blog.nus.edu.sg · web viewengraftment syndrome (es) may occur immediately before or at the time of...
TRANSCRIPT

SUPPORTIVE CARE GUIDELINESVIVA-ASIA BMT
VABMT Supportive Care Guidelines 2013 Page 1 of 38

CONTENTS
Topic Page
1. Engraftment Syndrome 32. Acute Graft Versus Host Disease 53. Blood Transfusion Guidelines 104. Hepatic Veno-occlusive Disease 165. Renal Complications 206. Transplant Associated Microangiopathy 247. Hemorrhagic Cystitis 288. Monitoring EBV Viral Load and Management of PTLD 329. Idiopathic Pneumonia Syndrome 35
VABMT Supportive Care Guidelines 2013 Page 2 of 38

VIVA-ASIA BMT
Engraftment Syndrome
I. Introduction:
Engraftment syndrome (ES) may occur immediately before or at the time of neutrophil engraftment. At the time of neutrophil recovery following cytotoxic chemotherapy, a spiralling production and release of cytokines and products of neutrophil degranulation and oxidative metabolism occurs leading to local and systemic tissue injury. The pathophysiologic mechanism of ES is multifactorial and may involve prominent cellular interactions of T cells, monocytes and other effector cells, complement activation and pro-inflammatory cytokines production and realease.
ES is more commonly seen in myeloablative allogeneic HSCT, in patients receiving cytokines, those experiencing rapid absolute neutrophil count (ANC) recovery, and those receiving grafts containing large numbers of mononuclear or CD34+ cells
II. Clinical Features:
The clinical manifestations of ES include fever, rash and non-cardiogenic pulmonary edema. This usually occurs within 96 hrs of neutrophil engraftment but may also occur before this time. The following diagnostic criteria has been proposed for the diagnosis of ES:
A. Major Criteria1. Temperature > 38.3oC with no identifiable infection2. Erythroderma > 25% body surface area, not attributable to medication3. Diffuse non-cardiogenic pulmonary edema, diffuse pulmonary infiltrates and hypoxi
B. Minor Criteria1. Total bilirubin > 2 mg/dL (36 umol/L), AST, ALT > 2x normal2. Renal insufficiency3. Weight gain > 2.5% baseline body weight4. Transient encephalopathy unexplained by other causes
A diagnosis of ES is established by the presence of ALL 3 MAJOR CRITERIA or 2 MAJORAND 1 OR MORE MINOR CRITERIA.
III. Management:
A. Measures to treat this complication should be instituted once the diagnosis of engraftment syndrome is strongly considered
1. All patients should have strict daily input and output monitoring2. Weights should be determined twice daily. Efforts to maintain negative fluid
balance during engraftment include the use of diuretics as well as fluid restriction 3. Monitor renal and hepatic function closely
VABMT Supportive Care Guidelines 2013 Page 3 of 38

4. Mild ES may not require treatment as the clinical symptoms resolve with hematologic recovery and discontinuation of growth factors
5. Patients who develop engraftment syndrome may have growth factors discontinued if ANC is greater than 500
6. For patients with progressive or symptomatic ES, particularly with pulmonary involvement, methylprednisolone, typically at a beginning dose of 2 mg/kg per day intravenously or as per treatment for acute GVHD should be started
7. Empiric antibiotic coverage should be started8. Continue antiviral and antifungal prophylaxis9. Refer to the Pediatric Intensive Care Unit early if close monitoring and possible
ventilator support is anticipated
IV. References:
1. Foncillas MA, Diaz MA, Sevilla J, Gonzalez VM, Fernandez-Plaza S, Madero L. Engraftment syndrome emerge as the main cause of transplant related mortality in pediatric patients receiving autologous peripheral blood progenitor cell transplantation. J Ped Hematol Oncol 26 (8): 492-6, 2004.
2. Madero L, Vicent MG, Sevilla J, Prudencio M, Rodriguez F, Diaz MA. Engraftment syndrome in children undergoing autolgous peripheral blood progenitor cell transplantation. Bone Marrow Transplant 30(6): 355-8, 2002.
3. Mossad S, Kalaycio M, Sobecks R, Pohlman B, Andresen S, Avery R, Rybicki L, Jarvis J, Bolwell B. Steroids prevent engraftment syndrome after autologous blood and marrow transplantation without increasing the risk of infection. Bone Marrow Transplant 35(4): 373-81, 2005.
4. Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant (27)9: 893-8, 2001.
VABMT Supportive Care Guidelines 2013 Page 4 of 38

VIVA-ASIA BMT
Acute Graft versus Host Disease
I. Introduction
Acute graft versus host disease (aGVHD) continues to be a major cause of morbidity and mortality in patients receiving allogeneic hematopoietic stem cells. Historically, aGVHD has been defined as occurring before 100 days post transplant; however, late onset aGVHD has now been recognized in patients receiving peripheral blood stem cells, those who received reduced intensity conditioning therapies and donor lymphocyte infusions. The incidence reported from pediatric studies ranges between 19-85%.
The clinical triad of aGVHD consists of inflammatory dermatitis, enteritis and hepatitis aGVHD. These manifestations reflect T cell activation resulting in the generation of cytotoxic lymphocytes and cytokines and eventually leading to tissue damage.
Risk factors for development of GVHD include histoincompatibility, stem cell source, increased donor or patient age, sex mismatch, CMV positivity, type of GVHD prophylaxis.
II. Diagnosis
The diagnosis of aGVHD is clinicohistopathologic. and is highly suspected when the combination of rash, diarrhea and jaundice/hepatitis occur around the period of engraftment. Histopathologic evaluation helps to corroborate the diagnosis, document extent of organ involvement and also helps rule out other conditions that may cause similar clinical manifestations particularly when the diagnosis is unclear or when only a single target organ is involved.
Tissue biopsies of the skin, upper/lower gastrointestinal tract are recommended and should be done as much as possible but should not delay treatment when patients manifest classical features of aGVHD.
III. Classification
The degree of severity of aGVHD can be based on extent of organ involvement and overall grade. Classification includes the modified Glucksberg-Seattle criteria or the International Bone Marrow Transplant Registry Severity Index as shown below
A. Glucksberg-Seattle Criteria for Staging aGVHD
Skin Gut Upper GI Liver
Stage 0 No rash <10 ml/kg/d (child)<500 ml/d (adult)
-- Bilirubin <2 mg/dL
VABMT Supportive Care Guidelines 2013 Page 5 of 38

Stage I Rash < 25% BSA 10-19.9 ml/kg/d (child)500-1000 ml/d (adult)
Severe nausea,vomiting
Bilirubin 2.1- mg/dL
Stage 2 Rash 25-50% BSA 20-30 ml/kg/d (child)1001-1500ml/d (adult)
-- Bilirubin 3.1-6 mg/dL
Stage 3 Rash >50%, generalized erythroderma
>30 ml/kg/d (child)>1500 ml/d (adult)
-- Bilirubin 6.1-15 mg/dL
Stage 4 3 plus bullae and desquamation
Severe abdominal pain +/- ileus, frank blood or melena
-- > 15 mg/dL
B. Grading of Severity of aGVHD based on the Keystone Consensus and CIBMTR criteria
Grade Skin Liver GI Upper GIConsensus
I 1-2 0 0 0II 3 1 1 1III - 2-3 2-4 -IV 4 4
IBMTRA 1 0 0 0B 2 1-2 1-2 1C 3 3 3 -D 4 4 4 -
Grade IIa GVH (Mc Donald et al): rash < 50% BSA, anorexia/nauseas/emesis or diarrhea< 20 mgl/kg/day, no liver involvement
IV. Treatment
A. Principles of treatment1. In deciding whether to begin systemic treatment, both extent/severity of diseas as well
as rate of progression should be considered. In patients with mild or localized skin involvement, systemic corticosteroids may be avoided.
2. Prompt institution of systemic treatment in patients with clinically significant aGVHD (grade II or greater) is important as survival has been show to correlate directly with response to initial treatment
3. The use of corticosteroids continues to be the standard first-line systemic treatment. 4. Indications for instituting second line systemic therapy may include any of the following:
a. Progression after days of starting first line therapyb. No improvement /persistence of manifestations of grade III aGVHD after 1 wk of
treatment or of grade II aGVHD after 2 wks of treatmentc. Inability to tolerate first line treatment using corticosteroids
VABMT Supportive Care Guidelines 2013 Page 6 of 38

