xpd-dependent activation of apoptosis in response to triplex
TRANSCRIPT

XPD-dependent activation of apoptosis in responseto triplex-induced DNA damageMeetu Kaushik Tiwari and Faye A Rogers
Department of Therapeutic Radiology Yale University School of Medicine New Haven CT 06520 USA
Received November 28 2012 Revised May 20 2013 Accepted May 26 2013
ABSTRACT
DNA sequences capable of forming triplexes areprevalent in the human genome and have beenfound to be intrinsically mutagenic Consequentlya balance between DNA repair and apoptosis iscritical to counteract their effect on genomic integ-rity Using triplex-forming oligonucleotides to syn-thetically create altered helical distortions wehave determined that pro-apoptotic pathways areactivated by the formation of triplex structuresMoreover the TFIIH factor XPD occupies acentral role in triggering apoptosis in response totriplex-induced DNA strand breaks Here we showthat triplexes are capable of inducing XPD-inde-pendent double strand breaks which result in theformation of cH2AX foci XPD was subsequently re-cruited to the triplex-induced double strand breaksand co-localized with cH2AX at the damage siteFurthermore phosphorylation of H2AX tyrosine 142was found to stimulate the signaling pathway ofXPD-dependent apoptosis We suggest that thismechanism may play an active role in minimizinggenomic instability induced by naturally occurringnoncanonical structures perhaps protectingagainst cancer initiation
INTRODUCTION
An intricate balance between DNA repair and apoptosishas evolved to protect the integrity of the human genomeagainst the potentially devastating effects of endogenousand exogenous genotoxins Decisions to activate eitherpathway in response to DNA damage minimize the like-lihood of genomic instability which can lead to mutagen-esis and ultimately to carcinogenesis Although theregulatory mechanisms and signaling pathwayscontrolling DNA repair and apoptosis are wellcharacterized the driving forces responsible for makingthe ultimate choice between DNA repair and cell
survival or apoptotic cell death in response to genotoxicstress remain unclear Key proteins that contribute tocellular survival by acting in DNA repair can become exe-cutioners in the face of excess DNA damage Studiessuggest that some proteins required for efficient nucleotideexcision repair (NER) may also play a role in apoptosis(1) The XPD protein has been identified as having twoprimary functions in NER (i) stabilization of the tran-scription factor complex TFIIH and (ii) 50 30 helicasefunction (2) In addition to its function in NER transcrip-tion and possibly cell cycle regulation XPD is alsorequired for p53-mediated apoptosis (3ndash5)The NER pathway occupies an important position in
the recognition and repair of a wide array of helix-distorting lesions Previous studies have shown thathigh-affinity DNA-binding molecules can create helicaldistortions upon binding to duplex DNA that stronglyprovoke NER-dependent repair (67) However it wasunknown whether formation of these structures caused asevere enough alteration in the DNA double helix totrigger activation of apoptotic pathways Triplex DNAis formed when triplex-forming oligonucleotides (TFOs)bind as third strands in a sequence-specific mannerwithin the major groove of duplex DNA at polypurinestretches These molecules provide a means to experimen-tally create bulky helical distortions that are subject toNER and afford an opportunity to evaluate cellular re-sponses to increasing levels of structurally induced DNAdamageThe human genome includes DNA sequence patterns
that can adopt a variety of alternative structures inaddition to the B-conformation described by Watsonand Crick (8) For example H-DNA (triplex) formationis favored by sequences that contain mirror repeatsymmetry and occurs at purinepyrimidine tracts (9ndash11)Naturally occurring sequences capable of formingH-DNA are found in the human genome as frequentlyas 1 in every 50 000 base pairs (12) Formation of thesestructures cause severe genomic alterations and representan endogenous source of genotoxic stress (1213) Forinstance the H-DNA forming sequence located in thepromoter region of the c-myc gene has been implicated
To whom correspondence should be addressed Tel +1 203 737 3658 Fax +1 203 737 6309 Email fayerogersyaleedu
Published online 2 August 2013 Nucleic Acids Research 2013 Vol 41 No 19 8979ndash8994doi101093nargkt670
The Author(s) 2013 Published by Oxford University PressThis is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (httpcreativecommonsorglicensesby-nc30) which permits non-commercial re-use distribution and reproduction in any medium provided the original work is properly cited For commercialre-use please contact journalspermissionsoupcom
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
in the translocation of the gene in Burkittrsquos lymphoma(14) Because the triplex region found in endogenousH-DNA is similar in structure to intermolecular triplexesformed by TFOs they represent an excellent model tostudy the molecular pathways that determine cellularfate in response to endogenous sources of genotoxic stressIn the present work we have determined that the for-
mation of triplex structures can indeed provoke apoptoticresponses and reveal an XPD-dependent mechanism thatmodulates survivalapoptotic decisions in response tostructurally induced DNA damage In conjunction withthe use of an established cell line and a transgenicmouse model containing multiple chromosomal targetsites sequence-specific TFOs were used to syntheticallycreate altered helical structures Our studies demonstratethat triplex-induced double strand breaks (DSBs) canstimulate cells to activate apoptosis both in vitro andin vivo Although knockdown of XPD did not modulatethe extent of triplex-induced DSBs its depletion resultedin a decrease in triplex-induced apoptosis Further inves-tigation determined that XPD is recruited to the H2AXserine 139 phosphorylation site and its presence is requiredfor the phosphorylation of the H2AX tyrosine 142residue which has been shown to be an essential post-translational modification for the recruitment of pro-apoptotic factors to the tail of gH2AX These resultsidentify a new role for XPD in addition to its previouslyreported requirement for p53-mediated apoptosis inregulating cellular fate decisions Our findings suggestthat XPD-dependent apoptosis plays a key role inpreserving genomic integrity in the presence of excessivestructurally induced DNA damage
MATERIALS AND METHODS
Oligonucleotides
Oligonucleotides were synthesized with a 30-amino-modifier C7 CPG (Glen Research) by the MidlandCertified Reagent Company Inc and purified by RP-HPLC The sequence of the TFO AG30 used in ourstudies along with its target site are depicted inFigure 1A The G-rich TFO was also synthesized withthe guanines in A8G30 replaced with 7-deaza-8-aza-guanine (PPG G Glen Research) as indicated inFigure 1F Third strand binding of the TFO to duplexDNA was measured by gel mobility assays as previouslydescribed (15) The control oligonucleotide MIX30 amixed base 30-mer has the following sequence 50-AGTCAGTCAGTCAGTCAGTCAGTCAGTCAG-30Labeled oligonucleotides were synthesized with 50-rhoda-mine modifications using rhodamine phosphoramidite
Cells lines and transfections
C127 cells were obtained from ATCC The mouse epithe-lial cell line AV16 containing 100 randomly integratedchromosomal copies of the triplex target site was derivedfrom the parental C127 cell line and target site copynumber was determined using quantitative DNA dotblot analysis as previously described (16) A mouse fibro-blast cell line with the supFG1 vector chromosomally
integrated and deficient in XPA was derived from trans-genic mice carrying the supFG1 vector as a transgeneand targeted disruptions in the NER gene XPA Asimilar cell line containing the supFG1 vector wasderived from wild-type mice and used for comparison
Cells were seeded in six-well plates at a density of2 105 cells per well the day before transfection Cellswere transfected with 2 mg of AG3O or MIX30 usingOligofectamine (Invitrogen) transfection reagentTransfection was performed as per manufacturerrsquos in-structions siRNA directed against XPD GAPDH andnontarget controls (ON-Target plus SMARTpoolreagents Dharmacon) were transfected into AV16 cellsusing Dharmafect-1 transfection reagent (Dharmacon) ac-cording to the manufacturerrsquos instructions Western blotanalysis was used to confirm knockdown of protein
AV16 cells were used to generate XPD cells usingshRNA Briefly lentivirus shRNA vectors for XPDknockdown were obtained from Sigma-Aldrich (XPD-18 TRCN0000071118 XPD-19 TRCN0000071119XPD-58 TRCN0000338058) AV16 cells were transducedwith Lentiviral expression constructs either for nontargetshRNA or GFP shRNA or one of the three differentshRNA targeting XPD (ERCC2) Stable clones expressingthe shRNA were established via puromycindihydrochloride selection (2 mgml) AV16 clone XPD-19-1 stably expressing shRNA XPD-19 was used in themutagenesis experiments
Metaphase chromosome spreads
AV16 cells were transfected with 2 mg of rhodamine-labeled MIX30 or AG30 Twenty-four hours posttransfec-tion cells were treated for 5 h with Colcemid (01 mgml)Cells were then collected and washed once with PBS Tothe cell pellet a 75mMKCl solution was added for 20 minat 37C Cell pellets were then resuspended in Carnoyrsquosfixative solution (75 methanol 25 acetic acid) After10min incubation at room temperature the cells werepelleted and resuspended in an additional 500 ml ofCarnoyrsquos fixative solution Cells were dropped from aheight onto glass slides and mounting medium withDAPI (Prolong Gold antifade reagent Invitrogen) wasadded to each slide Pictures were taken of 50ndash60 meta-phase spreads using an Axiovert 200 microscope (CarlZeiss Micro Imaging Inc)
Survival assays
Cell survival was assayed either by visualization of mono-layer growth or by colony formation To quantify survivalby monolayer growth cells were seeded at a defineddensity in either 6 or 12 well dishes and treated with theTFO as previously described Cells were stained withcrystal violet 24 48 and 72 h posttreatment for monolayervisualization To assay for cell survival by clonogenicsurvival cells were treated with 2 mg of AG3O orMIX30 for 48 h and then seeded at 250ndash500 cells perwell Colonies were washed with 09 saline solutionand stained with crystal violet 10ndash14 days later Coloniesconsisting of gt50 cells were counted Colony formationwas normalized to plating efficiency of the nontreated
8980 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
cells Errors bars in the survival analysis are based onthree independent experiments
Apoptosis analysis
Cells were analyzed by flow cytometry 24 h posttreatmentusing the Annexin V-FITCPI apoptosis detection kit (BDPharmingen) according to the manufacturerrsquos protocolThe apoptotic rate was calculated as the combined per-centage of early apoptotic and late apoptotic cells Dataanalysis was performed using FlowJo software
Western analysis
Floating and adherent cells were collected cell pellets werelysed with RIPA buffer (150mM NaCl 01 SDS andinhibitors) and 30ndash50 mg of total protein per sample wasresolved by SDS-PAGE Proteins were detected by a
standard immunoblot protocol using the followingprimary antibodies cleaved PARP cleaved caspase 3phospho-p53 (serine 15) phospho-H2AX (serine 139)(Cell Signaling Technology Inc Danvers MA)phospho-H2AX (tyrosine 142) (Millipore CorporationBillerica MA) XPD (BD Biosciences San Jose CA)tubulin (clone B-512 Sigma St Louis MO) Each experi-ment was carried out a minimum of three times and rep-resentative Western blots are shown
Immunofluorescence
Cells seeded onto UV-irradiated coverslips were treatedfor 24 h and samples were prepared under reduced light aspreviously described (17) Cells were incubated with thefollowing antibodies rabbit anti-gH2AX antibody (CellSignaling) and FITC-conjugated F(ab0)2 fragment
Figure 1 Analysis of triplex-induced cell death (A) Schematic for the generation of synthetic triplex DNA structures Triplex structures were createdusing a 30mer TFO AG30 which has been shown to bind sequence-specifically to the polypurine target sequence (B) Images of a nondenaturedmetaphase chromosome spread generated from AV16 cells treated with either 2 mg of rhodamine-labeled AG30 or the control oligonucleotideMIX30 (C) Quantification of AG30-induced chromosomal foci Fifty to sixty metaphase chromosome spreads were analyzed per treatmentPlt 005 (D) The established mouse cell line (AV16) was engineered to contain randomly integrated chromosomal triplex target sites Cells weretreated with 2mg of TFO and stained with crystal violet 48 h posttreatment Monolayer growth assays demonstrate a decrease in cell survival that isproportional to an increase in triplex formation (E) Survival by colony formation of AV16 cells following TFO treatment (meanplusmnSEM n=3)(F) Structure of the natural guanine base compared with the modified base 7-deaza-8aza-guanine (PPG) Sequence of the PPG-substituted 30-merTFO A8G30 compared with AG30 G represents PPG (G) Gel mobility shift assay of triplex formation The target duplex was end-labeled andincubated with increasing concentrations of the indicated TFO followed by native polyacrylamide gel electrophoresis (H) Monolayer growth assayreveal similar reduction in cell growth following treatment with either 2 mg of AG30 or A8G30
Nucleic Acids Research 2013 Vol 41 No 19 8981
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
donkey anti-rabbit IgG (H+L) (Molecular Probes Inc)and then stained with 100 ngml DAPI (Sigma) Imageswere captured using an Axiovert 200 microscope (CarlZeiss Micro Imaging Inc)
Neutral comet assay
Cells were collected 24 h posttreatment and neutral cometassays were performed according to the manufacturerrsquosprotocol (Trevigen Inc) with one adjustment of35 105 cellsml for each single cell suspension Cometswere visualized using an Axiovert 200 microscope andanalyzed with Comet ScoreTM software (TriTek Corp)Approximately 100ndash150 randomly selected nonover-lapping cells were analyzed per experiment Results wereexpressed as mean tail moment
In vivo analysis of triplex-induced DNA strand breaks
AV mice were derived from the CD1 background (CharlesRiver Laboratories Wilmington MA) and were generatedas previously described (18) DNA dot blot analysis con-firmed the AV founder mouse to carry 50 copies of thetriplex target site in its genome AV mice or CD1 controlmice (14 days old) were treated by intraperitoneal (ip)injection with PBS MIX30 or AG30 (50mgkg) Threemice were used per treatment group Mice were sacrificed6 h after treatment and tissue samples were collectedSpleen tissue was collected and fixed in 4 para-formaldehyde overnight at 4C embedded in paraffinand cut into sections for evaluation by immunohisto-chemistry Cut sections were stained for phospho-H2AX(Cell Signaling) and activated caspase 3 (Abcam) andanalyzed by microscopy All sections were analyzed andquantified by counting 12 randomly selected sections ofthe same sample The number of cells positive foractivated caspase 3 and gH2AX were manually countedon digital images of the specimens The differences in thepercentage of positive cells were analyzed by one-wayANOVA and Tukey test as posthoc Representative depic-tion of immunohistochemistry is shown Animal studieswere approved and performed according to the guidelinesof the Institutional Animal Care and Use Committee ofYale University
Coimmunoprecipitation
AV16 cells were transfected with AG30 or MIX30 and24 h posttransfection cells were lysed in IP lysis buffer(Thermo Scientific) To observe the interaction of XPDwith gH2AX cell lysates were immunoprecipitated withpolyclonal rabbit antibody gH2AX (Santa Cruz) or rabbitIgG (Jackson Immunoresearch Lab) using protein AGbeads (Santa Cruz) at 4C for 90 min The immunopre-cipitated complex was analyzed by immunoblotting
Cell cycle analysis and cH2AX
AV16 cells were collected at 6 and 24 h following transfec-tion with either MIX30 or AG30 After washing once withPBS the cells were fixed in 1 paraformaldehyde for15min on ice Cells were centrifuged and fixed in 70ethanol at 20C for 2 h The cells were then washed
with BSA-T-PBS (1 wv Bovine Serum Albumin and02 vv Triton X-100 in PBS) and incubated withgH2AX antibody (Cell Signaling) in BSA-T-PBS over-night at 4C After washing the cells were incubatedwith anti-rabbit IgG Fab2 Alexa 488 (Molecular Probes)at room temperature for 1 h in the dark Cells were washedand the pellet resuspended and incubated at room tem-perature in PI staining solution (PIRNase solution BD)for 15 min Cells were analyzed by flow cytometry
Mutagenesis assay
The mouse cell lines were established with multiplechromosomally integrated copies of the recoverable supFG1 shuttle vector carrying the supFG1 reportergene Following 48ndash72 h of TFO treatment genomicDNA was isolated and incubated with in vitro packagingextracts for shuttle vector rescue and reporter geneanalysis as previously described (1619) Briefly functionalsupFG1 genes suppress the nonsense mutations in the hostbacteria b-galactosidase gene yielding blue plaques in thepresence of IPTG and X-Gal If however a mutationoccurs in the supFG1 gene the amber mutation will notbe suppressed and the resulting plaque will be whiteMutation frequency was calculated by dividing thenumber of colorless mutant plaques by the total numberof plaques counted Experiments were done in triplicateand standard errors were calculated for the mutationfrequencies as indicated by the error bars
Statistical analysis
Differences in the mean number of gH2AX focicell thenumber of apoptotic cells and tail moment were analyzedby one-way ANOVA and Tukey test as posthoc All stat-istical analyses were performed using Graphpad Prismsoftware Plt 0001 Plt 001 Plt 005
RESULTS
Triplex-induced cell death
Using an assay to measure the induction of repair synthe-sis research has determined that TFOs create a helicaldistortion upon binding to duplex DNA that stronglyprovokes DNA repair (67) Additional studies have con-firmed the importance of the NER pathway in the recog-nition and repair of TFO-induced DNA alterations (720)To investigate the potential for helical distortions toinduce apoptosis in cells we generated a mouse epithelialcell line (AV16) with 100 copies of randomly integratedchromosomal triplex target sites (Figure 1A) (16) Triplexstructures were synthetically generated using the TFOAG30 which is designed to specifically bind to thepolypurine target site (Figure 1A) Through the use of arestriction protection assay AG30-induced triplex forma-tion has been previously detected at a chromosomallyintegrated target site (21) To confirm chromosomalbinding of AG30 in our cell line we preparednondenatured metaphase chromosome spreads fromAV16 cells that had been treated Johnson et al have pre-viously described a method for detecting third-stand
8982 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
binding to nondenatured fixed metaphase spreads (22) Todetect triplex formation in vivo we modified their tech-nique by treating the cells prior to generation of the meta-phase chromosome spreads AV16 cells were treated witheither 2 mg of rhodamine-labeled AG30 or the controlmixed sequence oligonucleotide MIX30 which cannotbind as a third strand to the target polypurinepolypyrimidine sites in the AV16 cells Twenty-fourhours posttreatment cells were treated for 5 h withColcemid and then collected and prepared asnondenatured fixed metaphase chromosome spreads ac-cording to the protocol previously described (22) Tominimize background fluorescence created by thepresence of unbound oligonucleotides and nonspecificbinding events multiple washes were incorporatedduring the preparation of the metaphase spreads It hasbeen previously established that the DNA ofnondenatured fixed metaphase spreads remains in aduplex state (22) Thus the generation of chromosomalAG30-foci under the conditions of this assay representsthird strand binding to fixed chromosomes with intactDNA double helix As shown in Figure 1B several rhoda-mine-AG30 chromosomal foci were detected 24 hposttransfection in the AV16 cell-derived metaphasespreads in contrast to the rhodamine-MIX30 treatedcells These results provide evidence for third strandbinding by AG30 to multiple chromosomal sites with45 of AV16 cells treated with rhodamine-AG30 beingpositive for 5 chromosomal foci per cell (Figure 1C)compared with 10 of cells treated with MIX30 Thebinding of non-triplexndashforming oligonucleotides likeMIX30 to DNA requires single stranded targets as aresult one would not expect to see high levels of nonspe-cific chromosomal interactions with the MIX30 controlThis is consistent with the fact that this mixed sequenceoligonucleotide cannot form Hoogsteen or reverseHoogsteen triplexes even at high concentrations in vitroAlthough there may be some nonspecific retention ofMIX30 leading to low levels of foci formation this isjust background in the assay Treatment of the parentalcell line (C127) which lacks the polypurinepolypyrimidine target site with rhodamine-AG30 furtherconfirms the specificity of third strand binding(Supplementary Figure S1) Analysis of C127 cell-derived metaphase spreads revealed that 13 of cellswere positive for 5 chromosomal foci per cell followingtreatment with either MIX30 or AG30 These results aresupportive of our conclusions because both MIX30 andAG30 treatment of C127 cells resulted in the same nonspe-cific background levels The specific foci formation byAG30 in this assay provides evidence for third strandbinding and triplex formation in the treated AV16 cellsThe generation of rhodamine-AG30 foci likely representsseveral third strand binding events in proximity and sothese results support the formation of multiple triplexstructures following AG30 treatment of AV16 cells Tothis end the results from these studies provide evidencethat AG30-foci formation represents sequence-specificbinding to its polypurine target site
