bi/ch 422/622 outline
TRANSCRIPT

1
BI/CH 422/622OUTLINE:Protein Degradation (Catabolism)
DigestionInside of cellsProtein turnover
UbiquitinActivation-E1Conjugation-E2Ligation-E3
Proteosome
Amino-Acid DegradationAmmonia
freetransamination-mechanism to know
Urea Cycle – dealing with the nitrogen5 Steps
Carbamoyl-phosphate synthetaseOrnithine transcarbamylaseArginino-succinate synthetaseArginino-succinaseArginase
EnergeticsUrea Bi-cycleUrea Cycle – dealing with the nitrogenFeeding the Urea Cycle
Glucose-Alanine CycleFree Ammonia
GlutamineGlutamate dehydrogenase
Overall energetics
OUTLINE:Amino-Acid Degradation
Dealing with the carbonSeven Families
1. ADENQ2. RPH
oxidaseone-carbon metabolism
THFSAM
3. GSCPLP uses
4. MT – one carbon metabolism5. FY – oxidase vs oxygenase6. KW –7. BCAA – VIL
Convergence with Fatty acid-odd chainOverview
Fates of the 29 nitrogen atoms in 20 AA:
9 ammonia18 transamination2 urea
Amino Acid DegradationA. Concepts
1. Convergent2. ketogenic/glucogenic3. Reactions seen before
B. Transaminase (A,D,E) / Deaminase (Q,N) FamilyC. Related to biosynthesis (R,P,H; C,G,S; M,T)
1.Glu Familya. Introduce oxidases/oxygenasesb. Introduce one-carbon metabolism (1C)
2.Pyruvate Familya. PLP reactions
3. a-Ketobutyric Family (M,T)a. 1-C metabolism
D. Dedicated1. Aromatic Family (F,Y)
