deubiquitinase function of arterivirus papain-like protease 2
TRANSCRIPT

Deubiquitinase function of arterivirus papain-likeprotease 2 suppresses the innate immuneresponse in infected host cellsPuck B. van Kasterena, Ben A. Bailey-Elkinb,1, Terrence W. Jamesb,1, Dennis K. Ninabera, Corrine Beugelinga,Mazdak Khajehpourc, Eric J. Snijdera, Brian L. Markb,2,3, and Marjolein Kikkerta,2,3
aMolecular Virology Laboratory, Department of Medical Microbiology, Center of Infectious Diseases, Leiden University Medical Center, 2333 ZA, Leiden,The Netherlands; and Departments of bMicrobiology and cChemistry, University of Manitoba, Winnipeg, MB, Canada R3T 2N2
Edited by Peter Palese, Mount Sinai School of Medicine, New York, NY, and approved January 17, 2013 (received for review October 23, 2012)
Protein ubiquitination regulates important innate immune responses.The discovery of viruses encoding deubiquitinating enzymes (DUBs)suggests they remove ubiquitin to evade ubiquitin-dependent anti-viral responses; however, this has never been conclusively demon-strated in virus-infected cells. Arteriviruses are economically importantpositive-stranded RNA viruses that encode an ovarian tumor (OTU)domain DUB known as papain-like protease 2 (PLP2). This enzymeis essential for arterivirus replication by cleaving a site withinthe viral replicase polyproteins and also removes ubiquitin fromcellular proteins. To dissect this dual specificity,which relies on a singlecatalytic site, we determined the crystal structure of equine arteritisvirus PLP2 in complex with ubiquitin (1.45 Å). PLP2 binds ubiquitinusing a zinc finger that is uniquely integrated into an exceptionallycompact OTU-domain fold that represents a new subclass of zinc-dependent OTUDUBs. Notably, the ubiquitin-binding surface is dis-tant from the catalytic site, which allowed us to mutate this surfaceto significantly reduce DUB activity without affecting polyproteincleavage. Viruses harboring such mutations exhibited WT replica-tion kinetics, confirming that PLP2-mediated polyprotein cleavagewas intact, but the loss of DUB activity strikingly enhanced innateimmune signaling. Compared with WT virus infection, IFN-β mRNAlevels in equine cells infected with PLP2 mutants were increasedby nearly an order of magnitude. Our findings not only establishPLP2 DUB activity as a critical factor in arteriviral innate immuneevasion, but the selective inactivation of DUB activity also opensunique possibilities for developing improved live attenuated vac-cines against arteriviruses and other viruses encoding similar dual-specificity proteases.
interferon-stimulated gene 15 | ISG15 | +RNA
The synthesis and posttranslational cleavage of polyproteins isa common genome expression strategy used by positive-
stranded (+)RNA viruses of eukaryotes. It is used to cope withthe consequences of cytoplasmic replication and the limitationsof the eukaryotic translation machinery, which essentially pre-clude the use of (nuclear) RNA splicing and polycistronic mRNAs,respectively (1). The critical cleavage of these viral polyproteinsinto their functional subunits is mediated by internal virus-encoded proteases (2–5), many of which have been found to alsotarget cellular substrates to promote virus replication or subverthost antiviral responses. Well-known examples of such dual-spec-ificity proteases are the poliovirus 2A and hepatitis C virus NS3/4Aenzymes that, in addition to the viral polyprotein, target host cellproteins involved in translation and innate immune signaling,respectively (6–10).Arteriviruses are +RNA viruses that, together with the corona-
and roniviruses, belong to the orderNidovirales and include equinearteritis virus (EAV) and porcine reproductive and respiratorysyndrome virus (PRRSV). EAV is the family prototype and cancause abortion in pregnant mares, pneumonia in neonatal foals,and influenza-like illness in adult horses (11). PRRSV ranks amongthe most important swine pathogens, and infections are charac-
terized by reproductive failure in sows and severe respiratory dis-ease in young pigs (12). As in all nidoviruses, the synthesis andcleavage of the replicase polyproteins (pp1a and pp1ab) are criticalfirst steps in arterivirus infection. They are encoded by the 5′-proximal three-quarters of the 13- to 16-kb arterivirus genomeand are the precursors of the nonstructural proteins (nsps) re-quired for genome replication and transcription. In the case ofEAV, at least 13 nsps are produced when the replicase poly-proteins are cleaved by three virus-encoded proteases (13, 14),one of which is a papain-like protease (PLP2) located in theN-terminal region of nsp2 (15, 16). This enzyme cleaves thensp2jnsp3 junction in pp1a and pp1ab, an event that is essentialfor virus replication because an EAV PLP2 active site mutant waspreviously found to be nonviable (17). In addition to this criticalrole in viral polyprotein maturation, arterivirus PLP2 was pro-posed to contribute to the evasion of host innate immune re-sponses by removing ubiquitin (Ub) from cellular targets (18). Ubis an 8-kDa protein moiety that can be covalently attached tolysine residues of target proteins in a number of structurally dif-ferent configurations, either by monoubiquitination or through
Significance
Many viruses encode proteases that cleave both viral and hostsubstrates. Arteriviruses encode such a dual-specificity pro-tease (PLP2) that removes ubiquitin from cellular proteins in-volved in host immunity. Based on a 3D structure of PLP2, weengineered the protease to have diminished deubiquitinatingactivity without affecting its activity toward its viral substrate.Viruses expressing such engineered proteases displayed a sig-nificantly weakened ability to evade host immune responses.This result demonstrates a crucial role for PLP2 in arterivirusimmune evasion and opens new possibilities for developingimproved attenuated virus vaccines against economicallyimportant arteriviruses and other viruses encoding similardual-specificity proteases.
