a role for slp-76 lck slp-76 binding to p56
TRANSCRIPT

of April 6, 2018.This information is current as
TCR/CD3 Signaling Complexin CD4-Induced Desensitization of the
: A Role for SLP-76lckSLP-76 Binding to p56
Ralf Sanzenbacher, Dieter Kabelitz and Ottmar Janssen
http://www.jimmunol.org/content/163/6/31431999; 163:3143-3152; ;J Immunol
Referenceshttp://www.jimmunol.org/content/163/6/3143.full#ref-list-1
, 40 of which you can access for free at: cites 79 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 1999 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

SLP-76 Binding to p56lck: A Role for SLP-76 in CD4-InducedDesensitization of the TCR/CD3 Signaling Complex1
Ralf Sanzenbacher, Dieter Kabelitz, and Ottmar Janssen2
Nonreceptor protein tyrosine kinases and associated substrates play a pivotal role in Ag receptor stimulation of resting cells andin the initiation of activation-induced cell death (AICD) of preactivated T cells. CD4-associated p56lck has been implicated not onlyin the activation of primary T cells, but also in the inhibition of T cell responses. We have previously shown that CD41 T cell clonescan be rescued from AICD when surface CD4 is engaged before the TCR stimulus. In this study, we show that prevention of AICDis associated with a CD4-dependent inhibition of TCR-triggered tyrosine phosphorylation of the Src homology 2 domain-con-taining leukocyte protein of 76 kDa (SLP-76) and Vav. We provide evidence for a SLP-76 interaction with Src homology 3 domainsof p56lck and identify amino acids 185–194 of SLP-76 as relevant docking site. In view of the multiple functions of p56lck andSLP-76/Vav in the initiation of TCR/CD3/CD4 signaling, we propose a model for the CD4-dependent inhibition of TCR signalingand AICD of preactivated T cells. Our data suggest that preformed activation complexes of adapter proteins and enzymes in thevicinity of the CD4/p56lck complex are no longer available for the TCR signal when CD4 receptors are engaged before TCRstimulation. The Journal of Immunology,1999, 163: 3143–3152.
T he cell death pathway referred to as activation-inducedcell death (AICD3) can be initiated in preactivated matureT lymphocytes by religation of the Ag receptor with Ag,
superantigen, or mAb against the TCR/CD3 complex (1–3). AICDis based on a protein tyrosine kinase (PTK)-dependent up-regula-tion of Fas ligand (FasL, Apo-1L, CD95L) surface expression tosubsequently trigger the Fas (CD95, Apo-1) death signaling cas-cade (4–8). Thus, AICD of mature T cells seems to require pri-mary and secondary activation to result in surface expression ofboth the death receptor and the corresponding death factor.
Although AICD has been extensively studied over the pastyears, only a few reports have analyzed the potential modulation ofAICD by coligation of other T cell surface molecules. For exam-ple, ligation of CD28 has been demonstrated to inhibit subsequentapoptosis by TCR/CD3 ligation in murine and human T cells (9,10). We have reported that preligation of CD4 molecules on T cellclones with mAb or gp120 of HIV significantly inhibits AICD(11). The inhibition was associated with a diminished TCR-in-duced tyrosine phosphorylation, reduced levels of CD3-inducibleFasL mRNA and FasL surface expression, and a block of TCR-stimulated production of IFN-g and IL-2.
In the present study, we obtained evidence for a biochemicallink between CD4 engagement and negative regulation of TCR/CD3 stimulation and AICD. We identified the product of the pro-
tooncogene Vav (12) and the Vav-associated multifunctional SH2domain-containing leukocyte protein of 76 kDa (SLP-76; Refs.13–15) as components of TCR/CD3 signaling cascade that are dif-ferentially phosphorylated upon TCR ligation in CD4-stimulatedvs unstimulated T cell clones.
The three functional domains of SLP-76 mediate binding toSH2-containing proteins, including Vav (acidic N terminus whenphosphorylated), to SH3-containing molecules such as Grb-2 (pro-line-rich central part), and tyrosine-phosphorylated molecules suchas SLAP-130 (5FYB) and pp62 (C-terminal SH2 domain) (15).More recently, studies with SLP-deficient T cells and SLP-762/2
mice revealed a profound role of this adapter protein in T celldevelopment and activation (16–18). Thus, SLP-76 couples TCR-associated PTKs to PLC-g1-induced signaling cascades, and isabsolutely required for normal T cell development and function.Interestingly, the expression of SLP-76 is restricted to T lympho-cytes. In B cells, BLNK or SLP-65, an adapter protein with a verysimilar overall structure, but a relatively low homology to SLP-76,fulfills the same role in linking the B cell receptor signals toPLC-g1 activation (19, 20).
To our knowledge, a direct association of SLP-76 with the TCR/CD3/CD4-complex or the respectivesrc-related kinases p56lck orp59fyn has not been demonstrated. We show that SLP-76 does notonly interact with Grb-2 in a SH3-mediated fashion, but also di-rectly, and selectively binds to SH3 domains of p56lck. With apeptide competition strategy, we map the binding site to a proline-rich stretch located between amino acids 185 and 194 of SLP-76,which is different from the sequence that has been shown to me-diate Grb-2 binding. Most important, we are able to coprecipitatesubstantial amounts of p56lck with SLP-76 from lysates of untrans-fected Jurkat cells.
Our results suggest that upon ligation of CD4, crucial compo-nents of the TCR/CD3 signaling cascade (i.e., SLP-76 and Vav)are sequestered to the CD4-p56lck complex via SH3-mediated in-teractions. As a consequence, we observe a transient TCR/CD3desensitization of anti-CD4-pretreated cells and provide a model toexplain the reduced TCR-dependent cytokine production andAICD described before (11).
Department of Immunology, Paul-Ehrlich-Institute, Langen, Germany
Received for publication February 24, 1999. Accepted for publication July 7, 1999.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work forms part of the Ph.D. thesis of R.S.2 Address correspondence and reprint requests to Dr. Ottmar Janssen, Institute forImmunology, Christian-Albrechts-University, Brunswiker Strasse 4, D-24105 Kiel,Germany. E-mail address: [email protected] Abbreviations used in this paper: AICD, activation-induced cell death; FasL, Fasligand; PLC, phospholipase C; PTK, protein tyrosine kinase; SEA, staphylococcalenterotoxin A superantigen; SH2 and SH3 domain, Src homology 2 and 3 domain,respectively; SLP-76, SH2 domain-containing leukocyte protein of 76 kDa; TCL,total cell lysate.
Copyright © 1999 by The American Association of Immunologists 0022-1767/99/$02.00
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Materials and MethodsT cell clones and lines
The IL-2-dependent CD31CD41CD51 T cell clone D894/25 has been de-scribed (3, 11). Clone cells were restimulated periodically with irradiatedPBMC and EBV-transformed lymphoblastoid cell lines in the presence ofPHA (0.5 mg/ml; Wellcome, Burgwedel, Germany). Three days after re-stimulation, the cells were washed extensively and further expanded in thepresence of human rIL-2 (10 U/ml; EuroCetus, Frankfurt, Germany) forseveral days before use in the assays. Dead cells were removed by Ficollgradient centrifugation when necessary. For biochemical analysis, we alsoused leukemic Jurkat cells (clone E6.1; American Type Culture Collection(ATCC), Manassas, VA) and PHA-stimulated peripheral blood T lympho-cyte populations. To this end, PBMC were cultured with PHA (0.5mg/ml)for 3 days and further expanded in rIL-2-containing medium. All cells weregrown at 37°C in a humidified atmosphere with 6% CO2. Culture mediumwas RPMI 1640 with 10% (v/v) FBS (Biochrom, Berlin, Germany), anti-biotics (penicillin at 100 U/ml and streptomycin at 100mg/ml), L-glutamine(2 mM), and HEPES buffer solution (10 mM).
Antibodies and reagents
For stimulation of the TCR/CD3 complex, we used anti-CD3 mAb OKT3(mouse IgG2a; Cilag, Sulzbach, Germany) or staphylococcal enterotoxinsuperantigens from Toxin Technologies (Sarasota, FL). Anti-CD3 mAbwere cross-linked with rabbit anti-mouse IgG secondary Abs (Jackson Im-munoResearch, West Grove, PA) when indicated. For ligation or immu-noprecipitation of CD4, mAb OKT4 (mouse IgG2b, hybridoma fromATCC) and 5F8 (mouse IgG1, generated in our laboratory) were used.Anti-CD5 mAb UCHT2 (provided by the Sixth International LeukocyteTyping Workshop) was utilized as a control in some experiments. Theanti-SLP-76 sheep polyclonal antiserum and anti-pTyr mAb 4G10 werepurchased from Upstate Biotechnology (Lake Placid, NY). Anti-c-Cbl (C-15, rabbit polyclonal antiserum), anti-Vav (C-12, rabbit polyclonal anti-serum), and anti-Sam68 (7-1, mAb) were obtained from Santa Cruz Bio-technology (Santa Cruz, CA). The anti-p56lck antiserum was raised againsta lck-specific peptide (residues 39–64) (22), and mAb 4/215 (IgG1)against the SH3lckfusion protein (this laboratory). Anti-GST mAb B11F8(this laboratory) was used for far Western blotting with GST fusion pro-teins. For Western blotting with ECL detection (Amersham, Braunschweig,Germany), HRP-conjugated secondary reagents (anti-IgG) from Rockland(Gilbertsville, PA; rabbit anti-sheep) and Amersham (donkey anti-rabbit,rabbit anti-mouse) were used. The expression and purification of GST fu-sion proteins containing the SH2 and/or SH3 domains of p56lck andp59fyn(T) have been described elsewhere (21, 22).
