challenges to therapy for peroxisome assembly disorders nancy braverman, ms, md mcgill...
TRANSCRIPT

Challenges to therapy for peroxisome assembly
disorders
Nancy Braverman, MS, MDMcGIll University-MCH-RI
March 9 2010HGEN 171-575

Properties of peroxisomes
• Spherical, single membrane bound, • Diameter = 0.2 - 1 µm, several hundred/cell• All eukaryotes

Peroxisomes originate from ER membranes and by fission of
existing peroxisomes
adapted from Annu Rev Genet. 2000;34:623-652. Sacksteder KA, Gould SJ.
NEXT >>Click to view animation >>
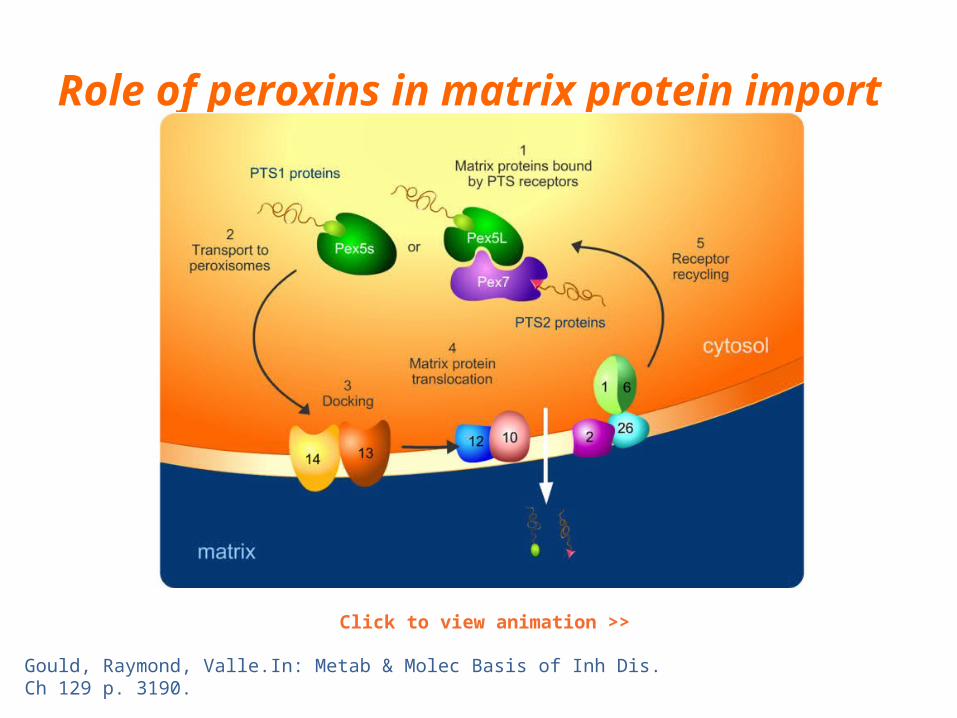
Role of peroxins in matrix protein import
Click to view animation >>
Gould, Raymond, Valle.In: Metab & Molec Basis of Inh Dis. Ch 129 p. 3190.

Enzymatic pathways in peroxisomes
• Fatty acid oxidation (VLCFA, PA)• H2O2 detoxification (catalase)• Docohexanoic acid (DHA) synthesis• Bile acid synthesis• Plasmalogen (ether phospholipid) synthesis • Cholesterol synthesis • Glyoxylate detoxification• Lysine catabolism (pipecolic acid)

The 3 major metabolic pathways in peroxisomes

Properties of peroxisomal matrix proteins
• Contain Peroxisome Targeting Sequences (PTS)
• Imported as oligomers/fully assembled proteins• Can have dual localizations in mitochondria,
cytosol
C - terminal (-SKL)Most matrix proteinsReceptor is PEX5
-SKL
PTS1 PTS2
N - terminal (-R/KLX5 Q/HL-)
Presequence cleaved internally3 enzymes only: Thiolase, PhyH, AGPSReceptor is PEX7
R/KLX5Q/HL-SKL

