bispecific antibodies for cancer therapy - freepchames.free.fr/chames and baty co ddd 2009.pdf · 2...
TRANSCRIPT

1
Current Opinion in Drug Discovery & Development 2009 12(2):
© Thomson Reuters (Scientific) Ltd ISSN 1367-6733
Bispecific antibodies for cancer therapy
Patrick Chames* & Daniel Baty
Address
Institut de Biologie Structurale et Microbiologie, Laboratoire d'Ingénierie des Systèmes Macromoléculaires,
CNRS UPR 9027, GDR 2352, 31 chemin Joseph Aiguier, 13402 Marseille Cedex 20, France
Email: [email protected]
*To whom correspondence should be addressed
The 2 last decades have seen the emergence of monoclonal antibodies as therapeutics. Nine monoclonal
antibodies have been approved for cancer therapy. However the efficiency of these molecules is far from
optimal, and antibody engineering is actively used to improve them. Because of their ability to simultaneously
bind two different antigens, bispecific antibodies are unique, and their wide potential as retargeting reagent has
been demonstrated over the years. However their use as therapeutics has been restrained by manufacturing
challenges. Several new recombinant formats have changed the situation. Innovative molecules have led to
impressive preclinical and clinical results, and hold great promise. This review presents an overview of the most
promising candidates.
Keywords Bispecific, cancer therapy, immunotherapy, retargeting, single domain antibodies
Abbreviations
ADCC antibody-dependent cell-mediated cytotoxicity, bsAb bispecific antibody, BiTE bispecific T-cell engager,
dAb domain antibody, Db diabody, DC dendritic cell, FcR Fc receptor, IFN interferon, Ig immunoglobulin, IL
interleukin, PBMC peripheral blood mononuclear cell, scFv single chain Fv, TaFv tandem scFv

2
Introduction
Monoclonal antibodies (mAbs) are endowed with exquisite specificities, and since their discovery in 1975, they
have raised many expectations for the development of novel treatments, particularly in cancer therapy [1].
However, all of these molecules had to be extensively optimized and engineered before they were able to
deliver approved therapeutic molecules [2]. Nine mAbs are currently approved for cancer therapy, and
hundreds are at different stages of clinical development [3]. Despite the enthusiasm for developing mAbs for
therapy, most mAbs show limited efficacy, and so there is great potential for further improvement in this
research area. Most mAbs function either by directly blocking a ligand or a receptor, sometimes leading to
apoptosis, or as an adaptor molecule capable of recruiting effector cells from the immune system through
interaction between their Fc portion and various Fc receptor (FcR) bearing cells. Consequently, considerable
efforts are spent in the optimization of the Fc/FcR interaction [4].
Bispecific antibodies: Concepts, formats and limitations
However, as hypothesized in 1985, one solution for many mAb shortcomings might be the creation of bispecific
antibodies (bsAbs), which would be capable of the simultaneous binding of two different targets and thus also
capable of delivering a great variety of payloads to cancer cells [5]. BsAbs, however, do not exist in nature.
Despite numerous attempts and various proposed formats, the production BsAbs in significant quantities
remains, to a certain extent, a challenge. Three main approaches to the large-scale production of bsAbs have
been described. The first was the quadroma technology, involving a somatic fusion of two different hybridoma
cell lines [6]. Because of the random pairing of the two heavy and two lights chains of the antibody, the use of
sophisticated purification procedures to isolate the bsAb was required. This inefficient, time- and resource-
consuming approach led to the production of small amounts of bsAb that, although were sufficient for
characterization, were insufficient for clinical requirements. Numerous studies have demonstrated the potential
of such bsAbs but have also highlighted important shortcomings, including the high immunogenicity of such
murine molecules and a high toxicity, which was attributed to the presence of an Fc portion capable of
stimulating various FcR-bearing cells [7]. As a consequence, quadromas are no longer favored, with one
notable exception: Lindhofer et al demonstrated that the fusion of a murine immunoglobulin (Ig)G2a-producing
hybridoma and a rat IgG2b-producing hybridoma may lead to preferential heavy/light chain pairing and easy
separation of the homo- and heterodimers of heavy chains [8]. This approach can thus efficiently produce
bispecific forms of whole IgG. Moreover, the resulting hybrid Fc portion of the resulting antibody has been
shown to efficiently bind to activating human FcRs [9]. These molecules, called triomabs, are thus considered to
be trispecific, and have produced exciting results in clinical trials (see below) [10].
The second approach that was developed to produce bsAbs is based on the use of heterobifunctional chemical
reagents. The three major problems associated with chemical cross-linking are: (i) product homogeneity,

3
leading to poor yields and complicated purification steps; (ii) possible inactivation of the molecules or poor
stability; and (iii) toxicity associated with the presence of Fc portions. Nevertheless, such molecules, such as
CD20Bi and Her2Bi, have been evaluated in the clinic [11]. To decrease toxicity, F(ab') can be produced by
enzymatic digestion and mild reduction of the two parental IgGs, followed by heterodimerization via thiol-
reactive heterobifunctional reagents. Such artificial bispecific F(ab')2 have been extensively studied, and some
molecules have reached the clinic, although none of them have been approved so far. [12].
The implementation of recombinant antibody techniques has revolutionized the bsAb field, leading to a third
approach. More precisely the ability to use single-chain FV (scFv) fragments (antibody variable domains linked
by a flexible peptide linker) has been a source of inspiration [13]. Two formats have been intensively studied.
Tandem scFvs (TaFv) consist of two scFv fragments linked via an extra peptide linker. This 50-kDa molecule,
which is difficult to produce in Escherichia coli, is well expressed by mammalian cells and is expected to confer
a good flexibility to each scFv fragment. This format has been used to create bispecific T-cell engager molecules
(BiTE), which are extremely potent bsAbs that have shown impressive results in clinical trials [14].
