Çukurova Ünİversİtesİ fen bİlİmlerİ enstİtÜsÜlibrary.cu.edu.tr/tezler/8026.pdf ·...
TRANSCRIPT

ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
YÜKSEK LİSANS TEZİ
Mehtap KURTULUŞ
LİGNOSELÜLOZİK MATERYALLERDEN TERMOKATALİTİK İŞLEMLE
SUDA ÇÖZÜNDÜRÜLEN POLİSAKKARİTLERİN MOLEKÜLER
YAPILARININ İNCELENMESİ
KİMYA ANABİLİM DALI
ADANA, 2010

ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
LİGNOSELÜLOZİK MATERYALLERDEN TERMOKATALİTİK İŞLEMLE
SUDA ÇÖZÜNDÜRÜLEN POLİSAKKARİTLERİN MOLEKÜLER YAPILARININ İNCELENMESİ
Mehtap KURTULUŞ
YÜKSEK LİSANS TEZİ
KİMYA ANABİLİM DALI
Bu Tez ../../2010 Tarihinde Aşağıdaki Jüri Üyeleri Tarafından Oybirliği/Oyçokluğu ile Kabul Edilmiştir. ……………….................... .…………………………….. ……...................................... Yrd.Doç.Dr. Sibel IRMAK Prof.Dr. Oktay ERBATUR Yrd.Doç.Dr. Belgin GÖZMEN DANIŞMAN ÜYE ÜYE Bu Tez Enstitümüz Kimya Anabilim Dalında hazırlanmıştır. Kod No:
Prof. Dr. İlhami YEĞİNGİL Enstitü Müdürü
Bu Çalışma Ç. Ü. Bilimsel Araştırma Projeleri Birimi Tarafından Desteklenmiştir. Proje No: FEF2009YL60 Not: Bu tezde kullanılan özgün ve başka kaynaktan yapılan bildirişlerin, çizelge ve fotoğrafların
kaynak gösterilmeden kullanımı, 5846 sayılı Fikir ve Sanat Eserleri Kanunundaki hükümlere tabidir.

I
ÖZ
YÜKSEK LİSANS TEZİ
LİGNOSELÜLOZİK MATERYALLERDEN TERMOKATALİTİK İŞLEMLE SUDA ÇÖZÜNDÜRÜLEN POLİSAKKARİTLERİN MOLEKÜLER
YAPILARININ İNCELENMESİ
Mehtap KURTULUŞ
ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
KİMYA ANABİLİM DALI Danışman :Yrd. Doç. Dr. Sibel IRMAK Yıl: 2010, Sayfa: 91 Jüri :Yrd. Doç. Dr. Sibel IRMAK :Prof. Dr. Oktay ERBATUR :Yrd. Doç. Dr. Belgin GÖZMEN
Bu çalışmanın amacı, bazı temsili lignoselülozik biyokütlelerin APR (Sulu Faz Reformlama) yöntemi ile gazlaştırılması sırasındaki selülozik, hemiselülozik ve ligninik davranışlarını incelemektir. Buğday samanı ve kenaf sapı bu çalışma için seçilmiş önemli lignoselülozik materyallerdir. Biyokütlelerden selülozik, hemiselülozik ve ligninik fraksiyonları izole edebilmek ve saflaştırabilmek amacıyla ilgili standart metodlar kullanılarak kapsamlı işlemler uygulanmıştır. Her fraksiyonun saflık derecesi izole edilen örneklerle ticari kaynaklardan temin edilen standartların karşılaştırılmasıyla ortaya konmuştur. Ayrıca her fraksiyonun yapısı SEM ve XRD metodlarıyla incelenmiştir. Buğday samanı ve kenaf sapından izole edilen tüm fraksiyonlar APR yöntemiyle iki farklı yaklaşıma göre incelenmiştir. Öncelikle lignoselülozik fraksiyonlar hiçbir ön işleme tabi tutulmadan APR yöntemiyle doğrudan gazlaştırılmıştır. Bu fraksiyonlar ilk olarak sulu faz içerisinde ısıl işlemlerle ılımlı sıcaklık ve basınç koşulları altında gazlaştırılmıştır. İkinci yaklaşımda ise subkritik su ile hidroliz ön işleminden geçirilmiş ve polisakkarit bakımından zengin sulu çözelti APR yöntemiyle gazlaştırılmıştır. Buğday samanı ve kenaf sapından izole edilen selüloz ve hemiselüloz fraksiyonlarının doğrudan gazlaştırma sonuçları ile hidroliz sonrası gazlaştırma sonuçları kıyaslandığında en yüksek hidrojen veriminin doğrudan gazlaştırma deneylerine ait olduğu görülmektedir. Başlangıç selüloz materyalinin kabaca üçte biri gaz haline (H2, CH4, C2H6) transfer olmaktadır. Kalan kısım ise hala çözünmemiş organik yapılar içeren sulu çözeltide bulunmaktadır. Hidrojen gazı üretimi için selülozun en etkili fraksiyon olduğu gözlenirken lignin fraksiyonunda ise hiç hidrojen gazı üretimi tespit edilememiştir.
Anahtar Kelimeler: Sulu faz reformlama, lignoselülozik materyal, selüloz, hemiselüloz, lignin.

II
ABSTRACT
MSc THESIS
EXAMINATION OF MOLECULAR STRUCTURES OF POLYSACCHARIDES SOLUBILIZED BY THERMOCATALYTIC
TREATMENT OF LIGNOCELLULOSIC MATERIALS
Mehtap KURTULUŞ
ÇUKUROVA UNIVERSITY INSTITUTE OF NATURAL AND APPLIED SCIENCES
CHEMISTRY DEPARTMENT
Supervisor : Asist. Prof. Dr. Sibel IRMAK Year: 2010, Pages: 91 Jury : Asist. Prof. Dr. Sibel IRMAK : Prof. Dr. Oktay ERBATUR : Asist. Prof. Dr. Belgin GÖZMEN
The aim of the work was to understand the behaviour of the cellulosic, hemicellulosic and ligninic fractions of some representative lignocellulosic biomass material during gasification via “Aqueous Phase Reforming: APR”. Wheat straw and kenaf stalk were selected as the important representatives of various lignocellulosic material. Both materials were subjected to exhaustive treatments by using the relevant standart methods to isolate and purify the cellulosic, hemicellulosic and ligninic fractions. Purity of each fraction was tested and confirmed by comparing the FTIR spectra of the samples produced and the standart samples received from commercial sources. The structures of each fraction were also examined via SEM and XRD methods. All the fractions obtained from both wheat straw and kenaf stalk were subjected to APR via two different approaches. While in the first approach, the materials to be gasified were directly subjected to APR without any pretreatment, they were preliminary solubilized in aqueous medium via catalytic thermal treatment under relatively mild temperature and pressure conditions and the aqueous solutions containing polysaccharide-derived material were subjected to APR in the second approach. Direct APR treatment of the cellulosic and hemicellulosic fractions of both the wheat straw and the kenaf stalk gave higher yields of hydrogen when compared with the corresponding results of the second approach. Roughly one third of the hydrogen content of the initial cellulose material was transferred into gaseous form (H2, CH4, C2H6) while the rest was still in the structure of the organic products dissolved in aqueous solution. It was shown that cellulose was the most effective fraction for hydrogen gas production whereas no hydrogen gas was obtained from the ligninic fraction. Keywords: Aqueous phase reforming, lignocellulosic materials, cellulose, hemicellulose, lignin.

III
TEŞEKKÜR
Tez konumda bana çalışma olanağı sağlayan ve tez çalışmam süresince bana
yol gösteren, yardımlarını esirgemeyen sevgili danışmanım Yrd. Doç. Dr. Sibel
IRMAK’a, her konuda bana gerekli destek ve anlayışı gösteren değerli hocam Prof.
Dr. Oktay ERBATUR’a teşekkürlerimi sunarım. Yardımları ve görüşleriyle tezime
katkıda bulunan sevgili hocam Öğr. Gör. Dr. Arif HESENOV’a teşekkür ederim.
Tezimin tüm aşamalarında bana yardımcı olan çalışma arkadaşlarım Bahar
Meryemoğlu, Burçak Kaya, Tuğba Balın, Ayşe Bilen, Okan İçten ve İlker Öztürk’e,
katkılarından dolayı Uzman Serkan Karaca’ya, ayrıca deneysel çalışmalarım
sırasında zaman zaman olanaklarından yararlandığım Organik Kimya Araştırma
laboratuvarı çalışma grubuna teşekkür ederim. Ayrıca maddi destek sağlayan
TÜBİTAK’a teşekkür ederim.
Tüm öğrenim hayatım boyunca maddi, manevi büyük fedakarlıklar yaparak
benim bu noktaya gelmemi sağlayan annem Meryem KURTULUŞ, babam Mehmet
KURTULUŞ ve biricik kardeşim Meltem KURTULUŞ’a sonsuz teşekkürlerimi
sunarım.

IV
İÇİNDEKİLER SAYFA
ÖZ........................................................................................................................ I
ABSTRACT...........................................................................…......................... II
TEŞEKKÜR...............................................……................................................ III
İÇİNDEKİLER………………………………………………………….......... IV
SİMGELER VE KISALTMALAR………………………………………….. VII
ÇİZELGELER DİZİNİ..................................................................................... VIII
ŞEKİLLER DİZİNİ.................................................................…...................... IX
1.GİRİŞ.......................................................................................………............ 1
1.1. Lignoselülozik Biyokütle Kaynakları ve Özellikleri…………………… 2
1.1.1. Selüloz ..……………………………………………..................... 4
1.1.2 Hemiselüloz………………………..……………………………... 5
1.1.3. Lignin……………..……………………………………................ 6
1.2. Buğday Samanının Özellikleri………………………………….............. 9
1.3. Kenafın Özellikleri………………………………….……………........... 10
1.4. Termokimyasal Dönüşüm Prosesleri…………………………………… 11
1.4.1. Doğrudan Yakma …………………….......................................... 13
1.4.2. Gazlaştırma……………………………………………………… 14
1.5. Sulu Faz Reformlaması (Aqueous Phase Reforming, (APR)) ile
Hidrojen Gazı Üretimi………………………………………………… 16
1.6. APR’de Kullanılan Organik Materyalin Niteliğinin Hidrojen
Oluşumuna Etkisi……………………………………………………….. 20
1.7. Şekerlerden Hidrojen Üretimi……………………………………........... 20
1.8. Biyokütlenin Hidrolizi ve Lignoselülozik Materyallere Uygulanan Ön
İşlemler…………………………………………………………………. 21
1.8.1. Fiziksel Ön İşlemler……………………………………………… 22
1.8.1.1. Mekanik Parçalama …………………………………….. 22
1.8.1.2. Piroliz…………………………………………………… 22
1.8.2. Fizikokimyasal Ön İşlemler……………………………………… 22
1.8.2.1. Buhar Patlaması (Steam Explosion)…………………..... 22

V
1.8.2.2. Amonyak Lif Patlaması (Ammonia Fiber
Explosion,AFEX)………………………………………... 23
1.8.2.3. CO2 Patlaması (CO2 Explosion)………………………… 24
1.8.2.4. Subkritik Su ile Hidroliz………………………………… 24
1.8.3. Kimyasal Ön İşlemler……………………………………………. 25
1.8.3.1. Ozon…………………………………………………….. 25
1.8.3.2. Asit ile Hidroliz…………………………………………. 25
1.8.3.3. Alkali ile Hidroliz……………………………………….. 26
1.8.3.4. Oksidatif Yolla Ligninin Uzaklaştırılması……………… 27
1.8.3.5. Organosolv Prosesi……………………………………… 27
1.8.4. Biyolojik Ön İşlemler……………………………………………. 28
1.8.4.1. Enzimatik Hidroliz……………………………………… 28
1.9. Moleküler Kütle Belirleme Teknikleri…………………………………. 29
1.9.1. Boyut Eleme Kromatografisi (SEC)…………………………..... 29
1.9.2. Polimerlerin Molekül Ağırlıklarının GPC Yöntemi ile
Belirlenmesi……………………………………………………… 29
2. ÖNCEKİ ÇALIŞMALAR............................................................................. 33
3. MATERYAL VE METOT............................................................................ 41
3.1. Materyal………………………………………………………………… 41
3.1.1. Kullanılan Kimyasal Maddeler…………………………………... 41
3.1.2. Kullanılan Araç ve Gereçler……………………………..…......... 42
3.2. Metot………………………………………………………………........ 43
3.2.1. Lignoselülozik Materyallerin Selüloz İçeriğinin
Saptanması.……………………………………………………… 43
3.2.2. Lignoselülozik Materyallerin Hemiselüloz İçeriğinin Saptanması 44
3.2.3. Lignoselülozik Materyallerin Lignin İçeriğinin Saptanması........ 44
3.2.4. Hidroliz Deneyleri….………………………………………......... 45
3.2.5. Hidroliz Çözeltilerinin TOC, Monoşeker ve Polisakkarit
Analizleri…………………………………………………............ 45
3.2.6. Hidroliz Çözeltilerinin Gazlaştırılması ve Gaz Analizleri….….… 46
3.2.7. FT-IR Analizleri …………………………………………………. 47

VI
3.2.8.Yüzey Analizleri………………………………………………….. 48
3.2.9. GC/MS Analizleri……………………….………………………... 48
4. BULGULAR VE TARTIŞMA...................................................................... 49
4.1. Buğday Samanı ve Kenaf’tan Selüloz, Hemiselüloz ve Lignin
İzolasyonu……………………………………………………………… 49
4.1.1. FT-IR Analizleri………………………………………………….. 49
4.1.2. SEM Analizleri…………………………………………………... 54
4.1.3. XRD Analizleri…………………………………………………... 56
4.1.4. GC-MS Analizleri………………………………………………... 58
4.1.5. Selüloz Fraksiyonunun TFA ile Hidrolizinin
İncelenmesi……............................................................................. 58
4.1.6. Lignoselülozik Fraksiyonların Subkritik Suda Hidrolizinin
İncelenmesi………………………………………………………. 59
4.2. Lignoselülozik Materyallerden İzole Edilen Fraksiyonların Sulu Faz
Reformlama ile Gazlaştırılması………………………………………... 62
4.2.1. Doğrudan Gazlaştırma…………………………………………… 62
4.2.2. Hidroliz Sonrası Gazlaştırma…………………………………….. 63
4.2.3. Ksilanın Gazlaştırılması…………………………………….......... 65
4.3. TOC Analizleri………………………………….…………………….. 67
4.4. Gazlaştırma Sonrası Çözeltinin Polisakkarit Analizleri………………... 70
5. SONUÇLAR VE ÖNERİLER................….........................................…..... 75
KAYNAKLAR.............................................................................…….............. 77
ÖZGEÇMİŞ...............................................................................................…..... 91

VII
SİMGELER VE KISALTMALAR
Acac : Asetilasetonat
Pt-Akt. Karbon : Platin Destekli Aktif Karbon
APR : Aqueous Phase Reforming (Sulu Faz Reformlama)
BW : Beechwood (kayın ağacı)
C : Karbon
FT-IR : Fourier Transformed Infrared
GC : Gas Chromatography (Gaz Kromatografisi)
GC-MS : Gas Chromatography- Mass Spectrometry (Gaz Kromatografisi- Kütle
Spektrometri)
GPC : Gel Permeation Chromatography (Boyut Eleme Kromatografisi)
HPLC : High Performance Liquid Chromatography (Yüksek Performanslı Sıvı
Kromatografisi)
Co : Kobalt
Cu : Bakır
Ni : Nikel
Pt : Platin
OS : Oat spelts (yulafxbuğday çaprazlanması ile oluşturulmuş bir tahıl)
SEM : Scanning Electron Microscopy (Taramalı Elektron Mikroskop)
TCD: Thermal Conductivity Detector (Termal İletkenlik Dedektör)
TEM : Tranmission Electron Microscopy (Geçirimli Elektron Mikroskop)
TFA : Trifloro Asetik Asit
TOC : Toplam Organik Karbon
TGA : Termogravimetrik Analiz
XRD : X-Ray Diffraction (X-Işını Kırınımı)
WGS : Water-gas shift reaction (Su-gaz dönüşüm reaksiyonu)

VIII
ÇİZELGELER DİZİNİ SAYFA
Çizelge 1.1. Bazı lignoselülozik maddeler ve bileşenleri……………… 3
Çizelge 2.1. Selüloz, ksilan ve lignin içeren karışımların gazlaştırılma-
sı sonucu oluşan gaz kompozisyonları……………………. 39
Çizelge 3.1. Buğday samanı ve kenafın nem, kül, selüloz ve içerikleri... 41
Çizelge 4.1. Buğday samanından izole edilen selüloz fraksiyonunun TFA
ile hidrolizi sonucu elde edilen polisakkaritlerin molekül
dağılımı……………………………………………………. 59
Çizelge 4.2. Lignoselülozik fraksiyonların subkritik suda hidrolizinin so-
nucu oluşan polisakkaritler ve hidrolizatların ve TOC
içerikleri……………………………………………………. 60
Çizelge 4.3. Buğday samanı lignoselüloziklerinin katalizörsüz gazlaştırıl-
ması sonucu oluşan gaz ürünleri……………………………. 62
Çizelge 4.4. Buğday samanı lignoselüloziklerinin katalizörlü gazlaştırıl-
ması sonucu oluşan gaz ürünleri……………………………. 62
Çizelge 4.5. Lignoselülozik fraksiyonların katalizörsüz olarak gazlaştırıl-
ması sonucu oluşan gaz ürünleri……………………………. 64
Çizelge 4.6. Lignoselülozik fraksiyonların katalizör varlığında gazlaştırıl-
ması sonucu oluşan gaz ürünleri………………………………… 65
Çizelge 4.7. Standart ksilanların katalizörsüz ve katalizörlü olarak gazlaş-
tırılması sonucu oluşan gaz ürünleri……………………….... 66
Çizelge 4.8. Lignoselülozik fraksiyonların katalizörlü ve katalizörsüz ola-
rak gazlaştırılması sonucu çözeltilerin polisakkarit içerikleri.. 71

IX
ŞEKİLLER DİZİNİ SAYFA
Şekil 1.1. Selülozun yapısı…………………………………………………. 4
Şekil 1.2. Hemiselüloz Yapısı…………………………………………….... 6
Şekil 1.3. Lignini oluşturan yapılar (a) koniferil alkol; (b) p-kumaril alkol;
(c) şiringil alkol…………………...…………………………….. 7
Şekil 1.4. Lignininin Önerilen Yapısı …………………...…………………. 8
Şekil 1.5. Hasat sonrası kenaf………………………………………………. 11
Şekil 1.6. Etilen glikolden "Sulu Faz Reformlama" ile H2 eldesinde seçiciliği
belirleyecek olası tepkime süreçleri ….………………………....... 18
Şekil 1.7. Fruktozun sulu faz reformlama mekanizması…………………….. 21
Şekil 1.8. Polistiren yapısı…………………………………………………… 30
Şekil 1.9. Dekstran genel yapısı…………………………………...………… 31
Şekil 3.1. Gazlaştırmada kullanılacak yüksek basınç reaktörü………............ 47
Şekil 4.1. Standart selüloz ve buğday samanından izole edilen selülozun
İnfrared spektrumları………………………………………........... 50
Şekil 4.2. Standart selüloz ve kenaftan izole edilen selülozun
infrared spektrumları………………………………………........... 50
Şekil 4.3. Buğday samanı ve kenaf sapından izole edilen hemiselüloz fraksi-
yonlarının FT-IR spektrumları……………………………………. 51
Şekil 4.4. Kayın ağacı ve yulafxbuğdaydan izole edilen standart ksilanlar
ile kenaf ve buğday samanından izole edilen hemiselüloz fraksi-
yonlarının infrared spektrumları………………………………….. 52
Şekil 4.5. Buğday samanından ve kenaftan izole edilen lignin fraksiyonları-
nın FT-IR spektrumları………………………………………….... 53
Şekil 4.6. SEM görüntüleri(a)Buğday samanınından izole edilen selüloz,
(b) standart selüloz (c) Kenaftan izole edilen selüloz…………….. 54
Şekil 4.7. SEM görüntüleri (a,b) Buğday samanından izole edilen hemise-
lüloz, (c) Kenaftan izole edilen hemiselüloz, (d) Standart ksilan… 55

X
Şekil 4.8. SEM görüntüleri (a) Buğday samanından izole edilen lignin, (b)
referans görüntü olarak kullanılan lignin (c) Kenaftan izole edilen
lignin……………………………………………………………... 56
Şekil 4.9. Standart ve izole selüloza ait XRD spektrumları………………... 57
Şekil 4.10. Hemiselüloz ve lignin fraksiyonlarına ait XRD spektrumları…… 57
Şekil 4.11. Selüloz fraksiyonunun TFA ile hidrolizi sonucu oluşan karbohid-
ratlar (a) Standart selülozun hidroliz çözeltisinin GPC kromatog-
ramı (b) İzole selülozun hidroliz çözeltisinin GPC kromatogramı.. 58
Şekil 4.12. Buğday samanından izole edilen fraksiyonların (a) Selüloz,(b)
Hemiselüloz, (c) Lignin, hidroliz çözeltilerinin GPC kromatog-
ramları…………………………………………………………….. 60
Şekil 4.13. Kenaftan izole edilen fraksiyonların (a) Selüloz, (b) Hemiselüloz,
(c) Lignin, hidroliz çözeltilerinin GPC kromatogramları……….. 61
Şekil 4.14. Buğday samanı ve kenaf sapından izole edilen hemiselüloz fraksi-
yonlarının Pt katalizörü varlığında oluşan gaz kompozisyonlarının
standart ksilanlarla karşılaştırılması……………………………… 67
Şekil 4.15. Buğday samanı ve kenaf selüloz fraksiyonlarının gazlaştırma
öncesi ve sonrası TOC sonuçları…………………………………. 67
Şekil 4.16. Buğday samanı ve kenaftan izole edilen hemiselüloz hidrolizat-
larının gazlaştırma öncesi ve sonrası TOC içerikleri…………….. 68
Şekil 4.17. Buğday samanı ve kenaftan izole edilen lignin fraksiyonlarının
gazlaştırma öncesinde ve sonrasında TOC sonuçları…………….. 69
Şekil 4.18. Buğday samanı fraksiyonlarının doğrudan ve hidroliz sonrası
gazlaştırma deneyleri sonrası TOC içerikleri…………………….. 69
Şekil 4.19. Buğday samanı lignoselüloziklerinin GPC kromatogramları (a)
Buğday samanından izole edilen selüloz, (b) Buğday samanından
izole edilen hemiselüloz, (c) Buğday samanından izole edilen lignin 72
Şekil 4.20. Kenaf sapı lignoselüloziklerinin GPC kromatogramları (a) Kenaf
sapından izole edilen selüloz, (b) Kenaf sapından izole edilen
hemiselüloz, (c) Kenaf sapından izole edilen lignin……..…………. 73

XI
Şekil 4.21. Standart ksilanların GPC kromatogramları (a) Kayın ağacı
(Beechwood) standart ksilanı, (b)Yulafxbuğday (Oat spelts)
standart ksilanı…………………………………………………....... 73

1. GİRİŞ Mehtap KURTULUŞ
1
1. GİRİŞ
Enerji, günümüzde insan hayatının vazgeçilmez bir parçası ve dünyadaki
sürdürülebilir kalkınma çabalarının en önemli araçlarından biridir. Dünyadaki nüfus
artışı, sanayileşme ve bilimsel faaliyetlerin gelişmesi ile enerjiye olan ihtiyaç her
geçen gün artmaktadır. İhtiyaç duyulan bu enerjinin büyük bir kısmı fosil yakıtlar
olarak nitelendirdiğimiz petrol, kömür ve doğal gazdan karşılanmaktadır. Ancak bu
enerji kaynakları rezervlerinin kısıtlı olması ve çevre üzerindeki olumsuz etkileri
dünya genelinde bu enerji kaynaklarının en uygun şekilde kullanımını ve yeni enerji
teknolojilerinin gerekliliğini açıkça ortaya koymuştur. Bu durum, enerjiyi gelişmiş ve
gelişmekte olan ülkeler için en önemli ve çözümlenmesi gerekli bir sorun haline
getirmiştir. Ülkemiz, enerji ihtiyacının büyük bir kısmını fosil yakıtlardan
sağlamaktadır. Başta petrol olmak üzere bu enerji kaynaklarının önemli bir kısmı
ithal ürünler olup, ülke ekonomisine olan yükü ve çevreye yaptığı olumsuz etkileri
her geçen gün artmaktadır. Ülkemizin kalkınması, sanayileşmesi ve gelişmesi enerji
üretimiyle doğru orantılıdır. Bu nedenle enerji sektöründe temel amaç, gelişen
ekonominin ve artan nüfusun enerji gereksinimlerini sağlıklı, güvenilir, sürekli,
kesintisiz ve en ekonomik maliyetle karşılayabilmektir. O nedenle, petrol ve doğal
gaz gibi ithal yakıtlara olan bağımlılığımızın azaltılması için yenilenebilir enerji
kaynakları arayışlarını hızlandırmamız gerekmektedir (Ültanır, 1996).
Ülkemiz, su (hidroelektrik, jeotermal, deniz enerjisi), güneş, rüzgar ve
biyokütle olarak sınıflandırabileceğimiz yeni-yenilenebilir enerji kaynakları
açısından oldukça iyi bir potansiyele sahiptir. Ülkemizde yeni ve yenilenebilir enerji
kaynakları üretimi, toplam kömür üretiminden sonra ikinci en yüksek üretime
sahiptir. Bu kaynakların yaklaşık üçte ikisini biyokütle oluşturmaktadır. Geri kalan
üçte birlik yenilenebilir enerji kaynağının da büyük çoğunluğunu hidroelektrik enerji
meydana getirmektedir. Görüldüğü gibi ülkemiz için biyokütlenin büyük bir önemi
vardır. Bu durum uygun süreçler geliştirilerek biyokütlenin enerji kaynağı olarak
değerlendirilmesinde yapılacak olan çalışmaların önemini de arttırmaktadır (Türkiye
Enerji Raporu, 2003).

1. GİRİŞ Mehtap KURTULUŞ
2
Dünya nüfusunun sürekli artması enerjiye olan talebi de artırmaktadır. Bunun
sonucu olarak fosil yakıtların sınırlı olan kaynaklarının hızla azalarak tükeneceği
endişesi ve yüksek kalitede enerji ihtiyacından kaynaklanan zehirli ve kalitesiz
atıkların büyük çevre problemlerine yol açması bilim adamlarını yeni enerji
kaynakları konusunda araştırmalar yapmak zorunda bırakmıştır (Lodhi, 1987). Bu
sebeple yenilenebilir enerji kaynaklarından biyokütlenin yeri ve önemi tartışılmazdır.
Yüksek karbonlu polimerik yapıya sahip olan biyokütle materyalleri genel anlamda
depolimerizasyonla parçalanarak veya CO ve H2 gibi indirgenlerle reaksiyona
sokularak yeni kimyasal ürünlere dönüştürülmeye elverişli hammadde kaynaklarıdır
(Demirbaş, A., 1996).
Biyokütle enerjisi, yetiştiriciliğe dayalı olduğu için yenilenebilir, çevre dostu,
yerli ve yerel bir kaynak olarak önem kazanmaktadır (Sayigh, 1999). Biyokütle
doğrudan yakılarak veya çeşitli süreçlerle yakıt kalitesi attırılıp, mevcut yakıtlara
eşdeğer özelliklerde alternatif biyoyakıtlar (kolay taşınabilir, depolanabilir ve
kullanılabilir yakıtlar) elde edilerek enerji teknolojisinde değerlendirilebilir
(Çetinkaya ve ark., 2003). Son zamanlarda biyokütle kaynaklarından enerji kaynağı
olarak yararlanmak artan bir ilgi sözkonusudur (Islam ve ark., 2005; Çulcuoğlu ve
ark., 2005).
Başarılı bir kimyasal işlemin uygulanabilmesi için lignoselülozik biyokütle
kaynakları ve özelliklerini daha yakından incelemek gerekmektedir.
1.1. Lignoselülozik Biyokütle Kaynakları ve Özellikleri
Dünya yıllık bitki ve tarımsal artık miktarı yaklaşık olarak 2.273.080.000
tondur. Türkiye'de ise her yıl 36.940.000 ton tarımsal artık elde edilmekte olup
bunun 18 milyon ton kadarı buğday sapı, 8 milyon tonu arpa sapı, 2.5 milyon tonu
mısır sapı, 3 milyon tonu pamuk sapı, 2.5 milyon tonu ayçiçeği sapı, 200 bin tonu
pirinç sapı, 240 bin tonu çavdar sapı, 300 bin tonu tütün sapı, 2 milyon tonu kendir-
kenevir, 200 bin tonu göl kamışı oluşturmaktadır (T.C. Başbakanlık Devlet İstatistik
Enstitüsü Yayınları, Tarımsal Yapı ve Üretimi, Ankara, 1995).