Genetic disorders of peroxisomes
• Multiple enzyme deficiencies: Peroxisomal Biogenesis Disorders (PBD) – Zellweger spectrum disorder (ZSD) (~1/60,000) – Rhizomelic chondrodysplasia punctata spectrum
(RCDP)(~1/100,000)• Single enzyme deficiencies
– X-linked adrenoleukodystrophy (X-ALD) (~1/20,000)– 3-methyl-CoA racemase deficiency– Adult Refsum disease– Hyperoxaluria Type I

Some single enzyme deficiencies can mimic PBDs
• VLCFA oxidation → Zellweger spectrum disorder– Acyl-CoA oxidase – D-Bifunctional protein (hydratase/dehydrogenase)
• Plasmalogen biosynthesis → RCDP spectrum – DHAPAT (RCDP2)– ADHAPS (RCDP3)
• Some PBDs mimic SEDs → – Adult Refsum disease causes PEX7 deficiency

Develop therapies targeted to the metabolic defects
• Phytanic acid restriction• Reduction in VLCFA
dietary reductionenhance VLCFA omega oxidationreduce VLCFA synthesis
• Supplementation with DHA, bile acids, plasmalogens
A--------->B

Develop therapies targeted to the molecular defects
• Enhance activity of a defective PEX protein-improve protein folding
• Bypass the need for a specific PEX protein-upregulate a partner PEX protein
• Induce peroxisome proliferation• Enzyme/PEX protein replacement therapy ?• Liver/stem cell transplant ?• Gene therapy ? • Manipulate the intestinal microbiome?
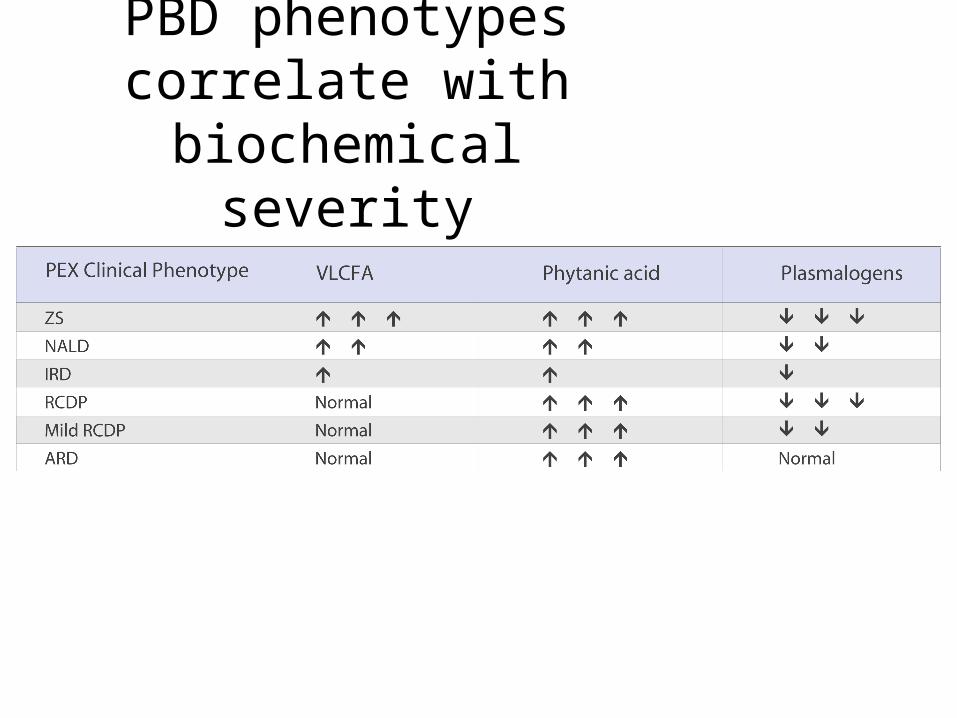
PBD phenotypes correlate with biochemical severity
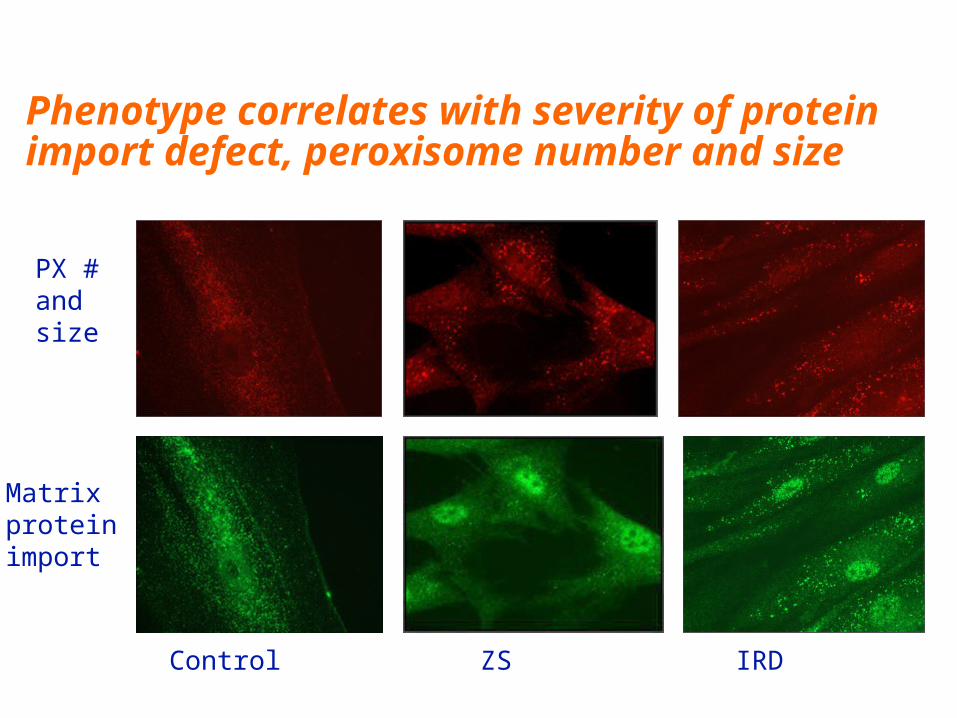
Control ZS
PX #and size
Matrix protein import
IRD
Phenotype correlates with severity of protein import defect, peroxisome number and size