a. oxidases/oxygenases2. a-Ketoadipic Family (K,W)3. Branched-chain Family (V,I,L)
E. Convergence with Fatty Acids: propionyl-CoA
29 N
The SEVEN (7) Families
2
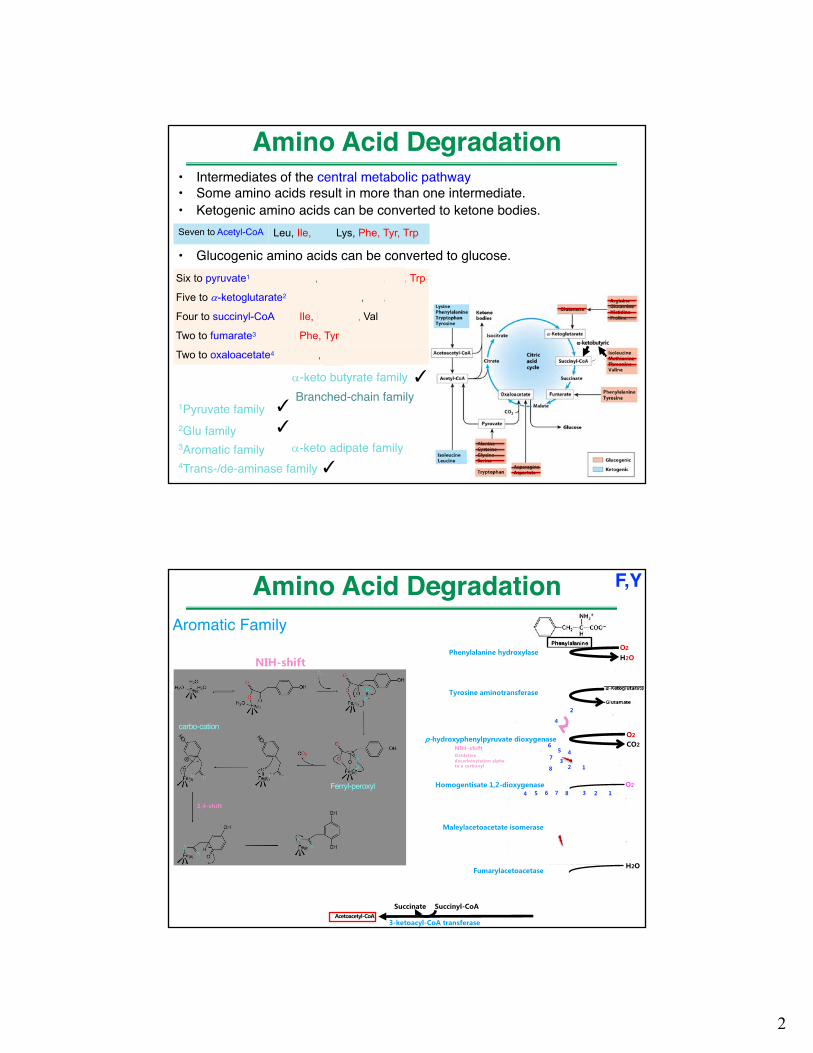
• Intermediates of the central metabolic pathway• Some amino acids result in more than one intermediate.• Ketogenic amino acids can be converted to ketone bodies.
• Glucogenic amino acids can be converted to glucose.Six to pyruvate1 Ala, Cys, Gly, Ser, Thr, Trp
Five to a-ketoglutarate2 Arg, Glu, Gln, His, Pro
Four to succinyl-CoA Ile, Met, Thr, Val
Two to fumarate3 Phe, Tyr
Two to oxaloacetate4 Asp, Asn
Seven to Acetyl-CoA Leu, Ile, Thr, Lys, Phe, Tyr, Trp
Amino Acid Degradation
2Glu family3Aromatic family4Trans-/de-aminase family
1Pyruvate family
a-ketobutyric
✓
✓
✓
a-keto butyrate family
a-keto adipate family
✓Branched-chain family
Amino Acid DegradationAromatic Family
NIH-shift
–OH
–OH
_
Ferryl-peroxylOO
O
O
Acetoacetate
2,4-shift
Phenylalanine hydroxylase
Tyrosine aminotransferase
p-hydroxyphenylpyruvate dioxygenase
Homogentisate 1,2-dioxygenase
Maleylacetoacetate isomerase
Fumarylacetoacetase
123
56
7
8
124 35 6 7 8
F,Y
O2
H2O
O2
CO2NIH-shiftOxidative decarboxylation alpha to a carbonyl
4
carbo-cation
2
4
O2
H2O
Acetoacetyl-CoA3-ketoacyl-CoA transferase
Succinyl-CoASuccinate
⊕

