v9 from protein complexes to networks and back
DESCRIPTION
V9 From Protein Complexes to Networks and back. Protein interaction could be defined in a number of ways (1) Proteins that form permanent supracomplexes = „protein machines“ (2) Proteins that bind each other transiently (signal transduction, bioenergetics ... ) - PowerPoint PPT PresentationTRANSCRIPT

9. Lecture WS 2006/07
Softwarewerkzeuge 1
V9 From Protein Complexes to Networks and back
Protein interaction could be defined in a number of ways
(1) Proteins that form permanent supracomplexes = „protein machines“
(2) Proteins that bind each other transiently
(signal transduction, bioenergetics ... )
(3) Co-regulated expression of genes/proteins
(4) Proteins participating in the same metabolic pathways
(5) Proteins sharing substrates
(6) Proteins that are co-localized
Techniques: Experimental methods + computational methods.

9. Lecture WS 2006/07
Softwarewerkzeuge 2
1 Protein-Protein Complexes
It has been realized for quite some time that cells don‘t work by random
diffusion of proteins,
but require a delicate structural organization into large protein complexes.
Which complexes do we know?

9. Lecture WS 2006/07
Softwarewerkzeuge 3
RNA Polymerase II
RNA polymerase II is the
central enzyme of gene
expression and synthesizes all
messenger RNA in
eukaryotes.
Cramer et al., Science 288, 640 (2000)

9. Lecture WS 2006/07
Softwarewerkzeuge 4
RNA processing: splicesome
Structure of a cellular editor that "cuts and pastes" the first draft of RNA straight
after it is formed from its DNA template. It has two distinct, unequal halves
surrounding a tunnel. The larger part appears to contain proteins and the short
segments of RNA, while the smaller half is made up of proteins alone. On one
side, the tunnel opens up into a cavity, which the researchers think functions as
a holding space for the fragile RNA waiting to be processed in the tunnel itself.
Profs. Ruth and Joseph Sperlinghttp://www.weizmann.ac.il/

9. Lecture WS 2006/07
Softwarewerkzeuge 5
Protein synthesis: ribosome
The ribosome is a complex
subcellular particle composed of
protein and RNA. It is the site of
protein synthesis,
http://www.millerandlevine.com/chapter/12/cryo-em.html
Model of a ribosome with a
newly manufactured protein
(multicolored beads) exiting
on the right.

9. Lecture WS 2006/07
Softwarewerkzeuge 6
Signal recognition particle
40S small ribosomal subunit
(yellow) 60S large ribosomal
subunit (blue), P-site tRNA
(green), SRP (red).
Halic et al. Nature 427, 808 (2004)
Cotranslational translocation of proteins across or into membranes is a vital process in all kingdoms of life. It requires that the translating ribosome be targeted to the membrane by the signal recognition particle (SRP), an evolutionarily conserved ribonucleoprotein particle. SRP recognizes signal sequences of nascent protein chains emerging from the ribosome. Subsequent binding of SRP leads to a pause in peptide elongation and to the ribosome docking to the membrane-bound SRP receptor. SRP shows 3 main activities in the process of cotranslational targeting: first, it binds to signal sequences emerging from the translating ribosome; second, it pauses peptide elongation; and third, it promotes protein translocation by docking to the membrane-bound SRP receptor and transferring the ribosome nascent chain complex (RNC) to the protein-conducting channel.

9. Lecture WS 2006/07
Softwarewerkzeuge 7
Nuclear Pore ComplexA three-dimensional image of the
nuclear pore complex (NPC),
revealed by electron microscopy.
A-B The NPC in yeast.
Figure A shows the NPC seen
from the cytoplasm while figure B
displays a side view.
C-D The NPC in vertebrate
(Xenopus).
http://www.nobel.se/medicine/educational/dna/a/transport/ncp_em1.htmlThree-Dimensional Architecture of the Isolated Yeast Nuclear Pore Complex: Functional and Evolutionary Implications, Qing Yang, Michael P. Rout and Christopher W. Akey. Molecular Cell, 1:223-234, 1998
NPC is a 50-100 MDa protein assembly that
regulates and controls trafficking of
macromolecules through the nuclear envelope.
9. Lecture WS 2006/07
Softwarewerkzeuge 8
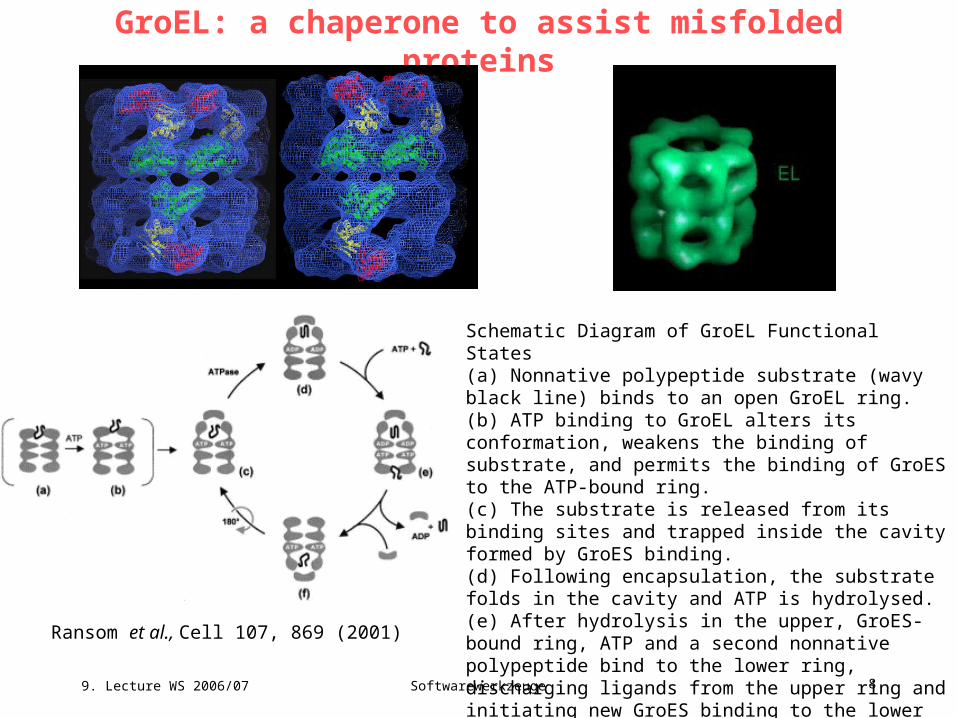
GroEL: a chaperone to assist misfolded proteins
Schematic Diagram of GroEL Functional States(a) Nonnative polypeptide substrate (wavy black line) binds to an open GroEL ring. (b) ATP binding to GroEL alters its conformation, weakens the binding of substrate, and permits the binding of GroES to the ATP-bound ring. (c) The substrate is released from its binding sites and trapped inside the cavity formed by GroES binding. (d) Following encapsulation, the substrate folds in the cavity and ATP is hydrolysed. (e) After hydrolysis in the upper, GroES-bound ring, ATP and a second nonnative polypeptide bind to the lower ring, discharging ligands from the upper ring and initiating new GroES binding to the lower ring (f) to form a new folding active complex on the lower ring and complete the cycle.
http://people.cryst.bbk.ac.uk/~ubcg16z/chaperone.html
Ransom et al., Cell 107, 869 (2001)

