a practical and general approach to the late-stage
TRANSCRIPT

doi.org/10.26434/chemrxiv.7550117.v1
A Practical and General Approach to the Late-Stage Functionalization ofComplex SulfonamidesPatrick Fier, Kevin M. Maloney
Submitted date: 04/01/2019 • Posted date: 07/01/2019Licence: CC BY-NC-ND 4.0Citation information: Fier, Patrick; Maloney, Kevin M. (2019): A Practical and General Approach to theLate-Stage Functionalization of Complex Sulfonamides. ChemRxiv. Preprint.
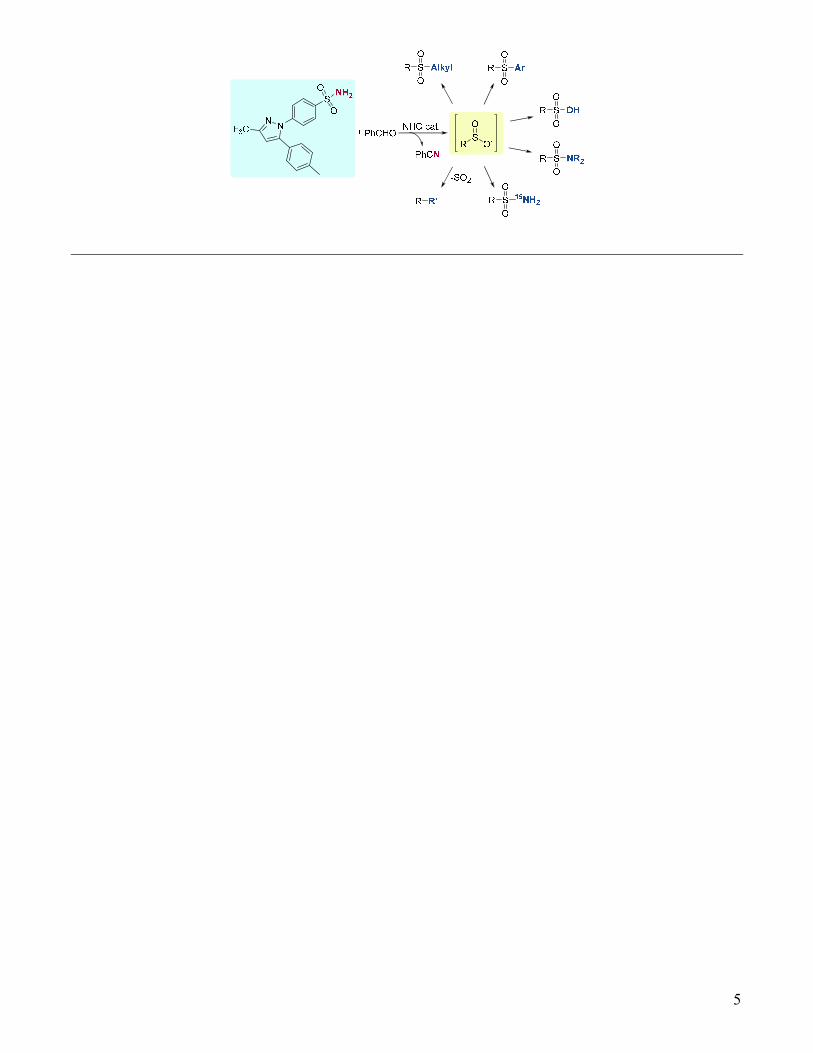
Herein we describe the development and application of a method for the mild, late-stage conversion ofprimary sulfonamides to several other other functional groups. These reactions occur via initial reductivedeamination of sulfonamides to sulfinates via an NHC-catalyzed reaction of transiently formedN-sulfonylimines. The method described here is tolerant of nearly all common functional groups, asexemplified by the late-stage derivatization of several complex pharmaceutical compounds. Based on theprevalence of sulfonamide-containing drugs and building blocks, we have developed a method to enablesulfonamides to be applied as versatile synthetic handles for synthetic chemsitry.
File list (2)
download fileview on ChemRxivFier Maloney Sulfonamide Paper - Final.pdf (224.65 KiB)
download fileview on ChemRxivFier Maloney Sulfonamide Supporting Information.pdf (1.18 MiB)