Author contributions: E.J.S., B.L.M., and M. Kikkert designed research; P.B.v.K., B.A.B.-E.,T.W.J., D.K.N., and C.B. performed research; P.B.v.K., B.A.B.-E., T.W.J., M. Khajehpour,E.J.S., B.L.M., and M. Kikkert analyzed data; and P.B.v.K., B.A.B.-E., M. Khajehpour,E.J.S., B.L.M., and M. Kikkert wrote the paper.
Conflict of interest statement: The authors have filed a provisional patent applicationthat relates to some aspects of this work.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank,www.pdb.org (PDB ID code 4IUM).1B.A.B.-E. and T.W.J. contributed equally to this work.2B.L.M. and M.K. contributed equally to this work.3To whom correspondence may be addressed. E-mail: [email protected] [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218464110/-/DCSupplemental.
E838–E847 | PNAS | Published online February 11, 2013 www.pnas.org/cgi/doi/10.1073/pnas.1218464110

the formation of polyubiquitin chains (19, 20). The effects of ubiq-uitination can range from targeting substrates for proteasomaldegradation to initiating signaling cascades and—importantly—they can be reversed by deubiquitinating enzymes (DUBs), whichthus allow for negative regulation of Ub-activated processes (21–24). The latter include the innate immune signaling cascadestriggered by invading RNA viruses (25, 26), which ultimately leadto the transcription of genes encoding IFN-β and other proin-flammatory cytokines (27, 28).Arterivirus PLP2 and a protease domain found in the unrelated
nairovirus Crimean-Congo hemorrhagic fever virus (CCHFV)were first identified as potential members of the ovarian tumordomain-containing (OTU) superfamily of DUBs on the basis ofcomparative sequence analysis (29). Several laboratories, in-cluding our own, subsequently confirmed that arterivirus PLP2sindeed have DUB activity that may be used to remove Ub frominnate immune signaling factors to suppress the induction of anantiviral state (18, 30, 31). The potential benefits of this strategyare highlighted by the fact that proteases from virus groups asdiverse as arteri-, corona-, nairo-, picorna-, hepadna-, and her-pesviruses have all been implicated in DUB-based innate immuneevasion (18, 32–37). Thus far, however, direct evidence linkingDUB activity to the suppression of innate immune responses invirus-infected cells has not been reported for any of theseproteases.Because the DUB activity of arterivirus PLP2 depends on the
same active site mediating the critical nsp2jnsp3 cleavage, it hasnot been possible to independently study the role of PLP2 inpolyprotein processing and immune evasion in the context ofvirus infection. Here we present the crystal structure of EAVPLP2 in complex with Ub at 1.45-Å resolution. The complexreveals a distinctly compact conformation compared with otherOTU superfamily members and the incorporation of a zinc fingerwithin the OTU fold. Given these features, arterivirus PLP2represents a unique subclass of zinc-dependent OTU DUBs.Importantly, the PLP2 active site is distant from its Ub-bindingsurface, allowing for the introduction of mutations in this regionthat dramatically reduced DUB activity yet did not affectnsp2jnsp3 cleavage. Compared with WT EAV, viruses carryingthese mutations elicited a significantly enhanced innate immuneresponse in primary equine cells while displaying WT replicationkinetics. Taken together, our results demonstrate that PLP2DUB activity indeed mediates innate immune suppression dur-ing arterivirus infection. The ability to selectively inactivate thePLP2 DUB function may thus contribute to the engineering ofimproved live attenuated vaccines against arteriviruses and othervirus families encoding proteases with similar dual specificities.
