cosmo-rs: from quantum chemistry to cheminformaticsinfochim.u-strasbg.fr/chemoinformatics/pdf... ·...
TRANSCRIPT

Binary mixture of Butanol(1) and Heptane (2)
at 50° C
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0x1 Mole fraction of 1-butanol (1)
ln(
)
1-Butanol (calculated)n-Heptane (calculated)1-Butanol (experiment)n-Heptane (experiment)
Andreas KlamtCOSMOlogic GmbH&Co.KG, Leverkusen, Germanyand Inst. of Physical Chemistry, University of Regensburg, Germany
COSMO-RS: From quantum chemistry
to Cheminformatics

gas phase
latitudes ofsolvation
water
alkanes
horizon ofCOSMO-RS
horizon of gas-phase methods
solid
phase
Thermophys ical data prediction methods
Quantum Chemistrywith dielectric
solvation models like PCM
or COSMO
quantumchemistry
-OH
-OCH3
-C(=O)H
-CarH-CarH -C
arH-Car
-Car -Car
Group contribution methodsUNIFAC, CLOGP, LOGKOW, fingerprints,.. etc.
simple, well explored solvents
fitted parameters: CLOGP:~ 1500UNIFAC: ~5000 +50% gaps
≠
MD / MCforce-fieldsimulations
MD/MC
softbiomatter

Dielectric Continuum Solvation Models (CSM) Dielectric Continuum Solvation Models (CSM)
- Born 1920, Kirkwood 1934, Onsager1936
- Rivail, Rinaldi et al. - Katritzky, Zerner et al.- Cramer, Truhlar et al. (AMSOL)- Tomasi et al. (PCM) - Orozco et al. - Klamt, Schüürmann (COSMO) e.g. DMol3/COSMO and others
- promising results for solvents water, alkanes, and a few other solvents
- empirical finding: cavity radii should be about 1.2 vdW-radii
solute molecule embedded in a dielectric continuum,self-consistent inclusion of solvent polarisation
(screening charges) into MO-calculation (SCRF)
But CSMs are basically wrong and give a poor, macroscopic description of the solvent !
Density Functional Theory (DFT)is appropriate level of QC!COSMO almost as fast as gasphase!programs: DMol3, Turbomole,
Gaussian98_release2001up to 25 atom:< 24 h on LINUX PC
COSMO = COnductor-like Screening Model,just a (clever) variant of dielectric CSMs

Why are Continuum Solvation Models Why are Continuum Solvation Models wrong for polar molecules in polar solvents? wrong for polar molecules in polar solvents?
-only electronic polarizibility-homogeneously distributed-linear response up to very high fieldsdielectric continuum theory should be reasonably applicable
-discrete permanant dipoles -mainly reorientational polarizibility-linear response requires Ereor << kT- typically Ereor ~ 8 kcal/mol !!!no linear response, no homogenityno similarity with dielectric theory

gas phase
latitudes ofsolvation
water
acetone
alkanes
horizon ofCOSMO-RS
horizon of gas-phase methods
solid
statebridge ofsymmetry
How to come to the latitudes of solvation?
QM/MMCar-Parrinello
-OH
-OCH3
-C(=O)H
-CarH-CarH -C
arH-Car
-Car -Car
Group contribution methodsUNIFAC, CLOGP, LOGKOW, etc.
Quantum Chemistrywith dielectric
solvation models like COSMO
or PCM
MD / MCsimulations
native home of computational chemistry
COSMO-RS
state of ideal screening home of COSMOlogic

1) Put molecules into ‚virtual‘ conductor (DFT/COSMO)COSMO-RS:COSMO-RS:
++++++
____ _
σ '
σ
σ >> 0σ ' << 0(1)
(2) hydrogen bond
electrostat. misfit
ideal contact
3) Remove the conductor on molecular contact areas (stepwise) and ask for the energetic costs of each step.
2) Compress the ensemble to approximately right density
(3) specificinteractions
2)'(2
')',( σσασσ += effmisfit aG
}',0min{)()',( 2hbhbeffhb TcaG σσ σσσ +=
In this way the molecular interactions reduce to pair interactions of surfaces!

