cytoplasmic mislocalization of brca1 caused by cancer-associated mutations in the brct domain
TRANSCRIPT

www.elsevier.com/locate/yexcr
Experimental Cell Research 293 (2004) 14–21
Cytoplasmic mislocalization of BRCA1 caused by cancer-associated
mutations in the BRCT domain
Jose Antonio Rodriguez,1 Wendy W.Y. Au, and Beric R. Henderson*
Westmead Institute for Cancer Research, University of Sydney, Westmead Millennium Institute at Westmead Hospital, Westmead, NSW 2145, Australia
Received 3 June 2003, revised version received 17 September 2003
Abstract
BRCA1 is inactivated by gene mutations in >50% of familial breast and ovarian cancers. BRCA1 is primarily a nuclear protein,
although others previously reported cytoplasmic staining in breast tumor cells. In this study, we demonstrate the cytoplasmic
mislocalization of BRCA1 caused by a subgroup of clinically relevant cancer mutations. We show that mutations that disrupt or delete the
C-terminal BRCT domains, but not other regions of BRCA1, caused significant relocalization of BRCA1 from nucleus to cytoplasm. Two
of the BRCT mutations tested (M1775R and Y1853X) are known to adversely affect BRCA1 protein folding and nuclear function. The
BRCT mutations reduced BRCA1 nuclear import by a mechanism consistent with altered protein folding, as indicated by the restoration
of nuclear staining by more extensive C-terminal deletions. Furthermore, we observed increased cytoplasmic staining of both the ectopic
and endogenous forms of the BRCA1-5382insC mutant (deleted BRCT domain) in HCC1937 breast cancer cells. Unlike wild-type
BRCA1, the BRCA1-5382insC mutant failed to form DNA damage-inducible foci when targeted to the nucleus by BARD1. We propose
that BRCT mutations alter nuclear targeting of BRCA1, and that this may contribute to the inhibition of nuclear DNA repair and
transcription function.
D 2003 Elsevier Inc. All rights reserved.
Keywords: Nuclear transport; Breast cancer; BRCA1 mutations; BRCT domains; DNA repair
Introduction
The BRCA1 tumor suppressor gene is mutated in the
germ-line of women who suffer a genetic predisposition to
breast and ovarian cancer [1,2]. BRCA1 mutations are
found in approximately 50% of patients with inherited
breast cancer, and up to 90% of families with breast and
ovarian cancer susceptibility [1,2]. The BRCA1 protein
comprises 1863 amino acids [1], and has proposed roles
in DNA repair, transcriptional activation, cell cycle regula-
tion and apoptosis [3–5]. BRCA1 has two main types of
protein interaction domains that are frequently targeted by
genetic mutations: an N-terminal RING finger domain and
two tandem BRCT domains at the C-terminal end. The
0014-4827/$ - see front matter D 2003 Elsevier Inc. All rights reserved.
doi:10.1016/j.yexcr.2003.09.027
* Corresponding author. Westmead Millennium Institute, Darcy Road,
PO Box 412, Westmead, NSW 2145, Australia. Fax: +61-2-9845-9102.
E-mail address: [email protected] (B.R. Henderson).1 Present address: Department ofMedical Oncology, Academic Hospital
Vrije Universiteit Amsterdam, 1081HV, Amsterdam, The Netherlands.
RING domain mediates an enzymatic ubiquitin E3 ligase
activity when BRCA1 forms a stable heterodimer with its
binding partner, BARD1 [6,7]. The C-terminal BRCT
domains are thought to mediate the transcriptional activity
of BRCA1 [8,9]. As a tumor suppressor, inherited mutations
affect certain vital functions of BRCA1. The most frequent
published mutations occur within the BRCT domains and
these have been reported to adversely affect BRCA1 nuclear
functions including DNA repair [10,11] and transcriptional
activity [9,12,13].
