uv-induced dna damage and repairbio.classes.ucsc.edu/bio105l/exercises/uv mutants/notes.pdf ·...
TRANSCRIPT

3/31/11
UV-Induced DNA Damage and Repair Bacteriologists discovered in the 19th Century that direct sunlight exposure was lethal to bacteria and other microorganisms. Subsequent studies showed the lethal action of sunlight to be primarily attributable to the UV portion of the spectrum between 250-260 nm. This corresponds to the Amax for the DNA bases (whereas the Amax for proteins is near 280 nm). UV irradiation produced by (germicidal lamps) is has been used a method for disinfection since the early 20th Century. UV absorbance (A260) is a common and simple assay for [DNA] in relatively pure samples (50 ug/ml = A = 1.0). A260 increases 40% in ss relative to ds DNA (hyperchromic shift). UV mutagenicity (as opposed to lethality) for bacteria was demonstrated in 1914 by V. Henri, 13 years before Muller’s demonstration of X-ray mutagenesis in Drosophila. Henri's discovery was not followed up until the advent of bacteriophage genetics in the 1940’s. Then, Demerec confirmed UV mutagenesis in E. coli by demonstrating a 103X enrichment of bacteriophage T1-resistant mutants in a population exposed to UV. DNA Damage Caused by Short Wave UV UV-induced DNA lesions primarily involve covalent crosslinking of two adjacent pyrimidines in the same strand. The best known, and most common photoproducts are cyclopyrimidine dimers (CPD’s) involving adjacent thymines (i.e. the so-called "thymine dimer"). Another photoproduct is a 6,4 linkage of adjacent pyrimidines.
3/31/11
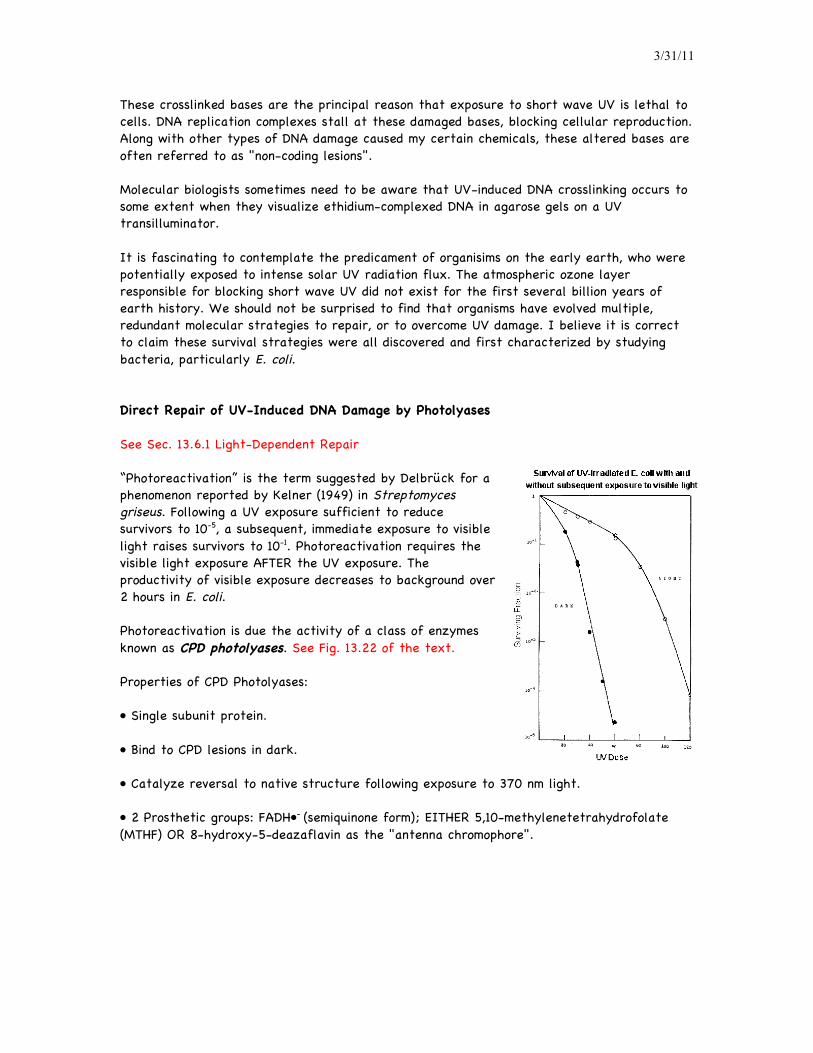
These crosslinked bases are the principal reason that exposure to short wave UV is lethal to cells. DNA replication complexes stall at these damaged bases, blocking cellular reproduction. Along with other types of DNA damage caused my certain chemicals, these altered bases are often referred to as "non-coding lesions". Molecular biologists sometimes need to be aware that UV-induced DNA crosslinking occurs to some extent when they visualize ethidium-complexed DNA in agarose gels on a UV transilluminator. It is fascinating to contemplate the predicament of organisims on the early earth, who were potentially exposed to intense solar UV radiation flux. The atmospheric ozone layer responsible for blocking short wave UV did not exist for the first several billion years of earth history. We should not be surprised to find that organisms have evolved multiple, redundant molecular strategies to repair, or to overcome UV damage. I believe it is correct to claim these survival strategies were all discovered and first characterized by studying bacteria, particularly E. coli. Direct Repair of UV-Induced DNA Damage by Photolyases See Sec. 13.6.1 Light-Dependent Repair “Photoreactivation” is the term suggested by Delbrück for a phenomenon reported by Kelner (1949) in Streptomyces griseus. Following a UV exposure sufficient to reduce survivors to 10-5, a subsequent, immediate exposure to visible light raises survivors to 10-1. Photoreactivation requires the visible light exposure AFTER the UV exposure. The productivity of visible exposure decreases to background over 2 hours in E. coli. Photoreactivation is due the activity of a class of enzymes known as CPD photolyases. See Fig. 13.22 of the text. Properties of CPD Photolyases:
• Single subunit protein. • Bind to CPD lesions in dark. • Catalyze reversal to native structure following exposure to 370 nm light. • 2 Prosthetic groups: FADH•- (semiquinone form); EITHER 5,10-methylenetetrahydrofolate (MTHF) OR 8-hydroxy-5-deazaflavin as the "antenna chromophore".

