biosimilar market: a new dynamic in pharmaceuticals · ©2013 waters corporation 2 “the...
TRANSCRIPT

©2013 Waters Corporation 1
Biosimilar Market:
A New Dynamic in
Pharmaceuticals
Confidential

©2013 Waters Corporation 2
“The development of the biosimilar market will
bring a new dynamic into the Pharma world.…
...with its very specific dynamics, this market
will differ significantly from those for
originator products and for ‘classic’ generics.
In general, biosimilars will be a higher-risk but
also higher-rewarded business compared to
classic generic drugs.”
Accenture Report: “Biosimilars – Emergence of a third market dynamic between original products and generics”
Confidential

©2013 Waters Corporation 3
Agenda
Definitions and Terminology
Regulatory Landscape
Market
– Size and Forecast
– Drivers and resistors
Stepwise Approach required for Biosimilars
Examples of Biosimilar Challenges
Confidential

©2013 Waters Corporation 4
Terminology
‘Bio-generics’
‘Bio-betters’
‘Follow-on Biologics’ ‘2nd Generation mAbs’
‘subsequent-entry
biologicals/biologics’ ‘me-too
biologicals/biologics’
‘noninnovator
proteins’
‘similar
biopharmaceuticals’
Confidential

©2013 Waters Corporation 5
Terminology: the term Biosimilars is now widely adopted
‘Bio-generics’
‘Bio-betters’
‘Follow-on Biologics’ ‘2nd Generation mAbs’
‘subsequent-entry
biologicals/biologics’ ‘me-too
biologicals/biologics’
‘noninnovator
proteins’
‘similar
biopharmaceuticals’
BIOSIMILARS
Confidential

©2013 Waters Corporation 6
Terminology: the term Biosimilars is now widely adopted
‘Bio-generics’
‘Bio-betters’
‘Follow-on Biologics’ ‘2nd Generation mAbs’
‘subsequent-entry
biologicals/biologics’ ‘me-too
biologicals/biologics’
‘noninnovator
proteins’
‘similar
biopharmaceuticals’
BIOSIMILARS
Referred to as Similar Biotherapeutic Product (SBPs)
by WHO
Referred to as Subsequent Entry Biologics (SBPs)
By Canada
Referred to as Biocomparables
By Mexico
Confidential

©2013 Waters Corporation 7
Definitions relating to Biosimilars
Biosimilar or Biosimilarity means:
“the biological product is highly similar to the reference product not
withstanding minor differences in clinically inactive components”
And
“there is no clinically meaningful differences between the
biological product and the reference product in terms of safety,
purity, and potency of the product”
Confidential

