caspase-3-mediated cleavage of phf-1 tau during …tres/papers/kang.2005.pdf · caspase-3-mediated...
TRANSCRIPT

www.elsevier.com/locate/ynbdi
Neurobiology of Disease 18 (2005) 450–458
Caspase-3-mediated cleavage of PHF-1 tau during apoptosis
irrespective of excitotoxicity and oxidative stress:
an implication to Alzheimer’s disease
Hyo Jung Kang,a,b Won Joo Yoon,a Gyeong Joon Moon,a Doo Yeon Kim,c Seonghyang Sohn,d
Hyuk Jae Kwon,d and Byoung Joo Gwagb,e,*
aDepartment of Neuroscience, Ajou University School of Medicine, Suwon, Kyungkido, KoreabDepartment of Pharmacology, Ajou University School of Medicine, Suwon, Kyungkido, KoreacNeurobiology of Disease Laboratory, Genetics and Aging Research Unit, Department of Neurology and Mass General Institute for Neurodegenerative
Diseases, Massachusetts General Hospital, Harvard Medical School, Charlestown, MA 01219, USAdLaboratory of Cell Biology, Ajou University School of Medicine, Suwon, Kyungkido, KoreaeCenter for the Interventional Therapy of Stroke and Alzheimer’s Disease, Ajou University School of Medicine, Suwon, Kyungkido, Korea
Received 9 March 2004; revised 13 November 2004; accepted 10 December 2004
Available online 25 January 2005
Excitotoxicity, oxidative stress, and apoptosis have been recognized as
routes to neuronal death in various neurological diseases. We examined
the possibility that PHF-1 tau, a substrate for various proteases, would
be selectively cleaved depending upon routes of neuronal death.
Cleavage form of PHF-1 tau was not observed in cortical cell cultures
exposed to excitotoxins or oxidative stress that cause neuronal cell
necrosis. PHF-1 tau was cleaved within 8 h following exposure of
cortical cell cultures to apoptosis-inducing agents. This cleavage was
blocked by inclusion of zDEVD-fmk, an inhibitor of caspase-3, and
accompanied by activation of caspase-3. Levels and cleavage of PHF-1
tau were markedly increased in AD brain compared with control.
Moreover, PHF-1 tau and active caspase-3 were colocalized mostly in
tangle-bearing neurons. The current findings suggest that PHF-1 tau is
cleaved by caspase-3 during apoptosis and neurodegenerative process
in AD.
D 2005 Elsevier Inc. All rights reserved.
Keywords: Alzheimer’s disease; Neurofibrillary tangles; Caspase-3;
Apoptosis; Tau; Glutamate
Introduction
Accumulating evidence suggests that oxidative stress, excitotox-
icity, and apoptosis comprise the major routes of neuronal death in
0969-9961/$ - see front matter D 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.nbd.2004.12.004
* Corresponding author. Department of Pharmacology, Ajou University
School of Medicine, San 5, Woncheon, 442-749, Suwon, Kyungkido,
Korea. Fax: +82 31 219 5069.
E-mail address: [email protected] (B.J. Gwag).
Available online on ScienceDirect (www.sciencedirect.com).
Alzheimer’s disease (AD). Redox-active iron is accumulated in
extracellular senile plaques and intracellular neurofibrillary tangles
(NFTs) in AD (Smith et al., 1997). Reduced energy metabolism,
advanced glycation end products, and beta amyloid can produce
oxidative stress in AD (Castellani et al., 2001). A great deal of work
reports increase in oxidation products of lipid, DNA, and protein in
AD (Ando et al., 1998; Smith et al., 1991). Glutamate transporters
are deficient in AD, which can cause accumulation of glutamate at
the synaptic cleft (Keller et al., 1997; Masliah et al., 1996). Cortical
neurons treated with beta amyloid are highly vulnerable to
excitotoxicity (Koh et al., 1990; Mattson et al., 1992). This implies
that oxidative stress and excitotoxicity may contribute to neuro-
degeneration in AD.
Recently, apoptosis has been proposed as additional type of death
in AD based upon prominent DNA damage positive to terminal
transferase-mediated dUTP-biotin nick end labeling (TUNEL) and
activation of caspases, a family of cysteine proteases mediating
apoptosis in various types of cells (Smale et al., 1995; Su et al.,
2000). However, TUNEL-positive cells and activated caspases have
been reported following exposure to H2O2 and hypoxic ischemic
injury that can cause necrosis as well as apoptosis (Cole and Perez-
Polo, 2002; Moroni et al., 2001). Thus, it remains to be resolved if
apoptosis contributes to neuronal death in AD.
Evidence has been accumulated suggesting that excitotox-
icity, oxidative stress, and apoptosis are propagated through
distinctive signaling pathways (Gwag et al., 2002). Recently, we
have observed that PHF-1-positive phosphorylated tau (PHF-1
tau), a major constituent of paired helical filaments (PHF) in
AD, is cleaved by caspase-3 in the process of calyculin A-
induced neuronal cell apoptosis (Ko et al., 2000). The present
study was aimed to examine the possibility that PHF-1 tau
would be cleaved selectively in the process of neuronal cell

H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458 451
apoptosis. We additionally studied cleavage of PHF-1 tau and
activation of caspase-3 in AD to determine if apoptosis would
contribute to neuronal death in AD as additional route besides
excitotoxicity and oxidative stress.