Several years ago, Chen et al. [14] published controver-
sial findings which claimed that BRCA1 was mislocalized
almost exclusively to the cytoplasm in breast cancer tissues,
but remained nuclear in normal tissue and in other cancer
cell types. However, this finding was not validated by others
[15], and it was later concluded that antibody specificity was
the main problem, with most agreeing that BRCA1 in
general displayed a ‘‘primarily’’ nuclear localization in
different breast cancer cell lines and in histological breast
tumor tissue slices [16]. Nonetheless, BRCA1 subcellular
localization is well known to vary between nucleus and
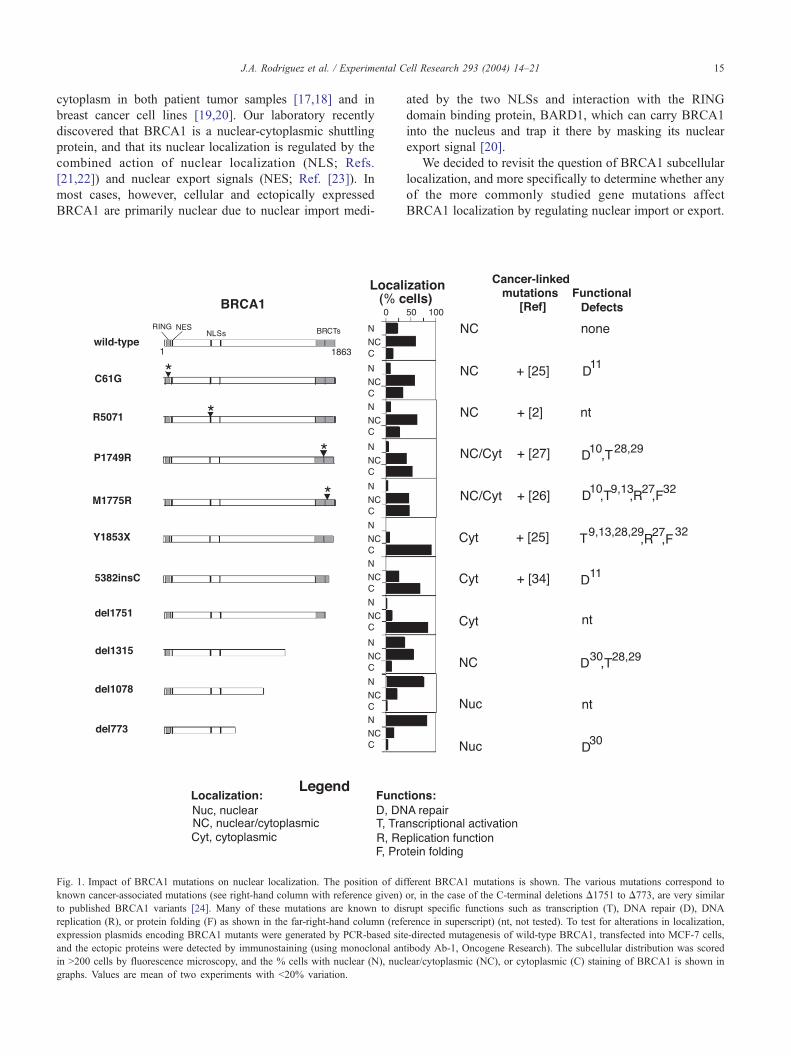
J.A. Rodriguez et al. / Experimental Cell Research 293 (2004) 14–21 15
cytoplasm in both patient tumor samples [17,18] and in
breast cancer cell lines [19,20]. Our laboratory recently
discovered that BRCA1 is a nuclear-cytoplasmic shuttling
protein, and that its nuclear localization is regulated by the
combined action of nuclear localization (NLS; Refs.
[21,22]) and nuclear export signals (NES; Ref. [23]). In
most cases, however, cellular and ectopically expressed
BRCA1 are primarily nuclear due to nuclear import medi-
Fig. 1. Impact of BRCA1 mutations on nuclear localization. The position of di
known cancer-associated mutations (see right-hand column with reference given)
to published BRCA1 variants [24]. Many of these mutations are known to dis
replication (R), or protein folding (F) as shown in the far-right-hand column (refe
expression plasmids encoding BRCA1 mutants were generated by PCR-based sit
and the ectopic proteins were detected by immunostaining (using monoclonal an
in >200 cells by fluorescence microscopy, and the % cells with nuclear (N), nuc
graphs. Values are mean of two experiments with <20% variation.
ated by the two NLSs and interaction with the RING
domain binding protein, BARD1, which can carry BRCA1
into the nucleus and trap it there by masking its nuclear
export signal [20].
We decided to revisit the question of BRCA1 subcellular
localization, and more specifically to determine whether any
of the more commonly studied gene mutations affect
BRCA1 localization by regulating nuclear import or export.
fferent BRCA1 mutations is shown. The various mutations correspond to
or, in the case of the C-terminal deletions D1751 to D773, are very similar
rupt specific functions such as transcription (T), DNA repair (D), DNA
rence in superscript) (nt, not tested). To test for alterations in localization,
e-directed mutagenesis of wild-type BRCA1, transfected into MCF-7 cells,
tibody Ab-1, Oncogene Research). The subcellular distribution was scored
lear/cytoplasmic (NC), or cytoplasmic (C) staining of BRCA1 is shown in

J.A. Rodriguez et al. / Experimental Cell Research 293 (2004) 14–2116
We demonstrate that out of the ten mutations tested, only the
five which target the BRCT domains altered BRCA1
localization, causing it to be excluded from the nucleus.
This nuclear exclusion was not due to increased nuclear
export, but to reduced nuclear import. Similar findings were
observed for both the overexpressed and endogenous forms
of the BRCT mutant, BRCA1(5382insC). Since the BARD-
dependent import pathway [20] is still active for the BRCT
mutants, their overall nuclear entry is reduced but not fully
negated. Our new findings may explain some of the dispar-
ity in the literature concerning BRCA1 localization and
movement.
Results
BRCA1 mutations within the BRCT domain cause
cytoplasmic mislocalization
To assess the impact of BRCA1 cancer mutations on
subcellular localization, we first used PCR-based muta-
genesis to create a series of single-site or truncated
BRCA1 mutant cDNAs corresponding to naturally occur-
ring breast and ovarian cancer-linked mutations [24–27].