T cell stimulation and preparation of cell lysates
To determine effects of CD4 ligation on TCR signals, 1.53 106 (total celllysates) or 20–503 106 (precipitations) clone cells per sample were in-cubated in the absence or presence of anti-CD4 (or anti-CD5) mAb at 10mg/ml for 1 h at37°C. In other experiments, respective numbers of Jurkatcells were used without CD4 engagement. Before TCR stimulation, thecells were washed twice in RPMI with 2% FBS. A total of 50ml of pre-warmed Ab solution with mAb OKT3 at 10mg/ml and cross-linking Absat 1mg/ml were added to the pellet, and incubation was performed at 37°Cfor the indicated intervals. Clone cells were also activated with SEA at 5ng/ml. Stimulation was stopped by adding 1 ml of cold PBS, quick spincentrifugation, aspiration of the PBS/Ab supernatant, and immediate lysisin 30 ml of Brij96 (Sigma, Deisenhofen, Germany) or Nonidet P-40 (FlukaChemie AG, Buchs, Switzerland) lysis buffer (1% (v/v) of detergent in 20mM Tris-HCl (pH 7.4), 150 mM NaCl, with protease and phosphataseinhibitors aprotinin (10mg/ml), leupeptin (10mg/ml), 1 mM PMSF, 1 mMsodium orthovanadate, 1 mM sodium pyrophosphate (all from Sigma), and10 mM sodium fluoride (Fluka)). Lysates remained on ice for 15–30 minbefore centrifugation at 4°C and 14,000 rpm for 7 min. Supernatants werethen transferred into fresh tubes for further analysis.
Immunoprecipitations, precipitations with fusion proteins, andWestern blotting
For precipitation with fusion proteins or immunoprecipitation, supernatantswere incubated for 90 min rotating at 4°C with 2–5mg of the respective Abor 20–50mg of GST fusion protein. A total of 60ml of a 50% slurry ofprotein A-Sepharose CL4B, protein G-Sepharose, or glutathione Sepharose4B beads (Pharmacia, Piscataway, NJ) was added, and the samples wererotated for an additional 30 min. The beads were pelleted, washed thrice incold lysis buffer, and boiled in sample buffer containing 2-ME. Total celllysates boiled with an equal volume of sample buffer or precipitates were
loaded onto SDS polyacrylamide gels. Separated proteins were transferredto nitrocellulose membranes (Hybond C-Extra; Amersham). Protein load-ing and efficiency of transfer were monitored with Ponceau S (Sigma). Theblots were blocked with 5% BSA (Sigma) for 1 h, and proteins were an-alyzed with the indicated primary and secondary Abs or with fusion pro-teins, anti-GST mAb, and HRP-conjugated anti-mouse Ig, and ECL detec-tion reagents.
Peptide competition assay
The peptides listed in Table I were synthesized on an AMS 422 peptidesynthesizer (Abimed, Langenfeld, Germany). All peptides were dissolvedin PBS and used at concentrations specified inResults. For competitionexperiments, 20mg of the respective fusion protein on beads was incubatedwith peptide for 10–15 min at 4°C with constant rotation. A total of 100mlof filtered cell lysates corresponding to 25–503 106 cells was then added,and incubation was prolonged for 10 min. The beads were then washedextensively and subjected to SDS-PAGE and Western blotting, as de-scribed. For ex vivo peptide competition of the SLP-76/lck interactions,filtered lysates of 4003 106 Jurkat cells were incubated overnight, rotatingat 4°C with 2 mM of peptides, followed by SLP-76 immunoprecipitationand anti-lckimmunoblot.
ResultsInhibition of TCR/CD3-induced tyrosine phosphorylation bypreceding CD4 engagement
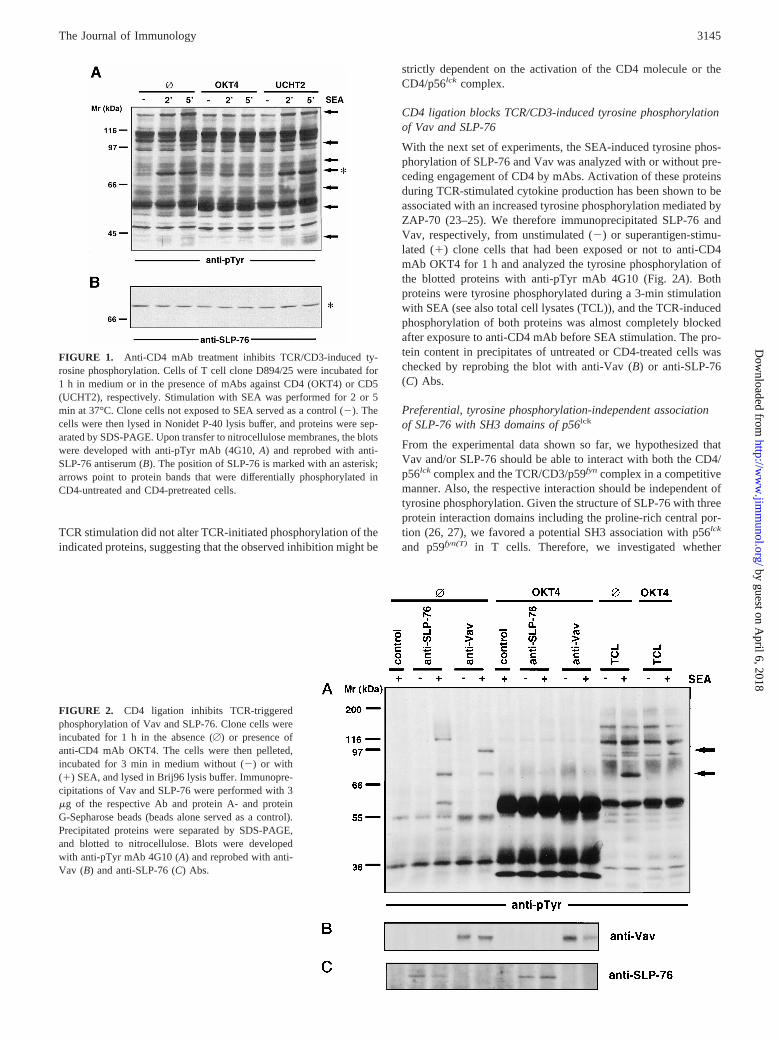
CD41CD31 T cells (clone D894/25) were incubated for 1 h in thepresence or absence of anti-CD4 (OKT4) or anti-CD5 (UCHT2)mAbs before stimulation with SEA for 2 or 5 min. As shown inFig. 1A, a selective reduction of the level of tyrosine phosphory-lation of polypeptides with approximate m.w. of 40–44, 48–50,62–64, 76, 80, 95, and 145–150 kDa (arrows) was observed inNonidet P-40 lysates of CD4-treated cells. Preligation of CD4 alsoinhibited SEA-induced cytokine production and AICD in 894/25cells (11). Time-course experiments revealed that CD4 ligationwas most effective in reducing the subsequent superantigen-in-duced phosphorylation when CD4 engagement was initiated atleast 30 min before the TCR stimulation (not shown). To identifyproteins that may be involved in the CD4-mediated down-regula-tion of the SEA-induced TCR response, we first focused on thepolypeptide of 76 kDa. Recently, the SH2 domain-containing leu-kocyte protein of 76 kDa (SLP-76) has been described as an es-sential transducer of TCR signals to IL-2 gene activation in concertwith Vav, Grb-2, and ZAP-70 (23–25). We therefore reprobed thesame blot shown in Fig. 1A with an anti-SLP antiserum and visu-alized equal amounts of SLP-76 protein loaded in each lane (Fig.1B). The overlay of the two films strongly suggested that the 76-kDa band in fact represented SLP-76. Of note, CD5 ligation before
Table I. Peptides used for competition assays to determine SLP-76/SH3interaction sitesa
PeptideCorresponding
to Amino Acids Amino Acid Sequence
SLP-76 peptidesA 488–497 G L R G K E D F L SB 84–93 R K P Q V P R F P EC 143–152 A D Y E P P P S N DD 182–191 T P Q Q P P VP P QE 185–194 Q P P V P P Q R PMF 195–204 A A L P P P P A G RG 206–215 H S P L P P P Q T NH 283–292 Q K P P L P P T T EI 348–357 K P S P M N P L P SK 400–411 L P L P N K P R P P S P
c-Cbl peptidesL 542–551 L P P P P P P D R PM 489–498 P Q A S L P P V P P
a Proline residues characteristic for potential SH3 binding sites are highlighted inbold.
3144 SLP-76/p56lck INTERACTION REGULATES TCR SIGNALING
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
TCR stimulation did not alter TCR-initiated phosphorylation of theindicated proteins, suggesting that the observed inhibition might be
strictly dependent on the activation of the CD4 molecule or theCD4/p56lck complex.
