depolymerization of pp review article
TRANSCRIPT

JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 1
Journal of Polymer & Composites ISSN: 2321-2810(online), ISSN: 2321-8525(print)
Volume 4, Issue 2
www.stmjournals.com
Depolymerization of Polypropylene: A Systematic Review
Parag Kulkarni*, Ruchika Pache Department Polymer and Surface Engineering, Institute of Chemical Technology, Mumbai-400019,
Maharashtra, India
Abstract Polypropylene have become an indispensable ingredient of human life and most widely used
thermoplastic in commodity applications. Polymerization of propylene is carried out by using
Ziegler-Natta or Metallocene catalysts which results in high molecular weights and
crystalline polypropylene which are not required for several applications. Reduction in
molecular weight of polypropylene in conventional reactors is uneconomical and complicated.
Therefore to overcome this problem researchers and technologists proposed a post reactor
operation to reduce molecular weight (depolymerization) of polypropylene. To induce
modification in polypropylene different techniques of depolymerization are proposed. In this
review article, we have discussed various types of polypropylene depolymerization methods
with their mechanisms like oxidative, thermal, high energy radiation and chemical (free
radical induced) depolymerization along with their effect on thermal, rheological, crystalline
and chemical properties of the polypropylene.
Keywords: Polypropylene, molecular weight, depolymerization, thermal depolymerization,
rheology
*Author for Correspondence E-mail: [email protected]
INTRODUCTION Polyolefins are the most widely used polymers
for manufacturing of commodity plastics
article. They are manufactured usually by
using transition metal catalysts and have very
high molecular weights. Polypropylene is an
enormously adaptable member of the
polyolefin family, which is due to the
prochiral nature of the propylene monomer.
Highly crystalline polypropylene is built up by
the chain growth polymerization of propylene,
a gaseous compound obtained by the thermal
cracking of ethane, propane and butane or
naptha fraction of petroleum. In 1951
scientists at Phillips Robert L Banks et al.
found that one of their processes of converting
propylene into gasoline led to the formation of
whitish substance called crystalline
polypropylene. Ziegler modified the
equipment and prepared polypropylene in
1954. In 1954 Natta managed to synthesize
polypropylene, followed shortly by Ziegler.
The first industrially important crystalline,
high molecular weight polypropylene was
synthesized by Natta in 1955 from organo-
metallic catalysts based on titanium and
aluminum [1]. Polypropylene was developed
by scientists at Phillips, Hoechst, Montecatini,
Hercules, Farbwerke-Hoechst, by Ziegler and
Natta and in separate development by standard
oil of Indiana, Du Pont in 1957 [2].
Polypropylene has been widely used as
injection molding plastics as well as in many
extruded forms including film, fiber and split
yarns [3].
Polypropylene is one of the world’s most
important thermoplastic materials and it is
used in numerous applications in the plastics
industry. It has desirable properties such as
low-cost, high-melting point, low density,
appropriate process ability, high-strength, high
stiffness, hinge properties, easy to handle and
excellent chemical resistance [4]. Its
applications have expanded continuously for
the last decade. Since commodity plastics do
not require extremely high mechanical
properties, molecular weights and crystallinity
always, highly crystalline isotactic
polypropylene has been reported to be having
very high melt viscosity with consequently
poor flow properties and deficient in impact
strength at low temperatures, mainly owing to
its relatively high Tg and large spherulite
dimensions. It’s moderately elevated glass
transition temperature results in it being too

Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 2
brittle and hard at application temperatures
below 0ºC. These high molecular weights of
polypropylene give resistance to flow at
elevated temperatures and hence operations of
shaping by compression molding or injection
molding or extrusion are unnecessarily
complex. The toughness and rheological
properties of polypropylene can be enhanced
by reducing the molecular weight, molecular
weight distribution, introduction of branching
in polymer backbone and controlling average
size of the spherulites in depolymerization.
Control of molecular weight in the reactor,
during the process of polymerization is
difficult. The process parameters are set and
any modification in the process like addition
of a chain transfer agent would result in
resetting the process, which in turn is very
tedious, difficult and economically not
feasible. To achieve the diversity in
polypropylene grades suitable for the different
applications the molecular weight and
molecular weight distribution (MWD) must be
tailor-made to fit the performance
requirements of each application.
DEPOLYMERIZATION OF
POLYPROPYLENE Depolymerization leads to changes in
molecular properties such as molecular
weight, molecular weight distribution, thermal
properties, and crystallisability, etc. which are
considered to be more beneficial in mechanical
properties. This foregoing defects and
deficiencies can be overcome by
depolymerization process for reducing the
molecular weight of the polypropylene.
Polypropylene is depolymerized to achieve
easy processing for high-quality products in
injection molding and fiber spinning with
enhancing mechanical properties [5–7].
Depolymerization is the reversion of a
polymer to a low molecular weight polymer
fragments or splitting of polymers into
molecules [8]. Reverse of the propagation step
in chain polymerization is depolymerization or
unzipping, which is characterized by reduction
in molecular weight of the polymer to low
molecular weight polymer fragments [9].
Depolymerization of polymer may be brought
about by physical factor such as heat, light,
mechanical stress, high energy radiation, and
ultrasonication or by chemical agent such as
initiator, oxygen, ozone etc. Depolymerization
of the polymer is subdivided according to its
various modes of initiation. There are
oxidative, thermal, high energy radiation and
chemical (free radical induced) modes for
depolymerization of polypropylene. Lanrence
has patented a process which gives a
procedure of controlled thermal
depolymerization of a crystalline polymer of
an aliphatic mono-a-olefin which comprises
treating the polymer as slurry with water,
containing oxygen and including free radicals,
at a temperature of 60 to 90oC and recovering
from the slurry, a polymer of increased melt
index [10]. The depolymerization of 16.7
weight % slurry of isotactic polypropylene
dispersed in water was carried out in presence
of free radical initiator and it was found that in
the presence of 0.2% benzoyl peroxide at
95°C, the melt flow index increased to 13 from
9 after 1 h of reaction. Similarly in the
presence of 0.3% AIBN the melt flow index
increased from 0.1 to 1.2 after 2 h of reaction.
It is important to note that this reduction in
polymer is brought about by molecular chains
scission of polypropylene. Hans et al. gave a
process for reduction in border viscosities of
the polymer, by treating polypropylene in
fluidized bed, in presence of oxygen, within
temperature range of 100–130°C. Powdered
polypropylene, of border viscosity 6, was
treated in an eddy furnace for 40 min and at
temperature of 130°C. The resulting polymer
had a border viscosity of 3 [11]. The Dow
Chemical company has patented a process for
reduction in molecular weight of isotactic
polypropylene to improve its proccessability
[12]. The process comprises of thermally
depolymerizing polypropylene at temperatures
of 160 to 300°C, in the presence of small
amount of depolymerizing agent such as
bromine or bromine containing compound (to
result in a bromide radical). The treated
polypropylene had a melt index improved by
2.2. Davis et al. subjected polypropylene
samples to various degrees of thermal
depolymerization [13]. The molecular weight
distributions of these samples were
experimentally determined and compared with
those expected theoretically for random
scission of the polymer chains. The
comparison confirmed that the chain breakage
was predominately random and also indicated
that determination of molecular weight by
viscosity average molecular weight were

Journal of Polymer & Composites
Volume 4, Issue 2
ISSN: 2321-2810(online), ISSN: 2321-8525(print)
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 3
adequate for use in evaluating hypothesized
depolymerization mechanisms. The most
common types of depolymerization occur
through chemical reactions that alter the
molecular weight of the polypropylene,
leading to a change in its mechanical, thermal,
chemical and rheological properties. Such
reactions include (a) chain scission, (b) cross-
linking, (c) modification of branched chains,
or (d) a combination of all of these reactions.