ResultsEAV PLP2 Adopts a Compact OTU-Domain Fold with a Unique IntegralZinc Finger. Previously, EAV PLP2 was identified and character-ized by a combination of bioinformatics analysis and site-directedmutagenesis, and two residues in particular were implicated incatalysis: Cys270 and His332 (16). Throughout this paper, aminoacid numbers refer to the sequence of full-length EAV pp1a. Thecrystal structure of EAV PLP2 (residues 261–392; 13.6 kDa) wasdetermined as a covalent complex with the mechanism-based in-hibitor Ub(1–75)-3-bromopropylamine (Ub–3Br) (38, 39). Becausethe conservation of multiple cysteine residues and their dem-onstrated importance for protease function suggested that PLP2could bind zinc (16), the crystal structure of the complex was de-termined by a multiwavelength anomalous dispersion (MAD)phasing experiment using X-ray diffraction data collected over thezinc absorption edge (Table S1). The resulting electron density maprevealed residues 261–387 of PLP2 bound to a complete Ub mol-ecule and allowed a model of the complex to be built and refined(Rwork= 0.16, Rfree= 0.18) to 1.45-Å resolution (Fig. 1 A and B).
The protease adopts a compact, two-domain fold with a shal-low Ub-binding surface that directs the C terminus of the boundUb molecule (the distal Ub in a Ub dimer) toward a solventexposed active site that includes Cys270 and His332 (Fig. 1 A andB). Domain I of PLP2 (residues 267–307 and 365–387) consistsof a three-helix bundle (α1, α2, α4) packed against a two-stranded antiparallel sheet (β2↑ β6↓). Domain II centers ona four-stranded β-sheet (β1↑ β5↓ β4↑ β3↑) and an α-helix (α3)that together pack against helices α1 and α2 of domain I. Do-main II comprises the majority of the Ub-binding surface, whichis stabilized by four cysteine residues (Cys319, 349, 354, and 356)that coordinate a zinc ion with tetrahedral geometry (Fig. 1C).Their arrangement forms a C4 zinc finger that resembles a C-terminal type zinc necklace motif (40). A large insertion betweenpositions C1 (Cys319) and C2 (Cys349), which includes His332,appears to extend the stabilizing effect of the zinc fingerthroughout much of domain II. A fifth cysteine (Cys344) is lo-cated near the zinc ion but does not coordinate with it, consistentwith other zinc necklace motifs that have been described (40)and with previous findings showing that a Cys344 to alaninemutation had no effect on catalytic activity of PLP2 (16). Threeof the cysteines (Cys319, 349, and 354) are fully conserved inarteriviruses, and mutational analysis of these residues andCys356 demonstrated zinc binding to be essential for catalyticactivity (16). Given its distance from the active site, however(∼25 Å; Fig. 1B), the zinc atom appears to play a structural roleas opposed to participating in catalysis. Expression of PLP2 inEscherichia coli grown in the absence of zinc (M9 medium)yielded insoluble protein, supporting the idea that the role of thezinc finger is structural and is likely required for correct foldingof the protease.Consistent with OTU DUBs and papain-like cysteine pro-
teases in general, the PLP2 active site contains a catalytic cys-teine nucleophile (Cys270) and histidine (His332) residue, alongwith an asparagine (Asn263) that hydrogen bonds with the im-idazole ring of His332 (Fig. 1D). As expected, the side chain ofCys270 is covalently coupled to the C terminus of Ub via the 3-propylamine (3CN) modification, mimicking the acyl-enzymeintermediate step of the catalytic reaction and confirming theidentity of Cys270 as the catalytic nucleophile.Fold analysis of the PLP2 structure using the DALI server (41)
revealed that its closest structural homologs indeed belong to theOTU superfamily of DUBs (29, 42). The most significant matcheswere to yeast Otu1 (38) (Z-score: 5.1) and the viral OTU pro-tease from CCHFV (43–45) (Z-score: 4.6; Fig. 1E), followed byOtubain1 from human (46) and Caenorhabditis elegans (47) (Z-scores: 4.1 and 4.0, respectively) and the OTU domain of humanDUBA (48) (Z-score: 3.9). The sequence identity of the PLP2regions that aligned with these OTU proteases was low (rangingfrom 9% for yeast Otu1 to 21% for human Otubain1); however,they accounted for ∼60% of the total PLP2 structure and su-perposed well with the equivalent regions in the above proteins,with an average rmsd of ∼2.8 Å. The greatest structural similaritybetween PLP2 and members of the OTU superfamily occurs atthe active site and adjoining channel that binds the C-terminalRLRGG-tail of Ub.
