determination of total mercury by vapor generation in situ
TRANSCRIPT

Chem. Anal. (Warsaw), 53, 905 (2008)
Determination of Total Mercury by Vapor GenerationIn Situ Trapping Flame Atomic Absorption Spectrometry
by Henryk Matusiewicz* and Magdalena Krawczyk
Poznañ University of Technology, Department of Analytical Chemistryul. Piotrowo 3, 60-965 Poznañ, Poland
Keywords: Mercury; Reference materials; In situ trapping; Hydride generation; Flameatomic absorption spectrometry
The analytical performance of non-chromatographic coupled hydride generation, integratedatom trap (HG–IAT) atomizer flame absorption spectrometry (FAAS) systems were evalu-ated for the determination of total mercury in environmental samples. Mercury, using forma-tion of mercury vapors were atomized in air-acetylene flame-heated IAT. A new design ofvapor generation integrated atom trap flame atomic absorption spectrometry (VG–IAT–FAAS) hyphenated technique that would exceed the operational capabilities of existingarrangements was investigated. This novel approach enables to decrease the detection limitdown to low pg mL–1 levels. The concentration detection limit, defined as 3 times the blankstandard deviation was 0.4 ng mL–1. For a 120 s in situ pre-concentration time (samplevolume of 2 mL), sensitivity enhancement compared to flame AAS, was 750 folds for Hg,using vapor generation-atom trapping technique. The sensitivity can be further improved byincreasing the collection time. The precision, expressed by RSD, was 9.3% (n = 6) for Hg.Reference and real sample materials were analyzed. The accuracy of the method was veri-fied by the use of certified reference materials and by aqueous standard calibrationtechnique.The measured Hg content, in reference materials, were in satisfactory agreementwith the certified values. The hyphenated technique was applied for mercury determina-tions in coal fly ash, sewage and water.
Opracowano metodê oznaczania rtêci pozwalaj¹c¹ na zwiêkszenie mo¿liwoœci analitycznychp³omieniowej absorpcyjnej spektrometrii atomowej (FAAS) przez po³¹czenie technikigenerowania wodorków (HG) oraz systemu zintegrowanego ³¹cz¹cego nasadkê szczelinow¹z kolektorem rurkowym (IAT). Pary rtêci ulega³y atomizacji w systemie zintegrowanymogrzewanym za pomoc¹ p³omienia powietrze-acetylen. Zaproponowano i zbadano now¹technikê sprzê¿on¹ VG–IAT–FAAS, która zwiêksza mo¿liwoœci analityczne wczeœniejzastosowanych systemów. Opracowana metoda pozwala na obni¿enie granicy wykrywalnoœci
* Corresponding author. E-mail: [email protected] to Professor Rajmund Dybczyñski on the occasion of his 75th birthday.

906 H. Matusiewicz and M. Krawczyk
The detection of mercury at trace levels is a complex analytical task because ofits specific physical and chemical properties and is one of the most difficult analyticalproblems in atomic absorption spectrometry (AAS). The most sensitive mercury reso-nance line lies in the vacuum UV region and is therefore not suitable for conventionalspectrometers. The less sensitive wavelength at 253.7 nm is the only available alter-native. However, the detection limit of conventional flame AAS (FAAS) is not suffi-ciently low to accurately determine of mercury, at trace levels, because of the limitedatom number density in the light path associated with poor nebulization and atomiza-tion. A FAAS technique gives only a characteristic concentration of 5 mg L–1 for 1%absorption and a detection limit of 0.2 mg L–1 at 253.7 nm and therefore, it is of littleuse for the determination of mercury [1]. Additionally chemical separation of mer-cury from the sample matrix is difficult to its high volatility. Therefore, in most casesa pre-concentration and vapor and hydride generation procedures, before the elementcan be determined, is essential. Critical reviews [2, 3] have focused on the metho-dologies for the determination at trace levels of mercury. Of all the methods for thedetermination of mercury, atomic absorption spectrometry and atomic fluorescencespectrometry (AFS) especially after a cold vapor generation technique and hydridegeneration step, is the most useful and sensitive technique. This focused on the use ofthree reductants, SnCl2, Na(K)BH4 and HCOOH and provided information on LODand tolerance to interferences.
It is well established that significant improvements in the limits of detection ofFAAS and graphite furnace atomic absorption spectrometry (GF–AAS) may beachieved by chemical vapor generation, mostly hydride generation (HG) and in-ato-mizer (silica or graphite tube) trapping (pre-concentration) as reviewed [4–6] for thedetermination of As, Bi, Ge, In, Pb, Sb, Se, Sn, Te and Tl (as well as Hg).
Atom trapping approaches that involve the collection and in situ pre-concentra-tion of mercury using a dual „bent” tube atom trap (Au was used as a coating mate-rial) was first demonstrated by Ellis and Roberts [7]. Sensitivity of the technique was0.0598 mg L–1 for a 2 min collection. A comparison of the atom trapping techniqueand cold vapor method was carried out and the results were found to be in closeagreement.
do poziomu pg mL–1. Stê¿eniowa granica wykrywalnoœci, zdefiniowana jako trzykrotnawartoœæ odchylenia standardowego œlepej próby, wynosi³a 0.4 ng mL–1. Wspó³czynnikzatê¿ania Hg wynosi³ 750, po zatê¿aniu próbki o objêtoœci 2 mL, przez 120 s za pomoc¹techniki sprzê¿onej VG-IAT. Czu³oœæ metody mo¿na poprawiæ przez wyd³u¿enie czasuzatê¿ania. Precyzja, wyra¿ona jako wzglêdne odchylenie standardowe wynosi³a 9.3%(n = 6). Dok³adnoœæ opracowanej metody sprawdzono oznaczaj¹c Hg w certyfikowanychmateria³ach odniesienia standardow¹ technik¹ kalibracyjn¹ . Otrzymane wyniki by³y zgodnez wartoœciami certyfikowanymi rtêci. Opracowan¹ technikê sprzê¿on¹ zastosowano dooznaczania rtêci w popio³ach lotnych, œciekach i wodzie.