9. Lecture WS 2006/07
Softwarewerkzeuge 9
proteasome
The proteasome is the central
enzyme of non-lysosomal protein
degradation. It is involved in the
degradation of misfolded proteins
as well as in the degradation and
processing of short lived regulatory
proteins.The 20S Proteasome
degrades completely unfoleded
proteins into peptides with a
narrow length distribution of 7 to
13 amino acids.
http://www.biochem.mpg.de/xray/projects/hubome/images/rpr.gifLöwe, J., Stock, D., Jap, B., Zwickl, P., Baumeister, W. and Huber, R. (1995). Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science 268, 533-539.
9. Lecture WS 2006/07
Softwarewerkzeuge 10
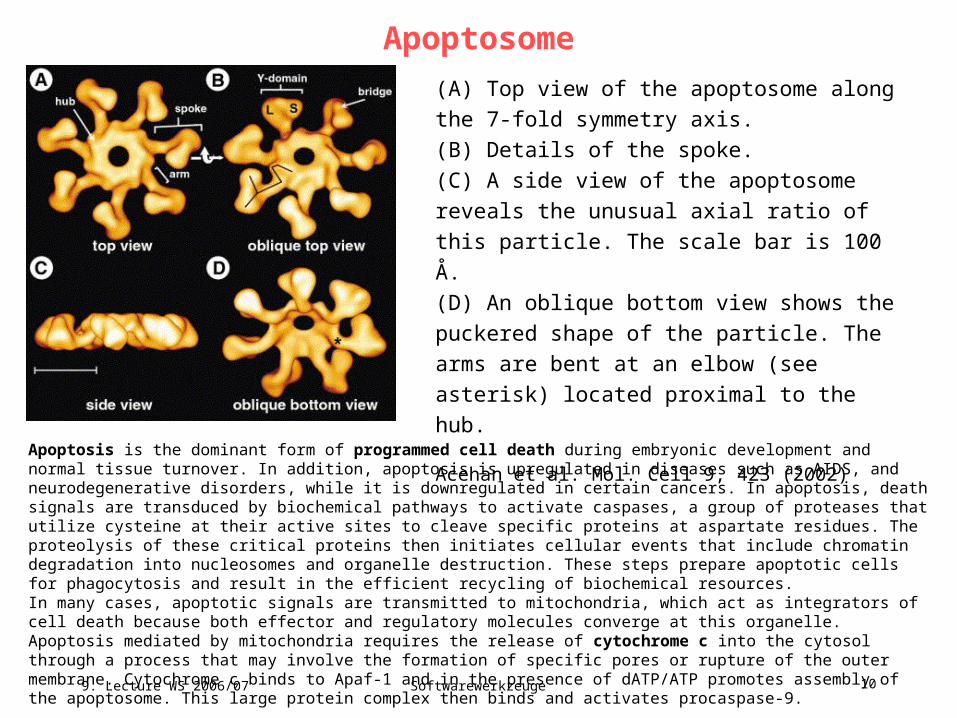
Apoptosome
(A) Top view of the apoptosome along the 7-fold
symmetry axis.
(B) Details of the spoke.
(C) A side view of the apoptosome reveals the
unusual axial ratio of this particle. The scale bar is
100 Å.
(D) An oblique bottom view shows the puckered
shape of the particle. The arms are bent at an
elbow (see asterisk) located proximal to the hub.
Acehan et al. Mol. Cell 9, 423 (2002)
Apoptosis is the dominant form of programmed cell death during embryonic development and normal tissue turnover. In addition, apoptosis is upregulated in diseases such as AIDS, and neurodegenerative disorders, while it is downregulated in certain cancers. In apoptosis, death signals are transduced by biochemical pathways to activate caspases, a group of proteases that utilize cysteine at their active sites to cleave specific proteins at aspartate residues. The proteolysis of these critical proteins then initiates cellular events that include chromatin degradation into nucleosomes and organelle destruction. These steps prepare apoptotic cells for phagocytosis and result in the efficient recycling of biochemical resources.In many cases, apoptotic signals are transmitted to mitochondria, which act as integrators of cell death because both effector and regulatory molecules converge at this organelle. Apoptosis mediated by mitochondria requires the release of cytochrome c into the cytosol through a process that may involve the formation of specific pores or rupture of the outer membrane. Cytochrome c binds to Apaf-1 and in the presence of dATP/ATP promotes assembly of the apoptosome. This large protein complex then binds and activates procaspase-9.