The agent(s) initiating the depolymerization
process defines the type of depolymerization.
A review of the different types of
depolymerizations and their initiating agents
are given in Table 1.
Table 1: Initiating Agents and
Depolymerization Methods.
Initiating Agent Depolymerization Type
Heat Thermal
Oxygen, Ozone Oxidative
X-rays, γ-rays, electron
beam radiation High energy radiation
Free radical induced Chemical
The chemical reactions in depolymerization
process are comparatively similar, with barely
trivial differences owing to variations in the
initiation mechanism [14].
Depolymerization Mechanism of
Polypropylene Depolymerization of polypropylene is usually
done by means of free radical chain reaction
mechanism shown in Figure 1 consisting of
the steps of: (i) initiation (Eq. (1)), (ii)
propagation (Eq. (2)) and (iii) termination
(Eq. (3)). Throughout the steps, heat-
facilitated hydrogen abstraction from the
tertiary carbons may lead to the formation of
tertiary alkyl radicals shown in Figure 1. In
PP, tertiary radicals are formed mostly, due to
the poorer dissociation energy of a tertiary C-
H bond (ca. 373 kJ.mol-1 at 25ºC), compared
to that of a secondary C-H bond (ca.
394 kJ.mol-1 at 25ºC) [19]. During the later
propagation step, formed tertiary radicals will
follow separate reaction paths. The reaction
paths followed by tertiary radicals are depicted
in schemes.
Termination of polymer reactions involving
radicals or macroradicals may take place [20].
Recombination is influenced by cage effects,
steric control, mutual diffusion and the
molecular dynamics of the polymer matrix
[21–23].
Oxidative Depolymerization Free radical depolymerization of
polypropylene cannot be controlled, to avoid
such problems in depolymerization; oxidation
on neat polymer is to work with solutions and
initiator begun by Dulog, Radlman and Kerns
[24]. In oxidative depolymerization, the
formation of free radical sites on the chain
backbone by attack of molecular oxygen,
ozone etc. is done [9]. The free-radical chain
reactions depicted in Figure 2 describe
oxidative depolymerization of polypropylene
[20, 25], consisting of individual steps of
initiation, propagation and termination.
During the initiation stage of oxidative
depolymerization, initiating agent stimuli such
as heat or UV radiation are significant for the
growth of radicals. Cleavage of a polymer
chain is done, due to the abstraction of a
hydrogen atom from the polymer backbone
(reaction 2.1).
Fig. 1: Depolymerization Mechanism of Polypropylene.

Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 4
Polymer chain scission is frequently caused by
severe deformation of a polymer. In the
propagation steps, the radicals formed during
initiation, react with molecular oxygen and are
changed into macro-alkyl peroxyl radicals
(reaction 2.2). Then abstraction of a hydrogen
atom from another polymer molecule gives
macro-hydroperoxides and more alkyl radicals
(reaction 2.3). Hydroperoxides formed through
this step of reaction might be cleaved
homolytically to give up radical. These kinds
of radicals are capable to abstract hydrogen
form adjacent polymer chains ensuing to the
growth of more radicals accountable for
initiating depolymerization.
Free radicals produced through the first two
steps may also go through further reactions.
Such reactions are identified as β-scission
(reaction 2.5), reduce the molecular weight of
chains and also persuade the crystallinity of
polymers [26]. Considering as amorphous
regions of semi-crystalline polypropylene
depolymerized faster than the crystalline phase
[27–29], the scission of polymer backbone,
dependable for relating two adjoining
crystalline areas, will cause a rapid devastation
of the physical and mechanical properties of a
polypropylene. Elevated mobility of oxygen in
non-crystalline material leads to propagation
within amorphous regions. The termination
reactions are reliant on the molecular structure
of the polymer as well as the existing
depolymerization conditions and termination
(reactions 2.7, 2.8) prevails [20]. Though, in
the case of oxygen starvation alkyl radical
dominate and bimolecular termination
reactions are of superior importance. This
leads to cross-linking which is evidenced by a
rise in molecular weight. Oxidative cycle of
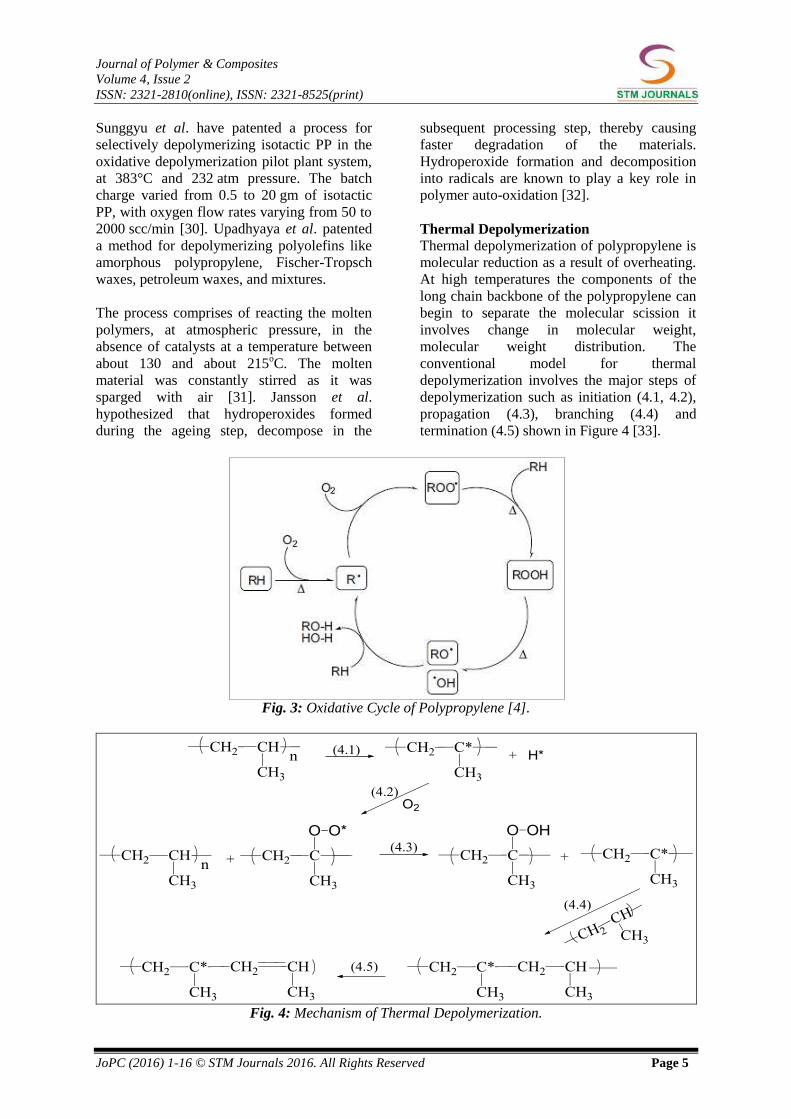
polypropylene is shown in Figure 3 [4].
Fig. 2: Oxidative Free Radical Depolymerization of Polypropylene.
