wgs assembly and reads clustering zemin ning production software group informatics division
TRANSCRIPT

WGS Assembly WGS Assembly and Reads Clustering and Reads Clustering
Zemin NingZemin Ning
Production Software Group
Informatics Division

Outline of the Talk:
Whole Genome Shotgun Sequencing Insert Sizes Repeats in the Genomes Kmer Words Hashing and Distribution Relational Matrix Profile of unique kmer words Phusion Steps How to run Phusion – parameter selections

Clone-by-Clone Sequencing– ADV. Easy assembly– DIS. Build library & physical map; redundant sequencing
Whole Genome Shotgun (WGS)– ADV. No mapping, no redundant sequencing– DIS. Difficult to assemble and resolve repeats
WGS Sequencing:
The WGS method begins by fragmenting the genome into many pieces of various sizes. This fragmentation can be done in several ways, including physically shaking the DNA and cutting it with restriction enzymes. Depending on the size of the resulting fragment, various hosts are used to clone these regions.

Whole Genome Shotgun Sequencing
cut many times at random
genome
forward-reverse paired reads
plasmids (2 – 10 Kbp)
cosmids (40 Kbp) known dist
~500 bp~500 bp

Automatic Sequencing
W hole genomeBAC/cosm id clone
f in a l con sen sus seq u en ce
Finishingq u a lity
b o th s ta n ds covera geg a p f illing
Partial Assem blyco n tigs
DNA sequencingra n d om clo n es
Clone libraryp U C 18
Sm all fragm ents1 .0 - 2 .0 kb
DNA fragm entationso n ic d is rup tion
n e bu liza tion
W hole genomeBAC/cosm id clone

Base Calling - Phred
Idealized traces would
consist of evenly spaced,
nonoverlapping peaks.
Real traces deviate from
this ideal due to imper-
fections of the
sequencing
reactions, of gel electro-
phoresis, and of trace
processing.
The first 50 or so peaks
and peaks over 500 or so
are particularly noisy.
Quality:high – noambiguities
medium – someambiguities
Poor – low confidence

Historical ContextHistorical Context1995: H.influenzae sequenced using TIGR by Craig Venter. H. influenzae is the first free living organism to be sequnced. It has roughly 2 million base pairs. The sequencing used a shotgun method that assembled 25,000 fragments of 500 bp each.1997:
Whole Genome Shotgun paper written by Weber & Meyers. This is the first time that a shotgun method has been suggested for sequencing the human genome. By this time, the public Human Genome Project has already started using a clone-by-clone method.1997:
Phil Green writes review against WGS.1998:
Celera founded. Celera entered into a competition with the public Human Genome Project to sequence the human genome first. Celera’s main advantage was using the Whole Genome Shotgun method, which had a chance of failing, but if successful would produce faster results.

1999: Fly genome (180Mbs) sequenced by Celera using the Celera
assembler. The genome is available by subscription to Celera’s database2001:
Human Genome published. The genome was sequenced using data from the public Human Genome Project and Celera. The public effort used the clone-by-clone method, while Celera used the Whole Genome Shotgun method. Celera gives access to the genome throughsubscription to the database. The results from the public project are free to access.2001:
Mouse Genome sequenced by Celera using the Whole Genome Shotgun method. It is made available by Celera on a subscription basis.2002:
The Mouse Genome published. Whitehead’s ARACHNE and Sanger’s Phusion were involved.

Whole Genome Assemblers
TIGR Assembler G.G. Sutton et al., Genome Sci Technol 1, 9-19 (1995)
PHRAP P. Green (1996)
Celera Assembler
CAP3 X. Huang, A. Madan, Genome Res 9, 868-877 (1999)
RePS J. Wang et al. Genome Res 12, 824-831 (2002)
Phusion (Sanger) J.C. Mullikin, Z. Ning, Genome Res 13, 81-90 (2003)
Arachne (Whitehead/MIT)
Euler (UCSD, USC) P.A. Pevzner, H. Tang, M.S. Waterman, RECOMB (2001)
most assemblers follow the same approach:
overlap – layout - consensus

Unique and Repetitive DNA SectionsUnique and Repetitive DNA Sections
DepthDepthUnique SectionUnique Section
DepthDepth
Repetitive SectionRepetitive Section
AA X’ X’ B B X’’ X’’ C C