1. GİRİŞ Mehtap KURTULUŞ
3
Tarımsal artıkların ucuzluğu, atmosferdeki karbondioksit gazını kullanarak
oluşma nitelikleriyle enerji üretiminde kullanıldığında, atmosferdeki sera gazı
artışına katkıda bulunmayışı ve gıda maddesi olarak insanlar tarafından
tüketilmeyenler sınıfına girdiği için lignoselülozik biyokütlenin enerji alanında
değerlendirilmesi cazip görünmektedir.
Yüksek bitkilerin hücre duvarları lignoselüloz içerir. Ligninin ayrılması
durumunda geriye polisakkarit türevi kalır. Bitki hücresindeki polisakkaritlere
haloselüloz da denir. Haloselülozlar selülozlar ve hemiselülozlardan oluşur.
Haloselüloz hidroliz edilirse C6 ve C5 şekerleri, üronik asitler ve asetil gruplar elde
edilir. C6 şekerleri glikoz, mannoz ve galaktozdur. C5 şekerleri ise başlıca ksiloz ve
arabinozdur. Her bir bileşiğin oranı bitki kaynağına göre değişir (Beyatlı, 1996).
Lignoselülozik doğal kaynakların temel bileşenleri selüloz, hemiselülozlar,
ligninler, özütlenebilir maddeler ve inorganiklerdir. Doğada selüloz; çeşitli nişasta,
pektin ve hemiselüloz gibi polisakkaritlere bağlı olarak bulunur. Hemiselülozlar ise
galaktoz, mannoz, ksiloz, arabinoz ve diğer şekerlerle; üronik asitlerin polimerleri ve
heteropolimerlerini içerirler. Bunlara ek olarak, doğadaki hemen hemen her selüloz,
selüloz-lignin karışımı halinde bulunur. Çizelge 1.1’de doğadaki bazı lignoselülozik
maddeler ile bileşenleri görülmektedir.
Çizelge 1.1. Bazı lignoselülozik maddeler ve bileşenleri (Parisi, 1989)
Biyokütle
Selüloz (%)
Hemiselüloz (%)
Lignin (%)
Kozalaklı ağaçlar 40-50 20-30 25-35
Şeker kamışı 40 30 20
Yaprakları dökülen ağaçlar
40-50 30-40 15-20
Mısır koçanı 45 35 15
Buğday sapı 30 50 15

1. GİRİŞ Mehtap KURTULUŞ
4
1.1.1. Selüloz
Bitki dünyasında en fazla bulunan ve en basit yapıya sahip olan, aynı
zamanda hücre duvarı yapısında yer alan yapısal polisakkaritlerin en önemlilerinden
birisi selülozdur (Anonim, 1969; Eriksson ve ark., 1990).
Selüloz, glukoz ünitelerinin β-1,4 bağları ile bağlanması sonucu oluşmuş bir
homopolimerdir. Selüloz molekülünün büyüklüğü (polimerizasyon derecesi) bitki
hücresinin duvarında bulunan ikincil duvarda her molekülde 500’den daha az glukoz
biriminin bulunmasına bağlı olarak değişir (Ljungdal ve Eriksson, 1985). Selülozun
yapısı Şekil 1.1’de gösterilmiştir.
Şekil 1.1. Selülozun yapısı (Valenzuela, 2006)
Selüloz; bütün bitki, ot ve ağaçların temel yapı taşıdır. Selülozun en önemli
görevi bitkilere sağlamlık, diklik ve destek sağlamaktır. Doğada saf halde bulunmaz.
Odunun ağırlıkça %40’ını, ketenin %60-85’ini pamuk liflerinin %85-90’ını selüloz
oluşturur (Johansson, 1999). Genellikle selülozun bitki hücre duvarındaki oranı hücre
tipine ve evresine göre değişmektedir. Örneğin; birincil duvarın kuru ağırlığının
%20-40’ı selülozdan oluşurken ikincil duvarın %40-60’ı selülozdan meydana
gelmektedir (Nugzar, 1997). Pamuk tohumunun ikincil duvarının %100’ü selülozdur.
İkincil hücre duvarı mikrofibrilleri birincil hücre duvarı mikrofibrillerine göre daha
yoğundur ve daha çok selüloz kristalleri içerir. Selüloz doğada hemen hemen hiçbir
zaman tek başına bulunmaz. Genellikle diğer bitkisel maddelerle beraber bulunur. Bu
selülozun doğal ortamda parçalanmasını etkilemektedir. Selüloz fibrilleri öncelikle
hemiselüloz, pektin ve proteinlerin dahil olduğu diğer polimerlerin matriksine
gömülmüş haldedir. Selüloz, hücre duvarına turgor basıncına dayanabilecek
gerilebilir bir kuvvet verir. Eğer hücre duvarındaki su lignin ile değiştirilirse yüksek
bir kuvvet elde edilir.

1. GİRİŞ Mehtap KURTULUŞ
5
Doğada birkaç çeşit selüloz bulunmaktadır. Bunların hepsi de endüstri
açısından önemlidir fakat değişik amaçlar için kullanılırlar. Selüloz türleri
birbirinden a,b,d harfleriyle ayırt edilir. A- selüloz, pamuktaki selüloz türüdür. Bütün
türler arasında en önemli olanıdır. “Hemi-selüloz” adını alan b-selüloz ve d-selüloz
ise asitler ve bazlara karşı daha az dayanıklı moleküller dallanmış halde ve daha
kolay kopabilme özelliğine sahiptir (Anonim, 1984).
1.1.2. Hemiselüloz
Lignoselülozik maddelerin selülozdan sonraki en önemli bileşenleri
hemiselülozlardır. Selüloz gibi kristalin bir yapıya sahip değildir. Hemiselülozlar,
odundaki selüloz olmayan başlıca polisakkaritlerdir (Şekil 1.2). Hücre çeperindeki
polisakkaritlerin %20-35 ‘ini oluşturmaktadırlar. Odunun üç ana bileşeni arasında
ısıya en duyarlı olanı hemiselülozlardır ve 200-260 ºC arasında bozunurlar (Gunnar,
2000). Hemiselüloz ve selüloz odundaki holoselülozu oluşturur. Hemiselülozlar,
selülozdan bazı özellikleri ile ayrılır (Yoon ve ark., 2005). Odunun diğer
elemanlarından ayrıldıktan sonra seyreltik alkali çözeltisinde ve kaynayan suda
çözünebilirler. Hemiselülozların kimyasal yapısı hakkında bugün çok az şey
bilinmektedir. Ama şu açıkça bilinmektedir ki hemiselülozlar selülozdan daha
heterojendir (Robert ve ark., 2001).
Hemiselüloz polimerleri (DP (Degree of Polimerization): 150-250) oldukça
amorf ve düzensiz dallanmalara sahiptir; düz zincirler şeklinde düzenlenmiş selüloza
göre reaksiyonlara daha duyarlıdır. Hemiselülozlar kendilerini oluşturan şeker
birimlerine göre; ksilanlar, mannanlar, arabinoksanlar, glikomannanlar ve
glikoksilanlar şeklinde isimlendirilirler (Mutlu, 1990).
Ksilanlar, hemiselülozik yapı içinde nicelik açısından önemli yer tutarlar.
Kara bitkilerinin ligninli dokularındaki hemiselülozların temel bileşenini
oluştururlar. Olgunlaşmış odunların % 20-25’i, otların % 15-20’si ve yumuşak
odunların önemli bir kısmı ksilanlar ve glikomannanlardan oluşur. Tahıl sapları ile
tohum kabuklarının da, kuru ağırlık olarak % 20-30’u ksilanlardır (Aspinal, 1970).
Ksilanlar, selülozla birleşik halde bulundukları gibi, ligninle de etkileşim

1. GİRİŞ Mehtap KURTULUŞ
6
içindedirler. Polisakkaritlerin hemiselüloz grubunun temel bileşenini oluşturan
ksilanlar, bitkilerden alkali çözeltilerle özütlenebilirler (Whistler, 1953).
Şekil 1.2. Hemiselüloz yapısı (http://chemistry.umeche.maine.edu/CHY431/Wood14.html)
1.1.3. Lignin
Bitkide kök ve gövdenin odunsu yapısını oluşturan madde olarak da bilinir.
“Odunun özü” de denen su geçirmez bir yapıya sahiptir. Yaşlanmış ölü hücrelerin
selüloz çeperleri üzerinde birikerek bitkiyi uygun olmayan çevre şartlarından korur
(Martinez ve ark,. 2001). Lignin bir glikozit olup kolayca glukoz ve aromatik bir
alkole ayrıştırılabilmektedir. Bu glikozit koniferin olarak adlandırılır. Bu bileşikten
türeyen alkole de buna uygun olarak koniferil alkol denilmiştir (Strayer ve ark.,
2002). Potasyum permanganat ile ligninin oksidasyonu sonucu hemipin asitleri ve
türevleri meydana gelmektedir (Sfountoulakis, 2002). İğne yapraklı ağaç odunları
lignininden esas itibari ile “guayasil” kalıntısı taşıyan parçalanma ürünleri elde
edilmesine karşılık, yapraklı ağaç odunu lignininden yukarıdaki ürünlerin yanı sıra
aynı seri içinde “şiringil” kalıntısı taşıyan ürünlerde elde edilmektedir (Elke ve ark.,
1997).
Lignin bir karbonhidrat olmamakla beraber fonksiyonları bakımından
karbonhidratlara yakın bir maddedir. Hücrede sekonder çeper yapısına büyük oranda
iştirak eder. Hücre çeperini oluşturan selüloz misellerin arasını amorf lignin doldurur
ve böylece dokuda odunlaşma meydana gelir (Hirofimi ve ark., 1999). Çam

1. GİRİŞ Mehtap KURTULUŞ
7
ağaçlarının iğnelerinde yoğun miktarda bulunan ligninin, çürüyüp toprağa karışması
uzun bir zaman aldığından çam ağaçlarının altında birikir. Bu biriken maddeler yavaş
yavaş çözündükçe toprakta asit birikmesi olur. Ayrıca alt tabakadaki bitkiler oluşan
bu iğne yumağının altında kaldığı için ışık alamayarak çürürler (Breen, 1999).
Parçalanma ürünlerinden anlaşılmaktadır ki; ligninin temel yapı taşı bir
aromatik çekirdek ile bir propan zincirinden oluşmaktadır (Guiraud ve ark., 1998).
Burada molekülün bazı yerlerinde çeşitli fonksiyonel gruplar bulunmaktadır. Bu
guruplar sayesinde çeşitli diğer birimlere bağlanabilme olasılıkları ortaya
çıkmaktadır (Adosinda ve ark., 2002). Ligninin temel yapı taşı veya temel birimi
fenil propan olarak adlandırılmaktadır. Fenil propan üyeleri çok çeşitli tarzlarda
birbirlerine bağlanarak lignini meydana getirirler (Adosinda ve ark., 2002).
Lignin kimyasal olarak polisakkaritlere bağlı olarak bulunur. Bunca
çalışmalara rağmen lignin hakkında yeterli bilgi elde edilememiştir. Bunun nedeni
elde etme esnasında; özütleme aşamasında maddenin doğasının bozulmasıdır. Bu
yüzden kimyacılar odun özünü (lignini) doğada bulunduğu biçimiyle elde
edememekte, asıl madde yerine türevlerini incelemek zorunda kalmaktadırlar.
Lignin; kağıt üretiminde kükürtdioksit, sodyum sülfit ya da sodyum hidroksit gibi
maddeler yardımı ile odun hamurundan ayrılır. Ayrılan bu lignin kendisinden
yararlanılacak uygun bir kimyasal teknolojinin yokluğu nedeni ile çoğunlukla yakılır.
Ligninin birimleri Şekil 1.3’te gösterilmiştir.
Şekil 1.3. Lignini oluşturan yapılar a) koniferil alkol; b) p-kumaril alkol; c) şiringil alkol (Valanzuela, 2006)

1. GİRİŞ Mehtap KURTULUŞ
8
Ligninin kimyasal yapısını incelediğimizde birbirine yakın üç aromatik
bileşikten meydana geldiğini görürüz. Bu maddeler koniferil alkol, sinapil alkol ve p-
kumaril alkoldür.
Lignin asitlerle kolayca hidroliz olmaz. Bu alkoller içinde koniferil alkol esas
bileşen olup, kozalaklı ağaçların lignininde %90, yayvan yapraklı ağaçların
lignininde ise %50 oranında koniferil alkol bulunur (Chrestini ve ark., 1998).
Ligninin tek karbon/enerji kaynağı olarak mikroorganizmalar tarafından
kullanılamaması standart zenginleştirme yöntemleri ile lignini degrade eden
mikroorganizmaların izolasyonunu güçleştirmektedir (Crawfort, 1981). Buğday
samanına ait ligninin kimyasal yapısı için bir önerme Şekil 1.4’ te gösterilmiştir.
Şekil 1.4. Buğday samanı lignininin önerilen yapısı (Sun ve ark., 1997).

1. GİRİŞ Mehtap KURTULUŞ
9
1.2. Buğday Samanının Özellikleri
Buğday (Triticum aestium), dünyada en çok yetiştirilen bitkilerin başında
gelir. Yıllık dünya buğday üretimi yaklaşık 627 milyon tondur ve Türkiye buğday
üretiminde dünyada 7. sırayı almaktadır. Türkiye'de 2005 yılında 10 milyon hektar
alanda buğday ekimi yapılmış olup, 21 milyon ton üretim sağlanmıştır. Buğdaydan
elde edilecek saman, hasat edilen buğdayın türüne, iklime ve ziraai koşullara bağlı
olmakla birlikte ortalama 1 kg buğday eldesine karşılık 1,3 kg saman açığa
çıkmaktadır (Montane ve ark., 1998).
Buğday sapı, bağlantı yerlerinden boğumlarla ayrılmış, dik ve silindir
şeklinde gövdelerdir. Saplar genelde altı iç-boğuma sahip olup cinslerine, iklime ve
toprağın durumuna bağlı olarak 0,5 ile 1,5 metre arasında uzunluğa ulaşırlar.
Lignoselülozik lif yapıları, dolayısı ile odunu andıran buğday sapları gibi tahıl sapları
tarihsel olarak kağıt hamuru ve kağıt yapımında yaygın olarak kullanılmıştır. Fakat
Kuzey Amerika ve Avrupa’nın büyük bir kısmında odundan kağıt hamuru üretimi
çok ekonomik duruma geldiği için buğday saplarının kağıt endüstrisindeki kullanımı
zarar görmüş ve azalmıştır. Çoğu Asya, Güney Amerika ve Doğu Avrupa ülkeleri
hala tahıl saplarını kağıt hamuru üretiminde kullanmaktadırlar (Misra, 1983).
Türkiye kaynak olarak çok büyük buğday ve diğer tahıl sapları, kendir sapları ile
diğer tarımsal atık potansiyeline sahiptir.
Morfolojik karakterleri açısından buğday saplarından elde edilen lifler odun
liflerine kıyasla daha heterojendir. Odunla kıyaslandığında, buğday sapları hemen
hemen aynı miktarda holoselüloza sahip olmalarına rağmen çok daha az alfa-selüloza
sahiptirler. Pentozan miktarı fazla olmakla birlikte lignin miktarı odundan biraz daha
azdır. Sonuç olarak, buğday sapları kimyasal içerik bakımından yapraklı ağaçlara
daha fazla benzemektedir.
Buğday sapları %70-75 oranında holoselüloz içerir ki bunun yaklaşık olarak
yarısı (%35) alfa-selülozdur. Holoselülozlar bitki dokularındaki suda çözünmeyen
karbonhidratlar olarak tanımlanırlar ve alfa-selüloz (ya da basitce selüloz) ile
hemiselülozdan oluşurlar.

1. GİRİŞ Mehtap KURTULUŞ
10
1.3. Kenafın Özellikleri
Kenaf (Hibiscus cannabius L.) Malvaceae ailesine ait, kökeni Afrika olan ve
yıllık yetişen bir elyaf bitkisidir (Sameshima, 1995). Endüstride kullanılan ideal bir
lif kaynağıdır. 4000 yıllık tarihi vardır. Pamuk ve bamya familyası ile aynı
familyadadır. Amerikanın birçok bölgesinde iyi bir şekilde yetişmektedir. Ağaçları
kesmeden kağıt üretmenin bir yoludur. Birim alandaki karbondioksiti absorplama
hızı ağaçlara kıyasla 4-5 kat daha fazla olduğundan global sera etkisini önleyici
önemli bir bitki olarak görülmektedir (Nakamura ve ark., 2001). Kenaf dünyada
genellikle lif üretimi (halat ipi vb.) ve kağıt sanayinde (Nielsen, 2004) kullanılmakla
birlikte son yıllarda kaba yem açığının kapatılması amacıyla ruminant beslemede
yem kaynağı olarak da kullanılmaktadır. Kenaf ruminant beslemede yeşil ve kuru ot
olarak kullanıldığı gibi silaj, pelet ve küp formunda kullanımına rastlanmaktadır.
Ayrıca su tutma özelliği nedeniyle kanatlı yetiştiriciliğinde, altlık materyali olarak da
kullanılmaktadır.
Kenaf bitkisi ılımlı bölgelerde yetişir ve 4-5 ay içinde 4-5 m boyuna
ulaşabilecek kadar hızlı gelişmektedir. Olgunluğa 150 gün içinde ulaşabilmekte ve
bu süre sonunda tüm bitki hasat edilmektedir. Çiçeklenme sezonu 3-4 hafta veya
daha fazla sürer ve her bir çiçeğin bir günlük ömrü vardır. Yani bir günde açar ve
aynı gün dökülür. Hasatta, tüm kenaf bitkileri mekanik lif seperator (ayırıcı aygıt) ile
aynı pamuk çırçırındaki gibi işlenir. Kenaf bitki çiçeklerinin yetişme sezonu
sonunda, açan çiçekler düştükten sonra geride bir tohum kabuğu bırakırlar. Hemen
hemen bütün Amerika bölgelerinde bu tohumlar olgunlaşmaz, bunun sebebi kenafın
Afrika kökenli olmasıdır. Çimlenme noktasına erişebilmek için 60-90 gün serbest
don ıslahı yapılması gerekir. Bu demektir ki kenaf bitkisini Amerika genelinde
yabani bir ot gibi yetiştiremezsiniz. Ayrıca bitkinin gelecek yıllarda geliştiricilerin
kullanması için uygun tohum erzakı temin edilir. Birçok araştırmalar tohum
geliştirme alanında Vision Paper gibi öncü şirketler tarafından yapılmaktadır.
Ülkemizde Tire’de yapılan adaptasyon çalışmalarından olumlu sonuçlar alınmıştır.
Kenaf, yaklaşık %42 oranında selüloz ve %14-15 oranında lignin içerir
(Ververis ve ark., 2004). Şekil 1.5’te hasat sonrası kenaf gösterilmiştir.

1. GİRİŞ Mehtap KURTULUŞ
11
Şekil 1.5. Hasat sonrası kenaf
1.4. Termokimyasal Dönüşüm Prosesleri
Termokimyasal dönüşüm proseslerinin iki basit yaklaşımı vardır. Birincisi
biyokütlenin gazlaştırılması ve hidrokarbona dönüşümünü sağlamaktır. İkincisi ise,
yüksek sıcaklık pirolizi, yüksek basınç sıvılaştırması, ultra piroliz ya da süperkritik
ekstraksiyon ile direkt olarak sıvılaştırmaktır. Bu prosesler, atık biyokütleyi enerjice
zengin yararlı ürünlere dönüştürebilmektedir. Dönüşüm prosesinin seçimi
biyokütlenin kalitesine, tipine, istenilen enerjiye ve kullanılacak gereçlere, çevresel
standartlara, ekonomik koşullara ve projenin spesifik faktörlerine bağlıdır.
Farklı termokimyasal dönüşüm prosesleri; yanma, gazlaştırma, sıvılaştırma,
hidrojenleme ve pirolizi içerir. Piroliz halen gelişen bir yöntem olmasına rağmen
biyokütleyi oksijen yokluğunda termal bozundurmaya uğratarak direkt katı, sıvı ve
gazlara dönüştürebilmektedir. Tarım ülkeleri için piroliz, çok fazla biyokütle yan
ürünleri ile birlikte önemli ve etkili kullanım sunmaktadır.
Biyokütle biyokimyasal ve termokimyasal işlemler ile ticari yakıtlara
dönüştürülebilmektedir. Piroliz oksijensiz ortamda biyokütlenin termal
parçalanmasını gerektiren termokimyasal bir işlemdir (Gerçel, 2002; Brigwater ve
ark., 2002). Biyokütlenin pirolizi 350-550oC sıcaklıkta başlar ve 700oC sıcaklığa

1. GİRİŞ Mehtap KURTULUŞ
12
kadar çıkar. Bu da sıvı yağ, gazlar ve katı ürünler gibi yararlı ürünlerin oluşumunu
izler. Farklı durumlar farklı miktarlarda ürün oluşumunu sağlar.
Piroliz, organik materyallerin inert atmosfer ya da vakum ortamında ısı ile
bozulması olayıdır. Isıtma veya kısmi yanma olan piroliz, biyokütleden ikincil
yakıtların ve kimyasal ürünlerin üretiminde kullanılır. Pirolizin hammaddesi (girdisi)
odun, kömür, biyokütle atıkları ve yerel atıklar, ürünleri ise; gazlar, sıvılar
(yoğunlaştırılmış buharlar), tarlar, yağlar, katılar (char) ve küldür. Biyokütlenin
pirolizinden elde edilen gaz ürünlerin miktarı ve enerji değerlerinin sıcaklık ve
katalizörün cinsi ve miktarına göre değişmektedir (Çağlar ve Demirbaş, 2002).
Gazlaşma, ikincil yakıt gazların maksimum miktarda üretildiği bir piroliz türüdür.
Pirolizden elde edilen yakıt ürünler orijinal biyokütleden elde edilen yakıt ürünlerden
temizlik, kullanım ve nakliye bakımından çok daha uygundur. Elde edilen kimyasal
ürünler, diğer prosesler için kimyasal besleme stoğu olarak veya doğrudan kullanım
kolaylığı bakımından önemlidir. Randıman olarak, biyokütlenin doğrudan
yanmasından elde edilen ısı ile ikincil yakıt ürünlerin yanmasından elde edilen ısı
değerlerinin karşılaştırılmasında %80–90’a çıkan bir fark bulunmuştur (Twidell,
1986). Biyokütlenin termokimyasal dönüşüm yöntemlerinden en verimli ve en
ekonomik olanı pirolizdir ve özelikle sıvı hidrokarbon üretiminde en çok kullanılan
bir proses olarak dikkat çekmektedir (Soltej, 1988). Pirolizden sıvı ürünlerin elde
edilmesinde sıcaklık, katalizör ve diğer piroliz şartlarının etkisi oldukça büyüktür
(Çağlar ve Demirbaş, 2000; Çağlar ve Demirbaş, 2001). Biyokütlenin pirolizi üzerine
yapılan çalışmalar, en uygun verimde ve kaliteli ürünlerin elde edilmesi için işleme
şartlarının geliştirilmesi üzerinde odaklanmıştır (Bridgwater ve Bridge, 1991). Piroliz
esnasında işleme şartlarının değiştirilmesi meydana gelecek reaksiyonların yolunu da
değiştirmektedir. Bu değişiklik ürün dağılımını etkilemektedir. Özelikle uygulanan
prosesin kinetiği ana proses parametreleri olan; sıcaklık, residans zamanı, besleme
stoğunun bileşimi, partikül boyutu ve ısınma hızına bağlı olarak değişmektedir
(Maschio ve ark., 1992). Biyokütlenin katalitik pirolizinden elde edilen sıvı ürünlerin
miktarına ve dağılımına katalizörün etkisinin araştırıldığı çalışmalarda, katalizörün
ürün verimine doğrudan etki ettiği görülmektedir (Çağlar ve Demirbaş, 2000;
Demirbaş, 1998; Mansilla ve ark., 1998). Piroliz işlemlerinde nikel dolamit ve alkali,

1. GİRİŞ Mehtap KURTULUŞ
13
toprak alkali ve geçiş metali tuzları katalizör olarak kullanılmaktadır (Sutton ve ark.,
2001). Katalitik piroliz işlemlerinde genel olarak alkali metal karbonatları ve boraks
en yaygın olarak kullanılan katalizörlerdendir (Mudge ve ark., 1985). Ancak
katalizör olarak kullanılan Na, Li ve K karbonatları piroliz dönüşüm oranını
arttırırken sıvı üründen çok gaz ürün verimini arttırmaktadır (Manshchio, 1994). Sıvı
ürün veriminin nispeten arttığı piroliz işlemlerinde ise TiO2, ZnCl2, AlCl3, ZnO ve
Fe2O3 katalizörleri kullanılmıştır (Mansilla ve ark., 1998).
Lignoselülozik materyallerin pirolizi oldukça karmaşıktır. Çünkü
lignoselülozik materyallerin ana bileşenleri olan selüloz, lignin ve hemiselüloz
oldukça farklı reaktivite göstermektedirler. Sıcaklık ve dönüşüm parametrelerine
bağlı olarak, her bir bileşenin termal parçalanmasında farklı birçok reaksiyon
meydana gelmektedir. Bu durum materyalin özelliklerinde değişikliklere neden
olmaktadır. Biyokütle bileşenleri ve tüm biyokütle örneklerinde çok az miktarda
doğal olarak bulunan mineral maddeleri arasındaki etkileşimler, piroliz esnasında
meydana gelen sayısız reaksiyonu kataliz etmektedir (Mair ve Faix, 1999).
1.4.1. Doğrudan Yakma
Biyokütlenin doğrudan yakılarak enerji üretilmesi, bilinen en eski yöntem
olmasına karşın, son yıllarda verimi yükseltmek için yeni yakma sistemleri
geliştirilmektedir. Özellikle biyokütle ile çalışan termik santral yapımında akışkan
yataklı sistemler alışılagelmiş yakma sistemlerinin yerlerini almaktadır.
Hemen her türlü biyokütle kaynağını doğrudan yakmak olanaklıdır. Ancak,
nem oranı yükseldikçe elde edilen ısıl değer azalır. Yanma, biyokütle içindeki
yanabilir maddelerin hidrojenle hızlı kimyasal tepkimesi olarak tanımlanır. Örneğin
mısır, ayçiçeği sapları gibi tarım atıkları içindeki yanabilir maddeler, karbon,
hidrojen ve potasyum gibi bazı metalik elementlerdir. Bu kimyasal tepkime sonucu
ortaya çıkan atık maddeler ise, karbondioksit, su buharı ve bazı metal oksitlerdir.
Herhangi bir biyokütleyi yakmak mümkündür ancak pratikte yanma sadece kül
miktarı < %50 olan biyokütle için uygundur. Biyokütleyi yakma prosesi kullanan
yakma sistemine bağlı olarak farklı verimlerde ısı üretimine sebebiyet vermektedir.

