biolechnolouv and food science - urząd miasta Łodzicybra.p.lodz.pl/content/5223/biotechnology and...
TRANSCRIPT

Voluma15 Numbar 2 ZESlYlY NAUKOWE POLITECHNIKI ŁÓDZKIEJ CHEMIA SPOŻYWCZA I BIOTECHNOLOGIA
Biolechnolouv and
Food Science
Technical UniversilV oIlodz Poland lOOZ2011

Biotechnology and Food Science
Volume 75 Number 2 2011
CONTENTS
Agnieszka Kowalska-Baron, Preeti Choudhary, Denise Montes
The effect of immobilization in the PVA films on the fluorescence and phosphorescence lifetime of indole and its derivatives 3
Agnieszka Kowalska-Baron, Michaela Brychtova, Ivana Petrović, Igor Passos Sene, Paloma Quiñones, Jana Šogorkova
Ibuprofen-tyrosine (Val-Tyr, Val-Tyr-Val) interactions. Theoretical and experimental studies 15
Iwona Majak, Joanna Leszczyńska, Agata Łącka Immunoreactivity of chemically cross-linked gluten and hydrolysates of wheat flour 27
Elżbieta Sobiecka, Witold Sroczyński Fly ash vitrification as the effective physico-chemical waste stabilization method 35
Marta Słowianek, Iwona Majak Methods of allergen detection based on DNA analysis 39
Kinga Libiszewska Laktony jako związki biologicznie czynne 45
Danuta Czajkowska, Małgorzata Milner-Krawczyk, Marta Kazanecka Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 55
Elżbieta Sobiecka, Karolina Piątkowska, Danuta Posiła Ochrona bioróżnorodności roślin województwa łódzkiego 71
Radosław Wąchała, Tomasz Ramięga, Rita Pyć, Tadeusz Antczak Otrzymywanie włókien nanocelulozy 87

Biotechnology and Food Science ISSN 2084-0136
Journal appeared since 1955, as Zeszyty Naukowe Politechniki Łódzkiej:
Chemia Spożywcza (1955-1979) Technologia i Chemia Spożywcza (1980-1997)
Chemia Spożywcza i Biotechnologia (1998-2010)
Editor-in-Chief: Danuta Kalemba
Editors: Elżbieta Łodyga-Chruścińska, Małgorzata Piotrowska, Anna Sykuła-Zając
Scientific Board
Wojciech Ambroziak, Maria Koziolkiewicz, Józef Kula, Ewa Nebesny, Krzysztof Śmigielski
Cover design: Krzysztof Kozieł
© Copyright by Faculty of Biotechnology and Food Sciences Technical University of Lodz 2011
TECHNICAL UNIVERSITY OF LODZ PRESS 90-924 Lodz, 223 Wólczańska Street
phone//fax: +48 42 684 07 93 e-mail: [email protected]
www.wydawnictwa.p.lodz.pl
Edition 100 copies Printed by
Offset printing „Quick-Druk” s.c. 90-562 Lodz, 11 Łąkowa Street

Biotechnology and Food Science Research article
Biotechnol Food Sci 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
The effect of immobilization in the PVA films on the fluorescence and phosphorescence lifetime of indole
and its derivatives
Agnieszka Kowalska-Baron,1* Preeti Choudhary,2 Denise Montes2
1 Institute of General Food Chemistry, Faculty of Biotechnology and Food Science, Technical University of Lodz, 90-924 Lodz, Poland
2 Department of Natural Sciences, University of Houston-Downtown, 77002, Houston, USA
Abstract: This work is devoted to study how immobilization in the PVA films affects the fluorescence and phosphorescence lifetime of indole and its derivatives. The obtained results indicated that immobilization of the studied indoles in the PVA matrix, which leads to the increased microrigidity of the environment around the indole moiety, results in the increase of singlet and triplet excited state lifetime of the studied compounds. Most probably, the enhancement of the rigidity of the environment near the chromophore reduces the rates of the non-radiative deactivation pathways, which leads to the increase of excited state lifetimes of the studied compounds.
Keywords: indole; N-acetyl-L-tryptophanamide (NATA); tryptophan; fluorescence and phosphorescence lifetime.
Introduction The room temperature tryptophan fluorescence and phosphorescence can be
observed from proteins in aqueous solution, therefore fluorescence and phosphorescence techniques can be used in the study of protein structure and dynamics. Steady state luminescence studies provide the overall information as they contain intensity-weighted average information of the emitting chromophore, while time-resolved studies may provide information on the population distribution of the excited states of the emitting species. Therefore, a combination of steady state and time-resolved luminescence measurements of the tryptophan fluorescence in proteins can provide details of the local environment of the protein and its dynamics [1].
It is well known that fluorescence lifetime of tryptophan in proteins depends on the local environment of indole moiety and can vary from a few to tens of nanoseconds. Therefore, fluorescence techniques offers a possibility to study protein conformational changes in the above mentioned nanosecond time scale. Many studies have demonstrated that in almost all cases proteins containing a

Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
4
single tryptophan residue exhibit multiexponential fluorescence decay kinetics [1]. Most probably, the heterogenous intensity decay of tryptophan in proteins is due to the existence of different rotameric conformations of the tryptophan side chain. It has been suggested that tryptophan can populate a number of low energy conformations, as a result of rotation around the Cα-Cβ and/or the Cβ-Cγ bonds, with each conformer exhibiting a distinct fluorescence lifetime. This model was firstly proposed by Szabo [2] to explain the biexponential fluorescence decay of pure tryptophan in aqueous solution and was subsequently shown to be applicable to small tryptophan containing peptides. The distinct decay times of different tryptophan rotamers most probably reflect interactions between the excited indole moiety and the unique surrounding amino acid residues and peptide groups of the protein conformation [3].
Recently, thanks to the rapid development of the instrumentation, which allows to register the low intensity phosphorescence at ambient temperature, the room temperature tryptophan phosphorescence (RTTP) methodology has been increasingly applied in the protein dynamics studies. The above mentioned RTTP techniques permits the monitoring of much slower processes, extending the observation time scale from the nanosecond range of fluorescence up to microsecond to second range. Additional potential of a triplet state as a structural probe comes from the drastic dependence of the tryptophan phosphorescence lifetime on the differences in the rigidity (microviscosity) of the indole environment [4, 5]. Therefore, the photophysics of triplet state of indole and its derivatives, tryptophan and NATA (N-acetyl-L-tryptophanamide); the latter considered as the standard reference compound for tryptophan in proteins by its mimicking of the amino acid’s attachment in the backbone chain, in different media have been recently widely studied.
The results of a great number of previously performed studies have shown that the phosphorescence lifetime of unquenched tryptophan in proteins at ambient temperature ranges from 0.2 ms for a solvent exposed tryptophan residue to 4 s for that one which is deeply buried within protein interior [5, 6], while tryptophan luminescence from dry protein films was found to be of 10-15 seconds [4]. It has been observed that protein folding, which involves internalization of solvent exposed tryptophan residues, is accompanied with the increase in the lifetime of room temperature phosphorescence for tryptophan residues on the surface of protein.
Due to the fact, that the similarity is expected between room temperature tryptophan phosphorescence lifetime of the tryptophan residues which are exposed to solvent and the lifetime of free tryptophan in solution, it is of a great importance to determine the lifetime of the latter. Although many studies have been devoted to study photophysics of triplet state of aqueous indole and its derivatives, the mechanism of the triplet state decay is still unclear. The most significant ambiguity is that concerning the lifetimes of free indole and its derivatives in solution.

The effect of immobilization in the PVA films…
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
5
According to Fischer [7], the triplet-state lifetime of aqueous deoxygenated indole and several its derivatives, determined using sensitive photon-counting techniques, was approximately 40 μs, which is close to the previous reports by Bent and Hayon [8] (12 μs for indole, 14 μs for tryptophan) and Tsentalowich [9] (12.5 μs for tryptophan) based on flash photolysis, but much shorter than the millisecond long lifetime observed by Strambini [6] (about 5 ms for indole (5 μM)) using similar photon-counting techniques.
As far as NATA is concerned, the average phosphorescence lifetime of aqueous NATA at 20°C determined by Fischer [7] was about 39 μs. In contrast to the result obtained by Fischer, the study of Strambini [10] using similar photon counting technique reported that in 10 mM Tris the phosphorescence lifetime of NATA was around 2 ms. In 2004 Strambini reexamined the lifetime of NATA and estimated the value of the NATA (5 μM) triplet state lifetime in low-purity water to be about 5 ms at 20°C. Strambini [6] explained that the latter higher value is the result of lowered sample contamination, which was obtained applying purer fresh water supply and more effective degassing procedures. Banks and Kerwin [11] also have found millisecond long (2 ms) phosphorescence lifetime of NATA in 1 mM Tris solution of pH = 7.
The above mentioned considerations show that the reported phosphorescence lifetime of aqueous indole derivatives vary from μs to ms. It seems that the main sources of this discrepancy are, on one hand, the effect of triplet state quenching by residual oxygen, impurities or photoproducts in solution, and on the other hand, the formation of triplet state in non-radiative reactions of radical species, which lead to the increase in the observed radiative lifetime [6, 7, 9].
Previous studies have shown that the increased rigidity and viscosity of the environment surrounding the indole moiety results in the increase of phosphores-cence lifetime of indole and its derivatives. For example, the lifetime of the triplet states of indole and its derivatives earlier measured in rigid media (frozen solutions) was found to be several seconds [12]. At low temperature, about 12 K, the phosphorescence lifetime of NATA and L-tryptophan in 60% glycerol/H20 mixtures and in the trehalose/sucrose film at pH = 7 was determined to be about 5 seconds [13].
As far as we know there is no study on the photophysics of indole, tryptophan and NATA immobilized in the PVA matrix. Poly(vinyl alcohol) (PVA) is a polymer in which hydroxyl groups of carbone chain backbone are attached to methane carbons. These OH groups can be a source of hydrogen bonding and hence assist the formation of polymer complexes. Over recent years polyvinyl alcohol (PVA) polymers have attracted attention due to their variety of applications. Poly(vinyl alcohol) (PVA) has been developed for various biomedical applications such as artificial pancreas, hemodialysis and implantable medical materials [14].
While the lifetime of aqueous indole derivatives (indole, NATA, tryptophan) is still under debate, the main aim of this study is to examine how the changes in microrigidity caused by immobilization of the studied indoles in the PVA matrix

Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
6
affects the fluorescence and phosphorescence lifetime of indole and its derivatives (L-tryptophan, NATA).
Experimental Materials
Indole, NATA (N-acetyl-L-tryptophanamide) and L-tryptophan were of the highest purity grade available from commercial sources. Indole, NATA and L-tryptophan were from Sigma-Aldrich and were used without further purification. All absorption and fluorescence measurements in solutions were performed in water at 20°C.
Poly(vinyl alcohol) purchased from Sigma-Aldrich was used as is. PVA powder of 10 g was dissolved in distilled water of 100 ml at 90°C. Immobilization of the indoles were obtained simply by mixing 1ml of stock solutions of the studied indoles (10-3 M) with 9 ml of room temperature PVA solution to obtain 10-4 M concentration of the studied indoles in the PVA solution. 5 ml of the solution was poured on a horizontal plastic circular plate of the radius 3 cm. After it was dried at room temperature in 10 days, a PVA film about 0.1 cm thick was obtained.
Methods
Absorption measurements Absorption spectra were recorded in a Nicolet Evolution 300 (Thermo Electron
Corporation) UV-Vis spectrophotometer. The measurements were performed at room temperature (20°C).
Steady-state fluorescence measurements Steady-state fluorescence measurements were performed with a FluoroMax2
(Jobin Yvon Spex) spectrofluorimeter using an excitation wavelength of 290 nm. All measurements were performed in a standard quartz cuvette at 20°C.
Time-resolved fluorescence measurements Fluorescence lifetime measurements were carried out with a FL900CDT
time-correlated single photon counting fluorimeter from Edinburgh Analytical Instruments. The excitation and emission wavelengths were set to 290 and 360 nm, respectively. The fluorescence emission decays were monitored at a 90° angle to excitation at 20°C. The instrument response function was recorded by collecting scattered light from a LUDOX silica suspension.
Data acquisition and analysis were performed using the software provided by Edinburgh Analytical Instrumentation. Time-resolved data results were analyzed according to the multi-exponential decay law:

The effect of immobilization in the PVA films…
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
7
where i and i are preexponential factor and decay lifetime of component I, respectively. The parameters describing the decay function were extracted from experimental data by a non-linear least squares convolution process. The goodness of the fit of fluorescence curves was judged by the reduced χ2 value.
Phosphorescence measurements Phosphorescence measurements were made on homemade system. The heart
of this system was 800 MHz gated photon counters with 32 bit counter resolution and down to 250 ns time per channel. (PMS-400A, Becker&Hickl GmbH). Emission was excited by UV xenon flash lamp with 400 ns pulses, light output stability 1.9% p-p and repetition rate up to 100 Hz. Intensity of light drops down 1000 times after 30 microseconds. (L9455-01, Hamamatsu). There were two moods of wave-length selection by monochromator or interference filters. Due to the low energy passing through the sample acquisition was stopped at 100 000 sweeps. The system was calibrated on aqueous solution of TbCl3. The lifetime was 426.5 microsecond and very well agreed with literature data (427 microsecond).
Since molecular oxygen is known to be a strong quenching agent as energy is easily transferred from the excited triplet state of the molecule to the triplet ground state of O2, molecular oxygen has to be efficiently removed from the sample. For phosphorescence measurements in PVA films, O2 removal was achieved by the application of moderate vacuum and inlet of ultrapure nitrogen. The pre-purified nitrogen gas was further purified by passing through an oxygen-trapping filter. For the measurements in solutions this degassing procedure was additionally accompanied by the addition of 0.3 ml of 0.1 M Na2SO3 as an O2 scavenger [15]. The sample (indole solution (10-4 M) with Na2SO3 or PVA film) was placed in quartz cuvette, which was connected to the N2/vacuum line by tubing. Five cycles of deoxygenation were performed. After deoxygenation, the cuvette was moved into the phosphorimeter, while remaining attached to the tubing and allowed to equilibrate to 30°C before taking the measurements. The background emission was determined by measurements carried out before deoxygenation of the sample and was normally subtracted from the phosphorescence decay.
All phosphorescence decays, after subtraction of the background, were analyzed in terms of a sum of exponential components by a nonlinear least squares fitting algorithm using the software provided by Edinburgh Analytical Instrumentation.
Results and Discussion Determination of quantum yield of indole and NATA
The fluorescence quantum yield is defined as the ratio of photons absorbed to photons emitted through fluorescence. The quantum yield of indole and NATA in water was determined using the comparative method of Williams et al. [16], in which tryptophan with known quantum yield value of 0.14 [2] was applied as a standard sample.
Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
8
It is known that the lowest energy absorption band of indoles originates from two close-lying electronic states with perpendicularly oriented transition dipole moments. These transitions, labeled as 1La (4.77eV) and 1Lb (4.37 eV), by analogy with the Platt scheme, differ substantially by their permanent dipole moments [17]. The 1La state has a larger static dipole moment than its ground state and thus it is more sensitive to solvation [18]. At longer wavelengths (>250 nm) both the 1La and 1Lb states contribute to the absorption spectrum of indoles in water, and around 260 nm, the contribution of the 1La state is dominant [18].
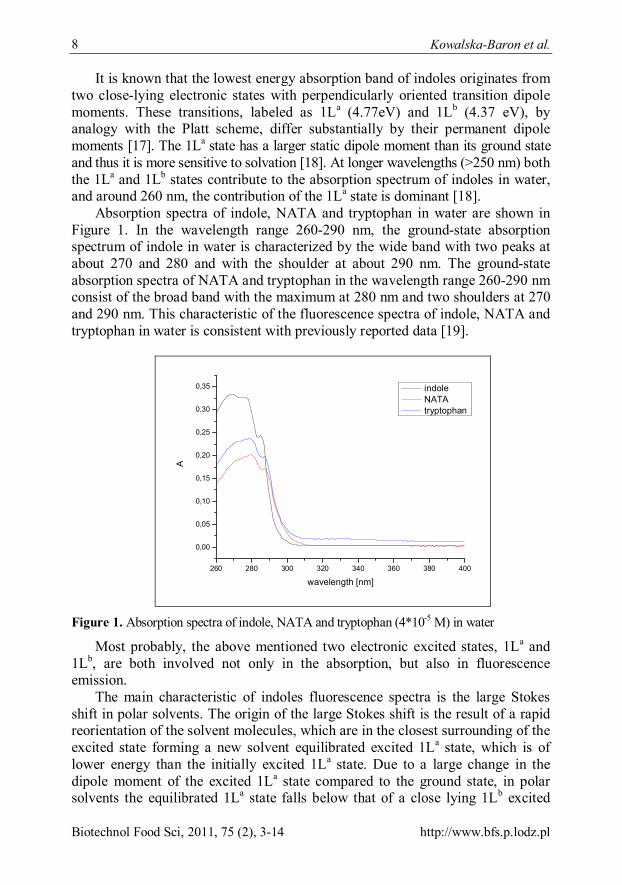
Absorption spectra of indole, NATA and tryptophan in water are shown in Figure 1. In the wavelength range 260-290 nm, the ground-state absorption spectrum of indole in water is characterized by the wide band with two peaks at about 270 and 280 and with the shoulder at about 290 nm. The ground-state absorption spectra of NATA and tryptophan in the wavelength range 260-290 nm consist of the broad band with the maximum at 280 nm and two shoulders at 270 and 290 nm. This characteristic of the fluorescence spectra of indole, NATA and tryptophan in water is consistent with previously reported data [19].
260 280 300 320 340 360 380 400
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
A
wavelength [nm]
indole NATA tryptophan
Figure 1. Absorption spectra of indole, NATA and tryptophan (4*10-5 M) in water
Most probably, the above mentioned two electronic excited states, 1La and 1Lb, are both involved not only in the absorption, but also in fluorescence emission.
The main characteristic of indoles fluorescence spectra is the large Stokes shift in polar solvents. The origin of the large Stokes shift is the result of a rapid reorientation of the solvent molecules, which are in the closest surrounding of the excited state forming a new solvent equilibrated excited 1La state, which is of lower energy than the initially excited 1La state. Due to a large change in the dipole moment of the excited 1La state compared to the ground state, in polar solvents the equilibrated 1La state falls below that of a close lying 1Lb excited
The effect of immobilization in the PVA films…
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
9
state. It has also been proposed that the large Stokes shift in polar hydrogen-bonding solvents is due to the formation of a solvent-indole(s) exciplexes. A third alternative for the large Stokes shift is that it may be due to emission from a solvated Rydberg state of indoles [20].
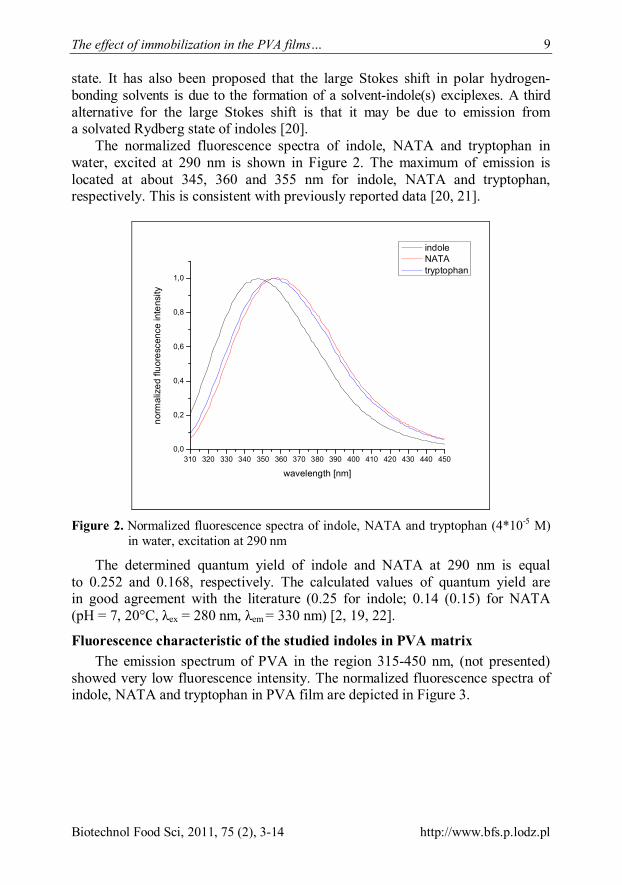
The normalized fluorescence spectra of indole, NATA and tryptophan in water, excited at 290 nm is shown in Figure 2. The maximum of emission is located at about 345, 360 and 355 nm for indole, NATA and tryptophan, respectively. This is consistent with previously reported data [20, 21].
310 320 330 340 350 360 370 380 390 400 410 420 430 440 4500,0
0,2
0,4
0,6
0,8
1,0
norm
aliz
ed fl
uore
scen
ce in
tens
ity
wavelength [nm]
indole NATA tryptophan
Figure 2. Normalized fluorescence spectra of indole, NATA and tryptophan (4*10-5 M)
in water, excitation at 290 nm
The determined quantum yield of indole and NATA at 290 nm is equal to 0.252 and 0.168, respectively. The calculated values of quantum yield are in good agreement with the literature (0.25 for indole; 0.14 (0.15) for NATA (pH = 7, 20°C, λex = 280 nm, λem = 330 nm) [2, 19, 22].
Fluorescence characteristic of the studied indoles in PVA matrix The emission spectrum of PVA in the region 315-450 nm, (not presented)
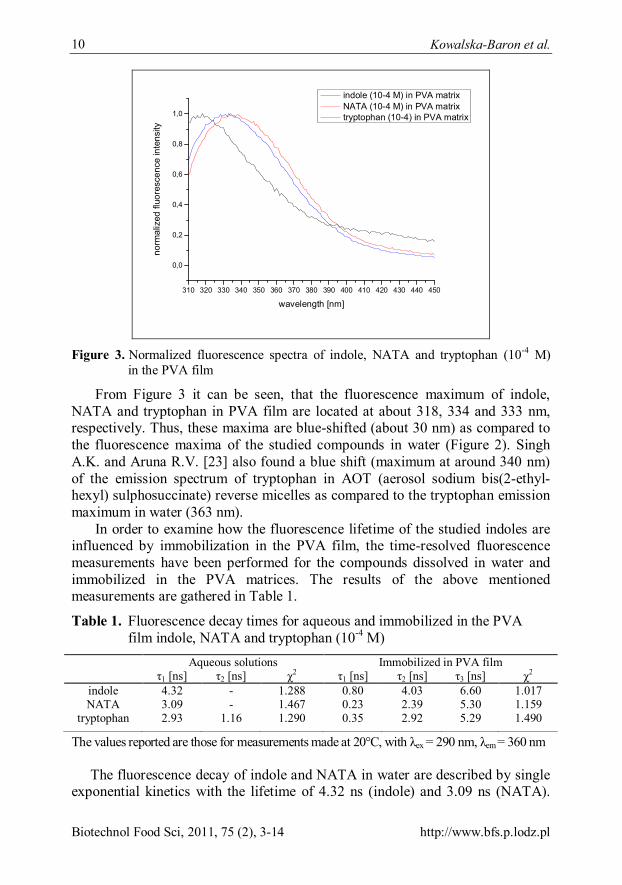
showed very low fluorescence intensity. The normalized fluorescence spectra of indole, NATA and tryptophan in PVA film are depicted in Figure 3.
Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
10
310 320 330 340 350 360 370 380 390 400 410 420 430 440 450
0,0
0,2
0,4
0,6
0,8
1,0
norm
aliz
ed fl
uore
scen
ce in
tens
ity
wavelength [nm]
indole (10-4 M) in PVA matrix NATA (10-4 M) in PVA matrix tryptophan (10-4) in PVA matrix
Figure 3. Normalized fluorescence spectra of indole, NATA and tryptophan (10-4 M)
in the PVA film
From Figure 3 it can be seen, that the fluorescence maximum of indole, NATA and tryptophan in PVA film are located at about 318, 334 and 333 nm, respectively. Thus, these maxima are blue-shifted (about 30 nm) as compared to the fluorescence maxima of the studied compounds in water (Figure 2). Singh A.K. and Aruna R.V. [23] also found a blue shift (maximum at around 340 nm) of the emission spectrum of tryptophan in AOT (aerosol sodium bis(2-ethyl-hexyl) sulphosuccinate) reverse micelles as compared to the tryptophan emission maximum in water (363 nm).
In order to examine how the fluorescence lifetime of the studied indoles are influenced by immobilization in the PVA film, the time-resolved fluorescence measurements have been performed for the compounds dissolved in water and immobilized in the PVA matrices. The results of the above mentioned measurements are gathered in Table 1.
Table 1. Fluorescence decay times for aqueous and immobilized in the PVA film indole, NATA and tryptophan (10-4 M)
Aqueous solutions Immobilized in PVA film τ1 [ns] τ2 [ns] χ2 τ1 [ns] τ2 [ns] τ3 [ns] χ2
indole 4.32 - 1.288 0.80 4.03 6.60 1.017 NATA 3.09 - 1.467 0.23 2.39 5.30 1.159
tryptophan 2.93 1.16 1.290 0.35 2.92 5.29 1.490
The values reported are those for measurements made at 20°C, with λex = 290 nm, λem = 360 nm The fluorescence decay of indole and NATA in water are described by single
exponential kinetics with the lifetime of 4.32 ns (indole) and 3.09 ns (NATA).
The effect of immobilization in the PVA films…
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
11
The obtained results are in reasonable agreement with those (4.8 ns (indole), pH = 7, 20°C, λex = 280 nm, λem = 330 nm) and 3.0 ns ((NATA), pH = 7, 20°C, λex = 280 nm, λem = 330 nm) measured by Szabo and coworkers [2] and Qiang and Seeger (2.85 ± 0.05 ns for NATA) [24].
Our results indicated that the fluorescence of tryptophan in water obeys double exponential decay kinetics with the two components about 3 ns and 1 ns. Szabo and Rayner [2] also demonstrated that the fluorescent decay of aqueous solutions of tryptophan could be resolved into two exponentially decaying components, 3.25±0.03 ns and 0.65±0.14 ns (pH = 7, 20°C, λex = 280 nm, λem = 360 nm). The authors demonstrated that the dual exponential decay of fluorescence of aqueous tryptophan originates from two or more rotamers along Cα-Cβ bond of L-tryptophan which has different configurations of alanyl side chain in reference to the indole nucleus. The rotamer with the ammonium group closest to the indole ring has been assigned as the 3 ns component, while the rotamer with the carboxylate group closest to the indole ring (the α-ammonium group is farthest away from the indole ring) is the short-lived (0.6 ns) component.
The fluorescence decay of indole, NATA and tryptophan immobilized in PVA film may be described by three exponential kinetics with the longer-lived component of about 5 ns (NATA, tryptophan) or 6.60 ns (indole), which may be attributed to the studied indoles immobilized in the PVA matrices. It may be concluded that the immobilization of the studied indoles in the PVA matrices leads to the increase of fluorescence lifetime. Zelent and coworkers [25] also reported that the changes in microviscosity of NATA leads to the increase of singlet state lifetime (the fluorescence lifetime of NATA in propylene glycol at 20°C has been determined to be about 5.3 ns).
Phosphorescence lifetime of the studied indoles in PVA matrix In order to verify how the immobilization of the studied indoles affects the
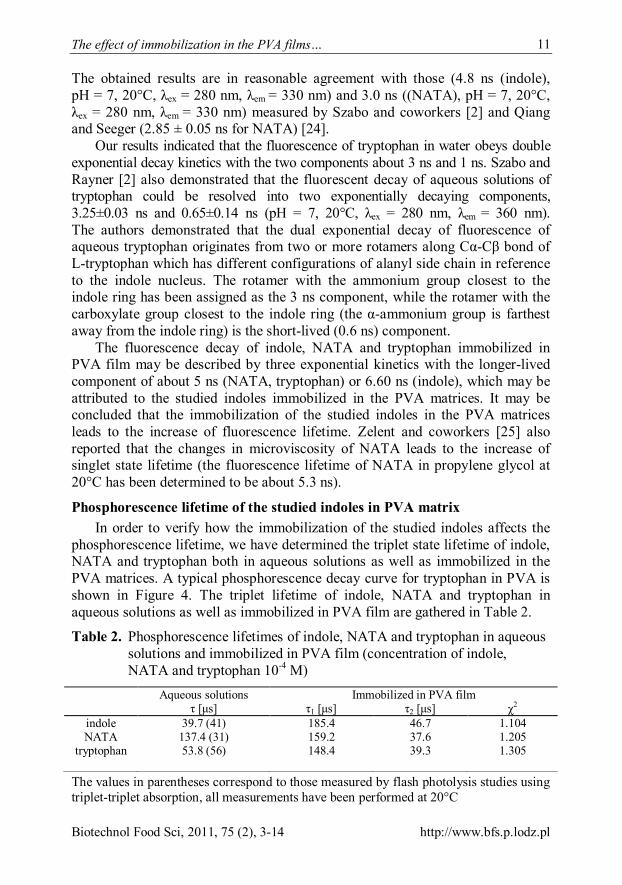
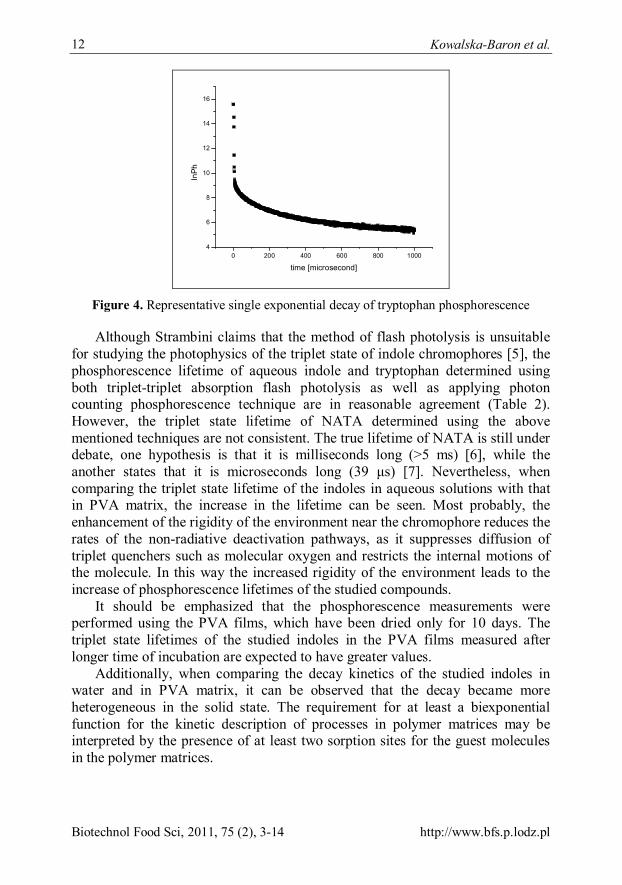
phosphorescence lifetime, we have determined the triplet state lifetime of indole, NATA and tryptophan both in aqueous solutions as well as immobilized in the PVA matrices. A typical phosphorescence decay curve for tryptophan in PVA is shown in Figure 4. The triplet lifetime of indole, NATA and tryptophan in aqueous solutions as well as immobilized in PVA film are gathered in Table 2.
Table 2. Phosphorescence lifetimes of indole, NATA and tryptophan in aqueous solutions and immobilized in PVA film (concentration of indole, NATA and tryptophan 10-4 M)
Aqueous solutions Immobilized in PVA film τ [μs] τ1 [μs] τ2 [μs] χ2
indole 39.7 (41) 185.4 46.7 1.104 NATA 137.4 (31) 159.2 37.6 1.205
tryptophan 53.8 (56) 148.4 39.3 1.305
The values in parentheses correspond to those measured by flash photolysis studies using triplet-triplet absorption, all measurements have been performed at 20°C
Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
12
0 200 400 600 800 10004
6
8
10
12
14
16
lnPh
time [microsecond]
Figure 4. Representative single exponential decay of tryptophan phosphorescence
Although Strambini claims that the method of flash photolysis is unsuitable for studying the photophysics of the triplet state of indole chromophores [5], the phosphorescence lifetime of aqueous indole and tryptophan determined using both triplet-triplet absorption flash photolysis as well as applying photon counting phosphorescence technique are in reasonable agreement (Table 2). However, the triplet state lifetime of NATA determined using the above mentioned techniques are not consistent. The true lifetime of NATA is still under debate, one hypothesis is that it is milliseconds long (>5 ms) [6], while the another states that it is microseconds long (39 μs) [7]. Nevertheless, when comparing the triplet state lifetime of the indoles in aqueous solutions with that in PVA matrix, the increase in the lifetime can be seen. Most probably, the enhancement of the rigidity of the environment near the chromophore reduces the rates of the non-radiative deactivation pathways, as it suppresses diffusion of triplet quenchers such as molecular oxygen and restricts the internal motions of the molecule. In this way the increased rigidity of the environment leads to the increase of phosphorescence lifetimes of the studied compounds.
It should be emphasized that the phosphorescence measurements were performed using the PVA films, which have been dried only for 10 days. The triplet state lifetimes of the studied indoles in the PVA films measured after longer time of incubation are expected to have greater values.
Additionally, when comparing the decay kinetics of the studied indoles in water and in PVA matrix, it can be observed that the decay became more heterogeneous in the solid state. The requirement for at least a biexponential function for the kinetic description of processes in polymer matrices may be interpreted by the presence of at least two sorption sites for the guest molecules in the polymer matrices.

The effect of immobilization in the PVA films…
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
13
Conclusions In this work the fluorescence and phosphorescence techniques have been
applied in order to study the influence immobilization in PVA matrixes on the singlet and triplet excited state lifetime of indole and its derivatives. The results indicated that the maximum of fluorescence for all the studied indoles in the PVA matrix is approximately 30 nm blue shifted in comparison to the fluorescence maxima of the studied compounds in aqueous solutions.
Moreover, the obtained results showed that the immobilization in PVA matrix leads to the increase of both fluorescence and phosphorescence lifetime of the studied indoles, which may suggest that exploiting protective screening effect of organized media restricts the internal motion of the molecules (which minimizes self-quenching) and suppresses diffusion of triplet quenchers, which all together result in the increase of phosphorescence lifetime of the studied indoles.
Acknowledgements The authors would like to thank Prof. S. Wysocki, head of the Institute of
General Food Chemistry, who founded the project and provided the equipment.
References 1. Lakowicz JR, Protein fluorescence. In: Principles of fluorescence spectroscopy. 2nd
ed. Kluwer Academic/Plenum Publishers, New York, USA 1999, pp. 446-485. 2. Szabo AG, Rayner D.M, Fluorescence decay of tryptophan conformers in aqueous
solution. J Am Chem Soc 1980, 101: 554-563. 3. Millar DP. Time-resolved fluorescence spectroscopy. Curr Opin Struct Biol 1996,
6:637-642. 4. Strambini EG, Strambini GB, Tryptophan phosphorescence as a monitor of protein
conformations in molecular films. Biosens Bioelectron 2000, 15:483-490. 5. Strambini GB, Gonnelli M, Tryptophan phosphorescence in fluid solution. J Am
Chem Soc 1995, 117:7646-7651. 6. Strambini GB., Kerwin BA, Mason BD. Gonelli M, The triplet-state lifetime of
indole derivatives in aqueous solution. Photochem Photobiol 2004, 80:462-470. 7. Fischer CJ, Gafni A, Steel DG, Schauerte JA, The triplet-state lifetime of indole in
aqueous and viscous environments: significance to the interpretation of room temperature phosphorescence in proteins. J Am Chem Soc 2002, 124:10359-10366.
8. Bent DV, Hayon E, Excited state chemistry of aromatic amino acids and related peptides. III. Tryptophan. J Am Chem Soc 1975, 97:2612-2619.
9. Tsentalowich Y, Snytnikova OA, Sagdeev RZ, Properties of excited states of aqueous tryptophan. J Photochem Photobiol A Chem 2004, 162:371-379.
10. Strambini GB, Gonnelli M. Tryptophan phosphorescence in fluid solutions. J Am Chem Soc 1995, 117:7646-7651.
11. Banks DD, Kerwin BA, A deoxygenation system for measuring protein phosphorescence. Anal Biochem 2004, 324:106-114.
12. Strambini GB, Gonelli M, The indole nucleus triplet-state lifetime and its dependence on solvent microviscosity. Chem Phys Lett 1985, 115:196-204.

Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 3-14 http://www.bfs.p.lodz.pl
14
13. Dashnau JL, Zelent B, Vanderkooi JM, Tryptophan interactions with glycerol/ water and trehalose/sucrose cryosolvents: infrared and fluorescence spectroscopy and ab initio calculations. Biophys Chem 2005, 114:71-83.
14. Rung-Shu C, Yi-Jane C, Min-Huey C, Tai-Horng Y, The behavior of rat tooth germ cells on poly(vinyl alcohol). Act Biomater 2009, 5:1064-1074.
15. Kuijt J, Ariese F, Brinkman UAT, Gooijer C, Room temperature phosphorescence in the liquid state as a tool in analytical chemistry. Anal Chim Acta 2003, 488:135-171.
16. Williams ATR., Winfield SA, Miller JN, Relative fluorescence quantum yields using a computer controlled luminescence spectrometer. Analyst 1983, 108:1067-1068.
17. Somers KRF, Kryachko ES, Ceulemans A, Theoretical study of indole: protonation, indolyl radical, tautomers of indole, and its interaction with water. Chem Phys 2004, 301:61-79.
18. Kamath SD, Kartha VB, Mahato KK, Dynamics of L-tryptophan in aqueous solution by simultaneous laser induced fluorescence (LIF) and photoacoustic spectroscopy (PAS). Spectrochim Act A 2008, 70:187-19.
19. Ryuzi K, Dependence of photoionization quantum yield of indole and tryptophan in water on excitation wavelength. J Photochem Photobiol A 2007, 189:211-217.
20. Feng S, Zong W, Liu R, Chai J, Liu Y, Micro-environmental influences on the fluorescence of tryptophan. Spectrochim Act A, 2010, 76:142-145.
21. Sau AK, Mitra S, Steady state and picosecond time-resolved fluorescence studies on native, desulpho and deflavo xanthine oxidase. Biochim Biophys Act 2000, 1481:273-282.
22. Kirby EP, Steiner RF, The influence of solvent and temperature upon the fluorescence of indole derivatives. J Phys Chem 1970, 74:4480-4490.
23. Singh AK, Aruna RV, Fluorescence studies of tryptophan and tryptophan-retinol Shiff base in reverse micellar matrix. J Photochem Photobiol A 1995, 89:247-250.
24. Qiang L, Seeger S, Label-free detection of protein interactions using deep UV fluorescence lifetime microscopy. Anal Biochem 2007, 367:104-110.
25. Zelent B, Kuśba J, Gryczynski I, Johnson MJ, Lakowicz JR, Time-resolved and steady state fluorescence quenching of N-acetyl-L-tryptophanamide by acrylamide and iodide. Biophys Chem 1998, 73:53-75.

Biotechnology and Food Science Research article
Biotechnol Food Sci 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
Ibuprofen-tyrosine (Val-Tyr, Val-Tyr-Val) interactions. Theoretical and experimental studies
Agnieszka Kowalska-Baron1*, Michaela Brychtova2, Ivana Petrović3, Igor Passos Sene4, Paloma Quiñones5, Jana Šogorkova6
1 Institute of General Food Chemistry, Faculty of Biotechnology and Food Science, Technical University of Lodz, 90-924 Lodz, Poland
2 Faculty of Technology, Tomas Bata University in Zlin, 762 72, Zlín, Czech Republic
3 Faculty of Food Technology, The University of Josip Juraj Strossmayer, 31000, Osijek, Croatia
4 Faculty of Food Engineering, State University of Campinas (UNICAMP), 13035-388, Campinas, Brazil
5 Instituto Tecnológico de Durango, Chemical and Biochemical Department, 1803, Victoria de Durango, México
6 Institute of Chemical Technology, Faculty of Food and Biochemical Technology, 166 28, Prague, Czech Republic
Abstract: In this work, the interactions between ibuprofen and tyrosine as well selected tyrosine-containing oligopeptides (Val-Tyr, Val-Tyr-Val) have been studied using both experimental (absorption and fluorescence spectroscopy) and theoretical (the PM3 method) techniques. The experimentally obtained values of association constant together with free Gibbs energy of association for the studied tyrosine-ibuprofen and Val-Tyr-; Val-Tyr-Val-ibuprofen complexes have been determined. The obtained results indicated that the mechanism of action of ibuprofen is most probably based on the hydrogen bonding interaction which involves hydroxyl group of tyrosine.
Keywords: ibuprofen; hydrogen bonding interactions; PM3 calculations
Introduction The influence of drugs on human body is measured by their pharmacokinetics
and pharmacodynamics. The former is the study of drug processing in the body which includes: absorption, distribution, metabolism and excretion of the drug in the human body. The latter is a study about mechanism of action of the drug on the human body [1]. Ibuprofen, 2-[4-(2-methylpropyl)phenyl] propanoic acid, (Figure 1), is a drug from the group of non-steroidal anti-inflammatory drugs (NSAID). This compound is usually used as analgesic, antipyretic and anti-

Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
16
rheumatic [2]. Ibuprofen is used to reduce fever, inflammation and sensation pain, as most probably, this drug works by inhibiting the action of prostaglandins [1].
Figure 1. Molecular structure of ibuprofen
Prostaglandins are chemicals that cause inflammation and contribute to the brains perception of pain [3]. Ibuprofen reduces fever by blocking prostaglandin synthesis in the hypothalamus, a structure in the brain that regulates body temperature. The accurately mechanism of action of ibuprofen and other NSAID is not completely known. It is very likely that limitation of prostaglandin synthesis due to cyclooxygenase (COX) inhibition is involved in it.
Cyclooxygenases (COX) are enzymes which could be found in three isoforms of COX1, COX2 and COX3, have an active role in the production of prostaglandins. COX1 catalyzes the normal production of prostaglandins in the body. The second isoform has a role in production of prostaglandins in inflammatory cells. COX 3 has been found to be selectively inhibited by paracetamol, phenacetin and antipyrine. There are two active sites on COX1: cyclooxygenase and a peroxidase. COX1 catalyzes the production of prostaglandins by removing a hydrogen atom from arachidonic acid and transferring it to Tyr-385 in the active site of COX1. A hydrogen bond between Tyr-348-Tyr385 is crucial for this activity [3]. Any interruption has an influence in changing prostaglandins synthesis. Due to the role that tyrosine residues have in the biological activity of COX1, tyrosine and some tyrosine-containing oligopeptides (dipeptide (Val-Tyr), tripeptide (Val-Tyr-Val)) became also main object in this study.
Tyrosine is one of the 20 basic amino acids. It is extremely reactive because of its hydrophilic side chain. Tyrosine is also an essential amino acid. It has a role in the intercellular transport, synthesis of hormones, and biologically active substances such as adrenaline, noradrenaline and dopamine [3]. Additionally, tyrosine is the largest constituent of all residues at the active site of cyclooxygenase.
The knowledge of the intermolecular interactions between tyrosine residue and ibuprofen might shed light into the complex mechanism of action of ibuprofen. Therefore, the aim of this study is to investigate the molecular interactions between ibuprofen and tyrosine residue in aqueous solution. This

Ibuprofen-tyrosine interactions…
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
17
aim will be obtained using both experimental (absorption and fluorescence spectroscopy) and theoretical (semi-empirical PM3) methods.
Experimental Materials
Ibuprofen, L-valine-L-tyrosine, L-valine-L-tyrosine-L-valine and L-tyrosine used in experimental part of this project were commercial products produced by Sigma-Aldrich. Dipeptide, tyrosine and tripeptide were dissolved in distilled water. Because of very poor solubility of ibuprofen in water, the stock solution of ibuprofen (0,05 M) was prepared in ethanol. All reagents were least analytical grade and were used without further purification.
Experimental methods Nicolet Evolution 300 UV-Vis spectrophotometer from Thermo Electron
Corporation with resolution 0,5 nm and range 0-6 was used in absorption measurements. Spectrofluorometer Generic Fluoromax-4P from Horiba Jobin Yvon was used in steady state measurements. Solutions were placed into a 10 mm quartz cuvette. During the measurements, temperature was constant (25 °C). In titration experiments the concentration of tyrosine, dipeptide and tripeptide was kept constant (5*10-5 M) while varying the concentration of ibuprofen.
Computational methods The geometries of tyrosine, dipeptide, tripeptide (in zwitterionic form) and
ibuprofen (anion) as well as tyrosine (dipeptide, tripeptide)-ibuprofen complexes have been optimized applying the PM3 method implemented in Gaussian 03 program. We have also performed frequency calculations of the above mentioned systems at the same level of theory. The analysis of the PM3 calculated frequencies have been performed to verify whether the optimized structures correspond to the minima. Harmonic oscillator approximation has been used in the vibration calculations.
The ground state interaction energies (ΔE) of the interacting systems have been calculated using the supermolecule approach [4] as the electronic energy difference between the complex and the isolated molecules (tyrosine, di- and tri-peptide, ibuprofen):
∆퐸 = 퐸 − (퐸 ( , ) + 퐸 )
The entalphy of association was calculated using the following equation from the data listed in the output of Gaussian calculation without scaling their values:
∆퐻 = 퐻 − (퐻 ( , + 퐻 )
All calculations have been performed in the gas phase with the use of Gaussian 03 program [5].
Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
18
Results and Discussion UV-Vis spectroscopy
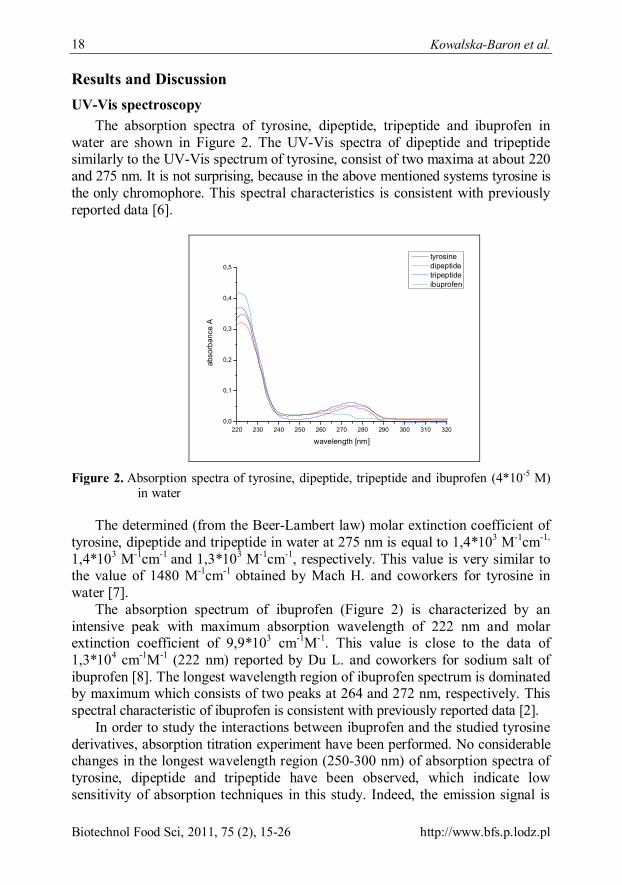
The absorption spectra of tyrosine, dipeptide, tripeptide and ibuprofen in water are shown in Figure 2. The UV-Vis spectra of dipeptide and tripeptide similarly to the UV-Vis spectrum of tyrosine, consist of two maxima at about 220 and 275 nm. It is not surprising, because in the above mentioned systems tyrosine is the only chromophore. This spectral characteristics is consistent with previously reported data [6].
220 230 240 250 260 270 280 290 300 310 3200,0
0,1
0,2
0,3
0,4
0,5
abso
rban
ce A
wavelength [nm]
tyrosine dipeptide tripeptide ibuprofen
Figure 2. Absorption spectra of tyrosine, dipeptide, tripeptide and ibuprofen (4*10-5 M)
in water
The determined (from the Beer-Lambert law) molar extinction coefficient of tyrosine, dipeptide and tripeptide in water at 275 nm is equal to 1,4*103 M-1cm-1,
1,4*103 M-1cm-1 and 1,3*103 M-1cm-1, respectively. This value is very similar to the value of 1480 M-1cm-1 obtained by Mach H. and coworkers for tyrosine in water [7].
The absorption spectrum of ibuprofen (Figure 2) is characterized by an intensive peak with maximum absorption wavelength of 222 nm and molar extinction coefficient of 9,9*103 cm-1M-1. This value is close to the data of 1,3*104 cm-1M-1 (222 nm) reported by Du L. and coworkers for sodium salt of ibuprofen [8]. The longest wavelength region of ibuprofen spectrum is dominated by maximum which consists of two peaks at 264 and 272 nm, respectively. This spectral characteristic of ibuprofen is consistent with previously reported data [2].
In order to study the interactions between ibuprofen and the studied tyrosine derivatives, absorption titration experiment have been performed. No considerable changes in the longest wavelength region (250-300 nm) of absorption spectra of tyrosine, dipeptide and tripeptide have been observed, which indicate low sensitivity of absorption techniques in this study. Indeed, the emission signal is

Ibuprofen-tyrosine interactions…
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
19
directly observed, enhanced and registered, while in absorption spectroscopy the difference between the incident and transmitted light intensity is measured. Due to low sensitivity of absorption technique in the study of intermolecular interactions between tyrosine derivatives and ibuprofen, the fluorescence spectroscopy will be used to determine association constant of tyrosine (dipeptide, tripeptide)-ibuprofen complexes.
Fluorescence spectroscopy The steady-state fluorescence spectrum of tyrosine (excited at 280 nm) in
water (not shown) shows the maximum at 303 nm, which is consistent with previously reported data [9]. Fluorescence spectra of dipeptide (Figure 3) and tripeptide (not shown) in water show maximum at 309 nm. This red shift of 6 nm with respect to tyrosine fluorescence maximum may suggest that tyrosine emission is very sensitive to the surrounding medium (the presence of valine residue in the studied di- and tripeptide). Fluorescence intensity of ibuprofen (5*10-6 M-1*10-4 M) in water (not shown) is very low with the maximum at about 340 nm, which is consistent with previously reported data [2].
Complex formation between Tyr (dipeptide, tripeptide) (represented by M) and ibuprofen (represented by L) proceeds according to:
[푀] + [퐿] ⇆ [푀퐿]
The association constant (Ka) of the complex can be expressed as:
퐾 =[푀]
[푀][퐿] eq. 2
[M] and [L] is the concentration of free (non-bonded in complex), tyrosine (dipeptide, tripeptide) and ibuprofen; respectively and can be expressed as:
[푀] = [푀] − [푀퐿]
[퐿] = [퐿] − [푀퐿] eq. 3
where [M]0 and [L]0 is the analytical concentration of tyrosine (dipeptide, tripeptide) and ibuprofen, respectively; [ML] is the concentration of the complex. Assuming that [퐿] ⋙ [퐿] ([퐿] → ∞) [퐿] = [퐿] , and inserting equation 3 to 2 we obtain:
[푀퐿] =퐾 . [푀] [퐿]1 + 퐾 . [퐿] eq. 4
It is known that the fluorescence can be expressed as:
퐹 = 훼 . 휙 . 휀 . 푐 eq. 5

Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
20
where: α – apparatus factor φi – fluorescence quantum yield of i εi – molar extinction coefficient of i ci – concentration of i Under the condition that 퐴 ≤ 0,1, the fluorescence intensity of the mixture of tyrosine (dipeptide, tripeptide) and ibuprofen (Fmix) can be written as:
퐹 = 훼 . 휙 . 휀 [푀] + 훼 . 휙 . 휀 [퐿] + 훼 . 휙 . 휀 [푀퐿] eq. 6
Inserting equation 3 to equation 6 and taking into consideration that 휙 ≅ 0 we obtain:
퐹 = 훼 . { 휙 . 휀 [푀] + (휙 . 휀 − 휙 . 휀 )[푀퐿]}
eq. 7
In the absence of ibuprofen, the fluorescence intensity is equal to:
퐹 = 훼 . 휙 . 휀 [푀] eq. 8
If [퐿] → ∞, all tyrosine is incorporated in complexes formation, therefore we can write:
퐹 = 훼 . 휙 . 휀 [푀] eq. 9
Inserting eq. 8 and eq. 9 to eq. 7 we obtain:
퐹 = 퐹 + [푀퐿]퐹
[푀] −퐹
[푀] eq. 10
Inserting eq. 4 to eq. 10 and converting, we finally come to the following formula:
퐹 − 퐹퐹 − 퐹 = 퐾 [퐿] eq. 11
In this way the slope of : 퐹 − 퐹퐹 − 퐹 = 푓[퐿]
enables us to determine Ka of tyrosine–ibuprofen, dipeptide–ibuprofen and tripeptide–ibuprofen complexes.
Ibuprofen-tyrosine interactions…
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
21
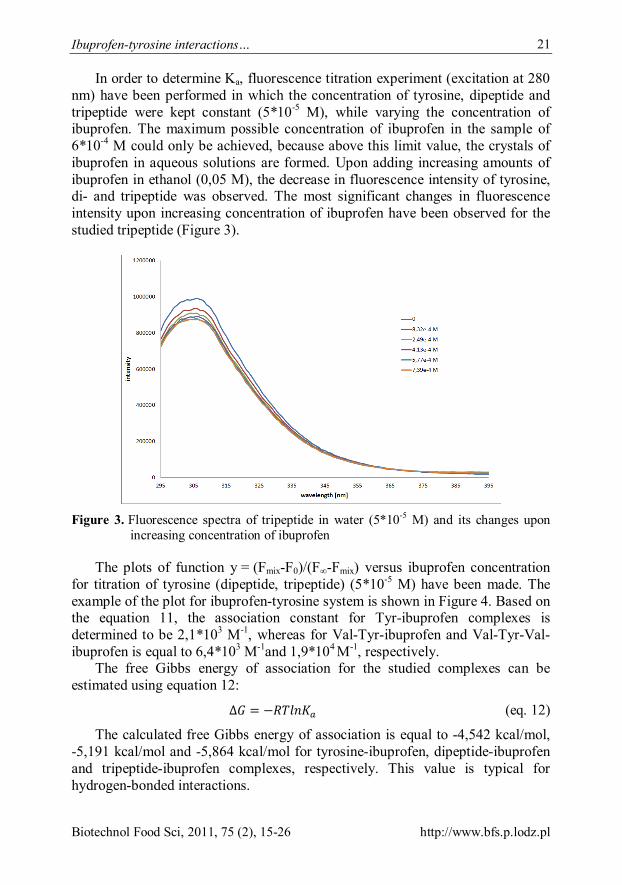
In order to determine Ka, fluorescence titration experiment (excitation at 280 nm) have been performed in which the concentration of tyrosine, dipeptide and tripeptide were kept constant (5*10-5 M), while varying the concentration of ibuprofen. The maximum possible concentration of ibuprofen in the sample of 6*10-4 M could only be achieved, because above this limit value, the crystals of ibuprofen in aqueous solutions are formed. Upon adding increasing amounts of ibuprofen in ethanol (0,05 M), the decrease in fluorescence intensity of tyrosine, di- and tripeptide was observed. The most significant changes in fluorescence intensity upon increasing concentration of ibuprofen have been observed for the studied tripeptide (Figure 3).
Figure 3. Fluorescence spectra of tripeptide in water (5*10-5 M) and its changes upon
increasing concentration of ibuprofen
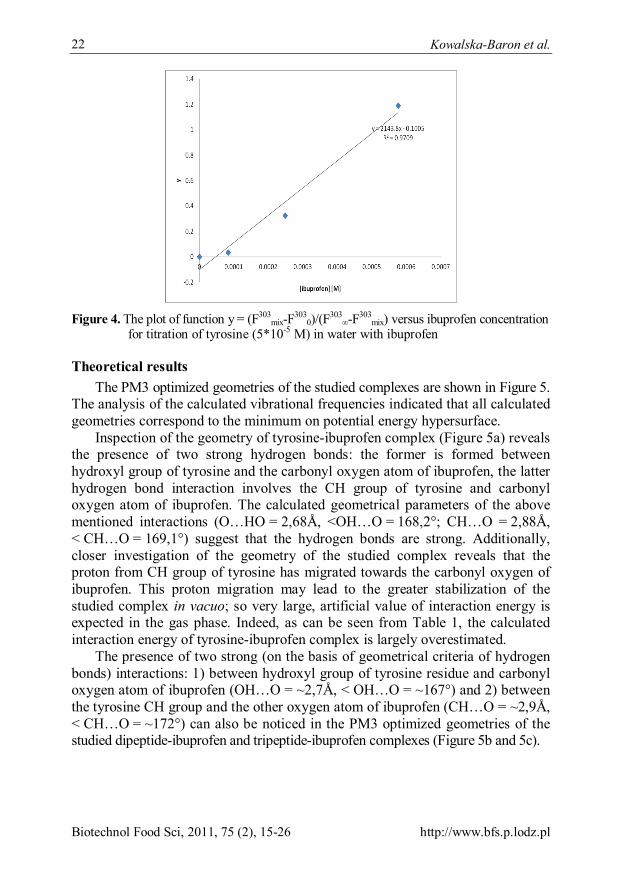
The plots of function y = (Fmix-F0)/(F∞-Fmix) versus ibuprofen concentration for titration of tyrosine (dipeptide, tripeptide) (5*10-5 M) have been made. The example of the plot for ibuprofen-tyrosine system is shown in Figure 4. Based on the equation 11, the association constant for Tyr-ibuprofen complexes is determined to be 2,1*103 M-1, whereas for Val-Tyr-ibuprofen and Val-Tyr-Val-ibuprofen is equal to 6,4*103 M-1and 1,9*104 M-1, respectively.
The free Gibbs energy of association for the studied complexes can be estimated using equation 12:
∆퐺 = −푅푇푙푛퐾 (eq. 12)
The calculated free Gibbs energy of association is equal to -4,542 kcal/mol, -5,191 kcal/mol and -5,864 kcal/mol for tyrosine-ibuprofen, dipeptide-ibuprofen and tripeptide-ibuprofen complexes, respectively. This value is typical for hydrogen-bonded interactions.
Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
22
Figure 4. The plot of function y = (F303
mix-F3030)/(F303
∞-F303mix) versus ibuprofen concentration
for titration of tyrosine (5*10-5 M) in water with ibuprofen
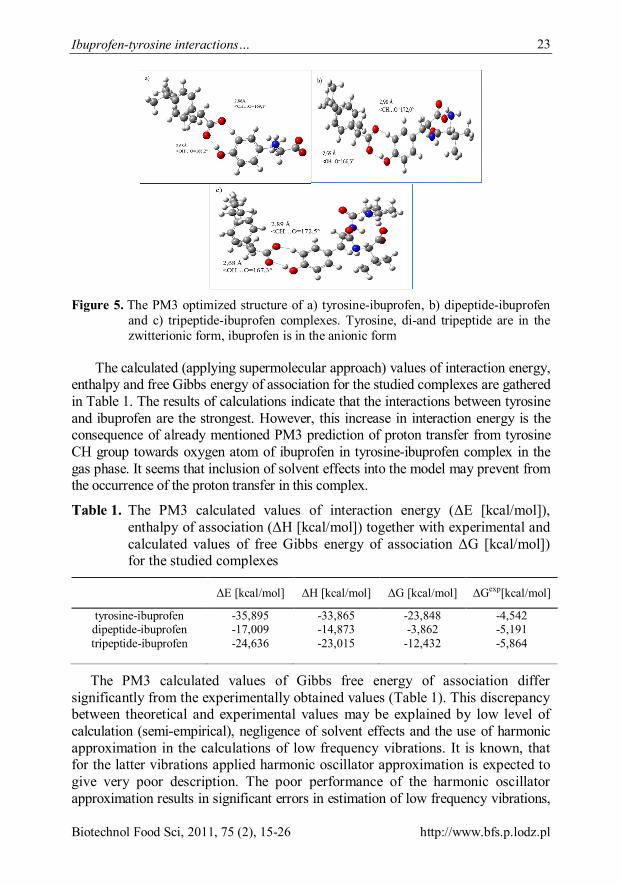
Theoretical results The PM3 optimized geometries of the studied complexes are shown in Figure 5.
The analysis of the calculated vibrational frequencies indicated that all calculated geometries correspond to the minimum on potential energy hypersurface.
Inspection of the geometry of tyrosine-ibuprofen complex (Figure 5a) reveals the presence of two strong hydrogen bonds: the former is formed between hydroxyl group of tyrosine and the carbonyl oxygen atom of ibuprofen, the latter hydrogen bond interaction involves the CH group of tyrosine and carbonyl oxygen atom of ibuprofen. The calculated geometrical parameters of the above mentioned interactions (O…HO = 2,68Å, <OH…O = 168,2°; CH…O = 2,88Å, < CH…O = 169,1°) suggest that the hydrogen bonds are strong. Additionally, closer investigation of the geometry of the studied complex reveals that the proton from CH group of tyrosine has migrated towards the carbonyl oxygen of ibuprofen. This proton migration may lead to the greater stabilization of the studied complex in vacuo; so very large, artificial value of interaction energy is expected in the gas phase. Indeed, as can be seen from Table 1, the calculated interaction energy of tyrosine-ibuprofen complex is largely overestimated.
The presence of two strong (on the basis of geometrical criteria of hydrogen bonds) interactions: 1) between hydroxyl group of tyrosine residue and carbonyl oxygen atom of ibuprofen (OH…O = ~2,7Å, < OH…O = ~167°) and 2) between the tyrosine CH group and the other oxygen atom of ibuprofen (CH…O = ~2,9Å, < CH…O = ~172°) can also be noticed in the PM3 optimized geometries of the studied dipeptide-ibuprofen and tripeptide-ibuprofen complexes (Figure 5b and 5c).
Ibuprofen-tyrosine interactions…
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
23
Figure 5. The PM3 optimized structure of a) tyrosine-ibuprofen, b) dipeptide-ibuprofen
and c) tripeptide-ibuprofen complexes. Tyrosine, di-and tripeptide are in the zwitterionic form, ibuprofen is in the anionic form
The calculated (applying supermolecular approach) values of interaction energy, enthalpy and free Gibbs energy of association for the studied complexes are gathered in Table 1. The results of calculations indicate that the interactions between tyrosine and ibuprofen are the strongest. However, this increase in interaction energy is the consequence of already mentioned PM3 prediction of proton transfer from tyrosine CH group towards oxygen atom of ibuprofen in tyrosine-ibuprofen complex in the gas phase. It seems that inclusion of solvent effects into the model may prevent from the occurrence of the proton transfer in this complex.
Table 1. The PM3 calculated values of interaction energy (ΔE [kcal/mol]), enthalpy of association (ΔH [kcal/mol]) together with experimental and calculated values of free Gibbs energy of association ΔG [kcal/mol]) for the studied complexes
ΔE [kcal/mol] ΔH [kcal/mol] ΔG [kcal/mol] ΔGexp[kcal/mol]
tyrosine-ibuprofen -35,895 -33,865 -23,848 -4,542 dipeptide-ibuprofen -17,009 -14,873 -3,862 -5,191 tripeptide-ibuprofen -24,636 -23,015 -12,432 -5,864
The PM3 calculated values of Gibbs free energy of association differ significantly from the experimentally obtained values (Table 1). This discrepancy between theoretical and experimental values may be explained by low level of calculation (semi-empirical), negligence of solvent effects and the use of harmonic approximation in the calculations of low frequency vibrations. It is known, that for the latter vibrations applied harmonic oscillator approximation is expected to give very poor description. The poor performance of the harmonic oscillator approximation results in significant errors in estimation of low frequency vibrations,
Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
24
which leads to very large errors in calculated entropies, which in turn means large errors in free energy. Therefore, the calculated ΔG of the association is not reliable to predict reasonable stabilities of the system studied [10].
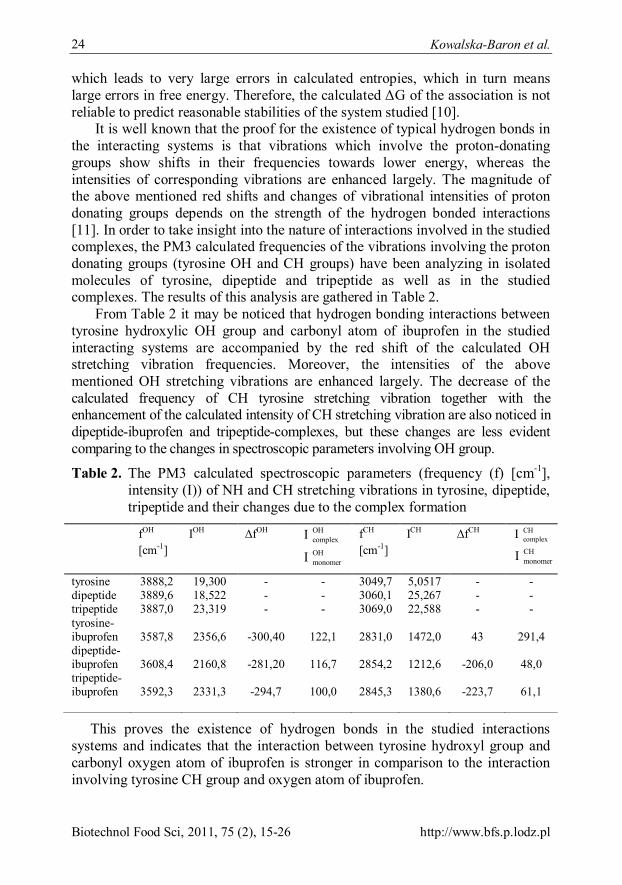
It is well known that the proof for the existence of typical hydrogen bonds in the interacting systems is that vibrations which involve the proton-donating groups show shifts in their frequencies towards lower energy, whereas the intensities of corresponding vibrations are enhanced largely. The magnitude of the above mentioned red shifts and changes of vibrational intensities of proton donating groups depends on the strength of the hydrogen bonded interactions [11]. In order to take insight into the nature of interactions involved in the studied complexes, the PM3 calculated frequencies of the vibrations involving the proton donating groups (tyrosine OH and CH groups) have been analyzing in isolated molecules of tyrosine, dipeptide and tripeptide as well as in the studied complexes. The results of this analysis are gathered in Table 2.
From Table 2 it may be noticed that hydrogen bonding interactions between tyrosine hydroxylic OH group and carbonyl atom of ibuprofen in the studied interacting systems are accompanied by the red shift of the calculated OH stretching vibration frequencies. Moreover, the intensities of the above mentioned OH stretching vibrations are enhanced largely. The decrease of the calculated frequency of CH tyrosine stretching vibration together with the enhancement of the calculated intensity of CH stretching vibration are also noticed in dipeptide-ibuprofen and tripeptide-complexes, but these changes are less evident comparing to the changes in spectroscopic parameters involving OH group.
Table 2. The PM3 calculated spectroscopic parameters (frequency (f) [cm-1], intensity (I)) of NH and CH stretching vibrations in tyrosine, dipeptide, tripeptide and their changes due to the complex formation
fOH [cm-1]
IOH ΔfOH OHcomplexI
OHmonomerI
fCH [cm-1]
ICH ΔfCH CHcomplexI
CHmonomerI
tyrosine 3888,2 19,300 - - 3049,7 5,0517 - - dipeptide 3889,6 18,522 - - 3060,1 25,267 - - tripeptide 3887,0 23,319 - - 3069,0 22,588 - - tyrosine-ibuprofen 3587,8 2356,6 -300,40 122,1 2831,0 1472,0 43 291,4 dipeptide-ibuprofen 3608,4 2160,8 -281,20 116,7 2854,2 1212,6 -206,0 48,0 tripeptide-ibuprofen 3592,3 2331,3 -294,7 100,0 2845,3 1380,6 -223,7 61,1
This proves the existence of hydrogen bonds in the studied interactions systems and indicates that the interaction between tyrosine hydroxyl group and carbonyl oxygen atom of ibuprofen is stronger in comparison to the interaction involving tyrosine CH group and oxygen atom of ibuprofen.

Ibuprofen-tyrosine interactions…
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
25
Additionally, the vibrational analysis reveal that the interaction involving CH group of tyrosine and ibuprofen oxygen atom in the tyrosine-ibuprofen complex in vacuo is not a typical hydrogen bond (as the result of complex formation blue shift instead of red shift of tyrosine CH stretching frequency has been predicted by the PM3 method).
In conclusions, the interactions between ibuprofen and tyrosine as well selected tyrosine-containing oligopeptides (Val-Tyr, Val-Tyr-Val) have been studied using both experimental (absorption and fluorescence spectroscopy) and theoretical (the PM3 method) techniques. The experimentally obtained values of association constant together with free Gibbs energy of association for the studied tyrosine-ibuprofen and Val-Tyr-; Val-Tyr-Val-ibuprofen complexes have been determined as the results of fluorescence titration experiment of the studied tyrosine derivatives with ibuprofen. The obtained values suggest that, assuming that the complexes of 1:1 stoichiometry are only formed, the intermolecular interactions involved in ibuprofen/tripeptide and ibuprofen/dipeptide are stronger in comparison to the interactions between tyrosine and ibuprofen.
Theoretical calculations failed to provide reasonable values of free Gibbs energy of association. The reason for this is the poor performance of applied harmonic oscillator approximation in the calculations of low-frequency vibrations. However, the results of PM3 calculations revealed the nature of interactions involved in ibuprofen/Tyr (Val-Tyr, Val-Tyr-Val) systems. The above mentioned interactions are mainly based on the formation of strong hydrogen bond between the tyrosine hydroxyl group and carbonyl oxygen atom of ibuprofen. Additionally, the presence of the interaction incorporating tyrosine ring CH-group, which is close to tyrosine hydroxyl group, enhanced the stability of the studied interacting systems. The results obtained in this study may lead to the conclusion that the mechanism of action of ibuprofen is most probably based on the hydrogen bonding interaction which involves hydroxyl group of tyrosine.
Acknowledgements The authors would like to thank Prof. S. Wysocki, head of the Institute of
General Food Chemistry, who funded the project and provided the equipment. We acknowledge IAESTE for the opportunity to gain invaluable work experience.
References 1. Flanagan RJ. Guidelines for the interpretation of analytical toxicology results and unit
of measurement conversion factors. Ann Clinn Biochem 1998, 35: 261-266. 2. Jasińska A, Ferguson A, Mohamed WS, Szreder T, The study of interactions between
ibuprofen and bovine serum albumin. Zesz Nauk PŁ Chem Spoż Biotechnol 2009, 73:15-24.
3. Tito A, Jimenez-Lopez C, Kowalska A, Wysocki S. A study of the intermolecular interactions of tolmetin/N-acetyl-L-tyrosine ethyl ester complex. Spectrochim Acta A 2009, 72:1000-1006.

Kowalska-Baron et al.
Biotechnol Food Sci, 2011, 75 (2), 15-26 http://www.bfs.p.lodz.pl
26
4. Piela L. Oddziaływania międzymolekularne. In: Idee chemii kwantowej, PWN, Warszawa, Polska 2003, pp. 706-718.
5. Frisch MJ et al. Gaussian 03, Revision D01 2004, Gaussian Inc., Wallingford, CT. 6. Yang JZ, Zhang ZF. Studies on the absorption spectra of aqueous L-tyrosine and its
dissociation equilibria. Talanta 1998, 45:947-950. 7. Mach H, Middaugh CR, Lewis RV. Statistical determination of the average values of
the extinction coefficients of tryptophan and tyrosine in native proteins. Anal Biochem 1992, 200:74-80.
8. Du L, Liu X, Huang W, Wang E. A study on the interaction between ibuprofen and bilayer liquid membrane. Electrochim Acta 2006, 51:5754-5760.
9. Guzow K, Szabelski M, Rzeska A, Karolczak J, Sulowska H, Wiczk W. Photophysical properties of tyrosine at low pH range. Chem Phys Lett 2002, 362:519-526.
10. Cramer CJ. Thermodynamic properties. In: Essentials of Computational Chemistry. Theories and Models, 2nd ed., Wiley, Chichester, England 2006, pp. 355-377.
11. Yilgor E, Yilgor I, Yurtsever E. Hydrogen bonding and polyurethane morphology. I. Quantum mechanical calculations of hydrogen bond energies and vibrational spectroscopy of model compound. Polymer 2002, 43:6551-6559.