5. Although there are numerous agents that have been used for second-line treatment of aGVHD, no single agent or combination of agents has yet been established as the treatment of choice for these patients. Practice may continue to vary between institutions and practice may change as new information is obtained from ongoing clinical studies.
B. Mild aGVHD (skin)1. Grade I skin involvement may not immediately require additional systemic therapy2. Aside from optimising levels of calcineurin inhibitors, topical steroid therapy may
added3. Topical agents that can be used may include the following
a. Triamcinolone 0.1% or tacrolimus 0.1% to bodyb. Hydrocortisone 1% to facec. The Bristish (BSCH) Guidelines suggest the following topical steroid preparations as
suitable for use in aGVHD
Steroid Strength
Very Potent Potent Moderately Potent
Mildly Potent
Example Dermovate(Clobetasol)
Betnovate(Betamethasone
)
Eumovate 1% Hydrocortisone
Face Should be generally avoided
Twice daily x 4-12 wks
Twice daily x6-12 wks
Twice dailyLong term use acceptable
Body Twice daily x4-12 wks
Twi ce dailyLong term therapy may be acceptable
Palms and Soles
Twice dailyMay be used under occlusion to enhance efficacy. Long term therapy may be appropriate
Twice dailyLong term therapy may be appropriate
* Dermatology supervision is advised when using potent steroids on the face
4 Psoralen –UVA (PUVA) 2-3 times/wk5. Consider starting systemic steroids if symptoms worsen 3-4 days after starting topical
or if no improvement noted 1 week after starting topical therapy
C. Mild Gastrointestinal aGVHD: Grad IIa1. Defined by McDonald et al as follows:
VABMT Supportive Care Guidelines 2013 Page 7 of 38

a. Rash < 50% BSAb. Anorexia, nausea, vomiting or diarrhea < 20 ml/kg/dayc. No liver involvement
2. The use of nonabsorbale steroids such as oral budesonideand beclomethasone has been studied in adults but the its role in children has not yet been fully established.
3. Lower doses of methylprednisolone s at 1 mg/kg/day may be started with and increased if no response after 4 days of therapy
D. Grade II-IV1. First line therapy for consists of methylprednisolone at 2 mg/kg/day or prednisone at
2-2.5 mg/kg/day in addition to optimising calcineurin inhibitor levels2. Non-absorbable steroids such as budesonide and beclomethasone may be considered
in addition to steroids for gastrointestinal involvement 3. Consider addition of topic steroids for skin involvement4. Considering addition second line agent if no improvement or with progression of
symptoms
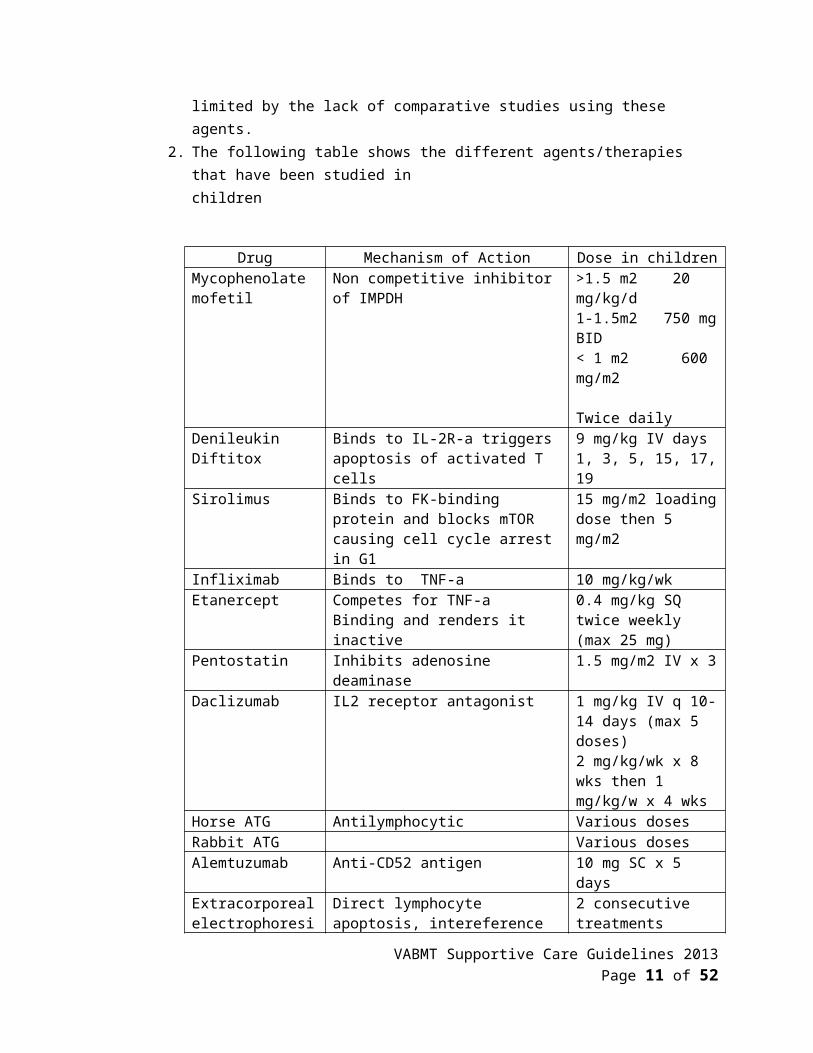
E. Alternative therapy for steroid resistant aGVHD1. Various agents have been used to date as second- or third- line therapy for patients
who do not respond or are unable to tolerate systemic corticosteroids. The efficacy of these therapies are limited by the lack of comparative studies using these agents.
2. The following table shows the different agents/therapies that have been studied inchildren
Drug Mechanism of Action Dose in childrenMycophenolate mofetil
Non competitive inhibitor of IMPDH >1.5 m2 20 mg/kg/d1-1.5m2 750 mg BID< 1 m2 600 mg/m2 Twice daily
Denileukin Diftitox Binds to IL-2R-a triggers apoptosis of activated T cells
9 mg/kg IV days 1, 3, 5, 15, 17, 19
Sirolimus Binds to FK-binding protein and blocks mTOR causing cell cycle arrest in G1
15 mg/m2 loading dose then 5 mg/m2
Infliximab Binds to TNF-a 10 mg/kg/wkEtanercept Competes for TNF-a
Binding and renders it inactive0.4 mg/kg SQ twice weekly (max 25 mg)
Pentostatin Inhibits adenosine deaminase 1.5 mg/m2 IV x 3Daclizumab IL2 receptor antagonist 1 mg/kg IV q 10-14
days (max 5 doses)2 mg/kg/wk x 8 wks then 1 mg/kg/w x 4 wks
Horse ATG Antilymphocytic Various dosesRabbit ATG Various doses
VABMT Supportive Care Guidelines 2013 Page 8 of 38

Alemtuzumab Anti-CD52 antigen 10 mg SC x 5 daysExtracorporeal electrophoresis
Direct lymphocyte apoptosis, intereference with dendritic cell maturation, modulation of cytokine production, expansion of Tregs
2 consecutive treatments weekly or every other week then taper based on response
V. References:
1. Martin P, Rizzo J, Wingard J, et al. First – and second-line systemic treatment of acute graft versus host disease: Recommendations of the American Society of Blood and Marrow Transplantation. Biol Blood Marrow Transplant 18:110-1163, 2012.2. Dignan F, Clark A, Amrolia P, et al. Diagnosis and management of acute graft-versus-host disease. Br J Haematol 158(1) 30-45, 2012.3. Carpenter P, MacMillan M. Management of acute graft versus host disease in children. Pediatr Clin N Am 57(1): 273-295, 2010.4. Jacobson D. Acute graft versus host disease in children. Bone Marrow Transplant 41: 215- 221, 2008.5. Hamidieh A, Hadjibabaieh M, Ghehi M, et al. Long term follow up of children treated with daclizumab for steroid refractory gastrointestinal GVHD in a prospective study. Pediatr Transplant 16(6): 664-669, 2012.6. Teachey D, BIkert B, Bunin M. Daclizumab for children with corticosteroid refractory graft versus host disease. Bone Marrow Transplant 37(1): 95-99, 2006.7. Yang J, CheukD, Ha S, et al. Infliximab for steroid refractory or dependent gastrointestinal acute graft versus host disease in children after allogeneic hematopoietic stem cell
transplantation. Pediatr Transplant 16(7):771-778, 2012.8. Alousi M, Daniel W, Brent L, et al. Etanercept, mycophenolate, denileukin, or pentostatin plus corticosteroids for acute graft versus host disease a randomized phase 2 trial from the Blood and Marrow Transplant Clincial Trials Network. Blood 114 511-517, 2009.9. Sleight B, Chan K, Braun T, et al. Infliximab for GVHD therapy in children. Bone Marrow Transplant 40(5): 474-480, 2007.10. Perotti C, Del Fante C, Tinelli C, et al. Extracorporeal photochemotherapy in graft-versus-
host disease: a longitudinal study on factors influencing the response and survival in pediatric patients. Transfusion 50: 1359-1369, 2010.
11. Benito A, Furlong T, Martin P, et al. Sirolimus (Rapamycin) for the treatment of steroid- refractory acute graft-versus-host disease. Transplantation 72(12): 1924-1929, 2001.12. Muller I, Kordowich S, Holwarth C, et al. Application of multipotent mesenchymal stromal cells in pediatric patients following allogenetic stem cell transplantation. Blood Cells
Mol Dis 40(1): 25-32, 2008.13. Prasad V, Lucas K, Klelner G, et al. Efficacy and safety of ex vivo cultured adult human mesenchymal stem cells (ProchymalTM) in pediatric patients with severe refractory
VABMT Supportive Care Guidelines 2013 Page 9 of 38