Journal of Polymer & Composites
Volume 4, Issue 2
ISSN: 2321-2810(online), ISSN: 2321-8525(print)
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 5
Sunggyu et al. have patented a process for
selectively depolymerizing isotactic PP in the
oxidative depolymerization pilot plant system,
at 383°C and 232 atm pressure. The batch
charge varied from 0.5 to 20 gm of isotactic
PP, with oxygen flow rates varying from 50 to
2000 scc/min [30]. Upadhyaya et al. patented
a method for depolymerizing polyolefins like
amorphous polypropylene, Fischer-Tropsch
waxes, petroleum waxes, and mixtures.
The process comprises of reacting the molten
polymers, at atmospheric pressure, in the
absence of catalysts at a temperature between
about 130 and about 215oC. The molten
material was constantly stirred as it was
sparged with air [31]. Jansson et al.
hypothesized that hydroperoxides formed
during the ageing step, decompose in the
subsequent processing step, thereby causing
faster degradation of the materials.
Hydroperoxide formation and decomposition
into radicals are known to play a key role in
polymer auto-oxidation [32].
Thermal Depolymerization Thermal depolymerization of polypropylene is
molecular reduction as a result of overheating.
At high temperatures the components of the
long chain backbone of the polypropylene can
begin to separate the molecular scission it
involves change in molecular weight,
molecular weight distribution. The
conventional model for thermal
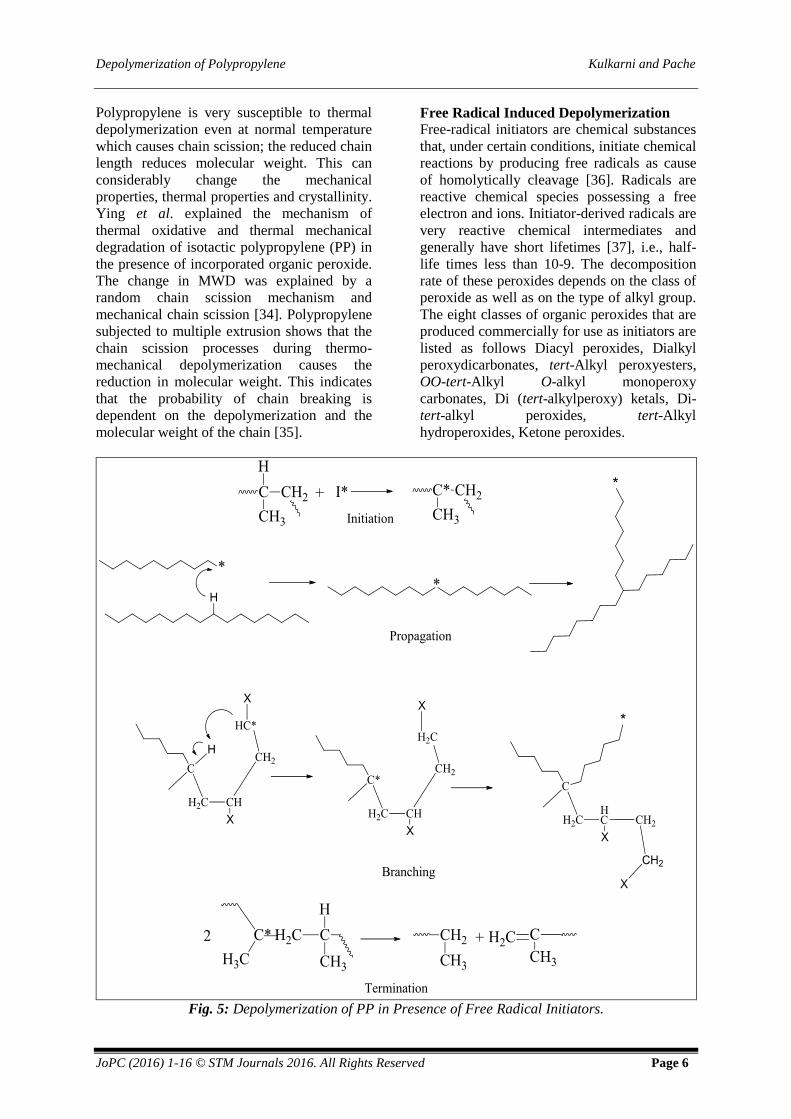
depolymerization involves the major steps of
depolymerization such as initiation (4.1, 4.2),
propagation (4.3), branching (4.4) and
termination (4.5) shown in Figure 4 [33].
Fig. 3: Oxidative Cycle of Polypropylene [4].
Fig. 4: Mechanism of Thermal Depolymerization.
Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 6
Polypropylene is very susceptible to thermal
depolymerization even at normal temperature
which causes chain scission; the reduced chain
length reduces molecular weight. This can
considerably change the mechanical
properties, thermal properties and crystallinity.
Ying et al. explained the mechanism of
thermal oxidative and thermal mechanical
degradation of isotactic polypropylene (PP) in
the presence of incorporated organic peroxide.
The change in MWD was explained by a
random chain scission mechanism and
mechanical chain scission [34]. Polypropylene
subjected to multiple extrusion shows that the
chain scission processes during thermo-
mechanical depolymerization causes the
reduction in molecular weight. This indicates
that the probability of chain breaking is
dependent on the depolymerization and the
molecular weight of the chain [35].
Free Radical Induced Depolymerization
Free-radical initiators are chemical substances
that, under certain conditions, initiate chemical
reactions by producing free radicals as cause
of homolytically cleavage [36]. Radicals are
reactive chemical species possessing a free
electron and ions. Initiator-derived radicals are
very reactive chemical intermediates and
generally have short lifetimes [37], i.e., half-
life times less than 10-9. The decomposition
rate of these peroxides depends on the class of
peroxide as well as on the type of alkyl group.
The eight classes of organic peroxides that are
produced commercially for use as initiators are
listed as follows Diacyl peroxides, Dialkyl
peroxydicarbonates, tert-Alkyl peroxyesters,
OO-tert-Alkyl O-alkyl monoperoxy
carbonates, Di (tert-alkylperoxy) ketals, Di-
tert-alkyl peroxides, tert-Alkyl
hydroperoxides, Ketone peroxides.
Fig. 5: Depolymerization of PP in Presence of Free Radical Initiators.

Journal of Polymer & Composites
Volume 4, Issue 2
ISSN: 2321-2810(online), ISSN: 2321-8525(print)
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 7
The Saule et al. studied of the modification of
polypropylene based on the decomposition of
unsaturated peroxides which helped to
decrease the molecular weight of the polymer.
Mechanisms for the branching and the
depolymerization of the polypropylene were
also proposed [38]. To optimize desired
processing capabilities and the end use
properties the controlled degradation of
polypropylene (PP) using peroxides is
proposed by Iedema et al.
The author performed a series of controlled
degradation experiments on a twin-screw
extruder with polypropylene of varying MW
under addition of various amounts of initiator
and developed a model which was adapted to
the geometry of the extruder entrance and the
peroxide feed practice. Effect of thermal
heterogeneities, residence time distribution,
micromixing on molecular weight distribution
was studied [39]. Balke et al. presents a new
kinetic model for the free radical initiated
degradation of polypropylene in the reactive
extrusion. The peroxide used was 2,5-
Dimethyl-2,5-di(t-butylperoxy) hexane. The
resulting product is known to provide superior
processing properties; this is an example of the
production of a specialty polymer by chemical
modification of a commodity polymer [40].
Possible reactions mechanism of
polypropylene in presence of free radical
initiators or organic peroxide is shown in
Figure 5 [41].