PMP70 PTS1/PTS2 CATALASE
37
30
Mild PBD, PEX1-G843D/G843D

PEX1-G843D/ G843D, expressing GFP-PTS1 reporter
No Treatment
30 oC
Glycerol 5%
37 oC
TMAO200 uM
37 oC
No Treatment37 oC


Disorder-to-order conformational transitions in protein structure and its relationship to disease

• Lower temperature• Chaperone (protein or drug)
– Nonspecific chemical chaperone– Pharmacologic chaperone
Enzyme substrate
Protein ligand (protein kinase and kinase inhibitor)
Vitamin cofactor


Conformational changes of p97 AAA ATPase during its ATPase cycle
Bind and hydrolyze ATP generating chemical energy that is converted into motion of the molecule.
Motion used to pull PEX5 out of the membrane for another round of import

Role of peroxins in matrix protein import
Click to view animation >>
Gould, Raymond, Valle.In: Metab & Molec Basis of Inh Dis. Ch 129 p. 3190.

High throughput chemical screen• Cells incubated in chemicals 2 days (2000
compounds)• Negative control: media alone• Positive control: TMAO and glycerol

3 chemicals rescued import
• Data indicated that they can non-competitively bind to the ATP binding sites of proteins
• Potential pharmacologic chaperones!