907Determination of total mercury by vapor generation in situ trapping FAAS
In situ trapping permits a significant enhancement in sensitivity for batch andcontinuous hydride generation approaches used in the ultra-trace determination ofvolatile hydride species. Due to its importance, in situ trapping, which allows thecoupling of hydride generation to integrated atom trap (IAT) [8–13] was chosen forthis study.
An analytical system was developed to trap and pre-concentrate Bi from the vaporphase stream. Bismuthine formed by sodium tetrahydroborate reduction was trappedon a tungsten coil previously heated to 270°C. The analyte species were re-volatili-zed by increasing the coil temperature to 1200°C and then transported to an exter-nally heated silica T-tube by using a mixture of argon and hydrogen as the carrier gas[14]. Korkmaz et al. [15] investigated the nature of re-volatilization from atom trapsurfaces in flame by AAS. Analytes Au, Bi, Cd, Mn and Pb were trapped on a water-cooled, U-shaped silica trap or a slotted silica tube trap and re-volatilized by organicsolvent aspiration. They concluded that, although heating was not necessarily associ-ated with re-volatilization, direct contact between the flame and the active silica sur-face was required. Recently [16] the analytical performance of three trap systems(water-cooled U-shaped silica trap, water-cooled U-shaped silica trap combined withslotted silica tube and slotted tube trap) for flame AAS were evaluated for determina-tion of Cd and Pb in waters. Guo and Guo [17] reported SeH2 collection at gold wireheated to 200°C situated in a quartz tube atomizer (AFS or AAS detection) withseparate inlet for argon. A successful trapping of PbH4 in a bare quartz tube wasannounced by Korkmaz et al. [18], who also suggested that the same trap could beused also for other hydrides. Recently, a preliminary evaluation of quartz tube trap forcollection of SbH3 and for volatilization of trapped analyte with subsequent atomiza-tion in a multiple microflame quartz tube atomizer for AAS was presented [19]. Kratzerand Dedina extended their investigation of stibine trapping in quartz tube traps [20]to stibine collection (and subsequent analyte atomization) in conventional quartz tubeatomizers [19]. They employed the simplest possible experimental arrangement; justthe commercially available externally heated quartz tube atomizer without any trapor additional heating device. Further, a modification of the externally heated quartztube atomizer, making possible in situ trapping of bismuthine and subsequent analyteatomization for Bi was described [21]. Krejèi et al. [22] investigated the collection ofSb and Bi on a molybdenum foil strip situated in a laboratory-made quartz T-tube,single-slot burner head, following hydride generation. Recently [23], the analyticalperformance of a miniature quartz trap coupled with electrochemical hydride genera-tor for antimony determination was described. A portion of the inlet arm of the conven-tional quartz tube atomizer was used as an integrated trap medium for on-line pre-concentration of electrochemically generated hydrides. Very recently [24], a novelquartz device has been designed to trap arsine and selenium hydride and subsequentlyto volatilize the collected analyte and atomize it for AAS detection. The device is

908 H. Matusiewicz and M. Krawczyk
actually the multiple microflame quartz-tube atomizer with inlet arm modified toserve as the trap and to accommodate the oxygen-delivery capillary used to combusthydrogen during the trapping step. Ertas et al. [25] demonstrated in situ trapping oflead hydride species on the inner walls of a flame-heated quartz tube under highlyoxidizing flame conditions followed by the release of trapped species by introducingMIBK.
In this work we described a conceptually similar approach of trapping mercuryvapor (mercury vapor was generated in a laboratory built hydride generators) usingan integrated atom trap system for flame AAS that is applicable to determination oftotal Hg in real and certified reference materials. The hydride generation techniquebrings the FAAS method closer to the detection limits of HG–GF–AAS (in situ trap-ping technique) [26]. Finally, in order to check the accuracy of the proposed system,analysis of certified reference materials were performed. The hyphenated techniquewas satisfactorily applied to different types of samples such as coal fly ash, sewageand water.
EXPERIMENTAL
Spectrometer
A Carl Zeiss Jena (Jena, Germany) Model AAS3 flame atomic absorption spectrometer [27] equippedwith a 10 cm air-acetylene burner head assembly and an IBM–PC compatible computer was used throughoutthe study. The sampling rate for the PMT signal was 10 Hz. Signals were processed with in-house software(Turbo Pascal Version 7) to extract the transient peak heights, area and peak time. NARVA Hg hollowcathode lamp was used as the radiation source. No background correction was required in this mode ofoperation. Operating parameters of the AAS instrument are summarized in Table 1 after appropriate optimi-zation.
Table 1. Instrumental operating conditions for determination of mercury by IAT–FAAS
(Continuation on the next page)

909Determination of total mercury by vapor generation in situ trapping FAAS
Table 1. (Continuation)
a Nebulizer uptake rate, 5 mL min–1. Air flow rate 475 L h–1, acetylene flow rate 50 L h–1 (fuel-lean flame);10 cm slot burner.
Hydride generation systems
Hydride generation was accomplished in the batch (discontinuous) mode and in a continuous modeusing a manually controlled two channel peristaltic pump (Gilson, Model Minipuls 3, France). A mass flowcontroller with a precision pressure gauge (Models ERG 500 and ERG 2000, power supply Model ERG 2M,DHN, Warsaw, Poland) were used to regulate the purge and transfer gas flow rates accurately and repro-ducibly.
Even the hydride generation cells served as gas-liquid separators, in an attempt to completely reducewater vapor, a quartz gas-liquid separator identical to that described earlier [28] was installed betweenthe chemifolds and the nebulizer/spray chamber.
Batch hydride generation. Analyte vapors were generated in a laboratory-made Pyrex cell (batchsystem, the total volume of the glass cell was about 80 mL) [29] into which SnCl2 was introduced witha peristaltic pump. The evolved vapors were stripped from the solution and swept into the air-acetylenenebulizer/burner-IAT system with an air purge gas. The cell assembly and the sequence of operations havebeen described in a previous paper [29].
Continuous-flow hydride generation. Vapor generation was accomplished in the continuous-flowmode using a system similar to that described previously [30].
The operating conditions for batch and continuous-flow vapor generation atomic absorption spectro-metry are summarized in Table 2. A schematic diagram of the batch and continuous-flow VG–IAT–FAASsystem with in situ pre-concentration in the IAT unit is shown in Figure 1.