By reducing the length of the peptide linker between variable domains so that the domains cannot assemble, it
is possible to force the pairing of domains from two different polypeptides, leading to compact bsAbs called
diabodies (Dbs) [15]. These molecules can be expressed at high yields in bacteria, and have been shown by
crystallography experiments to adopt several conformations [16]. The Db format has been improved further by
the addition of an extra peptide linker between the two polypeptides in order to further decrease the number of
homodimers formed, leading to a fragment called a single chain diabody (scDb) [1]. Numerous studies have
demonstrated the potency of these formats, although the reduced flexibility of the two binding sites might
prove to be a limitation in some cases.
In addition to TaFv and Db, several other scFv-based formats have been proposed which use various
heterodimerization motifs including the human CH1/Cκ domains and CH3 domain pairs that have been elegantly
engineered to favor heterodimerization [17]. More recently, an approach called Dock-and-Lock was used to
create trivalent bispecific molecules [18]; this method relies on the natural interaction between the 44 C-
terminal residues of the regulatory unit of human protein kinase A, which spontaneously dimerize and bind to a
17-residue motif derived from the A-kinase anchoring protein. This trimerization motif has been stabilized by
the introduction of cysteine residues into both domains, leading to the formation of covalent disulfide bonds.
Such 150-kDa trivalent molecules have been successfully used for pretargeting strategies (see below) [18].
Recently, a new format consisting of fusions of an extra variable domain to each N-terminus of an IgG (ie, VH1-
VH2-CH1-Fc and VL1-VL2-CL) was described [19]. The resulting bispecific molecules, called dual-variable-
domain antibodies, have been shown to fold correctly and to exhibit pharmacokinetics similar to those of the

4
parental mAbs [19]. Steric constraints have to be expected for one of the binding sites, although this antibody
format has been shown to successfully target soluble targets.
Finally, recent years have seen the emergence of domain antibodies (dAbs), which were first engineered from
mouse VH domains [20], and later from human variable domains [21]. DAbs are also found in nature in some
isotypes of camelids and sharks [22,23]. These fragments are compact, extremely stable, and do not need
domain pairing, and so represent ideal building units for the generation of more complex molecules including
multispecific or multivalent molecules. For example, bispecific tandem dAbs can be produced efficiently using
flexible linkers and such molecules have demonstrated excellent production yields and stability [24].
Interestingly, their modularity has been used to create a linker-free bsAb by directly fusing two llama-derived
dAbs to a human CH1/Cκ heterodimerization motif, thereby creating a Fab-like bispecific antibody fragment
termed bsFab [Cornillon, A. et al, unpublished]. These bispecific molecules are easily produced, extremely
stable, and do not suffer from the usual limitations experienced by linker-based molecules, including production
issues, poor stability, aggregation that can favor immunogenicity, and proteolytic degradation.
Pharmacokinetics of bispecific antibodies
Several of the methods for producing bsAbs described above generate bispecific antibody fragments of a small
size (eg, 30 kDa for TaFv). Although a small size is clearly an advantage in terms of tumor penetration, it is
also a potential problem because small molecules administered through intravenous injection are rapidly
eliminated from the circulation by renal clearance. Moreover, several bsAb formats do not possess an Fc
portion, and thus do not bind to the neonatal Fc receptor (FcRn), which is a receptor expressed by endothelial
cells and responsible for the very long serum half-life of IgGs [25].
Several solutions have been designed to overcome the rapid clearance of of bsAbs. Addition of PEG to bsAbs
has been shown to improve the serum half-life [26], but this often alters the binding affinities of bsAbs [27],
which might affect tumor targeting. Two other methodologies designed to reduce the clearance of bsAbs are
based on the interaction between human serum albumin (HSA) and FcRn. HSA, as is the case with IgG, enables
recycling of the bsAb by endocytosis, thereby leading to reduced renal clearance and extended serum half-life.
Indeed, a direct fusion of a bsAb to HSA can significantly improve its kinetics, as has been demonstrated for
anti-CEA x CD3 Dbs [28]. The fusion of a Fab fragment to HSA-binding peptides has been shown to increase
the serum half-life of the Fab fragment by 10- to 15-fold, leading to superior in vivo efficacy of the fragment
compared to the full IgG [29]. This general strategy has not yet been applied to bsAb but should be applicable
with success to TaFv and Db. Similarly, small dAbs (llama and human) that bind strongly to serum albumin
have been isolated. These dAbs have an extended serum half-life matching that of serum albumin itself [30],
and can be fused to other antibody fragments to create bsAbs with favorable kinetics [31].

5
New strategies enabled by bispecific antibodies
(Sub) Dual targeting
The inhibition of signaling pathways is one of the main modes of action of therapeutic antibodies. Combinations
of small-molecule-based treatments can lead to additive or even synergistic effects [32], block redundant
signaling pathways and minimize the possibility of escape from therapy, but are often not adopted in the clinic
because of issues with high toxicity. However, human or humanized mAbs are often very well tolerated and are
thus very good candidates for combination therapies. Consequently, several companies are actively testing
combinations of mAbs, including Genentech Inc with trastuzumab (anti-Her2) plus bevacizumab (anti-VEGF),
and Immunomedics Inc with epratuzumab (anti-CD22) plus rituximab (anti-CD20) [33]. However, the
combination of therapeutic antibodies faces several limitations, including intellectual property issues and the
high costs involved in research, manufacturing and regulatory affairs. An elegant answer to these difficulties
might be the simultaneous inactivation of two targets by a single (bispecific) antibody, as exemplified by two
studies [34,35] that successfully simultaneously targeted epidermal growth factor (EGF) and insulin-like growth
factor (IGF) receptors, demonstrating that simultaneous targeting is more efficient than monotherapies against
the same targets.