Materials and methods
Human brain tissue
Autopsy brain tissue from the cortex of five neuropatholog-
ically confirmed AD cases and three age-matched cases with no
AD changes or any other neurodegenerative condition, e.g.,
Pick’s disease, Parkinsons disease, Dementia with Lewy Bodies,
Progressive Supranuclear Palsy (Table 1). Each brain underwent
exhaustive neuropathological analysis. Human brain tissues used
in this study were provided by Boston University Alzheimer’s
Disease Center (Dr. Ann C. Mckee).
Cell culture
Cortical cells were prepared from fetal ICR mice (E15) and
mechanically triturated. Dissociated cells were plated on 24 well
plates (5 hemispheres/plate, approximately 105 cells/well) in a
plating medium consisting of Eagle’s minimal essential media
(MEM, Earle’s salts, 11090-081) supplemented with 5% horse
serum, 5% fetal bovine serum, 2 mM glutamine and 21 mM
glucose. Proliferation of non-neuronal cells was halted by adding
cytosine arabinoside (final concentration 10 AM) at 7–9 days in
vitro (DIV 7–9) when astrocytes became confluent. Cultures were
then fed twice a week with plating medium lacking fetal serum.
Cultures were maintained at 378C in a humidified 5% CO2
atmosphere.
Transmission electron microscopic study
Cultures were fixed in Karnovsky’s fixative solution (1%
paraformaldehyde, 2% glutaraldehyde, 2 mM calcium chloride,
100 mM cacodylate buffer, pH7.4) for 2 h, washed with cacodylate
buffer, and postfixed in 1% osmium tetroxide and 1.5% potassium
ferrocyanide for 1 h. Cells were then stained en bloc in 0.5% uranyl
acetate, dehydrated through a graded ethanol series and embedded
in Poly/Bed 812 resin (Pelco, CA, USA). Cells were sectioned
using Reichert Jung Ultracut S (Leica, Cambridge, UK). After
staining cells with uranyl acetate and lead citrate, cells were
observed and photographed under Zeiss EM 902 A.
Table 1
Case demographics
Case Group Age/sex
(years)
PMI
(h)
Neur
diagn
1 Control 66/M 8.5 Norm
2 Control 85/M 19.0 Norm
3 Control 73/M 17.0 Norm
4 AD 80/M 8.5 AD
5 AD 80/M 7.0 AD
6 AD 70/M 3.0 AD
7 AD 79/M 7.0 AD
8 AD 83/M 12.0 AD
Note. PMI, postmortem interval.
Western blot analysis
Cultured cells were lysed in a lysis buffer containing 50 mM
Tris–HCl pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5%
deoxycholic acid, 0.1% SDS, 1 mM PMSF, and 100 Ag/ml
leupeptin. Brain tissues were lysed in a lysis buffer containing
10 mM Tris–HCl pH 7.5, 50 mM NaCl, 1% Triton X-100, 30 mM
sodium pyrophosphate, 50 mM NaF, 5 AM ZnCl2, 2 mM PMSF,
and 100 Ag/ml leupeptin, 10 Ag/ml pepstatin A, and 1 mM DTT.
Lysates were centrifuged at 13,000 � g for 10 min, the
supernatants collected, subjected to electrophoresis on a 12%
SDS-polyacrylamide gel, and transferred to a nitrocellulose
membrane. The blot was incubated in 5% nonfat dry milk for
30 min, reacted with primary antibodies for overnight at 48C, andthen incubated with a biotinylated anti-mouse or rabbit secondary
antibody (1:1000) for 2 h. For internal controls, polyclonal
antibody for actin (Sigma, St. Louis, MO, USA, 1:1000) was
probed on the same blot. Signals were detected using the
VECTASTAIN ABC kit (Vector Lab., Burlingame, CA, USA)
and luminol as an enhanced chemiluminescence substrate (Amer-
sham, Buckinghamshire, UK), and then analyzed using Kodak X-
Omat film or an image analyzer LAS1000 (Fuji Photo Film Co.,
Ltd.). The PHF-1 tau monoclonal antibody recognizes tau
phosphorylated at Ser-396 and Ser-404 (a generous gift from
Dr. P. Davis, Department of Pathology, Albert Einstein College of
Medicine, Bronx, NY, USA, 1:1000). The Tau-5 monoclonal
antibody recognizes the tau at 200–233 amino acids independent
on phosphorylation state (a generous gift from Dr. Lester I Binder,
Northwestern University School of Medicine, Chicago, IL, USA,
1:5000). The rabbit polyclonal antibody for caspase-3 recognizes
active caspase-3 of 17–20 kDa (Cell Signaling, Beverly, MA,
USA, 1:1000) or pro caspase-3 of 32 kDa (Sigma, St. Louis, MO,
USA, 1:1000).
Immunohistochemistry
Brain sections were incubated in 10% normal horse serum
for 1 h, reacted with mouse monoclonal antibody recognizing
PHF-1 (1:200), rabbit polyclonal antibody recognizing active
caspase-3 (Cell Signaling, Beverly, MA, USA, 1:100) overnight,
and then reacted with FITC- or Texas-red-labeled secondary
antibodies (Vector, Burlingame, CA, USA, 1:200). To stain
nuclei, Hoechst (Molecular Probes, Eugene, OR, USA) was
applied for 10 min at RT. Finally, sections were washed with
distilled water, air dried, and mounted with Vectashield (Vector,
Burlingame, CA, USA). All images were collected and analyzed
opathological
osis
Braak and
Braak stage
Cause of death
al 0 Sepsis
al 0 Sepsis
al 0 Unknown
VI Pneumonia
VI Congestive heart failure
VI Urosepsis
VI Carcinoma of stomach
VI Pneumonia

H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458452
with a fluorescence microscopy (Zeiss, Germany) equipped with
the REAL-14k precision digital camera (Apogee Instruments,
Tucson, AZ, USA) and ImagePro Plus Plug-in.