Water
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
-0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020σ [e/A2]
pwat
er(s
) (am
ount
of s
urfa
ce)
Screening charge distribution on molecular surface reduces to "σ-profile"
COSMO-RS COSMO-RS For an efficient statistical thermodynamics reduce the ensemble of
molecules to an ensemble of pair-wise interacting surface segments !
✪

0
5
10
15
20
25
-0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020
σ [e/A2]
pX ( σ)
Water
Methanol
Acetone
Benzene
Chloroform
Hexane
Screening charge distribution on molecular surface reduces to "σ-profile"
A. Klamt, J. Phys. Chem., 99 (1995) 2224COSMO-RS COSMO-RS For an efficient statistical thermodynamics reduce the ensemble of
molecules to an ensemble of pair-wise interacting surface segments !(same approximation as is UNIFAC)

-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Mole fraction of acetone (1)
ln( γ
)
Acetone (calculated)
Chloroform (calculated)
Acetone (experiment, Rabinovichet al.)Chloroform(experiment,Rabinovich et al.)Aceton (experiment, Apelblat etal.)Chloroform (experiment, Apelblatet al.)
0
5
10
15
20
25
-0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020
σ [e/A2]
pX ()
Acetone
Chloroform
Because their Because their σσ-profiles are -profiles are almost complementary!almost complementary!
Why do acetone and chloroform Why do acetone and chloroform like each other so much?like each other so much?

• Replace ensemble of interacting molecules by an ensemble S of interacting pairs of surface segments• Ensemble S is fully characterized by its σ-profile pS(σ)
( pS(σ) of mixtures is additive! -> no problem with mixtures! )• Chemical potential of a surface segment with charge density σ is exactly(!) described by:
( )µ σ σ σσ σ µ σ
S Sint SkT d p
EkT
( ) ln ' ' exp( , ') ( ')
= − −−
∫
chemical potential of solute X in S:
( ) ( ) SS
XXS AkTpd lnλσµσσµ −= ∫
activity coefficients → arbitrary liquid-liquid equilibria
Statistical ThermodynamicsStatistical Thermodynamics
combinatorial contribution:solvent size effects
σ-potential:affinity of solvent forspecific polarity σ combX
S,γ

σσ-profiles -profiles and and
σσ-potentials of -potentials of representative liquidsrepresentative liquids
-0.50
-0.30
-0.10
0.10
0.30
0.50
0.70
-0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020σ [e/A2]
µX ( σ) [
kJ/m
ol A
2 ]
Water
Methanol
Acetone
Benzene
Chloroform
Hexane
hydrophobicity
affinity for HB-donors
affinity for HB-acceptors
0
5
10
15
20
25
-0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020
σ [e/A2]
pX ( σ)
Water
Methanol
Acetone
Benzene
Chloroform
Hexane

a) DGhydr (in kcal/mol)
-11 -10 -9 -8 -7 -6 -5 -4 -3 -2 -1 0 1 2 3
-2
-1
0
1
2
b) log Pvapor (in bar)-4 -3 -2 -1 0 1 2
-2
-1
0
1
2
c) log KOctanol/Water-2 -1 0 1 2 3 4 5 6
-2
-1
0
1
2
d) log KHexane/Water-5 -4 -3 -2 -1 0 1 2 3 4 5 6 7
-2
-1
0
1
2
e) log KBenzene/Water
-4 -3 -2 -1 0 1 2 3 4 5-2
-1
0
1
2
f) log KEther/Water-3 -2 -1 0 1 2 3
-2
-1
0
1
2
alkanes alkenes alkines alcohols ethers carbonyls esters aryls diverse amines amides N-aryls nitriles nitro chloro water
Results of parametrization based on DFT (DMol3: BP91, DNP-basis
650 data17 parametersrms = 0.41 kcal/mol
A. Klamt, V. Jonas, J. Lohrenz, T. Bürger, J. Phys. Chem. A, 102, 5074 (1998)
meanwhile:COSMOtherm5.0 with Turbomole BP91/TZVPrms = 0.36 kcal/mol
Res
idua
ls

