chap 19 thermodynamics.ppt - bakersfield college b1b spring 2013/chap_19...consider the laws of...
TRANSCRIPT

1/16/2013
1
Thermodynamics
1
http://www.chem.purdue.edu/gchelp/howtosolveit/howtosolveit.html

1/16/2013
2
• Study of energy changes and flow of energy
• Answers several fundamental questions:
– Is it possible for reaction to occur?
– Will reaction occur spontaneously (without outside
interference) at given T?
– Will reaction release or absorb heat?
• Tells us nothing about time frame of reaction
– Kinetics
• Two major considerations—must be balanced
– Enthalpy changes, ∆H (heats of reaction)
• Heat exchange between system and surroundings
– Nature's trend to randomness or disorder
Thermodynamics
2

1/16/2013
3
Examples
of
Spontaneous Reactions
3

1/16/2013
4
2 NH3 (g) + CO2 (g) → NH2CONH2 (aq) + H2O (l)
Consider this Reaction
Concerning this reaction:
1. Does this reaction naturally occur as written?
2. Will the reaction mixture contain sufficient
amount of product at equilibrium?
4

1/16/2013
5
We can answer these questions with
heat measurements only!!!
1. We can predict the natural direction.
How?
2. We can determine the composition of the
mixture at equilibrium.
5

1/16/2013
6
Consider the Laws of Thermodynamics
1st Law: The change in internal energy of a
system ∆U, equals q + w
∆E = ∆U = q + w
2nd Law: The total entropy of a system and its
surroundings increases for a spontaneous
process.
3rd Law: A substance that is perfectly crystalline
at 0 K has an entropy of zero.
You can’t win
You cant break even.
You can’t quit the game
6

1/16/2013
7
• Internal energy, E or U
– System's total energy
– Sum of KE and PE of all particles in system
– or for chemical reaction
– ∆E + energy into system
– ∆E −−−− energy out of system
systemsystemsystem PEKEE )()( +=
initialfinal EEE −−−−====∆∆∆∆
reactantsproducts EEE −−−−====∆∆∆∆
Review of First Law of Thermodynamics
7
1/16/2013
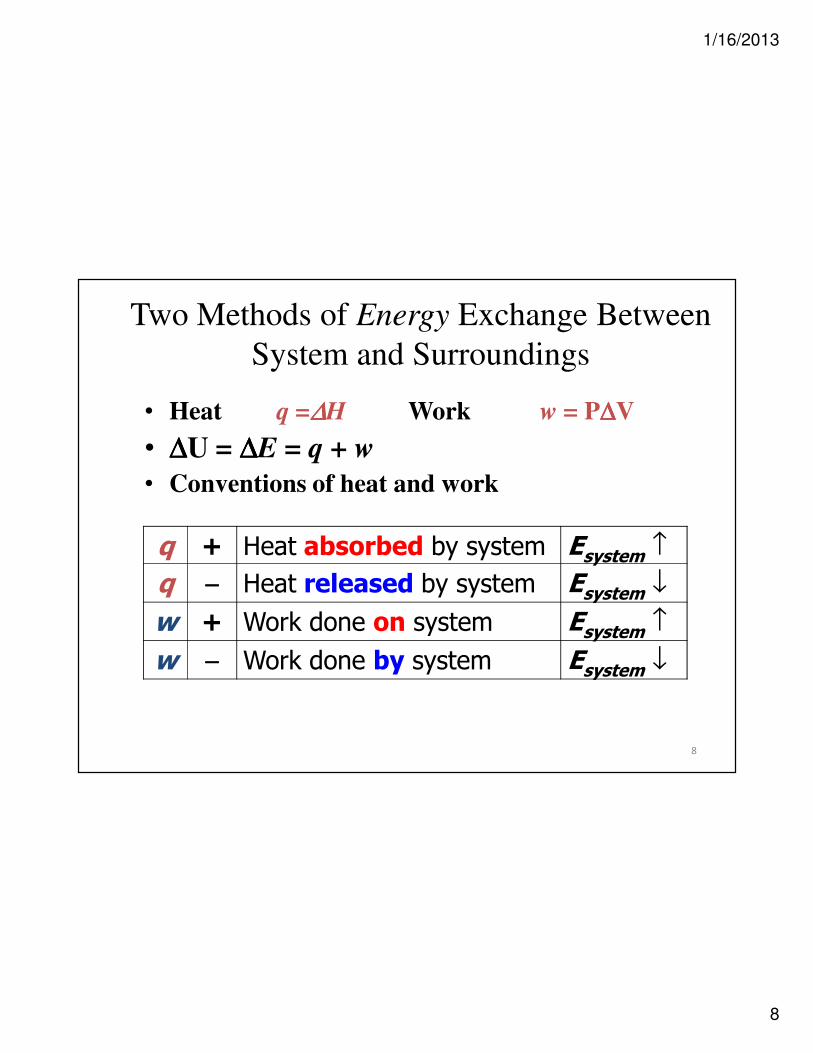
8
q + Heat absorbed by system Esystem ↑
q −−−− Heat released by system Esystem ↓
w + Work done on system Esystem ↑
w −−−− Work done by system Esystem ↓
• Heat q =∆∆∆∆H Work w = P∆∆∆∆V
• ∆∆∆∆U = ∆∆∆∆E = q + w
• Conventions of heat and work
Two Methods of Energy Exchange Between
System and Surroundings
8

1/16/2013
9
∆U(Internal Energy) is a state function:
Most often we are interested in the change:
Internal Energy = ∆Ε = ∆U = Uf - Ui
q = Energy that moves in and out of a system
w = Force x distance = F x d = P∆V
9

1/16/2013
10
• Energy can neither be created nor destroyed
• It can only be converted from one form to another
– KE ↔ PE
– Chemical ↔ Electrical
– Electrical ↔ Mechanical
• E and ∆E are state functions
– Path independent
– ∆∆∆∆E = -∆∆∆∆U = q + w
First Law of Thermodynamics
systemsystemsystem PEKEE )()( +=10

1/16/2013
11
VPq)VP(qE ∆∆∆∆−−−−====∆∆∆∆−−−−++++====∆∆∆∆
1. Electrical
2. Pressure-volume or P∆∆∆∆V
– w = −−−− P∆∆∆∆V
• Where P = external pressure
– If P∆∆∆∆V only work in chemical system, then
Work in Chemical Systems
11
1/16/2013
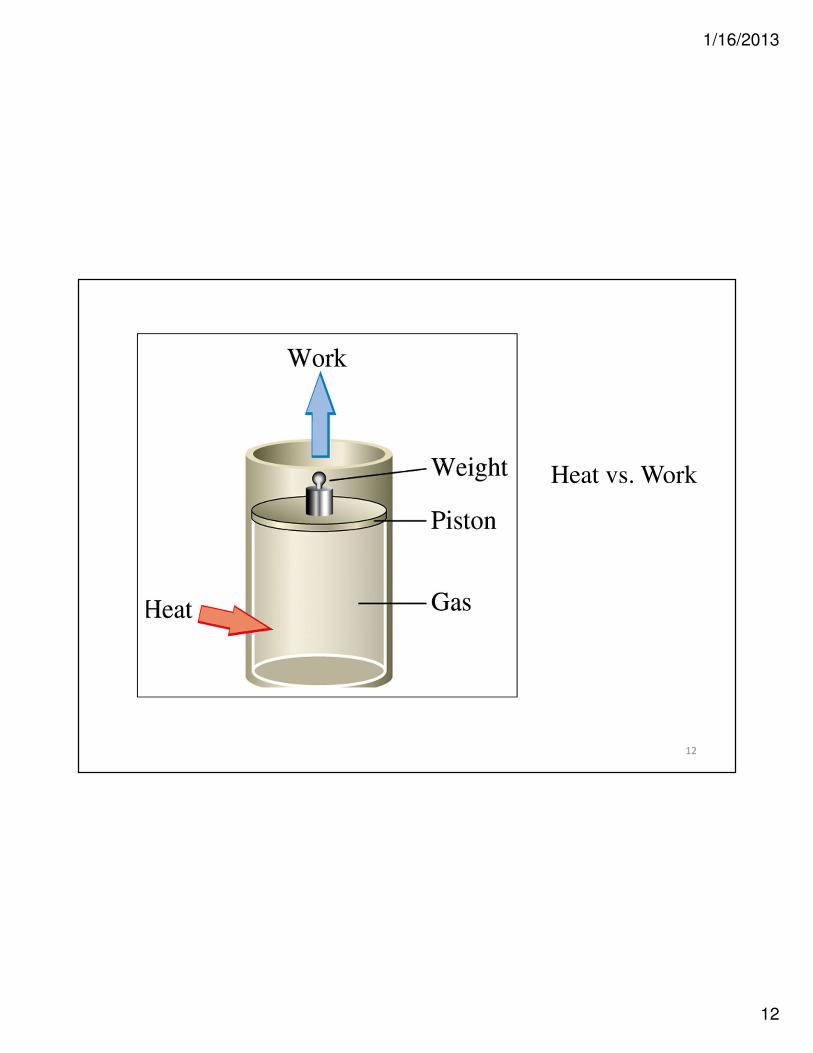
12
Heat vs. Work
12

1/16/2013
13
Showing work is P∆V
w = - F x h
= - F x ∆V/A
= -F/A x ∆V
= -P∆V
13

1/16/2013
14
Reaction done at constant V
∆V = 0
P∆V = 0, so
∆E = qV
Entire E change due to heat absorbed or
lost
Heat at Constant Volume
14

1/16/2013
15
Heat at Constant Pressure• More common
• Reactions open to atmosphere
– Constant P
• Enthalpy
– H = E + PV
• Enthalpy change
– ∆H = ∆E + P∆V
• Substituting in first law for ∆E gives
– ∆H = (q −−−− P∆V) + P∆V = qP
– ∆H = qP
• Heat of reaction at constant pressure15