CD4 ligation blocks TCR/CD3-induced tyrosine phosphorylationof Vav and SLP-76
With the next set of experiments, the SEA-induced tyrosine phos-phorylation of SLP-76 and Vav was analyzed with or without pre-ceding engagement of CD4 by mAbs. Activation of these proteinsduring TCR-stimulated cytokine production has been shown to beassociated with an increased tyrosine phosphorylation mediated byZAP-70 (23–25). We therefore immunoprecipitated SLP-76 andVav, respectively, from unstimulated (2) or superantigen-stimu-lated (1) clone cells that had been exposed or not to anti-CD4mAb OKT4 for 1 h and analyzed the tyrosine phosphorylation ofthe blotted proteins with anti-pTyr mAb 4G10 (Fig. 2A). Bothproteins were tyrosine phosphorylated during a 3-min stimulationwith SEA (see also total cell lysates (TCL)), and the TCR-inducedphosphorylation of both proteins was almost completely blockedafter exposure to anti-CD4 mAb before SEA stimulation. The pro-tein content in precipitates of untreated or CD4-treated cells waschecked by reprobing the blot with anti-Vav (B) or anti-SLP-76(C) Abs.
Preferential, tyrosine phosphorylation-independent associationof SLP-76 with SH3 domains of p56lck
From the experimental data shown so far, we hypothesized thatVav and/or SLP-76 should be able to interact with both the CD4/p56lck complex and the TCR/CD3/p59fyn complex in a competitivemanner. Also, the respective interaction should be independent oftyrosine phosphorylation. Given the structure of SLP-76 with threeprotein interaction domains including the proline-rich central por-tion (26, 27), we favored a potential SH3 association with p56lck
and p59fyn(T) in T cells. Therefore, we investigated whether
FIGURE 1. Anti-CD4 mAb treatment inhibits TCR/CD3-induced ty-rosine phosphorylation. Cells of T cell clone D894/25 were incubated for1 h in medium or in the presence of mAbs against CD4 (OKT4) or CD5(UCHT2), respectively. Stimulation with SEA was performed for 2 or 5min at 37°C. Clone cells not exposed to SEA served as a control (2). Thecells were then lysed in Nonidet P-40 lysis buffer, and proteins were sep-arated by SDS-PAGE. Upon transfer to nitrocellulose membranes, the blotswere developed with anti-pTyr mAb (4G10,A) and reprobed with anti-SLP-76 antiserum (B). The position of SLP-76 is marked with an asterisk;arrows point to protein bands that were differentially phosphorylated inCD4-untreated and CD4-pretreated cells.
FIGURE 2. CD4 ligation inhibits TCR-triggeredphosphorylation of Vav and SLP-76. Clone cells wereincubated for 1 h in the absence (A) or presence ofanti-CD4 mAb OKT4. The cells were then pelleted,incubated for 3 min in medium without (2) or with(1) SEA, and lysed in Brij96 lysis buffer. Immunopre-cipitations of Vav and SLP-76 were performed with 3mg of the respective Ab and protein A- and proteinG-Sepharose beads (beads alone served as a control).Precipitated proteins were separated by SDS-PAGE,and blotted to nitrocellulose. Blots were developedwith anti-pTyr mAb 4G10 (A) and reprobed with anti-Vav (B) and anti-SLP-76 (C) Abs.
3145The Journal of Immunology
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
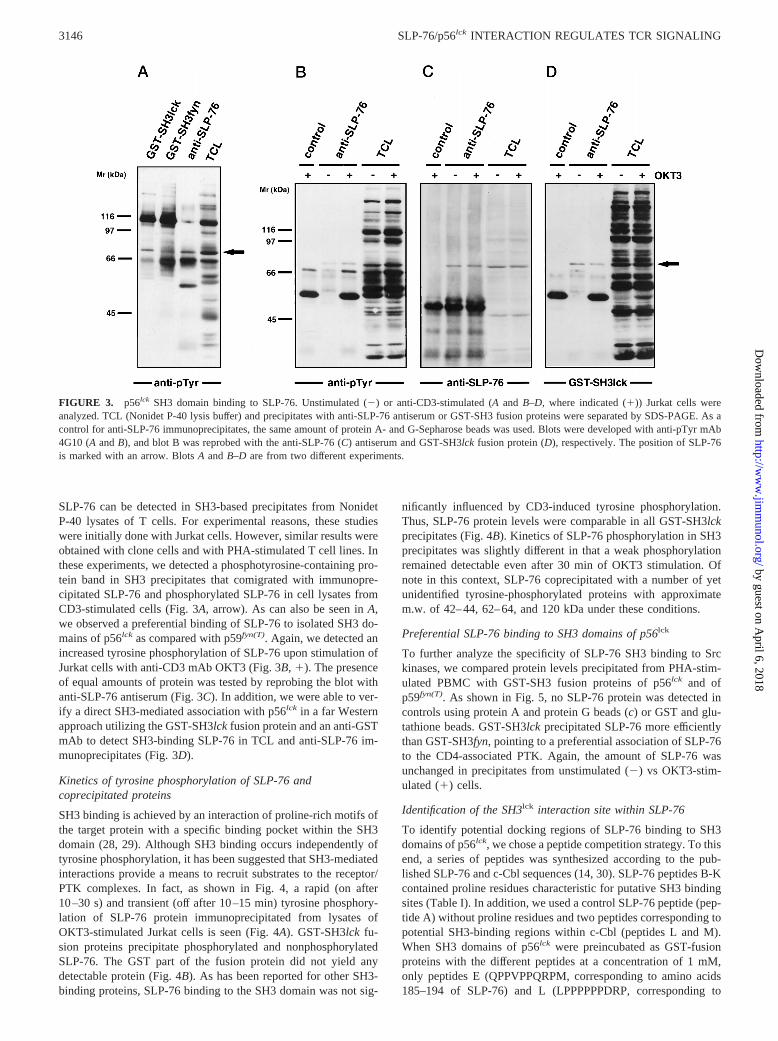
SLP-76 can be detected in SH3-based precipitates from NonidetP-40 lysates of T cells. For experimental reasons, these studieswere initially done with Jurkat cells. However, similar results wereobtained with clone cells and with PHA-stimulated T cell lines. Inthese experiments, we detected a phosphotyrosine-containing pro-tein band in SH3 precipitates that comigrated with immunopre-cipitated SLP-76 and phosphorylated SLP-76 in cell lysates fromCD3-stimulated cells (Fig. 3A, arrow). As can also be seen inA,we observed a preferential binding of SLP-76 to isolated SH3 do-mains of p56lck as compared with p59fyn(T). Again, we detected anincreased tyrosine phosphorylation of SLP-76 upon stimulation ofJurkat cells with anti-CD3 mAb OKT3 (Fig. 3B, 1). The presenceof equal amounts of protein was tested by reprobing the blot withanti-SLP-76 antiserum (Fig. 3C). In addition, we were able to ver-ify a direct SH3-mediated association with p56lck in a far Westernapproach utilizing the GST-SH3lck fusion protein and an anti-GSTmAb to detect SH3-binding SLP-76 in TCL and anti-SLP-76 im-munoprecipitates (Fig. 3D).
Kinetics of tyrosine phosphorylation of SLP-76 andcoprecipitated proteins
SH3 binding is achieved by an interaction of proline-rich motifs ofthe target protein with a specific binding pocket within the SH3domain (28, 29). Although SH3 binding occurs independently oftyrosine phosphorylation, it has been suggested that SH3-mediatedinteractions provide a means to recruit substrates to the receptor/PTK complexes. In fact, as shown in Fig. 4, a rapid (on after10–30 s) and transient (off after 10–15 min) tyrosine phosphory-lation of SLP-76 protein immunoprecipitated from lysates ofOKT3-stimulated Jurkat cells is seen (Fig. 4A). GST-SH3lckfu-sion proteins precipitate phosphorylated and nonphosphorylatedSLP-76. The GST part of the fusion protein did not yield anydetectable protein (Fig. 4B). As has been reported for other SH3-binding proteins, SLP-76 binding to the SH3 domain was not sig-
nificantly influenced by CD3-induced tyrosine phosphorylation.Thus, SLP-76 protein levels were comparable in all GST-SH3lckprecipitates (Fig. 4B). Kinetics of SLP-76 phosphorylation in SH3precipitates was slightly different in that a weak phosphorylationremained detectable even after 30 min of OKT3 stimulation. Ofnote in this context, SLP-76 coprecipitated with a number of yetunidentified tyrosine-phosphorylated proteins with approximatem.w. of 42–44, 62–64, and 120 kDa under these conditions.
Preferential SLP-76 binding to SH3 domains of p56lck
To further analyze the specificity of SLP-76 SH3 binding to Srckinases, we compared protein levels precipitated from PHA-stim-ulated PBMC with GST-SH3 fusion proteins of p56lck and ofp59fyn(T). As shown in Fig. 5, no SLP-76 protein was detected incontrols using protein A and protein G beads (c) or GST and glu-tathione beads. GST-SH3lckprecipitated SLP-76 more efficientlythan GST-SH3fyn, pointing to a preferential association of SLP-76to the CD4-associated PTK. Again, the amount of SLP-76 wasunchanged in precipitates from unstimulated (2) vs OKT3-stim-ulated (1) cells.