910 H. Matusiewicz and M. Krawczyk
Figu
re 1
.Sc
hem
atic
dia
gram
of t
he V
G–I
AT–F
AA
S sy
stem

911Determination of total mercury by vapor generation in situ trapping FAAS
Table 2. Optimized operating conditions for determination of Hg by VG–IAT–FAAS
Atom trapping techniques
Three designs of atom trap were investigated. Since the atom trap systems (trapping medium) wasdescribed in detail in previous papers [8, 31] this will not be discussed again here, but briefly summarizedonly.
A double-slotted quartz tube (STAT) was installed on a standard 10 cm air-acetylene burner. The designpermits changeover from analysis with the IAT [8] to that for a conventional flame in a few seconds.
A water-cooled single silica tube (WCAT) atom trap was arranged, as previously described [8] andmounted on a 10 cm burner in such a manner in order to permit the system to be vertically and laterallyadjusted to the flame. The tubes were of an O.D. 3 mm and an I.D. of 1 mm for water cooling.
An integrated atom trap (IAT) was designed and constructed in this laboratory [8] it consisted ofa combination of a WCAT and a STAT [8]. The IAT system was mounted over an air-acetylene burner ona mounting bracket, which permitted calibrated movement both vertically and horizontally.
A modified cooling system was used for cooling the water [9]. The continuously flowing cooling waterkept the surface of the silica tube at the temperature below 100°C. This allowed the analyte atoms to con-dense on the surface of the tube.

912 H. Matusiewicz and M. Krawczyk
The single silica and slotted quartz tubes were coated with palladium to prevent devitrification andto improve the silica surface properties (increasing the surface area) and adsorption efficiency by continuousaspiration via the burner nebulizer of 0.1% palladium solution, for 5 min; then tubes were conditioned forseveral times in the flame. The tubes were re-coated after approximately 100 runs.
Gases and reagents
Compressed air and argon gases of N–50 purity (99.999%) obtained from BOC GAZY (Poznañ,Poland) were employed as the carrier gas (air) for the nebulizer/burner unit and purification agent, respec-tively, without further purification. Compressed medical purity acetylene (Cezal, Poznañ, Poland) was usedas the source of air-acetylene flame.
Standard solutions of Hg were prepared from a 1000 mg L–1 Hg atomic absorption standards (Titri-sol grade, Merck, Darmstadt, Germany). All working standard solutions of Hg were prepared daily to pre-vent any possible species change, by diluting appropriate aliquots of the stock solution in high-purity water.
Sodium tetrahydroborate(III) and potassium tetrahydroborate(III) used as reducing solutions, were pre-pared daily, or more frequently if required, by dissolving proper amounts of NaBH4 and KBH4 (pellets)(Alfa Inorganics, Ward Hill, USA) in high-purity water and stabilizing with 0.1% (m/v) NaOH (Suprapur,Merck, Darmstadt, Germany) solution to decrease its rate of decomposition, and was used without filtration.
Tin(II) chloride solution: a 1% (m/v) solution of SnCl2 in 2 mol L–1 HCl was used. The solutionwas gently sparged for 30 min with argon in order to minimize the Hg0 content. Only clear, colorless reduc-ing solution was used.
The quartz tubes were coated with 1000 mg L–1 Au, Pd and Pt solutions (Titrisol grade, Merck, Darmstadt,Germany). The mixtures of Pd (700 mg L–1) or Pt (700 mg L–1) and Au (300 mg L–1) were prepared beforeuse.
All mineral acids (HNO3, HCl, HF) and hydrogen peroxide 30% (v/v) of the highest quality (Suprapur,Merck, Darmstadt, Germany) were used. High-purity water: deionized water (model DEMIWA 5 ROSA,Watek, Czech Republic), and doubly distilled water (quartz apparatus, Bi18, Heraeus, Hanau, Germany)were used throughout the experiments.
Certified reference materials and samples
Validation of the method described in this work was performed using three certified reference mate-rials. The following materials were chosen: SRM 1633a (Coal Fly Ash) NIST, IAEA/W-4 (Simulated FreshWater) and GBW 07601 (Human Hair) from National Research Center for CRM’s, Beijing, China.The certified reference values are available for mercury for assessment of the method accuracy. All solidreference materials were used as bottled, without further grinding and sieving.
The following real samples were used in this study: the untreated waste water and coal fly ash wassampled from Poznañ Coal-fired Power Plant in Poland.
To ensure homogeneity, it was necessary to grind the real, solid sample in an agate pestle and mortar bymanual grinding of coal fly ash and by a vibrational mixer mill Model S (Testchem, Pszów, Poland) equippedwith 30 mL grinding chamber and rod (6 cm diameter), all made of tungsten carbide.
Microwave digestion system
A laboratory-built prototype of high pressure-temperature focused microwave heated digestion system,equipped with a closed TFM–PTFE vessel (30 mL internal volume) [32] was employed for wet-pressuresample digestion.

913Determination of total mercury by vapor generation in situ trapping FAAS
Method development
The whole analytical procedure consists of various steps: (1) closed wet digestion of the samples,(2) generation of the Hg vapors and its in situ trapping (collection) in an IAT system, (3) flame atomizationof collected vapors and (4) measurement by FAAS.
Microwave-assisted high pressure Teflon bomb digestion. Preparation of all standards and diges-tions of all samples were conducted under typical laboratory conditions. The microwave-assisted pressu-rized digestion technique was used for biological and environmental samples [32].
Approximately 500 mg of powdered inorganic reference material and sample (Coal Fly Ash,) wereplaced in the TFM–PTFE vessel („bomb”) of the microwave digestion system and moistened by 1 mL of30% H2O2; then 3 mL of concentrated HNO3 and 1 mL of concentrated HF were added. The samples wereheated for 15 min at 150 W. After dissolution, the clear digested solution was transferred into 10 mL cali-brated flask and diluted to volume with water. When working with organic material (Human Hair), approxi-mately 250 mg of sample was first moistened by 1 mL of 30% H2O2, then 3 mL of concentrated HNO3 wasused. 10 mL of sewage sample was moistened by 1 mL of 30% H2O2, then 3 mL of concentrated HNO3 wasadded. The samples were heated for 10 min at 100 W. Before further analysis these were appropriatelydiluted depending on the concentration level of the element. In all cases, a corresponding blank was alsoprepared according to the above microwave-assisted digestion procedure.
Simplex optimization procedure. In the optimization that was done in this study, the one-factor-at-a--time method was used to obtain a satisfactory condition for the analysis. It took this method a long time,however, not only to find the optimization condition but also to determine whether or not an optimizationcondition better than the one that was found existed. To avoid these drawbacks the simplex optimizationapproach was undertaken in this study to establish, for mercury, the best conditions for vapors generation,transport and atomization. The parameters optimized are listed in Table 3, along with the ranges over whichoptimization experiments were possible and conducted. In practice, the ranges were judiciously selected foreach parameter in turn, taking into account the practical problems of maintaining a stable absorbance signal.
Table 3. Optimum operating conditions for VG–IAT–FAAS measurement of Hg obtained by simplex andunivariate methods
Simplex optimization experiments were performed using a software package obtained from the Univer-sity of Plymouth. The optimization was carried out using aqueous standard solutions of element (mercury)