9. Lecture WS 2006/07
Softwarewerkzeuge 11
Experiment
Start from 232 purified complexes from Tandem Affinity Purification (TAP) strategy.
Select 102 that gave samples most promising for EM from analysis of gels and
protein concentrations.
Take EM images.
Theory
Make list of components.
Assign known structures of individual proteins.
Assign templates of complexes-If complex structure available for this pair- if complex structure available for homologous protein- if complex structure available for structurally similar protein (SCOP)
2 Aim: generate structures of protein complexes
Bettina Böttcher (EM)Rob Russell (Bioinformatics)

9. Lecture WS 2006/07
Softwarewerkzeuge 12
How transferable are interactions?interaction similariy (iRMSD) vs. %
sequence identity for all the available
pairs of interacting domains with
known 3D structure.
Curve shows 80% percentile (i.e. 80%
of the data lies below the curve), and
points below the line (iRMSD = 10 Å)
are similar in interaction. Aloy et al. Science, 303, 2026 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 13
Bioinformatics Strategy
Illustration of the methods and concepts
used. How predictions are made within
complexes (circles) and between them
(cross-talk). Bottom right shows two
binary interactions combined into a three-
component model
Aloy et al. Science, 303, 2026 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 14
Successful models of yeast complexes
(A) Exosome model on PNPase fit into
EM map.
(B) RNA polymerase II with RPB4
(green)/RPB7 (red) built on
Methanococcus jannaschii equivalents,
and SPT5/pol II (cyan) built with IF5A.
(C and D) Views of CCT (gold) and
phosphoducin 2/VID27 (red) fit into EM
map.
(E) Micrograph of POP complex, with
particle types highlighted.
(F) Ski complex built by combination of
two complexes.
Aloy et al. Science, 303, 2026 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 15
Future?
Structural genomics (X-ray) may soon generate enough templates of individal
folds.
Structural genomics may be expanded to protein complexes.
Interactions between proteins of the same fold tend to be similar when the
sequence identity is above approximately 30% (Aloy et al.).
Hybrid modelling of X-ray/EM will not be able to answer all questions- problem of induced fit- transient complexes cannot be addressed by these techniques
Essential to combine large variety of hybrid + complementary methods
Russell et al. Curr. Opin. Struct. Biol. 14, 313 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 16
3 Bioinformatic identification of interface patches
Statistical analysis of protein-protein interfaces in crystal structures of
protein-protein complexes: residues at interfaces have significantly different
amino acid composition that the rest of the protein.
predict protein-protein interaction sites from local sequence information ?
Conservation at protein-protein interfaces: interface regions are more conserved
than other regions on the protein surface
identify conserved regions on protein surface e.g. from solvent accessibility
Patterns in multiple sequence alignments: Interacting residues on two binding partners
often show correlated mutations (among different organisms) if being mutated
identify correlated mutations
Structural patterns: surface patterns of protein-protein interfaces: interface often
formed by hydrophobic patch surrounded by ring of polar or charged residues.
identify suitable patches on surface if 3D structure is known

9. Lecture WS 2006/07
Softwarewerkzeuge 17
4 NOXClass: Distinguish Permanent / Transient Complexes
Aim:
(1) distinguish different types of biological interactions (X-ray structures of protein-
protein complexes).
(2) develop automatic classification scheme.
Zhu, Domingues, Sommer, Lengauer, BMC Bioinformatics 7, 27 (2006),

9. Lecture WS 2006/07
Softwarewerkzeuge 18
Dataset

9. Lecture WS 2006/07
Softwarewerkzeuge 19
Interface properties considered
Zhu, Domingues, Sommer, Lengauer, BMC Bioinformatics 7, 27 (2006),
9. Lecture WS 2006/07
Softwarewerkzeuge 20
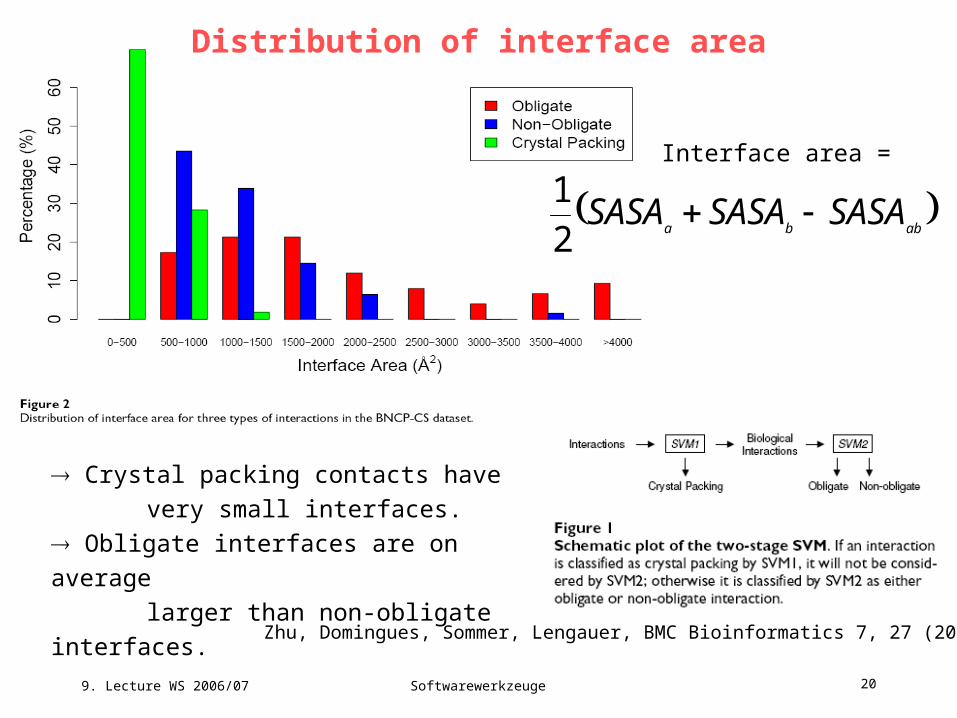
Distribution of interface area
Crystal packing contacts have
very small interfaces.
Obligate interfaces are on average
larger than non-obligate interfaces.
Interface area =
abba
SASASASASASA 2
1
Zhu, Domingues, Sommer, Lengauer, BMC Bioinformatics 7, 27 (2006),

9. Lecture WS 2006/07
Softwarewerkzeuge 21
Dataset
ba
SASASASA ,min
Area InterfaceRatio Area Interface
The distributions of obligate and non-obligate interfaces are quite similar, but
very different from crystal packing contacts.
Zhu, Domingues, Sommer, Lengauer, BMC Bioinformatics 7, 27 (2006),

9. Lecture WS 2006/07
Softwarewerkzeuge 22
Hydrophobic residues (FLIV) contribute twice as much to obligate interfaces as
to crystal packing contacts.
Aromatic residues (FWY) tend to be more abundant in biological interfaces.Zhu, Domingues, Sommer, Lengauer, BMC Bioinformatics 7, 27 (2006),

9. Lecture WS 2006/07
Softwarewerkzeuge 23
Good Performance
Zhu, Domingues, Sommer, Lengauer, BMC Bioinformatics 7, 27 (2006),

9. Lecture WS 2006/07
Softwarewerkzeuge 24
5 Correlated mutations at interface
Pazos, Helmer-Citterich, Ausiello, Valencia J Mol Biol 271, 511 (1997):
correlation information is sufficient for selecting the correct structural arrangement of
known heterodimers and protein domains because the correlated pairs between the
monomers tend to accumulate at the contact interface.
Use same idea to identify interacting protein pairs.

