androgenreceptoristhekeytranscriptionalmediatorofthe tumor suppressor spop … · spop mutants...
TRANSCRIPT

Tumor and Stem Cell Biology
AndrogenReceptor Is theKeyTranscriptionalMediator of theTumor Suppressor SPOP in Prostate Cancer
Chuandong Geng1,2, Kimal Rajapakshe2, Shrijal S. Shah1,2, John Shou1,2, Vijay Kumar Eedunuri3,Christopher Foley1,2, Warren Fiskus1,2, Mahitha Rajendran1,2, Sue Anne Chew1,2, Martin Zimmermann1,2,Richard Bond1,2, Bin He1,2, Cristian Coarfa2, and Nicholas Mitsiades1,2,4
AbstractSomatic missense mutations in the substrate-binding pocket of the E3 ubiquitin ligase adaptor SPOP are
present in up to 15% of human prostate adenocarcinomas, but are rare in other malignancies, suggesting aprostate-specific mechanism of action. SPOP promotes ubiquitination and degradation of several proteinsubstrates, including the androgen receptor (AR) coactivator SRC-3. However, the relative contributions thatSPOP substrates may make to the pathophysiology of SPOP-mutant (mt) prostate adenocarcinomas areunknown. Using an unbiased bioinformatics approach, we determined that the gene expression profile ofprostate adenocarcinoma cells engineered to express mt-SPOP overlaps greatly with the gene signatureof both SRC-3 and AR transcriptional output, with a stronger similarity to AR than SRC-3. This findingsuggests that in addition to its SRC-3–mediated effects, SPOP also exerts SRC-3–independent effects thatare AR-mediated. Indeed, we found that wild-type (wt) but not prostate adenocarcinoma–associatedmutants of SPOP promoted AR ubiquitination and degradation, acting directly through a SPOP-bindingmotif in the hinge region of AR. In support of these results, tumor xenografts composed of prostateadenocarcinoma cells expressing mt-SPOP exhibited higher AR protein levels and grew faster than tumorscomposed of prostate adenocarcinoma cells expressing wt-SPOP. Furthermore, genetic ablation ofSPOP was sufficient to increase AR protein levels in mouse prostate. Examination of public human prostateadenocarcinoma datasets confirmed a strong link between transcriptomic profiles of mt-SPOP andAR. Overall, our studies highlight the AR axis as the key transcriptional output of SPOP in prostateadenocarcinoma and provide an explanation for the prostate-specific tumor suppressor role of wt-SPOP.Cancer Res; 74(19); 5631–43. �2014 AACR.
IntroductionThe androgen receptor (AR) is a critical driver of prostate
adenocarcinoma pathophysiology, regulating proliferation,metabolism and migration, and is also a validated therapeutictarget (1). The importance of the AR axis in prostate adenocar-cinoma is further illustrated by the frequent overexpres-
sion, especially in the state of castration-resistant prostatecancer (CRPC), of steroidogenic enzymes that lead topersistenceof intratumoral androgens (2–7), aswell asAR itself (2, 8–12), andits coactivators (8, 10, 13, 14). Additional mechanisms of non-canonical ARactivation, includingARmutations (15–19), ligand-independent AR splice variants (20–24) and cytokine-inducedligand-independent activation (25), as well as epigenetic dysre-gulation ofmiRNAs that control ARhomeostasis (26), contributeto CRPC progression and further highlight the critical impor-tance of the AR axis in prostate adenocarcinoma. There iscompelling evidence that even taxanes, the only family ofcytotoxic chemotherapeutics that has ever demonstrated anoverall survival benefit in prostate adenocarcinoma, exert anti-cancer activity in CRPC by inhibiting the AR axis (27–29).
Whole-exome sequencing studies recently discovered thatthe E3 ubiquitin ligase adaptor speckle-type POZ protein(SPOP) is frequently affected by somatic nonsynonymous pointmutations in prostate adenocarcinoma (30–32). SPOP harborsan N-terminal MATH (Meprin and Traf Homology) domainthat recruits substrate proteins, and a C-terminal BTB (Bric-a-brac/Tramtrack/Broad complex) domain that interacts withCullin 3 (Cul3) and Rbx1 to promote substrate ubiquitination.SPOP, via its MATH domain, binds to and promotes the
1Department of Medicine, Baylor College of Medicine, Houston, Texas.2Department ofMolecular andCellular Biology, Baylor College ofMedicine,Houston, Texas. 3Adrienne Helis Malvin Medical Research Foundation,New Orleans, Louisiana. 4Center for Drug Discovery, Baylor College ofMedicine, Houston, Texas.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corrected online August 9, 2019.
C. Geng, K. Rajapakshe, and S.S. Shah contributed equally to this article.
CorrespondingAuthors:NicholasMitsiades, Department ofMedicine andMolecular and Cellular Biology, Baylor College of Medicine, One BaylorPlaza, Suite R407, MS: BCM187, Houston, TX 77030. Phone: 713-798-2205; Fax: 713-798-6677; E-mail: [email protected]; andCristian Coarfa,One Baylor Plaza, Suite 450A-10, MS: BCM130, Houston, TX 77030.Phone: 713-798-7938; Fax: 713-790-1275; E-mail: [email protected].
doi: 10.1158/0008-5472.CAN-14-0476
�2014 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 5631
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

ubiquitination of several substrates, including the deathdomain–associated protein Daxx (33), the phosphatase Puc,the transcriptional regulator Ci/Gli (34), the variant histoneMacroH2A (35), and the key AR coactivator Steroid ReceptorCoactivator (SRC)-3 (36, 37). All SPOP mutations reported inprostate adenocarcinoma affect conserved residues in thesubstrate-binding pocket (30–32), suggesting that they mod-ify substrate specificity and can drive the accumulation ofseveral proteins with roles in prostate adenocarcinomapathophysiology. We previously reported that wt-SPOP playsa critical tumor suppressor role in prostate adenocarcinomacells and promotes the turnover of SRC-3 protein, thussuppressing its capacity to function as an AR coactivator.This tumor suppressor effect is abrogated by the prostateadenocarcinoma–associated SPOP mutations (36). SRC-3promotes prostate adenocarcinoma cell proliferation andsurvival (38), cell migration and invasiveness (39), anddevelopment of CRPC (40). However, the relative contribu-tion of the various reported SPOP substrates to the patho-physiology of SPOP-mutant (mt) prostate adenocarcinomahas not been fully elucidated.
In the present study, we examined the global gene expres-sion profiles of AR(þ) prostate adenocarcinoma cells engi-neered to express wild-type (wt)-SPOP or three different mt-SPOPs. The gene signatures of all mt-SPOPs exhibited highoverlap with each other and with the gene signature ofandrogen-treated prostate adenocarcinoma cells. Gene SetEnrichment Analysis (GSEA) revealed that while the tran-scriptomic footprint of mt-SPOP enriches for signatures ofboth SRC-3 and AR transcriptional output, it matches moreclosely to AR than to SRC-3. This suggested that, in additionto SRC-3–mediated effects, SPOP also exerts important SRC-3–independent/AR-mediated functions. Our biochemicalstudies revealed that wt-SPOP can bind AR directly andpromote its ubiquitination and degradation. This activity isabrogated by the prostate adenocarcinoma–associatedSPOP mutations, leading to AR stabilization and increasedcell proliferation. Xenografts of prostate adenocarcinomacells expressing mt-SPOP expressed more AR protein andgrew faster in immunocompromised mice than thoseexpressing wt-SPOP. Genetic ablation of Spop resulted inincreased AR protein levels in the mouse prostate. In addi-tion, examination of several publicly available human pros-tate adenocarcinoma datasets confirmed the link betweenthe transcriptomic outputs of mt-SPOPs and AR. Our studiesidentify the AR axis as the key transcriptional output of thetumor suppressor SPOP in prostate adenocarcinoma andprovide an explanation why mutations in the substrate-binding pocket of SPOP occur frequently and in a prostateadenocarcinoma–specific manner.
Materials and MethodsReagents and antibodies
Enzalutamide (MDV3100) was kindly provided from Med-ivation. The antibodies used were: mouse monoclonal anti-FlagM2 (Sigma), anti-SPOP (Abcam), rabbit polyclonal anti-AR(Cell Signaling Technology), rabbit anti-AR (Santa CruzBiotechnology), anti-b-actin (Sigma), mouse anti-Flag-HRP
(Sigma), rat anti-HA-HRP (Roche), anti-rabbit IgG-HRP, andanti-rat IgG-HRP (Sigma), respectively.