(Sub) T-cell retargeting
Perhaps the most obvious applications of bsAb are in T-cell retargeting. Cytotoxic T-cells are considered the
most potent killer cells in the immune system. These T-cells are abundant, can efficiently proliferate upon
activation, can kill multiple times [36], and can efficiently infiltrate tumors but do not express Fcγ receptors.
The concept of applying the novel bsAb methodology to enable T-cells to kill tumor cells more potently emerged
in the 1980s [5]. BsAbs directed against both a tumor marker and CD3 have the potential to redirect and
activate any circulating T-cells against tumor cells. However, T-cells have a major drawback. Without a
secondary signal provided by the interaction between CD28 and one of its ligands, such as B7, T-cells are not
fully activated and may even become anergic [37]. The first anti-CD3 bsAbs were therefore applied in
combination with anti-CD28 antibodies, leading to mixed results [38]. Other alternatives are being explored,
such as the massive ex vivo expansion (> 300 x 109) and activation of patient's T-cells using low concentrations
of anti-CD3 and interleukin (IL)-2, followed by reinjection of these polyclonally activated T-cells decorated with
an anti-CD3 bsAb [11]. Interestingly, activated T-cells armed with Her2/neu bsAbs have been demonstrated to
be resistant to activation-induced cell death and are able to proliferate and kill tumor cells repeatedly [39].
Surprisingly, a small recombinant bsAb format appears to bypass the need for costimulation. Baeuerle and
collaborators are developing a murine TaFv BiTE [40]. An anti-CD19 x CD3 BiTE exhibited very efficient

6
inhibition of tumor growth in subcutaneous leukemic human B-cell lymphoma xenograft models at low
concentrations and low effector-to-target ratios [41]. Similar impressive results, including the eradication of
tumors from both a human colon cancer cell line and ovarian cancer metastasis in immunodeficient mice, have
been obtained with an anti-EpCAM x CD3 BiTE [42]. In immunocompetent mice, a BiTE directed against the
murine CD3 led to a strong antitumor effect in the absence of costimulation [43]. The proposed explanation for
the outstanding efficacy of this BiTE in the absence of costimulation involves the highly efficient formation of
immunologic synapses due to the very short linker (five residues) connecting the two scFv fragments [44]. In
the last year, the significant potential of these molecules has been clearly demonstrated in clinical trials [14].
Blinatumomab (MT-103, Micromet Inc/MedImmune), an anti-CD3 x CD19 BiTE, was shown to induce partial and
complete regression in non–Hodgkin's lymphoma patients at extremely low doses (15 µg/m²/day) equating to
approximately five orders of magnitude below the serum levels reported for efficacious amounts of rituximab
(375 mg/m²/week). All of the seven patients treated with 60 µg/m²/day of blinatumomab experienced tumor
regression. A clinical trial targeting solid tumors has also been initiated by Micromet with another BiTE, MT-110
(anti-EpCAM x CD3) [45].
However, T-cell stimulation via CD28 has been shown to be highly efficient and self sufficient. rM28, a TaFv
dimer directed to a melanoma-associated proteoglycan (NG2) and to the costimulatory CD28 molecule on
human T-cells induced pronounced T-cell activation in peripheral blood mononuclear cell (PBMC) preparations
without additional T-cell receptor (TCR)/CD3 stimulation being required via supra-agonistic T-cell activation.
Interestingly, cytokines produced by CD28-stimulated T-cells were demonstrated to activate natural killer (NK)
cells, which subsequently significantly contribute to tumor cell lysis [46,47]. The safety and efficiency of
intralesional application of the murine bispecific TaFv rM28 and autologous PBMCs in patients with stage III/IV
metastatic melanoma and unresectable metastasis are currently being studied in a phase I/II clinical trial.
Perhaps the most unexpected bsAb successes have been the rat/mouse chimeric full IgG anti-CD3 bsAbs, which
are named triomabs and were designed by Lindhofer and collaborators. The resulting chimeric Fc portion of the
triomabs was shown to efficiently interact with activating human FcRs (FcγRI and FcγRIII) but not inhibitory
FcRs (FcγRIIB), thereby achieving the goal of several academic and industrial laboratories using human Fc
engineering. Furthermore, triomabs were shown to be capable of activating dendritic cells (DCs), inducing NK-
dependent antibody-dependent cell-mediated cytotoxicity (ADCC), and stimulating tumor cell phagocytosis by
macrophages [9,48]. Consequently, the chimeric Fc adds two crucial functions to the anti-CD3 x target bsAb:
additive tumor killing capabilities through the recruitment of macrophages and NK cells, and, most importantly,
the efficient costimulation of T-cells through direct contact with accessory cells such as macrophages and DCs
(B7/CD28, CD40/CD40L, LFA3/CD2) or cytokine secretion (IL-2, IL-6, IL-12). The only limitation of these
exciting bsAb molecules resides in their immunogenicity, which precludes repetitive injection of these bsAbs in
large quantities. Nevertheless, triomabs have exhibited impressive preclinical and clinical results, with

7
reductions of up to 5 log in the number of tumor cells in ovarian carcinoma patients, despite the use of low
doses of bsAb to prevent reversible adverse effects [49]. Three of these chimeric bsAb molecules,
catumaxomab (anti EpCAM x CD3), ertumaxomab (anti Her2 x CD3) and FBT-A05 (Lymphomun; anti CD20 x
CD3), are under development by Fresenius Biotech GmbH and under preregistration (catumaxomab) or in
phase II clinical trials (phase both ertumaxomab and FBT-A05).