Pipeline for new drugs 5-10 yrs…• Develop HT assays to screen chemical libraries for compounds that
recover target function• Best to start with ‘in vivo’ assay, several complementary assays• Confirm ‘hits’ from the screening assay • Study structure-function relationships to develop best ‘lead’
compounds• Evaluate mechanisms of recovery• Asses pharmacokinetics: half-life, metabolism, excretion, recovery in
the brain, toxicity, tissue pathology (rodents)• Assay efficacy: animal models• Approval of drug for clinical trials or off-use label• Ensure drug supply, design and approval of clinical trial• Funding for clinical trial

Intracellular distribution of AGT, a protein with an N-terminal MTS & C-terminal PTS1

Primary hyperoxaluria type 1
• 15-20% European and North American population has Pro11Leu missense substitution– Decreased AGT stability– Decreased enzymatic activity– Enhances effect of additional mutations that are
predicted to be innocuous in its absence – Redirects AGT to mitochondria
• Gly170Arg folding delay promotes mitochondrial import

Protein evolution depends on diet
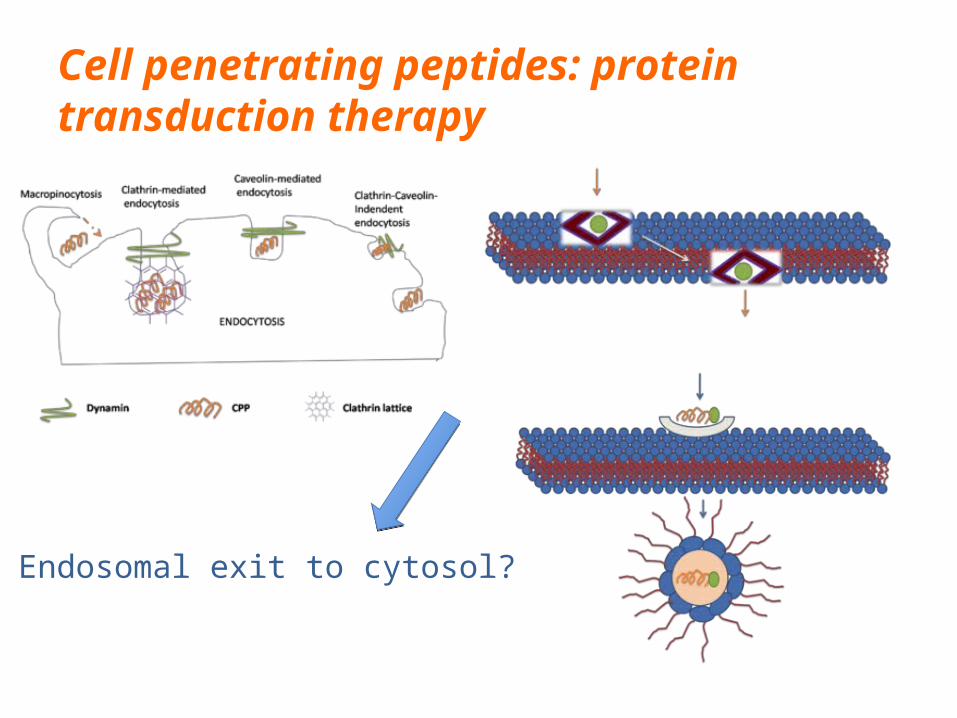
Cell penetrating peptides: protein transduction therapy
Endosomal exit to cytosol?


Role of peroxins in matrix protein import
Click to view animation >>
Gould, Raymond, Valle.In: Metab & Molec Basis of Inh Dis. Ch 129 p. 3190.

Understanding the pathophysiology may reveal other targets for therapy
Selective inactivation of PEX5 gene in neural cells Pex5-loxP x Nestin-cre (neurons, oligodendrocytes and astrocytes)
Pex5-loxP x CNPase-cre (oligodendrocyte) Abnormal compaction of myelin Axonal damage and transport defects Reactive astrocytosis and microgliosis CD8+ T helper cells and increased cytokines