3/31/11
Nucleotide Excision Repair (see Fig. 13.24 of the text) Many versions of DNA nucleotide excision repair (NER) mechanisms are known, with specificity for various types of lesion. Excision repair of TT dimers in E. coli was the first such mechanism characterized.
The UvrABC "exinuclease" complex binds to DNA specifically in the vicinity of pyrimidine dimers and makes 2 single strand cuts in the dimer-containing strand on either side of the lesion. With the aid of UvrD (helicase II), a short fragment containing the dimer is released. The resulting gap in the DNA helix is repaired by a generic scenario involving DNA Polymerase I and DNA ligase. NER of pyrimidine dimers in E. coli is accomplished by the sequential activity of 7 proteins. First, a UvrA-UvrB complex scans the DNA, with the UvrA subunit recognizing distortions in the helix caused by pyrimidine dimers. When the complex recognizes such a distortion, the UvrA subunit leaves and is replaced by a UvrC protein. UvrB cleaves a phosphodiester bond in the dimer-containing strand 4 nucleotides on the 3' side of the DNA damage, and UvrC cleaves a phosphodiester bond 8 nucleotides on the 5' side. Next, DNA helicase II (sometimes called UvrD) removes the dimer-containing fragment by actively breaking the hydrogen bonds
3/31/11
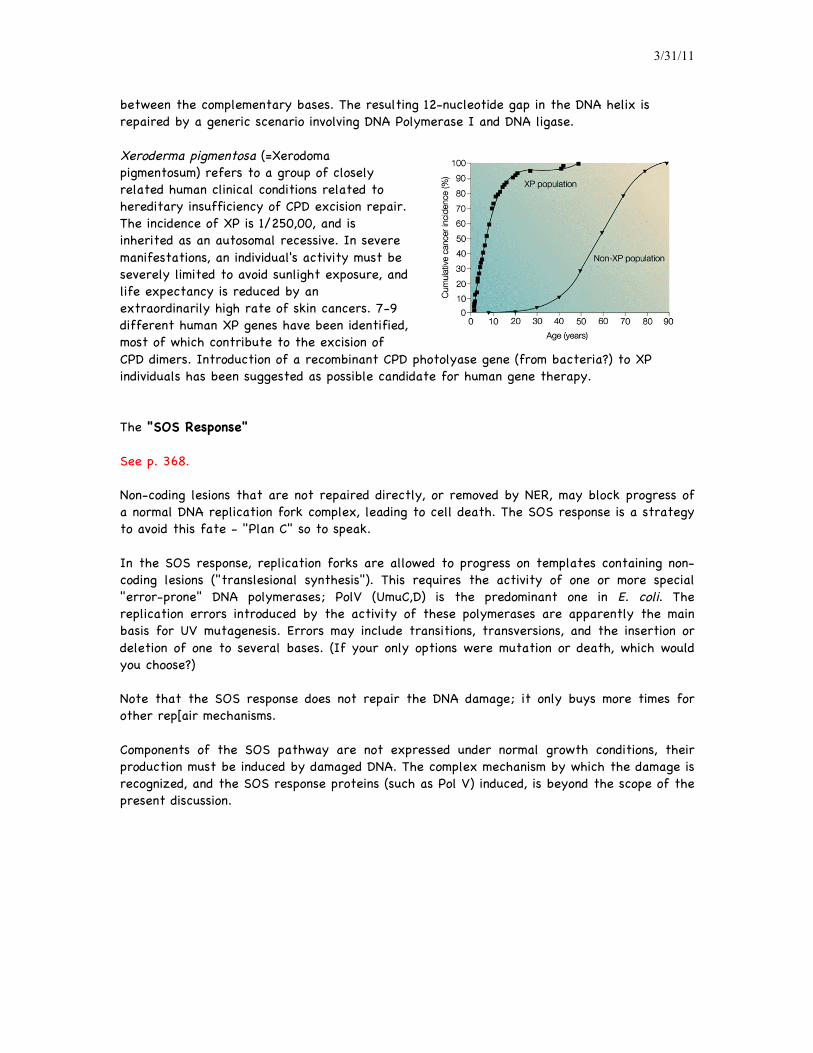
between the complementary bases. The resulting 12-nucleotide gap in the DNA helix is repaired by a generic scenario involving DNA Polymerase I and DNA ligase. Xeroderma pigmentosa (=Xerodoma pigmentosum) refers to a group of closely related human clinical conditions related to hereditary insufficiency of CPD excision repair. The incidence of XP is 1/250,00, and is inherited as an autosomal recessive. In severe manifestations, an individual's activity must be severely limited to avoid sunlight exposure, and life expectancy is reduced by an extraordinarily high rate of skin cancers. 7-9 different human XP genes have been identified, most of which contribute to the excision of CPD dimers. Introduction of a recombinant CPD photolyase gene (from bacteria?) to XP individuals has been suggested as possible candidate for human gene therapy.