(Sub) FcγR-bearing cell retargeting
Although T-cells are very attractive effector cells, NK cells have also been shown to be very efficient at
destroying tumor cells. ADCC is thought to be one of the main modes of action of therapeutic antibodies, as has
been demonstrated by the association of functionally relevant FcγRIIIA polymorphisms with clinical responses to
rituximab [50]. NK cells are known to mediate essentially all of the ADCC measured in vitro, using PBMCs as
effector cells. Of note is the fact that neutrophils, the most numerous leukocyte type, express FcγRIIIB (which is
extremely homologous to FcγRIIIA), but cannot be activated to induce ADCC through this receptor. In vitro,
neutrophils do not interfere with ADCC efficiency [51]. NK cells have the advantage over T-cells in that pre-
activation is not required, because NK cells constitutively exhibit cytolytic functions [52]. However, NK cells
represent less than 10% of lymphocytes, and very small numbers of NK cells are found in direct contact with
tumor cells, suggesting that NK cells poorly infiltrate tumors [13]. Many ongoing studies aim to optimize NK cell
killing through engineering of the Fc/FcγRIIIA interaction [4]. Indeed, therapeutic antibodies suffer from several
limitations, not only the influence of FcγR polymorphisms as described earlier, but also the influence of the
glycans that are post-translationally added to the IgG CH2 domain, competition with high concentrations of
circulating IgG for binding to FcγRIIIA, and co-activation of inhibitory receptors such as FcγRIIB decreasing the
overall immune response [2]. As postulated in the early 1990s, one way to overcome all of these limitations
would be to redirect and activate NK cells using anti-target x FcγRIIIA bispecific antibodies that targeted an
epitope not involved in IgG binding [53,54]. The potential of this approach has been validated in clinical trials
[55,56]. However, significant human anti-mouse responses prevented further clinical evaluation of these
quadromas. One of these promising bsAbs (HRS-3/A9) was converted to a bispecific Db format, and the
resulting molecule was shown to induce tumor lysis by NK cells more efficiently than the parental molecules
[52], and is expected be less immunogenic than quadroma derived because of the absence of the Fc portion.
Yet this antibody fragment still carries the framework sequences of murine origin. The clinical potential of bsAb-
based NK cell retargeting still has to be demonstrated. For this goal, the authors of this article have isolated
activating anti-FcγRIIIA dAbs [57] and anti-CEA dAbs [Behar G et al, unpublished results] that have been used
to produce anti-CEA x FcγRIIIA bsFab molecules. These stable and efficiently produced bsAbs, capable of
inducing very potent ADCC at sub-picomolar concentrations [Cornillon et al, unpublished results], are currently
undergoing preclinical studies.

8
A third class of interesting effector cells is the neutrophils. These cells are of special interest because several
studies support their in vitro and in vivo role in tumor cell lysis and tumor rejection [58-61]. Moreover,
neutrophils represent by far the most abundant FcR-expressing leukocyte subset in blood, and their numbers
can be increased by treatment with granulocyte colony-stimulating factor (G-CSF). FcαRI is constitutively
expressed on myeloid effector cells, including neutrophils, monocytes, macrophages, eosinophils and DCs, but
not on non-cytolytic cell populations [62]. Because FcαRI is capable of efficiently activating immature
neutrophils [63], targeting this receptor by the use of bsAbs has considerable potential that should be explored
more extensively in the future.
Finally, FcγRI (CD64) has also been targeted by bsAbs. This receptor is constitutively expressed by monocytes
and macrophages. Moreover, the expression of FcγRI can be induced at the surface of neutrophils upon
stimulation with interferon (IFN)γ or G-CSF. Most notably, two bsAbs, constructed by the chemical conjugation
of Fab' fragments, have been extensively studied for the treatment of advanced breast cancer (anti-Her2 x
CD64) and head and neck cancer (anti-EGFR x CD64). Both of these bsAbs were tested in phase I clinical trials
[12]. The molecules were well tolerated, but unfortunately did not lead to consistent antitumor activity. The
lack of efficacy was suggested to be due to both an effector-to-target ratio and a bsAb concentration that were
too low. An anti-EpCAM x CD64 bsAb also demonstrated efficient carcinoma cell killing using G-CSF- and IFNγ-
stimulated neutrophils [64], supporting the concept that activated neutrophils might represent efficient tumor
killing cells.
(Sub) Pretargeting
Radioimmunotherapy approaches have several advantages over more conventional treatments for cancer. One
of the most attractive advantages is the bystander effect. In a particular tumor, cells often tend to lose antigen
expression to escape from therapy. However, radiolabeled antibodies bind to antigen-positive tumor cells and
can irradiate cells in their close proximity (bystander cells), including antigen-negative tumor cells, tumor
stromal fibroblasts and tumor vasculature. Unfortunately, and despite promising results obtained during the last
25 years with directly labeled antibodies, few convincing positive clinical results have been achieved, mostly
because IgG remains in the circulation for days and causes unacceptable toxicities to normal tissues [65].
Almost 20 years ago one method proposed to resolve this problem involved the separation of the antibody
targeting moiety from the radiolabeled effector [66]. Antibody fragments rapidly clear from the circulation and
often show better tumor penetration and faster maximum tumor uptake than whole antibodies, but overall lead
to very poor total tumor uptake and an insufficient radiation dose. In the two-step pretargeting method, a bsAb
targeting the tumor would be injected in the first step. Once optimal tumor uptake and clearance of the bsAb
from the circulation had occurred, the second step would involve the injection of a small radiolabeled effector
that would be captured at the tumor site by the bsAb and rapidly cleared from the rest of the system, thereby

9
sparing the organism from excessive doses of radiation. Although the potential of the bsAb pretargeting
technology was shown to be successful in both preclinical and clinical experiments, the poor production yield
caused by the use of chemical constructs has so far limited the expansion of this approach as a clinically
available therapy [67].