Biotechnology and Food Science Research article
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
Immunoreactivity of chemically cross-linked gluten and hydrolysates of wheat flour
Iwona Majak, Joanna Leszczyńska, Agata Łącka*
Institute of General Food Chemistry, Technical University of Lodz, 90-924 Łódź, Poland *[email protected]
Abstract: The immunoreactivity of gluten and wheat flour proteins cross-linked with chosen chemical reagents was investigated. Native proteins and flour hydrolysates subject to enzymatic proteolysis with collagenase and subtilisin were studied. Determination of immunoreactivity was performed with noncompetitive ELISA method with coeliac patients’ sera. The lowest immunoreactivity values were obtained during cross-linking of wheat flour hydrolyzates with polyethyleneimine, below 5% of the values for non-modified flour.
Keywords: wheat; gluten; cross-linking; ELISA
Introduction Cereals are the base of human diet all over the world. However, consumption
of wheat or products enriched with wheat proteins can manifest with different undesirable reactions of the organism. Among the most prevalent symptoms of food allergies are: urticaria, atopic dermatitis (AD) and wheat-dependent exercise-induced anaphylaxia (WDEIA) [1]. Apart from food allergies, disorders related with food intolerance of gluten, known as coeliac disease (CD), can occur. Consumption of gluten causes small intestine disorders resulting in destruction of epithelium and inflammatory response. The strongest response is evoked by peptides obtained as a result of digestion of one of the gluten fractions of wheat proteins [2].
One of the methods applied to decrease the immunoreactivity of proteins is the formation of covalent, intra- and intermolecular, bonds between them, so called cross-linking. It allows for covering of the molecular epitopes structure, which previously were presented on the surface of the allergogenic protein and therefore, the depletion of their recognition of antibodies. It is an interesting method, because the created bonds can not be hydrolysed in human intestinal tract by the digestive enzymes. What is more, the cross-linked proteins may favour an enhancement of organoleptic or technological properties of the initial product.
The aim of this work was to find an appropriate chemical reagent for covalent cross-linking of gluten proteins and wheat hydrolyzates.

Majak et al.
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
28
Experimental Reagents
Gluten (Sigma) and wheat flour type 500 from “Kruszynek” mill, containing 18.8% of gluten, 0.51% ash and 14.9% moisture content were chemically modified. To 1g of gluten or wheat flour 2ml of distilled water and 2% or 10% in regard to substrate (gluten or flour) mass of chemical reagent were added. The modifications were performed at the temperature of 37°C on a shaker with an incubation time of 4 hours. After the termination of the reaction all samples were centrifuged and washed twice with distilled water and centrifuged again. Supernatant was used for further analysis.
As cross-linking reagents were used: diethanolamine (D-8885), glutaraldehyde (G-7526), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) (E-6383), poliethylenimine (P-3143), 4-vinylpyridine (V-3877), polyethyleneglycol PEG 3350 (P-3640) and PEG 1450 (P-5402), D-sorbitol (S-1876), formaldehyde (F-1635), polyvinyl alcohol (P-8136, all of the above from Sigma) glycerol (analytically pure) (POCh, Poland).
Enzymatic modifications Enzymatic modifications were carried out accordingly to chemical modifications.
Two proteolytic enzymes were used: collagenase from Clostridium histolyticum (EC 3.4.21.3) and subtilisin from Bacillus licheniformis (EC 3.4.21.62). Both enzymes were chosen basing on previous research [3]. The substrate (gluten or flour) was incubated with enzyme for 18 hours at 37oC on a shaker, the weight ratio of enzyme to substrate was 1:100. The enzymatic reaction was terminated by 5 min heating in boiling temperature.
Immunoreactivity analysis To estimate the immunoreactivity of gliadin fraction isolated from modified flour
or gluten the indirect enzyme-linked immunosorbent assay (ELISA) technique was used, in which two human sera containing antigliadin antibodies and monoclonal antibodies against human antiglobuline IgG conjugated with an alkaline phosphatase were used [4]. Antibodies from both sera did not recognize proteins of albumin and globulin fractions contained in the wheat flour (data not shown).
Proteins extracts were isolated according to the Osborne’s procedure [5]. Microtitre plates EB 92029330 (Labsystem, Helsinki, Finland) were coated
overnight at 4C with 100 μl of the antigen solution (100 times diluted extracts) in 0.1 M carbonate buffer (pH 9.6), containing about 1.5 mg of protein. Plates were washed and free binding sites were blocked by incubation of the plates with a 3% solution of low fat milk in phosphate buffer (pH 7.2), containing 0.1% Tween-20 for 2 h. This was followed by removal of buffer solution, rinsing the plates five times and further incubation with human sera containing antigliadin antibodies diluted with phosphate buffer for 1 h at room temperature. The plates
Immunoreactivity of cross-linked gluten and hydrolysates of wheat flour
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
29
were washed again and 100 μl of 1000-fold diluted solution of anti-IgG antibodies conjugated with an alkaline phosphatase from mouse, clone GG-5 (Sigma, A 2064) was added. After incubation of the plates for 1 h and rinsing with phosphate buffer the bound phosphatase activity was determined by reaction with p-NPP with Multiscan MC reader at 405 nm. A 0.1% milk solution in phosphate buffer (pH 7.2), containing 0.05% Tween-20 was used for all washings and dilutions of antibodies.
The residual immunoreactivity of probes was estimated in ratio to untreated wheat flour or gluten.
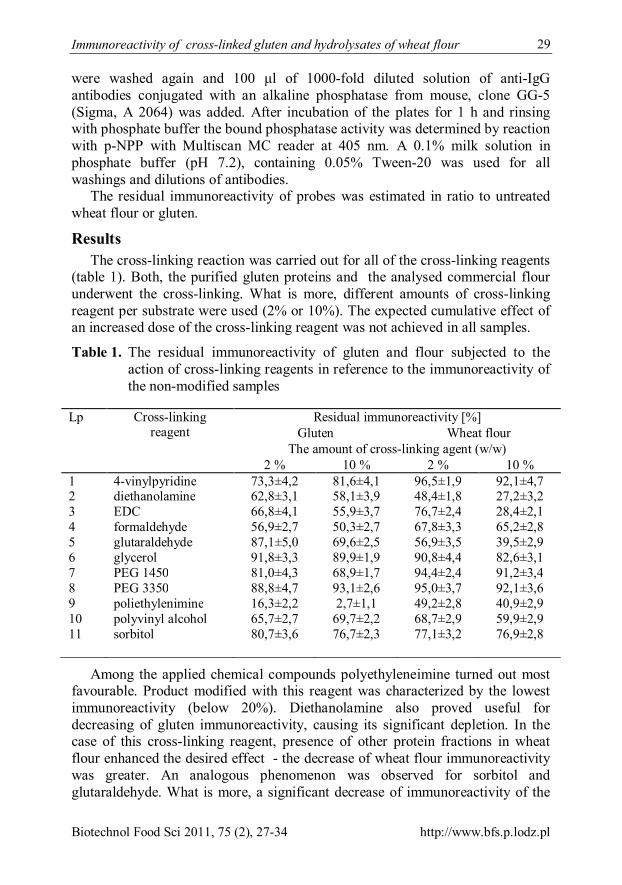
Results The cross-linking reaction was carried out for all of the cross-linking reagents
(table 1). Both, the purified gluten proteins and the analysed commercial flour underwent the cross-linking. What is more, different amounts of cross-linking reagent per substrate were used (2% or 10%). The expected cumulative effect of an increased dose of the cross-linking reagent was not achieved in all samples.
Table 1. The residual immunoreactivity of gluten and flour subjected to the action of cross-linking reagents in reference to the immunoreactivity of the non-modified samples
Lp Cross-linking reagent
Residual immunoreactivity [%] Gluten Wheat flour
The amount of cross-linking agent (w/w) 2 % 10 % 2 % 10 %
1 4-vinylpyridine 73,3±4,2 81,6±4,1 96,5±1,9 92,1±4,7 2 diethanolamine 62,8±3,1 58,1±3,9 48,4±1,8 27,2±3,2 3 EDC 66,8±4,1 55,9±3,7 76,7±2,4 28,4±2,1 4 formaldehyde 56,9±2,7 50,3±2,7 67,8±3,3 65,2±2,8 5 glutaraldehyde 87,1±5,0 69,6±2,5 56,9±3,5 39,5±2,9 6 glycerol 91,8±3,3 89,9±1,9 90,8±4,4 82,6±3,1 7 PEG 1450 81,0±4,3 68,9±1,7 94,4±2,4 91,2±3,4 8 PEG 3350 88,8±4,7 93,1±2,6 95,0±3,7 92,1±3,6 9 poliethylenimine 16,3±2,2 2,7±1,1 49,2±2,8 40,9±2,9 10 polyvinyl alcohol 65,7±2,7 69,7±2,2 68,7±2,9 59,9±2,9 11 sorbitol 80,7±3,6 76,7±2,3 77,1±3,2 76,9±2,8
Among the applied chemical compounds polyethyleneimine turned out most favourable. Product modified with this reagent was characterized by the lowest immunoreactivity (below 20%). Diethanolamine also proved useful for decreasing of gluten immunoreactivity, causing its significant depletion. In the case of this cross-linking reagent, presence of other protein fractions in wheat flour enhanced the desired effect - the decrease of wheat flour immunoreactivity was greater. An analogous phenomenon was observed for sorbitol and glutaraldehyde. What is more, a significant decrease of immunoreactivity of the

Majak et al.
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
30
analysed proteins (from 70 to 30%) in reference to unmodified sample was obtained with 1-ethyl-3(3-dimethylamino-propyl)carbodiimide (EDC).
In the second part of the experiment a reaction of plasteinisation was conducted, that is an enzymatic hydrolysis of gluten proteins to polypeptides with a subsequent cross-linking reaction with chosen cross-linking reagents. All of proteins contained in wheat flour were subject to plasteinisation due to the possibility of reaggregation of polypeptides, and derived from albumin and globulin hydrolysis. The hydrolysis was conducted paralelly with two proteases: subtilisine and collagenase, chosen on the basis of previous research on decreasing of wheat gliadins immunoreactivity through enzymatic modifications [3]. To the subsequent cross-linking only those reagents were chosen, which were characterized by the greatest decrease of gluten IR (dithyleneamine, EDC, polyethyleneimine) or those approved as food additives (glycerol, sorbitol, PEG 1450, PEG 3350). The obtained results are presented in table 2.
Table 2. The residual immunoreactivity of wheat flour subjected to the action of protease (collagenase, subtilisin) and cross-linking reagents (10%) in relation to native sample immunoreactivity
Cross-linking reagent Residual immunoreactivity [%]
collagenase subtilisin
diethanolamine 33,8±1,9 3,5±1,6 EDC 19,1±1,8 6,1±1,8 glycerol 31,7±2,1 39,2±2,0 PEG 1450 41,6±2,3 78,7±2,3 PEG 3350 40,7±2,4 61,1±2,4 poliethylenimine 4,6±2,3 3,3±1,5 sorbitol 37,4±2,6 42,3±2,4 none 29,3±1,9 70,3±2,6
The combination of enzymatic hydrolysis and chemical cross-linking allowed for significant decreasing of wheat flour gliadins' immunoreactivity down to about 3%, when subtilisin was used as the hydrolysing enzyme. The application of collagenase as the hydrolysing enzyme also proved effective (the remaining immunoreactivity of 4,6%), but only when the cross-linking reagent was polyethylenimine. In other cases the remaining immunoreactivity of the gliadins modified with cross-linking as well as plasteinisation was of the same order of magnitude. Surprising results were obtained for two reagents: PEG 1450 and PEG 3350, for which the flour samples exhibited lower immunoreactivity after hydrolysis than after the subsequent cross-linking reaction.

Immunoreactivity of cross-linked gluten and hydrolysates of wheat flour
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
31
Discussion Cross-linking as a method of decreasing of allergenic proteins immuno-
reactivity is relatively little investigated, although it gives a possibility of blocking contact of the antibodies present in the organism with epitopes of immunogenic polypeptides contained in food, especially ingredients of those food products that cannot be entirely eliminated from diet due to their common application in production technology of a wide variety of foods. One of the main components presenting such character is wheat gluten, which is responsible for both, food allergies as well as immunological reactions connected with its intolerance (coeliac disease).
Up to this time, researchers put the main emphasis on the enzymatic, and therefore strictly oriented, cross-linking of gluten proteins. Cross-linking of prolamines is a beneficial phenomenon in technology of wheat flour processing, enhancing the physico-chemical parameters of the obtained bread, regardless of the initial half-products quality [6, 7, 8]. A number of enzymes allowing for cross-linking of gluten proteins has been known, however, an impact of cross-linking on the immunoreactivity of protein was investigated only for one of them – transglutaminase (TG).
Gluten proteins cross-linked with TG express much lower immunoreactivity towards IgE class antibodies (coming from allergy sufferers) [9], however similar or even higher towards IgG [10] and IgA [11] class antibodies (both classes from coeliac patients) than the native protein, owing to which, gluten modified with this enzyme should not be intended for people with gluten intolerance [12]. A significant problem is, therefore, finding of another gluten proteins cross-linking method suitable for both, allergy sufferers and patients with coeliac disease.
The change of cross-linked with chemical reagents proteins' immunoreactivity was analysed mainly for cross-linking with polyethyleneglycols (PEG). PEGylation of proteins caused depletion of immunological response of antibodies to cow's milk proteins: α-lactoalbumin and β-lactoglobulin. The proteins expressed residual immunoreactivity below 0,1% of the initial milk immunoreactivity in in vitro tests [13].
Cross-linking reagents used in this work, forming covalent bonds between protein molecules, were chosen basing on the analysis of literature reports concerning cross-linking of proteins, both for food products production as well as biodegradable plastics obtained from protein materials, used as packing materials. So far, only four out of 11 analysed cross-linking reagents have been legally admitted as food additive (glycerol, sorbitol, PEG 1450 and PEG 3350).
An addition of polyols and amines to gluten proteins was considered as plastifier in biodegradable protein films production for packing materials. Among the large group of investigated compounds the best mechanical properties of the films were obtained for low molecular weight amines: diethanolamine and triethanolamine, and glycerol [14]. In that research an addition of plastifiers exceeds 20% in reference to the gluten proteins. In our research we applied lower

Majak et al.
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
32
amounts of chemical reagents, because protein cross-linking can take place at low concentrations of cross-linking reagent, e.g. gluten proteins cross-linked with glutaraldehyde lost bands below 250 kDa on SDS electroforegrams already at concentration of 0,05% [15], however the cross-linking required longer reaction time.
Too high temperatures of protein cross-linking (above 50°C) have a significant influence on the time of reaction, however, in such conditions a simultaneous reaction of polymerisation of the cross-linking reagent alone can proceed [15], and most of all, an independent of the reagent used intermolecular regrouping of disulphide bonds between glutenin residues [16] or gliadins integration to glutenins polymers [17]. Therefore, the cross-linking reactions were carried out below 40°C, in neutral environment, because at such conditions, at minimum gluten proteins solubility in water, the cross-linking reagent is in contact only with aminoacids on the surface of proteins, which in turn favours creation of intermolecular bonds [18], without formation of intramolecular bonds in glutenins' subunits.
The differences between the immunoreactivity of gluten and flour subject to cross-linking can be explained with competitiveness of other proteins present in flour (albumins, globulins) as substrates for cross-linking. Only glutaraldehyde, diethanolamine and sorbitol caused a stronger depletion of protein immunoreactivity in flour than in purified gluten. It points to the lack of substrate competition of the remaining fractions present in flour or attachment of those proteins to gluten in regions recognized by antibodies.
Low values of the remaining gluten immunoreactivity after cross-linking with EDC at the applied quantities of cross-linking reagent (0.13 i 0.64 mmol EDC/g gluten) are surprising, because research on gluten cross-linking a molar ratio 4:1 of EDC to carboxyl groups in proteins was recognized as optimal [18], while at ratios below 0.1:1 no significant changes pointing to cross-linking were observed [19]. Since 1g of gluten contains about 0.4 mmol of free carboxylic groups, then the applied in this work amounts of EDC are far from optimal for cross-linking (the ratio EDC : COOH was 0.325:1 and 1.6:1), however, even in this quantity EDC has a significant impact on gluten immunoreactivity.
The next step of the research was based on plasteinisation, that is hydrolysis of native wheat flour protein with two chosen non-digestive enzymes (subtilisin, collagenase) and a following reaction of chemical cross-linking. Differences in enzymatic hydrolysates of wheat flour immunoreactivity were connected with substrate diversity of the enzymes used. Collagenase is an enzyme hydrolysing peptide bond on the side of carboxylic residue neighbouring proline residue. It is significant, because majority of epitopes recognized by anti-gluten antibodies contain proline residues, which hamper hydrolysis by digestive enzymes. Subtilisin is a nonspecific enzyme, which destructive action towards gluten was confirmed in an earlier study [3, 20]. Both enzymes proved to be a suitable choice for protein hydrolysis before cross-linking, allowing for binding of inert

Immunoreactivity of cross-linked gluten and hydrolysates of wheat flour
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
33
polypeptide fragments from hydrolysis of other wheat protein fractions to immunogenic polypeptides. Persistence of such bonds during chemical treatment and digestion requires separate research. The only inconvenience of the process turned out to be the fact, that the mass distributions of the plasteinised proteins was not coordinated with the obtained immunoreactivity values (data not shown).
The above research is purely cognitive, however, there are two reasons allowing to hope, that flour modified with chemically induced cross-links will not be toxic. The first one arises from the dose of the reagent; an application of such a small concentration should not leave reaction residues. Secondly, the chosen chemical compounds are applied for production of gluten films used as an easily biodegradable packing material, not polluting the environment. These are not circumstances sufficient to conclude about their usefulness or uselessness for food purposes, however, a research on the toxicity of the chosen cross-linking reagents and emerging cross-linking reaction products is required before they are admitted for consumption.
References 1. Battais F, Courcoux P, Popineau Y, Kanny G, Moneret-Vautrin DA, Denery-Papini S.
Food allergy to wheat: differences in immunoglobulin E-binding proteins as a function of age or symptoms. J Cereal Sci 2005, 42:109-117.
2. Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297:2275-2279.
3. Leszczyńska J, Łącka A, Pytasz U, Lukamowicz J, Szemraj J, Lewiński A. Influence of proteolysis on gliadin immunoreactivity. Pol J Food Nutr. Sci 2002, 11(SI2):145-148.
4. Leszczyńska J, Łącka A, Szemraj J, Lukamowicz J, Żegota H. The influence of gamma irradiation on the immunoreactivity of gliadin and wheat flour. Eur Food Res Technol 2003, 217:143-147.
5. Osborne TB. The proteins of the wheat kernel. Carnegie Institute, Washington DC, Publ. No. 84, p.1907.
6. Larre C, Denery-Papini S, Popineau Y, Deshayes G, Desserme C, Lefebvre J. Biochemical analysis and rheological properties of gluten modified by transglutaminase. Cereal Chem 2000, 77:121-127.
7. Bonet A, Caballero PA, Gomez M, Rosell CM. Microbial transglutaminase as a tool to restore the functionality of gluten from insect-damaged wheat. Cereal Chem 2005, 82:425-430.
8. Caballero PA, Bonet A, Rosell CM, Gomez M. Effect of microbial transglutaminase on the rheological and thermal properties of insect damaged wheat flour. J Cereal Sci 2005, 42:93-100.
9. Watanabe M, Ikezawa Z, Arai S. Fabrication and quality evaluation of hypoallergenic wheat products. Biosci Biotechnol Biochem 1994, 58:388-390.
10. Leszczyńska J, Łącka A, Bryszewska M, The use of transglutaminase in the reduction of immunoreactivity of wheat flour. Food Agric Immunol 2006, 17:105-113.
11. Berti C, Roncoroni L, Falini ML, Caramanico R, Dolfini E, Bardella MT, Elli L, Terrani C, Forlani F. Celiac-related properties of chemically and enzymatically modified gluten proteins. J Agric Food Chem 2007, 55:2482-2488.

Majak et al.
Biotechnol Food Sci 2011, 75 (2), 27-34 http://www.bfs.p.lodz.pl
34
12. Gerrard JA, Sutton KH. Addition of transglutaminase to cereal products may generate the epitope responsible for coeliac disease. Trends Food Sci Technol 2005, 16:510-512.
13. Wróblewska B, Jędrychowski L. Effect of conjugation of cow milk whey protein with polyethylene glycol on changes in their immunoreactive and allergic properties. Food Agric Immunol 2002, 14:155-162.
14. Irissin-Mangata J, Bauduin G, Boutevin B, Gontard N. New plasticizers for wheat gluten films. Europ Polymer J 2001, 37:1533-1541.
15. Reddy N, Tan Y, Li Y, Yang Y. Effect of glutaraldehyde crosslinking conditions on the strength and water stability of wheat gluten fibers. Macromol Mater Eng 2008, 293:614-620.
16. Lagrain B, Goderis B, Brijs K, Delcour JA. Molecular basis of processing wheat gluten toward biobased materials. Biomacromolecules 2010, 11:533-541.
17. Lagrain B, Thewissen BG, Brijs K, Delcour JA. Mechanism of gliadin-glutenin cross-linking during hydrothermal treatment. Food Chem 2008, 107:753-760.
18. Tropini V, Lens JP, Mulder WJ, Silvestre F. Cross-linking of wheat gluten using a water-soluble carbodiimide. Cereal Chem 2000, 77:333-338.
19. Tropini V, Lens JP, Mulder WJ, Silvestre F. Wheat gluten films cross-linked with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide. Ind. Crop Prod 2004, 20:281-289.
20. Leszczyńska J, Bryszewska M, Łącka A, Wolska K, Żegota H. Immunoreactivity reduction of wheat flour proteins modified by the treatment with subtilisin, Pol J Food Nutr Sci 2008, 58:339-344.

Biotechnology and Food Science Research article
Biotechnol Food Sci 2011, 75 (2), 35-38 http://www.bfs.p.lodz.pl
Fly ash vitrification as the effective physico-chemical waste stabilization method
Elzbieta Sobiecka,* Witold Sroczyński
Institute of General Food Chemistry, Technical University of Lodz, 90-924 Lodz *corresponding author: [email protected]
Abstract: In this paper, the physico-chemical stabilization method of electrical power plant fly ash of the Province of Lodz in central Poland was studied. The chemical composition and leachability of metals were determined in the investigated samples. The ashes were vitrified in a thermal plasma process and their chemical stability was tested to determine whether the ashes can be disposed safely into the environment, specifically onto surface soil. The heavy metal leachability tests proved that the products after vitrification were more stable than non-vitrified waste making it more suitable for disposal into the environment.
Keywords: electrical power plant waste, fly ash, vitrification, solidification/ stabilization
Introduction The used of a coal as the source of classic energy has created the pollution
problems of gas emission to the atmosphere as well as the fly ash appearance after the process of heat production from power electrical plant. Fly ashes and their disposal is a current issue affecting the global environment.
The Lodz Province in central Poland, as the big agglomeration with a population of 2.6 million inhabitants, was not aware of this problem. In 2009, 13732.4 tons of wastes were created in this region where 5074.8 tons came from the thermal processes of power plants [1]. The most popular method of their utilization (almost 90%) is a landfill storage.
The fly ash as the final product of a hard coal and a lignite combustion, contains various elements (also heavy metals) which cause the harmful effect to the human health and the environment. Coal is the most popular type of the classic source energy in Poland which means that after the burning processes the fly ashes appeared in a huge amount. It causes that fly ash requires a special treated as the hazardous waste listed with the code 10.01.02 [2]. This type of waste cannot be storage as the raw material directly on the land. It is important to find the effective method which transforms the fly ashes to the inert or less toxic products.
One of the solidification/stabilization (S/S) method used for the fly ash utilization is a thermal plasma stabilization [3,4]. This process is an effective method that provides to the environmental neutrally glassy product called

36 Sobiecka and Sroczyński
Biotechnol Food Sci 2011, 75 (2), 35-38 http://www.bfs.p.lodz.pl
vitrificate [5-9]. This technology is more effective than classic combustion because it guarantees a complete decomposition of organic substances, also pathogens and a significant reduction of the material volume about 75% of the incinerated waste.
The aim of the study was to demonstrate the advantage of electric power plant fly ash vitrification process as the one of the most effective physico-chemical method of waste stabilization.
Experimental Materials
The fly ash samples came from an electrical power plant Belchatow of the Lodz Province. The samples were collected after two months of the mixture chimney filter fly ashes.
Methods Chemical composition of fly ash
The ash chemical composition was determined by the Atomic Absorption Spectrometry – Graphite Furnace AAS-GF (air-acetylene flame) according to the BS DD CEN/TS 14429 concerning the leachability tests of wastes [10]. The ash composition of particular radicals was shown as the oxides. Metal leachability Ash. Metal leachability analyses were done according to the procedure which was described previously by Cedzynska et al. [11]. The chosen metals (Cd, Cr, Cu, Ni, Pb, Zn, Fe, Al, Mn) concentrations in water extracts were measured by Atomic Absorption Spectrometry Graphite Furnace AAS-GF (air-acetylene flame). The GBC 932 Plus spectrometer with a GF 3000 graphite furnace was used. Vitificates. Metal leachability analyses of vitrificates (stabilised ash forms) were done according to the EN 12457-2:2000 [12] concerned granulated wastes and sludge leaching tests analyses. Vitrification
The plasma vitrification process was used for stabilisation of fly ashes. Plasma ,,Little JetArc” device, designed and constructed in the Institute of Electrical Apparatus Technical University of Lodz, was used for the vitrification [13]. In this device plasma was generated directly in a DC arc forming between the graphite crucible melting chamber acting as the anode and a graphite rod cathode. The processes of ash stabilization were run in the atmosphere of argon during 300s with a delivered power of 1400 W.
Results and Discussion The results of the chemical analyses of fly ash (Table 1) indicated that silica,
calcium, and aluminium are the primary constituents found in the waste samples under investigation. These three components in the forms of oxides, make up
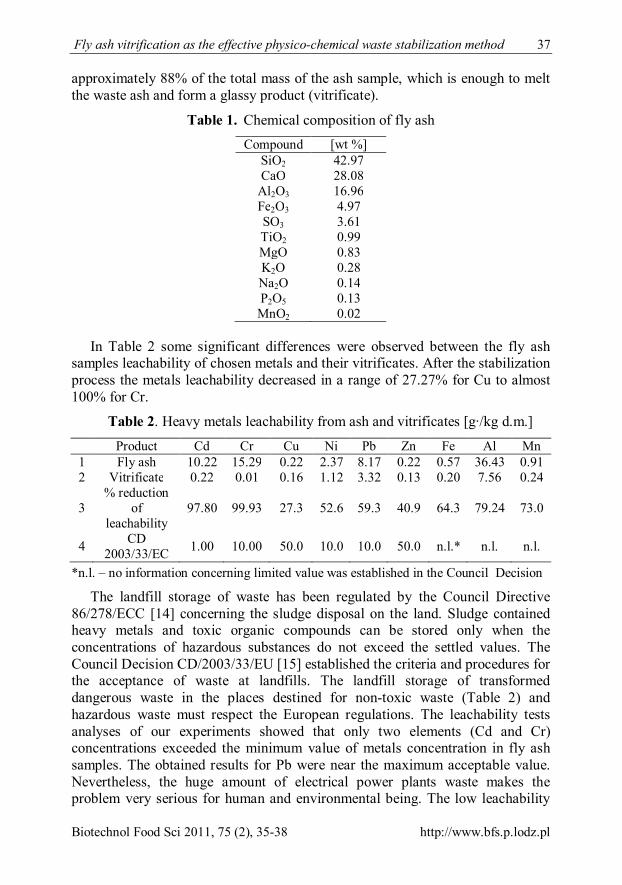
Fly ash vitrification as the effective physico-chemical waste stabilization method 37
Biotechnol Food Sci 2011, 75 (2), 35-38 http://www.bfs.p.lodz.pl
approximately 88% of the total mass of the ash sample, which is enough to melt the waste ash and form a glassy product (vitrificate).
Table 1. Chemical composition of fly ash Compound [wt %]
SiO2 42.97 CaO 28.08
Al2O3 16.96 Fe2O3 4.97 SO3 3.61 TiO2 0.99 MgO 0.83 K2O 0.28 Na2O 0.14 P2O5 0.13 MnO2 0.02
In Table 2 some significant differences were observed between the fly ash samples leachability of chosen metals and their vitrificates. After the stabilization process the metals leachability decreased in a range of 27.27% for Cu to almost 100% for Cr.
Table 2. Heavy metals leachability from ash and vitrificates [g∙/kg d.m.]
Product Cd Cr Cu Ni Pb Zn Fe Al Mn 1 Fly ash 10.22 15.29 0.22 2.37 8.17 0.22 0.57 36.43 0.91 2 Vitrificate 0.22 0.01 0.16 1.12 3.32 0.13 0.20 7.56 0.24
3 % reduction
of leachability
97.80 99.93 27.3 52.6 59.3 40.9 64.3 79.24 73.0
4 CD 2003/33/EC 1.00 10.00 50.0 10.0 10.0 50.0 n.l.* n.l. n.l.
*n.l. – no information concerning limited value was established in the Council Decision
The landfill storage of waste has been regulated by the Council Directive 86/278/ECC [14] concerning the sludge disposal on the land. Sludge contained heavy metals and toxic organic compounds can be stored only when the concentrations of hazardous substances do not exceed the settled values. The Council Decision CD/2003/33/EU [15] established the criteria and procedures for the acceptance of waste at landfills. The landfill storage of transformed dangerous waste in the places destined for non-toxic waste (Table 2) and hazardous waste must respect the European regulations. The leachability tests analyses of our experiments showed that only two elements (Cd and Cr) concentrations exceeded the minimum value of metals concentration in fly ash samples. The obtained results for Pb were near the maximum acceptable value. Nevertheless, the huge amount of electrical power plants waste makes the problem very serious for human and environmental being. The low leachability

38 Sobiecka and Sroczyński
Biotechnol Food Sci 2011, 75 (2), 35-38 http://www.bfs.p.lodz.pl
of heavy metals from vitrificates qualifies this physico-chemical method of stabilization to the one of the most effective process of waste utilization. Finally the environmental friendly products have appeared and in such form the wastes could be stored on a land without any harmful effects to the soil.
The European regulations are very strict regarding heavy metals concentrations of storage wastes. The high temperature plasma technology utilization as one of the stabilization methods is a proper tool in a waste hierarchy requirements of Council Directive ED/2008/98/EC [16]. The vitrification process used in stabilization of fly ash creates a good alternative methods of hazardous wastes utilization and their disposal among the available offers.
The fly ash wastes management is an important issue in the worldwide environment, therefore their proper collection and disposal are important as they can directly impact the health risks. The electrical power plant waste management plan should also include the special technologies which are required for their utilization.
Thermal plasma vitrification is an effective method of neutralizing fly ashes from electrical power plants which makes a possibility to obtain the environmentally harmless products with a significant reduction of volume.
References 1. Provincial Inspectorate for Environmental Protection of Lodz. Wastes in Report about
environmental condition in Lodz Province, Ed. AGRI s.c., Wroclaw 2010, pp. 94-210. 2. Law of Environmental Protection. Dz. U. Nr 62 poz. 627, in Polish 2001. 3. Freeman HM, Solidification and stabilization technology in standard handbook of
hazardous waste treatment and disposal. McGraw-Hill Editor, 2nd Edition 1997, section 7.31-7.41.
4. Shi C, Spence R. Designing of cement-based formula for solidification/stabilization of hazardous, radioactive and mixed wastes. Critical reviews. Environ Sci Techn 2004, 34:391-417.
5. Cedzynska K, Kolacinski Z, Izydorczyk M, Sroczynski W. Thermal plasma treatment of waste incinerator ash. in: Progress in Plasma Processing of Materials. ed. P. Fauchais and J. Amouroux, Begell House inc. 1999,pp. 707-711.
6. Pisciella P, Crisucci S, Karamanov A, Pelino M. Chemical durability of glasses obtained by vitrification of industrial wastes. Waste Managem 2001, 21:1-9.
7. Park YJ, Heob J. Vitrification of fly ash from municipal solid waste incinerator. J Haz Mat 2002, B91:83-93.
8. Gomez E, Amutha Rani D, Cheeseman CR, Deegan D, Wise M, Boccaccini AR. Thermal plasma technology for the treatment of wastes: A critical review. J Haz Mat 2009, 161:614-626.

Biotechnology and Food Science Review article
Biotechnol Food Sci 2011, 75 (2), 39-44 http://www.bfs.p.lodz.pl
Methods of allergen detection based on DNA analysis
Marta Słowianek,* Iwona Majak
Institute of General Food Chemistry, Technical University of Lodz, 90-924, Lodz, Poland * [email protected]
Abstract: Many allergens, such as hazelnut, peanut, charlock, celery, sesame, lupine, walnut, almond, macadamia nut, hickory, pistachio, wheat gliadins, may be present in food products, however, undeclared or as unintentional additives. Due to the growing number of allergic reactions, it is crucial to have fast, reliable methods of allergen detection in processed food products. This review summarizes the recent methods of allergen detection in food products based on PCR reactions, namely PCR-ELISA, Real-time PCR, PCR-PNA-HPLC, Duplex PCR and Multiplex Real-time PCR, describing their principles, applications, detection limits, drawbacks and advantages.
Keywords: PCR allergens, hazelnut, peanut, charlock, celery, sesame lupine, walnut, almond, macadamia nut, hickory, pistachio, wheat
Introduction Many allergens, such as hazelnut, peanut, charlock, celery, sesame, lupine,
walnut, almond, macadamia nut, hickory, pistachio, wheat gliadins, may be present in food products, however, undeclared or as unintentional additives. Due to the growing number of allergic reactions, it is crucial to have fast, reliable methods of allergen detection in processed food products.
This review summarizes the recent methods of allergen detection in food products based on PCR reactions, namely PCR-ELISA, Real-time PCR, PCR-PNA-HPLC, Duplex PCR and Multiplex Real-time PCR, describing their principles, applications, detection limits, drawbacks and advantages. PCR techniques are promising in the field of allergen detection on the level surpassing immunodetection.
PCR techniques rely on the detection of a defined DNA sequence. The DNA fragment is amplified with DNA polymerase which requires a pair of short primer fragments in order to recognize the required DNA sequence characteristic for a given allergen source.
Methods based on detection of specific DNA sequences Methods applying detection of coding sequences of allergenic proteins allow
direct analysis of allergenic food components. The detection of allergenic protein is performed through amplification of its specific DNA fragment in PCR reaction [1]. The PCR reaction allows obtaining qualitative results pointing to the presence of

40 Słowianek and Majak
Biotechnol Food Sci, 2011, 75 (2), 39-44 http://www.bfs.p.lodz.pl
sequence coding for given allergen. Semi quantitative determination can be obtained with PCR-ELISA reaction, while precise quantitative determination is granted by Real-Time PCR [2].
The PCR method was successfully applied to the detection of DNA coding for flour gliadins and main allergen of hazelnut. Such determinations are highly sensitive, free from cross-reactions and able to detect amounts of allergen source admixture less than 0.001% [3].
Hazelnut is a potential food allergen present in pastries, confectionery and ice cream. Its undeclared amounts can be present as an unintentional food contamination. Specie-specific amplification of gene fragment - Cor a 1.0401 – an allergen of hazelnut, enables to detect its admixtures in food products in quantity of 0.001% (w/w) [4].
Dovicovicova et al. suggested a specie-specific PCR for celery detection through the amplification of mannitol dehydrgenase gene. The technique is specific towards four celery species with the detection limit ≦ 1.53 ng DNA and 0.1% (w/w) in meat products [5].
Assessing PCR techniques for the detection of allergen DNA the problem of DNA degradation during technological processing of foods should be taken into account. Its reflection is found in research of many scientists who analyze the impact of food processing on the detection levels of allergen DNA. The works of Koeppel et al., who investigated processed oat products (cereals and oatmeals) regarding their wheat gliadin content, are worth of attention.
In the research PCR and ELISA techniques were compared, in which it was demonstrated, that despite the standing out sensitivity of PCR method, only one out of eight samples gave a positive result. However, all samples contained between 1 and 2 mg/100g of gliadin detectable with ELISA technique, which points to high degradation of DNA in processing [6].
The influence of parameters of food processing on transgenic soy and corn identification in bakery products with PCR techniques was also researched by Moser et al. [7] They concluded that the DNA of 0.5% addition of transgenic corn was undetectable in processed product (bread and cake). However, the DNA of soy (0.3%) was detected in white toast bread after each technological step.
Similarly as in the case of ELISA technique, there are several variants and modifications of PCR technique, as: PCR-ELISA, Real-time PCR, PCR–PNA-HPLC, Duplex PCR and Multiplex real-time PCR.
PCR-ELISA In the PCR-ELISA method the detection of an allergen does not require gel
electrophoresis. The amplified DNA fragment is labeled with biotin or digoxygenin, thanks to which it can be easily determined with ELISA test [8, 1]. This combination applies the high specificity of detection based on DNA with the simplicity and low cost of ELISA. Holzhauser et al. demonstrated that PCR-ELISA is an important tool for allergen monitoring. With this method they

Methods of allergen detection based on DNA analysis 41
Biotechnol Food Sci, 2011, 75 (2), 39-44 http://www.bfs.p.lodz.pl
determined hazelnut allergens in processed foods at a level below 0.001% (w/w). The high specificity of PCR-ELISA towards hazelnut and high stability of DNA causes that this method is very useful and advisable for monitoring of this allergen in food products [9].
Real-Time PCR The Real-Time PCR is commonly used for allergen detection in foods as:
peanuts, celery, charlock, sesame, lupine, cashew, walnut, pistachios, pecan nuts, almond and macadamia nuts [10]. Development of real-time PCR techniques with Taq-Man probes enables to carry out specific detection of trace amounts of peanuts in food [1]. Analysis conducted with this method allows for detection of undeclared amounts of peanuts in 13% of samples from 0.0001 to 0.0074% (w/w) [12]. Works describing application of real-time PCR with Taq-Man probe for hazelnut [13], pecan nut [14], and walnut [7] in food with detection limit of 0.01% in standard samples of biscuit were published. The research covers DNA isolation by chaotropic extraction of solid phase and secondary PCR with fluorescent Taq-Man probe and primers specific for the gene of hazelnut, pecan nut or walnut. The intrinsic detection limit of this method obtained for hazelnut is 13 pg of hazelnut DNA in confectioner's and bakery products (0.01% hazelnut content) [13], 1 pg of pecan nut DNA (0.01% pecan nut content) [14], for walnut – 0.24 ng DNA (0.01% walnut content). In the case of walnut there were two samples of biscuits detected with an undeclared presence of walnut in the total of 13 samples [7].
Real-Time PCR was also applied for detection of sesame traces in food. Sesame was revealed in all samples with declared sesame content (crackers, sesame biscuits, shortbread) with an exception of sesame oil, however, sesame was not detected in samples which could contain it or where it was not declared. The determination did not give cross-reactions with 17 similar food components [16]. Also, the presence of mannitol dehydrogenase gene was detected in food with this method. The method was tested on over 50 samples of foods with detection limit 0.0005-0.001% (w/w) in standard samples of sausage [17]. Mustorp et al. presented a quantitative and sensitive Real-Time PCR method applying Taq-Man probes able to detect additions of 0.01-0.001% (w/w) of celery and 0.005% of mustard and sesame in food [18].
PCR-PNA-HPLC PCR–PNA-HPLC technique is a modification of PCR using probes labeled
with peptide nucleic acid (PNA) and detection with high performance liquid chromatography (HPLC). The applications of this method for detection of hazelnut in vegetable products allowed determining 5 pg of hazelnut DNA PCR-PNA-HPLC method enables effective detection of potentially hidden allergens even in products with undeclared presence of hazelnut or as possible contamination [19].