Results
Selective cleavage of PHF-1 tau during apoptosis
We first examined if PHF-1 tau would be cleaved in the
process of apoptosis by staurosporine, a non-specific protein
kinase inhibitor known to cause apoptosis in various types of
cells as well as calyculin A. Cortical cell cultures exposed to 100
nM staurosporine or 10 nM calyculin A revealed apoptotic
features of degenerating neurons within 8 h that was evident by
cell body shrinkage, aggregated condensation of nuclear chro-
matin, and collapse of nuclear membrane prior to plasma
membrane (Figs. 1A–1C). As previously reported (Ko et al.,
2000), PHF-1 tau was cleaved within 8 h following exposure of
cortical cell cultures to calyculin A. PHF-1 tau was also cleaved
in cortical cell cultures exposed to staurosporine for 8–12 h
(Fig. 1D). We also observed cleavage of PHF-1 tau in the
process of neuronal cell apoptosis induced by BAPTA-AM, an
intracellular calcium chelator, or serum deprivation (data not
shown). This implies that PHF-1 tau is cleaved in the process of
neuronal cell apoptosis. Size of PHF-1 tau cleaved by
staurosporine or other apoptosis-inducing agents was similar to
that of caspase-3-mediated cleavage of PHF-1 tau by calyculin,
suggesting that treatment with pro-apoptotic agents induces
proteolytic cleavage of PHF-1 tau through activation of
Fig. 1. Cleavage of PHF-1 tau during neuronal apoptosis by staurosporine or ca
following sham control (A) or exposure of cortical cell cultures (DIV 11) to 100 n
evident by cell body shrinkage and aggregated condensation of nuclear chroma
mitochondria. Scale bar denotes 2.5 Am. (D) Western blot analysis of PHF-1 tau in c
calyculin A for indicated points of time.
caspase-3. This was supported by blockade of PHF-1 tau
cleavage in the presence of 100 AM zDEVD, an inhibitor of
caspase-3 (Fig. 2A). Caspase-3-mediated cleavage of tau was
observed using tau-5 antibody, a monoclonal antibody recogniz-
ing 45–68 kDa tau, in cortical cell cultures exposed to
staurosporine (Fig. 2B). Furthermore, the active form of
caspase-3 was observed within 4–8 h after exposure of cortical
cell cultures to staurosporine or calyculin A (Fig. 2C).
We next examined if neuronal death by excitotoxicity or
oxidative stress would be accompanied by cleavage of PHF-1 tau.
As previously reported (Gwag et al., 1995), cortical cell cultures
underwent widespread neuronal cell necrosis within 2 h following
exposure to low doses of excitotoxins, 30 AM NMDA or 50 AMkainite, that was evident by marked swelling of cell body and
mitochondria, scattering condensation of the nuclear chromatin,
and early fenestration of plasma membrane (Figs. 3A–3C). PHF-1
tau was not cleaved in cortical cell cultures exposed to excitotoxins
up to 2 h (Fig. 3D). As most cortical neurons treated with
excitotoxins for 2 h revealed lysis of plasma membrane, exposure
time longer than 2 h was avoided.
Oxidative stress was induced by exposing cortical cell cultures
to Fe2+ or menadione that produces hydroxyl radical or superoxide,
respectively. Most cortical neurons revealed marked cell body
swelling within 8 h after exposure to Fe2+ or menadione. The
degenerating neurons were accompanied by ultrastructural changes
reminiscent of necrosis that was observed in neurons undergoing
excitotoxic degeneration (Figs. 4A–4C). Cleavage of PHF-1 tau
was not observed in the process of neuronal necrosis following
exposure of cortical cell cultures to Fe2+ or menadione for up to 8 h
(Fig. 4D).