Applications to Phase Diagrams and AzeotropesApplications to Phase Diagrams and Azeotropes
Binary mixture of Butanol(1) and Heptane (2)
at 50° C
0.0
0.2
0.4
0.6
0.8
1.0
0.0 0.2 0.4 0.6 0.8 1.0x
y CalculatedExperiment
Binary mixture of Butanol(1) and Heptane (2)
at 50° C
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0x1 Mole fraction of 1-butanol (1)
ln(
)
1-Butanol (calculated)n-Heptane (calculated)1-Butanol (experiment)n-Heptane (experiment)
Binary mixture of ethanol (1) and benzene (2)
at 25° C
0.0
0.2
0.4
0.6
0.8
1.0
0.0 0.2 0.4 0.6 0.8 1.0x
yCalculatedExperiment
Binary Mixture of 1-butanol (1) and water
at 60° C
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0x
y
CalculatedExperiment
miscibility gap
November 2002: COSMOtherm wins the VLE prediction contest of Nat. Inst. of Standards (NIST)
and American Inst. of Chem. Engineers (AICHE)

sigma-profiles
0
2
4
6
8
10
12
14
-0.02 -0.01 0 0.01 0.02screening charge density [e /A²]
vanillin
w ater
acetone
sigma-potential
-0.2
-0.15
-0.1
-0.05
0
0.05
0.1
-0.02 -0.01 0 0.01 0.02
Chemical Structure
Quantum ChemicalCalculation with COSMO
(full optimization)
σ-profiles of compounds
other compounds
ideally screened moleculeenergy + screening charge distribution on surface
DFT/COSMO COSMOtherm
σ-profile of mixture
σ-potential of mixture
Fast Statistical Thermodynamics
Equilibrium data:activity coefficientsvapor pressure,solubility,partition coefficients
Phase Diagrams
Database of COSMO-files
(incl. all common solvents)
Flow Chart of COSMO-RS Binary Mixture of
Butanol and Water at 60° C
0.0
0.2
0.4
0.6
0.8
1.0
0.0 0.2 0.4 0.6 0.8 1.0x
y Calculated
Experiment
miscibility gap

gas phase
latitudes ofsolvation
water
acetone
alkanes
horizon ofCOSMO-RS
horizon of gas-phase methods
solid
statebridge ofsymmetry
How to come to the latitudes of solvation?
QM/MMCar-Parrinello
-OH
-OCH3
-C(=O)H
-CarH-CarH -C
arH-Car
-Car -Car
Group contribution methodsUNIFAC, CLOGP, LOGKOW, etc.
Quantum Chemistrywith dielectric
solvation models like COSMO
or PCM
MD / MCsimulations
native home of computational chemistry
COSMO-RS
state of ideal screening home of COSMOlogic

Extension of COSMOtherm to multi-conformations
COSMOtherm can treat a compound as a set of several conformers- each conformer needs a COSMO calculation - conformational population is treated consistently
according to total free energy of conformers (by external self-consistency loop)
Unfortunately, many molecules have more than one relevant conformation

σ -profiles of glycerol conformers
0
2
4
6
8
10
12
14
-0.02 -0.015 -0.01 -0.005 0 0.005 0.01 0.015 0.02σ
h2oglycerol4_cosmoc01glycerol2_cosmoc02glycerol3_cosmoc04glycerol1_cosmoc05glycerol0_cosmoc03glycerol3_cosmoc05glycerol3_cosmoc03
z
Conformational effects for glycerollowest COSMO conformerall 3 donors are bound in one 6-ring and two 5-rings,also least polar conformer39% in octane 9% in acetone
2nd COSMO conformer Ecosmo=+0.37 kcal/mol Ediel =+2 kcal/mol1 free donor, two bound in one 6-ring and one 5-rings 16% in octane 8% in acetone
7th COSMO conformer Ecosmo=+1.3 kcal/mol Ediel =+3.3 kcal/mol2 free donors, one bound in strong 6-ring(represents ~4 similar conformations) 2% in octane41% in acetone
partition coefficient between acetone and octone:
logKAO = -3.3 (lowest conformer)logKAO = -4.0 (conformer ensemble)
difference of 0.7 log-units ≈ 1 kcal/mol
Conclusions:
- Conformational effects can be important for the detailed understanding of phase equilibria
- In most cases one conformation dominates in all phases
- Effects are especially large for molecules with sub-optimal intramolecular HBs in solvents having strong HB acceptors, but a deficit of HB-donors.
-Tautomers can be considered as a kind of conformers.
-Unfortunately the DFT level of QC is not always reliable regarding the energy differences between conformers and even more between tautomers. Energy corrections may be required.