914 H. Matusiewicz and M. Krawczyk
determined. Net S/B ratio was taken as the criterion of merit. Some preliminary univariate experiments(searches) were performed prior to the simplex optimization in order to establish the boundaries of thevalues of each parameter. Three measurements for each variables were conducted at the factor of interest.Between each experiment, a blank corrective experiment was run to ensure stable and repeatable results.
The optimum conditions obtained from this procedure were then used to run standard mercury solutionsand quantify the mercury present in the samples.
Vapor generation and procedure for in situ trapping. The procedures for vapor generation of samplesfrom trapping to atomization are outlined below and were not conducted in a clean laboratory environment.Vapor generation was accomplished using two different hydride generators, the continuous unit andthe batch system (cell) [30]. In brief, the mercury vapors were generated continuously or in batch mode andwere introduced into the IAT system by the carrier gas (air) during the vapor-trapping step of the atomizertemperature program only; this step was 120 s in duration in all experiments.
Total mercury. After preparation of solid samples, all the mercury present in the sample will be obtai-ned in solution in the +2 oxidation state (all species will be converted to Hg(II) as the main oxidation product).
Continuous-flow generation measurements of volatile mercury vapors (Hg0) were studied usingthe system shown schematically in Figure 1. 2 mL aliquot sample: 0.2 mL volume of 32% HCl solution and1.8 mL volume of water were placed in a quartz vessel. The PVC/PTFE transfer line from the reaction cellwas placed in the nebulizer/burner system. The Hg sample was being continuously introduced at a rate of1 mL min–1 to merge with a 1.0% (m/v) solution of SnCl2 (flow rate 1.0 mL min–1). The merging solutionfeeds the gas-liquid separator to the IAT system. Mercury vapor that evolved was transferred (carrier airflow rate, 100 mL min–1) to the IAT system, where analyte species (Hg0) were trapped and collected ontothe palladium-coated quartz tube surface. A continuous flow of cooling-water permitted Hg volatile speciesto condense on the surface of the tube during vapors collection. The liquid phase was being continuouslyremoved to waste after neutralization with 0.1% NaOH solution.
Vapor generation was also accomplished using the hydride generation batch system as described previo-usly [30]. In brief, the mercury vapors were generated in batch mode and were introduced into the IATsystem by the carrier gas (air) during the mercury vapor-trapping step of the atomizer temperature programonly; this step was 120 s in duration in all experiments.
Mercury vapors were generated from 10 mL volume of samples (batch mode). The SnCl2 solu-tion (1.0% m/v) was pumped for 60 s and the vapors that evolved were transferred (carrier air flow rate,125 mL min–1) to the IAT system, where they were collected onto the palladium-coated quartz tube.The merging solution feeds the gas-liquid separator to the IAT system. A continuous flow of cooling waterpermitted analyte vapors (atoms) to condense on the surface of the tube during vapors collection; a further60 s air purge of the generator completed the transfer process.
Flame atomization of collected vapors. After collection, vacuum water pump was then turned onto rapidly remove the cooling water from the quartz tube. The tube rapidly heated in the flame, generatinga transient atomic absorption signal as the analyte atoms were released from the surface. Finally, the analyteswere atomized for 7 s at feasible temperature of about 1330°C.
VG–IAT–FAAS analysis. After completion of the vapor generation and collection stage, the analytewas vaporized and atomized for 7 s by heating the quartz tube up to ca 1330°C. The integrated transientabsorbance signals peak area were measured for the Hg line. Both peak height and peak area signals wererecorded. Peak area absorbance signals were used for calculations. A simplex optimization approach wasundertaken to establish, the best conditions for vapors generation, transport, in situ trapping and vaporiza-