There are several common processes for
supplying the energy required to variety
radicals from initiators thermal and radiation
[36]. Formerly produced, radicals experience
two basic types of reactions, propagation
reactions and termination reactions. The
initiator molecule is denoted by I and the free
radical produced is shown by I*. In a
propagation reaction, a radical reacts to form a
covalent bond and to create a new radical. The
three most ordinary propagating reactions are
atom abstraction, ß-scission, and addition to
carbon–carbon double bonds or aromatic
rings. The polypropylene radicals produced
will further react with other polymer
molecules. The main importance of shift to
polymer is chain branching which frequently
occurs in two ways such as:
i. Hydrogen might be abstracted from sites
on the backbone thus transferring radical
movement and initiating a long chain
branch.
ii. Short chain branches possibly will be
fashioned by intra-molecular atom transfer
(backbiting). In this case the propagating
radical abstracts backbone hydrogen as of
a close by carbon.
In a termination reaction, two radicals
interrelate in a mutually vicious reaction in
which both radicals fashioned covalent bonds
and reaction ceases. Termination of the
polymer radicals can also be done by
recombination, where two polymer radicals
formed would merge.
i. In which combination of two tertiary
radicals would effect in cross-linking,
which is slightest liable in case of
polypropylene.
ii. Amalgamation of two secondary, or a
secondary and tertiary radical would give
a polymer molecule or by
disproportionate.
In the provisions of the free radical
depolymerization theory, intermolecular
transfer of the radical was found to be the
leading process. An arch linking the degree of
depolymerization to the intrinsic viscosity was
obtained from calculations based on a random
depolymerization. With this arch, intrinsic
viscosities measured as a function of
depolymerization time could be used to gain
the time dependence of the degree of
depolymerization [13].
Depolymerization by High Energy
Radiation Depolymerization of polypropylene can be
done by means of irradiation methods, such as
electron beams, ultrasonic, ultraviolet (UV)
and γ-rays, which have been favored to modify
the structure and properties of polypropylene
besides using chemical initiators and free
radicals.
Electron beams, ultrasonic, ultraviolet
radiation and γ-rays energy which possibly
will cause bond scission by free radical
formation for higher doses than 60 kGy for
long period depolymerization occurs. The

Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 8
depolymerization of polypropylene resulting
from experience to high energy radiation is an
important subject for many reasons. The
mechanism of high energy induced
depolymerization is a free radical, one similar
to the general mechanism of depolymerization
as shown in Figure 6.
Fig. 6: Mechanism of High Energy Induced Depolymerization PP.
In case of PP irradiation it has been
established that the PP undergoes essentially
chain scission. Polymers are increasingly more
exposed to radiation in their service where
depolymerization may cause [42]. Radiation-
induced depolymerization has been well
reviewed [43–46]. High energy radiation is
defined as all forms of radiation with energies
much higher than those of chemical bonds. It
includes both electromagnetic radiation (x-
rays, γ-rays, etc.) and particulate beams (α-
and β-particles, electrons, neutrons, etc.). The
series of energies is exceptionally broad and
superior to chemical bond energies. Radiation
is absorbed by interaction with atomic nuclei
and electron clouds. He et al. developed UV
initiation based reactive procedure for
depolymerization of polypropylene for the
production of controlled rheology
polypropylene, benzophonene has been used
as the photo initiator. At low-irradiation time
and low-levels of photo initiator molecular
weight distribution was noticeably tailored.
After depolymerization, MFR improved with
drop off in viscosity and elasticity with
benzophonene concentration as predictable.
Crystallinity levels and crystallization
temperatures of the tailored polypropylene
were lower than those of virgin polypropylene
because of the reduced molecular weight and
narrower MWDs [47]. Radiation induced
depolymerization caused by chain scission was
successfully used. Controlling the degree of
degradation, uniform molecular weight is
friendly process are the beneficial effects of
using radiation technology. When polymeric
materials are irradiated by ionizing radiation,
they are divided into two types, degradation
(chain scission) and chain link (crosslinking).
Studying the effect of hydrogen peroxide
and/or γ irradiation on the degradation process
of Na-alginate was investigated. It was found
that the molecular weight of the polymer
decreases by using gamma radiation or H2O2.
However, combining both γ radiation and
H2O2 accelerates the degradation rate [48]. The
influence of three hindered amine light
stabilizers (HALS) and two EVA copolymers
on the radiation degradation processes in
isotactic polypropylene (iPP) was investigated
by Zimek et al. The process consequences
degradation, oxidation, release of harmful
gaseous products, etc. as the polymer is known
as one of the most degradable upon experience
to ionizing radiation. Polypropylene properties
are changed even before exposure to electron
beam due to structural modifications caused
due to chain scission initiated by additional
components [49]. The range of energies is
extremely broad and higher than chemical
bond energies. Radiation is absorbed by
interaction with atomic nuclei and electron
clouds. The radiation interaction with the
electron clouds of molecules consequences in
the transfer of energy to the molecules to form
ions, with the elimination of a secondary
electron [50, 51]. Further ionization and
excitation of nearby molecules is caused by
these secondary electrons having enough
kinetic energy. The immediate effect of
absorption of high energy radiation is
construction of energetic or excited species,
including trapped electrons, ions, and radical
ions, results in fragmentation to provide free
radicals. In polypropylene, scission of main-
chain C-C bonds gives radical pairs. The
scission of C-H bonds leads to comparatively
stable radicals and formation of molecular
hydrogen. The primary radical formed by
irradiation might not be stable. For example,
irradiation of polypropylene with γ-radiation at
doses of 25, 50, 100 and 150 kGy produces the
secondary radical [52]. In the absence of
oxygen, carbon-centered radicals might

Journal of Polymer & Composites
Volume 4, Issue 2
ISSN: 2321-2810(online), ISSN: 2321-8525(print)
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 9
undergo cross-linking, by recombination,
chain scission, β-elimination depending on the
radical structure. Polymers can consequently
be divided into two main groups, those which
react to radiation by cross-linking and those
which drop molecular weight by chain scission
[53]. Ultrasonic irradiation has been recently
looked upon as a new technique for
degradation of polymer compounds, mainly
due to the fact that the reduction in the
molecular weight is simply by splitting the
most susceptible chemical bond without
causing any changes in the chemical nature of
the polymer. Detailed analysis of cavitations
generated ultrasound radiation used for
molecular weight reduction of polypropylene
has been done by Desai et al. The effect of
various operating parameters including initial
concentration of the polymer, power density
into the system and type of solvent, on the
extent of depolymerization of polypropylene
are investigated. Cavitations results in liquid
turbulence associated with liquid shear and
generation of highly reactive free radicals. The
shear force leads to the rupture of chemical
bonds of the polypropylene, ultrasonic sound
waves produce a permanent reduction in
viscosity [54].
CHARACTERIZATIONS TO STUDY
DEPOLYMERIZATION As it has been mentioned before,
polypropylene has extreme importance
commercially, but regrettably it is also less
susceptible to attack by heat, oxygen,
radiation, and free radical initiator. It is very
essential to study the depolymerization process
i.e., to set up the mechanisms throughout
oxidative depolymerization, the products
formed as well as the influence of molecular
weight and structure on the properties of
material. A full understanding of
depolymerization is of great significance,
since without this, successful stabilization
approaches and superior methods of lifetime
prediction would not have been possible. It is,
consequently, essential to consider the
characterization techniques employed to study
the depolymerization process. Analytical
chemistry field possesses a conventional
techniques that have been used for the purpose
of studying polypropylene depolymerization,
some of which include high temperature gel
permeation chromatography (GPC), Fourier
transform infrared spectroscopy (FTIR),
differential scanning calorimetry (DSC) and
viscometry. A brief overview on the use of
some of these techniques for the purpose of
studying polyolefin depolymerization will now
be presented.