„Conformational analysis of cyclic acidic α-amino acids in aqueous solution - an evaluation of
different continuum hydration models."by Peter Aadal Nielsen, Per-Ola Norrby, Jerzy W. Jaroszewski, and Tommy Liljefors(private comm., Ph.D. thesis)Method Solvent rms rms (4 points) Max Dev Model (kJ/mol) (kJ/mol) (kJ/mol)AM1 SM5.4A 4.6 5.6 9.2PM3 SM5.4P 13.6 16.2 20.5AM1 SM2.1 7.4 9.0 16.7HF/6-31+G* C-PCM 3.1 3.8 5.9HF/6-31+G* PB-SCRF 4.7 5.8 8.8AMBER* GB/SA 13.2 16.2 24.3MMFF GB/SA 18.5 19.9 31.4
BP-DFT/TZVP COSMO-RS 2.2 2.6 4.8COSMO-RS was evaluated as a blind test !!!

Water Solubility log(xH2O)calculated with COSMOtherm
-13
-12
-11
-10
-9
-8
-7
-6
-5
-4
-3
-2
-1
0
1
-13 -12 -11 -10 -9 -8 -7 -6 -5 -4 -3 -2 -1 0 1Calculated
Expe
rimen
t
DGfus < 0
DGfus > 0
McFarland Test Set
questionable
R2= 0.90 rms=0.66n = 150
logSXS = (µX
X-µXS+ min(0,∆ Gfus))/1.365
∆ GXfus = 0.54 µX
water - 0.18*NXringatom +0.0029*volume
Dataset taken from Jorgensen and Duffy (BOSS)
Stable model: No changes required for pesticides!A.Klamt, F. Eckert, M. Hornig, M. Beck, and T. Bürger: J. Comp. Chem. 23, 275-281 (2002)

COSMOtherm prediction of drug solubility in diverse solvents(blind test performed with Merck&Co., Inc., Rahway, NJ, USA)
all predictions are relative to ethanol
solvents:
ButanolTriethylamineAcetonitrileEthanolAcetoneChlorobenzeneTolueneHeptaneMethanolEthyl AcetateDMF2-Propanol1-PropanolWater
triethylamine
heptane

Free energy of Hydration [kcal/mol] for Ions
-120
-110
-100
-90
-80
-70
-60
-50
-120 -110 -100 -90 -80 -70 -60 -50
Experiment
Cal
cula
ted
Ionic Free Energies of Hydrationby COSMOtherm-Ion-Extension
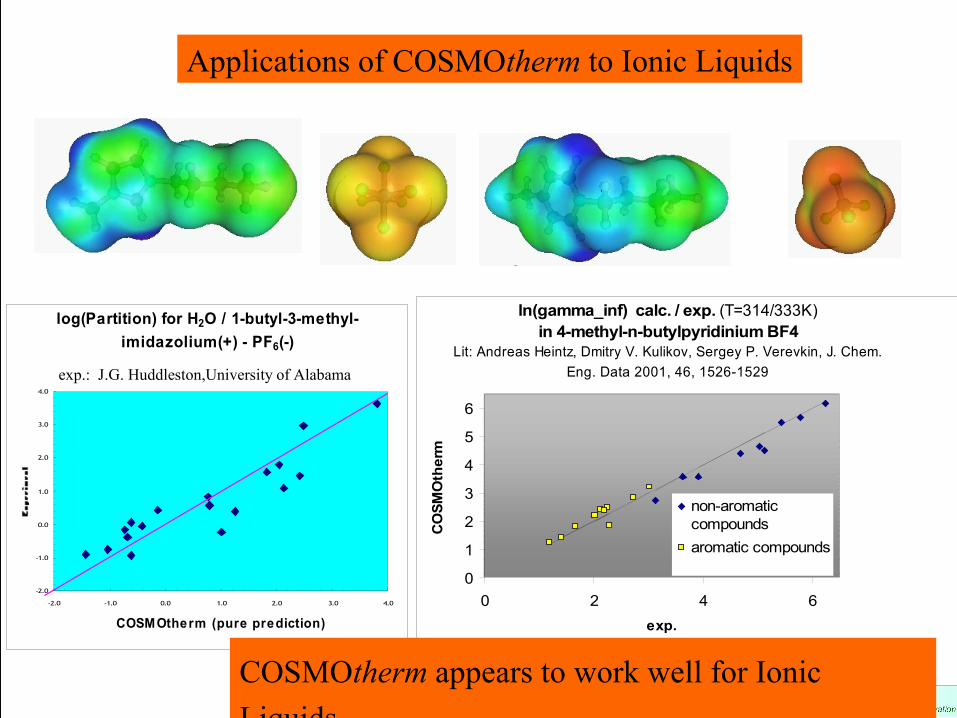
ln(gamma_inf) calc. / exp. (T=314/333K)in 4-methyl-n-butylpyridinium BF4
Lit: Andreas Heintz, Dmitry V. Kulikov, Sergey P. Verevkin, J. Chem. Eng. Data 2001, 46, 1526-1529
0
1
2
3
4
5
6
0 2 4 6exp.
CO
SMO
ther
m
non-aromaticcompoundsaromatic compounds
Applications of COSMOtherm to Ionic Liquids
log(Partition) for H2O / 1-butyl-3-methyl-imidazolium(+) - PF6(-)
-2.0
-1.0
0.0
1.0
2.0
3.0
4.0
-2.0 -1.0 0.0 1.0 2.0 3.0 4.0
COSMOtherm (pure prediction)
exp.: J.G. Huddleston,University of Alabama
COSMOtherm appears to work well for Ionic Liquids

