visualizing protein structures. genetic information, stored in dna, is conveyed as proteins
Post on 20-Dec-2015
217 views
TRANSCRIPT

Visualizing Protein Structures

Genetic information, stored in DNA, is conveyed as proteins

Genetic information, stored in DNA, is conveyed as proteins

The immediate product of translation is the primary protein
structure

General Amino Acid Structure
Cα
H
R
COOHH2N

List of Amino Acids and Their Abbreviations
Nonpolar (hydrophobic)
amino acid 3 letter code 1 letter code
glycine Gly G
alanine Ala A
valine Val V
leucine Leu L
isoleucine Ile I
methionine Met M
phenylalanine Phe F
tryptophan Trp W
proline Pro P

Polar (hydrophilic)
serine Ser Sthreonine Thr Tcysteine Cys Ctyrosine Tyr Y
asparagine Asn Nglutamine Gln Q
Electrically Charged (negative and hydrophilic)
aspartic acid Asp Dglutamic acid Glu E
Electrically Charged (positive and hydrophilic)
lysine Lys Karginine Arg Rhistidine
OthersX = unknown
His H
* = STOP


General Amino Acid Structure
Cα
H
R
COOHH2N

Peptide Bond Formation

Peptides have rotatable bonds of defined lengths
Note- all proteins have polarity- N termini; C termini

The ‘protein-folding problem’.• Proteins -- hundreds of thousands of different
ones -- are the biochemical molecules that make up cells, organs and organisms. Proteins put themselves together, in a process termed "folding." How they do that is called "the protein-folding problem," and it may be the most important unanswered question in the life sciences.
WHY??

The primary sequence dictates the secondary and tertiary structure of the protein

Protein Structure

Two questions
• Can you change the 3o (tertiary) sequence without changing the 1o (primary) sequence?
• Can you change the 1o (primary) sequence without changing the 3o (tertiary) sequence?

What is known about protein folding?
•

Secondary Structures are dominated by:
• 1) helix
• 2) -sheet

helical structure is a very regular structure (3.6 amino acids/turn)

-sheet: anti-parallel

-sheet: parallel

Hydrogen BondingAnd Secondary Structure
alpha-helix beta-sheet

Hydrogen Bonding• One of the most important stabilizing forces
in protein structure!
• Both -helix and -sheet are dependent on H-bonding.

Protein Folding is progressive?
1° - first
2°- second
3° - third

Formation of tertiary structure
The tertiary structure (or conformation) is the way alpha -helixes and beta -pleated sheets fold in respect to each other.
Amino acids which are very distant in the primary structure might be close in the tertiary one because of the folding of the chain.

Structure Stabilizing Interactions(Factors governing 3° structure)
• Noncovalent– Van der Waals forces (transient, weak electrical
attraction of one atom for another)– Hydrophobic (clustering of nonpolar groups)– Hydrogen bonding– Salt bridges
• Covalent– Disulfide bonds

Hydrophobic and Hydrophilic Interactions:
• Hydrophilic amino acids are those whose sidechains offer hydrogen bonding partners to the surrounding water molecules.

• Hydrophobic amino acids:
• Tend to internalize in water.
• Tend to externalize in a membrane
• Hydrophilic amino acids:
• Tend to externalize in water.
• Tend to internalize in a membrane