©2013 Waters Corporation 8
“Alternative” biologics are NOT Biosimilars
“Non-Comparable biologics” (not highly similar to a Approved Reference Product)– not approved in highly regulated markets– are NOT biosimilars Confidential
©2013 Waters Corporation 9
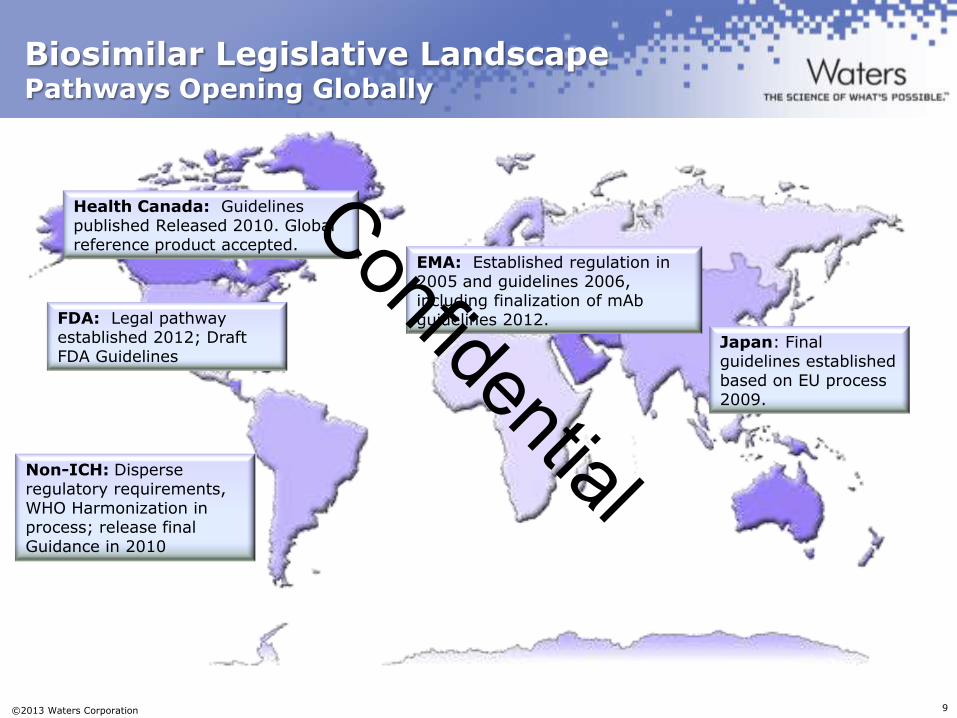
Biosimilar Legislative Landscape Pathways Opening Globally
FDA: Legal pathway established 2012; Draft FDA Guidelines
EMA: Established regulation in 2005 and guidelines 2006, including finalization of mAb guidelines 2012.
Health Canada: Guidelines published Released 2010. Global reference product accepted.
Japan: Final guidelines established based on EU process 2009.
Non-ICH: Disperse regulatory requirements, WHO Harmonization in process; release final Guidance in 2010
Confidential

©2013 Waters Corporation 10
In June 2012 the European Medicines Agency finalized two documents with guidance on how to develop medicines containing monoclonal – Guideline on similar biological medicinal products
containing monoclonal antibodies – non-clinical and clinical issues
– Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use
Public consultation period ran from November 2010 to May 2011 with final versions adopted in May 2012.
Both guidelines in effect on December 1, 2012.
In July 2013, the first two monoclonal antibody biosimilars were approved in Europe – Remsima and Inflectra
– Both contain the same known active substance, infliximab.
Europe and monoclonal antibodies
Confidential

©2013 Waters Corporation 11
WHO Guidelines
“Guidelines on Evaluation of Similar
Biotherapeutic Products (SBPs)” was
developed and adopted by the 60th
meeting of the WHO Expert Committee on
Biological Standardization in 2009.
WHO has published guidelines on the
evaluation of biosimilars in order to
facilitate the global harmonization for both
regulatory bodies and the pharmaceutical
industry
WHO has recommended EU legislation/
guidelines as a WW model
– EU legislation for Biosimilars in place since
early 2000’s, first guidelines adopted in 2006.
Confidential

©2013 Waters Corporation 12
Status of Biosimilar Legislation and Guidance
China: No clear pathway, favoring local products
Hong Kong: No clear pathway
Russia: No guidelines yet
India: Guidelines established and implemented August 15, 2012; alternative pathway for local products
S. Korea: Guidelines established similar to EU Guidelines; alternative pathway for local products
Turkey: Adopted EU Guidelines
Switzerland: Adopted EU Guidelines
New Zealand: Biosimilar to follow EU guidelines
Australia: Adopted overarching Guidelines similar to EU, and adopted product-specific EU Guidelines
Saudi Arabia: Guidelines Established
Malaysia: Guidelines established
Singapore: Guidelines Established
Malaysia: Guidelines established
Argentina Guidance established in 2008
Brazil Guidance established for biosimilars in 2010
Chile Draft Guidance in 2011
Mexico Guidance effective April 2012, called biocomparables
Confidential