42 Słowianek and Majak
Biotechnol Food Sci, 2011, 75 (2), 39-44 http://www.bfs.p.lodz.pl
Duplex PCR Reactions of Duplex PCR type was applied to simultaneous detection of
hazelnut and peanut in native and processed food with two specific pairs of primers and allowed for detection of less than 50 pg DNA in final product [20]. The method was also used for detection of two species of wheat in pasta dried in high temperature, with detection limit of 0.2% (w/w) [21].
Multiplex real-time PCR Multiplex real-time PCR allows to amplify several DNA fragments
simultaneously by application of several pairs of primers [21]. This method was used to determine DNA of eight allergens at the same time: hazelnut, peanut, celery, soy, egg, milk, almond and sesame. The test exhibits high specificity and sensitivity in the range of 0.01%, lower for egg and milk due to low content of DNA [22].
Advantages and disadvantages of methods based on PCR Analysis with PCR techniques displays high potential due to its speed,
efficiency and simplicity [22]. The technique is reliable, highly specific and sensitive, with noted detection limits less than 10mg/kg of almonds, hazelnuts, soy, milk or peanuts [23]. It allows minimizing the cross-reactivity and avoiding false-positive results by choosing suitable primers differentiating between two closely related DNA sequences [24].
The main disadvantage is DNA degradation during food processing and the dependence of detection limit on the amount and purity of the matrix [24]. What is more, techniques based on PCR do not detect components responsible for allergic reaction, only the specie-specific DNA [25]. Therefore, presence of DNA coding for a protein in food sample does not necessarily signify the presence of the allergen, but only about the origination of the product from a characteristic genus, specie in the case of contamination [8]. The main limitation of methods based on DNA is the fact, that the particular allergenic protein is not directly determined, only the DNA from the given source, and in consequence, there is an possibility of false-negative or false-positive results for the presence of allergens in food [24]. These methods are not recommended for detection of allergens in food products with high protein and low DNA content, e.g. eggs. Consequently, these methods are a good choice where the protein content is low, e.g. in celery [26]. Despite that, PCR techniques are promising in the field of allergen detection on the level surpassing immunodetection.
The advantage of PCR techniques over methods based on protein detection is the efficient extraction of DNA in denaturing conditions, more effective than during protein extraction from food matrices, and DNA stability over geographical and seasonal changes, while protein concentration in food products depends on many factors as genus, specie or growth conditions [1, 26]. PCR techniques rely on the

Methods of allergen detection based on DNA analysis 43
Biotechnol Food Sci, 2011, 75 (2), 39-44 http://www.bfs.p.lodz.pl
simple and well defined DNA sequence [24]. Methods basing on PCR are fast and can be set up even in few days, if the analyzed sequence of DNA is known [24].
Processes of food preparation differently influence the DNA, which can be easily degraded during processing, leading to false results [2]. Köppel et al. managed to detect contaminations below 0.1% (w/w) with PCR techniques, while ELISA test was about 10 times less sensitive [6].
Summary Basing on the literature review it can be concluded that there exists a wide
range of research tools presenting different sensitivity for allergen detection in food products. Most frequently used techniques are based on Real-Time PCR. However, there is still a need to enhance reliability of the available analytical methods and develop new standardized methods for food industry in order to ensure safety of the consumers prone to allergic reactions. The new or improved tests have to be faster, more sensitive (lower detection limit), more precise (lower limit of quantitative determination) and more specific for similar allergenic proteins, they should enable to unequivocally identify allergens.
References 1. Mafra I, Ferreira I, Oliveira PP, Beatriz M. Food authentication by PCR-based
methods: review. Eur Food Res Technol 2008, 227:649-665. 2. Poms RE, Klein CL, Anklam E. Methods for allergen analysis in food: a review.
Food Add Contam 2004, 21: 1-31. 3. Besler M, Determination of allergens in foods. Trends Anal Chem 2011, 11:662-672. 4. Holzhauser T, Wangorsch A, Vieths S. Polymerase chain reaction (PCR) for
detection of potentially allergenic hazelnut residues in complex food matrixes. Eur Food Res.Technol 2000, 211:360-365.
5. Dovicovicova L, Olexová L, Pangallo D, Siekel P, Kuchta T. Polymerase chain reaction (PCR) for the detection of celery (Apium graveolens) in food. Eur Food Res Technol 2004, 218:493-495.
6. Kὂppel E, Stadler M, Lüthy J, Hȕbner P. Detection of wheat contamination in oats by polymerase chain reaction (PCR) and enzyme-linked immunosorbent assay (ELISA). Z Lebensm Unters Forsch A 1998, 206:399-403.
7. Moser MA, Kniel B, Schmitz-Winnenthal J. Influence of processing conditions on the analytical determination of genetically modified ingredients of baked products. Getreide Mehl Brot 1999, 53:334-341.
8. Kirsch S, Fourdrilis S, Dobson R, Scippo ML, Maghuin-Rogister G, De Pauw E. Quantitative methods for food allergens: a review. Anal Bioanal Chem 2009, 395: 57-67.
9. Holzhauser T, Stephan O, Vieths S. Detection of potentially allergenic hazelnut (Corylus avellana) residues in food: a comparative study with DNA PCR-ELISA and protein sandwich-ELISA. J Agric Food Chem 2002, 50:5808-5815.
10. D’Andrea M, Coisson JD, Travaglia F, Garino C, Arlorio M. Development and Validation of a SYBR-Green I Real-Time PCR Protocol To Detect Hazelnut (Corylus avellana L.) in Foods through Calibration via Plasmid Reference Standard. J Agric Food Chem 2009, 57:11201-11208.

44 Słowianek and Majak
Biotechnol Food Sci, 2011, 75 (2), 39-44 http://www.bfs.p.lodz.pl
11. Arlorio M, Coïsson JD, Cereti E, Travaglia F, Capasso M, Martelli A. Polymerase chain reaction (PCR) of puroindoline b and ribosomal/puroindoline b multiplex PCR for the detection of common wheat (Triticum aestivum) in Italian pasta. Eur Food Res Technol 2003, 216:253-258.
12. Stephan O, Vieths S. Development of a real-time PCR and a sandwich ELISA for detection of potentially allergenic trace amounts of peanut (Arachis hypogaea) in processed foods. J Agric Food Chem 2004, 52:3754-3760.
13. Piknová L, Pangallo D, Kuchta T. A novel real-time polymerase chain reaction (PCR) method for the detection of hazelnuts in food. Eur Food Res Technol 2007, 226:1155-1158.
14. Brězná B, Kuchta T. A novel real-time polymerase chain reaction method for the detection of pecan nuts in food. Eur Food Res Technol 2008, 226:1113-1118.
15. Brězná B, Hudecová L, Kuchta T. A novel real-time polymerase detection of walnuts in food. Eur Food Res Technol 2006, 223:373-377.
16. Schoringhumer K, Cichna-Markl M. Development of a real-time PCR method to detect potentially allergenic sesame (Sesamum indicum) in food. J Agric Food Chem 2007, 55:10540-10547.
17. Hupfer C, Waiblinger HU, Busch U. Development and validation of a real-time PCR detection method for celery in food. Eur Food Res Technol 2007, 225:329-335.
18. Mustorp S, Engdahl-Axelsson C, Svensson U, Holck A. Detection of celery (Apium graveolens), mustard (Sinapis alba, Brassica juncea, Brassica nigra) and sesame (Sesamum indicum) in food by real-time PCR. Eur Food Res Technol 2008, 226:771-778.
19. Germini A, Scaravelli E, Lesignoli F, Sforza S, Corradini R, Marchelli R. Polymerase chain reaction coupled with peptide nucleic acid high-performance liquid chromatography for the sensitive detection of traces of potentially allergenic hazelnut in foodstuffs. Eur Food Res Technol 2005, 220:619-624.
20. Rossi S, Scaravelli E, Germini A, Corradini R, Fogher C, Marchelli R. A PNA-array platform for the detection of hidden allergens in foodstuffs. Eur Food Res Technol 2006, 223:1-6.
21. Raszka A, Ziembińska A, Wiechetek A. Metody i techniki biologii molekularnej w biotechnologii środowiskowej. Środowisko czasopismo techniczne 2009, 2: 102-114.
22. Köppel R, Dvorak V, Zimmerli F, Breitenmoser A, Eugster A, Waiblinger HU. Two tetraplex real-time PCR for the detection and quantification of DNA from eight allergens in food. Eur Food Res Technol 2010, 230:367-374.
23. Poms RE, Anklam E, Kuhn M. Polymerase chain reaction techniques for food allergen detection. JAOAC 2004, 87:1391-1397.
24. Besler M. Determination of allergens in foods. Trends Anal Chem 2001, 20, 11: 662-672.
25. Blais BW, Gaudreault M, Phillippe LM. Multiplex enzyme immunoassay system for the simultaneous detection of multiple allergens in foods. Food Control 2003, 14:43-47.
26. Van Hengel AJ, Food allergen detection methods and the challenge to protect food-allergic consumers: review. Anal Bioanal Chem 2007, 389:111-118.

Biotechnology and Food Science Review article
Biotechnol Food Sci 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
Laktony jako związki biologicznie czynne
Kinga Libiszewska Instytut Podstaw Chemii Żywności, Politechnika Łódzka, 90-924 Łódź, Polska
Streszczenie: Wśród produktów naturalnych aktywnych biologicznie licznie reprezentowane są laktony. Cząsteczki te wykazują niezwykłe zróżnicowanie budowy – od pierścieni 4-6 członowych do struktur makrocyklicznych i szerokie spektrum aktywności. Niniejszy przegląd literatury prezentujący różnorodną aktywność biologiczną laktonów obejmuje głównie publikacje, które ukazały się w ciągu ostatnich pięciu lat.
Słowa kluczowe: β-lakton, α-metyleno-γ-butyrolakton, aktywność biologiczna.
Wstęp Laktony należą do dobrze znanych związków organicznych. Wykazują one
różnorodne działanie biologiczne, np. właściwości przeciwnowotworowe, przeciwwirusowe, przeciwbakteryjne, przeciwzapalne i inne. Są wśród nich także liczne substancje zapachowe i smakowe. W ostatnich latach nasila się tendencja do poszukiwania substancji aktywnych w przyrodzie. Stosując testy biologiczne, wyodrębnia się związki czynne, identyfikuje, a następnie opracowuje metody syntezy, które pozwalają modyfikować strukturę wiodącą, określić zależność właściwości biologicznych od budowy związku i zaprojektować nowe analogi o silniejszym lub odmiennym działaniu.
Aktywność przeciwnowotworowa Właściwości przeciwnowotworowe wykazują laktony makrocykliczne,
oraz liczne związki bicykliczne o wieloczłonowym pierścieniu skondensowanym z pierścieniem α-metyleno-γ-butyrolaktonu.
Laktony makrocykliczne stanowią fragment epotilonów stosowanych w leczeniu nowotworów piersi, jajników, jąder oraz trzustki. Epotilony są wtórnymi metabolitami glebowych miksobakterii Sporangium cellulosum. Leki te indukują agregację tubuliny, formację multipolarnych wrzecion, a w kon-sekwencji powodują zatrzymanie mitozy w fazie G2. Takie właściwości wykazują epotilon A (1) oraz B (2) [1-3]. Antybiotykami o działaniu cyto-statycznym zawierającymi ugrupowanie laktonowe są aktynomycyny wytwarzane przez kultury Actinomycetes antibioticus. Działanie aktynomycyn polega na wywołaniu zmian w cyklu komórkowym, powodujących spowolnienie przecho-dzenia komórek przez fazę S i zatrzymanie podziału w fazie G2. Takie właściwości wykazuje np. aktynomycyna D (3) [4,5]. Antybiotyk ten zbudowany

Libiszewska
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
46
jest z dwóch cyklopentapeptydów, w których wiazanie laktonowe łączy grupę hydroksylową treoniny z grupą karboksylową waliny.
Właściwości przeciwnowotworowe wykazują także germakranolidy, w których α-metyleno-γ-butyrolakton jest skondensowany z nienasyconym lub nasyconym dziesięcioczłonowym ketonem. Do tej grupy należą kalealakton C (4) izolowany z Calea urticifolia oraz germakranolid 5 wyodrębniony z nasion Carpesium triste, wykazujące aktywność cytostatyczną wobec komórek ludzkiej białaczki. W leczeniu nowotworów układu krwionośnego oraz jelita grubego aktywność wykazują inne germakranolidy michaolid A (6) oraz krassumolid A (7), wyizolowane z koralowców rodziny Labophytum. W obydwu przypadkach mechanizm toksyczności dla komórek nowotworowych jest obecnie badany [6].
O
O
OH OO
S
NOH
1
NH
O
ONH
O
N
O N
O
O
N
NH
O
O
NH
O
N
O
NO
ON
O
N
O
NH2
O O
3
O
O
OH OO
S
NOH
2
O
O
O
O
OAcOOH
4
OO
O
O
OO
O
OH
5
O
O
OH
OAc
OAc
O
6
O OOH
7
Inhibicja enzymów Podstawione laktony o pierścieniu czteroczłonowym wykazują często
zdolność hamowania enzymów. Wymienić tu można lipstatynę (8) wyizolowaną ze Streptomyces toxytricini oraz jej nasycony analog tetrahydrolipstatynę (9). Oba związki są inhibitorami lipazy trzustkowej, co sprawia iż znajdują zasto-sowanie w leczeniu otyłości [7,8]. Są składnikami Orlistatu®, preparatu na otyłość, który blokuje enzymy trawienne, w wyniku czego tłuszcze z pożywienia
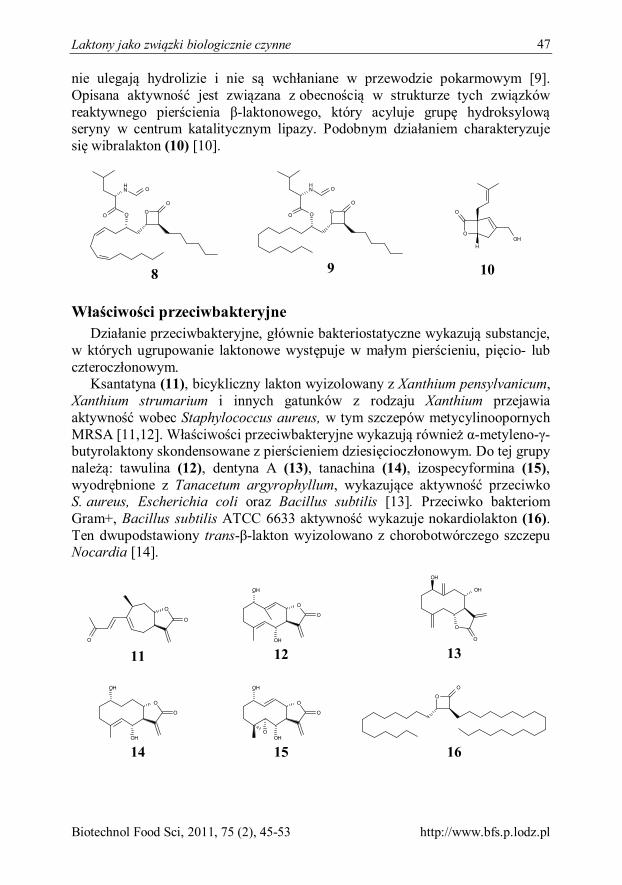
Laktony jako związki biologicznie czynne
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
47
nie ulegają hydrolizie i nie są wchłaniane w przewodzie pokarmowym [9]. Opisana aktywność jest związana z obecnością w strukturze tych związków reaktywnego pierścienia β-laktonowego, który acyluje grupę hydroksylową seryny w centrum katalitycznym lipazy. Podobnym działaniem charakteryzuje się wibralakton (10) [10].
OO
OO
NH
O
8
O
O
OO
NH
O
9
O
O
OHH
10
Właściwości przeciwbakteryjne Działanie przeciwbakteryjne, głównie bakteriostatyczne wykazują substancje,
w których ugrupowanie laktonowe występuje w małym pierścieniu, pięcio- lub czteroczłonowym.
Ksantatyna (11), bicykliczny lakton wyizolowany z Xanthium pensylvanicum, Xanthium strumarium i innych gatunków z rodzaju Xanthium przejawia aktywność wobec Staphylococcus aureus, w tym szczepów metycylinoopornych MRSA [11,12]. Właściwości przeciwbakteryjne wykazują również α-metyleno-γ-butyrolaktony skondensowane z pierścieniem dziesięcioczłonowym. Do tej grupy należą: tawulina (12), dentyna A (13), tanachina (14), izospecyformina (15), wyodrębnione z Tanacetum argyrophyllum, wykazujące aktywność przeciwko S. aureus, Escherichia coli oraz Bacillus subtilis [13]. Przeciwko bakteriom Gram+, Bacillus subtilis ATCC 6633 aktywność wykazuje nokardiolakton (16). Ten dwupodstawiony trans-β-lakton wyizolowano z chorobotwórczego szczepu Nocardia [14].
O
O
O
11
OH
OH
O
O
12
OH
OH
O
O 13
OH
OH
O
O
14
OH
OH
O
O
O 15
O
O
16

Libiszewska
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
48
Aktywność przeciwwirusowa Właściwości przeciwwirusowe wykazują skondensowane układy policykliczne
zawierające pierścień α-metyleno-γ-butyrolaktonowy. Należą do nich centa-urepensyna (chlorohyssopifolina A) (17), chlorojaneryna (18), 13-acetylo-solstitialina A (19), wyizolowane z nadziemnych części Centaurea solstitialis. Stosowane są głównie w leczeniu infekcji wirusowych górnych dróg oddecho-wych, chorobach układu pokarmowego oraz opryszczki wywoływanej przez HPV 2 (wirus opryszczki narządów płciowych) [15].
O
O
O
O
OH
ClOH
OH
Cl
H
H
17
O
O
O
OOH
OH
Cl
OHH
H
18
O
O
OH
OH
O
O
H
H
H
19
Działanie przeciwmalaryczne Malaria jest groźną chorobą zakaźną przenoszoną przez komary widliszki,
a wywoływaną przez pierwotniaki rodzaju Plasmodium. Cierpi na nią wielu mieszkańców krajów tropikalnych, szczególnie południowo-wschodniej Azji oraz zachodniej Afryki. Właściwości przeciwmalaryczne wykazuje artemizynina (20), seskwiterpenowy lakton występujący w Artemisia annua [16-18].
Spośród wielu hipotez dotyczących mechanizmu działania artemizyniny, żadna nie została w pełni potwierdzona. Wiadomo jedynie, że istotnym ze względu na działanie przeciwmalaryczne elementem struktury artemizyniny jest obecność układu nadtlenkowego. Co więcej, wiadomo, że wpływ na aktywność tego laktonu wywierają jony Fe2+ pochodzące z hemoglobiny. Jedna z hipotez zakłada, że w wyniku reakcji zachodzących między jonami Fe2+ i artemizyniną, powstaje znaczna ilość reaktywnych form tlenu, co w konsekwencji prowadzi do „przeciążenia” zabezpieczeń antyoksydacyjnych pasożyta [16-19].
O
O
H
H
O
O
O
20
Właściwości przeciwzapalne Niektóre laktony wykazują właściwości przeciwzapalne. Należy do nich
seskwiterpenowy lakton helenalina (21), wyodrębniona z Arnica Montana i Arnica chamissonis ssp. foliosa. Przez wiele lat ekstrakt alkoholowy z arniki zawierający helenalinę oraz jej pochodne był stosowany w tradycyjnej medycynie

Laktony jako związki biologicznie czynne
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
49
w leczeniu krwiaków, chorób reumatycznych oraz powierzchownych stanów zapalnych skóry.
Helenalina jest inhibitorem czynnika transkrypcyjnego NF-κB, który odgrywa kluczową rolę w regulacji odpowiedzi immunologicznej na infekcje. Lakton ten wpływa na białka NF-κB obecne w komórkach T i powoduje uruchomienie mitochondrialnego szlaku apoptozy oraz zatrzymanie cyklu komórkowego w fazie G2/M, co w konsekwencji przyczynia się do znacznego osłabienia odpowiedzi immunologicznej [20].
O
HO
O
OH 21
Aktywność przeciwpasożytnicza Wywoływane przez pasożyty choroby, takie jak trypanosomoza czy leiszmanioza
są głównym zagrożeniem zdrowia mieszkańców krajów rozwijających się i przy-czyniają się do ogromnej liczby zgonów rocznie.
Aktywność przeciw świdrowcom z rodziny Trypanosoma wykazuje wspomnia-na już wcześniej helenalina (21), natomiast psylostachyina (22) i peruwina (23) stosowane są na wiciowce z rodzaju Leishmania [21-23].
O
OOH
O O 22
O
O
OH
O
23
Zapach i smak Dobrze znanymi naturalnie występującymi chiralnymi związkami zapacho-
wymi są γ- i δ-laktony. Podczas gdy γ-laktony są ważnymi składnikami zapacho-wymi wielu owoców, δ-laktony nadają także zapach produktom mlecznym oraz innym produktom należącym do żywności fermentowanej [24].
Do najbardziej znanych oraz najczęściej wykorzystywanych w przemyśle spo-żywczym δ-laktonów należy między innymi δ-nonalakton (24). Charakteryzuje się on kremowym, słodkim, kokosowym i mlecznym zapachem. Występuje przede wszystkim w owocach tropikalnych, a jego niewielką ilość można znaleźć m. in. w maśle oraz mleku.
Podobnymi właściwościami odznacza się δ-dekalakton (25), o kremowym, słodkim, mlecznym, kokosowo-brzoskwiniowym aromacie, występujący głównie w owocach mango, kokosie oraz malinie. δ-Undekalakton (26) natomiast charakteryzuje się kremowym, woskowym, owocowym, kokosowo–brzoskwi-
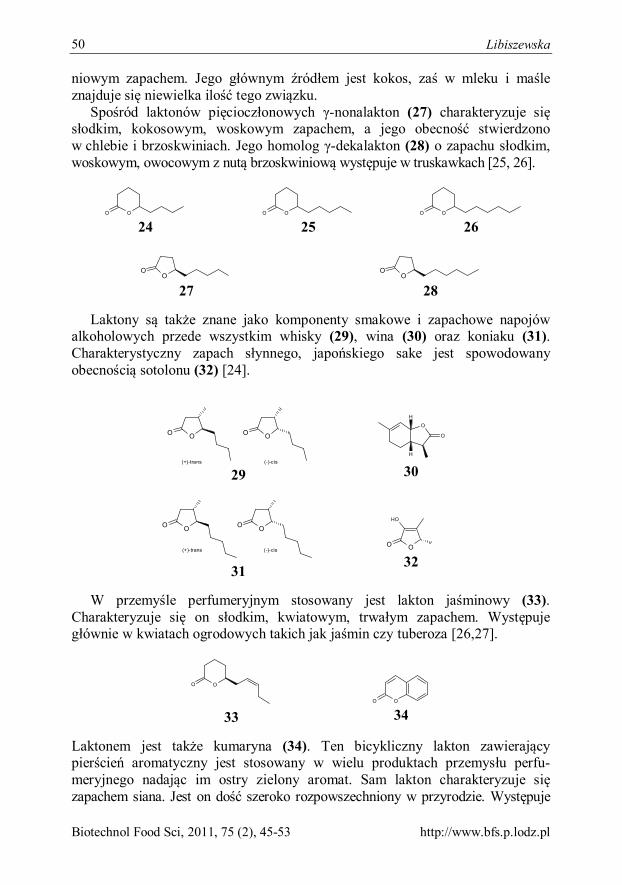
Libiszewska
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
50
niowym zapachem. Jego głównym źródłem jest kokos, zaś w mleku i maśle znajduje się niewielka ilość tego związku.
Spośród laktonów pięcioczłonowych γ-nonalakton (27) charakteryzuje się słodkim, kokosowym, woskowym zapachem, a jego obecność stwierdzono w chlebie i brzoskwiniach. Jego homolog γ-dekalakton (28) o zapachu słodkim, woskowym, owocowym z nutą brzoskwiniową występuje w truskawkach [25, 26].
OO 24
OO 25
OO 26
OO
27
OO
28 Laktony są także znane jako komponenty smakowe i zapachowe napojów
alkoholowych przede wszystkim whisky (29), wina (30) oraz koniaku (31). Charakterystyczny zapach słynnego, japońskiego sake jest spowodowany obecnością sotolonu (32) [24].
OO OO
(+)-trans (-)-cis 29
O
O
H
H 30
OO OO
(+)-trans (-)-cis
31
OO
OH
32
W przemyśle perfumeryjnym stosowany jest lakton jaśminowy (33).
Charakteryzuje się on słodkim, kwiatowym, trwałym zapachem. Występuje głównie w kwiatach ogrodowych takich jak jaśmin czy tuberoza [26,27].
OO
33
OO 34
Laktonem jest także kumaryna (34). Ten bicykliczny lakton zawierający pierścień aromatyczny jest stosowany w wielu produktach przemysłu perfu-meryjnego nadając im ostry zielony aromat. Sam lakton charakteryzuje się zapachem siana. Jest on dość szeroko rozpowszechniony w przyrodzie. Występuje

Laktony jako związki biologicznie czynne
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
51
głównie w takich roślinach jak: tonka wonna, nastrzyk lekarski, marzanka wonna oraz turówka wonna. Prócz perfumerii, kumaryna znalazła szerokie zastosowanie w medycynie, głównie w kardiologii jako składnik leków rozszerzających naczynia krwionośne. Podobne właściwości wykazują również pochodne kumaryny: furanokumaryny i piranokumaryny. Sama kumaryna jest bowiem wycofywana ze składu leków, jak i produktów spożywczych z powodu toksycznego działania na wątrobę [28].
Prócz laktonów mono- i bicyklicznych na uwagę zasługują również laktony wielkopierścieniowe. Jednym z ważniejszych przedstawicieli tej grupy jest egzaltolid (35).
O O
35
Jego obecność nadaje kompozycji zapach piżmowy, słodki, balsamiczny. Występuje on w olejkach roślinnych (np. olejku arcydzięglowym). Wielo-pierścieniowe laktony są jednocześnie cennymi składnikami zapachowymi i utrwalaczami wielu kompozycji perfumeryjnych.
Podsumowanie Przyroda jest niewyczerpanym źródłem substancji biologicznie czynnych.
Chemia produktów naturalnych przeżywa obecnie renesans, gdyż dzięki postępowi w technikach separacji, metodach analitycznych oraz testach biolo-gicznych możliwe jest wyodrębnianie substancji obecnej w badanym materiale w niewielkich ilościach. Dostępność związku umożliwia badanie mechanizmu działania oraz zależności aktywności biologicznej od struktury. Zaprezentowane w artykule przykłady pozwalają sądzić, że reaktywne ugrupowanie laktonowe odgrywa kluczową rolę w oddziaływaniu z innymi biocząsteczkami (enzymami, receptorami), którego skutkiem jest obserwowana czynność biologiczna.
Literatura 1. Zhu B, Panek JS. Total synthesis of epothilone A. Org Lett 2000, 2:2575-2578. 2. Altmann KH. Recent developments in the chemical biology of epothilones. Curr
Pharm Design 2005, 11: 1595-1613 3. Goodin S, Kane MP, Rubin EH. Epothilones: mechanism of action and biologic
activity. J. Clin. Oncol. 2004, 22:2015-2025 4. Koba M. Zależność aktywności biologicznej aktynomycyn od ich budowy
chemicznej na przykładzie modyfikacji struktury aktynomycyny D. Post Hig Med Dośw 2005, 59:276-282.
5. Koba M, Konopa J. Aktynomycyna D i mechanizmy jej działania. Post Hig Med Dośw 2005, 59:290-298.
6. Kitson RAR, Millemaggi A, Taylor RJK. The renaissance of α-methylene-γ-butyrolactones: new synthetic approaches. Angew Chem Int Ed 2009, 48:9426-9451.

Libiszewska
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
52
7. Venukadasula PKM, Chegondi R, Maitra S, Hanson PR. A concise, phosphate – mediated approach to the total synthesis of (-)-Trtrahydrolipstatin. Org Lett 2010, 12:1556-1559.
8. Ghosh AK, Shurrush K, Kulkarni S. Asymmetric synthesis of anti-aldol segments via a nonaldol route: synthetic applications to statines and (-)-Tatrahydrolipstatin. J Org Chem 2009, 74:4508-4518.
9. Yadav JS, Rao KV, Prasad AR. A chiron approach to (-)-Tetrahydrolipstatin. Synthesis 2006, 22:3888-3894
10. Liu D, Wang F, Liao T, Tang J, Steglich W, Zhu H, Liu J. Vibralactone: a lipase inhibitor with an unusual fused β-lactone produced by cultures of the Basidiomycete Boreostreum vibrans. Org Lett 2006, 8:5749-5752.
11. Yokoe H, Yoshida M, Shishido K. Total synthesis of (-)-Xanthatin. Tetrahedron Lett 2008, 49:3504-3506.
12. Matsuo K, Ohtsuki K, Yoshikawa T, Shisho K, Yokotani-Tomita K, Shinto M. Total synthesis of xanthanolides. Tetrahedron 2010, 66:8407-8419.
13. Goven N, Jakupovic J, Topal S. Sesquiterpene lactones with antibacterial activity from Tanacetum argyrophyllum ar. Argyrophyllum. Phytochemistry 1990, 29:1467-1469.
14. Kumaraswamy G, Markondaiah B. Proline-catalyzed stereoselective synthesis of natural and unnatural nocardiolactone. Tetrahedron 2008, 64:5861-5865.
15. Ozcelik B, Gürbüz I, Karaoglu T, Yeşiloda E. Antiviral and antimicrobial activities of three sesquiterpene lactones from Centaurea solstitialis L. ssp. Solstitialis. Microbiol Res 2009, 164:545-552.
16. Bharati A, Sabat SC. A spctrophotometric assay for quantification of artemisinin Talanta 2010, 82:1033-1037.
17. Nibret E, Wink M. Volatile components of four Ethiopian Artemisia species extracts and their in vivo antitrypanosomal and cytotoxic activities. Phytomedicine 2010, 17:369-374.
18. Woerdenbag HJ, Luers JFJ, van Uden W, Pras N, Malingre TM, Alfermann AW. Production of the new antimalarial drug artemisinin in shoot cultures of Artemisia annua L. Plant Cell Tiss Org 1993, 32:247-257.
19. Krishna S, Uhlemann A, Haynes R. Artemisinins: mechanisms of action and potential for resistance. Drug Resist Update 2004, 7:233-244.
20. Berges C, Fuchs D, Opel G, Daniel V, Naujokat C. Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms. Mol Immun 2009, 46:2892-2901.
21. Schmidt T, Brun R, Willuhn G, Khalid S. Anti-trypanosomal activity of helenalin and some structurally related sesquiterpene lactones. Planta Med 2002, 68:750-751.
22. Schmidt TJ, Nour AMM, Khalid SA, Kaiser M, Brun R. Quantitative structure – antiprotozoal activity relationships of sesquiterpene lactones. Molecules 2009, 14: 2063-2076.
23. Sulsen VP, Frank FM, Cazorla SI, Anesini CA, Machiodi EL, Freixa B, Vila R, Muschietti LV, Martino VS. Trypanocidal and leishmanicidal activities of sesquiterpene lactones from Ambrosia tenuifolia Sprengel (Asteraceae). Antimicrob Agents Chemother 2008, 52:2415-2419.
24. Brenna E, Fuganti C, Serra S. Enantioselective perception of chiral odorants. Tetrahedron Asymmetr 2003, 14:1-42.
25. Mosandl A, Gunther C. Stereoisomeric flavor compounds. 20.l Structure and properties of γ-lactone enantiomers. J Agric: Food Chem 1989, 37:413-418.

Laktony jako związki biologicznie czynne
Biotechnol Food Sci, 2011, 75 (2), 45-53 http://www.bfs.p.lodz.pl
53
26. Obara R, Szumny A, Wzorek A, Szmigiel-Pieczewska M, Białońska A, Ciunik Z, Wawrzeńczyk C. Lactones 31. Synthetic odoriferous unsaturated γ-lactones. Flavour Fragr J 2008, 23:416-425.
27. Kiyota H, Higashi E, Koike T, Oritani T. Syntheses and odor descriptions of jasmine lactone and cis-jasmone analogues bearing cyclopropane moieties. Flavour Fragr J 2001, 16:175-179.
28. Scotter MJ, Roberts DPT, Rees GO. Development and single – laboratory validation of an HPLC method for the determination of coumarin in foodstuffs using internal standardization and solid – phase extraction cleanup. Anal Methods 2011, 3: 414-419.
Lactones as biologically active compounds
Abstract: Among natural, biologically active products, numerous lactones can be found. These molecules exhibit highly diversified structures-starting from 4-6 membered rings, up to macrocyclic structures, and show a wide spectrum of activities. This review covers mainly publications from the last five years and presents different bioactivities of lactones

80 Artur Wirowski

Biotechnology and Food Science Review article
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie
Danuta Czajkowska,* Małgorzata Milner-Krawczyk, Marta Kazanecka
Wydział Chemiczny, Instytut Biotechnologii, Zakład Technologii i Biotechnologii Środków Leczniczych, Politechnika Warszawska, 00-664 Warszawa
Streszczenie: W pracy przedstawiono przegląd danych literaturowych dotyczących budowy i właściwości kwasu hialuronowego (HA). Wskazano miejsca występowania tego związku w organizmie człowieka oraz pełnione przez niego funkcje. Omówiono sposoby otrzymywania związku z materiału zwierzęcego oraz przy wykorzystaniu procesów biotechnologicznych. Wskazano cechy dodatnie i ujemne preparatów otrzymanych dwoma ww. sposobami. W części artykułu dotyczącej wytwarzania kwasu przez bakterie zwrócono szczególną uwagę na modyfikacje genomu bakterii fermentacji mlekowej oraz szczepów Escherichia coli i Bacillus subtilis, pod kątem poprawy produktywności kwasu. Polegały one na wprowadzeniu genów kodujących enzymy: syntazę hialuronową, UDP-glukozo-dehydrogenazę oraz UDP-glukozo-pirofosforylazę. Przedstawiono metody i warunki hodowli bakterii oraz uzyskiwane wydajności HA. Omówiono również możliwości zastosowania związku oraz jego pochodnych w leczeniu różnego typu schorzeń, w diagnostyce medycznej, w medycynie estetycznej i kosmetyce oraz w farmacji przy otrzymywaniu docelowych leków powoli uwalniających substancje aktywne.
Słowa kluczowe: kwas hialuronowy, właściwości, funkcja, zastosowanie
Budowa i charakterystyka kwasu hialuronowego Kwas hialuronowy (HA) należy do grupy związków określanych terminem
glikozaminoglikany (GAG). Są to polisacharydy, zbudowane z wielu powtarzających się jednostek disacharydów. Cząsteczka disacharydu HA składa się z cząsteczki kwasu D-glukuronowego i cząsteczki N-acetyloglukozaminy połączonych naprzemiennie wiązaniami β-(1-4) i β-(1-3) glikozydowymi (rysunek 1). Kwas hialuronowy jest naturalnym związkiem, mającym taką samą budowę chemiczną zarówno u człowieka, jak i u innych kręgowców oraz bakterii. W organizmach żywych występuje zazwyczaj w postaci soli sodowej – hialuronianu sodu [1, 2].