A Practical and General Approach to the Late-Stage Functionalization of Complex Sulfonamides
Patrick S. Fier* and Kevin M. Maloney*
Department of Process Research and Development, Merck & Co., Inc., Rahway, New Jersey 07065, United States
ABSTRACT: Herein we describe the development and application of a method for the mild, late-stage conversion of primary sulfonamides to several other other functional groups. These reactions occur via initial reductive deamination of sulfonamides to sulfinates via an NHC-catalyzed reaction of transiently formed N-sulfonylimines. The method described here is tolerant of nearly all common functional groups, as exemplified by the late-stage derivatization of several complex pharmaceutical compounds. Based on the prevalence of sulfonamide-containing drugs and building blocks, we have developed a method to enable sulfonamides to be applied as versatile synthetic handles for synthetic chemsitry.
Sulfonamides and related sulfur-based functional groups are of paramount importance in drug discovery and development. Since the first sulfa antibiotic, Prontosil, was introduced to the market in the 1930’s, sulfonamides and other sulfur-containing drugs have become pervasive in medicines spanning all therapeutic areas (Figure 1).1 In fact, a recent analysis revealed that sulfur is the 5th most common element in FDA-approved drugs, just behind the mainstays of organic chemistry: C, H, N, and O.1 Based on the prevalence of S-containing drugs, methods that enable the late-stage interconversion of common sulfur functional groups will have a meaningful impact in drug discovery.2
Figure 1. Representative sulfur-containing drugs
Of the many sulfur-containing drugs, nearly 30% contain a sulfonamide moiety (Figure 1).1 As such, numerous methods have been developed to prepare sulfonamides from a wide-array of starting materials.3 Yet, analogous methods for the conversion of sulfonamides into other functional groups, specifically in the context of complex molecule diversification, are essentially
unknown. Such general methods, as exemplified in eq. 1, could have an immediate impact in the discovery of new biologically active compounds through late-stage functionalization,4 given the prevalence of sulfonamide-containing molecules in compound libraries across pharmaceutical and agrochemical companies. For instance, Merck’s building block library contains similar numbers of sulfonamides and arylboronic acids, yet sulfonamides are almost exclusively considered to be terminal functional groups rather than synthetic handles. To address the lack of late-stage functionalization methods for sulfonamides, we report here a general method to readily convert complex drug-like sulfonamides to a variety of common functional groups.
R SO
OAlkyl
R SO
OAr
R SO
ONR2
R SO
OOH
R SO
O15NH2R'R
(1)R SO
ONH2
Generally inaccessible functionalgroups from sulfonamides
Pervasive sulfonamide-containingdrugs and building blocks
For the conversion of sulfonamides to other functional groups,
we proposed that the most versatile method would occur via initial formation of a sulfinate salt. This proposal was based on the ease of functionalization of sulfinates via electrophilic trapping or through loss of SO2, classes of reactions which typically occur under mild conditions.5 To achieve this goal, we proposed that primary sulfonamides could be converted to sulfinate salts via the intermediacy of N-sulfonylimines that would form via condensation with aldehydes, ultimately liberating a nitrile as the stoichiometric byproduct (Figure 2).
Figure 2. Strategy for the late-stage functionalization of sulfonamides
This hypothesis was tested with benzenesulfonamide as a model substrate under a multitude of reaction conditions, comprising a wide array of aldehydes, bases, and solvents (see

Supporting Information). Yet, across this diverse set of reaction conditions, only trace amounts of the sulfinate product was observed (Figure 3A). In considering the elimination of the sulfinate leaving group, we hypothesized that an NHC catalyst could lower the energy barrier and facilitate the reaction (Figure 3B).6 This proposal was largely inspired by pioneering work involving NHC-catalyzed reactions of α-reducible aldehydes that form acylazolium species (Figure 3C).7 In the presence of benzaldehyde, a mild base (K2CO3), and 3 mol % of triazole-based NHC precatalyst 1, benzenesulfonamide was converted to benzenesulfinate in quantitative yield, along with the formation of benzonitrile as the byproduct (see Supporting Information for additional details).
Figure 3. Discovery of a method for the functionalization of sulfonamides via NHC-catalyzed reductive N-S bond cleavage
The NHC-catalyzed deamination was investigated further, and a general set of reaction conditions were identified that could be applied to all classes of primary sulfonamides (Scheme 1). The reaction conditions involve heating a mixture of the sulfonamide substrate in the presence of a slight excess of benzaldehyde and potassium carbonate with 3 mol % of 1 in ethanol. During the course of the reaction, a portion of the benzaldehyde undergoes benzoin condensation.8 However, as the benzoin reaction is reversible, this side process is of little consequence. Of particular practical importance is that the reactions can be set up on the benchtop without the exclusion of air or moisture, are run at high concentrations (1 M in EtOH), use only simple reagents, and form innocuous byproducts.
Scheme 1. Establishing substrate scope with respect to steric and electronic properties of sulfonamide 2
aConversion of sulfonamide to sulfinate based on UPLC area percent at
210 nm or 1H NMR spectroscopy. Percent conversion compared within 2% of the assay yields of benzonitrile with a calibrated UPLC instrument.
Having validated the deamination reaction on aromatic and aliphatic sulfonamides with varying electronic and steric properties, we turned our attention to the functionalization of drug-like molecules, along with enabling the in situ functionalization of the sulfinate salts. Conditions were identified that enable the crude mixtures from the deamination reactions to be directly converted to methyl sulfones by treatment with methyl iodide (Scheme 2), or to sulfonic acids via oxidation with H2O2 (Scheme 3). The deamination step tolerates carboxylic acids, free amines, basic heterocycles, halides, non-primary sulfonamides, and several other common functional groups. While primary and secondary amino groups can reversibly condense with benzaldehyde, such imines do not react further, ultimately resulting in exclusive selectivity for the functionalization of primary sulfonamides in the presence of other amino functionality. Thus, the high selectivity for functionalizing primary sulfonamides underscores the value of this approach for predictable, high-yielding late-stage diversification.4
Scheme 2. Conversion of complex sulfonamides to methyl sulfonesa