9. Lecture WS 2006/07
Softwarewerkzeuge 25
Correlated mutations at interface
Correlated mutations evaluate the similarity in variation patterns between positions in
a multiple sequence alignment.
Similarity of those variation patterns is thought to be related to compensatory
mutations.
Calculate for each positions i and j in the sequence a rank correlation coefficient (rij):
Pazos, Valencia, Proteins 47, 219 (2002)
lkjjkl
lkiikl
lkjjkliikl
ij
SSSS
SSSS
r
,
2
,
2
,
where the summations run over every possible pair of proteins k and l in the multiple
sequence alignment.
Sikl is the ranked similarity between residue i in protein k and residue i in protein l.
Sjkl is the same for residue j.
Si and Sj are the means of Sikl and Sjkl.

9. Lecture WS 2006/07
Softwarewerkzeuge 26
i2h method
Schematic representation of the i2h method.
A: Family alignments are collected for two
different proteins, 1 and 2, including
corresponding sequences from different
species (a, b, c, ).
B: A virtual alignment is constructed,
concatenating the sequences of the probable
orthologous sequences of the two proteins.
Correlated mutations are calculated.
C: The distributions of the correlation values
are recorded. We used 10 correlation levels.
The corresponding distributions are
represented for the pairs of residues internal
to the two proteins (P11 and P22) and for the
pairs composed of one residue from each of
the two proteins (P12). Pazos, Valencia, Proteins 47, 219 (2002)

9. Lecture WS 2006/07
Softwarewerkzeuge 27
Predictions from correlated mutationsResults obtained by i2h in a set of 14 two domain
proteins of known structure = proteins with two
interacting domains. Treat the 2 domains as different
proteins.
A: Interaction index for the 133 pairs with 11 or more
sequences in common. The true positive hits are
highlighted with filled squares.
B: Representation of i2h results, reminiscent of those
obtained in the experimental yeast two-hybrid system.
The diameter of the black circles is proportional to the
interaction index; true pairs are highlighted with gray
squares. Empty spaces correspond to those cases in
which the i2h system could not be applied, because they
contained <11 sequences from different species in
common for the two domains.
In most cases, i2h scored the correct pair of protein
domains above all other possible interactions.Pazos, Valencia, Proteins 47, 219 (2002)

9. Lecture WS 2006/07
Softwarewerkzeuge 28
6 Coevolutionary Analysis
Idea: if co-evolution is relevant, a ligand-receptor pair should occupy related
positions in phylogenetic trees.
Goh & Cohen, 2002 showed that within correlated phylogenetic trees,
the protein pairs that bind have a higher correlation between their phylogenetic
distance matrices than other homologs drawn drom the ligand and receptor
families that do not bind.
Other Idea: analyze occurrence of proteins that can functionally substitute for
another in various organisms.
Detect analogous enzymes in thiamin biosynthesis

9. Lecture WS 2006/07
Softwarewerkzeuge 29
Detect analogous enzymes in thiamin biosynthesis Gene names are applied according to the first gene
described from a group of orthologs.
Solid black arrows represent known or proposed
reaction steps and dashed black arrows indicate
unknown reactions. In addition, significant
anticorrelations in the occurrence of genes across
species (red arrows), and relevant in silico predicted
protein-protein interactions (blue dashed arrows) are
illustrated.
Distinct precursors have been proposed for different
species (indicated in gray). Genes with orthologous
sequences in eukaryotes and prokaryotes are in
green; genes assumed to be prokaryote-specific are
black. Interestingly, significant 'one-to-one'
anticorrelations usually involve a prokaryote-specific
and a 'ubiquitous' gene.
Abbreviations: AIR, 5-aminoimidazole ribonucleotide;
Cys, cysteine; Gly, glycine; His, histidine; HMP, 2-
methyl-4-amino-5-hydroxymethylpyrimidine; THZ, 4-
methyl-5- -hydroxyethylthiazole; Tyr, tyrosine; Vit. B6,
Vitamin B6.
Morett et al. Nature Biotech 21, 790 (2003)

9. Lecture WS 2006/07
Softwarewerkzeuge 30
THI-PP biosynthesis pathway: analogous genesNegatively correlating gene
occurrences are highlighted using the
same colors. Species having at least
two genes with a role unique to THI-
PP biosynthesis are predicted to
possess the functional pathway. The
column 'STRING score' shows the
most significant interaction for each
gene, predicted using the STRING
server. Predicted interaction partners
are listed in the column 'Interact. with'.
COG id: „id in groups of orthologous
proteins server“
(a) Essential THI-PP biosynthesis
enzymes, which are unique to the
pathway.
(b) Essential THI-PP biosynthesis
enzymes, which have been implicated
in more than one biological process.
The thiO gene, suggested to play a
role in the pathway, was also added to
that list.
(c) Proteins predicted in silico to be
involved in the pathway.
Morett et al. Nature Biotech 21, 790 (2003)
4 analogies detected:thiE can be replaced by MTH861thiL by THI80thiG by THI4thiC by tenA

9. Lecture WS 2006/07
Softwarewerkzeuge 31
Interpretation
Proteins that functionally substitute eachother
have anti-correlated distribution pattern across organisms.
allows discovery of non-obvious components of pathways
and function prediction of uncharacterized proteins
and prediction of novel interactions.
Morett et al. Nature Biotech 21, 790 (2003)