lyculin A. (A–C) Electron photomicrographs of cortical neurons taken 8 h
M staurosporine (B) or 10 nM calyculin A (C). Note the apoptotic neurons
tin. Abbreviations: PM, plasma membrane; NM, nuclear membrane; MI,
ortical cell cultures (DIV 10–12) exposed to 100 nM staurosporine or 10 nM
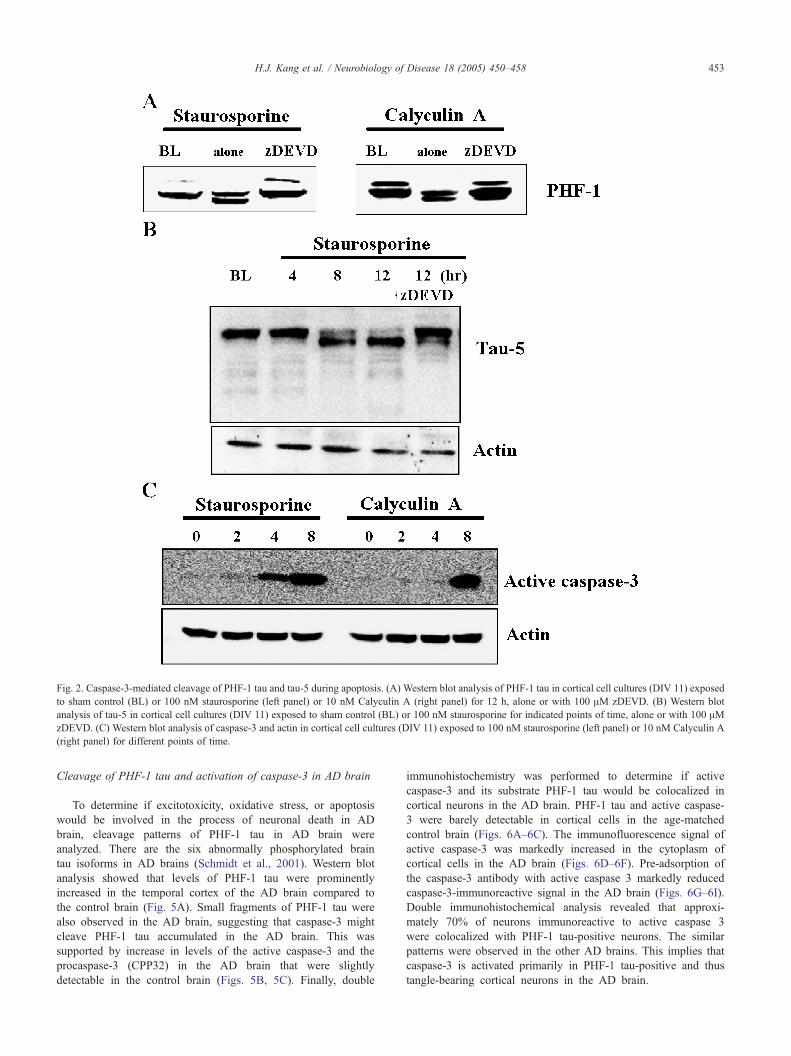
Fig. 2. Caspase-3-mediated cleavage of PHF-1 tau and tau-5 during apoptosis. (A) Western blot analysis of PHF-1 tau in cortical cell cultures (DIV 11) exposed
to sham control (BL) or 100 nM staurosporine (left panel) or 10 nM Calyculin A (right panel) for 12 h, alone or with 100 AM zDEVD. (B) Western blot
analysis of tau-5 in cortical cell cultures (DIV 11) exposed to sham control (BL) or 100 nM staurosporine for indicated points of time, alone or with 100 AMzDEVD. (C) Western blot analysis of caspase-3 and actin in cortical cell cultures (DIV 11) exposed to 100 nM staurosporine (left panel) or 10 nM Calyculin A
(right panel) for different points of time.
H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458 453
Cleavage of PHF-1 tau and activation of caspase-3 in AD brain
To determine if excitotoxicity, oxidative stress, or apoptosis
would be involved in the process of neuronal death in AD
brain, cleavage patterns of PHF-1 tau in AD brain were
analyzed. There are the six abnormally phosphorylated brain
tau isoforms in AD brains (Schmidt et al., 2001). Western blot
analysis showed that levels of PHF-1 tau were prominently
increased in the temporal cortex of the AD brain compared to
the control brain (Fig. 5A). Small fragments of PHF-1 tau were
also observed in the AD brain, suggesting that caspase-3 might
cleave PHF-1 tau accumulated in the AD brain. This was
supported by increase in levels of the active caspase-3 and the
procaspase-3 (CPP32) in the AD brain that were slightly
detectable in the control brain (Figs. 5B, 5C). Finally, double
immunohistochemistry was performed to determine if active
caspase-3 and its substrate PHF-1 tau would be colocalized in
cortical neurons in the AD brain. PHF-1 tau and active caspase-
3 were barely detectable in cortical cells in the age-matched
control brain (Figs. 6A–6C). The immunofluorescence signal of
active caspase-3 was markedly increased in the cytoplasm of
cortical cells in the AD brain (Figs. 6D–6F). Pre-adsorption of
the caspase-3 antibody with active caspase 3 markedly reduced
caspase-3-immunoreactive signal in the AD brain (Figs. 6G–6I).
Double immunohistochemical analysis revealed that approxi-
mately 70% of neurons immunoreactive to active caspase 3
were colocalized with PHF-1 tau-positive neurons. The similar
patterns were observed in the other AD brains. This implies that
caspase-3 is activated primarily in PHF-1 tau-positive and thus
tangle-bearing cortical neurons in the AD brain.

Fig. 3. PHF-1 tau is not cleaved during excitotoxicity. (A–C) Electron photomicrographs of cortical neurons taken 2 h following sham control (A) or exposure
of cortical cell cultures (DIV 12) to 30 AM NMDA (B) or 50 AM Kinate (C). Note the necrotic neurons evident by cell body swelling and scattering
condensation of nuclear chromatin. Abbreviations: PM, plasma membrane; NM, nuclear membrane; MI, mitochondria. Scale bar denotes 2.5 Am. (D) Western
blot analysis of PHF-1 tau in cortical cell cultures (DIV 10–12) exposed to 30 AM NMDA or 50 AM Kinate for indicated points of time.
H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458454
The present study suggests that caspase-3-mediated cleavage of
PHF-1 tau is selective in the process of neuronal apoptosis and
possibly contributes to apoptotic degeneration of tangle-bearing
neurons in AD.