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
56
Rysunek 1. Budowa disacharydu kwasu hialuronowego [2]
Kwas hialuronowy wykazuje wiele właściwości, które mają istotne znaczenie w licznych zastosowaniach. Jedną z nich jest zdolność wiązania dużych ilości wody [1]. Jest to możliwe dzięki anionowej budowie kwasu, której efektem jest przyciąganie różnorodnych kationów, np. Ca2+. Związek charakteryzuje się wysoką aktywność osmotyczną, a ta z kolei umożliwia przyciąganie znacznych ilości wody na drodze osmozy. Dla przykładu, kiedy stężenie kwasu hialuronowego w roztworze osiągnie poziom 0,1 mg/ml, jedna cząsteczka kwasu jest w stanie związać nawet 1 litr płynu. Nie uwzględniając ograniczeń przestrzennych, spowo-dowałoby to 1000-krotne powiększenie jej objętości. W naturalnym środowisku, jakim jest macierz zewnątrzkomórkowa, takie pęczniejące cząstki kwasu napierają na zewnątrz, wywołując ciśnienie turgorowe, które wzrasta proporcjonalnie do ilości pobranej wody. Ponadto HA ma interesujące właściwości lepkosprężyste, wynikające z jego polimerowego charakteru. Cecha kwasu hialuronowego określana terminem „biozgodność” wynika z tego, że naturalnie występuje on w skórze, więc w bardzo niskim stopniu wywołuje niekorzystne reakcje organizmu. Może być całkowicie wchłaniany przez organizm, co jest niewątpliwą zaletą podczas stosowania go jako implantu w medycynie kosmetycznej.
Kwas hialuronowy w organizmach zwierzęcych jest syntetyzowany w błonie komórkowej. W procesie tym bierze udział syntaza hialuronianowa usytuowana po wewnętrznej stronie błony. Uczestniczy ona w naprzemiennym łączeniu reszt kwasu glukuronowego i N-acetyloglukozaminy [3]. Nowo zsyntetyzowany polimer jest transportowany do przestrzeni pozakomórkowej. Syntaza hialuronianowa charakteryzuje się wysoką aktywnością. Jest ona w stanie spolimeryzować około 100 cząsteczek monosacharydów na sekundę w warunkach in vitro. U ssaków stwierdzono występowanie trzech syntaz hialuronowych: Has 1, Has 2 i Has 3. Pierwsza z nich katalizuje polimeryzację małych ilości HA o masie cząsteczko-wej 2x106 Da; druga większych ilości tego kwasu o tej samej wielkości cząstek, natomiast trzecia, najaktywniejsza, polimeryzuje duże ilości HA o wielkości cząstek do 2x105 Da – rysunek 2 [3, 4].

Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 57
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
Rysunek 2. Kwas hialuronowy syntetyzowany przez syntazy hialuronowe występujące
u ssaków Has1, Has2, Has2 [4]
Występowanie i funkcje kwasu hialuronowego w organizmie ludzkim Szacuje się, że u dorosłego człowieka o masie ciała 70 kg, znajduje się około
15 g HA w postaci hialuronianu sodu [4]. Początkowo uważano, że HA jest jedynie wypełniaczem tkanki łącznej. Późniejsze badania, ujawniły jednak, że może być on połączony z komórkami za pomocą receptorów znajdujących się na ich powierzchni i pośredniczyć w wielu ważnych procesach fizjologicznych.
Największa ilość kwasu hialuronowego znajduje się w macierzy zewnątrz-komórkowej (ECM) tkanek łącznych. Występuje on w jednym z komponentów ECM, we frakcji proteoglikanów. Niemalże połowa kwasu hialuronowego znajdującego się w ciele człowieka występuje w skórze, a większość jego jest odnajdywana w przestrzeni międzykomórkowej. Szacuje się, że w ludzkiej skórze właściwej jest 200-500 g/ml HA, a w naskórku 100 g/ml. Kwas hialuronowy, poza pełnieniem funkcji macierzy, w której są zatopione komórki, odgrywa rolę związku wiążącego wodę, wpływając na objętość i ściśliwość skóry. Może on zwiększać oporność tkanki na stres mechaniczny poprzez wiązanie i zatrzymywanie wody w przestrzeniach międzykomórkowych. Odgrywa także rolę w wyłapywaniu wolnych rodników, powstających na skutek promieniowania ultrafioletowego [5, 6].
Kolejnym miejscem występowania kwasu hialuronowego w organizmie ludzkim jest chrząstka [5]. Związek ten występuje tu jako ważny strukturalnie element macierzy, której sieć kolagenowa wypełniona jest proteoglikanami z HA. Kwas hialuronowy bierze również udział w chondrogenezie. Niewielkie fragmenty tego związku indukują aktywację czynników transkrypcyjnych w chondrocytach.
Kwas hialuronowy odgrywa ponadto istotną rolę w mazi stawowej [5, 6]. W płynie stawowym torebki stawowej, występuje znaczna ilość HA (1400-

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
58
3600 g/ml) o dużej masie cząsteczkowej. Występuje on w formie niezwiązanej z białkami. Zapewnia on niezbędne nawilżenie stawów. Służy jako amortyzator, redukujący tarcie między poruszającymi się kośćmi, przez co zmniejsza się zużywanie się stawów.
Kwas hialuronowy występuje również w ciałku szklistym oka [7]. Substancja żelowa znajdująca się w tym organie, jest wyspecjalizowanym typem wysoce uwodnionej zewnątrzkomórkowej macierzy. Składa się ona ze wzajemnie powiązanych sieci glikozaminoglikanów i włókien kolagenowych. Zawartość glikozaminoglikanów (a wśród nich kwasu hialuronowego), a także ich konformacja, są odpowiedzialne za odpowiednią strukturę i funkcje ciałka szklistego. Stężenie kwasu hialuronowego w ciałku szklistym wynosi około 190-320 g/ml, z czego 45-77% to HA o dużej masie cząsteczkowej, większej niż 1000 kDa. Właśnie ten rodzaj polimeru zapewnia lepkosprężystość, cechę niezbędną w ciałku szklistym. Kwas hialuronowy współtworzy także film łzowy [7].
Omawiany związek odgrywa istotną rolę w biomechanicznych właści-wościach strun głosowych [8]. Wpływa on na lepkość, przepływ tkankowy, osmozę, amortyzację wstrząsów oraz gojenie się ran w tym organie. Jest to szczególnie ważne w przypadku strun, które są nieustannie narażone na urazy spowodowane wibracjami. Istotne znaczenie kwasu hialuronowego we właściwym funkcjonowaniu strun głosowych zostało udowodnione w doświadczeniach Chana i in. [9]. Naukowcy oceniali próbki strun głosowych, w których z zewnątrz-komórkowej macierzy usuwano hialuronian sodu za pomocą enzymu hialuronidazy. Brak HA powodował spadek elastyczności tkanek, sztywność oraz znaczny wzrost lepkości (o 70%). W efekcie powodowało to niewłaściwe działanie strun głosowych. Powyższe badania potwierdziły skuteczność stosowania kwasu hialuronowego jako chirurgicznego implantu strun głosowych.
Kwas hialuronowy występuje także w nerkach. Duże ilości HA stwierdzono w wewnętrznej części rdzenia, w tzw. brodawce nerkowej, natomiast bardzo niewielkie stężenie w korze [1]. Ta różnica ma istotne znaczenie w wytwarzaniu bardziej lub mniej stężonego moczu. Kwas hialuronowy wpływa na homeostazę, dystrybucję i przepływ wody oraz wspólnie z wazopresyną reguluje reabsorpcję wody do rdzenia nerkowego.
Wysoką zawartość kwasu hialuronowego o dużej masie cząsteczkowej odnotowano także w tkance łącznej pępowiny ludzkiej (4100 g/ml). Zapobiega on zerwaniu się połączenia pomiędzy matką a płodem [5]. Związek ten jest również obecny w mózgu, a także w moczu i surowicy krwi [5].
Badania Volpi i in. [4] wskazały na udział HA w wielu procesach związanych z owulacją i zapłodnieniem. Występujący w dużych ilościach kwas hialuronowy tworzy elastyczną, gąbczastą i odkształcalną macierz, która ułatwia uwalnianie oocytu podczas owulacji. Po owulacji, komórki pęcherzyka jajnikowego otacza-jącego oocyt pozostają osadzone w sieci kwasu hialuronowego. Zaobserwowano także związek pomiędzy zdolnością plemników do penetracji roztworu o dużej lepkości wynikającej z obecności HA a ruchliwością plemników i efektywnością

Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 59
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
zapłodnienia. Itano [10] wskazał także na istotną rolę kwasu hialuronowego w rozwoju embrionalnym człowieka.
Stern i in. [11] stwierdzili, że funkcje biologiczne kwasu w organizmie ludzkim zależą od wielkości cząsteczek polimeru. Fragmenty kwasu o dużej masie cząsteczkowej wykazują działanie immunosupresyjne i przeciwzapalne. Hamują również fagocytozę, wpływają na integralność tkanek, chroniąc je przed uszkodzeniami i apoptozą. Taki rodzaj kwasu hialuronowego uczestniczy także w procesach owulacji, zapłodnieniu i embriogenezie. Podczas procesów zapalnych powstaje nadzwyczajna forma wielkocząsteczkowego kwasu, silnie usieciowana, tworząca kompleksy z licznymi włóknami w miejscach zapalnych [12]. W ten sposób tworzy się tarcza ochronna i zapewniona jest niepodzielność tkanek w pobliżu miejsca zapalnego. W samym miejscu zapalnym wielkocząsteczkowe łańcuchy kwasu hialuronowego zostają zdegradowane do mniejszych (wielkości 4-6 sacharydów) przez działanie reaktywnych form tlenu i hialuronidazy. Drobno-cząsteczkowe fragmenty kwasu mogą przyłączać się do receptorów komórek odpornościowych, aktywując je i wpływając na szereg procesów, takich jak indukcja ekspresji cytokininy. Kwas hialuronowy o dużej masie molowej gro-madzi się także w pobliżu uszkodzeń w układzie nerwowym. Jest to obserwowane szczególnie w przypadku stwardnienia rozsianego [11]. Polimery o średniej wielkości, złożone z 25-50 disacharydów, wykazują działanie przeciwzapalne, stymulują procesy immunologiczne i naczyniotwórcze [5]. Oligosacharydy mniejsze od ww. są antyapoptotyczne i mają związek z białkami szoku termicznego [13].
Zaobserwowano zależność pomiędzy agresywnością nowotworów a zawartością kwasu hialuronowego w tkankach położonych w ich pobliżu [6]. Stwierdzono, że w przypadku występowania nowotworu złośliwego, w tkankach położonych w pobliżu guza obserwuje się nadmierną produkcję kwasu hialuronowego o niewielkich masach cząsteczkowych. Zwiększa się również jego stężenie w surowicy krwi. HA jest wypełniaczem przestrzeni, ale może również otwierać drogę dla komórek nowotworowych, co ma związek z przerzutami, np. raka piersi czy jelita grubego. Kwas hialuronowy oddziałuje z powierzchniowymi receptorami komórek rakowych, zwiększając ich przeżywalność i inwazyjność.
Zastosowanie kwasu hialuronowego i jego pochodnych Kwas hialuronowy jest wykorzystywany w medycynie, farmacji, w chirurgii
plastycznej i w kosmetyce. Jednym z głównych zastosowań kwasu hialuronowego jest jego użycie w chirurgii ortopedycznej [5, 14]. W chorobach artretycznych, takich jak osteoartretyzm czy reumatoidalne zapalenie stawów, objętość mazi stawowej wzrasta, co prowadzi do obniżenia stężenia kwasu. Ponadto wielko-cząsteczkowy HA jest rozkładany przez reaktywne formy tlenu, które redukują jego lepkość i upośledzają jego nawilżające i amortyzujące właściwości. Struktura stawu ulega zniszczeniu, zmienione tkanki są rozpoznawane przez układ immunologiczny jako ciało obce, co powoduje, że choroba staje się schorzeniem ogólnoustrojowym, wpływającym na funkcjonowanie całego organizmu. Walka z chorobą polega na uzupełnieniu ubytków w mazi stawowej

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
60
za pomocą iniekcji dostawowych. Stwierdzono, że podawanie HA łagodzi ból i objawy choroby zwyrodnieniowej stawów. Ponadto następuje odnowienie lepko-sprężystych właściwości płynu stawowego, stymulacja produkcji endogennych cząsteczek kwasu oraz powstrzymanie rozkładu chrząstki.
Kwas hialuronowy jest używany ponadto przy wielu zabiegach oku-listycznych [5]. Najczęściej stosuje się preparaty będące połączeniem kwasu hialuronowego z hydroksypropylometylocelulozą [15]. Wykorzystuje się go przy implantacji rogówki, wszczepianiu soczewki wewnątrzgałkowej, operacjach zaćmy i jaskry oraz laserowej chirurgii oka. Kwas hialuronowy jest również składnikiem kropli do oczu używanych w „zespole suchego oka” [4]. Omawiany związek jest bardzo podobny do mucyny, głównego składnika łez. Użyteczność HA opiera się na nawilżaniu oka, stanowieniu bariery ochronnej przed szkodliwymi czynnikami środowiska. Istotny jest fakt, że nie jest on usuwany z powierzchni oka nawet podczas mrugnięć.
HA wykorzystuje się także w otolaryngologii [5]. W tym zastosowaniu używa się preparaty zawierające usieciowany kwas, gdyż zapewnia to przedłużenie czasu jego przebywania w organizmie, nawet do roku. Istnieją możliwości zastosowania HA do przyśpieszenia gojenia się ran błony bębenkowej ucha, jako bioimplantu do chirurgicznej naprawy ubytków strun głosowych, regeneracji zranionych lub zabliźnionych fałd, rekonstrukcji krtani i głośni [9].
Preparaty kwasu hialuronowego o dużej masie cząsteczkowej, stosowane miejscowo, wspomagają gojenie się świeżych ran skóry oraz są przydatne przy przewlekłych stanach zranienia lub oparzeniach [5]. Ze względu na właściwości antyoksydacyjne, HA służy jako składnik przeciwzapalny w materiałach opatrunkowych. W połączeniu z dekspentanolem, jest używany jako preparat silnie nawilżający i regenerujący. Żele na bazie HA są stosowane przy leczeniu owrzodzenia jamy ustnej, przyspieszają gojenie, redukując ból, objawy zapalenia i dyskomfort [16].
Interesującym kierunkiem jest wykorzystanie oligosacharadów HA do opóźniania wzrostu guza, a nawet jego apoptozy [17]. Podczas takiej terapii, komórki nowotworowe stają się bardziej wrażliwe na działanie leków, niż w przypadku terapii bez HA. Wprowadzenie takiej formy leczenia, pozwala na zmniejszenie ilości leków używanych w chemioterapii.
Ciekawym zastosowaniem HA w inżynierii tkankowej jest odbudowa chrząstki, bazująca na żelu opartym na fibrynie i HA [18]. Stanowi on dobrą substancję transportującą chondrocyty do uszkodzonego fragmentu tkanki.
Najczęściej produkty bazujące na kwasie hialuronowym są wykorzystywane w chirurgii plastycznej jako implanty poprawiające wygląd skóry [5, 19]. Umożliwiają one bezpieczne wypełnianie zmarszczek i wklęsłych blizn na twarzy. W kosmetyce stosuje się żele HA całkowicie lub częściowo nasycone wodą [20]. Pierwsze z nich nie pobierają wody z otaczających je tkanek; drugie natomiast chłoną wodę z otoczenia, zwiększając swoją objętość, a tym samym dając efekt wolumizacji. Żele HA są bardziej efektywne niż te z kolagenem, gdyż

Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 61
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
charakteryzują się większą elastycznością. Co więcej, implanty takie można apli-kować poprzez wstrzykiwanie, nie trzeba wszczepiać ich operacyjnie, co eliminuje ryzyko infekcji. Jedną z wielu korzyści w tego typu zabiegach jest to, że nie ma okresu rekonwalescencji. Pacjent po zabiegu może praktycznie od razu przystąpić do codziennych czynności. Kwas hialuronowy obecnie jest stosowany do poprawy wyglądu skóry twarzy, fałd nosowo-wargowych, łuków brwiowych, kości policzkowych, powiększenia ust, drobnych korekcji nosa czy podbródka [21].
Kwas hialuronowy, po wstrzyknięciu do skóry, jest stosunkowo szybko degradowany przez hialuronidazy i wolne rodniki, które naturalnie tam wystę-pują [2]. Ponadto związek ten, nawet o wysokiej masie cząsteczkowej, ma słabe właściwości biomechaniczne. Podejmowane są zatem próby ulepszenia właściwości HA poprzez proces sieciowania [22]. W procesie tym używa się środków sieciujących, które tworzą połączenia pomiędzy cząsteczkami kwasu hialuronowego. Środki te mają różne grupy funkcyjne, przez co reagują w odmienny sposób z kwasem hialuronowym. Zazwyczaj do sieciowania stosuje się formaldehyd, aldehyd glutarowy, epoksydy, diwinylosulfon, karbodiimidy. Biorąc pod uwagę biostabilność nowo powstałych preparatów, stwierdzono, że kwas hialuronowy usieciowany karbodiimidami może przetrwać in vivo 15 dni, usieciowany epoksydami powyżej 3 miesięcy, natomiast sieciowany HA z wiązaniami estrowymi degraduje szybciej niż jakikolwiek inny preparat. Niekiedy jednak pojedyncze sieciowanie jest niewystarczające dla stabilności produktu. Z tego względu opracowano technologie podwójnego sieciowania – najpierw poprzez tworzenie wiązań eterowych, a potem estrowych [22]. Drugie wiązania poprawiają bio-stabilność poprzez zwiększenie stopnia usieciowania, zwiększają także oporność zmodyfikowanego produktu na trawienie hialuronidazami oraz na działanie wolnych rodników poprzez utrudnienie docierania tych czynników do łańcuchów polimerów.
Właściwości kwasu hialuronowego jako biopolimeru zostały również wyko-rzystane do otrzymywania preparatów farmaceutycznych, których celem jest dostarczenie leku w określone miejsce w organizmie i jego powolne uwolnienie [23]. Opracowano metody produkcji hydrożeli, w których obecność grup hydroksylowych i sieciowanie umożliwia ich sprzężenie z lekiem. Innym rozwiązaniem jest tworzenie mikrokapsułek z HA zawierających lek. Yadav i in. [23] wskazali, że w tkankach nowotworowych czy w tkankach ze stanem zapalnym, występuje nadekspresja receptorów kwasu hialuronowego. Sprzyja to zwiększeniu wiązania kompleksu HA-lek i ma istotny wpływ na efektywność leczenia.
W medycynie klinicznej, kwas hialuronowy jest stosowany jako marker do diagnozowania wielu chorób, takich jak reumatoidalne zapalenie stawów, nowotwory, schorzenia wątroby [5]. Według badań Geramizadeh i in. [24] określenie poziomu HA w surowicy krwi może być przydatne do oceny stopnia zwłóknienia wątroby u pacjentów z zapaleniem wątroby typu B. U osób z HBV, zawartość HA waha się na poziomie 73,4 ± 124,8 ng/ml, przy normie 20,4 ± 15,5 ng/ml. U pacjentów z rozległą marskością wątroby poziom HA

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
62
oscyluje wokół wartości 309.7 ± 143.5 ng/ml, a u tych z łagodną w granicach 24,7 ± 31.9 ng/ml. HA może być stosowany jako biochemiczny marker w diagnostyce atrezji dróg żółciowych powodowanej powszechnie przez przedłu-żoną żółtaczkę wieku niemowlęcego [25]. Kishida i in. [26] w swoich badaniach udowodnili, że omawiany związek może być również wskaźnikiem obecności stanu zapalnego w szyjce macicy lub w błonie płodowej, które mogą być przyczyną przedwczesnego porodu. Badania Kogana i in. [6] wskazały też na możliwość szybkiego zdiagnozowania obecności nowotworu złośliwego u człowieka.
Otrzymywanie kwasu hialuronowego z organizmów zwierzęcych oraz metodami biotechnologicznymi
Jak dotąd podstawowym sposobem otrzymywania HA jest jego izolacja z różnych części organizmów zwierzęcych, tj. grzebieni kogucich, gałek ocznych kręgowców, skóry rekina, pępowiny. W procesie tym otrzymuje się głównie wielkocząsteczkowy HA. Średnia masa molowa powszechnie dostępnych ekstrakcyjnych preparatów HA z tkanek zwierzęcych, ma wielkość od kilkuset tysięcy Da do około 2,5 MDa. Grzebienie kogucie, to tkanka zwierzęca o największej zawartości HA (7500 g/ml) [5]. Kang i in. [27] opisali efektywną metodę otrzymywania HA z materiału zwierzęcego, w której można było uzyskać z 500 g grzebieni kogucich 500 mg suchego kwasu hialuronowego, natomiast Amagai i in. [28] – metodę otrzymywania kwasu z gałek ocznych tuńczyka wielkookiego Thunnus obesus. Źródłem kwasu hialuronowego może być również ludzka pępowina [29].
Proces pozyskiwania kwasu hialuronowego ze źródeł zwierzęcych ma kilka istotnych wad [4]. HA występuje naturalnie w organizmach zwierzęcych jako kompleks z innymi biopolimerami, na przykład białkami. Związki te, będąc zanieczyszczeniem preparatów HA mogą wykazywać działanie alergenne. I tak Andre i in. [30], prowadząc badania na grupie 4320 pacjentów, odnotowali wystąpienie nadwrażliwości u 34 osób (w 16 przypadkach zmiany wystąpiły niemal bezpośrednio po wstrzyknięciu HA, u pozostałych osób pojawiły się z pewnym opóźnieniem). Obserwowane reakcje zapalne w pobliżu miejsc wstrzyknięcia kwasu, to: opuchlizna, zaczerwienienie, ropnie, guzki, ziarnina typu ciała obcego. Ponadto w próbkach kwasu mogą znajdować się śladowe ilości kationów metali, co może stanowić potencjalne ryzyko zmniejszenia masy cząsteczkowej HA, a w konsekwencji zmianę jego właściwości. Obecność w preparatach pozostałości kwasów nukleinowych doprowadza czasami do indukcji cytokinin zapalnych [4]. Z tych powodów, po każdym procesie produkcji istnieje konieczność przeprowadzania specjalnych procedur oczyszczających, mających na celu otrzymanie czystego kwasu. Stosuje się różnorodne metody, takie jak: traktowanie próbek proteazami; wytrącanie kwasu hialuronowego, np. chlorkiem cetylopirydyny; oczyszczanie na membranie ultrafiltracyjnej; wytrącanie HA bez rozpuszczalnika i liofilizacja.

Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 63
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
Rysunek 3. Schemat powstawania kwasu hialuronowego u bakterii z rodzaju
Streptococcus [31]
Ze względu na wyżej wymienione niedoskonałości kwasu hialuronowego otrzymywanego ze źródeł zwierzęcych, coraz większą uwagę zwraca się na metody bazujące na hodowli odpowiednich szczepów bakterii. Kwas hialuronowy wyizolo-wano po raz pierwszy z chorobotwórczych bakterii z rodzaju Streptococcus [31]. Występował on w grubej otoczce tych mikroorganizmów. Badając metabolizm tych drobnoustrojów, Chong i in. [31] stwierdzili, że kwas D-glukuronowy i N-acetylo-glukozamina (składniki disacharydu HA) powstawały w komórkach bakterii odpowiednio z glukozo-6-fosforanu i fruktozo-6-fosforanu. W procesie tym uczestniczyło wiele enzymów: fosfoglukomutaza, UDP-glukozo-dehydrogenaza, UDP-glukozo-pirofosforylaza, syntaza HA, amidotransferaza, acetylotransferaza, mutaza i pirofosforylaza (rysunek 3). Na zsyntetyzowanie 1 mola HA były zużywane 4 mole ATP.

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
64
Ze względu na to, że w procesach biotechnologicznych nie mogą być wykorzystywane bakterie chorobotwórcze, podjęto próby znalezienia drobno-ustrojów mających status GRAS, które można byłoby wykorzystać w procesach biosyntezy kwasu hialuronowego. Szczególną uwagę zwrócono na niepatogenne bakterie fermentacji mlekowej.
Izawa i in. [32] dokonali skriningu 46 szczepów bakterii Streptococcus thermophilus wyizolowanych z produktów spożywczych. Przeprowadzili hodowlę bakterii na odtłuszczonym mleku i określili ilość wyprodukowanego HA. Zdolność wytwarzania biopolimeru odnotowano u sześciu szczepów. Największą ilość kwasu (8 mg/l) wytwarzał szczep YIT2084. Po jego hodowli w optymalnych warunkach (temp. 35°C, 100 obr/min, pH 6,8) na pożywce z mleka z peptydami sojowymi jako źródłem azotu uzyskano 208 mg HA/l. Chie i Lee [33] podjęli próbę opracowania procesu otrzymywania HA przy wykorzystaniu szczepów bakterii fermentacji mlekowej modyfikowanych genetycznie. W tym celu użyli bakterii Lactococcus lactis, do genomu których wprowadzali geny syntazy HA i UDP-glukozo-dehydrogenazy. Geny odpowie-dzialne za syntezę tych enzymów uzyskano z genomu Streptococcus equi subsp. zooepidemicus. System ekspresji ww. genów w bakteriach L. lactis znajdował się pod kontrolą nizyny (naturalnej bakteriocyny, wytwarzanej przez te bakterie). Przeprowadzono dwuetapową hodowlę rekombinata LL-NA z genem has oraz podwójnego rekombinata LL-NAB z genem has i genem kodującym UDP-glukozo-dehydrogenazę. W pierwszym etapie namnażano biomasę na pożywce zawierające 0,5% w/v glukozy. W drugim etapie, bakterie hodowano w pożywce z 1% w/v glukozy, do której w pewnym momencie dodawano nizynę w celu zaindukowania ekspresji wstawionych genów. Po zakończeniu hodowli, w wa-riancie doświadczenia z bakteriami LL-NA zawartość HA wynosiła około 0,08 g/l, natomiast w wariancie z rekombinantem LL-NAB była ponad 8-krotnie wyższa. Otrzymane wyniki potwierdziły fakt, że poziom ekspresji genu kodującego UDP-glukozo-dehydrogenazę u L. lactis jest niski. Zatem zasadne jest wprowadzenie dodatkowego genu kodującego UDP-glukozo-dehydrogenazę ze Streptococcus.
Próbą udoskonalenia wyżej opisanej metody produkcji HA było wpro-wadzenie do genomu bakterii L. lactis, obok dwóch opisanych wcześniej genów, także genu kodującego UDP-glukozo-pirofosforylazę [34]. Szczep SJR2 zawierał gen hasA (syntaza HA) i hasB (UDP-glukozo-dehydrogenaza), natomiast szczep SJR3 gen hasA, hasB oraz hasC (UDP-glukozo-pirofosforylaza). Po 15 godzinnej hodowli obu rekombinantów uzyskano w przypadku rekombinata SJR2 około 100 mg HA/l, natomiast w przypadku SJR3 nieco ponad 200 mg HA/l. Prasad i in. [34] przeprowadzili hodowlę dwóch wyżej wymienionych szczepów w bioreaktorze. Najlepsze wyniki zawartości HA (1,8 g/l) uzyskano w hodowli napowietrzanej szczepu SJR3.
W chwili obecnej najczęściej wykorzystywanymi szczepami do produkcji HA są naturalne szczepy S. equi i S. equi subsp. zooepidemicus [4]. W warunkach

Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 65
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
tlenowych, w temperaturze hodowli w zakresie 36-40°C oraz przy pH obojętnym, ilość wytwarzanego HA wahała się w granicach 3-6 g/l. Wydajność ta zależy od uzyskanej masy cząsteczkowej kwasu. Czas hodowli jest ograniczony ze względu na rosnącą lepkość cieczy pohodowlanej, wynikającą z właściwości samego kwasu. Przeprowadzono wiele prób mających na celu zwiększenie ilości wytwarzanego kwasu hialuronowego w hodowlach bakterii S. zooepidemicus. Liu i in. [35] badali wpływ dodawania n-dodekanu na ilość wyprodukowanego HA. Badacze stwierdzili, że przy stężeniu n-dodekanu wynoszącym 5% następuje wzrost produkcji kwasu hialuronowego o 30%. Związek ten wpływał również na wielkość masy cząsteczkowej HA. Wzrosła ona z poziomu (1,30,1)106 Da do (1,90,2)106 Da, gdy stężenie n-dodekanu wynosiło 3%, a następnie zmalała do 1,6106 Da przy stężeniu 5%. Zwiększenie ilości wytwarzanego kwasu hialuronowego przez S. zooepidemicus można było także osiągnąć poprzez zastosowanie w hodowli przerywanego stresu alkalicznego [36]. W tego typu hodowli, pH było utrzymywane na poziomie 7,0 przez pierwsze 6 godzin, po czym było szybko podwyższane do wartości pH 8,0 na 0,5 godziny, następnie do wartości pH 8,5 na 1 godz. i do pH 9 na 1,5 godz. Ostatecznie powracano do pH 7 na 1 godz. przed zakończeniem procesu fermentacji. Stosując opisany sposób hodowli udało się uzyskać wzrost produkcji kwasu hialuronowego z poziomu 50,1 g/l (kontrola) do 6,50,2 g/l.
Ze względu na konieczność hodowli bakterii kwasu mlekowego, w tym także z rodzaju Streptococcus na drogich pożywkach, zaczęto poszukiwać mniej wymagających szczepów drobnoustrojów. Jednym z obiecujących, potencjalnych kandydatów do produkcji kwasu hialuronowego są Gram-dodatnie bakterie Bacillus subtilis [5, 37]. Ich genom został dobrze zbadany, co ułatwia mani-pulację genetyczną. Ponadto zaletą tych szczepów jest łatwość hodowli na dużą skalę oraz brak wytwarzania egzo- i endotoksyn. Co więcej, B. subtilis nie pro-dukuje hialuronidazy, która mogłaby zniszczyć syntetyzowany przez bakterie HA.
W procesie otrzymywania kwasu hialuronowego wykorzystano szczep B. subtilis RB161 niosący gen hasA ze Streptococcus equisimilis [5]. Tak skonstruowany szczep wytwarzał zewnątrzkomórkowo HA o masie cząsteczko-wej w zakresie 1 MDa. Produkcja HA przez te bakterie była ekonomiczna, ponieważ rosły one na minimalnej, a więc taniej pożywce. Widner i in. [37] opisali hodowlę szczepu B. subtilis RB161 metodą okresową z zasilaniem pod kątem uzyskiwania HA. Przeprowadzili ją w bioreaktorze o pojemności 3 l, na minimalnej pożywce, zawierającej sacharozę jako źródło węgla. Ilość HA zwiększała się do około 25 h hodowli i osiągała poziom około 1,0 g/l. Zwiększe-nie produkcji kwasu hialuronowego przez B. subtilis można było uzyskać poprzez koekspresję enzymu UDP-glukozo-dehydrogenazy ze Streptococcus z syntazą HA, co powodowało dwukrotny wzrost ilości wytworzonego kwasu [38]. Przepro-wadzono również badania wpływu genu kodującego bakteryjną hemoglobinę, pochodzącego z Vitreoscill na wzrost i produkcję HA przez te bakterie [38].

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
66
Po 30 h hodowli, obserwowano zwiększenie populacji komórek o 25% oraz produkcji kwasu hialuronowego o 100%. Wydajność HA wynosiła 1,8 g/l
Kolejną bakterią wykorzystywaną do wytwarzania kwasu hialuronowego była Escherichia coli [39, 40]. Mao i in. [40] opisali szczep JM109, do którego wprowadzili gen pmHas kodujący syntazę HA, (pochodzący z bakterii Pasteurella multocida) oraz gen kfiD kodujący UDP-glukozo-dehydrogenazę (pochodzący ze szczepu K5 E. coli). Szczepem wyjściowym, poddawanym modyfikacjom genetycznym, był szczep E. coli JM109/pBQ niewytwarzający HA. Początkowo ww. autorzy przeprowadzali hodowle wstrząsane w kolbach. Stosując rekombinowany szczep JM109/pHK, uzyskali w czasie 24 godzin hodowli kwas o masie cząsteczkowej 1,5 MDa w ilości 0,55 g/l. W kolejnym etapie przeprowadzili hodowlę bakterii w 1 l bioreaktorze zawierającym 500 ml bulionu Terrific, do którego dodatkowo wprowadzano 100 µg/ml ampicyliny i 100 µg/ml kanamycyny. Pierwszy etap hodowli trwał 4 godziny, pH było utrzymywane na poziomie 6,8, temperatura na poziomie 37°C. Po tym okresie, w celu wywołania ekspresji genów pmHas i kfiD, do pożywki dodawano izopro-pylotiogalaktozyd (IPTG), glukozę i fosforan amonu, w ilościach, odpowiednio 1 mM, 50 g/l i 10 g/l pożywki. Temperaturę obniżono do 30°C, aby umożliwić lepszą ekspresję zrekombinowanych białek. Po 14 godzinach dodawano drugą porcję pożywki o tym samym składzie. Wprowadzono również różne ilości glukozaminy (do stężenia 25 g/l) i fosfomycyny (do stężenia 200 µg/ml). Wyniki doświadczeń wykazały, że w hodowli z okresowym zasilaniem można było otrzymać w cieczy 2,0-3,8 g/l kwasu hialuronowego (0,027 g HA/g glukozy). Wydajność rekombinantów E. coli była jednak 2-3 razy niższa niż naturalnych producentów kwasu hialuronowego Streptococcus (0,065-0,088 g HA/g glukozy). Produktywność jest lepsza w warunkach tlenowych i zwiększa się o 35% przy utrzymywaniu rozpuszczalności tlenu na poziomie 10%. Synteza kwasu hialuronowego jest powiązana z namnażaniem biomasy. Zbyt duża ilość biomasy może powodować trudności w dostarczaniu składników odżywczych do wszystkich bakterii, co powoduje zahamowanie wytwarzania kwasu hialuronowego. Oddzielenie od siebie dwóch procesów: namnażania biomasy i produkcji HA, umożliwia uzyskanie lepszych wydajności produktu. Użycie do inhibicji wzrostu bakterii fosfomycyny powoduje zwiększenie produkcji HA o 70%. Dodatek glukozaminy (skrócenie szlaku biosyntezy HA) przyczynia się do wzrostu produkcji kwasu o 37%. Do modyfikacji bakterii E. coli używano również genów kodujących syntazę kwasu hialuronowego pochodzących ze S. pyogenes lub S. equisimilis [39]. Uzyskano mniejsze wydajności kwasu (odpowiednio: 48 mg/l z 48-godzinnej hodowli i 190 mg/l z 24-godzinnej hodowli).
Metody pozyskiwania kwasu hialuronowego z wykorzystaniem mikro-organizmów mają kilka wad [5]. Niektóre z nich to: ryzyko mutacji w szczepach bakteryjnych, możliwość wytwarzania różnych toksyn, pirogenów i immunogenów. Te powody przyczyniają się do tego, że kwas hialuronowy pochodzący z grze-bieni kogucich jest nadal preferowanym preparatem służącym do leczenia

Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 67
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
człowieka w przypadkach, gdy preparat jest wstrzykiwany do oka czy stawu kolanowego. Obecność niekorzystnych substancji wymusza konieczność zastoso-wania procedur mających na celu oczyszczenie preparatów. W chwili obecnej wykorzystuje się wiele różnych sposobów oczyszczania kwasu hialuronowego pochodzenia mikrobiologicznego, podczas których zwraca się szczególną uwagę na to, aby nie uległ on degradacji [4]. Po zakończeniu hodowli, otrzymuje się lepką ciecz pofermentacyjną, którą rozcieńcza się wodą i odfiltrowuje lub odwirowuje w celu oddzielenia biomasy bakteryjnej. HA jest wydzielany zewnątrzkomórkowo, więc znajduje się w cieczy pohodowlanej, którą poddaje się kilku etapom oczyszczania. Dla przykładu, Rangaswamy i Jain [41] lepką zawiesinę zawierającą 5 g HA/l rozcieńczali wodą i wirowali przez 20 min w temp. 4°C. Kwas hialuronowy obecny w cieczy wytrącali 2-propanolem, a następnie zawieszali w octanie sodu. Roztwór ten w temperaturze pokojowej wytrząsali z żelem krzemionkowym (2%) przy 150 obr/min przez 2 godziny i wirowali przez 20 min w 4oC. Następnie roztwór przepuszczali przez filtr węglowy i po 5-krotnym rozcieńczeniu oczyszczali za pomocą ultrafiltracji w trybie diafiltracji. Ostatnim etapem była filtracja sterylizująca, w której wykorzystywali filtry o wielkości porów 0,22 μm. W powyższej metodzie 96% zanieczyszczeń było usuwanych poprzez działanie żelem krzemionkowym i przy wykorzystaniu filtrów węglowych. Dalsze postępowanie pozwalało uzyskać produkt z 0,06% białek w stosunku do HA. Opisana powyżej metoda była prosta, efektywna, dawała wysoką wydajność oczyszczania próbek kwasu hialuronowego, a preparaty HA otrzymane w ten sposób ze względu na swoją czystość mogły być używane w medycynie.
Podsumowanie Niedoskonałości ekstrakcyjnego kwasu hialuronowego otrzymywanego z ma-
teriału zwierzęcego i jednocześnie olbrzymie możliwości zastosowania omawianego polimeru wskazują na potrzebę kontynuacji badań nad otrzymywaniem i oczy-szczaniem kwasu hialuronowego przy wykorzystaniu bakterii. Dalsze prace zmierzające do opracowania opłacalnej technologii produkcji kwasu powinny być ukierunkowane na doskonalenie szczepów poprzez świadomą modyfikację ich genomów oraz doskonalenie metod hodowli niwelujących negatywne za-leżności między ilością namnożonej biomasy a produkcją HA.
Literatura 1. Rügheimer L. Hyaluronan: A matrix component. Proc AIP Conf 2008, 1049:126-132. 2. Kablik J, Monheit GD, Yu L, Chang G, Gershkovich J. Comparative physical
properties of hyaluronic acid dermal fillers. Dermatol Surg 2009, 35 Suppl 1:302-312. 3. Daroszewski J, Rybka J, Gamian A. Glikozaminoglikany w patogenezie i diagnostyce
oftalmopatii Gravesa. Postepy Hig Med Dosw 2006, 60:370-378.