9. Lecture WS 2006/07
Softwarewerkzeuge 32
7 Construct complete network of gene association
Network reconstructions have largely focused on physical protein interaction and so
represent only a subset of biologically important relations.
Aim: construct a more accurate and extensive gene network by considering functional,
rather than physical, associations, realizing that each experiment, whether genetic,
biochemical, or computational, adds evidence linking pairs of genes, with associated error
rates and degree of coverage.
In this framework, gene-gene linkages are probabilistic summaries representing functional
coupling between genes. Only some of the links represent direct protein-protein interactions;
the rest are associations not mediated by physical contact, such as regulatory, genetic, or
metabolic coupling, that, nonetheless, represent functional constraints satisfied by the cell
during the course of the experiments.
Working with probabilistic functional linkages allows many diverse classes of
experiments to be integrated into a single coherent network which enables the linkages
themselves to be more reliably
Lee, ..., Marcotte, Science 306, 1555 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 33
Method for integrating functional genomics data
Benchmark functional genomics data sets for their
relative accuracies.
Several raw data sets already have intrinsic
scoring schemes, indicated in parentheses (e.g.,
CC, correlation coefficients; P, probabilities, and
MI, mutual information scores).
These data are rescored with LLS, then integrated
into an initial network (IntNet).
Additional linkages from the genes’ network
contexts (ContextNet) are then integrated to create
the final network (FinalNet), with È34,000 linkages
between 4681 genes (ConfidentNet) scoring
higher than the gold standard (small-scale assays
of protein interactions).
Hierarchical clustering of ConfidentNet defined
627 modules of functionally linked genes spanning
3285 genes (‘‘ModularNet’’), approximating the set
of cellular systems in yeast.
Lee, ..., Marcotte, Science 306, 1555 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 34
Scoring scheme for linkages
Unified scoring scheme for linkages is based on a Bayesian statistics approach.
Each experiment is evaluated for its ability to reconstruct known gene pathways
and systems by measuring the likelihood that pairs of genes are functionally
linked conditioned on the evidence, calculated as a log likelihood score:
P(L|E) and P(L|E) : frequencies of linkages (L) observed in the given
experiment (E) between annotated genes operating in the same pathway and in
different pathways
P(L) and P(L): the prior expectations (i.e., the total frequency of linkages
between all annotated yeast genes operating in the same pathway and operating
in different pathways).
Scores > 0 indicate that the experiment tends to link genes in the same pathway,
with higher scores indicating more confident linkages.
Lee, ..., Marcotte, Science 306, 1555 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 35
Benchmarks
As scoring benchmarks, the method was tested against two primary annotation
references:
(1) the Kyoto-based KEGG pathway database and
(2) the experimentally observed yeast protein subcellular locations determined by
genome-wide green fluorescent protein (GFP)–tagging and microscopy.
KEGG scores were used for integrating linkages, with the other benchmark
withheld as an independent test of linkage accuracy.
Cross-validated benchmarks and benchmarks based on the Gene Ontology (GO)
and COG gene annotations provided comparable results.
Lee, ..., Marcotte, Science 306, 1555 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 36
Functional inference from interaction networks
Benchmarked accuracy and extent of functional genomics data sets and the integrated networks. A critical point is the comparable performance of the networks on distinct benchmarks, which assess the tendencies for linked genes to share (A) KEGG pathway annotations or (B) protein subcellular locations.x axis: percentage of protein-encoding yeast genes provided with linkages by the plotted data;y axis: relative accuracy, measured as the of the linked genes’ annotations on that benchmark. The gold standards of accuracy (red star) for calibrating the benchmarks are smallscale protein-protein interaction data from DIP. Colored markers indicate experimental linkages; gray markers, computational. The initial integrated network (lower black line), trained using only the KEGG benchmark, has measurably higher accuracy than any individual data set on the subcellular localization benchmark; adding context-inferred linkages in the final network (upper black line) further improves the size and accuracy of the network.
Lee, ..., Marcotte, Science 306, 1555 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 37
Features of integrated networks
At an intermediate degree of clustering that maximizes cluster size and functional coherence, 564 (of
627) modules are shown connected by the 950 strongest intermodule linkages.
Module colors and shapes indicate associated functions, as defined by Munich Information Center for
Protein Sequencing (MIPS), with sizes proportional to the number of genes, and connections inversely
proportional to the fraction of genes linking the clusters.
Lee, ..., Marcotte, Science 306, 1555 (2004)
9. Lecture WS 2006/07
Softwarewerkzeuge 38
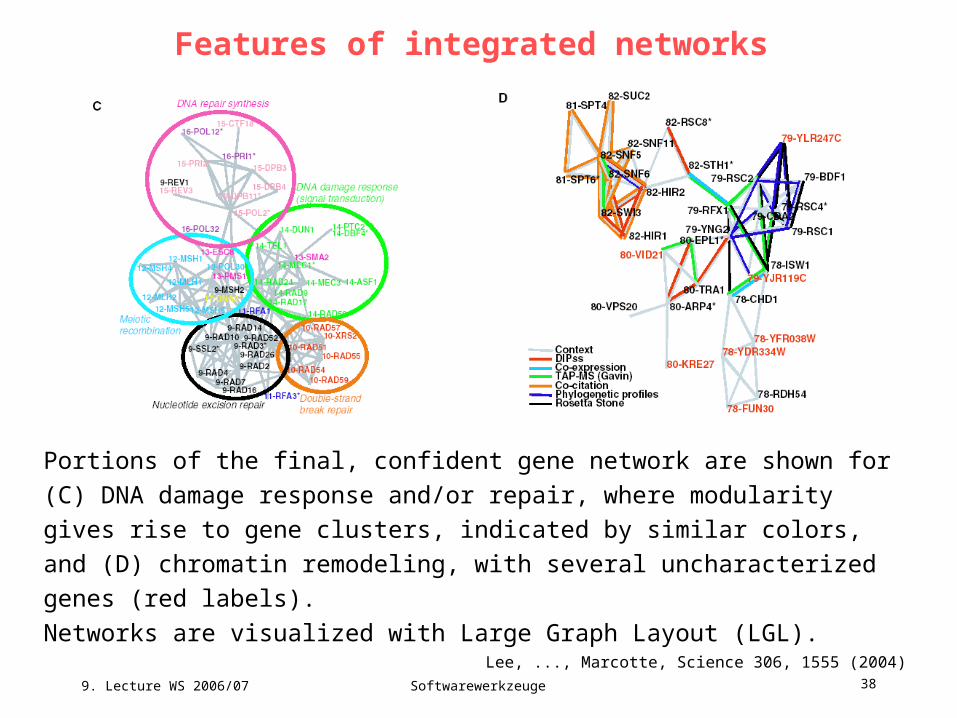
Features of integrated networks
Portions of the final, confident gene network are shown for (C) DNA damage
response and/or repair, where modularity gives rise to gene clusters, indicated by
similar colors, and (D) chromatin remodeling, with several uncharacterized
genes (red labels).
Networks are visualized with Large Graph Layout (LGL).
Lee, ..., Marcotte, Science 306, 1555 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 39
zusätzliche Folien (nicht benutzt)

