x-linked lymphoproliferative disease due to sap/sh2d1a ..._2011.pdfvere immune dysregulation and...
TRANSCRIPT

Zurich Open Repository andArchiveUniversity of ZurichMain LibraryStrickhofstrasse 39CH-8057 Zurichwww.zora.uzh.ch
Year: 2011
X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: amulticenter study on the manifestations, management and outcome of the
disease
Booth, C; Gilmour, K C; Veys, P; et al; Pachlopnik Schmid, Jana
Abstract: X-linked lymphoproliferative disease (XLP1) is a rare immunodeficiency characterized by se-vere immune dysregulation and caused by mutations in the SH2D1A/SAP gene. Clinical manifestationsare varied and include hemophagocytic lymphohistiocytosis (HLH), lymphoma and dysgammaglobuline-mia, often triggered by Epstein-Barr virus infection. Historical data published before improved treatmentregimens shows very poor outcome. We describe a large cohort of 91 genetically defined XLP1 patientscollected from centers worldwide and report characteristics and outcome data for 43 patients receivinghematopoietic stem cell transplant (HSCT) and 48 untransplanted patients. The advent of better treat-ment strategies for HLH and malignancy has greatly reduced mortality for these patients, but HLH stillremains the most severe feature of XLP1. Survival after allogeneic HSCT is 81.4% with good immunereconstitution in the large majority of patients and little evidence of posttransplant lymphoproliferativedisease. However, survival falls to 50% in patients with HLH as a feature of disease. Untransplantedpatients have an overall survival of 62.5% with the majority on immunoglobulin replacement therapy, butthe outcome for those untransplanted after HLH is extremely poor (18.8%). HSCT should be undertakenin all patients with HLH, because outcome without transplant is extremely poor. The outcome of HSCTfor other manifestations of XLP1 is very good, and if HSCT is not undertaken immediately, patientsmust be monitored closely for evidence of disease progression.
DOI: https://doi.org/10.1182/blood-2010-06-284935
Posted at the Zurich Open Repository and Archive, University of ZurichZORA URL: https://doi.org/10.5167/uzh-59078Journal ArticlePublished Version
Originally published at:Booth, C; Gilmour, K C; Veys, P; et al; Pachlopnik Schmid, Jana (2011). X-linked lymphoproliferativedisease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management andoutcome of the disease. Blood, 117(1):53-62.DOI: https://doi.org/10.1182/blood-2010-06-284935

doi:10.1182/blood-2010-06-284935Prepublished online October 6, 2010;2011 117: 53-62
Choo, Joanne Smart, Peter D. Arkwright and Hubert B. GasparKanegane, Kim E. Nichols, I. Celine Hanson, Neena Kapoor, Elie Haddad, Morton Cowan, Sharon Bonanomi, Christina Peters, Krzysztof Kalwak, Srdjan Pasic, Petr Sedlacek, Janez Jazbec, HirokazuAlessandro Plebani, Annarosa Soresina, Andrea Finocchi, Claudio Pignata, Emilia Cirillo, Sonia Rieber, Brigitte Strahm, Henrike Ritterbusch, Arjan Lankester, Nico G. Hartwig, Isabelle Meyts,Schmid, Sylvain Latour, Genevieve de Saint-Basile, Michael Albert, Gundula Notheis, Nikolaus Marina Cavazzana-Calvo, Alain Fischer, Despina Moshous, Stephane Blanche, Jana PachlopnikPaul T. Heath, Colin G. Steward, Owen Smith, Anna O'Meara, Hilary Kerrigan, Nizar Mahlaoui, Claire Booth, Kimberly C. Gilmour, Paul Veys, Andrew R. Gennery, Mary A. Slatter, Helen Chapel, the diseasemulticenter study on the manifestations, management and outcome of X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a
http://bloodjournal.hematologylibrary.org/content/117/1/53.full.htmlUpdated information and services can be found at:
(4674 articles)Immunobiology � (3397 articles)Clinical Trials and Observations �
Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Washington DC 20036.by the American Society of Hematology, 2021 L St, NW, Suite 900, Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly
only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom

CLINICAL TRIALS AND OBSERVATIONS
X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: amulticenter study on the manifestations, management and outcome of the diseaseClaire Booth,1 Kimberly C. Gilmour,1 Paul Veys,1 Andrew R. Gennery,2 Mary A. Slatter,2 Helen Chapel,3 Paul T. Heath,4
Colin G. Steward,5 Owen Smith,6 Anna O’Meara,6 Hilary Kerrigan,6 Nizar Mahlaoui,7 Marina Cavazzana-Calvo,7
Alain Fischer,7 Despina Moshous,7 Stephane Blanche,7 Jana Pachlopnik Schmid,7 Sylvain Latour,8
Genevieve de Saint-Basile,8 Michael Albert,9 Gundula Notheis,9 Nikolaus Rieber,9 Brigitte Strahm,10 Henrike Ritterbusch,11
Arjan Lankester,12 Nico G. Hartwig,13 Isabelle Meyts,14 Alessandro Plebani,15 Annarosa Soresina,15 Andrea Finocchi,16
Claudio Pignata,17 Emilia Cirillo,17 Sonia Bonanomi,18 Christina Peters,19 Krzysztof Kalwak,20 Srdjan Pasic,21
Petr Sedlacek,22 Janez Jazbec,23 Hirokazu Kanegane,24 Kim E. Nichols,25 I. Celine Hanson,26 Neena Kapoor,27
Elie Haddad,28 Morton Cowan,29 Sharon Choo,30 Joanne Smart,30 Peter D. Arkwright,31 and Hubert B. Gaspar1
1Center of Immunodeficiency, Molecular Immunology Unit, Institute of Child Health, London, United Kingdom; 2Institute of Cellular Medicine, Newcastle University,Newcastle upon Tyne, United Kingdom; 3Department of Clinical Immunology, Nuffield Department of Medicine, University of Oxford and Oxford Radcliffe Hospitals, Oxford,United Kingdom; 4St George’s Hospital, London, United Kingdom; 5Bone Marrow Transplant Unit, Royal Hospital for Children, Bristol, United Kingdom; 6Department ofHaematology and Oncology, Our Lady’s Children’s Hospital, Dublin, Ireland; 7Unite d’Immuno-Hematologie et Rhumatologie Pediatrique, Hopital Necker-Enfants Malades,Assistance Publique–Hopitaux de Paris, Paris, France; 8Inserm U678, Hopital Necker-Enfants Malades, Paris, France; 9Department of Pediatric Hematology/Oncologyand Infection/Immunity, Dr von Haunersches Kinderspital, Munich, Germany; 10Pediatric Hematology and Oncology, Center for Pediatric and Adolescent Medicine,University of Freiburg, Freiburg, Germany; 11Center for Chronic Immunodeficiency, University Medical Center Freiburg, Freiburg, Germany; 12Department of Pediatrics,Division of Immunology, Haematology, Oncology, Bone Marrow Transplantation and Autoimmune Diseases, Leiden University Medical Center, Leiden, The Netherlands;13Department of Paediatric Infectious Disease and Immunology, Erasmus Medical Center, Sophia Children’s Hospital, Rotterdam, The Netherlands; 14Department ofPaediatrics, University Hospital Leuven, Leuven, Belgium; 15Department of Paediatrics, University of Brescia, Brescia, Italy; 16Department of Pediatrics, Unit ofImmunoinfectivology, Children’s Hospital Bambino Gesu, Tor Vergata University, Rome, Italy; 17Department of Pediatrics, Federico II University, Naples, Italy; 18ClinicaPediatrica dell’Universita di Milano-Bicocca, Centro Trapianto di Midollo Osseo, Ospedale San Gerardo, Monza, Italy; 19Bone Marrow Transplantation Unit, St AnnaChildren’s Hospital, Vienna, Austria; 20Department of Paediatric Haematology and Oncology, Medical University of Wroclaw, Wroclaw, Poland; 21Departments of PaediatricImmunology, Pathology, and Transfusion Medicine, Mother and Child Health Institute Dr Vukan Cupic, Belgrade, Serbia; 22Department of Paediatric Haematology andOncology, University Hospital Motol, Charles University, Prague, Czech Republic; 23Division of Oncology and Hematology, Department of Pediatrics, Medical Center,Ljubljana, Slovenia; 24Department of Paediatrics, Graduate School Of Medicine, University of Toyama, Toyama, Japan; 25Division of Oncology, Children’s Hospital ofPhiladelphia, Philadelphia, PA; 26Department of Pediatrics, Texas Children’s Hospital, Baylor College of Medicine, Houston, TX; 27Division of Research Immunology/BoneMarrow Transplantation, Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, CA; 28Department of Pediatrics, andMicrobiology and Immunology, Centre hospitalier universitaire Sainte-Justine, Universite de Montreal, Montreal, QC; 29Pediatric Blood and Marrow Transplant Division,University of California San Francisco Children’s Hospital, San Francisco, CA; 30Department of Allergy and Immunology, Royal Children’s Hospital, Parkville, Australia; and31University of Manchester, Royal Manchester Children’s Hospital, Manchester, United Kingdom
X-linked lymphoproliferative disease(XLP1) is a rare immunodeficiency charac-terized by severe immune dysregulationand caused by mutations in the SH2D1A/SAP gene. Clinical manifestations arevaried and include hemophagocytic lym-phohistiocytosis (HLH), lymphoma anddysgammaglobulinemia, often triggeredby Epstein-Barr virus infection. Historicaldata published before improved treat-ment regimens shows very poor out-come. We describe a large cohort of 91 ge-netically defined XLP1 patients collectedfrom centers worldwide and report char-
acteristics and outcome data for 43 pa-tients receiving hematopoietic stem celltransplant (HSCT) and 48 untransplantedpatients. The advent of better treatmentstrategies for HLH and malignancy hasgreatly reduced mortality for these pa-tients, but HLH still remains the mostsevere feature of XLP1. Survival afterallogeneic HSCT is 81.4% with good im-mune reconstitution in the large majorityof patients and little evidence of posttrans-plant lymphoproliferative disease. How-ever, survival falls to 50% in patients withHLH as a feature of disease. Untrans-
planted patients have an overall survivalof 62.5% with the majority on immuno-globulin replacement therapy, but the out-come for those untransplanted after HLHis extremely poor (18.8%). HSCT shouldbe undertaken in all patients with HLH,because outcome without transplant isextremely poor. The outcome of HSCT forother manifestations of XLP1 is very good,and if HSCT is not undertaken immedi-ately, patients must be monitored closelyfor evidence of disease progression.(Blood. 2011;117(1):53-62)
Introduction
X-linked lymphoproliferative disease (XLP) is a rare primaryimmunodeficiency first described in 1975 by Purtilo1 and character-
ized by severe immune dysregulation often after viral infection(typically with Epstein-Barr virus [EBV]). Since XLP was first
Submitted June 10, 2010; accepted September 19, 2010. Prepublished online asBlood First Edition paper, October 6, 2010; DOI 10.1182/blood-2010-06-284935.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 USC section 1734.