aYields shown for reactions performed on 1.0 mmol scale, sulfinate
intermediate not isolated. For experimental details, see the Supporting Information.
High selectivity was also observed during the conversion of the sulfinate intermediates to methyl sulfones. This observation is due to the fact that methyl iodide reacts more rapidly with sulfinate salts than with other nucleophilic groups.9 Similarly, selective oxidation of the sulfinates to sulfonic acids could be carried out with excellent functional group tolerance (Scheme 3). In these cases, the pure sulfonic acid product could be isolated via crystallization by simply adding water and aqueous HCl.
While most of the sulfonamide substrates used throughout this work are widely studied drugs, many of the sulfone and sulfonic acid products prepared are novel. This is significant, as it serves to highlight the fact that sulfonamides have traditionally been synthetic dead-ends, and calls attention to the difficulty and lack of structure-activity relationship (SAR) studies at S(VI).
Scheme 3. Conversion of complex sulfonamides to sulfonic acidsa
aYields shown for reactions performed on 1.0 mmol scale, sulfinate
intermediate not isolated. For experimental details, see the Supporting Information.
To further validate the utility of this late-stage functionalization method, we demonstrated that the crude sulfinate salts generated in situ could be diverted to several different functional groups (Scheme 4). Sulfonamide deamination followed by treatment of the sulfinate with I2 in the presence of an amine offers a convenient method to interconvert sulfonamides.10a,b Beyond being a useful approach to SAR studies on small scale, this deamination/amination sequence was readily scaled to prepare decagram quantities of compound 6d. Similarly, using 15NH4OH as the amine source enables rapid access to 15N-labeled sulfonamide drugs directly from the 14N parent molecule. The ability to selectively and completely exchange 14N for 15N on a drug-like molecule without resorting to multi-step syntheses is valuable for labeled compound synthesis to support pharmacological studies in drug discovery.11
Scheme 4. Late-stage diversification of Celebrex (celecoxib)a
S
NNF3C
O
O
N
NNF3C
OS
NNF3C
NH2O
S
NNF3C
O
O
S
NNF3C
O
O
15NH2
a,b
a,f
a,c
a,d
a,e
10 g scale
2d
6d74%
7d81%
8d88%
9d75%
10d66%
S
NNF3C
O
ON
CF3
SO2