9. Lecture WS 2006/07
Softwarewerkzeuge 40
1. Methods for the structural characterization of macromolecular assemblies
Russell et al. Curr. Opin. Struct. Biol. 14, 313 (2004)
(a) Electron diffraction map and 3D X-ray protein structure. X-ray provides atomic-resolution structures. (b) 3D protein structure and plot showing chemical shifts determined by NMR. NMR spectroscopy extracts distances between atoms by measuring transitions between different nuclear spin states within a magnetic field. These distances are then used as restraints to build 3D structures. NMR spectroscopy also provides atomic-resolution structures, but is generally limited to proteins of about 300 residues. It plays an increasingly important role in studying interaction interfaces between structures determined independently. (c) EM micrograph and 3D reconstruction of a virus capsid. EM is based on the analysis of images of stained particles. Different views and conformations of the complexes are trapped and thus thousands of images have to be averaged to reconstruct the three-dimensional structure. Classical implementations were limited to a resolution of 20 Å. More recently, single-particle cryo techniques, whereby samples are fast frozen before study, have reached resolutions as high as approximately 6 Å. EM provides information about the overall shape and symmetry of macromolecules. (d) Slice images and rendered surface of a ribosome-decorated portion of endoplasmic reticulum. In electron tomography, the specimen studied is progressively tilted upon an axis perpendicular to the electron beam. A set of projection images is then recorded and used to build a 3D model. This technique can tackle large organelles or even complete cells without perturbing their physiological environment. It provides shape information at resolutions of approximately 30 Å. (e) Yeast two-hybrid array screen and small network of interacting proteins. Interaction discovery comprises many different methods whose objective is to determine spatial proximity between proteins. These include techniques such as the two-hybrid system, affinity purification, FRET, chemical cross-linking, footprinting and protein arrays. These methods provide very limited structural information and no molecular details. Their strength is that they often give a quasi-comprehensive list of protein interactions and the networks they form.
9. Lecture WS 2006/07
Softwarewerkzeuge 41
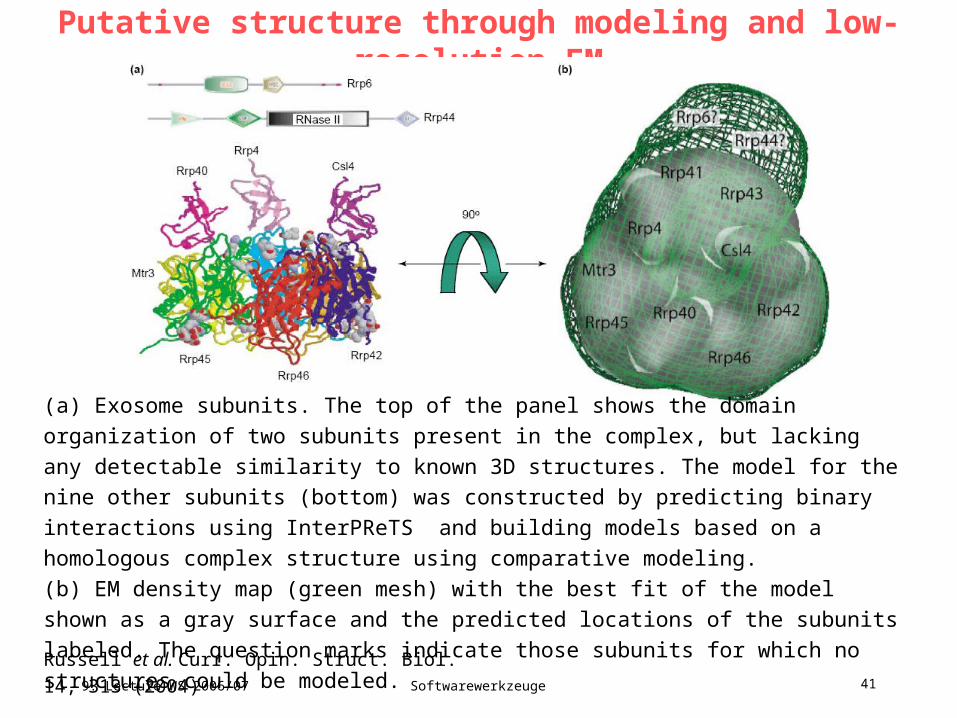
Putative structure through modeling and low-resolution EM
Russell et al. Curr. Opin. Struct. Biol. 14, 313 (2004)
(a) Exosome subunits. The top of the panel shows the domain organization of two subunits
present in the complex, but lacking any detectable similarity to known 3D structures. The
model for the nine other subunits (bottom) was constructed by predicting binary interactions
using InterPReTS and building models based on a homologous complex structure using
comparative modeling.
(b) EM density map (green mesh) with the best fit of the model shown as a gray surface
and the predicted locations of the subunits labeled. The question marks indicate those
subunits for which no structures could be modeled.
9. Lecture WS 2006/07
Softwarewerkzeuge 42
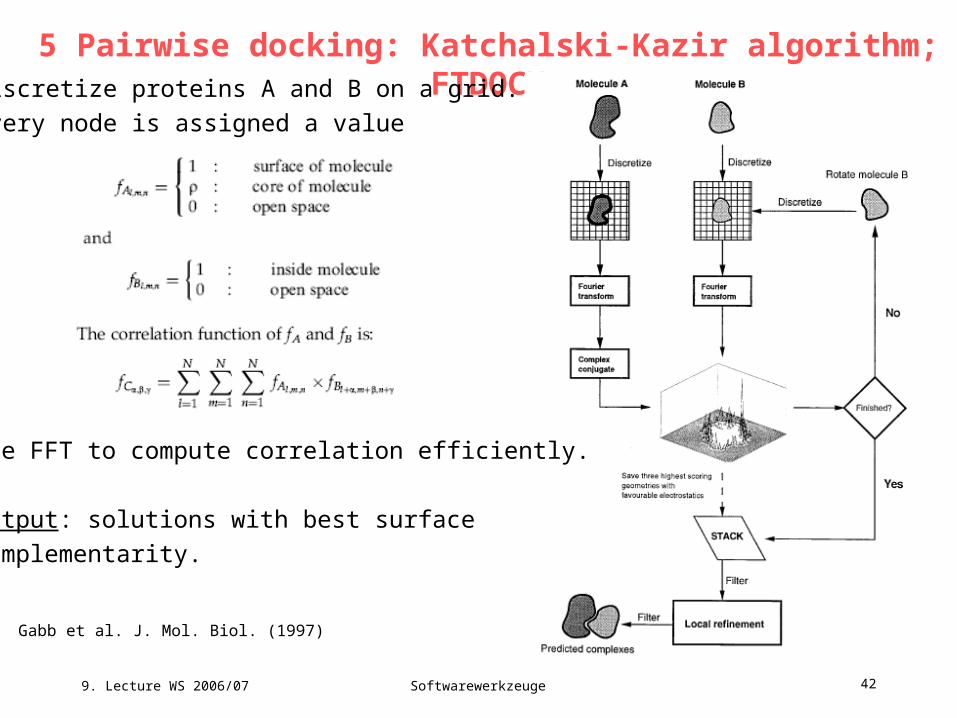
5 Pairwise docking: Katchalski-Kazir algorithm; FTDOCK
Gabb et al. J. Mol. Biol. (1997)
Discretize proteins A and B on a grid.
Every node is assigned a value
Use FFT to compute correlation efficiently.
Output: solutions with best surface
complementarity.