Fig. 4. PHF-1 tau is not cleaved during free radical neurotoxicity. (A–C) Electron p
exposure of cortical cell cultures (DIV 12) to 50 AM Fe2+ (B) or 20 AM menadione
condensation of nuclear chromatin. Abbreviations: PM, plasma membrane; NM, n
blot analysis of PHF-1 tau in cortical cell cultures (DIV 10–12) to 50 AM Fe2+ o
Discussion
We demonstrate that PHF-1 tau is cleaved in neurons selectively
undergoing apoptosis induced by staurosporine, calyculin A, or
hotomicrographs of cortical neurons taken 8 h following sham control (A) or
(C). Note the necrotic neurons evident by cell body swelling and scattering
uclear membrane; MI, mitochondria. Scale bar denotes 2.5 Am. (D) Western
r 20 AM menadione for indicated points of time.

Fig. 5. Cleavage of PHF-1 tau and activation of caspase-3 in Alzheimer’s disease brain. (A) Western blot analysis of PHF-1 tau (top panel) and tau-5 (bottom
panel) in the temporal cortex obtained from the postmortem brain of 3 age-matched controls (CT) and 5 Alzheimer’s disease (AD) patients. (B–D) Western blot
analysis of active caspase-3 (B), procaspase-3(C), and actin (D) in the temporal cortex of 3 control group (CT) and 5 Alzheimer’s disease (AD) patients.
H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458 455
serum deprivation, but not necrosis induced by excitotoxicity or
oxidative stress in cultured cortical neurons. Cleavage of PHF-1 tau
depends upon activation of caspase-3. Cleaved PHF-1 tau and
activated caspase-3 were observed in the temporal cortex of the AD
brain and appeared to be colocalized to a great extent, suggesting
that activation of caspase-3 may contribute to cleavage of PHF-1 tau
and neuronal apoptosis in AD brain through mechanisms irrespec-
tive of excitotoxicity and oxidative stress.
The hyperphoshorylated forms of tau in the paired helical
filaments constitute the major component of neurofibrillary tangles
in AD brain (Goedert, 1993; Trojanowski and Lee, 1995). The
monoclonal antibody PHF-1 tau recognizes the phosphorylated
forms of tau at Ser396 or Ser404 that is sensitive to calpain or
caspase-3 (Han et al., 2001; Ko et al., 2000; Yang and Ksiezak-
Reding, 1995). Asp-Met-Val-Asp at the carboxy-terminal region of
PHF-1 tau is cleaved by caspase-3 during apoptosis induced by
calyculin A, a selective inhibitor of Ser/Thr phosphatase I and IIA
(Chung et al., 2001; Ko et al., 2000). We also found that PHF-1 tau
was cleaved in the course of neuronal apoptosis following exposure
to staurosporine as well as serum deprivation. Pro-apoptotic
cleavage of PHF-1 tau was accompanied by activation of caspase-
3 and blocked by a selective inhibitor of caspase-3. This suggests
that neuronal apoptosis is accompanied by caspase-3-mediated
cleavage of PHF-1 tau.
Excess activation of ionotropic glutamate receptors appears to
cause neuronal cell death exclusively through necrosis (Dessi et al.,
1993; Gwag et al., 1995). The ultrastructural features of degenerat-
ing cortical neurons following exposures to low doses of NMDA or
kainate confirmed necrotic degeneration evident by cell body
swelling, scattering condensation of nuclear chromatin, and early
fenestration of plasma membrane. While tau proteins were sensitive
to the calcium-dependent protease calpain that was shown to be
activated by administration of the excitotoxins (Johnson et al., 1989;
Yang and Ksiezak-Reding, 1995), PHF-1 tau was not cleaved in
cortical cell cultures undergoing excitotoxic necrosis after exposure
to NMDA or kainate. This is consistent with previous findings that
phosphorylated forms of tau including PHF-1 tau are extremely
resistant to proteolytic cleavage by calpain (Litersky and Johnson,
1995; Yang and Ksiezak-Reding, 1995).
Oxidative stress has been reported to cause neuronal cell death
through apoptosis or necrosis. Ultrastructural analysis of degenerat-
ing cortical neurons demonstrates that necrosis is a predominant
pattern of neuronal cell death in cortical cell cultures exposed to
prooxidants, Fe2+ or menadione as previously reported (Ryu et al.,
1999; Won et al., 2000). PHF-1 tau was not cleaved in the course of
free radical-induced neuronal cell necrosis. Thus, PHF-1 tau appears
to be cleaved by caspase-3 selectively during neuronal cell
apoptosis.
Occurrence of apoptosis in AD has been supported by beta
amyloid-induced apoptosis in cultured neurons, DNA fragmenta-
tion and TUNEL-positive neurons in AD brain, and induction of
apoptosis in neurons overexpressing mutant presenilin 1 (Chui et
al., 1999; Guo et al., 1997; Kim et al., 1997; Loo et al., 1993;
Weihl et al., 1999). In the present study, we have found that PHF-1
tau abundantly accumulated in the AD brain is present as cleaved
fragments as well as full isoforms. While further study will be

Fig. 6. Colocalization of active caspase-3 and PHF-1 tau in AD. Fluorescence photomicrographs of control (top panel) and AD (middle and bottom panels)
brain sections after double immunolabeling with antibodies for PHF-1 tau (A, D and G, red) and active caspase-3 (B and E, green). In bottom panel, active
caspase-3 antibody was pre-absorbed with active caspase-3 prior to immunolabeling (H). The merged fluorescence photomicrographs showed that pre-
adsorption of the caspase-3 antibody markedly reduced caspase-3-immunoreactive signal in colocalization of PHF-1 tau in AD brain (F and I). Double-labeled
sections were stained with Hoechst dye labeling (blue) (C). Arrow indicates a PHF-1 tau and active caspase-3 -positive neuron (yellow).
