarthur

Makingsenseofonegenomerequiresplacingitinapopulationcontext
vs
• more than one million genomes and exomes have been sequenced worldwide
• …yet many are inaccessible for ethical, political and technical reasons

ExomeAggregationConsortium(ExAC):aggregatingandcalling92,000exomes
Subset of 60,706 “reference” samples:• high-quality exomes• unrelated individuals• consent for public data sharing• free of known severe pediatric disease
All data reprocessed
with BWA/Picard
Joint calling across all samples
with GATK 3 Haplotype
Caller

Laramie Duncan
1000 Genomes ESP ExAC
010000
20000
30000
40000
50000
60000
Sample Size (N) and Ancestral Diversity1000 Genomes, ESP, ExAC
Indi
vidu
als
with
Exo
me
Seq
uenc
e D
ata East Asian
South AsianEuropeanMiddle EasternAfricanNative American ancestryDiverse Other
World proportions
World PopulationScaled to ExAC height
1000 Genomes
ESP ExAC World
Biggerandmorediverse
LatinoAfricanEuropeanSouth AsianEast AsianOther

Catalogueofprotein-codingvariation• Largestevercollectionofhumanprotein-codinggeneticvariants– over10millionvariants:onevariantevery
6basepairs;mostarerareandnovel
singleton 2-10 alleles 0.01% 0.1% 1% ≥10%
0%
10%
20%
30%
40%
50%
60%
ExAC global allele frequency
Prop
ortio
n of
ExA
C a
llele
s
novelother dbSNPin 1kG or ESP

Publicdatarelease• Allvariantsandpopulationfrequenciesarepubliclyavailable:
exac.broadinstitute.org
KonradKarczewski
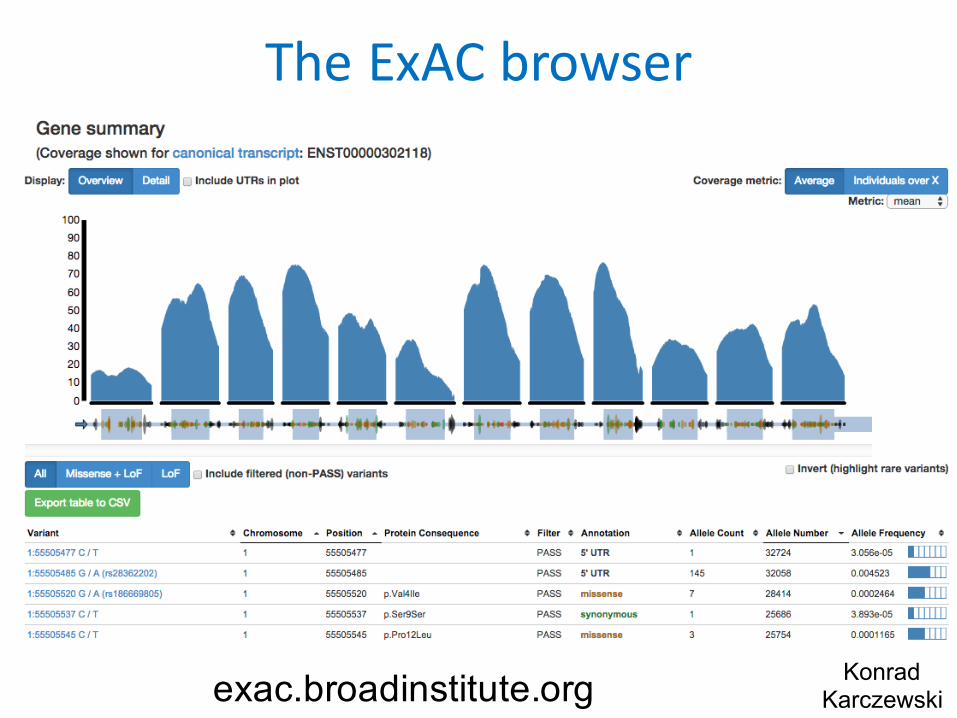
TheExAC browser
KonradKarczewskiexac.broadinstitute.org

Caveats
• ExACsampleinclusionisopportunistic:mostsampleshavelimitedphenotypedata,aren’tconsentedforrecontact
• Severepediatric diseasecasesaredepletedfromthedataset,butnotabsent