acute graft-versus-host disease in a compassionate use study. Biol Blood Marrow Transplant
17(4): 534-541, 2011.
VIVA-ASIA BMT
Blood Transfusion Guidelines for Children undergoing Hematopoietic Stem Cell Transplantation:
I. Introduction:
Patients who undergo hematopoietic stem cell transplantation (HSCT) often require intensive blood component support and have unique transfusion requirements. Special considerations are given to donor-recipient blood group matching, chimerism state post-transplant, cytomegalovirus (CMV) status and the need for irradiated blood.
Transfusion thresholds for different components should be similar to those for patients withoncological conditions. The standard of blood transfusion services and blood banking may vary in different places. Local consensus in transfusion policies should be developed as some areas of transfusion medicine may be based more on clinical experience and practices rather than high level evidence-based studies.
II. Management of ABO Incompatibility:
ABO incompatibility in allogeneic HSCT is conventional divided into major and minor. ABO incompatibility is associated with an increased risk of hemolysis, delayed red cell engraftment, pure red cell aplasia and increased transfusion requirements.
A. Major Incompatibility
Donor Recipient
AB, A, B O
AB A or B
1. Stem cell processing:
a. For bone marrow, red cell depletion is requiredb. For cord blood, red cell depletion is normally not done before freezing due to
the concern about stem cell loss.
VABMT Supportive Care Guidelines 2013 Page 10 of 38
c. For peripheral blood stem cells (PBSC) processing, red cell depletion may or may not be done and the practice varies for different centres
2. Red cell transfusion:
a. First choice - recipient type till antibody titre falls to undetectable level, then can use donor type red cells
b. Second Choice- Type O
3. Platelet and Plasma
a. First choice –donor type (to minimize reaction)b. Second choice – Type AB
B. Minor Incompatibility
Donor Recipient
O AB, A, B
A or B AB
1. Stem Cell Processing:
a. For bone marrow, plasma depletion is required
2. Red cell transfusion:
a. First choice – donor type red cellsb. Second choice – Type O
3. Platelet and Plasma:
a. First choice - recipient type until recipient red cells are absent then can use donor type
b. Second choice – Type AB
C. Major and Minor Incompatibility
VABMT Supportive Care Guidelines 2013 Page 11 of 38

Donor Recipient
A B
B A
2. Stem Cell Processing:
a. For bone marrow, red cell and plasma depletion are required
3. Red Cell Transfusion:
a. First choice – Type O red cells until antibody titre falls to undetectable level, then use donor type
4. Platelet and Plasma:
a. First choice – Type AB until recipient red cells are absent then use donor type
Incompatibility Donor Recipient Red Cells Platelets FFPMajor ABO incompatibility 1st choice 2nd choice
A O O A O AB O O B O B
AB O O AB A ABAB A O/A AB A ABAB B O/B AB B AB
Minor ABO incompatibility
O A O A O AO B O B O BO AB O A O ABA AB O/A A O ABB AB O/B B O AB
Major and minor ABO incompatibility
A B O A B ABB A O B A AB
D. Transplant with Major Donor/Recipient ABO Incompatibility
VABMT Supportive Care Guidelines 2013 Page 12 of 38

A major incompatibility occurs when the transfusion of the donor marrow (unseparated) is highly likely to cause a fatal haemolytic reaction, eg. A donor into B, or O recipient; B donor into A, or O recipient. Measure IgM and IgG anti-A and/or anti-B as before transplant.
1. All major ABO incompatible donor marrows to be Dextran separated. (This should remove approximately 99% of the red cells).
2. Red cell count to be done on the marrow at the end of in vitro treatment. 3. Infuse the bone marrow, as for the other BMTs but taking more care especially if
residual red cells are obviously present. Test first urine passed post BM infusion for haemoglobin, and measure serum haptoglobins and free haemoglobin on day +1 if reaction occurs with marrow infusion.
4. Measure Anti-A and/or Anti-B (IgG + IgM) weekly. If titres very high (> 1:1024) and no peripheral blood recovery, examine bone marrow at +2/52.
5. If no engraftment, consider plasma exchange. Antibody should eventually be absorbed out by the developing bone marrow.
6. The recipient should in most cases receive blood of his/her own ABO type, until complete change over has occurred
III. General recommendations regarding Blood component therapy and special blood products:
A. Red Blood Cell
1. All red cells should be packed or plasma reduced. 2. Transfuse if hemoglobin level < 8 g/dl, or symptomatic, or bleeding 3. The hemoglobin in patients who are oxygen-dependent should be kept above 10 g/dl4. If engraftment is occurring and patient has reticulocytosis in the 2nd and 3rd week post
BMT, only transfuse if necessary. 5. Recipients having regular blood component support should have a weekly red cell
antibody screen. If transfusion requirements increase without obvious bleeding, perform a direct Coombs test.
6. If the donor is (major mismatch) ABO incompatible, measure appropriate anti-A and/or anti-B titres at regular intervals (IgM and IgG).
7. If donor is (minor mismatch) ABO incompatible e.g. O donor A recipient, occasionally mild hemolysis may occur in the first 3 weeks after BMT (anti-A production from donor lymphocytes).
B. Platelets
1. Random donor platelet units are used 2. Transfuse platelets if platelet count < 20 x 109/1 in well patient or < 30 x 109/1 if patient
has infection, bleeding or DIC.3. For invasive procedures, the platelet level to be kept above should depend on local
guidelines.4. An adequate number of units (if not bleeding, infected, and no DIC) should be given
according to platelet counts and local recommendations.
VABMT Supportive Care Guidelines 2013 Page 13 of 38

5. If no obvious increments at +24 hours, assess response to transfusions (if not bleeding, infected, and no DIC) by taking platelet count pre and 1 hour post-transfusion to confirm platelet refractoriness. The increment should be approximately 7 x 109/m2 for every unit transfused.
C. Granulocytes
1. Rarely indicated in case of proven septicemia with no response to antibiotics in a severely neutropenic patient (<0.1 x 109 /1)
2. Continue therapy for at least 5 days if instituted, unless counts improving and > 0.5 neutrophils
3. The number of granulocyte units for transfusion should be based on local guidelines.
D. Fresh Frozen Plasma and Cryoprecipitate
1. FFP is indicated at a volume of 10-15 ml/kg for:a. As replacement therapy for TTP when plasma exchange is undertakenb. Severe DICc. Coagulopathy due to liver disease
2. Cryoprecipitate transfusion is indicated in DIC when fibrinogen level is < 100 mg/dl
E. Gamma-irradiated Blood
1. All Blood cellular components (red cells, white cells and platelets) should be irradiated with 2500 cGy from time of conditioning (SCIDs to have irradiated blood from admission).
2. Irradiation to continue until 6 months post-BMT (allo) or when lymphocyte count is > 1x10^9/L without chronic GVHD.
3. Duration could be extended if patient is still on immuno-suppressive drug4. Irradiation of blood components required from 7 days before autologous stem cell
harvest. Irradiation to continue until 3 months post-BMT (auto)5. Patients on purine analogues (e.g. fludarabine)6. Patients with Hodgkin’s disease7. Patients with stage IV neuroblastoma8. Gamma irradiation of cellular blood products should be considered early (as soon as
the patient is considered a transplant candidate)
F. CMV Status and choice of blood products
1. CMV Sero-negative recipient and donora. Use sero-negative blood components. If such components are not available, use
Appropriate filters for red cell and platelet transfusion2. CMV seropositive recipient or seronegative recipient with positive donor
VABMT Supportive Care Guidelines 2013 Page 14 of 38

a. These recipients are not eligible for CMV screened blood components. Since they have a higher risk of getting CMV infections, they must be monitored regularly for CMV disease or reactivation
G. Leucocyte Depletion
The practice of leucocyte depletion varies in different places. In some countries, universal leucodepletion is organised. The indications agreed by most people are:
1. Pre-HSCT patients with severe aplastic anemia to reduce the like hood of graft failure2. To prevent recurrent febrile non-hemolytic transfusion reaction (FNHTR) during and
after transplantation3. As an alternative to CMV seronegative components
VABMT Supportive Care Guidelines 2013 Page 15 of 38