Using the Dock-and-Lock approach, Goldenberg and collaborators have produced trivalent bsAbs, which are
composed of two anti-MUC1 Fabs linked to Fab TF2 binding to the hapten HSG (histamine-succinyl-glycine).
This molecule was used in the pretargeting of 90Y-labeled hapten, and led to ratios of more than 100 to 1 in
tumor to non-tumor normal tissue and average tumor to blood ratios of 1000 to 1 in animal model studies. A
single injection of this molecule could potentially lead to complete regression of eight out of ten tumors,
demonstrating the high clinical potential of this efficiently produced bsAb [18].
The Holy Grail: BsAb-dependent induction of adaptive response?
As demonstrated by successful clinical trials, the possibility of efficiently retargeting effector cells to destroy
tumor cells seems to be within reach. However, if bsAb-based therapies could also be developed to stimulate
the involvement of the adaptive immune system, tumor killing T-lymphocytes could potentially be generated,
and vaccination could be established (ie, the provision of long-term immune response and memory cells that
could rapidly generate a new and efficient response in the case of metastasis development). The induction of an
adaptive response would be dependent on the uptake of tumor cell debris resulting from bsAb-dependent lysis,
and presentation of the debris by macrophages, B-cells and DCs, as complexes with major histocompatibility
complex (MHC) molecules. However, DCs normally present exogenous antigen through MHC class II molecules
whereas cytotoxic T-cell activation requires presentation through MHC class I molecules. Class I-based
presentation of exogenous antigen, a poorly described phenomenon called cross-presentation or cross-priming,
has already been described in some cases of tumor cells opsonized by antitumor mAbs [68,69]. Lum and
collaborators have reported an increase in antitumor activity in the endogenous immune cells of patients
treated with ex vivo amplified and activated T-cells retargeted by an anti-CD3 x target. These researchers
hypothesized that secretion of GM-CSF (granulocyte macrophage colony-stimulating factor), RANTES, IFNγ and
IL-2 upon engagement of retargeted T-cells would recruit endogenous T-cells and DCs, leading to vaccination
[11]. Triomabs may have a huge advantage in this respect. These molecules have been demonstrated to
retarget macrophages and DCs toward tumor cells and stimulate these leukocytes via binding to their Fcγ
receptors. In fact, a mouse model study demonstrated that an anti-human EpCAM x murine CD3 triomab could
protect mice against a second tumor challenge that was initiated 144 days after the first challenge. This
protection was proved to be dependent on the presence of the chimeric Fc, as the F(ab')2 fragment of the same
mAb did not lead to long-term protection. Moreover, Linhofer and coworkers observed a strong correlation
between the induction of a humoral immune response with tumor-reactive antibodies and the survival of the

10
mice. [10,70]. These results strongly indicate that an induction of long-lasting antitumor immunity was
established in these animals and raises huge expectations for antibody-based cancer therapy.
Conclusion
Through dual binding, the retargeting of effector cells and radiolabeled haptens, a new generation of efficiently
produced bispecific antibodies have the potential to substantially improve antibody-based therapies in the
future, and might even lead to vaccination effects. As several of these molecules are in clinical trials at the
moment and hundreds are in preclinical trials, we should know in the near future whether the beginning of the
end of war against cancer is within reach.
Figure legend
Figure 1. Formats of antibodies.
A conventional antibody (light green for light chains, dark green for heavy chains; blue triangles indicate the
site of glycosylation) and the derived fragments (shaded areas represent the binding sites). Color orange
symbolizes a different specificity. Llama domain antibodies are depicted in mauve or blue.
(Adapted with permission from Chames and Baty © 2009 Chames and Baty)
bsAb bispecific antibody, bsFab bispecific Fab fragment, dAb domain antibody, scFv single-chain Fv fragment
References •• of outstanding interest • of special interest 1. Kohler G, Milstein C: Continuous cultures of fused cells secreting antibody of predefined
specificity. Nature (1975) 256(5517):495-497. 2. Chames P, Baty D: Therapeutic antibodies: Successes, limitations and future hopes. Br J
Pharmacol (2009): in press. 3. Reichert JM, Valge-Archer VE: Development trends for monoclonal antibody cancer
therapeutics. Nat Rev Drug Discov (2007) 6(5):349-356. 4. Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY: Optimizing engagement of the immune system
by anti-tumor antibodies: An engineer's perspective. Drug Discov Today (2007) 12(21-22):898-910.
•• Excellent review covering most aspects of Fc engineering. 5. Staerz UD, Kanagawa O, Bevan MJ: Hybrid antibodies can target sites for attack by T cells.
Nature (1985) 314:628-631. • Represents the first proof of concept of the bispecific antibody approach. 6. Cotton RG, Milstein C: Letter: Fusion of two immunoglobulin-producing myeloma cells. Nature
(1973) 244(5410):42-43. 7. Peipp M, Valerius T: Bispecific antibodies targeting cancer cells. Biochem Soc Trans (2002)
30(4):507-511. 8. Lindhofer H, Mocikat R, Steipe B, Thierfelder S: Preferential species-restricted heavy/light chain
pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol (1995) 155(1):219-225.

11
9. Zeidler R, Mysliwietz J, Csanady M, Walz A, Ziegler I, Schmitt B, Wollenberg B, Lindhofer H: The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer (2000) 83(2):261-266.
10. Morecki S, Lindhofer H, Yacovlev E, Gelfand Y, Ruf P, Slavin S: Induction of long-lasting antitumor
immunity by concomitant cell therapy with allogeneic lymphocytes and trifunctional bispecific antibody. Exp Hematol (2008).
• Demonstrates the possibility to efficiently vaccinate mice against cancer. 11. Lum LG, Davol PA, Lee RJ: The new face of bispecific antibodies: Targeting cancer and much
more. Exp Hematol (2006) 34(1):1-6. 12. Repp R, van Ojik HH, Valerius T, Groenewegen G, Wieland G, Oetzel C, Stockmeyer B, Becker W,
Eisenhut M, Steininger H, Deo YM et al: Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcgammaRI x anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. Br J Cancer (2003) 89(12):2234-2243.