These mutant BRCA1 cDNAs were cloned into the
mammalian expression vector, pFLAG-CMV2 (see Ref.
[23]), for transfection and expression in human breast
cancer cells. As summarized in Fig. 1, many of the
Fig. 2. Effect of blocking BRCA1 nuclear export on the localization of BRCA1 mu
MCF-7 cells showing predominantly nuclear (N), nuclear/cytoplasmic (NC), or cy
CRM1-dependent nuclear export. BRCA1 nuclear export was blocked by LMB tr
nuclear export signal (Leu86Ala/Ile90Ala, see Ref. [23]). Bars represent the mean
of transfected MCF-7 cells expressing each of the BRCA1 variants bearing an ac
BRCA1 mutations selected for testing were previously
reported to disrupt BRCA1 nuclear transcription
[8,9,28,29], DNA repair [10,11,30] or DNA replication
[31] activities, and some affect folding of the BRCA1
carboxy terminus [32].
When transiently expressed in MCF-7 cells and exam-
ined for localization by fluorescence microscopy, most
BRCA1 mutants displayed an altered subcellular distribu-
tion compared to that of wild-type BRCA1 (Fig. 1).
Interestingly, we tested a single point mutation (R507I)
that lies within the first BRCA1 nuclear localization signal
(503-KRKRRP, Ref. [21]), but this had only a very minor
effect on nuclear localization. By contrast, five different
BRCA1 constructs (P1749R, M1775R, Y1853X, 5382insC
and D1751) that contain single amino acid mutations or
short deletions (including removal of only the last 11 amino
acids in Y1853X) within the C-terminal tandem BRCT
domains, shifted BRCA1 from the nucleus to the cytoplasm
(Fig. 1). Similar results were observed in T47D breast
cancer cells, for both untagged and Yellow Fluorescent
Protein (YFP)-tagged versions of these constructs (data not
shown). Two of the C-terminal mutations (M1775R and
Y1853X) that restricted nuclear localization are identical to
mutations that disrupt BRCA1 C-terminal folding [32] (Fig.
1), suggesting that the conformational changes they elicit
might be deleterious to BRCA1 nuclear transport. Indeed,
the cytoplasmic mislocalization was specific to changes in
the BRCT region, as more extensive C-terminal deletions
tants C61G, D1751 and Y1853X. Graphs show the proportion of transfected
toplasmic (C) BRCA1 (wild-type or mutant) in the presence or absence of
eatment (6 ng/ml for 12 h) or by introducing an inactivating mutation in the
F SE of at least two experiments. Panels show confocal microscopy images
tive (NESwt) or inactive (NESmut) export signal.

Fig. 3. Nuclear-cytoplasmic distribution of endogenous BRCA1(5382insC).
Subcellular localization of the endogenous BRCA1(5382insC) mutant in
HCC1937 cells was determined by two approaches. (A) Immunofluor-
escence microscopy using two-well characterized primary monoclonal
antibodies, Ab1 (binds N terminus) and Ab4 (binds exon 11) (Oncogene
Research), and a fluorescein anti-mouse secondary antibody. (B) Western
blot analysis of full-length BRCA1 using Ab4, where the nuclear/
cytoplasmic (N/C) ratio was determined by densitometry. Reprobing the
blot with anti-Topoisomerase II antibody provided a fractionation control.
J.A. Rodriguez et al. / Experimental Cell Research 293 (2004) 14–21 17
had the opposite effect and caused a progressive increase in
the nuclear localization of BRCA1 (Fig. 1).
BRCT mutations reduce BRCA1 nuclear import
BRCA1 is a nuclear-cytoplasmic shuttling protein [23],
and contains sequences that facilitate both nuclear import
(NLS) and export (NES; reviewed in Ref. [33]). Conse-
quently, the increased cytoplasmic localization of BRCT-
targeted mutations could result from either a blockage to
nuclear import, or from enhanced nuclear export. To
distinguish between these possibilities, we evaluated the
change in localization of different BRCA1 mutants when
nuclear export was blocked. Inhibition of CRM1-depen-
dent nuclear export was achieved by treatment with the
export inhibitor leptomycin B (LMB; see Ref. [23]), or by
the introduction of a site-directed mutation into the
BRCA1 nuclear export signal (as first described for
wild-type BRCA1 in Ref. [23]). We found that blocking
nuclear export by either method caused wild-type BRCA1
to shift even more prominently to the nucleus of trans-
fected MCF-7 cells (Fig. 2). A similar nuclear shift was
observed for the N-terminal RING mutation, C61G. In
contrast, the BRCA1 C-terminal mutants D1751 and
Y1853X remained trapped in the cytoplasm, even when
nuclear export was inhibited (Fig. 2). These novel findings
reveal that the nuclear import of the BRCT mutants is
severely impaired.
A BRCT mutation affects nuclear localization of
endogenous BRCA1
Currently there is only one well-studied cell line,
HCC1937 [34], reported to express a form of BRCA1
mutated within the C-terminal BRCT domain. We exam-
ined mutant BRCA1 localization in HCC1937 cells by
immunofluorescence microscopy and by Western blotting
of fractionated extracts. Consistent with our transient
expression data, the localization of endogenous BRCA1-
5382insC was significantly more cytoplasmic than the
wild-type BRCA1 expressed in HBL-100 cells (Fig. 3).