VIVA-ASIA BMT
Hepatic Veno-occlusive Disease
I. Introduction:
Hepatic veno-occlusive disease (VOD) is a syndrome of hepatic dysfunction caused by damage of sinusoidal endothelial cells leading to subendothelial edema, RBC extravasation, fibrin deposition and occlusion of central venules.
A. Risk factors for the development of hepatic VOD include the following:1. Young age (especially <=2 years)2. Pre-existing liver disease (HBV, HCV, hepatic fibrosis, cirrhosis)3. Transplant for Thalassemia major, advanced malignancies, SCID4. Past history of VOD5. Previous abdominal irradiation6. Previous exposure to myeloablative regimen7. Pre-existing liver dysfunction (AST> 4x upper limit of normal, hyperbilirubinemia,
hypoalbuminemia)8. Allogeneic transplant, unrelated transplant, HLA-mismatched transplant, second or
subsequent transplant9. Use of myeloablative regimen 10. Use of conditioning regimen containing busulfan, cyclophosphamide, high dose TBI11. Exposure to dacarbazine, actinomycin D, Ara C, thioguanine, mercaptopurine,
terbinafine, urethane, and gemtuzumab ozogamicin (Mylotarg)12. Acute GVHD13. Delayed platelet engraftment14. Carriers of hemochromatosis gene C282Y allele, or glutathione S-transferase M1 null
allele.
B. Predictors for mortality include the following:1. Severe hepatic VOD2. Allogeneic transplant with non-sibling donors3. Severe hepatic GVHD4. Admission to ICU5. Weight gain >9%6. Presence of pleural effusion
VABMT Supportive Care Guidelines 2013 Page 16 of 38

7. D+21 bilirubin >200umol/L8. Peak bilirubin >300umol/L9. Rise of bilirubin >15umol/L/day in the first week after VOD onset
II. Diagnosis
Seattle criteria Modified Seattle criteria Baltimore criteria2 of 3 criteria within first month after HSCT
1. Jaundice2. Hepatomegaly AND
RUQ pain3. Ascites AND/OR
unexplained weight gain
2 of 3 criteria within 20 days of HSCT
1. Hyperbilirubinemia (T.bil >=34 umol/L)
2. Hepatomegaly OR RUQ pain of liver origin
3. Unexplained weight gain (>2% of baseline) because of fluid accumulation
Hyperbilirubinemia (T.bil >=34 umol/L) within 21 days of HSCT AND 2 of 3 criteria
1. Tender hepatomegaly2. Weight gain >5% from
baseline3. Ascites
III. Prevention
A. Avoid using high risk regimen in high risk patient.B. Use of intravenous instead of oral busulfan. C. Adjust dose of busulfan according to plasma concentration if available.D. Ursodeoxycholic acid 6mg/kg po BID for the first month in all allogeneic HSCT.E. Alternative prophylactic medications to be considered in high risk patients 1. Defibrotide: 10-40mg/kg/day IV (divided in 4 doses) 2. Heparin: 100units/kg/day IV or Low molecular weight heparin (enoxaparin): 1mg/kg SC
Q12H or Q24H 3. Danaparoid: 30 units/kg IV Q12H 4. Prostaglandin E1: 0.075-0.4microgram/kg/hour
5. Glutamine: 2g/m2 daily po
IV. Evaluation
A. Frequent liver function monitoringB. Ultrasound liver + Doppler studies: hepatomegaly, ascites, attenuated or reversed portal
venous blood flow with/without collateral circulation (late finding). Perfusion index >0.3 is suggestive of VOD. None of these findings is sensitive enough, but helps in excluding extrahapatic biliary obstruction and malignant infiltration.
VABMT Supportive Care Guidelines 2013 Page 17 of 38

C. Measuring transjugular hepatic venous pressure gradient (HVPG): only indicated when diagnosis is problematic and when potentially hazardous therapies are planned. HVPG >10mmHg is >90% specific and 60% sensitive for VOD
D. Consider liver biopsy
Early changes thickening of subintimal zone of central and sublobular venules, luminal narrowing, widening of subendothelial zone by
fragmented RBC, edema and fibrinogen/fibrin.
Intermediate changes subintimal lesions become fibrotic, with ingrowth of small vascular channels and centrilobular scarring
Late changes perivenular fibrosis, pericellular fibrosis, central-central fibrous bridges, cirrhosis
V. Management
A. Supportive care:1. Monitor body weight closely.2. Measure central venous pressure if available.3. Prevent and relieve fluid accumulation by fluid restriction and diuretics.4. Low dose dopamine (<3ug/kg/min) to maintain urine output.5. Consider vitamin K.6. Consider increase dose of ursodeoxycholic acid to maximum 10mg/kg po QID.
B. Specific treatment:1. Defibrotide: start with 10-20mg/kg/day IV (divided in 4 doses), can increase dose
gradually up to 60mg/kg/day. Effective in 35-55% of cases.2. Consider antithrombin III replacement if level <70%.3. Consider prostaglandin E1 infusion.4. Consider low dose heparin infusion.5. Consider glutamine and high dose vitamin E.6. Consider tissue plasminogen activator (tPA): improvement in <30% of cases, high risk of
hemorrhage.7. Transjugular intrahepatic portosystemic shunt (TIPS): decompresses portal circulation,
relieves ascites, but effect on outcome is controversial. 8. Liver transplantation: in selected cases in the absence of malignancy or other significant
organ impairment.
VI. References:
1. Helmy A. Review article: updates in the pathogenesis and therapy of hepatic sinusoidal obstruction syndrome. Aliment Pharmacol Ther 23: 11-25, 2006.
2. Cheuk DKL, Wang P, Lee TL, et al. Risk factors and mortality predictors of hepatic veno-occlusive disease after pediatric hematopoietic stem cell transplantation. Bone Marrow Transplant 40: 935-44, 2007.
VABMT Supportive Care Guidelines 2013 Page 18 of 38

3. Tay F, Tinmouth A, Fergusson D, et al. Systematic review of controlled clinical trials on the use of ursodeoxycholic acid for the prevention of hepatic veno-occlusive disease in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 13: 206-17, 2007.
4. Ho VT, Revta C, Richardson PG. Hepatic veno-occlusive disease after hematopoietic stem cell transplantation: update on defibrotide and other current investigational therapies. Bone Marrow Transplant 41: 229-37, 2008.
5. Sakaguchi H, Watanabe N, Muramatsu H, et al. Danaparoid as the prophylaxis for hepatic veno-occlusive disease after allogeneic hematopoietic stem cell transplantation in childhood hematological malignancy. Pediatr Blood Cancer 55: 1118-25, 2010.
6. Peres E, Kintzel P, Dansey R, et al. Early intervention with antithrombin III therapy to prevent progression of hepatic venoocclusive disease. Blood Coagul Fibrinolysis; 2008: 203-7.
VABMT Supportive Care Guidelines 2013 Page 19 of 38

VIVA-ASIA BMT
Renal Complications Following Hematopoietic Stem cell Transplantation
I. Introduction:
In the setting of HSCT, multiple conditions are associated with renal dysfunction. Pathogeneic factors include conditioning chemotherapy and irradiation, cytokine production ( released by recipient’s natural immunocyte in early stage due to tissue damage and late, by donor T lymphocyte initiated immunologic reaction) , microorgnism infection and its therapy, nephrotoxicity from GVHD prophylatic agents, and chronic graft versus host reaction.
The clinical features of renal injury after HSCT mainly include three types. 1) The majority of the patients are featured with decreased GFR (glomerular filtration rate), which is conventionally termed as ARF and CRF (acute and chronic renal failure) respectively, according the diagnostic time on an early (within 100 days) and later (100 days and thereafter) period after HSCT. 2) The second type is characterized by nephrotic syndrome, the onset of which is closely associated with cGVHD or changes of cGVHD medications. 3) The third group of symptons are actually the consequences due to systematic damage in microvessels, while the renal failure and urine abnormal are the local manifestation for MODF. HUS and/or TMA are the representives of this group.
II. Acute Renal Failure
A. Causes and risk factors of early renal dysfunction and ARF (~100 days):
1. Sepsis, hypovolemia, hypotension2. Conditioning regimen related injury 3. Drug nephrotoxicity: CSA, tacrolimus, amphotericin, aminoglycosides,
vancomycin, foscarnet, acyclovir, etc4. Cytokine/inflammation/endothelial damage: VOD, Engraftment syndrome5. Marrow infusion related toxicity6. Tumor lysis syndrome
In a report on early renal injury after HSCT, the most common reason for ARF in autologus HSCT is sepsis while that in allogeneic HSCT is CSA toxity and VOD. Low albumin level is the sole predictive factor for ARF and patient with sepsis and ARF implicate a poor survival in all patients studied.
VABMT Supportive Care Guidelines 2013 Page 20 of 38