H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458456
needed to determine whether and where human PHF-1 tau in AD is
cleaved by caspase-3, apoptosis-specific and caspase-3-dependent
cleavage of PHF-1 tau in murine cortical cell cultures suggests that
cleaved PHF-1 tau in AD is associated with activation of caspase-3
during apoptosis. In support of this, levels and activation of
caspase-3 are also increased in the AD brain (Shimohama et al.,
1999). Moreover, activated caspase-3 is observed in most of PHF-1
tau-positive neurons in the AD brain. Caspase-3-dependent
cleavage form of fodrin has been detected in neurofibillary
tangle-bearing neurons in AD (Rohn et al., 2001).
Hyperphophorylated forms of tau are accumulated in the AD
brain due to activation of protein kinases such as glycogen
synthase kinase-3h (GSK-3h) and cyclin-dependent kinase-5
(cdk5) or reduced activity of protein phosphatase 2A- and protein
phosphatase 1 (Geschwind, 2003; Godemann et al., 1999; Iqbal et
al., 1998; Pei et al., 1998; Yamaguchi et al., 1996). Tau-positive
neurofibrillary tangles are also observed in various dementing
diseases including progressive supranuclear palsy (PSP), cortico-
basal degeneration, Pick’s disease, and familial frontotemporal
dementia and Parkinsonism linked to chromosome 17 (FTDP-17)
(Spillantini and Goedert, 1998). Overexpression of mutant tau
derived from FTDP-17 causes accumulation of hyperphosphory-
lated tau and neuronal degeneration (Tanemura et al., 2001;
Tatebayashi et al., 2002; Vogelsberg-Ragaglia et al., 2000). Beta
amyloid fibrils that induce accumulation of PHF-1 tau cause
neuronal death in the presence of tau (Busciglio et al., 1995;
Rapoport et al., 2002). Blockade of caspase 3-mediated cleavage
of tau attenuates apoptotic cell death induced by staurosporine
and okadaic acid (Chung et al., 2001). Taken together, the
phosphorylation and caspase-3-mediated cleavage of tau likely
play a role in the formation of neurofibrillary tangles and
neurodegeneration.
The present study supports that apoptosis may contribute to
neurondegeneration in AD possibly through mechanisms involv-
ing activation of caspase-3 and cleavage of PHF-1 tau. The
apoptosis appears to be different from excitotoxicity and oxidative
stress that cause neuronal death primarily through necrosis and do
not cleave PHF-1 tau. Thus, apoptosis as well as excitotoxic and
oxidative necrosis may constitute additional route of neuronal
death in AD.
Acknowledgments
We thank Dr. Peter Davis (Albert Einstein College of Medicine)
for the PHF-1 antibody, Dr. Lester I Binder (Northwestern
University School of Medicine) for the Tau-5 antibody and Boston

H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458 457
University Alzheimer’s Disease Center (Dr. Ann C. Mckee) for
human brain tissues. This work was supported by a National
Research Laboratory grant and the 21st Century Frontier Research
Program (01610) (BJG).
References
Ando, Y., Brannstrom, T., Uchida, K., Nyhlin, N., Nasman, B., Suhr, O.,
Yamashita, T., Olsson, T., El Salhy, M., Uchino, M., Ando, M., 1998.
Histochemical detection of 4-hydroxynonenal protein in Alzheimer
amyloid. J. Neurol. Sci. 156, 172–176.
Busciglio, J., Lorenzo, A., Yeh, J., Yankner, B.A., 1995. beta-amyloid
fibrils induce tau phosphorylation and loss of microtubule binding.
Neuron 14, 879–888.
Castellani, R.J., Harris, P.L., Sayre, L.M., Fujii, J., Taniguchi, N., Vitek,
M.P., Founds, H., Atwood, C.S., Perry, G., Smith, M.A., 2001. Active
glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-
(carboxymethyl) lysine and hexitol-lysine. Free Radical Biol. Med. 31,
175–180.
Chui, D.H., Tanahashi, H., Ozawa, K., Ikeda, S., Checler, F., Ueda, O.,
Suzuki, H., Araki, W., Inoue, H., Shirotani, K., Takahashi, K., Gallyas,
F., Tabira, T., 1999. Transgenic mice with Alzheimer presenilin 1
mutations show accelerated neurodegeneration without amyloid plaque
formation. Nat. Med. 5, 560–564.
Chung, C.W., Song, Y.H., Kim, I.K., Yoon, W.J., Ryu, B.R., Jo, D.G., Woo,
H.N., Kwon, Y.K., Kim, H.H., Gwag, B.J., Mook-Jung, I.H., Jung,
Y.K., 2001. Proapoptotic effects of tau cleavage product generated by
caspase-3. Neurobiol. Dis. 8, 162–172.
Cole, K.K., Perez-Polo, J.R., 2002. Poly (ADP-ribose) polymerase
inhibition prevents both apoptotic-like delayed neuronal death and
necrosis after H(2)O(2) injury. J. Neurochem. 82, 19–29.