Following this validation we proceeded to examinewhether triplex formation was capable of inducing death
in cells with multiple TFO target sites The parental cellline C127 which lacks the target site served as a controlto assess the possibility of nonspecific oligonucleotideinteractions that could result in cell death Forty-eighthours after treatment monolayer growth assaysdemonstrated a decrease in cell growth that correlatedwith the formation of triplex structures suggestingthat DNA helical distortions can lead to cell death(Figure 1D) It is important to note that a decrease incell growth following TFO treatment was only observedin AV16 cells which have the potential to acquire multipletriplex structures and not in C127 cells which lack thetriplex-binding site In addition growth inhibition wasnot observed in cells that were treated with the controloligonucleotide MIX30 To further attribute a decreasein cell survival resulting from triplex formationclonogenic survival studies were performed using AV16cells As expected we found that only cells treated withAG30 experienced a decrease in cell survival (Figure 1E)As observed in the monolayer assay nonspecific toxicitywas not detected with either the transfection agent (mocktreatment) or MIX30 The results obtained from thesurvival assays establish that helical distortions inducedby the formation of triplex structures are capable ofinducing cell deathTo investigate the possibility that the observed cell
death could be attributed to G-quadruplex formation(23ndash25) A8G30 was designed with the same sequence asAG30 but with every third guanine substituted with themodified guanine base 7-deaza-8-aza-guanine (PPG or G)(Figure 1F) Studies have determined that substitution ofevery third guanine with PPG was sufficient to reduce self-association of TFOs containing long runs of guanines(26) To determine the relative binding affinities of theG-rich TFOs for the target duplex a gel mobility shiftassay was performed (Figure 1G) The Kd for each TFOwas estimated as the concentration of TFO at whichbinding was one-half maximal As shown in Figure 1Gusing buffer conditions that promote triplex formationboth TFOs bound to the target site with high affinity(Kd 1 109M) Treatment of AV16 cells with eitherAG30 or the PPG-substituted TFO A8G30 resulted in areduction in cell growth as observed by monolayer growthassay (Figure 1H) Taken together the inability of thecontrol oligonucleotide to induce cell death and the induc-tion of cell death after treatment with A8G30 show thatthe increase in cell death observed in the survival assays isa result of a specific and site-directed effect of AG30binding to the chromosomal target site
Triplex-induced apoptosis
Studies were then initiated to determine whether theincrease in cell death observed in the survival assays fol-lowing TFO treatment resulted from the activation of apro-apoptotic pathway AV16 cells were treated with amock transfection MIX30 or AG30 and analyzed forinduction of apoptosis 24 h posttreatment by detectionof Annexin V binding using flow cytometry Annexin Vbinding to exposed phosphatidylserine residues in the cellmembrane is an early marker of apoptosis and 23 of the
Nucleic Acids Research 2013 Vol 41 No 19 8983
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
AG30-treated cells were determined to be Annexin Vpositive as a result of triplex-induced helical distortions(Figure 2A) Western blot analysis of caspase-mediatedcleavage of Poly(ADP-ribose) polymerase (PARP) wasalso used to confirm triplex-induced apoptosis PARP anuclear DNA-binding protein that recognizes DNAstrand breaks is a substrate for caspase 3 and itscleavage is an early event in apoptotic response (27)Caspase-mediated cleavage of PARP was only detectedin cell lysates isolated from AG30-treated AV16 cells(Figure 2B) To confirm that the observed apoptosisresulted from triplex formation and not generation ofG-quadruplexes production of cleaved PARP wasanalyzed by Western blot analysis 24 h after transfectionwith either AG30 or A8G30 Similar levels of cleavedPARP were detected in cell lysates isolated from A8G30-treated AV16 cells compared with AG30 treatment sug-gesting that the observed apoptosis can be primarilyattributed to triplex formation (Figure 2C) Triplex-induced apoptosis was detected as early as 6 h and up to72 h post TFO treatment as determined by Western blotanalysis (Figure 2D)To test the impact of multiple triplex structure forma-
tion on the level of induced apoptosis AV16 cells wereexposed to increasing concentrations of AG30 As shownin Figure 2E the percentage of Annexin V positive cells
increased with higher concentrations of TFO treatmentTreatment with a low concentration of AG30 (50 nM)resulted in an Annexin V-positive population of 8which was slightly higher than that of background( 6) The percentage of Annexin V-positive cells plat-eaued at 60 following treatment with 200 nM ofAG30 Moreover no increase in the level of apoptoticcells was observed in the cells treated with increasing con-centrations of the control oligonucleotide further con-firming that the observed apoptosis can be attributed tothe formation of altered helical structures and not due tononspecific toxicity generated by the oligonucleotide itselfAs shown in Figure 2F clonogenic survival studies correl-ate with these results with the surviving cell fractiondecreasing with increasing AG30 concentrationCollectively these results support a mechanism that alter-ations to the DNA duplex structure created by the forma-tion of multiple triplex structures is capable of inducingapoptosis
Triplex-induced DSBs
To determine whether TFO-induced altered helical struc-tures can act as a fragile site resulting in DSBs we per-formed neutral comet assays Single cell lsquocometsrsquo wereobserved microscopically after separation of DNA frag-ments from the cells by electrophoresis (Figure 3A) Using
Figure 2 Induction of apoptosis via formation of triplex structures in AV16 cells (A) Annexin V binding to exposed phosphatidylserine residues24 h after treatment with 2mg of oligonucleotides (B) Western blot analysis of caspase-mediated cleavage of PARP Cells were collected and lysatesprepared 24 h posttreatment (C) Activation of apoptosis can be attributed to triplex formation rather than G-quadruplex formation as determinedby Western blot analysis of cleaved PARP following 2 mg treatment with AG30 or A8G30 (D) Time course of induced apoptosis by Western blotanalysis of cleaved PARP (E) Dose response of increasing concentrations of AG30 and its effect on apoptotic cell death 48 h posttreatment(F) Clonogenic survival after 48 h exposure of AV16 cells to AG30 or MIX30
8984 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
the lsquocomet tail momentrsquo as a measure of the extent ofDNA breakage we assessed the presence of DSBs result-ing from triplex formation As shown in Figure 3A wedetermined that AG30 treatment resulted in more DSBscompared with untreated and MIX30-treated cells
Histone variant H2AX becomes phosphorylated onserine 139 (gH2AX) in response to DNA damage thatinvolves formation of DSBs (28) and foci formation isfrequently used as a quantitative marker for DSBs in im-munofluorescence microscopy (29) The presence oftriplex-induced DSBs was also determined by co-stainingfor gH2AX and DAPI 24 h after treatment (Figure 3B)AV16 cells treated with AG30 resulted in the formation ofmore gH2AX nuclear foci compared with untreated cells(Figure 3C) Western blot analysis of gH2AX alsoconfirms the presence of H2AX S139 phosphorylation inonly the AG30-treated cells in agreement with the im-munofluorescence results (Figure 3D) These datasuggest that the formation of triplex structures in cellsthat contain multiple target sites generates substantialDSBs which may overwhelm the cellrsquos repair capacitycausing the initiation of an apoptotic response
To ensure that the presence of gH2AX foci was truly ahallmark of DSBs and not generated in the course ofDNA fragmentation during apoptosis we used amultiparameter cytometry assay (30ndash32) The presence oftriplex-induced DSBs was determined using flowcytometry by staining for gH2AX in the presence ofpropidium iodine gH2AX expression attributed toDSBs is cell cycle independent while high intensegH2AX expression in S-phase is associated with apoptosis(30) Cells were harvested 6 h and 24 h after treatment withAG30 Flow cytometry analysis of gH2AX expressionduring the cell cycle indicated increased levels of gH2AXin all phases of the cell cycle of AV16 cells 6 h afterexposure to AG30 (Figure 3E) This signal persisted forup to 24 h after TFO treatment and increased to 217(Figure 3E) suggesting that many sites marked by gH2AXfoci remained unrepaired Analysis of the flow cytometryprofiles indicates that gH2AX expression in AG30-treatedcells was significantly higher compared with mock-treatedand MIX30-treated cells (Figure 3F)
In Vivo generation of triplex-induced DSBs
To evaluate the potential for triplex DNA to induce DSBsin vivo we used a transgenic mouse model (AV mouse)with 50 copies of the triplex target sequencechromosomally integrated into its genome (Figure 4A)(18) Immunohistochemistry staining for gH2AX and theapoptosis marker activated caspase 3 was used to assessthe cellular response to in vivo triplex formation AV micewere administered a 50mgkg dose of MIX30 or AG30 viaip injection To investigate the extent of triplex-inducedDNA strand breaks we performed immunohistochemistrystaining of gH2AX as a marker for ongoing DNA damageon spleen tissue harvested 6 h posttreatment We observedlow levels of gH2AX staining in the spleens of mice thathad received ip doses of the control oligonucleotideMIX30 (Figure 4B and C) In contrast we found thatAG30 treatment triggered an increase in the percentage
of cells positive for gH2AX foci compared with thePBS- and MIX30-treated mice (Figure 4B and C) Tofurther substantiate the specificity of triplex-inducedgH2AX foci CD1 control mice which lack the triplextarget site were also administered a 50mgkg dose ofAG30 Immunohistochemistry analysis of spleen tissuerevealed no increase in the percentage of cells positive ofgH2AX foci above background (Supplementary FigureS2) An increase in the production of gH2AX foci solelyin the AV mice after AG30 treatment would suggest thepresence of triplex-induced DSBs Immunohistochemistrystaining for activated caspase 3 was then used to deter-mine whether the formation of triplex-induced gH2AXfoci could elicit an apoptotic response in vivo Micetreated with the control oligonucleotide MIX30 showedalmost a complete absence of activated caspase 3 stainingin their spleens 6 h following treatment (Figure 4B and D)However analysis of spleens from AV mice dosed withAG30 revealed that 26 of their spleen cells werepositive for activated caspase 3 staining (Figure 4B andD) Examination of spleen tissue samples obtained fromCD1 mice treated with AG30 determined a nonexistenceof activated caspase 3 6 h posttreatment (SupplementaryFigure S2) Altogether these results are consistent withthe interpretation that the formation of endogenoustriplex structures can result in DSBs which can in turnprompt the activation of apoptosis
NER deficiency results in increased apoptosis levels
Activation of apoptosis in response to DNA damageprovides a default mechanism that can be implementedto prevent clonal expansion of cells with unrepaireddamage As a result studies were initiated to investigatecellular response to triplex-induced DSBs under circum-stances where cells may be ineffective at repair XPA is akey NER factor responsible for verifying altered DNAconformations and is crucial for correct assembly of theremaining repair machinery around a lesion (33) Previousstudies have determined that TFOs were capable ofbinding to duplex DNA and creating altered helical struc-tures that strongly provoked XPA-dependent DNArepair To investigate whether triplex formation inducedDSBs in an XPA-dependent manner we treated XPA-pro-ficient and XPA-deficient mouse fibroblast cells with amock transfection MIX30 or AG30 Cells were harvested24 h following treatment and through the use of flowcytometry we evaluated the levels of triplex-inducedDNA strand breaks by staining for gH2AX Analysis ofgH2AX expression levels as a measure of double strandbreak formation indicated increased levels of gH2AX inboth the XPA-proficient and -deficient cells followingAG30 treatment (Figure 5A) Exposure to AG30increased gH2AX expression in XPA-proficient cells to22 and a similar expression level of 28 wasobserved in the XPA-deficient cells (Figure 5A)We then proceeded to evaluate induced apoptosis levels
as a result of triplex-induced DNA strand breaks in XPA-deficient cells XPA-deficient cells were treated with amock transfection MIX30 or AG30 and analyzed forthe induction of apoptosis Following 24 h administration
Nucleic Acids Research 2013 Vol 41 No 19 8985
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

in the translocation of the gene in Burkittrsquos lymphoma(14) Because the triplex region found in endogenousH-DNA is similar in structure to intermolecular triplexesformed by TFOs they represent an excellent model tostudy the molecular pathways that determine cellularfate in response to endogenous sources of genotoxic stressIn the present work we have determined that the for-
mation of triplex structures can indeed provoke apoptoticresponses and reveal an XPD-dependent mechanism thatmodulates survivalapoptotic decisions in response tostructurally induced DNA damage In conjunction withthe use of an established cell line and a transgenicmouse model containing multiple chromosomal targetsites sequence-specific TFOs were used to syntheticallycreate altered helical structures Our studies demonstratethat triplex-induced double strand breaks (DSBs) canstimulate cells to activate apoptosis both in vitro andin vivo Although knockdown of XPD did not modulatethe extent of triplex-induced DSBs its depletion resultedin a decrease in triplex-induced apoptosis Further inves-tigation determined that XPD is recruited to the H2AXserine 139 phosphorylation site and its presence is requiredfor the phosphorylation of the H2AX tyrosine 142residue which has been shown to be an essential post-translational modification for the recruitment of pro-apoptotic factors to the tail of gH2AX These resultsidentify a new role for XPD in addition to its previouslyreported requirement for p53-mediated apoptosis inregulating cellular fate decisions Our findings suggestthat XPD-dependent apoptosis plays a key role inpreserving genomic integrity in the presence of excessivestructurally induced DNA damage
MATERIALS AND METHODS
Oligonucleotides
Oligonucleotides were synthesized with a 30-amino-modifier C7 CPG (Glen Research) by the MidlandCertified Reagent Company Inc and purified by RP-HPLC The sequence of the TFO AG30 used in ourstudies along with its target site are depicted inFigure 1A The G-rich TFO was also synthesized withthe guanines in A8G30 replaced with 7-deaza-8-aza-guanine (PPG G Glen Research) as indicated inFigure 1F Third strand binding of the TFO to duplexDNA was measured by gel mobility assays as previouslydescribed (15) The control oligonucleotide MIX30 amixed base 30-mer has the following sequence 50-AGTCAGTCAGTCAGTCAGTCAGTCAGTCAG-30Labeled oligonucleotides were synthesized with 50-rhoda-mine modifications using rhodamine phosphoramidite
Cells lines and transfections
C127 cells were obtained from ATCC The mouse epithe-lial cell line AV16 containing 100 randomly integratedchromosomal copies of the triplex target site was derivedfrom the parental C127 cell line and target site copynumber was determined using quantitative DNA dotblot analysis as previously described (16) A mouse fibro-blast cell line with the supFG1 vector chromosomally
integrated and deficient in XPA was derived from trans-genic mice carrying the supFG1 vector as a transgeneand targeted disruptions in the NER gene XPA Asimilar cell line containing the supFG1 vector wasderived from wild-type mice and used for comparison
Cells were seeded in six-well plates at a density of2 105 cells per well the day before transfection Cellswere transfected with 2 mg of AG3O or MIX30 usingOligofectamine (Invitrogen) transfection reagentTransfection was performed as per manufacturerrsquos in-structions siRNA directed against XPD GAPDH andnontarget controls (ON-Target plus SMARTpoolreagents Dharmacon) were transfected into AV16 cellsusing Dharmafect-1 transfection reagent (Dharmacon) ac-cording to the manufacturerrsquos instructions Western blotanalysis was used to confirm knockdown of protein
AV16 cells were used to generate XPD cells usingshRNA Briefly lentivirus shRNA vectors for XPDknockdown were obtained from Sigma-Aldrich (XPD-18 TRCN0000071118 XPD-19 TRCN0000071119XPD-58 TRCN0000338058) AV16 cells were transducedwith Lentiviral expression constructs either for nontargetshRNA or GFP shRNA or one of the three differentshRNA targeting XPD (ERCC2) Stable clones expressingthe shRNA were established via puromycindihydrochloride selection (2 mgml) AV16 clone XPD-19-1 stably expressing shRNA XPD-19 was used in themutagenesis experiments
Metaphase chromosome spreads
AV16 cells were transfected with 2 mg of rhodamine-labeled MIX30 or AG30 Twenty-four hours posttransfec-tion cells were treated for 5 h with Colcemid (01 mgml)Cells were then collected and washed once with PBS Tothe cell pellet a 75mMKCl solution was added for 20 minat 37C Cell pellets were then resuspended in Carnoyrsquosfixative solution (75 methanol 25 acetic acid) After10min incubation at room temperature the cells werepelleted and resuspended in an additional 500 ml ofCarnoyrsquos fixative solution Cells were dropped from aheight onto glass slides and mounting medium withDAPI (Prolong Gold antifade reagent Invitrogen) wasadded to each slide Pictures were taken of 50ndash60 meta-phase spreads using an Axiovert 200 microscope (CarlZeiss Micro Imaging Inc)
Survival assays
Cell survival was assayed either by visualization of mono-layer growth or by colony formation To quantify survivalby monolayer growth cells were seeded at a defineddensity in either 6 or 12 well dishes and treated with theTFO as previously described Cells were stained withcrystal violet 24 48 and 72 h posttreatment for monolayervisualization To assay for cell survival by clonogenicsurvival cells were treated with 2 mg of AG3O orMIX30 for 48 h and then seeded at 250ndash500 cells perwell Colonies were washed with 09 saline solutionand stained with crystal violet 10ndash14 days later Coloniesconsisting of gt50 cells were counted Colony formationwas normalized to plating efficiency of the nontreated
8980 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
cells Errors bars in the survival analysis are based onthree independent experiments
Apoptosis analysis
Cells were analyzed by flow cytometry 24 h posttreatmentusing the Annexin V-FITCPI apoptosis detection kit (BDPharmingen) according to the manufacturerrsquos protocolThe apoptotic rate was calculated as the combined per-centage of early apoptotic and late apoptotic cells Dataanalysis was performed using FlowJo software
Western analysis
Floating and adherent cells were collected cell pellets werelysed with RIPA buffer (150mM NaCl 01 SDS andinhibitors) and 30ndash50 mg of total protein per sample wasresolved by SDS-PAGE Proteins were detected by a
standard immunoblot protocol using the followingprimary antibodies cleaved PARP cleaved caspase 3phospho-p53 (serine 15) phospho-H2AX (serine 139)(Cell Signaling Technology Inc Danvers MA)phospho-H2AX (tyrosine 142) (Millipore CorporationBillerica MA) XPD (BD Biosciences San Jose CA)tubulin (clone B-512 Sigma St Louis MO) Each experi-ment was carried out a minimum of three times and rep-resentative Western blots are shown
Immunofluorescence
Cells seeded onto UV-irradiated coverslips were treatedfor 24 h and samples were prepared under reduced light aspreviously described (17) Cells were incubated with thefollowing antibodies rabbit anti-gH2AX antibody (CellSignaling) and FITC-conjugated F(ab0)2 fragment
Figure 1 Analysis of triplex-induced cell death (A) Schematic for the generation of synthetic triplex DNA structures Triplex structures were createdusing a 30mer TFO AG30 which has been shown to bind sequence-specifically to the polypurine target sequence (B) Images of a nondenaturedmetaphase chromosome spread generated from AV16 cells treated with either 2 mg of rhodamine-labeled AG30 or the control oligonucleotideMIX30 (C) Quantification of AG30-induced chromosomal foci Fifty to sixty metaphase chromosome spreads were analyzed per treatmentPlt 005 (D) The established mouse cell line (AV16) was engineered to contain randomly integrated chromosomal triplex target sites Cells weretreated with 2mg of TFO and stained with crystal violet 48 h posttreatment Monolayer growth assays demonstrate a decrease in cell survival that isproportional to an increase in triplex formation (E) Survival by colony formation of AV16 cells following TFO treatment (meanplusmnSEM n=3)(F) Structure of the natural guanine base compared with the modified base 7-deaza-8aza-guanine (PPG) Sequence of the PPG-substituted 30-merTFO A8G30 compared with AG30 G represents PPG (G) Gel mobility shift assay of triplex formation The target duplex was end-labeled andincubated with increasing concentrations of the indicated TFO followed by native polyacrylamide gel electrophoresis (H) Monolayer growth assayreveal similar reduction in cell growth following treatment with either 2 mg of AG30 or A8G30