COSMOtherm first principle pKa prediction ( A. Klamt, et. al. J. Phys. Chem. A, Nov. 2003)
pKa = 0.59∆ Gdiss/(RTln10) +0.88
N=60 R2=0.978, rms=0.49
0.00
2.00
4.00
6.00
8.00
10.00
12.00
14.00
16.00
18.00
0 10 20 30 40
∆ Gdiss
pKa_
exp
all
alcohols
carboxylic acids
inorganic acids
subst. phenols
N-acids (uracils,imines)Linear (all)
Uracil
and others
trans-5-formyluracilthyminecis-5-formyluracil5-nitrouracil5-fluorouracilboricacidphosphoricacid2sulfurousacidnitrousacidhypoiodousacidhypobromousacidhypochlorousacid2,2,2-trichloroethanolethanolpentachlorophenolphenolcarbonicacid0fumaricacidmaleicacid3oxalicacid0benzoicacid
2,2-dimethylpropanoicacidn-pentanoicacidtrichloroaceticaciddichloroaceticacid0chloroaceticacidaceticacidformicacid
latest results for bases (pKb):similar rmsslope between 0.59 and 0.71

σ-Moment Approach
>±+<±
≅=
≥=≅
−−
−=∑
hbhb
hbdonacc
ii
m
ii
iSS
ifif
ff
andiforfwithfc
σσσσσσ
σσ
σσσσµ
0)()(
0)()()(
/1/2
2
XsoluteofmomentsdfpMwith
Mcdfcpdp
iXX
i
m
i
Xi
iS
m
ii
iS
XS
XXS
−==
≅≅=
∫
∑∫ ∑∫−=−=
σσσσ
σσσσσµσµ
)()(
)()()()(22
Now the chemical potential of a solute X in this matrix S is:
The coefficients can now be derived from experimental (log.) partition databy linear regression. => σ-moments are excellent QSAR-descriptors forgeneral partition behaviour of molecules. “The solvent space is approximately 5-dimensional!“Zissimos, et al.: ‘A comparison between the two general sets of linear free energy descriptors of Abraham and Klamt‘, J. Chem. Inf. Comput. Sci., 42, 1320-1331 (2002) σ -moment models for ADME proprties as
logBB, intestinal absorption, logHSA, …
-0.70
-0.50
-0.30
-0.10
0.10
0.30
0.50
0.70
-0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020
σ-po
tent
ial
WaterAcetoneHexane

σ -moment logBB regressionlogBB = 0.0046 area -0.017 sig2 -0.0029 sig3 +0.19
n = 103, r² = 0.71, rms = 0.40data from: "Modeling Blood-Brain Barrier Partitioning Using Topological Structure
Descriptors", Rose, Hall, Hall, and Kier, MDL-Whitepaper, 2003
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
-2.0 -1.5 -1.0 -0.5 0.0 0.5 1.0 1.5
calc.
exp.
minimum_COSMO_conf.CORINA_optimized

σ-moment logKHSA regressionlogKHSA = 0.0081 area -0.016 sig2 -0.013 sig3 +0.145 sigHacc+0.88n = 82, r² = 0.69, rms = 0.33data from: Kier, Hall, Hall, MDL-Whitepaper, 2002
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
-1.5 -1.0 -0.5 0.0 0.5 1.0 1.5
logK(HSA) [calc.]
logK
(HSA
) [ex
p.]
0.008115997
0.008115997-0.016419311