915Determination of total mercury by vapor generation in situ trapping FAAS
tion/atomization. Analytical blanks were also carried through the entire procedure outlined above, in orderto correct possible contaminants in the reagents that were used for the sample preparation. The mean blankvalue, if necessary, was substracted from the sample value after all calculations. The system was manuallyoperated during the experiments. Quantification of Hg was based on aqueous standard calibration curves(external calibration). All detection limits were calculated for raw unsmoothed data based on a 3σ criterionof the blank counts.
RESULTS AND DISCUSSION
The study included an investigation of the vapor generation with in situ trapping(pre-concentration, collection), the thermal vaporization-atomization into the FAAS,and its application to practical analysis. The optimization parameters affecting theefficiency of the vapor generation, trapping, atomization and analysis technique willbe discussed separately.
Effect of palladium on the trapping efficiency of mercury vapors
When mercury was determined without any stabilizing agents the sensitivity wasfound to be very low. Therefore, application of a modifier should be realized bycoating modifier on to the quartz tube. Initial experiments were carried out to evalu-ate the different coating materials. The gold, palladium, platinum and their alloyshave been used to improve the sensitivity of a mercury. These coating elements wereexamined for their suitability to determine Hg. It was thought that the gaseous mer-cury may become trapped in the matrix of the coating material allowing Hg to bedetermined. This did occur and improvement in sensitivity was observed (Fig. 2).A possible explanation is that enough amalgam is formed and therefore sufficient Hgis trapped for an increase in sensitivity to be observed. With all investigated metalsand alloys, the lowest sensitivity was observed in the presence of gold; the best sensi-tivity was achieved in the presence of Pd, when introduced into the quartz tube alone.Therefore, the trapping efficiency of vapors onto the Pd-coated quartz tube was foundto be optimum for Hg vapors.
Based on the above experiments it is supposed that palladium is not removedfrom the quartz surface and remains on the quartz tube surface acting as an efficientpermanent chemical modifier. The amount of Pd remaining on the surface decreasesrapidly with increasing number of firings up to about 10 firings. After 10 firingspalladium remained in the quartz tube as a permanent modifier (adsorber agent).The lifetime of the Pd coating was ca 100 quartz tube firings without significantchanges in reproducibility of Pd absorbances.
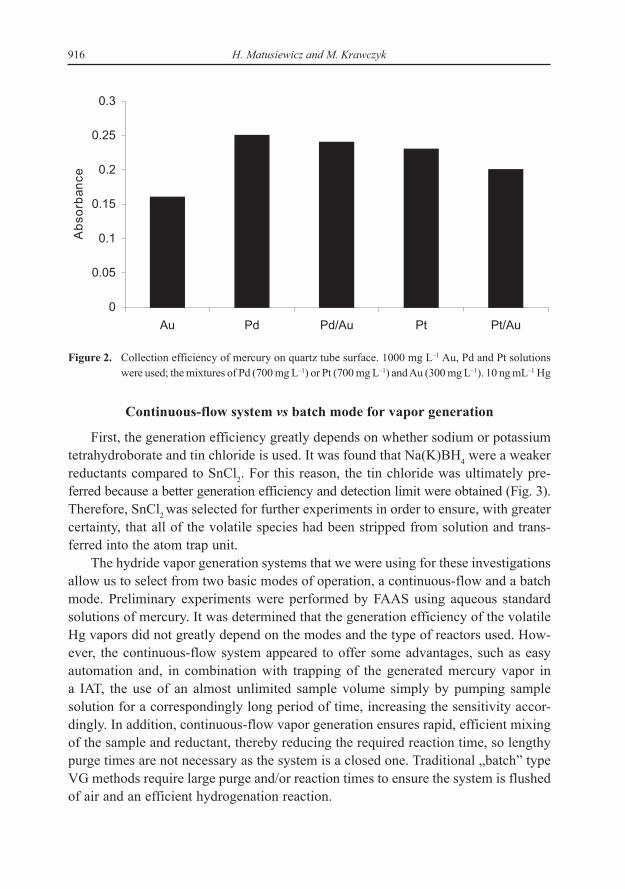
916 H. Matusiewicz and M. Krawczyk
Figure 2. Collection efficiency of mercury on quartz tube surface. 1000 mg L–1 Au, Pd and Pt solutionswere used; the mixtures of Pd (700 mg L–1) or Pt (700 mg L–1) and Au (300 mg L–1). 10 ng mL–1 Hg
Continuous-flow system vs batch mode for vapor generation
First, the generation efficiency greatly depends on whether sodium or potassiumtetrahydroborate and tin chloride is used. It was found that Na(K)BH4 were a weakerreductants compared to SnCl2. For this reason, the tin chloride was ultimately pre-ferred because a better generation efficiency and detection limit were obtained (Fig. 3).Therefore, SnCl2 was selected for further experiments in order to ensure, with greatercertainty, that all of the volatile species had been stripped from solution and trans-ferred into the atom trap unit.
The hydride vapor generation systems that we were using for these investigationsallow us to select from two basic modes of operation, a continuous-flow and a batchmode. Preliminary experiments were performed by FAAS using aqueous standardsolutions of mercury. It was determined that the generation efficiency of the volatileHg vapors did not greatly depend on the modes and the type of reactors used. How-ever, the continuous-flow system appeared to offer some advantages, such as easyautomation and, in combination with trapping of the generated mercury vapor ina IAT, the use of an almost unlimited sample volume simply by pumping samplesolution for a correspondingly long period of time, increasing the sensitivity accor-dingly. In addition, continuous-flow vapor generation ensures rapid, efficient mixingof the sample and reductant, thereby reducing the required reaction time, so lengthypurge times are not necessary as the system is a closed one. Traditional „batch” typeVG methods require large purge and/or reaction times to ensure the system is flushedof air and an efficient hydrogenation reaction.

917Determination of total mercury by vapor generation in situ trapping FAAS
Figure 3. Influence of SnCl2 ( ), NaBH4 ( ) and KBH4 ( ) concentration on the peak areasignals of Hgtot (5 ng mL–1) in batch system. The experimental conditions employed are detailedin the Experimental Section
Process blank
Blanks using the continuous mode system were determined using the same treat-ment procedure as for samples (microwave-assisted sample digestion procedure).Although these experiments were conducted in an ordinary laboratory, the detectablesource of the blank was determined experimentally to be the reductant solution and,in particular, the hydrochloric acid used to stabilize the tin chloride. Even thoughthe chemicals used were of the best quality available, trace amounts of the analytes inthe reagent were pre-concentrated in the silica tube during the in situ trapping, resul-ting in blank signals. Using the continuous mode system and SnCl2 as reductant,absolute blank of 0.6 ng for Hgtot, was achieved.
Simplex optimization of operational variables
The optimized vapor generation conditions are given in Table 1 and 2.The stability of the flame is obviously controlled by the gas flow rate, it is there-
fore essential to keep the flame stable in the section of the atomizer. The effects offlame conditions on the trapping and release of the mercury were studied by varyingthe fuel flow rate. The influence of the flame condition on the signal intensity wasinvestigated by fixing the air flow rate (475 L h–1) and altering the acetylene flowrate. The best sensitivity was obtained by using a 50 L h–1 of flow rate (lean flame) foracetylene. The absorbances of Hg was not very different for collecting or releasingin a lean, full-rich and stoichiometric flame.