3
Amino Acid DegradationAromatic Family
NIH-shift
–OH
–OH
_
Ferryl-peroxylOO
O
O
Acetoacetate
2,4-shift
Phenylalanine hydroxylase
Tyrosine aminotransferase
p-hydroxyphenylpyruvate dioxygenase
Homogentisate 1,2-dioxygenase
Maleylacetoacetate isomerase
Fumarylacetoacetase
123
56
7
8
124 35 6 7 8
F,Y
O2
H2O
O2
CO2NIH-shiftOxidative decarboxylation alpha to a carbonyl
4
carbocation
2
4
O2
H2O
Acetoacetyl-CoA3-ketoacyl-CoA transferase
Succinyl-CoASuccinate
⊕
Amino Acid Degradation
• In phenylketonuria (PKU) there is a buildup of Pheand phenylpyruvate
– Impairs neurological development leading to severe intellectual deficits
– Controlled by limiting dietary intake of Phe• In Alkatonuria there is a buildup of homogentisate,
which turns black on oxidation
Genetic Defects in Aromatic Family Degradation Lead to Disease
Aromatic Family
⊕⇀Alkaptonuria

4
Amino Acid Degradation
• In phenylketonuria (PKU) there is a buildup of Pheand phenylpyruvate
– Impairs neurological development leading to severe intellectual deficits
– Controlled by limiting dietary intake of Phe• In Alkatonuria there is a buildup of homogentisate,
which turns black on oxidation
Genetic Defects in Aromatic Family Degradation Lead to Disease
Aromatic Family
⊕⇀Alkaptonuria
Oxidases – use molecular oxygen as an electron acceptor, but no atoms into substrate
– e.g., cytochrome oxidase, proline oxidase – usually have water or hydrogen peroxide as product
Oxygenase* – use of molecular oxygen to put into the substrate– dioxygenases – use both atoms; e.g., cysteine dioxygenase
– mono-oxygenases – one atom in substrate, one atom as water
Amino Acid Degradation: Oxidases
*a.k.a.: hydroxylase, mixed-function oxygenase, “mixed-function oxidase”
Nomenclature
AH + BH2 + O2 à AOH + B + H2O
A(red) + O2 + 4H+ à A(ox) + 2H2O
RH + BH2 + O2 à ROH + B + H2O
A + O2 à AO2
• The enzyme is a cytochrome called P-450• The co-substrate electrons are from NADPH• There is Cytochrome P-450 reductase to funnel electrons from co-substrate

5
Amino Acid Degradation: OxidasesRH + BH2 + O2 à ROH + B + H2O
Example of P-450 reactions in adrenal gland:
1) Source of reducing electrons are most often NADPH (the co-substrate)
2) Other co-substratesa) FAD, FeS clusters, a-ketoglutarate (oxidative
decarboxylation to succinate)b) Others: tetrahydrobiopterin (Phe
hydroxylase)3) Major class are the P-450 heme hydroxlases
a) Expressed in liver and adrenal glandsb) Steroids & fatty acidsc) Xenobiotics (drugs)
i. Specificii. Non-specificiii. Inducibleiv. Drug-drug interactions
Hydroxylases
• Intermediates of the central metabolic pathway• Some amino acids result in more than one intermediate.• Ketogenic amino acids can be converted to ketone bodies.
• Glucogenic amino acids can be converted to glucose.Six to pyruvate1 Ala, Cys, Gly, Ser, Thr, Trp
Five to a-ketoglutarate2 Arg, Glu, Gln, His, Pro
Four to succinyl-CoA Ile, Met, Thr, Val
Two to fumarate3 Phe, Tyr
Two to oxaloacetate4 Asp, Asn
Seven to Acetyl-CoA Leu, Ile, Thr, Lys, Phe, Tyr, Trp
Amino Acid Degradation
2Glu family3Aromatic family4Trans-/de-aminase family
1Pyruvate family
F,Y
a-keto butyrate family
a-keto adipate family
Branched-chain family

6
Amino Acid Degradation K,W
a-Ketoadipic Family
2-Amino muconate e-semialdehyde
N-formyl-L-kynurenine
O
O
O
O
O
Alanine
O
OO
3-Hydroxyanthranilic acid 3,4-dioxygenase
Tryptophan 2,3-dioxygenase
Kynurenine formamidase
Kynurenine hydroxylase
Kynureninase
2-Amino 3-carboxyl-
muconate e-semialdehydedecarboxylase
Muconicacid
a-Amino muconate e-
semialdehyde dehydrogenase
a-Aminomuconate reductase
Amino Acid Degradation K,W
a-Ketoadipic Family
2-Amino muconate e-semialdehyde
N-formyl-L-kynurenine
O
O
O
O
O
Alanine
O
OO
Tryptophan 2,3-dioxygenase
Kynurenine formamidase
Kynurenine hydroxylase
Kynureninase
3-Hydroxyanthranilic acid 3,4-dioxygenase
2-Amino 3-carboxyl-
muconate e-semialdehydedecarboxylase
Muconicacid
a-Amino muconate e-
semialdehydedehydrogenase
a-Aminomuconate reductase

7
Amino Acid Degradation
Saccharopinedehydrogenase
a-Ketoadipic Family -removal of e-amino-oxidation of e-carbon
Lysine:a-ketoglutaratereductase
a-aminoadipate-d-semialdehydedehydrogenase
2-aminoadipate aminotransferase
PLPa-ketoglutarate
glutamate
Amino Acid Degradation
Saccharopinedehydrogenase
a-Ketoadipic Family -removal of e-amino-oxidation of e-carbon
Lysine:a-ketoglutaratereductase
a-aminoadipate-d-semialdehydedehydrogenase
2-aminoadipate aminotransferase
PLPa-ketoglutarate
glutamate