Cell cultureHuman cells lines were obtained from the ATCC via the
Tissue and Cell Culture Core Laboratory at Baylor College ofMedicine (Houston, TX), where they are regularly submittedfor cell line authentication (by STR profiling) and mycoplasmatesting, and passaged for fewer than 6months: Human Embry-onic Kidney 293T cells, cervical carcinoma HeLa cells, and PCDU145 cells were cultured in DMEM high glucose (Life Tech-nologies) with 10% FBS (Life Technologies) in a 5% CO2
incubator at 37�C; LNCaP and 22Rv1 cells were cultured inRPMI1640 (Life Technologies) supplemented with 10% FBS;LAPC4 cells were cultured in Iscove Modified Dulbecco'sMedium (Life Technologies) plus 15% FBS, 1 nmol/L R1881,and 2 mmol/L of L-glutamine; PC3 cells were cultured inDMEM/F12 (F-12 Nutrient Medium, Life Technologies) with10% FBS; and VCaP cells were maintained in DMEM highglucose (Life Technologies) with 10% FBS and 1 nmol/L R1881.The human CRPC cell line Abl [characterized by and obtainedfrom Dr. Zoran Culig, Innsbruck Medical University, Tyrol,Austria (19)], was maintained in RPMI1640 medium (LifeTechnologies) supplemented with 10% charcoal-stripped FBS.SRC-3 knockout (KO) cells (SRC-3�/�), generated using a zincfinger nuclease (ZFN) to knockout both SRC-3 alleles anddocumented to lack SRC-3 (41), were a generous gift from Dr.Bert W. O'Malley, Baylor College of Medicine (Houston, TX).Previously described (36) doxycycline-inducible Abl stabletransfectants (Abl-control vector, Abl-SPOPwt, Abl-SPOP-Y87N, Abl-SPOP-F102C, Abl-SPOP-S119N, Abl-SPOP-W131G,Abl-SPOP-F133L, and Abl-SPOP-F133V) were maintained inRPMI1640 supplemented with 10% tetracycline-tested FBS(Atlanta Biotech. Inc.) and 300 mg/mL G-418 (Life Technolo-gies). Additional prostate adenocarcinoma cell lines expres-sing, under doxycycline-inducible promoter, wt or mutantSPOP, were established from LNCaP and VCaP cells, as pre-viously (36).
Additional methods for bioinformatics analysis, in vitro andin vivo studies are presented in the Supplementary Methods.
ResultsThe F102C, F133V, and F133L SPOP mutants result insimilar transcriptomic responses in prostateadenocarcinoma cells
We performed gene expression profiling of Abl prostateadenocarcinoma cells engineered to express the prostateadenocarcinoma–associated SPOP mutants F102C, F133V,and F133L, or SPOP WT (or control vector). Gene expres-sion signatures were derived for each individual mutantagainst SPOP WT and for all three mutants combinedagainst SPOP WT (P < 0.05, fold change exceeding 4/3), aspresented in Fig. 1A. By comparing the three signatureswith respect to each other, we determined that the threeSPOP mutants tested produce highly similar transcriptomicresponses (Fig. 1A and Supplementary Fig. S1), which arevery distinct (essentially inverted) from the effects of thewt-SPOP (Fig. 1A).
Geng et al.
Cancer Res; 74(19) October 1, 2014 Cancer Research5632
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

111
pro
bes
(103
gen
es)
132
pro
bes
(117
gen
es)
q < 0.001
q < 0.001
q < 0.001
q < 0.001
q < 0.001
q < 0.001
q < 0.001
q < 0.001
q = 0.018
q = 0.014
q = 0.008
q = 0.019 q < 0.001
q < 0.001
q < 0.001
q < 0.001
NES = 1.38 NES = 2.11 NES = 2.07NES = 2.46
NES = 1.39 NES = 2.11 NES = 2.55NES = 2.69
NES = 2.42NES = 1.88NES = 1.47 NES = 2.84
NES = 1.35 NES = 2.29NES = 2.05 NES = 2.60
B-2.0 0.0 2.0A
DsiSRC3Down
siARDown
Androgenup
(Hieronymus et al.)
Androgenup
(Nelson et al.)
Normalized enrichment score
F102C vs. SPOPwt
F133V vs. SPOPwt
F133L vs. SPOPwt
Combinedmutationsvs. SPOPwt
En
rich
men
tsc
ore
En
rich
men
tsc
ore
En
rich
men
tsc
ore
En
rich
men
tsc
ore
C
AR
act
ivit
y sc
ore
(co
mp
ared
wit
h c
on
tro
l vec
tor)
F102C
F133L
F133V
Combined
0.0 0.5 1.0 1.5 2.0 2.5
NelsonResponse toAndrogen up
Wang responseto both androgenand forskolin up
Wangresponse to
androgen up
Yegnasubramanianprostate
cancer
Qihypoxia
201510
50
-5-10-15-20
WT
WT
Control
Control
vect
or
vect
or
F133L
F133L
F133V
F133V
F102C
F102C
Figure 1. Global gene expression profiling identifies the AR transcriptional output as the top enriched gene set in prostate adenocarcinoma cells expressingmutant SPOPs. A, hierarchical clustering of gene expression profiles of Abl prostate adenocarcinoma cells transfected with control vector, wt-SPOP, or theSPOP mutants F102C, F133V, F133L (genes differentially expressed, t test P < 0.05, fold change exceeding 4/3 times) demonstrates that all three SPOPmutants have highly similar effects on the prostate adenocarcinoma transcriptome, which are very distinct (essentially inverted) from the effects of the wt-SPOP. B, the topMolecular Signature Database (MSigDB) match for the mt-SPOP gene signature, out of 10,295 available gene sets analyzed in an unbiasedfashion, is the "NELSON_RESPOSE_TO_ANDROGEN_UP." Also, three of the top five chemical and genetic perturbation (CGP) gene sets in prostateadenocarcinoma correspond to androgen-induced transcriptomic responses. C, the AR activity score was calculated on the basis of a previously publishedgene signature of androgen-stimulated LNCaP cells (42), for Abl cells expressing control vector, wt-SPOP, or mt-SPOP. Compared with control vector, cellsexpressing wt-SPOP exhibit a lower AR activity score, whereas mt-SPOPs increase the AR activity score. D, GSEA revealed that SRC-3–upregulated genes(genes downregulated by SRC-3 siRNA) are significantly enriched in the mt-SPOP gene signatures. The values on the y-axis for each graph representenrichment scores (corresponding to themagnitude of the enrichments for each analysis). For each graph, the normalized enrichment score (NES, computedvia the GSEA analysis) and the significance of the enrichment (q¼ false discovery rate also computed via the GSEA analysis) are indicated. The NES scoresrange from 1.35 to 1.47 (all q < 0.02). However, we found that the mt-SPOP gene signatures show stronger enrichment, with NES ranging from 1.88 to 2.71(q < 0.001), for androgen-induced genes (genes downregulated by AR siRNA or induced by androgen treatment of prostate adenocarcinoma cells).
AR Axis as the Mediator of SPOP in Prostate Cancer
www.aacrjournals.org Cancer Res; 74(19) October 1, 2014 5633
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

GSEAusing theMolecular Signatures Database identifiesthe AR transcriptional output as the top enriched geneset in prostate adenocarcinoma cells expressing mutantSPOPs
We evaluated the transcriptomic responses induced by mt-SPOPs (for each mutant SPOP, genes were ranked by the foldchange between the mutant and the WT SPOP samples) in anunbiased comparisonwith the entireMolecular Signatures Data-base (MSigDB, containing 10,295 gene signatures), using adjustedq < 0.05 as our filtering criteria. The overall best match was"NELSON_RESPOSE_TO_ANDROGEN_UP," which correspondsto genes upregulated by androgen in prostate adenocarcinomacells (42). We further focused on the Chemical and GeneticPerturbations gene set collection (over 3,400 signatures), andfiltered the results for experiments in prostate adenocarcinomacells. Three of the top five gene sets correspond to androgen-induced signatures (Fig. 1B).Utilizing theNelson signature (42) tocalculate an AR activity score for each transfected prostateadenocarcinoma sample, we confirmed that, compared withcontrol vector, cells expressing wt-SPOP exhibit a lower ARactivity score, whereas mt-SPOPs (F102C, F133V, and F133L)increase the AR activity score (Fig. 1C). Similar results wereobtained using another androgen-induced signature as well (43)(Supplementary Fig. S2). We also expanded our analysis using awider panel of SPOPmutants (including also Y87C, Y87N, S119N,F125V, and W131G) and demonstrated by quantitative reverse-transcriptase polymerase chain reaction (qt-PCR) that they, too,have partially or completely lost the capacity of wt-SPOP tosuppress the expression of the AR-dependent genes SGK1,CAMKK2, ABCC4, HOMER2, SEPP1, and NKX3.1 (SupplementaryFig. S3). In combination with our prior report that wt-SPOP, butnot these 8 prostate cancer–associated mutants, suppresses theexpression of the AR-dependent genes KLK3 (PSA) and FKBP5(36), we have confirmed that all 8 SPOPmutants result in similargene expression profiles, specifically lacking the capacity of wt-SPOP to suppress AR transcriptional activity.
The gene signatures of mt-SPOPs enrich for genesregulated by direct AR antagonists
We next investigated, using the GSEA method, the enrich-ment of SPOP-regulated gene signatures for genes regulated bya collection of drugs (FDA-approved or experimental) used orproposed, respectively, for prostate adenocarcinoma treat-ment. The genes upregulated by mt-SPOPs (over wt-SPOPsamples) were highly enriched for genes downregulated bydirect AR antagonists, such as enzalutamide, bicalutamide,ARN-509, and compound 30. Inversely, the genes downregu-lated by mt-SPOPs were highly enriched for genes upregulatedby the AR antagonists (Supplementary Fig. S4). Other drugs,including several that have been proposed to have indirecteffects on AR (such asHDAC inhibitors, cardiac glycosides, anddocetaxel), enriched less robustly.
The gene signatures of mt-SPOPs enrich for genesinduced by SRC-3 and androgen
We investigated the enrichment of SRC-3–regulated genes inour mt-SPOP signatures using the GSEA method. SRC-3–dependent genes (genes downregulated by SRC-3 siRNA) are
significantly enriched in the mt-SPOP gene signatures, withnormalized enrichment scores (NES) ranging from 1.35 to 1.47(all q < 0.02, Fig. 1D). This confirms our previous report that theprostate adenocarcinoma–associated SPOPmutants posttran-slationally stabilize SRC-3 (36). We also evaluated the enrich-ment of androgen-induced genes, utilizing our own geneexpression signature from prostate adenocarcinoma cellstreated with AR siRNA (genes downregulated by AR siRNA)and previously published gene signatures from androgen-treated prostate adenocarcinoma cells (42–43). We discoveredthat androgen/AR-induced genes are highly enriched in theSPOP-mutant signatures, with NES ranging from 1.88 to 2.71(q < 0.001). The higher NES observed for the AR-dependentgenes, compared with the SRC-3–dependent genes, indicatethat SPOP mutants promote AR-mediated signaling thatextends beyond what can be explained purely by their impacton SRC-3 stabilization (Fig. 1D). This led us to examine inmoredetail the impact of SPOP on regulation of AR itself.