HowExACimprovesVUSanalysis
1. Filteringvariantsthataretoocommontobecausal
2. Comparisonswithlargecaseseriestoassesspenetrance
3. Identifyinggenes(orregions)thataredepleted forspecificclassesofvariation

Valueforrarediseasefiltering
• #variantsremaininginanexomeafterapplyinga0.1%filteracrossallpopulations
• Bothsizeandancestraldiversityincreasefilteringpower
Eric MinikelESP ExAC
East AsianSouth AsianLatinoAfricanEuropean
# va
riant
s re
mai
ning
afte
r filt
erin
g

• Manuallyreviewedsupportfor197reportedpathogenicvariantsat>1%inatleastoneExACpopulation– effectivelyallarespuriousclaims
• Therearehundreds ofExACindividualscarryingvariantsreportedtocausesevere,dominantpediatric disease
Applicationtoreportedpathogenicvariants
Anne O’Donnell Luria

UsingExACtoassesspenetrance• What explains these alleged dominant
disease variants in “healthy” people?– False positive assertions of pathogenicity– Undiagnosed disease– Somatic mosaicism– Incomplete penetrance?
• Prion diseases as a model– Severe, fatal adult diseases, lifetime risk ~1/10,000– 15% of cases are genetic, due to >60 known
dominant gain-of-function variants in PRNP– Virtually every case has PRNP sequenced– These mutations as a class are >30x more common
in ExAC than they should be given disease incidence!
Eric VallabhMinikel
Science Translational Medicine 8:322ra9 (2016)

Allelecountsin10,460sequencedcasesversus60,706controls
Cases reported to prion surveillance centers
Alle
le c
ount
in E
xAC
popu
latio
n co
ntro
ls
●
●●●●●●●●●●●●● ●●●●● ●●
●
●●●
●
●●●●●● ●
●
●●●●
●
●
●
●●●● ●●
●
●
●
●
●
●●
●
●●●●●
●
●●
0 100 200 300 400 500 600
0
1
2
3
4
5
6
7
8
9
10
P39L
P102LA117V
R148H
D178N
V180I
T188R
E196A
E200K
V203I
R208C
R208H
V210I
M232R ● reportedly pathogenic variantsegregation in multiple multigenerational families;spontaneous disease in mouse models
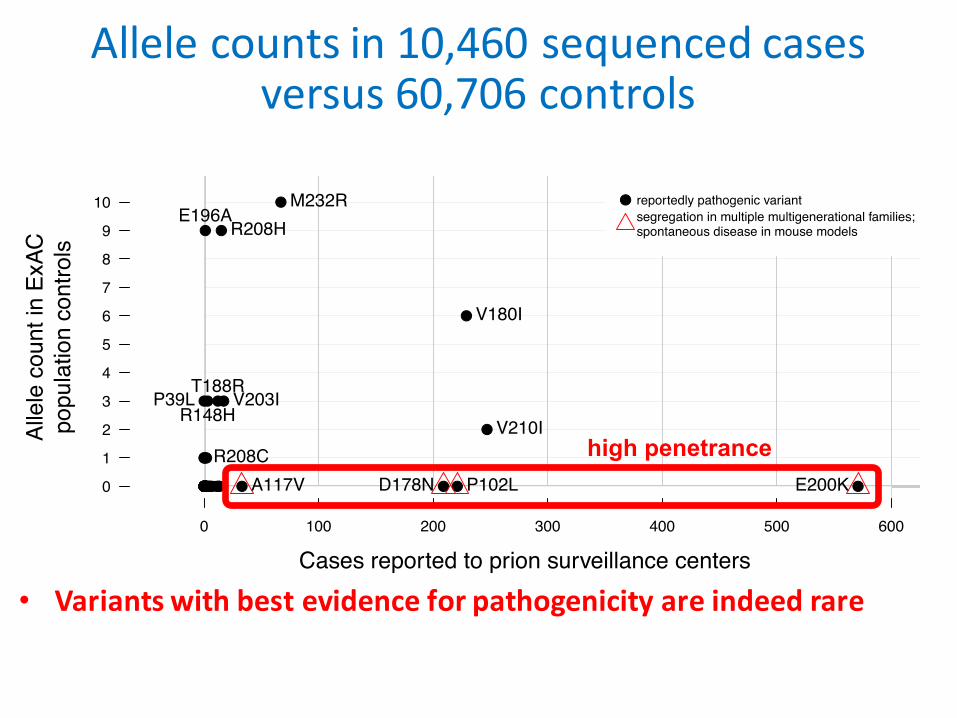
Allelecountsin10,460sequencedcasesversus60,706controls
• VariantswithbestevidenceforpathogenicityareindeedrareCases reported to prion surveillance centers
Alle
le c
ount
in E
xAC
popu
latio
n co
ntro
ls
●
●●●●●●●●●●●●● ●●●●● ●●
●
●●●
●
●●●●●● ●
●
●●●●
●
●
●
●●●● ●●
●
●
●
●
●
●●
●
●●●●●
●
●●
0 100 200 300 400 500 600
0
1
2
3
4
5
6
7
8
9
10
P39L
P102LA117V
R148H
D178N
V180I
T188R
E196A
E200K
V203I
R208C
R208H
V210I
M232R ● reportedly pathogenic variantsegregation in multiple multigenerational families;spontaneous disease in mouse models
high penetrance