aFor experimental details, see the Supporting Information. Reaction conditions: (a) standard deamination conditions; (b) piperidine, I2; (c) 15NH4OH, I2; (d) 2-chloro-5-trifluoromethylpyridine; (e) Xantphos/Pd, PhI; (f) XPhos/Pd, PhOTf.
The sulfinates generated in situ, as soft, anionic nucleophiles, can react with various alkyl electrophiles beyond methyl iodide, as well as electron deficient aryl halides in SNAr reactions (Scheme 4).9,10c Furthermore, the crude sulfinate salts can engage in cross-couplings with aryl electrophiles to provide aryl sulfones.10d Finally, the sulfinates can act as aryl nucleophiles, analogous to arylboronic acids, in cross-coupling reactions to form biaryl compounds through loss of SO2.
12 The latter reaction is particularly intriguing, as primary sulfonamides can now be thought of as precursors to aryl nucleophiles for cross-coupling. Overall, this diverse set of reactions showcases the remarkable breadth and utility of the deamination/sulfinate functionalization strategy reported here.
In conclusion, we have developed a general, reliable, and user-friendly approach to the late-stage functionalization of sulfonamides. These reactions occur with exceptional scope in regards to the steric and electronic properties of the sulfonamide, as well as the functionality contained within the molecule. The methods outlined here have been exemplified on several complex drug and drug-like sulfonamides and have been used to prepare novel derivatives that would otherwise require lengthy, de novo syntheses. Moreover, the value of this approach will continue to grow with the development of new methods for sulfinate functionalization. Having already witnessed a rapid uptake of this chemistry amongst our colleagues across drug discovery and development, we are confident that the work described here will have an immediate and tangible impact across synthetic chemistry, as primary sulfonamides can now be applied as versatile synthetic handles.
AUTHOR INFORMATION
Corresponding Authors
*E-mail: [email protected] *E-mail: [email protected]
ACKNOWLEDGMENTS
We would like to thank L. C. Campeau, Neil Strotman, and Tamas Benkovics (all employees of Merck) for helpful feedback on our manuscript.
REFERENCES
(1) (a) Lesch, J. E. The First Miracle Drugs: How the Sulfa Drugs Transformed Medicine; Oxford University Press, 2006. (b) Li, J. J.; Corey, E. J. Drug Discovery. Practices, Processes, and Perspectives; Wiley, 2013. (c) Scott, K. A.; Njardarson, J. T. Top. Curr. Chem. (Z) 2018, 376. (d) Smith, B. R.; Eastman, C. M.; Njardarson, J. T.; J. Med. Chem. 2014, 57, 9764. (e) Ilardi, E. A.; Vitaku, E.; Njardarson, J. T.; J. Med. Chem. 2014, 57, 2832.
(2) Gauthier Jr., D. R.; Yoshikawa, N. Org. Lett. 2016, 18, 5994. (3) For selected modern methods to prepare sulfonamides, see: (a)
Chen, Y.; Murray, P. R. D.; Davies, A. T.; Willis, M. C. J. Am. Chem. Soc. 2018, 140, 8781. (b) Shavnya, A.; Coffey, S. B.; Smith, A. C.; Mascitti, V. Org. Lett. 2013, 15, 6226. (c) Deeming, A. S.; Russell, C. J.; Willis, M. C. Angew. Chem., Int. Ed. 2016, 55, 747. (d) Johnson, M. W.; Bagley, S. W.; Mankad, N. P.; Bergman, R. G.; Mascitti, V.; Toste, F. D. Angew. Chem., Int. Ed. 2014, 53, 4404. (e) Zhu, H.; Shen, Y.; Deng, Q.; Huang, C.; Tu, T. Chem. - Asian J. 2017, 12, 706. (f) DeBergh, J. R.; Niljianskul, N.; Buchwald, S. L. J. Am. Chem. Soc. 2013, 135, 10638. (g) Du, B.; Wang, Y.; Sha, W.; Qian, P.; Mei, H.; Han, J.; Pan, Y. Asian J. Org. Chem. 2017, 6, 153. (h) Zhang, F.; Zheng, D.; Lai, L.; Cheng, J.; Sun, J.; Wu, J. Org. Lett. 2018, 20, 1167. (i) Tsai, A. S.; Curto, J. M.; Rocke, B. N.; Dechert-Schmitt, A.-M. R.; Ingle, G. K.; Mascitti, V. Org. Lett. 2016, 18, 508. (j) Caddick, S.; Wilden, J. D.; Judd, D. B. J. Am.
Chem. Soc. 2004, 126, 1024. (k) Moon, S.-Y.; Nam, J.; Rathwell, K.; Kim, W.-S. Org. Lett. 2014, 16, 338. (l) Frost, C. G.; Hartley, J. P.; Griffin, D. Synlett 2002, 2002, 1928.
(4) Cernak, T.; Dykstra, K. D.; Tyagarajan, S.; Vachal, P.; Krska, S. W. Chem. Soc. Rev. 2016, 45, 546.
(5) (a) Aziz, J.; Messaoudi, S.; Alami, M.; Hamze, A. Org. Biomol. Chem. 2014, 12, 9743. (b) Modha, S. G.; Mehta, V. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2013, 42, 5042. (c) Smith, J. M.; Dixon, J. A.; deGruyter, J. N.; Baran, P. S. J. Med. Chem. DOI: 10.1021/acs.jmedchem.8b01303.
(6) (a) Nolan, S. P. N-Heterocyclic Carbenes; Wiley-VCH, 2014. (b) Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307.
(7) Selected list of inspiring studies in NHC-catalyzed reaction development: (a) Chow, K. Y.-K.; Bode, J. W. J. Am. Chem. Soc. 2004, 126, 8126. (b) Reynolds, N. T.; Read de Alaniz, J.; Rovis, T. J. Am. Chem. Soc. 2004, 126, 9518. (c) DiRocco, D. A.; Oberg, K. M.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 6143. (d) Chiang, P.-C.; Kaeobamrung, J.; Bode, J. W. J. Am. Chem. Soc. 2007, 129, 3520. (e) He, M.; Struble, J. R.; Bode, J. W. J. Am. Chem. Soc. 2006, 128, 8418. (f) He, L.; Jian, T.-Y.; Ye, S. J. Org. Chem. 2007, 72, 7466. (g) Chen, D.-D.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2008, 73, 5578. (h) Jin, Z.; Xu, J.; Yang, S.; Song, B.-A.; Chi, Y. R. Angew. Chem. Int. Ed. 2013, 52, 12354. (i) Binanzer, M.; Hsieh, S.-Y.; Bode, J. W. J. Am. Chem. Soc. 2011, 133, 19698. (j) Chan, A.; Scheidt, K. A. Org. Lett. 2005, 7, 905. (k) Zeitler, K. Org. Lett. 2006, 8, 637. (l) Reynolds, N. T.; Rovis, T. J. Am. Chem. Soc. 2005, 127, 16406. (m) Kawanaka, Y.; Phillips, E. M.; Scheidt, K. A. J. Am. Chem. Soc. 2009, 131, 18028.
(8) Menon, R. S.; Biju, A. T.; Nair, V. Beilstein J. Org. Chem. 2016, 12, 444.
(9) The nucleophilicity of sulfinate ions have been quantified, see: Baidya, M.; Kobayashi, S.; Mayr, H. J. Am. Chem. Soc. 2010, 132, 4796.
(10) (a) Pan, X.; Gao, J.; Liu, J.; Lai, J.; Jiang, H.; Yuan, G. Green Chem. 2015, 17, 1400. (b) Yang, K.; Ke, M.; Lin, Y.; Song, Q. Green Chem. 2015, 17, 1395. (c) Maloney, K. M.; Kuethe, J. T.; Linn, K. Org. Lett. 2011, 13, 102. (d) Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Paris, L. M.; Bernini, R. J. Org. Chem. 2004, 69, 5608.
(11)Dean, D. C.; Filer, C. N.; McCarthy, K. E. Synthesis and Applications of Isotopically Labelled Compounds; Wiley, 2004.
(12) (a) Ortgies, D. H.; Hassanpour, A.; Chen, F.; Woo, S.; Forgione, P. Eur. J. Org. Chem. 2016, 2016, 408. (b) Zhou, C.; Liu, Q.; Li, Y.; Zhang, R.; Fu, X.; Duan, C. J. Org. Chem. 2012, 77, 10468. (c) Ortgies, D. H.; Barthelme, A.; Aly, S.; Desharnais, B.; Rioux, S.; Forgione, P. Synthesis 2013, 45, 694. (d) Markovic, T.; Rocke, B. N.; Blakemore, D. C.; Mascitti, V.; Willis, M. C. Org. Lett. 2017, 19, 6033. (e) Markovic, T.; Rocke, B. N.; Blakemore, D. C.; Mascitti, V.; Willis, M. C. Chem. Sci. 2017, 8, 4437.
5