918 H. Matusiewicz and M. Krawczyk
The water-cooled silica tube trap position was not optimized in our experiments,but was selected based upon previous experience [8]. The optimum position of thetrap tube (single silica tube and STAT) corresponded to the distance (gap) of 5 mmabove the burner, and the position of the silica tube corresponded to obscuration ofabout one-third of the light beam by the upper part of the tube. No significant diffe-rences were found in the absorbances when a coolant water flow rate of 1–4 L min–1
were used during the collection cycle of Hg vapors.The trapping time is one of the most important factors concerning the sensitivity
of VG–IAT–FAAS technique. In time-based pre-concentration system the sampleloading time value indicates the pre-concentration time of the method and reflects theenrichment factor. It was demonstrated (although not shown) that, although the rela-tionship is not in general linear, a longer trapping time increased the analytical signal.Although sensitivity is not linear with respect to trapping time the calibration graphsare linear for any single trapping time. A reasonable trapping time per sample ina routine laboratory would be about 2 min, in order to ensure, with greater certainty,that all of the Hg vapors had been stripped from the solution and transferred into theatomizer and as a compromise between medium sample consumption, high sensiti-vity and sufficient sampling frequency. This time was chosen to investigate the analy-tical performance of the VG–IAT–FAAS system with respect to linearity, sensitivity,precision and detection limit.
To optimize the sample and reductant flow rate, first the optimum flow for Hgwas estimated in the range of 0.5–3 mL min–1 (Fig. 4a). In this study a 1 mL min–1
sample and reductant flow rate was chosen.The continuous hydride generation system was optimized by varying the concen-
tration of SnCl2, HCl and the air carrier flow that regulate volatilization and transport.However, this information was available from a review [4] pertaining to trace ele-ment detection by atom trapping and in situ pre-concentration for FAAS and on ourown observations. All of the factors show more or less significant effect.
The efficiency of the generation of mercury vapors in hydrochloric acid andnitric acids was investigated. When using HNO3 the Hg signals were found to beabout 20 to 30% lower than those obtained with HCl which is the most appropriateacid to use. The effect of HCl concentration on the peak area absorbance is illustratedin Figure 4b, and 1 mol L–1 portion of HCl was chosen. The concentration of SnCl2has been recognized as one of the most critical variables in VG. Concentrations in therange 0.5–3.0% of SnCl2 were assayed using 3 mol L–1 HCl (Fig. 4c). The higher itsconcentration the higher the signal, so the faster should be the reaction and moreactive intermediates should be formed, but relative standard deviations also increased.On the other hand, higher SnCl2 concentrations than 1% (m/v) would result in a vio-lent reaction (more hydrogen generated) in the gas-liquid separator and eventuallylead to an unstable signal. High SnCl2 concentrations had to be avoided and an opti-
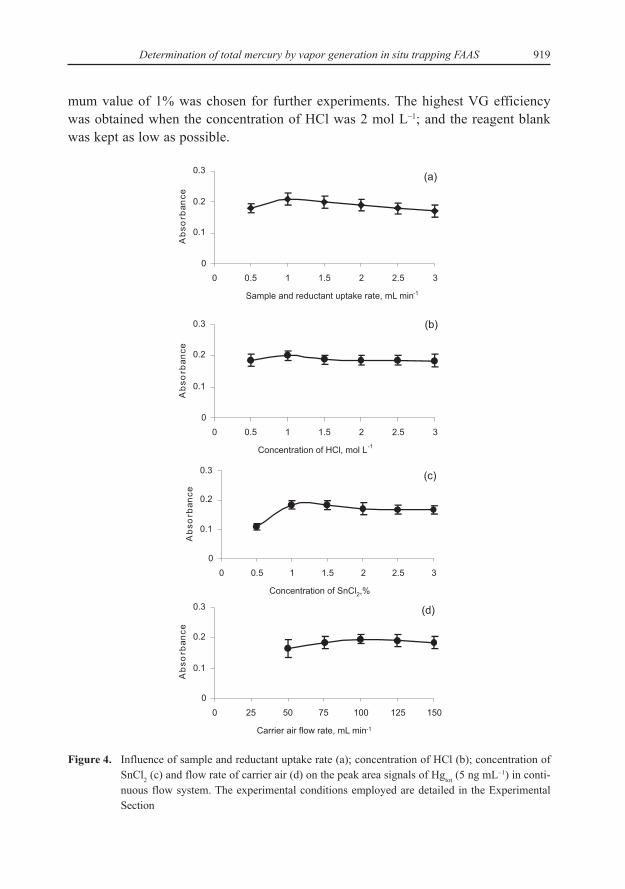
919Determination of total mercury by vapor generation in situ trapping FAAS
mum value of 1% was chosen for further experiments. The highest VG efficiencywas obtained when the concentration of HCl was 2 mol L–1; and the reagent blankwas kept as low as possible.
Figure 4. Influence of sample and reductant uptake rate (a); concentration of HCl (b); concentration ofSnCl2 (c) and flow rate of carrier air (d) on the peak area signals of Hgtot (5 ng mL–1) in conti-nuous flow system. The experimental conditions employed are detailed in the ExperimentalSection