B. Definition and Grading of ARF
1. ARF is defined as at least a doubling of pre-HSCT serum Cr in the first 3 months post -HSCT.
2. GradingGrade 0 decrement in CrCl < 25% from the baseline valuesGrade 1 < 2 fold increase in serum Cr to baseline ; but decrease
in CrCl > 25% baselineGrade 2 2 fold rise in serum Cr to baseline but no need for ≧
dialysisGrade 3 2 fold rise in serum Cr and need for dialysis≧
Grade 0 is the same as normal renal function whereas Grade 2 and 3 renal dysfunction are considered severe renal dysfunction or ARF.
C. Definition of CRF
1. CRF is defined as decline in CrCl persisting for 3 months as follows
CrCl <30mL/min./1.73m2 for more than 3 months old infant CrCl <44 mL/min./1.73m2 for more than 6monthsCrCl <70 mL/min.1.73m2 for more than 1 yearsCrCl <80 mL/min.1.73m2 for more than 2 years
III. Late renal disorders seen > 100 days after HSCT
A. CSA/Tacrolimus toxicity 1. CSA toxicity was one of the most common reason for renal dysfunction in
allogeneic HSCT patients (40%).B. BMT nephropathy
1. This is due to apoptosis ocurring in renal cells following radiation injury. Found in up to 25% of transplanted adults, this contributes to the most common reason for CRF or end stage renal disease with a very poor outcome in terms of survival. The form of acute radiation nephritis develops 6-12 months after irradiation, whereas chronic radiation nephritis develops years later.
C. Nephrotic syndrome1. Overall incidence is 1% as reported in a paper including 889 HSCT cases. Onset
ranges between 7-12 months after HSCT . It is considered as a renal targeted cGVHD.
2. Pathology shows that NS after HSCT comprises mainly membranous glomerulonephritis (MGN) in 2/3 patients and minimal change disease (MCD) in 1/4 patients.
3. Like other organ cGVHD, NS can be resolved by immunosuppression therapy, but relapse of maligancies have been seen in cases not long after remission of NS. There are cases that have shown a very good response with rituximab.
VABMT Supportive Care Guidelines 2013 Page 21 of 38

IV. Laboratory examination and monitoring parameters for renal dysfunction
A. 24 hour CrCl B. Urine analysisC. 24 hour urine protein quantitationD. Urine microprotein serialE. Renal ultrasonographyF. Daily blood pressure
V. Supportive Care measures to prevent renal dysfunction in patients undergoing HSCT
Since ARF in combination with other HSCT associated complications was reported to correlate to a poor survival in HSCT recipients, supportive care to protect the kidney should be emphasized in all transplant practice, and work-up to screen and monitor of kidney dysfunction and structure injury should be more detailed than current practices.
A. Prior to HSCT, renal function should be evaluated in a very careful and precise way using urinalysis, GFR and ultrasonography. 1. Recurrence of abnormality in urine analysis such as proteinuria or hematuria should be
further evaluated by serial urine microprotein or other examination to exclude a pre-HSCT injury in renal parenchyma. In late condition, nephrotoxic drug should avoid during HSCT.
B. Persistent high BP due to steroids or CSA should be managed to control to a acceptable range for high BP might have a negative impact in impaired renal function and accelerate the progress of CRF into end stage.
C. Urinalysis, GFR and ultrasonography should be monitored at 3, 6, and 12 months, then yearly thereafter rather than only examined before HSCT to screen cases at risk of late renal disease.
D. Drug level should be monitored weekly in patient receiving CSA or tacrolimus.1. For cyclosporine, the dose of which should be adjust to maintain [CSA] in 200-250 within
day 30 and afterward 150-2002. For tacrolimus, dose should be adjusted to maintain the level at 8-10. As voriconazole
arise the blood concentration of CSA and tarolimus, it is suggested that drug level be tested (within 3 days) after voriconazole administration.
E. During period of conditioning and donor stem cell infusion, vigorous hydration and alkalinization should be strictly adopted before and during marrow infusion to preventi regimen and marrow infusion related toxicity. In case of HSCT for CML/JMML/MDS, who without prior chemotherapy to alleviate to hypercellularity in marrow, measures to prevent tumor lysis syndrome should be implemented.
F. During peri-engraftment period, factors such as hypoalbuminemia or hypovolemia should be carefully avoided. Transfusions of PRBC, platelets and albumin should be given to maintain the HB>100g/dL, albumin level>30g/L
VI. Indications for renal replacement therapy (RRT) in HSCT
A. Volume overload refractory to diuretics, causing trouble in intravenous therapy
VABMT Supportive Care Guidelines 2013 Page 22 of 38

B. Persistent hyperkalemia (K+>6.5 mmol/L) unresponsive to medical managementC. Neurologic symptoms (altered mental status, seizures)
VII. Indications for renal biopsy
A. CRF persist for 6 months B. Decline in GFR within 3 months when nephrotoxic drug stop for 6 month
VIII. References:
1. Caliskan Y, Besisik SK, Sargin D, Ecder T. Early renal injury after myeloablative allogeneic and autologous hematopoietic cell transplantation. Bone Marrow Transplantation 38, 141–147, 2006.
2. Cohen EP. Radiation nephropathy after bone marrow transplantation. Kidney Int 58: 903–918, 2000. http://emedicine.medscape.com/article/243766-overview Updated: Jul 22, 2008
3. Reddy P, Johnson K, Uberti JP, Reynolds C, Silver S, Ayash L, Braun TM,, Ratanatharathorn V. Nephrotic syndrome associated with chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation Bone Marrow Transplantation 38: 351–357, 2006.
4. Brukamp K, Doyle AM, Bloom RD, Bunin N, Tomaszewski JE, Cizman B. Nephrotic Syndrome after Hematopoietic Cell Transplantation: Do Glomerular Lesions Represent Renal Graft-versus-Host Disease? Clin J Am Soc Nephrol 1: 685–694, 2006.
5. Kist-van Holthe JE, Bresters D, Ahmed-Ousenkova YM, Goedvolk CA, Abbink FC,Wolterbeek R, Bredius RG, Pauwels EK, van der Heijden AJ. Long-term renal function after hemopoietic stem cell transplantation in children Bone Marrow Transplantation 36: 605–610, 2005.
VABMT Supportive Care Guidelines 2013 Page 23 of 38

VIVA-ASIA BMT
Transplant-Associated Microangiopathy
I. Introduction
Transplant-associated thrombotic microangiopathy (TA-TMA) refers to a severe vascular damage related complication of HSCT. Clinically it encompasses a spectrum of syndromes that includes: (1) calcineurin inhibitor associated nephrotoxocity with microangiopathic hemolytic anemia (MAHA), (2)calcineurin inhibitor associated neurotoxicity with MAHA, (3) conditioning regimen associated hemolytic uremic syndrome and (4) fulminating multifactorial thrombotic microangiopathy.
The incidence of TA-TMA varies widely, ranging between 0.5-76%, mostly due to differences in diagnostic criteria. A retrospective review from St. Jude Children’s Research Hospital showed that hemolytic-uremic syndrome (HUS) developed in 9.6% of children transplanted between 1992-1999. Recently, in twenty children who received both sirolimus and tacrolimus, 23.8% developed TA-TMA. In the autologous transplant setting, a 30% prevalence in TA-TMA was reported in children with neuroblastoma.
The pathogenesis of TA-TMA is related to endothelial cell damage, leading to apoptosis, leukocyte adhesion, activation of platelets and thrombi formation. Multiple factors such as drug toxicity (use of calcineurin inhibitors, sirolimus), radiation, opportunistic infections, unrelated/mismatched donor grafts and graft versus host disease have been identified as associated with an increased risk of developing TA-TMA. In children, an increased risk of HUS was also seen with older donor age, use of ATG, and recipient CMV seronegativity. In contrast to classic thrombotic thrombocytopenic purpura (TTP), it is not associated with severe deficiency of ADAMTS13.
II. Diagnosis and Grading
A. Diagnostic Criteria
2005 BMT-CTN 1. RBC fragmentation and > schistocytes per high power field on peripheral film
2. concurrent increased serum LDH above institutional baseline
3. concurrent renal and/or neurologic dysfunction without
VABMT Supportive Care Guidelines 2013 Page 24 of 38

other explanation (doubling of serum creatinine from baseline or 50% decrease in creatinine clearance from baseline)
4. negative direct and indirect Coomb’s test
2007 EBMT-IWG 1. increased percentage (>4%) of schistocytes in the blood
2. de novo, prolonged or progressive thrombocytopenia (platelet count < 5 x 109/L oro a 50% or greater decrease from previous counts)
3. sudden and persistent decrease in LDH
4. decrease in hemoglobin concentration or Increased red blood cell transfusion requirement
5. decrease in serum haptoglobin concentration
B. Grading (Miano et al, 2008)
III. Prevention and early detection
A. Close monitoring ang maintenance of calcineurin inhibitor levels, LDH and creatinineB. Early index of suspicion – early evaluation of peripheral blood smear and other tests in
patients with rising/persistently elevated levels of the above
IV. Evaluation
A. CBC, peripheral blood smearB. LDHC. Serum haptoglobinD. Renal function testE. Coomb’s test
IV. Management
A. Initial management includes discontinuation of calcineurin inhibitor or dose reduction to maintain lowest possible therapeutic level
B. Alternative agents commonly used include mycophenolate mofetil, corticosteroid, azathioprine
VABMT Supportive Care Guidelines 2013 Page 25 of 38
Grade
1 Schistocytosis w/o clinical consequences2 Schistocytosis + creatinine x 1-33 Schistocytosis + creatinine x 34 Schistocytosis + renal failure requiring dialysis + encephalopathy