download fileview on ChemRxivFier Maloney Sulfonamide Paper - Final.pdf (224.65 KiB)

Supporting Information for:
A Practical and General Approach to the Late-Stage Functionalization of Complex Sulfonamides Patrick S. Fier and Kevin M. Maloney
S1

General experimental details Reagents and common substrates were purchased from commercial suppliers and used as received. Non-commercially available substrates were procured from Merck’s building block collection and used as received. NHC precatalyst 1 (CAS #862893-81-0) was purchased from Sigma Aldrich and used as received. NMR chemical shifts are reported in ppm and referenced to residual solvent peaks. Coupling constants are reported in hertz. General procedures for the conversion of sulfonamides To a screw-cap vial was added sulfonamide (2, 1.0 equiv), NHC precatalyst (1, 3 mol %), powdered potassium carbonate (2.0 equiv), EtOH (1.0 mL per mmol of 2), and benzaldehyde (1.2 equiv). The vial was sealed and heated at 78 oC with rapid stirring for 18 h. For cases where the sulfinate product is electron-rich and potentially prone to aerobic oxidation, the reactions were placed under a N2 atmosphere prior to heating. For cases where assay yields of the sulfinate salt were determined, the entire reaction mixture was diluted in a volumetric flask with 3:1 MeCN : 0.1% aqueous H3PO4 and the resulting solution was analyzed on an UPLC instrument calibrated to benzonitrile. Along with the assay yield of benzonitrile, the area percent of the starting sulfonamide and product sulfinate were used to determine the percent conversion, as displayed in Scheme 1. For cases where the product was not UV active, the reactions were also analyzed by 1H NMR spectroscopy. For cases where the sulfinate was converted to a methyl sulfone, the reaction mixture was diluted with DMF (4.0 mL per mmol of 2) and cooled in an ice bath. A freshly made solution of methyl iodide in DMF (0.5 M) was added dropwise to the crude sulfinate mixture. After complete conversion of the sulfinate, the reaction mixture was diluted with water and the product was extracted into DCM. The organic phase was dried over MgSO4 and the product was purified by silica gel chromatography. For cases where the sulfinate was converted to a sulfonic acid, the reaction mixture was concentrated to dryness and reconstituted in water (4.0 mL per mmol of 2). Tungstic acid (1 mol %) was added and the mixture was cooled in an ice bath. Aqueous hydrogen peroxide (30% w/w, 1.0-2.0 equiv) was added dropwise and the reaction was stirred at room temperature until complete conversion was observed by UPLC. The reaction mixture was acidified to pH 1 by the dropwise addition of 2 M aqueous HCl, during which time the sulfonic acid product precipitated from the reaction mixture. The solid was collected, washed with water, and dried under vacuum with a nitrogen sparge for 24 hours. For cases where the sulfinate was converted to other products, see the procedures described below to prepare products 6d – 10d.
S2

Summary of Reaction Development 96 reaction conditions were investigated in a high-throughput fashion for the base-mediated conversion of benzenesulfonamide to benzenesulfinate in the presence of an aldehyde. The 96 reactions consisted of an array of 4 bases (K2CO3, K3PO4, DBU, Hunig’s base), 6 solvents (THF, DMF, PhMe, EtOH, DMSO, IPAc), and 4 aldehydes (benzaldehyde, 4-trifluoromethylbenzaldehyde, 4-methoxybenzaldehyde, pivaldehyde) at 80 °C for 18 hours. Across all reactions, only trace quantities of benzensulfinate were observed. For NHC-catalyzed reactions, see below for a summary of the impact of different bases, solvents, and catalysts on the reaction of benzensulfonamide. While reactions in DMF were equally high yielding, EtOH was ultimately chosen as the solvent for Green Chemistry considerations and for the ability to concentrate end-of-reaction mixtures under mild vacuum. Reactions catalyzed by the phenyl variant of 1 performed similarly well to reactions with 1, however 1 has much greater commercial availability.
S3