1/16/2013
16
q = 165 J
P∆V = -92 J
∆U = q + w = (+165) + (-92) = +73 J16
1/16/2013
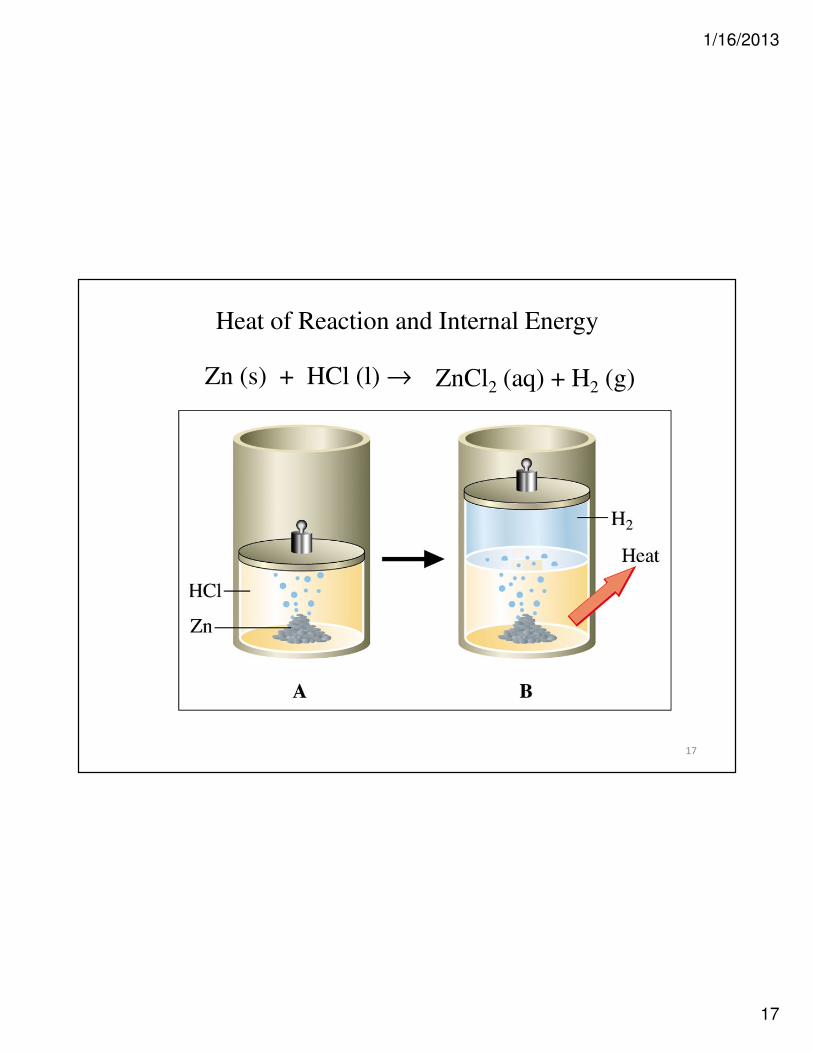
17
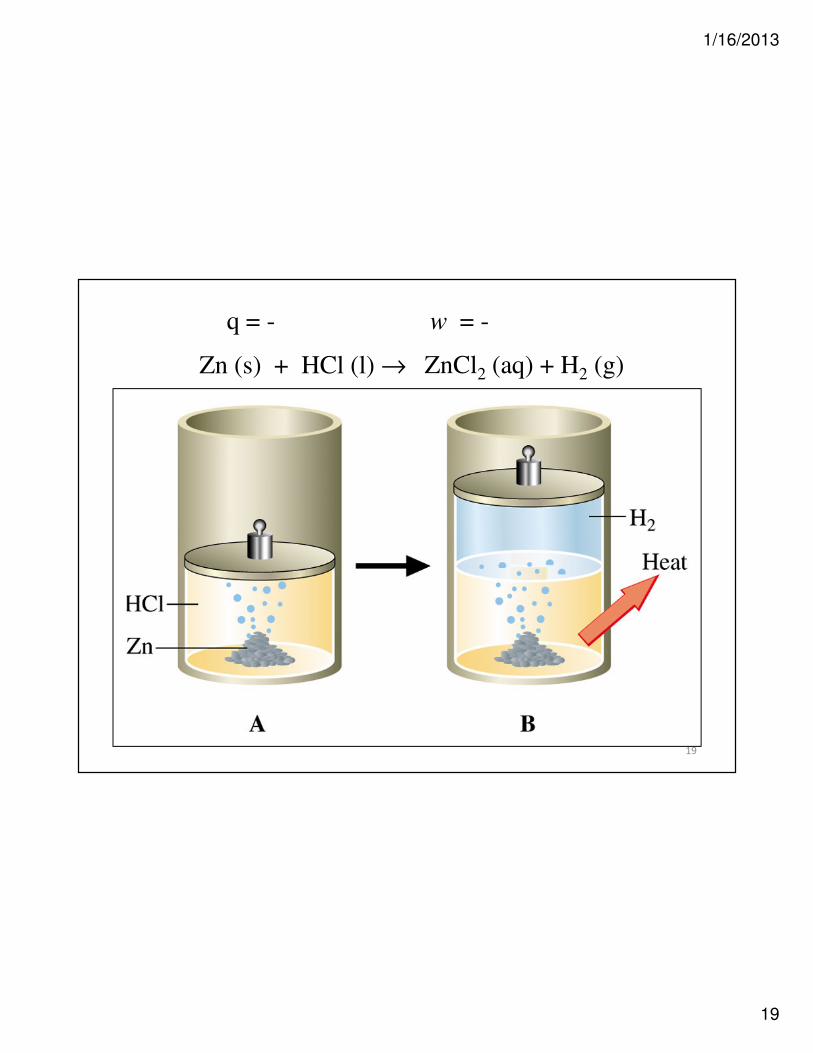
Heat of Reaction and Internal Energy
Zn (s) + HCl (l) → ZnCl2 (aq) + H2 (g)
17
1/16/2013
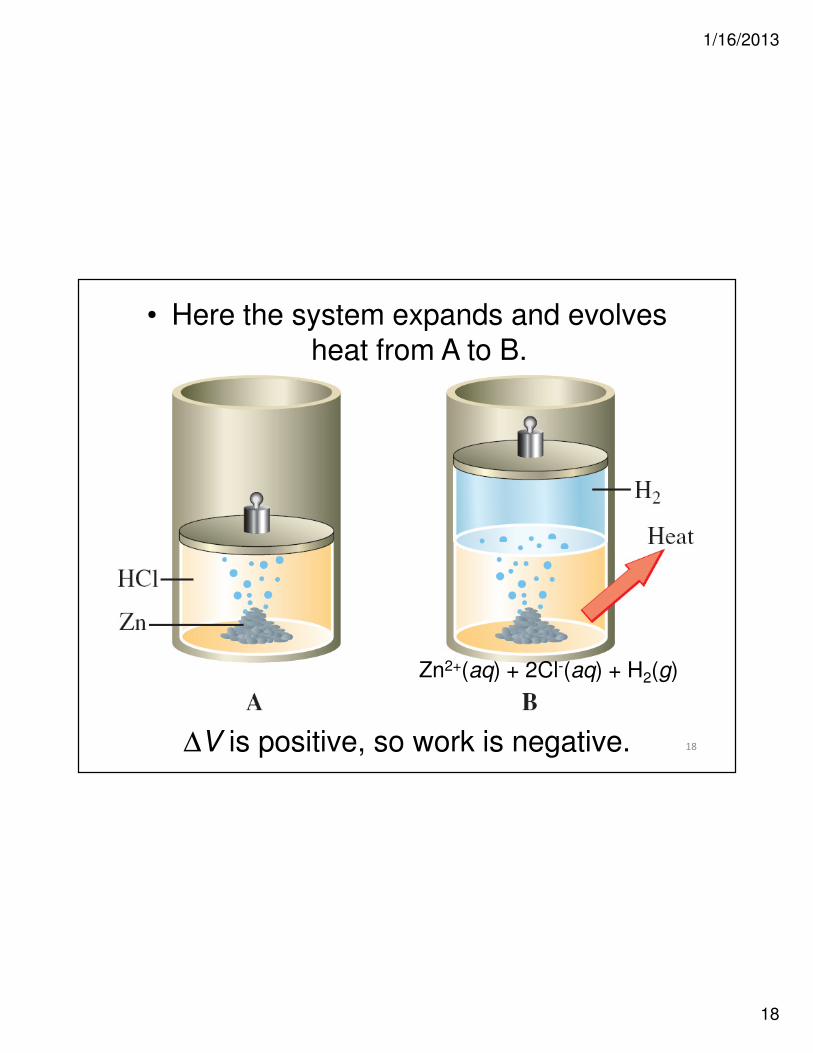
18
18
• Here the system expands and evolves
heat from A to B.
Zn2+(aq) + 2Cl-(aq) + H2(g)
∆V is positive, so work is negative.
1/16/2013
19
19
q = - w = -
Zn (s) + HCl (l) → ZnCl2 (aq) + H2 (g)
19

1/16/2013
20
w = -P∆V
= -(1.01 x 105 pa)(24.5 l)
= -1.01 x 105 pa)(24.5 x 10-3m3)
= -2.47 x 103 J
= -2.47 kJ
q = -152 kJ
∆U = -152 kJ + (-2.47 kJ) = -154.9 kJ
20

1/16/2013
21
Diagram and explain the change in internal energy
for the following reaction.
CH4 (g) + 2 O2 (g) → CO2 (g) + 2 H2O (l)
q = -890.2 kJ
w = P∆V = +(1.01 x 105 pa)(24.5 l)(2)
= +(1.01 x 105 pa)(24.5 x 10-3m3)(2)
= +4.95 kJ
∆U = -890.2 kJ + (+4.95 kJ) = -885.2 kJ
21

1/16/2013
22
Enthalpy and Enthalpy Change
Enthalpy is defined as qp
Enthalpy = H = U + PV
All are state functions.
∆H = Hf - Hi
∆H = (Uf + PVf) – (Ui + PVi) = (Uf –Ui) + P(Vf –Vi)
∆U = qp - P∆V
∆H = (qp - P∆V) + P∆V = qp
∆Hof = Σn∆Ho
f (products) - Σm∆Hof (reactants)
∆Hof = Standard enthalpy change (25oC) 22

1/16/2013
23
∆Hof = Σn∆Ho
f (products) - Σm∆Hof (reactants)
2 NH3 (g) + CO2 (g) → NH2CONH2 (aq) + H2O (l)
Calculate ∆Hof for the reaction in slide 15
∆Hof = for NH3 (g) = -45.9 kJ
= for CO2 (g) = -393.5 kJ
= for NH2CONH2 = -319.2 kJ
= for H2O (l) = -285.8 kJ
∆Ho = [(-319.2 – 285.8) – (-2 x 45.9 – 393.50)] kJ = -119.7
Since ∆Ho has a negative sign, heat is evolved
23

1/16/2013
24
Converting Between ∆E and ∆H For Chemical
Reactions
• ∆H ≠≠≠≠ ∆E
• Differ by ∆∆∆∆H −−−− ∆∆∆∆E = P∆∆∆∆V
• Only differ significantly when gases formed or
consumed
• Assume gases are ideal
• Since P and T are constant
P
nRTV ====
∆∆∆∆====∆∆∆∆
P
nRTV
∆∆∆∆====∆∆∆∆
P
RTnV
24

1/16/2013
25
Converting Between ∆E and ∆H For Chemical
Reactions
• When reaction occurs
– ∆V caused by ∆n of gas
• Not all reactants and products are gases
– So redefine as ∆ngas
Where ∆ngas = (ngas)products – (ngas)reactants
• Substituting into ∆H = ∆E + P∆V gives
– or
∆∆∆∆∗∗∗∗++++∆∆∆∆====∆∆∆∆
P
RTnPEH gas
RTnEH gas∆∆∆∆++++∆∆∆∆====∆∆∆∆
25

1/16/2013
26
Ex. 1. What is the difference between ∆H and ∆E for the following reaction at 25 °C?
2 N2O5 (g) →→→→ 4 NO2 (g) + O2 (g)
What is the % difference between ∆H and ∆E?
Step 1: Calculate ∆∆∆∆H using data (Table 7.2)
Recall
∆H° = (4 mol)(33.8 kJ/mol) + (1 mol)(0.0 kJ/mol) – (2 mol)(11 kJ/mol)
reactants)()( ooo
fproductsf HHH ∆∆∆∆−−−−∆∆∆∆====∆∆∆∆
)O(N2)(O)(NO4 5222oooo
fff HHHH ∆∆∆∆−−−−∆∆∆∆++++∆∆∆∆====∆∆∆∆
∆∆∆∆H° = 113 kJ 26

1/16/2013
27
Step 2: Calculate
∆ngas = (ngas)products – (ngas)reactants
∆ngas = (4 + 1 – 2) mol = 3 mol
Step 3: Calculate ∆∆∆∆E using
R = 8.31451 J/K·mol T = 298 K
∆E = 113 kJ –(3 mol)(8.31451 J/K·mol)(298 K)(1 kJ/1000 J)
∆E = 113 kJ – 7.43 kJ = 106 kJ
Ex. 1. (cont.)
27

1/16/2013
28
Ex. 1. finish
Step 4: Calculate % difference
Bigger than most, but still small
Note: Assumes that volumes of solids and liquids
are negligible
Vsolid ≈≈≈≈ Vliquid << Vgas
%.%kJ
kJ.66100
113
437difference % ====∗∗∗∗====
28

1/16/2013
29
Is Assumption that Vsolid ≈ Vliquid << Vgas Justified?
• Consider CaCO3(s) + 2H+(aq) →→→→ Ca2+(aq) + H2O(l) + CO2(g)
37.0 mL 2*18.0 mL 18 mL 18 mL 24.4 L
Volumes assuming each coefficient equal
number of moles
• So ∆V = ∆Vprod – ∆Vreac = 24.363 L ≈ 24.4 L
• Yes, assumption is justified
Note: If No gases are present reduces to
HE ∆∆∆∆≈≈≈≈∆∆∆∆29

1/16/2013
30
Learning Check• Consider the following reaction for picric acid:
8O2(g) + 2C6H2(NO2)3OH(ℓ) → 3 N2(g) + 12CO2(g) + 6H2O(ℓ)
• What type of reaction is it?
• Calculate ΔΗ°, ΔΕ°
8O2(g) + 2C6H
2(NO
2)
3OH(ℓ) → 3N2(g) + 12CO2(g) + 6H2O(ℓ)
ΔΗ°f
(kJ/mol) 0.00 3862.94 0.00 -393.5 -241.83
ΔΕ° = ΔH° – ΔngasRT = ΔH° – (15 – 8)mol*298K*
8.314×10–3kJ/(mol·K)
ΔH0 = 12mol(–393.5 kJ/mol) + 6mol(–241.83kJ/mol) +
6mol(0.00kJ/mol) – 8mol(0.00kJ/mol) – 2mol(3862.94kJ/mol)
ΔH0 = – 13,898.9 kJ
∆Ε° = –13,898.9 kJ – 29.0 kJ = – 13,927.9 kJ 30

1/16/2013
31
Your Turn!Given the following:
3H2(g) + N2(g) → 2NH3(g) ∆Ho = -46.19 kJ mol-1
Determine ∆E for the reaction.
A. -51.14 kJ mol-1
B. -41.23 kJ mol-1
C. -46.19 kJ mol-1
D. -46.60 kJ mol-1
• ∆H = ∆E + ∆nRT ∆E = ∆H - ∆nRT
• ∆E =-46.19 kJ mol – (∆NRT)
• -(-2 mol)(8.314 J K-1mol-1)(298K)(1 kJ/1000 J)
• ∆E = -41.23 kJ mol-131