Nucleic Acids Research 2013 Vol 41 No 19 8981
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
donkey anti-rabbit IgG (H+L) (Molecular Probes Inc)and then stained with 100 ngml DAPI (Sigma) Imageswere captured using an Axiovert 200 microscope (CarlZeiss Micro Imaging Inc)
Neutral comet assay
Cells were collected 24 h posttreatment and neutral cometassays were performed according to the manufacturerrsquosprotocol (Trevigen Inc) with one adjustment of35 105 cellsml for each single cell suspension Cometswere visualized using an Axiovert 200 microscope andanalyzed with Comet ScoreTM software (TriTek Corp)Approximately 100ndash150 randomly selected nonover-lapping cells were analyzed per experiment Results wereexpressed as mean tail moment
In vivo analysis of triplex-induced DNA strand breaks
AV mice were derived from the CD1 background (CharlesRiver Laboratories Wilmington MA) and were generatedas previously described (18) DNA dot blot analysis con-firmed the AV founder mouse to carry 50 copies of thetriplex target site in its genome AV mice or CD1 controlmice (14 days old) were treated by intraperitoneal (ip)injection with PBS MIX30 or AG30 (50mgkg) Threemice were used per treatment group Mice were sacrificed6 h after treatment and tissue samples were collectedSpleen tissue was collected and fixed in 4 para-formaldehyde overnight at 4C embedded in paraffinand cut into sections for evaluation by immunohisto-chemistry Cut sections were stained for phospho-H2AX(Cell Signaling) and activated caspase 3 (Abcam) andanalyzed by microscopy All sections were analyzed andquantified by counting 12 randomly selected sections ofthe same sample The number of cells positive foractivated caspase 3 and gH2AX were manually countedon digital images of the specimens The differences in thepercentage of positive cells were analyzed by one-wayANOVA and Tukey test as posthoc Representative depic-tion of immunohistochemistry is shown Animal studieswere approved and performed according to the guidelinesof the Institutional Animal Care and Use Committee ofYale University
Coimmunoprecipitation
AV16 cells were transfected with AG30 or MIX30 and24 h posttransfection cells were lysed in IP lysis buffer(Thermo Scientific) To observe the interaction of XPDwith gH2AX cell lysates were immunoprecipitated withpolyclonal rabbit antibody gH2AX (Santa Cruz) or rabbitIgG (Jackson Immunoresearch Lab) using protein AGbeads (Santa Cruz) at 4C for 90 min The immunopre-cipitated complex was analyzed by immunoblotting
Cell cycle analysis and cH2AX
AV16 cells were collected at 6 and 24 h following transfec-tion with either MIX30 or AG30 After washing once withPBS the cells were fixed in 1 paraformaldehyde for15min on ice Cells were centrifuged and fixed in 70ethanol at 20C for 2 h The cells were then washed
with BSA-T-PBS (1 wv Bovine Serum Albumin and02 vv Triton X-100 in PBS) and incubated withgH2AX antibody (Cell Signaling) in BSA-T-PBS over-night at 4C After washing the cells were incubatedwith anti-rabbit IgG Fab2 Alexa 488 (Molecular Probes)at room temperature for 1 h in the dark Cells were washedand the pellet resuspended and incubated at room tem-perature in PI staining solution (PIRNase solution BD)for 15 min Cells were analyzed by flow cytometry
Mutagenesis assay
The mouse cell lines were established with multiplechromosomally integrated copies of the recoverable supFG1 shuttle vector carrying the supFG1 reportergene Following 48ndash72 h of TFO treatment genomicDNA was isolated and incubated with in vitro packagingextracts for shuttle vector rescue and reporter geneanalysis as previously described (1619) Briefly functionalsupFG1 genes suppress the nonsense mutations in the hostbacteria b-galactosidase gene yielding blue plaques in thepresence of IPTG and X-Gal If however a mutationoccurs in the supFG1 gene the amber mutation will notbe suppressed and the resulting plaque will be whiteMutation frequency was calculated by dividing thenumber of colorless mutant plaques by the total numberof plaques counted Experiments were done in triplicateand standard errors were calculated for the mutationfrequencies as indicated by the error bars
Statistical analysis
Differences in the mean number of gH2AX focicell thenumber of apoptotic cells and tail moment were analyzedby one-way ANOVA and Tukey test as posthoc All stat-istical analyses were performed using Graphpad Prismsoftware Plt 0001 Plt 001 Plt 005
RESULTS
Triplex-induced cell death
Using an assay to measure the induction of repair synthe-sis research has determined that TFOs create a helicaldistortion upon binding to duplex DNA that stronglyprovokes DNA repair (67) Additional studies have con-firmed the importance of the NER pathway in the recog-nition and repair of TFO-induced DNA alterations (720)To investigate the potential for helical distortions toinduce apoptosis in cells we generated a mouse epithelialcell line (AV16) with 100 copies of randomly integratedchromosomal triplex target sites (Figure 1A) (16) Triplexstructures were synthetically generated using the TFOAG30 which is designed to specifically bind to thepolypurine target site (Figure 1A) Through the use of arestriction protection assay AG30-induced triplex forma-tion has been previously detected at a chromosomallyintegrated target site (21) To confirm chromosomalbinding of AG30 in our cell line we preparednondenatured metaphase chromosome spreads fromAV16 cells that had been treated Johnson et al have pre-viously described a method for detecting third-stand
8982 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
binding to nondenatured fixed metaphase spreads (22) Todetect triplex formation in vivo we modified their tech-nique by treating the cells prior to generation of the meta-phase chromosome spreads AV16 cells were treated witheither 2 mg of rhodamine-labeled AG30 or the controlmixed sequence oligonucleotide MIX30 which cannotbind as a third strand to the target polypurinepolypyrimidine sites in the AV16 cells Twenty-fourhours posttreatment cells were treated for 5 h withColcemid and then collected and prepared asnondenatured fixed metaphase chromosome spreads ac-cording to the protocol previously described (22) Tominimize background fluorescence created by thepresence of unbound oligonucleotides and nonspecificbinding events multiple washes were incorporatedduring the preparation of the metaphase spreads It hasbeen previously established that the DNA ofnondenatured fixed metaphase spreads remains in aduplex state (22) Thus the generation of chromosomalAG30-foci under the conditions of this assay representsthird strand binding to fixed chromosomes with intactDNA double helix As shown in Figure 1B several rhoda-mine-AG30 chromosomal foci were detected 24 hposttransfection in the AV16 cell-derived metaphasespreads in contrast to the rhodamine-MIX30 treatedcells These results provide evidence for third strandbinding by AG30 to multiple chromosomal sites with45 of AV16 cells treated with rhodamine-AG30 beingpositive for 5 chromosomal foci per cell (Figure 1C)compared with 10 of cells treated with MIX30 Thebinding of non-triplexndashforming oligonucleotides likeMIX30 to DNA requires single stranded targets as aresult one would not expect to see high levels of nonspe-cific chromosomal interactions with the MIX30 controlThis is consistent with the fact that this mixed sequenceoligonucleotide cannot form Hoogsteen or reverseHoogsteen triplexes even at high concentrations in vitroAlthough there may be some nonspecific retention ofMIX30 leading to low levels of foci formation this isjust background in the assay Treatment of the parentalcell line (C127) which lacks the polypurinepolypyrimidine target site with rhodamine-AG30 furtherconfirms the specificity of third strand binding(Supplementary Figure S1) Analysis of C127 cell-derived metaphase spreads revealed that 13 of cellswere positive for 5 chromosomal foci per cell followingtreatment with either MIX30 or AG30 These results aresupportive of our conclusions because both MIX30 andAG30 treatment of C127 cells resulted in the same nonspe-cific background levels The specific foci formation byAG30 in this assay provides evidence for third strandbinding and triplex formation in the treated AV16 cellsThe generation of rhodamine-AG30 foci likely representsseveral third strand binding events in proximity and sothese results support the formation of multiple triplexstructures following AG30 treatment of AV16 cells Tothis end the results from these studies provide evidencethat AG30-foci formation represents sequence-specificbinding to its polypurine target site
Following this validation we proceeded to examinewhether triplex formation was capable of inducing death
in cells with multiple TFO target sites The parental cellline C127 which lacks the target site served as a controlto assess the possibility of nonspecific oligonucleotideinteractions that could result in cell death Forty-eighthours after treatment monolayer growth assaysdemonstrated a decrease in cell growth that correlatedwith the formation of triplex structures suggestingthat DNA helical distortions can lead to cell death(Figure 1D) It is important to note that a decrease incell growth following TFO treatment was only observedin AV16 cells which have the potential to acquire multipletriplex structures and not in C127 cells which lack thetriplex-binding site In addition growth inhibition wasnot observed in cells that were treated with the controloligonucleotide MIX30 To further attribute a decreasein cell survival resulting from triplex formationclonogenic survival studies were performed using AV16cells As expected we found that only cells treated withAG30 experienced a decrease in cell survival (Figure 1E)As observed in the monolayer assay nonspecific toxicitywas not detected with either the transfection agent (mocktreatment) or MIX30 The results obtained from thesurvival assays establish that helical distortions inducedby the formation of triplex structures are capable ofinducing cell deathTo investigate the possibility that the observed cell
death could be attributed to G-quadruplex formation(23ndash25) A8G30 was designed with the same sequence asAG30 but with every third guanine substituted with themodified guanine base 7-deaza-8-aza-guanine (PPG or G)(Figure 1F) Studies have determined that substitution ofevery third guanine with PPG was sufficient to reduce self-association of TFOs containing long runs of guanines(26) To determine the relative binding affinities of theG-rich TFOs for the target duplex a gel mobility shiftassay was performed (Figure 1G) The Kd for each TFOwas estimated as the concentration of TFO at whichbinding was one-half maximal As shown in Figure 1Gusing buffer conditions that promote triplex formationboth TFOs bound to the target site with high affinity(Kd 1 109M) Treatment of AV16 cells with eitherAG30 or the PPG-substituted TFO A8G30 resulted in areduction in cell growth as observed by monolayer growthassay (Figure 1H) Taken together the inability of thecontrol oligonucleotide to induce cell death and the induc-tion of cell death after treatment with A8G30 show thatthe increase in cell death observed in the survival assays isa result of a specific and site-directed effect of AG30binding to the chromosomal target site
Triplex-induced apoptosis
Studies were then initiated to determine whether theincrease in cell death observed in the survival assays fol-lowing TFO treatment resulted from the activation of apro-apoptotic pathway AV16 cells were treated with amock transfection MIX30 or AG30 and analyzed forinduction of apoptosis 24 h posttreatment by detectionof Annexin V binding using flow cytometry Annexin Vbinding to exposed phosphatidylserine residues in the cellmembrane is an early marker of apoptosis and 23 of the
Nucleic Acids Research 2013 Vol 41 No 19 8983
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
AG30-treated cells were determined to be Annexin Vpositive as a result of triplex-induced helical distortions(Figure 2A) Western blot analysis of caspase-mediatedcleavage of Poly(ADP-ribose) polymerase (PARP) wasalso used to confirm triplex-induced apoptosis PARP anuclear DNA-binding protein that recognizes DNAstrand breaks is a substrate for caspase 3 and itscleavage is an early event in apoptotic response (27)Caspase-mediated cleavage of PARP was only detectedin cell lysates isolated from AG30-treated AV16 cells(Figure 2B) To confirm that the observed apoptosisresulted from triplex formation and not generation ofG-quadruplexes production of cleaved PARP wasanalyzed by Western blot analysis 24 h after transfectionwith either AG30 or A8G30 Similar levels of cleavedPARP were detected in cell lysates isolated from A8G30-treated AV16 cells compared with AG30 treatment sug-gesting that the observed apoptosis can be primarilyattributed to triplex formation (Figure 2C) Triplex-induced apoptosis was detected as early as 6 h and up to72 h post TFO treatment as determined by Western blotanalysis (Figure 2D)To test the impact of multiple triplex structure forma-
tion on the level of induced apoptosis AV16 cells wereexposed to increasing concentrations of AG30 As shownin Figure 2E the percentage of Annexin V positive cells
increased with higher concentrations of TFO treatmentTreatment with a low concentration of AG30 (50 nM)resulted in an Annexin V-positive population of 8which was slightly higher than that of background( 6) The percentage of Annexin V-positive cells plat-eaued at 60 following treatment with 200 nM ofAG30 Moreover no increase in the level of apoptoticcells was observed in the cells treated with increasing con-centrations of the control oligonucleotide further con-firming that the observed apoptosis can be attributed tothe formation of altered helical structures and not due tononspecific toxicity generated by the oligonucleotide itselfAs shown in Figure 2F clonogenic survival studies correl-ate with these results with the surviving cell fractiondecreasing with increasing AG30 concentrationCollectively these results support a mechanism that alter-ations to the DNA duplex structure created by the forma-tion of multiple triplex structures is capable of inducingapoptosis
Triplex-induced DSBs
To determine whether TFO-induced altered helical struc-tures can act as a fragile site resulting in DSBs we per-formed neutral comet assays Single cell lsquocometsrsquo wereobserved microscopically after separation of DNA frag-ments from the cells by electrophoresis (Figure 3A) Using
Figure 2 Induction of apoptosis via formation of triplex structures in AV16 cells (A) Annexin V binding to exposed phosphatidylserine residues24 h after treatment with 2mg of oligonucleotides (B) Western blot analysis of caspase-mediated cleavage of PARP Cells were collected and lysatesprepared 24 h posttreatment (C) Activation of apoptosis can be attributed to triplex formation rather than G-quadruplex formation as determinedby Western blot analysis of cleaved PARP following 2 mg treatment with AG30 or A8G30 (D) Time course of induced apoptosis by Western blotanalysis of cleaved PARP (E) Dose response of increasing concentrations of AG30 and its effect on apoptotic cell death 48 h posttreatment(F) Clonogenic survival after 48 h exposure of AV16 cells to AG30 or MIX30
8984 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
the lsquocomet tail momentrsquo as a measure of the extent ofDNA breakage we assessed the presence of DSBs result-ing from triplex formation As shown in Figure 3A wedetermined that AG30 treatment resulted in more DSBscompared with untreated and MIX30-treated cells
Histone variant H2AX becomes phosphorylated onserine 139 (gH2AX) in response to DNA damage thatinvolves formation of DSBs (28) and foci formation isfrequently used as a quantitative marker for DSBs in im-munofluorescence microscopy (29) The presence oftriplex-induced DSBs was also determined by co-stainingfor gH2AX and DAPI 24 h after treatment (Figure 3B)AV16 cells treated with AG30 resulted in the formation ofmore gH2AX nuclear foci compared with untreated cells(Figure 3C) Western blot analysis of gH2AX alsoconfirms the presence of H2AX S139 phosphorylation inonly the AG30-treated cells in agreement with the im-munofluorescence results (Figure 3D) These datasuggest that the formation of triplex structures in cellsthat contain multiple target sites generates substantialDSBs which may overwhelm the cellrsquos repair capacitycausing the initiation of an apoptotic response
To ensure that the presence of gH2AX foci was truly ahallmark of DSBs and not generated in the course ofDNA fragmentation during apoptosis we used amultiparameter cytometry assay (30ndash32) The presence oftriplex-induced DSBs was determined using flowcytometry by staining for gH2AX in the presence ofpropidium iodine gH2AX expression attributed toDSBs is cell cycle independent while high intensegH2AX expression in S-phase is associated with apoptosis(30) Cells were harvested 6 h and 24 h after treatment withAG30 Flow cytometry analysis of gH2AX expressionduring the cell cycle indicated increased levels of gH2AXin all phases of the cell cycle of AV16 cells 6 h afterexposure to AG30 (Figure 3E) This signal persisted forup to 24 h after TFO treatment and increased to 217(Figure 3E) suggesting that many sites marked by gH2AXfoci remained unrepaired Analysis of the flow cytometryprofiles indicates that gH2AX expression in AG30-treatedcells was significantly higher compared with mock-treatedand MIX30-treated cells (Figure 3F)
In Vivo generation of triplex-induced DSBs
To evaluate the potential for triplex DNA to induce DSBsin vivo we used a transgenic mouse model (AV mouse)with 50 copies of the triplex target sequencechromosomally integrated into its genome (Figure 4A)(18) Immunohistochemistry staining for gH2AX and theapoptosis marker activated caspase 3 was used to assessthe cellular response to in vivo triplex formation AV micewere administered a 50mgkg dose of MIX30 or AG30 viaip injection To investigate the extent of triplex-inducedDNA strand breaks we performed immunohistochemistrystaining of gH2AX as a marker for ongoing DNA damageon spleen tissue harvested 6 h posttreatment We observedlow levels of gH2AX staining in the spleens of mice thathad received ip doses of the control oligonucleotideMIX30 (Figure 4B and C) In contrast we found thatAG30 treatment triggered an increase in the percentage
of cells positive for gH2AX foci compared with thePBS- and MIX30-treated mice (Figure 4B and C) Tofurther substantiate the specificity of triplex-inducedgH2AX foci CD1 control mice which lack the triplextarget site were also administered a 50mgkg dose ofAG30 Immunohistochemistry analysis of spleen tissuerevealed no increase in the percentage of cells positive ofgH2AX foci above background (Supplementary FigureS2) An increase in the production of gH2AX foci solelyin the AV mice after AG30 treatment would suggest thepresence of triplex-induced DSBs Immunohistochemistrystaining for activated caspase 3 was then used to deter-mine whether the formation of triplex-induced gH2AXfoci could elicit an apoptotic response in vivo Micetreated with the control oligonucleotide MIX30 showedalmost a complete absence of activated caspase 3 stainingin their spleens 6 h following treatment (Figure 4B and D)However analysis of spleens from AV mice dosed withAG30 revealed that 26 of their spleen cells werepositive for activated caspase 3 staining (Figure 4B andD) Examination of spleen tissue samples obtained fromCD1 mice treated with AG30 determined a nonexistenceof activated caspase 3 6 h posttreatment (SupplementaryFigure S2) Altogether these results are consistent withthe interpretation that the formation of endogenoustriplex structures can result in DSBs which can in turnprompt the activation of apoptosis
NER deficiency results in increased apoptosis levels
Activation of apoptosis in response to DNA damageprovides a default mechanism that can be implementedto prevent clonal expansion of cells with unrepaireddamage As a result studies were initiated to investigatecellular response to triplex-induced DSBs under circum-stances where cells may be ineffective at repair XPA is akey NER factor responsible for verifying altered DNAconformations and is crucial for correct assembly of theremaining repair machinery around a lesion (33) Previousstudies have determined that TFOs were capable ofbinding to duplex DNA and creating altered helical struc-tures that strongly provoked XPA-dependent DNArepair To investigate whether triplex formation inducedDSBs in an XPA-dependent manner we treated XPA-pro-ficient and XPA-deficient mouse fibroblast cells with amock transfection MIX30 or AG30 Cells were harvested24 h following treatment and through the use of flowcytometry we evaluated the levels of triplex-inducedDNA strand breaks by staining for gH2AX Analysis ofgH2AX expression levels as a measure of double strandbreak formation indicated increased levels of gH2AX inboth the XPA-proficient and -deficient cells followingAG30 treatment (Figure 5A) Exposure to AG30increased gH2AX expression in XPA-proficient cells to22 and a similar expression level of 28 wasobserved in the XPA-deficient cells (Figure 5A)We then proceeded to evaluate induced apoptosis levels