© 2011 by The American Society of Hematology
53BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1
only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom

described, our understanding of the molecular and cellular patho-genesis of the disease has greatly improved. However, clinically, itis still difficult to determine optimal management and prognosis forpatients due to the variability of clinical presentation, lack ofgenotype-phenotype correlation, and rarity of the disease. Purtiloestablished an XLP registry in 1980, and by 1995 more than270 boys had been identified in 80 kindreds.2 To date this registryhas provided the only data on clinical phenotype and prognosis forthis patient group. Overall mortality in this group was 75%, with70% of boys succumbing before 10 years of age. However, currentoutcomes for XLP may be very different due to the availability ofunambiguous molecular diagnosis, improved viral monitoring, andthe improvement in treatment regimens for disease manifestations.
XLP affects 1 to 3 million boys,3,4 and most commonly presentsin childhood or early adolescence. Presentation may be acute in thecase of fulminant infectious mononucleosis (FIM)/hemophago-cytic lymphohistiocytosis (HLH) or lymphoma or less aggressivewith dysgammaglobulinemia or recurrent infections. Patients oftenmanifest more than one phenotype and may progress from onephenotype to another, for example presenting with hypogam-maglobulinemia and progressing to lymphoma, and differentclinical features are often present in families highlighting the lackof genotype-phenotype correlation. Other rare but well-describedpresenting features include aplastic anemia, vasculitis, and chronicgastritis.2,5-8 It is now known that the clinical syndrome of XLParises from 2 different genetic defects in SH2D1A (XLP1, by farthe most common and the focus of this report) and the BIRC/XIAPgene (XLP2). The gene responsible for XLP1 is the SH2D1A genefound on the X chromosome at position Xq25,9-11 which encodesthe cytoplasmic protein SAP (signaling lymphocyte activationmolecule or SLAM-associated protein). SAP is a key regulator ofnormal immune function in T cells,12-14 natural killer (NK)cells,15-18 NKT cells,19,20 and possibly B cells,21 and defects inthis protein lead to the varied immune defects described inXLP1 patients.20,22 Humoral defects seen in this disease are thoughtto arise from impaired CD4� T-cell interaction with B cells and notan intrinsic B-cell deficit.23
Although it has always been presumed that EBV infectionplays a crucial role in the development of clinical features inXLP1 patients, it is now clear that a proportion of boys are EBVnegative at presentation and remain so. Indeed, 10% of patientshave immunological abnormalities before any evidence of EBVexposure.4,24 XLP1 is therefore a disorder of immune dysregulationrather than a disorder specifically associated with EBV infection.
Before 1994, acute management of FIM and HLH includedantiviral medications, high-dose intravenous immunoglobulin (Ig),immunosuppressants, and other immune modulators such as inter-feron-�. These treatments proved disappointing25 and the XLPregistry data showed a survival of only 4% for boys presenting withthese manifestations. Improved chemotherapy regimes for lym-phoma and immunosuppressive protocols to treat HLH (includingrituximab) may reduce the mortality rate for XLP1 patients andallow stabilization before hematopoietic stem cell transplant(HSCT).26 Our report provides valuable outcome data collectedsince the introduction of current HLH treatment protocols, focusingon XLP1 patients with mutations in the SH2D1A gene.
Allogeneic HSCT remains the only curative option for XLP1 atpresent although large scale outcome studies are not available.Recently, Lankester et al reviewed 14 cases in the literature whohad undergone HSCT and found an overall survival of 71% (10/14)with little evidence of EBV reactivation and posttransplant lym-
phoproliferative disease.27 We describe here outcome data for amuch larger cohort of patients transplanted since 1997.
There is no consensus on whether clinically stable XLP1 patientsshould undergo HSCT as the natural history of the disease is so variable,even within the same family. Treatment and management of the diseaseis severely hampered by the lack of data of a large cohort of patients andpreviously published outcome data are based on historical data,which may represent patients with conditions other than XLP1 asinclusion was based on clinical and not genetic diagnosis. Also,little recent data exist for patients who remain untransplanted. Hence, wedescribe a large cohort of genetically defined XLP1 patients collectedfrom centers worldwide. The data presented will allow for bettercounseling of affected families regarding prognosis and managementoptions, particularly in relation to timing of transplant.
Methods
Data collection
Questionnaires regarding patient demographics, transplant characteristics,and outcome were sent to centers worldwide identified through theEuropean Society for Immunodeficiencies/European Bone Marrow Trans-plantation Registry, published case reports or centers known to performpediatric HSCT. Retrospective analysis was performed using data collectedfor 91 patients from 32 centers worldwide. The number of cases from eachcenter varied between 1 and 27 but was on average 1-2 cases. Patientsincluded in this study were born between 1941 and 2005; 63 were born in orafter 1990 (24 untransplanted patients and 39 transplanted patients). Onlypatients with a confirmed mutation in the SH2D1A gene were included inthis series. Patients with mutations in other XLP-associated genes such asXIAP/BIRC-4 were excluded, as were patients with abnormal SAPexpression but no confirmed mutation in SH2D1A. EBV status wasdetermined by polymerase chain reaction to avoid variable serology resultsin XLP1 patients and especially in those with dysgammaglobulinemia.Questionnaires offered reporting of FIM and HLH separately; thus, somecenters with experience in this area reported patient data accordingly, and itis presented as such.
Data in various forms from 11 patients have been previously pub-lished5,27-32 but standardized information was recollected in this study andadded to the series.
Management of HLH and lymphoma
Patients who presented with HLH were managed predominantly inaccordance with HLH 94 or HLH 2004 protocols. Additional or alternativetreatment included antiviral therapy (aciclovir, ganciclovir, or foscarnet,n � 6), high-dose intravenous immunoglobulin (n � 9), immunosuppres-sion (steroids, cyclosporine, and etoposide, n � 12), or anti-CD20 antibody(rituximab, n � 10). Intrathecal therapy was used where central nervoussystem involvement was suspected. Ten patients who proceeded to trans-plant received rituximab therapy before transplant, either as treatment forHLH or during conditioning.
Regimes for the treatment of lymphoma varied in line with appropriatenational guidelines (eg, COPAD [cyclophosphamide, vincristine, predni-sone, and doxorubicin]) study, Berlin-Frankfurt-Munster Group, Associazi-one Italiana Ematologia Oncologia Pediatrica, or United Kingdom Chil-dren’s Cancer Study Group guidelines) and only occasionally involvedsurgical management.
Statistical analysis
Kaplan-Meier curves were used to analyze survival figures. The log ranktest (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests were used to com-pare survival between different groups. Statistical analysis including hazardratio calculation was performed using GraphPad Prism Version 5.00 forWindows.
54 BOOTH et al BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom

Results
Data from 91 patients (64 pedigrees) in 32 centers worldwide wereincluded in this report. The overall survival of XLP1 patients was71.4% (65/91), and patients displayed a heterogeneous clinicalphenotype. Due to the heterogeneity of the group, data wereanalyzed according to presentation with HLH, EBV status, andwhether patients had received HSCT, allowing characterization ofoutcome after transplant.
Spectrum of XLP1 mutations
In keeping with previous publications, no genotype/phenotypecorrelation was evident, and the most frequently reported mutationinvolved the arginine residue at position 55 (exon 2) found in11 patients from 9 different families. Detailed genetic informationwas available for 62 patients (50 pedigrees; supplemental Table 1,available on the Blood Web site; see the Supplemental Materialslink at the top of the online article). Exon 2 had the most mutationswith missense mutations accounting for the majority but nonsense,frameshift, and splice site mutations were also reported. Large genedeletions (up to 11 Mb) including those involving the whole genewere identified in 5 families. Three of these larger deletions wereassociated with gastrointestinal symptoms of colitis and gastritis.Such symptoms were not found in patients with other mutationsapart from a patient with diarrhea as a feature (missense mutationexon 1, 62 T � C). In a further 29 patients, detailed genetic datawere not supplied but a SAP/SH2D1A gene defect was confirmedby the documenting center.