1. GİRİŞ Mehtap KURTULUŞ
14
Biyokütleden yakma prosesiyle ısı enerjisi elde etme işleminde en önemli kısım
kabuk içeriği oluşturmaktadır. Kabuk içeriği daha fazla ekstraktif madde nedeniyle
daha fazla enerji yaymaktadır (Fengel ve Wegener, 1984).
Odun ve odunsu biyokütle yanarak CO2 ve H2O’ya dönüşür (Klason ve ark.,
1910; Kollman, 1951): C42H60O28 + 43O2 → 42CO2 + 30H2O
Odun ve odunsu biyokütlenin direk yanması üzerine birçok araştırma vardır
(Browne, 1958; Robert ve Auston, 1981).
1.4.2. Gazlaştırma
Yenilenebilir biyokütle ve biyokütleden elde edilen yakıtlar çevresel fayda
sağlaması sebebiyle günümüz enerji kullanımında kolaylıkla fosil yakıtların yerine
geçebilecektir. Biyokütlenin gazlaştırılması; katı yakıtların ısıl çevirim teknolojisiyle
yanabilen bir gaza dönüştürülmesi işlemidir. Sınırlandırılmış oksijen, hava, buhar
veya bunların kombinasyonları reaksiyonu başlatmaktadır. Üretilen gaz
karbonmonoksit, karbondioksit, hidrojen, metan, su ve azotun yanı sıra kömür
parçacıkları, kül ve katran gibi artıkları da içermektedir. Üretilen gaz temizlendikten
sonra kazanlarda, motorlarda, türbinlerde ısı ve güç üretilmek üzere kullanılmaktadır.
Gazlaştırma tekniği ile biyokütleden, yüksek bir randıman alarak petrolle çalışan güç
ve ısı sağlayan tirbünlerde kullanılacak bir gaz yakıt elde edilebilir.
Biyokütle kaynaklarının temini fosil kaynaklardan daha pahalıdır. Fakat
biyokütle yenilenebilir bir kaynak olmasıyla tükenmekte olan fosil yakıtların yanında
sürdürülebilir küresel enerjinin önemli bir unsurudur. Buna ek olarak sera gazları
emisyonu ve karbon döngüsünü azaltıp, kırsal ekonominin gelişimiyle yeşil
endüstriyi desteklemektedir. Biyokültenin gazlaştırılması ile elde edilen gaz yakıt
doğal gazın kullanıldığı yerlerde küçük modifikasyonlar yapılarak kullanımı
yaygınlaştırılabilir ve gelecekte kolaylıkla doğal gazın kullanıldığı yerlerde enerjinin
büyük bir kısmı bu yakıttan sağlanabilir.
Biyokütleden gazlaştırılma ile elde edilen temizlenmiş gaz yakıt ısı ve buhar
üreten kazanlarda direk yakılarak veya Stirling motorlarda %20-30 verimle elektrik

1. GİRİŞ Mehtap KURTULUŞ
15
üretimi için kullanılabilmektedir. Basınçlı gazlaştırma tirbünlerinde ise %40 veya
daha fazla verimlilikte elektrik üretimi yapılabilmektedir.
Katı yakıt gazlaştırma, özellikle başta kömür, linyit, biyokütle ve katı atıklar
olmak üzere tüm katı yakıtları katı halden gaz haline dönüştüren temiz enerji
dönüşüm prosesidir. Hava ve/veya oksijen kontrollü olarak sisteme verilir ve böylece
redüksiyon koşullarının kalıcılığı sağlanır. Gazlaştırma vakumlu, atmosferik ve
basınçlı ortamda, gazlaştırıcı içinde gerçekleştirilir ve ürün CO ve H2 karışımından
oluşan ve syngaz olarak adlandırılan gazdır. Çıkan gaz temizlenir ve yüksek basınç
ve yüksek sıcaklıkta oksijen veya hava ile yakılarak enerji üretilir veya metanol,
amonyak, gübre gibi kimyasal maddelerle, benzin, dizel gibi sıvı yakıtların
üretiminde kullanılır. Katı atık ve katı yakıt esaslı elektrik gücü üretim teknolojileri
içinde gazlaştırma, düşük düzeyde en uygun hava emisyonlarına, katı atık ve atık su
değerlerine sahip en temiz teknolojidir. Yüksek enerji verimliliğinin nedeni, daha az
karbondioksit (CO2) emisyonlarıyla sonuçlanan katı yakıt gazlaştırmada aynı
miktarda enerji üretmek için daha az katı yakıt kullanılmasıdır. Gazlaştırma, yakma
teknolojilerine göre daha çevreci bir teknolojidir ve CO2, SO2, NOx emisyonları
bakımından çok daha avantajlıdır. Mevcut kükürt çoğunlukla, SO2’ye nazaran daha
kolay şekilde giderilebilen H2S şekline dönüşür. Gazlaştırma sırasında NOx, dioksin
ve furan problemleri oluşmamaktadır. British Gas Lurgi (BGL) gazlaştırıcısı ve ilgili
tesisleri enerji ve kimyasal maddelerin en yüksek verimde üretilmesini sağlayan en
önemli proseslerden biridir. Syngaz’dan kimyasal madde veya sıvı yakıt üretilmesi
durumunda üretim syngaz üretim tesisinden CO2 emisyonlarıyla
karşılaşılmamaktadır.
Gazlaştırma prosesinde biyokütle 800-900oC sıcaklık aralığında kısmı
oksidasyon ile yanıcı gaz karışımlarına dönüştürülür. (Demirbaş, 2001; White ve
Plasket, 1981; Othmer, K., 1980). Reaksiyonlar aşağıdaki gibidir:
C + O2 → CO2
C + 1/2O2 → CO
CO + 1/2O2 → CO
CO2 + C ↔ 2CO

1. GİRİŞ Mehtap KURTULUŞ
16
CO2 + 4H2 → CH4 + 2H2O
Son zamanlarda pamuk, odun, ağaç kabuğu, bataklık kömürü ve pirinç
gövdesi gibi selülozik materyallerin gazlaştırılması için atılımlar yapılmıştır. Genel
görüş bu materyallerin düşük sıcaklık pirolizinin uygulanacağı bunun cracking ile
gaz geliştirmenin takip edileceğini ve daha sonra bazı durumlarda kok oluşumuna
buhar ile muamele edileceği yönündedir. Bu yaklaşım genellikle oldukça önemli
miktarlardaki katılar ile CO2 bakımından zengin, gazlı ürünlerin ve sıvı ürünlerin
oluşumu ile sonuçlanır. Bir alternatif görüş, hidrojen ve CO oluşumu ve önemli
miktarlarda CO2 ve metan ile sonuçlanan selülozun direkt yüksek sıcaklık pirolizidir
(Tran ve Rai, 1978).
1.5. Sulu Faz Reformlaması (Aqueous Phase Reforming, APR) ile Hidrojen Gazı
Üretimi
Biyokütlelerin gazlaştırılmasında süperkritik ve subkritik suyun kullanılması
için yapılan araştırmalar yaklaşık otuz yıl geriye gitmesine rağmen (Bobleter ve
Consin, 1979; Model, 1985) sulu fazda reformlama (Aqueous Phase Reforming:
APR) tekniğinin geliştirilmesine ilişkin çalışmalar oldukça yenidir (Cortright ve ark.,
2002). Bu tekniği süperkritik ve subkritik su işlemlerinden ayıran özellik kullanılan
sıcaklık (200-250 ºC) ve basıncın (10-50 bar) oldukça düşük sayılabilecek değerlerde
olmasıdır. APR tekniği; sulu fazda, daha düşük sıcaklık ve basınçta gliserol, şeker
ve şeker alkolleri gibi biyokütle kaynaklı bileşikleri katalitik bozundurmaya
uğratarak hidrojen ağırlıklı gaz ürünü elde etmede kullanılmaktadır. Prosesin ılımlı
sıcaklık ve basınçta gerçekleşmesi önemli derecede enerji tasarrufu sağlarken,
karbohidratların yüksek sıcaklıklarda geri dönüşümlü reaksiyonlar aracılığı ile
karbonizasyonu önlenmektedir. Ayrıca, su-gaz dönüşüm reaksiyonu (Water-gas shift
reaction (WGS): CO+ H2O = CO2 + H2) ürünler lehinde ilerleyecek sıcaklık ve
basınçta gerçekleştiğinden ağırlıklı olarak hidrojen oluşmakta, CO’in son gaz
ürününün içindeki oranı ise düşük düzeylerde kalmaktadır (Davda ve ark., 2005).

1. GİRİŞ Mehtap KURTULUŞ
17
Karbonhidratların APR çalışması ile hidrojen üretiminin birçok avantajı
vardır:
• İlgilenilen oksijenli bileşikler yanıcı ve toksik değildir, depolanabilir ve
güvenle taşınılabilir.
• APR, su-gaz değişim reaksiyonlarının uygun olduğu sıcaklık ve basınçta
meydana gelir, bu tek kimyasal reaktör içinde düşük CO içerikli hidrojen gazı
üretimini mümkün kılar.
• APR düşük sıcaklıklarda meydana gelir ki bu karbonhidratların yüksek
sıcaklıklarda bozunmasına neden olan istenmeyen reaksiyonları minimize
eder.
• APR yönteminde biyokütle kurutulmadan kullanılabilir.
Antal ve ark. (1994) öncelikle buhar reformlama yöntemi ile hidrojen üretimi
için potansiyel hammadde olarak glukozu denemişlerdir. Bu reaksiyon sonucunda
hidrokarbon bakımından zengin gaz karışımı elde edilirken hidrojen verimi oldukça
düşük olmuştur. Dumesic ve çalışma arkadaşları, oksijenli hidrokarbonların
bozunması yoluyla hidrojen üretimi için yeni bir yöntem olan APR’yi
geliştirmişlerdir (Dumesic, Cortright, Davda, Huber, Shabaker; 2002, 2003, 2004,
2005). Bu yöntemle C:O oranı 1:1 olan biyokütle türevi oksijenli hidrokarbonları
(metanol, etilen glikol, gliserol, glukoz ve sorbitol) destekli metal katalizörler
kullanarak H2, CO, CO2 ve alkan gaz ürünlerine dönüştürebilmişlerdir.
C:O oranı 1:1 olan biyokütle türevi oksijenli hidrokarbonlardan APR
yöntemiyle hidrojen üretimi aşağıdaki gibi genel bir reaksiyonla gösterilebilir:
CxH2X+2Ox + XH2O (2X+1)H2 + XCO2 Reaksiyon termodinamik olarak H2 ve CO2 üretimine istemli olmasına rağmen
oluşan ürünlerin APR koşullarında ileri reaksiyonla alkan ürünlerine dönüşmesi
mümkündür:
CO2 + H2O CH4 + 2H2O

1. GİRİŞ Mehtap KURTULUŞ
18
Proses, şekerler, şeker alkolleri ve gliserol gibi suda çözünebilen oksijenli
bileşikler gerektirmektedir. Diğer biyokütle bileşenleri (selüloz, hemiselüloz, nişasta)
kullanıldığında, bunların suda çözünebilir hale dönüştürülmesi gerekmektedir.
Bu nedenle APR tekniği suda çözünebilen şeker ve polialkoller ile benzeri
materyalden özellikle hidrojen eldesine yönelik olarak geliştirilmekte olup (Davda ve
ark., 2005; Huber ve Dumesic, 2006) bu konuda iki adet USA patenti alınmıştır (US
Patent Appl. Nos: 0030170171 ve 0050207971).
APR ile suda çözündürülen ürünlerden H2 eldesi amaçlandığında Şekil 1.6’da
görüldüğü gibi I nolu süreç hakim kılınmalı, alkol ve alkanlara giden diğer süreçler
ise uygun katalizör ve proses koşulları kullanılarak en düşük düzeye indirilmelidir.
Şekil 1.6. Etilen glikolden APR ile H2 eldesinde seçiciliği belirleyecek olası tepkime süreçleri (Shabaker ve ark., 2004).
APR sürecinde oksijenli hidrokarbonların moleküler yapılarında C-C, C-H
ve/veya O-H bağlarının kırılması için ilgili kısımların katalizör yüzeylerine uygun
geometri ile adsorbe olmaları gerekmektedir. Moleküler bozunma sırasında oluşan
CO, katalizör yüzeyine yoğun olarak adsorplanırsa katalizörün aktivitesini düşürür.
Hidrojen oluşumuna seçici olan bir katalizör C-C bağlarının kırılmasını aktive
ederken, aynı zamanda su-gazı kayma tepkimesini de aktive ederek katalizör
yüzeyindeki CO’nın ayrılmasını sağlamalıdır. Ayrıca bu katalizörün C-O bağlarının
kırılmasını ve CO ile CO2’nin hidrojenasyonunu aktiflememesi gerekir. C-C bağ

1. GİRİŞ Mehtap KURTULUŞ
19
kırılması Pt, Pd, Rh, Sn, Ni, Co, Cu, Zn ve bunların kombinasyonlarından oluşan
metal yüzeylerinde gerçekleşebilmektedir (Davda ve ark., 2003).
Shabaker ve arkadaşları (2003) karbon siyahı, karbon ve alümina destekli Pt
katalizörlerinin etilen glikol çözeltisinin sulu faz reformlamasında hidrojen
üretiminde etkili katalizörler olduğunu bildirmişlerdir. Aktif karbon, alümina, silika,
zirkonia, TiO2 gibi destekler üzerine tutturulmuş çeşitli kıymetli metal katalizörleri
içinden karbon desteğine yüklenmiş Pt, hidrojen seçiciliği dolayısıyla verimi
açısından en iyi sonucu vermiştir (Huber ve Dumesic, 2005). APR’de kullanılan
metal katalizörlerin aktivitesi kullanılan destek materyaline oldukça bağlıdır (Huber
ve Dumesic, 2005; Meryemoglu ve ark., 2010). Genellikle, SiO2-Al2O3 gibi asidik
destekler daha çok alkan seçici davranırken, Al2O3 ve karbon gibi bazik/nötral
destekler H2 seçici olmaktadır. Çözelti pH’ı nötral olduğunda seçicilik artmaktadır
(Tanksale ve ark., 2010). Şekil 1.6’da görüldüğüi gibi dehidrojenasyon asit
oluşumuna neden olurken, pH’ın düşürülmesi daha fazla alkan oluşumuna neden
olmaktadır.
Buğday samanı hidrolizatının destekli değerli metal katalizörleri varlığında
sulu faz reformlaması çalışmasında hidrojen oluşumu açısından en iyi destek
maddesinin karbon olduğu gözlenmiştir (Meryemoglu ve ark, 2010). Sözkonusu
çalışmada kullanılan değerli metallerin hidrojen üretimindeki aktivite ve seçicilikleri
azalan sıraya göre şu şekilde olmuştur: Pt > Ru > Pd. Benzer sıralama etilen glikolün
silika destekli monometalik Pd, Pt, Ni, Ru, Rh ve Ir katalizörleri varlığında sulu faz
reformlaması çalışmasında da gözlenmiştir (Davda ve ark., 2003). Katalizörün H2-
seçiciliği Pt ve Pd ile birlikte Ni, Co ve Fe’nin alaşım olarak Al2O3 gibi seçiciliği
yüksek destek materyalleri üzerine hazırlanması ile arttırılabilir. Atomik oranları
1:1’den 1:9’a kadar olan PtNi ve PtCo katalizörlerinde etilen glikol reformlamanın
seçiciliğinin arttığı gözlenmiştir (Huber ve ark., 2006).
Metalik nikel ve aluminyumun eritilip soğutulmasının ardından Al’nin
ekstrakte edilmesiyle geriye kalan gözenekli ve nikel içeren atık “raney Ni” diye
adlandırılır (Lei ve ark., 2001). Raney nikel katalizörü APR’de hidrojen oluşumu ve
seçiciliği açısından Pt katalizöründen daha iyi aktivite göstermektedir (Meryemoglu
ve ark, 2010).

1. GİRİŞ Mehtap KURTULUŞ
20
1.6. APR’de Kullanılan Organik Materyalin Niteliğinin Hidrojen Oluşumuna
Etkisi
Hidrojen seçiciliği başlangıç çözeltisi olarak kullanılan materyaldeki
molekülün karbon sayısının artmasıyla azalmaktadır. 498 ve 538 K sıcaklıkta
Pt/Al2O3 katalizörü varlığında glukoz, sorbitol, gliserol, etilen glikol ve metanolle
yapılan APR deneyleri H2 seçiciliği açısından glukoz < sorbitol < gliserol < etilen
glikol < metanol şeklinde etkinlik göstermiştir (Cortright ve ark., 2002).
Alkan seçiciliği ise hidrojen seçiciliğinin tersi yönünde olmuştur. En yüksek
hidrojen verimi sorbitol, gliserol ve etilen glikolün sulu faz reformlamasında elde
edilmiştir. Bu moleküllerin yenilenebilir kaynaklardan türetilebiliyor olmasına
rağmen (Wang ve Deng 2000; Blanc ve ark., 2000) bol bulunması, daha basit ve
doğrudan elde edilebilmesi açısından glukozun bu amaç için kullanılması daha cazip
görülmektedir. Poliollerin kütlece % 1-10 arasındaki konsantrasyonları reformlama
yoluyla yüksek hidrojen veriminde benzer etkinliği gösterirken glukozun
konsantrasyonu kütlece %10’a gittikçe istenmeyen homojen reaksiyonların
gözlenmesinden dolayı hidrojen veriminin düştüğü gözlenmiştir (Eggleston ve
Vercellotti, 2000; Kabyemela ve ark., 1997).
1.7. Şekerlerden Hidrojen Üretimi
Şeker ve şeker alkollerinin sulu reformlaması ile hidrojen üretilebilmektedir
(Tanksale ve ark., 2006, 2007, 2008). Şekerler ve sorbitol gibi şeker alkolleri
doğrudan biyokütlenin hidrolizi sonucu üretilebilir (Bobleter, 1994; Yu ve ark.,
2008). Kompleks kimyasal yapılarından dolayı şekerlerin reformlaması, metanol ve
etilen glikolden daha karmaşıktır ve reaksiyonlarında paralel seçicilik durumları
sözkonusu olabilmektedir. Şekil 1.7’de fruktozun 200ºC’de Pt/Al2O3 katalizörü
varlığında sulu faz reformlamasının olası reaksiyon yollarını göstermektedir
(Tanksale ve ark., 2006). Hidrojenin harcanmasına neden olan reaksiyonlar
istenmeyen reaksiyonlardır, o nedenle kullanılan katalizörler WGS seçici olmalıdır.

1. GİRİŞ Mehtap KURTULUŞ
21
Literatürden bilindiği üzere, C-C bağ kırılması reformlama prosesinde
sınırlayıcı faktördür (Shabaker ve ark., 2004). Aktif metal bölgelerin eksikliğinde
(çalışılan tek materyal alüminada olduğu gibi) fruktoz molekülünün
dehidrojenasyonu C-C bağ kırılmasına neden olmaz (Tanksale ve ark., 2006).
Şekil 1.7. Fruktozun sulu faz reformlama mekanizması
1.8. Biyokütlenin Hidrolizi ve Lignoselülozik Materyallere Uygulanan Ön
İşlemler
Lignoselülozik kütlenin suda çözünmeyişi bu materyallerden yeterince
yararlanamama sorununu ortaya çıkarmaktadır. Teknolojik gelişmelere bağlı olarak
biyokütleden faydalanma şekli zamanla değişmiş ve karmaşık yeni prosesler
geliştirilmiştir. Lignoselülozik materyallere uygulanan ön işlemler uzun zamandır
bilinen yöntemlerdir (McMillan, 1994). Bu ön işlemlerin amacı, lignin ve
hemiselülozu bozundurmak, selüloz kristalliğini azaltmak ve gözenekliliği
arttırmaktır. Ön hazırlığın şu gereksinimleri karşılaması gerekir: (1) Enzimatik
hidroliz ile şeker oluşumunu veya akabinde şeker oluşturma yeteneğini hızlandırmak,
(2) Karbonhidratların kaybını ve bozunmasını önlemek, (3) Hidroliz ve fermentasyon
prosesleri sonrasında engelleyici yan ürünlerin oluşumunu önlemek ve (4) Makul
maliyette olmak.

1. GİRİŞ Mehtap KURTULUŞ
22
Lignoselülozik materyallerin ön işlemi için fiziksel, fizikokimyasal, kimyasal
ve biyolojik prosesler kullanılmaktadır.
1.8.1. Fiziksel Ön İşlemler
1.8.1.1. Mekanik Parçalama
Atık materyaller, selüloz kristalliğini azaltmak için yontma, öğütme ve ezme
(presleme) kombinasyonları ile parçalanabilirler. Materyallerin boyutu yontma
işleminden sonra genellikle 10-30 mm, öğütme ve ezme işlemlerinden sonra ise 0.2-2
mm arasındadır. Titreşimli top öğütmesinin amacı; selüloz kristalliğini kırarak
biyokütlenin sindirilebilirliğini geliştirmektir ve bu nedenle sıradan top
öğütmesinden daha etkili olmaktadır (Millet, 1976). Tarımsal malzemelerin mekanik
parçalanmasının zorluğu, son tanecik boyutu ve atık biyokütle karakteristiklerine
bağlıdır (Cadoche ve Lopez, 1989).
1.8.1.2. Piroliz
Piroliz de lignoselülozik materyallerin ön işlemleri için kullanılır.
Lignoselülozik materyaller 300ºC’den daha yüksek sıcaklıklarda işlem gördükleri
zaman, selüloz hızlıca ayrışarak gaz ürünleri ve kömür kalıntısı oluşturur (Kilzer ve
Broido, 1965; Shafizadeh ve Bradbury, 1979). Bu ayrışma çok yavaştır ve daha
düşük sıcaklıklarda daha az uçucu ürünler oluşturulur. Süreç, oksijenin varlığıyla
zenginleştirilebilir (Shafizadeh ve Bradbury, 1979). Çinko klorür veya sodyum
karbonat, bir katalizör olarak eklendiği zaman, saf selülozun ayrışması, daha düşük
bir sıcaklıkta meydana gelebilir (Shafizadeh ve Lai, 1975).
1.8.2. Fizikokimyasal Ön İşlemler
1.8.2.1. Buhar Patlaması (Steam Explosion)
Lignosellülozik materyallere uygulanan en yaygın önişlemdir (McMillan,
1994). Küçük parçalara ayrılmış biyokütle yüksek basınçtaki doygun buhar ile

1. GİRİŞ Mehtap KURTULUŞ
23
muamele edilir. Daha sonra basınç süratle azaltılarak materyallerin patlayıcı
dekompresyonu sağlanır.
Buhar patlaması genellikle 160-260ºC’de ve 0,69-4,83 MPa basınçta
başlatılır. Bu koşullarda birkaç dakika ya da daha az sürede tutulduktan sonra
materyal atmosferik basınca maruz bırakılır. Bu işlem yüksek sıcaklıktan dolayı
hemiselülozun parçalanmasına ve ligninin dönüşümüne sebep olur ve böylece selüloz
hidrolizi artar. Buhar patlaması ön işleminin etkinliği zamana, sıcaklığa, biyokütle
parçacıklarının büyüklüğüne ve nem içeriğine bağlı olarak değişir. Optimal
hemiselüloz solubilizasyonu ve hidrolizi ya yüksek sıcaklık-kısa zaman (270ºC, 1
dk) ya da düşük sıcaklık-uzun zaman (190ºC, 10 dk) uygulaması yapılarak
sağlanabilir. Yapılan çalışmalar düşük sıcaklık-uzun zaman koşullarının daha etkili
olduğunu göstermiştir.
Buhar patlaması ön işleminin avantajları, mekanik parçalama ile
karşılaştırıldığında % 70 daha az enerji gerektirmesidir (Holtzapple ve ark., 1989).
Bu yöntem, en ekonomik önişlem olarak ziraati atıklara ve sert odunlara başarıyla
uygulanmış ancak yumuşak odunlarda daha az etkin hidroliz elde edilmiştir (Clark
and Mackie, 1987).
1.8.2.2. Amonyak Lif Patlaması (Ammonia Fiber Explosion, AFEX )
AFEX, belli bir zaman periyodunda lignoselülozik materyallerin yüksek
sıcaklık ve basınçta sıvı amonyağa maruz bırakılıp ardından basıncın hızlıca
azaltıldığı fizikokimyasal ön işlemlerin bir çeşididir. Tipik bir AFEX prosesi 1-2 kg
amonyak/kg kuru biyokütle, 90ºC sıcaklık ve 30 dk süre koşullarında yapılır.
AFEX önişlemi çeşitli otsu bitkilerin sakkarizasyon işlemini önemli derecede
hızlandırmıştır. Alfalfa, arpa-buğday-pirinç samanı, tahıl kabuğu, mısır koçanı,
şehirsel katı atıklar gibi çeşitli lignosellülozik materyaller bu yöntemle hidroliz
edilebilmiştir (Vlasenko, 1997; Mes-Hartree, 1988). AFEX, asitli önişlemlere göre
hemisellülozü daha az çözündürür.
Yapılan bir çalışmada, buğday samanı ve alfalfa yapraklarının enzimatik
hidrolizi için buhar ve amonyak önişlemleri karşılaştırılmış ve bu biyokütlelerdeki

1. GİRİŞ Mehtap KURTULUŞ
24
hemiselüloz fraksiyonlarının AFEX ile çözünmediği ancak buhar patlamasında
çözündüğü görülmüştür (Mes-Hartree ve ark., 1988). Öte yandan %5 oranında lignin
içeren Bermuda otuna AFEX uygulaması ile selüloz ve hemiselülozun %90’nı
hidroliz edilmiştir (Holtzapple ve ark., 1991). AFEX prosesi lignin içeriği yüksek
olan biyokütlelere uygulandığında düşük hidroliz verimi elde edilmiştir.
1.8.2.3. CO2 Patlaması (CO2 Explosion)
Buhar ve amonyak lif patlaması yöntemine benzerdir. CO2 patlaması da
lignoselülozik maddelerin ön işlemlerinde kullanılır. Bu yöntemde karbonik asit
oluşumuyla hidroliz hızı arttırılır. Dale ve Moreira (1982) alfalfa bitkisine
karbondioksit patlaması ön işlemini (5,62 MPa basıncında 4 kg CO2/kg lif)
uygulamışlar ve enzimatik hidrolizde 24 saat boyunca serbest bırakılan teorik
glikozun %75i’ni elde etmişlerdir. Ürün verimliliği göreceli olarak buhar ve
amonyak patlamaları ön işlemleri ile karşılaştırıldığında düşük, enzimatik hidroliz ön
işlemi ile karşılaştırıldığında yüksektir.
Hidroliz verimi buhar ve amonyak explosion yöntemlerine göre düşüktür.
Recycle edilmiş kağıt karışımı ve şeker kamışı bagasının CO2 explosion ile hidrolizi,
buhar ve amonyak explosion yöntemleri ile karşılaştırıldığında amonyaktan daha
maliyetli olduğu ve biyolojik işlemleri inhibe edecek ürünlerin buhar explosionda
oluşurken CO2’de oluşmadığı gözlenmiştir (Zheng, 1998).
1.8.2.4. Subkritik Su ile Hidroliz
Lignoselülozik biyokütlelerin hidroliz işlemlerinde konsantre asit ya da baz
kullanılması çevre açısından hiç istenmeyen teknolojilerdir. Buna karşılık süperkritik
suda hidroliz son derece hızlı ve ek kimyasal gerektirmeyen bir işlemdir. Sıcaklık ve
basıncın ekonomik düşüncelerle daha düşük tutulması ile subkritik bölgede çalışmak
mümkündür. Su subkritik koşullarında (100ºC < T < 374,2ºC ve basınç, suyu sıvı
halde tutacak kadar yüksek), biyokütle hidrolizi için etkili bir tepkime ortamı yaratır.
Subkritik suyun, normal sudan daha düşük bir dielektrik sabiti ve daha yüksek
iyonlaşma çarpım sayısı (ion product number) vardır. Suyun sıcaklığı, oda

1. GİRİŞ Mehtap KURTULUŞ
25
sıcaklığından 250 ºC’ ye arttırıldığı zaman göreli dielektrik sabiti yaklaşık 80’den 27
civarına düşer, bu durum suya asetona benzer bir özellik kazandırır. Ayrıca, subkritik
suyun iyonlaşma ürünü sayısı sıcaklıkla artar. Bu nedenle hidroliz ve bozunma gibi
kimyasal reaksiyonlar subkritik ortamda herhangi bir katalizör olmadan
gerçekleşebilir. (Rogalinski, 2005).
1.8.3. Kimyasal Ön İşlemler
1.8.3.1. Ozon
Ozon; buğday samanı (Ben-Ghedalia ve Shefet, 1981), küspe, yeşil ot, fıstık,
çam (Neely,1984), pamuk samanı (Ben-Ghedalia ve Shefet,1983), kavak talaşı
(Vidal ve Molinier, 1988) gibi birçok lignoselülozik materyallerden lignin ve
hemiselülozu ayrıştırmada kullanılmıştır. Bu ayrıştırma aslında lignin ve
hemiselüloza karşı sınırlıdır, selülozu etkilemesi daha zordur. Ozonlama ön
işleminin, bazı avantajları vardır; (1) etkili olarak lignin bozundurulur (2) takiben
uygulanacak biyolojik süreçler için zehirli artıklar üretmez, (3) tepkimeler oda
sıcaklığı ve basıncında tamamlanır (Vidal ve Molinier, 1988). Ozon ön işlemi;
ligninin etkili uzaklaştırılması, toksik atıkların oluşmaması ve reaksiyonun oda
sıcaklığında ve atmosfer basıncında gerçekleşmesi açısından avantajlıdır ancak
yüksek miktarda ozon kullanımını gerektirmesi prosesi pahalı yapmaktadır.
1.8.3.2. Asit ile Hidroliz
Lignoselülozik materyallerin işlenmesinde konsantre H2SO4 ve HCl
kullanılmaktadır. Bunlar selüloz hidrolizinde etkili kimyasallardır ancak toksik,
korozif, tehlikeli olmaları ve korozyona dayanıklı reaktörlerin kullanılma
zorunluluğu dezavantaj oluşturur. Ekonomik açıdan hidrolizden sonra kuvvetli asit
geri kazanılmalıdır.
Seyreltik asit hidrolizi uygulaması ile de selüloz hidrolizi önemli derecede
sağlanabilmiştir. (Esteghlalian, 1997). Ancak, selülozun glukoza makul derecede
dönüşüm hızını elde etmek için yüksek sıcaklıklar gerekmektedir (McMillan, 1994).