C. Management of concurrent problems such as infection, GVHD, hepatic VOD are also important
D. Aside from the above, there has been no consensus as to best approach to therapy. Nevertheless, several agents have been tried and have demonstrated benefit as reported in small combined adult/pediatric series and a few pediatric cases, as follows:
Agent Administration Comment
Daclizumab 2mg/kg LD then 1mg/kg IV q wk
N=13 ; youngest patient 18 years old with CR
Defibrotide 30-50 mg/kg PO daily N=12; 3 pediatric patients all with CR
Rituximab 375 mg/m2 IV q wk x 4 N=5, 1 pediatric patient with response
E. Although plasma exchange has been widely employed, its efficacy in the treatment of TA-TMA is variable As it is also associated with significant adverse effects (i.e. infection, catheter thrombosis, bleeding, etc), it is no longer recommended.
V. References
1. Kojouri K, George JN. Thrombotic microangiopathy following allogeneic hematopoietic stem cell transplantation. Curr Op Oncol 19: 148-154, 2007.2. Hale G, Bowman L, Rochester R, et al. Hemolytic uremic syndrome after bone marrow transplantation: clinical characteristics and outcome in children. Biol Blood Marrow Transplant 11: 912-920, 2005.3. Rosenthal J, Pawlowska A, Bolotin E, et al. Transplant-associated thrombotic microangiopathy in pediatric patients treated with sirolimus and tacrolimus.
Pediatric Blood Cancer 57(1): 142-146, 2011.4. Laskin B, Goebel J, Davies S, et al. Early clinical indicators of transplant-associated thrombotic microangiopathy in pediatric neuroblastoma undergoing auto SCT. Bone Marrow Transplant 46(5):682-689, 2011.5. Uderzo C, Fumagalli M, De Lorenzo P, et al. Impact of thrombotic thrombocytopenic purpura on leukemic children undergoing bone marrow transplantation. Bone Marrow Transplant 26:1005-1009, 2000.6. Ho V, Cutler C, Carter S, et al. Blood and marrow transplant clinical trials network toxicity
committee consensus summary: thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 11: 571-575, 2005.
7. Ruutu T, Barosi G, Benjamin R, et al. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica 92: 95-100, 2007.
8. Miano M, Faraci M, Dini G, et al. Early complications following haematopoietic SCT in children. Bone Marrow Transplant 41: S39-S42, 2008.
VABMT Supportive Care Guidelines 2013 Page 26 of 38

9. Wolff D, Wilhelm S, Hahn J, et al. Replacement of calcineurin inhibitors with with daclizumab in patients with transplantation-associated microangiopathy or renal insufficiency associated with graft versus host disease. Bone Marrow Transplant 2006; 38: 445-451, 2006.
10. Au W, Ma E, Lee T, et al. Successful treatment of thrombotic microangiopathy after haematopoietic stem cell transplantation with rituximab. Br J Haematology 137: 475-478, 2007.
11. Corti P, Uderzo C, Tagliabue A, et al. Defibrotide as a promising treatment for thrombotic thrombocytopenic purpura in patients undergoing bone marrow transplantation. Bone Marrow Transplant 29; 542-543, 2002.
VABMT Supportive Care Guidelines 2013 Page 27 of 38

VIVA-ASIA BMT
Hemorrhagic Cystitis
I. Introduction:
Hemorrhagic cystitis refers to the diffuse inflammation of the urinary bladder resulting in sustained hematuria and lower urinary tract symptoms, in the absence of bacterial or fungal infection, bleeding diathesis, neoplasm or urinary calculi. The reported incidence in children following HSCT is variable, ranging 3.6-70% and may be a result of differences in definition and grading.
Pathophysiologic risk factors that have been identified as associated with hemorrhagic cystitis include: (a) direct toxicity to urotradiation induced microscopic obliterative endarteritis/mucosal ischemia and immunosuppression that may facilitate viral cytopathy and capillary fragility, (b) radiation induced microscopic obliterative endarteritis/mucosal ischemia and immunosuppression that may facilitate viral cytopathy, (c) viral cytopathic effects which may be exacerbated by graft versus host disease, and (d) other factors such as the use of unrelated donors and intensity of immunosuppression. Early onset (occurring 2-3 days after conditioning) is mostly attributed to the conditioning regimen whereas late onset (occurring more than 3 days after conditioning and within 6 months post transplant) is associated with decreased cellular immunity, graft versus host disease and viral infection (BK, adenovirus, CMV, JC virus).
II. Management:
A. Evaluate severity - although there are a variety of grading systems for classifying the severity of hemorrhagic cystitis, the Droller Grading system is the most commonly used
Table 1 Droller Grading System for Hemorrhagic Cystitis
Grade
1 Microscopic hematuria2 Macroscopic hematuria3 Macroscopic hematuria with small clots4 Gross hematuria with clots causing urinary tract
obstruction requiring instrumentation for clot evacuation
VABMT Supportive Care Guidelines 2013 Page 28 of 38

B. Laboratory studies
1. Send urine for urinalysis, urine cytology, urine culture (bacterial and fungal) and viral studies (serology and PCR)
2. Serum –viral serology and PCR for commonly associated viruses such as BK, adenovirus and JC virus
C. Diagnostic imaging
1. Ultrasonographic evaluation of the bladder and kidneys may be warranted for those patients with severe diseas
D. Intervention
1. Initial intervention for all patients includes hyperhydration and forced diuresis, correction of any concurrent coagulopathy by platelet or fresh frozen plasma transfusion as well as pain control using opiates and antispasmodics. Primary therapydirected to any identified viral or other microbial cause should likewise be given.
2. If there is no response or there is progression of symptoms, continuous bladder irrigation, cystoscopy with clot evacuation and electrocoagulation of bleeding sites as we well as systemic therapy may be employed,
3. Various systemic therapies may also be employed as shown in Table 2
Table 2. Systemic agents used in hemorrhagic cystitis
Agent Administration Action
Conjugated estrogen
12.5-50 mg IV BID2.5-5 mg PO BID-TID
Stabilization of microvasculature, promotes tissue repair
Ciprofloxacin 500 mg PO BID200 mg IV BID
Decrease BK viral replication, reactivation in adults
Cidofovir Various doses 3-5 mg/kg
Antiviral
Recombinant Factor VII
80 mcg/kg initial dose foll by up to two doses 120 mcg/kg given 3 hrs apart
Hemostatic agent
Sodium 100 mg PO TID Reduces inflammatory response of endothelium by
VABMT Supportive Care Guidelines 2013 Page 29 of 38

pentosan polysulfonate
then 100 mg QD
replacing depleted surface glycosminoglycans
Risperidone ? 5 HT2a antagonist(5HT2a receptor required for JCvirus
Hyerbaric oxygen
100% 1.4-3 atm over 1-2 hrs
Promotes capillary angiogenesis, tissue healing
1. For children with refractory hemorrhagic cystitis, the intravesical administration of the following may likewise be considered (Table 3)
Table 3. Intravesical Therapy
Agent Administration Action
Formalin 1-10% solution by gravity with pressure < 15 cm H2O over 15 min
Protein cross linking fixes bladder mucosa
1% Alum 1% solution, CBI rate 200 ml/hr
Protein precipitation, vasoconstriction, decrease capillary permeability
Silver nitrate 0.5-1% over 10-20 minutes Chemical coagulation of bleeding sitesProstaglandin Carboprost tromethamine
0.1-0.4mg%Regulates mucus productionContraction of smooth muscle of blood vesselsPlatelet aggregation
E-aminocaproic acid
5 g LD then 1 g/hr x 8 hrs (max 30 gm)
Prevents clot lysis by plasmininhibition
Cidofovir 5 mg/kg in 60 ml normal saline over 1 hr
Antiviral (vs. BK and Adeno) control of replication
Sodium hyaluronate
40 mg in 50 ml solution over 20 min.
Mechanical protection from urothelial damage;? inhibit viral replication
Fibrin glue Via air distension Sealant
III. References
1. Hale GA, Rochester RJ, Heslop HE, et al. Hemorrhagic cystitis after allogeneic bone marrow transplantation in children: clinical characteristics and outcome. Biol Blood Marrow Transpl 9: 698-705, 2003.
2. Decker DB, Karam JA, Wilcox DT. Pediatric hemorrhagic cystitis. J Pediatr Urol 5: 254 -264, 2009.
VABMT Supportive Care Guidelines 2013 Page 30 of 38