SPOP directly regulates AR protein stability and thisactivity is abrogated by the prostate adenocarcinoma–associated SPOP mutations
We transiently coexpressed in 293T cells AR and SPOP (WTor the prostate adenocarcinoma–associated SPOP mutants:SPOP-Y87N, SPOP-Y87C, SPOP-F102C, SPOP-S119N, SPOP-F125V, SPOP-W131G, SPOP-F133L, and SPOP-F133V). Immu-noblot analyses revealed that the expression of ARprotein itselfwas strongly suppressed in the presence of SPOP-wt, butnot by the prostate adenocarcinoma–associated SPOPmutants (Fig. 2A). We next determined whether wt-SPOP caninteractwithARprotein. Coimmunoprecipitation experimentsrevealed that SPOP-wt, but not the prostate adenocarcinoma–associated SPOP mutants, can interact with the AR protein(Fig. 2B). This suggested that AR is a substrate for SPOP-wt, andthat the mutations in the substrate-binding pocket of theMATH domain of SPOP abolish its interaction with AR. Inagreement, while the C-terminal fragment of wt-SPOP (a.a.172–374, containing the BTB domain) failed to bind to the ARprotein, the N-terminal fragment (a.a. 1–172) of wt-SPOP,containing the MATH domain and its substrate-binding pock-et, efficiently coimmunoprecipitated with AR protein in vitro(Fig. 2C). These observations indicate that SPOP-wt can bind toAR and that the AR–SPOP interaction is critically dependenton the SPOP substrate-binding cleft of the MATH domain. Tofurther dissect the impact of the tumor suppressor SPOP onARexpression in prostate adenocarcinoma cells, we examined ARprotein expression in Abl prostate adenocarcinoma cells engi-neered to express, under a tetracycline-inducible promoter,SPOP-wt or the prostate adenocarcinoma–associated SPOPmutants. Immunoblot analyses revealed that, upon inductionwith doxycycline, SPOP-wt, but not the prostate adenocarci-noma–associated SPOP mutants, significantly suppressedAR protein expression in Abl prostate adenocarcinoma cells(Fig. 3A). Of note, a subset of mutants (including F102C)increased AR protein expression above baseline (i.e., no exog-enous SPOP) levels, suggesting a possible gain-of-function"dominant-negative effect" of these SPOP mutants on thefunction of endogenous (wt) SPOP. Real-time qRT-PCR
Geng et al.
Cancer Res; 74(19) October 1, 2014 Cancer Research5634
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

revealed that SPOP-wt did not suppress the expression of ARmRNA in these cells, suggesting that the impact of SPOP on ARprotein levels is most likely posttranslational (SupplementaryFig. S5). Similar results were obtained from ligand-dependentLNCaP and VCaP cells (Supplementary Fig. S6).
To further explore the impact of SPOP on AR protein sta-bility, we used cycloheximide treatment and immunoblot ana-lyses to quantify the half-life of the AR protein in Abl cellsinduced to express SPOP-wt or the prostate adenocarcinoma–associated SPOP mutants (SPOP-Y87N, SPOP-Y87C, SPOP-F102C, SPOP-S119N, SPOP-F125V, SPOP-W131G, SPOP-F133L,and SPOP-F133V), respectively. In Abl cells transfected withthe control vector, the half-life of AR is approximately 500minutes (Fig. 3B). Expression of SPOP-wt destabilized anddramatically shortened the half-life of AR protein to approxi-mately 200 minutes. On the contrary, in Abl cells expressingmt-SPOPs, the half-life of AR protein is significantly extended, inparticular in the case of SPOP-F102C (Fig. 3B). We also con-firmed the interaction of AR protein and wt-SPOP in Abl cellsusing coimmunoprecipitation. In the immune complex pre-cipitated by the anti-AR antibody, bothARprotein andwt-SPOPwere detected (Fig. 3C). Similarly, AR protein was detected inthe immune complex precipitated by the SPOP-specific anti-body in Abl cells transfected with wt-SPOP (Fig. 3C). Impor-tantly, in Abl cells transfected with SPOP-F102C, SPOP did notcoimmunoprecipitate with AR in either condition, i.e., immu-noprecipitation with anti-AR or with anti-SPOP (Fig. 3C). Takentogether, these data provide direct evidence for the associationbetween AR protein and the E3 ubiquitin ligase adaptor SPOP(wt) in prostate adenocarcinoma cells.
SPOP promotes ubiquitination of AR protein and thisactivity is abrogated by the prostate adenocarcinoma–associated SPOP mutations
SPOP functions as an adaptor protein that facilitates therecruitment of substrates to the Cullin-3/RBX-1 E3 ligasecomplex to promote ubiquitination and degradation of itssubstrate proteins (36). To further dissect the functional roleof binding of SPOP on ubiquitination and degradation of ARprotein, we coexpressed AR (Flag-tagged) and HA-taggedubiquitin (Ub) together with SPOP-wt or prostate adenocar-cinoma–associated SPOPmutants (SPOP-F102C, SPOP-F133V,SPOP-F125V, SPOP-S119N, SPOP-Y87C, and SPOP-Y87N) orSPOP-C terminal fragment (lacking the MATH domain) in293T cells, and examined the levels of ubiquitin-conjugatedAR protein. As shown in Fig. 4A, the levels of ubiquinated ARprotein (immunoprecipitated by anti-Flag antibody) weresignificantly increased when SPOP-wt was also expressed,whereas expression of any prostate adenocarcinoma–associ-ated SPOP mutant effectively inhibited the accumulation ofUb-AR. Furthermore, the SPOP C-terminal fragment (a.a. 172–374) had no effect on the ubiquitination state of AR protein(Fig. 4A). These data are consistent with the fact that thisfragment lacks the MATH domain and did not bind to the ARprotein (Fig. 2C). We also coexpressed a Cullin-3 dominantnegative (DN) construct (CUL-DN, truncated Cullin-3 contain-ing the SPOP binding domain but defective in RBX-1 recruit-ment) together with SPOP-wt and AR in 293T cells andobserved that CUL-DN efficiently rescued the depletion of ARprotein levels caused by SPOP-wt (Fig. 4B). Collectively, theseobservations suggest that SPOP serves as an adaptor proteinfor the Cullin-3 E3 ubiquitin ligase complex to promote ubi-quitination of AR protein.
C
Vec
tor
Co
ntr
ol
SP
OP
-wt
SP
OP
-F10
2CS
PO
P-F
133V
SP
OP
-F13
3LS
PO
P-W
131G
SP
OP
-F12
5VS
PO
P-S
119N
SP
OP
-Y87
CS
PO
P-Y
87N
Input (IB: Anti-Flag (AR))
IP: Anti-Flag (AR)IB: Anti-HA (SPOP)
BV
ecto
r C
trl
Vector Flag-AR: - + + + + + + + + + +
Vector SPOP: -
AAR + + + + + + + +
S119N F125V W131G F133L+ -+ -+ -+ -SPOP +-+ + -+ -+ -
+ + + + + + + + + +
Y87C Y87NWT F133VF102C
AR
SPOPs
Actin
-
+ +Vector Flag-AR: -
Vector SPOP:
Input (IB: Anti-Flag (AR))
IP: Anti-Flag (AR)IB: Anti-HA (SPOP)
SP
OP
(aa1
-aa1
72)
SP
OP
(aa1
72-a
a374
)
Figure 2. Wt-SPOP, but not its prostate adenocarcinoma–associatedmutants, binds to and promotes degradation of AR protein. A, 293T cellswere cotransfected with pcDNA3-AR-FLAG and pcDNA3.1-HA-SPOP-wt or pcDNA3.1-HA-SPOP-mutant expression vectors. Forty-eight hoursafter transfection, cells were collected and the cell lysates were preparedand analyzed by immunoblotting for the expression of Flag-tagged AR(anti-Flag-HRP), HA-tagged SPOP (anti-HA-HRP), and b-actin. Theexpression of AR protein was strongly suppressed in the presence ofSPOP-wt, but not by its prostate adenocarcinoma–associated mutants.B, 293T cells were cotransfected with pcDNA3-AR-Flag and pcDNA3.1 (vector control) or pcDNA3.1-HA-SPOPwt, pcDNA3.1-HA-SPOP-F102C, pcDNA3.1-HA-SPOP-F133V, pcDNA3.1-HA-SPOP-F133L,pcDNA3.1-HA-SPOP-W131G, pcDNA3.1- HA-SPOP-F125V,pcDNA3.1-HA-SPOP-S119N, pcDNA3.1-HA-SPOP-Y87C, orpcDNA3.1-HA-SPOP-Y87N. The transfected cells were treated with250 nmol/L of the proteasome inhibitor bortezomib (PS341) for 8 morehours and the lysates were used for coimmunoprecipitation/immunoblotanalysis. Immunoblot analysis revealed that SPOP-wt, but not thePC-associatedSPOPmutants, can interactwith ARprotein. C, 293Tcellswere cotransfected with pcDNA3-AR-Flag and pcDNA3.1 (vectorcontrol), pcDNA3.1-HA-SPOP N-terminal (a.a. 1–172) residues, orpcDNA3.1-HA-SPOP C-terminal (a.a. 172–374) residues.Immunoprecipitation with anti-Flag antibody and immunoblottinganalysis were conducted as described in B and revealed that, while theC-terminal fragment of wt-SPOP (a.a.172-a.a.374, containing the BTBdomain) failed to bind AR protein, the N-terminal fragment (a.a. 1–172) ofwt-SPOP, containing the MATH domain and its substrate-bindingpocket, efficiently coimmunoprecipitated with AR protein in vitro.
