blosum scoring matrices - iitkhome.iitk.ac.in/~rsankar/courses/lec04.pdfdr. r. sankar, bse 633...
TRANSCRIPT

Dr. R. Sankar, BSE 633 (2020)
BLOSUM Scoring Matrices
Derived from UNGAPPED alignments
Based on observed amino acid substitutions in a large set of ~2000 conserved AA patterns (Blocks)
These BLOCKS are constructed from a protein sequence database PROSITE
They represent 500 families of related proteins (similar biochemical functions); act as signatures
Different type of sequence analysis; much larger data set than PAM matrices Henikoff & Henikoff (1992)

Dr. R. Sankar, BSE 633 (2020)
PROSITE Database of protein families and domains. By analyzing the constant and variable properties of groups of similar sequences, it is possible to derive a signature for a protein family or domain, which distinguishes its members from all other unrelated proteins. A protein signature can be used to assign a newly sequenced protein to a specific family of proteins and thus to formulate hypotheses about its function. PROSITE currently contains patterns and profiles specific for more than a thousand protein families or domains. Each of these signatures comes with documentation providing background information on the structure and function of these proteins.
Dr. R. Sankar, BSE 633 (2020)
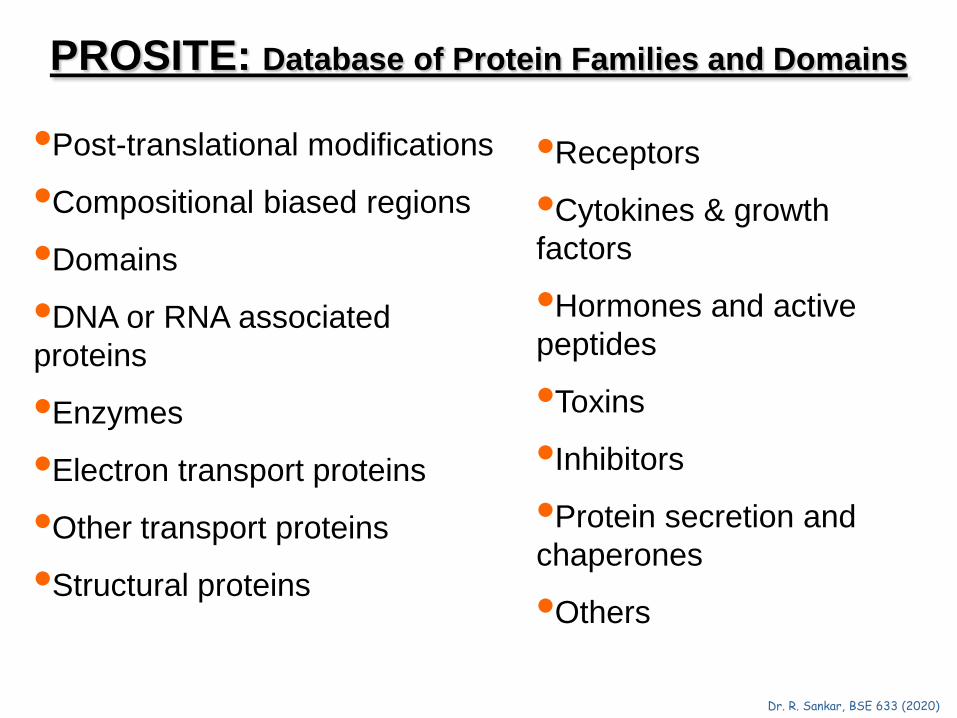
PROSITE: Database of Protein Families and Domains
•Post-translational modifications
•Compositional biased regions
•Domains
•DNA or RNA associated proteins
•Enzymes
•Electron transport proteins
•Other transport proteins
•Structural proteins
•Receptors
•Cytokines & growth factors
•Hormones and active peptides
•Toxins
•Inhibitors
•Protein secretion and chaperones
•Others

Dr. R. Sankar, BSE 633 (2020)