Identification of the SH3lck interaction site within SLP-76
To identify potential docking regions of SLP-76 binding to SH3domains of p56lck, we chose a peptide competition strategy. To thisend, a series of peptides was synthesized according to the pub-lished SLP-76 and c-Cbl sequences (14, 30). SLP-76 peptides B-Kcontained proline residues characteristic for putative SH3 bindingsites (Table I). In addition, we used a control SLP-76 peptide (pep-tide A) without proline residues and two peptides corresponding topotential SH3-binding regions within c-Cbl (peptides L and M).When SH3 domains of p56lck were preincubated as GST-fusionproteins with the different peptides at a concentration of 1 mM,only peptides E (QPPVPPQRPM, corresponding to amino acids185–194 of SLP-76) and L (LPPPPPPDRP, corresponding to
FIGURE 3. p56lck SH3 domain binding to SLP-76. Unstimulated (2) or anti-CD3-stimulated (A and B–D, where indicated (1)) Jurkat cells wereanalyzed. TCL (Nonidet P-40 lysis buffer) and precipitates with anti-SLP-76 antiserum or GST-SH3 fusion proteins were separated by SDS-PAGE. As acontrol for anti-SLP-76 immunoprecipitates, the same amount of protein A- and G-Sepharose beads was used. Blots were developed with anti-pTyr mAb4G10 (AandB), and blot B was reprobed with the anti-SLP-76 (C) antiserum and GST-SH3lckfusion protein (D), respectively. The position of SLP-76is marked with an arrow. BlotsA andB–D are from two different experiments.
3146 SLP-76/p56lck INTERACTION REGULATES TCR SIGNALING
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
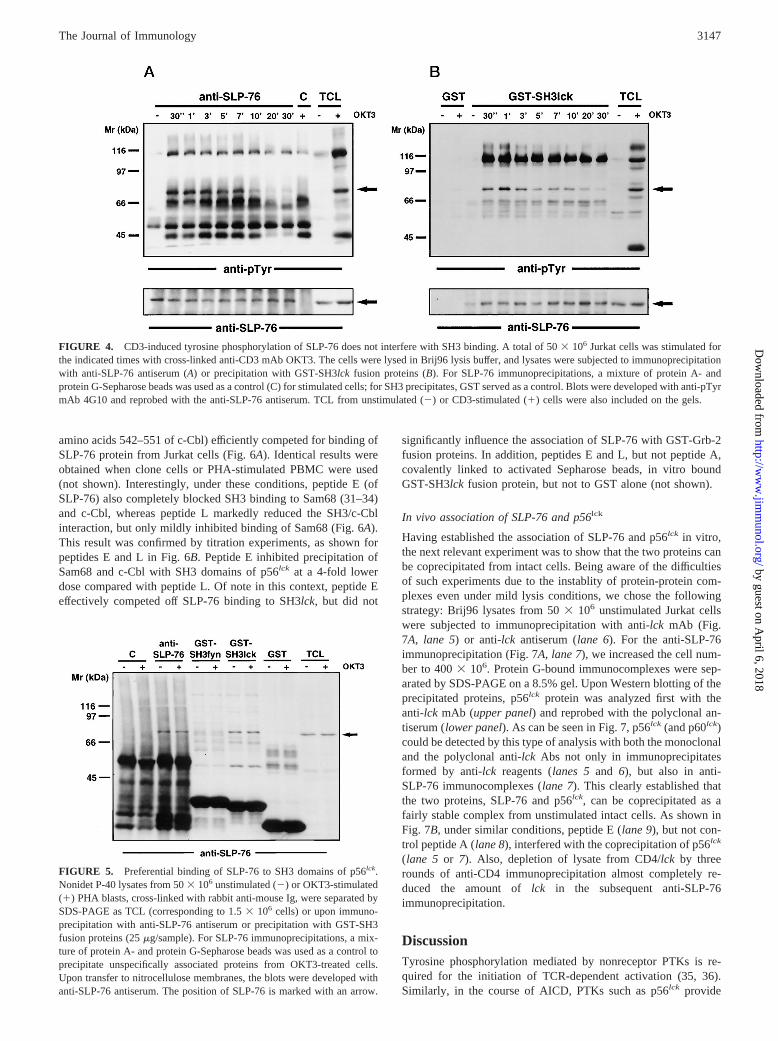
amino acids 542–551 of c-Cbl) efficiently competed for binding ofSLP-76 protein from Jurkat cells (Fig. 6A). Identical results wereobtained when clone cells or PHA-stimulated PBMC were used(not shown). Interestingly, under these conditions, peptide E (ofSLP-76) also completely blocked SH3 binding to Sam68 (31–34)and c-Cbl, whereas peptide L markedly reduced the SH3/c-Cblinteraction, but only mildly inhibited binding of Sam68 (Fig. 6A).This result was confirmed by titration experiments, as shown forpeptides E and L in Fig. 6B. Peptide E inhibited precipitation ofSam68 and c-Cbl with SH3 domains of p56lck at a 4-fold lowerdose compared with peptide L. Of note in this context, peptide Eeffectively competed off SLP-76 binding to SH3lck, but did not
significantly influence the association of SLP-76 with GST-Grb-2fusion proteins. In addition, peptides E and L, but not peptide A,covalently linked to activated Sepharose beads, in vitro boundGST-SH3lckfusion protein, but not to GST alone (not shown).
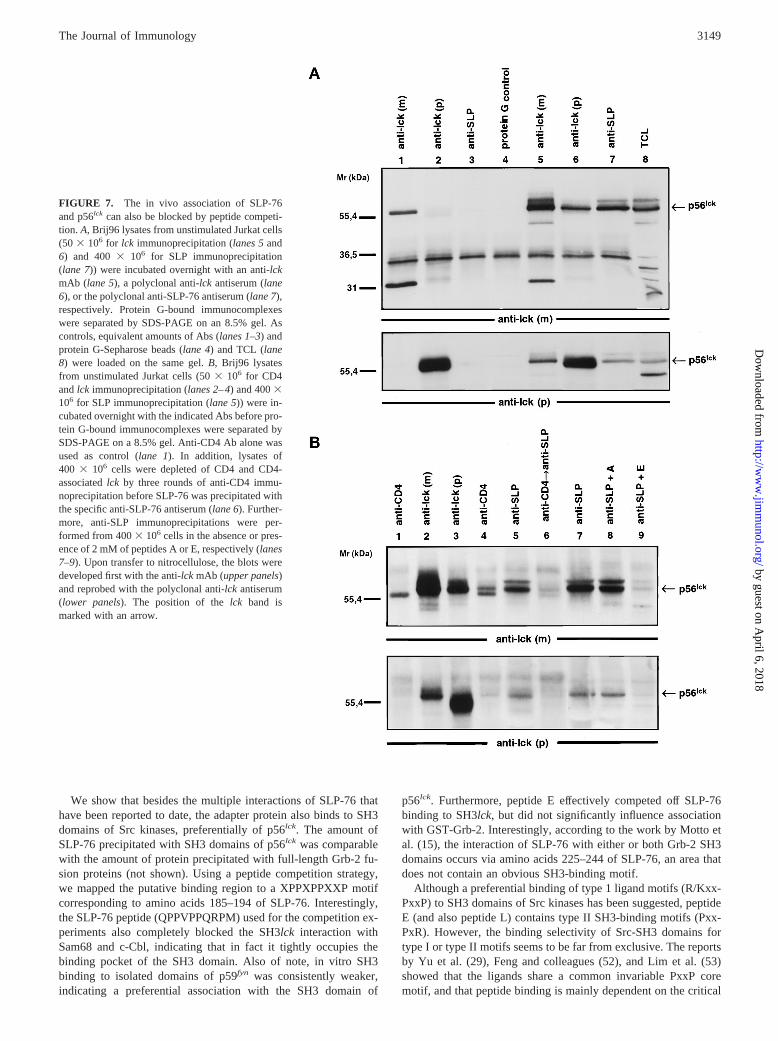
In vivo association of SLP-76 and p56lck
Having established the association of SLP-76 and p56lck in vitro,the next relevant experiment was to show that the two proteins canbe coprecipitated from intact cells. Being aware of the difficultiesof such experiments due to the instablity of protein-protein com-plexes even under mild lysis conditions, we chose the followingstrategy: Brij96 lysates from 503 106 unstimulated Jurkat cellswere subjected to immunoprecipitation with anti-lck mAb (Fig.7A, lane 5) or anti-lckantiserum (lane 6). For the anti-SLP-76immunoprecipitation (Fig. 7A, lane 7), we increased the cell num-ber to 4003 106. Protein G-bound immunocomplexes were sep-arated by SDS-PAGE on a 8.5% gel. Upon Western blotting of theprecipitated proteins, p56lck protein was analyzed first with theanti-lck mAb (upper panel) and reprobed with the polyclonal an-tiserum (lower panel). As can be seen in Fig. 7, p56lck (and p60lck)could be detected by this type of analysis with both the monoclonaland the polyclonal anti-lck Abs not only in immunoprecipitatesformed by anti-lck reagents (lanes 5 and 6), but also in anti-SLP-76 immunocomplexes (lane 7). This clearly established thatthe two proteins, SLP-76 and p56lck, can be coprecipitated as afairly stable complex from unstimulated intact cells. As shown inFig. 7B, under similar conditions, peptide E (lane 9), but not con-trol peptide A (lane 8), interfered with the coprecipitation of p56lck
(lane 5 or 7). Also, depletion of lysate from CD4/lck by threerounds of anti-CD4 immunoprecipitation almost completely re-duced the amount oflck in the subsequent anti-SLP-76immunoprecipitation.