1/16/2013
32
∆Hof = Σn∆Ho
f (products) - Σm∆Hof (reactants)
2 NH3 (g) + CO2 (g) → NH2CONH2 (aq) + H2O (l)
Calculate ∆Hof for the reaction in slide 15
∆Hof = for NH3 (g) = -45.9 kJ
= for CO2 (g) = -393.5 kJ
= for NH2CONH2 = -319.2 kJ
= for H2O (l) = -285.8 kJ
∆Ho = [(-319.2 – 285.8) – (-2 x 45.9 – 393.50)] kJ = -119.7
Since ∆Ho has a negative sign, heat is evolved
32
1/16/2013
33
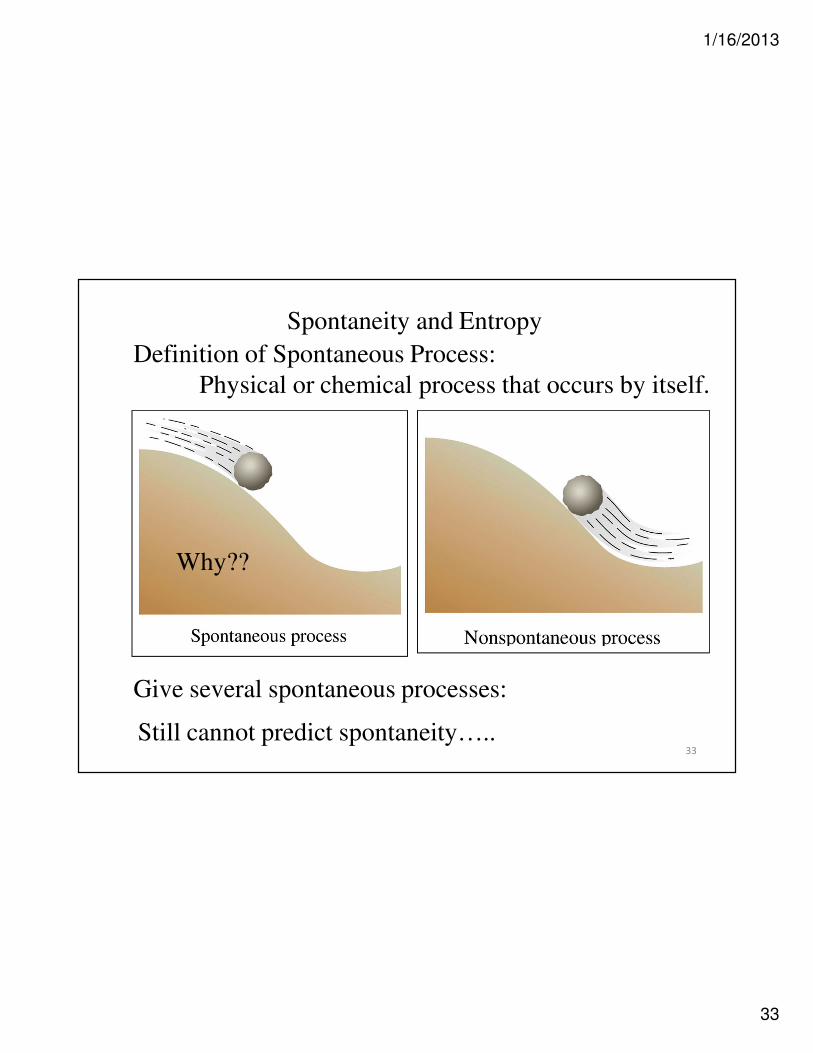
Spontaneity and Entropy
Definition of Spontaneous Process:
Physical or chemical process that occurs by itself.
Give several spontaneous processes:
Still cannot predict spontaneity…..
Why??
33

1/16/2013
34
Enthalpy Changes and Spontaneity
• What are relationships among factors that influence
spontaneity?
• Spontaneous Change
– Occurs by itself
– Without outside assistance until
finished
• Ex.
– Water flowing over waterfall
– Melting of ice cubes in glass
on warm day
34

1/16/2013
35
Nonspontaneous Change
• Occurs only with outside assistance
• Never occurs by itself:
– Room gets straightened up
– Pile of bricks turns into a brick wall
– Decomposition of H2O by electrolysis
• Continues only as long as outside assistance occurs:
– Person does work to clean up room
– Bricklayer layers mortar and bricks
– Electric current passed through H2O
35

1/16/2013
36
Entropy and the 2nd Law of Thermodynamics
2nd Law: The total entropy of a system and its
surroundings increases for a spontaneous
process.
Entropy = S = A thermodynamic quantity that is a
measure of how dispersed the energy
is among the different possible ways
that a system can contain energy.
Consider:
1. A hot cup of coffee on the table
2. Rock rolling down the side of a hill.
3. Gas expanding.
4. Stretching a rubber band.36

1/16/2013
37
Reaction Rate and Spontaneity
• ∆H indicates if reaction has tendency to occur
• Rate of reaction also plays role
– Some very rapid:
• Neurons firing in nerves in response to pain
• Detonation of stick of dynamite
– Some gradual:
• Erosion of stone
• Ice melting
• Iron rusting
– Some so slow, appear to be nonspontaneous:
• Gasoline and O2 at RT
• Many biochemical processes
1/16/2013
38
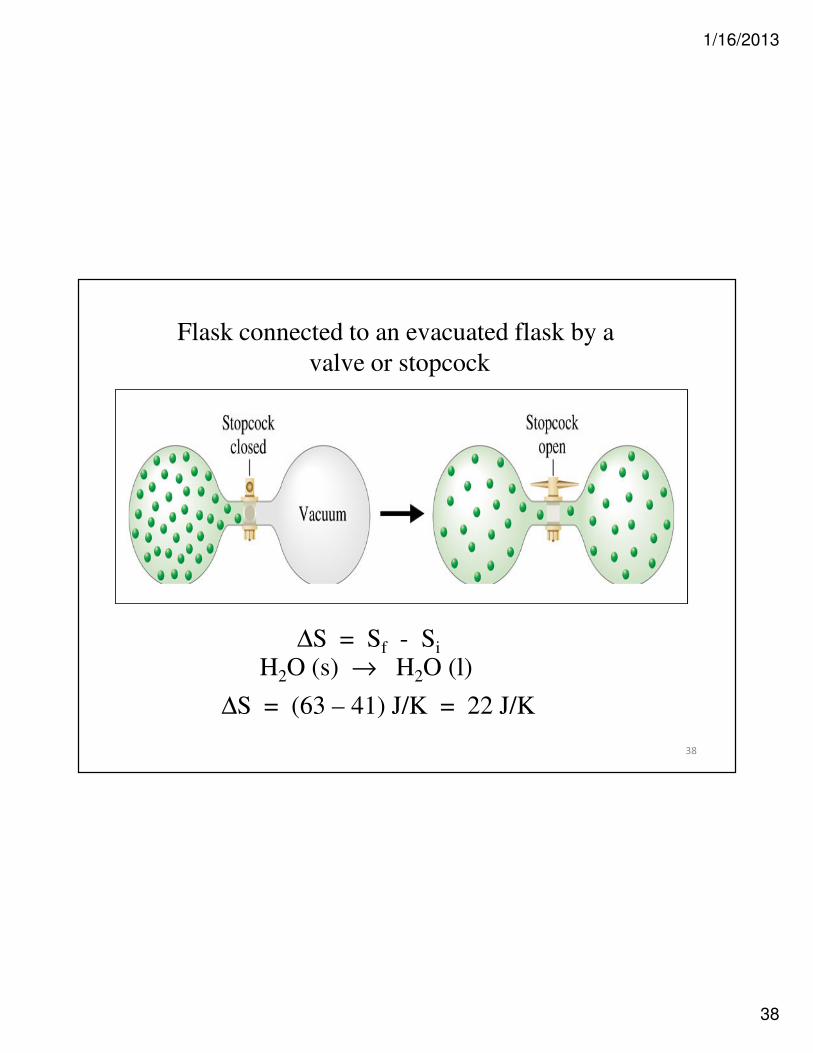
Flask connected to an evacuated flask by a
valve or stopcock
∆S = Sf - Si
H2O (s) → H2O (l)
∆S = (63 – 41) J/K = 22 J/K
38

1/16/2013
39
Direction of Spontaneous Change
• Many reactions which occur spontaneously are
exothermic:
– Iron rusting
– Fuel burning
• ∆H and ∆E are negative
– Heat given off
– Energy leaving system
• Thus, ∆H is one factor that influences spontaneity
Early thoughts were that exothermic reactions were spontaneous)
39

1/16/2013
40
Direction of Spontaneous Change
• Some endothermic reactions occur
spontaneously:
– Ice melting
– Evaporation of water from lake
– Expansion of CO2 gas into vacuum
• ∆H and ∆E are positive
– Heat absorbed
– Energy entering system
• Clearly other factors influence spontaneity
40

1/16/2013
41
Concept Check: You have a sample of solid iodine
at room temperature. Later you notice
that the iodine has sublimed. What
can you say about the entropy change
of the iodine?
41

1/16/2013
42
There are how many Laws of thermodynamics?
a. 1b. 2c. 3
d. 4
e. 5
42

1/16/2013
43
2nd Law: The total entropy of a system and its surroundings increases for a spontaneousprocess.
Process occurs naturally as a result of energy dispersalIn the system.
∆S = entropy created + q/T
∆S > q/T
For a spontaneous process at a given temperature, thechange in entropy of the system is greater than the heat divided by the absolute temperature.
43

1/16/2013
44
Entropy and Molecular Disorder
44

1/16/2013
45
Reactants
Products
Energy
Reaction Progress
Thermodynamic vs. Kinetics• Thermodynamics tells us:
– Direction of reaction
– Is it possible for reaction to
occur?
– Will reaction occur
spontaneously at
given T?
– Will reaction release
or absorb heat?
– Kinetics tells us:
– Speed of reaction
– Pathway between reactants
and products
• Domain of
Thermodynamics
(initial and final states)
Domain of Kinetics
(reaction pathway)
45

1/16/2013
46
Heat Transfer Between Hot and Cold Objects
• Consider system of two objects
– Initially one hot and one cold
– “Hot” = higher KEave of molecules = faster
– “Cold” = lower KEave of molecules = slower
• When they collide, what is most likely to occur?
– Faster objects bump into colder objects and transfer energy so…
– “Hot” objects cool down and slow down
– “Cold” objects warm up and speed up
– The reverse doesn’t occur

1/16/2013
47
Heat Transfer Between Hot and Cold Objects
Result:
• Heat flows spontaneously from hot to colder object
• Heat flows because of probable outcome of
intermolecular collisions
• Spontaneous processes tend to proceed from states
of low probability to states of higher probability
• Spontaneous processes tend to disperse energy

1/16/2013
48
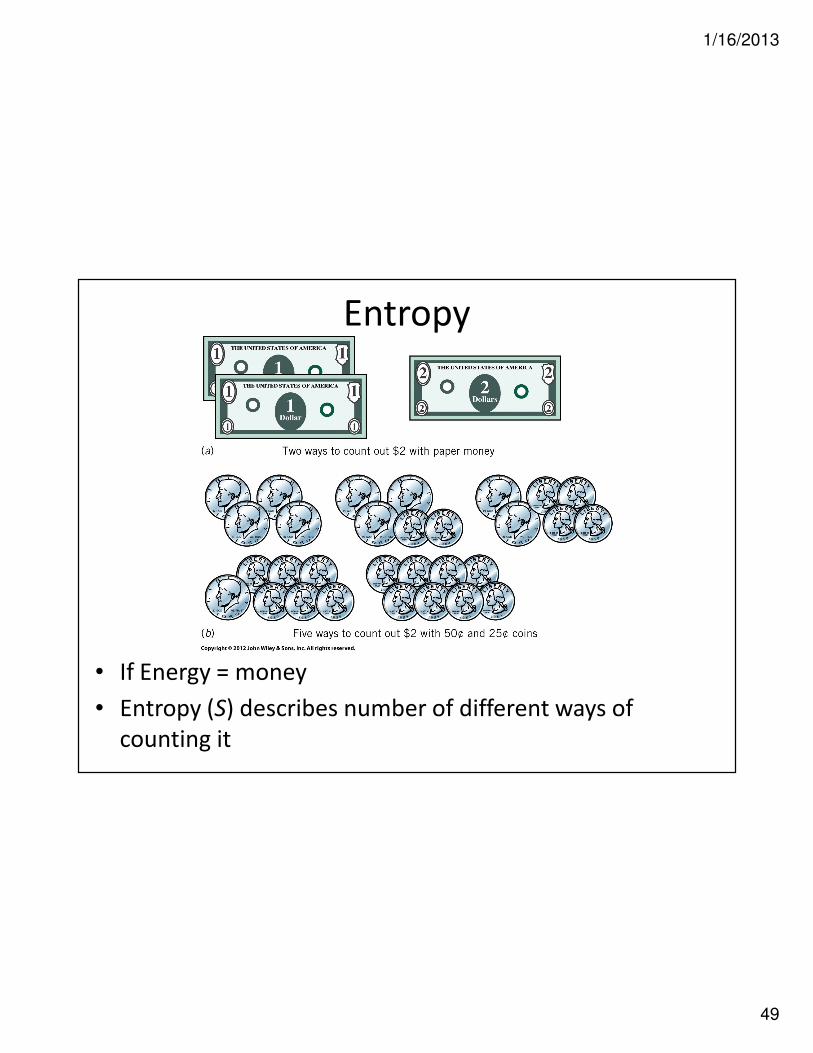
Entropy (symbol S)
• Thermodynamic quantity
• Describes number of equivalent ways that energy can be distributed
• Quantity that describes randomness of system
• Greater statistical probability of particular state means greater the entropy!
– Larger S, means more random and ∴∴∴∴ more probable
48
1/16/2013
49
Entropy
• If Energy = money
• Entropy (S) describes number of different ways of
counting it