AR Axis as the Mediator of SPOP in Prostate Cancer
www.aacrjournals.org Cancer Res; 74(19) October 1, 2014 5635
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

SPOPdirectly binds the ARprotein through an SBCmotifin the hinge region of AR
Because wt-SPOP (but not its prostate adenocarcinoma–associated mutants) can bind the AR coactivator SRC-3 (36),we determined whether SRC-3 mediates or facilitates theinteraction of wt-SPOP with AR. We coexpressed AR andSPOP-wt in 293T cells and HeLa cells (both express SRC-3)
and in a HeLa SRC-3 KO subclone, that lacks SRC-3 expression(41). We found that SPOP-wt could effectively suppress ARprotein levels and coimmunoprecipitate with AR in all threecell lines (Supplementary Fig. S7), suggesting that the inter-action between SPOP-wt and AR can occur even in the absenceof SRC-3 protein. This led us to examine whether SPOP-wt canbindARdirectly. SPOP recognizes and binds to its substrates at
WT Y87N Y87C F102C S119N F125V W131G F133L F133V
Dox − + + − + + − + + + −+ + + + − + − + ++ + −+ + −−
Abl-SPOPWT Abl-SPOPF102C
Inp
ut
IgG
An
ti-S
PO
P
Inp
ut
IgG
An
ti-S
PO
P
Anti-HA(SPOP)
Anti-AR
Inp
ut
IgG
An
ti-A
R
Inp
ut
IgG
An
ti-A
R
Abl-SPOPWT Abl-SPOPF102C
C
AR
pro
tein
leve
ls(%
of
corr
esp
on
din
g c
on
tro
l)
Mins after addition of cycloheximide
B
A
AR
SPOP
b-Actin
1.0 0.7 0.1 1.0 1.1 1.1 1.0 1.1 1.3 1.0 1.7 1.6 1.0 1.9 1.8 1.0 1.1 0.8 1.0 1.1 0.8 1.0 0.9 0.6 1.0 0.9 0.9
250
200
150
100
50
00 200 400 600
Vector control
SPOP WT
SPOP Y87C
SPOP Y87N
SPOP F102C
SPOP S119N
SPOP F125V
SPOP W131G
SPOP F133L
SPOP F133V
Figure 3. Ectopic expression of wt-SPOP posttranslationally regulates AR in PC cells. A, Abl prostate adenocarcinoma cells engineered to express,under a tetracycline-inducible promoter, SPOP-wt, or its prostate adenocarcinoma–associated mutants, were treated with 0, 50, or 500 ng/mL ofdoxycycline (Dox) for 72 hours (36). Following this, cells were collected and cell lysates were prepared. Immunoblot analyses were conducted for theexpression of AR, SPOP, and b-actin in the cell lysates. SPOP-wt, but not its prostate adenocarcinoma–associated mutants, significantly suppressedAR protein expression in Abl prostate adenocarcinoma cells. The numbers beneath the bands represent densitometry analysis performed onrepresentative blots from each cell line. B, Abl prostate adenocarcinoma cells engineered to express, under a tetracycline-inducible promoter, SPOP-wt, or the prostate adenocarcinoma–associated SPOP mutants, were treated with 200 ng/mL of doxycycline (Dox) for 24 hours. After Dox induction,cells were treated with 100 mg/mL cycloheximide for 12 more hours. Cells were collected at 0, 200, 500, and 700 minutes following cycloheximidetreatment and cell lysates were prepared. Immunoblot analysis was conducted for the expression levels of AR and b-actin in the cell lysates. Theimmunoblot signals of AR proteins were quantified and plotted as described in Materials and Methods. AR expression is plotted as percentage of therespective (time zero) controls. C, Abl-SPOPwt and Abl-SPOP-F102C cells were induced with Dox for 24 hours and treated with 250 nmol/L ofbortezomib (PS-341) for another 8 hours. At the end of treatment, cell lysates were prepared and utilized for immunoprecipitation with anti-HA antibodyor anti-AR antibody. Immunoblot analyses were conducted for the expression levels of HA-SPOP or AR protein in the immunoprecipitates and detectedassociation between AR protein and SPOP-wt (but not SPOP-F102C) in prostate adenocarcinoma cells.
Geng et al.
Cancer Res; 74(19) October 1, 2014 Cancer Research5636
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

a specific serine/threonine-rich peptide motif (SPOP BindingConsensus motif, SBC; ref. 35). Our bioinformatics analysisof AR protein sequence identified an SBCmotif within its hingeregion, 646-ASSTT-650 (Supplementary Fig. S8A), which islocated in the PEST sequence previously identified (44). Muta-tions in this motif segment have been frequently reportedin patients and are associated with various conditions, includ-ing partial androgen insensitivity syndrome (PAIS, A646D,and S648N), complete androgen insensitivity syndrome (CAIS,A646D), mild androgen insensitivity syndrome (MAIS, A646D),and prostate adenocarcinoma (S647F, S648N and T650A) (45).
We confirmed that point mutations in this AR SBC motif,specifically A646D, S647F, S648N, and STT648/649/650AAA,can abolish the affinity of AR for SPOP-wt upon coexpression in293T cells (Supplementary Fig. S8B). Moreover, ARv7, a nat-urally occurring C-terminal–truncated AR variant that lacksthe SBC motif, did not bind SPOP-wt upon coexpression in293T cells (Supplementary Fig. S8C). These data stronglysupport the functional involvement of this AR SBC motif tothe recognition and binding of AR protein by SPOP-wt.
Endogenous SPOP regulatesARprotein levels inprostateadenocarcinoma cells
SPOP is ubiquitously expressed in prostate adenocarcinomacell lines (36).Wenext examined the interaction of endogenousSPOP (wt) with AR protein and the role of SPOP in regulatingAR expression in a panel of prostate adenocarcinoma cells. Wefound that in LNCaP, Abl, VCaP, and LAPC4 prostate adeno-carcinoma cells, AR protein coimmunoprecipitated withendogenous SPOP (wt) by anti-SPOP–specific antibody (Sup-plementary Fig. S9A). As expected, no signal was found in AR-negative PC-3 and DU145 control prostate adenocarcinomacells. Moreover, by silencing endogenous SPOP by siRNAtransfection (three different SPOP siRNAs) in LNCaP and Ablcells, we induced significant increase of AR protein levels inthese cells (Supplementary Fig. S9B). The AR mRNA levels didnot change to a degree that could explain the increase in ARprotein (Supplementary Fig. S9C and S9D), suggesting that theincrease in AR protein expression is mainly posttranslational.These observations confirm that endogenous SPOP functionsas an adaptor for the E3 ligase Cullin-3/RBX-1 complex topromote AR protein ubiquitination and eventually, degrada-tion. We also examined the impact of silencing SPOP on ARprotein levels in 22Rv1 cells, that endogenously express bothfull-length (FL) AR and its splice variant ARv7. All three SPOPsiRNAs increased both the FL AR protein form (as expected) aswell as, unexpectedly, AR-v7 (Fig. 5A). Neither AR FL nor ARv7mRNA was increased by silencing SPOP (SupplementaryFig. S10), again supporting a posttranslational mechanism ofaction. Moreover, exogenous expression of SPOP-wt in 22Rv1cells suppressed the protein expression of both FL AR and AR-v7 (Fig. 5B). As these results apparently contradicted our priorfindings from transient expression of ARv7 in 293T cells(Supplementary Fig. S8C), we hypothesized that, in 22Rv1 cells,an indirect mechanism may allow the interaction of SPOP-wtwith ARv7, despite the absence of the SBC that we character-ized above (Supplementary Fig. S8). Specifically, we hypothe-sized that ARv7 may be able to interact with SPOP-wt viaheterodimerization with AR-FL. To explore this hypothesis, weexamined the in vitro recruitment of ARv7 to SPOP-wt with orwithout concurrent presence of AR-FL in 293T cells. In 293Tcells (that lack endogenous AR), when ARv7 is expressed in theabsence of AR-FL, its expression in unaffected by SPOP-wt(Fig. 5C) and it cannot interact with SPOP-wt (Fig. 5D). How-ever, ARv7 protein could interact with SPOP-wt when coex-pressed with AR-FL (Fig. 5D). This observation helps explainour findings in 22Rv1 cells and provides an alternative, indirectmechanism for SPOP-wt to regulate ubiquitination and pro-teasomal degradation of ARv7 in prostate adenocarcinoma
A
Vec
tor
Co
ntr
ol
SP
OP
-WT
SP
OP
-F10
2CS
PO
P-F
133V
SP
OP
-F12
5VS
PO
P-S
119N
SP
OP
-Y87
CS
PO
P-Y
87N
SP
OP
(aa1
72-a
a374
)
250 KDa
150 KDa
100 KDa
75 KDa
(Ubi)n-AR
293T cells
Input AR
AR + + ++ +-SPOP-wt
+--CUL3-DNAR
CUL3-DN
Actin
293T cells
B
Vector AR: - + + + + + + + + +Vector SPOP: -
Figure 4. Wt-SPOP promotes ubiquitination of AR protein in vitro and thisactivity is abrogated by the prostate adenocarcinoma–associated SPOPmutations. A, 293T cells were cotransfected with pcDNA3-HA-humanubiquitin (1 mg) and pcDNA3-AR-Flag (1 mg), together with same amount(1 mg) of pcDNA3.1 expression vectors for Wt-SPOP or its prostateadenocarcinoma–associated mutated variants (SPOP-F102C, SPOP-F133V, SPOP-F125V, SPOP-S119N, SPOP-Y87C, SPOP-Y87N) orSPOP-C terminal fragment (a.a. 172–374, lacking the MATH domain),respectively. Anti-Flag antibody was used to immunoprecipitate ARprotein and anti-HA-HRP antibody was used to visualize theubiquitinated ARby immunoblot analysis. To detect the ARprotein (input)in cell lysate samples, anti-AR antibody (Santa Cruz Biotechnology) wasused. The levels of ubiquitinated AR protein were significantly increasedwhen SPOP-wt was also expressed, whereas expression of any prostateadenocarcinoma–associated SPOP mutant effectively inhibited theaccumulation of Ub-AR. Furthermore, the SPOP C-terminal fragment(a.a. 172–374) had no effect on AR ubiquitination. B, overexpression ofCullin-3 dominant negative (DN) efficiently rescued the depletion of ARprotein levels caused by SPOP-wt. 293T cells were cotransfected withpcDNA3-AR-Flag (0.5 mg), and pcDNA3.1 (empty vector control) orpcDNA3.1-HA-SPOPwt, together with pcDNA 3 (empty vector control) orpcDNA 3-HA-CUL3-DN vector, respectively. The cells were collectedand lysed for immunoblot analysis to detect the expression of AR protein(Flag-tagged AR), Cullin-3 DN (HA-tagged CUL3-DN), and b-actin.