Allelecountsin10,460sequencedcasesversus60,706controls
• Variantswithbestevidenceforpathogenicityareindeedrare• Variantsrareincases,commonincontrolsmaybebenign
Cases reported to prion surveillance centers
Alle
le c
ount
in E
xAC
popu
latio
n co
ntro
ls
●
●●●●●●●●●●●●● ●●●●● ●●
●
●●●
●
●●●●●● ●
●
●●●●
●
●
●
●●●● ●●
●
●
●
●
●
●●
●
●●●●●
●
●●
0 100 200 300 400 500 600
0
1
2
3
4
5
6
7
8
9
10
P39L
P102LA117V
R148H
D178N
V180I
T188R
E196A
E200K
V203I
R208C
R208H
V210I
M232R ● reportedly pathogenic variantsegregation in multiple multigenerational families;spontaneous disease in mouse models
high penetrance
benign or low risk
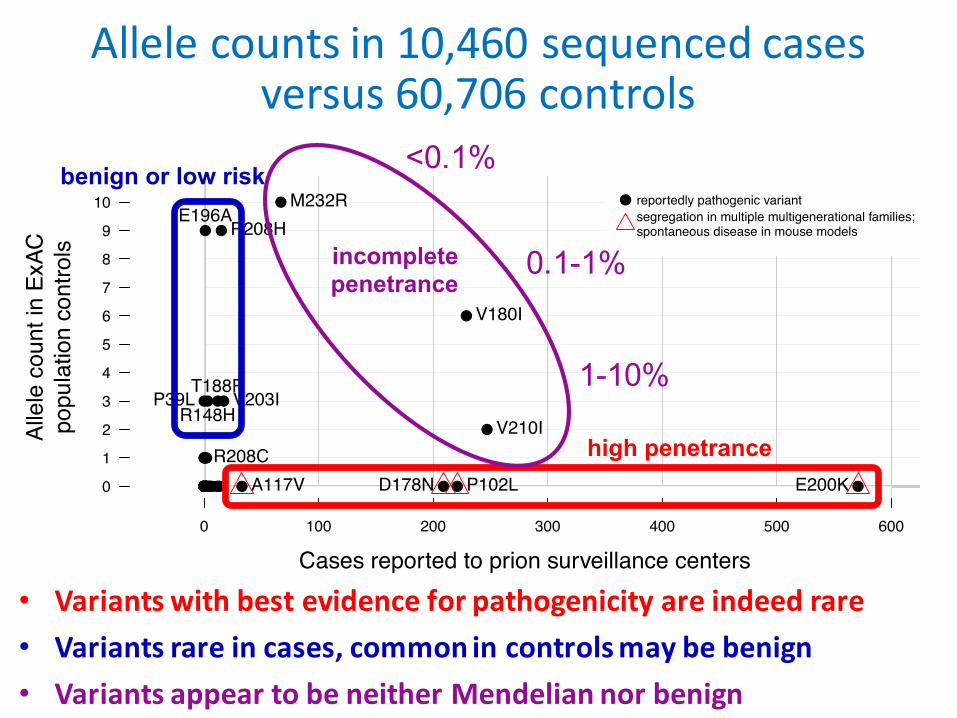
Allelecountsin10,460sequencedcasesversus60,706controls
• Variantswithbestevidenceforpathogenicityareindeedrare• Variantsrareincases,commonincontrolsmaybebenign• VariantsappeartobeneitherMendeliannorbenign
Cases reported to prion surveillance centers
Alle
le c
ount
in E
xAC
popu
latio
n co
ntro
ls
●
●●●●●●●●●●●●● ●●●●● ●●
●
●●●
●
●●●●●● ●
●
●●●●
●
●
●
●●●● ●●
●
●
●
●
●
●●
●
●●●●●
●
●●
0 100 200 300 400 500 600
0
1
2
3
4
5
6
7
8
9
10
P39L
P102LA117V
R148H
D178N
V180I
T188R
E196A
E200K
V203I
R208C
R208H
V210I
M232R ● reportedly pathogenic variantsegregation in multiple multigenerational families;spontaneous disease in mouse models
high penetrance
benign or low risk
incompletepenetrance
<0.1%
0.1-1%
1-10%