1/16/2013
50
Criterion for Spontaneity• Clear order to things
– Things get rusty spontaneously
• Don't get shiny again
– Sugar dissolves in coffee
• Stir more—it doesn't undissolve
– Ice →→→→ liquid water at RT
• Opposite does NOT occur
– Fire burns wood, smoke goes up chimney
• Can't regenerate wood
• Common factor in all of these:
– Increase in randomness and disorder of system
– Something that brings about randomness more
likely to occur than something that brings order

1/16/2013
51
Entropy, S
• Measure of randomness and disorder
• Measure of chaos
• State function
• Independent of path
• ∆S = Change in Entropy
• Also state function
• For chemical reaction
initialfinal SSS −−−−====∆∆∆∆
reactantsSSS products −−−−====∆∆∆∆51

1/16/2013
52
Entropy• Sproducts > Sreactants means ∆S +
– Entropy ↑'s
– Probability of state ↑'s
– Randomness ↑'s
– Favors spontaneity
• Sproducts < Sreactants means ∆S −−−−
– Entropy ↓'s
– Probability of state ↓'s
– Randomness ↓'s
– Does not favor spontaneity
• Any reaction that occurs with ↑ in entropy tends to occur spontaneously
52

1/16/2013
53
Effect of Volume on Entropy
• For gases, Entropy ↑ as Volume ↑
A. Gas separated from vacuum by partition
B. Partition removed
C. Gas expands to achieve more probable particle
distribution
• More random, higher probability, more positive S53
1/16/2013
54
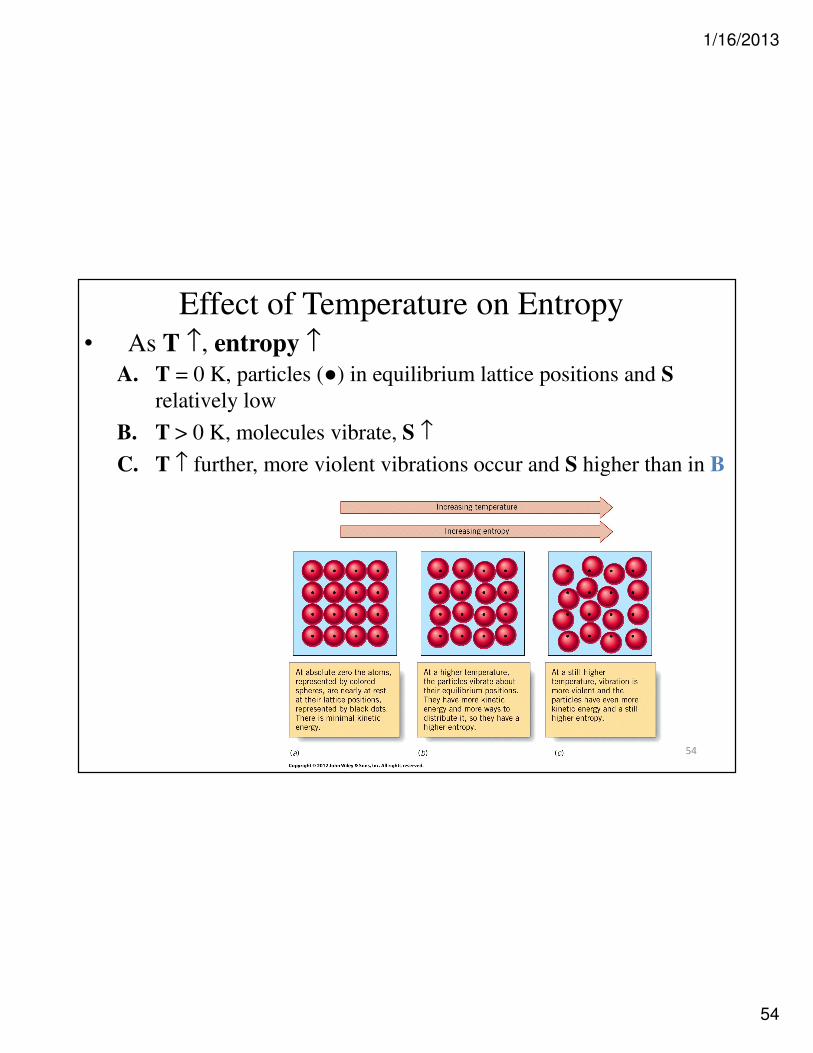
Effect of Temperature on Entropy• As T ↑, entropy ↑
A. T = 0 K, particles (●) in equilibrium lattice positions and S
relatively low
B. T > 0 K, molecules vibrate, S ↑
C. T ↑ further, more violent vibrations occur and S higher than in B
54

1/16/2013
55
Effect of Physical State on Entropy
• Crystalline solid very low S
• Liquid higher S, molecules can move freely
– More ways to distribute KE among them
• Gas highest S, particles randomly distributed throughout container
– Many, many ways to distribute KE
55
1/16/2013
56
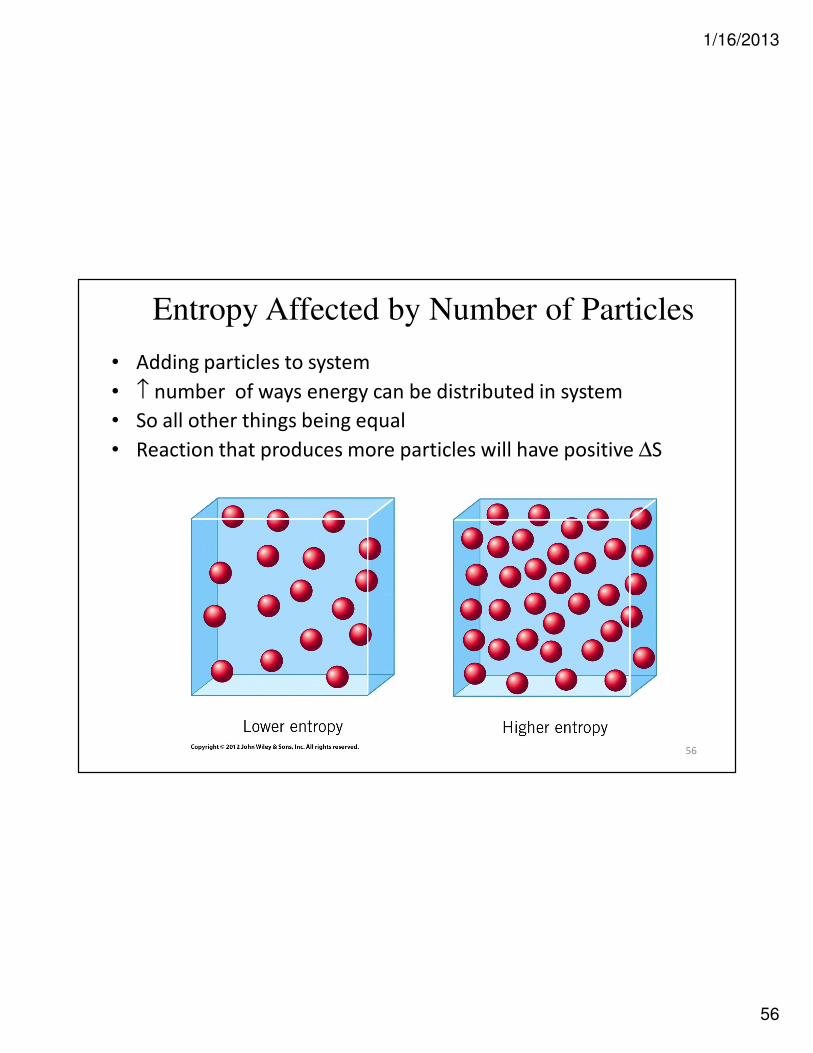
Entropy Affected by Number of Particles
• Adding particles to system
• ↑ number of ways energy can be distributed in system
• So all other things being equal
• Reaction that produces more particles will have positive ∆S
56

1/16/2013
57
Summary
• Larger V, greater ∆S
– Expansion of gas ∆S +
• Higher T, greater ∆S
– Higher T, means more KE in particles, move more, so random
distributions favored
• Ssolid < Sliquid << Sgas
– Solids more ordered than liquids, which are much more ordered
than gases
Reactions involving gases
• Simply calculate change in number of mole gas,
∆ngas
– If ∆ngas + , ∆S +
– If ∆ngas −−−− , ∆S −−−−57

1/16/2013
58
Entropy Change for a Reaction
∆So may be positive for reactions with the following:
1. The reaction is one in which a molecule is
broken into two or more smaller molecules.
2. The reaction is one in which there is an increase
in moles of gas.
3. The process is one in which a solid changes to
a liquid or a liquid changes to a gas.

1/16/2013
59
Did You Get It!
Which represents an increase in entropy?
A. water vapor condensing to liquid
B. carbon dioxide subliming
C. liquefying helium gas
D. proteins forming from amino acids
59

1/16/2013
60
Entropy Changes in Chemical Reactions
Ex. N2 (g) + 3 H2(g) → 2 NH3(g)
nreactant = 4 nproduct = 2
∆n = 2 – 4 = –2
Predict ∆Srxn < 0
Higher positional
probability
Lower positional
probability

1/16/2013
61
Entropy Changes in Chemical Reactions
Reactions without gases
• Simply calculate number of mole molecules
∆n = nproducts – nreactants
– If ∆n + , ∆S +
– If ∆n −−−− , ∆S −−−−
– More molecules, means more disorder
– Usually the side with more molecules, has less complex
molecules
• Smaller, fewer atoms per molecule

1/16/2013
62
Ex. 2 Predict Sign of ∆S for Following Reactions
CaCO3(s) + 2H+ (aq) →→→→ Ca2+(aq) + H2O(l) + CO2(g)
– ∆ngas = 1 mol – 0 mol = 1 mol
– ∴ ∆ngas +, ∆S +
2 N2O5 (g) →→→→ 4 NO2 (g) + O2 (g)
– ∆ngas = 4 mol + 1 mol – 2 mol = 3 mol
– ∴ ∆ngas +, ∆S +
OH−−−− (aq) + H+ (aq) →→→→ H2O (l)
– ∆ngas = 0 mol
– ∆n = 1 mol – 2 mol = –1 mol
– ∴ ∆n −−−− , ∆S −−−−

1/16/2013
63
Predict Sign of ∆S in Following:
• Dry ice → carbon dioxide gas
• Moisture condenses on a cool window
• AB → A + B
• A drop of food coloring added to a glass of water
disperses
• 2Al(s) + 3Br2(ℓ)→ 2AlBr3(s)
positive
positive
positive
negative
CO2(s)→ CO2(g)
H2O(g) → H2O(ℓ)
negative

1/16/2013
64
Which of the following has the most entropy at standard conditions?
A. H2O(ℓ)
B. NaCl(aq)
C. AlCl3(s)
D. Can’t tell from the information
Did You Get It?
Which reaction would have a negative entropy?
A. Ag+(aq) + Cl-(aq)→AgCl(s)
B. N2O4(g) → 2NO2 (g)
C. C8H18(l ) + 25/2 O2(g) → 8CO2(g) + 9H2O(g)
D. CaCO3(s) → CaO(s) + CO2(g)
64

1/16/2013
65
Both Entropy and Enthalpy Can Affect Reaction
Spontaneity
• Sometimes they work together
– Building collapses
– PE ↓ ∆H −−−−
– Stones disordered ∆S +
• Sometimes work against each other
– Ice melting (ice/water mix)
– Endothermic
• ∆H + nonspontaneous
– ↑ Disorder of molecules
• ∆S + spontaneous

1/16/2013
66
Which Prevails?
• Hard to tell—depends on temperature!
– At 25 °C, ice melts
– At −25 °C, water freezes
• So three factors affect spontaneity:
– ∆H
– ∆S
– T
66

1/16/2013
67
Second Law of Thermodynamics
• When a spontaneous event occurs, total entropy of
universe ↑’s
– (∆Stotal > 0)
• In a spontaneous process, ∆∆∆∆Ssystem can decrease as
long as total entropy of universe increases
– ∆∆∆∆Stotal = ∆∆∆∆Ssystem + ∆∆∆∆Ssurroundings
• It can be shown that
T
qS
gssurroundingssurroundin =∆

1/16/2013
68
2nd Law: The total entropy of a system and its surroundings increases for a spontaneousprocess.
Process occurs naturally as a result of energy dispersalIn the system.
∆S = entropy created + q/T
∆S > q/T
For a spontaneous process at a given temperature, thechange in entropy of the system is greater than the heat divided by the absolute temperature.
68

1/16/2013
69
Does This Make Sense?
• Yes?
• As heat added
– Disorder or entropy ↑, ∴ S ∝ q
• But how much ↑ T, ↑ S, depends on T at
which it occurs
– Low T, larger ↑ S
– High T, smaller ↑ S
• ∴ S ∝ 1/T

1/16/2013
70
What do you think?
When ice melts in your hand (assume your hand is 30o C),
A. the entropy change of the system is less then the entropy
change of the surroundings.
B.the entropy change of the surroundings is less than the
entropy change of the system.
C.the entropy change of the system equals the entropy
change of the surroundings.
• ∆Ssys = ∆H/273K ∆Ssurr = ∆H/303K
• ∆Ssys > ∆Ssurr