DiscussionTyrosine phosphorylation mediated by nonreceptor PTKs is re-quired for the initiation of TCR-dependent activation (35, 36).Similarly, in the course of AICD, PTKs such as p56lck provide
FIGURE 4. CD3-induced tyrosine phosphorylation of SLP-76 does not interfere with SH3 binding. A total of 503 106 Jurkat cells was stimulated forthe indicated times with cross-linked anti-CD3 mAb OKT3. The cells were lysed in Brij96 lysis buffer, and lysates were subjected to immunoprecipitationwith anti-SLP-76 antiserum (A) or precipitation with GST-SH3lck fusion proteins (B). For SLP-76 immunoprecipitations, a mixture of protein A- andprotein G-Sepharose beads was used as a control (C) for stimulated cells; for SH3 precipitates, GST served as a control. Blots were developed with anti-pTyrmAb 4G10 and reprobed with the anti-SLP-76 antiserum. TCL from unstimulated (2) or CD3-stimulated (1) cells were also included on the gels.
FIGURE 5. Preferential binding of SLP-76 to SH3 domains of p56lck.Nonidet P-40 lysates from 503 106 unstimulated (2) or OKT3-stimulated(1) PHA blasts, cross-linked with rabbit anti-mouse Ig, were separated bySDS-PAGE as TCL (corresponding to 1.53 106 cells) or upon immuno-precipitation with anti-SLP-76 antiserum or precipitation with GST-SH3fusion proteins (25mg/sample). For SLP-76 immunoprecipitations, a mix-ture of protein A- and protein G-Sepharose beads was used as a control toprecipitate unspecifically associated proteins from OKT3-treated cells.Upon transfer to nitrocellulose membranes, the blots were developed withanti-SLP-76 antiserum. The position of SLP-76 is marked with an arrow.
3147The Journal of Immunology
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

necessary and essential signals for the regulation of FasL expres-sion (37–39). Therefore, PTK inhibitors efficiently block CD3/TCR-triggered activation and AICD (40–42). An inhibitory mod-ulation of TCR-dependent kinase signaling cascades byengagement of costimulatory and/or accessory molecules could beexplained on the basis of negative signaling, e.g., by phosphatases,or by a recomposition of preformed signaling complexes in thevicinity of Src- or Syk-type PTKs associated with the correspond-ing surface receptors.
We have previously observed that CD4 stimulation of cloned Tcells with mAb or HIV-1 gp120 modified subsequent TCR signal-ing, resulting in a marked reduction of cytokine production andinhibition of AICD (11). This inhibition of TCR signaling wasassociated with reduced levels of tyrosine phosphorylation of anumber of proteins detected in whole cell lysates (11). We nowreport that two PTK substrates implicated in TCR-mediated signaltransduction, the product of the protooncogenevav (12, 43–45)and the associated SH2 domain-containing leukocyte protein of 76kDa (SLP-76) (14–18, 25–27), show this characteristic inhibitionof TCR-inducible tyrosine phosphorylation in clone cells pre-treated with anti-CD4 mAb. Our results demonstrate that CD4 li-gation can prevent the formation of signal-transducing unitsaround the TCR/CD3 complex leading to a desensitization of theAg receptor.
Recently, Jabado and colleagues also pointed to a defective for-mation of p21ras activity-regulating complexes, including PLC-g1,p120GAP, and other yet unidentified proteins upon CD4 ligandbinding (46). In addition, Goldman et al. suggested that CD4 li-gation may inhibit TCR signaling by sequestering p56lck to theactin-cytoskeleton (47). When they pretreated cells with gp120
1–4 h before stimulation, TCR-directed PTK activation was mark-edly reduced. Also, in gp120/anti-gp120-stimulated cells, p56lck
was found predominantly in association with the cytoskeleton inthe Nonidet P-40 detergent-insoluble cytoskeletal, but not in theNonidet P-40 detergent-soluble cytosolic fractions (47).
With Vav and SLP-76, we link two other key players in TCRsignal transduction to the CD4/p56lck complex. The multifunc-tional adapter protein SLP-76 was characterized as a molecule thatis phosphorylated by ZAP-70 upon TCR stimulation and subse-quently associates with SH2 domains of Vav via phosphotyrosyl-containing motifs in the N-terminal portion of SLP-76 (13, 23–25).The physical link between Vav and SLP-76 may also be differen-tially regulated by associated phosphatases including CD45 (48).In addition, a constitutive association of SLP-76 with Grb-2 SH3domains has been reported, mediated by the proline-rich centralpart (14, 15). With its C-terminal SH2 domain, SLP-76 also reactswith phosphoproteins including SLAP-130 (5FYB) and pp62 (27,49, 50).
Recent studies with SLP-76-deficient mice and SLP-76-defi-cient T cell lines revealed a very profound role of the adapterprotein in T cell development and activation (16–18). It wasshown that SLP-76 couples TCR-associated PTKs to PLC-g1-in-duced signaling cascades and that it is absolutely required for nor-mal T cell development and function. Interestingly, the expressionof SLP-76 is restricted to hemopoietic cells of monocyte, granu-locyte, and T lymphocyte lineage (51), whereas in B cells, SLP-65(or BLNK), an adapter protein with a very similar overall domainstructure but fairly low homology, fulfills the same role in linkingthe B cell receptor signals to PLC-g1 activation (19, 20).
FIGURE 6. Peptide competition reveals aa185–194 as SH3lck binding site of SLP-76. Pep-tides corresponding to stretches within theSLP-76 and c-Cbl molecules, as detailed in Ta-ble I, were used at 1 mM (A) or at varying con-centrations from 0.25–5 mM (B) in PBS. A totalof 20 mg of GST-SH3lck fusion protein on glu-tathione beads was incubated with the respec-tive peptide with constant rotation for 15 min at4°C. A total of 100ml of filtered Jurkat celllysates (253 106 cells lysed in 1% Brij96 lysisbuffer) was then added, and incubation was pro-longed for 10 min. The beads were washed, andprecipitated proteins were subjected to SDS-PAGE and Western blotting. Blots were devel-oped with the anti-SLP-76 antiserum and sub-sequently with anti-Sam68 mAb and anti-c-Cblantiserum. Equal loading of fusion protein waschecked in each experiment by Ponceau S stain-ing (data not shown).
3148 SLP-76/p56lck INTERACTION REGULATES TCR SIGNALING
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
We show that besides the multiple interactions of SLP-76 thathave been reported to date, the adapter protein also binds to SH3domains of Src kinases, preferentially of p56lck. The amount ofSLP-76 precipitated with SH3 domains of p56lck was comparablewith the amount of protein precipitated with full-length Grb-2 fu-sion proteins (not shown). Using a peptide competition strategy,we mapped the putative binding region to a XPPXPPXXP motifcorresponding to amino acids 185–194 of SLP-76. Interestingly,the SLP-76 peptide (QPPVPPQRPM) used for the competition ex-periments also completely blocked the SH3lck interaction withSam68 and c-Cbl, indicating that in fact it tightly occupies thebinding pocket of the SH3 domain. Also of note, in vitro SH3binding to isolated domains of p59fyn was consistently weaker,indicating a preferential association with the SH3 domain of
p56lck. Furthermore, peptide E effectively competed off SLP-76binding to SH3lck, but did not significantly influence associationwith GST-Grb-2. Interestingly, according to the work by Motto etal. (15), the interaction of SLP-76 with either or both Grb-2 SH3domains occurs via amino acids 225–244 of SLP-76, an area thatdoes not contain an obvious SH3-binding motif.
Although a preferential binding of type 1 ligand motifs (R/Kxx-PxxP) to SH3 domains of Src kinases has been suggested, peptideE (and also peptide L) contains type II SH3-binding motifs (Pxx-PxR). However, the binding selectivity of Src-SH3 domains fortype I or type II motifs seems to be far from exclusive. The reportsby Yu et al. (29), Feng and colleagues (52), and Lim et al. (53)showed that the ligands share a common invariable PxxP coremotif, and that peptide binding is mainly dependent on the critical
FIGURE 7. The in vivo association of SLP-76and p56lck can also be blocked by peptide competi-tion. A, Brij96 lysates from unstimulated Jurkat cells(50 3 106 for lck immunoprecipitation (lanes 5and6) and 4003 106 for SLP immunoprecipitation(lane 7)) were incubated overnight with an anti-lckmAb (lane 5), a polyclonal anti-lck antiserum (lane6), or the polyclonal anti-SLP-76 antiserum (lane 7),respectively. Protein G-bound immunocomplexeswere separated by SDS-PAGE on an 8.5% gel. Ascontrols, equivalent amounts of Abs (lanes 1–3) andprotein G-Sepharose beads (lane 4) and TCL (lane8) were loaded on the same gel.B, Brij96 lysatesfrom unstimulated Jurkat cells (503 106 for CD4andlck immunoprecipitation (lanes 2–4) and 4003106 for SLP immunoprecipitation (lane 5)) were in-cubated overnight with the indicated Abs before pro-tein G-bound immunocomplexes were separated bySDS-PAGE on a 8.5% gel. Anti-CD4 Ab alone wasused as control (lane 1). In addition, lysates of400 3 106 cells were depleted of CD4 and CD4-associatedlck by three rounds of anti-CD4 immu-noprecipitation before SLP-76 was precipitated withthe specific anti-SLP-76 antiserum (lane 6). Further-more, anti-SLP immunoprecipitations were per-formed from 4003 106 cells in the absence or pres-ence of 2 mM of peptides A or E, respectively (lanes7–9). Upon transfer to nitrocellulose, the blots weredeveloped first with the anti-lck mAb (upper panels)and reprobed with the polyclonal anti-lck antiserum(lower panels). The position of thelck band ismarked with an arrow.
