csci6904 genomics and biological computing seminar 1 – sequencing a genome in one step. projects...
TRANSCRIPT

CSCI6904
Genomics and Biological Computing
Seminar 1 – Sequencing a genome in one step.
Projects and paper presentations

Genetic engineering, four slides about it.
It is possible to cut and paste DNA into artificial constructs:• Mutate on demand• Make hybrid• Neutralize toxic genes by expressing them in two parts
Michael SmithUBC, Nobel 1993 in chemistry

Ctrl-X Ctrl-V on DNA
PrinciplePalindrome SequenceCut and leave “Sticky ends”Isolate on the basis of sizePaste somewhere useful
Why this exist?To systematically destroy
foreign DNA (Virus, parasites, etc…).
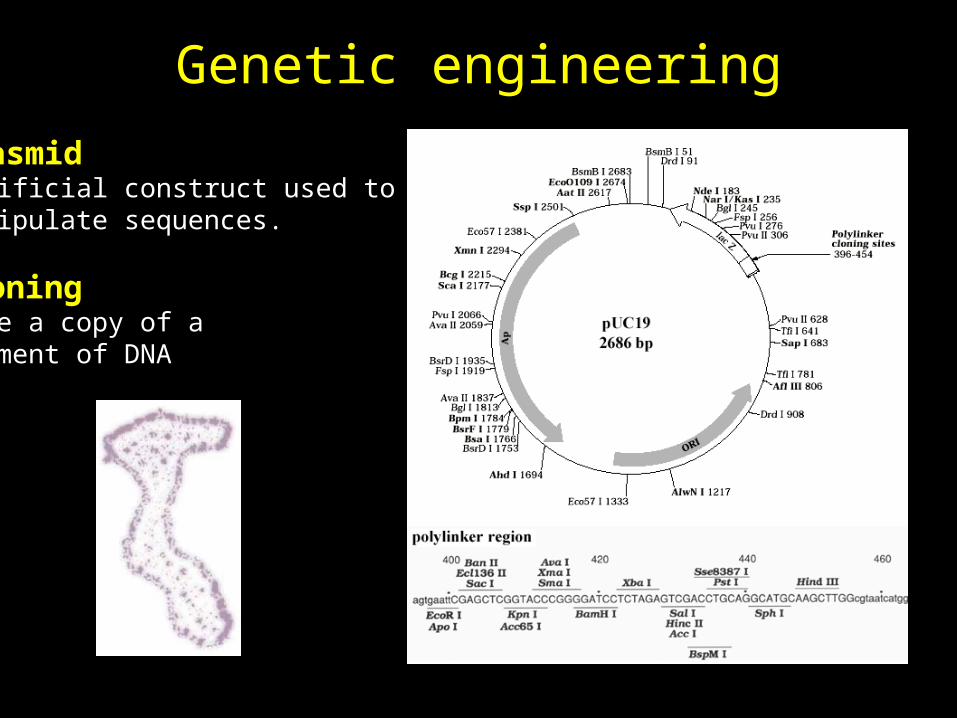
Genetic engineering
PlasmidArtificial construct used tomanipulate sequences.
CloningMake a copy of asegment of DNA

Genetic engineering
MutantSequence in which an Copy error is introduced duringDNA replication.
By the way…Everybody is a mutant
relative to their parents.

Genetic engineering
Site directed mutagenesisMutation can be introduced on demand by using artificial DNA fragments with one/a few error in them.

Genetic engineering
Example
1. Environmental Bioremediation1. PCB degrading bacteria2. Oil eating bacteria3. Plastic degrading bacteria maybe someday.
2. Genetically modified organisms1. Human insulin producing pigs.2. Antifreeze produce.3. Long shelf-life tomatoes.4. High-fat or specialized fat grains.
The bulk of the ethical issues may be largely due to a natural resistance to new technologies.

Polymerase Chain Reaction
PrincipleExponential amplification of a single piece of DNA.
•Forensic science -> DNA evidence.
•Rapid medical diagnistic
•Jurassic Park (dinosaure blood in amber samples)

DNA replication
Needs (in vitro):
-template DNA-Polymerase-Primers-Nucleotides in solution

Polymerase Chain Reaction
PrincipleExponential amplification
Taq

Paper presentation
Problem
Why this is a problem of interest.
Abstraction
Translate your problem into something that has nothing to do with biology anymore. Setting the “specifications” or the problem.
How it was done
Methods, results and conclusions.
Validation

GenoFrag
Problem
Need to amplify rapidly a whole genome to quickly identify the difference. This can be particularly useful in case of outbreak which cause is still unknown coming from a bacteria/virus not know to have such harmful characters.
Example of this would be:
E. coli contamination of watershed (Walkerton, June 2000).
Flesh-eating disease (S. aureus)
SARS
HIV
Influenza
General case of drug resistance.
GenoFrag
Abstraction
Need to be able to scan whole genomes very fast.
Assume that the target organism is very similar to something we already know the genomic sequence.
Target of interest: S. aureus. An ubiquitous, yet sometimes extremely harmful bacteria (mutation, inversion, deletion, transposons,…).
Technique: Long range PCR.
Problem: need a lot of oligonucleotide primer pairs to cover the entire sequence.