920 H. Matusiewicz and M. Krawczyk
The air carrier gas flow through the apparatus is one of the basic parametersinfluencing the transport of the Hg0 into the trap-flame system, but also the mixingeffect of vapor-forming reaction solutions, and thus it can affect the determination ofHg markedly. The vapors were stripped and trapped in the silica tube (atomizer) at airflow rates in the range of 50–150 mL min–1 with a 120 s collection time. It was evi-dent that the use of low carrier gas flow can successfully reduce high analyte lossescaused by sorption on the inner surfaces of the apparatus (transport tubing), and leadsto a slight improvement in the analytical signals. This may be connected with themore efficient separation of the vapors from the reaction solution. On the other hand,higher carrier gas flow rates (> 100 mL min–1) can result in a slightly decrease intrapping efficiency. The decrease observed is probably due to the diluting effect ofthe air flow. An air flow of 100 mL min–1 was therefore chosen as optimum and usedthroughout the experiments (Fig. 4d). In the atomization step, signal with regularshape was observed for trapping temperature and was not significantly influenced bysample introduction time in the tested range between 1 and 3 min. It should be stressedthat multiple absorption peaks were not observed.
The length of the transfer tubing was not optimized, but was selected based uponprevious experience [9–13]. Therefore, for practical reasons, a transport tubing lengthof 10 cm was selected for this study.
Validation of the method by analysis of certified reference materials
Validation of the technique proposed included analysis of three standard refe-rence materials (SRMs): coal fly ash, hair and water. These reference materials werechosen as they were the closest available to biological samples and are certified forthe analyte of interest to be determined in the environmental and biological samples.The matrix effects on the VG–IAT–FAAS analytical signals were evaluated by compa-ring the conventional calibration with the standard additions slopes. No significantdifferences were found between the slopes obtained by both calibration procedureswhen using VG–IAT–FAAS. Therefore, to determine total mercury in all samples bythis analytical technique, the conventional calibration mode was used. Results obtai-ned for the analysis of SRMs by VG–IAT–FAAS method using aqueous standardcalibration technique are summarized in Table 4. The short-term precision is expressedas the RSD of six replicate measurements of each sample. The results obtained byexternal calibration technique do agree with certified values for any reference mate-rial indicating that calibration against aqueous solution could produce accurateresults. No significant differences were found between experimental and certificatevalues. The concentrations reported for this element in reference materials are signi-ficantly higher than the detection limits that are attained for the measurement.
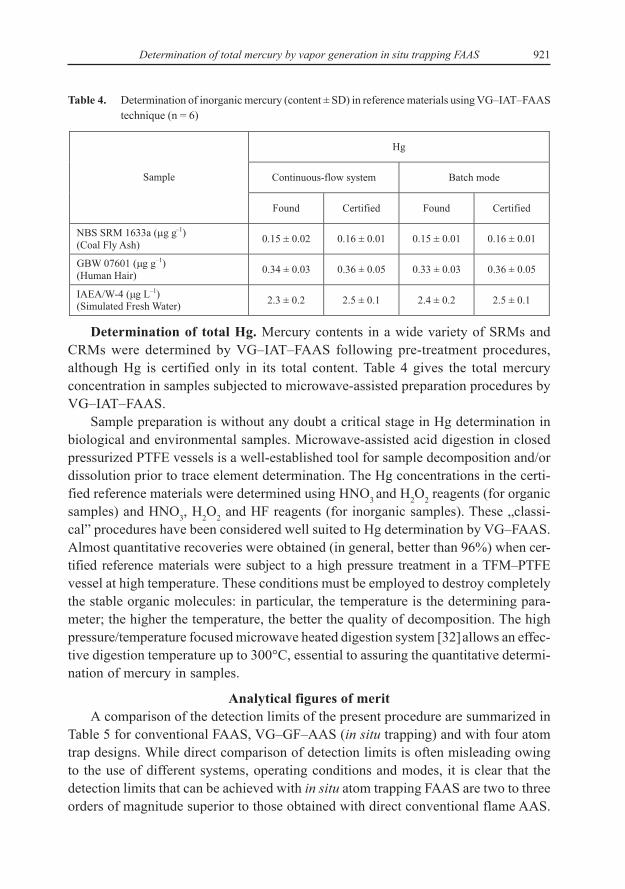
921Determination of total mercury by vapor generation in situ trapping FAAS
Table 4. Determination of inorganic mercury (content ± SD) in reference materials using VG–IAT–FAAStechnique (n = 6)
Determination of total Hg. Mercury contents in a wide variety of SRMs andCRMs were determined by VG–IAT–FAAS following pre-treatment procedures,although Hg is certified only in its total content. Table 4 gives the total mercuryconcentration in samples subjected to microwave-assisted preparation procedures byVG–IAT–FAAS.
Sample preparation is without any doubt a critical stage in Hg determination inbiological and environmental samples. Microwave-assisted acid digestion in closedpressurized PTFE vessels is a well-established tool for sample decomposition and/ordissolution prior to trace element determination. The Hg concentrations in the certi-fied reference materials were determined using HNO3 and H2O2 reagents (for organicsamples) and HNO3, H2O2 and HF reagents (for inorganic samples). These „classi-cal” procedures have been considered well suited to Hg determination by VG–FAAS.Almost quantitative recoveries were obtained (in general, better than 96%) when cer-tified reference materials were subject to a high pressure treatment in a TFM–PTFEvessel at high temperature. These conditions must be employed to destroy completelythe stable organic molecules: in particular, the temperature is the determining para-meter; the higher the temperature, the better the quality of decomposition. The highpressure/temperature focused microwave heated digestion system [32] allows an effec-tive digestion temperature up to 300°C, essential to assuring the quantitative determi-nation of mercury in samples.
Analytical figures of meritA comparison of the detection limits of the present procedure are summarized in
Table 5 for conventional FAAS, VG–GF–AAS (in situ trapping) and with four atomtrap designs. While direct comparison of detection limits is often misleading owingto the use of different systems, operating conditions and modes, it is clear that thedetection limits that can be achieved with in situ atom trapping FAAS are two to threeorders of magnitude superior to those obtained with direct conventional flame AAS.

922 H. Matusiewicz and M. Krawczyk
Tabl
e 5.
Com
paris
on o
f det
ectio
n lim
its (L
OD
)a for
mer
cury
usi
ng v
ario
us a
tom
trap
s (ng
mL–1
)
a2
min
col
lect
ion
time.
bD
etec
tion
limit
defin
ed a
s „3
blan
k” c
riter
ion
(n =
6).
cEn
hanc
emen
t (im
prov
emen
t) fa
ctor
.d
Enha
ncem
ent f
acto
r sho
ws t
he a
bilit
y of
the
anal
ytic
al sy
stem
rega
rdin
g pe
riod
of ti
me
requ
ired
for a
naly
sis:
X/m
inut
es o
f sam
plin
g (m
in–1
).e
Enha
ncem
ent f
acto
r sho
ws i
nflu
ence
of s
ampl
e vo
lum
e: X
/mill
ilite
rs o
f sam
ple
(mL–1
).f
Ref
. [26
], C
V–A
AS,
in si
tu c
once
ntra
tion,
for s
ampl
e vo
lum
e of
50
mL,
LO
D (n
g L–1
)g
Ref
. [7]
, FA
AS,
in si
tu c
once
ntra
tion,
2 m
in c
olle
ctio
n tim
e.

923Determination of total mercury by vapor generation in situ trapping FAAS
The analytical performance characteristics were evaluated for mercury. The limitof detection (LOD) were obtained by use of optimized operating conditions. Since nodetection limits obtained by identical techniques are available, the results are compa-red by the quartz atom trap and flame atomic absorption spectrometry (QAT–AAS)(in situ trapping) and by the CV–GF–AAS (in situ trapping) technique (Tab. 5).The detection limits of the developed procedure are evidently better, as comparedwith other atom traps. Detection limits obtained with a 120 s collection time werein the ppt (pg mL–1) range and are the best ones; VG–IAT–FAAS procedure affordedsignificant improvements in the detection limit with respect to those reported forQAT–AAS (59.8 ng mL–1; in situ trapping) methodology for the determination of thisvapor forming trace element [7]. The achieved limit of detection (LOD): 0.4 ng mL–1
for Hg (sensitivity enhancement compared to FAAS is 750 folds), is at least severalorders of magnitude worse to the LOD achieved for Hg in situ trapping in graphitefurnace [26]. Six replicate measurements of the total procedure (reagent) blank solu-tion were carried out and the relative standard deviation (RSD) of the backgroundvalues for the raw, unsmoothed data were calculated. Precision was in the range of9.3% (as peak area); this reflects the cumulative imprecision of all of the samplehandling, vapor generation, trapping, atomization and detection steps. The peak heightprecision was always slightly, but significantly, worse.
Total mercury determination in selected real samples
Finally, in order to evaluate the usefulness of the proposed method in determin-ing total mercury contents in some real samples were analyzed using the optimizedexperimental conditions (Tab. 6). In all cases, the calibration was achieved using theaqueous standard calibration curves. The proposed method was validated by spikingthe samples with known amount of Hg(II). The recoveries from spiked solutionswere varied in the range 92–96%. The precision of replicate determinations is typi-cally better than 10% RSD.
Table 6. Inorganic mercury concentration in real samples (n = 6)