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
68
4. Volpi N, Schiller J, Stern R, Soltes L. Role, metabolism, chemical modifications and applications of hyaluronan. Curr Med Chem 2009, 16:1718-1745.
5. Kogan G, Soltes L, Stern R, Gemeiner P. Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol Lett 2007, 29:17-25.
6. Kogan G, Soltes L, Stern R, Schiller J, Mendichi R. Hyaluronic acid: Its function and degradation in in vivo systems. Stud Nat Prod Chem 2008, 34:789-882.
7. Theocharis DA, Skandalis SS, Noulas AV, Papageorgakopoulou N, Theocharis AD. Karamanos NK, Hyaluronan and chondroitin sulfate proteoglycans in the supramolecular organization of the mammalian vitreous body. Connect Tissue Res 2008, 49:124-128.
8. Ward PD, Thibeault SL, Gray SD. Hyaluronic acid:its role in voice. J Voice 2002, 16:303-309.
9. Chan RW, Gray SD, Titze IR. The importance of hyaluronic acid in vocal fold biomechanics. Otolaryngol Head Neck Surg 2001, 124:607-614.
10. Itano N. Simple primary structure, complex turnover regulation and multiple roles of hyaluronan. J Biochem 2008, 144:131-137.
11. Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information-rich system. Eur J Cell Biol 2006, 85:699-715.
12. Krasiński R, Tchórzewski H. Hialuronian jako czynnik regulujący proces zapalenia. Postepy Hig Med Dosw 2007, 61:683-689.
13. Xu H, Ito T, Tawada A, Maeda H, Yamanokuchi H, Isahara K, Yoshida K, Uchiyama Y, Asari A. Effect of hyaluronan oligosaccharides on the expression of heat shock protein 72. J Biol Chem 2002, 277:17308-17314.
14. Kalman DS, Heimer M, Valdeon A, Schwartz H, Sheldon E. Effect of a natural extract of chicken combs with a high content of hyaluronic acid (Hyal-Joint) on pain relief and quality of life in subjects with knee osteoarthritis:a pilot randomized double-blind placebo-controlled trial. Nutr J 2008, 7,3:1-9.
15. Maltese A, Borzacchiello A, Mayol L, Bucolo C, Maugeri F, Nicolais L, Ambrosio L. Novel polysaccharides-based viscoelastic formulations for ophthalmic surgery: rheological characterization. Biomaterials 2006, 27:5134-5142.
16. Lee JH, Jung JY, Bang D. The efficacy of topical 0.2% hyaluronic acid gel on recurrent oral ulcers: comparison between recurrent aphthous ulcers and the oral ulcers of Behcet's disease. J Eur Acad Dermatol Venereol 2008, 22:590-595.
17. Toole BP, Ghatak S, Misra S. Hyaluronan oligosaccharides as a potential anticancer therapeutic. Curr Pharm Biotechnol 2008, 9:249-252.
18. Park SH, Cui JH, Park SR, Min BH. Potential of fortified fibrin/hyaluronic acid composite gel as a cell delivery vehicle for chondrocytes. Artif Organs 2009, 33:439-447.
19. Raspaldo H. Volumizing effect of a new hyaluronic acid sub-dermal facial filler: a retrospective analysis based on 102 cases. J Cosmet Laser Ther 2008, 10:134-142.
20. Tezel A, Fredrickson GH. The science of hyaluronic acid dermal fillers. J Cosmet Laser Ther 2008, 10:35-42.
21. Andre P. New trends in face rejuvenation by hyaluronic acid injections. J Cosmet Dermatol 2008, 7:251-258.
22. Zhao X. Synthesis and characterization of a novel hyaluronic acid hydrogel. J Biomater Sci Polym Ed 2006, 17:419-433.
23. Yadav AK, Mishra P, Agrawal GP. An insight on hyaluronic acid in drug targeting and drug delivery. J Drug Target, 2008, 16:91-107.
24. Geramizadeh B, Janfeshan K, Saberfiroozi M. Serum hyaluronic acid as a noninvasive marker of hepatic fibrosis in chronic hepatitis B. Saudi J Gastroenterol. 2008, 14:174-177.

Kwas hialuronowy – charakterystyka, otrzymywanie i zastosowanie 69
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
25. Ukarapol N, Wongsawasdi L, Ong-Chai S, Riddhiputra P, Kongtawelert P. Hyaluronic acid: additional biochemical marker in the diagnosis of biliary atresia. Pediatr Int 2007, 49:608-611.
26. Kishida T, Yabushita H, Wakatsuki A, Zhuo L, Kimata K. Hyaluronan (HA) and serum-derived hyaluronan-associated protein (SHAP)-HA complex as predictive markers of cervical ripening in premature labor. Connect Tissue Res 2008, 49:105-108.
27. Kang DY, Kim WS, Heo IS, Park YH, Lee S. Extraction of hyaluronic acid (HA) from rooster comb and characterization using flow field-flow fractionation (FlFFF) coupled with multiangle light scattering (MALS). J Sep Sci 2010, 33:3530-3536.
28. Amagai I, Tashiro Y, Ogawa H. Improvement of the extraction procedure for hyaluronan from fish eyeball and the molecular characterization. Fisheries Sci 2009, 75:805-810.
29. Lago G, Oruna L, Cremata JA, Perez C, Coto G, Lauzan E, Kennedy JF. Isolation, purification and characterization of hyaluronan from human umbilical cord residues. Carbohyd Polym 2005, 62:321-326.
30. Andre P, Lowe NJ, Parc A, Clerici TH, Zimmermann U. Adverse reactions to dermal fillers: a review of European experiences. J Cosmet Laser Ther 2005, 7:171-176.
31. Chong BF, Blank LM, McLaughlin R, Nielsen LK. Microbial hyaluronic acid production. Appl Microbiol Biotechnol 2005, 66:341-351.
32. Izawa N, Hanamizu T, Iizuka R, Sone T, Mizukoshi H, Kimura K, Chiba K. Streptococcus thermophilus produces exopolysaccharides including hyaluronic acid. J Biosci Bioeng 2009, 107:119-123.
33. Chien LJ, Lee CK. Hyaluronic acid production by recombinant Lactococcus lactis. Appl Microbiol Biotechnol 2007, 77:339-346.
34. Prasad SB, Jayaraman G, Ramachandran KB. Hyaluronic acid production is enhanced by the additional co-expression of UDP-glucose pyrophosphorylase in Lactococcus lactis. Appl Microbiol Biotechnol 2010, 86:273-283.
35. Liu L, Yang H, Zhang D, Du G, Chen J, Wang M, Sun J. Enhancement of hyaluronic acid production by batch culture of Streptococcus zooepidemicus with n-dodecane as an oxygen vector. J Microbiol Biotechnol 2009, 19:596-603.
36. Liu L, Wang M, Du G, Chen J. Enhanced hyaluronic acid production of Streptococcus zooepidemicus by an intermittent alkaline-stress strategy. Lett Appl Microbiol 2008, 46:383-388.
37. Widner B, Behr R, Von Dollen S, Tang M, Heu T, Sloma A, Sternberg D, Deangelis PL, Weigel PH, Brown S. Hyaluronic acid production in Bacillus subtilis. Appl Environ Microbiol 2005, 71:3747-3752.
38. Chien LJ, Lee CK. Enhanced hyaluronic acid production in Bacillus subtilis by coexpressing bacterial hemoglobin. Biotechnol Prog 2007, 23:1017-1022.
39. Yu H, Stephanopoulos G. Metabolic engineering of Escherichia coli for biosynthesis of hyaluronic acid. Metab Eng 2008, 10:24-32.
40. Mao Z, Shin HD, Chen R. A recombinant E. coli bioprocess for hyaluronan synthesis. Appl Microbiol Biotechnol 2009, 84:63-69.
41. Rangaswamy V, Jain D. An efficient process for production and purification of hyaluronic acid from Streptococcus equi subsp. zooepidemicus. Biotechnol Lett 2008, 30:493-496.

Czajkowska i in.
Biotechnol Food Sci 2011, 75(2), 55-70 http://www.bfs.p.lodz.pl
70
Hyaluronic acid – characterization, production and application
Abstract: In this review, bibliography concerning the structure and the properties of hyaluronic acid (HA) as well as the localization and the functions of this compound in human organism were presented. The methods of HA preparations obtainment including the extraction of animal tissues and production in biotechnological processes were described. The advantages and disadvantages of above mentioned methods were pointed. In the section of this review dealt with HA microbiological production, modifications of the genomes of lactic acid bacteria as well as Escherichia coli and Bacillus subtilis strains to improve the HA productivity were shown. Consideration was taken to introduce hyaluronic synthases, UDP – glucose dehydrogenase and UDP-glucose pyrophosphorylase genes. Methods and culture conditions of bacteria as well as HA yield were presented. Applications of hyaluronic acid and its derivatives in therapy, diagnostics, aesthetic medicine as well as in cosmetology were presented. An insight on HA utilization in drug targeting and drug delivery was made.

Biotechnology and Food Science Review article
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
Ochrona bioróżnorodności roślin województwa łódzkiego
Elżbieta Sobiecka,1* Karolina Piątkowska,1 Danuta Posiła2
1 Institute of General Food Chemistry, Technical University of Lodz, 90-943, Lodz
2 XXXI High School of Ludwik Zamenhof, 93-236, Lodz *[email protected]
Streszczenie: W pracy zostały zebrane i uporządkowane informacje dotyczące biologicznej różnorodności świata roślinnego na terenie województwa łódzkiego. Część pracy poświęcono na omówienie zagrożeń równowagi ekosystemów występujących na terenie regionu. Szczegółowo przedstawione zostały formy ochrony bioróżnorodności roślin województwa jak również korzyści płynące z utrzymania równowagi biologicznej biocenoz.
Słowa kluczowe: bioróżnorodność, ekosystem, siedlisko
Wstęp Bioróżnorodność ma ogromne znaczenie dla ewolucji oraz trwałości układów
podtrzymujących życie w biosferze. Różnorodność biologiczną należy chronić, by móc z niej korzystać teraz i w przyszłości. Dzięki temu, zostaje utrzymana równowaga w przyrodzie. Im bardziej różnorodny jest układ przyrodniczy tym trudniej go zachwiać.
Cele przyświecające ochronie bioróżnorodności mają wymiar na wielu płasz-czyznach. Są one rozpatrywane i badane nie tylko w aspekcie przyrodniczym, ale również kulturowym, społecznym i ekonomicznym [1].
Ochrona różnorodności biologicznej to zachowanie genetycznej zmienności, zróżnicowania gatunków i populacji, a także zachowanie istotnych dla życia pro-cesów zachodzących w obrębie ekosystemów, włączając w to również wpływ klimatu [2].
Bioróżnorodność w Polsce Natura w Polsce różnorodnie reprezentuje świat istot żywych kontynentu
i dlatego ma wyjątkową wartość dla Europy [3]. Polska należy do krajów o dużym zróżnicowaniu przyrodniczym. Różnorodność gatunkowa roślin i zwierząt w tej strefie klimatycznej należy do najbogatszych w Europie.
Polska leży w strefie lasów mieszanych środkowoeuropejskich. Część północno-wschodnia znajduje się w strefie lasów mieszanych wschodnioeuropejskich, zaś część południowo-wschodnia należy do strefy stepów lesistych i lasów liściastych wschodnioeuropejskich. Roślinność w środkowej Polsce składa się z zespołów

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
72
naturalnych: leśnych, wodnych, szuwarowych, torfowiskowych oraz nieleśnych – półnaturalnych łąk oraz pastwisk. Razem w Polsce centralnej występuje ponad 150 typów fitocenoz.
Aktualnie w Polsce centralnej na terenach leśnych występuje około 1100 gatunków flory naczyniowej, 166 gatunków porostów, 134 gatunki mszaków i około 1000 gatunków grzybów. W większości są to rośliny pospolite, występu-jące na terenie całej Polski.
Bioróżnorodność województwa łódzkiego Środowisko przyrodnicze w województwie łódzkim charakteryzuje się dużym
zróżnicowaniem siedlisk. Sprzyja to bogactwu gatunkowemu flory i fauny, dla-tego też występuje na tym terenie wiele gatunków rzadkich, które są objęte ochroną, w tym zagrożone wyginięciem według Polskiej Czerwonej Księgi Zwierząt i Polskiej Czerwonej Księgi Roślin.
We florze województwa dominują gatunki roślin, które występują na obsza-rach strefy umiarkowanej półkuli północnej. Obecne są również gatunki, które pochodzą z odległych obszarów geograficznych np. gatunki wschodnio-europejsko-syberyjskie i pontyjskie, np. sasanka otwarta, szczodrzeniec ruski, dzwonecznik wonny oraz pluskwica europejska [4, 5].
Region województwa łódzkiego leży na pograniczu nizin i wyżyn co sprawia, że poza licznymi gatunkami niżowymi występują także gatunki górskie, m.in.: jodła pospolita, liczydło górskie, wroniec widlasty, kokoryczka okółkowa i wiele innych [6].
Przez teren województwa łódzkiego przebiega północna granica z trzema głównymi gatunkami drzew lasotwórczych: jodły, świerka oraz buka [7]. Obecnie w województwie łódzkim lasy dębowo-bukowe zajmują łącznie 5% powierzchni leśnej, dominują zaś siedliska grądów, grądy zdegenerowane sosną oraz monokultury sosnowe, świerkowe lub grabowe. Ponad 80% przestrzeni leśnej województwa łódzkiego zdominowane jest przez drzewostany sosnowe. W większości siedliska grądowe zajmują użytki rolne: uprawy pszenicy, żyta, łąki i pastwiska.
Zagrożenia dla różnorodności biologicznej województwa łódzkiego i formy jej ochrony
Powszechnym zagrożeniem naturalnych ekosystemów w Polsce są gatunki obce, które pojawiły się na obszarze naszego kraju poprzez migrację. Spośród tych gatunków do tej pory dla polskiej biocenozy zidentyfikowano najwięcej gatunków roślin (466) i bezkręgowców (348), wśród których najliczniejszą grupę stanowią stawonogi.
Chcąc przystąpić do ochrony bioróżnorodności na danym obszarze należy zacząć od wyznaczenia terenów chronionych, reintrodukować wiele gatunków, odtworzyć siedliska oraz objąć kontrolą lub wyeliminować uprzednio introdukowane gatunki. Efektywne strategie ochrony bioróżnorodności muszą być sprecyzowane

Ochrona bioróżnorodności roślin województwa łódzkiego
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
73
poprzez wyznaczenie jasnych celów. Strategie narodowe i globalne, muszą być odpowiednie dla potrzeb rządów, zaś strategie lokalne – dla władz lokalnych [2].
Poziom zagrożenia różnorodności flory województwa łódzkiego jest oceniany na 1/3 ogólnej liczby gatunków. Według wielu zestawień 405 gatunków roślin naczyniowych, zbiorowisk naturalnych i półnaturalnych jest w różnym stopniu zagrożonych. Z tej grupy 31 gatunków występuje w aktualnej wersji Polskiej Czerwonej Księgi Roślin [8]. Na terenie województwa łódzkiego występuje 5 gatunków roślin zaliczanych do grupy ważnych dla Wspólnoty Europejskiej wymienianych w Dyrektywie Siedliskowej. Są to: dzwonecznik wonny, starodub łąkowy, skalnica torfowa, lipiennik Loesela oraz sasanka otwarta. W Polsce występują także specjalne obszary ochrony, które wchodzą w skład europejskiej sieci ekologicznej Natura 2000.
Jednym z wyznaczników wartości roślin lokalnych jest obecność gatunków, które są objęte formalną ochroną prawną. W tej chwili obowiązuje rozporządzenie Ministra Środowiska w sprawie gatunków dziko występujących roślin objętych ochroną z dnia 9 lipca 2004 roku (Dz.U. z 2004, Nr 168, poz.1764). Z 500 gatunków objętych ochroną prawną, na terenie województwa występuje około 120 gatunków roślin naczyniowych, które są zaliczone do gatunków ściśle chronionych oraz 20 gatunków częściowo chronionych. W odniesieniu do kilku gatunków nie ma danych dotyczących miejsca ich występowania, dlatego trzeba przyjąć, że niektóre już całkowicie wymarły, np. skalnica torfowiskowa.
Na ratowanie chronionych i rzadkich gatunków oraz siedlisk przyrodniczych, duży wpływ ma również świadome planowanie zabiegów hodowlanych i gospo-darczych przez pracowników placówek naukowych podległych Ministerstwu Środowiska. Niezbędnym jest aby dysponowali oni pełnymi danymi, które dotyczą obszarów o unikatowych walorach przyrodniczych, które znajdują się na ich obszarach [4, 9, 10].
Ogromny wkład w odbudowę różnorodności biologicznej mają korytarze ekologiczne szczególnie dla zwierząt, które zamieszkują tereny zalesione, unikając otwartych przestrzeni. Mogą one migrować jedynie wzdłuż prawidłowo zalesio-nych obszarów o zwartej strukturze.
Ważnym elementem ochrony są działania, które zapobiegają postępującej synantropizacji fauny i flory oraz zjawisku neofityzmu (przenikania gatunków obcych). Przebieg niekontrolowanej inwazji i zadomawiania się obcych gatun-ków flory i fauny jest najważniejszą przyczyną zagrożenia naturalnej różno-rodności biologicznej [2].
W 2008 i 2009 roku, na terenie województwa łódzkiego był prowadzony monitoring określonych wskaźników. Akcja skupiła się na: określeniu liczby chronionych i zagrożonych gatunków fauny i flory, opracowaniu i wdrożeniu projektów ochrony wybranych gatunków, określeniu liczby utworzonych stref ochrony gatunków zagrożonych,

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
74
oszacowaniu powierzchni nowo utworzonych rezerwatów przyrody, zespołów przyrodniczo-krajobrazowych, obszarów chronionego krajobrazu oraz obszarów Natura 2000,
określeniu ilości pomników przyrody.
Rezerwaty przyrody Najwyższą formą ochrony w centralnej części Polski są rezerwaty. Według
danych z końca 2009 roku, na terenie województwa łódzkiego znajduje się aktualnie 89 rezerwatów przyrody o łącznej powierzchni 7427,10 ha. Obejmują one obszary w stanie naturalnym lub półnaturalnym, siedliska i ostoje przyrodnicze, a także siedliska zwierząt, roślin i grzybów oraz składniki i twory przyrody nieożywionej wyróżniające się szczególnymi wartościami naukowymi, przyrodniczymi, kulturowymi lub też walorami krajobrazowymi [7].
Ochrona rezerwatów jest zwykle podstawowym warunkiem zachowania rzadkich gatunków lub siedlisk zwłaszcza w czasach intensywnej urbanizacji. Są one potrzebne także dla zachowania potrzeb rekreacji jak również idealnie służą zachowaniu kulturowej wiedzy dotyczącej gatunków. Strefy te są stałym źródłem imigrantów i chronią rezerwat przed efektami brzeżnymi, maksymalizując ochronę gatunków przystosowanych do życia w centrum siedliska [9].
Najmłodszym rezerwatem na terenie województwa łódzkiego jest Gać Spalska o powierzchni 81,65 ha, utworzona w 2006 roku. Obejmuje część doliny rzeki Gać z licznymi źródłami i czterema zbiornikami [11, 12].
Parki krajobrazowe jako forma ochrony gatunkowej Ochrona gatunkowa zapewnienia przetrwanie i właściwy stan przyrody dziko
występującej, endemicznych gatunków roślin, grzybów i zwierząt oraz ich siedlisk, a także zachowanie różnorodności genetycznej i gatunkowej.
Kwestie ochrony gatunkowej regulują rozporządzenia Ministra Środowiska. Określają one gatunki roślin, grzybów i zwierząt, które podlegają ochronie ścisłej oraz częściowej, sposoby ich ochrony, w tym listę zakazów właściwych dla poszczególnych gatunków roślin i odstępstwa od tych zakazów. Wobec dziko występujących roślin chronionych obowiązuje wiele zakazów między innymi: zakaz zrywania, uszkadzania, niszczenia roślin oraz ich siedlisk i ostoi, stosowania środków chemicznych, dokonywania zmian stosunków wodnych, zdobywania i oferowania roślin do sprzedaży, niszczenie ściółki leśnej i wiele innych [4, 9]. Spełnienie powyższych wymagań zapewniają parki krajobrazowe.
Na obszarze województwa łódzkiego występuje 7 parków krajobrazowych. Łączna powierzchnia tych parków zajmuje około 178000 ha i obejmuje 9,8% województwa [7, 9].
Do głównych celów ochrony flory w Parku należy zagwarantowanie trwałości lokalnych populacji gatunków chronionych, regionalnie zagrożonych i rzadkich jak również zachowanie pełnej bioróżnorodności w kierunku wszystkich grup systematycznych, ochrona pul genowych tradycyjnych odmian roślin uprawnych, zmniejszenie procesu neofityzacji flory, zachowanie pełnego inwentarza zbiorowisk

Ochrona bioróżnorodności roślin województwa łódzkiego
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
75
roślinnych, naturalnych oraz antropogenicznych i zachowanie wszystkich ważnych dla środowiska przyrodniczego typów ekosystemów. Bolimowski Park Krajobrazowy
Park położony jest na pograniczu województw mazowieckiego i łódzkiego, utworzony został w 1986 roku. Zajmuje obszar o powierzchni 20512,32 ha, a otulina 3102,43 ha. W skład Bolimowskiego Parku Krajobrazowego wchodzą lasy Puszczy Bolimowskiej rozciągającej się pomiędzy Łowiczem, Skierniewicami, Bolimowem i Żyrardowem.
Świat roślinny Parku jest niezwykle bogaty. Spośród 2200 gatunków roślin naczyniowych Polski w Parku tym występuje niemal połowa. Są to gatunki charakterystyczne dla pogórza czy okolic nadmorskich, ale także dla Polski centralnej. W Parku Bolimowskim dominują lasy zajmujące około 70% powierzchni. Tworzą one kompleks różnorodnych drzewostanów sosnowych z domieszkami brzozy, osiki. Pojedynczo występują tu: dąb, lipa, grab, klon i jesion zaś obszary żyźniejsze porastają grądy. W podmokłych zagłębieniach wzdłuż cieków wodnych wykształciły się olsy.
Wśród różnorodnej flory ponad 40 gatunków jest objętych ochroną i ponad 100 uznanych za zagrożone i ginące. Należą do nich widłaki lub kolorowe storczyki. Wśród gatunków rzadkich i chronionych występują: zimoziół północny, wolffia bezkorzeniowa, którą znaleźć można w jednym ze starorzeczy Rawki, rosiczka okrągłolistna, konwalia majowa czy też trujący wawrzynek wilczełyko. Wśród bardzo rzadkich roślin można wymienić turówkę wonną za to dość licznie występuje bluszcz pospolity [13, 14]. Park Krajobrazowy Międzyrzecza Warty i Widawki
Park Krajobrazowy Międzyrzecza Warty i Widawki powstał w 1989 roku. Park ten znajduje się na terenie 9 gmin: Widawy, Burzenina, Konopnicy, Zapolic, Sędziejowic, Sieradza, Ostrówka, Zduńskiej Woli i Ruśca. Powierzchnia parku zajmuje 25330 ha. Pod ochroną znajdują się również ekosystemy należące do doliny rzek Warty, Grabi, Oleśnicy, Wierznicy, Widawki i Niecieczy.
Lasy w Parku stanowią tylko około 25% ogólnej powierzchni. Dominują sztucznie wprowadzone zbiorowiska borowe, a naturalne zespoły borów bagiennych, grądów i łęgów zachowały się w niedużych fragmentach. Małe zmiany w środowisku przyrodniczym, które wiążą się z działalnością człowieka przyczyniły się do wyjątkowej atrakcyjności tego terenu. Na terenie Parku stwierdzono występowanie około 600 gatunków roślin naczyniowych. Obszary są porośnięte licznym gatunkami lasów liściastych, borowych i mieszanych, występują tu rośliny związane z wilgotnymi łąkami, torfowiskami, szuwarami, wodami oraz innymi podtopionymi siedliskami, np.: rzęsa garbata, wolffia bezkorzeniowa, czermień błotna, zachylnik błotny, jeżogłówka najmniejsza, jaskier wielki czy strzałka wodna. Można też spotkać: rosiczkę okrągłolistną, borówkę bagienną, żurawinę błotną, wełnianki, bagno zwyczajne oraz liczne gatunki mchów. W niektórych punktach Parku istotnym elementem jego flory są także gatunki kserotermiczne.

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
76
Na obszarze Parku występuje około 60 gatunków roślin chronionych, w tym 30 gatunków jest pod ochroną ścisłą. Przykładami są: orlik pospolity, goździk pyszny, dziewięćsił bezłodygowy, aster gawędka, dzwonek syberyjski i boloński, rosiczka długolistna i okrągłolistna, kruszczyk błotny i szerokolistny, lilia złotogłów, kosaciec syberyjski, widłak jałowcowaty i widłak torfowy, paprotka zwyczajna, bagno zwyczajne, grzybienie północne, storczyk krwisty, męski i szerokolistny, sasanka łąkowa, pływacz drobny i średni.
Rośliny naczyniowe, które podlegają ochronie częściowej to: konwalia majowa, kopytnik pospolity, kocanki piaskowe, bobrek trójlistkowy, bluszcz pospolity, pierwiosnka wyniosła i lekarska, kalina koralowa oraz grążel żółty. Rośliny rzadkie stanowią 20% całej flory parku. Najliczniejszymi wśród nich są: ostrożeń krótkołodygowy, czyściec kosmaty, koniczyna dwukłosowa oraz pajęcznica gałęzista.
Obszar Parku wyróżnia się wyjątkowym otoczeniem, urozmaiconą rzeźbą terenu oraz licznymi obiektami, które są objęte ochroną pomnikową [15]. Park Krajobrazowy Wzniesień Łódzkich
Park Krajobrazowy Wzniesień Łódzkich został utworzony w 1996 roku. Obejmuje regiony Łodzi, Brzezin i Strykowa na obszarze 13767 ha.
Większa część terytorium okryta jest użytkami rolnymi (65%), lasy zaś zajmują 28% jego powierzchni. Na terenie Parku występują 3 rezerwaty: Rezerwat Las Łagiewnicki, Rezerwat Struga Dobieszkowska i Rezerwat Parowy Janinowskie oraz zespół przyrodniczo-krajobrazowy Górna Mrożyca. W Niesułkowie Kolonii znajduje się stanowisko dokumentacyjne oraz 52 pojedyncze drzewa będące pomnikami przyrody. Do najciekawszych drzew należą: lipa drobnolistna, dąb szypułkowy, wiąz szypułkowy oraz czereśnia.
Flora Parku jest ogromnie zróżnicowana. Stwierdzono występowanie 735 gatunków roślin naczyniowych. Występuje tu 71 gatunków, które są zaliczone do listy zagrożonych w skali regionu oraz gatunki znajdujące się w Polskiej Czerwonej Księdze Roślin, takie jak storczyk – żłobik koralowy. Na terenie Parku występuje 39 gatunków roślin prawnie chronionych w tym 24 podlegające ścisłej ochronie oraz 15 chronionych częściowo. Niektóre, bardzo rzadkie gatunki posiadają tylko klika stanowisk na terenie Parku, np. wroniec widlasty – gatunek reglowy, który towarzyszy jodle i bukowi.
Z wielu gatunków roślin nasiennych i paprotników, które są objęte ścisłą ochroną gatunkową należy zwrócić uwagę na: kosaciec syberyjski, kruszczyk błotny, kruszczyk szerokolistny, listera jajowata, lilia złotogłów, pełnik europejski, rosiczka okrągłolistna, storczyk szerokolistny oraz storczyk krwisty [11, 16]. Załęczański Park Krajobrazowy
Załęczański Park Krajobrazowy powstał w 1978 roku jako jeden z pierwszych parków w Polsce, w celu ochrony unikatowego krajobrazu jurajskich wapiennych ostańców będących środowiskiem wapieniolubnej fauny i flory, a także pięknego odcinka rzeki Warty, będącego najwartościowszym przyrodniczo w stosunku do

Ochrona bioróżnorodności roślin województwa łódzkiego
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
77
jej całego biegu. Obecnie powierzchnia Parku wynosi 14750 ha, a strefy ochronnej 12 010 ha.
Dużą część powierzchni Parku (około 49%) zajmują lasy. Dominują siedliska borowe. Spośród 21 jednostek roślinności leśnej oraz zaroślowej występują takie, które posiadają nadzwyczajną wartość naukową i krajobrazową.
Flora tego Parku liczy około 1200 gatunków: prawie 770 taksonów roślin naczyniowych, około 100 gatunków mszaków oraz około 80 gatunków porostów. Gatunki te należą do wielu grup siedliskowych, jednak najliczniej reprezentowane są rośliny, które są związane z siedliskami zmienionymi na skutek pracy człowieka. Większość Parku zajmują gatunki, które charakteryzują lasy liściaste i rośliny siedlisk łąkowych oraz mokradeł związanych z doliną Warty.
Obecnie na obszarze Załęczańskiego Parku Krajobrazowego, występuje 39 gatunków roślin naczyniowych, które podlegają ochronie w tym 25 jest objętych ochroną ścisłą. Na obszarze Parku zidentyfikowano około 30 gatunków roślin rzadkich w skali regionu lub kraju, gatunków górskich i występujących na granicy zasięgów geograficznych.
Dla zachowania cennych walorów przyrodniczych Parku utworzono na jego terenie 3 rezerwaty przyrody: rezerwat leśny „Dąbrowa” w Niżankowicach, rezerwat geologiczny „Węże” oraz rezerwat leśny „Bukowa Góra”. W granicach otuliny występują 2 rezerwaty przyrody: Rezerwat leśny „Stawiska” oraz rezerwat geologiczny „Szachownica”. W Parku ochroną pomnikową jest objętych 8 pomników przyrody z ciekawą geologią [17, 18]. Sulejowski Park Krajobrazowy
Sulejowski Park Krajobrazowy został utworzony w 1994 roku. Obejmuje dolinę rzeki Pilicy wraz z terenami przyległymi. Powierzchnia parku wynosi 17444 ha, otuliny zaś 38927 ha. Połowę obszaru tego Parku zajmują lasy, natomiast wody (wraz ze Zbiornikiem Sulejowskim) około 5%.
Na terenie Parku występuje 17 zespołów leśnych oraz zaroślowych i szereg zbiorowisk zastępczych. Charakterystycznymi dla tych obszarów są lasy higrofilne przede wszystkim łęgi jesionowo-olszowe i wierzbowe, olsy oraz wikliny nadrzeczne. Lasy mezofilne mieszane to przede wszystkim grądy rosnące na terenie Puszczy Pilickiej. Do lasów mieszanych należy także dąbrowa świetlista. Często spotykanym zespołem jest bór mieszany sosnowo-dębowy rzadziej wyżynny bór mieszany jodłowy. Bory sosnowe reprezentują zespoły: suboceaniczny bór świeży, bór chrobotkowy, bór wilgotny oraz bór bagienny. Obszary bardziej przekształcone, charakteryzują się rozwojem roślinności synantropijnej.
Na terenie Sulejowskiego Parku Krajobrazowego stwierdzono około 1000 gatunków roślin naczyniowych takich jak: widłaki (4 gatunki), storczyki (12 gatunków), długosz królewski, koniczyna łubinowata, zimoziół północny i kokoryczka okółkowa. Chronione są jodły pospolite, buki, świerki, klony i jawory.