The compound was prepared using the standard procedure on 1.0 mmol scale. The product was purified on silica gel with a gradient of 0-100% EtOAc in hexanes, and obtained as a white solid in 89% yield.
1H NMR (500 MHz, Acetonitrile-d3) δ 7.93 (d, J = 2.8 Hz, 2H), 7.87 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 8.3 Hz, 2H), 7.44 (dd, J = 8.9, 2.8 Hz, 1H), 7.06 (d, J = 8.9 Hz, 1H), 3.84 (s, 3H), 3.67 (d, J = 6.0 Hz, 2H), 3.07 (s, 3H), 3.02 (t, J = 6.9 Hz, 2H).
13C NMR (126 MHz, Acetonitrile-d3) δ 163.56, 156.33, 146.23, 139.01, 132.04, 130.67, 129.87, 127.28, 125.44, 123.45, 113.83, 56.11, 43.59, 40.35, 35.10.
ESI MS: 368.03 (M+H)
The compound was prepared using the standard procedure on 1.0 mmol scale. The product was purified on silica gel with a gradient of 0-40% EtOAc in hexanes, and obtained as a white solid in 94% yield.
1H NMR (500 MHz, Acetonitrile-d3) δ 8.40 (s, 1H), 8.30 (dd, J = 8.3, 1.5 Hz, 1H), 8.19 (d, J = 8.3 Hz, 1H), 3.39 – 3.28 (m, 4H), 3.18 (s, 3H), 1.57 (p, J = 7.6 Hz, 4H), 1.28 (hept, J = 7.3 Hz, 4H), 0.90 (t, J = 7.4 Hz, 6H).
13C NMR (126 MHz, Acetonitrile-d3) δ 144.57 – 144.40 (m), 144.27, 131.97, 131.34, 128.37 (q, J = 34.1 Hz), 127.83 (q, J = 6.7 Hz), 122.26 (q, J = 274.1 Hz), 47.92, 43.23, 30.49, 19.51, 12.93.
ESI MS: 416.17 (M+H)
S4

The compound was prepared using the standard procedure on 1.0 mmol scale. The product was purified on silica gel with a gradient of 0-80% EtOAc in hexanes, and obtained as a white solid in 81% yield.
1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 7.70 (d, J = 2.0 Hz, 1H), 7.54 (d, J = 1.9 Hz, 1H), 7.35 – 7.26 (m, 2H), 7.06 (t, J = 7.4 Hz, 1H), 6.88 – 6.82 (m, 2H), 5.27 (t, J = 5.7 Hz, 1H), 3.23 (s, 3H), 3.09 (q, J = 6.7 Hz, 2H), 1.39 (p, J = 7.2 Hz, 2H), 1.13 (h, J = 7.4 Hz, 2H), 0.79 (t, J = 7.4 Hz, 3H).
13C NMR (126 MHz, DMSO-d6) δ 166.72, 156.44, 143.13, 140.39, 135.05, 129.88, 129.25, 123.15, 116.83, 115.78, 115.75, 43.65, 42.45, 30.56, 19.79, 14.06.
ESI MS, negative ion detection: 362.31 (M-H)
The compound was prepared using the standard procedure on 1.0 mmol scale. The product was purified on silica gel with a gradient of 0-50% EtOAc in hexanes, and obtained as a white solid in 93% yield.
1H NMR (500 MHz, DMSO-d6) δ 8.06 – 7.99 (m, 2H), 7.65 – 7.59 (m, 2H), 7.22 (d, J = 11.9 Hz, 5H), 3.29 (s, 3H), 2.32 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ 145.85, 143.34 – 142.44 (two overlapping peaks), 141.08, 139.65, 129.93, 129.25, 128.73, 126.65, 125.73, 121.72 (q, J = 268.9 Hz), 107.10 – 106.52 (m), 43.75, 21.27.
ESI MS: 380.88 (M+H)
S5

The compound was prepared using the standard procedure on 1.0 mmol scale. The assay yield was determined to be 76% by diluting the entire reaction mixture into a volumetric flask with 3:1 MeCN:water and analyzing the resulting solution on a UPLC instrument calibrated to authentic 4e obtained from Alfa Aesar (CAS # 5470-49-5).
The compound was prepared using the standard procedure on 1.0 mmol scale. The product was purified on silica gel with a gradient of 0-10% MeOH in DCM, and obtained as a white solid in 85% yield.
1H NMR (500 MHz, DMSO-d6) δ 8.85 (d, J = 2.4 Hz, 1H), 8.51 (dd, J = 4.8, 1.6 Hz, 1H), 8.44 (d, J = 1.7 Hz, 1H), 8.15 (d, J = 2.4 Hz, 1H), 7.93 – 7.88 (m, 2H), 7.69 (dt, J = 7.9, 1.9 Hz, 1H), 7.57 – 7.52 (m, 2H), 7.38 – 7.33 (m, 1H), 3.25 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ 152.69, 150.47, 149.59, 148.25, 143.12, 140.78, 138.55, 137.51, 136.23, 134.67, 131.12, 130.91, 127.54, 123.60, 43.76.
ESI MS: 345.02 (M+H)
S6