3. Leung AY, Wong AS, Chan GC, et al. Urinary activation of polyoma BK virus but not adenovirus in paediatric and adolescent patients after hematopoietic stem cell transplantation. Bone Marrow Transplant 37: 621-623, 2006.
4. Cesaro S. Brugiolo A, Faraci M,e t al. Incidence and treatment of hemorrhagic cystitis in children given hematopoietic stem cell transplantation: A survey from the Italian association of pediatric hematology oncology-bone marrow transplantation group. Bone Marrow Transplant 32: 925-931, 2003.
5. Gorcynska E, Turkeiwicz D, Rybka K, et al. Incidence, clinical outcome, and management of virus induced hemorrhagic cystitis in children and adolescents after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 11: 797-804, 2005.
6. Droller MJ, Saral R, Santos G. Prevention of cyclophosphamide-induuced hemorrhagic cystitis. J Urol 1982; 20: 256-258, 1982.
7. Manikandan R, Kumar S, Dorairajan L. Hemorrhagic cystitis: A challenge to the urologist. Indian J Urol 2010; 20: 159-166, 2010.
8. Health JA, Mishra S, Mitchell S, et al. Estrogen as treatment of hemorrhagic cystitis in children and adolescents undergoing bone marrow transplantation. Bone Marrow Transplant 37: 523-526, 2006.
9. Purves JT, Graham ML, Ramakumar S. Application of fibrin glue to damaged bladder mucosa in a case of BK viral hemorrhagic cystitis. Urology 66: 641-643, 2005.
10. Ashrani AS, Gabriel DA, Gajewski JL, et al. Pilot study to test the efficacy and safety of activated recombinant factor VII (NovoSeven) in the treatment of refractory hemorrhagic cystitis following high dose chemotherapy. Bone Marrow Transplant 2006: 38: 825-828, 2006.
11. Miodosky M, Abdul-Hai A, Tsirigotis P, et al. Treatment of hematopoietic stem cell transplantation hemorrhagic cystitis with intravesical sodium hyaluronate. Bone Marrow Transplant 2006; 38: 507-511, 2006.
12. Bridges B, Donegan S, Badros A. Cidofovir bladder installation for the treatment of BK Hemorrhagic cystitis after allogeneic stem cell transplantation. Am J Hematol 81:535-537, 2008.13. Cesaro S, Hirsch H, Faraci M, et al. Cidofovir for BK virus-associated hemorrhagic cystitis: a retrospective study. Clin Infect Dis 49: 233-240, 2009.14. Leung A, Chan M, Yuen K, et al. Ciprofloxacin decreased polyoma BK virus load in patients
who underwent allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 40: 528- 537, 2005.
VABMT Supportive Care Guidelines 2013 Page 31 of 38

VIVA-ASIA BMT
Monitoring of Epstein-Barr Virus (EBV) Viral Load and Management of Post-transplant Lymphoproliferative Disease (PTLD)
I. Introduction:
Epstein-Barr virus (EBV) post-transplant lymphoproliferative disease (PTLD) is the result of outgrowth of EBV-infected B cells which is in turn due to an imbalance of latently infected B-cells and defective EBV-specific T-cell immunity. Post-transplant lymphoproliferative disease (PTLD) after HSCT is predominately derived from donor B cells and is typically occurs within the first 6 months after transplantation (1). In contrast, PTLD occuring after solid organ transplant is usually presented late and is of recipient in origin.
In the HSCT setting, the cumulative incidence of PTLD in 26,901 allogeneic HSCT recipients, was 0.2% with no T-cell depletion, 1.7% in patients receiving anti-thymocyte globulin (ATG) as in vivo T lymphocyte depletion for GVHD prophylaxis and / or treatment and 2.8% with selective T-cell ex vivo depletion (2). Hoegh-Petersen et al reported the incidence and mortality were up to 8.1% and 28% respectively in in vivo T-cell depleted HSCT recipients (3). Risk factors of developing PTLD include the degree of mismatch between donor and recipient, ex vivo or in vivo T cell depletion, and the degree and duration of immunosuppressants used to prevent and treat graft-versus-host diseases (GVHD) (4). Recently, Cheng et al also reported that PTLD can also complicate unrelated umbilical cord blood transplantation (5).
In HSCT recipient of T-cell depleted grafts, it was shown that an elevated EBV-DNA viral load was highly predictive of EBV-PTLD. Moreover, Rowe et al showed that EBV viral load in peripheral blood of transplant patients with PTLD was higher than transplant patients without PTLD and healthy EBV carriers (7). However, follow-up studies included a broader range of HSCT recipients, showed that only 50% of patients with elevated EBV-DNA subsequently developed PTLD (8)
PTLD is a heterogenous group of disease which is classified into 4 categories: (i) early lesions; (ii) polymorphic type; (iii) monomorphic type and (iv) classic Hodgkin lymphoma-type PTLD. The diagnosis is mostly relied on tissue biopsy. This disease typically occurs before reconstitution of EBV-specific cytotoxic T lymphocyte (CTL) response. It can, however, occur later in the most severely immunocompromised patients namely patients with severe acute or chronic graft-versus-host diseases who are on multiple immunosuppressants.
II. Evaluation:
VABMT Supportive Care Guidelines 2013 Page 32 of 38

A. Significant attention has been focused on laboratory assays for early diagnosis of PTLD for two reasons
1. clinical presentation is non-specific and is easily confused with infections or GVHD and has fundamental difference in treatment directions
2. it is a reasonable assumption that early treatment improves survival and this needs to be further proven by randomized controlled studies
B. Polymerase chain reaction (PCR) 1. The sensitivity of PCR assays for diagnosis of PTLD in patients presenting with clinical
signs and symptoms ranged between 78%-100% whereas the sensitivity was 50%-80% when used for preemptive diagnosis.
2. About 20-60% of HSCT recipients developed subclinical reactivation of EBV detectable by PCR without the clinical presentation of PTLD.
3. Timing of monitoring of quantitative EBV-DNAa. Once every 2 wks in the first 6 months of transplant
4. Levels of EBV reactivationa. Low 1000-9999 copies per 105 PBMCb. Mid 10000-19999 copies per 105 PBMCc. High > 20000 copies per 105 PBMC
C. Biopsy1. Immunohistochemical staining of biopsy materials for CD3, CD5, CD10, CD15, CD20,
CD21, CD23, CD30, CD43, CD68, CD79a, Kappa, Lambda, TdT, MIB1 and cytokeratin is recommended
2. Immunohistochemical staining for Epstein-Barr Virus latent membrane protein (EBV-LMP)
3. Fluorescence -in-situ hybridization for Epstein-Barr encoded RNA (EBER)
III. Management
Up till now, there is no standardized strategy or consensus for monitoring and preemptive treatment of reactivation of EBV infection in pediatric allogeneic haematopoietic stem cell transplant recipients.
A. Asymptomatic patients1. For patients who develop low level or mid-level elevation of EBV DNA who are
asymptomatic, modulation of immuosuppressive therapy (if feasible) is a reasonable approach
2. Pre-emptive treatment with 1-2 doses of anti-CD 20 antibody (Rituxan, Genentech, Inc., 375mg/m2 once weekly) can be considered in patients (i) with high level EBV reactivation; or (ii) more than or equal to 50% rise in EBV viral load noted by serial EBV viral load monitoring
B. Symptomatic patients with elevated EBV DNA1. Early imaging (CT of neck, thorax, abdomen, pelvis)2. Biopsy of involved sites if technically feasible
C. Patients with localized disease1. Reduction of immunosuppressive therapy if feasible
VABMT Supportive Care Guidelines 2013 Page 33 of 38