Clinical manifestations of XLP1
Table 1 shows the presenting features of disease as well as featuresof disease manifesting throughout the course of the condition. HLHremained the most common presenting feature (39.6%), althoughdysgammaglobulinemia was the manifestation seen most com-monly in patients during the course of the illness.
Although clinical features have remained similar to previouslypublished data,2 the survival associated with XLP1 is 71.4%, whichis significantly improved over historical survival of 25%. Thesurvival associated with different phenotypes has also changedsignificantly with mortality associated with HLH decreased from96% to 65%, lymphoproliferative disease from 35% to 8%, anddysgammaglobulinemia from 55% to 5%.
Twenty-two patients suffered from malignant lymphoprolifera-tive disease, with eighteen patients (81.8%) diagnosed with B-cellnon-Hodgkin lymphoma mainly of the abdomen and cervicalregion. In 5 patients the disease was recurrent, with 1 patientexperiencing a cerebral tumor. Only 1 patient was reported withcerebral T-cell lymphoma. Data on tumor histology is lacking in3 patients.
Immunological abnormalities at diagnosis
Details of immune function were available for 57 patients, althoughin some cases, data were only available after the onset of diseasemanifestations that may have influenced immunoglobulin andlymphocyte subset levels. Immunoglobulin levels were recordedin 49 patients, and 32 of these showed varying degrees ofabnormal immunoglobulin levels. Twelve children presented withneutropenia. Lymphocyte subset data were available for 47 pa-tients; 19 showed a reduced percentage of B cells, 26 showed lowNK cell numbers, and 12 had a reversed CD4:CD8 ratio.
Presentation with HLH
The mortality for patients presenting with HLH was 65.6%, with amedian age at presentation of 3 years 2 months (range 8 months to9 years). Of the 32 patients with HLH, 16 underwent transplant, ofwhom 8 survived (50%; Figure 1). Of those who did not receive atransplant, only 3 survived (18.8%), confirming previous reportsthat the prognosis for patients with HLH associated with a geneticdefect is extremely poor and that HSCT is necessary.
EBV status
EBV status was documented in 79 patients showing that 51 (64.6%)were EBV positive at presentation or diagnosis (Table 2 and supplemen-tal Figure 1). The median age of presentation in this group was4 years (range 8 months to 40 years), and the overall mortality was35.2% (18/51). There was no significant difference in mortality betweenpatients with (35.2%) and without (28.6%) documented EBV infection.
Table 1. Presenting symptoms and features of XLP1 patients withassociated mortality
Incidence Mortality
Presenting symptom
HLH 31.9% 65.5%
FIM 7.7% 14.3%
Lymphoma 14.3% 7.7%
Dysgammaglobulinemia 22% 5%
Family history of
XLP1 alone
16.5% 20%
Other 7.7% 14.3%
Features occurring at any time
HLH 35.2% 65.6%
FIM 9.9% 22.2%
Lymphoma 24.2% 9%
Dysgammaglobulinemia 50.5% 13%
Other 15.4% 28.6%
Figure 1. Outcome of patients with HLH during course of disease. Survival ofpatients who present with HLH—patients who remain untransplanted have a poorsurvival outcome with only 18.8% survival. By contrast the survival of those whoundergo transplant is higher at 50%.
OUTCOME OF PATIENTS WITH XLP1 AND SH2D1A MUTATIONS 55BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom
HLH/FIM was the most common feature in this group being seen in35 patients (68.6%), with lymphoma present in 10 patients (19.6%), anddysgammaglobulinemia in 19 (37.2%). Nine EBV-positive patients hada family history of XLP1, and two others had a family history suggestiveof an X-linked immunodeficiency. Of the 18 EBV-positive patients whodied, the majority (14/18) died within 2 months of presentation due todisease progression. Three died in the early posttransplant period ofinfective complications and disease progression, and 1 died duringtreatment for lymphoma.
Twenty-eight patients were EBV negative at presentation ordiagnosis. The median age of presentation for this group was3 years (range birth to 31 years). Family history of XLP1 was thepresenting feature for 12 patients, and a further 7 patients describeda family history suggestive of an X-linked immunodeficiency orlymphoma. There was a higher rate of dysgammaglobulinemia(51.8%) in this group. Lymphoma was present in 7 patients. Fewer
EBV negative patients presented with HLH/FIM, and this maysuggest that at least for this manifestation a viral trigger isimportant. Information was sought on other viral infectious agentsincluding cytomegalovirus and adenovirus, but data were notavailable for most patients. Other clinical features included aplasticanemia in 3 patients and vasculitis in 2 patients. The mortality forthis EBV negative group was 28.6% (8/28); 3 patients died shortlyafter presentation before HSCT with central nervous systemvasculitis (2) and HLH with enterococcal sepsis (1). One patientdied 11 years after presentation following a complex course, anda further 4 patients died in the early posttransplant period (de-scribed in Table 5).
HSCT for XLP1
HSCT was undertaken in 22 centers (range of patients/center: 1-7)between 1997 and 2009 (Table 3). Forty-six transplants wereperformed on 43 patients, and the median age at transplant was6.25 years (range 8 months to 19 years); 1 patient who hadundergone a haploidentical transplant received a CD34� selectedboost 1 year after initial transplant. One patient received anallogeneic HSCT to treat lymphoma before a diagnosis of XLP1was established. Most patients received bone marrow or peripheralblood stem cells, and only 2 patients received umbilical cordHSCT. Donor grafts were from human leukocyte antigen-matchedfamily donors in 14 cases, mismatched family donors or matchedunrelated grafts in 28 cases, and haploidentical donors in 4 cases.Half of the transplant procedures (23/46) were performed usingmyeloablative conditioning regimes including combinations of
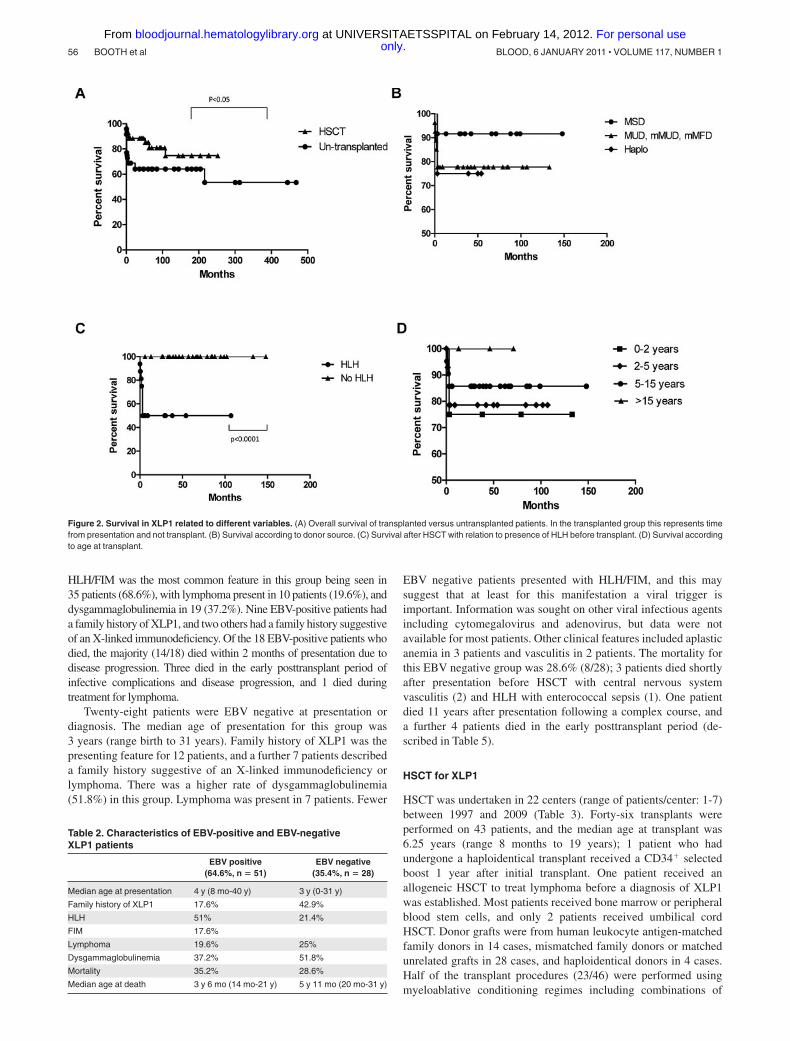
Figure 2. Survival in XLP1 related to different variables. (A) Overall survival of transplanted versus untransplanted patients. In the transplanted group this represents timefrom presentation and not transplant. (B) Survival according to donor source. (C) Survival after HSCT with relation to presence of HLH before transplant. (D) Survival accordingto age at transplant.