as a result of triplex-induced DNA strand breaks in XPA-deficient cells XPA-deficient cells were treated with amock transfection MIX30 or AG30 and analyzed forthe induction of apoptosis Following 24 h administration
Nucleic Acids Research 2013 Vol 41 No 19 8985
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

cells Errors bars in the survival analysis are based onthree independent experiments
Apoptosis analysis
Cells were analyzed by flow cytometry 24 h posttreatmentusing the Annexin V-FITCPI apoptosis detection kit (BDPharmingen) according to the manufacturerrsquos protocolThe apoptotic rate was calculated as the combined per-centage of early apoptotic and late apoptotic cells Dataanalysis was performed using FlowJo software
Western analysis
Floating and adherent cells were collected cell pellets werelysed with RIPA buffer (150mM NaCl 01 SDS andinhibitors) and 30ndash50 mg of total protein per sample wasresolved by SDS-PAGE Proteins were detected by a
standard immunoblot protocol using the followingprimary antibodies cleaved PARP cleaved caspase 3phospho-p53 (serine 15) phospho-H2AX (serine 139)(Cell Signaling Technology Inc Danvers MA)phospho-H2AX (tyrosine 142) (Millipore CorporationBillerica MA) XPD (BD Biosciences San Jose CA)tubulin (clone B-512 Sigma St Louis MO) Each experi-ment was carried out a minimum of three times and rep-resentative Western blots are shown
Immunofluorescence
Cells seeded onto UV-irradiated coverslips were treatedfor 24 h and samples were prepared under reduced light aspreviously described (17) Cells were incubated with thefollowing antibodies rabbit anti-gH2AX antibody (CellSignaling) and FITC-conjugated F(ab0)2 fragment
Figure 1 Analysis of triplex-induced cell death (A) Schematic for the generation of synthetic triplex DNA structures Triplex structures were createdusing a 30mer TFO AG30 which has been shown to bind sequence-specifically to the polypurine target sequence (B) Images of a nondenaturedmetaphase chromosome spread generated from AV16 cells treated with either 2 mg of rhodamine-labeled AG30 or the control oligonucleotideMIX30 (C) Quantification of AG30-induced chromosomal foci Fifty to sixty metaphase chromosome spreads were analyzed per treatmentPlt 005 (D) The established mouse cell line (AV16) was engineered to contain randomly integrated chromosomal triplex target sites Cells weretreated with 2mg of TFO and stained with crystal violet 48 h posttreatment Monolayer growth assays demonstrate a decrease in cell survival that isproportional to an increase in triplex formation (E) Survival by colony formation of AV16 cells following TFO treatment (meanplusmnSEM n=3)(F) Structure of the natural guanine base compared with the modified base 7-deaza-8aza-guanine (PPG) Sequence of the PPG-substituted 30-merTFO A8G30 compared with AG30 G represents PPG (G) Gel mobility shift assay of triplex formation The target duplex was end-labeled andincubated with increasing concentrations of the indicated TFO followed by native polyacrylamide gel electrophoresis (H) Monolayer growth assayreveal similar reduction in cell growth following treatment with either 2 mg of AG30 or A8G30
Nucleic Acids Research 2013 Vol 41 No 19 8981
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
donkey anti-rabbit IgG (H+L) (Molecular Probes Inc)and then stained with 100 ngml DAPI (Sigma) Imageswere captured using an Axiovert 200 microscope (CarlZeiss Micro Imaging Inc)
Neutral comet assay
Cells were collected 24 h posttreatment and neutral cometassays were performed according to the manufacturerrsquosprotocol (Trevigen Inc) with one adjustment of35 105 cellsml for each single cell suspension Cometswere visualized using an Axiovert 200 microscope andanalyzed with Comet ScoreTM software (TriTek Corp)Approximately 100ndash150 randomly selected nonover-lapping cells were analyzed per experiment Results wereexpressed as mean tail moment
In vivo analysis of triplex-induced DNA strand breaks
AV mice were derived from the CD1 background (CharlesRiver Laboratories Wilmington MA) and were generatedas previously described (18) DNA dot blot analysis con-firmed the AV founder mouse to carry 50 copies of thetriplex target site in its genome AV mice or CD1 controlmice (14 days old) were treated by intraperitoneal (ip)injection with PBS MIX30 or AG30 (50mgkg) Threemice were used per treatment group Mice were sacrificed6 h after treatment and tissue samples were collectedSpleen tissue was collected and fixed in 4 para-formaldehyde overnight at 4C embedded in paraffinand cut into sections for evaluation by immunohisto-chemistry Cut sections were stained for phospho-H2AX(Cell Signaling) and activated caspase 3 (Abcam) andanalyzed by microscopy All sections were analyzed andquantified by counting 12 randomly selected sections ofthe same sample The number of cells positive foractivated caspase 3 and gH2AX were manually countedon digital images of the specimens The differences in thepercentage of positive cells were analyzed by one-wayANOVA and Tukey test as posthoc Representative depic-tion of immunohistochemistry is shown Animal studieswere approved and performed according to the guidelinesof the Institutional Animal Care and Use Committee ofYale University
Coimmunoprecipitation
AV16 cells were transfected with AG30 or MIX30 and24 h posttransfection cells were lysed in IP lysis buffer(Thermo Scientific) To observe the interaction of XPDwith gH2AX cell lysates were immunoprecipitated withpolyclonal rabbit antibody gH2AX (Santa Cruz) or rabbitIgG (Jackson Immunoresearch Lab) using protein AGbeads (Santa Cruz) at 4C for 90 min The immunopre-cipitated complex was analyzed by immunoblotting
Cell cycle analysis and cH2AX
AV16 cells were collected at 6 and 24 h following transfec-tion with either MIX30 or AG30 After washing once withPBS the cells were fixed in 1 paraformaldehyde for15min on ice Cells were centrifuged and fixed in 70ethanol at 20C for 2 h The cells were then washed
with BSA-T-PBS (1 wv Bovine Serum Albumin and02 vv Triton X-100 in PBS) and incubated withgH2AX antibody (Cell Signaling) in BSA-T-PBS over-night at 4C After washing the cells were incubatedwith anti-rabbit IgG Fab2 Alexa 488 (Molecular Probes)at room temperature for 1 h in the dark Cells were washedand the pellet resuspended and incubated at room tem-perature in PI staining solution (PIRNase solution BD)for 15 min Cells were analyzed by flow cytometry
Mutagenesis assay
The mouse cell lines were established with multiplechromosomally integrated copies of the recoverable supFG1 shuttle vector carrying the supFG1 reportergene Following 48ndash72 h of TFO treatment genomicDNA was isolated and incubated with in vitro packagingextracts for shuttle vector rescue and reporter geneanalysis as previously described (1619) Briefly functionalsupFG1 genes suppress the nonsense mutations in the hostbacteria b-galactosidase gene yielding blue plaques in thepresence of IPTG and X-Gal If however a mutationoccurs in the supFG1 gene the amber mutation will notbe suppressed and the resulting plaque will be whiteMutation frequency was calculated by dividing thenumber of colorless mutant plaques by the total numberof plaques counted Experiments were done in triplicateand standard errors were calculated for the mutationfrequencies as indicated by the error bars
Statistical analysis
Differences in the mean number of gH2AX focicell thenumber of apoptotic cells and tail moment were analyzedby one-way ANOVA and Tukey test as posthoc All stat-istical analyses were performed using Graphpad Prismsoftware Plt 0001 Plt 001 Plt 005
RESULTS
Triplex-induced cell death
Using an assay to measure the induction of repair synthe-sis research has determined that TFOs create a helicaldistortion upon binding to duplex DNA that stronglyprovokes DNA repair (67) Additional studies have con-firmed the importance of the NER pathway in the recog-nition and repair of TFO-induced DNA alterations (720)To investigate the potential for helical distortions toinduce apoptosis in cells we generated a mouse epithelialcell line (AV16) with 100 copies of randomly integratedchromosomal triplex target sites (Figure 1A) (16) Triplexstructures were synthetically generated using the TFOAG30 which is designed to specifically bind to thepolypurine target site (Figure 1A) Through the use of arestriction protection assay AG30-induced triplex forma-tion has been previously detected at a chromosomallyintegrated target site (21) To confirm chromosomalbinding of AG30 in our cell line we preparednondenatured metaphase chromosome spreads fromAV16 cells that had been treated Johnson et al have pre-viously described a method for detecting third-stand
8982 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
binding to nondenatured fixed metaphase spreads (22) Todetect triplex formation in vivo we modified their tech-nique by treating the cells prior to generation of the meta-phase chromosome spreads AV16 cells were treated witheither 2 mg of rhodamine-labeled AG30 or the controlmixed sequence oligonucleotide MIX30 which cannotbind as a third strand to the target polypurinepolypyrimidine sites in the AV16 cells Twenty-fourhours posttreatment cells were treated for 5 h withColcemid and then collected and prepared asnondenatured fixed metaphase chromosome spreads ac-cording to the protocol previously described (22) Tominimize background fluorescence created by thepresence of unbound oligonucleotides and nonspecificbinding events multiple washes were incorporatedduring the preparation of the metaphase spreads It hasbeen previously established that the DNA ofnondenatured fixed metaphase spreads remains in aduplex state (22) Thus the generation of chromosomalAG30-foci under the conditions of this assay representsthird strand binding to fixed chromosomes with intactDNA double helix As shown in Figure 1B several rhoda-mine-AG30 chromosomal foci were detected 24 hposttransfection in the AV16 cell-derived metaphasespreads in contrast to the rhodamine-MIX30 treatedcells These results provide evidence for third strandbinding by AG30 to multiple chromosomal sites with45 of AV16 cells treated with rhodamine-AG30 beingpositive for 5 chromosomal foci per cell (Figure 1C)compared with 10 of cells treated with MIX30 Thebinding of non-triplexndashforming oligonucleotides likeMIX30 to DNA requires single stranded targets as aresult one would not expect to see high levels of nonspe-cific chromosomal interactions with the MIX30 controlThis is consistent with the fact that this mixed sequenceoligonucleotide cannot form Hoogsteen or reverseHoogsteen triplexes even at high concentrations in vitroAlthough there may be some nonspecific retention ofMIX30 leading to low levels of foci formation this isjust background in the assay Treatment of the parentalcell line (C127) which lacks the polypurinepolypyrimidine target site with rhodamine-AG30 furtherconfirms the specificity of third strand binding(Supplementary Figure S1) Analysis of C127 cell-derived metaphase spreads revealed that 13 of cellswere positive for 5 chromosomal foci per cell followingtreatment with either MIX30 or AG30 These results aresupportive of our conclusions because both MIX30 andAG30 treatment of C127 cells resulted in the same nonspe-cific background levels The specific foci formation byAG30 in this assay provides evidence for third strandbinding and triplex formation in the treated AV16 cellsThe generation of rhodamine-AG30 foci likely representsseveral third strand binding events in proximity and sothese results support the formation of multiple triplexstructures following AG30 treatment of AV16 cells Tothis end the results from these studies provide evidencethat AG30-foci formation represents sequence-specificbinding to its polypurine target site
Following this validation we proceeded to examinewhether triplex formation was capable of inducing death
in cells with multiple TFO target sites The parental cellline C127 which lacks the target site served as a controlto assess the possibility of nonspecific oligonucleotideinteractions that could result in cell death Forty-eighthours after treatment monolayer growth assaysdemonstrated a decrease in cell growth that correlatedwith the formation of triplex structures suggestingthat DNA helical distortions can lead to cell death(Figure 1D) It is important to note that a decrease incell growth following TFO treatment was only observedin AV16 cells which have the potential to acquire multipletriplex structures and not in C127 cells which lack thetriplex-binding site In addition growth inhibition wasnot observed in cells that were treated with the controloligonucleotide MIX30 To further attribute a decreasein cell survival resulting from triplex formationclonogenic survival studies were performed using AV16cells As expected we found that only cells treated withAG30 experienced a decrease in cell survival (Figure 1E)As observed in the monolayer assay nonspecific toxicitywas not detected with either the transfection agent (mocktreatment) or MIX30 The results obtained from thesurvival assays establish that helical distortions inducedby the formation of triplex structures are capable ofinducing cell deathTo investigate the possibility that the observed cell
death could be attributed to G-quadruplex formation(23ndash25) A8G30 was designed with the same sequence asAG30 but with every third guanine substituted with themodified guanine base 7-deaza-8-aza-guanine (PPG or G)(Figure 1F) Studies have determined that substitution ofevery third guanine with PPG was sufficient to reduce self-association of TFOs containing long runs of guanines(26) To determine the relative binding affinities of theG-rich TFOs for the target duplex a gel mobility shiftassay was performed (Figure 1G) The Kd for each TFOwas estimated as the concentration of TFO at whichbinding was one-half maximal As shown in Figure 1Gusing buffer conditions that promote triplex formationboth TFOs bound to the target site with high affinity(Kd 1 109M) Treatment of AV16 cells with eitherAG30 or the PPG-substituted TFO A8G30 resulted in areduction in cell growth as observed by monolayer growthassay (Figure 1H) Taken together the inability of thecontrol oligonucleotide to induce cell death and the induc-tion of cell death after treatment with A8G30 show thatthe increase in cell death observed in the survival assays isa result of a specific and site-directed effect of AG30binding to the chromosomal target site
Triplex-induced apoptosis
Studies were then initiated to determine whether theincrease in cell death observed in the survival assays fol-lowing TFO treatment resulted from the activation of apro-apoptotic pathway AV16 cells were treated with amock transfection MIX30 or AG30 and analyzed forinduction of apoptosis 24 h posttreatment by detectionof Annexin V binding using flow cytometry Annexin Vbinding to exposed phosphatidylserine residues in the cellmembrane is an early marker of apoptosis and 23 of the
Nucleic Acids Research 2013 Vol 41 No 19 8983
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
AG30-treated cells were determined to be Annexin Vpositive as a result of triplex-induced helical distortions(Figure 2A) Western blot analysis of caspase-mediatedcleavage of Poly(ADP-ribose) polymerase (PARP) wasalso used to confirm triplex-induced apoptosis PARP anuclear DNA-binding protein that recognizes DNAstrand breaks is a substrate for caspase 3 and itscleavage is an early event in apoptotic response (27)Caspase-mediated cleavage of PARP was only detectedin cell lysates isolated from AG30-treated AV16 cells(Figure 2B) To confirm that the observed apoptosisresulted from triplex formation and not generation ofG-quadruplexes production of cleaved PARP wasanalyzed by Western blot analysis 24 h after transfectionwith either AG30 or A8G30 Similar levels of cleavedPARP were detected in cell lysates isolated from A8G30-treated AV16 cells compared with AG30 treatment sug-gesting that the observed apoptosis can be primarilyattributed to triplex formation (Figure 2C) Triplex-induced apoptosis was detected as early as 6 h and up to72 h post TFO treatment as determined by Western blotanalysis (Figure 2D)To test the impact of multiple triplex structure forma-
tion on the level of induced apoptosis AV16 cells wereexposed to increasing concentrations of AG30 As shownin Figure 2E the percentage of Annexin V positive cells
increased with higher concentrations of TFO treatmentTreatment with a low concentration of AG30 (50 nM)resulted in an Annexin V-positive population of 8which was slightly higher than that of background( 6) The percentage of Annexin V-positive cells plat-eaued at 60 following treatment with 200 nM ofAG30 Moreover no increase in the level of apoptoticcells was observed in the cells treated with increasing con-centrations of the control oligonucleotide further con-firming that the observed apoptosis can be attributed tothe formation of altered helical structures and not due tononspecific toxicity generated by the oligonucleotide itselfAs shown in Figure 2F clonogenic survival studies correl-ate with these results with the surviving cell fractiondecreasing with increasing AG30 concentrationCollectively these results support a mechanism that alter-ations to the DNA duplex structure created by the forma-tion of multiple triplex structures is capable of inducingapoptosis
Triplex-induced DSBs
To determine whether TFO-induced altered helical struc-tures can act as a fragile site resulting in DSBs we per-formed neutral comet assays Single cell lsquocometsrsquo wereobserved microscopically after separation of DNA frag-ments from the cells by electrophoresis (Figure 3A) Using
Figure 2 Induction of apoptosis via formation of triplex structures in AV16 cells (A) Annexin V binding to exposed phosphatidylserine residues24 h after treatment with 2mg of oligonucleotides (B) Western blot analysis of caspase-mediated cleavage of PARP Cells were collected and lysatesprepared 24 h posttreatment (C) Activation of apoptosis can be attributed to triplex formation rather than G-quadruplex formation as determinedby Western blot analysis of cleaved PARP following 2 mg treatment with AG30 or A8G30 (D) Time course of induced apoptosis by Western blotanalysis of cleaved PARP (E) Dose response of increasing concentrations of AG30 and its effect on apoptotic cell death 48 h posttreatment(F) Clonogenic survival after 48 h exposure of AV16 cells to AG30 or MIX30
8984 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
the lsquocomet tail momentrsquo as a measure of the extent ofDNA breakage we assessed the presence of DSBs result-ing from triplex formation As shown in Figure 3A wedetermined that AG30 treatment resulted in more DSBscompared with untreated and MIX30-treated cells
Histone variant H2AX becomes phosphorylated onserine 139 (gH2AX) in response to DNA damage thatinvolves formation of DSBs (28) and foci formation isfrequently used as a quantitative marker for DSBs in im-munofluorescence microscopy (29) The presence oftriplex-induced DSBs was also determined by co-stainingfor gH2AX and DAPI 24 h after treatment (Figure 3B)AV16 cells treated with AG30 resulted in the formation ofmore gH2AX nuclear foci compared with untreated cells(Figure 3C) Western blot analysis of gH2AX alsoconfirms the presence of H2AX S139 phosphorylation inonly the AG30-treated cells in agreement with the im-munofluorescence results (Figure 3D) These datasuggest that the formation of triplex structures in cellsthat contain multiple target sites generates substantialDSBs which may overwhelm the cellrsquos repair capacitycausing the initiation of an apoptotic response
To ensure that the presence of gH2AX foci was truly ahallmark of DSBs and not generated in the course ofDNA fragmentation during apoptosis we used amultiparameter cytometry assay (30ndash32) The presence oftriplex-induced DSBs was determined using flowcytometry by staining for gH2AX in the presence ofpropidium iodine gH2AX expression attributed toDSBs is cell cycle independent while high intensegH2AX expression in S-phase is associated with apoptosis(30) Cells were harvested 6 h and 24 h after treatment withAG30 Flow cytometry analysis of gH2AX expressionduring the cell cycle indicated increased levels of gH2AXin all phases of the cell cycle of AV16 cells 6 h afterexposure to AG30 (Figure 3E) This signal persisted forup to 24 h after TFO treatment and increased to 217(Figure 3E) suggesting that many sites marked by gH2AXfoci remained unrepaired Analysis of the flow cytometryprofiles indicates that gH2AX expression in AG30-treatedcells was significantly higher compared with mock-treatedand MIX30-treated cells (Figure 3F)
In Vivo generation of triplex-induced DSBs
To evaluate the potential for triplex DNA to induce DSBsin vivo we used a transgenic mouse model (AV mouse)with 50 copies of the triplex target sequencechromosomally integrated into its genome (Figure 4A)(18) Immunohistochemistry staining for gH2AX and theapoptosis marker activated caspase 3 was used to assessthe cellular response to in vivo triplex formation AV micewere administered a 50mgkg dose of MIX30 or AG30 viaip injection To investigate the extent of triplex-inducedDNA strand breaks we performed immunohistochemistrystaining of gH2AX as a marker for ongoing DNA damageon spleen tissue harvested 6 h posttreatment We observedlow levels of gH2AX staining in the spleens of mice thathad received ip doses of the control oligonucleotideMIX30 (Figure 4B and C) In contrast we found thatAG30 treatment triggered an increase in the percentage