Dr. R. Sankar, BSE 633 (2020)
Dr. R. Sankar, BSE 633 (2020)
Dr. R. Sankar, BSE 633 (2020)
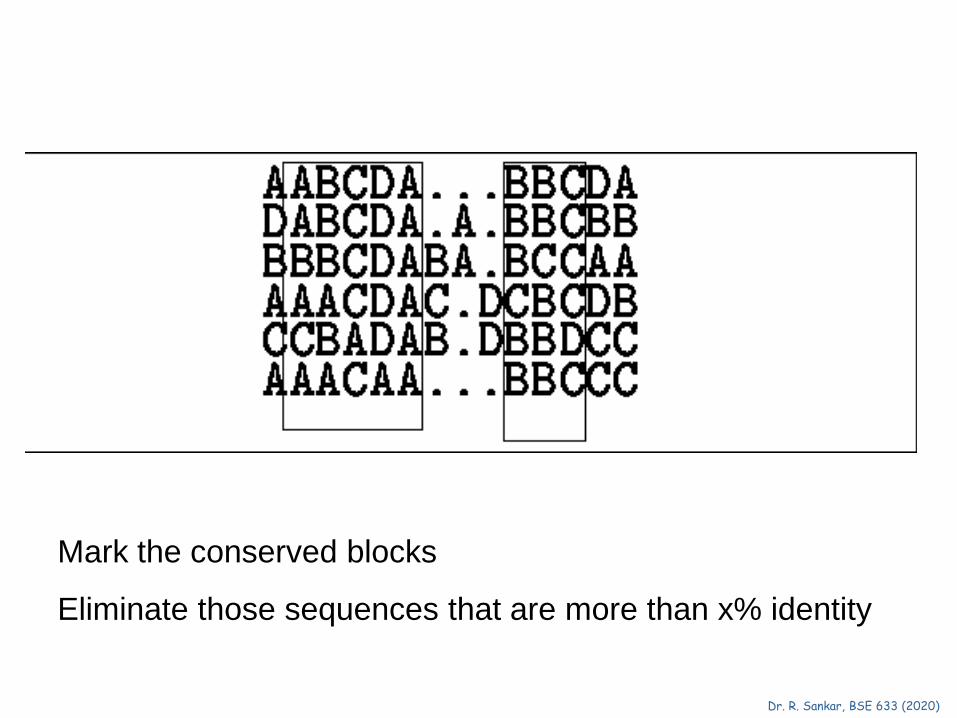
Mark the conserved blocks
Eliminate those sequences that are more than x% identity

Dr. R. Sankar, BSE 633 (2020)
Constructing a BLOSUM matr. 1. Counting mutations

Dr. R. Sankar, BSE 633 (2020)
2. Tallying mutation frequencies

Dr. R. Sankar, BSE 633 (2020)
3. Matrix of mutation probs.

Dr. R. Sankar, BSE 633 (2020)
4. Calculate abundance of each residue (Marginal prob)

Dr. R. Sankar, BSE 633 (2020)
5. Obtaining a BLOSUM matrix

Dr. R. Sankar, BSE 633 (2020)
If we expect to find a and b aligned together in homologous sequences more often than we expect
them to occur by chance (pab>fa fb), then the odds ratio is greater than one and the score is positive.

Dr. R. Sankar, BSE 633 (2020)
BLOSUM62 amino acid substitution matrix

Dr. R. Sankar, BSE 633 (2020)

Dr. R. Sankar, BSE 633 (2020)
PAM & BLOSUM Matrices
PAM60, PAM80, PAM120, PAM250: represent a level of 60%, 80%, 120% and 250% of change expected
250% level of change seems very large; sequences with this level of divergence still have about 20% similarity
BLOSUM60, BLOSUM80: derived from patterns that were 60% and 80% identical respectively
PAM – based on mutational model of evolution
BLOSUM: based on all amino acid changes observed in an aligned region from a related family of proteins
This is regardless of overall similarity between protein sequences

Dr. R. Sankar, BSE 633 (2020)
Choice of Scoring Matrix

Dr. R. Sankar, BSE 633 (2020)
Other scoring matrices
Simple identity; match or mismatch
Genetic code changes
Based on chemical similarity, molecular volume, polarity, hydrophobicity
Based on structurally aligned 3D structures
Based on 400X400 dipeptide substitution matrix
Transmembrane proteins

Dr. R. Sankar, BSE 633 (2020)
Gap Penalty
Gap opening penalty (g)
Gap extension penalty (r)
Gap score wx = g + rx (affine gap penalty)
wx = g + (r-1)x
x = length of the gap
When do you use penalties for end gaps?

Dr. R. Sankar, BSE 633 (2020)
Affine Gap Penalties • In nature, a series of k indels often come as a single
event rather than a series of k single nucleotide events:
Normal scoring would give the same score for both alignments
This is more likely.
This is less likely.
ATA__GGC ATGATCGC
ATA_G_GC ATGATCGC

Dr. R. Sankar, BSE 633 (2020)
End Gap Penalties No end gap penalty
Gaps will be liberally placed at the ends of alignments by DP
Usually best to include gap penalty for global alignments
Will not affect local alignment
Sequences of same length: Include end gap penalty
Sequences of unknown homology/different lengths:
May be better not to include end gap penalty
One sequence contained in the other: Reasonable to include end gap penalty for the shorter sequence
However, at least once test by including end-gap penalty

Dr. R. Sankar, BSE 633 (2020)
High mismatch and gap penalties:
local alignments
longest region of exact matches
Mismatch penalty > 2 * match:
gap penalty becomes a decisive parameter
Mismatch penalty < 2* gap penalty
large number of possible alignments
Different scenarios in alignments

Dr. R. Sankar, BSE 633 (2020)
Optimal combinations of scoring matrices and gap penalties
1.Some scoring matrices are superior to others at finding related proteins based on sequence or structure
2.Gap penalties that for a given scoring matrix are adjusted to produce a local alignments are the most suitable
3.Significance of alignment scores should be estimated to identify related sequences

Dr. R. Sankar, BSE 633 (2020)