1. GİRİŞ Mehtap KURTULUŞ
26
Yüksek sıcaklık hemiselüloz şekerlerinin dekompozisyon hızını ve aynı zamanda
teçhizat korozyonunu arttırır. Yüksek sıcaklık ve düşük maruziyet zamanında glukoz
verimi maksimum olmakta ancak bu bile teorik glukoz veriminin %50’si ila %60’ı
arasına düşmektedir.
Şeker degradasyonunu azaltmak için iki aşamalı hidroliz prosesi
geliştirilmiştir. İlk aşamada ılımlı koşullarda (170-190°C) hemiselüloz fraksiyonu
hidroliz edilir ardından daha sert koşullarda (200-230°C) selüloz hidrolizi
gerçekleştirilir. İki aşamalı asit hidrolizi ile yumuşak odundan %70-98 verimle
ksiloz, galaktoz, mannoz ve arabinoz elde edilebilmiştir fakat glükoz verimi yine
düşük olmaktadır (%50).
Genel olarak, selülozun asitlerle hidrolize edilmesinde en büyük sorun, yoğun
hidrojen bağları ile sıkışık haldeki kristalin bölgelerin asitlerle reaksiyonunun, amorf
bölgelere ve hemiselülozlara göre daha zor olması olarak ifade edilebilir. Bu
nedenle, asitlerle selülozun hidroliz edilmesi, kristallik derecesi ile yakından
ilgilidir. Asitlerin selülozu parçalaması (hidroliz etmesi) konsantrasyona bağlı olarak
genellikle iki aşamada olur. İlk aşamada asitler, kolayca ulaşabildiği amorf bölgeleri
parçalar ve uzaklaştırır. Amorf bölgesi uzaklaşan selüloz hidroselüloz olarak
isimlendirilir. Bu nedenle bozulmadan kalan selülozun kristallik derecesi artar.
Derişik asitlerin kullanılması ve reaksiyon süresinin uzatılması sonucu selüloz
monomerik yapı taşı olan glikoza dönüşebilir. Tipik olarak, odunların asitlerle
hidrolizasyona uğratılmasıyla selülozdan %90 saflıkta glikoz elde edilebilir.
Seyreltik asit uygulaması selüloz hidrolizini önemli derecede arttırır ancak
buhar ve amonyak uygulamasından daha pahalıdır ve enzimatik hidroliz ve
fermentasyon prosesinden önce pH’ın nötral yapılması gerekmektedir.
1.8.3.3. Alkali ile Hidroliz
Bazı bazlar da lignozelülozik materyallerin ön işlemleri için kullanılabilir ve
bazik ön işlemin etkisi kullanılan materyalin lignin içeriğine bağlıdır (Fan ve ark.,
1987; McMillan, 1994). Alkali ile hidroliz mekanizmasında çaprazbağlı ksilan
hemiselülozlardaki ve diğer bileşenlerdeki (lignin ve diğer hemiselülozlar)

1. GİRİŞ Mehtap KURTULUŞ
27
intermoleküler ester bağlarının sabunlaştığı düşünülmektedir. Lignoselülozik
materyallerin porozitesi çapraz bağların uzaklaştırılmasıyla artar (Tarcow ve Fiest,
1969).
Seyreltik NaOH uygulaması, lignoselülozik materyallerin şişmesine, iç yüzey
alanının artmasına, polimerizasyon derecesinin azalmasına, kristalliğin azalmasına,
lignin-karbohidrat arasındaki yapısal bağların kırılmasına ve lignin yapısının
bozulmasına sebep olmaktadır (Fan ve ark., 1987). Sert odunun NaOH ile
bozundurulması lignin içeriği azaldıkça artmıştır. Ancak, seyreltik NaOH
uygulaması lignin içeriği %26’dan fazla olan yumuşak odunlarda etkili olamamıştır.
Yüzde 10-18 oranında düşük lignin içeriğine sahip samanların hidrolizinde de NaOH
etkili olmuştur. Baz ile hidroliz prosesinde amonyak ta lignini uzaklaştırmak için
kullanılmaktadır. Amonyağın tekrar dönüşümünün sağlandığı ve 170 ºC’de % 2,5-20
amonyak konsantrasyonunda ve 1 saat işlem şeklinde uygulanan bir proseste mısır
koçanının lignini %60-80 azaltılmış; aynı işlemle switchgrass bitkisinin lignini ise %
65-85 uzaklaştırılmıştır.
1.8.3.4. Oksidatif Yolla Ligninin Uzaklaştırılması
H2O2 lignoselülozik materyallerin ön işleminde enzimatik hidrolizin
hassasiyetini oldukça artırmaktadır. Ligninin biyolojik bozunması H2O2 varlığında
peroksidaz enzimi ile katalizlenir (Azzam, 1989). Şeker kamışı bagasının hidrojen
peroksitle etkileştirilmesi sonucunda enzimatik hidroliz önemli derecede
arttırılmıştır. % 2 H2O2 kullanılarak 30 ºC’de 8 saat işlem sonucunda, hemiselülozun
büyük bir kısmı, ligninin ise yaklaşık % 50’si solubilize olmuştur. Daha sonra
uygulanan selülazla sakkarizasyon işleminde (45 ºC, 24 saat) selülozdan %95
verimle glukoz elde edilmiştir.
1.8.3.5. Organosolv Prosesi
Bu yöntemde, lignin iç yapısı ve hemiselüloz bağları inorganik asit
katalizörleri varlığında (HCl veya H2SO4) organik solvent veya sulu organik solvent
kullanılarak kırılır. Bu amaçla metanol, etanol, etilen glikol ve tetrahidrofurfuril alkol

1. GİRİŞ Mehtap KURTULUŞ
28
kullanılır (Chum, 1988; Thring, 1990). Organosolv prosesinde okzalik asit,
asetilsalisilik ve salisilik asit gibi organik asitler de kullanılabilir (Sarkanen, 1980).
Yüksek sıcaklıklarda (185 ºC’nin üzerinde) çalışılacaksa delignifikasyon için
katalizör ilavesine gerek yoktur (Sarkanen, 1980; Aziz ve Sarkanen, 1989). Asit
ilavesiyle genellikle yüksek verimde ksiloz elde edilebilir. Enzimatik hidroliz ve
fermentasyon aşamalarında inhibisyon etkisi yaratacağından kullanılan solvent işlem
sonunda reaktörden alınmalıdır. Maliyeti düşürmek açısından solvent gerekirse
recycle edilebilir.
1.8.4. Biyolojik Ön İşlemler
Biyolojik ön işlem proseslerinde atık materyallerdeki lignin ve hemiselülozu
parçalamak için kahverengi-, beyaz- ve yumuşak- küf mantarları kullanılır (Schurz,
1978). Kahverengi küf mantarları, esas olarak selüloza atak ederken, beyaz ve
yumuşak küf mantarları selüloz ve lignine atak eder.
Lignoselülozik materyallerin biyolojik önişlemlerinde, beyaz-rot fungiler en etkili
Basidiomycetes (küf mantarları/şapkalı mantarlar)’dır (Fan et al., 1987). Yapılan bir
çalışmada, buğday samanının 19 beyaz-rot fungisiyle önişlem etkinliği araştırılmış
ve samanın %35’i Pleurotus ostreatus ile 5 haftada indirgen şekerlere
dönüştürülebilmiştir. Benzer dönüşüm Phanerochaete sordida 37 ve Pycnoporus
cinnabarinus 115 ile 4 haftada elde edilmiştir (Hatakka, 1983).
1.8.4.1. Enzimatik Hidroliz
Selülozun enzimatik hidrolizi oldukça spesifik olan selüloz enzimleri
tarafından gerçekleştirilir (Beguin ve Aubert, 1994). Enzimatik hidroliz asidik ve
bazik hidroliz ile karşılaştırıldığında verimi düşüktür. Enzim hidrolizi genellikle
ılımlı koşullarda (pH 4.8 ve sıcaklık 45-50oC) yürütülür ve bir korozyon problemi
oluşturmaz (Duff and Murray, 1996). Selüloz zincirleri selüloz enzimleri tarafından
glikoz moleküllerine kırılabilir. Polifenol oksidaz, lakkaz ve kinon indirgeyici
enzimler de lignini parçalayabilmektedir. Biyolojik önişlemlerin avantajları olarak

1. GİRİŞ Mehtap KURTULUŞ
29
düşük enerji ihtiyacı ve ılımlı çevre koşulları sayılabilir. Ancak, birçok biyolojik
proseste hidroliz hızı oldukça düşüktür.
1.9. Moleküler Kütle Belirleme Teknikleri
1.9.1. Boyut Eleme Kromatografisi (SEC)
Boyut Eleme Kromatografisi (Size Exlusion Chromatography; SEC), farklı
büyüklükteki moleküllerin birbirinden ayrılması esasına dayanır. Büyük moleküller
önce elue olurken, küçük moleküller daha geç kolondan çıkarlar. Endüstriyel
polimerlerin karakterizasyonu için kullanıldığında “Jel Geçirgenlik Kromatografisi”
(Gel Permeation Chromatography; GPC) adını alırken, proteinler ve polisakkaritler
gibi biyolojik polimerler için kullanıldığında “Jel Filtrasyon Kromatografisi” (Gel
Filtration Chromatography; GFC) adını da alabilmektedir. Boyut eleme
kromatografisinde ideal dolgu malzemeleri analit molekülleri ile adsorptif bir
etkileşme göstermezler. Bundan dolayı SEC’in yönlendirici mekanizması dolgu
materyalinin gözeneklerinde analit moleküllerinin sterik dışlanmalarıdır. Ayırma
aralığı bir kolonun gözenek hacmi ile sınırlı olduğundan, dolgunun spesifik boşluk
hacmi (doldurma yoğunluğu ile birlikte) bir dolgunun performansını belirler. Farklı
dolguların seçicilikleri, dolgunun ortalama gözenek büyüklüğü ve gözenek
büyüklüğü değişimi tarafından belirlenir. Sulu çözeltilerdeki polar polimerlerin SEC
uygulamalarında dolgu malzemesi olarak metakrilat bazlı polar dolgular kullanılır.
Silikadan türevlendirilmiş “diol” dolgu malzemeleri de proteinler ve diğer polar
polimerlerin ayırımlarında kullanılmaktadır (Column Handbook for Size Exclusion
Chromatography, 1999).
1.9.2. Polimerlerin Molekül Ağırlıklarının GPC Yöntemi ile Belirlenmesi
Polisakkaritler (dekstranlar, selüloz, hemisellüloz v.b) farmakoloji, gıda ve
tekstil alanında kullanılan önemli bileşenlerdir. Bu moleküllerin farklılığını ve
olağanüstü karmaşıklığını anlatabilecek bir tek genel bir teori bulunmamasına
rağmen polisakkaritler bütün biyolojik sistemlerde temel rol oynarlar (Varki, 1993).

1. GİRİŞ Mehtap KURTULUŞ
30
GPC, hidrodinamik hacmi elüsyon hacmiyle ilişkendiren üniversal
kalibrasyon görüşüne dayalı (Grubisic ve ark., 1967) hesaplamalarla molekül ağırlığı
dağılımı yapar. Hidrodinamik hacim, çözeltideki etkin polimer boyutunu dikkate alır
ve bunun farklı kimyasal kompozisyona sahip polimerlerin elüsyon hacmiyle lineer
olduğunu kabul eder. Bu kabul özellikle analiz edilecek spesifik polimerlerin bir
standardı olmadığında faydalı olmaktadır.
Önceki çalışmalarda polisakkarit ve lignin türevi polimerlerin moleküler
kütlesini belirlemede kullanılan iki tür standardın olduğu görülmektedir. Bunlar
polistiren ve dekstrandan ibaret olan kitlerdir. Polistirenin yapısı şekil 1.8’de
gösterilmiştir.
Şekil 1.8. Polistiren yapısı
Striegel ve Timpa (1995), eklem bacaklıların üzerini kaplayan örtü şeklindeki
kabuğun yapısını oluşturan, bitkiler, funguslar ve bira mayasının hücre duvarında
bulunan, N-asetil glikozaminden meydana gelmiş, kimyasal olarak nötr bir
polisakkarit olan sitini (chitin) lityum klorürlü aprotik solvent N,N-dimetilasetamit
içerisinde (Me2NAc-LiCl) çözerek bu polisakkaritin çözünürlük parametrelerini ve
fiziksel özelliklerini belirlemişlerdir. Moleküler kütlesini belirlemede polistiren
standartları kullanmışlardır.
Fahmi ve ark. (2008), düşük lignin içerikli Lolium Festuca otunun pirolizi ve
piroliz sonrası oluşan sıvının stabililitesi ve kalitesini belirlemek için analizler
yapmışlardır. Ligninden türeyen yüksek molekül ağırlıklı bileşenleri polistiren
standartları kullanılarak GPC ile belirlemişlerdir. Bu amaçla 161-72200 g/mol molar
kütlelerine sahip polistiren standartları refraktif indeks ve UV fotometre (280 nm)
olmak üzere iki dedektör kullanılarak analiz edilmiştir. Data değerlendirilmesi

1. GİRİŞ Mehtap KURTULUŞ
31
ortalama molar kütle sayısı (Mn), ortalama molar kütle dağılımı (Mw) ve refraktif
indeksten hesaplanan polidispersitiy indeksi sayısı (PD=Mw/Mn) verileri
kullanılarak GPC software ile yapılmıştır.
Dekstranlar ise farmokoloji, fotografik, tarım ve gıda sanayi gibi çeşitli
alanlarda kullanılmaktadır. Dekstranın yapısı şekil 1.9’da gösterilmiştir.
Şekil 1.9. Dekstran genel yapısı
Dekstranların bu kadar çok yönlü kullanımını sağlayan özellikleri:
• Doğaldır ve suda çözünebilir,
• Kolayca filtrelenebilir,
• Biyolojik olarak uygundur,
• Uzun süre kararlılığını koruyabilmektedir.
Dekstran α-D-1,6-glukoz bağlı glukanlardan oluşmuştur. Dekstran
biyopolimerinin ana zincirine 1-3 yan zincirleriyle bağlanmıştır. Dallanma derecesi
yaklaşık %5 ‘tir. Dallanmalar daha çok 1-2 glukoz birimlerinden oluşmuştur.
Dekstran fraksiyonlarının molekül ağırlıkları 1000 Da ile 2 000 000 Da
arasında olabilmektedir.
Dekstran 5, dekstran 10 gibi gösterimler moleküler kütlenin 1000 Da’na
bölündüğünü işaret eder. Örneğin 10 moleküler kütlesi 10 000 Da anlamına gelir.
Dekstran fraksiyonları suda ve elektrolit çözeltilerde çözünebilir, kararlı, berrak
çözeltiler oluştururlar. pH değişiminden belirgin bir şekilde etkilenmezler. Dekstran
fraksiyonlarıyla çalışılırken derişik çözeltilerin hazırlanması (>%50) önerilir. GPC
yönteminde moleküler kütleyi belirlemede kullanılan 10 adet dekstran standardı
mevcuttur. Bu standartlar 1000 Da’dan 670 000 Da’na kadar olan moleküler kütleyi

1. GİRİŞ Mehtap KURTULUŞ
32
kapsamaktadır. GPC standartları Mw (ağırlıkça moleküler kütlelerinin ortalaması),
Mn (sayıca moleküler kütlelerinin ortalaması), Mp (piklerin moleküler kütlelerinin
ortalaması) değerlerini içeren özellikleriyle verilir.

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
33
2. ÖNCEKİ ÇALIŞMALAR
Lv ve ark., (2009), hem doğal ve hem de asit ile etkileştirilmiş biyokütleyi
alkali çözeltiler kullanmadan piroliz ve gazlaştırma işlemlerine tabii tutmuşlar ve
sonuçlarını termogravimetrik (TGA) yöntem ile incelemişlerdir. Bunun için fixed-
bed tipi reaktör kullanmışlardır. Bu çalışmada altı farklı biyokütle tipi ve
biyokütledeki selüloz ve lignin bileşenlerini incelemişlerdir. Yapılan TGA
deneylerinde biyokütlenin piroliz ve gazlaştırma sonuçlarının biyokütle türüne ve
bileşenlerinin oranlarına bağlı olduğunu bulmuşlardır. Biyokütle pirolizi sırasında
selüloz bileşimi arttıkça tar ve gaz ürünlerinde artış gözlenirken karbonlaşma
ürünleri azalma göstermiştir. Selüloz bileşimi yüksek olduğu zaman piroliz hızı
artarken lignin bileşimi yüksek olduğu zaman düşmektedir.
Azadi ve ark., (2008), glukozun nikel (II) asetilasetonat, (Ni(acac)2), kobalt
(II) asetilasetonat, (Co(acac)2), demir (III) asetilasetonat, (Fe(acac)3) ve raney Ni
varlığında glukozun hidrotermal gazlaştırılmasını çalışmışlardır. Reaksiyon sıcaklığı
310-350°C ve reaksiyon basıncı 100-210 bar olarak seçilmiştir. Gaz miktarı üzerine,
glukozun kompozisyonun yanı sıra katalizör, sıcaklık, zaman, konsantrasyon ve sıvı
miktarının etkisi araştırılmıştır. Sonuçlar, organometalik bileşiklerin glukozun
bozunma hızının belli belirsiz kolaylaştırdığını göstermiştir. Reaksiyon hızındaki bu
artışın hammaddenin düşük konsantrasyonları (~0.06 M) ile karşılaştırıldığında daha
yüksek konsantrasyonlarda daha fazla olduğu söylenebilir. Homojen katalizörlere
karşın raney Ni katalizörü gaz verimini geliştirmiştir. Deney süresi 3 dakikadan 30
dakikaya çıkarıldığında gaz miktarının neredeyse 2 katına çıktığı gözlenmiştir fakat
30 dakikadan 60 dakikaya çıkıldığında önemli bir değişikliğin olmadığı görülmüştür.
Wen ve ark., (2008), gliserolden sulu faz reformlama yöntemi ile hidrojen
gazı üretimini çalışmışlardır. Pt, Ni, Co, ve Cu katalizörlerinin bu yöntemle
aktiviteleri ve kararlılıkları incelenmiştir. Destek materyalinin katalizörün aktivitesi
ve kararlılığına etkisini anlayabilmek için fixed-bed-flow tipi reaktör kullanmışlardır.
Hem işlem öncesi hem de işlem sonrasında katalizörler XRD ile karakterize
edilmiştir. Metal katalizörlerin aktivitesinin Co, Ni, Cu ve Pt sırasına göre arttığı
bulunmuştur. Pt katalizörünün farklı destek materyalleri üzerindeki etkisi şu şekilde

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
34
bulunmuştur: SAPO-11<AC(aktif karbon)˂HUSY˂SiO2˂MgO<Al2O3 Ayrıca ana
destek materyali kullanıldığı zaman yüksek aktivite ve daha yüksek hidrojen
konsantrasyonu gözlenmektedir.
Lv ve ark., (2007), biyokütlenin gazifikasyonundan metanol, dimetil eter gibi
sıvı yakıt sentezinde kullanılacak sentez gazı (syngas) üretimi için yöntem
geliştirmeye çalışmışlardır. Bu amaçla çam talaşı biyokütlesi kullanılmıştır.
Gazlaştırma işlemi akışkan ve sabit yataklı reaktörlerde dolomit ve nikel bazlı
katalizörler kullanılarak gerçekleştirilmiştir. Çam talaşı biyokütlesinin 1,87 ile 4,45
arasında değişen, oldukça yüksek H2/CO oranında gazlaştırılması sağlanmıştır. 750
°C’deki gazlaştırma işleminde katalizör miktarının arttırılması H2/CO oranın da
artmasına sebep olmuştur.
Wei ve ark., (2007), baklagiller samanı ve çam talaşı biyokütlelerinin 750 ila
850°C’de buhar gazifikasyonunu çalışmışlardır. Gazlaşmanın etkinliğine reaktör
sıcaklığının, buhar/biyokütle oranının ve katalizörün (dolomit, kireç taşı, olivine)
etkisi incelenmiş; oluşan gaz komposizyonu belirlenmiştir. Maksimum gaz verimi ve
hidrojen gazı konsantrasyonu buhar/biyokütle oranının 0,6 g/g olduğu koşullarda
gözlenmiştir. Gaz verimi ve H2 konsantrasyonu reaktör sıcaklığının arttırılmasıyla
artmış, bunun yanında katran, kömürleşmiş materyal (char), CO ve CH4
konsantrasyonları azalmıştır. Dolomit katalizörü katran oluşumunu yıkması
açısından iyi performans göstermiş ve kısa süreli katalizör-buhar temasında gaz
üretimi artmıştır.
Gray ve ark., (2007), 180 °C suda ticari ksilanın ve mısır hasatından sonra
kalan atık biyokütlenin hidrolizinden elde edilen oligomerlerin çözünürlüğünü
araştırmışlardır. Huş ağacı ve “oat spelt” ksilanları 80°C’den 26 °C’ye
soğutulduğunda yüksek polimerleşme derecesine sahip oligomerlerin çöktüğü
gözlenmiştir. Çözelti 80 °C’de 10 seyreltme faktörü ile seyreltildiğinde dahi 26
°C’ye soğutulduğunda yine çökeltiler oluşmuştur. Daha düşük hemiselüloz
fraksiyonu içermesinden dolayı mısır atık biyokütlesinden elde edilen hidroliz
çözeltisinde ticari ksilanlara göre daha az yüksek polimerleşme derecesine sahip
oligomerlerin çöktüğü gözlenmiştir. Düşük polimerleşme derecesine sahip
oligomerlerin yüksek polimerleşme derecesine sahip olanlara oranı

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
35
karşılaştırıldığında çözünen oligomerlerin ortalama polimerleşme derecesinin
reaksiyon arttıkça azaldığı saptanmıştır.
Hsieh, Tseng ve ark. (2006), araştırmalarında Ganoderma lucidum’un
polisakkaritlarinin farklı ortamlardaki oluşumunu ve hücre büyümesini anlamaya
çalışmışlardır. Bu çalışmada, üç farklı derişimde karbon, azot, fosfat, magnezyum
kaynağı ve çözünmüş oksijen Ganoderma lucidum’u geliştirmek ve hücre
büyümesini, derişimini ve polisakkaritlerin molekül ağırlığını değerlendirmek için
kullanılmıştır. Glukoz derişimi 60 g/l’den 20 g/l’ye düştüğü zaman polisakkarit
derişimi 1,79 g/l’den 0,91 g/l’ e düşmüştür. En yüksek spesifik polisakkarit oluşumu
0,299 g/g hücreli 60 g/l glukozda bulunmuştur. En düşük molekül ağırlığı ise karbon
kaynaklı çalışmada bulunmuştur. Azot kaynağı altında hücrenin derişimi düşük iken,
hem polisakkarit oluşumu hem de spesifik polisakkarit oluşumu en yüksek çıkmıştır.
Polisakkaritin en düşük molekül ağırlığı azot kaynağı altında bulunmuştur. Hem
fosfat hem de magnezyum kaynaklı çalışma düşük hücre büyümesini göstermiştir.
Hem düşük polisakkarit oluşumu hem de polisakkaritin düşük molekül ağırlığı fosfat
kaynaklı işlemde elde edilmiştir. Magnezyum kaynaklıda polisakkarit oluşumu düşük
iken, moleküler kütle daha yüksek çıkmıştır. En iyi polisakkarit oluşumu, ilk beş
günlük gelişim için, yeterli oksijen desteği ile sağlanırken, sonrasında diğer beş
günlük gelişim için oksijen miktarı değiştirilmiştir. Polisakkaritin en yüksek molekül
ağırlığı fermantasyon prosesinde oksijen sınırlaması ile sağlanmıştır. Bu çalışmada
fermantasyondaki glukoz iyon değiştirici kolona sahip kırılma indisi dedektörlü bir
HPLC sistemi ile belirlenmiştir. Polisakkaritlerin molekül ağırlıkları emniyet ve
analitik kolonlu boyut-eleme kolonları kullanılarak GPC ile belirlenmiştir.
Buğday samanı farklı oranlarda asetik asit, formik asit, etanol, metanol ve su
karışımlarında % 0,1 HCl katalizörü varlığında 85°C’de ısıtıldığında hemiselüloz ve
lignin yapıları ayrılabilmektedir (Xu ve ark., 2006). Elde edilen filtrattan hemiselüloz
% 95 etanolle çökerken çökmeyen kısım lignin fraksiyonunu oluşturmaktadır.
Formik asit-asetik asit-su (30/60/10; v/v/v) karışımı ile buğday samanındaki orjinal
ligninin % 94’ü, hemiselülozun ise % 76,5’i elde edilmiştir. Hemiselüloz
fraksiyonunun asit hidrolizi yaklaşık % 74 oranında ksiloz vermiştir.

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
36
Tanksale ve ark., (2006), glukoz ve fruktozdan meydana gelen sukroz
disakkariti ile glukoz ve fruktoz monoşeker karışımını Pt/Al2O3 katalizörü varlığında
batch tipi bir reaktörde 185, 200 ve 220 ºC’de 25-30 bar basınç altında ayrı ayrı
gazlaştırmışlardır. Hidrojen gazı oluşumu sıcaklık arttıkça artmıştır. Glukoz ve
fruktoz monoşeker karışımından hidrojen oluşumu sukroza göre daha yüksek
olmuştur. Sukrozda iki monoşekeri bağlayan C-O bağının oluşu gazlaştırma hızını
azaltmıştır.
Williams ve Onwudili (2006), selüloz, nişasta ve glukoz model bileşikleri ile
cassava atık biyokütlesinin subkritik ve süperkritik su ortamındaki gazifikasyonunu
batch tipi bir reaktörde çalışmışlardır. Üretilen temel gazların karbon dioksit, karbon
monoksit, hidrojen, metan ve diğer hidrokarbonlardan oluştuğu bildirilmiştir.
Selülozun kömürleşmesinin nişasta ve glukoza göre daha fazla gerçekleştiği, karbon
monoksit ve C1-C4 hidrokarbonların daha yüksek verimde oluştuğu gözlenmiştir.
Buna karşılık glukozun gazifikasyonu ile en yüksek verimde hidrojen elde edilmiştir.
Cassava biyokütle atığı nişasta ile benzer oranda kömürleşme göstermiş, ancak
hidrojen verimi daha az olmuştur.
Miyazawa ve ark. (2005), agar, nişasta ve ksilan gibi polisakkaritleri, küçük
bir batch reaktörde karbondioksit ile ve karbondioksitsiz hidrotermal koşullarda
mono- ve oligosakkaritlerini elde etmek için hidroliz etmişlerdir. Hidroliz sonrasında
ele edilen reaksiyon ürünlerinin moleküler kütle dağılımı GPC ile belirlenmiştir.
Analiz sonuçlarına göre, polisakkarit hidrolizatlarının moleküler kütle dağılımları,
karbondioksit yüklemesinin arttırılmasıyla daha düşük molekül ağırlıklı kütlelere
kayma göstermiştir. Örneğin, 200 °C’de nişastanın hidrolizinden oluşan glukozun
verimi, 15 dakikalık reaksiyon süresince karbondioksit yüklemesinin arttırılmasıyla
%3,7’den %53’e artmıştır Su ve karbondioksitten oluşan karbonik asit, yüksek
sıcaklık ve yüksek basınçlı suya göre daha düşük pH’da görülmüştür. Bu çalışma,
karbondioksit mevcudiyetinde hidrotermal koşullar altında polisakkaritlerin
hidroliziyle nötralizasyon ve ayırmada ticari asit ve baz kullanmadan mono- ve
oligosakkaritleri üretmek için çevre dostu bir yöntem olarak önerilmiştir.
Sun ve ark., (2005), asidik dioksan/su ve dimetil sülfoksit kullanarak buğday
samanından lignin ve hemiselüloz fraksiyonlarını ekstrakte etmişlerdir. Bu yöntemle

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
37
samandaki orjinal hemiselülozun % 16,9’sı ekstrakte edilebilmiştir. İzole edilen
hemiselüloz fraksiyonlarındaki toplam ksiloz oranı yaklaşık % 63 iken glukoz % 14
ve arabinoz % 11’tür. Lignin fraksiyonunun alkali nitrobenzen oksidasyonu ile
bozundurulması sonucunda ağırlıklı olarak syringaldehit ve vanillin olan fenolik asit
ve aldehit karışımı elde edilmiştir.
Yoshida ve arkadaşlarının (2004) yaptığı çalışmada lignin, selüloz, ve
bunların karışımı süperkritik su ortamında 673 K, 25 Mpa basınçta nikel katalizörü
varlığında gazlaştırılmıştır. Karışımda yumuşak odun lignini olduğu zaman
gazlaştırma verimi düşük olmuştur. Selüloz ve yumuşak odun lignininin reaksiyonu
sonucu oluşan katranlı ürünler katalizörü deaktive ederek gazlaştırma veriminde
düşüşe neden olmuştur. Öte yandan sert odun ve otsu materyal lignini daha kolay
gazlaşmıştır.
Sinag ve ark., (2004), süperkritik su içinde 1-3 K/dk ısıtma hızlarında kütlece
% 5’lik glukozun hidroprolizini çalışmışlardır. Sonuçlar, yüksek verimde H2, CH4 ve
CO2’in yüksek ısıtma hızında elde edileceğini göstermiş ve bu koşullarda CO
oluşumu düşük olmuştur.
Osada ve ark., (2004), lignini 673 K (400°C) ve 30 Mpa basınçta rutenyum
katalizörü varlığında tamamen metan ve karbondioksite gazlaştırmışlardır. Alkil
fenoller ve formaldehit gibi ara bileşiklerin hemen ayrışmasının gazlı ürünlerden
yüksek verimler elde etmek için önemli olduğunu bildirmişlerdir. Dahası da, lignin
gazlaştırmasındaki gaz ürün veriminin, 673 K’de rutenyum katalizörü üzerinde, artan
su yoğunluğu ile arttığını gözlemlemişlerdir. Bunun nedeni, düşük molekül ağırlıklı
fragmentlerin oluşum hızının artan su yoğunluğu ile artmasıdır.
Liu ve Wyman (2003), mısır hasatından sonra kalan biyokütledeki (mısır
koçanı, sapı, yaprağı) ksilan, lignin ve toplam kütlenin çözünürlüğünde sıkıştırılmış
suyun ve akış hızının etkisini incelemişlerdir. Bu amaçla 180, 200 ve 220 °C
sıcaklıkta çalışılmıştır. Hemiselülozun çözünürlüğü özellikle yüksek sıcaklıklarda
artan akış hızı ile artmıştır. Tüm koşullarda ligninin %30’undan fazlası
uzaklaştırılamazken; yüksek akış hızlarında orijinal Klason ligninin %75’i açığa
çıkmış ve ksilan ile ligninin uzaklaşması birbiriyle paralellik göstermiştir. Sıcaklığın
arttırılması ligninin biyokütleden açığa çıkmasını hızlandırmış ve bu durum yüksek

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
38
akış hızlarında daha etkin olmuştur. Ancak, sıcaklığın aşırı arttırılması çöken bir
takım ürünlerin oluşmasına sebep olmuş ve ligninin uzaklaştırılması etkin
olamamıştır.
Dumesic ve ark., (2002), ilk kez model biyokütle kaynaklı oksijenli
bileşiklerin sulu faz reformlamasından bahsetmişlerdir. Akışkan tip reaktör içinde,
gazlaştırma ya da pirolizden daha düşük sıcaklıklarda (yaklaşık 500 K) hidrojen
üretim kapasitesini ispatlamışlardır.
Cortright ve ark., (2002), platin bazlı katalizör kullanarak APR prosesinde
227 °C’e yakın sıcaklıkta şekerlerden hidrojen üretilebileceğini göstermişlerdir.
Etilen glikol gibi birçok biyokütleden çıkarılan materyaller su içinde çözünebilir ve
hidrojen prosesi için kullanılabilirdir.
Yoshida ve Matsumura (2002), selüloz ve lignin karışımını nikel katalizörü
varlığında süperkritik suda 673 K (400 °C) sıcaklıkta gazlaştırmışlardır ve ana gaz
ürünleri olarak CH4 ve CO2 elde etmişlerdir. Lignin fraksiyonunun gazlaştırmayı
engellediğini söylemişlerdir.
Soykan ve ark., (2001) (GMA-Alkil Metakrilat) kopolimerlerinin ortalama
molekül ağırlıklarının tayini için bir çalışma yapmışlardır. Mol olarak % 50:% 50
oranlarında hazırlanan GMA-Alkil metakrilat kopolimerlerinin ortalama molekül
ağırlıkları, THF çözücüsü kullanılarak Jel Geçirgenlik Kromatografisi (GPC) (Gel
Permation Chromatography) ile belirlenmiştir. Ortalama molekül ağırlığı değerleri,
Universal Kalibrasyon yöntemiyle standart olarak polistiren kullanılarak
hesaplanmıştır (çözücü akış hızı: 1.0 ml/dk). Birleşmeyle veya ayrı ayrı sonlanmayla
ilerleyen radikalik polimerizasyonlarda teorik olarak heterojenlik indisi değerleri 1,5-
2,0 arasındadır. Kopolimerizasyonlarda heterojenlik indisi ayrı zamanda zincir
transferi reaksiyonlarına da bağlıdır.
Yoshida ve Matsumura (2001), standart selüloz, ksilan ve lignin (izole
edildikleri kaynaklar belirtilmemiş) karışımlarının 673 K ve 25 Mpa süperkritik su
ortamında nikel katalizörü kullanarak ve 20 dk süredeki gazlaştırma etkinliklerini
incelemişlerdir. Karışımın lignin içeriğinin oluşan gazın miktarını ve
kompozisyonunu belirgin şekilde değiştirdiği gözlenmiştir. Lignin içeriği arttıkça
oluşan gaz miktarı ve bu gaz karışımındaki hidrojen komposizyonu azalmıştır.