AR Axis as the Mediator of SPOP in Prostate Cancer
www.aacrjournals.org Cancer Res; 74(19) October 1, 2014 5637
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
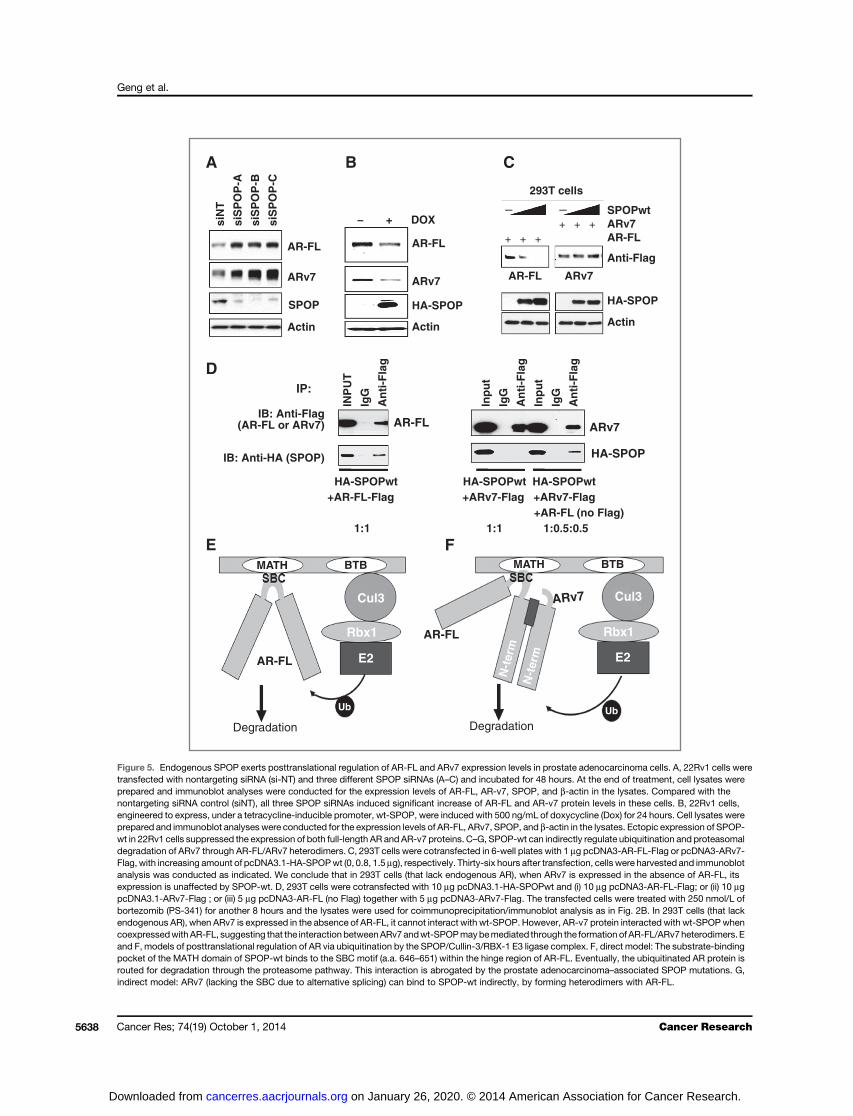
siN
T
siS
PO
P-A
siS
PO
P-B
siS
PO
P-C
BA
HA-SPOP
AR-FL
ARv7
Actin
+ DOX-
E F
HA-SPOPwt HA-SPOPwt HA-SPOPwt +AR-FL-Flag +ARv7-Flag +ARv7-Flag
+AR-FL (no Flag)1:1 1:1 1:0.5:0.5
AR-FL ARv7
INP
UT
IgG
An
ti-F
lag
Inp
ut
IgG
An
ti-F
lag
Inp
ut
IgG
An
ti-F
lag
IP:
IB: Anti-HA (SPOP)
IB: Anti-Flag(AR-FL or ARv7)
HA-SPOP
D
SPOP
AR-FL
ARv7
Actin
SPOPwtARv7AR-FL+ + +
−−
ARv7AR-FL
Anti-Flag
+ + +
293T cells
HA-SPOP
Actin
C
Degradation Degradation
AR-FL
ARv7
AR-FL
MATH BTB MATH BTB
Cul3 Cul3
Rbx1 Rbx1
E2
Ub Ub
E2
N-t
erm
N-t
erm
Figure 5. Endogenous SPOP exerts posttranslational regulation of AR-FL and ARv7 expression levels in prostate adenocarcinoma cells. A, 22Rv1 cells weretransfected with nontargeting siRNA (si-NT) and three different SPOP siRNAs (A–C) and incubated for 48 hours. At the end of treatment, cell lysates wereprepared and immunoblot analyses were conducted for the expression levels of AR-FL, AR-v7, SPOP, and b-actin in the lysates. Compared with thenontargeting siRNA control (siNT), all three SPOP siRNAs induced significant increase of AR-FL and AR-v7 protein levels in these cells. B, 22Rv1 cells,engineered to express, under a tetracycline-inducible promoter, wt-SPOP, were induced with 500 ng/mL of doxycycline (Dox) for 24 hours. Cell lysates wereprepared and immunoblot analyses were conducted for the expression levels of AR-FL, ARv7, SPOP, and b-actin in the lysates. Ectopic expression of SPOP-wt in 22Rv1 cells suppressed the expression of both full-length AR and AR-v7 proteins. C–G, SPOP-wt can indirectly regulate ubiquitination and proteasomaldegradation of ARv7 through AR-FL/ARv7 heterodimers. C, 293T cells were cotransfected in 6-well plates with 1 mg pcDNA3-AR-FL-Flag or pcDNA3-ARv7-Flag, with increasing amount of pcDNA3.1-HA-SPOPwt (0, 0.8, 1.5 mg), respectively. Thirty-six hours after transfection, cells were harvested and immunoblotanalysis was conducted as indicated. We conclude that in 293T cells (that lack endogenous AR), when ARv7 is expressed in the absence of AR-FL, itsexpression is unaffected by SPOP-wt. D, 293T cells were cotransfected with 10 mg pcDNA3.1-HA-SPOPwt and (i) 10 mg pcDNA3-AR-FL-Flag; or (ii) 10 mgpcDNA3.1-ARv7-Flag ; or (iii) 5 mg pcDNA3-AR-FL (no Flag) together with 5 mg pcDNA3-ARv7-Flag. The transfected cells were treated with 250 nmol/L ofbortezomib (PS-341) for another 8 hours and the lysates were used for coimmunoprecipitation/immunoblot analysis as in Fig. 2B. In 293T cells (that lackendogenous AR), when ARv7 is expressed in the absence of AR-FL, it cannot interact with wt-SPOP. However, AR-v7 protein interacted with wt-SPOPwhencoexpressedwithAR-FL, suggesting that the interactionbetweenARv7andwt-SPOPmaybemediated through the formation ofAR-FL/ARv7heterodimers.Eand F, models of posttranslational regulation of AR via ubiquitination by the SPOP/Cullin-3/RBX-1 E3 ligase complex. F, direct model: The substrate-bindingpocket of the MATH domain of SPOP-wt binds to the SBC motif (a.a. 646–651) within the hinge region of AR-FL. Eventually, the ubiquitinated AR protein isrouted for degradation through the proteasome pathway. This interaction is abrogated by the prostate adenocarcinoma–associated SPOP mutations. G,indirect model: ARv7 (lacking the SBC due to alternative splicing) can bind to SPOP-wt indirectly, by forming heterodimers with AR-FL.
Geng et al.
Cancer Res; 74(19) October 1, 2014 Cancer Research5638
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

cells, despite the absence of the SBC, and possibly through theformation of AR-FL/ARv7 heterodimers (Fig. 5E and F).