924 H. Matusiewicz and M. Krawczyk
CONCLUSION
This work is a continuation of previous efforts to employ an integrated silicatubes as a trap for species volatilized in a hydride generation system act, in a sense,like an enrichment device [8–13, 30, 31]. The results presented in this work confirmthe idea that the present hyphenated technique using a continuous mode vapor gene-ration gas phase in situ trapping on an integrated silica tubes trap, followed by atomi-zation in acetylene-air flame with simultaneous direct thermal heating of the ato-mizer, can be used for the determination of trace amounts of total mercury (Hgtot)in certified reference materials and real samples. Following the trapping stage,the performance of the device and related problems are quite similar to the case ofhydride generation-graphite furnace atomization (in situ trapping) HG–GF–AAS.The detection limit of this VG–IAT–FAAS system for Hg is considerably improvedcompared with those reported for measurements of Hg by any flame AAS approachand was significantly lower than that for QAT–AAS (59.8 ng mL–1; in situ trapping).However, there are still many unknowns regarding mechanisms of trapping (collec-tion, pre-concentration) and re-volatilization (atomization). This very simple and cheaptechnique constitutes an attractive alternative to VG-in situ trapping-GF–AAS sys-tem at significantly lower cost. Although, the achieved very low concentration detec-tion limit for Hg, 0.4 ng mL–1, is worse than those for the in situ trapping of Hg vaporsin a commercial graphite furnace: 0.628 ng L–1 [26]. The determinations were equiva-lent within the precision of the methods. It is expected that detection limits could befurther improved with the use of high purity reagents. However, in most of the casesblank values and/or saturation of trap surface by analyte and interferent species puta practical limit to sample size and sampling period; therefore, the enhancement cannever be infinitely improved.
The proposed experimental speciation approach, offers interesting perspectiveand good prospects for the determination of other hydride forming elements, in therange of pg mL–1, e.g., Ge and Sn. This is subject of on-going research. A simple AAflame spectrometer, the low cost, easy operation, and high sensitivity of the presentsystem make it very attractive for laboratories not equipped with any graphite fur-nace apparatus. Although time required for each measurement is longer than thatrequired for the classic hydride generation technique.
Acknowledgements
Financial support by the State Committee for Scientific Research (KBN), Poland, Grant No. 4 T09A 1030 is gratefully acknowledged.

925Determination of total mercury by vapor generation in situ trapping FAAS
REFERENCES
1. Welz B. and Sperling M., Atomic Absorption Spectrometry, Wiley-VCH, Weinheim 1999.2. Clevenger W.L., Smith B.W. and Winefordner J.D., Crit. Rev. Anal. Chem., 27, 1 (1997).3. Wu L., Zheng Ch., Ma Q., Hu Ch. and Hou X., Appl. Spectrosc. Rev., 42, 79 (2007).4. Matusiewicz H., Spectrochim. Acta, Part B, 52, 1711 (1997).5. Matusiewicz H. and Sturgeon R.E., Spectrochim. Acta, Part B, 51, 377 (1996).6. Dìdina J., Spectrochim. Acta, Part B, 62, 846 (2007).7. Ellis L.A. and Roberts D.J., J. Anal. At. Spectrom., 11, 1063 (1996).8. Matusiewicz H. and Kopras M., J. Anal. At. Spectrom., 12, 1287 (1997).9. Matusiewicz H. and Krawczyk M., Anal. Sci., 22, 249 (2006).
10. Matusiewicz H. and Krawczyk M., Microchem. J., 83, 17 (2006).11. Matusiewicz H. and Krawczyk M., J. Braz. Chem. Soc., 18, 304 (2007).12. Matusiewicz H. and Krawczyk M., Spectrochim. Acta, Part B, 62, 309 (2007).13. Matusiewicz H. and Krawczyk M., J. Anal. At. Spectrom., 23, 43 (2008)14. Cankur O., Ertaº N. and Ataman O.Y., J. Anal. At. Spectrom., 17, 603 (2002).15. Korkmaz D., Kumser S., Ertaº N., Mahmut M. and Ataman O.Y., J. Anal. At. Spectrom., 17, 1610
(2002).16. Ertaº N., Korkmaz D.K., Kumser S. and Ataman O.Y., J. Anal. At. Spectrom., 17, 1415 (2002).17. Guo X-m. and Guo X-w., J. Anal. At. Spectrom., 16, 1414 (2001).18. Korkmaz D.K., Ertaº N. and Ataman O.Y., Spectrochim. Acta, Part B, 57, 571 (2002).19. Korkmaz D., Dìdina J. and Ataman O.Y, J. Anal. At. Spectrom., 18, 255 (2004).20. Kratzer J. and Dìdina J., Spectrochim. Acta, Part B, 60, 859 (2005).21. Kratzer J. and Dìdina J., J. Anal. At. Spectrom., 21, 208 (2006).22. Krejèi P., Doèekal B. and Hrušovská Z., Spectrochim. Acta, Part B, 61, 444 (2006).23. Menemenlioglu I., Korkmaz D. and Ataman O.Y., Spectrochim. Acta, Part B, 62, 40 (2007).24. Kratzer J. and Dìdina J., Anal. Bioanal. Chem., 388, 793 (2007).25. Ertas N., Arslan Z. and Tyson J.F., J. Anal. At. Spectrom., 23, 223 (2008).26. Yan X-P. and Ni Z-M., Anal. Chim. Acta, 272, 105 (1993).27. Matusiewicz H., J. Anal. At. Spectrom., 4, 265 (1989).28. Veber M., Èujes K. and Gomišèek S., J. Anal. At. Spectrom., 9, 285 (1994).29. Sturgeon R.E., Willie S.N. and Berman S.S., Anal. Chem., 57, 2311 (1985).30. Matusiewicz H., Kopras M. and Sturgeon R.E., Analyst, 122, 331 (1997).31. Matusiewicz H., Sturgeon R., Luong V. and Moffatt K., Fresenius’ J. Anal. Chem., 340, 35 (1991)
(and references cited therein).32. Matusiewicz H., Anal. Chem., 66, 751 (1994).
Received May 2008Revised November 2008
Accepted November 2008