©2013 Waters Corporation 13
There are five well recognized principles with regard to the assessment of biosimilar products:
– The generic approach is not appropriate for biosimilars
– Biosimilar products should be similar to the reference in terms of quality, safety, efficacy
– A step-wise comparability approach is required that indicates the similarity of the Biosimilar to Reference Biological Product in terms of quality is a prerequisite for the reduction of non-clinical and clinical data submitted
– The assessment of a biosimilar is based on a case-by-case approach for different classes of products
– The importance of pharmacovigilance is stressed
No significant difference in the general concept and basic principles of all guidelines
If Biosimilar candidates shown to be “highly similar” at the analytical level, this can result in streamline pre-clinical and clinical studies If the product attributes of Biosimilar candidate deviate from the reference, this can increase pre-clinical and clinical studies requirements.
Confidential

©2013 Waters Corporation 14
Comparing EMA and FDA - Similarities
Definition of Biosimilar: Similarity is assessed by structural and functional
comparisons to an approved reference product
Stepwise Approach to Demonstrating Biosimilarity: Starting with detailed
structural characterization of the proposed biosimilar and its reference product,
followed by preclinical then clinical characterizations, makes allowances for
product-specific differences in analytical techniques and clinical endpoints
Confidential

©2013 Waters Corporation 15
EMA Guidance Today
Confidential

©2013 Waters Corporation 16
Comparing EMA and FDA
FDA
– Case –by-case
o 3 very general draft guidelines
• Scientific Considerations in Demonstrating
Biosimilarity to a Reference Product
• Quality Considerations in Demonstrating
Biosimilarity to a Reference Protein Product
• Biosimilars: Questions and Answers Regarding
Implementation ofSp the Biologics Price
Competition and Innovation Act of 2009
– Reference product that was previously
licensed by FDA, but does provide the
provision to use data derived from animal or
clinical studies comparing a proposed
product to a non-US-licensed product.
Adequate data is required to justify
relevance.
EMA
– Class-by-Class
o Guidance for the various classes of
biopharmaceuticals as well as general
guidelines
o Specific guidelines include:
• Recombinant erthroepoetin
• Recombinant human insulin
• Granulocyte-colony stimulating factor
• Interferon
• Monoclonal Antibodies
– No provision for non-EMA licensed
reference products in current guidance
– As of Sept 2012, EMA announced its
intent to revise guidance to accept
biosimilar reference product sourced
outside Europena Economic Area. Agency
expects to release draft for public
consultation in early 2013.
Confidential

©2013 Waters Corporation 17
WHO Guidance on Reference Product
National regulatory bodies have required the use of a
nationally licensed reference product for licensing of
generic medicines.
WHO recognizes that this practice may not be feasible
for countries lacking nationally-licensed Reference
Biological Products (RBPs).
WHO provides considerations for choosing a RBP
Note: Only Canada explicitly allows the use of a non-Canadian
reference product.
Confidential

©2013 Waters Corporation 18
FDA
– Interchangeability means automatic substitution which refers to the practice of dispensing one medicine instead of another equivalent and interchangeable medicine at the pharmacy level without requiring consultation with the prescriber.
– FDA did not cover the issue in its recently issued draft guidances – except in the Q&A document:
Q. I.14. Can an applicant obtain a determination of interchangeability
between its proposed product and the reference product in an original 351(k) application?
A. I.14. (Proposed Answer): Yes. Under the BPCI Act, FDA can make a determination of interchangeability in a 351(k) application or any supplement to a 351(k) application. An interchangeable product must be shown to be biosimilar to the reference product and meet the other standards described in section 351(k)(4) of the PHS Act. At this time, it would be difficult as a scientific matter for a prospective biosimilar applicant to establish interchangeability in an original 351(k) application given the statutory standard for interchangeability and the sequential nature of that assessment. FDA is continuing to consider the type of information sufficient to enable FDA to determine that a biological product is interchangeable with the reference product.
Automatic interchangeability cannot be applied to all biosimilars in the US. Have to meet the higher standard of interchangeability
EMA
– Interchangeability refers to the
“doctor’s ability to prescribe a
biosimilar in place of the reference
product”
– Substitutability is to allow
“pharmacists to dispense a
biosimilar rather than the reference
drug”.
o Substitutability is clearly a more
contentious issue
EMA has been silent about
interchangeability, but has made
statements opposing the idea of
automatic substitution of biosimilars
(for example, at the pharmacy level)
Comparing EMA and FDA
Confidential