High Temperature Gel Permeation
Chromatography According to hydrodynamic volume high
temperature gel permeation chromatography
(HTGPC) separates molecules and is the
preferred method for the determination of
molecular weight and molecular weight
distribution. During the depolymerization of
polypropylene, changes in average molecular
weight and molecular weight distribution
(MWD) are often observed [55–57].
Depending on the polymer and the
depolymerization methods, many radical
reactions can take place. In the case of
polyolefin’s, branching and recombination
reactions predominate at lower temperatures,
yielding a long chain branching, which leads
to an increase in the average molecular weight
of the material [58]. In case of polypropylene,
depolymerization almost exclusively by means
of chain scission, leads to a reduction of its
average molecular weight [59]. Chain scission
during depolymerization consequences in
continuous breaking of polymer chains
yielding chains of shorter lengths. The average
result is that the number of short chains
increases with depolymerization time and is
accompanied by a broadening of the molecular
weight distribution.
The molecular weight curve obtained from
GPC measurements, as a result, shifts towards
the region of lower molar mass when chain
scission is the dominant depolymerization
mechanism [60–62]. As depolymerization
proceeds and chain scissions continue, the
molecular weight curve will exhibit bimodality
with the portion of highly depolymerized short
molecules appearing as a narrow distribution
on the lower molecular weight side of the
original material. David et al. proposed an
Eq. (A) for determining the number of chain
scissions in degraded samples [63], by shaping
the numerical average of the initial and final
number-average molecular weights.

Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 10
𝑛𝑅 =𝑀 𝑛𝑜
𝑀𝑛𝑓− 1 (A)
Where,
𝑛𝑅is the number of chain scission,
𝑀 𝑛𝑜 is initial number average molecular
weight,
𝑀𝑛𝑓is final number average molecular weight.
High temperature gel permeation
chromatography can be used to record
molecular weight changes over a cross-section
of a thick polymer sample by means of
microtoming [64] or layer-by-layer milling
[65]. Computer-aided study method called
molecular weight distribution computer
analysis (MWDCA) has also proven to be
useful for the comparison of depolymerization
rates in polypropylene [66–68]. This technique
uses molecular weight data, obtained by
evaluation of experimental GPC molecular
weight distribution profiles with computer
simulations for determining the scission or
crosslink. Shyichuk investigated chain scission
and crosslink concentrations in photo-
degraded LDPE, polypropylene and ethylene-
propylene copolymer.
Differential Scanning Calorimetry Differential scanning calorimetry is an
additional technique frequently used to obtain
information on the depolymerization behavior
of polymers [69, 70]. Profile of melt
endotherms and changes in the peak
temperatures and as well as the glass transition
temperature, can provide information on the
susceptibility of different crystalline phases or
arrangements to depolymerization [71]. DSC
is very useful in determining the oxidative
induction time (OIT) [72–74], oxidative
temperature (Tox) [75] and thermal stability
degree of depolymerization of polymers under
high temperature conditions of polymers. Rosa
et al. investigated the influence of several
parameters on DSC statistics and found that
sample preparation (shape and size), oxygen
flow and heating rate of the experiment had a
considerable influence on the data obtained
[70].
Fourier Transform Infrared Spectroscopy For studying out the chemical changes brought
about by polymer depolymerization IR
spectroscopy is one of the most accepted
techniques. It has been used for qualitative as
well as quantitative characterization of
depolymerization products [76–80]. The
depolymerization route creates the formation
of different functional groups which are
strongly reliant on the chemical structure of
the polymer. The main chemical species
detectable by infrared spectroscopy are
hydroxyl and carbonyl groups [81]. The
configuration of these groups usually leads to
visible changes in infrared spectrum,
appearing in the regions of 1850–1550 and
3700–3200 cm-1
, respectively [82–84].
Moreover, change in the FTIR absorption of
depolymerization products in PE and PP can
also be experimental and used for studying the
difference in their oxidative depolymerization
mechanisms [85]. From the hydroxyl and
carbonyl groups, depolymerization products
such as peroxides, alcohols, can be identified
by means of derivatisation reactions [86].
Derivatisation methods for this point were first
applied to polyolefins by Carlsson et al. [87].
FTIR has also been sensible in determining
changes in unsaturation of PE during
depolymerization [88]. The degree of
depolymerization of polypropylene is
determined on the basis of their carbonyl index
[89–91]. The bands at 1892, 974, 2720 and
840 cm-1
have all been used as reference bands
for determining the carbonyl index in
polypropylene [89–92].
Viscometry
Viscometry is a useful technique for
determining the polymer molecular weight.
The molecular weight obtained by this
technique is the viscosity averaged molecular
weight. The increase in the viscosity imparted
by the macromolecules in solution is a direct
function of the hydrodynamic volume and,
hence, the molecular weight of the
macromolecule. The relationship between the
intrinsic viscosity and the viscosity average
molecular weight is given by the semi-
empirical Mark-Houwink Eq. (B) [9]. [𝜂] = 𝐾(𝑀𝑉)⍺ (B)
Where η is intrinsic viscosity, K and ⍺ are
constants for a polymer-solvent system at a
given temperature and Mv is viscosity average
molecular weight. The η, values for the
polypropylene solution were calculated by the
one point intrinsic viscosity Eq. (C).
[𝜂] =(2(ηsp−ln ηr))
0.5
𝐶 (C)

Journal of Polymer & Composites
Volume 4, Issue 2
ISSN: 2321-2810(online), ISSN: 2321-8525(print)
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 11
Where ηr and ηsp are relative and specific
viscosities respectively, C is concentration of
polymer solution [93]. The viscometric studies
of polymer solutions as a means of molecular
characterization of polymers are well
recognized and widely practiced because of
simplicity in terms of experimental approach
and the apparatus needed. Dilute solution
viscosity can be conveniently measured in
capillary viscometers such as Ostwald type or
the Ubbelohde type [9]. Shenoy et al.
dissolved the depolymerized polypropylene
samples in xylene at 100°C and the viscosity
measurement at 85°C was carried out by using
the Ubbelohde viscometer. Relative (ηr) and
specific viscosities (ηsp) respectively were
calculated using standard formulae to
determine the viscosity average molecular
weight [94]. For PP-xylene system at 85°C the
Mark Houwink constants are K=9.6×10–3
and
α=0.63 [100].
EFFECTOF MOLECULAR WEIGHT
REDUCTION Melt Flow Index
The melt flow index (MFI) of the samples is
considered as a critical parameter in polymer
processing and industrial designs. MFI of a
polyolefin resin refers to the rate at which it
extrudes from a capillary die under a standard
set of conditions. The MFI is reflected by the
average dimensions of the molecules in a resin
and their entanglements with one another so it
depends on molecular characteristics (Mw and
MWD) and branching characteristics, short
chain branching (SCB), of the sample. Fazeli
et al. have showed that the MFI of the samples
is related to their branching characteristics.