9. Lecture WS 2006/07
Softwarewerkzeuge 43
2. Yeast 2-Hybrid Screen
Data on protein-protein
interactions from
Yeast 2-Hybrid Screen.
One role of bioinformatics is to
sort the data.

9. Lecture WS 2006/07
Softwarewerkzeuge 44
Protein cluster in yeast
Schwikowski, Uetz, Fields, Nature Biotech. 18, 1257 (2001)
Cluster-algorithm
generates one large
cluster for proteins
interacting with each
other based on
binding data of
yeast proteins.

9. Lecture WS 2006/07
Softwarewerkzeuge 45
Annotation of function
Schwikowski, Uetz, Fields, Nature Biotech. 18, 1257 (2001)
After functional annotation:
connect clusters of
interacting proteins.
9. Lecture WS 2006/07
Softwarewerkzeuge 46
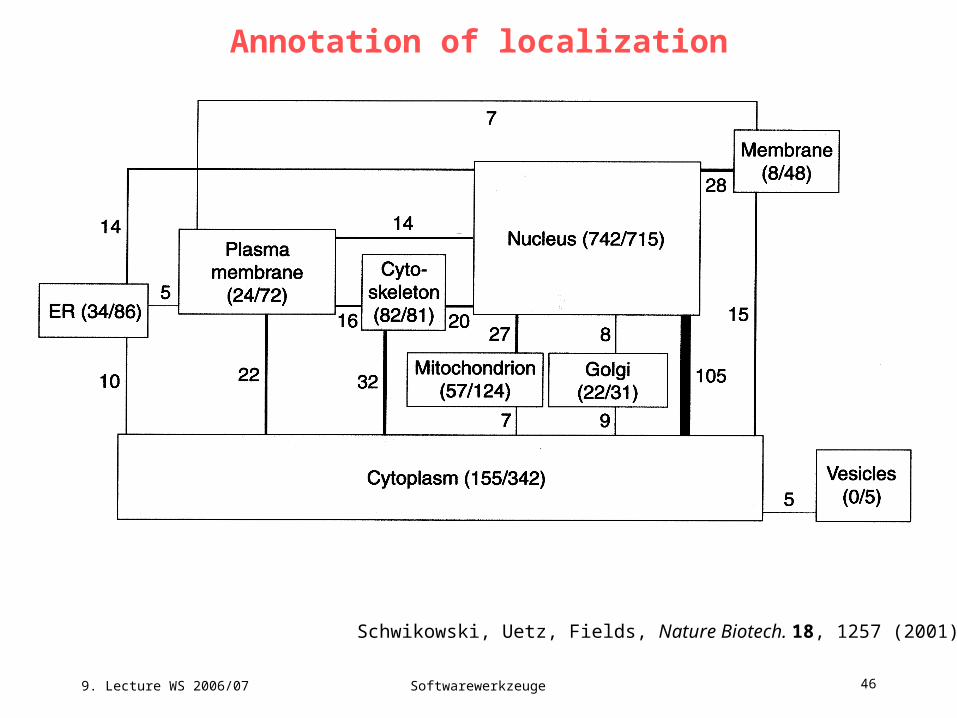
Annotation of localization
Schwikowski, Uetz, Fields, Nature Biotech. 18, 1257 (2001)
9. Lecture WS 2006/07
Softwarewerkzeuge 47
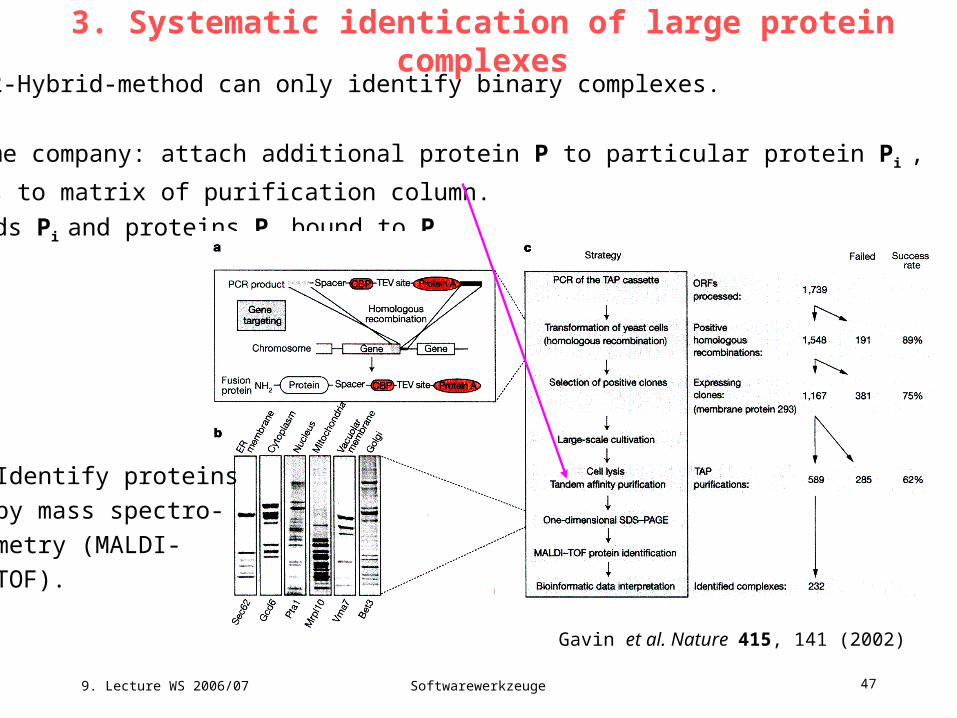
3. Systematic identication of large protein complexesYeast 2-Hybrid-method can only identify binary complexes.
Cellzome company: attach additional protein P to particular protein Pi ,
P binds to matrix of purification column.
yields Pi and proteins Pk bound to Pi .
Gavin et al. Nature 415, 141 (2002)
Identify proteins
by mass spectro-
metry (MALDI-
TOF).