©2013 Waters Corporation 19
“Interchangeability”
The debate on substitutability and interchangeability of ultimately addresses the question of how similar biosimilars are or should be in order to qualify for substitutability and interchangeability
The main parties and stakeholders in this debate are
– Originators
– Generics/biosimilars industries and their respective trade associations
– Drug regulatory agencies & WHO
– Insurers/payers
– Doctors & pharmacists
– Patients
A consequence of this is that companies will need to invest significantly in marketing to assure that their biosimilars are used
Confidential

©2013 Waters Corporation 20
Comparing EMA and FDA
FDA
– Requirement for Safety and Efficacy Trials: “As a scientific matter, comparative safety and
effectiveness data will be necessary to support a
demonstration of biosimilarity if there are residual
uncertainties about the biosimilarity of the two
products based on structural and functional
characterization, animal testing, human PK and
PD data, and clinical immunogenicity
assessment. A sponsor may provide a scientific
justification if it believes that some or all of the
these comparisons on clinical safety or effectiveness
are not necessary.”
– Exclusivity Period:
o A section (k) application may not be filed until 4
years after reference product approval.
o A biosimilar may not be approved until 12 years
after reference product approval”
EMA
– Requirement for Safety and Efficacy
Trials: “Usually comparative clinical trials will
be necessary to demonstrate clinical
comparability between the biosimilar and the
reference medicinal product”
– Exclusivity Period:”8 +2+1:
o A biosimilar application may not be filed until 8
years after the reference product approval.
o A biosimilar may not be approved until 10
years after reference approval.
o The market exclusivity may be extended by an
additional year if the reference product sponsor
obtains approval for a second significant
indication during the data exclusivity period.
Confidential

©2013 Waters Corporation 21
International Non-proprietary Name (INN) and Biosimilars in Europe
The European Commission’s Directive 2012/52/EU of 20 December 2012 – Requires that Member States of the EU recognize medical prescriptions issued in
other Member States
– To enable this recognition, the Directive states that ‘in order to enable the correct identification of products’ the INN or common name should be used.
However, in the case of biological products, the Directive makes an exception – In contrast to small-molecule drugs, biological products are required to
use the brand name.
– This is stated in the Directive as necessary in order ‘to ensure clear identification’ of the biological product because of the ‘special characteristics’ of these products.
Biosimilars manufacturers would of course prefer that biological products were prescribed by INN – Allows for possible substitution in the future
– Enables biosimilars to grab a bigger market share.
– Prescribing by brand requires much more intensive marketing by biosimilars manufacturers in order to increase brand awareness and encourage doctors to prescribe and patients to request biosimilars.
Source: Europa
Confidential

©2013 Waters Corporation 22
Biologics Market
Sales of biologicals have almost doubled from
US$63.8 billion in 2006 to US$124.6 billion in
2012.
The global biologics market ~$150B today
and expected to be $252B by 2017
By 2014, 7 out of 10 of the top brands will be
biologics
Majority of companies have biologics in their
pipeline
Many biologics are focused on unmet needs
PhRMA Report Medicines in Development Biotechnology2011
Confidential

©2013 Waters Corporation 23
EU vs US Patent Expiry Dates
Sources: http://www.gabionline.net/Biosimilars/Research/US-54-billion-worth-of-biosimilar-patents-expiring-before-2020 http://www.genengnews.com/insight-and-intelligenceand153/biosimilars-10-drugs-to-watch/77899804/?page=1 http://www.terripinn.com/biosimilar
Confidential

©2013 Waters Corporation 24
Expiry dates for major patents on best-selling biologicals
Confidential