of cells positive for gH2AX foci compared with thePBS- and MIX30-treated mice (Figure 4B and C) Tofurther substantiate the specificity of triplex-inducedgH2AX foci CD1 control mice which lack the triplextarget site were also administered a 50mgkg dose ofAG30 Immunohistochemistry analysis of spleen tissuerevealed no increase in the percentage of cells positive ofgH2AX foci above background (Supplementary FigureS2) An increase in the production of gH2AX foci solelyin the AV mice after AG30 treatment would suggest thepresence of triplex-induced DSBs Immunohistochemistrystaining for activated caspase 3 was then used to deter-mine whether the formation of triplex-induced gH2AXfoci could elicit an apoptotic response in vivo Micetreated with the control oligonucleotide MIX30 showedalmost a complete absence of activated caspase 3 stainingin their spleens 6 h following treatment (Figure 4B and D)However analysis of spleens from AV mice dosed withAG30 revealed that 26 of their spleen cells werepositive for activated caspase 3 staining (Figure 4B andD) Examination of spleen tissue samples obtained fromCD1 mice treated with AG30 determined a nonexistenceof activated caspase 3 6 h posttreatment (SupplementaryFigure S2) Altogether these results are consistent withthe interpretation that the formation of endogenoustriplex structures can result in DSBs which can in turnprompt the activation of apoptosis
NER deficiency results in increased apoptosis levels
Activation of apoptosis in response to DNA damageprovides a default mechanism that can be implementedto prevent clonal expansion of cells with unrepaireddamage As a result studies were initiated to investigatecellular response to triplex-induced DSBs under circum-stances where cells may be ineffective at repair XPA is akey NER factor responsible for verifying altered DNAconformations and is crucial for correct assembly of theremaining repair machinery around a lesion (33) Previousstudies have determined that TFOs were capable ofbinding to duplex DNA and creating altered helical struc-tures that strongly provoked XPA-dependent DNArepair To investigate whether triplex formation inducedDSBs in an XPA-dependent manner we treated XPA-pro-ficient and XPA-deficient mouse fibroblast cells with amock transfection MIX30 or AG30 Cells were harvested24 h following treatment and through the use of flowcytometry we evaluated the levels of triplex-inducedDNA strand breaks by staining for gH2AX Analysis ofgH2AX expression levels as a measure of double strandbreak formation indicated increased levels of gH2AX inboth the XPA-proficient and -deficient cells followingAG30 treatment (Figure 5A) Exposure to AG30increased gH2AX expression in XPA-proficient cells to22 and a similar expression level of 28 wasobserved in the XPA-deficient cells (Figure 5A)We then proceeded to evaluate induced apoptosis levels
as a result of triplex-induced DNA strand breaks in XPA-deficient cells XPA-deficient cells were treated with amock transfection MIX30 or AG30 and analyzed forthe induction of apoptosis Following 24 h administration
Nucleic Acids Research 2013 Vol 41 No 19 8985
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

donkey anti-rabbit IgG (H+L) (Molecular Probes Inc)and then stained with 100 ngml DAPI (Sigma) Imageswere captured using an Axiovert 200 microscope (CarlZeiss Micro Imaging Inc)
Neutral comet assay
Cells were collected 24 h posttreatment and neutral cometassays were performed according to the manufacturerrsquosprotocol (Trevigen Inc) with one adjustment of35 105 cellsml for each single cell suspension Cometswere visualized using an Axiovert 200 microscope andanalyzed with Comet ScoreTM software (TriTek Corp)Approximately 100ndash150 randomly selected nonover-lapping cells were analyzed per experiment Results wereexpressed as mean tail moment
In vivo analysis of triplex-induced DNA strand breaks
AV mice were derived from the CD1 background (CharlesRiver Laboratories Wilmington MA) and were generatedas previously described (18) DNA dot blot analysis con-firmed the AV founder mouse to carry 50 copies of thetriplex target site in its genome AV mice or CD1 controlmice (14 days old) were treated by intraperitoneal (ip)injection with PBS MIX30 or AG30 (50mgkg) Threemice were used per treatment group Mice were sacrificed6 h after treatment and tissue samples were collectedSpleen tissue was collected and fixed in 4 para-formaldehyde overnight at 4C embedded in paraffinand cut into sections for evaluation by immunohisto-chemistry Cut sections were stained for phospho-H2AX(Cell Signaling) and activated caspase 3 (Abcam) andanalyzed by microscopy All sections were analyzed andquantified by counting 12 randomly selected sections ofthe same sample The number of cells positive foractivated caspase 3 and gH2AX were manually countedon digital images of the specimens The differences in thepercentage of positive cells were analyzed by one-wayANOVA and Tukey test as posthoc Representative depic-tion of immunohistochemistry is shown Animal studieswere approved and performed according to the guidelinesof the Institutional Animal Care and Use Committee ofYale University
Coimmunoprecipitation
AV16 cells were transfected with AG30 or MIX30 and24 h posttransfection cells were lysed in IP lysis buffer(Thermo Scientific) To observe the interaction of XPDwith gH2AX cell lysates were immunoprecipitated withpolyclonal rabbit antibody gH2AX (Santa Cruz) or rabbitIgG (Jackson Immunoresearch Lab) using protein AGbeads (Santa Cruz) at 4C for 90 min The immunopre-cipitated complex was analyzed by immunoblotting
Cell cycle analysis and cH2AX
AV16 cells were collected at 6 and 24 h following transfec-tion with either MIX30 or AG30 After washing once withPBS the cells were fixed in 1 paraformaldehyde for15min on ice Cells were centrifuged and fixed in 70ethanol at 20C for 2 h The cells were then washed
with BSA-T-PBS (1 wv Bovine Serum Albumin and02 vv Triton X-100 in PBS) and incubated withgH2AX antibody (Cell Signaling) in BSA-T-PBS over-night at 4C After washing the cells were incubatedwith anti-rabbit IgG Fab2 Alexa 488 (Molecular Probes)at room temperature for 1 h in the dark Cells were washedand the pellet resuspended and incubated at room tem-perature in PI staining solution (PIRNase solution BD)for 15 min Cells were analyzed by flow cytometry
Mutagenesis assay
The mouse cell lines were established with multiplechromosomally integrated copies of the recoverable supFG1 shuttle vector carrying the supFG1 reportergene Following 48ndash72 h of TFO treatment genomicDNA was isolated and incubated with in vitro packagingextracts for shuttle vector rescue and reporter geneanalysis as previously described (1619) Briefly functionalsupFG1 genes suppress the nonsense mutations in the hostbacteria b-galactosidase gene yielding blue plaques in thepresence of IPTG and X-Gal If however a mutationoccurs in the supFG1 gene the amber mutation will notbe suppressed and the resulting plaque will be whiteMutation frequency was calculated by dividing thenumber of colorless mutant plaques by the total numberof plaques counted Experiments were done in triplicateand standard errors were calculated for the mutationfrequencies as indicated by the error bars
Statistical analysis
Differences in the mean number of gH2AX focicell thenumber of apoptotic cells and tail moment were analyzedby one-way ANOVA and Tukey test as posthoc All stat-istical analyses were performed using Graphpad Prismsoftware Plt 0001 Plt 001 Plt 005
RESULTS
Triplex-induced cell death
Using an assay to measure the induction of repair synthe-sis research has determined that TFOs create a helicaldistortion upon binding to duplex DNA that stronglyprovokes DNA repair (67) Additional studies have con-firmed the importance of the NER pathway in the recog-nition and repair of TFO-induced DNA alterations (720)To investigate the potential for helical distortions toinduce apoptosis in cells we generated a mouse epithelialcell line (AV16) with 100 copies of randomly integratedchromosomal triplex target sites (Figure 1A) (16) Triplexstructures were synthetically generated using the TFOAG30 which is designed to specifically bind to thepolypurine target site (Figure 1A) Through the use of arestriction protection assay AG30-induced triplex forma-tion has been previously detected at a chromosomallyintegrated target site (21) To confirm chromosomalbinding of AG30 in our cell line we preparednondenatured metaphase chromosome spreads fromAV16 cells that had been treated Johnson et al have pre-viously described a method for detecting third-stand
8982 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
binding to nondenatured fixed metaphase spreads (22) Todetect triplex formation in vivo we modified their tech-nique by treating the cells prior to generation of the meta-phase chromosome spreads AV16 cells were treated witheither 2 mg of rhodamine-labeled AG30 or the controlmixed sequence oligonucleotide MIX30 which cannotbind as a third strand to the target polypurinepolypyrimidine sites in the AV16 cells Twenty-fourhours posttreatment cells were treated for 5 h withColcemid and then collected and prepared asnondenatured fixed metaphase chromosome spreads ac-cording to the protocol previously described (22) Tominimize background fluorescence created by thepresence of unbound oligonucleotides and nonspecificbinding events multiple washes were incorporatedduring the preparation of the metaphase spreads It hasbeen previously established that the DNA ofnondenatured fixed metaphase spreads remains in aduplex state (22) Thus the generation of chromosomalAG30-foci under the conditions of this assay representsthird strand binding to fixed chromosomes with intactDNA double helix As shown in Figure 1B several rhoda-mine-AG30 chromosomal foci were detected 24 hposttransfection in the AV16 cell-derived metaphasespreads in contrast to the rhodamine-MIX30 treatedcells These results provide evidence for third strandbinding by AG30 to multiple chromosomal sites with45 of AV16 cells treated with rhodamine-AG30 beingpositive for 5 chromosomal foci per cell (Figure 1C)compared with 10 of cells treated with MIX30 Thebinding of non-triplexndashforming oligonucleotides likeMIX30 to DNA requires single stranded targets as aresult one would not expect to see high levels of nonspe-cific chromosomal interactions with the MIX30 controlThis is consistent with the fact that this mixed sequenceoligonucleotide cannot form Hoogsteen or reverseHoogsteen triplexes even at high concentrations in vitroAlthough there may be some nonspecific retention ofMIX30 leading to low levels of foci formation this isjust background in the assay Treatment of the parentalcell line (C127) which lacks the polypurinepolypyrimidine target site with rhodamine-AG30 furtherconfirms the specificity of third strand binding(Supplementary Figure S1) Analysis of C127 cell-derived metaphase spreads revealed that 13 of cellswere positive for 5 chromosomal foci per cell followingtreatment with either MIX30 or AG30 These results aresupportive of our conclusions because both MIX30 andAG30 treatment of C127 cells resulted in the same nonspe-cific background levels The specific foci formation byAG30 in this assay provides evidence for third strandbinding and triplex formation in the treated AV16 cellsThe generation of rhodamine-AG30 foci likely representsseveral third strand binding events in proximity and sothese results support the formation of multiple triplexstructures following AG30 treatment of AV16 cells Tothis end the results from these studies provide evidencethat AG30-foci formation represents sequence-specificbinding to its polypurine target site
Following this validation we proceeded to examinewhether triplex formation was capable of inducing death
in cells with multiple TFO target sites The parental cellline C127 which lacks the target site served as a controlto assess the possibility of nonspecific oligonucleotideinteractions that could result in cell death Forty-eighthours after treatment monolayer growth assaysdemonstrated a decrease in cell growth that correlatedwith the formation of triplex structures suggestingthat DNA helical distortions can lead to cell death(Figure 1D) It is important to note that a decrease incell growth following TFO treatment was only observedin AV16 cells which have the potential to acquire multipletriplex structures and not in C127 cells which lack thetriplex-binding site In addition growth inhibition wasnot observed in cells that were treated with the controloligonucleotide MIX30 To further attribute a decreasein cell survival resulting from triplex formationclonogenic survival studies were performed using AV16cells As expected we found that only cells treated withAG30 experienced a decrease in cell survival (Figure 1E)As observed in the monolayer assay nonspecific toxicitywas not detected with either the transfection agent (mocktreatment) or MIX30 The results obtained from thesurvival assays establish that helical distortions inducedby the formation of triplex structures are capable ofinducing cell deathTo investigate the possibility that the observed cell
death could be attributed to G-quadruplex formation(23ndash25) A8G30 was designed with the same sequence asAG30 but with every third guanine substituted with themodified guanine base 7-deaza-8-aza-guanine (PPG or G)(Figure 1F) Studies have determined that substitution ofevery third guanine with PPG was sufficient to reduce self-association of TFOs containing long runs of guanines(26) To determine the relative binding affinities of theG-rich TFOs for the target duplex a gel mobility shiftassay was performed (Figure 1G) The Kd for each TFOwas estimated as the concentration of TFO at whichbinding was one-half maximal As shown in Figure 1Gusing buffer conditions that promote triplex formationboth TFOs bound to the target site with high affinity(Kd 1 109M) Treatment of AV16 cells with eitherAG30 or the PPG-substituted TFO A8G30 resulted in areduction in cell growth as observed by monolayer growthassay (Figure 1H) Taken together the inability of thecontrol oligonucleotide to induce cell death and the induc-tion of cell death after treatment with A8G30 show thatthe increase in cell death observed in the survival assays isa result of a specific and site-directed effect of AG30binding to the chromosomal target site
Triplex-induced apoptosis
Studies were then initiated to determine whether theincrease in cell death observed in the survival assays fol-lowing TFO treatment resulted from the activation of apro-apoptotic pathway AV16 cells were treated with amock transfection MIX30 or AG30 and analyzed forinduction of apoptosis 24 h posttreatment by detectionof Annexin V binding using flow cytometry Annexin Vbinding to exposed phosphatidylserine residues in the cellmembrane is an early marker of apoptosis and 23 of the
Nucleic Acids Research 2013 Vol 41 No 19 8983
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
AG30-treated cells were determined to be Annexin Vpositive as a result of triplex-induced helical distortions(Figure 2A) Western blot analysis of caspase-mediatedcleavage of Poly(ADP-ribose) polymerase (PARP) wasalso used to confirm triplex-induced apoptosis PARP anuclear DNA-binding protein that recognizes DNAstrand breaks is a substrate for caspase 3 and itscleavage is an early event in apoptotic response (27)Caspase-mediated cleavage of PARP was only detectedin cell lysates isolated from AG30-treated AV16 cells(Figure 2B) To confirm that the observed apoptosisresulted from triplex formation and not generation ofG-quadruplexes production of cleaved PARP wasanalyzed by Western blot analysis 24 h after transfectionwith either AG30 or A8G30 Similar levels of cleavedPARP were detected in cell lysates isolated from A8G30-treated AV16 cells compared with AG30 treatment sug-gesting that the observed apoptosis can be primarilyattributed to triplex formation (Figure 2C) Triplex-induced apoptosis was detected as early as 6 h and up to72 h post TFO treatment as determined by Western blotanalysis (Figure 2D)To test the impact of multiple triplex structure forma-
tion on the level of induced apoptosis AV16 cells wereexposed to increasing concentrations of AG30 As shownin Figure 2E the percentage of Annexin V positive cells
increased with higher concentrations of TFO treatmentTreatment with a low concentration of AG30 (50 nM)resulted in an Annexin V-positive population of 8which was slightly higher than that of background( 6) The percentage of Annexin V-positive cells plat-eaued at 60 following treatment with 200 nM ofAG30 Moreover no increase in the level of apoptoticcells was observed in the cells treated with increasing con-centrations of the control oligonucleotide further con-firming that the observed apoptosis can be attributed tothe formation of altered helical structures and not due tononspecific toxicity generated by the oligonucleotide itselfAs shown in Figure 2F clonogenic survival studies correl-ate with these results with the surviving cell fractiondecreasing with increasing AG30 concentrationCollectively these results support a mechanism that alter-ations to the DNA duplex structure created by the forma-tion of multiple triplex structures is capable of inducingapoptosis
Triplex-induced DSBs
To determine whether TFO-induced altered helical struc-tures can act as a fragile site resulting in DSBs we per-formed neutral comet assays Single cell lsquocometsrsquo wereobserved microscopically after separation of DNA frag-ments from the cells by electrophoresis (Figure 3A) Using
Figure 2 Induction of apoptosis via formation of triplex structures in AV16 cells (A) Annexin V binding to exposed phosphatidylserine residues24 h after treatment with 2mg of oligonucleotides (B) Western blot analysis of caspase-mediated cleavage of PARP Cells were collected and lysatesprepared 24 h posttreatment (C) Activation of apoptosis can be attributed to triplex formation rather than G-quadruplex formation as determinedby Western blot analysis of cleaved PARP following 2 mg treatment with AG30 or A8G30 (D) Time course of induced apoptosis by Western blotanalysis of cleaved PARP (E) Dose response of increasing concentrations of AG30 and its effect on apoptotic cell death 48 h posttreatment(F) Clonogenic survival after 48 h exposure of AV16 cells to AG30 or MIX30
8984 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
the lsquocomet tail momentrsquo as a measure of the extent ofDNA breakage we assessed the presence of DSBs result-ing from triplex formation As shown in Figure 3A wedetermined that AG30 treatment resulted in more DSBscompared with untreated and MIX30-treated cells
Histone variant H2AX becomes phosphorylated onserine 139 (gH2AX) in response to DNA damage thatinvolves formation of DSBs (28) and foci formation isfrequently used as a quantitative marker for DSBs in im-munofluorescence microscopy (29) The presence oftriplex-induced DSBs was also determined by co-stainingfor gH2AX and DAPI 24 h after treatment (Figure 3B)AV16 cells treated with AG30 resulted in the formation ofmore gH2AX nuclear foci compared with untreated cells(Figure 3C) Western blot analysis of gH2AX alsoconfirms the presence of H2AX S139 phosphorylation inonly the AG30-treated cells in agreement with the im-munofluorescence results (Figure 3D) These datasuggest that the formation of triplex structures in cellsthat contain multiple target sites generates substantialDSBs which may overwhelm the cellrsquos repair capacitycausing the initiation of an apoptotic response
To ensure that the presence of gH2AX foci was truly ahallmark of DSBs and not generated in the course ofDNA fragmentation during apoptosis we used amultiparameter cytometry assay (30ndash32) The presence oftriplex-induced DSBs was determined using flowcytometry by staining for gH2AX in the presence ofpropidium iodine gH2AX expression attributed toDSBs is cell cycle independent while high intensegH2AX expression in S-phase is associated with apoptosis(30) Cells were harvested 6 h and 24 h after treatment withAG30 Flow cytometry analysis of gH2AX expressionduring the cell cycle indicated increased levels of gH2AXin all phases of the cell cycle of AV16 cells 6 h afterexposure to AG30 (Figure 3E) This signal persisted forup to 24 h after TFO treatment and increased to 217(Figure 3E) suggesting that many sites marked by gH2AXfoci remained unrepaired Analysis of the flow cytometryprofiles indicates that gH2AX expression in AG30-treatedcells was significantly higher compared with mock-treatedand MIX30-treated cells (Figure 3F)
In Vivo generation of triplex-induced DSBs
To evaluate the potential for triplex DNA to induce DSBsin vivo we used a transgenic mouse model (AV mouse)with 50 copies of the triplex target sequencechromosomally integrated into its genome (Figure 4A)(18) Immunohistochemistry staining for gH2AX and theapoptosis marker activated caspase 3 was used to assessthe cellular response to in vivo triplex formation AV micewere administered a 50mgkg dose of MIX30 or AG30 viaip injection To investigate the extent of triplex-inducedDNA strand breaks we performed immunohistochemistrystaining of gH2AX as a marker for ongoing DNA damageon spleen tissue harvested 6 h posttreatment We observedlow levels of gH2AX staining in the spleens of mice thathad received ip doses of the control oligonucleotideMIX30 (Figure 4B and C) In contrast we found thatAG30 treatment triggered an increase in the percentage