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
39
Sözkonusu çalışmada selüloz ve ksilanın bozunması sonucu oluşan bazı ara
ürünlerin ligninle reaksiyona girdiği belirtilmiştir. Farklı miktarlarda selüloz, ksilan
ve lignin içeren karışımların gazlaştırılması sonucu oluşan gaz kompozisyonları
aşağıdaki çizelgede gösterilmiştir.
Çizelge 2.1. Selüloz, ksilan ve lignin içeren karışımların gazlaştırılması sonucu oluşan gaz kompozisyonları
Selüloz:ksilan:lignin oranı
Gaz miktarı (mmol/g reaktant) H2 CO2 CH4 C2H6 C3H8
4:1:1 5,33 9,51 1,12 0,21 0,08 1:4:1 3,64 9,24 1,03 0,19 0,08 1:1:4 1,69 6,62 0,90 0,09 - 1:1:1 2,52 8,49 0,87 0,14 0,06
Aynı araştırmacılar, gerçek biyokütle örneklerinin süperkritik su
gazlaştırmasını da araştırmışlar ve biyokütleden yüksek dönüşüm ile hidrojen elde
edilebileceğini göstermişlerdir
Schmieder ve arkadaşları (2000), samanın gazlaştırılmasını çalışmışlardır.
120 dakikada kesikli tip reaktör ile 500 °C sıcaklık ve 35MPa basınçta alkali
katalizörler varlığında tamamen gazlaştırılmanın olduğunu H2 ve CO2 ürünlerinin
elde edilmesinde başarıya ulaşıldığını söylemişlerdir.
Sasaki ve arkadaşları (2000), selülozun subkritik ve süperkritik su ortamında
(25 MPa, 320-400 °C ve 0,05-10,0 sn) hidrolizini ve bozunmasını çalışmışlardır.
Selülozun dönüşümü 350 °C’nin üzerinde belirgin derecede artmıştır. 400 °C’de
selülozun %100 dönüşümü gerçekleşmiş ve hidroliz ürünü verimi %76,5 olmuştur.
Düşük sıcaklıklarda suda çözünmeyen ve yüksek polimerleşme derecesine sahip
oligomerler 400 °C ve 25 MPa’da çözünebilmiştir.
Yu ve arkadaşları (1993), biyokütlenin düşük konsantrasyonda gazlaştırlması
gerektiğini, çünkü yüksek konsantrasyonlarda ayrışan ürünlerin polimerizasyonunun
meydana geldiğini bildirmişlerdir. Bunun yanında, biyokütlenin daha yüksek
konsantrasyonunun ekonomik olarak gazlaştırılması gerekmektedir. Bu sebepten Xu
ve arkadaşları (1996), süperkritik su içinde yüksek konsantrasyonlu biyokütle
çözeltisinin yüksek sıcaklıkta gazlaştırmasını çalışmışlar ve bu amaçla aktif karbonu

2. ÖNCEKİ ÇALIŞMALAR Mehtap KURTULUŞ
40
kullanmışlardır. Hidrojen verimi düşük olmasına rağmen, biyokütle yüksek
konsantrasyonlarda bile tamamen gaza dönüşebilmiştir.
Antal ve ark., (1991), odunun hidrotermal prosesini çalışmışlar ve
mekanizmalarını karbonhidrat bileşenlerinden bozunan ürünler olarak önermişlerdir.

3. MATERYAL VE METOT Mehtap KURTULUŞ
41
3. MATERYAL VE METOT
3.1. Materyal
Çalışmada lignoselülozik biyokütle olarak buğday samanı ve kenaf sapı
kullanılmıştır. Buğday samanı Adana’daki lokal satış noktalarından, kenaf ise
Çukurova Üniversitesi Ziraat Fakültesi’nden temin edilmiştir. Bu materyaller
kullanılmadan önce parçalayıcıda öğütülmüştür.
Öğütülen materyallerin nem ve kül içerikleri standart metodlarla saptanmıştır.
CHN ve ısıl değer analizleri ise TÜBİTAK-MAM’da yaptırılmıştır. Bu analizlere
ilişkin sonuçlar Çizelge 3.1’de verilmiştir.
Çizelge 3.1. Buğday samanı ve kenafın nem, kül ve selüloz içerikleri
Materyal Nem (%) Kül (%) CHN (%)* Isıl değer (cal/g)
Buğday samanı 9,72 6,31
C: 46,48 H: 5,45 N: 0,33
O: 38,66 S: 0
Üst ısıl değer: 4150 Alt ısıl değer: : 3884
Kenaf 10,40 3,12
C: 48,60 H: 5,68 N: 0,13
O: 41,34 S: 0
Üst ısıl değer: 4219 Alt ısıl değer: 3939
* Örneklerin S değeri ölçülmemiş ve sıfır (0) kabul edilmiştir. Oksijen (O) farktan hesaplanmıştır.
3.1.1. Kullanılan Kimyasal Maddeler
• Dekstran standartları: Polisakkaritlerin moleküler kütle dağılımlarını
belirlemede standart olarak kullanılmıştır,
• Sodyum hidroksit: HPLC analizlerinde hareketli fazın hazırlanmasında,
• Aseton: Deney sonrasında reaktördeki organik maddeleri temizlemek için,
• Yüksek saflıkta oksijen gazı: TOC (Toplam Organik Karbon) analizlerinde,
• Potasyum ftalat: TOC analizinde standart olarak,
• Argon gazı: GC’de taşıyıcı gaz,

3. MATERYAL VE METOT Mehtap KURTULUŞ
42
• Helyum gazı: GC’de taşıyıcı gaz,
• Kuru hava: GC analizi için,
• Karbondioksit: Hidrolizde deneylerinde basınç oluşturmada,
• Asetonitril: HPLC analizlerinde mobil faz olarak,
• Pt %5 on Activated Carbon Gazlaştırmada deneylerinde katalizör olarak,
• Dietil eter: Hidroliz çözeltilerinde mevcut organik maddelerin GC-MS
analizleri için ekstraksiyon çözücüsü olarak,
• Na2SO4: Ekstrakte işleminden sonra kalan suyu tutmak için kullanılmıştır.
• Monoetanolamin (NH2CH2CH2OH): Kaynama noktasının yüksek oluşu
(~170°C) ve çözücü etkisinin iyi olaması nedeniyle lignoselülozik
materyallerin parçalanmasında kullanılmıştır.
• H2SO4: İzole edilen selülozun renginin ağartılması için kullanılmıştır.
• NaOCl: İzole edilen selülozun renginin ağartılması için kullanılmıştır.
• CH3COOH: Selüloz izolasyonu sırasında fazla bazik çözeltiyi nötralize etmek
için kullanılmıştır.
• KOH: Hemiselüloz eldesinde lignoselülozik materyallerin kuvvetli bir bazla
parçalanması amacıyla kullanılmıştır.
• Etanol: Hemiselüloz izolasyonu sırasında hemiselülozu çöktürebilmek
amacıyla kullanılmıştır.
3.1.2. Kullanılan Araç ve Gereçler
• Blender (Parçalayıcı)
• 500 mL hacimli Parr 4575 Model HP/HT yüksek basınç reaktörü
• 100 mL hacimli Parr 4590 Model Micro Bench top reaktör
• 260D Model ISCO yüksek basınç pompası
• Döner buharlaştırıcı (Rotary evaporatörü)
• Diyaliz kesesi (Membra-Cell Dialysis Tubing, MWCO 3500)
• Santrifüj
• Ultrasonik titreştirici

3. MATERYAL VE METOT Mehtap KURTULUŞ
43
• Vorteks karıştırıcı
• Pastör pipeti
• Mikro pipet
• Cam malzemeler (Deney tüpü, balon joje, rodajlı balon, mezür, beher, erlen,
pipet vb.)
• Kapaklı ve septumlu viyal (1,5 ml)
• Shimadzu 10VP serisi Yüksek Performanslı Sıvı Kromatografisi Cihazı
(High Performance Liquid Chromatography, HPLC)
• Waters Ultrahydrogel 250 (MWR: 1 kda. - 80 kda ve 300 mm x 7,8 mm x 250
A° ebatlarında) ve 2000 (MWR: 50 kda.-5000 kda ve 300 mm x 7,8 mm x
2000 A° ebatlarında) kolonları
• 0,45 ve 0,20 μm’lik filtre
• Varian 450-GC Gaz kromatografisi cihazı
• Varian MolSieve 5A 1m x 1/8" Ultimetal GC kolonu
• Varian Shincarbon 100/120 Mesh 2m x 1/16" x 1,0 mm GC kolonu
• Fourier Transform Infrared (FTIR) cihazı
• Thermo Finnigan GC-MS Cihazı
3.2. Metot
3.2.1. Lignoselülozik Materyallerin Selüloz İçeriğinin Saptanması
Buğday samanı ve kenafın selüloz fraksiyonları Foyle ve ark. (2007)’nın
kullandığı metot ile belirlenmiştir. Bu metoda göre 3 g kuru lignoselülozik materyal
(buğday samanı/kenaf) 100 ml monoetanolamin ile karıştırılarak 170 ºC’de 3 saat
geri soğutucu altında kaynatılmıştır. Soğutulan örneğe 100 ml su ilave edildikten
sonra süzülmüştür. Katı kısım 60 ºC su ile yıkandıktan sonra içerisinde 75 ml su olan
behere alınmıştır. Üzerine 10 ml %10 H2SO4 ve 10 ml 24 g/L NaOCl ilave edilerek
oda sıcaklığında 5 dk bekletilmiştir. Karışım süzüldükten sonra kalıntı 15 ml soğuk
su ve 15 ml %3 Na2SO3 ile yıkanmıştır. 50 ml su içerisine alınan katı örnek üzerine
50 ml %6 Na2SO3 eklendikten sonra kaynamış su içerisinde 20 dk inkübe edilmiştir.

3. MATERYAL VE METOT Mehtap KURTULUŞ
44
Soğutulan karışım süzülmüş ve elde edilen katı karışım olan selüloz sırasıyla 150 ml
kaynamış su, 50 ml soğuk su, 25 ml 10’luk CH3COOH, 50 ml soğuk su, 150 ml
kaynamış su, içerisinde iki damla NH4OH olan 75 ml soğuk su ve son olarak 200 ml
kaynamış su ile yıkanmıştır. Katı 150ºC’de kurutulup selüloz miktarı olarak
tartılmıştır.
3.2.2. Lignoselülozik Materyallerin Hemiselüloz İçeriğinin Saptanması
Buğday samanı ve kenafın hemiselüloz fraksiyonları Sun ve ark. (2002)’nın
kullandığı yönteme göre belirlenmiştir. Bu yönteme göre lignoselülozik materyaller
öncelikle (2:1 v/v) oranında toluen/etanol karışımı ile muamele edilerek varsa
organik bileşenlerden uzaklaştırılmıştır. Wax kısmı uzaklaştırılan materyalden 3 g
alınarak 100 ml 0,5 M KOH ile 35°C’de 2,5 saat ekstrakte edilmiştir. Karışım
süzüldükten sonra süzüntü kısmı alınarak 6 M asetikasit ile pH 5,5’e nötralize
edilmiştir. Bu işlemden sonra çözelti hacmi yarıya inene kadar konsantre edilmiştir.
Elde edilen çözelti %95’lik etanol ile 24 saat muamele edilerek çöktürülmüştür.
Karışım santrifüjlenerek katı kısım tekrar %75’lik etanol ile yıkanmıştır. Sonrasında
katı bir kez de suyla yıkanmıştır. Tekrar santrifüj yapılmıştır. Katı bu kez 1’e 4
hacim oranında %95’lik etanol ile 12 saat çöktürülmüştür. Santrifüjle alınan katı
kısım 40°C’de kurutulmuştur.
3.2.3. Lignoselülozik Materyallerin Lignin İçeriğinin Saptanması
Buğday samanı ve kenafın lignin içeriklerinin bulunması için Yaşar ve
arkadaşlarının (2002) belirlediği metod kullanılmıştır. Bu metoda göre 10 g biyokütle
150 ml %72’lik H2SO4 çözeltisi ile 24 saat oda sıcaklığında muamele edilmiştir. 24
saat sonunda çözelti üzerine 1850 ml saf su eklenerek 2 saat süreyle kaynatılmıştır.
Koloidal asidik çözelti su trompu ile pH nötr olana kadar süzülmüştür. Elde edilen
katı 100°C’de kurutulur.

3. MATERYAL VE METOT Mehtap KURTULUŞ
45
3.2.4. Hidroliz Deneyleri
Buğday samanının hidrolizi subkritik su (100ºC < T < 374,2ºC ; P: Suyu sıvı
fazda tutacak kadar yüksek, 217,76 atm < P ) ortamında gerçekleştirilmiştir. Buğday
samanı ve kenaf biyokütlelerinden izole edilen selüloz, hemiselüloz ve lignin
fraksiyonları ayrı ayrı hidroliz edilmiştir. Biyokütle fraksiyonlarından 4 g alınarak ve
200 ml su eklenerek 500 mL’lik reaktör (Parr model) içine yerleştirilmiştir. Daha
sonra reaktörün üst kısmına grafit gasket yerleştirildikten sonra manometreli,
karıştırıcılı ve valfli kapak kısmı reaktörün üstüne yerleştirilip vidalı split ring ile
sabitleştirilmiştir. Reaktör ısıtıcının içerisine yerleştirildikten sonra ısıl çifti
(termocouple) kapak kısmındaki yuvasına daldırılmış ve reaktörün içindeki havanın
minimize edilmesi için reaktöre 3 kez 1000 psi CO2 basılıp boşaltılmıştır. Reaktörün
sıcaklığı oda sıcaklığından başlayarak 250°C yükseltilmiş ve bu sıcaklığa
ulaşıldığında reaktördeki toplam gaz basıncı 2500 psi olacak şekilde CO2 gazı
basılmıştır. 2500 psi CO2 atmosferinde ve 250°C sıcaklıkta biyokütle 1000 rpm hızla
karıştırılarak 45 dakika süre ile hidroliz deneyleri gerçekleştirilmiştir. Deney
tamamlandıktan sonra reaktör buzlu su içerisine daldırılarak hızlı bir şekilde oda
sıcaklığına kadar soğutulmuştur. Reaktörün suya daldırılması ile tepkimenin sona
erdiği kabul edilmiştir.
3.2.5. Hidroliz Çözeltilerinin Toplam Organik Karbon, Monoşeker ve
Polisakkarit Analizleri
Hidroliz sonrası elde edilen çözeltiler 0,2 µm naylon filtrelerden geçirildikten
sonra toplam organik karbon (TOC) içerikleri Tekmar-Dohrmann Apollo 9000
karbon analizörü kullanılarak saptanmıştır. TOC analizi için ayrılan örnek dışındaki
tüm çözelti 25-30 ml’ye deriştirildikten sonra santrüfüjlenmiş ve berrak çözelti
deiyonize suda 3 gün diyaliz edilmiştir (Serva Membra-Cell Dialysis Tubing,
MWCO: 3500 Da). Diyaliz işlemi sonrasında diyaliz kesesinin içinde kalan çözelti
belirli bir hacme kadar deriştirildikten sonra polisakkarit içeriğine bakılmıştır.
Deriştirilen diyaliz kesesi dışındaki çözeltide ise monoşekerlere bakılmıştır.

3. MATERYAL VE METOT Mehtap KURTULUŞ
46
Polisakkaritlerin ortalama molekül kütleleri yüksek performanslı Jel
Geçirgenlik Kromatografisi (Gel Permeation Chromatography, GPC) ile Shimadzu
10VP serisi HPLC cihazı kullanılarak belirlenmiştir. Kalibrasyon eğrisi molekül
ağırlıkları 4400, 9900, 21400, 43500, 124000, 196000, 277000 ve 401000 Da olan
dekstran standartları kullanılarak oluşturulmuştur. İzole edilen polisakkaritlerin
analizlerinde, Waters Ultrahydrogel 250 (MWR: 1 kDa-80 kDa ve 300 mm x 7,8
mm; 250 A°) ve 2000 (MWR: 50 kDa-5000 kDa ve 300 mm x 7,8 mm; 2000 A°)
kolonları molekül büyüklüklerine göre büyükten küçüğe doğru seri olarak bağlanıp
HPLC cihazının donanımında bulunan fırın içerisine yerleştirilerek kullanılmıştır.
Analizler analitik modda, refraktif indeks (Shimadzu RID-10A) dedektör ile 70°C
fırın sıcaklığında yapılmıştır. Mobil faz olarak 0,001 M NaOH çözeltisi
kullanılmıştır. Elüsyon 0,7 ml/dk akış hızında izokratik olarak 37 dakikada
gerçekleştirilmiştir.
Monoşeker analizleri ise SupelcosilTM LC-NH2 kolonu kullanılarak 40°C
kolon sıcaklığında, su/asetonitril (25:75) mobil fazı kullanılarak 0,6 ml/dk. akış
hızında yapılmıştır. Analizlerde Sigma’dan temin edilen glukoz, galaktoz, mannoz,
ramnoz, arabinoz, ksiloz, fruktoz monosakkarit standartları kullanılmıştır.
3.2.6. Hidroliz Çözeltilerinin Gazlaştırılması ve Gaz Analizleri
Hidroliz sonucu elde edilen çözelti süzüldükten sonra bundan 50 ml hacimde
örnekler alınarak 100 ml hacimli Parr 4590 model mikro bench (Şekil 3.1) reaktör
içerisine konulmuştur. 0,12 g aktif karbon destekli %5 Pt katalizörü eklendikten
sonra reaktör kapatılmış ve içerisindeki havanın minimize edilmesi için argon gazı
basılıp boşaltılmıştır. Gazlaştırma deneyleri 250ºC’de katalizörlü ve katalizörsüz
olarak 1000 rpm hızla karıştırılarak 3 saat boyunca gerçekleştirilmiştir. Reaktörün
sıcaklığı oda sıcaklığından başlayarak 250°C’ ye yükseltilmiştir. Deney süresi
reaktörün istenen sıcaklığa (250 ºC) ulaşması anından itibaren başlatılmıştır. Deney
tamamlandıktan sonra reaktör su içerisine daldırılarak hızlı bir şekilde oda
sıcaklığına kadar soğutulmuş ve reaktörün suya daldırılması ile tepkimenin sona
erdiği kabul edilmiştir. APR deneyleri en az iki paralel olacak şekilde çalışılmıştır.

3. MATERYAL VE METOT Mehtap KURTULUŞ
47
Şekil 3.1. Gazlaştırmada kullanılacak yüksek basınç reaktörü
Reaktör içerisindeki gazın hacmi gaz büreti ile ölçüldükten sonra
kompozisyonları, birinden argon diğerinden helyum taşıyıcı gazların geçtiği iki
kanallı ve iki termal iletkenlik dedektörlü Varian-450 Gaz kromatograf ile
belirlenmiştir. Hidrojen dışındaki gazların (C1-C5) saptandığı kanalda Shincarbon
100/120 mesh 2m×1/16”×1mm kolon; hidrojen kanalında ise MolSieve 5A 1m×1/8”
Ultimetal kolon kullanılmıştır. GC analiz programı, 40ºC’de 3 dk bekleyip 8ºC/dk ile
230ºC’ye yükselme ve bu sıcaklıkta 5 dk bekleme şeklinde toplam 31,75 dk süreden
oluşmaktadır. Dedektör sıcaklığı 230ºC, filament sıcaklığı ise 300ºC olarak
çalışılmıştır. Standart gaz karışımı olarak Elite Gaz Teknolojileri İnş. Tur. San. Tic.
Ltd. Şti.’den (İstanbul, Türkiye) sağlanan içerisinde % mol olarak sırasıyla 1,8 ±
0,036 asetilen, 4,0 ± 0,08 etilen, 3,9 ± 0,078 etan, 4,9 ± 0,098 metan, 16,0 ± 0,32
karbonmonoksit, 22,0 ± 0,44 karbondioksit ve balans sağlayacak miktarda hidrojen
(47,4 ± 0,474) bulunan gaz karışımı kullanılmıştır.
3.2.7. FT-IR Analizleri
Buğday samanı ve kenaftan izole edilen selüloz, hemiselüloz ve lignin
fraksiyonlarının saflık derecelerini belirlemek için Spectrum RXI FT-IR (Fourier

3. MATERYAL VE METOT Mehtap KURTULUŞ
48
Transformed Infrared, Perkin Elmer) spektroskopi sistemi kullanılarak 400-4000 cm-
1 aralığında infrared spektrumları alınmıştır. Katı haldeki 1 mg numune yaklaşık 100
mg potasyum bromür (KBr) ile havanda iyice karıştırılmış ve 10000-15000 psi’lik
basınç uygulanarak saydam bir disk haline getirilmiştir. KBr referans alınarak her bir
örneğin infrared spektrumları alınmıştır.
3.2.8. Yüzey Analizleri
Lignoselülozik biyokütlelerden izole edilen fraksiyonların taramalı elektron
mikroskop (SEM) ile görüntüleri alınarak yapısal ve morfolojik özellikleri
incelenmiştir. SEM analizleri Leo-Evo 40 scanning electron microscope cihazı ile
İnönü Üniversitesi Araştırma Merkez Laboratuvarı’nda yaptırılmıştır. Buğday
samanı biyokütlesinden izole edilen selüloz fraksiyonunun kristal indeks değerini
belirlemek için ise X-Ray Diffraction (XRD) analizleri yapılmıştır. Bu analizler de
İnönü Üniversitesi Araştırma Merkez Laboratuvarı’nda yaptırılmıştır. XRD
analizlerinde 40 KW ve 40 mA koşullarında bakır Kα (λ=1.541 Å) ışını
kullanılmıştır. Numune 2°/dk ile 2θ=5°’ten 40°’ye kadar taranmıştır.
3.2.9. GC/MS Analizleri
Buğday samanı ve kenaftan izole edilen ligninin parçalanması sonucu açığa
çıkan ürünler GC-MS ile saptanmıştır. Bu amaçla hidroliz deneyi sonucu elde edilen
tüm çözelti 100 ml’ye konsantre edilmiş ve bu çözelti dietil eter ile ekstrakte
edildikten sonra Na2SO4 kolonundan geçirilmiştir. Ekstrakt deriştirildikten sonra
Thermo Finnigan GC-MS ile Thermo TR-5MS kapiler kolon (60 m x 0.25 mm ID x
0.25 µm) kullanılarak analiz edilmiştir. GC-MS analiz şartları şu şekilde
oluşturulmuştur: 60ºC’de 1 dk bekleyip 5ºC/dk ile 280ºC’ye yükselme ve bu
sıcaklıkta 9 dk bekleme. “Solvent delay” 11 dakika ve inlet sıcaklığı 220°C.
Enjeksiyonlar 1 µl numune miktarında 1:15 split oranında gerçekleştirilmiştir. Yapı
tanımlamalarında NIST 2002 kütle spektral kütüphanesi kullanılmıştır.

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
49
4. BULGULAR VE TARTIŞMA
4.1. Buğday Samanı ve Kenaf’tan Selüloz, Hemiselüloz ve Lignin İzolasyonu
Buğday samanı ve kenaf sapından selüloz izolasyonu için Foyle ve ark.
(2007)’nın, hemiselüloz izolasyonu için Sun ve ark. (2002)’nın, lignin izolasyonu
için ise Yaşar ve ark. (2002)’nın önerdiği yöntemler kullanılmıştır (Bakınız 3.2.
Metot). Bu yöntemlere göre buğday samanı ve kenaf materyallerinden elde edilen
selüloz, hemiselüloz ve lignin fraksiyonları FT-IR, SEM, XRD ve GC-MS
analizlerinden en az ikisi ile karakterize edilmiştir. Sonrasında lignoselülozik
fraksiyonlar gazlaştırma deneylerinde kullanılmış, oluşan gaz hacimleri ve gaz
kompozisyonları incelenmiştir.
4.1.1. FT-IR Analizleri
Lignoselülozik biyokütlelerden elde edilen selülozlar ile standart selülozun
Fourier transform infrared spektroskopi (FT-IR) ile spektrumu alınıp nitel olarak
karşılaştırma yapılmıştır. Şekil 4.1 ve Şekil 4.2 buğday samanından ve kenaftan
izole edilen selüloz ile standart selülozların infrared spektrumlarını göstermektedir.
Spektrumlarda görüldüğü gibi 3347-3374 cm-1’de gözlenen geniş ve şiddetli band
O-H titreşimlerinden kaynaklanmaktadır. Bu band selüloz matriksinde tekrarlayan
birimler arasında hidrojen bağlarıyla bir arada bulunan O-H gruplarına aittir. 2890-
2915 cm-1 arasındaki band C-H; 1160-1000 cm-1’deki ise C-O titreşimlerinden
kaynaklanmaktadır. 1109 cm-1’deki band selülozdaki glukoz halkalarının
gerilmelerine aittir. 898 cm-1’de gözlenen band ise selülozdaki β-glikozidik
bağlardan kaynaklanır (Sun ve ark., 1999 ; Corredor, 2008). 1641 cm-1 deki band
C=O fonksiyonel grubuna aittir. Bu band eğer 1700 cm-1’e doğru kaymış olsa idi
hemiselülozik yapıya ait C=O olarak düşünülebilirdi (Wissel ve ark., 2008). 1370-
1375 cm-1’de gözlenen band selülozdaki C-H gerilmelerinden ileri gelmektedir
(Corredor ve ark., 2008).