With increasing the degree of branching
(SCB/100°C), the MFI of the sample goes
through a minimum. The increase in SCB will
cause more entanglements between the chains
(inter-molecular entanglement) and therefore
will impede the flow [101]. However, after the
minimum point, increase in the SCB/100°C
will cause the chains to have a more compact
molecular profile (more intra-molecular
entanglements instead of inter-molecular
ones), so the chains will cause less hindrance
to the flow of other chains. In other words, the
degree of branching (number of branching per
100°C atoms of main chain) and the amount of
branching (the number of chains which have
the same degree of branching), have the same
effect on MFI. The melting point of a-form
iPP is strongly influenced by the
stereoregularity [102, 103]. Melting points in
the 160–168°C range are typical for
commercial homopolymer samples under
normal analysis conditions.
Melt Strength
The melt strength of a polymer is defined as
the maximum force at which a molten thread
can be drawn under standard conditions before
it breaks. High values of MS are desired in
processes where the material is stretched in its
molten state, such as in film blowing,
thermoforming, or foaming. Lagendijk et al.
have shown how the melt strength is enhanced
by the presence of strain hardening in
elongational viscosity [104]. Increasing
average molecular mass of a polymer results in
higher shear viscosity, as well as higher melt
strength. The melt strength also increases
when the molecular mass distribution (MWD)
becomes broader. Ghijsels et al. have
demonstrated how the melt strength increases
much more dramatically than shear viscosity
upon the addition of long chain branches on
the polymer backbone [105–107]. De Maio
and Dong have studied the effect of chain
structure on melt strength of polypropylene.
Comparison among several linear and
branched polypropylenes obtained by electron
beam irradiation has shown that the melt
strength of branched can be up to 10 times
higher than that of linear PP with the same
MFI [108]. Gotsis et al. found that the
enhancement of the melt strength is related to
the increase of the weight average number of
long chain branches per molecule [109].
Rheology The molecular architecture of the polymer i.e.
short and long chain branching and MWD
affects the rheology of the melt. In a thorough
review on the effect of long chain branching
(LCB) on the linear viscoelasticity of
polyolefins, Vega et al. showed that the
introduction of LCB induces higher levels of
elasticity than broadening of the MWD of a
melt of linear chains of similar molecular
weight [110]. On the other hand, it is current
understanding that short chain branching
(SCB) cannot cause large increases in

Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 12
elasticity. Yet, Vega et al. reported that SCB
resulted in higher zero shear rate viscosities,
higher relaxation times, higher values of the
elastic storage modulus and higher activation
energies of flow compared to linear polymers
[111]. Those authors surmised that the number
of entanglements per branch is a decisive
parameter that influences shear and
elongational behavior. Much less research has
been done on the extensional properties of PP
melts because appropriate samples have been
difficult to obtain. Hingmann and Marczinke
found that branched PP samples showed
distinct strain hardening and concluded that a
few branches on the chain had an enormous
effect on the extensional behavior of the melt
[112].
Crystallinity Wide angle X-ray scattering (WAXS) patterns
of iPP, sPP, and aPP are shown in Figure 7
[113]. The regular molecular structure of iPP
and sPP readily enables crystallization of the
chains, leading to well defined crystalline
reflections differing in unit cell symmetry. aPP
lacks a regular molecular structure, and does
not crystallize [114]. Brucker et al. have
shown that the isotactic polypropylene can
crystallize into three different crystal forms
depending on the temperature, pressure and
mechanical stress state: monoclinic a-,
orthorhombic γ- and hexagonal ß-forms [115,
116]. The γ-crystals are formed only under
high pressure in high molar mass
homopolymer polypropylene. Polypropylene
samples with low molar mass or low tacticity
and polypropylene copolymers crystallize
partially in γ-form. The degree of crystallinity
varies between 0 for a completely amorphous
material (such as aPP) and 1 for a completely
crystalline material. The degree of crystallinity
plays a critical role in determining properties
of polypropylene. Commonly measured
properties such as; modulus, yield stress,
oxygen-moisture barrier resistance, and
hardness increase with increasing crystallinity.
In addition to tacticity, crystallinity generally
increases with decreasing molecular weight
(increased chain mobility), and is promoted by
slower cooling rates from the melt. Busico et
al. have explained how the density of iPP in
the a-form varies between the limit of 100%
amorphous (ϱa=0.850 to 0.855 g/cm3) and
100% crystalline (ϱc=0.936 to 0.946 g/cm3)
[117].
Fig. 7: Wide-Angle X-Ray Scattering (WAXS) Patterns of iPP, sPP, and aPP [113].

Journal of Polymer & Composites
Volume 4, Issue 2
ISSN: 2321-2810(online), ISSN: 2321-8525(print)
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 13
Balta-Calleja and Vonk indicated a method for
evaluating crystallinity as a number by
numerical means from the relationship of the
peak area to the total area [118]. Jinghua et al.
investigated the nonisothermal crystallization
kinetics of linear and long chain branched
polypropylene (LCB PP) by differential
scanning calorimetry (DSC) at various cooling
rates. It was shown that LCB has the role of
heterogeneous nucleating agent and
accelerates the crystallization process of PP.
Also the activation energy of LCB PPs are
higher than that of linear PP, indicating that
the presence of LCB baffles the transfer of
macromolecular segments from PP melt to the
crystal growth surface [119]. Furthermore, the
crystal morphology of linear PP and LCB PPs
was observed through polarized optical
microscopy (POM), and fine spherulites were
observed for LCB PPs. De Nicola et al. and
Wang et al. have independently investigated
the effect of branching on the crystallization
behaviors of PP resins [120, 121]. It was
observed that the introduction of branching in
the PP resins increases crystal nuclei density.
This promotes faster crystallization, and,
hence, higher crystallization temperatures
were observed for branched materials as
compared to linear materials. The cooling rate
was fixed to be 10°C/min. It was observed that
the branching of PP chains significantly
promoted the crystallization kinetics of the PP
resins by increasing the crystallization
temperature about 20°C.
Chemical Properties
iPP is soluble in high boiling aliphatic and
aromatic hydrocarbons at high temperature.
The high chemical resistance of iPP results in
exceptional stain resistance, and has led to the
use of iPP in automobile batteries. iPP has
outstanding resistance to water and other
inorganic environments. iPP resists most
strong mineral acids and bases, but like other
polyolefins is subject to attack by oxidizing
agents including 98% sulfuric acid and 30%
hydrochloric acid at high temperature
(13<100°C), and fuming nitric acid (ambient
temperature) [122]. PP reacts with oxygen in
several ways, causing chain scission and
brittleness that is associated with the loss in
molecular weight. This action is promoted by
high temperatures, light, or mechanical stress.
Several scientists have independently
demonstrated treatment of polypropylene with
peroxides which has led to controlled rheology
resins with reduced molecular weight and
narrow polydispersity relative to polymerized
product from Ziegler-Natta catalysts [123–
125]. These resins are used in some fiber
spinning and injection molding applications.
The creation of radical sites along the polymer
backbone, most often through peroxide-based
initiation, is also an essential condition for
many fictionalization/grafting chemistries.
CONCLUSION It is concluded from the foregone discussion
that understanding about the mechanism can
go a long way in helping the researchers and
the technologists to induce the different types
of depolymerization methods in the
polypropylene. This depolymerization can
further be enhanced by the addition of the
initiator, additives in the polypropylene and by
understanding the various factors such as
mechanical, physical, and chemical which are
responsible for this depolymerization. It is also
concluded from this discussion that
polypropylene depolymerization could be
enhanced its properties with modification in
structure.