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
78
Na terenie Parku istnieje 11 rezerwatów zajmujących 624 ha. Planuje się także utworzenie 5 rezerwatów o łącznej powierzchni 140 ha [19]. Spalski Park Krajobrazowy
Park ten został utworzony w 1995 roku. Jego powierzchnia wynosi 13110 ha, zaś obszar otuliny 24134 ha. Spalski Park Krajobrazowy leży w środkowej części dorzecza Pilicy na granicy Niżu Środkowoeuropejskiego i Wyżyn Polskich.
Przeważają tu tereny leśne, ale swój udział mają także użytki rolne. Rozległe lasy stanowią Puszczę Pilicką, w której dominują: sosny, dęby i świerki, spotyka się także lasy mieszane i liściaste. Duże zróżnicowanie Parku umożliwia bytowanie różnorodnej flory. W Parku tym występuje ponad 800 gatunków roślin naczyniowych, do których należą paprotniki i rośliny nasienne w tym 19 gatunków roślin, które podlegają ochronie całkowitej i 11 gatunków objętych ochroną częściową np. widłaki, pluskwica europejska, bluszcz pospolity, wawrzynek wilczełyko, czy rosiczka okrągłolistna oraz 50 gatunków rzadkich lub zagrożonych regionalnie. Uwagę należy zwrócić na gatunki górskie: wroniec widlasty, żywiec dziewięciolistny, trzcinnik owłosiony oraz jodła pospolita.
Na obszarze Parku i jego otuliny istnieje 5 rezerwatów o łącznej powierzchni 497 ha: Żądłowice, Konewka, Spała, Sługocice i Jeleń. Szereg obiektów jest objętych indywidualną formą ochrony. Występują tu 23 użytki ekologiczne oraz 134 pomniki przyrody. W najbliższym czasie planowane jest utworzenie nowych rezerwatów: Gaci, Studzianki i Ceteńki.
Na terenie Parku są wyznaczone obszary NATURA 2000, do których należą obszar specjalnej ochrony ptaków – Dolina Pilicy i obszary ochrony siedlisk, zajmujące ten sam fragment Doliny Pilicy i Lasów Spalskich [20]. Przedborski Park Krajobrazowy
Przedborski Park Krajobrazowy został powołany w 1988 roku. Zajmuje on 16640 ha, a sama otulina 14490 ha. Park obejmuje fragment Pasma Przedborsko-Małogoskiego w Górach Świętokrzyskich.
Park ten charakteryzuje się występowaniem zróżnicowanych biotopów mokradłowych i kserotermicznych z odpowiednimi gatunkami roślin. Flora naczyniowa liczy około 960 gatunków. W Przedborskim Parku Krajobrazowym znajduje się 70 gatunków chronionych i zagrożonych, wśród których znajdują się: salwinia pływająca, cis pospolity, pełnik europejski, rosiczka długolistna, parzydło leśne i liczydło górskie. W parku znajduje się 30 gatunków roślin naczyniowych, które są zaliczane do gatunków zagrożonych w Polsce, a 5 z nich jest umieszczonych w Polskiej Czerwonej Księdze Roślin (wisienka stepowa, sasanka wiosenna, wierzba borówkolistna, miłek szkarłatny oraz fiołek torfowy). Wśród lasów dominują lasy bukowe reprezentowane przez buczynę storczykową. Najlepiej zachowane buczyny, występują w rezerwacie Bukowa Góra. Zaś w rezerwacie Czarna Rózga występuje wyżynny bór mieszany jodłowy (jedlina świętokrzyska). Licznie występują gatunki ciepłolubne takie jak: aster gawędka, zawilec wielkokwiatowy oraz dziewięćsił bezłodygowy [21].

Ochrona bioróżnorodności roślin województwa łódzkiego
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
79
Obszary chronionego krajobrazu Obszary chronionego krajobrazu to tereny chronione ze względu na oryginalny
krajobraz o zróżnicowanych ekosystemach. Obszary te są wartościowe pod względem zaspokajania potrzeb związanych z turystyką oraz wypoczynkiem lub pełnioną funkcją korytarzy ekologicznych. Organem właściwym, który wyznacza likwidację bądź zmiany granic powierzchni chronionego krajobrazu jest sejmik województwa.
Powierzchnia obszarów chronionego krajobrazu w województwie łódzkim wynosi 246534,80 ha. W 2009 roku powstał nowy obszar – Piliczański Obszar Chronionego Krajobrazu. Został wyznaczony według rozporządzenia Nr 8/2009 Wojewody Łódzkiego 24 marca 2009 roku, a jego powierzchnia wynosi 46340,00 ha. Jest to region położony na terenie powiatów: piotrkowskiego, opoczyńskiego oraz radomszczańskiego. Ochronie podlega zróżnicowany, bogaty krajobraz, będący atrakcją turystyczną oraz pełniący funkcję korytarza eko-logicznego.
Obszary chronionego krajobrazu na terenie województwa łódzkiego prawnie usankcjonowane to obszary: Bolimowsko-Radziejowicki z doliną środkowej Rawki, Brąszewicki, Doliny Widawki, Doliny Przysowy, Doliny Prosny, Pradolina Warszawsko-Berlińska, Górnej Rawki, Mrogi i Mrożycy, obszar Nadwarciański, Puczniewski, Środkowej Grabi, Przedborski, Dolina Miazgi pod Andrespolem, Dolina Wolbórki, Dolina Chojnatki oraz obszar Piliczański i Dolina Bzury [7].
Indywidualne formy ochrony przyrody Do indywidualnych form ochrony przyrody zaliczane są: stanowiska doku-
mentacyjne, pomniki przyrody, użytki ekologiczne oraz zespoły przyrodniczo-krajobrazowe. Stanowiska dokumentacyjne
Na obszarze województwa łódzkiego znajdują się cztery stanowiska dokumentacyjne. Są to pojedyncze twory przyrody żywej oraz nieożywionej lub ich skupiska posiadające szczególną wartość przyrodniczą, naukową, kulturową, historyczną lub krajobrazową oraz odznaczające się indywidualnymi, charakterystycznymi cechami takimi, jak okazałe rozmiary drzew, wodospady, wywierzyska, jary czy jaskinie. W 2008 roku zostało ustanowione stanowisko dokumentacyjne „Groty Nagórzyckie”. Przedmiotem ochrony jest wzgórze piaskowcowe oraz pobliskie podziemne wyrobiska w postaci grot oraz sztolni.
W województwie łódzkim zostało utworzonych pięć zespołów przyrodniczo-krajobrazowych, których powierzchnia wynosi 3200,75 ha. W 2007 roku ustanowiono w Poddębicach „Poddębicki Zespół Przyrodniczo-Krajobrazowy” obejmujący bulwar nad Nerem, zabytkowy Park Miejski oraz obiekty sportowe w Poddębicach o łącznej powierzchni 5,77 ha.

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
80
Użytki ekologiczne W województwie łódzkim występuje około 800 użytków ekologicznych
o powierzchni ponad 1500 ha. Użytki stanowią pozostałości ekosystemów, które mają na celu zachowanie różnorodności biologicznej – naturalne zbiorniki śródpolne oraz śródleśne, zbiorniki wodne, oczka wodne, kępy krzewów i drzew, bagna, torfowiska, wydmy, starorzecza, kamieńce, siedliska przyrodnicze oraz obszary rzadkich lub chronionych gatunków fauny i flory, ich ostoje oraz miejsca rozmnażania lub przebywania. Pomniki przyrody
Na obszarze województwa znajdują się również pomniki przyrody będące unikatowymi, pojedynczymi tworami przyrody żywej oraz nieożywionej lub ich skupiskami posiadającymi szczególną wartość przyrodniczą, naukową, kulturową, historyczną lub krajobrazową. Na terenie województwa łódzkiego, jest ich obecnie około 4000 (na dzień 31 grudnia 2009 roku). Jest to o ponad 400 więcej niż w 2007 roku [7, 9, 11].
Ogród botaniczny z palmiarnią w Łodzi Ogrody botaniczne są to instytucje posiadające udokumentowane kolekcje
żywych roślin do celów badań naukowych, ochrony, ekspozycji i edukacji. Na świecie jest około 1850 ogrodów botanicznych, w tym 621 występuje w Europie. W Polsce w tej chwili istnieje 31 instytucji o charakterze ogrodów botanicznych, a 8 kolejnych jest w fazie planowania i organizowania.
Ogród botaniczny w Łodzi należy do największych w Polsce i zajmuje po-wierzchnię 67 ha. W ogrodzie tym, rośnie około 3400 gatunków, odmian i taksonów roślin gruntowych oraz około 2000 taksonów roślin szklarniowych. Rośliny gruntowe zostały posadzone w dziewięciu działach ekspozycyjnych zajmujących powierzchnię około 64 ha.
Ogród japoński zajmuje powierzchnię w przybliżeniu 2 ha. Przeznaczony jest do prezentacji roślin pochodzących z Japonii, Chin i innych krajów Dalekiego Wschodu. Wśród różnorodnej flory można wyróżnić: metasekwoję chińską (Metasequoia glyptostroboides), magnolie japońskie: magnolię gwieździstą (Magnolia stellata), magnolię purpurową (Magnolia liliiflora), pigwowiec japoński (Chaenomeles japonica), kalinę japońską (Viburnum plicatum), żywotnikowiec japoński (Thujopsis dolabrata) oraz wiśnię piłkowaną (Prunus serrulata) w wielu odmianach.
Dział systematyki roślin zielnych zajmuje obszar o powierzchni 1,5 ha. Uprawiane na terenie tego działu rośliny posadzono na rabatach porządkując je w rodziny botaniczne zgodnie z systemem Englera. Zgromadzona kolekcja liczy 400 taksonów, która należy do ponad 50 rodzin. Występują zarówno gatunki rodzime np. miłek wiosenny Adonis vernalis, jak również obcego pochodzenia tytoń długokwiatowy (Nicotiana longiflora) czy tojeść amerykańska (Asclepias Syriach).
Kolejnym działem jest Alpinarium, które zajmuje powierzchnię 4,5 ha. U podnóży różnorodnych formacji skalnych rosną drzewa i krzewy. Na uwagę zasługują krzewy iglaste – głównie sosny (Pinus sp.), świerki (Picea sp.), cisy

Ochrona bioróżnorodności roślin województwa łódzkiego
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
81
(Taxus sp.), modrzewie (Larix sp.) i jałowce (Juniperus sp.). Występuje tu także świerk serbski (Picea omorica) oraz świerk kaukaski (Picea orientalis). Poza drzewami i krzewami rosną byliny, głównie formy niskie i poduszkowate.
Dział biologii i morfologii roślin zajmuje powierzchnię około 7 ha. Należące tu kolekcje umożliwiają dogłębne poznanie świata roślin np. morfologię pędów, liści, różnice w pokrojach drzew i krzewów, sposoby zapylania kwiatów. Rośnie tam 280 gatunków i odmian drzew i krzewów oraz ponad 440 taksonów roślin zielnych. Kolekcja ta posiada wiele roślin cebulowych, jak: cebulice (Scilla sp.), czosnek (Allium sp.), hiacynty (Hyacinthus sp.), ranniki (Eranthis sp.).
Dział kolekcji roślin ozdobnych obejmuje obszar 2,1 ha. Gromadzi on kolekcję róż wielokwiatowych, roślin iglastych i wrzosowatych. Kolekcja róż obejmuje nie tylko odmiany hodowlane, zachwycające barwą, zapachem oraz kształtem kwiatów, ale także róże gatunkowe, rosnące na naturalnych stanowiskach w Europie. Występują także: różanecznik katawbijski (Rhododendron catawbiense), akebia pięciolistkowa (Akebia quinata), glicynia kwiecista (Wisteria floribunda).
Dział zieleni parkowej zajmuje powierzchnię około 9 ha. Charakteryzuje się dużą różnorodnością lip, z których można wyróżnić: lipę drobnolistną (Tilia mordata), lipę szerokolistną (Tilia platyphyllos) i lipę srebrzystą (Tilia tomentosa). Dział wypoczynkowy dekorują ponadto: kwitnące gęsiówki (Arabis sp.), żółte smagliczki (Alyssum sp.), żagwiny (Aubrieta sp.), floksy (Phlox sp.) i goździki (Dianthus sp.).
Dział roślin leczniczych i przemysłowych zajmuje obszar 5,7 ha. Zgroma-dzono tu kolekcje około 230 gatunków roślin szeroko stosowanych w zioło-lecznictwie oraz stanowiących surowiec dla przemysłu farmaceutycznego, kosmetycznego i spożywczego. Rośliny posadzono według oddziaływania farmakologicznego surowców zielarskich, np. rośliny moczopędne, żółciopędne, powlekające, regulujące przemianę materii, pobudzające trawienie. Wyróżnić można tu krzewy o właściwościach leczniczych, takie jak: dąb szypułkowy (Quercus robur), lipa szerokolistna (Tilia platyphyllos), berberys pospolity (Berberis vulgaris), aronia czarnoowocowa (Aronia melanocarpa), pigwowiec japoński (Chaenomeles japonica) czy sosna zwyczajna (Pinus sylvestris).
Dział flory polskiej (9,2 ha) jest urozmaiconym terenem, na którym prezentowane są gatunki flory rodzimej ze szczególnym uwzględnieniem roślin chronionych i zagrożonych wyginięciem. W zbiornikach wodnych występuje wiele gatunków roślin wodnych i szuwarowych, takich jak: włosienicznik wodny (Batrachium aquatile) czy grzybieńczyk wodny (Limnanthemum nymphoides).
Arboretum jest największym działem o powierzchni 18,7 ha. Jest to najmłodszy dział w Łódzkim Ogrodzie Botanicznym, w którym wprowadza się nowe nasadzenia. Z najciekawszych drzew, można wyróżnić: cypryśnik błotny (Taxodium distichum), iglicznia trójcierniowa (Gleditsia triacanthos), różne gatunki surmii (katalp) (Catalpa sp.) oraz wiązowiec południowy (Celtis australis) o złocistożółtych liściach jesienią [22].

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
82
Obok odmian roślin zgromadzonych w różnych działach ogrodu należy również pamiętać o kolekcji roślin szklarniowych liczącej około 2000 gatunków. Najliczniej reprezentowane są: storczykowate (Orchidaceae), kaktusowate (Cactaceae), obrazkowate (Araceae), begoniowate (Begoniaceae). Większość tych roślin znajduje się w Palmiarni, która składa się z trzech pawilonów przeznaczonych dla roślin o odmiennych wymaganiach klimatycznych. Pawilon pierwszy to roślinność twardolistna występująca głównie w krajach śródziemno-morskich, należy tu między innymi daktylowiec (Phoenix sp.), oleander (Oleander sp.), oliwka (Olea sp.), wawrzyn (Laurus sp.).
Pawilon drugi jest przeznaczony dla roślinności strefy równikowej i pod-równikowej, wśród której zgromadzono takie rośliny jak: storczyki z rodzajów falenopsis (Phalaenopsis sp.), ananasy (Ananas sp.), fikusy (Ficus sp.), różne gatunki palm oraz paproci drzewiastych.
Pawilon trzeci jest miejscem przeznaczonym dla roślinności pustyń i pół-pustyń obejmujących między innymi Saharę, część półwyspu Arabskiego oraz centralną część Australii. Tutaj eksponowana jest kolekcja kaktusów, między innymi z rodzajów (Cereus sp.), agaw (Agave sp.), aloesów (Aloe sp.) i gruboszy (Crassula sp.) [23].
W ostatnim stuleciu zaobserwowano masowe wymieranie dzikich roślin. Szacuje się, że na świecie zagrożonych jest już 20% gatunków roślin wyższych. Najbardziej niepokojąca jest sytuacja w obszarze strefy tropikalnej. Na ziemiach polskich w ciągu ostatnich 150 lat wyginęło około 40 paprotników i roślin kwiatowych [24]. Dlatego też w kolekcji roślin szklarniowych Ogrodu Botanicznego zgromadzono ponad 560 gatunków roślin objętych konwencją CITES. Najwięcej wśród nich jest kaktusów (około 240 gatunków) i storczyków (około 240 gatun-ków) [23].
Najważniejszymi zadaniami ogrodów botanicznych jest szerzenie wiedzy ogrodniczej i botanicznej, prowadzenie kolekcji roślinnych dla celów badawczych i dydaktycznych, pełnienie roli rezerwatów, w których mogą być zachowane gatunki rzadkie, ginące i chronione. Są to także atrakcyjne obiekty wypoczynkowo-rekreacyjne. Jednym z priorytetowych zadań ogrodów botanicznych stało się zakładanie banków genów oraz kolekcji klonów roślin w celu zachowania maksymalnego zróżnicowania gatunkowego i zabezpieczenia roślin, które są najbardziej zagrożone przed bezpowrotnym wyginięciem. W 2001 roku na terenie ogrodu zostało otwarte laboratorium, które namnaża materiał roślinny z gatunków chronionych lub zagrożonych wyginięciem. Innym zadaniem jest także rozmnażanie roślin rzadkich lub trudnych do mnożenia tradycyjnymi metodami ogrodniczymi, jak np. paproć drzewiasta (Cyathea cooperi) [22].
Arboretum Szkoły Głównej Gospodarstwa Wiejskiego w Rogowie Wśród wielu ogrodów botanicznych, najliczniejszą grupę stanowią arboreta,
zwane również ogrodami dendrologicznymi. Są to ogrody specjalistyczne, w których uprawiane są prawie wyłącznie krzewy i drzewa. Arboretum w Rogo-wie to jeden z największych tego typu ogrodów w Polsce mający charakter parku

Ochrona bioróżnorodności roślin województwa łódzkiego
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
83
leśnego. Na tym obszarze występuje jedna z najbogatszych kolekcji drzew i krze-wów Europy Środkowo-Wschodniej. W skład Arboretum wchodzą trzy działy: kolekcje dendrologiczne, leśne powierzchnie doświadczalne, alpinarium.
Kolekcje dendrologiczne zajmują obecnie powierzchnię 22 ha i są położone w centralnej części Arboretum. Obejmują około 2400 gatunków drzew i krzewów pochodzących prawie wyłącznie ze strefy klimatu umiarkowanego półkuli północnej (Europa, Azja, Ameryka Północna). Większość odmian ozdobnych, które stanowią jedną trzecią tej liczby występuje wśród roślin iglastych oraz w rodzinie wrzosowatych (między innymi azalie i różaneczniki). Gatunki iglaste są reprezentowane przez ponad 410 gatunków i odmian. Licznie występują gatunki sosny, świerka, jodły, modrzewiów, cyprysów, cisów, żywotników, rosną tu także rzadkie w Polsce modrzewniki (Pseudolarix), kuningamia (Cunninghamia), głowocisy (Cephalotaxus), cedrzyńce (Calocedrus) oraz żywotnikowce (Thujopsis). Rogowskie Arboretum posiada jedną z naj-bogatszych w Europie kolekcji klonów obejmującą 140 gatunków i odmian. Jest to „narodowa kolekcja” klonów w Polsce [25].
Leśne powierzchnie doświadczalne są to niewielkie powierzchniowo drzewostany złożone prawie wyłącznie z drzew obcego pochodzenia. Jest ich obecnie 150 z 75 gatunkami drzew zajmującymi łącznie 18 ha. Celem zakładania tych powierzchni, szczegółowo udokumentowanych, jest badanie dynamiki wzrostu, zdrowotności, zmienności i produkcyjności obcych gatunków drzew dla dokonania wstępnej oceny ich przydatności dla gospodarki leśnej. Przeważają tu drzewa północnoamerykańskie w tym liczne gatunki szybko rosnące, takie jak: daglezja zielona (Pseudotsuga menziesii), żywotnik olbrzymi (Thuja plicata), dąb czerwony (Quercus rubra) oraz jodła olbrzymia (Abies grandis). Uprawiane są także gatunki jodeł (Abies sp.), świerków (Picea sp.) i modrzewi (Larix sp.).
Alpinarium jest najmniejszym działem ogrodu o powierzchni 1,4 ha. Występuje tutaj 700 gatunków i odmian roślin pochodzących z gór całego świata. Rosną tu także krzewy i drzewa z terenów górzystych np. strzeliste świerki serbskie (Picea omorika), kształtne limby (Pinus cembra), srebrzyste jodły kali-fornijskie (Abies concolor), jak również wiele gatunków kwiatów charakte-rystycznych dla terenów górskich [25, 26].
Do najważniejszych zadań Arboretum należy prowadzenie działalności edukacyjnej. Dotyczy to nie tylko programów zajęć dydaktycznych, ale także edukacji i popularyzacji wiedzy botanicznej. Arboretum Rogowskie opiera się na bogatej dokumentacji kolekcji roślinnych, prowadzi działalność naukową. Prace te dotyczą ekologii roślin drzewiastych i dendrologii stosowanej. Arboretum dysponuje znaczą rezerwą terenu umożliwiającą stały rozwój kolekcji i zakładania leśnych powierzchni doświadczalnych. Miejsce to wydaje corocznie katalog nasion przeznaczonych na wymianę i utrzymuje kontakty z ponad 400

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
84
ogrodami botanicznymi w różnych częściach świata. Sprowadzone nasiona są podstawowym źródłem uzyskiwania nowych i rzadkich gatunków roślin [26].
Korzyści płynące z utrzymania bioróżnorodności Idea różnorodności biologicznej zakłada, że cenne jest całe bogactwo życia.
Oznacza to, że nie ma gatunków, które są zbędne i niepotrzebne. Każdy najmniejszy owad jest ogromnie ważnym elementem w wielkim łańcuchu życia.
Korzyści utrzymania bioróżnorodności roślin obejmują wiele kategorii, z których najpowszechniejszą stanowi dostarczanie przez ekosystemy bogactwa pożywienia między innymi: nasion, jadalnych bulw i korzeni czy owoców.
Wiele gatunków roślin stanowi naturalny materiał wykorzystywany w bu-downictwie (liście palmowe, trzcina, drewno), albo do wyrobu ubrań (włókna roślinne, liście). Kolejną ważną rolą jest dostarczanie substancji leczniczych, z których korzysta medycyna ludowa, a także firmy farmaceutyczne.
Rośliny biorą czynny udział w oczyszczaniu powietrza z pyłów, pochłanianiu dwutlenku węgla i regeneracji tlenu. Systemy korzeniowe roślin pobierają nadmiar substancji chemicznych zanieczyszczających glebę lub wodę. We wszystkich strefach klimatycznych obszary leśne na stokach górskich pełnią bardzo ważną rolę w zatrzymywaniu wody z silnych i gwałtownych opadów, chroniąc leżące niżej tereny przed powodziami oraz spływem gleby.
Rośliny biorą udział w krążeniu pierwiastków w przyrodzie oraz tworzeniu i utrzymywaniu jakości gleby. W tym przypadku ważną rolę stanowią organizmy zapylające, bez których byłoby niemożliwe uzyskiwanie plonów. W ostatnich latach w Polsce zmniejszyła się znacznie liczebność pszczół hodowlanych, co wpłynęło na obniżenie plonów sadowników.
Wielkie bogactwo płynące z funkcjonowania ekosystemów wiąże się z ludz-kimi potrzebami estetyki dotyczącej przyrody oraz czerpanej z niej inspiracji twórczej.
W roku 2000 Organizacja Narodów Zjednoczonych rozpoczęła inicjatywę – Milenijna Ocena Ekosystemów. Z ogłoszonego w 2005 raportu wynika, że 2/3 zagwarantowanych funkcji ekosystemów na Ziemi zanika lub też jest w stanie zagrożenia. W ramach ogólnoświatowej inicjatywy, która została podjęta w wyniku tych doniesień Unia Europejska wzięła odpowiedzialność za ocenę regionu europejskiego. Europejska Agencja Środowiska podjęła decyzję, że nowa ocena ma zostać przeprowadzona w 2015 roku. Wszystkie kroki mające na celu ochronę i utrzymanie różnorodności biologicznej w poszczególnych regionach państw członkowskich, również w województwie łódzkim, przyczynią się do wzrostu rozwoju ekosystemów i zmniejszenia zagrożenia ich zaniku.

Ochrona bioróżnorodności roślin województwa łódzkiego
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
85
Literatura 1. Kozłowski S. Ekorozwój. Wyzwanie XXI wieku. PWN, Warszawa, Polska 2000. 2. Mackenzie A, Ball AS, Virdee SR. Ekologia. PWN, Warszawa, Polska 2007, s. 356. 3. Czyżewska K, Hereźniak J. Biodiversity in relation to vegetation zones in Europe.
Polish Botanical Society, University of Lodz, Lodz-Warsaw 2005. 4. Wojewódzki Inspektorat Ochrony Środowiska w Łodzi oraz jego delegatury
w Piotrkowie Trybunalskim, Sieradzu i Skierniewicach. Raport o Stanie Środowiska w województwie łódzkim w 2004 roku. Łódź, Polska 2005.
5. Witosławski P. Atlas of distribiution of vascular plants in Łódź. Lodz University Press, Łódź 2006.
6. Jakubowska-Gabara J, Jost-Jakubowska B. Element górski we florze Polski Środkowej. Frgam. Flor. Geobot. Polonica 1978, 24, ss. 259-272.
7. Departament Rolnictwa i Ochrony Środowiska. Raport za lata 2008-2009 z wykonania „Programu ochrony środowiska województwa łódzkiego na lata 2008-2011”. Łódź, Polska 2010.
8. Kaźmierczakowa R, Zarzycki K. Polska Czerwona Księga Roślin. Paprotniki i rośliny kwiatowe. PAN, Kraków, Polska 2001.
9. Wojewódzki Inspektorat Ochrony Środowiska w Łodzi oraz jego delegatury w Piotrkowie Trybunalskim, Sieradzu i Skierniewicach. Raport o stanie środowiska w województwie łódzkim w 2007 roku. Łódź, Polska 2008.
10. Jakubowska-Gabara J, Kucharski L. Ginące i zagrożone gatunki flory naczyniowej zbiorowisk naturalnych i półnaturalnych Polski Środkowej. Frag. Flor. Geobot. Ser. Polonica 1999; 6, ss. 55-74.
11. Stankiewicz M, Zaborowska A, Andrzejewski H. Ochrona przyrody w woje-wództwie łódzkim. Dyrekcja Parku Krajobrazowego Wzniesień Łódzkich 2009, http://www.pkwl.pl
12. Rąkowski G. Rezerwaty przyrody w Polsce środkowej: monografia. Dział Wydawnictw Instytutu Ochrony Środowiska, Warszawa, Polska 2006.
13. Kiczyńska A. Aktualizacja planu ochrony Bolimowskiego Parku Krajobrazowego. Narodowa Fundacja Ochrony Środowiska – Operat Generalny 2006.
14. Jakubowska-Gabara J, Markowski J. Bolimowski Park krajobrazowy. RCEE, Łódź, Polska 2002.
15. Lisek S. 2012, Urząd Marszałkowski w Łodzi, Departament Rolnictwa i Ochrony Środowiska: http://www.przyroda.lodzkie.pl/przyroda/topMenu/ochrona_przyrody/parki_krajobrazowe/miedzyrzecza_warty_i_widawki/
16. Kurowski J. Park Krajobrazowy Wzniesień Łódzkich. Wydawnictwo Eko-Wynik, Łódź, Polska 1998.
17. Gara K. Załęczański Park Krajobrazowy. Arw ProArt, Sieradz, Polska 2008. 18. Czyżewska K. Załęczański Park Krajobrazowy: przewodnik. Arw ProArt, Sieradz,
Polska 1992. 19. Kurowski J. Sulejowski Park Krajobrazowy – środowisko przyrodniczo-geograficzne.
Wydawnictwo ZNPK, Moszczenica, Polska 1998. 20. http://www.znpk.com.pl/spalski_pk/flora 21. http://stud.ics.p.lodz.pl/~collina/znpk/ppk/ 22. Praca zbiorowa pod redakcją Tadeusza Kurzaca. Ogród Botaniczny w Łodzi.
Przewodnik. Łódź, Polska, 2008.

Sobiecka i in.
Biotechnol Food Sci 2011, 75 (2), 71-86 http://www.bfs.p.lodz.pl
86
23. Praca zbiorowa pod redakcją Tadeusza Kurzaca: Palmiarnia Ogrodu Botanicznego w Łodzi. Przewodnik. Łódź, Polska 2009.
24. Praca zbiorowa pod redakcją Tadeusza Kurzaca: Przyroda Ogrodu Botanicznego w Łodzi. Oficyna wydawniczo – reklamowa Sagalara, Łódź, Polska 1996.
25. Tumiłowicz J, Banaszczak P. Arboretum w Rogowie. MULTICO Oficyna Wydawnicza, Warszawa, Polska 2000.
26. http://arboretum.sggw.pl/alpinarium.html 2004.
The protection of plant biodiversity in Lodz Province
Abstract: The aim of the work was to review the information concerning the biological diversity of flora in the Lodz Province. The appearance of danger in ecosystems homeostasis were described. Part of the work focus on the various forms of the biodiversity plants protection in the Province well as advantages of the biological sustainability of biocenosis.

Biotechnology and Food Science Review article
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
Otrzymywanie włókien nanocelulozy
Radosław Wąchała,* Tomasz Ramięga, Rita Pyć, Tadeusz Antczak Instytut Biochemii Technicznej, Politechnika Łódzka, 90-924, Łódź, Polska *[email protected]
Streszczenie: Nanotechnologia należy do nauk technologicznych, obejmują-cych otrzymywanie oraz wykorzystywanie nowych nanomateriałów, takich jak nanoceluloza. Nanowłókna celulozy można otrzymywać dwoma sposobami top-down oraz buttom-up. Obecnie stosuje się głównie pierwszą metodę, w której zawiera się obróbka mechaniczna, chemiczna oraz enzymatyczna. Najlepsze efekty daje kombinacja utleniania chemicznego połączona z rozdrobnieniem mechanicznym, która pozwala na otrzymanie włókien o średnicy kilku nanometrów. Najbardziej przyjazną środowisku oraz przyszłościową okazuje się hydroliza enzymatyczna, która nie wymaga dużych nakładów finansowych i nie powoduje powstawania substancji trudnych do utylizacji. Otrzymane nanowłókna posiadają wiele potencjalnych zastosowań ze względu na właściwości reologiczne, takie jak wysoki moduł Young’a i odporność na rozciąganie przy niskiej rozszerzalności cieplnej.
Słowa kluczowe: nanoceluloza, hydroliza enzymatyczna, hydroliza chemiczna, nanowłókna, nanostruktury celulozy
Nanotechnologia – historia i definicje Nanotechnologię „wynalazła” natura. Ten rodzaj technologii „wybrała i dosko-
naliła” przyroda przez 5,5 miliarda lat, budując, atom po atomie, środowisko życia na Ziemi. Rozwój nauki i technologii w XX wieku, niewątpliwa zasługa ludzi, stworzył podstawy teoretyczne i praktyczne, dzięki którym stało się możliwe zajrzenie w strukturę materii i dostrzeżenie jej elementów w skali nano- (10-9 metra).
Za twórcę idei nanotechnologii uważa się Richarda P. Feynmana, który w roku 1959 podał pomysł stworzenia i wykorzystania nowatorskich technologii w skali rzędu kilkudziesięciu atomów [1].
Termin Nanotechnologia został użyły po raz pierwszy przez Norio Taniguchi w 1974 roku na określenie precyzji wytwarzania elementów maszyn z dokładnością mniejszą od 1 μm [2].
Jedynym ze stosowanych obecnie kryteriów pozwalających zdefiniować nanotechnologię jest rozmiar wytwarzanych obiektów, które powinny mieć przynajmniej jeden wymiar nie większy niż 100 nm. W procesie wytwarzania właściwości fizyczne i chemiczne, tych obiektów, powinny dać się kontrolować oraz musi istnieć możliwość budowania z nich większych obiektów [3].

Wąchała i in.
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
88
Metody otrzymywania nanostruktur Zarówno materia nieożywiona jak i ożywiona zawiera w swym składzie,
wykonane z atomów nanostruktury, które powielane i uporządkowane są budulcem środowiska naturalnego. Odkrycie fulerenów (nowej odmiany alotropowej węgla), poznanie amorficznej krzemionki – posiadającej uporządkowane nanopory czy wydobycie z żywych komórek silników molekularnych i uruchomienie ich poza komórką, a także wiele innych przykładów dowodzą tego, iż natura jest niewyczerpalnym źródłem różnych nanocząstek [4].
Obecnie nanotechnologia wytwarza nowe produkty, wykorzystując metody fizyko-chemiczne oraz coraz odważniej biotechnologiczne. Rozróżniamy dwie, ogólne metody wytwarzania nanomateriałów, które w jęz. angielskim określane są jako procesy top-down i bottom-up [5].
Metoda bottom-up zakłada budowę w skali molekularnej od podstaw przy użyciu atomów, molekuł i nanocząsteczek jako substratów. Zmieniając wielkość poszczególnych cegiełek budulca, kontrolując ich cechy powierzchniowe oraz ustalając w sposób zaplanowany warunki wytwarzania nanomateriałów można otrzymać produkty o pożądanych właściwościach. Do tego celu wykorzystuje się technologie wywodzące się z chemii i fizyki [7].
Metoda top-down polega na rozdrobnieniu materiału makroskopowego meto-dami: mechanicznymi (np. mielenie), chemicznymi (np. częściowa hydroliza kwasami lub zasadami), enzymatycznymi (np. traktowanie enzymami rozkłada-jącymi celulozę, hemicelulozę, pektyny i ligniny) i fizycznymi np. za pomocą technik wykorzystujących zogniskowaną wiązkę jonów Focused Ion Beam (FIB) lub laserów o dużej mocy [6]. Procesy te, w porównaniu z metodą bottom-up, są znacznie prostsze, gdyż głównie opierają się na podziale materiału makro-skopowego na mniejsze części lub na miniaturyzacji procesu podziału ciał stałych.
Jeśli w obu wymienionych metodach osiągniemy ten sam stopień kontroli nad prowadzonymi procesami, to można je określić wspólnym terminem inżynierii mikrostrukturalnej. Procesy biologiczne, biorąc pod uwagę skalę przestrzenną, są zjawiskami pośrednimi pomiędzy tymi metodami [8,9].
Obecnie aparatura pomiarowa wykorzystywana w nanotechnologii oraz maszyny służące do wytwarzania nanocząstek z każdym rokiem są doskonalsze i tańsze. Literatura fachowa i popularna dosłownie zasypuje nas informacjami naukowymi i użytkowymi, a laboratoria badawcze i przemysł dostarczają na rynek coraz bogatszy asortyment wytworzonych nano-produktów.
Otrzymywanie nanowłókien Nanowłóknami nazywamy włókna o średnicy mniejszej niż 100 nm, która jest
kilka tysięcy razy mniejsza do średnicy ludzkiego włosa, dlatego oglądać je można wyłącznie pod mikroskopem elektronowym. Wyjątkowe, często nie do opisania prawami tradycyjnej fizyki, są także ich właściwości. Nanowłókna mają dużą powierzchnię właściwą, 1 m2 tych włókien waży zaledwie 0,1-1 grama [10]. Włókna takie są w przeważającej mierze otrzymywane metodą bottom-up

Otrzymywanie włókien nanocelulozy
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
89
z użyciem elektroprzędzenia lub formowania na mokro z roztworów polimerów np. włókna formowane z polimerów syntetycznych, takich jak: poli(alkohol winylowy), poli(kwas mlekowy) [11-12] lub ich mieszaniny oraz z przetworzonych surowców naturalnych np. celulozy, chitozanu, keratyny, skrobi lub ich miesza-niny [13-22].
Metoda top-down również może być wykorzystana do otrzymania nano-włókien. W tym przypadku np. naturalne włókna roślinne zawarte w biomasie rozdrabnia się do rozmiarów nano- z zastosowaniem metod termicznych, chemicznych [23-25] lub biotechnologicznych wykorzystujących procesy biorafinacji [26].
Nanowłókna otrzymywane z celulozy Celuloza jest materiałem szeroko rozpowszechnionym w przyrodzie i można
pozyskać ją z wielu źródeł naturalnych, takich jak drewno, bawełna i biomasa roślinna [27]. Celuloza jest także syntetyzowana przez algi i niektóre bakterie. Innym interesującym źródłem celulozy są tuniki zwierząt morskich – osłonic [28, 29].
Włókna celulozy, mając jeden z wymiarów mniejszy niż 100 nm łączą w sobie cechy materiałów makroskopowych ze specyficznymi właściwościami nanomateriałów. Należą one do naturalnych włókien, których właściwości funkcjonalne determinowane są przez strukturę nanofibryli. Większa powierzchnia nanocelulozy w stosunku do naturalnej makrocelulozy powoduje silniejsze interakcje z otaczającymi cząstkami np. silnie wiąże własne i inne nanocząsteczki [30, 31].
Otrzymywanie nanowłókien celulozowych metodą bottom-up W metodzie tej wykorzystuje się do tego celu np. technikę elektroprzędzenia
[32, 35]. Nanowłókno jest wytwarzane z roztworów celulozy regenerowanej, które wyjściowo mogą także zawierać stałe nanocząsteczki [36].
Otrzymywanie nanowłókien celulozowych metodą top-down W metodzie top–down wykorzystuje się metody rafinacji o charakterze:
fizycznym [37-40], chemicznym [23, 38, 41] oraz biorafinację [26] bądź kom-binacje tych metod [28]. Polegają one na usunięciu z komórek roślinnych składników innych niż celuloza, takich jak: pektyny, hemicelulozy, ligniny i minerały. Następnie poprzez odpowiednią obróbkę włókien celulozowych można zwiększyć dostępność grup hydroksylowych, zmienić stopień krystaliczności, rozwinąć powierzchnię wewnętrzną, zerwać wiązania wodorowe, co powoduje wzrost reaktywności celulozy [42, 43]. Prowadzony w odpowiednich warunkach proces może doprowadzić do rozdzielenia włókien celulozowych na makro i mikrofibryle [28, 44, 45].