Identifyinggeneswithsignificantdepletionofvariation
• Usingamutationalmodelwecanpredictthenumberofvariantsinagivenfunctionalclassweshouldexpecttoseeineachgene inagivennumberofpeople(Samocha etal.2014NatGenet 46:944–950)
Obs
erve
d
Expected
Synonymous Missense Loss-of-function
0 500 1000 1500
050
010
0015
00
Expected Number of Synonymous Variants
Obs
erve
d N
umbe
r of S
ynon
ymou
s Va
riant
s
R = 0.9778
0 500 1000 1500 2000 2500 3000
050
010
0015
0020
0025
0030
00
Expected Number of Missense Variants
Obs
erve
d N
umbe
r of M
isse
nse
Varia
nts
R = 0.9482
0 50 100 150 200 250 300 350
050
100
150
200
250
300
350
Expected Number of Loss−of−Function Variants
Obs
erve
d N
umbe
r of L
oss−
of−F
unct
ion
Varia
nts
R = 0.5866
Expected Expected
Kaitlin Samocha

Empiricalidentificationofgenessubjecttostronghumanconstraint
0.97
0.33
0.03
1.02
0.55
0.01
• Overall we discover 2,651 genes with a high probability of LoF-intolerance (pLI > 0.95)
• Contains almost all known HI genes, but >75% have no known disease phenotype

Movingbeyondthegene:identifyingregionalmissenseconstraint
1 100 200 300 400 500 600 700 800 900 1000amino acid
protein kinase domain ATP binding site serine/threonine kinase domain signal peptidase serine active site
******** ***
*******
******
*******
******** *** *
constrained
• CDKL5mutationscausesevereinfantileseizures• ConstraintwouldallowustocorrectlyprioritizetheN-terminalregion,evenwithnopriorcausalmutations
• Overall,81% ofClinVar missenseinsevereHIgenesfallwithinthe14% mostconstrainedsequence

Whatnext?• Moresamples:willhavedatafrom~120Kexomesinnextrelease
• Movingtogenomes: testrunon5,500genomes;aimingfor20Kthisyear
• Genotype-basedrecall:movingfromgenotypetophenotypeinasubsetofExACsamples+othercohorts

What’sneeded?• Bigger,bettersamples:harmonized,centralizedrepositoriesofvariantslinkedethicallytophenotypes
• Regulatorysupportfordataaggregationandreuse – commondiseasesamplesaregreatcontrolsforrare
• Increasedfocusonsequencingsamplesconsentedforrecontact,deeperphenotypinganddatasharing
• Large,uniformlyascertainedcaseseriesforrarediseases toassesspenetrance

Daniel MacArthurDavid AltshulerDiego ArdissinoMichael BoehnkeMark DalyJohn DaneshRoberto ElosuaGad GetzChristina HultmanSekar KathiresanMarkku LaaksoSteven McCarroll
Mark McCarthyRuth McPhersonBenjamin NealeAarno PalotieShaun PurcellDanish SaleheenJeremiah ScharfPamela SklarPatrick SullivanJaakko TuomilehtoHugh WatkinsJames Wilson
ExAC Principal Investigators
Monkol LekEric MinikelKonrad KarczewskiAnne O’Donnell LuriaAndrew HillJames WareBeryl CummingsTaru TukiainenKarol EstradaDaniel BirnbaumBen WeisburdBrett ThomasIrina Armean
My Lab
Kaitlin SamochaLaramie DuncanMark DalyBen Neale
ATGU
exac.broadinstitute.orgBroad Genomics and Data Sciences Platforms
Menachem FromerDoug Ruderfer
ExAC analystsEric BanksTim FennellKathleen TibbettsNamrata GuptaStacey Donnelly
Broad Institute