Table 2. Characteristics of EBV-positive and EBV-negativeXLP1 patients
EBV positive(64.6%, n � 51)
EBV negative(35.4%, n � 28)
Median age at presentation 4 y (8 mo-40 y) 3 y (0-31 y)
Family history of XLP1 17.6% 42.9%
HLH 51% 21.4%
FIM 17.6%
Lymphoma 19.6% 25%
Dysgammaglobulinemia 37.2% 51.8%
Mortality 35.2% 28.6%
Median age at death 3 y 6 mo (14 mo-21 y) 5 y 11 mo (20 mo-31 y)
56 BOOTH et al BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom
busulfan 12-20 mg/kg, cyclophosphamide 50-200 mg/kg, and totalbody irradiation 5-12 Gy. The other half of procedures usednonmyeloablative conditioning regimes consisting of fludarabine(30 mg/kg), melphalan (70-140 mg/kg), busulphan (4-12 mg/kg),or total body irradiation (3-5 Gy). Twenty-six patients receivedadditional serotherapy with alemtuzumab, anti-thymocyte globu-lin, anti-CD3 antibody, and anti-CD20 antibody (rituximab). Graft-versus-host disease (GVHD) prophylaxis regimes differed betweencenters, but mostly involved combinations of cyclosporin withmethotrexate, mycophenolate mofetil, steroids, and tacrolimus.T-cell depletion of the graft was used in 1 case.
Outcome for XLP1 patients who received allogeneic HSCTwas good with 81.4% surviving the procedure (35/43) with a medianfollow up of 52 months. The majority of these patients (28/35 survivors)required no ongoing immunoglobulin replacement therapy. Tables 3 and4 highlight details of transplanted patients, and Figure 2 describessurvival according to several factors.
Sixteen patients were diagnosed with HLH before transplantand 12 patients had some form of lymphoproliferative disease(lymphoma). Only 51.2% of the cohort had documented evidenceof EBV infection (by polymerase chain reaction) with survival
rates in EBV� patients similar to those without EBV infection(75% vs 80%). Most patients experienced some delay from firstsymptoms to diagnosis (average delay 2 years 7 months) but once adiagnosis of XLP1 was established time to transplant was generallyless than 1 year. Median age at transplant was 6.25 years with arange of 8 months to 19 years.
Univariate analysis was performed to identify the major riskfactors for survival after HSCT. The most important risk factor wasprior HLH, which significantly decreased the survival outcome to50%. A previous diagnosis of lymphoma had a near significanteffect, but other variables were not shown to have a significanteffect including importantly, previous evidence of EBV infection,the age at transplant, donor type, or the conditioning regime. It isalso important to note that only patients who had HLH at somepoint before or during transplant died. Conversely, all patientswithout HLH (n � 27) survived the transplant procedure.
Half of the patients underwent a nonmyeloablative conditioningregime before HSCT and this did not impact on survival (nonmyelo-ablative vs myeloablative, 78.9% vs 82.9%) or long-term chimer-ism. More than 90% of patients achieved full donor chimerism, and
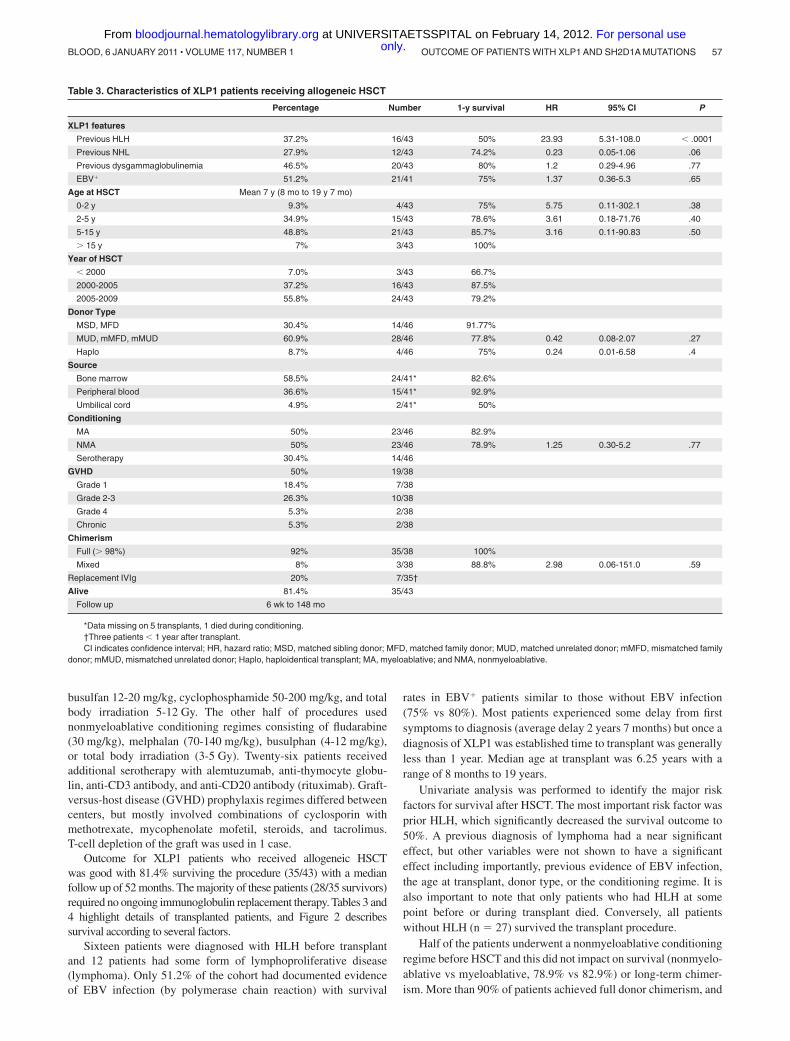
Table 3. Characteristics of XLP1 patients receiving allogeneic HSCT
Percentage Number 1-y survival HR 95% CI P
XLP1 features
Previous HLH 37.2% 16/43 50% 23.93 5.31-108.0 � .0001
Previous NHL 27.9% 12/43 74.2% 0.23 0.05-1.06 .06
Previous dysgammaglobulinemia 46.5% 20/43 80% 1.2 0.29-4.96 .77
EBV� 51.2% 21/41 75% 1.37 0.36-5.3 .65
Age at HSCT Mean 7 y (8 mo to 19 y 7 mo)
0-2 y 9.3% 4/43 75% 5.75 0.11-302.1 .38
2-5 y 34.9% 15/43 78.6% 3.61 0.18-71.76 .40
5-15 y 48.8% 21/43 85.7% 3.16 0.11-90.83 .50
� 15 y 7% 3/43 100%
Year of HSCT
� 2000 7.0% 3/43 66.7%
2000-2005 37.2% 16/43 87.5%
2005-2009 55.8% 24/43 79.2%
Donor Type
MSD, MFD 30.4% 14/46 91.77%
MUD, mMFD, mMUD 60.9% 28/46 77.8% 0.42 0.08-2.07 .27
Haplo 8.7% 4/46 75% 0.24 0.01-6.58 .4
Source
Bone marrow 58.5% 24/41* 82.6%
Peripheral blood 36.6% 15/41* 92.9%
Umbilical cord 4.9% 2/41* 50%
Conditioning
MA 50% 23/46 82.9%
NMA 50% 23/46 78.9% 1.25 0.30-5.2 .77
Serotherapy 30.4% 14/46
GVHD 50% 19/38
Grade 1 18.4% 7/38
Grade 2-3 26.3% 10/38
Grade 4 5.3% 2/38
Chronic 5.3% 2/38
Chimerism
Full (� 98%) 92% 35/38 100%
Mixed 8% 3/38 88.8% 2.98 0.06-151.0 .59
Replacement IVIg 20% 7/35†
Alive 81.4% 35/43
Follow up 6 wk to 148 mo
*Data missing on 5 transplants, 1 died during conditioning.†Three patients � 1 year after transplant.CI indicates confidence interval; HR, hazard ratio; MSD, matched sibling donor; MFD, matched family donor; MUD, matched unrelated donor; mMFD, mismatched family
donor; mMUD, mismatched unrelated donor; Haplo, haploidentical transplant; MA, myeloablative; and NMA, nonmyeloablative.
OUTCOME OF PATIENTS WITH XLP1 AND SH2D1A MUTATIONS 57BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom
those with a mixed or falling chimerism remained well with 1patient still receiving replacement immunoglobulin.
Data were also collected on common posttransplant com-plications such as GVHD, infectious complications and toxicityattributable to chemotherapy. Half of the patients (50%) sufferedfrom some form of GVHD; the majority of cases were grade1-3 affecting the skin, liver, and gut. Two patients suffered grade4 disease (of skin and liver), and 1 of these children died. Only2 patients went on to develop chronic GVHD (see Table 3). Onepatient experienced both veno-occlusive disease and renal toxicitydue to conditioning (busulfan, cyclophosphamide, and antithymo-cyte globulin), and this patient succumbed shortly after a haploiden-tical transplant.
In 3 patients with mixed chimerism in peripheral bloodmononuclear cells, this remained stable in all but 1 patient, inwhom it fell from 92% to 5%. However, this patient remains well3 years posttransplant and does not require replacement immuno-globulin therapy. From this series, there is little evidence of viralreactivation posttransplant. Thirty-five patients are alive with5 suffering some long-term effects including EBV viremia (man-aged with rituximab), bronchiectasis, autoimmune disease, chronicpsoriasis, and neutropenia.