Unlike the ectopically expressed 5382insC mutant, how-
ever, which showed partial nuclear staining in only 30% of
transfected cells, the endogenous mutant displayed partial
nuclear staining in most cells. This may partly reflect its
association with endogenous BARD1, given that BARD1
can act as a BRCA1 nuclear chaperone independent of the
BRCA1 NLSs [20]. In support of this possibility, we found
that co-expression of a BARD1 expression plasmid caused
ectopically expressed BRCA1(5382insC) to shift strongly
into the nucleus (Fig. 4A), as recently described for wild-
type BRCA1 [20]. These observations indicate that
BRCA1 nuclear import is reduced, but not abolished, by
BRCT mutations, and are consistent with the reduced
nuclear accumulation of mutant BRCA1 previously ob-
served in some breast tumors [18].
Nuclear localization of BRCT mutants is not increased by
proteasome blockade
Some BRCT mutants are known to display altered folding
and turnover [32]. To exclude the possibility that reduced
nuclear staining of these mutants was due to degradation in
the nuclear compartment, we treated transfected MCF-7 cells
with the proteasome inhibitor MG132, and compared the
distribution of wild-type or mutant BRCA1. As shown in
Fig. 4, using a concentration of MG132 effective in stabi-
lizing endogenous beta-catenin (data not shown), we ob-
served a general increase in YFP-BRCA1 staining but did
not observe a strong increase in nuclear staining of the BRCT

Fig. 4. Cytoplasmic localization of BRCA1 C-terminal mutants is not due to nuclear degradation. MCF-7 cells were transfected with wild-type or mutant
BRCA1 plasmids and treated for 16 h with 20 AM MG132 (a proteasome inhibitor) before fixation and mounting. The subcellular distribution of the YFP
fusions was scored (shown for wild-type and 5382insC mutant) and found to be very similar to that of untreated cells (see Fig. 1).
J.A. Rodriguez et al. / Experimental Cell Research 293 (2004) 14–2118
mutants. The same result was seen in three separate experi-
ments. This suggests that the cytoplasmic localization ob-
served is due more to reduced nuclear import than to nuclear
degradation of BRCA1.
BRCT mutations disrupt BRCA1 intranuclear localization
Within the nucleus, cellular BRCA1 co-localizes with
BARD1 and specific DNA repair factors in S-phase-associ-
ated dots [35,36] and redistributes into DNA repair-associat-
ed nuclear foci following DNA damage [37]. DNA damage-
induced foci are presumed to be intranuclear sites for DNA
repair [11,37]. Unlike wild-type BRCA1, the cellular form of
BRCA1(5382insC) expressed in HCC1937 cells does not
form nuclear foci before [10] or after DNA damage [38].
However, this observation has not yet been validated by
transfection experiments using the exogenous form of the
mutant, presumably because the transiently expressed mutant
protein displays such poor nuclear staining (Fig. 1). To
circumvent this problem, we co-expressed the BRCA1(5382-
insC) mutant with BARD1 to ensure its nuclear entry (see
Fig. 5A), and because BARD1 promotes recruitment of
BRCA1 to nuclear foci [20].
YFP fusions of wild-type and 5382insC mutant BRCA1
were used to distinguish the ectopic protein from endoge-
nous BRCA1. When co-expressed in asynchronous MCF-7
cells, both ectopic BRCA1 and BARD1 co-localized in
distinct ‘‘S-phase’’ dots, and following treatment with the
DNA damaging agent, methyl methanesulfonate, redistrib-
uted to form many discrete foci as previously observed for
cellular BRCA1 and BARD1 [35–37]. We scored the
proportion of BRCA1-positive nuclei that displayed foci
before and after DNA damage, and found that wild-type
BRCA1 formed >10 foci in over 40% of transfected cells
(Fig. 5B). In striking contrast, the 5382insC mutant dis-
played a severe loss in its ability to form nuclear foci, even
when targeted to the nucleus by BARD1 (Fig. 5B). This is
the first direct demonstration that BRCT mutations adversely
affect both BRCA1 nuclear localization and recruitment to
DNA repair-associated nuclear foci.
Discussion
In this study, we identified a specific subset of C-
terminal mutations (those localized to the BRCT domain)
that cause the partial or near-complete nuclear exclusion of
BRCA1. Similar findings were made for both transiently
expressed and endogenous BRCA1. The observed cyto-
plasmic mislocalization may partly explain why BRCT
mutations reduce BRCA1 nuclear DNA repair [10,11],
replication [27] and transcriptional activities [8,9,28,29].