1/16/2013
71
Law of Conservation of Energy
• Says q lost by system must be gained by surroundings
– qsurroundings = −−−− qsystem
• If system at constant P, then
– qsystem = ∆H
• So
– qsurroundings = −−−− ∆Hsystem
• and
T
H
T
qS
systemgssurroundingssurroundin
∆−==∆

1/16/2013
72
Thus Entropy for Entire Universe is
Multiplying both sides by T we get
T∆∆∆∆Stotal = T∆∆∆∆Ssystem – ∆∆∆∆Hsystem
or
T∆∆∆∆Stotal = – (∆∆∆∆Hsystem – T∆∆∆∆Ssystem)
• For reaction to be spontaneous
– T∆∆∆∆Stotal > 0 (+)
So,
(∆∆∆∆Hsystem – T∆∆∆∆Ssystem) < 0
– (–) for reaction to be spontaneous
T
HSS
systemsystemtotal
∆−∆=∆

1/16/2013
73
Standard Entropy (Absolute Entropy): Entropy
value for the standard state of a species.
See Table B-13 p. B-17 and Table B-16 p. B-28
Entropy values of substances must be positive.
So must be >0 but Ho can be plus or minus
(Why?)
How about ionic species?
So for H3O+ is set at zero.
73

1/16/2013
74
Predict The Entropy sign for the following reactions:
a. C6H12O11 (s) → 2CO2 (g)+ C2H5OH (l)
b. 2 NH3 (g) + CO2 (g) → NH2CONH2 (aq) + H2O (l)
c. CO (g) + H2O (g) → CO2 (g) + H2 (g)
Exercise 13.2 p 578
d. Stretching a rubber band
74

1/16/2013
75
Calculating ∆So for a reaction
∆So = Σn∆So (products) - Σm∆So (reactants)
Calculate the entropy change for the following reaction
At 25 oC.
2 NH3 (g) + CO2 (g) → NH2CONH2 (aq) + H2O (l)
So 2 x 193 214 174 70
∆So = Σn∆So (products) - Σm∆So (reactants)
∆So= [(174 + 70) - (2x 193 + 214)] J/K= -356 J/K
See exercise 13.3 p 584 and problems 8-12 and 23-2675

1/16/2013
76
Third Law of Thermodynamics • At absolute zero (0 K),
– Entropy of perfectly ordered, pure crystalline
substance is zero
• S = 0 at T = 0 K
• Since S = 0 at T = 0 K
– Define absolute entropy of substance at higher
temperatures
• Standard entropy, S°
– Entropy of 1 mole of substance at 298 K (25°C) and
1 atm pressure
– S°= ∆S for warming substance from 0 K to 298 K
(25°C)76

1/16/2013
77
Consequences of Third Law
1. All substances have positive entropies as they are more
disordered than at 0 K
– Heating ↑↑↑↑ randomness
– S°is biggest for gases—most disordered
2. For elements in their standard states
– S°≠ 0 (but ∆Hf°= 0)
• Units of S°⇒ J/(mol·K)
Standard Entropy Change
– To calculate ∆∆∆∆S°for reaction, do Hess's Law type calculation
– Use S°rather than entropies of formation
∑∑ −=∆ )reactants()products( ooo SmSnS77

1/16/2013
78
Learning Check
Calculate ∆S0 for the following:
• CO2(s) → CO2(g)
187.6 213.7 S0 (J/mol·K)
•CaCO3(s) → CO2(g) + CaO(s)
92.9 213.7 40 S0 (J/mol·K)
• ∆S0 = (213.7 – 187.6) J/mol·K
• ∆S0 = 26.1 J/mol·K
•∆S0 = (213.7 +40 – 92.9) J/mol·K
•∆S0 = 161 J/mol·K78

1/16/2013
79
Ex. 3. Calculate ∆∆∆∆S°for reduction of aluminum oxide by
hydrogen gas
Al2O3 (s) + 3 H2 (g) →→→→ 2 Al (s) + 3 H2O (g)
Substance S° (J/ K·mol)
Al (s) 28.3
Al2O3 (s) 51.00
H2 (g) 130.6
H2O (g) 188.7
∑∑ −=∆ ooo
reactantsSnSnS rproductsp
]3[
]32[
)(2)(32
)(2)(
oo
ooo
gs
gs
HOAl
OHAl
SS
SSS
+−
+=∆
79

1/16/2013
80
Ex. 3
⋅+
⋅−
⋅+
⋅=∆
Kmol
130.6J*mol 3
Kmol
J51.00*mol 1
Kmol
J188.7*mol 3
Kmol
J28.3*mol 2oS
∆S°= 56.5 J/K + 566.1 J/K – 51.00 J/K – 391.8 J/K
∆S°= 179.9 J/K
80

1/16/2013
81
Gibbs Free Energy
• Would like one quantity that includes all
three factors that affect spontaneity of
a reaction
• Define new state function
• Gibbs Free Energy
– Maximum energy in reaction that is "free" or
available to do useful work
G ≡≡≡≡ H – TS
• At constant P and T, changes in free energy
∆∆∆∆G = ∆∆∆∆H – T∆∆∆∆S
81

1/16/2013
82
Gibbs Free Energy
∆∆∆∆G < 0 Spontaneous process
∆∆∆∆G = 0 At equilibrium
∆∆∆∆G > 0 Nonspontaneous
∆∆∆∆G = ∆∆∆∆H – T∆∆∆∆S
• G ⇒ state function
– Made up of T, H and S = state functions
– Has units of energy
– Extensive property
• ∆∆∆∆G = Gfinal – Ginitial
82

1/16/2013
83
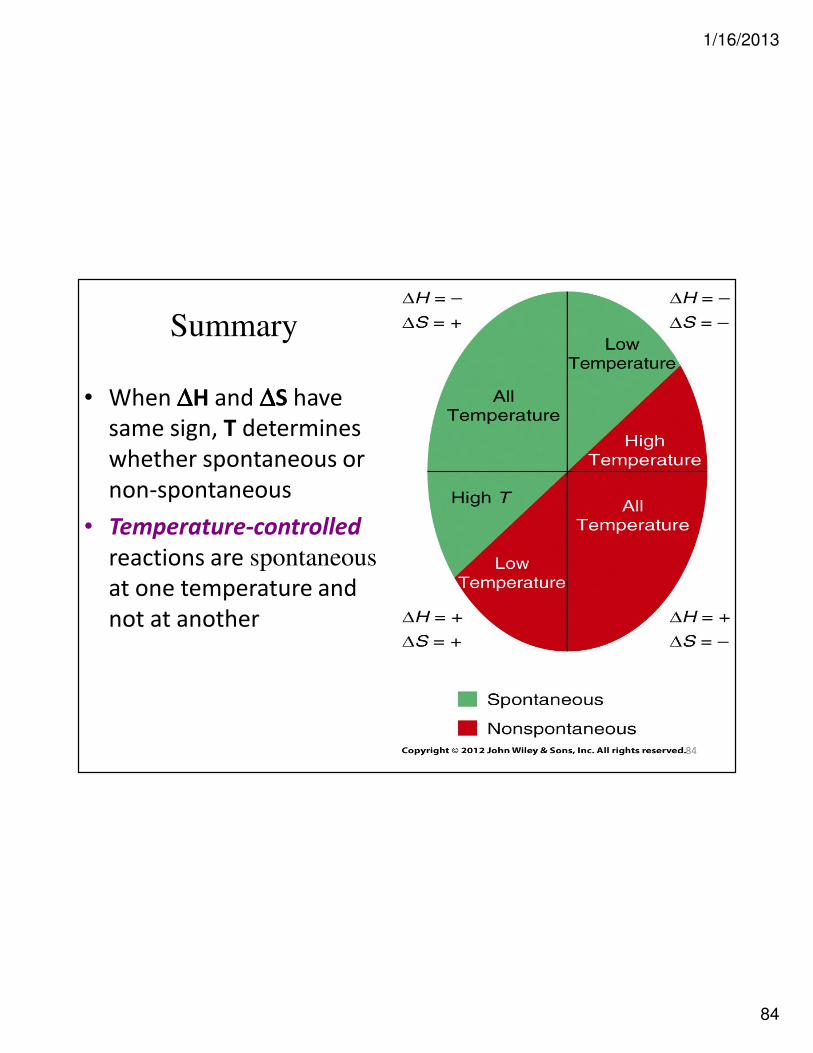
Criteria for Spontaneity?
• At constant P and T, process spontaneous only if it is
accompanied by ↓↓↓↓ in free energy of system
∆∆∆∆H ∆∆∆∆S Spontaneous?
– + ∆∆∆∆G = (–) – [T(+)] = – Always, regardless of T
+ – ∆∆∆∆G = (+) – [T(–)] = + Never, regardless of T
+ + ∆∆∆∆G = (+) – [T(+)] = ? Depends; spontaneous at
high T, –∆∆∆∆G
– – ∆∆∆∆G = (–) – [T(–)] = ? Depends; spontaneous at
low T, –∆∆∆∆G
83
1/16/2013
84
Summary
• When ∆∆∆∆H and ∆∆∆∆S have
same sign, T determines
whether spontaneous or
non-spontaneous
• Temperature-controlled
reactions are spontaneous
at one temperature and
not at another
84

1/16/2013
85
Free Energy Concept
∆Ho – T∆So Can Serve as a criteria for Spontaneity
2 NH3 (g) + CO2 (g) → NH2CONH2 (aq) + H2O (l)
∆Ho = -119.7 kJ ∆So = -365 J/K = -0.365 kJ/K
∆Ho – T∆So = (-119.7 kJ) – (298 K) x (-0.365kJ/K
= -13.6 kJ
∆Ho – T∆So is a negativity quantity, from which we can
conclude that the reaction is spontaneous
under standard conditions.
G = H – T S
85

1/16/2013
86
Free Energy and Spontaneity
Free Energy: Thermodynamic quantity defined by
the equation G = H - TS
∆G = ∆H - T∆S
If you can show that ∆G for a reaction at a given
temperature and pressure is negative, you can
predict that the reaction will be spontaneous…
86

1/16/2013
87
Standard Free Energy Changes
Standard Conditions:
1 atm pressure
1 atm partial pressure
1 M concentration
Temperature of 25 oC or 298 K
Standard free energy is free-energy change that takes
place when reactants in their standard states are
converted to products in their standard states.
∆Go = ∆Ho - T∆So
87

1/16/2013
88
Standard Free Energy Changes
• Standard Free Energy Change, ∆G°
– ∆G measured at 25 °C (298 K) and 1 atm
• Two ways to calculate, depending on what data is
available
•Method 1. ∆∆∆∆G°= ∆∆∆∆H°– (298.15 K)∆∆∆∆S°
Ex. 4. Calculate ∆∆∆∆G°for reduction of aluminum oxide by
hydrogen gas
Al2O3(s) + 3 H2(g) →→→→ 2 Al(s) + 3H2O(g)
88

1/16/2013
89
Ex. 4. Method 1
Al2O3 (s) + 3 H2 (g) →→→→ 2 Al (s) + 3 H2O (g)
Step 1: Calculate ∆∆∆∆H°for reaction using o
fH∆∆∆∆Substance (kJ/mol)
Al (s) 0.0
Al2O3 (s) –1669.8
H2 (g) 0.0
H2O (g) –241.8
reactantsHnproductsHnH frfp ∑∑ ∆−∆=∆ ooo
]3[
]32[
)(Hf)(OAlf
)O(Hf)Al(f
232
2
gs
gs
HH
HHH
oo
ooo
∆+∆−
∆+∆=∆
o
fH∆∆∆∆
89

1/16/2013
90
Ex. 4. Method 1 Step 1 (∆H°)
++++
−−−−−−−−
−−−−++++
====
mol
kJ. mol*
mol
kJ. mol*
mol
kJ. mol*
mol
kJ. mol*∆H
0003
816691
82413
0002o
∆H°= 0.0 kJ – 725.4 kJ – 0.00 kJ – (– 1669.8 kJ)
∆H°= 944.4 kJ
90

1/16/2013
91
Ex. 4 Method 1
Step 2: Calculate ∆S° see Ex. 4
∆S°= 179.9 J/K
Step 3: Calculate
∆G°= ∆∆∆∆H°– (298.15 K)∆∆∆∆S°
∆G°= 944.4 kJ – (298 K)(179.9 J/K)(1 kJ/1000 J)
∆G°= 944.4 kJ – 53.6 kJ = 890.8 kJ
– ∆G°= +
– ∴ not spontaneous
91

1/16/2013
92
Method 2
• Use Standard Free Energies of Formation
• Energy to form 1 mole of substance from its
elements in their standard states at 1 atm and 25
°C
o
fG∆∆∆∆
reactantsGnproductsGnG frfp ∑∑ ∆−∆=∆ ooo
92

1/16/2013
93
Ex. 4. Method 2
Calculate ∆∆∆∆G°for reduction of aluminum oxide
by hydrogen gas.
Al2O3 (s) + 3 H2 (g) →→→→ 2 Al (s) + 3 H2O (g)
]3[
]32[
)(Hf)(OAlf
)O(Hf)Al(f
232
2
gs
gs
GG
GGG
oo
ooo
∆+∆−
∆+∆=∆
Substance (kJ/mol)
Al (s) 0.0
Al2O3 (s) –1576.4
H2 (g) 0.0
H2O (g) –228.6
o
fG∆∆∆∆
93