0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60 70 80 90 100
PIA calculated by COSMO-KIA
PIA
exp
.
training set: n=38,rms=12.5%high quality test set: n=107,rms=12.8%questionable test set:n=24, rms=22%
COSMO-RS for Percentage Intestinal Absorption (PIA)Klamt, Diedenhofen, Connolly*, Jones* (submitted) *) GlaxoSmithKline
1.370.117M-0.113M-0.0024M-0.0053M-0.0040Mlog donacc320 +=KIA

-1
0
1
2
3
4
5
6
7
-1 0 1 2 3 4 5 6 7
COSMO-KOC
exp
. lo
gK
oc
Training Set rms=0.63
Test Set rms=0.72
Linear (Training Setrms=0.63)
Prediction of Soil SorptionJournal of Environmental Toxicology and Chemistry, in print

Adsorption to Activated Carbon
y = 0.9277x + 0.1379R2 = 0.9281
-10
-5
0
5
10
15
-10 -5 0 5 10 15
ln[He(exp.)]
ln[H
e(th
eor.)
]
Fluid Phase (23 Adsorptives)
Gasphase (15 Adsorptives)
Linear (Fluid Phase (23Adsorptives))
[Mehler, Peukert (TU München), Klamt; to be published]

solvent1
solvent2
solvent3
Free Energies relevant for Reactions
sum ofeducts
transitionstate (TS)
sum ofproducts
∆Greact
⇒equilibrium constant
∆Gactivation
⇒ kinetic constant
localisation of transition state often complicated: In this work gas-phase TS have been localised using techniques provided in Gaussian98 (DFT: B3LYP, 6-31G*)after that: single-point DFT/COSMO withTURBOMOLE (BP91-TZVP)
DFT is not reliable for TS energiesbut the solvent shifts should be reliable.
Calculation of the solvent dependence of ∆Greact is straightforward with COSMO-RS. Successful applications have been reported by industrial users (Dr. Franke, Degussa AG; Dr. Lohrenz, Bayer AG)

COSMOmic: Simulation of molecules in micelles and membranes
Concept:-define layers of membrane (shells of micelle)
-get probability to find a certain atom of surfactant in each layer (e.g. from MD)-convert this into a σ-profile p(σ,r) for each layer r using the COSMO-file of the surfactant-use COSMOtherm to calculate µ(σ,r) considering each layer as a liquid mixture-now calculate the chemical potential of a solute X in a certain postion and orientation by summing the chemical potentials of its segments in the respective layer. -sample the chemical potentials all positions and orientations of X
-construct a total partition sum and get the probability to find the solute in a certain depth and orientation.
-also get the average volume expansion in each layer- get a kind of micelle or membrane-water partition coefficient
o
o
o
o o
o
The tool COSMOmic facilitates all the previous steps together with COSMOthermPerspective: self-consistent treatment of new surfactants; CMC prediction

COSMOfrag: A fast shortcut of COSMOtherm suited for HTS-ADME prediction
1) large database of precalculated drug-like compounds (about 45000)2) for new compound find most similar fragments in database3) compose COSMO surface from surface fragments (write a meta-file)4) do usual COSMOtherm: solubility, partition properties
advantages: -about 1 sec. per compound!-you can add your typical inhouse structures to database -simple refinement of calculations
COSMOfrag ports COSMO-RS to Cheminformatics!

COSMOfrag:statistics and examples
Prediction of Soil Sorption Coefficients with COSMOfrag
0
1
2
3
4
5
6
0 1 2 3 4 5 6COSMOfrag
exp.
dat
a
Trainingsset rms=0.72 (0.63)
Tes tset rms=0.81(0.72) -10
-8
-6
-4
-2
0-10 -8 -6 -4 -2 0
log(xH2O) [meta]
log(
x H2O
) [ex
p.]
Water Solubility with COSMOfrag
Dataset of Jorgensen and Duffyrms: 0.71 (0.66)

Ligand – Recptor Binding
0
5
10
15
20
25
30
35
40
45
50
-0.03 -0.02 -0.01 0 0.01 0.02 0.03
Retinal
Bacteriorhodopsinbinding pocket
σ−profiles of the binding pocket of bacteriorhodopsin and retinal
Retinal
Mouth of the Retinal binding pocket
Meanwhile we can approximately treat enzymes and receptor pockets.The goal is to describe ligand receptor binding (incl. desolvation) based
COSMO polarization cahrge densities σ.