Fig. 5. BRCA1(5382insC) can be targeted to the nucleus by BARD1 but
does not form nuclear foci. (A) Nuclear entry of the C-terminal cancer
mutant (5382insC) was stimulated by BARD1 co-expression in MCF-7
cells (as described in Ref. [20] for wild-type BRCA1). Staining of
transiently expressed BRCA1(5382insC) and BARD1 is described in
Methods. (B) Comparison of BRCA1 nuclear focus formation. YFP
fusions of BRCA1 were co-transfected with a BARD1 expression vector
into MCF-7 cells, and BRCA1 nuclear foci were then scored before or
after a 1-h treatment with 0.01% MMS. The proportion of cells displaying
<10 or >10 BRCA1 nuclear foci was determined by fluorescence
microscopy (100� oil immersion objective) and graphed. Values shown
are mean F SE of two independent experiments scoring about 100
BRCA1-positive cells per sample. Representative cell images were
collected using a SPOT digital camera and are shown in the bottom
panel. Of the images shown for wild-type BRCA1, �MMS images are
typical of those with 1–10 foci, whereas +MMS images shown are typical
of DNA damaged cells with >10 foci.
J.A. Rodriguez et al. / Experimental Cell Research 293 (2004) 14–21 19
Significantly, even when the mutant BRCA1(5382insC)
was forced into the nucleus by the co-expression of
BARD1, it differed from wild-type BRCA1 in that it was
not recruited to nuclear foci which are thought to be the
sites of DNA repair. Therefore, altered intranuclear traf-
ficking may further contribute to the reduced DNA repair
activity of BRCT-mutated forms of BRCA1 [10,11].
Our findings predict enhanced cytoplasmic staining of
BRCA1 in familial breast cancers that carry similar BRCT
mutations. There are a few published immunohistochemical
reports observing increased cytoplasmic BRCA1 in tumors
from patients with BRCA1 germ-line mutations (e.g. Refs.
[14,17,18,39]); however, most have lacked suitable controls
for antibody cross-reactivity and/or classification of the
mutations involved. Furthermore, it is important to distin-
guish between full-length BRCA1 and the exon 11-splice
variant form, as we have done here by using the exon 11-
specific antibody Ab4 (see Fig. 3), because the shorter
splice variants often lack the NLSs and are therefore
intrinsically more cytoplasmic.
The abnormal subcellular distribution of the BRCT
mutants correlates with their ability to disrupt BRCA1 C-
terminal folding [32]. Interestingly, some BRCT mutations
are known to reduce BRCA1 expression in tumors [18], and
we also observed a decreased expression of these mutants in
transient assays. The expression level of both wild-type and
mutant BRCA1 was moderately increased following treat-
ment with the proteasome inhibitor, MG132. However, the
BRCT mutants did not relocalize back to the nucleus in
MG132-treated cells (Fig. 4), suggesting that their misloc-
alization does not reflect nuclear degradation.
It is important to note that the cytoplasmic mislocaliza-
tion observed was specific to changes in the BRCT region,
and that more extensive deletions of the carboxyl terminus
actually had the opposite effect, causing a progressive
increase in the nuclear localization of BRCA1 (Fig. 1).
We interpret these findings to suggest that C-terminal
deletion beyond the tandem BRCT repeats, which pack
together in a defined head-to-tail arrangement [32], at least
partially restores correct folding in the remainder of the
protein including accessibility of the central nuclear local-
ization signals. Put more simply, we propose that BRCT
mutations impair accessibility and function of the BRCA1
NLS. Indeed, the strong cytoplasmic shift caused by the
BRCT mutations Y1853X, 5382insC and D1751 was
similar to that previously observed in the same system
for NLS-mutated BRCA1 [20]. The mutations did not
seem to prevent the alternative RING/BARD1-dependent
nuclear import pathway [20], as co-expression of BARD1
caused almost complete relocalization of mutant BRCA1 to
the nucleus. In conclusion, it will be of interest to deter-
mine whether the altered localization of BRCT mutants
results in a gain of function, especially given that BRCA1
has previously been shown to associate with centrosomes
[40].
Methods
Cell culture and transfection
MCF-7, T47D human breast cancer cells and HBL100
immortalized human breast epithelial cells were maintained

J.A. Rodriguez et al. / Experimental Cell Research 293 (2004) 14–2120
in Dulbecco’s modified Eagle’s media (DMEM) supple-
mented with 10% fetal calf serum (FCS). HCC1937 cells
were grown in RPMI and 10% FCS. All cells were grown at
37jC in a humidified 5% CO2 atmosphere. Cells were
seeded onto sterile glass coverslips and transfected at 50–
60% confluency with 1–2 Ag of plasmid DNA using Lip-
ofectamine Reagent (Life Technologies) according to the
manufacturer’s instructions. At 6 h post-transfection, the
transfection mix was removed and replaced with DMEM
containing 10% FCS. Cells were fixed and processed 30
h post-transfection for fluorescence microscopy. When re-
quired, transfected cells were treated with LMB at a final
concentration of 6 ng/ml for 12 h before fixation.
Plasmid construction
A series of mutations, most of which correspond exactly
to published cancer-linked mutations, were introduced into
the BRCA1 cDNAwithin the expression vector pF-BRCA1
[23]. The C61G mutant was previously described [20]. The
introduction of all other point mutations and truncating
mutations involved a combination of PCR mutagenesis
and cloning, and specific details of the individual cloning
strategies and primer sequences are available upon request.