13. Muller D, Kontermann RE: Recombinant bispecific antibodies for cellular cancer
immunotherapy. Curr Opin Mol Ther (2007) 9(4):319-326. 14. Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler
M, Einsele H et al: Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science (2008) 321(5891):974-977.
•• Describes the most potent clinical results obtained to date with antitumor antibodies. 15. Holliger P, Prospero T, Winter G: "Diabodies": Small bivalent and bispecific antibody fragments.
Proc Natl Acad Sci USA (1993) 90(14):6444-6448. 16. Lawrence LJ, Kortt AA, Iliades P, Tulloch PA, Hudson PJ: Orientation of antigen binding sites in
dimeric and trimeric single chain Fv antibody fragments. FEBS Lett (1998) 425(3):479-484. 17. Fischer N, Leger O: Bispecific antibodies: Molecules that enable novel therapeutic strategies.
Pathobiology (2007) 74(1):3-14. 18. Goldenberg DM, Rossi EA, Sharkey RM, McBride WJ, Chang CH: Multifunctional antibodies by the
Dock-and-Lock method for improved cancer imaging and therapy by pretargeting. J Nucl Med (2008) 49(1):158-163.
19. Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu RR, Santora L,
Davis-Taber R et al: Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol (2007) 25(11):1290-1297.
20. Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G: Binding activities of a repertoire of single
immunoglobulin variable domains secreted from Escherichia coli. Nature (1989) 341(6242):544-546.
21. Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM: Domain antibodies: Proteins for
therapy. Trends Biotechnol (2003) 21(11):484-490. 22. Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N,
Hamers R: Naturally occurring antibodies devoid of light chains. Nature (1993) 363(6428):446-448.
23. Greenberg AS, Avila D, Hughes M, Hughes A, McKinney EC, Flajnik MF: A new antigen receptor
gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature (1995) 374(6518):168-173.
24. Els Conrath K, Lauwereys M, Wyns L, Muyldermans S: Camel single-domain antibodies as
modular building units in bispecific and bivalent antibody constructs. J Biol Chem (2001) 276(10):7346-7350.
25. Roopenian DC, Akilesh S: FcRn: The neonatal Fc receptor comes of age. Nat Rev Immunol (2007)
7(9):715-725. 26. Holliger P, Hudson PJ: Engineered antibody fragments and the rise of single domains. Nat
Biotechnol (2005) 23(9):1126-1136. 27. Kubetzko S, Balic E, Waibel R, Zangemeister-Wittke U, Pluckthun A: PEGylation and
multimerization of the anti-p185HER-2 single chain Fv fragment 4D5: Effects on tumor targeting. J Biol Chem (2006) 281(46):35186-35201.

12
28. Muller D, Karle A, Meissburger B, Hofig I, Stork R, Kontermann RE: Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J Biol Chem (2007) 282(17):12650-12660.
29. Dennis MS, Jin H, Dugger D, Yang R, McFarland L, Ogasawara A, Williams S, Cole MJ, Ross S, Schwall
R: Imaging tumors with an albumin-binding Fab, a novel tumor-targeting agent. Cancer Res (2007) 67(1):254-261.
• Demonstrates the possibility to significantly extend the half-life of antibody fragments in a very simple and efficient way. 30. Holt LJ, Basran A, Jones K, Chorlton J, Jespers LS, Brewis ND, Tomlinson IM: Anti-serum albumin
domain antibodies for extending the half-lives of short lived drugs. Protein Eng Des Sel (2008) 21(5):283-288.
31. Roovers RC, Laeremans T, Huang L, De Taeye S, Verkleij AJ, Revets H, de Haard HJ, van Bergen en
Henegouwen PM: Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR nanobodies. Cancer Immunol Immunother (2007) 56(3):303-317.
• The first report to describe the successful use of untagged dAbs for the in vivo treatment of solid tumours. 32. Takimoto CH, Awada A: Safety and anti-tumor activity of sorafenib (Nexavar) in combination
with other anti-cancer agents: A review of clinical trials. Cancer Chemother Pharmacol (2008) 61(4):535-548.
33. Leonard JP, Coleman M, Ketas J, Ashe M, Fiore JM, Furman RR, Niesvizky R, Shore T, Chadburn A,
Horne H, Kovacs J et al: Combination antibody therapy with epratuzumab and rituximab in relapsed or refractory non-Hodgkin's lymphoma. J Clin Oncol (2005) 23(22):5044-5051.
34. Lu D, Zhang H, Ludwig D, Persaud A, Jimenez X, Burtrum D, Balderes P, Liu M, Bohlen P, Witte L, Zhu
Z: Simultaneous blockade of both the epidermal growth factor receptor and the insulin-like growth factor receptor signaling pathways in cancer cells with a fully human recombinant bispecific antibody. J Biol Chem (2004) 279(4):2856-2865.
35. Lu D, Zhang H, Koo H, Tonra J, Balderes P, Prewett M, Corcoran E, Mangalampalli V, Bassi R, Anselma
D, Patel D et al: A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J Biol Chem (2005) 280(20):19665-19672.
36. Hoffmann P, Hofmeister R, Brischwein K, Brandl C, Crommer S, Bargou R, Itin C, Prang N, Baeuerle
PA: Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer (2005) 115(1):98-104.
37. Howland KC, Ausubel LJ, London CA, Abbas AK: The roles of CD28 and CD40 ligand in T cell
activation and tolerance. J Immunol (2000) 164(9):4465-4470. 38. Manzke O, Titzer S, Tesch H, Diehl V, Bohlen H: CD3 x CD19 bispecific antibodies and CD28
costimulation for locoregional treatment of low-malignancy non-Hodgkin's lymphoma. Cancer Immunol Immunother (1997) 45(3-4):198-202.