Eight patients did not survive after HSCT (see Table 5). Sevenpatients who died presented with HLH before HSCT (4/7 EBV�)compared with 8 of 35 survivors, but HLH was a feature of diseasein all 8 nonsurvivors. The majority of nonsurvivors were � 3 yearsold (5/8), and conditioning regime did not appear to play a role as5/8 patients received a full myeloablative regime. The main causeof death in this group was sepsis, but disease progression accountedfor 2 deaths. The 2 children dying with disease progression wentinto transplant with active disease; 1 died during conditioning andthe other 3 days after HSCT. One further patient died 3 weeks afterHSCT (7 months after presentation) from veno-occlusive disease(VOD), multiorgan failure, and renal toxicity attributable tochemotherapy. The remaining 5 patients died of sepsis (2 pseu-domonal sepsis, 1 parainfluenza III infection, 1 with disseminatedadenoviral infection, and 1 with EBV and fungal infection) within3 months of HSCT.
Untransplanted patients
Data were available for 48 patients who did not receive HSCT(Table 6); 30 are alive, 4 of whom are actively awaiting transplant,and 3 who refused HSCT. One patient had received an autologousHSCT before diagnosis with XLP1, and this patient’s data were
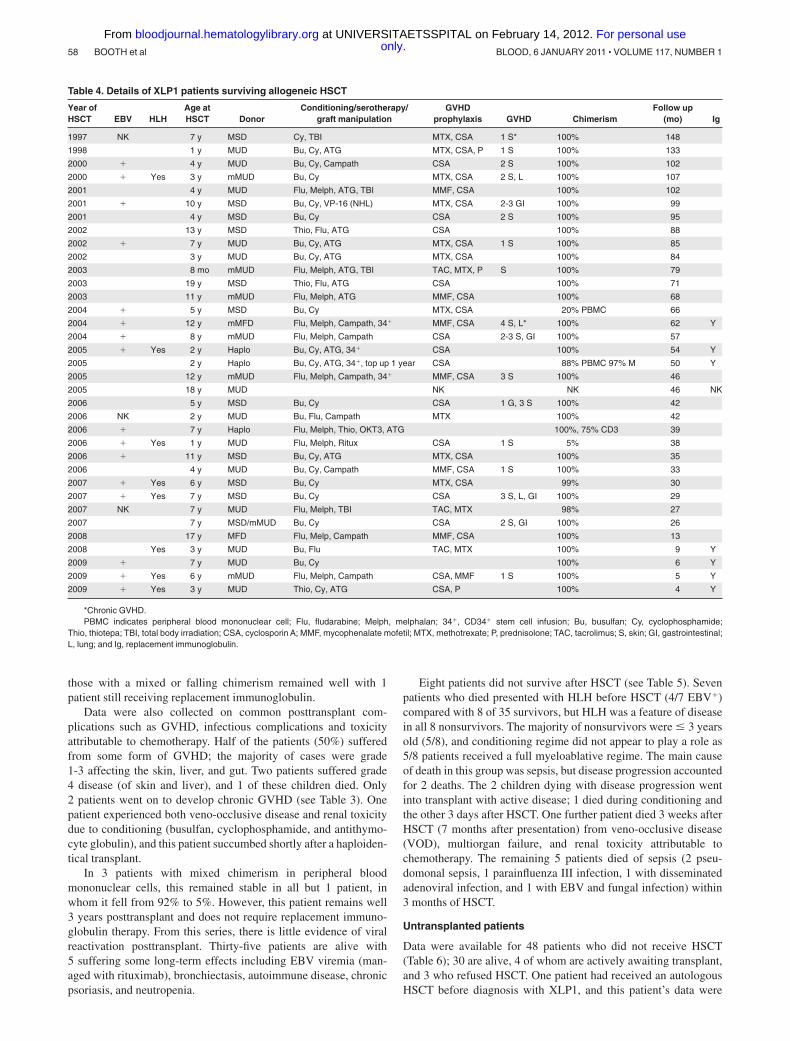
Table 4. Details of XLP1 patients surviving allogeneic HSCT
Year ofHSCT EBV HLH
Age atHSCT Donor
Conditioning/serotherapy/graft manipulation
GVHDprophylaxis GVHD Chimerism
Follow up(mo) Ig
1997 NK 7 y MSD Cy, TBI MTX, CSA 1 S* 100% 148
1998 1 y MUD Bu, Cy, ATG MTX, CSA, P 1 S 100% 133
2000 � 4 y MUD Bu, Cy, Campath CSA 2 S 100% 102
2000 � Yes 3 y mMUD Bu, Cy MTX, CSA 2 S, L 100% 107
2001 4 y MUD Flu, Melph, ATG, TBI MMF, CSA 100% 102
2001 � 10 y MSD Bu, Cy, VP-16 (NHL) MTX, CSA 2-3 GI 100% 99
2001 4 y MSD Bu, Cy CSA 2 S 100% 95
2002 13 y MSD Thio, Flu, ATG CSA 100% 88
2002 � 7 y MUD Bu, Cy, ATG MTX, CSA 1 S 100% 85
2002 3 y MUD Bu, Cy, ATG MTX, CSA 100% 84
2003 8 mo mMUD Flu, Melph, ATG, TBI TAC, MTX, P S 100% 79
2003 19 y MSD Thio, Flu, ATG CSA 100% 71
2003 11 y mMUD Flu, Melph, ATG MMF, CSA 100% 68
2004 � 5 y MSD Bu, Cy MTX, CSA 20% PBMC 66
2004 � 12 y mMFD Flu, Melph, Campath, 34� MMF, CSA 4 S, L* 100% 62 Y
2004 � 8 y mMUD Flu, Melph, Campath CSA 2-3 S, GI 100% 57
2005 � Yes 2 y Haplo Bu, Cy, ATG, 34� CSA 100% 54 Y
2005 2 y Haplo Bu, Cy, ATG, 34�, top up 1 year CSA 88% PBMC 97% M 50 Y
2005 12 y mMUD Flu, Melph, Campath, 34� MMF, CSA 3 S 100% 46
2005 18 y MUD NK NK 46 NK
2006 5 y MSD Bu, Cy CSA 1 G, 3 S 100% 42
2006 NK 2 y MUD Bu, Flu, Campath MTX 100% 42
2006 � 7 y Haplo Flu, Melph, Thio, OKT3, ATG 100%, 75% CD3 39
2006 � Yes 1 y MUD Flu, Melph, Ritux CSA 1 S 5% 38
2006 � 11 y MSD Bu, Cy, ATG MTX, CSA 100% 35
2006 4 y MUD Bu, Cy, Campath MMF, CSA 1 S 100% 33
2007 � Yes 6 y MSD Bu, Cy MTX, CSA 99% 30
2007 � Yes 7 y MSD Bu, Cy CSA 3 S, L, GI 100% 29
2007 NK 7 y MUD Flu, Melph, TBI TAC, MTX 98% 27
2007 7 y MSD/mMUD Bu, Cy CSA 2 S, GI 100% 26
2008 17 y MFD Flu, Melp, Campath MMF, CSA 100% 13
2008 Yes 3 y MUD Bu, Flu TAC, MTX 100% 9 Y
2009 � 7 y MUD Bu, Cy 100% 6 Y
2009 � Yes 6 y mMUD Flu, Melph, Campath CSA, MMF 1 S 100% 5 Y
2009 � Yes 3 y MUD Thio, Cy, ATG CSA, P 100% 4 Y
*Chronic GVHD.PBMC indicates peripheral blood mononuclear cell; Flu, fludarabine; Melph, melphalan; 34�, CD34� stem cell infusion; Bu, busulfan; Cy, cyclophosphamide;
Thio, thiotepa; TBI, total body irradiation; CSA, cyclosporin A; MMF, mycophenalate mofetil; MTX, methotrexate; P, prednisolone; TAC, tacrolimus; S, skin; GI, gastrointestinal;L, lung; and Ig, replacement immunoglobulin.
58 BOOTH et al BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom
analyzed as though untransplanted. Less detailed information wasavailable for this set of patients compared with those receivingHSCT. This may be because some patients died before EBV statusand immune function could be established and any first symptomsmay not have been recognized as a manifestation of XLP1. Fromdata available, median age at presentation was 5 years, and delay indiagnosis ranged from a few weeks to 32 years.
Presentation was highly variable but as expected includedHLH/FIM, dysgammaglobulinemia, and recurrent infection. Moreunusual presentations included 1 patient with central nervoussystem vasculitis, intracranial hemorrhage and myocardial fibrosis,and peripheral eosinophilia. The course of XLP1, both temporaland clinical, was extremely variable without any apparent correla-tion to family history or genetic mutation.