Immunofluorescence microscopy
Immunostaining was carried out as described [20]. Cells
expressing YFP-tagged proteins were fixed in 3.7% forma-
lin/PBS for 15 min at room temperature, washed and then
mounted for direct detection of the autofluorescent protein.
Untagged ectopic BRCA1 was detected by immunofluores-
cence using monoclonal antibodies Ab-1 and Ab-4 (Onco-
gene Research), which recognise an epitope in the amino
terminus and the central portion of BRCA1, respectively.
BRCA1-bound antibody was detected with fluorescein iso-
thiocyanate-conjugated goat anti-mouse secondary antibody
(Sigma). BARD1 was detected with polyclonal antibody
699D [20] diluted 1:800 in blocking solution, followed by
biotin-conjugated secondary antibodies (Santa Cruz) and
Texas Red–avidin D (Vector Laboratories). Cell nuclei were
counterstained with the chromosome dye Hoechst 33285
(Sigma). The subcellular localization of each ectopic protein
was determined by scoring cells using an Olympus BX40
epifluorescence microscope, and the proportion of cells
displaying nuclear foci was determined by scoring nuclei
using a 100� objective. Digital images were collected using
a SPOT camera or an Optiscan confocal microscope.
Cell fractionation and Western blotting
HCC1937 and HBL100 cells were separated into nuclear
and cytoplasmic fractions using the NE-PER extraction kit
(Pierce) according to the manufacturer’s instructions. Pro-
tein concentrations in nuclear and cytoplasmic fractions
were determined with the Bio-Rad dye-binding assay. Cell
extracts were denatured, separated by 8% SDS-polyacryl-
amide gel electrophoresis and transferred to PVDF mem-
branes. The Western blot filters were blocked in blocking
buffer (1% fetal calf serum, 5% dried milk in phosphate-
buffered saline containing 0.1% Tween 20) and probed with
the primary antibody. BRCA1 was detected with either the
monoclonal antibody Ab-1 diluted 1:200 (data not shown)
or Ab-4 diluted 1:200, followed by incubation with the
horseradish peroxidase-conjugated secondary antibody
(1:1000). As a fractionation control, the blot was also
probed with Topoisomerase II antibody Ab1 (Santa Cruz
Biotech). Blotted proteins were visualized using the ECL
detection system (Amersham Pharmacia Biotech). Rainbow
colour markers (Amersham Pharmacia Biotech) were used
as molecular size standards.
Acknowledgments
We thank Dr. Jeff Holt and Richard Baer for providing
BRCA1 and BARD1 plasmids and antibodies. This work
was supported in part by grants to BRH from the National
Health and Medical Research Council of Australia and the
CureCancer Australia Foundation.
References
[1] Y. Miki, J. Swensen, D. Shattuck-Eidens, P.A. Futreal, K. Ahrshman,
S. Tavigian, Q. Liu, C. Cochran, L.M. Bennett, W. Ding, et al., A
strong candidate for the breast cancer susceptibility gene BRCA1,
Science 266 (1994) 66–71.
[2] F.J. Couch, B.L. Weber, Mutations and polymorphisms in the familial
early-onset breast cancer (BRCA1) gene. Breast Cancer Information
Core, Hum. Mutat. 8 (1996) 8–18.
[3] Y. Chen, W.H. Lee, H.K. Chew, Emerging roles of BRCA1 in tran-
scriptional regulation and DNA repair, J. Cell Physiol. 1 (1999)
385–392.
[4] R. Scully, D.M. Livingston, In search of the tumour-suppressor func-
tions of BRCA1 and BRCA2, Nature 408 (2000) 429–432.
[5] D.P. Harkin, J.M. Bean, D. Miklos, Y.-H. Song, V.B. Truong, C.
Englert, F.C. Christians, L.W. Ellisen, S. Maheswaran, J.D. Oliner,
D.A. Haber, Induction of GADD45 and JNK/SAPK-dependent
apoptosis following inducible expression of BRCA1, Cell 97 (1999)
575–586.
[6] R. Baer, T. Ludwig, The BRCA1/BARD1 heterodimer, a tumor sup-
pressor complex with ubiquitin E3 ligase activity, Curr. Opin. Gen.
Dev. 12 (2002) 86–91.
[7] P.S. Brzovic, J.R. Keeffe, H. Nishikawa, K. Miyamoto , D. Fox III,
M. Fukuda, T. Ohta, R. Klevit, Binding and recognition in the assem-
bly of an active BRCA1/BARD1 ubiquitin – ligase complex, Proc.
Natl. Acad. Sci. U. S. A. 100 (2003) 5646–5651.
[8] M.S. Chapman, I.M. Verma, Transcriptional activation by BRCA1,
Nature 382 (1996) 678–679.
[9] A.N.A. Monteiro, A. August, H. Hanafusa, Evidence for a transcrip-
tional activation function of BRCA1 C-terminal region, Proc. Natl.
Acad. Sci. U. S. A. 93 (1996) 13595–13599.
[10] R. Scully, S. Ganesan, K. Vlaskova, J. Chen, M. Socolovsky, D.M.
Livingston, Genetic analysis of BRCA1 function in a defined tumor
cell line, Mol. Cell 4 (1999) 1093–1099.