COSMOsimbio-isoster search based on s-profilesexamples by Dr. M. Thormann, Morphochem AG
If the physiological distribution and the drug-receptor binding are governed by the COSMO σ-profiles, it is reasonable to use these for drug-similarity searching:
- search for molecules with maximum similarity of σ-profilesin order to find molecules with similar interactions, but different chemistry
-search is only based on surface polarity (σ) and not on structure
scaffold hopping
- either search over full COSMO-files of COSMOfrag-DB (48000 compounds)-screen millions of candidate compounds using the COSMOfrag method -Refine your search by explicit COSMO calculations on the most similar ~500 compds.
Lit: M. Thormann, A. Klamt, M. Hornig and M. Almstetter, "COSMOsim: Bioisosteric Similarity Based on COSMO-RS σ-Profiles”, J. Chem. Inf. Model. 46, (2006).
A.Bender, A. Klamt, K. Wichmann, M. Thormann, and R.C. Glen, „Molecular Similarity Searching Using COSMO Screening Charges (COSMO/3PP)“, in M.R. Berthold et al. (Eds.): CompLife 2005, LNBI 3695, pp. 175–185, 2005.Springer, Berlin Heidelberg 2005

Example 1: propionic acid
0.676719GXSEIQGKPCC1CC1C(=O)O
0.680418HLKLSJLHIOCCS
0.681717CUOCJIGKIOC(=O)C1CCC1
0.688516UMBRJEKLIOc1csnn1
0.691915HOMSZUGLICC(=O)C=NO
0.697814FFBMJKDKIOCCC(=O)O
0.698313EZHYEWAJICC(=NO)C
0.704112JMAKWZALIClc1nnn[nH]1
0.705211CZWYICCKICC(=O)O
0.710910WOJBMNDKVCC(O)C(=O)O
0.71719NBAKLRQLIOc1nnns1
0.72338HTYYARCJZCC(O)C#N
0.72697SDLNNSMIAOCC1CO1
0.74876DGWQYNDKICC(C)C(=O)O
0.75845VGZSDPDLICC=CC(=O)O
0.7654WCMTTAFLICC(=C)C(=O)O
0.7913RGQGEAHMICC=CC(=O)O
0.79962IAVMXKDKICCCC(=O)O
0.81691ITPZMBCLIOC(=O)C=C
10ZFQCMUCKICCC(=O)O propionic acid similars
-2
0
2
4
6
8
10
12
-0.03 -0.02 -0.02 -0.01 -0.01 0 0.01 0.01 0.02 0.02 0.03
p7p8p9p12p13p0p15
p7
p15p13p12
p9p8

Example 2: Metabotropic Glutamate Receptor Ligands
Synthesis and Pharmacology of Metabotropic Glutamate Receptor LigandsGrube-Jörgensen et al., ISMC 2004P239Drugs of the Future 2004 (29) Suppl. A: XVIIIth Symposium on MEDICINAL CHEMISTRY
A B C D a b c dA 1.000 0.711 0.666 0.697 0.396 0.440 0.459 0.488B 0.711 1.000 0.852 0.835 0.406 0.487 0.459 0.530C 0.666 0.852 1.000 0.857 0.378 0.461 0.455 0.507D 0.697 0.835 0.857 1.000 0.357 0.437 0.403 0.492a 0.396 0.406 0.378 0.357 1.000 0.665 0.679 0.642b 0.440 0.487 0.461 0.437 0.665 1.000 0.742 0.792c 0.459 0.459 0.455 0.403 0.679 0.742 1.000 0.700d 0.488 0.530 0.507 0.492 0.642 0.792 0.700 1.000
Tanimotoprime coefficients for COSMOsim matrixGlu (A), ibotenic acid (B), and thioibotenic acid (C) areknown mGluR agonists.D is novel and does also show mGluR agonist activity with mGluR subtype specificity most similar to that of C. The querie of d to our inhouse database containing > 2.000.000 sigma profiles employing the Tanimotoprime coefficient retrieves b at rank 3 with a similarity of 0.792.(M. Thormann, Morphochem, 2004)
0
2
4
6
8
10
12
14
16
-0.02 -0.015 -0.01 -0.005 0 0.005 0.01 0.015 0.02
abcd