PLP2 Zinc Finger Motif Plays a Central Role in Ub Binding. ArterivirusPLP2 and nairovirus OTU enzymes differ from eukaryotic OTUDUBs in that they also remove the Ub-like antiviral protein IFNstimulated gene 15 (ISG15) from target proteins, a process alsoknown as deISGylation (18, 49). ISG15 conjugation has beenpostulated to interfere with proper viral protein function, pos-sibly through steric hindrance, although the exact mechanismunderlying its antiviral activity is unknown (50). For CCHFVOTU, cross-reactivity with ISG15 arises primarily from a uniqueβ-hairpin on the Ub-binding surface. The hairpin modifies thesurface so that the viral enzyme binds the β-grasp folds of Ub and
van Kasteren et al. PNAS | Published online February 11, 2013 | E839
MICRO
BIOLO
GY
PNASPL
US

the C-terminal Ub-like domain of ISG15 in an orientation that isrotated nearly 75° with respect to that observed for Ub bound toa representative eukaryotic OTU DUB from yeast (Otu1) (43,44). Surprisingly, the β-hairpin is absent in EAV PLP2 andreplaced by helix α3 of the zinc finger motif (Fig. 2A). However,in keeping with the role of the β-hairpin in CCHFV OTU, res-idues of helix α3 bind to the hydrophobic Ile44 patch of Ub,a site commonly targeted by Ub-binding proteins (51), and theyalso assist in positioning Ub in a rotated manner equivalent tothat observed for CCHFV OTU (Fig. 2 B and C).
Structure-Guided Decoupling of PLP2 Deubiquitinase and PolyproteinCleavage Activities. Given the distance of helix α3 from the PLP2active site (Fig. 1B), we hypothesized that mutations could beintroduced into this region of the Ub-binding surface that wouldselectively disrupt PLP2 DUB activity without affecting EAVpolyprotein cleavage at the nsp2jnsp3 junction (presumablyRLIGG↓WIY). Although this sequence closely resembles the C-terminal tail of Ub (RLRGG↓), we postulated that the nsp2 se-quence immediately upstream of the nsp2jnsp3 junction does notadopt a Ub-like fold and that the majority of the PLP2 Ub-
Cys319
Cys349
Cys356
Cys354
B
F
β2β1
N
C
β2
α7
α1 β1
β6
β5β4
β3
α5
α6α2
α3 α4β7 N
Cα5
β4
β6α3
α4
α6
α1 α2β2
β5
β3
β1
G H
NC
α1
β1
β4
β5β6
Zn β3 α2
β2
α4
α3
Asn263
His332
3CN
2.9Å
4.4Å
EAV PLP2
CCHFV OTU
Yeast OTU1
Domain II Domain I
Activesite
EAV PLP2
Ubiquitin
A
β5β4β3
β6
α3α2
α1
α4
C4 Zn Finger
β5
β4
β3
β6
α3
α1
α2
α4
β2β1
EAV PLP2
Ubiquitin
Domain II
Domain I
D
E
90°
C
Cys270
Fig. 1. Structure of the EAV PLP2-Ub complex and superposition with yeast OTU1 and CCHFV OTU. (A) Structure of EAV PLP2 (blue) bound to Ub (orange)showing the two-domain fold. (B) 90° rotation of complex shown in A. (C) Electron density for the C4 zinc finger motif. Blue density is a maximum-likelihoodweighted 2Fo-Fc map contoured at 1.0σ. Green density about the zinc atom (gray) is a Fo-Fc omit map contoured at 5.0σ. (D) The catalytic triad of the EAV PLP2active site. Blue density is a maximum-likelihood weighted 2Fo-Fc map contoured at 1.0σ. The cysteine nucleophile (Cys270) is covalently linked to Ub via the3CN linker, which replaces Gly76 of Ub. Asn263, which orients the imidazole ring of His332, occurs in two alternate conformations. (E) Superposition of EAVPLP2 (blue), CCHFV OTU (cyan), and yeast OTU1 (red). PLP2 shares a conserved core of two central helices and a four-stranded β-sheet with CCHFV OTU andyeast OTU1. Topology diagrams for EAV PLP2, CCHFV OTU, and yeast OTU1 are shown in F–H, respectively. The region that is conserved among the enzymes isoutlined (dashed box). Structural images were prepared using PyMOL (84).
E840 | www.pnas.org/cgi/doi/10.1073/pnas.1218464110 van Kasteren et al.