Wąchała i in.
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
90
Otrzymywania nanocelulozy z wykorzystaniem metod mechanicznych Obróbka mechaniczna biomasy celulozowej polega na fizycznym rozdzie-
leniu włókien roślinnych w celu otrzymania produktu cechującego się rozmiarem rzędu kilku, kilkunastu lub kilkudziesięciu nanometrów. Spośród stosowanych metod mechanicznych można wyróżnić następujące techniki [30, 46]:
mielenie, mielenie kriogeniczne, homogenizację wysokociśnieniową, działanie ultradźwiękami.
Mogą być one stosowane pojedynczo lub łączone w bardziej skomplikowane procesy technologiczne. W zależności od specyfiki poszczególnych technologii oraz zadanych parametrów, wymiary oraz budowa wytworzonych nanowłókien mogą się znacznie pomiędzy sobą różnić [46].
Wstępne rozdrobnienie poprzez mielenie zazwyczaj jest stosowane jako proces poprzedzający homogenizację, ponieważ powoduje jedynie odsłonięcie fibryli w zewnętrznej powłoce wiązek włókien celulozowych, niewystarczająco rozdrabniając substrat. Powoduje ono nieodwracalne zmiany w strukturze i wielkości cząstek, wpływa na właściwości powierzchni fibryli, co może być korzystne przy dalszym wykorzystaniu produktu. Jednocześnie operacja ta może wpłynąć negatywnie na morfologię włókien, niszczyć strukturę mikrofibryli, zmniejszając masę molową celulozy oraz stopień jej krystaliczności [46].
Proces mielenia może być prowadzony np. w młynku tarczowym, w którym zawiesina podlega wymuszonemu przepływowi pomiędzy tarczami młynka posiadającymi na powierzchni specyficzne wyżłobienia, dzięki czemu celuloza poddawana jest powtarzającym się cyklom działania sił i naprężeń [46].
Po mieleniu włókna celulozy ulegają częściowej, powierzchniowej fibrylacji, jednak rdzenie włókien pozostają bez zmian. Jakkolwiek zmiana ta nie prowadzi do powstania nanocelulozy, może mieć dodatni wpływ na właściwości masy jako wypełniacza kompozytów, zwiększając powierzchnię oddziaływania z matrycą. Wprowadzenie homogenizacji jako następnego kroku, skutkuje rozszczepieniem poprzednio oddzielonych włókien i utworzeniem niewielkich agregatów, których wielkość zmniejsza się wraz ze wzrostem powtórzeń procesu homogenizacji [47].
Usprawnieniem mielenia jest odmiana nazywana mieleniem kriogenicznym, w której przed główną obróbką mechaniczną próbka poddawana jest zamrażaniu przy użyciu ciekłego azotu [46]. Podczas zamrożenia w komórkach rozdrabnia-nego materiału tworzą się kryształy lodu, które pod wpływem wysokiego ciśnie-nia w komórkach rozrywają matrycę od środka i uwalniają mikrofibryle [48]. Następnie zamrożona masa jest dodatkowo rozdrabniana mechanicznie np. za pomocą ostrzy wykonanych z tytanu co przyczynia się do pogłębienia procesu rozdrobnienia biomasy [48, 49]. Zastosowanie obróbki chemicznej jako następ-nego kroku sprawia, że biomasa zawiera głównie materiał celulozowy oraz nie-wielkie ilości ligniny oraz hemiceluloz. Niemal 60% włókien pozyskanych w ten sposób ma średnicę mieszczącą się w przedziale 30-120 nm [49].

Otrzymywanie włókien nanocelulozy
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
91
Podczas homogenizacji wysokociśnieniowej (100-200 MPa) wodna zawie-sina, wstępnie rozdrobnionej celulozy, zostaje poddana działaniu wysokiego ciśnienia. Zmniejszenie wymiarów materiału celulozowego następuje wskutek działania sił zgniatających występujących podczas uderzania o siebie cząstek, sił poprzecznych działających w strefie homogenizacji oraz sił kawitacyjnych powstałych w wyniku zmian prędkości cząstek [50]. Wraz ze wzrostem czasu procesu homogenizacji karboksymetylocelulozy malała długość i średnica wiązek włókien. Wzrastała natomiast przezroczystość folii wytworzonej z tego materiału [51].
Traktowanie ultradźwiękami (w środowisku kwaśnym lub zasadowym) przy-czynia się bezpośrednio do rozerwania wiązań glikozydowych w łańcuchu celu-lozy. Pod działaniem wysokiej częstotliwości rzędu 20 kHz następuje tworzenie, rozrost oraz pękanie mikropęcherzyków w wodnym roztworze, co powoduje rozszczepienie powierzchni włókien. Dzięki procesowi kawitacji energia ultra-dźwięków rzędu 10-100 kJ * mol-1, czyli bliska energii wiązania wodorowego, przenoszona jest na łańcuch polimerowy [52, 53].
Szerokość włókien celulozowych syntezowanych przez bakterie Acetobacter xylinum jest zawarta w przedziale 110-140 nm, jednak już po 15 minutach dzia-łania ultradźwiękami o częstotliwości rzędu 20 kHz zmniejsza się ich wielkość do 70-110 nm. Dalsze traktowanie ultradźwiękami prowadzi do zmniejszenia szerokości wstążek celulozowych nawet do 40-70 nm [52].
Wykorzystanie kwasów i zasad do otrzymywania nanocelulozy Działanie kwasami i zasadami na materiały ligninocelulozowe może się od-
bywać w różnoraki sposób w zależności od planowanych do osiągnięcia rezultatów. Może, na przykład, w celu zwiększenia powierzchni materiału ligninocelulozowego i zwiększenia podatności polisacharydów na hydrolizę rozpoczynać się nama-czaniem w roztworze wodorotlenku sodu [2].
Hydroliza kwasowa w temperaturze 60-80oC powoduje rozkład hemicelulozy. Wykonane po niej traktowanie zasadami nieorganicznymi w temperaturze 60-80oC pomaga rozerwać wiązania strukturalne pomiędzy ligniną, a węglowodanami, co powoduje uwolnienie ligniny. Rozpuszczeniu ulega lignina oraz pozostałości hemicelulozy i pektyny. Ponieważ w toku obróbki chemicznej celuloza może ulec niepożądanej degradacji, stosuje się roztwory zasad stężeniu poniżej 18% [54, 55]. Po obróbce chemicznej nanowłókna są wciąż związane ze składnikami strukturalny-mi ściany komórkowej, konieczne jest więc dalsze mechaniczne przetwarzanie [56].
Hydrolizę kwasową można przeprowadzić przy użyciu roztworów mocnych kwasów nieorganicznych, na przykład kwasu solnego lub kwasu siarkowego. Po obróbce chemicznej zawartość celulozy w biomasie wzrasta do 94-95%, podczas gdy łączna zawartość pozostałych składników spada do wartości poniżej 5%. Wieloetapowa obróbka chemiczna prowadzi do coraz większego oczyszczenia i rozdzielenia się biomasy na mniejsze fibryle [56]. Hydroliza kwasowa wpływa głównie na stopień polimeryzacji oraz stabilność cieplną nanowłókien, która jest to bardzo ważnym parametrem informującym o temperaturze przetwórstwa

Wąchała i in.
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
92
nanocząstek [55]. Produkt otrzymany przy użyciu tylko roztworu kwasu siarkowego ma niższą stabilność cieplną niż biomasa wyjściowa, której temperatura degradacji wynosi 180oC. Jest to spowodowane pojawianiem się podczas tego typu hydrolizy na powierzchni nanocelulozy grup siarczanowych, które „odwadniają” nanostrukturę. Grupy te jednak zmieniają ładunek powierzchnio-wy cząsteczek, utrudniając tym samym ich aglomerację, co jest w tym przypadku zjawiskiem pożądanym [55]. Dodatek kwasu solnego podczas hydrolizy kwasem siarkowym znacznie poprawia stabilność cieplną nanostruktur celulozowych (temp. degradacji sięga tu 300oC). Spowodowane jest to przyłączaniem się mniejszej ilości grup siarczanowych do powierzchni celulozy na rzecz jonów chlorkowych. Jony te jednak łatwo ulegają wymywaniu, co może skutkować ponowną aglomeracją nanocząsteczek celulozy. Dobierając odpowiednio warunki hydrolizy kwasowej, można zhydrolizować obszary amorficzne celulozy, przy jednoczesnym zachowaniu regionów krystalicznych [54, 55].
Wraz ze wzrostem stężenia zasady powyżej 17%, poprawia się wydajność oczyszczania włókien z niecelulozowych składników. Z drugiej strony jednak wyższe stężenie wodorotlenku sodu skutkuje produktem o niższym stopniu polimeryzacji. Niezależnie od stężenia zasady nanowłókna posiadają średnicę rzędu 6-10 nm oraz długość 80-170 nm [55].
Utlenianie katalizowane za pomocą TEMPO Utlenianie celulozy przy użyciu rodnika 2,2,6,6-tetrametylopiperydyno-1-
oksylu (TEMPO) jako katalizatora, umożliwia formowanie grup karboksylowych w pozycji C6 z grup hydroksylowych merów obecnych na powierzchni włókien. Ponieważ zdysocjowane grupy karboksylowe mają ładunek ujemny, występują siły odpychające pomiędzy mikrofibrylami. Dzięki temu nanowłókna celulozy o średnicy 3-4 nm mogą powstać za pomocą prostej obróbki mechanicznej utlenionego materiału. Reakcja może być przeprowadzana w środowisku zasadowym lub kwaśnym z dodatkiem różnych reagentów w roli utleniaczy. Folie tworzone z celulozy utlenionej w procesie katalizowanym przez TEMPO cechują się dobrą przezroczystością, stabilnością cieplną oraz niską prze-puszczalnością tlenu. Zarówno TEMPO jak i jego analogi są rozpuszczalne w wodzie [57].
Produkt utleniania celulozy katalizowanego przez TEMPO jest, w prze-ciwieństwie do celulozy natywnej, odporny na hydrolizę kwasową, a podatny na zasadową. Jest on także odporny na depolimeryzację prowadzoną przez typowe celulazy. Może ulec natomiast biodegradacji katalizowanej poprzez enzymy produkowane przez mikroorganizmy występujące w środowisku naturalnym, np. bakterie z rodzaju Brevundimonas [58].
Metoda eksplozyjno-parowa Metoda eksplozyjno-parowa polega na nasycaniu biomasy parą wodną pod
zwiększonym ciśnieniem ok. 2 MPa, a następnie na gwałtownej redukcji tego ciśnie-nia do 0,1 MPa, dzięki czemu surowiec ulega mechanicznej defibrylacji [59, 60].

Otrzymywanie włókien nanocelulozy
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
93
W trakcie przeprowadzania obróbki materiału metodą eksplozyjno-parową lignina ulega depolimeryzacji, a hemiceluloza częściowej hydrolizie. Wynikiem tego jest wymywanie cukrów i związków fenolowych do roztworu. Hydroliza wiązań eterowych w ligninie oraz autohydroliza wiązań glikozydowych w hemicelulozie jest katalizowana przez kwas octowy tworzący się w wysokiej temperaturze z grup acetylowych obecnych w hemicelulozie [59].
Metoda ta powoduje spadek stopnia polimeryzacji celulozy. Tylko 3% włókien ma średnicę powyżej 70 nm, 19% poniżej 30 nm, średnica zaś aż 64% nanowłókien mieści się w przedziale 30-50 nm [60].
Do zalet metody eksplozyjno-parowej należy zaliczyć niewielki negatywny wpływ na środowisko, brak konieczności stosowania substancji chemicznych oraz niskie zużycie energii oraz koszty urządzeń w porównaniu do homogenizacji wysokociśnieniowej. Główną wadą natomiast jest nieznaczne (ok. 3%) obniżenie stopnia polimeryzacji celulozy w tym procesie [59, 60].
Otrzymywanie nanocelulozy metodami enzymatycznymi Do wytwarzania nanocelulozy można wykorzystać także enzymy. W etapie
wstępnym np. po degradacji mechanicznej masy drzewnej lub innej biomasy można wykorzystać enzymy do usunięcia materiałów niecelulozowych (pektyn, hemiceluloz, lignin). Można także, w dalszym etapie, w celu otrzymania nano-włókien użyć celulaz, które, degradując przede wszystkim regiony amorficzne a nie krystaliczne celulozy, ułatwiają rozdzielanie włókien. Taka obróbka umożliwia otrzymanie mniejszych cząstek nanocelulozy [61, 62]. Zastosowanie, obok obróbki mechanicznej, dodatkowej hydrolizy enzymatycznej do procesu wytwarzania nanowłókien umożliwia rozdzielenie fibryli na nanofibryle o średni-cach mniejszych niż uzyskiwane przy użyciu tylko samych metod mechanicznych. Hydroliza enzymatyczna, w porównaniu do hydrolizy kwasowej pozwala na otrzymanie nanowłókien o długości powyżej 200 nm, cechujących się większą liczbą połączeń pomiędzy nanofibrylami. Są to cechy pożądane w przypadku nanocelulozy stosowanej jako wzmacniacza komponentów. Ponadto hydroliza enzymatyczna, poprzedzająca mielenie lub homogenizację, pozwala na obniżenie zużycia energii w procesie, a co za tym idzie na wzrost oszczędności [63].
Zaletą hydrolizy enzymatycznej, w przeciwieństwie do hydrolizy kwasowej, jest fakt iż na powierzchni włókna nie ma związanych jonów np. siarczanowych. Tak otrzymana nanoceluloza jest materiałem biokompatybilnym i może zna- leźć zastosowanie do otrzymywania produktów biomedycznych oraz farma-ceutycznych [63].
Hydroliza alkaliczna w porównaniu z enzymatyczną daje dużo gorsze efekty ze względu na duży spadek stopnia polimeryzacji podczas procesu oraz bardzo drastyczną degradację celulozy przy nieznacznym usuwaniu substancji balastowych.

Wąchała i in.
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
94
Zastosowanie nanoczasteczek celulozowych Nanowłókna celulozowe otrzymywane z biomasy roślinnej są szczególnie
pożądaną grupą produktów. Wprost nieograniczona dostępność surowca, jego biodegradowalność i biokompatybilność ze światem ożywionym sprawia, iż wiele laboratoriów prowadzi badania nad opracowaniem technologii pozyskiwania tego produktu [27-29, 65].
Wzmacnianie środków klejących Głównym celem dodatku „wzmacniaczy” do środków klejących stosowanych
do łączenia elementów drewnianych jest ich zdolność do wzmacniania i usztywniania kompozytu poprzez efektywne przenoszenie ich rozkładu naprężeń. Poza substancją wiążącą stosuje się dodatki do klejów w postaci wypełniaczy, które w zależności od rodzaju zastosowanego materiału, wpływają na poszczególne właściwości spoiw [66]. Najczęściej stosowanymi klejami do tych materiałów są żywice mocznikowo-foarmaldehydowe (UF), popularne ze wzglądu na swoją niską cenę. UF cechują się kruchością i tendencją do tworzenia mikropęknięć, co wpływa niekorzystnie na ich właściwości mechaniczne.
Dodatek nieprzetworzonej masy celulozowej do kleju UF nie wpływa na wytrzymałość regionu łączenia, natomiast zastosowanie nanocelulozy jako wypełniacza poprawia w znaczący sposób odporność badanych kompozytów na ścinanie z 10,3±0,9 MPa dla czystego UF do 13,8±1,4 MPa. Ponadto w środku klejącym zawierającym dodatek nanocelulozy nie występują mikropęknięcia [67].
Przezroczyste nanokompozyty celulozowe Ponieważ obiekty o średnicy mniejszej niż jedna dziesiąta długości fali
światła widzialnego nie powodują rozpraszania światła, nanoceluloza może być rozważana jako dobry materiał do wzmacniania przezroczystych tworzyw sztucznych. Przezroczyste nanokompozyty o zawartości nanocelulozy 7,4% wykazują wytrzymałość mechaniczną porównywalną do stali miękkiej [68].
Przezroczysty papier Proces produkcji tzw. „papieru z nanowłókien celulozowych” jest niemal
identyczny jak papieru tradycyjnego, a produkt różni się jedynie wielkością włókien i porowatością. Taki papier jest półprzezroczysty ze względu na powierzchniowe rozpraszanie światła, jednak pod wpływem polerowania papierem ściernym o ziarnistości 4000 oraz 15000 uzyskuje się produkt transparentny, o podatności na zginanie równej tradycyjnemu papierowi. Produkt taki cechuje się współczynnikiem rozszerzalności cieplnej równym 8,5 ppmK (wartość porównywalna do osiąganej przez szkło), modułem Young’a wielkości 13 GPa i wytrzymałością na rozciąganie rzędu 223 MPa [69].

Otrzymywanie włókien nanocelulozy
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
95
Aerożele Aerożele zawierające nanocelulozę cechują się porowatością 98% i gęstością
0,02 g/cm3. W przeciwieństwie do większości aerożeli, cechujących się kruchością, wykazują dużą elastyczność. Odkształcenie materiału przy ściskaniu wynosi około 70% [70].
Daje to nowe możliwości zastosowań tego typu elastycznych powłok do izolacji termicznej w licznych urządzeniach wojskowych i kosmicznych. Stoso-wanie takich areożeli może zminimalizować pękanie powłok poszyć statków kosmicznych podczas nagłych zmian temperatur.
Areożele zawierające celulozę idealnie nadaję się do produkcji obuwia turystycznego, gdyż zapewniają bardzo dobrą elastyczność oraz izolację termiczną przy minimalnej masie i grubości [70].
Kompozyty celulozowe W poszukiwaniu materiałów przyjaznych środowisku zwraca się coraz
większą uwagę na nadające się do recyklingu, biodegradowalne kompozyty składające się tylko z jednego rodzaju substancji. Przykładem może być kompozyt celulozowy, który może być otrzymywany dwiema metodami [67]: impregnacja matrycy celulozowej za pomocą włókien celulozowych, selektywne rozpuszczanie powierzchniowe.
Takie metody pozwalają otrzymać materiały przewyższające swoimi właści-wościami inne kompozyty [67].
W procesie selektywnego rozpuszczania powierzchniowego włókna celulozowe są częściowo rozpuszczane, np. w roztworze chlorku litu w dimetyloacetamidzie (DMAc), a następnie tworzona jest matryca dla kompozytu wzmacnianego niezmienionymi rdzeniami włókien celulozowych [71].
Kompozyt celulozowy otrzymany z celulozy bakteryjnej poprzez selektywne rozpuszczanie powierzchniowe charakteryzuje się wartością modułu Younga równą 18 GPa (polipropylen 1,5-2,5 GPa) i wytrzymałością na rozerwanie 411 MPa (polipropylen 20-38MPa) [72]. Metoda ta zatem pozawala na otrzymanie materiału o bardzo ciekawych właściwościach reologicznych.
Podsumowanie Niektórzy uczeni z rezerwą odnoszą się do codziennego obcowania człowieka
z produktami nanotechnologii. Dostrzegają możliwe ryzyko wynikające z poten-cjalnej toksyczności nanocząsteczek, zwłaszcza tych niedegradowanych w orga-nizmie człowieka. Cząsteczki te mogą dostawać się poprzez płuca, przez skórę w przypadku bezpośredniego kontaktu oraz przez układ pokarmowy. Losy nanocząsteczek trafiających do środowiska nie są w pełni znane. Istnieje ryzyko ich akumulacji w organizmach żywych (także roślinach uprawnych) i negatywnego wpływu na ekosystem [73, 74]. Przeprowadzone dotychczas badania wykazują, że niektóre nanocząsteczki, przedostając się do organizmów żywych drogami oddechowymi, wywołują niekorzystne zmiany oraz objawy chorobowe [73].

Wąchała i in.
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
96
Jednakże w przypadku nanocząsteczek celulozy należy domniemywać, że znajdowały się one od zawsze w powietrzu. Ich źródłem była przyroda zielona, a dodatkowym efektorem pożary i inne kataklizmy.
Należy pamiętać, iż rozwój nauk technologicznych może zostać spowolniony, wręcz zahamowany z racji braku społecznego przyzwolenia. Z tego powodu, obok znacznych nakładów finansowych na nanotechnologię i niewątpliwego postępu naukowego potrzebna jest szeroka akcja informacyjna promująca przyszłe, możliwe do osiągnięcia cele bez pominięcia ewentualnych, koniecznych do poniesienia kosztów.
Jednym z celów programu badań naukowych POIG 01.01.02-10-123/09 „Zastosowanie biomasy do wytwarzania polimerowych materiałów przyjaznych środowisku” realizowanego w latach 2010-2013 jest wytworzenie nanowłókien celulozowych z biomasy roślinnej. Zamierzenia te są realizowane w dwóch pakietach zadaniowych PZ 2.1. „Preparaty wieloenzymowe do ukierunkowanej degradacji biomasy roślinnej” i PZ 3.1. „Wytwarzanie nanowłókien celulo-zowych z surowców roślinnych”. Projekty te zmierzają do wytworzenia w procesie enzymatycznym, chemicznym i fizyko-chemicznym na bazie odpowiedniej biomasy (np. len, konopie, rzepak lub inne) nanowłókien celulozowych, które będą następnie przetworzone w innowacyjne, biodegradowalne materiały kompozytowe.
W naszym przekonaniu wytworzone nanowłókna będą stanowić rzetelny punkt wyjścia do produkcji całej gamy użytecznych materiałów polimerowych.
Literatura 1. Feynman P. There's Plenty of Room at the Bottom (transkrypcja wykładu z dnia
29 grudnia 1959), http://www.zyvex.com/nanotech/feynman.html. 2. NanoWord Newsletter nr 1. http://www.nanoword.net/library/nwn/1.html. 3. Poole P, Owens J. Introduction to nanotechnology. John Wiley & Sons 2003. 4. Gianella A, Jarzyna A, Mani V, Ramachandran S, Calcagno C, Tang J, Kann B, Dijk R,
Thijssen VL, Griffioen W, Storm G, Fayad A, Mulder M. Multifunctional Nanoemulsion Platform for Imaging Guided Therapy Evaluated in Experimental Cancer. ACS Nano 2011, 5:4422-4433.
5. Nano acceleration network. http://nanotechnology.espaces.com/nano_products.html. 6. Kelsall W, Hamley W, Geoghegan M. Nanoscale Science and Technology. John
Wiley & Sons 2005. 7. Kelsall W, Hamley W, Geoghegan M, Kurzydłowski K. Nanotechnologie, PWN,
Warszawa 2008. 8. Balzani V. Nanoscience and nanotechnology. Pure Appl Chem 2008, 80:1631-1650. 9. Nano acceleration network. http://nanotechnology.espaces.com/nano_products.html.
10. European Commission Nanotechnology Research needs on nanoparticles. Proc workshop held in Brussels, 25-26.01.2005.
11. Schaefer K, Thomas H, Dalton P, Moeller M. Nano-fibres for filter materials. http://www.scribd.com/doc/30357529/Nano-Fibres-for-Filter-Materials.
12. Yamashita Y, Ko F, Miyake H, Higashiyama A. Establishment of nano fiber preparation technique for nanocomposite. 16th Int Conf Composite Materials 2007.

Otrzymywanie włókien nanocelulozy
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
97
13. Fan Y, Fukuzumi H, Saito T, Isogai A. Comparative characterization of aqueous dispersions and cast films of different chitin nanowhiskers/nanofibers. Int J Biol Macromol 2012, 50:69-76.
14. Watthanaphanit A, Supaphol P, Tamura H, Tokura S, Rujiravanit R. Fabrication, structure, and properties of chitin whisker-reinforced alginate nanocomposite fibers. J Appl Polym Sci 2008, 110:890-899.
15. Fan Y, Saito T, Isogai A. TEMPO-mediated oxidation of β-chitin to prepare individual nanofibrils. Carbohydr Polym 2009, 77:832-838.
16. Fan Y, Saito T, Isogai A. Individual chitin nano-whiskers prepared from partially deacetylated α-chitin by fibril surface cationization. Carbohydr Polym 2010, 79: 1046-1051.
17. Cardamone M, Martin J. Keratin coatings for wool: shrinkproofing and nanoparticle delivery. Macromol Symp 2008, 272:161-166.
18. Martin J, Cardamone M, Irwin L, Brown M. Keratin capped silver nanoparticles-synthesis and characterization of a nanomaterial with desirable handling properties. Colloids Surf B Biointerfaces 2011, 54-61.
19. Xing Z, Yuan J, Chae W, Kang I, Kim S. Keratin nanofibers as a biomaterial. Int Conf Nanotechnology and Biosensors, Singapore, 2011, 2:120-124.
20. Tonin C, Aluigi A, Varesano A, Vineis C. Keratin-based nanofibres, in: nanofibers. InTech 2010, 139-158.
21. Adomavičiūtė E, Milašius R, Žemaitaitis A, Bendoraitienė J, Leskovšek M, Demšar A. Methods of forming nanofibres from bicomponent PVA/Cationic starch solution. Fib Text East Eur 2009, 17:29-33.
22. Ayse A. Biocomposites from wheat straw nanofibers: morphology, thermal and mechanical properties. Comp Sci Techn 2008, 68:557-565.
23. Fujisawa F, Okita Y, Fukuzumi H, Saito T, Isogai A. Preparation and characterization of TEMPO-oxidized cellulose nanofibril films with free carboxyl groups. Carbohyd Polym 2011, 84:579-583.
24. Isogai A, Saito T, Fukuzumi H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3:71-85.
25. Iwamoto S, Kai W, Isogai T, Saito T, Isogai A, Iwata T. Comparison study of TEMPO-analogous compounds on oxidation efficiency of wood cellulose for preparation of cellulose nanofibrils. Polym Degrad Stabil 2010, 95:1394-1398.
26. Hayashi N, Kondo T, Ishihara M, Enzymatically produced nano-ordered short elements containing cellulose Ib crystalline domains. Carbohyd Polym 2005, 61: 191-197.
27. Khalil A, Bhat A, Yusra I. Green composites from sustainable cellulose nanofibrils: A review. Carbohydr Polym 2011, 87:963-979.
28. Eichhorn S, Dufresne A. Review: current international research into cellulose nanofibres and nanocomposites. J. Mater. Sci. 2010, 45:1-33.
29. Siró I, Plackett D. Microfibrillated cellulose and new nanocomposite materials: a review. Cellulose 2010, 17:459-494.
30. Cheng Q, Wang S, Rials T, Lee S. Physical and mechanical properties of polyvinyl alcohol and polypropylene composite materials reinforced with fibril aggregates isolated from regenerated cellulose fibers. Cellulose, 2007, 14:593-602.
31. Klemm D, Schumann D, Kramer F, Heßler N, Koth D, Sultanova B. Nanocellulose materials–different cellulose, different functionality. Macromol Symp 2009, 280:60-71.

Wąchała i in.
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
98
32. Hubbe M, Rojas O, Lucia L, Sain M. Cellulosic nanocomposites: a review. Bioresources 2008, 3:929-980.
33. Nukavarapu S, James R, Hogan M, Laurencin C. Recent patents on electrospun biomedical nanostructures: an overview. Rec Patents on Biomed Eng 2008, 1:68-78.
34. Cengiz F, Krucińska I, Gliścińska E, Chrzanowski M, Göktepe F. Comparative analysis of various electrospinning methods of nanofibre formation. Fib Text East Eur 2009, 17:13-19.
35. Chiung H, Lin K, Lu T, Lou L, Chao C. Evaluation of the electrospinning manufacturing process based on the preparation of pva composite fibres. Fib Text East Eur 2009, 17:34-37.
36. Dabirian F, Hosseini A. Novel method for nanofibre yarn production using two differently charged nozzles. Fib Text East Eur 2009, 17:45-47.
37. Sójka-Ledakowicz J, Lewartowska J, Kudzin M, Jesionowski T, Siwińska-Stefańska K, Krysztafkiewicz A. Modification of textile materials with micro- and nano-structural metal oxides. Fib Text East Eur 2008, 16:112-116.
38. Syverud K, Chinga-Carrasco G, Toledo J, Toledo P. A comparative study of eucalyptus and pinus radiata pulp fibres as raw materials for production of cellulose nanofibrils. Carbohyd Polym 2011, 84:1033-1038.
39. Seydibeyoglu Ö, Oksman K. Novel nanocomposites based on polyurethane and micro fibrillated cellulose. Comp. Sci Techn 2008, 68:908-914.
40. Lee Y, Chun S, Kang I, Park J. Preparation of cellulose nanofibrils by high-pressure homogenizer and cellulose-based composite films. J Ind Eng Chem 2009, 15:50-55.
41. Siró I, Plackett D, Hedenqvist M, Ankerfors M, Lindström T. Highly transparent films from carboxymethylated microfibrillate cellulose: the effect of multiple homogenization steps on key properties. J Appl Polym Sci 2011, 119:2652-2660.
42. Yu C, Huimin T. Effects of cellulase on the modification of cellulose. Carbohyd Res 2002, 337:1291-1296.
43. Hu Z, Foston M, Ragauskas A. Comparative studies on hydrothermal pretreatment and enzymatic saccharification of leaves and internodes of alamo switchgrass. Biores Techn 2011, 102:7224-722.
44. Zuluaga R, Putaux J, Cruz J, Vélez J, Mondragon I, Gañán P. Cellulose microfibrils from banana rachis: effect of alkaline treatments on structural and morphological features. Carbohyd Polym 2009, 76:51-59.
45. Bhattacharya D, Germinario L, Winter W. Isolation, preparation and characterization of cellulose microfibers obtained from bagasse. Carbohyd. Polym. 2008, 73:371-377.
46. Siró I, Plackett D. Microfibrillated cellulose and new nanocomposite materials: a review. Cellulose 2010, 17:459-494.
47. Nakagaito A, Yano H. The effect of morphological changes from pulp fiber towards nano-scale fibrillated cellulose on the mechanical properties of high-strength plant fiber based composites. Appl Phys A 2004, 78:547-552.
48. Wang B, Sain M. Dispersion of soybean stock-based nanofiber in a plastic matrix. Polym Int 2007, 56:538-546.
49. Alemdar A, Sain M. Isolation and characterization of nanofibers from agricultural residues-wheat straw and soy hulls. Biores Technol 2008, 99:1664-1671.
50. Lee S, Chun S, Kang I, Park J. Preparation of cellulose nanofibrils by high-pressure homogenizer and cellulose-based composite films. J Ind Eng Chem 2009, 15:50-55.

Otrzymywanie włókien nanocelulozy
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
99
51. Siró I, Plackett D, Hedenqvist M, Ankerfors M, Lindström T. Highly transparent films from carboxymethylated microfibrillated cellulose: the effect of multiple homogenization steps on key properties. J Appl Polym Sci 2011, 119:2652-2660.
52. Tischer P, Sierakowski R, Westfahl H, Tischer C. Nanostructural reorganization of bacterial cellulose by ultrasonic treatment. Biomacromolecules 2010, 11:1217-1224.
53. Zhao P, Fenga X, Gao H. Ultrasonic technique for extracting nanofibers from nature materials. Appl Phys Lett 2007, 90:073112-1-073112-2.
54. Corrêa C, Teixeira E, Pessan L, Mattoso C. Cellulose nanofibers from curaua fibers. Cellulose 2010, 17:1183-1192.
55. Kumar S, Upadhyaya J, Negi Y. Preparation of nanoparticles from corn cobs by chemical treatment methods. Bioresources, 2010, 5:1292-1300.
56. Bhatnagar A, Sain M. Processing of cellulose nanofiber-reinforced composites. J Reinf Plast Comp 2005, 24:1259-1268.
57. Iwamoto S, Kai W, Isogai T, Saito T, Isogai A, Iwata T. Comparison study of TEMPO-analogous compounds on oxidation efficiency of wood cellulose for preparation of cellulose nanofibrils. Polym Degrad Stabil 2010, 95:1394-1398.
58. Isogai A, Saito T, Fukuzumi H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71-85.
59. Cherian M, Pothan A, Nguyen-Chung T, Mennig G, Kottaisamy M, Sabu T. A novel method for the synthesis of cellulose nanofibril whiskers from banana fibers and characterization. J Agric Food Chem 2008, 56:5617-5627.
60. Kaushik A, Singh M. Isolation and characterization of cellulose nanofibrils from wheat straw using steam explosion coupled with high shear homogenization. Carbohydr. Res. 2011, 346:76-85.
61. Hubbe M, Rojas O, Lucia L, Sain M. Cellulosic nanocomposites: a review. BioResources 2008, 3:929-980.
62. Janardhnan S, Sain M. Isolation of cellulose microfibrils – an enzymatic approach. Bioresources, 2006, 2:176-188.
63. Pääkkö M, Ankerfors M, Kosonen H, Nykänen A, Ahola S, Österberg M, Ruokolainen J, Laine J, Larsson P, Ikkala O, Lindström T. Enzymatic hydrolysis combined with mechanical shearing and high-pressure homogenization for nanoscale cellulose fibrils and strong gels. Biomacromolecules 2007, 8:1934-1941.
64. Satyamurthy P, Jain P, Balasubramanya R, Vigneshwaran N. Preparation and characterization of cellulose nanowhiskers from cotton fibres by controlled microbial hydrolysis. Carbohydr Polym 2001, 83:122-129.
65. Chen W, Yu H, Liu Y, Chen P, Zhang M, Hai Y. Individualization of cellulose nanofibers from wood using high-intensity ultrasonication combined with chemical pretreatments. Carbohyd Polym 2011, 83:1804-1811.
66. Vick B. Adhesive bonding of wood materials, wood handbook: wood as an engineering material. U.S. Department of Agriculture, Forest Service, Forest Products Laboratory, Madison, 1999, 9-1-9-24.
67. Eichhorn S, Dufresne A, Aranguren M, Marcovich N, Capadona J, Rowan S, Weder C, Thielemans W, Roman M, Renneckar S, Gindl W, Veigel S, Keckes J, Yano H, Abe K, Nogi M, Nakagaito A, Mangalam A, Simonsen J, Benight A, Bismarck A, Berglund L, Peijs T. Review: current international research into cellulose nanofibres and nanocomposites, J Mater Sci 2010, 45:1-33.

Wąchała i in.
Biotechnol Food Sci 2011, 75 (2), 87-100 http://www.bfs.p.lodz.pl
100
68. Yano H, Sugiyama J, Nakagaito A, Nogi M, Matsuura T, Hikita M, Handa K. Optically transparent composites reinforced with networks of bacterial nanofibers, Adv Mater 2005, 17:153-155.
69. Nogi M, Iwamoto S, Nakagaito A, Yano H. Optically transparent nanofiber paper. Adv Mater 2009, 21:1595-1598.
70. Pääkkö M, Vapaavuori J, Silvennoinen R, Kosonen H, Ankerfors M, Lindström T, Berglund L, Ikkala O. Long and entangled native cellulose I nanofibers allow flexible aerogels and hierarchically porous templates for functionalities. Soft Matter 2008, 4:2492-2499.
71. Soykeabkaew N, Arimoto N, Nishino T, Peijs T. All-cellulose composites by surface selective dissolution of aligned ligno-cellulosic fibres, Compos Sci Technol 2008, 68:2201-2207.
72. Soykeabkaew N, Sian C, Gea S, Nishino T, Peijs T. All-cellulose nanocomposites by surface selective dissolution of bacterial cellulose. Cellulose 2009, 16:435-444.
73. http://www.nanotec.org.uk/finalReport.htm. Nanoscience and nanotechnologies: opportunities and uncertainties. Royal Society and Royal Academy of Engineering. 2004, 35-50.
Preparation of cellulose nanofibers Abstract: Nanotechnology is the science of technology, including the receiptand use new nanomaterials, such as nanocellulose. Cellulose nanofibers can be prepared in two different ways: top-down and bottom-up. Currently researchers use the first method, which includes mechanical, chemical and enzymatic treatments. The best results are given by a combination of chemical oxidation and mechanical fragmentation, which allows to obtain nanofibers with a diameter of several nanometers. The most environmentally friendly and prospective is enzymatic hydrolysis, which does not require large money and does not cause production of substances which are difficult to dispose. Nanofibers have many potential applications due to rheological properties such as high Young’s modulus and tensile strength.

Biotechnology and Food Science
Biotechnol Food Sci 2011, 75 (2), 101-102 http://www.bfs.p.lodz.pl
We would like to express our gratitude for the Reviewers of the articles printed in Biotechnology and Food Science Volume 75, 2011.
Wojciech Ambroziak Tadeusz Antczak Jan Iciek Janina Kamińska Józef Kula Joanna Leszczyńska Elżbieta Łodyga-Chruścińska Andrzej Okruszek Magdalena Sikora Beata Smolińska Krzysztof Śmigielski Marianna Turkiewicz Stanisław Wysocki Alicja Zawadzka Dorota Żyżelewicz