COSMO-RS: From Quantum Chemistry to Cheminformatics
• The quantum-chemically derived surface polarization charge densities σ provide a novel and very rich description of molecular interactions in liquids and pseudo-liquids phases, combing electrostatics, hydrogen bonding and “hydrophobic interactions“ in one picture.
• COSMO-RS provides a novel, extremely fast and efficient way to do thermodynamics based on σ-profiles.
• drug solubility and many important ADME properties can be calculated with COSMO-RS
• Quantum chemical DFT/COSMO calculations are reasonably feasible for a few hundred or thousand drug-like molecules.
• COSMOfrag derives approximate s-profiles for druglike compouds in a second.
• COSMOsim enables drug-similaity screening based on σ-profiles------------------------------------Outlook: Ligand recepor binding based on σ-profiles

gas phase
latitudes ofsolvation
water
acetone
alkanes
horizon ofCOSMO-RS
horizon of gas-phase methods
solid
statebridge ofsymmetry
Hope you enjoyed the trip to the latitudes of solvation!
QM/MMCar-Parrinello
-OH
-OCH3
-C(=O)H
-CarH-CarH -C
arH-Car
-Car -Car
Group contribution methodsUNIFAC, CLOGP, LOGKOW, etc.
Quantum Chemistrywith dielectric
solvation models like COSMO
or PCM
MD / MCsimulations
native home of computational chemistry
COSMO-RS
state of ideal screening home of COSMOlogic
For references see: www.cosmologic.de
or read my book (Elsevier, 2005)COSMO-RS: From Quantum Chemistry to
Fluid Phase Thermodynamics and Drug Design
We are looking for an excellent cheminforatics expert to join our
team!

COSMO-RS for Drug-Design and -Development• water solubility of drugs, • Solvent Screening: relative solubilities of drugs in various solvents and mixtures• partition behaviour between almost arbitrary phases (blood-brain, intestinal absorption, BCF, ...)•. pKa prediction• visualization of partition coefficients and solubility as surface properties• one descriptor (σ) for entire interactions - electrostatics
- hydrogen bonding- lipophilicity/hydrophobicity
=> useful property for MFA- chemical potential of crystal surfaces in solution (morphology of drugs)•.identification of binding sites from σ-hotspots• surface integral scoring function for docking, including desolvation - extension to membrane and micelle partitioning
------------------------------------COSMOfrag: σ-profiles built from similar fragments out of 30000 compound database brings COSMO-RS in to the range of 5 sec./compound => applicable to HTS

Ideas for drug drug-receptor binding with COSMOtherm
-we need the σ-profile of the receptor once (QM/MM? not yet solved)
- we simply have the σ-profile of the ligands (even from COSMOfrag)
Idea 1: generate scoring function from COSMO-RS surface interaction modelIdea 2: consider receptor pocket as a kind of pseudo-liquid (overestimated receptor flexibility, but may be interesting)
Both simply include desolvation

Sigma profiles of Enzymescalculated with linear-scaling AM1/COSMO
(MOZYME in MOPAC2002)
Some common features:• Large charge
distribution in the region around σ = 0.
• Carbonyl oxygen between 0.01 and 0.02.
• Charged side chains in the outer regions (σ<-0.02 andσ>0.02)
0
100
200
300
400
500
600
700
800
900
1000
-0.03 -0.02 -0.01 0 0.01 0.02 0.03
BacteriorhodopsinBarnaseIsomeraseBPTICrambinPapainHIV-1 Protease

Bacteriorhodopsin and Retinal
0
5
10
15
20
25
30
35
40
45
50
-0.03 -0.02 -0.01 0 0.01 0.02 0.03
Retinal
Bacteriorhodopsinbinding pocket
Sigma profiles of the binding pocket of bacteriorhodopsin and retinal
Retinal
Mouth of the Retinal binding pocket

A few shots of the binding pocket

Amino Acids: Sigma profiles on two computational levels
0
2
4
6
8
10
12
14
-0.03 -0.02 -0.01 0 0.01 0.02 0.03
Alanine, AM1Alanine, BP/SVPGlutamic acid, AM1Glutamic acid, BP/SVPHistidine, AM1Histidine, BP/SVP
-COOH
-COOH
N lone pair
Alanine
Glutamicacid
Histidine