Disulfide Bridge

Disulfide Bridge – Linking Distant Amino Acids

Structure Stabilizing Interactions(Factors governing 3° structure)
• Noncovalent– Van der Waals forces (transient, weak electrical
attraction of one atom for another)– Hydrophobic (clustering of nonpolar groups)– Hydrogen bonding– Salt bridges
• Covalent– Disulfide bonds
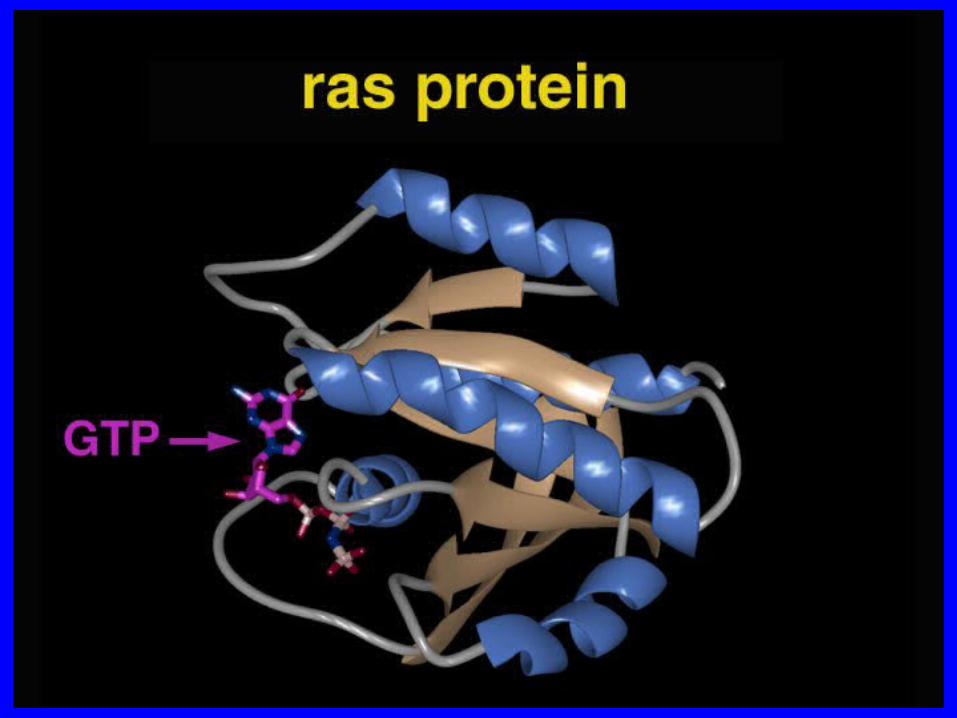


• The transformation happens quickly and spontaneously. It takes only a fraction of a second for a floppy chain of beads to fold into the shape it will keep for the rest of its working life.
• How does that happen? How do the linear -- and, in some sense, one-dimensional -- structures of proteins carry the information that tells them to take on permanent three-dimensional shapes? Is it possible to study a protein chain and predict the folded shape it will take?
• That is the protein-folding problem.

DNA sequencing information predictions of the primary amino
acid sequence.
Needed- Software that will convert the 1o sequence to its corresponding 3o sequence.
Needed- Software that will describe a 1o sequence that will generate a particular 3o sequence.

Structure classification:• Finding proteins that have similar chemical
architectures.
This involves developing a representation of how units of secondary structure come together to form ‘domains’*.
• *compact regions of structure within the large protein structure.



The End

• WHY IS PROTEIN FOLDING SO DIFFICULT TO UNDERSTAND?
• It's amazing that not only do proteins self-assemble -- fold -- but they do so amazingly quickly: some as fast as a millionth of a second. While this time is very fast on a person's timescale, it's remarkably long for computers to simulate. In fact there is a 1000 X gap between the simulation timescales (nanoseconds) and the times at which the fastest proteins fold (microseconds).

A Glimpse of the Holy Grail?• The prediction of the native conformation of a
protein of known amino acid sequence is one of the great open questions in molecular biology and one of the most demanding challenges in the new field of bioinformatics. Using fast programs and lots of supercomputer time, Duan and Kollman (1) report that they have successfully folded a reasonably sized (36-residue) protein fragment by molecular dynamics simulation into a structure that resembles the native state. At last it seems that the folding of a protein by detailed computer simulation is not as impossible as most workers in the field believe.

Proteins from Scratch:• Not long ago, it seemed inconceivable that proteins
could be designed from scratch. Because each protein sequence has an astronomical number of potential conformations, it appeared that only an experimentalist with the evolutionary life span of Mother Nature could design a sequence capable of folding into a single, well-defined three-dimensional structure. But now, on page 82 of this issue, Dahiyat and Mayo (1) describe a new approach that makes de novo protein design as easy as running a computer program. Well almost.

Progress in the ‘protein-folding problem’?
• When proteins fold, they don’t try ever possible 3D conformation. Protein folding is an orderly process (i.e. there are molecular shortcuts involved).

Success in protein-folding?
Given the primary sequence of a protein, the success rate in predicting the proper 3D structure of a protein shows strong correlation, to the % of the protein that showed similarity to proteins of known structure.

The primary sequence dictates the secondary and tertiary structure of the protein