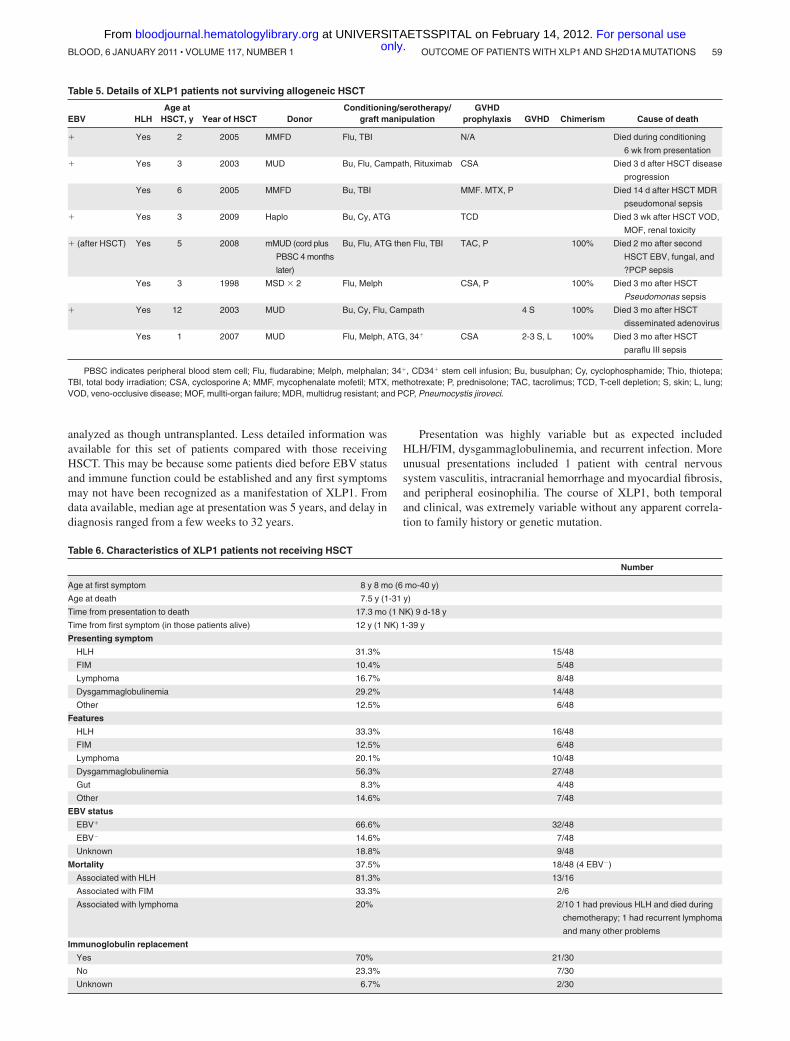
Table 5. Details of XLP1 patients not surviving allogeneic HSCT
EBV HLHAge at
HSCT, y Year of HSCT DonorConditioning/serotherapy/
graft manipulationGVHD
prophylaxis GVHD Chimerism Cause of death
� Yes 2 2005 MMFD Flu, TBI N/A Died during conditioning
6 wk from presentation
� Yes 3 2003 MUD Bu, Flu, Campath, Rituximab CSA Died 3 d after HSCT disease
progression
Yes 6 2005 MMFD Bu, TBI MMF. MTX, P Died 14 d after HSCT MDR
pseudomonal sepsis
� Yes 3 2009 Haplo Bu, Cy, ATG TCD Died 3 wk after HSCT VOD,
MOF, renal toxicity
� (after HSCT) Yes 5 2008 mMUD (cord plus
PBSC 4 months
later)
Bu, Flu, ATG then Flu, TBI TAC, P 100% Died 2 mo after second
HSCT EBV, fungal, and
?PCP sepsis
Yes 3 1998 MSD � 2 Flu, Melph CSA, P 100% Died 3 mo after HSCT
Pseudomonas sepsis
� Yes 12 2003 MUD Bu, Cy, Flu, Campath 4 S 100% Died 3 mo after HSCT
disseminated adenovirus
Yes 1 2007 MUD Flu, Melph, ATG, 34� CSA 2-3 S, L 100% Died 3 mo after HSCT
paraflu III sepsis
PBSC indicates peripheral blood stem cell; Flu, fludarabine; Melph, melphalan; 34�, CD34� stem cell infusion; Bu, busulphan; Cy, cyclophosphamide; Thio, thiotepa;TBI, total body irradiation; CSA, cyclosporine A; MMF, mycophenalate mofetil; MTX, methotrexate; P, prednisolone; TAC, tacrolimus; TCD, T-cell depletion; S, skin; L, lung;VOD, veno-occlusive disease; MOF, mullti-organ failure; MDR, multidrug resistant; and PCP, Pneumocystis jiroveci.
Table 6. Characteristics of XLP1 patients not receiving HSCT
Number
Age at first symptom 8 y 8 mo (6 mo-40 y)
Age at death 7.5 y (1-31 y)
Time from presentation to death 17.3 mo (1 NK) 9 d-18 y
Time from first symptom (in those patients alive) 12 y (1 NK) 1-39 y
Presenting symptom
HLH 31.3% 15/48
FIM 10.4% 5/48
Lymphoma 16.7% 8/48
Dysgammaglobulinemia 29.2% 14/48
Other 12.5% 6/48
Features
HLH 33.3% 16/48
FIM 12.5% 6/48
Lymphoma 20.1% 10/48
Dysgammaglobulinemia 56.3% 27/48
Gut 8.3% 4/48
Other 14.6% 7/48
EBV status
EBV� 66.6% 32/48
EBV� 14.6% 7/48
Unknown 18.8% 9/48
Mortality 37.5% 18/48 (4 EBV�)
Associated with HLH 81.3% 13/16
Associated with FIM 33.3% 2/6
Associated with lymphoma 20% 2/10 1 had previous HLH and died during
chemotherapy; 1 had recurrent lymphoma
and many other problems
Immunoglobulin replacement
Yes 70% 21/30
No 23.3% 7/30
Unknown 6.7% 2/30
OUTCOME OF PATIENTS WITH XLP1 AND SH2D1A MUTATIONS 59BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom

As with transplanted patients the significant mortality associ-ated with HLH is evident in untransplanted patients (81.3%).Presentation or manifestation of HLH (n � 15 and 16, respec-tively) was associated with a rapid decline and death within6 weeks, especially in patients less than 5 years of age. Of the48 patients, 32 did not have manifestations of HLH, and in thisgroup 5 died, thereby giving a survival of 84.4% with a meanfollow-up in this group of 11.6 years. For those untransplantedpatients who survive, 70% received replacement immunoglobulintherapy, with few suffering from long-term complications. Only5 patients have recorded complications, including 1 with recurrentinfection, 1 with neutropenia, 1 with bronchiectasis, and 2 boyswith gastrointestinal disease and growth delay.
Supplemental Table 2 compares the demographics betweentransplanted and untransplanted patients. No significant differenceswere seen between the 2 populations other than mortality, whichwas twice as high in the untransplanted cohort (P � .05). Age ofdeath was lower in transplanted patients and may reflect the moresevere course that may have led to the need for HSCT.
Discussion
This report summarizes data on 91 patients from 64 familiesworldwide with a genetic diagnosis of XLP1 and provides informa-tion on outcome with and without allogeneic HSCT using currenttreatment protocols (summarized in Figure 3). This report is thefirst large-scale analysis of XLP1 patients since the report by theXLP1 registry in 1995 and has for the first time gathered patientswho have confirmed SAP/SH2D1A mutations. Therefore thisreport represents a genetically homogeneous cohort and avoidspossible phenotypic variability through inclusion of other patientswith genetic defects such as XIAP/BIRC 4 mutations.
The clinical features of the disease are similar to those reportedby the XLP1 Registry, with HLH and FIM remaining the mostcommon and most lethal complication. With the advent of moreaccessible genetic screening and mutation analysis confirming thediagnosis, more patients have been diagnosed early on the basis offamily history and increased awareness of the disease has also ledto patients being diagnosed after presentation with immune dysregu-lation and more unusual presenting features such as vasculitis.
A diagnosis of XLP1 is still a difficult one to make, and it ispossible that some patients mistakenly fall under the umbrella ofcommon variable immunodeficiency, although previous geneticscreening studies suggest that the incidence of XLP1 patients in
common variable immunodeficiency cohorts is low.33 It is alsopossible that there are older individuals who present in adulthoodand have not been identified and included in this study, and thismay result in a bias in the method of data collection as the majorityof centers approached to contribute data were specialist pediatriccenters. For example, a recent case report describes a 41-year-oldman who presented with an EBV-induced central nervous systemB-cell lymphoma and absent B cells.34 The oldest surviving patientfrom this cohort presented at the age of 7 years with recurrentinfections and hypogammaglobulinemia, but remains well withouttransplant and is receiving replacement immunoglobulin therapy at46 years of age.
The prognosis for XLP1 has greatly improved since 1995, whenSeemayer et al2 reported an overall survival of 25% survival with71.4% of patients in this cohort alive at the time of data analysis.Indeed, the mortality in untransplanted patients was lower than weexpected, with 62.5% surviving, including 3 boys who presentedwith HLH, but the mortality in this group secondary to HLHremains high at 81.3%. It is also interesting to note that aconsiderable mortality of 28.6% is seen in EBV-negative patientswho do not receive HSCT and is related to HLH, sepsis, andvasculitis, suggesting that underlying immunological abnormalitiesin XLP1, and not only EBV-driven disease, can be fatal. Few complica-tions from recurrent infection and immune dysregulation were reported,suggesting that early diagnosis and good supportive care with replace-ment immunoglobulin and prophylactic antibiotics can improve theoutcome for untransplanted patients. Although over 60% of patientssurvive without HSCT, it will be important to follow patients carefully,since there is the potential for more severe manifestations to arise, andthe options for transplant should be explored.