©2013 Waters Corporation 25
Bisimilars in the US 2013
Guidance’s are not final
Abbott’s citizens petition to the FDA
States’ decisions for substitution – Legislation which permits pharmacists to dispense a biosimilar that has been
licensed by FDA as interchangeable
– With the following provisions:
o Unless the prescriber indicates such substitution is not authorized or the patient
insists on dispensing of the prescribed biological product.
o Requires any pharmacist who dispenses an interchangeable biosimilar to inform
the patient prior to dispensing the biosimilar, provide notification of the
substitution to the prescriber (Notification can range for 24 hr to 10 days)
o Requires records of the brand name or the product name and name of the
manufacturer of the biosimilar on the record of dispensing and the prescription
label (records can range from 2-10 years)
– To date of the 17 states that have considered this bill, only North Dakota has
passed this intact, and Virginia and Utah with subset clauses. For 6 the decision is
still pending, 2 are reviewing further, 6 have rejected the provisions
Confidential
©2013 Waters Corporation 26
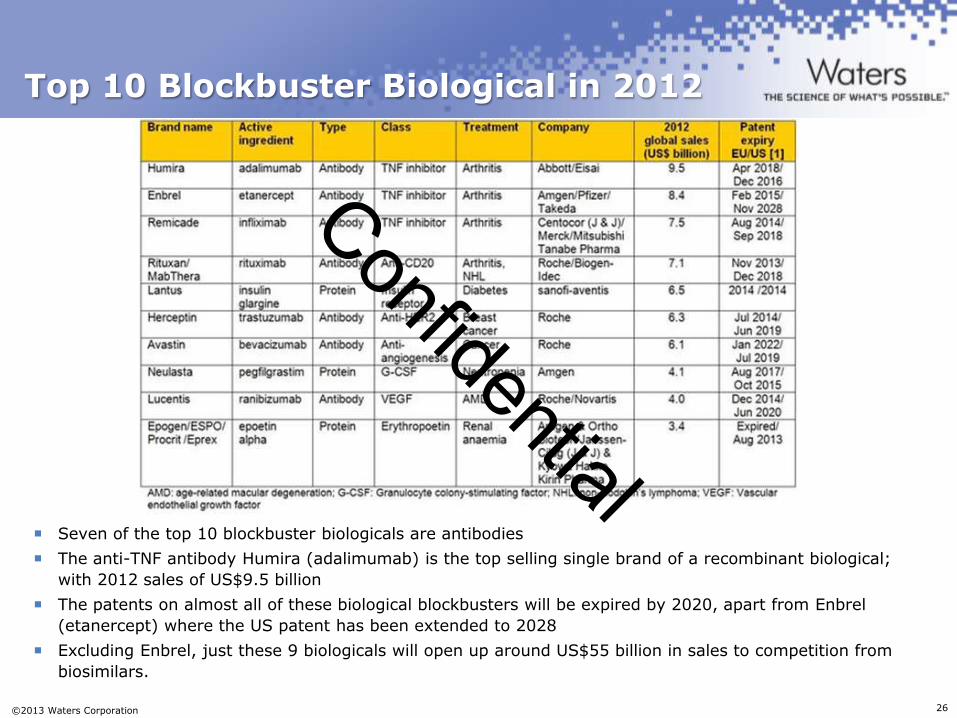
Top 10 Blockbuster Biological in 2012
Seven of the top 10 blockbuster biologicals are antibodies
The anti-TNF antibody Humira (adalimumab) is the top selling single brand of a recombinant biological;
with 2012 sales of US$9.5 billion
The patents on almost all of these biological blockbusters will be expired by 2020, apart from Enbrel
(etanercept) where the US patent has been extended to 2028
Excluding Enbrel, just these 9 biologicals will open up around US$55 billion in sales to competition from
biosimilars.
Confidential

©2013 Waters Corporation 27
Biosimilars Market Forecast 2010 to 2017
In 2010, the combined biosimilars market size for the US and five major
European markets was $172M
In 2015, the market should be between $2B and $4.8B
– Datamonitor estimated that market will grow from $243m in 2010 to $3.7bn
in 2015. *
– $1.9-2.0B by 2015**
– $4.8B***
By 2017, US$3,987 million****
Annual growth rate of 56.7% expected from 2010 to 2017
*Datamonitor ** IMS Health, December 2011 *** Global Industrial Analysts **** Frost & Sullivan
Confidential