of cells positive for gH2AX foci compared with thePBS- and MIX30-treated mice (Figure 4B and C) Tofurther substantiate the specificity of triplex-inducedgH2AX foci CD1 control mice which lack the triplextarget site were also administered a 50mgkg dose ofAG30 Immunohistochemistry analysis of spleen tissuerevealed no increase in the percentage of cells positive ofgH2AX foci above background (Supplementary FigureS2) An increase in the production of gH2AX foci solelyin the AV mice after AG30 treatment would suggest thepresence of triplex-induced DSBs Immunohistochemistrystaining for activated caspase 3 was then used to deter-mine whether the formation of triplex-induced gH2AXfoci could elicit an apoptotic response in vivo Micetreated with the control oligonucleotide MIX30 showedalmost a complete absence of activated caspase 3 stainingin their spleens 6 h following treatment (Figure 4B and D)However analysis of spleens from AV mice dosed withAG30 revealed that 26 of their spleen cells werepositive for activated caspase 3 staining (Figure 4B andD) Examination of spleen tissue samples obtained fromCD1 mice treated with AG30 determined a nonexistenceof activated caspase 3 6 h posttreatment (SupplementaryFigure S2) Altogether these results are consistent withthe interpretation that the formation of endogenoustriplex structures can result in DSBs which can in turnprompt the activation of apoptosis
NER deficiency results in increased apoptosis levels
Activation of apoptosis in response to DNA damageprovides a default mechanism that can be implementedto prevent clonal expansion of cells with unrepaireddamage As a result studies were initiated to investigatecellular response to triplex-induced DSBs under circum-stances where cells may be ineffective at repair XPA is akey NER factor responsible for verifying altered DNAconformations and is crucial for correct assembly of theremaining repair machinery around a lesion (33) Previousstudies have determined that TFOs were capable ofbinding to duplex DNA and creating altered helical struc-tures that strongly provoked XPA-dependent DNArepair To investigate whether triplex formation inducedDSBs in an XPA-dependent manner we treated XPA-pro-ficient and XPA-deficient mouse fibroblast cells with amock transfection MIX30 or AG30 Cells were harvested24 h following treatment and through the use of flowcytometry we evaluated the levels of triplex-inducedDNA strand breaks by staining for gH2AX Analysis ofgH2AX expression levels as a measure of double strandbreak formation indicated increased levels of gH2AX inboth the XPA-proficient and -deficient cells followingAG30 treatment (Figure 5A) Exposure to AG30increased gH2AX expression in XPA-proficient cells to22 and a similar expression level of 28 wasobserved in the XPA-deficient cells (Figure 5A)We then proceeded to evaluate induced apoptosis levels
as a result of triplex-induced DNA strand breaks in XPA-deficient cells XPA-deficient cells were treated with amock transfection MIX30 or AG30 and analyzed forthe induction of apoptosis Following 24 h administration
Nucleic Acids Research 2013 Vol 41 No 19 8985
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

binding to nondenatured fixed metaphase spreads (22) Todetect triplex formation in vivo we modified their tech-nique by treating the cells prior to generation of the meta-phase chromosome spreads AV16 cells were treated witheither 2 mg of rhodamine-labeled AG30 or the controlmixed sequence oligonucleotide MIX30 which cannotbind as a third strand to the target polypurinepolypyrimidine sites in the AV16 cells Twenty-fourhours posttreatment cells were treated for 5 h withColcemid and then collected and prepared asnondenatured fixed metaphase chromosome spreads ac-cording to the protocol previously described (22) Tominimize background fluorescence created by thepresence of unbound oligonucleotides and nonspecificbinding events multiple washes were incorporatedduring the preparation of the metaphase spreads It hasbeen previously established that the DNA ofnondenatured fixed metaphase spreads remains in aduplex state (22) Thus the generation of chromosomalAG30-foci under the conditions of this assay representsthird strand binding to fixed chromosomes with intactDNA double helix As shown in Figure 1B several rhoda-mine-AG30 chromosomal foci were detected 24 hposttransfection in the AV16 cell-derived metaphasespreads in contrast to the rhodamine-MIX30 treatedcells These results provide evidence for third strandbinding by AG30 to multiple chromosomal sites with45 of AV16 cells treated with rhodamine-AG30 beingpositive for 5 chromosomal foci per cell (Figure 1C)compared with 10 of cells treated with MIX30 Thebinding of non-triplexndashforming oligonucleotides likeMIX30 to DNA requires single stranded targets as aresult one would not expect to see high levels of nonspe-cific chromosomal interactions with the MIX30 controlThis is consistent with the fact that this mixed sequenceoligonucleotide cannot form Hoogsteen or reverseHoogsteen triplexes even at high concentrations in vitroAlthough there may be some nonspecific retention ofMIX30 leading to low levels of foci formation this isjust background in the assay Treatment of the parentalcell line (C127) which lacks the polypurinepolypyrimidine target site with rhodamine-AG30 furtherconfirms the specificity of third strand binding(Supplementary Figure S1) Analysis of C127 cell-derived metaphase spreads revealed that 13 of cellswere positive for 5 chromosomal foci per cell followingtreatment with either MIX30 or AG30 These results aresupportive of our conclusions because both MIX30 andAG30 treatment of C127 cells resulted in the same nonspe-cific background levels The specific foci formation byAG30 in this assay provides evidence for third strandbinding and triplex formation in the treated AV16 cellsThe generation of rhodamine-AG30 foci likely representsseveral third strand binding events in proximity and sothese results support the formation of multiple triplexstructures following AG30 treatment of AV16 cells Tothis end the results from these studies provide evidencethat AG30-foci formation represents sequence-specificbinding to its polypurine target site
Following this validation we proceeded to examinewhether triplex formation was capable of inducing death
in cells with multiple TFO target sites The parental cellline C127 which lacks the target site served as a controlto assess the possibility of nonspecific oligonucleotideinteractions that could result in cell death Forty-eighthours after treatment monolayer growth assaysdemonstrated a decrease in cell growth that correlatedwith the formation of triplex structures suggestingthat DNA helical distortions can lead to cell death(Figure 1D) It is important to note that a decrease incell growth following TFO treatment was only observedin AV16 cells which have the potential to acquire multipletriplex structures and not in C127 cells which lack thetriplex-binding site In addition growth inhibition wasnot observed in cells that were treated with the controloligonucleotide MIX30 To further attribute a decreasein cell survival resulting from triplex formationclonogenic survival studies were performed using AV16cells As expected we found that only cells treated withAG30 experienced a decrease in cell survival (Figure 1E)As observed in the monolayer assay nonspecific toxicitywas not detected with either the transfection agent (mocktreatment) or MIX30 The results obtained from thesurvival assays establish that helical distortions inducedby the formation of triplex structures are capable ofinducing cell deathTo investigate the possibility that the observed cell
death could be attributed to G-quadruplex formation(23ndash25) A8G30 was designed with the same sequence asAG30 but with every third guanine substituted with themodified guanine base 7-deaza-8-aza-guanine (PPG or G)(Figure 1F) Studies have determined that substitution ofevery third guanine with PPG was sufficient to reduce self-association of TFOs containing long runs of guanines(26) To determine the relative binding affinities of theG-rich TFOs for the target duplex a gel mobility shiftassay was performed (Figure 1G) The Kd for each TFOwas estimated as the concentration of TFO at whichbinding was one-half maximal As shown in Figure 1Gusing buffer conditions that promote triplex formationboth TFOs bound to the target site with high affinity(Kd 1 109M) Treatment of AV16 cells with eitherAG30 or the PPG-substituted TFO A8G30 resulted in areduction in cell growth as observed by monolayer growthassay (Figure 1H) Taken together the inability of thecontrol oligonucleotide to induce cell death and the induc-tion of cell death after treatment with A8G30 show thatthe increase in cell death observed in the survival assays isa result of a specific and site-directed effect of AG30binding to the chromosomal target site
Triplex-induced apoptosis
Studies were then initiated to determine whether theincrease in cell death observed in the survival assays fol-lowing TFO treatment resulted from the activation of apro-apoptotic pathway AV16 cells were treated with amock transfection MIX30 or AG30 and analyzed forinduction of apoptosis 24 h posttreatment by detectionof Annexin V binding using flow cytometry Annexin Vbinding to exposed phosphatidylserine residues in the cellmembrane is an early marker of apoptosis and 23 of the
Nucleic Acids Research 2013 Vol 41 No 19 8983
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
AG30-treated cells were determined to be Annexin Vpositive as a result of triplex-induced helical distortions(Figure 2A) Western blot analysis of caspase-mediatedcleavage of Poly(ADP-ribose) polymerase (PARP) wasalso used to confirm triplex-induced apoptosis PARP anuclear DNA-binding protein that recognizes DNAstrand breaks is a substrate for caspase 3 and itscleavage is an early event in apoptotic response (27)Caspase-mediated cleavage of PARP was only detectedin cell lysates isolated from AG30-treated AV16 cells(Figure 2B) To confirm that the observed apoptosisresulted from triplex formation and not generation ofG-quadruplexes production of cleaved PARP wasanalyzed by Western blot analysis 24 h after transfectionwith either AG30 or A8G30 Similar levels of cleavedPARP were detected in cell lysates isolated from A8G30-treated AV16 cells compared with AG30 treatment sug-gesting that the observed apoptosis can be primarilyattributed to triplex formation (Figure 2C) Triplex-induced apoptosis was detected as early as 6 h and up to72 h post TFO treatment as determined by Western blotanalysis (Figure 2D)To test the impact of multiple triplex structure forma-
tion on the level of induced apoptosis AV16 cells wereexposed to increasing concentrations of AG30 As shownin Figure 2E the percentage of Annexin V positive cells
increased with higher concentrations of TFO treatmentTreatment with a low concentration of AG30 (50 nM)resulted in an Annexin V-positive population of 8which was slightly higher than that of background( 6) The percentage of Annexin V-positive cells plat-eaued at 60 following treatment with 200 nM ofAG30 Moreover no increase in the level of apoptoticcells was observed in the cells treated with increasing con-centrations of the control oligonucleotide further con-firming that the observed apoptosis can be attributed tothe formation of altered helical structures and not due tononspecific toxicity generated by the oligonucleotide itselfAs shown in Figure 2F clonogenic survival studies correl-ate with these results with the surviving cell fractiondecreasing with increasing AG30 concentrationCollectively these results support a mechanism that alter-ations to the DNA duplex structure created by the forma-tion of multiple triplex structures is capable of inducingapoptosis
Triplex-induced DSBs
To determine whether TFO-induced altered helical struc-tures can act as a fragile site resulting in DSBs we per-formed neutral comet assays Single cell lsquocometsrsquo wereobserved microscopically after separation of DNA frag-ments from the cells by electrophoresis (Figure 3A) Using
Figure 2 Induction of apoptosis via formation of triplex structures in AV16 cells (A) Annexin V binding to exposed phosphatidylserine residues24 h after treatment with 2mg of oligonucleotides (B) Western blot analysis of caspase-mediated cleavage of PARP Cells were collected and lysatesprepared 24 h posttreatment (C) Activation of apoptosis can be attributed to triplex formation rather than G-quadruplex formation as determinedby Western blot analysis of cleaved PARP following 2 mg treatment with AG30 or A8G30 (D) Time course of induced apoptosis by Western blotanalysis of cleaved PARP (E) Dose response of increasing concentrations of AG30 and its effect on apoptotic cell death 48 h posttreatment(F) Clonogenic survival after 48 h exposure of AV16 cells to AG30 or MIX30
8984 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
the lsquocomet tail momentrsquo as a measure of the extent ofDNA breakage we assessed the presence of DSBs result-ing from triplex formation As shown in Figure 3A wedetermined that AG30 treatment resulted in more DSBscompared with untreated and MIX30-treated cells
Histone variant H2AX becomes phosphorylated onserine 139 (gH2AX) in response to DNA damage thatinvolves formation of DSBs (28) and foci formation isfrequently used as a quantitative marker for DSBs in im-munofluorescence microscopy (29) The presence oftriplex-induced DSBs was also determined by co-stainingfor gH2AX and DAPI 24 h after treatment (Figure 3B)AV16 cells treated with AG30 resulted in the formation ofmore gH2AX nuclear foci compared with untreated cells(Figure 3C) Western blot analysis of gH2AX alsoconfirms the presence of H2AX S139 phosphorylation inonly the AG30-treated cells in agreement with the im-munofluorescence results (Figure 3D) These datasuggest that the formation of triplex structures in cellsthat contain multiple target sites generates substantialDSBs which may overwhelm the cellrsquos repair capacitycausing the initiation of an apoptotic response
To ensure that the presence of gH2AX foci was truly ahallmark of DSBs and not generated in the course ofDNA fragmentation during apoptosis we used amultiparameter cytometry assay (30ndash32) The presence oftriplex-induced DSBs was determined using flowcytometry by staining for gH2AX in the presence ofpropidium iodine gH2AX expression attributed toDSBs is cell cycle independent while high intensegH2AX expression in S-phase is associated with apoptosis(30) Cells were harvested 6 h and 24 h after treatment withAG30 Flow cytometry analysis of gH2AX expressionduring the cell cycle indicated increased levels of gH2AXin all phases of the cell cycle of AV16 cells 6 h afterexposure to AG30 (Figure 3E) This signal persisted forup to 24 h after TFO treatment and increased to 217(Figure 3E) suggesting that many sites marked by gH2AXfoci remained unrepaired Analysis of the flow cytometryprofiles indicates that gH2AX expression in AG30-treatedcells was significantly higher compared with mock-treatedand MIX30-treated cells (Figure 3F)
In Vivo generation of triplex-induced DSBs
To evaluate the potential for triplex DNA to induce DSBsin vivo we used a transgenic mouse model (AV mouse)with 50 copies of the triplex target sequencechromosomally integrated into its genome (Figure 4A)(18) Immunohistochemistry staining for gH2AX and theapoptosis marker activated caspase 3 was used to assessthe cellular response to in vivo triplex formation AV micewere administered a 50mgkg dose of MIX30 or AG30 viaip injection To investigate the extent of triplex-inducedDNA strand breaks we performed immunohistochemistrystaining of gH2AX as a marker for ongoing DNA damageon spleen tissue harvested 6 h posttreatment We observedlow levels of gH2AX staining in the spleens of mice thathad received ip doses of the control oligonucleotideMIX30 (Figure 4B and C) In contrast we found thatAG30 treatment triggered an increase in the percentage
of cells positive for gH2AX foci compared with thePBS- and MIX30-treated mice (Figure 4B and C) Tofurther substantiate the specificity of triplex-inducedgH2AX foci CD1 control mice which lack the triplextarget site were also administered a 50mgkg dose ofAG30 Immunohistochemistry analysis of spleen tissuerevealed no increase in the percentage of cells positive ofgH2AX foci above background (Supplementary FigureS2) An increase in the production of gH2AX foci solelyin the AV mice after AG30 treatment would suggest thepresence of triplex-induced DSBs Immunohistochemistrystaining for activated caspase 3 was then used to deter-mine whether the formation of triplex-induced gH2AXfoci could elicit an apoptotic response in vivo Micetreated with the control oligonucleotide MIX30 showedalmost a complete absence of activated caspase 3 stainingin their spleens 6 h following treatment (Figure 4B and D)However analysis of spleens from AV mice dosed withAG30 revealed that 26 of their spleen cells werepositive for activated caspase 3 staining (Figure 4B andD) Examination of spleen tissue samples obtained fromCD1 mice treated with AG30 determined a nonexistenceof activated caspase 3 6 h posttreatment (SupplementaryFigure S2) Altogether these results are consistent withthe interpretation that the formation of endogenoustriplex structures can result in DSBs which can in turnprompt the activation of apoptosis
NER deficiency results in increased apoptosis levels
Activation of apoptosis in response to DNA damageprovides a default mechanism that can be implementedto prevent clonal expansion of cells with unrepaireddamage As a result studies were initiated to investigatecellular response to triplex-induced DSBs under circum-stances where cells may be ineffective at repair XPA is akey NER factor responsible for verifying altered DNAconformations and is crucial for correct assembly of theremaining repair machinery around a lesion (33) Previousstudies have determined that TFOs were capable ofbinding to duplex DNA and creating altered helical struc-tures that strongly provoked XPA-dependent DNArepair To investigate whether triplex formation inducedDSBs in an XPA-dependent manner we treated XPA-pro-ficient and XPA-deficient mouse fibroblast cells with amock transfection MIX30 or AG30 Cells were harvested24 h following treatment and through the use of flowcytometry we evaluated the levels of triplex-inducedDNA strand breaks by staining for gH2AX Analysis ofgH2AX expression levels as a measure of double strandbreak formation indicated increased levels of gH2AX inboth the XPA-proficient and -deficient cells followingAG30 treatment (Figure 5A) Exposure to AG30increased gH2AX expression in XPA-proficient cells to22 and a similar expression level of 28 wasobserved in the XPA-deficient cells (Figure 5A)We then proceeded to evaluate induced apoptosis levels
as a result of triplex-induced DNA strand breaks in XPA-deficient cells XPA-deficient cells were treated with amock transfection MIX30 or AG30 and analyzed forthe induction of apoptosis Following 24 h administration
Nucleic Acids Research 2013 Vol 41 No 19 8985
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
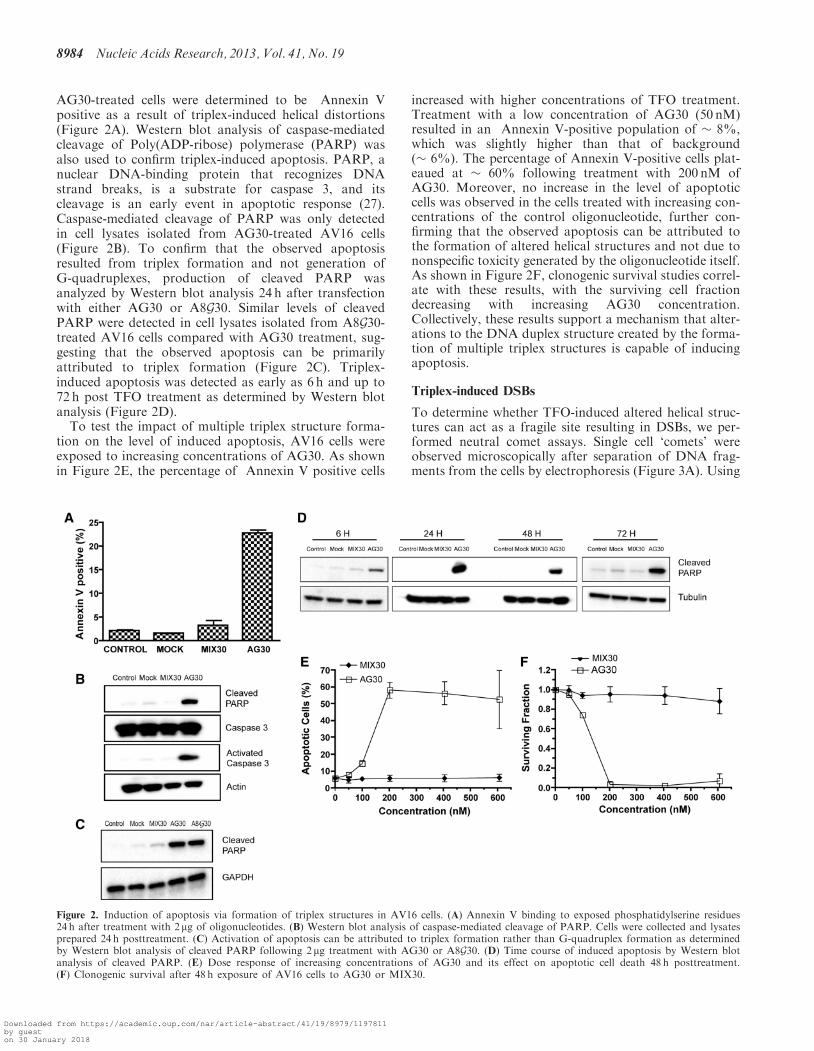
AG30-treated cells were determined to be Annexin Vpositive as a result of triplex-induced helical distortions(Figure 2A) Western blot analysis of caspase-mediatedcleavage of Poly(ADP-ribose) polymerase (PARP) wasalso used to confirm triplex-induced apoptosis PARP anuclear DNA-binding protein that recognizes DNAstrand breaks is a substrate for caspase 3 and itscleavage is an early event in apoptotic response (27)Caspase-mediated cleavage of PARP was only detectedin cell lysates isolated from AG30-treated AV16 cells(Figure 2B) To confirm that the observed apoptosisresulted from triplex formation and not generation ofG-quadruplexes production of cleaved PARP wasanalyzed by Western blot analysis 24 h after transfectionwith either AG30 or A8G30 Similar levels of cleavedPARP were detected in cell lysates isolated from A8G30-treated AV16 cells compared with AG30 treatment sug-gesting that the observed apoptosis can be primarilyattributed to triplex formation (Figure 2C) Triplex-induced apoptosis was detected as early as 6 h and up to72 h post TFO treatment as determined by Western blotanalysis (Figure 2D)To test the impact of multiple triplex structure forma-
tion on the level of induced apoptosis AV16 cells wereexposed to increasing concentrations of AG30 As shownin Figure 2E the percentage of Annexin V positive cells
increased with higher concentrations of TFO treatmentTreatment with a low concentration of AG30 (50 nM)resulted in an Annexin V-positive population of 8which was slightly higher than that of background( 6) The percentage of Annexin V-positive cells plat-eaued at 60 following treatment with 200 nM ofAG30 Moreover no increase in the level of apoptoticcells was observed in the cells treated with increasing con-centrations of the control oligonucleotide further con-firming that the observed apoptosis can be attributed tothe formation of altered helical structures and not due tononspecific toxicity generated by the oligonucleotide itselfAs shown in Figure 2F clonogenic survival studies correl-ate with these results with the surviving cell fractiondecreasing with increasing AG30 concentrationCollectively these results support a mechanism that alter-ations to the DNA duplex structure created by the forma-tion of multiple triplex structures is capable of inducingapoptosis
Triplex-induced DSBs