The compound was prepared using the standard procedure on 1.0 mmol scale. The assay yield was determined to be 90% by diluting the entire reaction mixture into a volumetric flask with 3:1 MeCN:water and analyzing the resulting solution on a UPLC instrument calibrated to authentic 4g obtained from Combi Blocks (CAS # 25195-52-2).
name (4h) The compound was prepared using the standard procedure on 1.0 mmol scale. The product was purified on silica gel with a gradient of 0-10% MeOH in DCM, and obtained as a white solid in 77% yield.
1H NMR (500 MHz, DMSO-d6) δ 9.49 (s, 1H), 8.70 (d, J = 1.8 Hz, 1H), 7.85 (d, J = 6.0 Hz, 1H), 7.83 – 7.75 (m, 2H), 7.46 (d, J = 1.0 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 6.89 (dd, J = 8.8, 1.7 Hz, 1H), 5.77 (d, J = 6.0 Hz, 1H), 4.07 (s, 3H), 3.51 (s, 3H), 3.17 (s, 3H), 2.63 (s, 3H), 2.56 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ 162.87, 159.75, 156.22, 147.44, 142.33, 140.30, 139.21, 133.14, 132.65, 128.55, 123.50, 122.34, 120.06, 119.99, 118.45, 114.44, 97.29, 43.80, 38.53, 37.85, 19.41, 9.88.
ESI MS: 437.04 (M+H)
S7

The compound was prepared using the standard procedure on 1.0 mmol scale and isolated as a white solid after direct precipitation from the reaction mixture in 91% yield.
1H NMR (500 MHz, DMSO-d6) δ 11.37 (s, 1H), 8.61 (s, 1H), 8.39 (s, 1H), 7.50 (s, 2H), 7.24 (s, 1H), 7.08 (s, 1H), 6.92 (s, 1H), 3.76 (s, 3H), 3.11 (hept, J = 6.9 Hz, 1H), 1.21 (d, J = 6.9 Hz, 6H).
13C NMR (126 MHz, DMSO-d6) δ 159.82, 153.99, 153.07, 144.22, 141.41, 135.55, 132.58, 125.60, 119.48, 111.31, 56.48, 27.21, 23.31.
ESI MS, negative ion detection: 353.08 (M-H)
The compound was prepared using the standard procedure on 1.0 mmol scale and isolated as a white solid after direct precipitation from the reaction mixture in 88% yield.
1H NMR (500 MHz, DMSO-d6) δ 8.31 (s, 1H), 6.99 (s, 1H).
13C NMR (126 MHz, DMSO-d6) δ 171.52, 161.54, 137.94, 137.33, 131.35, 118.89, 111.25.
ESI MS, negative ion detection: 251.01 (M-H)
S8

The compound was prepared using the standard procedure on 1.0 mmol scale and isolated as a white solid after direct precipitation from the reaction mixture in 88% yield.
1H NMR (500 MHz, DMSO-d6) δ 9.55 (s, 1H), 8.24 (s, 1H), 7.54 (d, J = 7.8 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 3.8 Hz, 2H), 3.89 (s, 3H), 2.69 (d, J = 5.7 Hz, 1H), 2.56 (d, J = 5.8 Hz, 1H), 1.32 (s, 3H), 0.87 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ 170.28, 147.94, 146.89, 138.43, 129.57, 128.28, 125.83, 124.32, 123.24, 120.43, 112.79, 56.54, 35.93, 32.99, 29.27, 22.06, 20.48.
S
NN
F3C
O
O N
6d
1-((4-(5-(p-tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)sulfonyl)piperidineChemical Formula: C22H22F3N3O2S
Exact Mass: 449.1385
The NHC-catalyzed deamination step was performed under the standard conditions with 10.0 grams of celecoxib (26.2 mmol) The crude sulfinate mixture was concentrated to dryness and reconstituted in 50 mL of water. Piperidine (1.5 equiv) was added, followed by iodine (1.0 equiv). The mixture was stirred vigorously at room temperature for 18 hours (procedure adapted from Green Chem. 2015, 17, 1400). The product was extracted into dichloromethane, and purified on silica gel with a gradient of 0-10% MeOH in DCM to afford 6d as a white solid in 74% yield
1H NMR (500 MHz, DMSO-d6) δ 7.81 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.20 (q, J = 8.1, 6.4 Hz, 5H), 2.92 (s, 4H), 2.32 (s, 3H), 1.53 (s, 4H), 1.45 – 1.33 (m, 2H).
13C NMR (126 MHz, DMSO-d6) δ 145.84, 143.23 – 142.33 (two overlapping peaks), 139.63, 136.23, 129.87, 129.20, 129.05, 126.89, 125.68, 121.74 (q, J = 268.9 Hz), 106.57, 47.04, 25.08, 23.30, 21.28.
S9

S
NN
F3C
O
O
15NH2
4-(5-(p-tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzenesulfonamide-15NChemical Formula: C17H14F3N2
15NO2SExact Mass: 382.0729
7d
The NHC-catalyzed deamination step was performed under the standard conditions with 0.7 mmol of celecoxib. The crude sulfinate mixture was concentrated to dryness and reconstituted in 5 mL of water. 15NH4OH (20 equiv) was added, followed by iodine (1.0 equiv). The mixture was stirred vigorously at room temperature for 18 hours (procedure adapted from Green Chem. 2015, 17, 1395). The assay yield was determined to be 90% by diluting the entire reaction mixture into a volumetric flask with 3:1 MeCN:water and analyzing the resulting solution on a UPLC instrument calibrated to authentic celecoxib obtained from Sigma Aldrich.
ESI MS: 383.12 (M+H), unlabeled celecoxib was not observed in the mass spectrum
S10