©2013 Waters Corporation 28
Market Drivers and Resistors
Economic Drivers: The governments and/or payers have to pay more
every day for medicines when they are trying to control their budgets
– Poor economy
– Health budget reductions and profit margins
– Biosimilar price around 70-80% of orginator
Legal and Commercial Drivers: Patent expiries is the most critical issue
in biosimilar development. Upcoming patent expiries of monoclonal antibody
drugs will boost biosimilar sales in US and Europe
– Patent expiry
– Uncontested sub-classes of biologics with expired patents
– Co-development and co-commercialization opportunities
Confidential

©2013 Waters Corporation 29
Cost of Biopharmaceutical Treatment Today
Confidential

©2013 Waters Corporation 30
Market Drivers and Resistors
Clinicial Drivers: Although the topic is clinical, the benefits are related to
economic policy of biosimilars – cheaper medicines will be used more widely
and will help more people to see the benefits
– Lower cost may allow long-term treatment which can potentially translate in
improved survival, quality of life and disability outcomes
– Biologics might be increasingly used as initial treatment
Scientific and Regulatory Drivers: Decades of scientific advances since
the first biologic and the efforts of the regulatory bodies to provide guidance will
be a benefit to biosimilar manufacturers
– Advances in analytical techniques
– Regulatory experience with approvals and interactions with the industry
– WHO guideline and US biosimilar legislation now in place
Confidential

©2013 Waters Corporation 31
Market Drivers and Resistors
Commercial Resistors: Biotechnological manufacturing is definitely more
difficult and expensive than that for small molecule drug. There will be high
level of competitions with so many players in this market.
– Requires strong manufacturing capabilities
– Outsourced manufacturing can be accompanied with greater risk and
uncertainties over price flexibility
– Opposition from innovators
– Competitions (over 150 biosimilar players globally)
– Cost of clinical studies may be higher than originally anticipated
– No experience in branded products
Confidential

©2013 Waters Corporation 32
Key players in Biosimilars
Confidential

©2013 Waters Corporation 33
Market Drivers and Resistors
Legal Resistors: Several innovators are interest in biosimilar business and
may try to control market. Interchangeability, substitution, exclusivity to the first biosimilar, etc are still unclear
– Unclear exclusivity
– Lack of time on the market
– Threat of acquisition by innovators or larger generic companies
Clinical and Regulatory Resistors: Perception and acceptance regarding
Biosimilars among prescribers and patients is not the same in every country. And originators are development new biologics to change the treatment regime is a threat to biosimilars
– Uncertainty over long-term safety risks
– Poor level of understanding from prescribers and patients
– Regulatory and pharmacovigilance burden
– Lack of regulatory consistency and globally harmonized approach
– Potential changes in treatment paradigm
Confidential

©2013 Waters Corporation 34
High-level Overview – Comparison of generics, biosimilars and biologics (Source Accenture Research)
Confidential

©2013 Waters Corporation 35
Complexity
Confidential

©2013 Waters Corporation 36
Amino Acid Substitution
N- and C-terminal modifications
Mismatched S-S bonds
Folding
Truncation
Aggregation
Multimer Dissociation
Denaturation
Acetylation
Fatty acylation
Deamidation
Oxidation
Carbamylation
Carboxylation
Formylation
γ- Carboxyglutamylation
O-linked Glycosylation
N-linked Glycosylation
Methylation
Phosphorylation
Sulphation
PEGylation
Protein Heterogeneity
Confidential

©2013 Waters Corporation 37
Proteins Modifications Impact different proteins differently
e.g., Methionine oxidation inhibits activity of some protein, but
not others
e.g., Deamidation may increase immunogenicity in one protein
but not another
Confidential

©2013 Waters Corporation 38
Which attributes matter and which don’t?
Understanding relationship between protein
attributes and clinical safety & efficacy profile
aids ability to predict clinical similarity
Confidential

©2013 Waters Corporation 39
More Analytical Data Required
CMC Strategy Forum 2012, E. Shacter
Confidential