Repetitive Contig and Read PairsRepetitive Contig and Read Pairs
DepthDepth
DepthDepthDepth Depth
Grouped Reads by PhusionGrouped Reads by Phusion
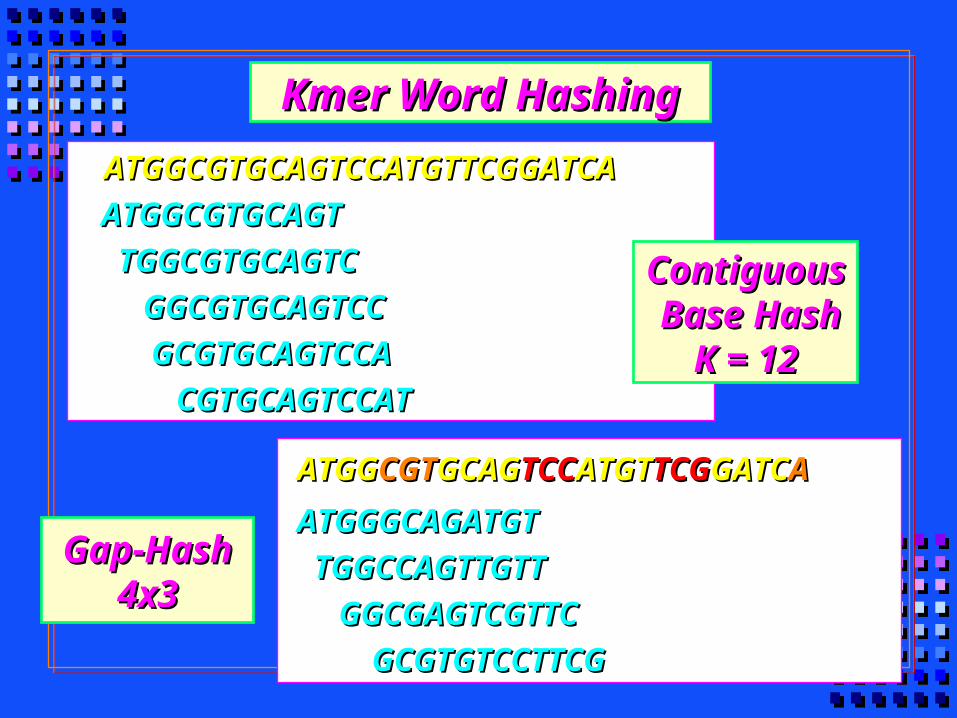
Gap-HashGap-Hash4x34x3
ATGGGCAGATGTATGGGCAGATGT
TGGCCAGTTGTTTGGCCAGTTGTT
GGCGAGTCGTTCGGCGAGTCGTTC
GCGTGTCCTTCGGCGTGTCCTTCG
ATGGATGGCGTCGTGCAGGCAGTCCTCCATGTATGTTCGTCGGATCGATCAA
ATGGCGTGCAGTATGGCGTGCAGT
TGGCGTGCAGTCTGGCGTGCAGTC
GGCGTGCAGTCCGGCGTGCAGTCC
GCGTGCAGTCCAGCGTGCAGTCCA
CGTGCAGTCCATCGTGCAGTCCAT
ATGGCGTGCAGTCCATGTTCGGATCAATGGCGTGCAGTCCATGTTCGGATCA
ContiguousContiguous Base HashBase Hash
K = 12K = 12
Kmer Word HashingKmer Word Hashing

Word use distribution for the mouse sequence data at ~7.5 foldWord use distribution for the mouse sequence data at ~7.5 fold
Useful Region
Poisson Curve
Real Data Curve

Sorted List of Each k-Mer and Its Read Indices
ACAGAAAAGC 10h06.p1cACAGAAAAGC 12a04.q1cACAGAAAAGC 13d01.p1cACAGAAAAGC 16d01.p1cACAGAAAAGC 26g04.p1cACAGAAAAGC 33h02.q1cACAGAAAAGC 37g12.p1cACAGAAAAGC 40d06.p1cACAGAAAAGG 16a02.p1cACAGAAAAGG 20a10.p1cACAGAAAAGG 22a03.p1cACAGAAAAGG 26e12.q1cACAGAAAAGG 30e12.q1cACAGAAAAGG 47a01.p1c
High bits Low bits
64 -2k64 -2k 2k2k