REFERENCES 1. Natta G, Pino P, Corradini P. J Am
Chem Soc. 1955; 77.
2. Seymour RB. History of Polyolefins. In:
Vasile C, Seymour RB, editors.
Handbook of Polyolefins. New York:
Marcel Dekker; 1993; 1p.
3. Qicong Ying, Yong Zhao, Yong Liu.
Makromol Chem. 1991; 192.
4. Denis Bertin, Marie Leblanc, Marque
Sylvain RA, et al. Polym Degrad Stabil.
2010; 95.
5. Imperial Chemical Industries Ltd. Germ
Pat. 1210562. Mar 17, 1961.
6. Chemische Werke Hls AG. French Pat.
1377951. Nov 6, 1964.
7. Caver Hill R, Taylor GW. Polymer.
1965; 6.
8. Gooch Jan W. Encyclopedic Dictionary
of Polymers. 2nd Edn. Springer; 2011.
9. Gowarikar VR, Vishwanathan NV,
Sreedhar Jayadev. Polymer Science.

Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 14
Reprint Edn. India: New Age
Publishers; 1994.
10. Laurence Stephen Rayner. Imperial
Chemical Industries Ltd. US Pat.
3232918. Feb 1, 1966.
11. Boehm Hans, Henglein Friedrich,
Schmidt-Thomee, et al. BASF AG, DE
1201552. Sep 1965.
12. Saunders Frank Linwood. Dow
Chemical Company, DE 1221798. Jul
1966.
13. Thomas Davis E, Robert Tobias L,
Peterli Elizabeth B. J Poly Sci. 1962; 56.
14. Wood DL, Luongo JP. Mod Plast. 1961;
38.
15. Williams SD, Yoo HJ, Drickman MR.
US Patent 5820981. 1998.
16. Lagendijk RP, Hogt AH,
Buijtenhuijsand A, et al. Polymer. 2001;
42.
17. Sugimoto M, Tanaka T, Masubuchi Y,
et al. J Appl Polym Sci. 1999; 73.
18. De Nicola Jr., Anthony J. US Patent
5047446. Sep 01, 1991.
19. Gonzalez-Gonzalez VA, Neira-
Velazquez G, Angulo-Sanchez JL.
Polym Degrad Stabil. 1998; 60.
20. Al-Malaika S. Adv Polym Sci. 2004;
169.
21. Garton A, Carlsson DJ, Wiles DM. J
Polym Sci Polym Chem Ed. 1978; 16.
22. Garton A, Carlsson DJ, Wiles DM.
Macro. 1979; 12.
23. Al-Malaika S. Polym Plast Technol Eng.
1988; 27.
24. Dale Van Sickle E. J Polym Sci. 1972;
10(a-1).
25. George GA, Celina M, Hamid SH.
Handbook of Polymer Degradation. 2nd
Edn. New York: Marcel Dekker Inc.;
2000.
26. Rabello MS, White JR. Polymer. 1997;
38.
27. Blais P, Carlsson DJ, Wiles DM. J
Polym Sci. 1972; 10(A1).
28. Schlick S, Kruczala K. JCT Res. 2005;
2.
29. Knight JB, Calvert PD, Billingham NC.
Polymer. 1985; 26.
30. Lee Sunggyu, Gencer Mehmet A,
Fullerton Kathy L, et al. US Pat.
5386055. Jan 31, 1995.
31. Upadhyaya Janardan D. US Pat.
4624993. Nov 25, 1986.
32. Anna Jansson, Kenneth Moller, Thomas
Hjertberg. Polym Degrad Stabil. 2004;
84.
33. Thermal Degradation of Polymers a
Primer. Pop Plast Packag. 2013; 12.
34. Qicong Ying, Yong Zhao, Yong Liu.
Makromol Chem. 1991; 192.
35. Sebastiaao V, Canevarolo. Polym
Degrad Stabil. 2000; 709.
36. Myers Terry N, Mark Herman F.
Encyclopedia of Polymer Science and
Technology. 3rd Edn. John Wiley and
Sons; 2004.
37. Griller D, Ingold KU. Acc Chem Res.
1976; 9.
38. Myriam Saule, Laurence Moine, Marie
Degueil-Castaing, et al.
Macromolecules. 2005; 38.
39. Iedema PD, Remerie NK, Vanderhamb
BM, et al. Chem Eng Sci. 2011; 66.
40. Balke ST, Suwanda D, Lew R. J Polym
Sci: Part C, Polym Lett. 1987; 25.
41. Imoto Minoro, Takemoto Kiichi, Kono
Masatsugu. Angew Makromol Chem.
1967; 1.
42. Clough RL, Gillen KT. Nuc Techno.
1982; 59.
43. David C. Degrad Polym. 1975; 14.
44. Schnabel WS, Jellinek HHG.
Degradation and Stabilization of
Polymers. Amsterdam: Elsevier; 1978.
45. Schnabel WS. Polymer Degradation:
Principles and Practical Applications.
Munich: Hanser; 1981.
46. Ivanov VS. Radiation Chemistry of
Polymers. Amsterdam: VSP; 1992.
47. Guangjian He, Costas Tzoganakis.
Polym Eng Sci. 2011.
48. Hegazy EA, Abdel-Rehim H, Diaa DA,
et al. Controlling of Degradation Effects
in Radiation Processing of Polymers.
International Atomic Energy Agency;
2009.
49. Zimek Z, Przybytniak G, Rafalski A, et
al. International Atomic Energy Agency;
2009.
50. Swallow A. Radiation Chemistry.
London: Longman; 1973.
51. Spinks JWT, Woods RJ. An Introduction
to Radiation Chemistry. New York:
John Wiley and Sons, Inc.; 1976.
52. Krupa I, Luyt AS. Polym Degrad Stabil.
2001; 72.

Journal of Polymer & Composites
Volume 4, Issue 2
ISSN: 2321-2810(online), ISSN: 2321-8525(print)
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 15
53. Bilingham NC. Degradation. In: Mark
HF, editor. Encyclopedia of Polymer
Science and Technology. Vol. 6. John
Wiley and Sons; 2005.
54. Vaibhav Desai, Shenoy Mohan A,
Gogate Parag R. Chem Eng J. 2008;
140.
55. Fayolle B, Audouin L, Verdu J. Polym
Degrad Stab. 2002; 75.
56. Gonzalez-Gonzalez VA, Neira-
Velazquez G, Angulo-Sanchez JL.
Polym Degrad Stab. 1998; 60.
57. Lew R, Suwanda D, Balke ST. J Appl
Polym Sci. 1988; 35.
58. Yadav A, Vimal KK, Singh R. J Polym
Composites. 2016; 4(1).
59. Ying Q, Zhao Y, Liu Y. Macromol
Chem. 1991; 192.
60. Alam MS, Nakatani H, Goss BGS, et al.
J Appl Polym Sci. 2002; 86.
61. Manabe N, Yokota H, Suzuki S, et al. J
Appl Polym Sci. 2006; 100.
62. Nakatani H, Manabe N, Yokota Y, et al.
Polym Int. 2007; 56.
63. David C, Trojan M, Daro A, et al.
Polym Degrad Stab. 1992; 37.
64. Girois S, Audouin L, Verdu J, et al.
Polym Degrad Stab. 1996; 51.
65. Turton TJ, White JR. Polym Degrad
Stab. 2001; 74.
66. Shyichuk AV, White JR, Craig IH, et al.
Polym Degrad Stab. 2005; 88.
67. Shyichuk AV, White JR. J Appl Polym
Sci. 2000; 77.