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
50
Şekil 4.1. Standart selüloz ve buğday samanından izole edilen selülozun infrared spektrumları
Şekil 4.2. Standart selüloz ve kenaftan izole edilen selülozun infrared spektrumları
440 400 300 200 150 100 4560,
6
7
7
8
8
9
9
10
10
11
11
12
12
13
13
14
145,
cm-1
%T
İzole selüloz
Standart selüloz
3374,79
2918,55
1643,83
1372,23
1164,36
1060,33
898,01611,87
1429,10
1109,63
1313,74
3347,61
2900,48
1641,39
1431,83
1373,55
1282,29
1166,17
1114,39
1058,861032,70
669,58 613,12
559,37
4400 4000 3000 2000 1500 1000 45035
40
50
60
70
80
90
100
110
120
130
140
150 154,
cm-1
%T İzole selüloz
Standart selüloz
3355,76
2899,71
2358,792138,63
1647,971431,17
1372,471319,76
1165,11
1112,781061,09
897,12 668,9617,87
3347,61
2900,48
2364,16
1641,40
1431,831373,55
1282,29
1166,17
1114,391058,86
1032,70
669,5613,12
559,37

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
51
Buğday samanı ve kenaf biyokütlelerinden izole edilen hemiselülozların FT-
IR spektrumları Şekil 4.3’te gösterilmiştir.
Şekil 4.3. Buğday samanı ve kenaf sapından izole edilen hemiselüloz fraksiyonlarının FT-IR spektrumları
Spektrumlarda görüldüğü gibi 3412-3406 cm-1’de O-H, 2920 cm-1 civarında
ise C-H gerilmelerine ait bandlar görülmektedir. 1610-1650 cm-1’de gözlenen
bandlar C=O fonksiyonel grubuna aittir. Bu bandın 1700 cm-1’lerden daha düşük
bölgede yer alması yapının su absorplamış olmasından kaynaklanmaktadır (Sun ve
ark., 2004; Buranov ve Mazza, 2010). 1740 cm-1’de bir bandın gözlenmemiş olması
asetil, uranik ve ferulik ester gruplarına ait ester bağlarının hemiselüloz yapısından
tamamen kırılmış olduğunu göstermektedir. 1510 cm-1 civarında görülen zayıf
bandlar hemiselüloz yapısında çok az miktarda bulunan lignine aittir (Sun ve ark.,
2004; Buranov ve Mazza, 2010). Hemiselüloz yapısındaki -CH2 gerilme
titreşimlerine ait bandlar 1420 cm-1’de gözlenmektedir. Tipik ksilan hemiselüloz
yapısına ait bandlar 1175 ve 1000 cm-1 arasında gözlenir ve C-O, C-C ve C-O-C
bağlarına ait gerilme ve bükülme bandları 1040 cm-1 civarında yoğunlaşır (Sun ve
ark., 2004; Buranov ve Mazza, 2010).
4400 4000 3000 2000 150 1000 451,9
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
101,1
cm-1
%T
Kenaftan izole hemiselüloz
Buğday samanı hemiselüloz
3412,16
2921,56
2359,53
1607,38
1417,69
1250,41
1045,02
897,62652,40
1506,01
1319,65
3406,21
2919,61
2135,98
1651,88
1513,52
1418,02
1325,25
1254,02
1042,57
896,38652,92

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
52
Hemiselülozda gözlenen 895 cm-1 deki keskin bandlar şeker birimleri
arasındaki β-glukozidik bağların karakteristik pikidir (Gupta ve ark.,1987).
Hemiselülozlar kendilerini oluşturan şeker birimlerine göre; ksilanlar,
mannanlar, arabinoksanlar, glikomannanlar ve glikoksilanlar şeklinde
isimlendirilirler (Mutlu, 1990). Ksilanlar, hemiselülozik yapı içinde nicelik
açısından önemli yer tutan fraksiyonlardır. Lignoselülozik biyokütlelerden izole
edilen hemiselüloz fraksiyonlarını standart ksilan yapılarıyla karşılaştırabilmek için
kayın ağacı (beechwood) ve yulafxbuğday (oat spelt)’dan (Oat spelt: Ortadoğu
orjinli, Doğu-Avrupa’da popüler olan ve buğdaydan daha yüksek protein içeriğine
sahip bir tür tahıl olan spelt ile yulafın çaprazlanması ile oluşturulmuş bir tahıl)
izole edilen standart ksilanların da infrared spektrumları alınmıştır. Şekil 4.4’te
standart ksilanların FT-IR spekrumları izole ettiğimiz hemiselüloz fraksiyonları ile
karşılaştırmalı olarak verilmiştir.
Şekil 4.4. Kayın ağacı ve yulafxbuğdaydan izole edilen standart ksilanlar ile kenaf ve buğday samanından izole edilen hemiselüloz fraksiyonlarının infrared spektrumları
4400, 4000 3000 2000 1500 1000 450,0
-1,7
5 10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
96,7
cm-1
%T Ksilan Beechwood
Ksilan Oat Spelts
B.Samanı Hemiselüloz
Kenaf Hemiselüloz
3419,1
2914,4
2361,48 2131,6
1613,72
1412,6
1319,11252,74
1164,6
1044,25 984,88
896,33
780,04
654,97
612,37536,71

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
53
Görüldüğü üzere kayın ağacı ve yulafxbuğday’dan izole edilen ksilan
standartları birbirine oldukça benzemektedir. Buğday samanı ve kenaftan elde
ettiğimiz hemiselüloz fraksiyonları da bu iki standart hemiselüloz fraksiyonları ile
benzerlik göstermektedir. Ancak, gözlenen küçük farklılıklar ksilan hemiselüloz
yapısına özgü bandlar olan 1175 ve 1000 cm-1 arasında ve C-O, C-C ve C-O-C
bağlarına ait gerilme ve bükülme bandlarının yoğunlaştığı 1040 cm-1’de belirgindir
(Sun ve ark., 2004; Buranov ve Mazza, 2010). Bunlar da izole ettiğimiz hemiselüloz
yapılarının sadece ksilandan ibaret olmadığını göstermektedir.
Buğday samanı ve kenaf sapından izole edilen lignin fraksiyonlarının FT-IR
spektrumları Şekil 4.5’te gösterilmiştir. Lignin örneklerinin IR spektrumlarında
3390-3420 cm-1’de O-H, 2930 cm-1 civarında ise aromatik C-H gerilmelerine ait
bandlar görülmektedir. 2920-2930 cm-1’deki band alifatik (-CH-) gerilmelerinden
kaynaklanmaktadır. 1710 cm-1’ deki bandlar C=O’ya aittir. 1608, 1490, 1460 cm-
1’de gözlenen bandlar ligninin aromatik iskelet yapısından kaynaklanan karakteristik
bandlardır (Lateef ve ark, 2009).
Şekil 4.5. Buğday samanından ve kenaftan izole edilen lignin fraksiyonlarının FT-IR spektrumları
4400 4000 3000 2000 1500 1000 450 cm-1
%T
Kenaf Lignin
Bugday Samani Lignin
3432,02
2924,56
2387,94
1710,851608,44
1496,141462,63
1211,70
1108,831018,65
579,83
3815,44
3404,56
2933,71
1709,021608,09
1499,401463,37
1424,011204,66
798,9
595,5
75
70
65
60
55
50
45
40
35
30
25
20
15
10

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
54
Yaklaşık 1652-1505 cm-1’de görülen bandlar C=C yapısına aittir. 1357
cm1’deki band -CH3’de bulunan alifatik C-H gerilmelerinden kaynaklanır (Zhao ve
ark, 2009). 1108 cm-1’de lignin için karakteristik olan p-hidroksi fenil propan H
birimine ait band; 1211 cm-1’de ise G birimine ait band görülmektedir (Zhao ve ark,
2009; Pan ve ark, 1999; Shukry ve ark, 2008).
Buğday samanı ve kenaftan izole edilen ligninlerin infrared spektrumlarında
gözlenen bazı farklılıklar bu lignoselülozik materyallerden elde edilen lignin
fraksiyonlarının farklı yapılarda olduğunu göstermektedir.
4.1.2. SEM Analizleri
Buğday samanından ve kenaftan izole edilen selüloz fraksiyonlarının SEM
görüntüleri Şekil 4.6’da verilmiştir. SEM görüntülerinden görüldüğü gibi selüloz
fraksiyonları mikrofibrillerden oluşan bir yapıya sahiptir.
Şekil 4.6. SEM görüntüleri (a) Buğday samanınından izole edilen selüloz, (b) standart selüloz (Haensel ve ark., 2009) (c) Kenaftan izole edilen selüloz
(a) (b)
(c)

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
55
Şekil 4.7. SEM görüntüleri (a,b) Buğday samanından izole edilen hemiselüloz, (c) Kenaftan izole edilen hemiselüloz,(d) Standart ksilan (Haimer ve ark., 2008)
Hemiselüloz fraksiyonlarının SEM görüntüleri selülozdan oldukça farklıdır
(Şekil 4.7). Hemiselüloz yapıları az gözenekli bir yapıya sahiptir (Haimer ve ark.,
2008) ve küçük partiküllerin varlığı göze çarpmaktadır. Bu partiküller safsızlıklara,
başlangıçta oluşan daha büyük partiküllerin parçalanması ya da kısmen daha küçük
hemiselüloz fraksiyonlarının çökmesine bağlı olarak oluşmuştur.
İzole edilen lignin fraksiyonlarının SEM görüntüleri Şekil 4.8’de verilmiştir.
Standart ligninin SEM görüntüsünde bir takım damlacıklar görülmektedir. Bazı
lignoselülozik materyallerin hidrolizi sonucu yüzeyde benzer damlacıklara
rastlanmış ve bunların lignin içerdiği saptanmıştır (Selig ve ark., 2007; Micic ve
ark., 2001).
(c) (d)
(a) (b)

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
56
Şekil 4.8. SEM görüntüleri (a) Buğday samanından izole edilen lignin, (b) Referans görüntü olarak kullanılan lignin (c) Kenaftan izole edilen lignin
4.1.3. XRD Sonuçları
Buğday samanınından izole edilen lignoselülozik materyallerin XRD
analizlerine ilişkin spektrumlar Şekil 4.9 ve Şekil 4.10’da verilmiştir. XRD
datalarından kristal indeks değeri hesaplanırken 4.1’deki denklem kullanılmıştır.
100002
002 xI
IICrI am−= (4.1.)
Bu bağıntıda I002; 2θ=22.4º’de 002 düzlemindeki kristal alan kırılma
yoğunluğunu; Iam ise 2θ=18.7º’da amorf bölgenin kırılma yoğunluğunu ifade
etmektedir (Segal ve ark., 1959; Kim ve Holtzapple, 2006).
Lignoselülozik materyallerden sadece selüloz kristalin bir yapıya sahiptir.
İzole edilen selüloza ait kristal indeks değeri %57,84; standart selülozunki ise
%79,76 olarak hesaplanmıştır.
(a) (b)
(c)

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
57
Selüloz
0
50
100
150
200
250
0 5 10 15 20 25 30 35 40 45
2 Theta
Inte
nsity
Standart Selüloz
0
100
200
300
400
500
600
0 10 20 30 40 50
2 Theta
Inte
nsity
Şekil 4.9. Standart ve izole selüloza ait XRD spektrumları
Değerlerin bu şekilde farklılık göstermesi, standart selülozun oldukça
kristallin bir yapıya sahipken izole selülozda amorf yapıların standarda göre daha
fazla olduğu anlaşılmaktadır. İzole edilen selülozun düşük kristal özellik göstermesi
selülozun izolasyon işlemi esnasında dekristallizasyonundan ve böylece daha amorf
bir yapıya dönüşmesinden kaynaklanmaktadır. Bu durum muhtemelen bir dizi
ekstraksiyon işleminden oluşan izolasyon işleminde kullanılan kimyasalların
etkisinden dolayıdır. Benzer şekilde kenaf lignoselülozik biyokütlesinin subkritik su
ile etkileştirilmesi sonucu da selüloz fraksiyonlarının amorf hale dönüşmesinden
dolayı kristal indeks değerinin azaldığı gözlenmiştir (Öztürk ve ark., 2010).
Hemiselüloz
0
10
20
30
40
50
60
5 10 15 20 25 30 35 40
2 Theta
Inte
nsity
Lignin
020406080
100120140160180
0 10 20 30 40 50
2 Theta
Inte
nsity
Şekil 4.10. Hemiselüloz ve lignin fraksiyonlarına ait XRD spektrumları
Hemiselüloz ve lignin yapıları kristalin yapı göstermediğinden; 2θ=22.4º’de
002 düzlemindeki kristal alan kırılma yoğunluğu gözlenememiştir. Bu durum bu
yapıların selüloz fraksiyonundan tamamen izole bir şekilde saflaştırılabildiğini
göstermektedir.
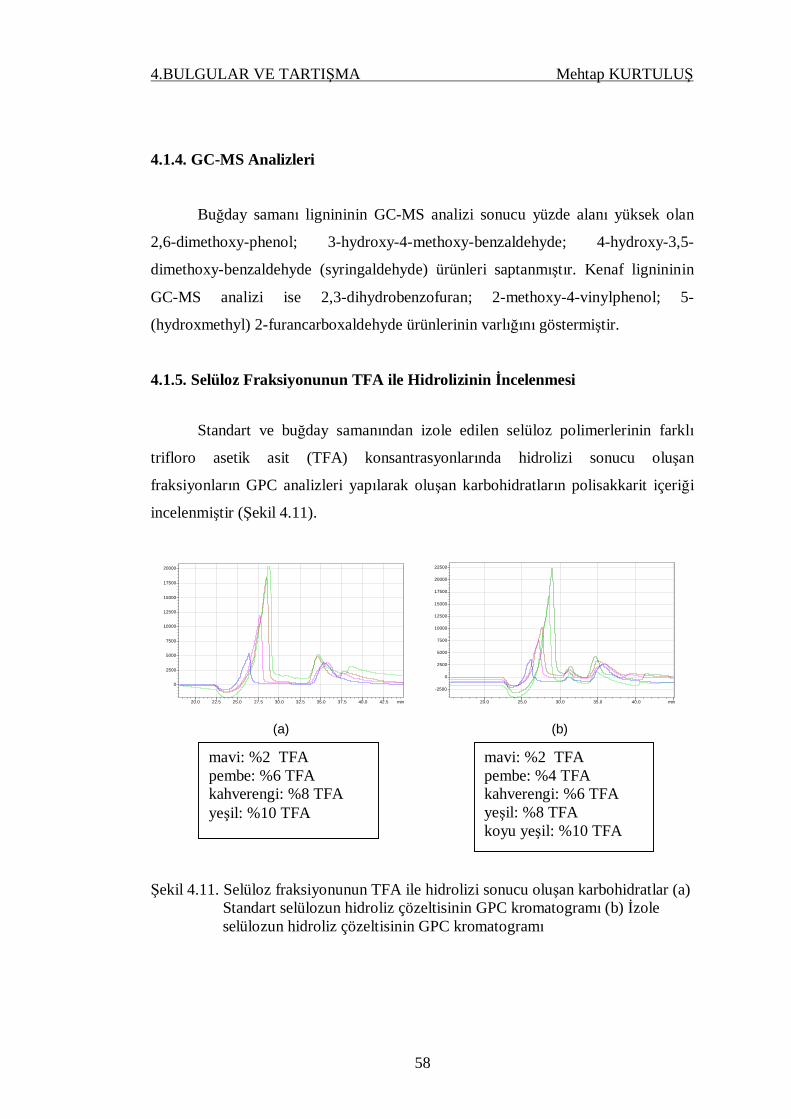
4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
58
4.1.4. GC-MS Analizleri
Buğday samanı lignininin GC-MS analizi sonucu yüzde alanı yüksek olan
2,6-dimethoxy-phenol; 3-hydroxy-4-methoxy-benzaldehyde; 4-hydroxy-3,5-
dimethoxy-benzaldehyde (syringaldehyde) ürünleri saptanmıştır. Kenaf lignininin
GC-MS analizi ise 2,3-dihydrobenzofuran; 2-methoxy-4-vinylphenol; 5-
(hydroxmethyl) 2-furancarboxaldehyde ürünlerinin varlığını göstermiştir.
4.1.5. Selüloz Fraksiyonunun TFA ile Hidrolizinin İncelenmesi
Standart ve buğday samanından izole edilen selüloz polimerlerinin farklı
trifloro asetik asit (TFA) konsantrasyonlarında hidrolizi sonucu oluşan
fraksiyonların GPC analizleri yapılarak oluşan karbohidratların polisakkarit içeriği
incelenmiştir (Şekil 4.11).
20.0 22.5 25.0 27.5 30.0 32.5 35.0 37.5 40.0 42.5 min
0
2500
5000
7500
10000
12500
15000
17500
20000
20.0 25.0 30.0 35.0 40.0 min
-2500
0
2500
5000
7500
10000
12500
15000
17500
20000
22500
(a) (b)
Şekil 4.11. Selüloz fraksiyonunun TFA ile hidrolizi sonucu oluşan karbohidratlar (a) Standart selülozun hidroliz çözeltisinin GPC kromatogramı (b) İzole selülozun hidroliz çözeltisinin GPC kromatogramı
mavi: %2 TFA pembe: %4 TFA kahverengi: %6 TFA yeşil: %8 TFA koyu yeşil: %10 TFA
mavi: %2 TFA pembe: %6 TFA kahverengi: %8 TFA yeşil: %10 TFA

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
59
Standart selüloz ve izole selülozun trifluoro asetik asit ile hidrolizi benzer
GPC kromatogramlarını vermiştir. Hidroliz sonrası oluşan polisakkarit
fraksiyonlarının moleküler ağırlığının orijinal selülozdan biraz fazla çıkması
aglomerizasyonun gerçekleştiğini göstermektedir (Çizelge 4.1.). Nitekim Zhao ve
arkadaşları (2007), selülozun 150ºC sıcaklık ve farklı sülfürik asit
konsantrasyonlarında aglomerizasyonun gerçekleştiğini ve aglomerizasyonun asit
gücüyle orantılı olarak arttığını gözlemlemişlerdir. Bu durumun mikrofibriller
arasındaki asit katalizli intermoleküler yüzey dehidratasyonunun bir sonucu olduğu
bildirilmiştir.
Çizelge 4.1. Buğday samanından izole edilen selüloz fraksiyonunun TFA ile hidrolizi sonucu elde edilen polisakkaritlerin molekül ağırlık dağılımı
Asit Yüzdeleri
Standart Selüloz İzole Selüloz Elüsyon Zamanı
(dk)
Moleküler Ağırlık
(Da) Elüsyon Zamanı
(dk) Moleküler
Ağırlık (Da)
%2 TFA 26,405 7191 26,160; 31,181 8209; 59
%4 TFA 27,522 3610
27,167; 31,057 4576; 73
%6 TFA 27,725 3126 27,597; 31,111 3427; 67
%8 TFA 28,505 1671 28,494; 31,302 1688; 48
%10 TFA 28,768 1314
28,960; 31,358 1093; 44
4.1.6. Lignoselülozik Fraksiyonların Subkritik Suda Hidrolizinin İncelenmesi
Her bir lignoselülozik fraksiyondan (selüloz, hemiselüloz ve lignin) 4,30’ar
gram alınarak 200 ml su içinde 250ºC’de 2500 psi CO2 basıncında 45 dakika
boyunca ayrı ayrı hidroliz edilmiştir. Bu hidroliz işlemi sonucu elde edilen
çözeltilerin polisakkarit ve TOC içerikleri Çizelge 4.2’de; GPC analizlerine ait
kromatogramlar ise Şekil 4.12 ve Şekil 4.13’te verilmiştir.

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
60
Çizelge 4.2. Lignoselülozik fraksiyonların subkritik suda hidrolizinin sonucu oluşan polisakkaritler ve hidrolizatların TOC içerikleri
Örnekler
Ortalama Mol Kütleleri (Da)
TOC (ppm)
Buğday Samanı
Selüloz 75631; 29815 5256 ± 0,8
Hemiselüloz 78404; 30474 4180 ± 1,3
Lignin 77867; 35022; 14945 595 ± 0,3
Kenaf
Selüloz 72698; 28238 5216 ± 0,24
Hemiselüloz 79986; 31862; 14404 4985 ± 0,83
Lignin 60174; 29385 606,5 ± 0,08
Şekil 4.12. Buğday samanından izole edilen fraksiyonların (a) Selüloz, (b) Hemiselüloz, (c) Lignin, hidroliz çözeltilerinin GPC kromatogramları
(a) (b)
(c)
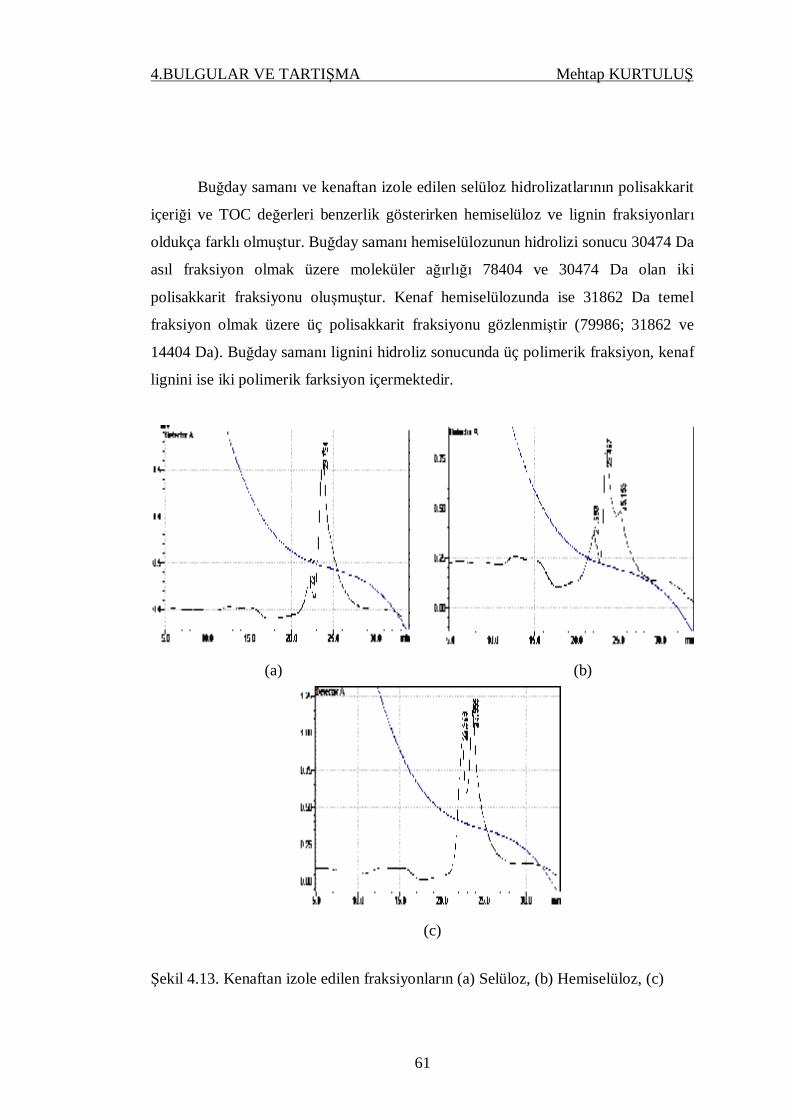
4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
61
Buğday samanı ve kenaftan izole edilen selüloz hidrolizatlarının polisakkarit
içeriği ve TOC değerleri benzerlik gösterirken hemiselüloz ve lignin fraksiyonları
oldukça farklı olmuştur. Buğday samanı hemiselülozunun hidrolizi sonucu 30474 Da
asıl fraksiyon olmak üzere moleküler ağırlığı 78404 ve 30474 Da olan iki
polisakkarit fraksiyonu oluşmuştur. Kenaf hemiselülozunda ise 31862 Da temel
fraksiyon olmak üzere üç polisakkarit fraksiyonu gözlenmiştir (79986; 31862 ve
14404 Da). Buğday samanı lignini hidroliz sonucunda üç polimerik fraksiyon, kenaf
lignini ise iki polimerik farksiyon içermektedir.
Şekil 4.13. Kenaftan izole edilen fraksiyonların (a) Selüloz, (b) Hemiselüloz, (c)
(a) (b)
(c)

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
62
Lignin, hidroliz çözeltilerinin GPC kromatogramları
4.2. Lignoselülozik Materyallerden İzole Edilen Fraksiyonların Sulu Faz
Reformlama ile Gazlaştırılması
4.2.1. Doğrudan Gazlaştırma
Buğday samanından izole edilen selüloz, hemiselüloz ve lignin
fraksiyonlarının katalizörsüz ve katalizörlü yapılan doğrudan gazlaştırma sonuçları
Çizelge 4.3’te ve Çizelge 4.4’te verilmiştir.
Çizelge 4.3. Buğday samanı lignoselüloziklerinin katalizörsüz gazlaştırılması sonucu oluşan gaz ürünleri
Buğday Samanı Oluşan
Gaz Hacmi(ml)
Gaz Bileşimi (%mol)
H2 CO2 CO CH4 C2H6
Selüloz 73,5 ± 45,9 - 83,6±9,2 13,4±4,9 3,0±1,2 -
Hemiselüloz 52,0 ± 2,8 - 60,6±3,6 39,4±3,6 - -
Lignin 38,1 ± 11,2 - 93,4±0,6 6,6±0,6 - -
Çizelge 4.4. Buğday samanı lignoselüloziklerinin katalizörlü gazlaştırılması sonucu oluşan gaz ürünleri
Buğday Samanı
Oluşan Gaz Hacmi
(ml)
Gaz Bileşimi (%mol)
H2 CO2 CO CH4 C2H6
Selüloz 479,5±2,1 21,5±4,9 64,5±9,9 0,2±0,4 7,6±4,3 6,2±3,3
Hemiselüloz 120,5±10,6 17,3±0,2 71,2±1,4 8,9±0,9 2,5±0,2 -
Lignin 38,1±1,4 - 80,3±6,3 10,8±1,2 3,7±0,1 -
1,071 g lignoselülozik materyalin 50 ml su içinde Pt içeriği 6 mg olacak
şekilde 0,12 g aktif karbon destekli %5 Pt katalizörü varlığında gazlaştırılması