Mutant SPOPpromotes in vivoprostate adenocarcinomagrowth in immunocompromised miceWe xenografted subcutaneously, in the flank of SCID-Beige
male mice, Abl cells engineered to express, under a tetracy-cline-inducible promoter, wt-SPOP or SPOP-F102C. The micewere fed with water containing doxycycline (200 mg/mL)starting the day after cell injection, and kept on doxycyclinefor the duration of the experiment. We observed that Abl-SPOP-F102C xenografts grew significantly faster (Fig. 6A, P <0.0001) and expressed more AR protein (Fig. 6B) than Abl-SPOP-WT xenografts, supporting the role of mt-SPOP as aprostate adenocarcinoma oncogene. There was no differencein the % tumor take rate between the SPOP alleles (100% inboth cases); there was only delay in the growth of the xeno-grafts expressing wt compared with the mt-SPOP.
Genetic ablation of Spop results in increased AR proteinlevels in the mouse prostate
Prostates of 9-month-old hemizygous (Spopþ/�) C57/BL6mice expressed more AR than SPOP wild-type littermates,providing further evidence that endogenous SPOP regulatesAR expression in vivo (Fig. 6C).
Oncogenic effect of mt-SPOP in the context of AR axisinhibition
LNCaP cells expressing the prostate adenocarcinoma–associated SPOP mutants proliferated faster than cellsexpressing wild-type SPOP. This was observed under regulargrowth conditions (medium supplemented with 10% FBS,which contains basal androgen levels), as well as underandrogen deprivation (medium supplemented with 10%CSS). However, the separation of the growth curves wasmore pronounced under androgen deprivation conditions,consistent with a more significant contribution of mt-SPOPto cell proliferation under androgen deprivation conditions(Supplementary Fig. S11A and S11B). Similarly, our datasuggest that the presence of SPOP mutations may contributeto partially decreased sensitivity to enzalutamide (Supple-mentary Fig. S11C).
Correlation of SPOP signature score with AR activityscore in prostate adenocarcinoma patient cohorts
Finally, we investigated the relationship between the tran-scriptional output of the SPOP pathway and the androgenicactivity in prostate adenocarcinoma specimens, using previ-ously published datasets from four large patient cohorts. Weapplied the established method of computing a gene signaturez-score for each sample, as previously described (8), by add-ing z-scores for upregulated genes and subtracting z-scoresfor downregulated genes. Specifically, we computed a sum ofz-scores for all the genes in the SPOPmutation gene signatures(F133V, F133L, F102C, and combined mutants), and for the allthe genes in two androgen-induced signatures: Hieronymusand colleagues (AR activity score 1; ref. 43) and Nelson andcolleagues (AR activity score 2; ref. 42). In all four patientcohorts, we found a strong positive correlation between theSPOP signature score and the AR activity scores (Fig. 7 andSupplementary Fig. S12).
DiscussionThe somatic heterozygous missense mutations in the sub-
strate-binding pocket of SPOP, that frequently occur in humanprostate adenocarcinomas, are rare to absent in other malig-nancies (endometrial carcinomas harbor SPOPmutations, butnot in the substrate-binding pocket). This observation suggeststhat the role of SPOP mutations in prostate adenocarcinomainvolves a change in SPOP affinity for a prostate-specificsubstrate. Several substrates have been reported for SPOP,including the death domain–associated protein Daxx (33), thephosphatase Puc, the transcriptional regulator Ci/Gli (34), thevariant histone MacroH2A (35), and the key AR coactivatorSteroid Receptor Coactivator (SRC)-3 (36–37), yet none of themis restricted to the prostate and it is unclear how they could
1.0 1.1 3.3 3.0
AR
C57/BL6
Spop+/-
1 2 1 2
SPOP
b-Actin
b-Actin
C
1.0 1.02 0.45 0.57
A
SP
OP
WT
AR
HA (SPOP)
SP
OP
F10
2C
B
1.0 5.6
4221
P < 0.0001
F102CWT
F102C
WT
P < 0.0001
Spop+/+
2,500
2,000
1,500
1,000
500
0
Days after implantation
Tum
or
volu
me
(mm
3 )
Figure 6. SPOP regulates AR protein expression in vivo. A and B, mutantSPOP promotes AR protein and prostate adenocarcinoma growth in vivoin immunocompromised mice. A, two million Abl cells engineered toexpress, under a tetracycline-inducible promoter, wt-SPOP or SPOP-F102CweremixedwithMatrigel and subcutaneously injected in the flankof male SCID-Beige mice (5 mice per cohort). The mice were providedwith water containing doxycycline (200 mg/mL) starting one day after cellinjection and kept for the duration of the experiment. The y-axis depictstumor volume inmm3. Comparison betweenwt-SPOP andSPOP-F102Cwas analyzed by unpaired t test. B, tumors frommice described in Awereexcised and cell lysates were prepared. Immunoblot analyses wereconducted for the expression levels of AR, HA-tagged SPOP, and b-actinin the tumor lysates. The numbers beneath the bands representdensitometry analysis conducted to quantify the expression of AR.C, genetic ablation of Spop results in increased AR protein levels in themouse prostate. Immunoblot analyses were conducted for murine AR,SPOP, and b-actin in the prostates of 9-month-old wild-type andhemizygous Spop (Spopþ/�) C57/BL6 mice. The numbers beneath thebands represent densitometry analysis to quantify AR and SPOPexpression in the prostate lysates.
AR Axis as the Mediator of SPOP in Prostate Cancer
www.aacrjournals.org Cancer Res; 74(19) October 1, 2014 5639
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

explain a prostate-specific mechanism of action for SPOPmutations. Through its participation in the AR axis, SRC-3 isa plausible candidate for this role, but it also is an importantoncogene in other malignancies (including breast carcinomas;refs. 46, 47) where the SPOP substrate-binding pocket is notmutated. Therefore, the relative contribution of the reportedSPOP substrates to the pathophysiology of SPOP-mutant (mt)prostate adenocarcinoma remains unknown.
For this reason, we interrogated, by microarray, the tran-scriptomic profiles of AR(þ) prostate adenocarcinoma cellsengineered to express wild-type (wt)-SPOP or three differentmt-SPOPs (F133V, F133L, and F102C). First, the gene signa-tures of all mt-SPOPs overlapped highly with each other,
suggesting that the transcriptional footprint of the threemutants tested is fairly identical. This is in agreement withexperimental data from our group (Figs. 2 and 3 and ref. 36)that all prostate adenocarcinoma–associated SPOP mutationsdisrupt SPOP binding to substrate, and with bioinformaticspredictions of the impact of thesemutations on SPOP function(30). An unbiased search for gene sets that are enriched ingenes upregulated by mt-SPOP revealed that the gene signa-tures most closely related to those of mt-SPOPs were signa-tures derived from androgen treatment of prostate adenocar-cinoma cells (42). Moreover, a secondary, candidate-basedGSEA analysis across a panel of gene signatures derived fromtreating prostate adenocarcinoma cells with various drugs
Cai et al., GSE32269 dataset (primary cancer specimens)
R = 0.96P < 0.001
R = 0.94P < 0.001
R = 0.95P < 0.001
R = 0.93P < 0.001
R = 0.90P < 0.001
R = 0.87P < 0.001
R = 0.91P < 0.001
R = 0.94P < 0.001
AR
act
ivit
y sc
ore
2
Combined SPOP mutation score
SPOP F102C scoreSPOP F133L scoreSPOP F133V score
R = 0.84P < 0.001
R = 0.86P < 0.001
R = 0.87P < 0.001
R = 0.86P < 0.001
R = 0.75P < 0.001
R = 0.83P < 0.001
R = 0.79P < 0.001
R = 0.75P < 0.001
A
B
Cai et al., GSE32269 dataset (metastatic cancer specimens)
AR
act
ivit
y sc
ore
1A
R a
ctiv
ity
sco
re 2
AR
act
ivit
y sc
ore
1
Combined SPOP mutation score
SPOP F102C scoreSPOP F133L scoreSPOP F133V score
Figure 7. SPOP gene signature scores in human prostate adenocarcinoma specimens correlate strongly with androgen receptor activity. A and B, wecomputed a sum of z-scores for all the genes in the SPOPmutation gene signatures (F133V, F133L, F102C, and combined mutation signature) and for all theandrogen-dependent genes [per two androgen-response datasets: Hieronymus and colleagues (AR activity score 1; ref. 43) and Nelson and colleagues(AR activity score 2; ref. 42)]. A, relative distribution of SPOP gene signature scores and androgen activity scores over primary prostate adenocarcinomasamples from the Cai and colleagues cohort (GSE32269; R range: 0.87–0.94, all comparisons P < 0.001; ref. 51). B, relative distribution of SPOP mutationscores and androgen activity scores over metastatic prostate adenocarcinoma samples from the Cai and colleagues cohort (GSE32269; R range: 0.75–0.87,all comparisons P < 0.001; ref. 51).
Geng et al.