[11] Q. Zhong, C.-F. Chen, S. Li, Y. Chen, C.-C. Wang, J. Xiao, P.-L.
Chen, Z.D. Sharp, W.-H. Lee, Association of BRCA1 with the
hRAD50–hMRE11–p95 complex and the DNA damage response,
Science 285 (1999) 747–750.
[12] F. Hayes, C. Cayanan, D. Barilla, A.N. Monteiro, Functional assay

J.A. Rodriguez et al. / Experimental Cell Research 293 (2004) 14–21 21
for BRCA1: mutagenesis of the COOH-terminal region reveals crit-
ical residues for transcription activation, Cancer Res. 60 (2000)
2408–2411.
[13] J. Vallon-Christersson, C. Cayanan, K. Haraldsson, N. Loman, J.T.
Bergthorsson, K. Brondum-Nielsen, A.M. Gerdes, P. Moller, U.
Kristoffersson, H. Olsson, A. Borg, A.N. Monteiro, Functional anal-
ysis of BRCA1 C-terminal missense mutations identified in breast
and ovarian cancer families, Hum. Mol. Genet. 10 (2001) 353–360.
[14] Y. Chen, C.-F. Chen, D.J. Riley, D.C. Allred, P.-L. Chen, D.V. Hoff,
C.K. Osborne, W.-H. Lee, Aberrant subcellular localization of
BRCA1 in breast cancer, Science 270 (1995) 789–791.
[15] R.S. Scully, S. Ganesan, M. Brown, J.A.D. Caprio, S.A. Cannistra,
J. Feunteun, S. Schnitt, D.M. Livingston, Location of BRCA1 in
human breast and ovarian cancer cells, Science 272 (1996) 123–125.
[16] C.A. Wilson, L. Ramos, M.R. Villasenor, K.H. Anders, M.F. Press,
K. Clarke, B. Karlan, J.-J. Chen, R. Scully, D. Livingston, R.H. Zuch,
M.H. Kanter, S. Cohen, F.J. Calzone, D.J. Slamon, Localization of
human BRCA1 and its loss in high-grade, non-inherited breast carci-
nomas, Nat. Genet. 21 (1999) 236–240.
[17] E.M. Jarvis, J.A. Kirk, C.L. Clarke, Loss of nuclear BRCA1 expres-
sion in breast cancers is associated with a highly proliferative tumor
phenotype, Cancer Genet. Cytogenet. 101 (1998) 109–115.
[18] J. Taylor, M. Lymboura, P.E. Pace, R.P.A. Hern, A.J. Desai, S.
Shousha, R.C. Coombes, S. Ali, An important role for BRCA1 in
breast cancer progression is indicated by its loss in a large proportion
of non-familial breast cancers, Int. J. Cancer 79 (1998) 334–342.
[19] J.E. Thomas, M. Smith, B. Rubinfeld, M. Gutowski, R.P. Beck-
man, P. Polakis, Subcellular localisation and analysis of apparent
180-kDa and 220-kDa proteins of the breast cancer susceptibility
gene, BRCA1, J. Biol. Chem. 271 (1996) 28630–28635.
[20] M. Fabbro, J.A. Rodriguez, R. Baer, B.R. Henderson, BARD1 indu-
ces BRCA1 intranuclear foci formation by increasing RING-depen-
dent BRCA1 nuclear import and inhibiting BRCA1 nuclear export,
J. Biol. Chem. 277 (2002) 21315–21324.
[21] C.-F. Chen, S. Li, Y. Chen, P.-L. Chen, Z.D. Sharp, W.-H. Lee, The
nuclear localization sequences of the BRCA1 protein interact with the
importin-a subunit of the nuclear transport signal, J. Biol. Chem. 271
(1996) 32863–32868.
[22] S. Thakur, H.B. Zhang, Y. Peng, H. Le, B. Carroll, T. Ward, J. Yao,
L.M. Farid, F.J. Couch, R.B. Wilson, B. Weber, Localization of
BRCA1 and a splice variant identifies the nuclear localization signal,
Mol. Cell. Biol. 17 (1997) 444–452.
[23] J.A. Rodriguez, B.R. Henderson, Identification of a functional nuclear
export signal in BRCA1, J. Biol. Chem. 275 (2000) 38589–38596.
[24] P.D. Stenson, E.V. Ball, M. Mort, A.D. Phillips, J.A. Shiel, N.S.
Thomas, S. Abeysinghe, M. Krawczak, D.N. Cooper, Human Gene
Mutation Database (HGMD): 2003 update, Hum. Mutat. 21 (2003)
577–581.
[25] L.S. Friedman, E.A. Ostermeyer, C.I. Szabo, P. Dowd, E.D. Lynch,
S.E. Rowell, M.-C. King, Confirmation of BRCA1 by analysis of
germline mutations linked to breast and ovarian cancer in ten families,
Nat. Genet. 8 (1994) 399–404.
[26] D. Shattuck-Eidens, M. McClure, J. Simard, F. Labrie, S. Narod, F.
Couch, K. Hoskins, B. Weber, L. Castilla, M. Erdos, L. Brody, L.