2. Trial of anti-CD20 antibody (Rituxan, Genentech, Inc., 375mg/m2 once weekly for 4 weeks)
3. Surgery may be considered in selected casesD. Patients with high grade or extensive disease
1. Trial of anti-CD 20 antibody (Rituxan, Genentech, Inc., 375mg/m2 once weekly for 4 weeks )
2. Chemotherapy should be seriously considered in extensive diseaseE. Patients who progress after initial maneuvers
1. Donor T-cell infusion 2. EBV-specific T cell infusion.
IV. References:
1. Sirvent N, Reviron D, de Lamballerie X et al. First report of Epstein-Barr virus lymphoproliferative disease after cord blood transplantation. Bone Marrow Transplant 25(1): 120-121, 2000.
2. Landgren O, Gilbert ES, Rizzo JD, et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 113 :4992-5001, 2009.
3. Hoegh-Petersen M, Goodyear D, Geddes MN, et al. High incidence of post transplant lymphoproliferative disorder after antithymocyte globulin-based conditioning and ineffective prediction by day 28 EBV-specific T lymphocyte counts. Bone Marrow Transplant. 2010 Nov 8. [Epub ahead of print]
4. Gottschalk S, Rooney CM, Heslop HE. Posttransplant lymphoproliferative disorders. Ann Rev Med 56:29-44, 2005.
5. Cheng FWT, Lee V, KF To et al. Post-transplant EBV-related Lymphoproliferative Disorder Complicating Umbilical Cord Blood Transplantation in Patients of Adrenoleukodystrophy. Pediatr Blood Cancer 53:1329-1331, 2009.
6. Elstrom RL, Andreadis C, Aqui NA, et al. Treatment of PTLD with rituximab or chemotherapy. Am J Transplant 6:569-576, 2006.
7. Rowe DT, Qu L, Reyes J et al. Use of quantitative competitive PCR to measure Epstein-Barr virus genome load in the peripheral blood of pediatric transplant patients with lymphoproliferative disorders. J Clin Microbiol 35:1612-1615, 1997.
8. Weinstock DM, Ambrossi GG, Brennan C, et al. Preemptive diagnosis and treatment of Epstein-Barr virus asspciated post transplant lymphoproliferative disorder in hematopoietic stem cell transplant: an approach in development. Bone Marrow Transplant 37:539-546, 2006.
9. Omar H, Hagglund H Gustafsson-Jernberg A, et al. Targeted monitoring of patients at high risk of post-transplant lymphoproliferative disease by quantitative Epstein-Barr virus polymerase chain reaction. Transpl Infect Dis 11: 393-399, 2009.
10. Heslop HE. How I treat EBV lymphoproliferation. Blood 114:4002-4008, 2009.11. Fellner MD, Durand K, Correa M et al. A semiquantitative PCR method (SQ-PCR) to measure
Epstein-Barr virus (EBV) load: its application in transplant patients. J Clin Oncol 28:323-330, 2003.
12. Ganne V, Siddiqi N, Kamaplath B, et al. Humanized anti-CD20 monoclonal antibody (Rituximab) treatment for post-transplant lymphoproliferative disorder. Clin Transplant17:417-422, 2003.
VABMT Supportive Care Guidelines 2013 Page 34 of 38

VIVA-ASIA BMT
Idiopathic Pneumonia Syndrome
I. Introduction:
Idiopathic pneumonia syndrome (IPSS).refers to severe acute non-infectious lung injury complicating hematopoietic stem cell transplantation. It occurs in 3-15% of HSCT patients. In a recebt retrospective report from Japan, they reported an incidence of 8% in 251 children transplanted between 1990-2009. Other reported incidence A multivariate anyalysis showed that high risk underlying disease and a busulfan containing regimen were significant risk factors for the development of IPSS in the reported children. Other studies have identified other risk factors such as myeloablative conditioning with total body irradiation, graft versus host disease, increased patient age, diagnosis of acute leukemia or myelodysplastic syndrome. In both children and adults, mortality ranges from 50-80% even with aggressive therapy.
Potential causes of IPSS include conditioning regimen related pulmonary toxicity, release of inflammatory cytokines such as TNF-, immune cell-mediated injury and occult pulmonary infections.
II. Diagnosis:
A. Clinical featurse and definition:1. The clinical spectrum of IPSS encompasses several conditions characterized by injury to
pulmonary insterstitial, vascular, alveolar tissue following HSCT, in the absence of infection, cardiac, renal or iatrogenic causes.
2. Clinical conditions that have been included under the classification of IPSS includea. Acute insterstitial pneumonitisb. Acute respiratory distress syndromec. Delayed pulmonary toxicity syndromed. Peri-engraftment respiratory distress syndromee. Noncardiogenic caplillary leak syndromef. Diffuse alveolar hemorrhageg. Cryptogenic organizing pneumonia/bronchiolitis obliterans organizing pneumoniah. Bronchiolitis oblieterans syndrome
3. The American Thoracic Society Committee on Idiopathic Pneumonia syndrome in 2011 updated the the definition of IPSS as shown below:
I. Evidence of widespread alveolar injury a. Multilobar infiltrates on routine chest radiographs or computed tomography
VABMT Supportive Care Guidelines 2013 Page 35 of 38

b. Symptoms and sings of pneumonia (cough, dyspnea, tachypnea, rales) c. Evidence of abnormal pulmonary physiology 1. increased alveolar to arterial oxygen difference 2. new or increased restrictive pulmonary function test abnormalityII. Absence of active lower respiratory tract infection based upon: a. Bronchoalveolar lavage negative for significant bacterial pathogens including acid-fast bacilli, Nocardia, and Legionella species
b. Bronchoalveolar lavage negarive for pathogenic nonbacterial microorganisms:: 1. routine culture for viruses and fungi 2. shell vial culture for CMV and respiratory RSV 3. cytology for CMV inclusion, fungi, and Pneumocystis jirovecii (carinii) 4. direct fluorescence staining with antibodies against CMV, RSV, HSV, VZV, Influenza virus, parainfluenza virus, adenovirus, and other organisms c. Other organisms/tests to also consider: 1. polymerase chain reaction for human metapneumovirus, rhinovirus, corona- Virus, and HHV6 2. polymerase chain reaction for Chlamydia, Mycoplasma, and Aspergillus Species d. transbronchial biopsy if condition of patient permits
III. Absence of cardiac dysfunction, acute renal failure, or iatrogenic fluid overload as etiology of pulmonary dysfunction
4. Once suspected, a well coordinated multispecialty evaluation is warranted in order optimize care.
a. Chest X-ray and CTb. Bronchoscopy for BAL c. Cultures, cytopathology and direct fluorescence staining
III. Management:
A. Aggressive respiratory supportB. Evaluate fluid balance, renal function and cardiac outputC. Start broad spectrum antimicrobialsD. Aggressive transfusion support particularly for patients with pulmonary/alveolar
hemorrhageE. Start intravenous corticosteroids i.e.. methylprednisolone 2- 4 mg/kg/dayF. Consider aminocaproic acid for diffuse alveolar hemorrhageG. Consider addition of etanercept 0.4 mg/kg (max 25 mg) twice weekly up to
a maximum of 8 doses
IV. References:
1. Panoskaltis-Mortari A, Griese M, Madtes DK, et al. An official American Thoracic Society statement Noninfectious lung injury after hematopoietic stem cell transplantation Idiopathic pneumonia syndrome. Am J Respir Crit Care Med 183: 1262-1279, 2011.
VABMT Supportive Care Guidelines 2013 Page 36 of 38

2. Sakaguchi H, Takahashi Y, Watanabe N, et al. Incidence, clinical features, and risk factors of idiopathic pneumonia syndrome following hematopoietic stem cell transplantation in children. Pediatr Blood Cancer 8 780-84, 2012.
3. Fukuda T, Hackman R, Guthrie K, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regiments for allogeneic stem cell transplantation. Blood 102(8): 2777-2785, 2003.
4. Yanik G, Ho V, Levine J, et al. The impact of soluble tumor necrosis factor etanercept on the treatment of idiopathic pneumonia syndrome after allogeneic hematopietic stem cell transplantation. Blood 112(8): 307-3081, 2008.
5. Frangoul H, Koyama T and Domm J. Etanercept for treatment of idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Blood 113(8):2868, 2009.
VABMT Supportive Care Guidelines 2013 Page 37 of 38

Contributors
Topic 2011-2012 2013
1 Blood Transfusion Support Dr. SY Ha (HK) 2 Mucositis Dr. GCF Chan (HK)3 Nutritional Support4 Infection
a. prophylaxisb. neutropenic feverc. infection treatment
5. Engraftment Syndrome Dr. PL Tan (Sing) 6. GVHD
a. Prophylaxisb. Acute GVHDc. Chronic GHVD
Dr. MLU del Rosario (Phil)
7. Hepatic VOD Dr. DKL Cheuk, SY HA (HK) 8. Hemorrhagic Cystitits Dr MLU del Rosario (Phil) 9. Leukoencephalopathy10. Thrombotic microangiopathy Dr. MLU del Rosario (Phil) 11. Pulmonary complications
a. Idiopathic Pneumonia Syndrome Dr. MLU del Rosario (Phil) 12. Renal complications Dr. Changying Luo (Shanghai, CH) 13. Cardiac complications14. Post transplant lymphoproliferative
disordersDr.FWT Cheng (HK)Dr. ACW Lee (Sing)
VABMT Supportive Care Guidelines 2013 Page 38 of 38