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
63
sonucu oluşan gaz hacmi katalizörsüz deneye kıyasla belirgin şekilde artmıştır.
Katalizörsüz yapılan deneylerde oluşan gaz hacimleri 38-75 ml arasında değişirken,
aktif karbon üzerine tutturulmuş Pt katalizörünün kullanıldığı deneylerde lignin
hariç 120-479 ml arasında değişen bir gaz karışımı elde edilmiştir. Ligninin
katalizörlü ve katalizörsüz gazlaştırılması gaz hacminde bir artışa sebep olmamıştır.
Gaz miktarlarındaki bu farklılıklar lignoselülozik materyallerin yapı farklılığından
ve kullanılan reformlama katalizörünün bu yapılara olan seçiciliğinden
kaynaklanmaktadır.
Katalizörlü yapılan deneylerde gaz kompozisyonu da oldukça farklı
olmuştur. Katalizörsüz yapılan deneylerde hidrojen gazı oluşumu gözlenmezken
selüloz ve hemiselüloz fraksiyonlarıyla katalizörlü yapılan deneylerde H2, CO, CO2
ve CH4’ten oluşan bir gaz karışımı elde edilmiştir. Selülozla yapılan deneyde bu
bileşimlere ilaveten C2H6 da oluşmuştur. Ligninin katalizörlü gazlaştırılmasında
katalizörsüzdeki gibi hidrojen oluşumu gözlenmemiş ancak CO ve CO2 yanında
%3,7 oranında CH4 oluşumu gerçekleşmiştir.
Sulu faz reformlama deneylerinde Pt katalizörlünün iyi performans
gösterdiği önceki çalışmalardan bilinmektedir (Davda ve ark., 2003). Pt, hem C-C
hem de C-O bağ kopmasını katalizlemekte ve reformlama boyunca H2, CO, CO2,
CH4 gibi gazlar yanında diğer alkanlar da oluşabilmektedir. Sonuçlardan aktif
karbon destekli %5 Pt katalizörünün selülozun gazlaştırılmasında hemiselüloza göre
daha iyi performans gösterdiği görülmektedir. Bu fraksiyonun gazlaştırılması
sonucu daha fazla gaz hacmi elde edilmiş ve bu gaz karışımının hidrojen
kompozisyonu daha iyi olmuştur. Bu gaz karışımında CO miktarının oldukça düşük
olması CO + H2O ↔ H2 + CO2 su-gaz dönüşüm (water-gas shift) reaksiyonunun
daha kolay aktive edildiğini göstermektedir.
Alifatik hidrokarbon (CH4 ve C2H6) oluşumu en fazla selüloz fraksiyonunun
kullanıldığı deneyde gözlenmiştir. Bu fraksiyonun katalizörsüz yapılan gazlaştırma
deneyinde bile %3 oranında CH4 oluşumuna rastlanmıştır.
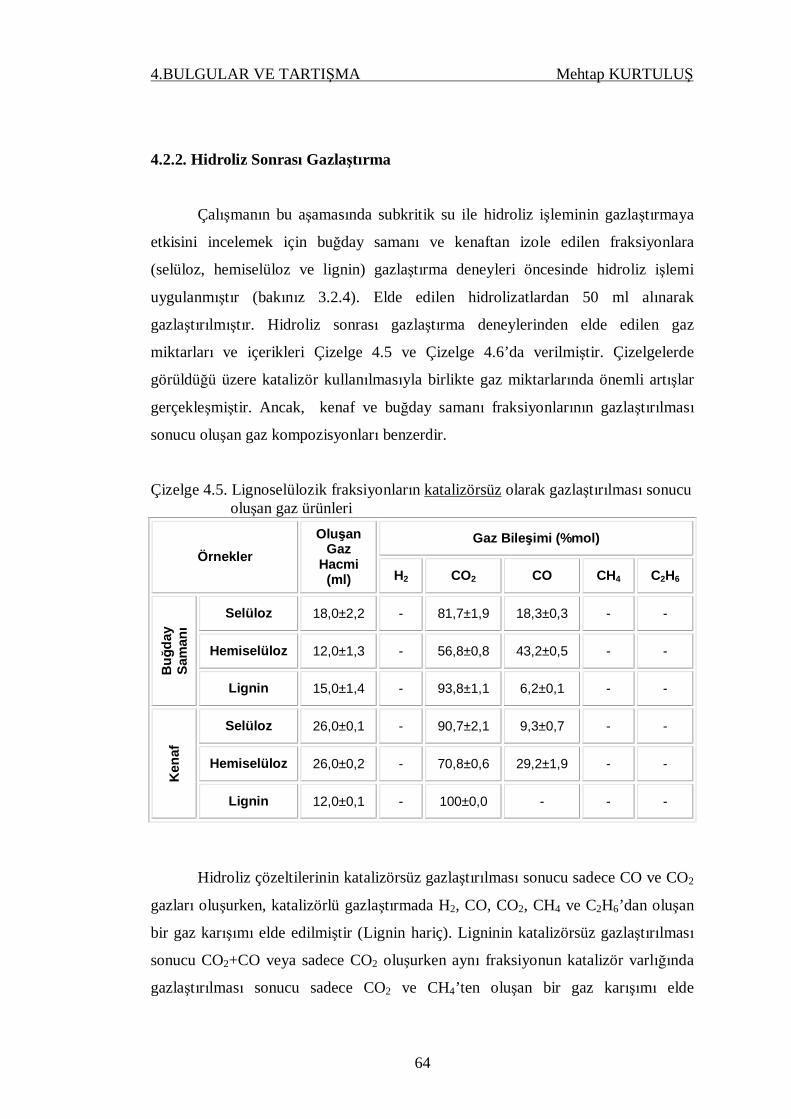
4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
64
4.2.2. Hidroliz Sonrası Gazlaştırma
Çalışmanın bu aşamasında subkritik su ile hidroliz işleminin gazlaştırmaya
etkisini incelemek için buğday samanı ve kenaftan izole edilen fraksiyonlara
(selüloz, hemiselüloz ve lignin) gazlaştırma deneyleri öncesinde hidroliz işlemi
uygulanmıştır (bakınız 3.2.4). Elde edilen hidrolizatlardan 50 ml alınarak
gazlaştırılmıştır. Hidroliz sonrası gazlaştırma deneylerinden elde edilen gaz
miktarları ve içerikleri Çizelge 4.5 ve Çizelge 4.6’da verilmiştir. Çizelgelerde
görüldüğü üzere katalizör kullanılmasıyla birlikte gaz miktarlarında önemli artışlar
gerçekleşmiştir. Ancak, kenaf ve buğday samanı fraksiyonlarının gazlaştırılması
sonucu oluşan gaz kompozisyonları benzerdir.
Çizelge 4.5. Lignoselülozik fraksiyonların katalizörsüz olarak gazlaştırılması sonucu oluşan gaz ürünleri
Örnekler Oluşan
Gaz Hacmi
(ml)
Gaz Bileşimi (%mol)
H2 CO2 CO CH4 C2H6
Buğ
day
Sam
anı
Selüloz 18,0±2,2 - 81,7±1,9 18,3±0,3 - -
Hemiselüloz 12,0±1,3 - 56,8±0,8 43,2±0,5 - -
Lignin 15,0±1,4 - 93,8±1,1 6,2±0,1 - -
Ken
af
Selüloz 26,0±0,1 - 90,7±2,1 9,3±0,7 - -
Hemiselüloz 26,0±0,2 - 70,8±0,6 29,2±1,9 - -
Lignin 12,0±0,1 - 100±0,0 - - -
Hidroliz çözeltilerinin katalizörsüz gazlaştırılması sonucu sadece CO ve CO2
gazları oluşurken, katalizörlü gazlaştırmada H2, CO, CO2, CH4 ve C2H6’dan oluşan
bir gaz karışımı elde edilmiştir (Lignin hariç). Ligninin katalizörsüz gazlaştırılması
sonucu CO2+CO veya sadece CO2 oluşurken aynı fraksiyonun katalizör varlığında
gazlaştırılması sonucu sadece CO2 ve CH4’ten oluşan bir gaz karışımı elde

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
65
edilmiştir. Bu durum buğday samanı ve kenaf lignini için aynı olmuştur. Katalizör
kullanılmasıyla birlikte en fazla gaz oluşumu selüloz fraksiyonunda gerçekleşirken
hidrojen gazı verimi açısından en iyi fraksiyon hemiselüloz olmuştur. Bu durum
çalışılan her iki lignoselülozik materyal için de benzerdir.
Çizelge 4.6. Lignoselülozik fraksiyonların katalizör varlığında gazlaştırılması sonucu oluşan gaz ürünleri
Örnekler Oluşan
Gaz Hacmi
(ml)
Gaz Bileşimi (%mol)
H2 CO2 CO CH4 C2H6
Buğ
day
Sam
anı
Selüloz 121,5±4,9 20,7±3,5 67,6±5,7 1,8±0,5 8,0±0,7 1,9±0,3
Hemiselüloz 63,5±0,7 27,0±4,0 58,8±6,2 0,7±0,9 10,7±1,1 2,8±0,1
Lignin 19,0±1,4 - 92,8±2,8 - 7,2±2,8 -
Ken
af
Selüloz 140,0±4,9 21,1±4,7 66,9±8,2 1,7±0,6 8,7±2,4 1,6±0,3
Hemiselüloz 111,0±4,2 24,0±2,3 68,0±3,9 - 6,3±1,2 1,7±0,4
Lignin 19,0±1,4 - 91,7±2,3 - 8,3±2,3 -
Literatürde lignin, selüloz ve hemiselüloz yapılarının sulu faz reformlama ile
gazlaştırılmasına ilişkin bir çalışma mevcut değidir. Ancak, Yoshida ve Matsumura
(2001), farklı miktarlarda standart selüloz, ksilan ve ligninden (izole edildikleri
kaynaklar belirtilmemiş) oluşan karışımların 673 K ve 25 Mpa süperkritik su
ortamında nikel katalizörü kullanarak ve 20 dk süredeki gazlaştırma etkinliklerini
incelemişlerdir. Karışımın lignin içeriğinin oluşan gazın miktarını ve
kompozisyonunu belirgin şekilde değiştirdiği gözlenmiştir. Lignin içeriği arttıkça
oluşan gaz miktarı ve bu gaz karışımındaki hidrojen komposizyonu belirgin şekilde
azalmıştır.

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
66
4.2.3. Ksilanın Gazlaştırılması
Ksilan iskelet yapısı yaklaşık % 80’i ksiloz ünitelerine C-2 ve/veya C-3 ile
bağlı arabinoz veya glukoüronik asitten oluşan monomerik yan zincirden ve
arabinoz, ksiloz ve bazende galaktoz ünitelerinden oluşan oligomerik yan zincirden
oluşur. Yumuşak odun heteroksilanında arabinofuranosil birimleri p-kumarik asit ve
ferulik asitle esterleşmiştir. Sert odun ksilanında ksiloz birimlerinin % 60-70’i
asetilleşmiştir. Sert odun ksilanının polimerizasyon derecesi (150-200) yumuşak
odun ksilanınkinden (70-130) yüksektir (Obembe, 2006). Bu çalışmada buğday
samanı ve kenaf sapından izole edilen hemiselüloz fraksiyonlarını belirli
standartlarla karşılaştırabilmek için kayın ağacından (sert odun) (Beechwood, BW)
ve yulafxbuğdaydan (otsu) (oat spelts, OS)’ten izole edilen ve sırasıyla Sigma ve
Serva firmalarından temin edilen ksilanlar standart model fraksiyonlar olarak
kullanılmıştır. Standart ksilanların hidroliz işleminden sonra katalizörsüz ve
katalizörlü (aktif karbon destekli %5 Pt katalizörü) olarak gazlaştırılması sonucu
oluşan gaz miktarları ve gaz kompozisyonları Çizelge 4.7’de verilmiştir.
Çizelge 4.7. Standart ksilanların katalizörsüz ve katalizörlü olarak gazlaştırılması sonucu oluşan gaz ürünleri
APR Deneyleri Oluşan
Gaz Hacmi
(ml)
Gaz Bileşimi (%mol)
H2 CO2 CO CH4 C2H6
Katalizörsüz BW 18,0±1,6 - 66,5±5,7 33,5±1,4 - -
OS 12,0±2,4 - 47,3±3,3 52,7±4,7 - -
% 5 Pt-Akt. Karbon
BW 72,0±0,1 25,8±0,1 65,3±0,1 - 6,2±0,1 2,8±0,1
OS 98,0±0,1 22,9±0,2 70,2±0,1 - 5,1±0,4 1,9±0,1
BW ve OS fraksiyonlarının katalizör varlığında gazlaşırılması sonucu
buğday samanı ve kenaf hemiselülozunda oluşan gazlara rastlanmıştır (H2, CO2, CH4
ve C2H6) ancak bunların % mol miktarları biraz farklılık göstermiştir.

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
67
Şekil 4.14’te buğday samanı ve kenaftan izole edilen hemiselüloz
fraksiyonları ile BW ve OS ksilanlarının katalizör (aktif karbon destekli %5 Pt)
varlığında gazlaştırma sonuçları karşılaştırmalı olarak gösterilmiştir. Görüldüğü
üzere CO2 gazı tüm hemiselüloz fraksiyonlarında en yüksek % mol bileşimine
sahiptir. En fazla H2 gazı buğday samanından izole edilen hemiselülozun
gazlaştırılması sonucu elde edilmiştir (%27). Diğer gazların oranları incelendiğinde
önemli bir değişiklik gözlenmemiştir.
0
10
20
30
40
50
60
70
80
Buğday Samanı Kenaf Beechwood Oat Spelts
%m
ol
H2CO2COCH4C2H6
Şekil 4.14. Buğday samanı ve kenaf sapından izole edilen hemiselüloz fraksiyonlarının Pt katalizörü varlığında gazlaştıılması sonucu oluşan gaz kompozisyonlarının standart ksilanlarla karşılaştırılması
4.3. Toplam Organik Karbon Analizleri
Buğday samanı ve kenaftan izole edilen selüloz fraksiyonlarının gazlaştırma
öncesi ve gazlaştırma sonrası elde edilen çözeltilerindeki toplam organik karbon
içerikleri Şekil 4.15’te gösterilmiştir.
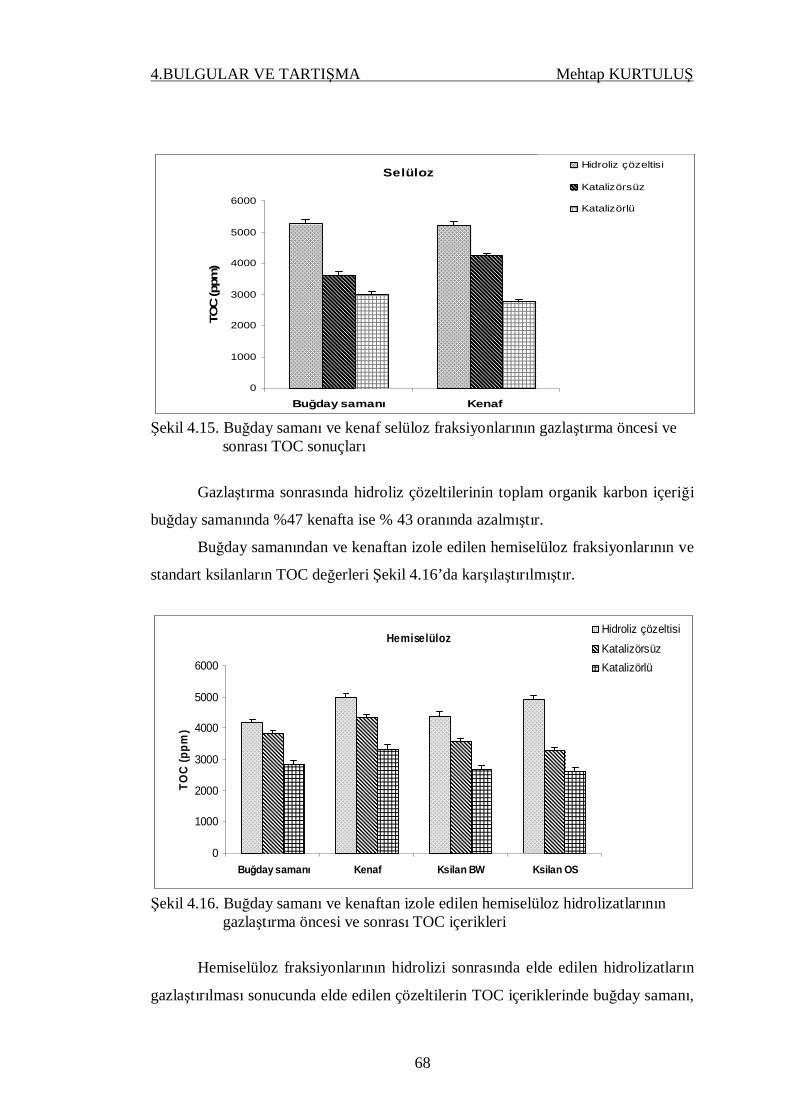
4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
68
Selüloz
0
1000
2000
3000
4000
5000
6000
Buğday samanı Kenaf
TOC
(ppm
)
Hidroliz çözeltisi
Katalizörsüz
Katalizörlü
Şekil 4.15. Buğday samanı ve kenaf selüloz fraksiyonlarının gazlaştırma öncesi ve sonrası TOC sonuçları
Gazlaştırma sonrasında hidroliz çözeltilerinin toplam organik karbon içeriği
buğday samanında %47 kenafta ise % 43 oranında azalmıştır.
Buğday samanından ve kenaftan izole edilen hemiselüloz fraksiyonlarının ve
standart ksilanların TOC değerleri Şekil 4.16’da karşılaştırılmıştır.
Hemiselüloz
0
1000
2000
3000
4000
5000
6000
Buğday samanı Kenaf Ksilan BW Ksilan OS
TOC
(ppm
)
Hidroliz çözeltisi
KatalizörsüzKatalizörlü
Şekil 4.16. Buğday samanı ve kenaftan izole edilen hemiselüloz hidrolizatlarının gazlaştırma öncesi ve sonrası TOC içerikleri
Hemiselüloz fraksiyonlarının hidrolizi sonrasında elde edilen hidrolizatların
gazlaştırılması sonucunda elde edilen çözeltilerin TOC içeriklerinde buğday samanı,

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
69
kenaf, kayın ağacı-ksilan ve yulafxbuğday-ksilan için sırasıyla yaklaşık %32, %33,
%39 ve %47 azalma gözlenmiştir. Buğday samanı ve kenafın hemiselüloz
fraksiyonlarınn TOC azalışı selülozdan daha az olmuştur.
Lignin fraksiyonlarına ait TOC değerleri Şekil 4.17’de verilmiştir. Ligninin
başlangıçtaki hidroliz çözeltileri ile katalizörsüz gazlaştırma deneylerinden elde
edilen çözeltilerin TOC değerleri karşılaştırıldığında çok önemli bir değişikliğin
olmadığı gözlenmiştir. Ancak her iki biyokütle için de başlangıçtaki hidroliz
çözeltileri ile katalizörlü gazlaştırma deneylerinden elde edilen çözeltilerin TOC
değerleri karşılaştırıldığında önemli oranda bir azalmanın olduğu gözlenmiştir.
Lignin fraksiyonlarının hidrolizi sonrasında elde edilen hidrolizatların
gazlaştırılması sonucunda elde edilen çözeltilerin TOC içeriklerinde buğday samanı,
ve kenaf için sırasıyla %47 ve %57 oranında azalma gerçekleşmiştir.
Lignin
0
100
200
300
400
500
600
700
Buğday samanı Kenaf
TOC
(ppm
)
Başlangıç Çözeltisi
Kör
Katalizörlü
Şekil 4.17. Buğday samanı ve kenaftan izole edilen lignin fraksiyonlarının gazlaştırma öncesinde ve sonrasında TOC sonuçları
Selüloz, hemiselüloz ve lignin fraksiyonları karşılaştırıldığında TOC
değerlerindeki azalmanın en fazla ligninde gerçekleştiği görülmektedir. Bu
fraksiyonun gazlaştırılması sonucu sadece CO2 (%92-93) ve CH4 (%7-8)’ten
meydana gelen karbonlu ürünlerin oluşması TOC değerlerindeki bu azalmayı
desteklemektedir.

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
70
0
500
1000
1500
2000
2500
3000
3500
4000
4500
Selüloz Hemiselüloz Lignin
TOC
(ppm
)
Doğrudan gazlaştırma
Hidroliz sonrası gazlaştırma
Şekil 4.18. Buğday samanı fraksiyonlarının doğrudan ve hidroliz sonrası gazlaştırma deneyleri sonrası TOC içerikleri
Şekil 4.18’de hidrolizin gazlaştırma üzerine etkisini inceleyebilmek için
buğday samanı fraksiyonlarının doğrudan ve hidroliz sonrası gazlaştırma işleminden
sonraki TOC değerleri karşılaştırılmıştır. Hidroliz işleminin selülozun APR sonrası
TOC içeriğine çok fazla etki etmediği görülmektedir. Ancak hemiselüloz ve lignin
fraksiyonlarında hidrolizin etkisi daha belirgindir. Her iki fraksiyonda da hidroliz
sonrası gazlaştırma TOC değerleri azalmıştır.
TOC’daki azalma gaz ürünlere dönüşümden dolayı olacağı gibi APR’den
sonra çözeltide gözlenen ve saptanamayan az miktardaki çözünmez durumdaki
karbonik materyalden dolayı da olabilir.
4.4. Gazlaştırma Sonrası Çözeltilerin Polisakkarit İçerikleri
Hidroliz işlemi uygulanmış lignoselülozik hidrolizatların gazlaştırılması
işleminden sonra kalan çözeltide polisakkarit analizleri yapılmıştır.
Katalizörlü ve katalizörsüz ortamda 250°C’de yapılan gazlaştırma deneyleri
sonunda çözeltilerin polisakkarit içerikleri Çizelge 4.8’de verilmiştir. Gazlaştırmada
kullanılan çözeltideki polisakkarit bileşimi ile kıyaslandığında buğday samanının
selülozunun APR deneyi sonrası daha küçük molekül ağırlıklı polisakkarit
fraksiyonlarına dönüştüğünü göstermektedir (Çizelge 4.8 ve Şekil 4.19a). Benzer

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
71
durum hemiselüloz ve lignin fraksiyonlarında da gözlenmiştir (Çizelge 4.8 ve Şekil
4.19 b ve c).
Hemiselüloz fraksiyonunda büyük molekül ağırlıklı polisakkaritlerin miktarı
iyice azalırken küçük molekül ağırlıklı polisakkarit fraksiyonunun baskın olduğu
görülmektedir Şekil 4.19b). Lignin fraksiyonunda oluşan fraksiyonlarda ise küçük
molekül ağırlıklı fraksiyonun miktarı azalırken daha büyük ağırlıklı olanın özellikle
katalizörlü deneyde iyice baskın hale geldiği gözlenmiştir (Şekil 4.19c).
Çizelge 4.8. Lignoselülozik fraksiyonların katalizörlü ve katalizörsüz olarak gazlaştırılması sonucu çözeltilerin polisakkarit içerikleri
Lignoselülozik Fraksiyonlar
APR Öncesi Hidroliz Çözeltisi
Polisakkarit Fraksiyonu, Da
APR Sonrası Polisakkarit Fraksiyonu
Katalizörsüz, Da
APR Sonrası Polisakkarit Fraksiyonu
Katalizörlü, Da
Buğday Samanı
Selüloz 75631; 29815
66933; 26431; 14542
64379; 29089;
19377
Hemiselüloz 78404; 30474
65044; 27354; 15615
64965; 30247;
14794
Lignin
77867; 35022; 14945
67814; 42656 61376; 31703
Kenaf
Selüloz 72698; 28238
7079; 29746; 17928
78895; 33964
Hemiselüloz
79986; 31862; 14404
79112; 31988;
16119
73636; 30882;
16882
Lignin 60174; 29385 64424; 32177 72259; 32671
Standart Ksilan
BW 65252; 21095 73745; 31447 78137; 33454
OS 81255; 33833 72435; 27614 78285; 32016

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
72
(a) (b)
(c) Şekil 4.19. Buğday samanı lignoselüloziklerinin GPC kromatogramları (a) Buğday samanından izole edilen selüloz, (b) Buğday samanından izole edilen hemiselüloz, (c) Buğday samanından izole edilen lignin
Kenaf selülozu katalizör varlığında gazlaştırıldığında polisakkarit derişiminin
oldukça azaldığı gözlenmiştir (Şekil 4.20 a ve c). Standart ksilan fraksiyonlarının
gazlaştırılmasında ise daha önce çok az olan polisakkarit bileşiminin katalizörlü ve
katalizörsüz her iki durumda genellikle molekül ağırlığında artışa sebep olduğu
gözlenmiştir (Çizelge 4.8; Şekil 4.20 a ve b).

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
73
(a) (b)
(c) Şekil 4.20. Kenaf sapı lignoselüloziklerinin GPC kromatogramları (a)Kenaf sapından izole edilen selüloz, (b) Kenaf sapından izole edilen hemiselüloz, (c) Kenaf sapından izole edilen lignin
(a) (b) Şekil 4.21. Standart ksilanların GPC kromatogramları (a) Kayın ağacı (Beechwood) standart ksilanı, (b)Yulafxbuğday (Oat Spelts) standart ksilanı

4.BULGULAR VE TARTIŞMA Mehtap KURTULUŞ
74

5. SONUÇLAR VE ÖNERİLER Mehtap KURTULUŞ
75
5. SONUÇLAR VE ÖNERİLER
• İzole edilen selüloza ait kristal indeks değeri %57,84; standart selülozunki ise
%79,76 olarak hesaplanmıştır. Değerlerin bu şekilde farklılık göstermesi,
standart selülozun oldukça kristallin bir yapıya sahipken izole selülozda
amorf yapıların standarda göre daha fazla olduğu anlaşılmaktadır.
• Yapılan XRD analizi sonucunda hemiselüloz ve lignin yapıları kristalin yapı
göstermediğinden; 2θ=22.4º’de 002 düzlemindeki kristal alan kırılma
yoğunluğu gözlenememiştir. Bu durum bu yapıların selüloz fraksiyonundan
tamamen izole bir şekilde saflaştırılabildiğini göstermiştir.
• Standart selüloz ve izole selülozun trifluoro asetik asit ile hidrolizi benzer
GPC kromatogramlarını vermiştir. Hidroliz sonrası oluşan polisakkarit
fraksiyonlarının moleküler ağırlığının orijinal selülozdan biraz fazla çıkması
aglomerizasyonun gerçekleştiğini göstermiştir.
• Buğday samanı ve kenaftan izole edilen selülozun hidrolizatların polisakkarit
içeriği ve TOC değerleri benzerlik gösterirken hemiselüloz ve lignin
fraksiyonları oldukça farklı olmuştur.
• Buğday samanından izole edilen selüloz fraksiyonun doğrudan ve hidroliz
sonrası APR yöntemiyle gazlaştırılması sonucu benzer gaz kompozisyonları
elde edilmiştir.
• Lignoselülozik materyallerden izole edilen fraksiyonların gazlaştırılması
sonucu oluşan gaz miktarları farklılık göstermiştir. Bu farklılıkların
lignoselülozik materyallerin yapı farklılığından ve kullanılan reformlama
katalizörünün bu yapılara olan seçiciliğinden kaynaklandığı düşünülmektedir.
• Doğrudan gazlaştırma sonuçlarından aktif karbon destekli %5 Pt
katalizörünün selülozun gazlaştırılmasında hemiselüloza göre daha iyi
performans gösterdiği görülmektedir. Selülozun gazlaştırılması sonucu daha
fazla gaz hacmi elde edilmiş ve bu gaz karışımının hidrojen kompozisyonu
daha iyi olmuştur.

5. SONUÇLAR VE ÖNERİLER Mehtap KURTULUŞ
76
• Hidroliz sonrası gazlaştırma deneylerinde katalizör kullanılmasıyla birlikte en
fazla gaz oluşumu selüloz fraksiyonunda gerçekleşirken hidrojen gazı verimi
açısından en iyi fraksiyon hemiselüloz olmuştur.
• Gazlaştırmada kullanılan çözeltideki polisakkarit bileşimi ile kıyaslandığında
buğday samanının selülozu APR deneyi sonrası daha küçük molekül ağırlıklı
polisakkarit fraksiyonlarına dönüşmüştür. Benzer durum hemiselüloz ve
lignin fraksiyonlarında da gözlenmiştir.

77
KAYNAKLAR
ADOSINDA, M., MARTINS, M., FERREIRA, ISABEL, C., 2002. Biodegradation
of bioaccessible textile azo dyes by Phanerochaete chrysosporium. Journal of
Biotechnology, 89, 91-98.
ANONİM, 1984. Difco Manuel, Dehydrate Culture Media and Reagents for
Microbiology. Detroit, Michigan USA, 1155.
ANTAL, M.J., LEESOMBOON, T., MOK, W. S., 1991. Carbohydrate Research,
217, 71-85.
ASPINAL, G. O., 1970 . Polysaccharides, Pergamon Press Headington Hill Hall,
Oxford, 431-438.
AZADI, P., KHODADADI, A.A., MORTAVAZI, Y., FARNOOD, R., 2008.
Hydrothermal gasification of glucose using Raney nickel and homogeneous
organometallic catalysts. Fuel Processing Technology,90, 145-151.
AZZAM, A.M., 1989. Pretreatment of cane bagasse with alkaline hydrogen peroxide
for enzymatic hydrolysis of cellulose and ethanol fermentation. Journal of
Enviromental Science and Health B, 24, 421–433.
BEGUIN, P., AUBERT, J.P., 1994. The biological degradation of cellulose. FEMS
Microbiological. Reviews, 13, 25–58.
BEN-GHEDALIA, D., SHEFET, G., 1983. Chemical treatments for increasing the
digestibility of cotton straw. J. Agrictural Science, 100, 393–400.
BEYATLI, Y., 1996. Biyoteknoloji ve Biyoprotein Üretimi, Kükem Derneği, 19, 23.
BOBLETER, O., CONSIN R., 1979. Degradation of popular lignin by hydrothermal
treatment. Cellulose Chemistry and Technology. 13, 583-593.
BOBLETER O.,1994. Hydrothermal degradation of polymers derived from plants,
Progress in Polymer Science, 19, 797-841.
BREEN, A., SIGLETON, F.L., 1999. Fungi in lignocellulose and biopulping.
Current Opinion in Biotechnology, 10, 252-258.
BRIDGWATER, A.V., BRIDGE, S.A., 1991. A Review of Biomass Pyrolysis and
Pyrolysis Technologies, Elsevier Applied Science, London.

78
BROWNE, F. L., 1958. Theories of the Combustion of Wood and its Control, Rept
No. 2136 Forest Products Lab., Madison,WI.
BURANOV, A.U., MAZZA, G., 2010. Extraction and characterization of
hemicelluloses from flax shives by different methods. Carbohydrate
Polymers, 79, 17–25.
CADOCHE, L., LOPEZ, G.D., 1989. Assessment of size reduction as a preliminary
step in the production of ethanol from lignocellulosic wastes. Biological
Wastes, 30, 153–157.
CHRESTINI, C., SERMANNI, G.G., ARGYROPOULOS, D.S., 1998. Structural
modifications induced during biodegradation of wheat lignin by Lentinula
edodes. Bioorganic & Medicinal Chemistry, 6, 957-973.
CHUM, H.L., JOHNSOON, D.K., BLACK, S., 1988. Organosolv pretreatment for
enzymatic hydrolysis of poplars: 1. enzyme hydrolysis of cellulosic residues.
Biotechnology and Bioengergy, 31, 643–649.
CLARK, T.A., MACKIE, K.L., 1987. Steam explosion of the soft-wood Pinus
radiata with sulphur dioxide addition. I. Process optimization. J.
Wood Chemical Technology, 7, 373–403.
CORREDOR, D.Y., 2008. Pretreatment and enzymatic hydrolysis of lignocellulosic
biomass. National University of Colombia, 2000 MS. Kansas State
University, 2005.
CORTRIGHT, R.D., DAVDA, R.R., DUMESIC, J.A., 2002. Hydrogen from
catalytic reforming of biomass-derived hydrocarbons in liquid water, Nature,
418, 964-967.
CRAWFORD, D.L., BARDER, M.J., POMETTO, A.L., III., CRAWFORD, R. L.,
1982. Chemistry of softwood lignin degradation of softwood lignin
degradation by Streptomyces viridoendosporus. Archives of Microbiology,
131, 140-145.
ÇAĞLAR, A., DEMİRBAŞ, A., 2000. Conversion of Cotton Cocoon Shell to Liquid
Products by Pyrolysis, Energy Conversion and management, 41, 1749 –
1756.