©2013 Waters Corporation 40
Structural Analysis
Generally, such tests include the following comparisons of the
drug substances of the proposed product and reference product:
– Primary structures, such as amino acid sequence
– Higher order structures, including secondary, tertiary, and
quaternary structure (including aggregation)
– Enzymatic post-translational modifications, such as glycosylation and
phosphorylation
– Other potential variants, such as protein deamidation and oxidation
– Intentional chemical modifications, such as PEGylation sites and
characteristics
Guidance for Industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER)
Confidential

©2013 Waters Corporation 41
Functional Analysis
The pharmacologic activity of protein products can be evaluated by in vitro and/or in vivo functional assays. These assays may include, but are not limited to, – Bioassays
– Biological assays
– Binding assays
– Enzyme kinetics.
A functional evaluation comparing a proposed product to the reference product are used to: – Support ”that there are no clinically meaningful differences between the proposed
product and the reference product”
– Scientifically justify a selective and targeted approach to animal and/or clinical testing
– Provide additional evidence that the MOA of the two products is the same to the extent the MOA of the reference product is known.
– Provide additional data to support results from structural analysis
– Investigate the consequences of observed structural differences, and explore structure activity relationships.
Confidential

©2013 Waters Corporation 42
Bioequivalence
The regulatory agencies realize that testing the bioequivalence of biosimilars differs from that of the standard generics.
Small Molecule Drugs – Testing is required for orally ingested products because they maybe absorbed into
the bloodstream at different rates or in different amounts
– Generic drug manufacturers to prove their formulation exhibits bioequivalence to the innovator product
– FDA requires the bioequivalence of the generic product to be between 80% and 125% of that of the innovator product
Biologics – Route of administration is usually via injection directly into bloodstream
o Two injected products are generally bioequivalent in terms of bioavailability
– But they may behave differently once in the body
o The manufacturers of biosimilars must fulfill the quality, efficacy and safety requirements
o The bioequivalence testing procedures for biosimilars are to be performed against the originator product as a control and include preclinical and clinical testing
o Due to varying nature of the biopharmaceutical drugs, many of the concepts discussed in these guidelines may have to be adapted on a case-by-case basis.
Confidential

©2013 Waters Corporation 43
General comparison of requirement
Confidential

©2013 Waters Corporation 44
General comparison of requirement
Confidential

©2013 Waters Corporation 45
Rapid Comparison of a Biosimilar candidate to an innovator
mAbs 2:4, 1-16; July/August 2010; © 2010 Landes Bioscience Confidential

©2013 Waters Corporation 46
Collaboration
Company X is recognized for a proven track record in generic
small molecule pharmaceuticals.
They wished to expand to produce biosimilar antibodies.
They collaborated with Waters to exploit UPLC and TofMS
technologies to support this new effort.
They were expecting only minor differences from the
innovator company mAb.
Graphic: http://en.wikipedia.org/wiki/File:Antibody2.JPG
= ?
Confidential

©2013 Waters Corporation 47
Qtof in Biopharma
QTof
– Accurate mass of precursor and product ions
– Ability to quantitate (4 orders in-spectral range)
– Comprehensive CID fragmentation
– Ability to predict fragmentation patterns and automate processing
For Biologics:
– Complexity of samples and high degree of ambiguity needs more 'power'....
– Need for comprehensive characterization (TOF: 'unlimited' mass range; same platform for intact and peptides
– Solves complex problems ('automatically', if MS^E and processing is applied)
MSe
– Ability to apply a generic method without pre-knowledge
– Comprehensive fragmentation and quantification without precursor isolation
– No bias due to abundance (unlike DDA)
– Advantages of processing MS^E info automatically
– Saves time
Confidential

©2013 Waters Corporation 48
Deconvoluted Spectra of the Heavy Chain
Biosimilar Heavy chains shifted -32 Da consistent with intact data
Innovator mAb
“Biosimilar” mAb
Confidential