Dessi, F., Charriaut-Marlangue, C., Khrestchatisky, M., Ben-Ari, Y., 1993.
Glutamate-induced neuronal death is not a programmed cell death in
cerebellar culture. J. Neurochem. 60, 1953–1955.
Geschwind, D.H., 2003. Tau phosphorylation, tangles, and neurodegenera-
tion: the chicken or the egg? Neuron 40, 457–460.
Godemann, R., Biernat, J., Mandelkow, E., Mandelkow, E.M., 1999.
Phosphorylation of tau protein by recombinant GSK-3beta: pronounced
phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at
Ser262 in the repeat domain. FEBS Lett. 454, 157–164.
Goedert, M., 1993. Tau protein and the neurofibrillary pathology of
Alzheimer’s disease. Trends Neurosci. 16, 460–465.
Guo, Q., Sopher, B.L., Furukawa, K., Pham, D.G., Robinson, N.,
Martin, G.M., Mattson, M.P., 1997. Alzheimer’s presenilin mutation
sensitizes neural cells to apoptosis induced by trophic factor
withdrawal and amyloid beta-peptide: involvement of calcium and
oxyradicals. J. Neurosci. 17, 4212–4222.
Gwag, B.J., Lobner, D., Koh, J.Y., Wie, M.B., Choi, D.W., 1995. Blockade
of glutamate receptors unmasks neuronal apoptosis after oxygen-
glucose deprivation in vitro. Neuroscience 68, 615–619.
Gwag, B.J., Won, S.J., Kim, D.Y., 2002. Excitotoxicity, oxidative stress and
apoptosis in ischemic neuronal death. In: Lin, R.C.S. (Ed.), New
concepts in cerebral ischemia. CRC, Boca Raton, FL, pp. 79–112.
Han, K.S., Kang, H.J., Kim, E.Y., Yoon, W.J., Sohn, S., Kwon, H.J.,
Gwag, B.J., 2001. 1,2-bis(2-Aminophenoxy)ethane-N,N,NV,NV-tetra-acetic acid induces caspase-mediated apoptosis and reactive oxygen
species-mediated necrosis in cultured cortical neurons. J. Neurochem.
78, 230–239.
Iqbal, K., Alonso, A.C., Gong, C.X., Khatoon, S., Pei, J.J., Wang, J.Z.,
Grundke-Iqbal, I., 1998. Mechanisms of neurofibrillary degeneration
and the formation of neurofibrillary tangles. J. Neural Transm. Suppl.
53, 169–180.
Johnson, G.V., Jope, R.S., Binder, L.I., 1989. Proteolysis of tau by calpain.
Biochem. Biophys. Res. Commun. 163, 1505–1511.
Keller, J.N., Mark, R.J., Bruce, A.J., Blanc, E., Rothstein, J.D., Uchida, K.,
Waeg, G.,Mattson,M.P., 1997. 4-Hydroxynonenal, an aldehydic product
of membrane lipid peroxidation, impairs glutamate transport and
mitochondrial function in synaptosomes. Neuroscience 80, 685–696.
Kim, T.W., Pettingell, W.H., Jung, Y.K., Kovacs, D.M., Tanzi, R.E., 1997.
Alternative cleavage of Alzheimer-associated presenilins during apop-
tosis by a caspase-3 family protease. Science 277, 373–376.
Ko, H.W., Han, K.S., Kim, E.Y., Ryu, B.R., Yoon, W.J., Jung, Y.K., Kim,
S.U., Gwag, B.J., 2000. Synergetic activation of p38 mitogen-activated
protein kinase and caspase-3-like proteases for execution of calyculin
A-induced apoptosis but not N-methyl-d-aspartate-induced necrosis in
mouse cortical neurons. J. Neurochem. 74, 2455–2461.
Koh, J.Y., Yang, L.L., Cotman, C.W., 1990. Beta-amyloid protein increases
the vulnerability of cultured cortical neurons to excitotoxic damage.
Brain Res. 533, 315–320.
Litersky, J.M., Johnson, G.V., 1995. Phosphorylation of tau in situ: inhibition
of calcium-dependent proteolysis. J. Neurochem. 65, 903–911.
Loo, D.T., Copani, A., Pike, C.J., Whittemore, E.R., Walencewicz, A.J.,
Cotman, C.W., 1993. Apoptosis is induced by beta-amyloid in cultured
central nervous system neurons. Proc. Natl. Acad. Sci. U. S. A. 90,
7951–7955.
Masliah, E., Alford, M., DeTeresa, R., Mallory, M., Hansen, L., 1996.
Deficient glutamate transport is associated with neurodegeneration in
Alzheimer’s disease. Ann. Neurol. 40, 759–766.
Mattson, M.P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., Rydel,
R.E., 1992. beta-Amyloid peptides destabilize calcium homeostasis and
render human cortical neurons vulnerable to excitotoxicity. J. Neurosci.
12, 376–389.
Moroni, F., Meli, E., Peruginelli, F., Chiarugi, A., Cozzi, A., Picca, R.,
Romagnoli, P., Pellicciari, R., Pellegrini-Giampietro, D.E., 2001.
Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not
apoptotic neuronal death in experimental models of cerebral ischemia.
Cell Death Differ. 8, 921–932.
Pei, J.J., Gong, C.X., Iqbal, K., Grundke-Iqbal, I., Wu, Q.L., Winblad, B.,
Cowburn, R.F., 1998. Subcellular distribution of protein phosphatases
and abnormally phosphorylated tau in the temporal cortex from
Alzheimer’s disease and control brains. J. Neural Transm. 105, 69–83.