binding surface is therefore not required for its cleavage. To testour hypothesis, we used the crystal structure to select threepositions within the PLP2 Ub-binding surface (Thr312, Ile313,and Ile353) and engineered a panel of (combined) mutations(Fig. 2 B andC). Ile353 is located at the C-terminal end of helix α3next to C3 (Cys354) of the zinc finger motif. It projects directlyinto the Ile44 patch of Ub where it makes extensive van derWaalsinteractions with Ile44, Val70, and Leu8. Given that Ile353 islocated on the Ub-binding surface, we aimed to disrupt Ubbinding by introducing various other residues at this position,including large bulky residues such as arginine and tryptophan.Thr312 and Ile313 are located closer to the active site, where theymake additional hydrophobic interactions with Leu8, Leu71, andLeu73 of Ub. In an attempt to further disrupt the interactionbetween PLP2 and Ub, the mutations Thr312Ala and Ile313Valwere also combined with changes at position Ile353. Given theclose proximity of Ile313 to Leu73 of the RLRGG motif of Ub,mutation to valine was chosen to minimize adverse effects onnsp2jnsp3 cleavage.Before proceeding to infection experiments with mutant viruses,
we used ectopic expression of PLP2 and an in vitro enzymaticassay to characterize the effect of various mutations at the posi-tions described above on polyprotein processing and DUB activity.To this end, mutations were introduced into a mammalian ex-pression vector encoding a self-cleaving nsp2-3 polyprotein car-rying an N-terminal HA-tag. On expression of nsp2-3 in HEK293Tcells, WT PLP2 mediated efficient cleavage of the nsp2jnsp3junction (Fig. 3A). As expected, a PLP2 active site mutant (C270A/H332A) did not display any processing of the nsp2jnsp3 site, andonly the nsp2-3 precursor was detected. Seven single-site mutants,in which the Ub-binding surface was targeted by replacement ofThr312 or Ile353, displayed WT levels of nsp2jnsp3 cleavage,suggesting that their polyprotein processing activity was not no-tably affected. In addition, combinations of mutations at posi-tions Thr312, Ile313, and Ile353 were tested, with similar results(Fig. 3A). Longer exposure times did reveal some nsp2-3 pre-cursor, even in the case of WT PLP2, but this amount onlymarginally increased when two or three mutations were com-bined (Fig. 4B).Next, we assayed the effect of these mutations on PLP2 DUB
activity by transfecting mammalian cells with plasmids encodingFLAG-tagged Ub and nsp2-3 carrying WT or mutant PLP2.
FLAG-tagged ubiquitination of a wide range of cellular targetscould be visualized by Western blot analysis using an anti-FLAGantibody (Fig. 3B). As expected, expression of WT PLP2 stronglydecreased the accumulation of Ub-conjugates, whereas expressionof the active site mutant (C270A/H332A) had a negligible effect.Fig. 3B presents the results obtained with a selection of PLP2Ub-binding surface mutants with the most pronounced effect onDUB activity, some of which approached the level of the activesite mutant. In contrast, these Ub-binding surface mutations hadonly minor effects on the deISGylating activity of PLP2 (Fig. S1).To corroborate our findings from the expression system showing
that PLP2 DUB activity could be selectively removed withoutdisturbing nsp2jnsp3 cleavage, an in vitro activity assay ofrecombinant PLP2 produced in E. coli was performed using thefluorescently labeled substrates Ub-aminomethylcoumarin (Ub-AMC) or RLRGG-AMC, representing the C-terminal peptidemotif of Ub. By comparing the activity of PLP2 mutants againstUb-AMC (which requires the Ub-binding surface) versus theiractivity against RLRGG-AMC (which binds to the active site re-gion only), PLP2 mutants with a selective reduction in DUB ac-tivity could be identified. Indeed, compared with WT enzyme,mutants I353W and I353R exhibited ∼10- and 40-fold reductionsin specificity (kcat/Km) toward Ub, respectively; in contrast, theiractivity toward RLRGG-AMC was essentially unaltered (Table 1).Expression of PLP2 containing additional mutations at Thr312and Ile313 yielded insoluble protein in E. coli, preventing analysisof these mutants. Nevertheless, the results of the in vitro activityassay further confirmed the successful decoupling of the poly-protein processing and DUB activities of EAV PLP2 by specifi-cally targeting key residues of the Ub-binding surface.