The mortality associated with the different clinical phenotypeshas changed over time, with an improved survival for both HLH(34.5% vs 4%) and lymphoma (91% vs 35%).2 This most likelyreflects improved treatment strategies for both HLH (especially theuse of agreed protocols such as HLH 9435 and 200436) and malignancy.Although these figures represent survival with either HLH or lymphomaas features of XLP1 at any stage, they are very similar to the survivalseen if patients present with these features (44.5% and 92% forHLH/FIM and lymphoma, respectively). A mortality of 13% in patientswho exhibit dysgammaglobulinemia is associated with HLH, infection,vasculitis, and hemorrhage and highlights that although clinically thisphenotype may be milder, it is not an innocuous phenotype, andprogression to further fatal symptoms is not uncommon.
The outcome data following allogeneic HSCT from this reportis encouraging. The outcome data presented is the largest ever
Figure 3. Outcome of patients with SAP/SH2D1A mutations.
60 BOOTH et al BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom

gathered and shows that approximately 80% of patients survive theprocedure with complete cellular and humoral reconstitution in thelarge majority of cases. In this series, there is little evidence ofproblematic EBV reactivation adversely affecting transplant out-come and no increased incidence of long-term complicatingfeatures such as autoimmunity in comparison to transplant for otherconditions.37,38 Although donor chimerism in the majority ofpatients was complete, even low level chimerism in 2 patients with5% and 20% donor chimerism was associated with good immunerecovery. Conversely however, when the patients who requiredongoing immunoglobulin support are analyzed, all but 1 have100% donor engraftment. Further detailed lineage-specific analysisand study of T- and B-cell function in these patients is necessary todetermine why humoral function has not been established. Theavailability of a fully matched donor is associated with animproved survival outcome (approximately 92%), although withthe present low numbers this is not statistically significant.Haploidentical grafts show a good outcome in this cohort, but thenumbers are extremely low (only 4 transplants performed), andtherefore this information needs to be interpreted with caution.
The most important factor affecting survival after transplant is amanifestation of HLH, which significantly reduces survival to50%. Indeed all 8 patients who died had a complication of HLH atsome point in their clinical course. This may reflect the effects ofHLH itself or HLH chemotherapy and immunosuppression on thetransplant process, including increased organ related toxicity andincreased susceptibility to pathogens. In comparison to datareported on cohorts of patients undergoing transplant for HLHassociated with other gene defects (eg, perforin and munc 13-4)39-41
it appears that the outcome for HLH associated with XLP1 is worseand may relate to the multiple immune deficits associated with SAPdeficiency. By contrast all XLP1 patients who had no HLHmanifestations (n � 27) survived the HSCT procedure.
These data may now allow more informed recommendations tobe made regarding transplantation in XLP1. It is clear from thisreport that HLH in XLP1 has a very poor prognosis if leftuntransplanted. Therefore any individual with HLH as a manifesta-tion of XLP1 should undergo allogeneic HSCT.
For patients who are newly diagnosed because of a familyhistory but with no clinical features or for those who present withmanifestations other than HLH/FIM, the decision to transplant arelatively well child has been more challenging. An importantobservation from this report is that all patients (n � 27) who wentinto transplant without prior HLH survived the procedure incomparison to 84.4% survival for those who are untransplanted andhave not manifested with HLH. Since progression to HLH withouttransplant may occur at a later stage, there is a strong argument totransplant all individuals with a diagnosis of XLP1.
However, there is a counter argument to such a recommenda-tion. As with other immunodeficiencies, the data collected andpresented here may not give a complete picture of the natural
course of XLP1 and is a historical cohort study conducted beforethe advent of recent improved therapies. Further, milder patientsmay also remain undiagnosed having been labeled with a diagnosisof common variable immunodeficiency. It is also the case that HLHis most often seen in younger patients (median age of presentation3.2 years) and older individuals are less likely to manifest withHLH. There may also be reluctance on the part of families andphysicians to undertake a transplant in a well child given that, evenin the best-case scenario, there will be a certain mortality associ-ated with any allogeneic transplant procedure.
A more pragmatic recommendation would be to undertaketransplant in all patients presenting or manifesting with HLH.Similarly for newly diagnosed or young children without any HLH,if a well-matched donor is available, HSCT should be undertaken,since a manifestation of HLH may be catastrophic or may severelycompromise transplant outcome. For older individuals, we wouldstill recommend that HSCT be undertaken, but this decision totransplant should be based on available donor status, wellbeing ofthe patient, and the attitude of family and physician to the risk oftransplant. If HSCT is not undertaken immediately, it is recom-mended that a donor source is identified and that all patientsare followed very carefully in case of disease progression andonset of other manifestations, at which point HSCT could beperformed rapidly.
Acknowledgments
We thank the European Society for Immunodeficiencies registry,which helped to identify patients in the European database. Wethank the Inborn Errors Working Party of the European Group forBlood and Marrow Transplantation for initiating the study.
This work was supported by the Wellcome Trust (082159/2/07/2; C.B. is a Clinical Research Fellow).
Authorship
Contribution: C.B. designed the research, collected and acquireddata, analyzed the data, and wrote the manuscript; H.B.G. assumedoverall responsibility for the research, oversaw analysis, andrevised the manuscript; and all authors contributed clinical data andreviewed the manuscript before submission.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
For a complete list of Inborn Errors Working Party participants,please see the supplemental Appendix.
Correspondence: Hubert B. Gaspar, Center for Immunodefi-ciency, Molecular Immunology Unit, Institute of Child Health,30 Guilford St, London, WC1N 1EH, United Kingdom; e-mail:[email protected].
References
1. Purtilo DT, Cassel CK, Yang JP, Harper R.X-linked recessive progressive combined variableimmunodeficiency (Duncan’s disease). Lancet.1975;1(7913):935-940.
2. Seemayer TA, Gross TG, Egeler RM, et al.X-linked lymphoproliferative disease: twenty-fiveyears after the discovery. Pediatr Res. 1995;38(4):471-478.
3. Purtilo DT, Grierson HL. Methods of detection of newfamilies with X-linked lymphoproliferative disease.Cancer Genet Cytogenet. 1991;51(2):143-153.
4. Sumegi J, Huang D, Lanyi A, et al. Correlation ofmutations of the SH2D1A gene and Epstein-Barrvirus infection with clinical phenotype and out-come in X-linked lymphoproliferative disease.Blood. 2000;96(9):3118-3125.
5. Rougemont AL, Fournet JC, Martin SR, et al.Chronic active gastritis in X-linked lymphoprolifera-tive disease. Am J Surg Pathol. 2008;32(2):323-328.
6. Talaat KR, Rothman JA, Cohen JI, et al. Lympho-cytic vasculitis involving the central nervous sys-tem occurs in patients with X-linked lymphoprolif-
erative disease in the absence of Epstein-Barrvirus infection. Pediatr Blood Cancer. 2009;53(6):1120-1123.
7. Loeffel S, Chang CH, Heyn R, et al. Necrotizinglymphoid vasculitis in X-linked lymphoproliferativesyndrome. Arch Pathol Lab Med. 1985;109(6):546-550.
8. Kanegane H, Ito Y, Ohshima K, et al. X-linkedlymphoproliferative syndrome presenting withsystemic lymphocytic vasculitis. Am J Hematol.2005;78(2):130-133.
OUTCOME OF PATIENTS WITH XLP1 AND SH2D1A MUTATIONS 61BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom

9. Sayos J, Wu C, Morra M, et al. The X-linkedlymphoproliferative-disease gene product SAPregulates signals induced through the co-receptorSLAM. Nature. 1998;395(6701):462-469.
10. Coffey AJ, Brooksbank RA, Brandau O, et al.Host response to EBV infection in X-linked lym-phoproliferative disease results from mutations inan SH2-domain encoding gene. Nat Genet. 1998;20(2):129-135.
11. Nichols KE, Harkin DP, Levitz S, et al. Inactivatingmutations in an SH2 domain-encoding gene inX-linked lymphoproliferative syndrome. Proc NatlAcad Sci U S A. 1998;95(23):13765-13770.
12. Okano M, Gross TG. Epstein-Barr virus-associated hemophagocytic syndrome and fatalinfectious mononucleosis. Am J Hematol. 1996;53(2):111-115.
13. Dupre L, Andolfi G, Tangye SG, et al. SAP con-trols the cytolytic activity of CD8� T cells againstEBV-infected cells. Blood. 2005;105(11):4383-4389.
14. Sharifi R, Sinclair JC, Gilmour KC, et al. SAP me-diates specific cytotoxic T-cell functions inX-linked lymphoproliferative disease. Blood.2004;103(10):3821-3827.
15. Benoit L, Wang X, Pabst HF, Dutz J, Tan R.Defective NK cell activation in X-linked lym-phoproliferative disease. J Immunol. 2000;165(7):3549-3553.
16. Nakajima H, Cella M, Bouchon A, et al. Patientswith X-linked lymphoproliferative disease have adefect in 2B4 receptor-mediated NK cell cytotox-icity. Eur J Immunol. 2000;30(11):3309-3318.
17. Tangye SG, Phillips JH, Lanier LL, Nichols KE.Functional requirement for SAP in 2B4-mediatedactivation of human natural killer cells as revealedby the X-linked lymphoproliferative syndrome.J Immunol. 2000;165(6):2932-2936.