©2013 Waters Corporation 49
MIRROR MAP shows Difference Due to Unknown Peak in Biosimilar Sample
New, Unknown Peak in BIOSIMILAR sample at m/z 1872.96
Changes clear in mirror plot
Peptide T34-35 (missed cleavage) confirmed in INNOVATOR with MSE
Significant Differences are visually salient
INNOVATOR (chromatogram)
BIOSIMILAR (chromatogram)
INNOVATOR (spectrum)
BIOSIMILAR (spectrum)
Confidential

©2013 Waters Corporation 50
Sorting out the T35 differences
Drugbank Herceptin™ HC T35 sequence is DELTK
– Drugbank database entry DB00072
– www.drugbank.ca/drugs/DB00072
Genentech Herceptin™ HC T35 sequence is EEMTK
– Mass is 32 Da heavier.
– Consistent with the marketed product
– REF: Journal of Chromatography B, 752 (2001) 233–245; Identification of multiple
sources of charge heterogeneity in a recombinant antibody. Harris et al.
Result: Incorrect protein sequence (Asian allotype) was cloned and expressed by Company X. Impact: The biosimilar candidate is not the same protein as the innovator and would be considered a new drug. $$$
Confidential

©2013 Waters Corporation 51
The Importance of High Order Structure for Biosimilars – An
example of a Failed Submission
“…it appears as if the Marvel
insulin tends to be more rapidly
absorbed than the reference
insulin…. One reason for these
results may be that the three-
dimensional structure of the
two insulins were not
identical and that, subsequently,
the forces that keep the insulin
monomers together as an insulin
hexamer … are weaker with
Marvel insulin than with other
[insulins]”.
Confidential

©2013 Waters Corporation 52
Waters and Japanese Pharmaceuticals and
Medical Devices Agency (PMDA)Collaboration - Insulin Higher Order Structure
Produced for ASMS 2012 – study helped understand the
dissociative events of insulin oligomers
We determined two particular regions which exhibited higher
deuterium uptake in Lispro, which are likely to be involved in
hexamerization and dimerization
Confidential

©2013 Waters Corporation 53
A Real Biotech Example – Genzyme
Genzyme receives PDUFA date from FDA for its Biologics License Application for Lumizyme - 21. January 2010 23:52 http://www.news-medical.net/news/20100121/Genzyme-receives-PDUFA-date-from-FDA-for-its-Biologics-License-Application-for-Lumizyme.aspx
Change of reactor led to Change in Glycosylation Pattern
160L Reactor
4000L Reactor
4000L Reactor
Glycosylation Pattern B Glycosylation
Pattern A
160 L Reactor
Confidential

©2013 Waters Corporation 54
A Real Biotech Story – Genzyme (now Sanofi)
Genzyme receives PDUFA date from FDA for its Biologics License Application for Lumizyme - 21. January 2010 23:52 http://www.news-medical.net/news/20100121/Genzyme-receives-PDUFA-date-from-FDA-for-its-Biologics-License-Application-for-Lumizyme.aspx
Biotech company had to re-submit as a new product
Glycosylation B Not COMPARABLE to Glycosylation A
US: New Clinical Trials
US: Resubmission for New Product
(No guidelines were in place)
EU: (fast) Submission as Biosimilar
EU: Limited Clinical Trials
4000L Reactor
Glycosylation Pattern B
Glycosylation Pattern A
160 L Reactor
ESTIMATED COST > 40 M USD
Confidential

©2013 Waters Corporation 55
Implications for Biosimilars
How much “similarity” do we
need?
– The requirement to understand
relationship between protein
attributes and clinical safety &
efficacy profile aids ability to
predict clinical similarity
– Know which attributes matter and
which don’t
Thus the Stepwise and Totality of
the Evidence Approaches
– Combine rigorous physicochemical
data with functional studies
– Case-by-case evaluation of
potential clinical impact
Confidential

©2013 Waters Corporation 56
Portfolio of Solutions for Structural Analysis
UPLC
– Amino Acid Analysis
– Peptide Mapping
– Released Glycan Analysis
– Intact RP
– Intact SEC
– Intact IEX
UPLC/MS
– Peptide mapping
– Glycan Analysis
– Intact Analysis
– HDX
– HCPs
Confidential

©2013 Waters Corporation 57
Confidential