Ability of PLP2 to Suppress Innate Immune Signaling Depends on ItsDUB Activity. The effect of the various PLP2 Ub-binding surfacemutations on innate immune signaling was initially assessed inthe context of ectopic nsp2-3 expression using a luciferase-basedIFN-β promoter activity assay. For this, HEK293T cells weretransfected with a combination of plasmids encoding firefly lu-ciferase under control of the IFN-β promoter, renilla luciferaseas an endogenous control, constitutively active RIG-I(2CARD) toinduce innate immune signaling (52), and EAV nsp2-3 contain-ing either WT or mutant PLP2. In this assay, reporter gene ex-pression was reduced to ∼20% on coexpression of WT PLP2,
Leu73
Leu71
Leu8
Thr312
Ile353
Zn
Cys354Ile313
Leu69
Thr9
Arg72
Ubiquitin
Ile353
Thr312
Ile313
EAV PLP2
B C
EAV C4zinc finger
A CCHFV Ub
EAV Ub
CCHFVβ-hairpin
EAVHelix α3
Ile44
Activesite
Val70
Fig. 2. Ub-binding surface of EAV PLP2. (A) Superposition of EAV PLP2 (blue) and CCHFV OTU (cyan) in complex with Ub. Both enzymes grasp Ub in a similarorientation, with the C4 zinc finger motif of EAV PLP2 replacing the β-hairpin of CCHFV OTU. (B) EAV PLP2 (blue) bound to Ub (orange), showing the 612-Å2
Ub-binding surface. Residues targeted for mutational analysis to disrupt DUB activity are indicated by arrows. (C) Close-up of the EAV PLP2 Ub-bindingsurface. EAV PLP2 residue Ile353 forms van der Waals interactions with Ile44, Leu8, and Val70 of Ub, whereas residues Thr312 and Ile313 interact with anadditional hydrophobic patch on Ub (formed by residues Leu8, Val70, Leu71, and Ile73) closer to the active site of PLP2.
van Kasteren et al. PNAS | Published online February 11, 2013 | E841
MICRO
BIOLO
GY
PNASPL
US

whereas 80% of the level of the untreated control was retained onexpression of the PLP2 active site mutant (C270A/H332A; Fig.4A). Compared with WT PLP2, all mutants included in Fig. 4Awere significantly impaired in their inhibitory activity (P < 0.01),with mutants I353R, I353W, T312A/I313V/I353R, and T312A/I313V/I353W displaying inhibition levels similar to that of theactive site mutant (P> 0.05).Western blot analysis confirmed equalexpression of WT and mutant nsp2-3 (Fig. 4B). These resultsdemonstrated that, at least in the context of the nsp2-3 expressionsystem, the selective removal of PLP2 DUB activity significantlydisrupted its ability to suppress IFN-β promoter activity.
Arteriviruses Lacking PLP2 DUB Activity Elicit an Enhanced Host InnateImmuneResponse.Having identified mutations in PLP2 that reduceits ability to suppress Ub-mediated innate immune signaling (Fig.4A) without adversely affecting nsp2jnsp3 cleavage (Fig. 3A), wewere in a position to directly evaluate the importance of PLP2DUB activity for immune evasion during arterivirus infection. Thesix (combinations of) mutations used in Figs. 3B and 4 were in-troduced into an EAV full-length cDNA clone, and mutant viruseswere launched by electroporation of in vitro transcribed RNA intoBHK-21 cells. Immunofluorescence microscopy subsequentlyconfirmed expression of both nsp2 and the nucleocapsid (N)protein, followed by virus spread to initially untransfected cells.These findings indicated that viruses carrying these (combinationsof) mutations were replication competent.We next focused on the two mutants showing the greatest de-
crease in inhibitory activity in the IFN-β promoter activity assay:I353R and T312A/I313V/I353R (Fig. 4A). We first characterized
Fig. 3. Decoupling of the polyprotein processing and DUB activities of EAVPLP2. (A) HEK293T cells were transfected with plasmids encoding nsp2-3containing WT or mutant PLP2. After 16 h at 37 °C, cells were lysed, andresults were analyzed by Western blot. Proteolytic processing of thensp2jnsp3 junction by WT PLP2 resulted in the release of HA-tagged nsp2from the nsp2-3 precursor. (B) HEK293T cells were transfected with a com-bination of plasmids encoding nsp2-3 containing WT or mutant PLP2 andFLAG-Ub. Expression of FLAG-Ub leads to FLAG-tagged ubiquitination ofa wide range of cellular proteins, which can be visualized on Western blotusing an anti-FLAG antibody. Ub, ubiquitin.
Fig. 4. PLP2 Ub-binding surface mutations attenuate inhibition of IFN-βpromoter activation. (A) Luciferase-based reporter assay to assess the effectof various Ub-binding surface mutations on the inhibition of IFN-β promoteractivity by PLP2. HEK293T cells were transfected with a combination ofplasmids encoding firefly luciferase under control of the IFN-β promoter,renilla luciferase, RIG-I(2CARD), and nsp2-3 containing WT or mutant PLP2.Results were obtained in three independent experiments. Error bars repre-sent SDs, and P values are relative to WT. (B) Lysates obtained in each of thethree experiments used for A were mixed in a 1:1:1 ratio and analyzed byWestern blot for the expression of nsp2-3.
E842 | www.pnas.org/cgi/doi/10.1073/pnas.1218464110 van Kasteren et al.