39. Grabert RC, Cousens LP, Smith JA, Olson S, Gall J, Young WB, Davol PA, Lum LG: Human T cells
armed with Her2/neu bispecific antibodies divide, are cytotoxic, and secrete cytokines with repeated stimulation. Clin Cancer Res (2006) 12(2):569-576.
40. Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA: BiTEs: Bispecific antibody constructs
with unique anti-tumor activity. Drug Discov Today (2005) 10(18):1237-1244. 41. Dreier T, Baeuerle PA, Fichtner I, Grun M, Schlereth B, Lorenczewski G, Kufer P, Lutterbuse R,
Riethmuller G, Gjorstrup P, Bargou RC: T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3- bispecific single-chain antibody construct. J Immunol (2003) 170(8):4397-4402.
42. Schlereth B, Fichtner I, Lorenczewski G, Kleindienst P, Brischwein K, da Silva A, Kufer P, Lutterbuese
R, Junghahn I, Kasimir-Bauer S, Wimberger P et al: Eradication of tumors from a human colon cancer cell line and from ovarian cancer metastases in immunodeficient mice by a single-chain Ep-CAM-/CD3-bispecific antibody construct. Cancer Res (2005) 65(7):2882-2889.
43. Schlereth B, Kleindienst P, Fichtner I, Lorenczewski G, Brischwein K, Lippold S, da Silva A, Locher M,
Kischel R, Lutterbuse R, Kufer P et al: Potent inhibition of local and disseminated tumor growth in immunocompetent mouse models by a bispecific antibody construct specific for Murine CD3. Cancer Immunol Immunother (2006) 55(7):785-796.

13
44. Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA: Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol (2006) 43(6):763-771.
45. Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T,
Raum T, Kleindienst P et al: MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol (2006) 43(8):1129-1143.
46. Grosse-Hovest L, Hartlapp I, Marwan W, Brem G, Rammensee HG, Jung G: A recombinant bispecific
single-chain antibody induces targeted, supra-agonistic CD28-stimulation and tumor cell killing. Eur J Immunol (2003) 33(5):1334-1340.
47. Grosse-Hovest L, Wick W, Minoia R, Weller M, Rammensee HG, Brem G, Jung G: Supraagonistic,
bispecific single-chain antibody purified from the serum of cloned, transgenic cows induces T-cell-mediated killing of glioblastoma cells in vitro and in vivo. Int J Cancer (2005) 117(6):1060-1064.
48. Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, Lindhofer H: Simultaneous
activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol (1999) 163(3):1246-1252.
49. Burges A, Wimberger P, Kumper C, Gorbounova V, Sommer H, Schmalfeldt B, Pfisterer J, Lichinitser M,
Makhson A, Moiseyenko V, Lahr A et al: Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: A phase I/II study. Clin Cancer Res (2007) 13(13):3899-3905.
• Demonstrates the high potential of triomabs, murine/rat-quadroma derived bsAb. 50. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H: Therapeutic
activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood (2002) 99(3):754-758.
51. Weiner LM, Alpaugh RK, Amoroso AR, Adams GP, Ring DB, Barth MW: Human neutrophil
interactions of a bispecific monoclonal antibody targeting tumor and human FcγRIII. Cancer Immunol Immunother (1996) 42(3):141-150.
52. Arndt MA, Krauss J, Kipriyanov SM, Pfreundschuh M, Little M: A bispecific diabody that mediates
natural killer cell cytotoxicity against xenotransplantated human Hodgkin's tumors [In Process Citation]. Blood (1999) 94(8):2562-2568.
53. de Palazzo IG, Gercel-Taylor C, Kitson J, Weiner LM: Potentiation of tumor lysis by a bispecific
antibody that binds to CA19-9 antigen and the Fcγ receptor expressed by human large granular lymphocytes. Cancer Res (1990) 50(22):7123-7128.
54. Hombach A, Jung W, Pohl C, Renner C, Sahin U, Schmits R, Wolf J, Kapp U, Diehl V, Pfreundschuh M:
A CD16/CD30 bispecific monoclonal antibody induces lysis of Hodgkin's cells by unstimulated natural killer cells in vitro and in vivo. Int J Cancer (1993) 55(5):830-836.
55. Weiner LM, Clark JI, Davey M, Li WS, Garcia de Palazzo I, Ring DB, Alpaugh RK: Phase I trial of 2B1,
a bispecific monoclonal antibody targeting c-erbB- 2 and Fc gamma RIII. Cancer Res (1995) 55(20):4586-4593.
56. Hartmann F, Renner C, Jung W, da Costa L, Tembrink S, Held G, Sek A, Konig J, Bauer S, Kloft M,
Pfreundschuh M: Anti-CD16/CD30 bispecific antibody treatment for Hodgkin's disease: Role of infusion schedule and costimulation with cytokines. Clin Cancer Res (2001) 7(7):1873-1881.
57. Behar G, Siberil S, Groulet A, Chames P, Pugniere M, Boix C, Sautes-Fridman C, Teillaud JL, Baty D:
Isolation and characterization of anti-Fc{gamma}RIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Eng Des Sel (2008) 21(1):1-10.
• Describes the isolation of anti-FcγRIIIA dAbs capable of efficiently inducing ADCC without the use of an Fc portion. 58. Lichtenstein A, Kahle J: Anti-tumor effect of inflammatory neutrophils: Characteristics of in
vivo generation and in vitro tumor cell lysis. Int J Cancer (1985) 35(1):121-127. 59. Midorikawa Y, Yamashita T, Sendo F: Modulation of the immune response to transplanted
tumors in rats by selective depletion of neutrophils in vivo using a monoclonal antibody: Abrogation of specific transplantation resistance to chemical carcinogen-induced syngeneic tumors by selective depletion of neutrophils in vivo. Cancer Res (1990) 50(19):6243-6247.