1/16/2013
94
Ex. 4. Method 2
+
−−
−+
=
mol
kJ. mol*
mol
kJ. mol*
mol
kJ. mol*
mol
kJ. mol*∆G
0003
415761
62283
0002o
∆G°= 0.0 kJ – 685.8 kJ – 0.00 kJ – (– 1576.4 kJ)
∆G°= 890.6 kJ
Both methods same
within experimental error94

1/16/2013
95
∆Go as a Criterion for Spontaneity
∆Go = ∆Ho - T∆So
1. When ∆Go is a large negative number (more
negative than about – 10 kJ), the reaction is spontaneous
as written, and reactants transform almost entirely into
products when equilibrium is reached.
2. When ∆Go is a large positive number (more
positive than about + 10 kJ), the reaction is not
spontaneous as written, and reactants transform almost
entirely into products when equilibrium is reached.
3. When ∆Go has a small positive or negative value(less
than about 10 kJ), the reaction mixture gives an
equilibrium mixture with significant amounts of both
reactants and products.. 95

1/16/2013
96
Interpreting the Sign of ∆Go
Calculate ∆Ho and ∆Go for the following reaction
2 KClO3 (s) → 2 KCl (s) + 3 O2 (g)
Interpret the signs of ∆Ho and ∆Go
∆Hfo are as follows: KClO3 (s) = -397.7 kJ/mol
KCl (s) = -436.7 kJ/mol
O2 (g) = 0
∆Gfo are as follows: KClO3 (s) = -296.3 kJ/mol
KCl (s) = -408.8 kJ/mol
O2 (g) = 0
96

1/16/2013
97
2 KClO3 (s) → 2 KCl (s) + 3 O2 (g)
∆Hfo 2 x (-397.70) 2 x (-436.7) 0 kJ
∆Gfo 2 x (-296.3) 2 x (-408.8) 0 kJ
Then:
∆Ho = [2 x (-436.7) – 2 x (-397.7)] kJ = -78 kJ
∆Go = [2 x (-408.8) – 2 x (-296.3)] kJ = -225 kJ
The reaction is exothermic, liberating 78 kJ of heat. The
large negative value of ∆Go indicates that the equilibrium
is mostly KCl and O2.
97

1/16/2013
98
Spontaneous Reactions Produce Useful Work
• Fuels burned in engines to power cars or heavy machinery
• Chemical reactions in batteries
– Start cars
– Run cellular phones, laptop computers, mp3 players
• Energy not harnessed if reaction run in an open dish
– All energy lost as heat to surroundings
• Engineers seek to capture energy to do work
– Maximize efficiency with which chemical energy is
converted to work
– Minimize amount of energy transformed to unproductive
heat
98

1/16/2013
99
Thermodynamically Reversible
• Process that can be reversed and is always very close to
equilibrium
– Change in quantities is infinitesimally small
• Example - expansion of gas
– Done reversibly, it does most work on surroundings
99

1/16/2013
100
∆G = Maximum Possible Work
• ∆G is maximum amount of energy produced
during a reaction that can theoretically be
harnessed as work
– Amount of work if reaction done under reversible
conditions
– Energy that need not be lost to surroundings as heat
– Energy that is “free” or available to do work
100

1/16/2013
101
Ex. 5
Calculate ΔG° for reaction below at 1 atm and
25°C, given ΔH° = –246.1kJ/mol, ΔS° = 377.1
/(mol·K).
H2C2O4(s) + ½ O2(g) → 2CO2(g) + H2O(ℓ)
∆∆∆∆G25 = ∆∆∆∆H – T∆∆∆∆S
ΔG° =(–246.1 – 112.4)kJ/mol
ΔG° = –358.5kJ/mol
⋅−−=∆
kJ
J
molK
JK
mol
kJG
1000
11.377)298(1.246o
101

1/16/2013
102
You Try!Calculate ∆Go for the following reaction,
H2O2(l ) → H2O(l ) + O2(g)
given ∆Ho = -196.8 kJ mol-1 and ∆So = +125.72 J K-1
mol-1.
A. -234.3 kJ mol-1
B. +234.3 kJ mol-1
C. 199.9 kJ mol-1
D. 3.7 x 105 kJ mol-1
•∆Go = -196.8 kJ mol-1 – 298 K (0.12572 kJ K-1 mol-1)
•∆Go = -234.3 kJ mol
102

1/16/2013
103
System at Equilibrium
• Neither spontaneous nor nonspontaneous
• In state of dynamic equilibrium
• Gproducts= Greactants
• ∆G = 0
• Consider freezing of water at 0o C
• H2O(ℓ) � H2O(s)
– System remains at equilibrium as long as no heat added
or removed
– Both phases can exist together indefinitely
– Below 0oC, ∆G < 0 freezing spontaneous
– Above 0oC, ∆G > 0 freezing nonspontaneous
•Define equilibrium103

1/16/2013
104
Ex. 6
Calculate Tbp for reaction below at 1 atm and 25°C, given
ΔH° = 31.0kJ/mol, ΔS° = 92.9 J/(mol·K)
Br2(l) → Br2(g)
• For T > 334 K, ΔG < 0 and reaction is
spontaneous (ΔS° dominates)
• For T < 334 K, ΔG > 0 and reaction is
nonspontaneous (ΔH° dominates)
• For T = 334 K, ΔG = 0 and T = normal
boiling point
KKmolkJ
molkJ
S
HTbp 334
)/(0929.0
/0.31=
⋅≈
∆
∆≈
o
o
104

1/16/2013
105
Free energy change during reaction
105

1/16/2013
106
Free energy change during reaction
106

1/16/2013
107
Free Energy and Equilibrium Constant
Very important relation is the relation between
free energy and the equilibrium constant.
Thermodynamic Equilibrium Constant- the equilibrium
constant in which the concentration of gases are
expressed in partial pressures in atmospheres,
whereas the concentration of solutes in liquid
are expressed in molarities.
K = Kc for reactions involving only liquid solutions
K = Kp for reactions involving only gases
107

1/16/2013
108
Equilibrium Expression
Kc =
aA + bB ⇔ cC + dD
[C]c[D]d
[A]a[B]b
Solids and liquids considered unity (1)
108

1/16/2013
109
Write Kp and Kc
• Consider again
N2(g) + 3H2(g) ⇄ 2 NH3(g)
Kc = [NH3]
2
[N2][H2]3
109

1/16/2013
110
• In terms of partial pressures,
N2(g) + 3H2(g) ⇄ 2 NH3(g)
Kp = [PNH3]
2
[PN2][PH2]3
110

1/16/2013
111
• Kc and Kp are related.
• PV = nRT
• P = [n/V]RT
• Kp=Kc × (RT)∆n
• ∆n = (number of moles of product gas) –(number of moles of reactant gas)
• For this reaction, ∆n = -2.
N2(g) + 3H2(g) ⇔ 2 NH3(g)
111

1/16/2013
112
No Work Done at Equilibrium• ∆G = 0
• No “free” energy available to do work
• Consider fully charged battery
– Initially • All reactants, no products
• ∆G large and negative
• Lots of energy available to do work
– As battery discharges• Reactants converted to products
• ∆G less negative
• Less energy available to do work
– At Equilibrium• ∆G = Gproducts – Greactants = 0
• No further work can be done
• Dead battery112

1/16/2013
113
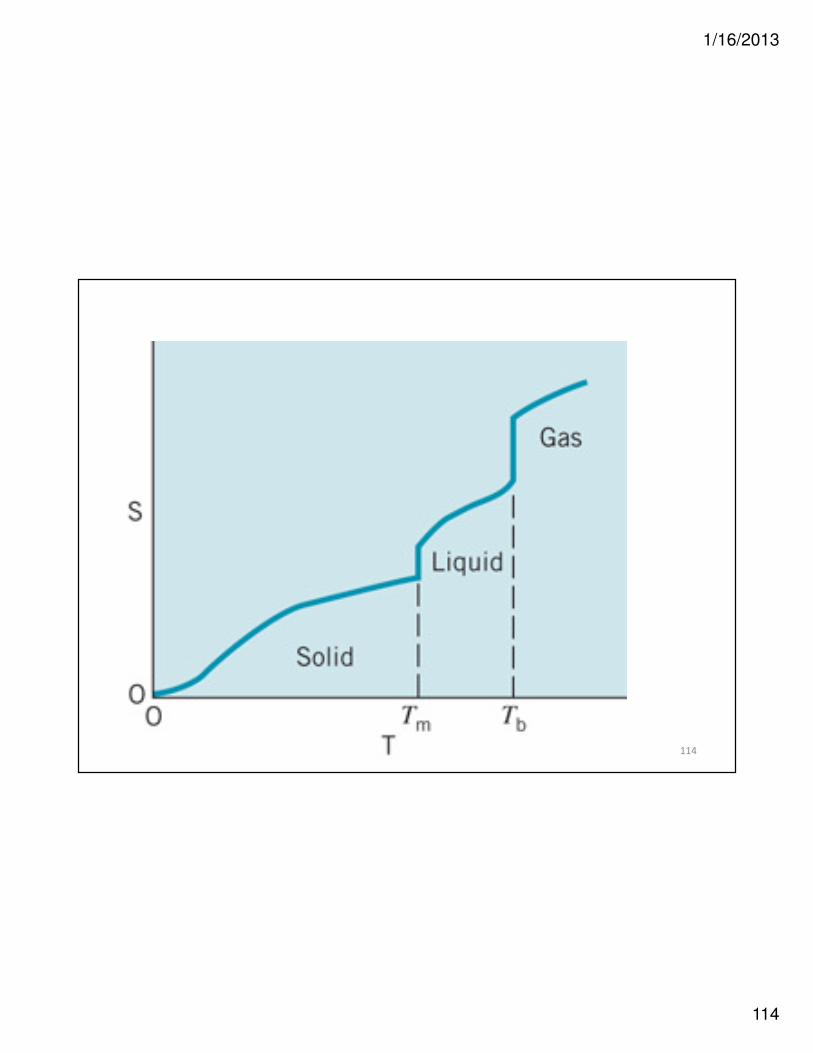
Phase Change = Equilibrium
• H2O(ℓ) ���� H2O(g)
• ∆G = 0 = ∆H – T∆S
• Only one temperature possible for phase
change at equilibrium
– Solid-liquid equilibrium
• Melting/freezing temperature (point)
– Liquid-vapor equilibrium
• Boiling temperature (point)
• Thus ∆H = T∆S and
• orT
HS
∆=∆
S
HT
∆
∆=
113
1/16/2013
114
114

1/16/2013
115
∆G°and Position of Equilibrium
• When ∆∆∆∆G°> 0 (positive)
– Position of equilibrium lies close to reactants
– Little reaction occurs by the time equilibrium is reached
– Reaction appears nonspontaneous
• When ∆∆∆∆G°< 0 (negative)
– Position of equilibrium lies close to products
– Mainly products exist by the time equilibrium is reached
– Reaction appears spontaneous
115

1/16/2013
116
∆G°and Position of Equilibrium
• When ∆∆∆∆G°= 0
– Position of equilibrium lies ~ halfway between products
and reactants
– Significant amount of both reactants and products
present at time equilibrium is reached
– Reaction appears spontaneous, whether start with
reactants or products
• Can Use ∆∆∆∆G°to Determine Reaction Outcome
– ∆G°large and positive
• No observable reaction occurs
– ∆G°large and negative
• Reaction goes to completion
116

1/16/2013
117
Learning Check
Ex. 7 Given that ΔH° = –97.6 kJ/mol,
ΔS° = –122 J/(mol·K), at 1atm and 298K,
will the following reaction occur spontaneously?
• MgO(s) + 2HCl(g) → H2O(ℓ) + MgCl2(s)
∆G° = ∆H° – T∆S°
= –97.6kJ/mol – 298K(–0.122 kJ/mol⋅K)
∆G° = –97.6kJ/mol +36.4kJ/mol
= –61.2 kJ/mol
117

1/16/2013
118
Effect of Temperature on ∆G°
• Reactions often run at T’s other that 298 K
• Position of equilibrium can change as ∆G°depends on T
– ∆G°= ∆H°−−−− T∆S°
• For T’s near 298 K, expect only very small changes in ∆H
and ∆S°
• For reaction at T, we can write:
ooo
TTT STHG ∆−∆=∆
ooo
298298 STHGT ∆−∆≈∆
118

1/16/2013
119
Ex. 8 Determining Effect of T on Spontaneity
• Calculate ∆∆∆∆G°at 25°C and 500°C for the Haber
process
N2 (g) + 3 H2 (g) ���� 2 NH3 (g)
• Assume that ∆∆∆∆H°and ∆∆∆∆S°do not change with T
• Solving Strategy
Step 1. Using data from Tables, calculate ∆∆∆∆H°and ∆∆∆∆S°
for the reaction at 25°C
– ∆∆∆∆H°= – 92.38 kJ
– ∆∆∆∆S°= – 198.4 J/K
119