18. Bottino C, Augugliaro R, Castriconi R, et al. Analysisof the molecular mechanism involved in 2B4-mediated NK cell activation: evidence that human2B4 is physically and functionally associated withthe linker for activation of T cells. Eur J Immunol.2000;30(12):3718-3722.
19. Nichols KE, Hom J, Gong SY, et al. Regulation ofNKT cell development by SAP, the protein defec-tive in XLP. Nat Med. 2005;11(3):340-345.
20. Chung B, Aoukaty A, Dutz J, Terhorst C, Tan R.Signaling lymphocytic activation molecule-associ-
ated protein controls NKT cell functions. J Immu-nol. 2005;174(6):3153-3157.
21. Ma CS, Pittaluga S, Avery DT, et al. Selectivegeneration of functional somatically mutatedIgM�CD27�, but not Ig isotype-switched,memory B cells in X-linked lymphoproliferativedisease. J Clin Invest. 2006;116(2):322-333.
22. Nichols KE, Ma CS, Cannons JL, Schwartzberg PL,Tangye SG. Molecular and cellular pathogenesis ofX-linked lymphoproliferative disease. Immunol Rev.2005;203:180-199.
23. Ma CS, Hare NJ, Nichols KE, et al. Impaired hu-moral immunity in X-linked lymphoproliferativedisease is associated with defective IL-10 pro-duction by CD4� T cells. J Clin Invest. 2005;115(4):1049-1059.
24. Gilmour KC, Cranston T, Jones A, et al. Diagnosisof X-linked lymphoproliferative disease by analy-sis of SLAM-associated protein expression. EurJ Immunol. 2000;30(6):1691-1697.
25. Okano M, Pirruccello SJ, Grierson HL, et al. Im-munovirological studies of fatal infectious mono-nucleosis in a patient with X-linked lymphoprolif-erative syndrome treated with intravenousimmunoglobulin and interferon-�. Clin ImmunolImmunopathol. 1990;54(3):410-418.
26. Trottestam H, Beutel K, Meeths M, et al. Treat-ment of the X-linked lymphoproliferative, Griscelliand Chediak-Higashi syndromes by HLH directedtherapy. Pediatr Blood Cancer. 2009;52(2):268-272.
27. Lankester AC, Visser LF, Hartwig NG, et al. Allo-geneic stem cell transplantation in X-linked lym-phoproliferative disease: two cases in one familyand review of the literature. Bone Marrow Trans-plant. 2005;36(2):99-105.
28. Strahm B, Rittweiler K, Duffner U, et al. RecurrentB-cell non-Hodgkin’s lymphoma in two brotherswith X-linked lymphoproliferative disease withoutevidence for Epstein-Barr virus infection. Br JHaematol. 2000;108(2):377-382.
29. Arkwright PD, Makin G, Will AM, et al. X linkedlymphoproliferative disease in a United Kingdomfamily. Arch Dis Child. 1998;79(1):52-55.
30. Amrolia P, Gaspar HB, Hassan A, et al. Non-myeloablative stem cell transplantation for con-genital immunodeficiencies. Blood. 2000;96(4):1239-1246.
31. Hugle B, Suchowerskyj P, Hellebrand H, et al.
Persistent hypogammaglobulinemia followingmononucleosis in boys is highly suggestive ofX-linked lymphoproliferative disease - report ofthree cases. J. Clin Immunol. 2004;24(5):515-522.
32. Nistala K, Gilmour KC, Cranston T, et al. X-linkedlymphoproliferative disease: three atypical cases.Clin Exp Immunol. 2001;126(1):126-130.
33. Eastwood D, Gilmour KC, Nistala K, et al. Preva-lence of SAP gene defects in male patients diag-nosed with common variable immunodeficiency.Clin Exp Immunol. 2004;137(3):584-588.
34. Hervier B, Latour S, Loussouarn D, et al. Anatypical case of X-linked lymphoproliferative dis-ease revealed as a late cerebral lymphoma.J Neuroimmunol. 2009;218(1-2):125-128.
35. Henter JI, Arico M, Egeler RM, et al. HLH-94: atreatment protocol for hemophagocytic lympho-histiocytosis. HLH study Group of the HistiocyteSociety. Med Pediatr Oncol. 1997;28(5):342-347.
36. Henter JI, Horne A, Arico M, et al. HLH-2004: di-agnostic and therapeutic guidelines for he-mophagocytic lymphohistiocytosis. Pediatr BloodCancer. 2007;48(2):124-131.
37. O’Brien TA, Eastlund T, Peters C, et al. Autoim-mune haemolytic anaemia complicating haema-topoietic cell transplantation in paediatric pa-tients: high incidence and significant mortality inunrelated donor transplants for non-malignantdiseases. Br J Haematol. 2004;127(1):67-75.
38. Sonici E, Bennato B, Bertoni E, et al. Autoimmu-nity after BMT in primary immunodeficiency dis-eases: single centre report of 184 children [ab-stract]. 36th Annual Meeting of the EuropeanGroup for Blood and Marrow Transplantation;2010.
39. Cooper N, Rao K, Gilmour K, et al. Stem celltransplantation with reduced-intensity condition-ing for hemophagocytic lymphohistiocytosis.Blood. 2006;107(3):1233-1236.
40. Yoon HS, Im HJ, Moon HN, et al. The outcome ofhematopoietic stem cell transplantation in Koreanchildren with hemophagocytic lymphohistiocyto-sis. Pediatr Transplant. 2010;20(2):438-45.
41. Ohga S, Kudo K, Ishii E, et al. Hematopoieticstem cell transplantation for familial hemophago-cytic lymphohistiocytosis and Epstein-Barr virus-associated hemophagocytic lymphohistiocytosisin Japan. Pediatr Blood Cancer. 2010;54(2):299-306.
62 BOOTH et al BLOOD, 6 JANUARY 2011 � VOLUME 117, NUMBER 1 only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom

Errata
Cappellini MD, Bejaoui M, Agaoglu L, et al. Iron chelation with deferasirox in adult andpediatric patients with thalassemia major: efficacy and safety during 5 years’ follow-up. Blood.2011;118(4):884-893.
On page 884 in the 28 July 2011 issue, there is an error in the affiliation of the first author (Cappellini). The word “Scientifico” ismisspelled. The affiliation should have read: “1Universita di Milano, Ca Granda Foundation Istituto di Ricovero e Cura a CarattereScientifico (IRCCS), Milan, Italy.” On pages 885, 887, 889, 891, and 893, there is an error in the running title of the article; the word“EFFICIENCY” should be “EFFICACY.” The running title should have read: “DEFERASIROX 5-YEAR EFFICACY AND SAFETY.”The errors were corrected in the online version, which now differs from the print version.
Booth C, Gilmour KC, Veys P, et al. X-linked lymphoproliferative disease due to SAP/SH2D1Adeficiency: a multicenter study on the manifestations, management and outcome of thedisease. Blood. 2011;117(1):53-62.
On page 53 of the 6 January 2011 issue, the 17th author’s last name was misspelled Pachlopnick-Schmid. The correct name is PachlopnikSchmid. The error has been corrected in the online version, which now differs from the print version.
Gregori S, Tomasoni D, Pacciani V, et al. Differentiation of type 1 T regulatory cells (Tr1) bytolerogenic DC-10 requires the IL-10–dependent ILT4/HLA-G pathway. Blood. 2010;116(6):935-944.
On page 935 in the 12 August 2010 issue, one of the affiliations of the third author (Valentina Pacciani) is incorrect. The second affiliationlisted as “Department of Pediatrics, Universita di Tor Vergata, Rome” should have been: “University Department of Pediatrics (DPUO),Bambino Gesu Children’s Hospital, Rome, Italy.” The correct byline and affiliations are shown. The error has been corrected in the onlineversion, which now differs from the print version.
Silvia Gregori,1 Daniela Tomasoni,1 Valentina Pacciani,1,2 Miriam Scirpoli,3 Manuela Battaglia,1,3 Chiara Francesca Magnani,1 EhudHauben,1 and Maria-Grazia Roncarolo1,4
1Department of Regenerative Medicine, Stem Cells, and Gene Therapy, San Raffaele Telethon Institute for Gene Therapy, Milan, Italy;2University Department of Pediatrics (DPUO), Bambino Gesu Children’s Hospital, Rome, Italy; 3Department of Immunology,Transplantation, and Infectious Diseases, Diabetes Research Institute, Milan, Italy; and 4Universita Vita-Salute San Raffaele, Milan, Italy
Gomes AL, Carvalho T, Serpa J, Torre C, Dias S. Hypercholesterolemia promotes bone marrowcell mobilization by perturbing the SDF-1:CXCR4 axis. Blood. 2010;115(19):3886-3894.
On pages 3888, 3889, and 3892 in the 13 May 2010 issue, there are errors in the color of the bars in the plots of several figure panels. InFigures 1C, 1D, 2B, 6B, and 6C, when parameters are compared between the 2 conditions, “Normal diet” on the left should be gray, and“High-cholesterol diet” on the right should be black. The corrected Figures 1, 2, and 6 are shown.
5060 BLOOD, 3 NOVEMBER 2011 � VOLUME 118, NUMBER 18
only.For personal use at UNIVERSITAETSSPITAL on February 14, 2012. bloodjournal.hematologylibrary.orgFrom