Cancer Res; 74(19) October 1, 2014 Cancer Research5640
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

(approved for prostate adenocarcinoma treatment or experi-mental agents) again confirmed that the transcriptional foot-prints of mt-SPOPs are highly anticorrelated with the signa-tures of AR antagonists: bicalutamide, enzalutamide(MDV3100), and ARN-509 (48). Specifically, genes upregulatedby the AR antagonists are downregulated bymt-SPOPs, where-as genes downregulated by the AR antagonists are upregulatedby mt-SPOPs. The gene signatures of mt-SPOPs had onlylimited or no overlap with the signatures of drugs that arenot direct AR antagonists. Collectively, these results providethe first evidence that ranks the AR axis as the top transcrip-tional output of the tumor suppressor SPOP in prostateadenocarcinoma.Wepreviously reported thatwt-SPOPbinds to and promotes
the turnover of SRC-3 protein, which suppresses the capacity ofSRC-3 to function as an AR coactivator, and the tumor sup-pressor effects of wt-SPOP are abrogated by the prostateadenocarcinoma–associated SPOPmutations (36). In the pres-ent studies, we determined the relative contribution of SRC-3versus AR to the transcriptional output of mt-SPOPs. Wesilenced SRC-3 or AR in LNCaP cells via siRNA and generatedcorresponding gene expression profiles. We also utilized twopublicly available signatures of androgen-treated prostateadenocarcinoma cells (42, 43). GSEA analysis revealed thatthe transcriptomic footprint of mt-SPOPs enriches for SRC-3–dependent genes, confirming the importance of SRC-3 that wedocumented in our prior study (36). Interestingly, we alsoidentified that the gene signatures of mt-SPOPs showed higherenrichment for genes regulated by AR and androgen than forgenes regulated by SRC-3. This raised the hypothesis that, inaddition to its SRC-3–mediated effects, SPOP also exertsimportant SRC-3–independent/AR-mediated functions. Thisled us to evaluate the impact of SPOP on AR itself.Our biochemical studies revealed that wt-SPOP can bind AR
directly and promote its ubiquitination and degradation. Weconfirmed these results in both androgen-dependent (LNCaP,VCaP, LAPC4) and androgen-independent (Abl, 22Rv1) cells.We identified a SBC motif in the hinge region of AR that isrecognized directly by SPOP. This binding activity is abrogatedby the prostate adenocarcinoma–associated SPOP mutations,leading to AR stabilization, AR-mediated signaling, andincreased cell proliferation. In agreement, we found that thealternatively spliced variant AR-v7, that lacks the SPOP-bind-ing hinge region, cannot directly bindwt-SPOPwhen expressedinto 293T cells. While our manuscript was in its final stage ofpreparation, An and colleagues. (49) also reported the presenceof an SBC motif in the hinge region of AR and found that wt-SPOP can regulate the expression of full-length AR but nottruncated variants that lack the hinge region. However, in ourextensive studies of 22Rv1 prostate adenocarcinoma cells, thatexpress both full-length AR and constitutively active variantAR-v7, we documented that three different SPOP siRNAsincreased the protein levels of both full-length AR and variantAR-v7. Moreover, ectopic expression of wt-SPOP suppressedthe protein levels of both full-length AR and variant AR-v7 in22Rv1 cells. This apparent contradiction was explained whenwe cotransfected both full-length AR and variant AR-v7 cDNA,together with SPOP-wt cDNA, into 293T cells. We found that
AR-v7 can be coimmunoprecipitated with wt-SPOP only if full-length AR is also present. Our findings suggest a model wherewt-SPOP can interact with AR-v7 via AR full-length/ AR-v7heterodimers (which have been previously proposed to exist inprostate adenocarcinoma; ref. 50). Moreover, our animal stud-ies demonstrated that xenografts of prostate adenocarcinomacells expressing mt-SPOP express more AR protein and growsignificantly faster in immunocompromised mice than pros-tate adenocarcinoma cells expressing wt-SPOP. We also foundthat the prostates of hemizygous Spop knockout mice expressmore AR protein compared to age-matched wild-type mice.Finally, examination of several publicly available human pros-tate adenocarcinoma specimen datasets further illustrated thestrong link between the transcriptomic outputs of mt-SPOPsand AR. Collectively, our data establish the role of mt-SPOP inAR regulation and prostate adenocarcinoma growth in vivo.We conclude that wt-SPOP is an important tumor suppressorin prostate cells, and mt-SPOP is an oncogenic driver inprostate adenocarcinoma. SPOP regulates the stability of twokey components of the AR axis: AR itself and its coactivatorSRC-3. We determined that the AR axis is the main transcrip-tional output of the tumor suppressor SPOP in prostate ade-nocarcinoma. Our studies provide an explanation why muta-tions in the substrate-binding pocket of SPOP occur frequentlyand in a prostate adenocarcinoma–specific manner andenhance our understanding of the pathophysiology of thiscommon prostate adenocarcinoma genotype. Induction ofexpression of wt-SPOP in prostate adenocarcinoma cells woulddeplete AR (full-length and constitutively active splice-variants)as well as its coactivator SRC-3, extinguish AR signaling andinhibit prostate adenocarcinoma growth, leading to significanttherapeutic implications. Taken together, these data support therationale to further explore the regulation of expression of thetumor suppressor SPOP in prostate adenocarcinoma.
Finally, our experimental results have raised the possibilitythat, while all prostate adenocarcinoma–associated SPOPmutants are associated with a loss of AR suppression (loss-of-function effect), certain SPOP mutants (e.g., F102C) mayactually enhance AR signaling above baseline (i.e., no exoge-nous SPOP) levels, thus exerting a "gain-of-function" oncogeniceffect. This phenomenon, which could be attributed to aputative "dominant-negative" effect of mutant SPOP on thefunction of wt-SPOP, may acquire particular importancebecause SPOP mutations are always heterozygous in prostatecancer specimens. Therefore, our data hint to possible func-tional differences between the various SPOP mutants regard-ing their oncogenic potential and even prognostic significance,which, obviously, will need to be validated in clinically anno-tated human prostate cancer specimens.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: C. Geng, V.K. Eedunuri, B. He, C. Coarfa, N. MitsiadesDevelopment of methodology: C. Geng, V.K. Eedunuri, B. He, N. MitsiadesAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): C. Geng, S.S. Shah, C. Foley, S.A. Chew, R. Bond,N. Mitsiades
AR Axis as the Mediator of SPOP in Prostate Cancer
www.aacrjournals.org Cancer Res; 74(19) October 1, 2014 5641
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Analysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): C. Geng, K. Rajapakshe, S.S. Shah, V.K. Eedunuri,W. Fiskus, C. Coarfa, N. MitsiadesWriting, review, and/or revision of themanuscript: C. Geng, K. Rajapakshe,W. Fiskus, R. Bond, C. Coarfa, N. MitsiadesAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): C. Geng, V.K. Eedunuri, M. Rajendran,M. Zimmermann, N. MitsiadesStudy supervision: C. Geng, B. He, C. Coarfa, N. MitsiadesOther: J. Shou (helpful discussion)
AcknowledgmentsThe authors acknowledge the joint participation by Adrienne Helis Malvin
Medical Research Foundation through its direct engagement in the continuousactive conduct of medical research in conjunction with Baylor College ofMedicine. The authors also acknowledge the assistance of the Genomic andRNA Profiling Shared Resource of the Dan L. Duncan Cancer Center (supportedby the NCI Cancer Center Support Grant P30CA125123).
Grant SupportThis work was supported by the Prostate Cancer Foundation (N. Mit-
siades), the Conquer Cancer Foundation of the American Society of ClinicalOncology Young Investigator and Career Development Awards (both to N.Mitsiades), and a Pilot/Feasibility Program of the Diabetes & EndocrinologyResearch Center (P30-DK079638) at Baylor College of Medicine (N. Mit-siades). N. Mitsiades is a Dan L. Duncan Scholar, a Caroline Wiess LawScholar, and a member of the Dan L. Duncan Cancer Center (supported bythe NCI Cancer Center Support Grant P30CA125123) and the Center for DrugDiscovery at Baylor College of Medicine.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received February 17, 2014; revised July 12, 2014; accepted August 13, 2014;published online October 1, 2014.
References1. Mitsiades N. A road map to comprehensive androgen receptor axis
targeting for castration-resistant prostate cancer. Cancer Res 2013;73:4599–605.
2. Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, et al.Distinct patterns of dysregulated expression of enzymes involved inandrogen synthesis and metabolism in metastatic prostate cancertumors. Cancer Res 2012;72:6142–52.
3. Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, et al.Intratumoral de novo steroid synthesis activates androgen receptor incastration-resistant prostate cancer and is upregulated by treatmentwith CYP17A1 inhibitors. Cancer Res 2011;71:6503–13.
4. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC,WoodCA, et al.Androgen levels increase by intratumoral de novo steroidogenesisduring progression of castration-resistant prostate cancer. CancerRes 2008;68:6407–15.
5. Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD,et al. Intraprostatic androgens and androgen-regulated geneexpression persist after testosterone suppression: therapeuticimplications for castration-resistant prostate cancer. Cancer Res2007;67:5033–41.
6. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF,Higano CS, et al. Maintenance of intratumoral androgens inmetastaticprostate cancer: a mechanism for castration-resistant tumor growth.Cancer Res 2008;68:4447–54.
7. Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB, et al.Activation of the androgen receptor by intratumoral bioconversion ofandrostanediol to dihydrotestosterone in prostate cancer. Cancer Res2011;71:1486–96.
8. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS,et al. Integrative genomic profiling of human prostate cancer. CancerCell 2010;18:11–22.
9. Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL,Visakorpi T. Amplification and overexpression of androgen receptorgene in hormone-refractory prostate cancer. Cancer Res 2001;61:3550–5.
10. Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, et al.A mechanism for androgen receptor-mediated prostate cancer recur-rence after androgen deprivation therapy. Cancer Res 2001;61:4315–9.
11. Gregory CW, Johnson RT Jr, Mohler JL, French FS, Wilson EM.Androgen receptor stabilization in recurrent prostate cancer is asso-ciated with hypersensitivity to low androgen. Cancer Res 2001;61:2892–8.
12. Waltering KK, HeleniusMA, SahuB,Manni V, LinjaMJ, JanneOA, et al.Increased expression of androgen receptor sensitizes prostate cancercells to low levels of androgens. Cancer Res 2009;69:8141–9.
13. Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE III, Erdem H,et al. Androgens modulate expression of transcription intermediaryfactor 2, an androgen receptor coactivator whose expression level
correlates with early biochemical recurrence in prostate cancer.Cancer Res 2006;66:10594–602.
14. O'Malley BW, Kumar R. Nuclear receptor coregulators in cancerbiology. Cancer Res 2009;69:8217–22.
15. Gaddipati JP, McLeod DG, Heidenberg HB, Sesterhenn IA, Finger MJ,Moul JW, et al. Frequent detection of codon 877 mutation in theandrogen receptor gene in advanced prostate cancers. Cancer Res1994;54:2861–4.
16. TaplinME, BubleyGJ, Ko YJ, Small EJ, UptonM, Rajeshkumar B, et al.Selection for androgen receptor mutations in prostate cancers treatedwith androgen antagonist. Cancer Res 1999;59:2511–5.
17. Marcelli M, Ittmann M, Mariani S, Sutherland R, Nigam R, Murthy L,et al. Androgen receptor mutations in prostate cancer. Cancer Res2000;60:944–9.
18. Steinkamp MP, O'Mahony OA, Brogley M, Rehman H, Lapensee EW,Dhanasekaran S, et al. Treatment-dependent androgen receptormutations in prostate cancer exploit multiple mechanisms to evadetherapy. Cancer Res 2009;69:4434–42.
19. Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, et al.Switch from antagonist to agonist of the androgen receptor bicaluta-mide is associated with prostate tumour progression in a new modelsystem. Br J Cancer 1999;81:242–51.
20. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicingof a novel androgen receptor exon generates a constitutively activeandrogen receptor that mediates prostate cancer therapy resistance.Cancer Res 2008;68:5469–77.
21. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al.Ligand-independent androgen receptor variants derived from splicingof cryptic exons signify hormone-refractory prostate cancer. CancerRes 2009;69:16–22.
22. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tanna-hill C, et al. Distinct transcriptional programs mediated by theligand-dependent full-length androgen receptor and its splicevariants in castration-resistant prostate cancer. Cancer Res2012;72:3457–62.
23. Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenicrearrangement and altered RNA splicing of the androgen receptor in acell-based model of prostate cancer progression. Cancer Res2011;71:2108–17.
24. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM.Androgen receptor splice variants mediate enzalutamide resistancein castration-resistant prostate cancer cell lines. Cancer Res2013;73:483–9.
25. Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H, et al.Interleukin-6 regulates prostate-specificprotein expression in prostatecarcinoma cells by activation of the androgen receptor. Cancer Res1998;58:4640–5.
26. Lin PC, Chiu YL, Banerjee S, Park K, Mosquera JM, Giannopoulou E,et al. Epigenetic repression of miR-31 disrupts androgen receptor
Geng et al.
Cancer Res; 74(19) October 1, 2014 Cancer Research5642
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

homeostasis and contributes to prostate cancer progression. CancerRes 2013;73:1232–44.
27. Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N.Tubulin-targeting chemotherapy impairs androgen receptor activity inprostate cancer. Cancer Res 2010;70:7992–8002.
28. Gan L, Chen S, Wang Y,Watahiki A, Bohrer L, Sun Z, et al. Inhibition ofthe androgen receptor as a novelmechanismof taxol chemotherapy inprostate cancer. Cancer Res 2009;69:8386–94.
29. Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, ZhouXK, et al. Taxane-induced blockade to nuclear accumulation of theandrogen receptor predicts clinical responses in metastatic prostatecancer. Cancer Res 2011;71:6019–29.
30. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theur-illat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 andMED12 mutations in prostate cancer. Nat Genet 2012;44:685–9.
31. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Siva-chenko AY, et al. The genomic complexity of primary human prostatecancer. Nature 2011;470:214–20.
32. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al.Diverse somatic mutation patterns and pathway alterations in humancancers. Nature 2010;466:869–73.
33. Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol JH, et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor ofDaxx for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem2006;281:12664–72.
34. ZhangQ,ShiQ,ChenY,YueT, Li S,WangB, et al.Multiple Ser/Thr-richdegrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3ubiquitin ligase. Proc Natl Acad Sci U S A 2009;106:21191–6.
35. Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M,et al. Structures of SPOP-substrate complexes: insights intomoleculararchitectures of BTB-Cul3 ubiquitin ligases. Mol Cell 2009;36:39–50.
36. GengC, He B, Xu L, Barbieri CE, Eedunuri VK, ChewSA, et al. Prostatecancer-associated mutations in speckle-type POZ protein (SPOP)regulate steroid receptor coactivator 3 protein turnover. Proc NatlAcad Sci U S A 2013;110:6997–7002.
37. LiC,Ao J, Fu J, LeeDF, Xu J, LonardD, et al. Tumor-suppressor role forthe SPOP ubiquitin ligase in signal-dependent proteolysis of theoncogenic co-activator SRC-3/AIB1. Oncogene 2011;30:4350–64.
38. Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, et al. SRC-3 isrequired for prostate cancer cell proliferation and survival. Cancer Res2005;65:7976–83.
39. Yan J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M, et al. Steroid receptorcoactivator-3/AIB1 promotes cell migration and invasiveness through
focal adhesion turnover and matrix metalloproteinase expression.Cancer Res 2008;68:5460–8.
40. Tien JC, Liu Z, Liao L, Wang F, Xu Y, Wu YL, et al. The steroid receptorcoactivator-3 is required for the development of castration-resistantprostate cancer. Cancer Res 2013;73:3997–4008.
41. Wang Y, Lonard DM, Yu Y, ChowDC, Palzkill TG,Wang J, et al. Bufalinis a potent small molecule inhibitor of the steroid receptor coactivatorsSRC-3 and SRC-1. Cancer Res 2014;74:1506–17.
42. Nelson PS, Clegg N, Arnold H, Ferguson C, BonhamM, White J, et al.The program of androgen-responsive genes in neoplastic prostateepithelium. Proc Natl Acad Sci U S A 2002;99:11890–5.
43. HieronymusH, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al.Gene expression signature-based chemical genomic prediction iden-tifies a novel class of HSP90 pathway modulators. Cancer Cell 2006;10:321–30.
44. Sheflin L, Keegan B, Zhang W, Spaulding SW. Inhibiting protea-somes in human HepG2 and LNCaP cells increases endogenousandrogen receptor levels. Biochem Biophys Res Commun 2000;276:144–50.
45. GottliebB,Beitel LK,NadarajahA,PaliourasM, TrifiroM. Theandrogenreceptor gene mutations database: 2012 update. Hum Mutat 2012;33:887–94.
46. Torres-ArzayusMI, Font deMora J, Yuan J, VazquezF,BronsonR,RueM, et al. High tumor incidence and activation of the PI3K/AKT pathwayin transgenic mice define AIB1 as an oncogene. Cancer Cell 2004;6:263–74.
47. Torres-Arzayus MI, Yuan J, DellaGatta JL, Lane H, Kung AL, BrownM. Targeting the AIB1 oncogene through mammalian target ofrapamycin inhibition in the mammary gland. Cancer Res 2006;66:11381–8.
48. CleggNJ,Wongvipat J, Joseph JD, TranC,OukS,DilhasA, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res2012;72:1494–503.
49. An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-lengthandrogen receptor by wild-type SPOP, but not prostate-cancer-asso-ciated mutants. Cell Rep 2014;6:657–69.
50. WatsonPA,ChenYF,BalbasMD,Wongvipat J, SocciND, Viale A, et al.Constitutively active androgen receptor splice variants expressed incastration-resistant prostate cancer require full-length androgenreceptor. Proc Natl Acad Sci U S A 2010;107:16759–65.
51. Cai C, Wang H, He HH, Chen S, He L, Ma F, et al. ERG inducesandrogen receptor-mediated regulation of SOX9 in prostate cancer.J Clin Invest 2013;123:1109–22.
www.aacrjournals.org Cancer Res; 74(19) October 1, 2014 5643
AR Axis as the Mediator of SPOP in Prostate Cancer
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Correction
Correction: Androgen Receptor Is the KeyTranscriptional Mediator of the TumorSuppressor SPOP in Prostate CancerChuandongGeng,KimalRajapakshe, Shrijal S. Shah, JohnShou,Vijay Kumar Eedunuri, Christopher Foley,Warren Fiskus,Mahitha Rajendran, Sue Anne Chew, Martin Zimmermann,Richard Bond, Bin He, Cristian Coarfa, and Nicholas Mitsiades
In the original version of this article (1), the duration of exposure to doxycycline(Dox) was incorrectly listed as 24 hours in the legend of Fig. 3A. The figure legend forFig. 3A has been corrected to indicate the exposure to doxycycline is 72 hours. Theerror has been corrected in the latest onlineHTML andPDF versions of the article. Theauthors regret this error.
Reference1. Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, et al. Androgen receptor is the key
transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 2014;74:5631–43.
Published first September 3, 2019.Cancer Res 2019;79:4552doi: 10.1158/0008-5472.CAN-19-1981�2019 American Association for Cancer Research.
CancerResearch
Cancer Res; 79(17) September 1, 20194552

2014;74:5631-5643. Cancer Res Chuandong Geng, Kimal Rajapakshe, Shrijal S. Shah, et al. Tumor Suppressor SPOP in Prostate CancerAndrogen Receptor Is the Key Transcriptional Mediator of the
Updated version
http://cancerres.aacrjournals.org/content/74/19/5631
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2014/10/08/74.19.5631.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/74/19/5631.full#ref-list-1
This article cites 51 articles, 38 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/74/19/5631.full#related-urls
This article has been cited by 13 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/74/19/5631To request permission to re-use all or part of this article, use this link
on January 26, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from