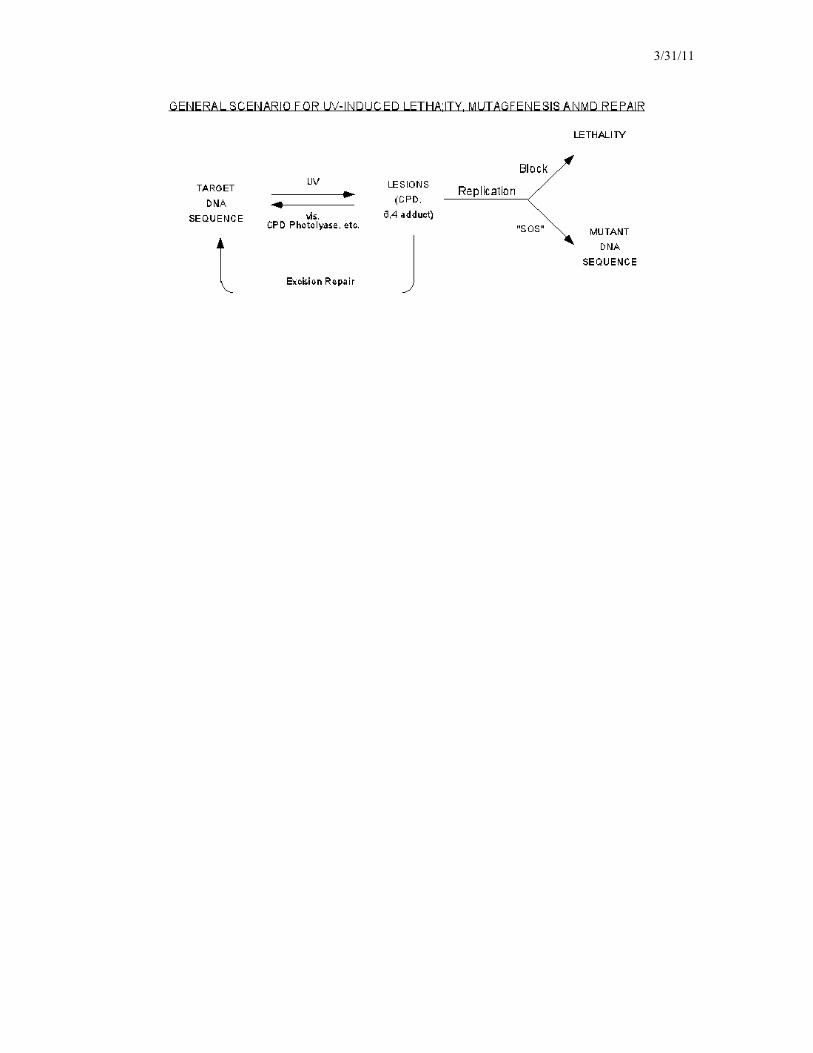
The "SOS Response" See p. 368. Non-coding lesions that are not repaired directly, or removed by NER, may block progress of a normal DNA replication fork complex, leading to cell death. The SOS response is a strategy to avoid this fate - "Plan C" so to speak. In the SOS response, replication forks are allowed to progress on templates containing non-coding lesions ("translesional synthesis"). This requires the activity of one or more special "error-prone" DNA polymerases; PolV (UmuC,D) is the predominant one in E. coli. The replication errors introduced by the activity of these polymerases are apparently the main basis for UV mutagenesis. Errors may include transitions, transversions, and the insertion or deletion of one to several bases. (If your only options were mutation or death, which would you choose?) Note that the SOS response does not repair the DNA damage; it only buys more times for other rep[air mechanisms. Components of the SOS pathway are not expressed under normal growth conditions, their production must be induced by damaged DNA. The complex mechanism by which the damage is recognized, and the SOS response proteins (such as Pol V) induced, is beyond the scope of the present discussion.
3/31/11