3149The Journal of Immunology
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

salt bridging of the arginine residue of the peptide and the con-served aspartate at position 99 of the Src-family SH3 domain, withthe residues in positions 3, 4, 6, and 7 of the peptide intercalatinginto the binding site and thus determining the binding orientationof the peptide. This was also confirmed by Morton and colleagues(54), who investigated ligand binding to the SH3 domain of Fyn.Based on the comparison of different peptides (including type I andtype II) for SH3 binding, they suggested that SH3 domains bind topolyproline peptides in a promiscuous manner, although the ligandwith the closest match to the class I consensus sequence boundwith highest affinity and in the predicted orientation. Also, theNMR of the SH3 domain oflck (55) confirmed the previous sug-gestions that peptide ligands can bind in two different orientationsto SH3 domains. This study also points to some unique featureswithin the SH3 domain of Lck, which could help to explain theobserved preference of SLP-76 for Lck compared with Fyn. Inview of the specific regulation of SH3 binding by the critical ar-ginine residue, it should also be mentioned that peptides D (aa182–191) and E (aa 185 to 194) are overlapping peptides that shareseven amino acids including the PxxP core. However, only theinhibitory peptide E contains an arginine residue in a position thatdetermines the type II-binding motif PxxPxR.
Most important, we were able to demonstrate that p56lck can becoprecipitated in an immunocomplex formed by anti-SLP-76 Absfrom unstimulated Jurkat cells, and that the coprecipitation can beabrogated by preincubation of the cell lysates with the competitorpeptide E. This established that the phosphorylation-independent(SH3-mediated) association detected and analyzed in vitro is alsooperational in vivo in intact cells.
The SH3 domain-mediated interaction of SLP-76 with p56lck
may help to explain the observations mentioned above (46, 47).Since we found a reduced tyrosine phosphorylation of SLP-76upon CD4 pretreatment, it seems likely that CD4 ligation resultedin a spatial dissociation from its kinase, ZAP-70. Thus, if p56lck isnot available for the phosphorylation of thez-chains, ZAP-70could not bind to the TCRz/CD3 complex to get activated. We arepresently analyzing whether the dissociation occurs due to seques-tering p56lck to the cytoskeleton, as has been suggested by Gold-man and colleagues (47), or by capping mechanisms that havebeen described for other systems. In fact, the influence of the CD4proximity to the TCR/CD3 complex had been correlated earlier toinhibitory or stimulatory signals provided by CD4 (56).
As a functional outcome of negative tuning of TCR responsesby CD4 engagement, the selective activation of CD4-positive na-ive but not memory cells has been suggested as a consequence ofMHC class II molecules on APC and CD4 ligation (57). In thecontext of thymocyte development, it was shown that CD4 en-gagement inhibited TCR expression and function in immatureCD41CD81 thymocytes (58). The effects of other ligands of CD4,including envelope proteins of HIV and the recently cloned IL-16,seem to be manyfold and very much depending on the investigatedsystem. For IL-16 as a natural ligand of CD4 (59, 60), a block ofCD3-dependent lymphocyte activation and proliferation has beenreported by Cruikshank and coworkers (61). In addition, IL-16seems to play a role in the regulation of HIV-1 infection and/orreplication (62–64). A protective effect of rIL-16 against AICDhas also been described, although the underlying mechanism needsfurther studies (64). The preparation of IL-16 that we used in ourprevious study was ineffective in our system. Moreover, CD4 li-gation in our hands affected CD95L but not CD95 expression (11),as has been suggested by others (64).
The multiple effects of cross-linking of CD4 by anti-CD4 mAbor gp120 of HIV on TCR-induced activation of resting vs preac-tivated cells have been extensively discussed in our previous report
(11). They range from costimulation of resting T cells to prolifer-ate (65, 66), priming of resting cells for AICD (67–70) and directinduction of apoptosis (71–76) to the inhibition of TCR-inducedactivation (46, 47, 77–80), and AICD (11). Taken together, thesestudies suggest that the observed differences in the CD4/TCRcross-talk between resting and activated cells very much dependon the investigated T cell population. Overall, resting cells seem tobe coactivated by CD4 engagement, whereas inhibitory effects ofCD4 ligation have been mainly observed in preactivated popula-tions. Although OKT4 does not bind to the gp120-binding region,it needs to be addressed in further studies whether the binding ofdifferent CD4 ligands or mAb to distinct functional epitopes withinthe CD4 molecule provokes different signals with regard to theinhibition of AICD (81).
In conclusion, SH3-mediated interactions of SLP-76 and asso-ciated molecules with the CD4/p56lck complex play a crucial rolein negative signaling through the CD4 molecule. In activated Tcells, CD4 ligation may lead to a removal of essential componentsof the TCR signaling complex and to a TCR desensitization withreduced responses in terms of proliferation, cytokine production,and AICD (see model in Fig. 8). Whether the removal of SLP-76-associated proteins from the TCR/CD3 complex is physical, e.g.,due to a phosphorylation-independent translocation of SLP-76/p56lck complexes to the cytoskeleton, is presently underinvestigation.
AcknowledgmentsWe thank Dr. Joachim Denner for peptide synthesis, and EuroCetus andCilag GmbH for providing essential reagents.
References1. Russell, J., C. L. White, D. Y. Loh, and P. Meleedy-Rey. 1992. Receptor-stim-
ulated death pathway is opened by antigen in mature T cells.Proc. Natl. Acad.Sci. USA 88:2151.
FIGURE 8. Model for the involvement of SLP-76 in CD4-induced de-sensitization of the TCR/CD3 signaling complex. We hypothesize that apreformed TCR/CD3/CD4 signaling complex exists in preactivated T cells.This complex would include PTKs, adapter proteins, and unknown down-stream elements associated in a tyrosine phosphorylation-independentmanner due to SH3-mediated interactions. A. TCR ligation without CD4engagement triggers PTKs associated with the TCR/CD3/CD4 complex.The result is a tyrosine phosphorylation-dependent activation and poten-tially AICD. B, Upon ligation of CD4, components of the preformed com-plex (i.e., SLP-76 and Vav) are removed from the TCR/CD3 complex. Thetransient TCR/CD3 desensitization results in an incomplete activation ca-pacity of TCR/CD3-triggered PTK-dependent pathways.
3150 SLP-76/p56lck INTERACTION REGULATES TCR SIGNALING
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

2. Janssen, O., S. Wesselborg, B. Heckl-O¨ streicher, K. Pechold, A. Bender,S. Schondelmaier, G. Moldenhauer, and D. Kabelitz. 1991. T cell receptor/CD3-signaling induces death by apoptosis in human T cell receptorgd1 T cells.J. Im-munol. 146:35.
3. Kabelitz, D., and S. Wesselborg. 1992. Life and death of a superantigen-reactivehuman CD41 T cell clone: staphylococcal enterotoxins induce death by apoptosisbut simultaneously trigger a proliferative response in the presence of HLA DR1
antigen-presenting cells.Int. Immunol. 4:1381.4. Nagata, S., and P. Golstein. 1995. The Fas death factor.Science 267:1449.5. Dhein, J., H. Walczak, C. Baeumler, K. M. Debatin, and P. H. Krammer. 1995.
Autocrine T-cell suicide mediated by APO-1/(Fas/CD95).Nature 373:438.6. Brunner, T., R. J. Mogil, D. LaFace, N. J. Yoo, A. Mahboubi, F. Echeverri,
S. J. Martin, W. R. Force, D. H. Lynch, C. F. Ware, and D. R. Green. 1995.Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-inducedapoptosis in T-cell hybridomas.Nature 373:441.
7. Ju, S. T., D. J. Panka, H. Cui, R. Ettinger, M. el-Khatib, D. H. Sherr,B. Z. Stanger, and A. Marshak-Rothstein. 1995. Fas(CD95)/FasL interactionsrequired for programmed cell death after T-cell activation.Nature 373:444.
8. Alderson, M. R., T. W. Tough, T. Davis-Smith, S. Braddy, B. Falk,K. A. Schooley, R. G. Goodwin, C. A. Smith, F. Ramsdell, and D. H. Lynch.1995. Fas ligand mediates activation-induced cell death in human T lymphocytes.J. Exp. Med. 181:71.
9. Radvanyi, L. G., Y. Shi, H. Vaziri, A. Sharma, R. Dhala, G. B. Mills, andR. G. Miller. 1996. CD28 costimulation inhibits TCR-induced apoptosis duringa primary T cell response.J. Immunol. 156:1788.
10. Groux, H., G. Torpier, D. Monte, Y. Mouton, A. Capron, and J. C. Ameisen.1992. Activation-induced death by apoptosis in CD41 T cells from human im-munodeficiency virus-infected asymptomatic individuals.J. Exp. Med. 175:331.