The NHC-catalyzed deamination step was performed under the standard conditions with 0.7 mmol of celecoxib. The crude sulfinate mixture was concentrated to dryness and reconstituted in 2 mL of DMAc. Tetra-n-butylammonium chloride (0.3 equiv) and 2-chloro-5-trifluoromethylpyridine (1.0 equiv) were added and the mixture was heated at 100 °C for 18 hours (procedure adapted from Org. Lett. 2011, 13, 102). The reaction mixture was diluted with water and the product was extracted into DCM. The crude material was purified on silica gel with a gradient of 0-20% EtOAc in hexanes to afford 8d as a white solid in 88% yield.
1H NMR (400 MHz, DMSO-d6) δ 9.19 – 9.14 (m, 1H), 8.67 – 8.59 (m, 1H), 8.46 (d, J = 8.3 Hz, 1H), 8.13 – 8.06 (m, 2H), 7.67 – 7.61 (m, 2H), 7.24 – 7.14 (m, 5H), 2.31 (s, 3H).
13C NMR (101 MHz, DMSO-d6) δ 161.09, 148.22 (m), 145.92, 143.85, 143.02 (q, J = 37.7 Hz), 139.70, 137.89 (m), 137.63, 130.66, 129.93, 129.24, 126.89, 125.62, 122.95, 106.94, 21.26. Not all distinct resononances could be observed due to overlapping peaks.
S11

The NHC-catalyzed deamination step was performed under the standard conditions with 0.7 mmol of celecoxib. The crude sulfinate mixture was concentrated to dryness. In a glovebox, the sulfinate mixture was reconstituted in 4 mL of PhMe, and tetra-n-butylammonium chloride (1.2 equiv), iodobenzene (1.2 equiv), Xantphos-Pd-G3 precatalyst (5 mol %) and Cs2CO3 were added. The vial was sealed and the mixture was heated at 80 °C for 18 hours under an inert atmosphere (procedure adapted form J. Org. Chem. 2004, 69, 5608). The reaction mixture was diluted with EtOAc and washed with water. The crude material was purified on silica gel with a gradient of 0-20% EtOAc in hexanes to afford 9d as a colorless oil in 75% yield.
1H NMR (500 MHz, DMSO-d6) δ 8.05 (d, J = 8.7 Hz, 2H), 8.02 – 7.97 (m, 2H), 7.73 (t, J = 7.4 Hz, 1H), 7.65 (t, J = 7.7 Hz, 2H), 7.61 – 7.55 (m, 2H), 7.23 – 7.16 (m, 5H), 2.31 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ 145.85, 143.36 – 142.46 (two overlapping peaks), 141.27, 140.99, 139.68, 134.50, 130.34, 129.92, 129.23, 129.18, 127.96, 126.93, 125.67, 121.67 (q, J = 268.9 Hz), 106.88, 21.27.
ESI MS: 443.27 (M+H)
S12

The NHC-catalyzed deamination step was performed under the standard conditions with 0.7 mmol of celecoxib. The crude sulfinate mixture was concentrated to dryness. In a glovebox, the sulfinate mixture was reconstituted in 3 mL of PhMe, phenyl triflate (1.2 equiv), and XPhos-Pd-G2 precatalyst (5 mol %) were added and the mixture was heated at 120 °C for 18 hours under an inert atmosphere (procedure adapted form J. Org. Chem. 2012, 77, 10468). The reaction mixture was diluted with EtOAc and washed with water. The crude material was purified on silica gel with a gradient of 0-20% EtOAc in hexanes to afford 9d as a colorless oil in 66% yield.
1H NMR (500 MHz, DMSO-d6) δ 7.78 (d, J = 8.5 Hz, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.48 (q, J = 7.2 Hz, 2H), 7.43 (dd, J = 10.2, 8.0 Hz, 3H), 7.26 – 7.19 (m, 4H), 7.17 (d, J = 4.5 Hz, 1H), 2.31 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ 145.42, 142.10 (m), 140.78, 139.31, 139.07, 138.61, 129.80, 129.52, 129.14, 128.67 – 125.96 (m), 125.24 – 120.00 (m), 106.06, 21.26. Not all distinct resononances could be observed due to overlapping peaks.
ESI MS: 379.21 (M+H)
S13

SO
O Me
O
NH
OMe
Cl
4a
S14

4b
SO
O
Bu2N
SO
O CH3F3C
S15

4cO OH
SO
O CH3O
HN
S16

4d
OS
NN
F3C
CH3O
S17

4f
N
N
SO
O CH3
Cl
S18

4h
SO
O CH3HNN
N
NNN
S19

5i
O
N
N
NH2
NH2
S
MeO
O
OH
O
S20

5kO
NHS
O
O OH
O
Cl
S21

6d
S
NN
F3C
O
O
N
S22

8d
S
NN
F3C
O
ON
CF3
S23

9d
S
NN
F3C
O
O
EtOAc
EtOAc
S24

10d
NN
F3C
S25

download fileview on ChemRxivFier Maloney Sulfonamide Supporting Information.pdf (1.18 MiB)