1 2 3 4 5 6 … j … N
3
1
4
2
6
5
i
N
227 0 0 0 0
R(i,j)
Relation Matrix: R(i,j) – number of kmer Relation Matrix: R(i,j) – number of kmer words shared between read i and read jwords shared between read i and read j
227 187 0 0 0 0 187 0 170 0
0 0 170 0 0
0 0 0 0 213
0 0 0 213 0
Group 1: (1,2,3,5)Group 1: (1,2,3,5)
Group 2: (4,6)Group 2: (4,6)
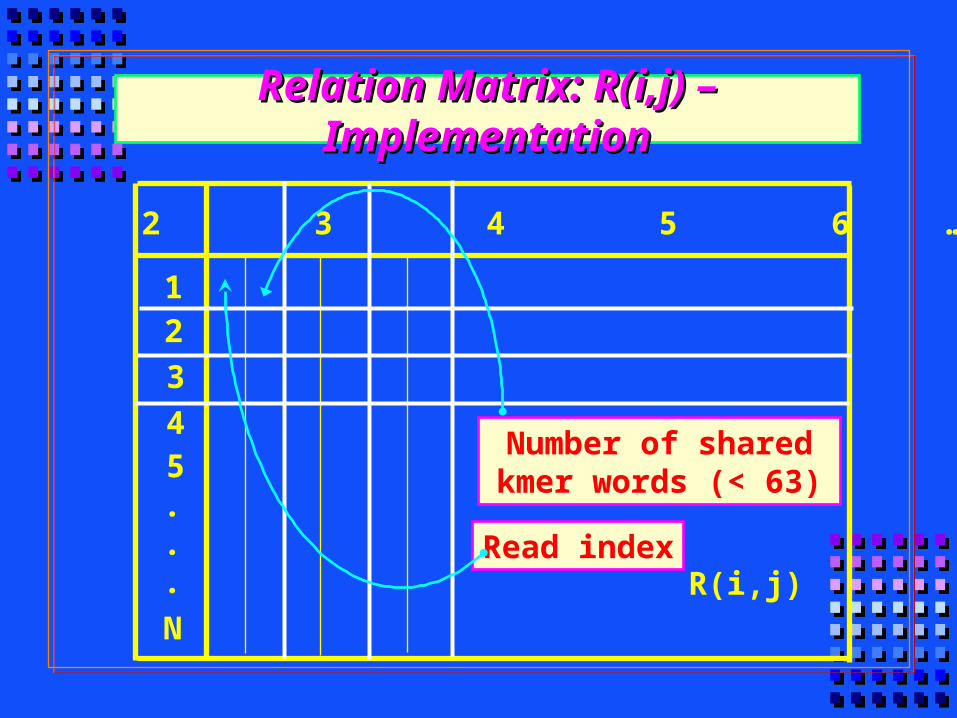
1 2 3 4 5 6 … j … 500
3
1
4
2
5
R(i,j)
Relation Matrix: R(i,j) – ImplementationRelation Matrix: R(i,j) – Implementation
Read index
Number of shared kmer words (< 63)
N
.
.
.

Phusion Iterations – Cutting The Weakest LinkPhusion Iterations – Cutting The Weakest Link

This graph shows the effect of k-mer on relative contig N50 size for C. briggsae assemblies. At k = 15, 4 ^ 15 is about 10 times the genome size.

Profile of Unique kmer Words Profile of Unique kmer Words
ATGGCGTGCAGTCCATTATGGCGTGCAGTCCATT TCGGATCATCCGTTAACGTTCGGATCATCCGTTAACGT
P=KmerP=P2
P=P1
Unique sequence Non-unique sequence
Quality values are reset over the readQuality values are reset over the read

Phusion StepsPhusion Steps
Hashing the kmer words; Calculate kmer words distribution; Get the list of kmer words – only use those occur 2-D
times; Combine the kmer words with read index; Sort the combined list; Build up relational matrix; Group the reads; Output.

Phusion command line for ZfishPhusion command line for Zfish
./phusion ./phusion ––kmer kmer 18 18
––depth depth 1313[-fill[-fill 6]6][-gap[-gap 5]5]-match-match 66-match2-match2 66-matrix-matrix 500500-break-break 11-set -set 1200012000
matesmatesfasta/fastq filesfasta/fastq files

Phusion2 ?Phusion2 ?

Acknowledgements:
Jim Mullkin Richard Durbin David Jaffe – Broad Institute