1/16/2013
120
Ex. 8 Determining Effect of T on Spontaneity
Step 2. Calculate ∆∆∆∆G°for the reaction at 25°C using
∆∆∆∆H°and ∆∆∆∆S°
N2 (g) + 3 H2 (g) ���� 2 NH3 (g)
– ∆∆∆∆H°= – 92.38 kJ
– ∆∆∆∆S°= – 198.4 J/K
• ∆∆∆∆G°= ∆∆∆∆H°– T∆∆∆∆S°
• ∆∆∆∆G°= –92.38 kJ – (298 K)(–198.4 J/K)
• ∆∆∆∆G°= –92.38 kJ+ 59.1 kJ = –33.3 kJ
• So the reaction is spontaneous at 25°C
J
kJ
1000
1
120

1/16/2013
121
Ex. 8 Determining Effect of T on Spontaneity
Step 3. Calculate ∆∆∆∆G°for the reaction at 500°C using
∆∆∆∆H°and ∆∆∆∆S°.
– T = 500°C + 273 = 773 K
– ∆∆∆∆H°= – 92.38 kJ
– ∆∆∆∆S°= – 198.4 J/K
• ∆∆∆∆G°= ∆∆∆∆H°– T∆∆∆∆S°
• ∆∆∆∆G°= –92.38 kJ – (773 K)(–198.4 J/K)
• ∆∆∆∆G°= –92.38 kJ+ 153 kJ = 61 kJ
• So the reaction is NOT spontaneous at 500°C
J
kJ
1000
1
121

1/16/2013
122
Ex. 8 Does this answer make sense?
• ∆∆∆∆G°= ∆∆∆∆H°– T∆∆∆∆S°
– ∆∆∆∆H°= – 92.38 kJ
– ∆∆∆∆S°= – 198.4 J/K
• Since both ∆∆∆∆H°and ∆∆∆∆S°are negative
• At low T
– ∆∆∆∆G°will be negative and spontaneous
• At high T
– T∆∆∆∆S°will become a bigger positive number and
– ∆∆∆∆G°will become more positive and thus eventually, at
high enough T, will become nonspontaneous
122

1/16/2013
123
Effect of Change in Pressure or Concentration on ∆G
• ∆G at nonstandard conditions is related to ∆G°at
standard conditions by an expression that includes
reaction quotient Q
• This important expression allows for any concentration
or pressure
• Recall:
QRTGG ln+∆=∆ o
x
y
Q][
][
reactants
products=
123

1/16/2013
124
Ex. 9 Calculating ∆G at Nonstandard conditions
• Calculate ∆∆∆∆G at 298 K for the Haber process
– N2 (g) + 3 H2 (g) ���� 2 NH3 (g) ∆G°= –33.3 kJ
• For a reaction mixture that consists of 1.0 atm N2, 3.0
atm H2 and 0.5 atm NH3
Step 1 Calculate Q
3
3
2
3
2
103.9)0.3)(0.1(
)50.0(
22
3 −×===HN
NH
PP
PQ
124

1/16/2013
125
Ex. 9 Calculating ∆G at Nonstandard conditions
Step 2 Calculate ∆∆∆∆G = ∆G°+ RT lnQ
∆∆∆∆G = –33.3 kJ/mol + (8.314J/K·mol)*(1kJ/1000J)* (298K)*ln(9.3 × 10–3)
= –33.3 kJ/mol + (2.479 kJ/mol)*ln(9.3 × 10–3)
= –33.3 kJ + (–11.6 kJ/mol) = – 44.9 kJ/mol
• At standard conditions all gases (N2, H2 and NH3) are at 1 atm of pressure
• ∆∆∆∆G becomes more negative when we go to 1.0 atm N2, 3.0 atm H2 and 0.5 atm NH3
• Indicates larger driving force to form NH3
– Preactants > Pproducts
125

1/16/2013
126
How K is related to ∆G°
• Use relation ∆∆∆∆G = ∆G°+ RT lnQ to derive relationship between K and ∆G°
• At Equilibrium
∆G = 0 and Q = K
• So 0 = ∆G°+ RT lnK
∆G°= −−−−RT lnK
Taking antilog (ex) of both sides gives
K = e−−−−∆G°/RT
126

1/16/2013
127
At Equilibrium• ∆G°= −−−−RT lnK and K = e−−−−∆G°/RT
• Provides connection between ∆G°and K
• Can estimate K’s at various T’s if know ∆G°
• Can get ∆G°in know K’s
Relationship between K and ∆G
Keq ∆∆∆∆G°°°° Reaction
> 1 − Spontaneous Favored
Energy released
< 1 + non-spontaneous Unfavorable
Energy needed
= 1 0 At Equilibrium127

1/16/2013
128
Ex. 10 Calculating ∆G° from K
• Ksp for AgCl(s) at 25°C is 1.8 × 10−10 Determine ∆G° for the process
• Ag+ (aq) + Cl−−−− (aq) →→→→ AgCl (s)
• Reverse of Ksp equation, so
• ∆G°= –RT lnK = –(8.3145J/K·mol)(298K)*ln(5.6 × 109)*(1kJ/1000J)
• ∆G°= –56 kJ/mol
• Negative ∆G°indicates precipitation will occur
9
10106.5
108.1
11×=
×==
−spK
K
128

1/16/2013
129
Ex. 11 Calculating K from ∆G°
• Calculate K at 25°C for the Haber process
N2 (g) + 3 H2 (g) ���� 2 NH3 (g)
∆G°= –33.3 kJ/mol = –33,300 J/mol
Step 1 Solve for exponent
Step 2 Take ex to obtain K
Large K indicates NH3 favored at RT
54.13/ 107 ×=== ∆− eeK RTG o
4.13)298)(/3145.8(
)/300,33(=
⋅
−−=
∆−
KmolKJ
molJ
RT
G o
RTGeK /o∆−=
129

1/16/2013
130
You Try!Calculate the equilibrium constant for the
decomposition of hydrogen peroxide at 298 K given
∆Go = -234.3 kJ mol.
A. 8.5 x 10-42
B. 1.0 x 10499
C. 3.4 x 10489
D. 1.17 x 1041
94.56 41
( 234,300 / )94.56
(8.3145 / )(298 )
1.17 x 10
G J mol
RT J K mol K
K e
−∆ − −= =
⋅
= =
o
130
1/16/2013
131
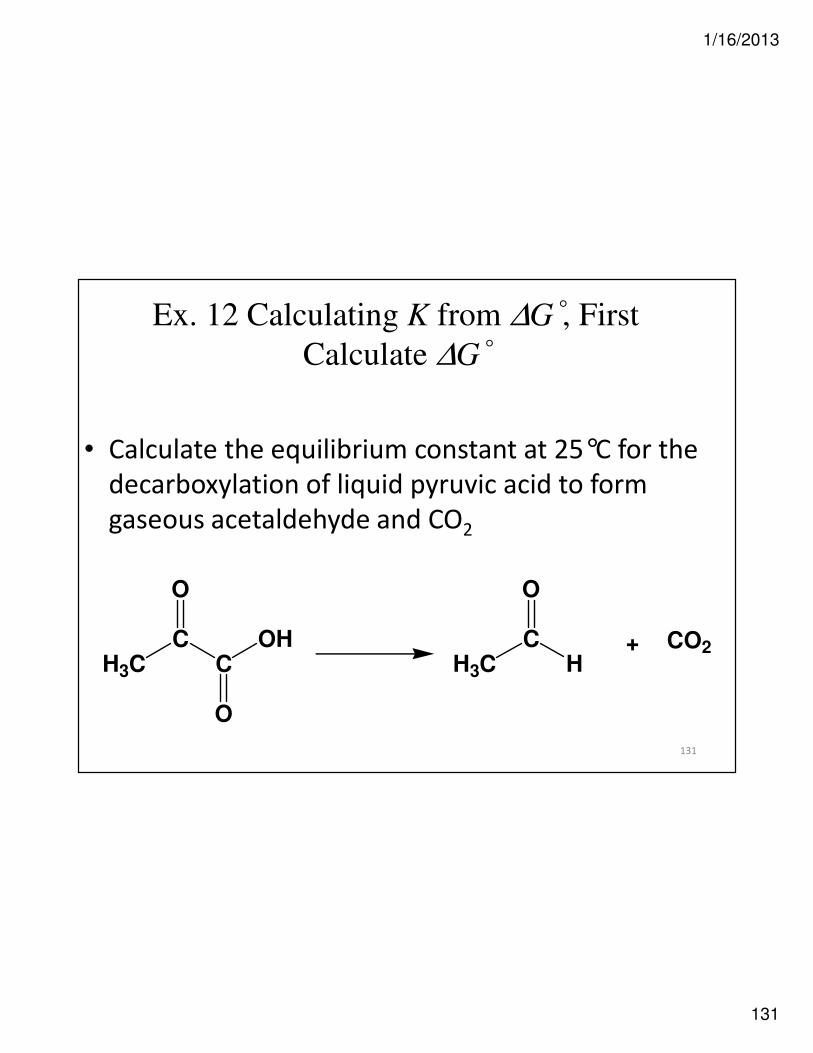
Ex. 12 Calculating K from ∆G°, First
Calculate ∆G°
• Calculate the equilibrium constant at 25°C for the
decarboxylation of liquid pyruvic acid to form
gaseous acetaldehyde and CO2
H3CC
C
O
OH
O
H3CC
O
H+ CO2
131

1/16/2013
132
First Calculate ∆G°from ∆Gf°
Compound ∆Gf°, kJ/mol
CH3COH −133.30
CH3COCOOH −463.38
CO2 −394.36
)()()( 323 COCOOHCHfCOfCOHCHf GGGG oooo ∆−∆+∆=∆
)38.463()36.394(30.133 −−−+−=∆ oG
kJG 28.64−=∆ o
132

1/16/2013
133
Next Calculate Equilibrium Constant
RTGeKo∆−=
94.251000
)298)(/314.8(
28.64−=×
−=
∆
kJ
J
KKJ
kJ
RT
Go
945.25)945.25( eeK == −−
K = 1.85 ×××× 1011
133

1/16/2013
134
Temperature Dependence of K
• ∆G°= –RT lnK = ∆H°– T∆S°
• Rearranging gives
• Equation for line
– Slope = – ∆H°/RT
– Intercept = ∆S°/R
• Also way to determine
K if you know ∆∆∆∆H°and ∆∆∆∆S°
R
∆S
T
1
R
∆Hln
oo
+
−=K
134

1/16/2013
135
Ex. 12 Calculate K given ∆H° and ∆S°
• Calculate K at 500 °C for Haber process
N2 (g) + 3 H2 (g) ���� 2 NH3 (g)
– Given ∆∆∆∆H°= – 92.38 kJ and ∆∆∆∆S°= – 198.4 J/K
• Assume that ∆∆∆∆H°and ∆∆∆∆S°do not change with T
R
S
RT
HK
oo ∆+
∆−=ln
)/314.8(
/4.198
)773)(/314.8(
380,92ln
KJ
KJ
KKJ
JK
−+
−−=
lnK = + 14.37 – 23.86 = – 9.49
K = e–9.49 = 7.56 × 10–5
135

1/16/2013
136
Calculation of ∆∆∆∆Go at Various Temperatures
Consider the following reaction:
CaCO3 (s) → CaO (s) + CO2 (g)
At 25 oC ∆Go = +130.9 and Kp = 1.1 x 10-23 atm
What do these values tell you about CaCO3?
What happens when the reaction is carried out at a
higher temperature?
136

1/16/2013
137
Calculating ∆∆∆∆Go and K at Various Temperatures
a. What is ∆Go at 1000oC for the calcium carbonate reaction?
CaCO3 (s) → CaO (s) + CO2 (g)
Is this reaction spontaneous at 1000oC and 1 atm?
b. What is the value of Kp at 1000oC for this reaction?
What is the partial pressure of CO2?
137

1/16/2013
138
Strategy for solution…
a. Calculate ∆Ho and ∆So at 25 oC using standard
enthalpies of formation and standard entropies.
Then substitute into the equation for ∆Gfo.
b. Use the ∆Gfo value to find K (=Kp )
138

1/16/2013
139
a. From Data Table you have the following:
CaCO3 (s) → CaO (s) + CO2 (g)
∆Hfo: -1206.9 -635.1 -393.5 kJ
So: 92.9 38.2 213.7 J/K
∆Ho = [(-635.1 – 393.5) – (-1206.9)] = 178.3 kJ
∆So = [(38.2 + 213.7) – (92.9)] = 159.0 J/K
∆GTo = ∆Ho –T∆So
= 178.3kJ – (1273 K)(0.1590kJ/K) = -24.1 kJ
∆Go is negative – reaction is spontaneous 139

1/16/2013
140
b. Substitute the values of ∆Go at 1273 K, which
equals -24.1 x 103 J, into the equation relating
ln K and ∆Go.
ln K = ∆Go
-RT=
-24.1 x 103
-8.31 x 1273= 2.278
K = Kp = e2.278 = 9.76
Kp = PCO2 = 9.76 atm
140

1/16/2013
141
Where does the reaction change from spontaneous
to non-spontaneous?
∆Go = 0 = ∆Ho - T∆So
Solve for T;
T = ∆Ho
∆So =178.3 kJ
0.1590 kJ/K
T = 1121 K = 848 oC
141

1/16/2013
142
142
• Amount of energy needed to break chemical
bond into electrically neutral fragments
• Useful to know
• Within reaction– Bonds of reactants broken
– New bonds formed as products appear
• Bond breaking
– 1st step in most reactions
– One of the factors that determines reaction rate
– Ex. N2 very unreactive due to strong N ≡ N bond
Bond Energy