the replication kinetics of these mutants in a time course ex-periment in primary equine lung fibroblasts (ELFs), which arederived from the natural host species of EAV and are likely tomaintain an intact innate immune response. ELFs were infectedwith WT or mutant EAV at a multiplicity of infection (m.o.i.) of0.5 or 5. The first-cycle replication kinetics as determined byreal-time quantitative reverse transcriptase PCR (qRT-PCR)measurement of viral genome RNA levels did not notably differbetween WT and mutant EAV (Fig. 5 A and B). In addition, viraltiters in cell culture supernatants harvested from infected ELFsat 24 h postinfection (hpi) revealed no significant differencebetween WT and mutant EAV (Fig. 5C). Finally, immunofluo-rescence microscopy of ELFs infected with m.o.i. 5 revealedexpression of nsp2 from 6 hpi onward, and by 9 hpi, expression ofN protein could be seen in all cells for both WT and mutantEAV (Fig. S2). These results demonstrated that the replicationkinetics of the PLP2 Ub-binding surface mutants and WT control
were essentially the same, in line with our previous conclusionthat cleavage of the nsp2jnsp3 site is hardly affected by thesemutations. At the same time, Western blot analysis showed that,compared with WT EAV, the DUB activity of both mutants wasseverely impaired during infection (Fig. 5D).Subsequently, we used real-time qRT-PCR to measure the
levels of mRNAs encoding IFN-β, the IFN-stimulated proteinMX1, and the proinflammatory cytokine IL8 and investigate theeffect of mutations I353R and T312A/I313V/I353R on innateimmune signaling during infection of ELFs. Initially, we infectedELFs with WT or mutant EAV at m.o.i. 5, but in this setup, IFN-βmRNA levels remained below the detection limit at all timepoints analyzed (from 3 to 12 hpi), suggesting that the innateimmune response triggered by EAV on initial infection is verylimited. Therefore, we next infected ELFs with WT or mutantEAV at m.o.i. 0.25, resulting in infection of ∼20% of cells. Wehypothesized that this would allow for IFN-β–mediated primingof uninfected cells as a result of paracrine signaling from cellsinfected during the first round. The expression of IFN-stimulatedgenes would then result in a more potent response during thesecond cycle of infection. Indeed, following such a low m.o.i. in-fection with WT EAV, low but detectable levels of IFN-β mRNAwere induced by 20 hpi (Fig. 6A). Interestingly, at both 20 and24 hpi, cells infected with either mutant showed significantlyincreased levels of IFN-β mRNA compared with WT virus-infectedcells (P < 0.05), with mutant T312A/I313V/I353R showing the mostpronounced difference. In addition, the levels of MX1 mRNAdiffered significantly between WT and mutant EAV-infectedcells at 24 hpi (Fig. 6B). In contrast, at 20 and 24 hpi, no sig-
Fig. 5. EAV PLP2 mutants display similar replication kinetics as WT virus. Equine lung fibroblasts were infected with WT or mutant EAV at m.o.i. 0.5 (A) or5 (B–D). (A and B) At the indicated time points, total RNA was isolated for qRT-PCR measurement of EAV genomic RNA levels. Results, which were obtained inthree independent experiments, were analyzed using the standard curve method and normalized against the relative quantities of GAPDH and β-actin mRNA.Error bars represent SDs. (C) At 24 hpi, cell culture supernatants were harvested, and virus titers were determined by plaque assay on ELFs. Results wereobtained in three independent experiments, and error bars represent SDs. (D) Cells were lysed at 10 hpi, and total ubiquitination was assessed byWestern blotanalysis. AU, arbitrary units; hpi, hours postinfection; N, nucleocapsid; pfu, plaque-forming units; Ub, ubiquitin.