To determine whether TFO-induced altered helical struc-tures can act as a fragile site resulting in DSBs we per-formed neutral comet assays Single cell lsquocometsrsquo wereobserved microscopically after separation of DNA frag-ments from the cells by electrophoresis (Figure 3A) Using
Figure 2 Induction of apoptosis via formation of triplex structures in AV16 cells (A) Annexin V binding to exposed phosphatidylserine residues24 h after treatment with 2mg of oligonucleotides (B) Western blot analysis of caspase-mediated cleavage of PARP Cells were collected and lysatesprepared 24 h posttreatment (C) Activation of apoptosis can be attributed to triplex formation rather than G-quadruplex formation as determinedby Western blot analysis of cleaved PARP following 2 mg treatment with AG30 or A8G30 (D) Time course of induced apoptosis by Western blotanalysis of cleaved PARP (E) Dose response of increasing concentrations of AG30 and its effect on apoptotic cell death 48 h posttreatment(F) Clonogenic survival after 48 h exposure of AV16 cells to AG30 or MIX30
8984 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
the lsquocomet tail momentrsquo as a measure of the extent ofDNA breakage we assessed the presence of DSBs result-ing from triplex formation As shown in Figure 3A wedetermined that AG30 treatment resulted in more DSBscompared with untreated and MIX30-treated cells
Histone variant H2AX becomes phosphorylated onserine 139 (gH2AX) in response to DNA damage thatinvolves formation of DSBs (28) and foci formation isfrequently used as a quantitative marker for DSBs in im-munofluorescence microscopy (29) The presence oftriplex-induced DSBs was also determined by co-stainingfor gH2AX and DAPI 24 h after treatment (Figure 3B)AV16 cells treated with AG30 resulted in the formation ofmore gH2AX nuclear foci compared with untreated cells(Figure 3C) Western blot analysis of gH2AX alsoconfirms the presence of H2AX S139 phosphorylation inonly the AG30-treated cells in agreement with the im-munofluorescence results (Figure 3D) These datasuggest that the formation of triplex structures in cellsthat contain multiple target sites generates substantialDSBs which may overwhelm the cellrsquos repair capacitycausing the initiation of an apoptotic response
To ensure that the presence of gH2AX foci was truly ahallmark of DSBs and not generated in the course ofDNA fragmentation during apoptosis we used amultiparameter cytometry assay (30ndash32) The presence oftriplex-induced DSBs was determined using flowcytometry by staining for gH2AX in the presence ofpropidium iodine gH2AX expression attributed toDSBs is cell cycle independent while high intensegH2AX expression in S-phase is associated with apoptosis(30) Cells were harvested 6 h and 24 h after treatment withAG30 Flow cytometry analysis of gH2AX expressionduring the cell cycle indicated increased levels of gH2AXin all phases of the cell cycle of AV16 cells 6 h afterexposure to AG30 (Figure 3E) This signal persisted forup to 24 h after TFO treatment and increased to 217(Figure 3E) suggesting that many sites marked by gH2AXfoci remained unrepaired Analysis of the flow cytometryprofiles indicates that gH2AX expression in AG30-treatedcells was significantly higher compared with mock-treatedand MIX30-treated cells (Figure 3F)
In Vivo generation of triplex-induced DSBs
To evaluate the potential for triplex DNA to induce DSBsin vivo we used a transgenic mouse model (AV mouse)with 50 copies of the triplex target sequencechromosomally integrated into its genome (Figure 4A)(18) Immunohistochemistry staining for gH2AX and theapoptosis marker activated caspase 3 was used to assessthe cellular response to in vivo triplex formation AV micewere administered a 50mgkg dose of MIX30 or AG30 viaip injection To investigate the extent of triplex-inducedDNA strand breaks we performed immunohistochemistrystaining of gH2AX as a marker for ongoing DNA damageon spleen tissue harvested 6 h posttreatment We observedlow levels of gH2AX staining in the spleens of mice thathad received ip doses of the control oligonucleotideMIX30 (Figure 4B and C) In contrast we found thatAG30 treatment triggered an increase in the percentage
of cells positive for gH2AX foci compared with thePBS- and MIX30-treated mice (Figure 4B and C) Tofurther substantiate the specificity of triplex-inducedgH2AX foci CD1 control mice which lack the triplextarget site were also administered a 50mgkg dose ofAG30 Immunohistochemistry analysis of spleen tissuerevealed no increase in the percentage of cells positive ofgH2AX foci above background (Supplementary FigureS2) An increase in the production of gH2AX foci solelyin the AV mice after AG30 treatment would suggest thepresence of triplex-induced DSBs Immunohistochemistrystaining for activated caspase 3 was then used to deter-mine whether the formation of triplex-induced gH2AXfoci could elicit an apoptotic response in vivo Micetreated with the control oligonucleotide MIX30 showedalmost a complete absence of activated caspase 3 stainingin their spleens 6 h following treatment (Figure 4B and D)However analysis of spleens from AV mice dosed withAG30 revealed that 26 of their spleen cells werepositive for activated caspase 3 staining (Figure 4B andD) Examination of spleen tissue samples obtained fromCD1 mice treated with AG30 determined a nonexistenceof activated caspase 3 6 h posttreatment (SupplementaryFigure S2) Altogether these results are consistent withthe interpretation that the formation of endogenoustriplex structures can result in DSBs which can in turnprompt the activation of apoptosis
NER deficiency results in increased apoptosis levels
Activation of apoptosis in response to DNA damageprovides a default mechanism that can be implementedto prevent clonal expansion of cells with unrepaireddamage As a result studies were initiated to investigatecellular response to triplex-induced DSBs under circum-stances where cells may be ineffective at repair XPA is akey NER factor responsible for verifying altered DNAconformations and is crucial for correct assembly of theremaining repair machinery around a lesion (33) Previousstudies have determined that TFOs were capable ofbinding to duplex DNA and creating altered helical struc-tures that strongly provoked XPA-dependent DNArepair To investigate whether triplex formation inducedDSBs in an XPA-dependent manner we treated XPA-pro-ficient and XPA-deficient mouse fibroblast cells with amock transfection MIX30 or AG30 Cells were harvested24 h following treatment and through the use of flowcytometry we evaluated the levels of triplex-inducedDNA strand breaks by staining for gH2AX Analysis ofgH2AX expression levels as a measure of double strandbreak formation indicated increased levels of gH2AX inboth the XPA-proficient and -deficient cells followingAG30 treatment (Figure 5A) Exposure to AG30increased gH2AX expression in XPA-proficient cells to22 and a similar expression level of 28 wasobserved in the XPA-deficient cells (Figure 5A)We then proceeded to evaluate induced apoptosis levels
as a result of triplex-induced DNA strand breaks in XPA-deficient cells XPA-deficient cells were treated with amock transfection MIX30 or AG30 and analyzed forthe induction of apoptosis Following 24 h administration
Nucleic Acids Research 2013 Vol 41 No 19 8985
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

the lsquocomet tail momentrsquo as a measure of the extent ofDNA breakage we assessed the presence of DSBs result-ing from triplex formation As shown in Figure 3A wedetermined that AG30 treatment resulted in more DSBscompared with untreated and MIX30-treated cells
Histone variant H2AX becomes phosphorylated onserine 139 (gH2AX) in response to DNA damage thatinvolves formation of DSBs (28) and foci formation isfrequently used as a quantitative marker for DSBs in im-munofluorescence microscopy (29) The presence oftriplex-induced DSBs was also determined by co-stainingfor gH2AX and DAPI 24 h after treatment (Figure 3B)AV16 cells treated with AG30 resulted in the formation ofmore gH2AX nuclear foci compared with untreated cells(Figure 3C) Western blot analysis of gH2AX alsoconfirms the presence of H2AX S139 phosphorylation inonly the AG30-treated cells in agreement with the im-munofluorescence results (Figure 3D) These datasuggest that the formation of triplex structures in cellsthat contain multiple target sites generates substantialDSBs which may overwhelm the cellrsquos repair capacitycausing the initiation of an apoptotic response
To ensure that the presence of gH2AX foci was truly ahallmark of DSBs and not generated in the course ofDNA fragmentation during apoptosis we used amultiparameter cytometry assay (30ndash32) The presence oftriplex-induced DSBs was determined using flowcytometry by staining for gH2AX in the presence ofpropidium iodine gH2AX expression attributed toDSBs is cell cycle independent while high intensegH2AX expression in S-phase is associated with apoptosis(30) Cells were harvested 6 h and 24 h after treatment withAG30 Flow cytometry analysis of gH2AX expressionduring the cell cycle indicated increased levels of gH2AXin all phases of the cell cycle of AV16 cells 6 h afterexposure to AG30 (Figure 3E) This signal persisted forup to 24 h after TFO treatment and increased to 217(Figure 3E) suggesting that many sites marked by gH2AXfoci remained unrepaired Analysis of the flow cytometryprofiles indicates that gH2AX expression in AG30-treatedcells was significantly higher compared with mock-treatedand MIX30-treated cells (Figure 3F)
In Vivo generation of triplex-induced DSBs
To evaluate the potential for triplex DNA to induce DSBsin vivo we used a transgenic mouse model (AV mouse)with 50 copies of the triplex target sequencechromosomally integrated into its genome (Figure 4A)(18) Immunohistochemistry staining for gH2AX and theapoptosis marker activated caspase 3 was used to assessthe cellular response to in vivo triplex formation AV micewere administered a 50mgkg dose of MIX30 or AG30 viaip injection To investigate the extent of triplex-inducedDNA strand breaks we performed immunohistochemistrystaining of gH2AX as a marker for ongoing DNA damageon spleen tissue harvested 6 h posttreatment We observedlow levels of gH2AX staining in the spleens of mice thathad received ip doses of the control oligonucleotideMIX30 (Figure 4B and C) In contrast we found thatAG30 treatment triggered an increase in the percentage
of cells positive for gH2AX foci compared with thePBS- and MIX30-treated mice (Figure 4B and C) Tofurther substantiate the specificity of triplex-inducedgH2AX foci CD1 control mice which lack the triplextarget site were also administered a 50mgkg dose ofAG30 Immunohistochemistry analysis of spleen tissuerevealed no increase in the percentage of cells positive ofgH2AX foci above background (Supplementary FigureS2) An increase in the production of gH2AX foci solelyin the AV mice after AG30 treatment would suggest thepresence of triplex-induced DSBs Immunohistochemistrystaining for activated caspase 3 was then used to deter-mine whether the formation of triplex-induced gH2AXfoci could elicit an apoptotic response in vivo Micetreated with the control oligonucleotide MIX30 showedalmost a complete absence of activated caspase 3 stainingin their spleens 6 h following treatment (Figure 4B and D)However analysis of spleens from AV mice dosed withAG30 revealed that 26 of their spleen cells werepositive for activated caspase 3 staining (Figure 4B andD) Examination of spleen tissue samples obtained fromCD1 mice treated with AG30 determined a nonexistenceof activated caspase 3 6 h posttreatment (SupplementaryFigure S2) Altogether these results are consistent withthe interpretation that the formation of endogenoustriplex structures can result in DSBs which can in turnprompt the activation of apoptosis
NER deficiency results in increased apoptosis levels
Activation of apoptosis in response to DNA damageprovides a default mechanism that can be implementedto prevent clonal expansion of cells with unrepaireddamage As a result studies were initiated to investigatecellular response to triplex-induced DSBs under circum-stances where cells may be ineffective at repair XPA is akey NER factor responsible for verifying altered DNAconformations and is crucial for correct assembly of theremaining repair machinery around a lesion (33) Previousstudies have determined that TFOs were capable ofbinding to duplex DNA and creating altered helical struc-tures that strongly provoked XPA-dependent DNArepair To investigate whether triplex formation inducedDSBs in an XPA-dependent manner we treated XPA-pro-ficient and XPA-deficient mouse fibroblast cells with amock transfection MIX30 or AG30 Cells were harvested24 h following treatment and through the use of flowcytometry we evaluated the levels of triplex-inducedDNA strand breaks by staining for gH2AX Analysis ofgH2AX expression levels as a measure of double strandbreak formation indicated increased levels of gH2AX inboth the XPA-proficient and -deficient cells followingAG30 treatment (Figure 5A) Exposure to AG30increased gH2AX expression in XPA-proficient cells to22 and a similar expression level of 28 wasobserved in the XPA-deficient cells (Figure 5A)We then proceeded to evaluate induced apoptosis levels
as a result of triplex-induced DNA strand breaks in XPA-deficient cells XPA-deficient cells were treated with amock transfection MIX30 or AG30 and analyzed forthe induction of apoptosis Following 24 h administration
Nucleic Acids Research 2013 Vol 41 No 19 8985
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
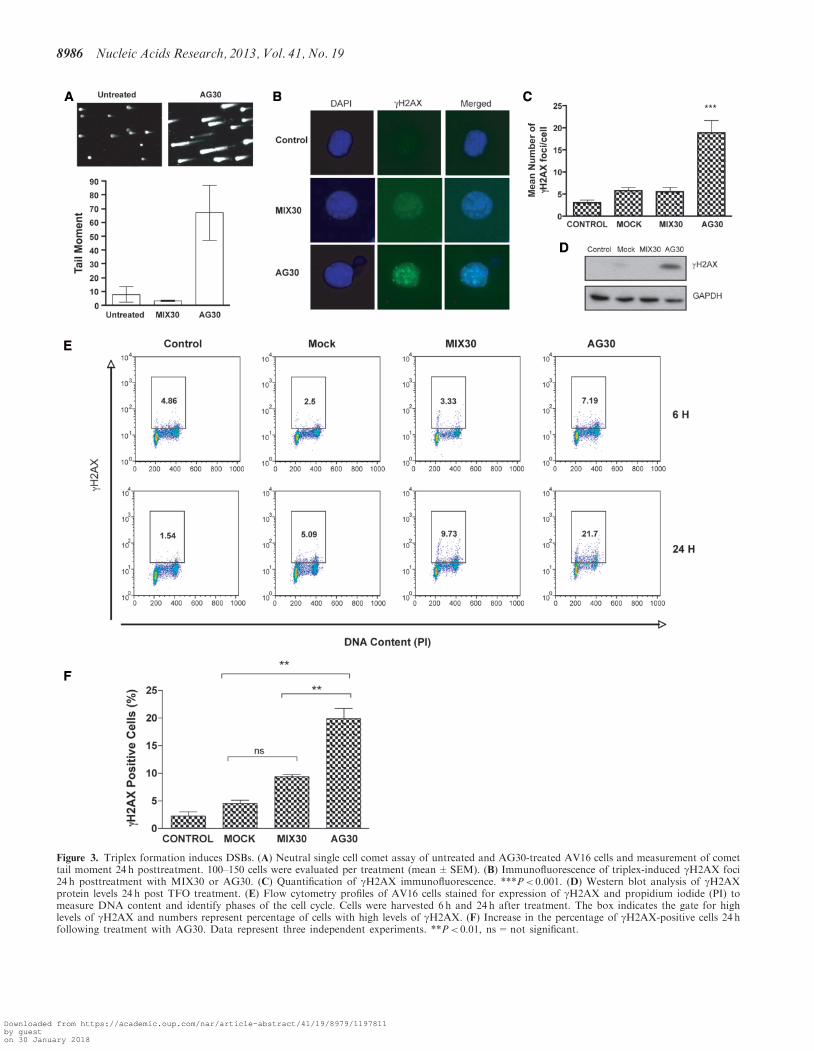
Figure 3 Triplex formation induces DSBs (A) Neutral single cell comet assay of untreated and AG30-treated AV16 cells and measurement of comettail moment 24 h posttreatment 100ndash150 cells were evaluated per treatment (meanplusmnSEM) (B) Immunofluorescence of triplex-induced gH2AX foci24 h posttreatment with MIX30 or AG30 (C) Quantification of gH2AX immunofluorescence Plt 0001 (D) Western blot analysis of gH2AXprotein levels 24 h post TFO treatment (E) Flow cytometry profiles of AV16 cells stained for expression of gH2AX and propidium iodide (PI) tomeasure DNA content and identify phases of the cell cycle Cells were harvested 6 h and 24 h after treatment The box indicates the gate for highlevels of gH2AX and numbers represent percentage of cells with high levels of gH2AX (F) Increase in the percentage of gH2AX-positive cells 24 hfollowing treatment with AG30 Data represent three independent experiments Plt 001 ns=not significant
8986 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

of AG30 55 of the deficient cells were determined to beAnnexin V positive compared with 32 of the AG30-treated NER-proficient cells (Figure 5B) Western blotanalysis of cleaved PARP also indicated an increase inapoptotic cell death in the absence of XPA (Figure 5C)These findings suggest that XPA is not required for acti-vation of apoptosis despite its importance in the repair oftriplex structures Furthermore we can conclude that aloss of XPA and possibly functional NER leads to anincrease in apoptosis in response to triplex-induced DSBs
XPD requirement for triplex-induced apoptosis
After determining that activation of apoptosis was acellular response to extensive triplex-induced DNAstrand breaks both in vitro and in vivo we were interestedin determining which proteins were involved in maintain-ing the switch from DNA repair to apoptosis Wehypothesized that a dual role NER protein like XPDwhich contributes to genomic stability by participatingin both repair and apoptosis may also aid in triggeringthe cell to activate a pro-apoptotic pathway in the
Figure 4 Triplex formation induced DSBs in transgenic mouse model (A) Schematic of transgenic mouse model AV which contains 50 copies ofthe triplex target site chromosomally integrated into its genome (B) Immunohistochemistry of spleen samples harvested from AV mice 6 hposttreatment with MIX30 or AG30 (50mgkg) (C D) Quantitation of immunohistochemical findings Plt 0001 Plt 001
Nucleic Acids Research 2013 Vol 41 No 19 8987
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

presence of excessive triplex-induced DNA DSBs AV16cells proficient or deficient for XPD were treated withAG30 and analyzed for the activation of apoptosis Asexpected monolayer growth assays demonstrated adecrease in cell survival in the XPD-proficient cells(Figure 6A) However a decrease in cell growth was notobserved following TFO treatment of the siRNA XPD-depleted cells where the level of cell growth was similar tothat of control cells To further attribute a role for XPD inactivating apoptosis in response to excessive DNA strandbreaks induced by triplex structures we also evaluated thelevel of Annexin V-positive cells AG30 treatment ofsiRNA XPD-depleted cells resulted in a significantdecrease in apoptosis (Plt 0001) (Figure 6B) Westernblot analysis of cleaved PARP also supports a reductionin triplex-induced apoptosis that is contingent on XPD(Figure 6C) To establish that the change in apoptosisdid not result from siRNA off-target effects we alsoevaluated cleaved PARP levels following AG30 treatmentin AV16 cells that had been transfected with controlsiRNAs This analysis revealed no reduction in thetriplex-induced apoptosis levels in cells that had beentreated with either nontarget or GAPDH siRNAcontrols compared with XPD-proficient cells(Supplementary Figure S3)XPD helicase mediates strand separation at the site of
the DNA lesion (34) In addition tumor suppressor p53 acentral component of apoptosis can bind to and inhibit itshelicase activity After AG30 treatment we determined byWestern blot that an increase in p53 protein levels corres-ponded to increased caspase 3-mediated cleavage ofPARP in XPD-proficient cells (Figure 6D) On the
contrary the relative amount of p53 protein remainedstable following TFO treatment of XPD-depleted cellscompared with untreated cells Upon DNA damagephosphorylation of p53 at serine 15 coordinatespolyphosphorylation maintains nuclear retention and sta-bilizes the protein through disruption of MDM2 binding(35ndash37) As determined by Western blot analysis triplex-induced DNA strand breaks resulted in increased phos-phorylation of serine 15 in XPD-proficient cells Howevera reduction in p53 phosphorylation at serine 15 wasobserved in the XPD-depleted cells (Figure 6E)
To investigate whether the differential induction ofapoptosis in XPD-proficient and -deficient cells was thehallmark of a differential induction of DNA damage weperformed neutral comet assays As shown in Figure 6Ftriplex formation induced the same degree of DSBs inXPD-proficient cells as it did in the XPD-depleted cellsverifying that triplex-induced DNA strand breaks arenot dependent on functional XPD The results fromthese studies support a mechanism where XPD is import-ant for activation of apoptosis and not required for theformation of triplex-induced DSBs
Recruitment of XPD to the DSB site
XPD has been implicated in our studies to occupy an im-portant role in activating apoptosis in response to triplex-induced DSBs To determine whether XPD proteininteracts with gH2AX foci we treated AV16 cells withMIX30 or AG30 Confocal microscopy analysis indicatesco-localization of XPD with gH2AX foci following treat-ment with the TFO AG30 (Figure 7A) Calculation of aco-localization coefficient using ImageJ demonstrates that
Figure 5 Role of XPA in the activation of triplex-induced apoptosis (A) Analysis of gH2AX expression levels as a measure of triplex-induced DSBsin XPA-proficient and XPA-deficient cells 24 h post AG30 treatment (B) Detection of Annexin V binding indicates an increase in apoptotic celldeath in the absence of XPA 24 h posttreatment with 2 mg of oligonucleotides (C) Western blot analysis of caspase-mediated cleavage of PARP as ameasure of triplex-induced apoptosis
8988 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
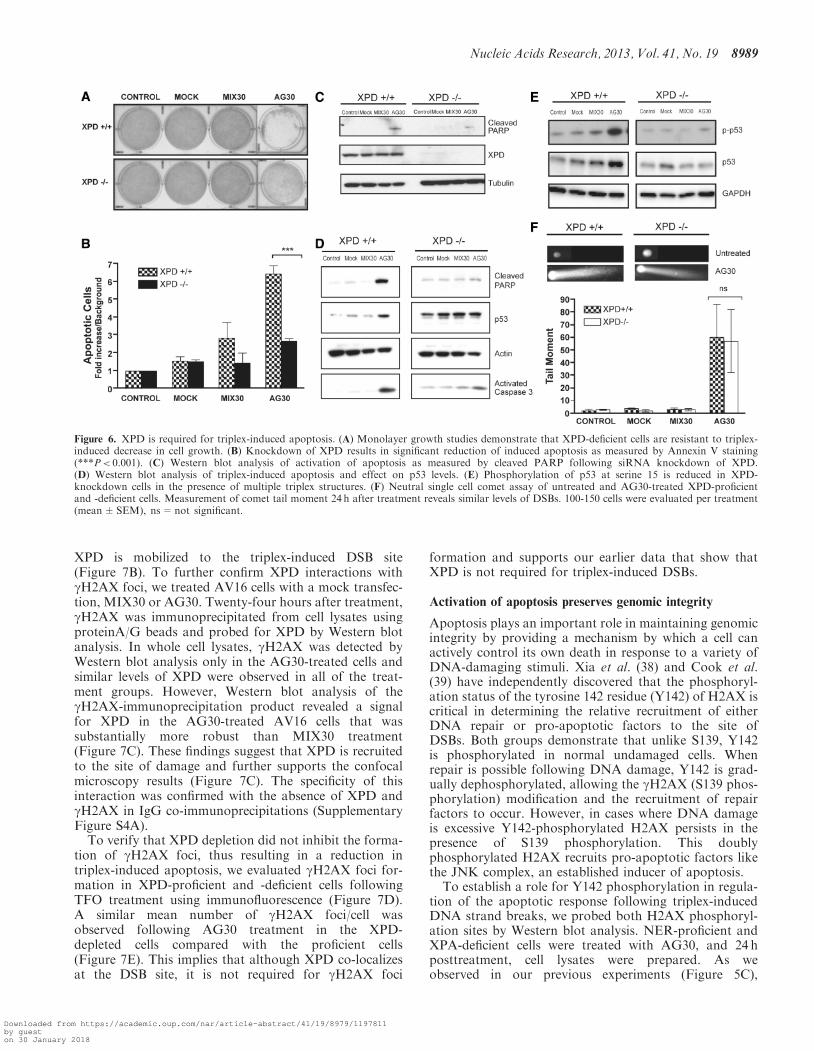
XPD is mobilized to the triplex-induced DSB site(Figure 7B) To further confirm XPD interactions withgH2AX foci we treated AV16 cells with a mock transfec-tion MIX30 or AG30 Twenty-four hours after treatmentgH2AX was immunoprecipitated from cell lysates usingproteinAG beads and probed for XPD by Western blotanalysis In whole cell lysates gH2AX was detected byWestern blot analysis only in the AG30-treated cells andsimilar levels of XPD were observed in all of the treat-ment groups However Western blot analysis of thegH2AX-immunoprecipitation product revealed a signalfor XPD in the AG30-treated AV16 cells that wassubstantially more robust than MIX30 treatment(Figure 7C) These findings suggest that XPD is recruitedto the site of damage and further supports the confocalmicroscopy results (Figure 7C) The specificity of thisinteraction was confirmed with the absence of XPD andgH2AX in IgG co-immunoprecipitations (SupplementaryFigure S4A)
To verify that XPD depletion did not inhibit the forma-tion of gH2AX foci thus resulting in a reduction intriplex-induced apoptosis we evaluated gH2AX foci for-mation in XPD-proficient and -deficient cells followingTFO treatment using immunofluorescence (Figure 7D)A similar mean number of gH2AX focicell wasobserved following AG30 treatment in the XPD-depleted cells compared with the proficient cells(Figure 7E) This implies that although XPD co-localizesat the DSB site it is not required for gH2AX foci
formation and supports our earlier data that show thatXPD is not required for triplex-induced DSBs
Activation of apoptosis preserves genomic integrity