68. Shyichuk AV, Stavychna DY, White JR.
Polym Degrad Stab. 2001; 72.
69. Olivares N, Tiemblo P, Gomez-Elvira
JM. Polym Degrad Stab. 1999; 65.
70. Rosa DS, Sarti J, Mei LHI, et al. Polym
Test. 2000; 19.
71. Elvira M, Tiemblo P, Gomez-Elvira JM.
Polym Degrad Stab. 2004; 83.
72. Camacho W, Karlsson S. Polym Degrad
Stab. 2002; 78.
73. Chang TC, Yu PY, Hong YS, et al.
Polym Degrad Stab. 2002; 77.
74. Groning M, Eriksson H, Hakkarainen
M, et al. Polym Degrad Stab. 2006; 9.
75. Ahlblad G, Gijsman P, Terselius B, et
al. Polym Degrad Stab. 2001; 73.
76. Gulmine JV, Janissek PR, Heise HM, et
al. Polym Degrad Stab. 2003; 79.
77. Edge M. Infrared Spectroscopy in
Analysis of Polymer Degradation. In:
Meyers RA, editor. Encycl Anal Chem.
Chichester: John Wiley and Sons Ltd.;
2000.
78. Gugumus F. Polym Degrad Stab. 1997;
55.
79. Nekhoroshev VP, Turov YP,
Nekhorosheva AV, et al. J Appl Chem.
2006; 79.
80. Costa L, Luda MP, Trossarelli L. Polym
Degrad Stab. 1997; 58.
81. Andreassen E, Karger-Kocsis J. Infrared
and Raman Spectroscopy of
Polypropylene. Dordrecht: Kluwer
Publishers; 1999.
82. Mani R, Singh RP, Sivaram S. Polym
Int. 1997; 44.
83. Rivaton A, Gardette JL, Mailhot B, et al.
Macromol Symp. 2005; 225.
84. Gugumus F. Polym Degrad Stab. 1999;
6.
85. Adams JH, Goodrich JE. J Polym Sci.
1970; Part A1(8).
86. Piton M, Rivaton A. Polym Degrd Stab.
1996; 53.
87. Carlsson DJ, Brousseau R, Zhang C, et
al. ACS Symp Ser. 1988; 364.
88. Kolbert AC, Didier JG, Xu L.
Macromolecules. 1996; 29.
89. Jansson A, Moller K, Gevert T. Polym
Degrad Stab. 2003; 82.
90. Miraftab M, Horrocks AR, Mwila J.
Polym Degrad Stab. 2002; 78.
91. Santos ASF, Agnelli JAM, Trevisan
DW, et al. Polym Degrad Stab. 2002;
77.
92. Rabello MS, White JR. Polym Degrad
Stab. 1997; 56.
93. Solomo O, Ciuta ZZ. J Appl Polym Sci.
1962; 6.
94. Shenoy MA, Mrinalini Patil. Polym Sci.
2010; 52.
95. Feng-Hua Su, Han-Xiong Huang. Polym
Eng Sci. 2009; 50(2).
96. Hamed Azizi, Ismaiel Ghasemi. Iran
Polym J. 2005; 14(5).
97. Manfred Raetlsch, Achim Hesse,
Harmut Bucka, et al. US Pat. 6136926.
Oct 24, 2000.
98. Horst David E, Michael Roth, Peter. US
Pat. 2007/0200272. Aug 30, 2007.

Depolymerization of Polypropylene Kulkarni and Pache
JoPC (2016) 1-16 © STM Journals 2016. All Rights Reserved Page 16
99. Gaylord Norman G. US Pat. 4506056.
Mar 19, 1985.
100. Brandrup J, Immergut EH, Grulke EA.
Polymer Handbook. New York: Wiley;
1999.
101. Fazeli N, Arabi H, Bolandi. Polym Test.
2006; 25.
102. Wunderlich B. Macromolecular Physics.
1980; 3.
103. Paukkeri R, Lehtinen A. Polymer. 1993;
34.
104. Lagendijk RP, Hogt AH, Buijtenhuijs A,
et al. Polymer. 2001; 42.
105. Ghijsels A, Ente JSM, Raadsen J. Int
Polym Process. 1990; 5.
106. Ghijsels A, Clippeleir De J. Int Polym
Process. 1994; 9.
107. Ghijsels A, Massardier CHC, Bradley
RM. Int Polym Process. 1997; 12.
108. De Maio VV, Dong D. Proceedings SPE
ANTEC. 1997; 43.
109. Gotsis AD, Zeevenhoven BLF, Hogt
AH. Polym Eng Sci. 2004; 44.
110. Vega J, Aguilar M, Peon J, et al. e-
Polymers. 2002; 46.
111. Vega JF, Santamaria A, Munoz-Escalera
A, et al. Macromolecules. 1998.
112. Hingmann R, Marczinke BL. J Rheol.
1994; 38.
113. Phillips RA, Wolkowicz MD, Moore
EP. Polypropylene Handbook. Munich:
Hanser; 1996.
114. Natta G, Corradini P. Del Nuovo
Cimento. 1960; 15.
115. Takodoro H. Structure of Crystalline
Polymers. USA: John Wiley and Sons;
1979.
116. Brucker S, Meille V, Petraccone, et al.
Prog Polym Sci. 1991; 16.
117. Busico V, Corradini P, Debiasio R, et
al. Macromolecules. 1994; 27.
118. Balta-Calleja FJ, Vonk CG. X-ray
Scattering of Synthetic Polymers.
Amsterdam: Elsevier; 1989.
119. Tian Jinghu, Yu Wei, Zhou Chixing. J
Appl Polym Sci. 2007; 104.
120. De Nicola AJ, Galombos AF,
Wolcowicz MD. Polym Mater Sci Eng.
1992; 67.
121. Wang X, Tzoganakis C, Rempel GL. J
Appl Polym Sci. 1996; 61.
122. Pro-fax Polypropylene Chemical
Resistance. Montell Polyolefins
Technical Bulletin. 1996; TL-101.
123. Zeichner G, Patel P. 2nd World
Congress of Chemical Engineering,
Montreal, P.Q., Canada. 1981.
124. Dziemianowicz TS, Cox WW. SPE
ANTEC. 1985; 85.
125. Gahleitner M, Wolfschwenger J,
Bachner C, et al. J Appl Polym Sci.
1996; 61.
Copyright Notice
Declaration & Copyright Transfer Form
I, the undersigned author(s) of the submitted
manuscript, hereby declare, that the above
manuscript which is submitted for publication
in the STM Journals(s), is not published
already in part or whole (except in the form of
abstract) in any journal or magazine for private
or public circulation, and, is not under
consideration of publication elsewhere.
I will not withdraw the manuscript after
1 week of submission as I have read the
author guidelines and will adhere to the
guidelines.
I have neither given nor will give this
manuscript elsewhere for publishing after
submitting in STM Journal(s).
I have read the original version of the
manuscript and am responsible for the
thought contents embodied in it. The work
dealt in the manuscript is my/our own, and
my/our individual contribution to this
work is significant enough to qualify for
authorship.
I also agree to the authorship of the article
in the following order:
Author’s name
1) Parag Rajabhau Kulkarni
2) RuchikaDhanrajPache
Cite this Article Parag Kulkarni, Ruchika Pache.
Depolymerization of Polypropylene: A
Systematic Review. Journal of Polymer
& Composites. 2016; 4(2): 1–16p.