8
• Intermediates of the central metabolic pathway• Some amino acids result in more than one intermediate.• Ketogenic amino acids can be converted to ketone bodies.
• Glucogenic amino acids can be converted to glucose.Six to pyruvate1 Ala, Cys, Gly, Ser, Thr, Trp
Five to a-ketoglutarate2 Arg, Glu, Gln, His, Pro
Four to succinyl-CoA Ile, Met, Thr, Val
Two to fumarate3 Phe, Tyr
Two to oxaloacetate4 Asp, Asn
Seven to Acetyl-CoA Leu, Ile, Thr, Lys, Phe, Tyr, Trp
Amino Acid Degradation
2Glu family3Aromatic family4Trans-/de-aminase family
1Pyruvate family
K,W
1.Transaminase/Deaminase Family2.Glu Family3.Pyruvate Family4.a-ketobutyric Family5.Aromatic Family6.a-ketoadipic Family7.Branched-chain Family
V,I,LAmino Acid Degradation•Degradation of branched-chain amino acids does
not occur in the liver• Leucine, isoleucine, and valine are oxidized for fuel
in muscle, adipose tissue, the kidneys, and the brain• Branched-chain amino acids are degraded to
succinyl-CoA, an important citric acid cycle intermediate.
3 steps
7 steps
CO2
5 steps
CO2
HMG-CoA Ketone Bodies
1234b-Hydroxy-methyl-glutaryl-CoA
(OMSGAP)
5
6

9
2
MethionineThreonine
a-Ketobutyrate
2
|COO –
2
LysineTryptophan
a-Ketoadipate
AlanineCysteineGlycineSerine
Pyruvate Acetyl-CoA
Amino Acid Degradation
GlutamateGlutamineHistidineProlineArginine
a-Ketoglutarate Succinyl-CoA
2Propionyl-CoA
|COO –
2
2
Glutaryl-CoA
a-Ketoisovalerate
a-Keto b-methylvalerate
a-Keto isocaproate
Isobutryl-CoA
a-Methybutryl-CoA
Isovaleryl-CoA
PLP
OH
OH
OH
|| O|| O
Dehydrogenase (FAD)
Hydrolase (CoASH)
Carboxylase (biotin)
HMG-CoA
Propionyl-CoAAcetyl-CoA +
a-methyl-acetoacetyl-CoA
Glutaconcyl-CoA Crotonyl-CoA
CO2
biotin
Hydratase (H2O)
Dehydrogenase (NAD+)
Thiolase (CoASH)
2Acetyl-CoACrotonyl-CoA
CH2
Methylmalonyl-CoA
CoASHCoASH
TPP
Lipoicacid
FAD
pyruvate dehydrogenase complex
a-ketoglutarate dehydrogenase complex
CO2
V,I,L
||O
biotin
OXIDATIVE DECARBOXYLATION
Amino Acid Degradation
2Propionyl-CoA
|COO –
2
2
Glutaryl-CoA
a-Ketoisovalerate
a-Keto b-methylvalerate
a-Keto isocaproate
Isobutryl-CoA
a-Methybutryl-CoA
Isovaleryl-CoA
PLP
TPP
Lipoicacid
AlanineCysteineGlycineSerine
Pyruvate Acetyl-CoA
GlutamateGlutamineHistidineProlineArginine
a-Ketoglutarate Succinyl-CoA
2
MethionineThreonine
a-Ketobutyrate
2
|COO –
2
LysineTryptophan
a-Ketoadipate
OH
OH
OH
|| O|| O
Dehydrogenase (FAD)
Dehydrogenase (NAD+)
Hydratase (H2O)
Hydrolase (CoASH)
Carboxylase (biotin)
CO2
HMG-CoA
a-methyl-acetoacetyl-CoA
Methylmalonyl-CoA
Propionyl-CoAAcetyl-CoA +
Glutaconcyl-CoA Crotonyl-CoA
CO2
biotin
Thiolase (CoASH)
2Acetyl-CoACrotonyl-CoA
CH2
CoASHCoASH
FAD
pyruvate dehydrogenase complex
a-ketoglutarate dehydrogenase complex
V,I,LOXIDATIVE DECARBOXYLATION