9. Lecture WS 2006/07
Softwarewerkzeuge 48
Analyis of protein complexes in yeast (S. cerevisae)
Gavin et al. Nature 415, 141 (2002)
Identify proteins by
scanning yeast protein
database for protein
composed of fragments
of suitable mass.
Here, the identified
proteins are listed
according to their
localization (a).
(b) lists the number of
proteins per complex.

9. Lecture WS 2006/07
Softwarewerkzeuge 49
Validation of methodology
Gavin et al. Nature 415, 141 (2002)
Check of the method: can the same
complex be obtained for different
choice of attachment point
(tag protein attached to different
coponents of complex)? Yes (see gel).
Method allows to identify components
of complex, not the binding interfaces.
Better for identification of interfaces:
Yeast 2-hybrid screen (binary interactions).
3D models of complexes are important
to develop inhibitors.
- theoretical methods (docking) - electron tomography

9. Lecture WS 2006/07
Softwarewerkzeuge 50
Potential errors in biochemical interaction discovery
Russell et al. Curr. Opin. Struct. Biol. 14, 313 (2004)
(a) Indirect interactions between
cyclin-dependent kinase regulatory
subunit (CKS) and cyclin A detected
by the Y2H system.
Several interactions between CKS
domains and cyclins were reported in
genome-scale two-hybrid studies.
However, analysis of 3D structures
suggests that the endogenous cyclin-
dependent kinase 2 (CDK2) probably
mediates the interaction, as
combining the CDK2–CKS and
CDK2–cyclin A structures places the
CKS and cyclin domains 18 Å apart.
(b) An example of an interaction that is not detected by any screen, possibly because molecular labels (e.g. affinity purification tags, or two-hybrid DNA binding or activation domains) are interfering with the interaction. The X-ray structure of the actin–profilin complex reveals that the actin C terminus (C-t) lies at the interaction interface (the other N and C termini are also labeled).

9. Lecture WS 2006/07
Softwarewerkzeuge 51
Cross talk between complexes
(Top) Triangles show
components with at least one
modelable structure and
interaction; squares, structure
only; circles, others.
Lines show predicted
interactions: thick lines imply a
conserved interaction interface;
red, those supported by
experiment.
(Bottom) Expanded view of
cross-talk between transcription
complexes built on by a
combination of two complexes.
Aloy et al. Science, 303, 2026 (2004)

9. Lecture WS 2006/07
Softwarewerkzeuge 52
3. In silico studies to predict protein protein contactsField of studying protein interactions is split into two areas:
(1) on the macro level: map networks of protein interactions
(2) on the micro level: understand mechanisms of interaction
to predict interaction sites
Growth of genome data stimulated a lot of research in area (1).
Fewer studies have addressed area (2).
Constructing detailed models of the protein-protein interfaces is important
for comprehensive understanding of molecular processes, for drug design and
for prediction the arrangement into macromolecular complexes.
Also: understanding (2) should facilitate (1).
Therefore, this lecture focusses on linking area (2) to area (1).

9. Lecture WS 2006/07
Softwarewerkzeuge 53
Analysis of interfaces
1812 non-redundant protein
complexes from PDB
(less than 25% identity).
Results don‘t change
significantly if NMR structures,
theoretical models, or
structures at lower resolution
(altogether 50%) are excluded.
Most interesting are the results
for transiently formed
complexes.
Ofran, Rost, J. Mol. Biol. 325, 377 (2003)
permanent
transient

9. Lecture WS 2006/07
Softwarewerkzeuge 54
Amino acid composition of interface types
The frequencies of all residues found in SWISS-PROT were used as background
when the frequency of an amino acid is similar to its frequency in SWISS-PROT, the
height of the bar is close to zero. Over-representation results in a positive bar, and
under-representation results in a negative bar. Ofran, Rost, J. Mol. Biol. 325, 377 (2003)

9. Lecture WS 2006/07
Softwarewerkzeuge 55
Pairing frequencies at interfacesred square: interaction occurs more
frequently than expected;
blue square: it occurs less frequently than
expected.
(A) Intra-domain: hydrophobic core is clear
(B) domain–domain,
(C) obligatory homo-oligomers,
(D) transient homo-oligomers,
(E) obligatory hetero-oligomers, and
(F) transient hetero-oligomers.
The amino acid residues are ordered
according to hydrophobicity, with isoleucine
as the most hydrophobic and arginine as the
least hydrophobic.
propensities have been successfully used
to score protein-protein docking runs. Ofran, Rost, J. Mol. Biol. 325, 377 (2003)

9. Lecture WS 2006/07
Softwarewerkzeuge 56
Predicted interactions for E. coli
Number of predicted interactions for E. coli.
The bars represent the number of
predicted interactions obtained from the
67,238 calculated pairs (having at least 11
homologous sequences of common
species for the two proteins in each pair),
depending on the interaction index cutoff
established as a limit to consider
interaction.
Pazos, Valencia, Proteins 47, 219 (2002)
Among the high scoring pairs are many cases of known interacting proteins.