Apoptosis plays an important role in maintaining genomicintegrity by providing a mechanism by which a cell canactively control its own death in response to a variety ofDNA-damaging stimuli Xia et al (38) and Cook et al(39) have independently discovered that the phosphoryl-ation status of the tyrosine 142 residue (Y142) of H2AX iscritical in determining the relative recruitment of eitherDNA repair or pro-apoptotic factors to the site ofDSBs Both groups demonstrate that unlike S139 Y142is phosphorylated in normal undamaged cells Whenrepair is possible following DNA damage Y142 is grad-ually dephosphorylated allowing the gH2AX (S139 phos-phorylation) modification and the recruitment of repairfactors to occur However in cases where DNA damageis excessive Y142-phosphorylated H2AX persists in thepresence of S139 phosphorylation This doublyphosphorylated H2AX recruits pro-apoptotic factors likethe JNK complex an established inducer of apoptosisTo establish a role for Y142 phosphorylation in regula-
tion of the apoptotic response following triplex-inducedDNA strand breaks we probed both H2AX phosphoryl-ation sites by Western blot analysis NER-proficient andXPA-deficient cells were treated with AG30 and 24 hposttreatment cell lysates were prepared As weobserved in our previous experiments (Figure 5C)
Figure 6 XPD is required for triplex-induced apoptosis (A) Monolayer growth studies demonstrate that XPD-deficient cells are resistant to triplex-induced decrease in cell growth (B) Knockdown of XPD results in significant reduction of induced apoptosis as measured by Annexin V staining(Plt 0001) (C) Western blot analysis of activation of apoptosis as measured by cleaved PARP following siRNA knockdown of XPD(D) Western blot analysis of triplex-induced apoptosis and effect on p53 levels (E) Phosphorylation of p53 at serine 15 is reduced in XPD-knockdown cells in the presence of multiple triplex structures (F) Neutral single cell comet assay of untreated and AG30-treated XPD-proficientand -deficient cells Measurement of comet tail moment 24 h after treatment reveals similar levels of DSBs 100-150 cells were evaluated per treatment(meanplusmnSEM) ns=not significant
Nucleic Acids Research 2013 Vol 41 No 19 8989
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

triplex-induced DNA strand breaks resulted in the activa-tion of apoptosis in both cell lines as determined by thepresence of cleaved PARP (Figure 8A) Although apop-tosis was observed in both XPA-proficient and XPA-deficient cells following TFO treatment slightly higherlevels of S139 phosphorylation was observed in theXPA-deficient cells suggesting the presence of moreDSBs In the case of the XPA-deficient cells an increasein the level of Y142 phosphorylation is also observed
compared with the XPA-proficient cells (Figure 8A)Tyrosine 142 phosphorylation is a prerequisite for recruit-ment of the proteins necessary for apoptosis These resultscorrespond with our observation that there is a 2-foldincrease in apoptotic cells in XPA-deficient cells comparedwith XPA-proficient following AG30 treatment
XPD-proficient and siRNA XPD-depleted cells werealso treated with AG30 and 24 h posttreatment celllysates were prepared As we observed in our previous
Figure 7 XPD is recruited to the gH2AX site (A) Confocal microscopy indicates co-localization of XPD with gH2AX foci (B) Co-localizationcoefficient calculated using NIH ImageJ software (Plt 005) (C) Co-immunoprecipitation of gH2AX with XPD by western blot analysis(D) Immunofluorescence studies of gH2AX foci formation in XPD++ and XPD cells 24 h post AG30 treatment (E) Western blot analysisof XPD protein levels in proficient and deficient cells Quantification of gH2AX foci formation per cell Sixty to seventy cells were evaluated pertreatment
8990 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
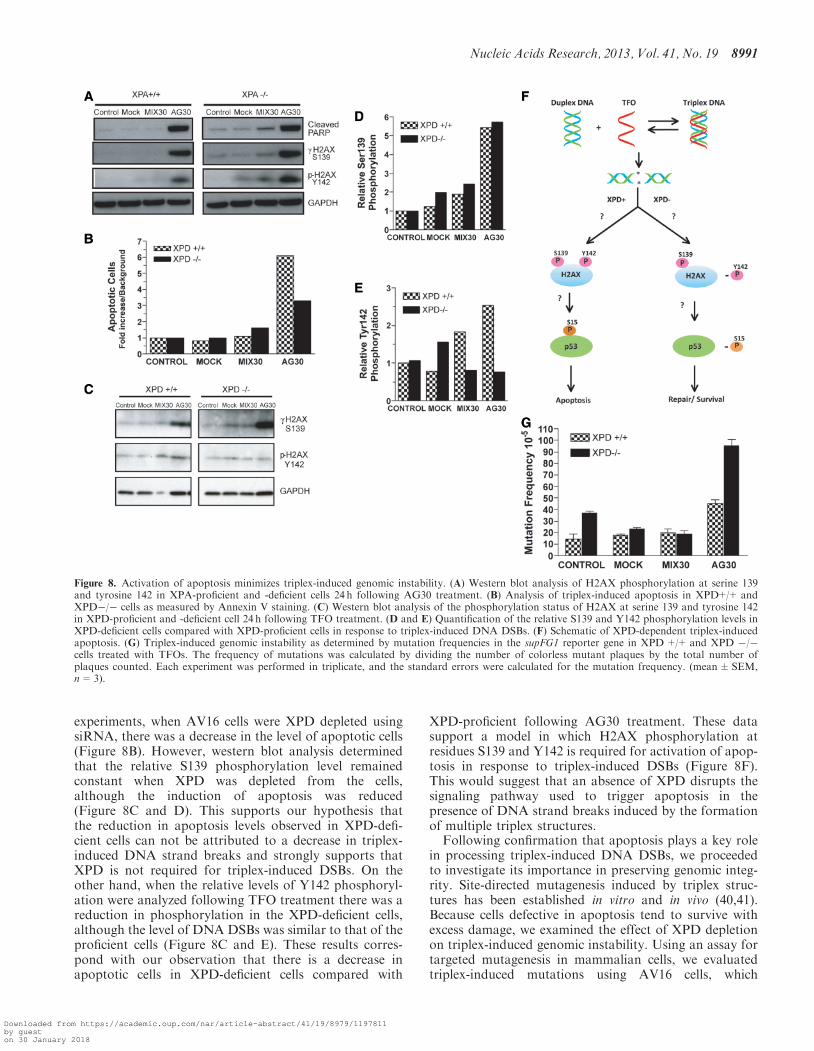
experiments when AV16 cells were XPD depleted usingsiRNA there was a decrease in the level of apoptotic cells(Figure 8B) However western blot analysis determinedthat the relative S139 phosphorylation level remainedconstant when XPD was depleted from the cellsalthough the induction of apoptosis was reduced(Figure 8C and D) This supports our hypothesis thatthe reduction in apoptosis levels observed in XPD-defi-cient cells can not be attributed to a decrease in triplex-induced DNA strand breaks and strongly supports thatXPD is not required for triplex-induced DSBs On theother hand when the relative levels of Y142 phosphoryl-ation were analyzed following TFO treatment there was areduction in phosphorylation in the XPD-deficient cellsalthough the level of DNA DSBs was similar to that of theproficient cells (Figure 8C and E) These results corres-pond with our observation that there is a decrease inapoptotic cells in XPD-deficient cells compared with
XPD-proficient following AG30 treatment These datasupport a model in which H2AX phosphorylation atresidues S139 and Y142 is required for activation of apop-tosis in response to triplex-induced DSBs (Figure 8F)This would suggest that an absence of XPD disrupts thesignaling pathway used to trigger apoptosis in thepresence of DNA strand breaks induced by the formationof multiple triplex structuresFollowing confirmation that apoptosis plays a key role
in processing triplex-induced DNA DSBs we proceededto investigate its importance in preserving genomic integ-rity Site-directed mutagenesis induced by triplex struc-tures has been established in vitro and in vivo (4041)Because cells defective in apoptosis tend to survive withexcess damage we examined the effect of XPD depletionon triplex-induced genomic instability Using an assay fortargeted mutagenesis in mammalian cells we evaluatedtriplex-induced mutations using AV16 cells which
Figure 8 Activation of apoptosis minimizes triplex-induced genomic instability (A) Western blot analysis of H2AX phosphorylation at serine 139and tyrosine 142 in XPA-proficient and -deficient cells 24 h following AG30 treatment (B) Analysis of triplex-induced apoptosis in XPD++ andXPD cells as measured by Annexin V staining (C) Western blot analysis of the phosphorylation status of H2AX at serine 139 and tyrosine 142in XPD-proficient and -deficient cell 24 h following TFO treatment (D and E) Quantification of the relative S139 and Y142 phosphorylation levels inXPD-deficient cells compared with XPD-proficient cells in response to triplex-induced DNA DSBs (F) Schematic of XPD-dependent triplex-inducedapoptosis (G) Triplex-induced genomic instability as determined by mutation frequencies in the supFG1 reporter gene in XPD++ and XPD cells treated with TFOs The frequency of mutations was calculated by dividing the number of colorless mutant plaques by the total number ofplaques counted Each experiment was performed in triplicate and the standard errors were calculated for the mutation frequency (meanplusmnSEMn=3)
Nucleic Acids Research 2013 Vol 41 No 19 8991
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

contain 100 copies of the supFG1 shuttle vector DNAin a chromosomal locus Through the use of packagingextracts the vector DNA can be isolated from genomicDNA into phage particles and subsequently analyzed forinduced mutations SupFG1 not only encodes an ambersuppressor tRNA whose function can be scored in indica-tor bacteria but also contains the AG30 triplex-bindingsite (16)AV16 and AV16 XPD-19-1 cells which stably expressed
XPD shRNA (Supplementary Figure S4B) were treatedwith a mock transfection MIX30 or AG30 and analyzedfor the induction of mutations 48 h posttreatment Weobserved a mutation frequency (45 105) in XPD-profi-cient cells following AG30 treatment that was 2-foldhigher than the frequency obtained from MIX30-treatedXPD++ cells (20 105) (Figure 8H) However AG30treatment (95 105) of XPD-deficient cells resulted in a5-fold increase in mutation frequency compared withXPD-deficient cells that received MIX30 treatment(19 105) (Figure 8H) The increase in mutation fre-quency observed in the XPD-deficient cells may beattributed in part to the cellsrsquo inability to activate apop-tosis Taken together these results position apoptosis asan important pathway in preserving genomic integrity inresponse to triplex-induced helical distortions
DISCUSSION
Cells are faced with the fundamental decision of activatingthe appropriate ratio of DNA repair and apoptosis inresponse to damage Our data suggest that the TFIIHprotein XPD is involved in maintaining the balancebetween these two outcomes in response to the formationof altered helical structures Thus we provide evidencethat the NER pathway is not only necessary for therepair of triplex structures but is also important in theactivation of pro-apoptotic pathways in response tohelical distorting DNA structures A key question existsas to how the cell determines when damage is excessiveand how this determination triggers the shift from repairto apoptosis The present study indicates that the absenceof XPD results in a decrease in phosphorylation of thetyrosine 142 residue of H2AX in addition to p53 Recentwork has determined that a balance between the kinaseactivity of WSTF and the phosphatase activity of Eyaproteins help to regulate cellular fate following DNAdamage (3839) When repair is possible Y142 must bede-phosphorylated by Eya to allow for S139 phosphoryl-ation and recruitment of repair proteins Otherwise Y142phosphorylation persists causing the cell to activate apop-tosis thus eliminating the cells with irreversible damageAlthough studies indicate that Y142 is gradually de-
phosphorylated after DNA damage it is possible thatY142 is re-phosphorylated after futile attempts to repairthe excessive DNA damage to facilitate apoptosis It istheoretically possible that if XPD is not present totrigger the switch to activate apoptosis this re-phosphor-ylation does not take place and the remaining DNAdamage response proteins necessary for apoptosis arenot recruited Chymkowitch et al (42) have recently
shown that the TFIIH complex is able to phosphorylatethe androgen receptor at position ARS515 via cdk7Additionally mutations in the C-terminal domain ofXPD were found to disturb the architecture of TFIIHleading to the dysregulation of cdk7-related phosphoryl-ation (4344) Taken together these findings along withour result that XPD co-localizes with gH2AX providesupport for an XPD-dependent apoptotic pathway
The XPD protein has been identified as having a role inNER transcription and possibly cell cycle HoweverXPD also exists in non-TFIIH complexes such asCAKndashXPD and MMXD and has function in othercellular processes including apoptosis Knockdown ofXPD did not reduce the intensity of triplex-inducedDSBs or gH2AX foci formation although a significantdecrease in apoptosis was observed It is apparent fromthe work presented that key proteins which contribute tocellular survival through their involvement in DNArepair also participate in the mechanism that shifts thecell from DNA repair to apoptosis
Intramolecular triplex DNA structures exist transientlyin genomic DNA and represent an endogenous source ofgenomic instability Naturally occurring sequencescapable of forming H-DNA are typically located in pro-moters and exons and are believed to be involved in theregulation of expression of several disease-linked genes(45ndash48) The human c-myc gene which is oftentranslocated and overexpressed in tumors contains anH-DNA forming sequence in its promoter (45) Manybreakpoints on the translocated c-myc gene are clusteredaround the H-DNA forming sequence in the promoterregion in Burkittrsquos lymphoma (14) Studies suggest thatnoncanonical structures result in fragile sites ormutation hotspots and can lead to DSBs and subsequenttranslocation of the gene The maintenance of a mechan-ism by which the cell can actively determine cellular fate inresponse to the formation of these structures may be ofcentral importance for avoiding progression to cancerbecause the default mechanism of apoptosis prevents ex-pansion of cells in which unrepaired damage would lead tomutation and to carcinogenesis Additionally we can alsohypothesize that XPD may be an integral component indetermining the fate of cells assaulted by other NER-recognized DNA damage including those induced byUV This study highlights the complexity of the balancebetween DNA repair and apoptosis in response to damageinduced by altered helical structures
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online
ACKNOWLEDGEMENTS
We are grateful to P Glazer J Sweasy D KidaneM Menezes and J Lloyd for helpful suggestions Wealso thank P Glazer for XPA ++ and XPA cellsand for the XPA antibody
8992 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

FUNDING
National Institutes of Health [K22 CA120049 K22CA120049-03S1 to FAR] American Cancer Society[IRG 58-012-51 to FAR] Kingsley Fellowship inMedical Research (to FAR) Funding for open accesscharge Institutional funds
Conflict of interest statement None declared
REFERENCES
1 BernsteinC BernsteinH PayneCM and GarewalH (2002)DNA repairpro-apoptotic dual-role proteins in five major DNArepair pathways fail-safe protection against carcinogenesis MutatRes 511 145ndash178
2 de LaatWL JaspersNG and HoeijmakersJH (1999)Molecular mechanism of nucleotide excision repair Genes Dev13 768ndash785
3 WangXW VermeulenW CoursenJD GibsonMLupoldSE ForresterK XuG ElmoreL YehHHoeijmakersJH et al (1996) The XPB and XPD DNA helicasesare components of the p53-mediated apoptosis pathway GenesDev 10 1219ndash1232
4 RoblesAI WangXW and HarrisCC (1999) Drug-inducedapoptosis is delayed and reduced in XPD lymphoblastoid celllines possible role of TFIIH in p53-mediated apoptotic celldeath Oncogene 18 4681ndash4688
5 WangXW YehH SchaefferL RoyR MoncollinVEglyJM WangZ FreidbergEC EvansMK TaffeBGet al (1995) p53 modulation of TFIIH-associated nucleotideexcision repair activity Nat Genet 10 188ndash195
6 WangG SeidmanMM and GlazerPM (1996) Mutagenesis inmammalian cells induced by triple helix formation andtranscription-coupled repair Science 271 802ndash805
7 RogersFA VasquezKM EgholmM and GlazerPM (2002)Site-directed recombination via bifunctional PNA-DNAconjugates Proc Natl Acad Sci USA 99 16695ndash16700
8 WatsonJD and CrickFH (1974) Molecular structure of nucleicacids a structure for deoxyribose nucleic acid JD Watson andFHC Crick Published in Nature number 4356 April 25 1953Nature 248 765
9 HtunH and DahlbergJE (1988) Single strands triple strandsand kinks in H-DNA Science 241 1791ndash1796
10 VoloshinON MirkinSM LyamichevVI BelotserkovskiiBPand Frank-KamenetskiiMD (1988) Chemical probing ofhomopurine-homopyrimidine mirror repeats in supercoiled DNANature 333 475ndash476
11 MirkinSM LyamichevVI DrushlyakKN DobryninVNFilippovSA and Frank-KamenetskiiMD (1987) DNA H formrequires a homopurine-homopyrimidine mirror repeat Nature330 495ndash497
12 SchrothGP and HoPS (1995) Occurrence of potentialcruciform and H-DNA forming sequences in genomic DNANucleic Acids Res 23 1977ndash1983
13 WangG and VasquezKM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells Proc NatlAcad Sci USA 101 13448ndash13453
14 SaglioG Grazia BorrelloM GuerrasioA SozziG SerraA diCellePF FoaR FerrariniM RoncellaS Borgna PignattiCet al (1993) Preferential clustering of chromosomal breakpoints inBurkittrsquos lymphomas and L3 type acute lymphoblastic leukemiaswith a t(814) translocation Genes Chromosomes Cancer 8 1ndash7
15 KnauertMP LloydJA RogersFA DattaHJ BennettMLWeeksDL and GlazerPM (2005) Distance and affinitydependence of triplex-induced recombination Biochemistry 443856ndash3864
16 RogersFA ManoharanM RabinovitchP WardDC andGlazerPM (2004) Peptide conjugates for chromosomal genetargeting by triplex-forming oligonucleotides Nucleic Acids Res32 6595ndash6604
17 StachelekGC DalalS DoniganKA Campisi HeganDSweasyJB and GlazerPM (2010) Potentiation of temozolomidecytotoxicity by inhibition of DNA polymerase beta is accentuatedby BRCA2 mutation Cancer Res 70 409ndash417
18 RogersFA LinSS HeganDC KrauseDS and GlazerPM(2012) Targeted gene modification of hematopoietic progenitorcells in mice following systemic administration of a PNA-peptideconjugate Mol Ther 20 109ndash118
19 GuntherEJ YeaskyTM GasparroFP and GlazerPM(1995) Mutagenesis by 8-methoxypsoralen and 5-methylangelicinphotoadducts in mouse fibroblasts mutations at cross-linkablesites induced by offoadducts as well as cross-links Cancer Res55 1283ndash1288
20 WangG ChenZ ZhangS WilsonGL and JingK (2001)Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclearextracts Nucleic Acids Res 29 1801ndash1807
21 MacrisMA and GlazerPM (2003) Transcription dependence ofchromosomal gene targeting by triplex-forming oligonucleotidesJ Biol Chem 278 3357ndash3362
22 JohnsonMD III and FrescoJR (1999) Third-strand in situhybridization (TISH) to non-denatured metaphase spreads andinterphase nuclei Chromosoma 108 181ndash189
23 SchwartzTR VastaCA BauerTL Parekh-OlmedoH andKmiecEB (2008) G-rich oligonucleotides alter cell cycleprogression and induce apoptosis specifically in OE19 esophagealtumor cells Oligonucleotides 18 51ndash63
24 QiH LinCP FuX WoodLM LiuAA TsaiYCChenY BarbieriCM PilchDS and LiuLF (2006) G-quadruplexes induce apoptosis in tumor cells Cancer Res 6611808ndash11816
25 DoNQ LimKW TeoMH HeddiB and PhanAT (2011)Stacking of G-quadruplexes NMR structure of a G-richoligonucleotide with potential anti-HIV and anticancer activityNucleic Acids Res 39 9448ndash9457
26 KutyavinIV LokhovSG AfoninaIA DempcyR GallAAGornVV LukhtanovE MetcalfM MillsA ReedMW et al(2002) Reduced aggregation and improved specificity of G-richoligodeoxyribonucleotides containing pyrazolo[34-d]pyrimidineguanine bases Nucleic Acids Res 30 4952ndash4959
27 ShahGM ShahRG and PoirierGG (1996) Different cleavagepattern for poly(ADP-ribose) polymerase during necrosis andapoptosis in HL-60 cells Biochem Biophys Res Commun 229838ndash844
28 RogakouEP PilchDR OrrAH IvanovaVS andBonnerWM (1998) DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139 J Biol Chem 2735858ndash5868
29 Fernandez-CapetilloO LeeA NussenzweigM andNussenzweigA (2004) H2AX the histone guardian of thegenome DNA Repair 3 959ndash967
30 HuangX OkafujiM TraganosF LutherE HoldenE andDarzynkiewiczZ (2004) Assessment of histone H2AXphosphorylation induced by DNA topoisomerase I and IIinhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin Cytometry A 58 99ndash110
31 CleaverJE (2011) gammaH2Ax biomarker of damage orfunctional participant in DNA repair lsquolsquoall that glitters is notgoldrsquorsquo Photochem Photobiol 87 1230ndash1239
32 HuangX HalickaHD TraganosF TanakaT KuroseA andDarzynkiewiczZ (2005) Cytometric assessment of DNA damagein relation to cell cycle phase and apoptosis Cell Prolif 38223ndash243
33 BattyDP and WoodRD (2000) Damage recognition innucleotide excision repair of DNA Gene 241 193ndash204
34 de BoerJ DonkerI de WitJ HoeijmakersJH and WeedaG(1998) Disruption of the mouse xeroderma pigmentosum group DDNA repairbasal transcription gene results in preimplantationlethality Cancer Res 58 89ndash94
35 KapoorM HammR YanW TayaY and LozanoG (2000)Cooperative phosphorylation at multiple sites is requiredto activate p53 in response to UV radiation Oncogene 19358ndash364
Nucleic Acids Research 2013 Vol 41 No 19 8993
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018
36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018

36 ShiehSY IkedaM TayaY and PrivesC (1997) DNAdamage-induced phosphorylation of p53 alleviates inhibition byMDM2 Cell 91 325ndash334
37 SteegengaWT van der EbAJ and JochemsenAG (1996) Howphosphorylation regulates the activity of p53 J Mol Biol 263103ndash113
38 XiaoA LiH ShechterD AhnSH FabrizioLA Erdjument-BromageH Ishibe-MurakamiS WangB TempstPHofmannK et al (2009) WSTF regulates the H2AX DNAdamage response via a novel tyrosine kinase activity Nature 45757ndash62
39 CookPJ JuBG TeleseF WangX GlassCK andRosenfeldMG (2009) Tyrosine dephosphorylation of H2AXmodulates apoptosis and survival decisions Nature 458 591ndash596
40 VasquezKM NarayananL and GlazerPM (2000) Specificmutations induced by triplex-forming oligonucleotides in miceScience 290 530ndash533
41 VasquezKM WangG HavrePA and GlazerPM (1999)Chromosomal mutations induced by triplex-formingoligonucleotides in mammalian cells Nucleic Acids Res 271176ndash1181
42 ChymkowitchP Le MayN CharneauP CompeE andEglyJM The phosphorylation of the androgen receptor by
TFIIH directs the ubiquitinproteasome process EMBO J 30468ndash479
43 KerielA StaryA SarasinA Rochette-EglyC and EglyJM(2002) XPD mutations prevent TFIIH-dependent transactivationby nuclear receptors and phosphorylation of RARalpha Cell109 125ndash135
44 CompeE DraneP LaurentC DiderichK BraunCHoeijmakersJH and EglyJM (2005) Dysregulation of theperoxisome proliferator-activated receptor target genes by XPDmutations Mol Cell Biol 25 6065ndash6076
45 KinniburghAJ (1989) A cis-acting transcription element of thec-myc gene can assume an H-DNA conformation Nucleic AcidsRes 17 7771ndash7778
46 PestovDG DaynA SiyanovaE GeorgeDL and MirkinSM(1991) H-DNA and Z-DNA in the mouse c-Ki-ras promoterNucleic Acids Res 19 6527ndash6532
47 BacollaA JaworskiA ConnorsTD and WellsRD (2001)Pkd1 unusual DNA conformations are recognized by nucleotideexcision repair J Biol Chem 276 18597ndash18604
48 BelotserkovskiiBP De SilvaE TornalettiS WangGVasquezKM and HanawaltPC (2007) A triplex-formingsequence from the human c-MYC promoter interferes with DNAtranscription J Biol Chem 282 32433ndash32441
8994 Nucleic Acids Research 2013 Vol 41 No 19
Downloaded from httpsacademicoupcomnararticle-abstract411989791197811by gueston 30 January 2018