10
• Intermediates of the central metabolic pathway• Some amino acids result in more than one intermediate.• Ketogenic amino acids can be converted to ketone bodies.
• Glucogenic amino acids can be converted to glucose.Six to pyruvate1 Ala, Cys, Gly, Ser, Thr, Trp
Five to a-ketoglutarate2 Arg, Glu, Gln, His, Pro
Four to succinyl-CoA Ile, Met, Thr, Val
Two to fumarate3 Phe, Tyr
Two to oxaloacetate4 Asp, Asn
Seven to Acetyl-CoA Leu, Ile, Thr, Lys, Phe, Tyr, Trp
Amino Acid Degradation
2Glu family3Aromatic family4Trans-/de-aminase family
1Pyruvate family
V,I,L
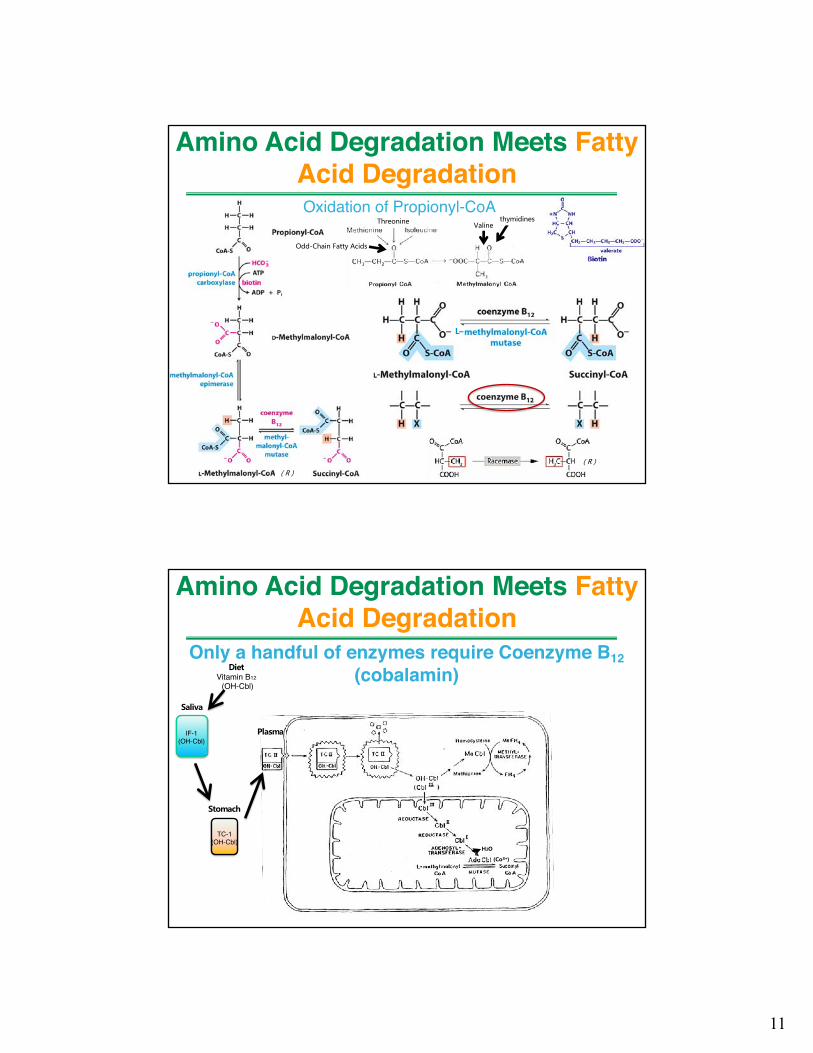
Amino Acid Degradation Meets Fatty Acid DegradationOxidation of Propionyl-CoA
( R )
( R )
L–
Threonine Valine
Odd-Chain Fatty Acids
thymidines
11
Amino Acid Degradation Meets Fatty Acid DegradationOxidation of Propionyl-CoA
( R )
( R )
L–
Threonine Valine
Odd-Chain Fatty Acids
thymidines
Only a handful of enzymes require Coenzyme B12(cobalamin)
Amino Acid Degradation Meets Fatty Acid Degradation
Vitamin B12(OH-Cbl)
TC-1(OH-Cbl)
IF-1(OH-Cbl)
Plasma
Saliva
Stomach
Diet
H2O
(Co3+)