GenoFrag
Problem with primers
25 nt long
Given G+C content
No character repeat longer than N
No “hairpins”
Correct Tm.
Self- and inter-
complementarity
Unique
(Representative of all
sub-species)
Sufficiently informative
Equally spaced
Maximally covering all
the dataset

GenoFrag
Problem with coverage
pairs must be roughly equally spaced (9-11Kbp).
Must overlap to some extent.
Minimize the number of pairs.
ALGORITHMS:
Shortest path problem.
Single Traverse Algorithm.

GenoFrag
Problem with primers
25 nt long
Given G+C content
No character repeat longer than N
No “hairpins”
Correct Tm.
Self- and inter-
complementarity
Unique
(Representative of all
sub-species)
Sufficiently informative
Equally spaced
Maximally covering all
the dataset

GenoFrag
The filtered potential primers are reasonably well distributed along the DNA sequence.

GenoFrag
Optimizing
SSP
- 10K fragments
- Maximal coverage
SITA - Equally sized fragments - Maximal coverage

GenoFrag
Graph abstraction of the problem

GenoFrag
Graph abstraction of the problemHence the linearity of the problem.
SSP graph

GenoFrag
Graph abstraction of the problemHence the linearity of the problem.
SITA graph

GenoFrag
Results
Performances
- 40s for 2.8 Mb on 1.6GHz PC
Failed when…
- Presence of large insertions**
- Less general use if omit to
use filter 7.
But otherwise:
- 1-step genome (or part of one) amplification seems possible.

Paper presentation
What to choose:
Try to do a search on a method or field of your choice and add bioinformatics in he query. There will be a selection of papers that will be related to something you will enjoy talking about.
Examples
Bioinformatic resources (database, interface, services)
Large scale projects (Folding@home, BlueGene, HGP, )
API for bioinformatics (Bioperl, Biopython, NCBI tk, etc…)
Machine learning applications (detection, prediction, method validation)
Parallelization computing in bioinformatics
Algorithm and application to a specific question
Theoretical papers and simulators *
Methods and applications *
*would require a bit more background knowledge.

Projects ideas
A protein contact map using Voronoi triangulation
Problem
Proteins have fairly complex 3D chain paths. Many structural bioinformatics methods require the knowledge of which characters are in contact with each other.
Such contact maps are usually implemented as applying a cutoff filter to a distance matrix between arbitrary chosen centroids.
A contact map based on whether there is a shared surface in a Voronoi diagram between two amino-acids would be a nice, general purpose bioinformatic tool.
References
http://bioinformatics.oupjournals.org/cgi/content/abstract/bth365v1

Projects ideas
A lattice-based protein folding simulatorProblem
Protein are computationally expensive to model using computational chemistry methods. Further, these models are empirical and have a rather limited scope which does NOT include protein folding.
There is a 2D/3D abstraction of protein chain that exist, fixing each amino acid to one vertex in a cubic lattice. With this simulation environment, seriously cool experiments can be performed, especially if the environment is efficient.
This type of simulation is actually simple, compared to the simulation environment of phyiscal systems.
References

Projects ideas
Finding phylogenetically informative sites using Matrix decomposition (PCA,ICA,…)
Problem
An in-house phylogenetic application is generating solution pools of phylogenetic trees. This creates matrices of trees columns and sites (datapoints as rows)
There is an interest to figure out which rows are the most informative at discriminating between trees, or whether it is possible to identify clusters of data points showing some level of dependence to each other (They are assumed to be independent). Identify such cluster could be used to identify regions of recombination, for example.
References

Projects ideas
Computing the shortest path of topological rearrangements between two binary tree topologies
Problem
Information about the optimization landscape in phylogeny is scarce. Drawing paths between solutions would allow to plot cross-section of the search space to access the shape of the search space in various locations.
References
Felsenstein, Inferring phylogenies, 2003, Sinauer Eds.

Projects ideas
Sequence Harvesters
Problem
Gathering sequence information from GeneBank is a time consuming task. Typically, one starts with a sequence of interest, query the database with BLAST, chose the laergest set of non-redundant sequences and paste them into a file. The sequence have to be renamed one at the time and checked for duplicates. This could be automated and run in a few seconds (instead of a few hours of fingerwork).
A suggested platform for this project would be Biopython.
References
See www.biopython.org
Projects ideas
Using XML for validating protein structures
Problem
Protein 3D structures are stored in files in the PDB format. This format is regularly abused. As a results, it is hard to parse directly the files. There are cases of missing information, duplicates of atoms, omission of labels, non-standard labels. This project would explore the use of the new PDBML format to use as data source instead of the PDB flat files.
References
See PDBML from www.pdb.org
The main ref isn’t available yet to Dalhousie...

Projects ideas
This is not a closed list!
Text Mining and conceptual Biology.
Gene expression and clustering.
Protein topologies and similarity detection.
Clustering 3D structures.
Sequence patterns near variable regions in proteins sequences.