11. Oberg, H.-H., R. Sanzenbacher, B. Lengl-Janben, T. Dobmeyer, S. Flindt,O. Janssen, and D. Kabelitz. 1997. Ligation of cell surface CD4 inhibits activa-tion-induced cell death of human T lymphocytes at the level of Fas-ligand ex-pression.J. Immunol. 159:5742.
12. Katzav, S., D. Martin-Zanca, and M. Barbacid. 1989. Vav, a novel human on-cogene derived from a locus ubiquitously expressed in hematopoietic cells.EMBO J. 8:2283.
13. Tuosto, L., F. Michel, and O. Acuto. 1996. p95vav associates with tyrosine-phos-phorylated SLP-76 in antigen-stimulated T cells.J. Exp. Med. 184:1161.
14. Jackman, J. K., D. G. Motto, Q. Sun, M. Tanemoto, C. W. Turck, G. A. Peltz,G. A. Koretzky, and P. R. Findell. 1995. Molecular cloning of SLP-76, a 76-kDatyrosine phosphoprotein associated with Grb2 in T cells.J. Biol. Chem. 270:7029.
15. Motto, D. G., S. E. Ross, J. Wu, L. R. Hendricks-Taylor, and G. A. Koretzky.1996. Implication of the Grb2-associated phosphoprotein SLP-76 in T cell re-ceptor-mediated interleukin 2 production.J. Exp. Med. 183:1937.
16. Pivniouk, V., E. Tsitsikov, P. Swinton, G. Rathbun, F. W. Alt, and R. S. Geha.1998. Impaired viability and profound block in thymocyte development in micelacking the adaptor protein SLP-76.Cell 94:229.
17. Clements, J. L., B. Yang, S. E. Ross-Barta, S. L. Eliason, R. F. Hrstka,R. A. Williamson, and G. A. Koretzky. 1998. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development.Science 281:416.
18. Yablonski, D., M. R. Kuhne, T. Kadlecek, and A. Weiss. 1998. Uncoupling ofnonreceptor tyrosine kinases from PLCg1 in a SLP-76-deficient T cell.Science281:413.
19. Wienands, J., J. Schweikert, B. Wollscheid, H. Jumaa, P. J. Nielsen, and M. Reth.1998. SLP-65: a new signaling component in B lymphocytes which requiresexpression of the antigen receptor for phosphorylation.J. Exp. Med. 188:791.
20. Fu, C., C. W. Turck, T. Kurosaki, and A. C. Chan. 1998. BLNK: a central linkerprotein in B cell activation.Immunity 9:93.
21. Prasad, K. V. S., O. Janssen, R. Kapeller, M. Raab, L. C. Cantley, and C. E. Rudd.1993. Src-homology 3 domain of protein kinase p59fyn mediates binding to phos-phatidylinositol 3-kinase in T cells.Proc. Natl. Acad. Sci. USA 90:7366.
22. Prasad, K. V. S., R. Kapeller, O. Janssen, J. S. Duke-Cohen, L. C. Cantley, andC. E. Rudd. 1993. Phosphatidylinositol 3-kinase and phosphatidylinositol 4-ki-nase binding to the CD4–p56lck complex: p56lck SH3 domain mediates bindingto PI-3 kinase but not to PI-4 kinase.Mol. Cell. Biol. 13:7708.
23. Wu, J., D. G. Motto, G. A. Koretzky, and A. Weiss. 1996. Vav and SLP-76interact and functionally cooperate in IL-2 gene activation.Immunity 4:593.
24. Wardenburg, J. B., C. Fu, J. K. Jackman, H. Flotow, S. E. Wilkinson,D. H. Williams, R. Johnson, G. Kong, A. C. Chan, and P. R. Findell. 1996.Phosphorylation of SLP-76 by the ZAP-70 protein tyrosine kinase is required forT-cell receptor function.J. Biol. Chem. 271:19641.
25. Raab, M., A. J. da Silva, P. R. Findell, and C. E. Rudd. 1997. Regulation ofVav-SLP-76 binding by ZAP-70 and its relevance to TCRz/CD3 induction ofinterleukin-2.Immunity 6:155.
26. Musci, M. A., D. G. Motto, S. E. Ross, N. Fang, and G. A. Koretzky. 1997. Threedomains of SLP-76 are required for its optimal function in a T cell line.J. Im-munol. 159:1639.
27. Koretzky, G. A. 1997. The role of Grb2-associated proteins in T cell activation.Immunol. Today 8:401.
28. Ren, R., B. J. Mayer, P. Cicchetti, and D. Baltimore. 1993. Identification of aten-amino acid proline-rich SH3 binding site.Science 259:1157.
29. Yu, H., J. K. Chen, S. Feng, D. C. Dalgarno, A. W. Brauer, and S. L. Schreiber.1994. Structural basis for the binding of proline-rich peptides to SH3 domains.Cell 76:933.
30. Blake, T. J., M. Shapiro, H. C. Morse III, and W. Y. Langdon. 1991. The se-quences of the human and mouse c-cbl protooncogenes show v-cbl was generated
by a large truncation encompassing a proline-rich domain and a leucine zipper-like motif. Oncogene 6:653.
31. Fukazawa, T., K. A. Reedquist, T. Trub, S. Soltoff, G. Panchamoorthy, B. Druker,L. Cantley, S. E. Schoelson, and H. Band. 1995. The SH3 domain-binding T celltyrosyl phosphoprotein p120: demonstration of its identity with the c-cbl pro-tooncogene product and in vivo complexes with Fyn, Grb-2 and phosphatidyl-inositol 3-kinase.J. Biol. Chem. 270:19141.
32. Taylor, S. J., and D. Shalloway. 1994. An RNA-binding protein associated withSrc through its SH2 and SH3 domains in mitosis.Nature 368:867.
33. Fumagalli, S., N. F. Totty, J. J. Hsuan, and S. A. Courtneidge. 1994. A target forSrc in mitosis.Nature 368:871.
34. Lock, P., S. Fumagalli, P. Polakis, F. McCormick, and S. A. Courtneidge. 1996.The human p62 cDNA encodes Sam68 and not the RasGAP-associated p62 pro-tein. Cell 84:23.
35. Rudd, C. E., O. Janssen, Y.-C. Cai, A. J. da Silva, M. Raab, and K. V. S. Prasad.1994. Two-step TCRz/CD3-CD4 and CD28 signaling in T cells: SH2/SH3 do-mains, protein-tyrosine kinases and lipid kinases.Immunol. Today 15:225.
36. Alberola-Ila, J., S. Takaki, J. D. Kerner, and R. Perlmutter. 1997. Differentialsignaling by lymphocyte antigen receptors.Annu. Rev. Immunol. 15:125.
37. Oyaizu, N., S. Than, T. W. McClosey, and S. Pahwa. 1995. Requirement ofp56lck in T-cell receptor/CD3-mediated apoptosis and Fas-ligand induction inJurkat cells. Biochem. Biophys. Res. Commun. 213:994.
38. Di Somma, M. M., S. Nuti, J. L. Telford, and C. T. Baldari. 1995. p56lck playsa key role in transducing apoptotic signals in T cells.FEBS Lett. 363:101.
39. Gonzalez-Garcia, A., L. R.-Borlado, E. Leonardo, I. Merida, C. Martinez-A., andA. C. Carrera. 1997. Lck is necessary and sufficient for Fas-ligand expression andapoptotic cell death in mature cycling T cells.J. Immunol. 158:4104.
40. June, C. H., M. C. Fletcher, J. A. Ledbetter, G. L. Schieven, J. N. Siegel,A. F. Phillips, and L. E. Samelson. 1990. Inhibition of tyrosine phosphorylationprevents T-cell receptor-mediated signal transduction.Proc. Natl. Acad. Sci. USA87:7722.
41. Migita, K., K. Eguchi, Y. Kawabe, A. Mizokami, T. Tsukada, and S. Nagataki.1994. Prevention of anti-CD3 monoclonal antibody-induced thymic apoptosis byprotein tyrosine kinase inhibitors.J. Immunol. 153:3457.
42. Oberg, H.-H., B. Lengl-Janben, M. J. Robertson, D. Kabelitz, and O. Janssen.1997. Differential role of tyrosine phosphorylation in the induction of apoptosisin T cell clones via CD95 or the TCR/CD3-complex.Cell Death Differ. 4:403.
43. Collins, T. L., M. Deckert, and A. Altman. 1997. Views on Vav.Immunol. Today18:221.
44. Bustelo, X. R., J. A. Ledbetter, and M. Barbacid. 1992. Product ofvavprotoon-cogene defines a new class of tyrosine protein kinase substrates.Nature 356:68.
45. Margolis, B., P. Hu, S. Katzav, W. Li, J. M. Oliver, A. Ullrich, A. Weiss, andJ. Schlessinger. 1992. Tyrosine phosphorylation ofvav protooncogene productcontaining SH2 domain and transcription factor motifs.Nature 356:71.
46. Jabado, N., A. Pallier, F. Le Deist, F. Bernard, A. Fischer, and C. Hivroz. 1997.CD4 ligands inhibit the formation of multifunctional transduction complexes in-volved in T cell activation.J. Immunol. 158:94.