Rapoport, M., Dawson, H.N., Binder, L.I., Vitek, M.P., Ferreira, A., 2002.
Tau is essential to beta-amyloid-induced neurotoxicity. Proc. Natl.
Acad. Sci. U. S. A. 99, 6364–6369.
Rohn, T.T., Head, E., Su, J.H., Anderson, A.J., Bahr, B.A., Cotman, C.W.,
Cribbs, D.H., 2001. Correlation between caspase activation and
neurofibrillary tangle formation in Alzheimer’s disease. Am. J. Pathol.
158, 189–198.
Ryu, B.R., Ko, H.W., Jou, I., Noh, J.S., Gwag, B.J., 1999. Phosphatidy-
linositol 3-kinase-mediated regulation of neuronal apoptosis and
necrosis by insulin and IGF-I. J. Neurobiol. 39, 536–546.
Schmidt, M.L., Zhukareva, V., Newell, K.L., Lee, V.M., Trojanowski, J.Q.,
2001. Tau isoform profile and phosphorylation state in dementia
pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. (Berl.)
101, 518–524.
Shimohama, S., Tanino, H., Fujimoto, S., 1999. Changes in caspase
expression in Alzheimer’s disease: comparison with development and
aging. Biochem. Biophys. Res. Commun. 256, 381–384.
Smale, G., Nichols, N.R., Brady, D.R., Finch, C.E., Horton, W.E.J., 1995.
Evidence for apoptotic cell death in Alzheimer’s disease. Exp. Neurol.
133, 225–230.
Smith, C.D., Carney, J.M., Starke-Reed, P.E., Oliver, C.N., Stadtman, E.R.,
Floyd, R.A., Markesbery, W.R., 1991. Excess brain protein oxidation
and enzyme dysfunction in normal aging and in Alzheimer disease.
Proc. Natl. Acad. Sci. U. S. A. 88, 10540–10543.
Smith, M.A., Harris, P.L., Sayre, L.M., Perry, G., 1997. Iron accumulation
in Alzheimer disease is a source of redox-generated free radicals. Proc.
Natl. Acad. Sci. U. S. A. 94, 9866–9868.
Spillantini, M.G., Goedert, M., 1998. Tau protein pathology in neuro-
degenerative diseases. Trends Neurosci. 21, 428–433.
Su, J.H., Nichol, K.E., Sitch, T., Sheu, P., Chubb, C., Miller, B.L.,
Tomaselli, K.J., Kim, R.C., Cotman, C.W., 2000. DNA damage and

H.J. Kang et al. / Neurobiology of Disease 18 (2005) 450–458458
activated caspase-3 expression in neurons and astrocytes: evidence for
apoptosis in frontotemporal dementia. Exp. Neurol. 163, 9–19.
Tanemura, K., Akagi, T., Murayama, M., Kikuchi, N., Murayama, O.,
Hashikawa, T., Yoshiike, Y., Park, J.M., Matsuda, K., Nakao, S., Sun,
X., Sato, S., Yamaguchi, H., Takashima, A., 2001. Formation of
filamentous tau aggregations in transgenic mice expressing V337M
human tau. Neurobiol. Dis. 8, 1036–1045.
Tatebayashi, Y., Miyasaka, T., Chui, D.H., Akagi, T., Mishima, K.,
Iwasaki, K., Fujiwara, M., Tanemura, K., Murayama, M., Ishiguro,
K., Planel, E., Sato, S., Hashikawa, T., Takashima, A., 2002. Tau
filament formation and associative memory deficit in aged mice
expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. U. S. A.
99, 13896–13901.
Trojanowski, J.Q., Lee, V.M., 1995. Phosphorylation of paired helical
filament tau in Alzheimer’s disease neurofibrillary lesions: focusing on
phosphatases. FASEB J. 9, 1570–1576.
Vogelsberg-Ragaglia, V., Bruce, J., Richter-Landsberg, C., Zhang, B.,
Hong, M., Trojanowski, J.Q., Lee, V.M., 2000. Distinct FTDP-17
missense mutations in tau produce tau aggregates and other pathological
phenotypes in transfected CHO cells. Mol. Biol. Cell 11, 4093–4104.
Weihl, C.C., Ghadge, G.D., Kennedy, S.G., Hay, N., Miller, R.J., Roos,
R.P., 1999. Mutant presenilin-1 induces apoptosis and downregulates
Akt/PKB. J. Neurosci. 19, 5360–5369.
Won, S.J., Park, E.C., Ryu, B.R., Ko, H.W., Sohn, S., Kwon, H.J., Gwag,
B.J., 2000. NT-4/5 exacerbates free radical-induced neuronal necrosis in
vitro and in vivo. Neurobiol. Dis. 7, 251–259.
Yamaguchi, H., Ishiguro, K., Uchida, T., Takashima, A., Lemere, C.A.,
Imahori, K., 1996. Preferential labeling of Alzheimer neurofibrillary
tangles with antisera for tau protein kinase (TPK) I/glycogen synthase
kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II.
Acta Neuropathol. (Berl.) 92, 232–241.
Yang, L.S., Ksiezak-Reding, H., 1995. Calpain-induced proteolysis of
normal human tau and tau associated with paired helical filaments. Eur.
J. Biochem. 233, 9–17.