Friedman, E. Ostermeyer, C. Szabo, M.-C. King, S. Jhanwar, K.
Offit, L. Norton, T. Gilewski, M. Lubin, M. Osborne, D. Black, M.
Boyd, M. Steel, M.S. Ingles, R. Haile, A. Lindblom, H. Olsson, A.
Borg, D.T. Bishop, E. Solomon, P. Radice, G. Spatti, S. Gayther, B.
Ponder, W. Warren, M. Stratton, Q. Liu, F. Fujimura, C. Lewis, M.
Skolnick, G. Goldgar, E. David, A collaborative survey of 80 muta-
tions in the BRCA1 breast and ovarian cancer susceptibility gene:
implications for presymptomatic testing and screening, J. Am. Med.
Assoc. 273 (1995) 535–541.
[27] S.A. Gayther, P. Harrington, P. Russell, G. Kharkevich, R.F.
Garkavtseva, B.A. Ponder, Rapid detection of regionally clustered
germ-line BRCA1 mutations by multiplex heteroduplex analysis.
UKCCCR Familial Ovarian Cancer Study Group, Am. J. Hum. Genet.
58 (1996) 451–456.
[28] K. Somasundaram, H. Zang, Y.-X. Zeng, Y. Houvras, Y. Peng, H.
Zhang, G.S. Wu, J. Licht, B.L. Weber, W.S. El-Deiry, Arrest of the
cell cycle by the tumour-suppressor BRCA1 requires the CDK-
inhibitor p21WAF-1/CIP1, Nature 389 (1997) 187–190.
[29] H. Zhang, K. Somasundaram, Y. Peng, H. Tian, H. Zhang, D. Bi,
B.L. Weber, W.S. El-Deiry, BRCA1 physically associates with p53
and stimulates its transcriptional activity, Oncogene 16 (1998)
1713–1721.
[30] S. Fan, R.-Q. Yuan, Y.X. Ma, Q. Meng, I.D. Goldberg, E.M. Rosen,
Mutant BRCA1 genes antagonize phenotype of wild-type BRCA1,
Oncogene 20 (2001) 8215–8235.
[31] Y.F. Hu, Z.L. Hao, R. Li, Chromatin remodeling and activation of
chromosomal DNA replication by an acidic transcriptional activation
domain from BRCA1, Genes Dev. 13 (1999) 637–642.
[32] R.S. Williams, R. Green, J.N.M. Glover, Crystal structure of the
BRCT repeat region from the breast cancer-associated protein
BRCA1, Nat. Struct. Biol. 8 (2001) 838–842.
[33] M. Fabbro, B.R. Henderson, Regulation of tumor suppressors by
nuclear-cytoplasmic shuttling, Exp. Cell Res. 282 (2003) 59–69.
[34] G.E. Tomlinson, T.T. Chen, V.A. Stastny, A.K. Virmani, M.A.
Spillman, V. Tonk, J.L. Blum, N.R. Schneider, I.I. Wistuba, J.W.
Shay, J.D. Minna, A.F. Gazdar, Characterization of a breast cancer
cell line derived from a germ-line BRCA1 mutation carrier, Cancer
Res. 58 (1998) 3237–3242.
[35] Y. Jin, X.L. Xu, M.-C.W. Yang, F. Wei, T.-C. Ayi, A.M. Bowcock,
R. Baer, Cell cycle-dependent colocalisation of BARD1 and BRCA1
proteins in discrete nuclear domains, Proc. Natl. Acad. Sci. U. S. A.
94 (1997) 12075–12080.
[36] R.S. Scully, J. Chen, A. Plug, Y. Xiao, D. Weaver, J. Feunteun,
T. Ashley, D.M. Livingston, Association of BRCA1 with Rad51
in mitotic and meiotic cells, Cell 88 (1997) 265–275.
[37] R. Scully, J. Chen, R.L. Ochs, K. Keegan, M. Hoekstra, J. Feunteun,
D.M. Livingston, Dynamic changes of BRCA1 subcellular location
and phosphorylation state are initiated by DNA damage, Cell 90
(1997) 425–435.
[38] X. Wu, J.H.J. Petrini, W.F. Heine, D.T. Weaver, D.M. Livingston,
J. Chen, Independence of R/M/N focus formation and the presence
of intact BRCA1, Science 289 (2000) 11a.
[39] K. Kashima, T. Oite, Y. Aoki, K. Takakuwa, H. Aida, H. Nagata,
M. Sekine, H.J. Wu, Y. Hirai, Y. Wada, K. Yamamoto, K. Hasegawa,
T. Sonoda, T. Maruo, I. Nagata, M. Ohno, M. Suzuki, I. Kobayashi,
K. Kuzuya, T. Takahashi, Y. Torii, K. Tanaka, Screening of BRCA1
mutation using immunohistochemical staining with C-terminal and
N-terminal antibodies in familial ovarian cancers, Jpn. J. Cancer Res.
91 (2000) 399–409.
[40] L.-C. Hsu, R.L. White, BRCA1 is associated with the centro-
some during mitosis, Proc. Natl. Acad. Sci. U. S. A. 95 (1998)
12983–12988.