79
ÇAĞLAR, A., DEMİRBAŞ, A., 2001. Conversion of Cotton Cocoon Shell to Liquid
Products by Supercritical Fluid Extraction and Low Pressure Pyrolysis in the
Presence of Alkalis, Energy Conversion and Management, 42, 1095 – 1104.
ÇAĞLAR, A., DEMİRBAŞ, A., 2002. Conversion of Cotton Cocoon Shell to
Hydrogen Rich Gaseous Products by Pyrolysis, Energy Conversion and
Management, 43, 489 –497.
ÇETİNKAYA, M., KARAOSMANOĞLU, F., 2003. Türkiye enerji profili ve. II.
Hidrojen Kongresi, (9 Temmuz 2003) Bildirileri, Filiz Karaosmanoğlu,
Merve Çetinkaya, Ertim Orkun (Editörler) Ankara, 25-40.
ÇULCUOĞLU, E., ÜNAY, E., ANGIN, D.,ŞENSÖZ, S., KARAOSMANOĞLU, F.
2005. Characteriza the Bio-Oil of Rapeseed Cake. Energy Sources, 27, 1217-
1223.
DALE, B.E., MOREIRA, M.J., 1982. A freeze-explosion technique for increasing
cellulose hydrolysis. Biotechnology Bioengergy Symposium, 12, 31–43.
DAVDA, R.R., SHABAKER, J.W., HUBER, G.W., CORTRIGHT, R.D.,
DUMESIC, J.A., 2003. Aqueous-phase reforming of ethylene glycol on
silica-supported metal catalysts. Applied Catalysis B, 43, 13–26.
DAVDA, R.R., DUMESIC, J.A., 2003. Catalytic reforming of oxygenated
hydrocarbons for hydrogen with low levels of carbon monoxide, Angewandte
Chemie Intetnational Edition, 42, 4068-4071.
DAVDA, R.R., DUMESIC, J.A., 2004. Renewable hydrogen by aqueous-phase
reforming of glucose, Chemical Communications, (Cambridge UK), 36–37.
DAVDA, R.R., SHABAKER, J.W., HUBER, G.W., CORTRIGHT, R.D.,
DUMESIC, J.A., 2005. A review of catalytic issues and process conditions
for renewable hydrogen and alkanes by aqueous-phase reforming of
oxygenated hydrocarbons over supported metal catalysts. Applied Catalysis
B: Environmental, 56, 171-186.
DEMİRBAŞ, A., 1996. Lignoselülozik Ürünlerin Katalitik Yöntemle Parçalanması,
Doğa dergisi, 10, 1- 25-33.
DEMİRBAŞ, A., 2001. Biomass resource fascilities and biomass conversion
processing for fuels and chemicals. Energy Convers Manage, 42, 1357–1378.

80
DUMESIC, J. A., DAVDA, R. R., ALCALA, R., SHABAKER, J. HUBER, G.,
CORTRIGHT, R. D., MAVRIKAKIS, M., 2002. Hydrogen Generation by
Catalytic Reforming of Oxygenated Hydrocarbonsin Liquid Water. Nature
418, 964-967.
EGGLESTON, G., VERCELLOTTI, J.R., 2000. Degradation of sucrose glucose and
fructose in concentrated aqueous solutions under constant pH conditions at
elevated temperature, Journal of Carbohydrate Chemistry, 19, 1305-1318.
ELKE, L., KLEEBERG, I., ZADRAZIL, F., 1997. Competition of Pleurotus sp. And
Dichomitus squalens with soil microorganisms during lignocellulose
decomposition.Bioresource Technology, 60, 95-99.
ESTEGHLALIAN, A., HASHIMOTO, A.G., FENSKE, J.J., PENNER, M.H., 1997.
ERIKSSON, K.E., BLANCHETTE, R.A., ANDER, P., 1990. Biodegradation of
lignin. In Microbial and enzymatic dagradation of wood and wood
components, 225-333. Springer-Verlag KG, Berlin.
FAHMI, R., BRIDGWATER, A.V., DONNISON, I., YATES, N., JONES, J.M.,
2008. The effect of lignin and inorganic species in biomass on pyrolysis oil
yields, quality and stability. Fuel 87, 1230–1240.
FAN, L.T., GHARPURAY, M.M., LEE, Y.H., 1987. In:Cellulose Hydrolysis
Biotechnology Monographs. Springer, Berlin, p. 57.
FENGEL D. WEGENER, G., 1984. Wood Chemistry, Ultrastructure, Reactions
Volume I., Walter de Gruyter, Berlin-New York.
FENGEL, D., WEGENER, G., 1989. Wood: chemistry, ultrastructure,
reactions.Walter de Gruyter: Berlin.
FOYLE, T., JENNINGS, L., MULCAHY, P., 2007. Compositional analysis of
lignocellulosic materials: Evaluation of methods used for sugar analysis of
waste paper and straw. Bioresource Technology, 98, 3026-3036.
GERÇEL, H.F., 2002. Production and Characterization of Pyrolysis Liquids From
Sunflower-Pressed Bagasse. BioresourceTechnology, 85, 113-117.
GRAY, M.C., CONVERSE, A.O., WYMAN, C.E., 2007. Solubilities of oligomer
mixtures produced by the hydrolysis of xylans and corn stover in water at 180
°C. Industrial Engineering Chemstry Research, 46, 2383-2391.

81
GRUBISIC, A., REMPP, P., BENOIT, H.A., Molecular characterization of
polysaccharides dissolved in Me2NAc-LiCl by gel-permeation
chromatography, Polymers Letters, 5 (1967) 753-759.
GUIRAUD, P., STEIMAN, R., AIT-LAYDI, L., MURANDI, F.S., 1998.
Degradation of phenolic and chloroaromatic compounds by coprinus spp,
Chemosphere, 38, 2775-2789.
GUPTA, S., MADAN, R.N., BANSAL, M.C., 1987. Chemical composition of Pinus
caribaea hemicellulose.Tappi Journal, 70,113-116.
HAENSEL, T., COMOUTH, A., LORENZ, P., AHMED, S., 2009. Schaefer
Pyrolysis of cellulose and lignin. Applied Surface Science, 255, 8183–8189.
HAIMER, E., WENDLAND, M., POTTHAST, A., ROSENAU, T., LIEBNER, F.,
2008. Precipitation of Hemicelluloses from DMSO/Water Mixtures Using
Carbon Dioxide as an Antisolvent. Journal of Nanomaterials.
HATTAKA, A.I., 1983. Pretreatment of wheat straw by white-rot fungi for
enzymatic saccharification of cellulose. Applied Microbiology and
Biotechnolgy, 18, 350–357.
HIGUCHI, T., CHANG, H.M., KIRK, T.K., 1981. Lignin biodegradation:
microbiology, chemistry, and potential applications; CRC Pres, Inc., Boca
Raton, FL.
HIROFIMI, H., TAKONORI, I., RYUICHIRO, K., 1999. Intracellular ferrireductase
involved in Mn(IV) reducing enzyme system to supply Mn(II) for lignin
biodegradation by white-rot fungus Phanerochaete sordida YK-624. Enzyme
and Microbial Technology, 30, 467-473.
HOLTZAPPLE, M.T., DAVISON, R.R., STUART, E.D., 1992. Biomass refining
process, US patent 5, 171, 592.
HSIEH, C., TSENG, M.H., LIU, C.J., 2006. Production of polysaccharides from
Ganoderma lucidum (CCRC 36041) under limitations of nutrients, Enzyme
Microb. Technol. 38, 109–117.

82
HUBER, G.W., DUMESIC, J.A. 2006. An overview of aqueous-phase catalytic
processes for production of hydrogen and alkanes in a biorefinery. Catalysis
Today, 111, 119-132.
http://commons.wikimedia.org/wiki/File:Lignin_structure.svg
http://chemistry.umeche.maine.edu/CHY431/Wood14.html
ISLAM, M.N., ALAMBEG, M. R., ISLAM, M.R., 2005. Pyrolytic oil f pyrolysis of
municipal solid waste and its characterization. Renewable Energy, 30, 413-
420.
JOHANSSON, E., KRANTZ-RULTCKER, C., ZHANG, B. X., 1999. Chlorination
and biodegradation of lignin . Soil Biology & Biochemistry, 1029-1032.
KILZER, F.J., BROIDO, A., 1965. Speculations on the nature of cellulose pyrolysis.
Pyrodynamics 2, 151–163.
KIM, S., HOLTZAPPLE, M.T., 2006. Effect of structural features on enzyme
digestibility of corn stover. Bioresource Technology 97, 583-591.
KLASON, P., HEIDENSTOM, G.V., NORLIA, E., 1910. Investigation into the
carbonization of wood. II. Dry distillation of pine,spruce, birch and beech
wood. Zeitschriftfur Angewandte Chemie 23, 1252-1257.
KOLLMANN, F., 1951. Technology of Wood and Wood-Base Products. Springer
Verlag, Berlin.
LATEEF, H., GRIMES, S., KEWCHAROENWONG, P., FEINBERG, B., 2009.
Separation and recovery of cellulose and lignin using ionic liquids: a process
for recovery from paper-based waste. Journal of Chemical Technology &
Biotechnology, 84, 12, 1818-1827.
LEI, H., SONG, Z., TAN, D., BAO, X., MU, X., ZONG, B., MIN, E., 2001.
Preparation of novel Raney-Ni catalysts and characterization by XRD, SEM
and XPS, Applied Catalysis A, 214, 69-76.
LIU, C., WYMAN, C.E., 2003. The effect of flow rate of compressed hot water on
xylan, lignin, and total mass removal from corn stover. Industrial Engineering
Chemstry Research, 42, 5409-5416.
LJUNGDAHL, L.G., ERIKSSON, K.E., 1985. Ecology of microbial Cellulose
Degradation . Adv. Microbiology Ecology, 8, 237-299.

83
LODHI, M.A.K., 1987. Hydrogen Production from Renewable Sources of Energy,
International Journal of Hydrogen Energy, 12, 7 461-468.
LV, D., XU, M., LIU, X., ZHAN, Z., LI, Z., YAO, H., 2009. Effect of cellulose,
lignin, alkali and alkaline earth metallic species on biomass pyrolysis and
gasification. Fuel Processing Technology.
LV, P., YUAN, Z., WU C., MA L., CHEN Y., TSUBAKI, N., 2007. Bio-syngas
production from biomass catalytic gasification, Energy Conversion and
Management, 48, 1132-1139.
MANSILLA, H.D., BAEZA, J., URZUA, S., MATURANA, G., VILLASENOR, J.,
DURAN, N., 1998. Acid-Catalysed Hydrolysis of Rice Hull: Evaluation of
Furfural Production, Bioresource Technology, 66 ,189-193.
MARTINEZ, A.T., CAMERERO, S., GUTIERREZ, A., 2001. Studies on wheat
lignin degradation by Pleurotus species using analytical pyrolysis. Journal of
Analytical and Applied Pyrolysis, 59, 401-411.
MASCHIO, G., KOUTOPANOS, C., LUCCHESI, A., 1992. Pyrolysis a Promising
Route for Biomass Utilization, Bioresource Technology, 42 , 219 – 231.
MASHCHIO, G., LUCCHES, A., STOPPATO, G., 1994. Production of Syngaz from
Biomass, Bioresource Technology, 48, 119-126.
MCMILLAN, J.D., 1994. Pretreatment of lignocellulosic biomass. In:Himmel, M.E.,
Baker, J.O., Overend, R.P. (Eds.), Enzymatic Conversion of Biomass for
Fuels Production. American Chemical Society, Washington, DC, pp.
292–324.
MEIER, D., FAIX, O., 1999. State of Theart of Applied Fast Pyrolysis of
Lignocellulosic Materials-A Review. Bioresource Technology, 68, 71-77.
MERYEMOĞLU B., HESENOV A., IRMAK S., ATANUR O.M., ERBATUR O.,
2010. Aqueous Phase Reforming of Biomass Using Various Types of
Supported Precious Metal and Raney Nickel Catalysts for Hydrogen
Production. Submitted to International Journal of Hydrogen Energy.
MES-HARTREE, M., DALE, B.E., CRAIG, W.K., 1988. Comparison of steam and
ammonia pretreatment for enzymatic hydrolysis of cellulose. Applied
Microbiology Biotechnology, 29, 462-468.

84
MICIC, M., BENITEZ, I., RUANO, M., MAVERS, M., JEREMIC, M., RADOTIC,
K., 2001. Probing the lignin nanomechanical properties and lignin-lignin
interactions using the atomic force microscopy. Chemical Physics Letters;
347, 41-5.
MILLET, M.A., BAKER, A.J., SCATTER, L.D., 1976. Physical and chemical
pretreatment for enhancing cellulose saccharification. Biotechnology
Bioengineering Symposium, 6, 125–153.
MISRA, D.K., 1983. Creal Straw. In: Pulp and paper manufacture. Volume 3.
Secondary Fibres and Agro-Based Pulping (Ed. F. Hamilton and B. Leopold).
TAPPI Press. Atlanta, Ga.
MIYAZAWA, T., FUNAZUKURI, T., 2005. Polysaccharide hydrolysis accelerated
by adding carbon dioxide under hydrothermal conditions. Biotechnology
Progress, 21, 1782–1785.
MODELL, M., 1985. Gasification and liquefaction of forest products in supercritical
water. In Overend, R.P., Milne, T.A., Mudge, L.K. (Eds.) Fundamentals of
Thermochemical Biomass Conversion, Elsevier Applied Science Publishers,
London, 95-119.
MONTANE, D., FARRIOL, X., SALVADO, J., JOLLEZ, P., CHORNET, E., 1998.
Application of steam explosion to the fractionation and rapid vapor-phase
alkaline pulping of wheat straw. Biomass and Bioenergy, 14, 261-276.
MUDGE, L.K., BAKER, E.G., MITCHELL, D.H., BROWN, M.D., 1985. Catalytic
Steam Gasification of Biomass for Methanol and Methane Production,
Journal of Solar Energy Engineering, 107,88-92.
MUTLU, S.F., 1990 . Ayçiçeği Bitkisinin Sap ve Tohum Kabuklarının Enzimatik
Yöntemlerle Şekere Dönüşümü, Doktora Tezi, Ankara Üniversitesi, Fen
Fakültesi, Kimya Bölümü, Ankara, 1-30.
NAKAMURA, Y., SAWADA, T., KOMATSU, A., SAWADA, H., KAWAMURA,
M., 2001. Degradation of kenaf core by steam explosion and saccharification
for useful utilization of biowaste, Journal Chemical Engineering. Japan, 34,
549-552.

85
NELLY, W.C., 1984. Factors affecting the pretreatment of biomass with gaseous
ozone. Biotechnology Bioengineering. 20, 59-65
NIELSEN, D.C., 2004. Kenaf Forage Yield and Quality under Varying Water
Availability.
NUGZAR, N., NUTSUBIDZE., SARKANEN, S., 1997. Concescutive
polymerization and depolymerization of kraft lignin by trametes cingulata.
Phytochemistry, 49, 1203-1212.
OSADA, M., SATO, T., WATANABE, M., ADSCHIRI, T., ARAI, K., 2004. Low
temperature catalytic gasification of lignin and cellulose with a ruthenium
catalyst in supercritical water. Energy and Fuels, 18, 327–333.
OTHMER, K., 1980. Encyclopedia of chemical technology, third ed, 11,347–60.
ÖZTÜRK, İ., IRMAK, S., HESENOV, A., ERBATUR, O., 2010. Hydrolysis of
kenaf (Hibiscus cannabinus L.) stems by catalytical thermal treatment in
subcritical water. Biomass and Bioenergy xx, 1-8. (Baskıda)
PAN, X. J., SANO, Y., HOLZFORSCHUNG., 1999. Atmospheric Acetic Acid
Pulping of Rice Straw IV: Physico-Chemical Characterization of Acetic Acid
Lignins from Rice Straw and Woods. Part 2. Chemical Structures, 53,6 590-
596 .
PARISI, F., 1989 . “Advances in lignocellulosic hydrolysis and in the utilisation of
the hydrolysates”, Advanses Biochemical Engineering, 38, 53-87.
RAMOS, L.P., 2003. The chemistry involved in the steam treatment of
lignocellulosic materials. Química Nova 26, 863-871.
ROBERT, J., AUSTON, F., 1982. In Proceedings 1981 International Conference on
Residential SolidFuels (edited by J. A. Copper and Malek), pp. 1089.
DEKKER, R.F.H., BARBOSA, A.M., 2001. The effect of lignin-related compounds
on the growth and production of laccases by the ascomycete , Botryosphaeria
sp. Enzyme and Microbial Technology 30, 374-380.
ROGALINSKI, T., DEL VALLE, J.M., ZETCL, C., BRUNNER, G., 2005. Food
Research International, 38, 2, 203-213.
SAMESHIMA, K., 1995. Kenaf and other non-wood pulp fiber usage development
for the preservation of global environment. Cell. Commun. 1, 2-5.

86
SARKANEN, K.V., 1980. Acid-catalyzed delignification of lignocellulosics in
organic solvents. Prog. Biomass Convers, 2, 127–144.
SASAKI, M., FANG, Z., FUKUSHIMA, Y., ADSCHIRI, T., ARAI, K., 2000.
Dissolution and hydrolysis of cellulose in subcritical and supercritical water.
Ind. Eng. Chem. Res. 39, 2883-2890.
SAYIGHY, A., 1999. Renewable energy-the way forward. Applied Energy, 64, 15-
30.
SCHMIEDER, H., ABELN, J., BOUKIS, N., DINJUS, E., KRUSE, A., KLUTH,
M., PETRICH,G., SADRI, E., and SCHACHT, M., 2000. Hydrothermal
gasification of biomass and organic wastes. The Journal Supercritical Fluids,
17, 145–153.
SCHURZ, J., 1978. In: Ghose, T.K. (Ed.), Bioconversion of Cellulosic Substances
into Energy Chemicals and Microbial Protein Symposium Proceedings,
IIT, New Delhi, 37.
SEGAL, L., CREELY, J.J., MARTIN, A.E.J., CONRAD, C.M., 1959. An empirical
method for estimating the degree of crystallinity of native cellulose using the
X-ray diffractometer. Textile Research Journal, 29, 786-794.
SELIG, M.J., VIAMAJALA, S., DECKER, S.R., TUCKER, M.P., HIMMEL, M.P.,
VINZANT, T.B., 2007. Deposition of lignin droplets produced during dilute
acid pretreatment of maize stems retards enzymatic hydrolysis of cellulose.
Biotechnol Prog; 23, 1333-9
SFOUNTOULAKIS, M., DOKIANAKIS, S.N., 2002. Removal of Phenolics in olive
mill wastewaters using the white-rot fungus Pleurotus ostreatus Water
Research 36, 4735-4744.
SHABAKER, J.W., DUMESIC, J.A., 2004. Batch aqueous phase reforming of
woody biomass. Industrial Engineering Chemistry Research, 4312, 3105-3112.
SHABAKER, J.W., HUBER G.W., DUMESIC J.A., 2004. Aqueous-phase
reforming of oxygenated hydrocarbons over Sn-modified Ni catalysts, J.
Catal, 222, 180-91.

87
SHAFIZADEH, F., LAI, Y.Z., 1975. Thermal degradation of 2-deoxy-Darabino-
hexonic acid and 3-deoxy-D-ribo-hexono-1,4-lactone. Carbohyd. Res. 42,
39–53.
SHUKRY, N., FADEL, S.M., AGBLEVOR, F.A., EL-KALYOUBİ, S.F.J., 2008.
Applied Polymer Science, 109, 434.
SINAG, A., KRUSE, A., RATHERT, J., 2004. Catalytic gasification of biomass
model compound in near-critical water. Industrial Engineering Chemistry
Research, 43, 2, 502-508.
SOLTEJ, E.J., 1988. Pyrolysis Oils from Biomass producing Analyzing and
Upgrading, 376. American Chemical Society symposium, Washington D. C.,
356 –365.
SOYKAN, C., İLTER, Z., 2001. Alkil metakrilatların glisidilmetakrilat ile
kopolimerlerinin sentezi ve karakterizasyonu, Erciyes Üniversitesi, Kayseri,
Fırat Üniversitesi, Elazığ, Kimya Bölümü, 17, 103-110.
STRAYER, R.F., FINGER, B.W., ALAZRKI, M.P., COOK, K., GARLAND, J.L.,
2002. Recovery of resources for advanced life support space applications:
effect of retention time on biodegradation of two crop residues in a fed-batch,
continuous stirred tank reaktor, 119-127.
STRIEGEL, A.M., TIMPA, J.D., 1995. Molecular characterization of
polysaccharides dissolved in Me2NAc-LiCl by gel-permeation
chromatography. Carbohydrate Research, 267, 271-290.
SUN, X.F., SUN, R.C., FOWLER, P., BAIRD, M.S., 2005. Extraction and
characterization of original lignin and hemicelluloses from wheat straw.
Agricultural Food Chemistry, 53, 860-870.
SUN, J.X., SUN, X.F., SUN, R.C., SU, Y.Q., 2004. Fractional extraction and
structural characterization of sugarcane bagasse hemicelluloses. Carbohydrate
Polymers 56, 195-204.
SUN, R.C., TOMKINSON, J., 2002. Characterizations of hemicelluloses obtained by
classical and ultrasonically assisted extractions from wheat straw,
Carbohydrate Polymers, 50, 263-271.

88
SUN, R., LAWTHER, J.M., BANKS, W.B., 1999. Fractional and structural
characterization of wheat straw hemicelluloses. Carbohydrrate Polymers 49,
415-423
SUN, R., LAWTHER, J.M., BANKS, W.B., 1997. A tentative chemical structure of
wheat straw lignin, Industrial Crops and Products, 6, 1-8.
SUTTON, D., KELLEHER, B., ROSS, J., 2001. Review of Literatura on Catalysts
for Biomass Gasification, Fuel Processing Technology, 3, 73, 155-173.
TANKSALE, A., BELTRAMINI, J.N., DUMESIC, J.A., LU, G.Q., 2008. Effect of
Pt and Pd promoter on Ni supported catalysts - A TPR/TPO/TPD and
microcalorimetry study, Journal Catalyst, 258, 366-377.
TANKSALE, A., WONG, Y., BELTRAMINI, J.N., LU, G.Q., 2007. Hydrogen
generation from liquid phase catalytic reforming of sugar solutions using
metal-supported catalysts, International Journal Hydrogen Energy, 32, 717–
724.
TANKSALE, A., BELTRAMINI, J.N., LU, G.Q., 2006. Reaction Mechanisms for
Renewable Hydrogen from Liquid Phase Reforming of Sugar Compounds.
Developments in Chemistry Enginneering and Mineral Processing, 14(1/2),
9-18.
T.C. BAŞBAKANLIK DEVLET İSTATİSTİK ENSTİTÜSÜ YAYINLARI,
Tarımsal Yapı ve Üretimi, Ankara, 1995
THRING, R.W., CHORENT, E., OVEREND, R., 1990. Recovery of a solvolytic
lignin: effects of spent liquor/acid volume ration, acid concentration and
temperature. Biomass 23, 289–305.
TRAN, D.O., RAI, C., 1978. Fuel 57, 293-298.
TÜRKİYE ENERJİ RAPORU 2003. Dünya Enerji Konseyi Türk Milli Komitesi,
Ankara, 101.
TWIDELL, J. W., WEIR, A. D., 1986. Renewable Energy Resources, Firs Published,
Cambridge Universty, Great Britain.
ÜLTANIR, M.Ö., 1996. 21. yüzyılın eşiğinde güneş enerjisi, Bilim ve Teknik
(TÜBİTAK), Ankara.

89
VALENZUELA, M.B., JONES, C.W., AGRAWAL, P.K., 2006. Energy Fuels 20
pp. 1744–1752.
VERVERIS, C., GEORGHIOU, K., CHRISTODOULAKIS, N., PANTAS, P.,
SANTAS, R., 2004. Fiber dimensions, lignin and cellulose content of various
plant materials and their suitability for paper production. Industrial Crops and
Products 19, 245-254.
VLASENKO, E.Y., DING, H., LABAVITCH, J.M., SHOEMAKER, S.P., 1997.
Enzymatic hydrolysis of pretreated rice straw. Bioresource Technol. 59,
109-119.
VIDAL, P.F., MOLINIER, J., 1988. Ozonolysis of lignin – improvement of in vitro
digestibility of poplar sawdust. Biomass 16, 1–17. Modeling and
optimization of the dilute-sulfuric-acid pretreatment of corn stover, poplar
and switchhgrass. Bioresoursce Technology, 59, 129–136.
WANG, Q., CAO, F., CHEN, Q., CHEN, C., 2005. Preparation of carbon micro-
spheres by hydrothermal treatment of methylcellulose sol. Material
Letters,59, 28, 3738- 3741.
WEI, L., XU S., ZHANG L., LIU C., ZHU H., LIU S., 2007. Steam gasification of
biomass for hydrogen-rich gas in a free-fall reactor, International Journal of
Hydrogen Energy 32, 24-31.
WEN, G., XU, Y., MA, H., XU, Z., TIAN, Z., 2008. Production of hydrogen by
aqueous-phase reforming of glycerol. International Journal of Hydrogen
Energy 33, 6657-6666.
WHISTLER, R.L., SMART, C.L., 1953 “Polysaccharide Chemistry”, Academic Pres
Inc. Publishers, New York, 114.
WHITE, L.P., PLASKET, L.G., 1981. Biomass as fuel. New York: Academic Press.
WILLIAMS, P.T., ONWUDILI, J., 2006. Subcritical and supercritical water
gasification of cellulose, starch, glucose, and biomass waste. Energy & Fuels,
20, 1259-1265.
WISSEL, H., MAYR, C., LUCKE, A., 2008. A new approach for the isolation of
cellulose from aquatic plant tissue and freshwater sediments for stable isotope
analysis. Organic Geochemistry 39, 1545-1561.

90
XU, Y., LI, K., ZHANG, M., 2007. Lignin precipitation on the pulp fibers in the
ethanol-based organosolv pulping. Colloids and Surfaces A: Physicochemical
Engineering Aspects 301, 255-263.
XU, F., LIU, C.F., GENG, Z.C., SUN, J.X., SUN, R.C., HEI, B.H., LIN, L., WU,
S.B., JE, J., 2006. Characterisation of degraded organosolv hemicelluloses
from wheat straw. Polymer Degradation and Stability 91, 1880-1886.
YAŞAR, S., 2002. Mischantus (Fil çimeni) Giganteus, Mischantus Goliath ve
Mischantus Silberfahne’de selüloz, hemiselüloz ve lignin miktarlarının
karşılaştırılması, Süleyman Demirel Üniversitesi Orman Fakültesi Dergisi, 2,
27-40.
YOON, J.J., KIM, Y.L., 2005. Degradation of crystalline cellulose by Brown-Rot
Basidiomycete Fomitopsis palustris. J. Microbial, 43, 487-492.
YOSHIDA, T., OSHIMA, Y., MATSUMURA, Y., 2004. Gasification of biomass
model compounds and real biomass in supercritical water Biomass
Bioenergy, 26,71-78.
YOSHIDA, T., MATSUMURA, Y., 2001. Gasification of cellulose, xylan, and
lignin mixtures in supercritical water. Industrial Engineering Chemistry
Research, 40, 5469–5474.
YU, D., AIHARA, M., ANTAL, M.J., 1993. Hydrogen production by steam
reforming glucose in supercritical water. Energy Fuels, 7, 5, 574-577.
YU, Y., LOU, X., WU, H., 2008. Some recent advances in hydrolysis of biomass in
hotcompressed water and its comparisons with other hydrolysis methods,
Energy Fuels, 22, 46-60.
ZHAO, X., DAI, L., LIU, D., 2009. Characterization and Comparison of Acetosolv
and Milox Lignin Isolated from Crofton Weed Stem. Journal of Applied
Polymer Science 114, 1295-1302.
ZHAO, H., KWAK, J.H., ZHANG, Z.C., BROWN, H.M., AREY, B.W.,
HOLLADAY, J.E., 2007. Studying cellulose fiber structure by SEM, XRD,
NMR and acid hydrolysis. Carbohydrate Polymers, 68, 235-241.
ZHENG, Y.Z., LIN, H.M., TSAO, G.T., 1998. Pretreatment for cellulose hydrolysis
by carbon dioxide explosion. Biotechnol. Prog. 14, 890–896.

91
ÖZGEÇMİŞ
1985 yılında Mersin’de doğdu. İlköğrenimini Cumhuriyet İlkokulu’nda, orta
ve lise öğrenimini Özel Türkmen Lisesi’nde tamamladı. 2004 yılında Kütahya
Dumlupınar Üniversitesi Kimya Bölümü’nü kazanıp, 2005 yılında Çukurova
Üniversitesi Kimya Bölümü’ne yatay geçiş yaptı. 2008 yılında bu bölümden mezun
oldu. Aynı yıl Çukurova Üniversitesi Fen Bilimleri Enstitüsü yüksek lisans
programına kaydoldu.