60. Seino K, Kayagaki N, Okumura K, Yagita H: Antitumor effect of locally produced CD95 ligand. Nat
Med (1997) 3(2):165-170.

14
61. Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P: The intriguing role of
polymorphonuclear neutrophils in antitumor reactions. Blood (2001) 97(2):339-345. 62. Van Spriel AB, van de Winkel JG: CD89 (FcαRI). J Biol Regul Homeost Agents (2001) 15(2):179-181. 63. Stockmeyer B, Dechant M, van Egmond M, Tutt AL, Sundarapandiyan K, Graziano RF, Repp R, Kalden
JR, Gramatzki M, Glennie MJ, van de Winkel JG et al: Triggering Fcα-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J Immunol (2000) 165(10):5954-5961.
64. Schweizer C, Strauss G, Lindner M, Marme A, Deo YM, Moldenhauer G: Efficient carcinoma cell
killing by activated polymorphonuclear neutrophils targeted with an Ep-CAMxCD64 (HEA125x197) bispecific antibody. Cancer Immunol Immunother (2002) 51(11-12):621-629.
65. Adams GP, Weiner LM: Radioimmunotherapy of solid tumors: From fairytale to reality. Cancer
Biother Radiopharm (2001) 16(1):9-11. 66. Chang CH, Sharkey RM, Rossi EA, Karacay H, McBride W, Hansen HJ, Chatal JF, Barbet J, Goldenberg
DM: Molecular advances in pretargeting radioimunotherapy with bispecific antibodies. Mol Cancer Ther (2002) 1(7):553-563.
67. Goldenberg DM, Chatal JF, Barbet J, Boerman O, Sharkey RM: Cancer Imaging and Therapy with
Bispecific Antibody Pretargeting. Update Cancer Ther (2007) 2(1):19-31. 68. Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV: Antitumor monoclonal antibodies
enhance cross-presentation of cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med (2002) 195(1):125-133.
69. Cioca DP, Deak E, Cioca F, Paunescu V: Monoclonal antibodies targeted against melanoma and
ovarian tumors enhance dendritic cell-mediated cross-presentation of tumor-associated antigens and efficiently cross-prime CD8+ T cells. J Immunother (2006) 29(1):41-52.
70. Ruf P, Lindhofer H: Induction of a long-lasting antitumor immunity by a trifunctional bispecific
antibody. Blood (2001) 98(8):2526-2534. • The first report to clearly demonstrate the possibility to vaccinate mice against cancer cells using a trifunctional antibody.
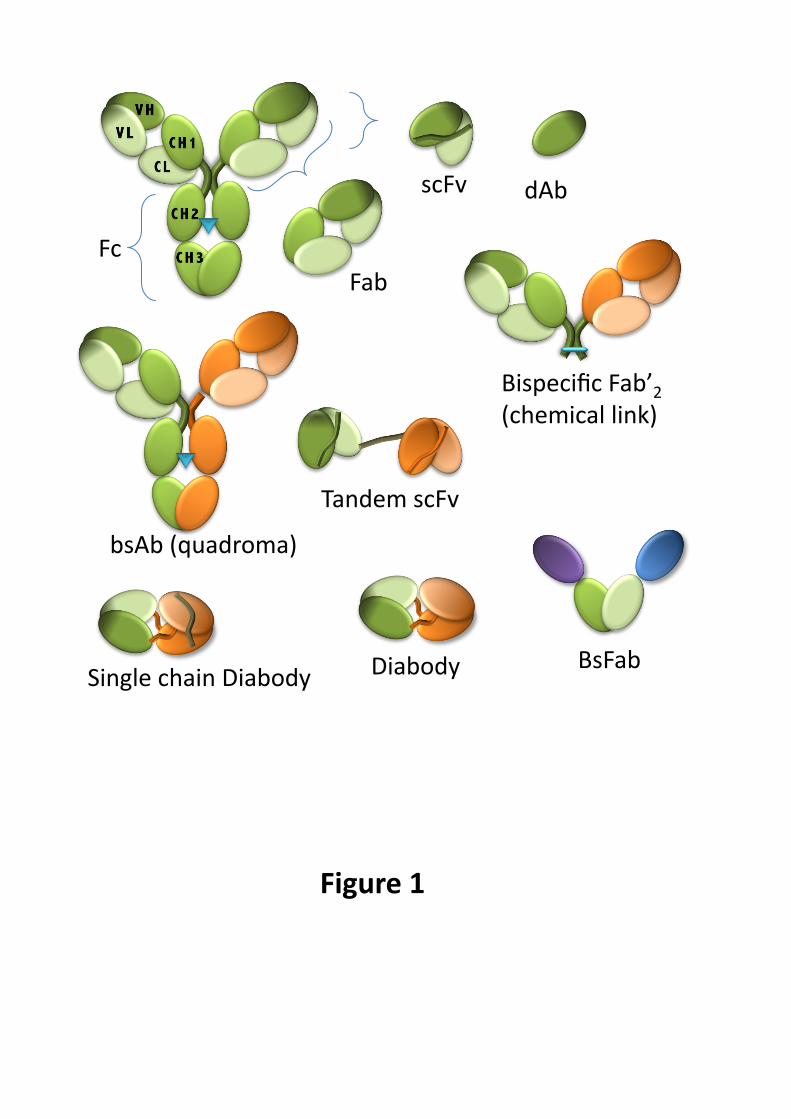
Figure1
BispecificFab’2(chemicallink)
TandemscFv
scFv
FabFc
SinglechainDiabodyBsFab
bsAb(quadroma)
dAb
Diabody