1/16/2013
143
143
Bond Energies
• Can be determined spectroscopically for simple
diatomic molecules
– H2, O2, Cl2
• More complex molecules, calculate using thermochemical data and Hess’s Law
– Use ∆H°formation enthalpy of formation
• Need to define new term
• Enthalpy of atomization or atomization energy,
∆∆∆∆Hatom
– Energy required to rupture chemical bonds of 1 mole of gaseous molecules to give gaseous atoms

1/16/2013
144
144
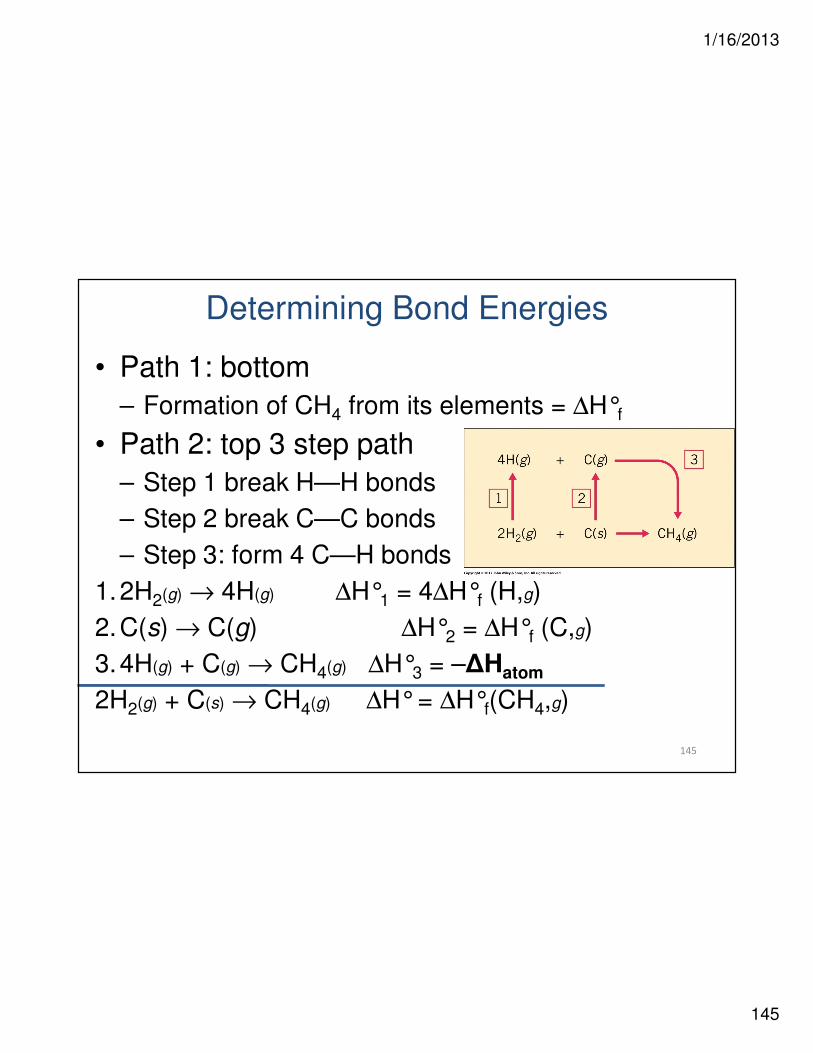
Determining Bond Energies
• Ex. CH4(g) →→→→ C(g) + 4 H(g)
• ∆Hatom = energy needed to break all bonds in molecule
• ∆Hatom /4 = average bond C—H dissociation energy in methane– D = bond dissociation energy
• Average bond energy to required to break all bonds in molecule
– How do we calculate this?
• Use ∆H°f for forming gaseous atoms from elements in their standard states
• Hess’s Law
1/16/2013
145
145
Determining Bond Energies
• Path 1: bottom
– Formation of CH4 from its elements = ∆H°f
• Path 2: top 3 step path
– Step 1 break H—H bonds
– Step 2 break C—C bonds
– Step 3: form 4 C—H bonds
1.2H2(g) → 4H(g) ∆H°1 = 4∆H°f (H,g)
2.C(s) → C(g) ∆H°2 = ∆H°f (C,g)
3.4H(g) + C(g) → CH4(g) ∆H°3 = –∆Hatom
2H2(g) + C(s) → CH4(g) ∆H°= ∆H°f(CH4,g)

1/16/2013
146
146
Calculating ∆Hatom and Bond Energy
∆H°f(CH4,g) = 4∆H°f(H,g) + ∆H°f(C,g) – ∆Hatom
• Rearranging gives
∆Hatom= 4∆H°f(H,g) + ∆H°f(C,g) –∆H°f(CH4,g)
• Look these up in Table 18.3, 6.2 or appendix C
∆Hatom= 4(217.9kJ/mol) + 716.7kJ/mol –(–74.8kJ/mol)
∆Hatom= 1663.1 kJ/mol of CH4
4
kJ/mol 1663.1energy bond =
= 415.8 kJ/mol of C—H bonds
1/16/2013
147
147
147
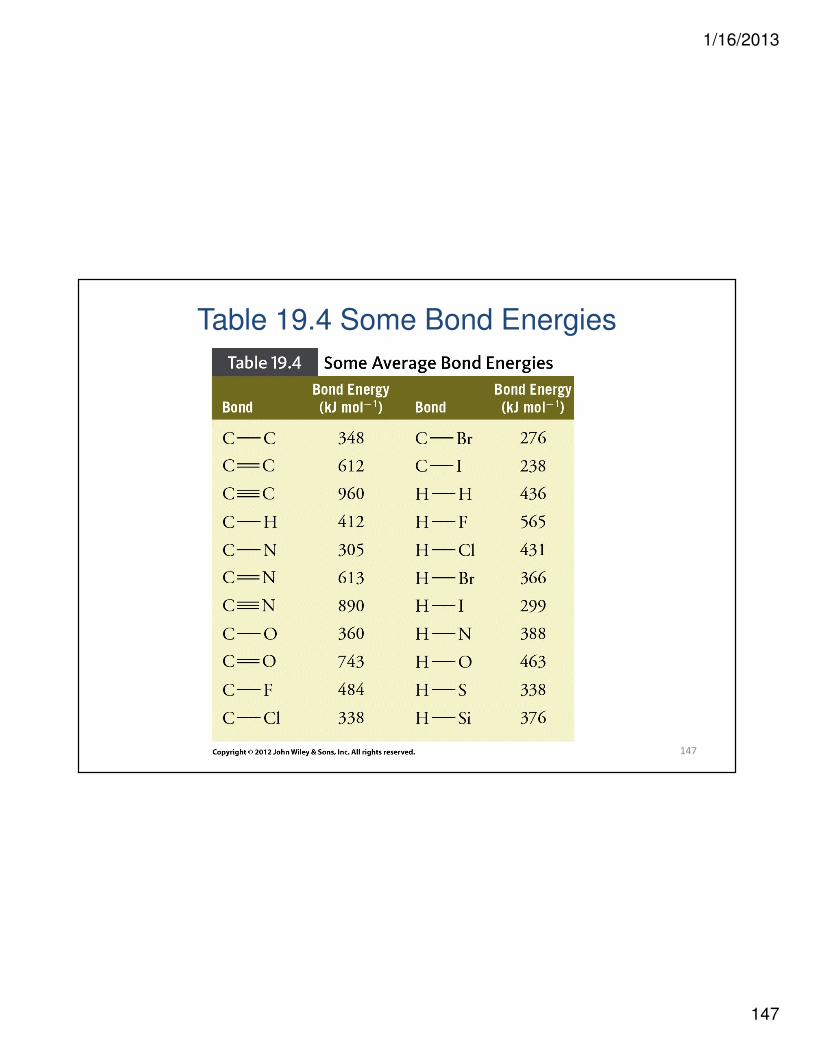
Table 19.4 Some Bond Energies
1/16/2013
148
148
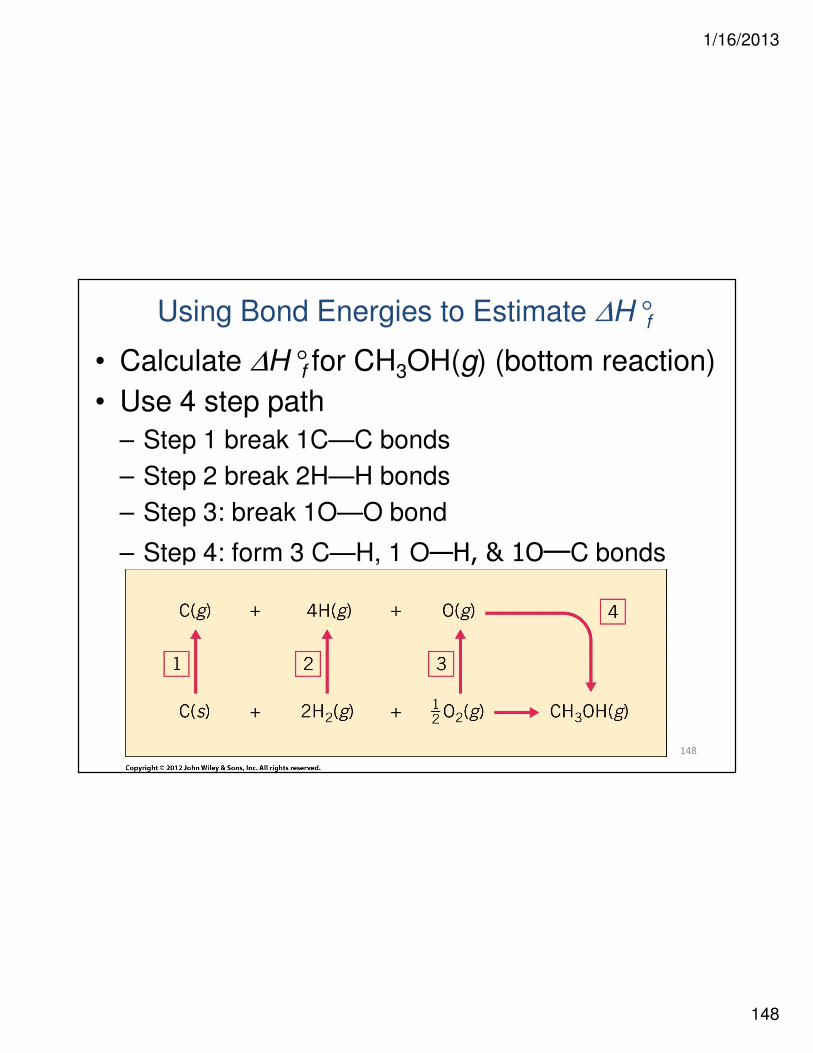
Using Bond Energies to Estimate ∆H°f
• Calculate ∆H°f for CH3OH(g) (bottom reaction)
• Use 4 step path
– Step 1 break 1C—C bonds
– Step 2 break 2H—H bonds
– Step 3: break 1O—O bond
– Step 4: form 3 C—H, 1 O—H, & 1O—C bonds

1/16/2013
149
149
Using Bond Energies
∆H°f(CH3OH,g) = ∆H°f(C,g) + 4∆H°f(H,g) + ∆H°f(O,g) – ∆Hatom (CH3OH,g)
• ∆H°f(C,g) + 4∆H°f(H,g) + ∆H°f(O,g) = {716.7 +(4*217.9) + 249.2}kJ = +1837.5 kJ
• ∆Hatom (CH3OH,g) = 3DC—H + DC—O + DO—H
= (3*412) + 360 + 463 = 2059 kJ
• ∆H°f(CH3OH,g) = +1837.5 kJ – 2059 kJ = – 222 kJ
• Experimentally find ∆H°f(CH3OH,g) = – 201 kJ/mol
• So bond energies give estimate within 10% of actual

1/16/2013
150
Chapter 13/19 Chemical Thermodynamics
1. Spontaneous Chemical and Physical Processes
2. Entropy and Disorder
3. Entropy and the Second Law of Thermodynamics
4. Standard-State Entropies of Reaction
5. The Third Law of Thermodynamics
6. Calculating Entropy Changes for Chemical Reactions
7. Gibbs Free Energy
8. The Effect of Temperature on the Free Energy of a Reaction
9. Beware of Oversimplification
10. Stand-State Free Energies of Reaction
11. Equilibria Expressed in Partial Pressures
12. Interpreting Stand-State Free Energy of Reaction Data
13. Relationship Between Free Energy and Equilibrium Constants
14. Temperature Dependence of Equilibrium Constants
15. Gibbs Free Energies of Formation and Absolute Entropies
16. Calculate ∆H with bond energies150

1/16/2013
151
Entropy Change for a Phase Transition
∆S > q/T (at equilibrium)
What processes can occur under phase
change at equilibrium?
Solid to liquidLiquid to gas
Solid to gas
151

1/16/2013
152
Solution:
∆S = ∆Hvap/T = (39.4 x 103 J/mol)/ 298 K= 132 j/(mol·K)
Entropy of Vapor = (216 + 132) J/(mol·K)= 348 J/(mol·K)
152

1/16/2013
153
153