47. Goldman, F., J. Crabtree, C. Hollenback, and G. A. Koretzky. 1997. Sequestra-tion of p56lck by gp120, a model for TCR desensitization.J. Immunol. 158:2017.
48. Onodera, H., D. G. Motto, G. A. Koretzky, and D. M. Rothstein. 1996. Differ-ential regulation of activation-induced tyrosine phosphorylation and recruitmentof SLP-76 to Vav by distinct isoforms of the CD45 protein-tyrosine phosphatase.J. Biol. Chem. 271:22225.
49. Musci, M. A., L. R. Hendricks-Taylor, D. G. Motto, M. Paskind, J. Kamens,C. W. Turck, and G. A. Koretzky. 1997. Molecular cloning of SLAP-130, anSLP-76-associated substrate of the T cell antigen receptor-stimulated protein ty-rosine kinases.J. Biol. Chem. 272:11674.
50. da Silva, A. J., Z. Li, C. De Vera, E. Canto, P. Findell, and C. E. Rudd. 1997.Cloning of a novel T-cell protein FYB that binds FYN and SH2-domain-con-taining leukocyte protein 76 and modulates interleukin 2 production.Proc. Natl.Acad. Sci. USA 94:7493.
51. Clements, J. L., S. E. Ross-Barta, L. T. Tygrett, T. J. Waldschmidt, andG. A. Koretzky. 1998. SLP-76 expression is restricted to hemopoietic cells ofmonocyte, granulocyte, and T lymphocyte lineage and is regulated during T cellmaturation and activation.J. Immunol. 161: 3880.
52. Feng, S., J. K. Cheng, H. Yu, J. A. Simon, and S. L. Schreiber. 1994. Two bindingorientations for peptides to the Src SH3 domain: development of a general modelfor SH3-ligand interactions.Science 266:1241.
53. Lim, W. A., F. M. Richards, and R. O. Fox. 1994. Structural determinants ofpeptide-binding orientation and of sequence specificity in SH3 domains. Nature372:375.
54. Morton, C. J., D. J. R. Pugh, E. L. J. Brown, J. D. Kahmann, D. A. C. Renzoni,and I. D. Campbell. 1996. Solution structure and peptide binding of the SH3domain from humanfyn. Structure 4:705.
55. Hiroaki, H., W. Klaus, and H. Senn. 1996. Determination of the solution structureof the SH3 domain of human p56lck tyrosine kinase. J. Biomol. NMR 8:105.
56. Ledbetter, J. A., C. H. June, P. S. Rabinovitch, A. Grossmann, T. T. Tsu, andJ. B. Imboden. 1988. Signal transduction through CD4 receptors: stimulatory vsinhibitory activity is regulated by CD4 proximity to the CD3/TCR receptor.Eur.J. Immunol. 18:525.
57. Farber, D. L., M. Luqman, O. Acuto, and K. Bottomly. 1995. Control of memoryCD4 T cell activation: MHC class II molecules on APCs and CD4 ligation inhibitmemory but not naive CD4 T cells.Immunity 2:249.
58. Nakayama, T., C. H. June, T. I. Munitz, M. Sheared, S. A. McCarthy,S. O. Sharrow, L. E. Samelson, and A. Singer. 1990. Inhibition of T cell receptorexpression and function in immature CD41CD81 cells by CD4.Science 249:1558.
3151The Journal of Immunology
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

59. Cruikshank, W. W., D. M. Center, N. Nisar, M. Wu, B. Natke, A. C. Theodore,and H. Kornfeld. 1994. Molecular and functional analysis of a lymphocyte che-moattractant factor: association of biologic function with CD4 expression.Proc.Natl. Acad. Sci. USA 91:5109.
60. Center, D. M., H. Kornfeld, and W. W. Cruikshank. 1996. Interleukin 16 and itsfunction as a CD4 ligand.Immunol. Today 17:476.
61. Cruikshank, W. W., K. Lim, A. C. Theodore, J. Cook, G. Fine, P. F. Weller, andD. M. Center. 1996. IL-16 inhibition of CD3-dependent lymphocyte activationand proliferation.J. Immunol. 157:5240.
62. Baier, M., A. Werner, N. Bannert, K. Metzner, and R. Kurth. 1995. HIV sup-pression by interleukin-16.Nature 378:563.
63. Zhou, P., S. Goldstein, K. Devadas, D. Tewari, and A. L. Notkins. 1997. HumanCD41 cells transfected with IL-16 cDNA are resistant to HIV-1 infection: inhi-bition of mRNA expression.Nat. Med. 3:659.
64. Idziorek, T., J. Khalife, O. Billaut-Mulot, E. Hermann, M. Aumencier,Y. Mouton, A. Capron, and G. M. Bahr. 1998. Recombinant human IL-16 inhibitsHIV-1 replication and protects against activation-induced cell death (AICD).Clin. Exp. Immunol. 112:84.
65. Anderson, P., M.-L. Blue, C. Morimoto, and S. F. Schlossman. 1987. Cross-linking of T3 (CD3) with T4 (CD4) enhances the proliferation of resting T lym-phocytes.J. Immunol. 139:678.
66. Oravecz, T., and M. A. Norcross. 1993. Costimulatory properties of the humanCD4 molecule: enhancement of CD3-induced T cell activation by human immu-nodeficiency virus type 1 through viral envelope glycoprotein gp120.AIDS Res.Hum. Retroviruses 9:945.
67. Newell, M. K., L. J. Haughn, C. R. Maroun, and M. H. Julius. 1990. Death ofmature T cells by separate ligation of CD4 and the T cell receptor for antigen.Nature 347:286.
68. Banda, N. K., J. Bernier, D. K. Kurahara, R. Kurrle, N. Haigwood, R.- P. Sekaly,and T. H. Finkel. 1992. Cross-linking CD4 by human immunodeficiency virusgp120 primes T cells for activation-induced apoptosis.J. Exp. Med. 176:1099.
69. Choy, E. H. S., J. Adjave, L. Forrest, G. H. Kingsley, and G. S. Panayi. 1993.Chimeric anti-CD4 monoclonal antibody cross-linked by monocyte Fcg receptormediates apoptosis of human CD4 lymphocytes.Eur. J. Immunol. 23:2676.
70. Algeciras, A., D. H. Dockrell, D. H. Lynch, and C. V. Paya.1998. CD4 regulatessusceptibility to Fas ligand- and tumor necrosis factor-mediated apoptosis.J. Exp.Med. 187:711.
71. Oyaizu, N., T. W. McCloskey, M. Coronesi, N. Chirmule, V. S. Kalyanaraman,and S. Pahwa. 1993. Accelerated apoptosis in peripheral blood mononuclear cells(PBMCs) from human immunodeficiency virus type-1 infected patients and inCD4 cross-linked PBMCs from normal individuals.Blood 82:3392.
72. Wang, Z.-Q., A. Dudhane, T. Orlikowsky, K. Clarke, X. Li, Z. Darzynkiewicz,and M. K. Hoffmann. 1994. CD4 engagement induces Fas antigen-dependentapoptosis of T cellsin vivo. Eur. J. Immunol. 24:1549.
73. Desbarats, J., J. H. Freed, P. A. Campbell, and M. K. Newell. 1996. Fas (CD95)expression and death-mediating function are induced by CD4 cross-linking onCD41 T cells.Proc. Natl. Acad. Sci. USA 93:11014.
74. Howie, S. E. M., A. J. Sommerfield, E. Gray, and D. J. Harrison. 1993. PeripheralT lymphocyte depletion by apoptosis after CD4 ligationin vivo: selective loss ofCD442 and ‘activating’ memory T cells.Clin. Exp. Immunol. 95:195.
75. Tian, H., R. Lempicki, L. King, E. Donoghue, L. E. Samelson, and D. I. Cohen.1996. HIV envelope-directed signaling aberrancies and cell death of CD41 Tcells in the absence of TCR co-stimulation.Int. Immunol. 8:65.
76. Corbeil, J., M. Tremblay, and D. D. Richman. 1996. HIV-induced apoptosisrequires the CD4 receptor cytoplasmic tail and is accelerated by interaction ofCD4 with p56lck. J. Exp. Med. 183:39.
77. Bank, I., and L. Chess. 1985. Perturbation of the T4 molecule transmits a negativesignal to T cells.J. Exp. Med. 162:1294.
78. Mittler, R. S., and M. K. Hoffmann. 1989. Synergism between HIV gp120 andgp120-specific antibody in blocking human T cell activation.Science 245:1380.
79. Dianzani, U., A. Shaw, M. Fernandez-Cabezuda, and C. A. Janeway Jr. 1992.Extensive CD4 cross-linking inhibits T cell activation by anti-receptor antibodybut not by antigen.Int. Immunol. 4:995.
80. Liegler, T. J., and D. P. Stites. 1994. HIV-1 gp120 and anti-gp120 induce reversibleunresponsiveness in peripheral CD4 T lymphocytes.J. Acquired Immune Defic.Syndr. 7:340.
81. Baldari, C. T., E. Milia, M. M. Di Somma, F. Baldoni, S. Valitutti, andJ. L. Telford. 1995. Distinct signaling properties identify functionally differentCD4 epitopes.Eur. J. Immunol. 25:1843.
3152 SLP-76/p56lck INTERACTION REGULATES TCR SIGNALING
by guest on April 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from