12
Complex Cobalt-Containing Compound: Coenzyme B12
Amino Acid Degradation Meets Fatty Acid Degradation
Dr. Kornberg: Lecture 16 02.27.17 (28:19-29:33) VitB-12 1.3 min
19641957
Nobel Prize
Amino Acid Degradation Meets Fatty Acid Degradation

13
Amino Acid Degradation: OverviewGlutamine
a-ketoadipic
a-ketobutyricPropionyl-CoA
Histidine Proline
Glu-semialdehyde
Arginine
Asparagine
Methionine
Threonine3 Steps
4 Steps 2 Steps2 Steps
5 Steps
SAMcycle
5/6 Steps
ValineIsoleucineLeucine2 Steps
5 Steps4 Steps
HMG-CoA3 Steps
Lysine
Tryptophan Alanine
Serine
Glycine
Cysteine
2 Steps 2 Steps
5 Steps
3 Steps
5 Steps
4 Steps3 Steps
glutaryl-CoA
THFcycle
1.Transaminase/Deaminase Family2.Glu Family3.Pyruvate Family4.a-ketobutyric Family5.Aromatic Family6.a-ketoadipic Family7.Branched-chain Family
Oxidative decarboxylation by a complex yielding NADHCO2 + NH4+
THF
TABLE 18-2 Some Human Genetic Disorders Affecting Amino Acid Catabolism
Medical condition
Approximate incidence (per 100,000 births) Defective process Defective enzyme Symptoms and effects
Albinism <3 Melanin synthesis from tyrosine
Tyrosine 3-monooxygenase(tyrosinase)
Lack of pigmentation; white hair, pink skin
Alkaptonuria <0.4 Tyrosine degradation Homogentisate 1,2-dioxygenase
Dark pigment in urine; late-developing arthritis
Argininemia <0.5 Urea synthesis Arginase Mental retardation
Argininosuccinic acidemia <1.5 Urea synthesis Argininosuccinase Vomiting; convulsions
Carbamoyl phosphate synthetase I deficiency
<0.5 Urea synthesis Carbamoyl phosphate synthetase I
Lethargy; convulsions; early death
Homocystinuria <0.5 Methionine degradation Cystathionine β-synthase
Faulty bone development; mental retardation
Maple syrup urine disease (branchedchain ketoaciduria)
<0.4 Isoleucine, leucine, and valine degradation
Branched-chain α-keto acid dehydrogenase complex
Vomiting; convulsions; mental retardation; early death
Methylmalonic acidemia <0.5 Conversion of propionyl-CoA to succinyl-CoA
Methylmalonyl-CoA mutase
Vomiting; convulsions; mental retardation; early death
Phenylketonuria <8 Conversion of phenylalanine to tyrosine
Phenylalanine hydroxylase
Neonatal vomiting; mental retardation
Amino Acid Degradation: Overview

14
• amino acids from protein are an important energy source in carnivorous animals
• the first step of AA catabolism is transfer of the NH3 via PLP-dependent aminotransferase usually to a-ketoglutarateto yield L-glutamate
• in most mammals, toxic ammonia is quickly recaptured into carbamoyl phosphate and passed into the urea cycle
• amino acids are degraded to pyruvate, acetyl-CoA, α-ketoglutarate, succinyl-CoA, and/or oxaloacetate
• amino acids yielding acetyl-CoA are ketogenic• amino acids yielding other end products are glucogenic• genetic defects in amino degradation pathways result in a
number of human diseases• amino acid catabolism is dependent on a variety of cofactors,
including THF, ado-Met (SAM), Cbl, biotin, and PLP
We learned that:Amino Acid Degradation: Overview
End of Material for Exam 3