Table 1. Effect of Ub-binding surface mutations on thesubstrate specificity of PLP2
PLP2 enzyme
Substrate
RLRGG-AMC(kcat/Km) (M
−1·s−1)Ub-AMC
(kcat/Km) (M−1·s−1)
WT 45 ± 11 17,000 ± 4,000I353R 65 ± 7 413 ± 177I353W 155 ± 12 1741 ± 850
van Kasteren et al. PNAS | Published online February 11, 2013 | E843
MICRO
BIOLO
GY
PNASPL
US
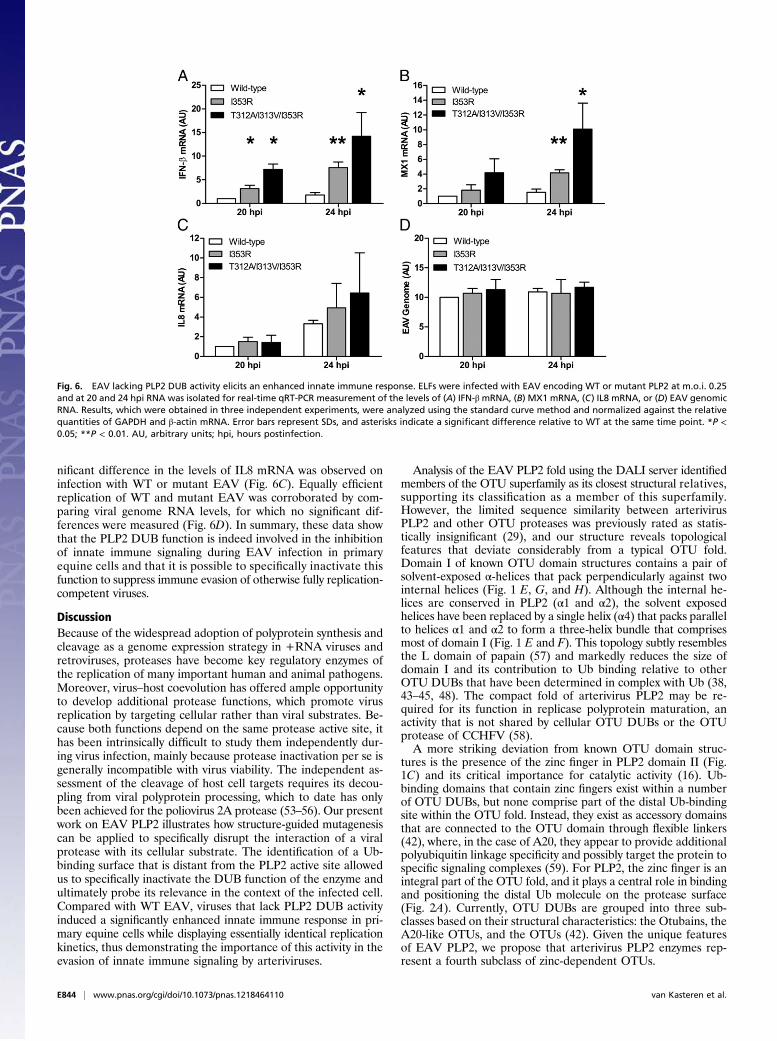
nificant difference in the levels of IL8 mRNA was observed oninfection with WT or mutant EAV (Fig. 6C). Equally efficientreplication of WT and mutant EAV was corroborated by com-paring viral genome RNA levels, for which no significant dif-ferences were measured (Fig. 6D). In summary, these data showthat the PLP2 DUB function is indeed involved in the inhibitionof innate immune signaling during EAV infection in primaryequine cells and that it is possible to specifically inactivate thisfunction to suppress immune evasion of otherwise fully replication-competent viruses.
DiscussionBecause of the widespread adoption of polyprotein synthesis andcleavage as a genome expression strategy in +RNA viruses andretroviruses, proteases have become key regulatory enzymes ofthe replication of many important human and animal pathogens.Moreover, virus–host coevolution has offered ample opportunityto develop additional protease functions, which promote virusreplication by targeting cellular rather than viral substrates. Be-cause both functions depend on the same protease active site, ithas been intrinsically difficult to study them independently dur-ing virus infection, mainly because protease inactivation per se isgenerally incompatible with virus viability. The independent as-sessment of the cleavage of host cell targets requires its decou-pling from viral polyprotein processing, which to date has onlybeen achieved for the poliovirus 2A protease (53–56). Our presentwork on EAV PLP2 illustrates how structure-guided mutagenesiscan be applied to specifically disrupt the interaction of a viralprotease with its cellular substrate. The identification of a Ub-binding surface that is distant from the PLP2 active site allowedus to specifically inactivate the DUB function of the enzyme andultimately probe its relevance in the context of the infected cell.Compared with WT EAV, viruses that lack PLP2 DUB activityinduced a significantly enhanced innate immune response in pri-mary equine cells while displaying essentially identical replicationkinetics, thus demonstrating the importance of this activity in theevasion of innate immune signaling by arteriviruses.
Analysis of the EAV PLP2 fold using the DALI server identifiedmembers of the OTU superfamily as its closest structural relatives,supporting its classification as a member of this superfamily.However, the limited sequence similarity between arterivirusPLP2 and other OTU proteases was previously rated as statis-tically insignificant (29), and our structure reveals topologicalfeatures that deviate considerably from a typical OTU fold.Domain I of known OTU domain structures contains a pair ofsolvent-exposed α-helices that pack perpendicularly against twointernal helices (Fig. 1 E, G, and H). Although the internal he-lices are conserved in PLP2 (α1 and α2), the solvent exposedhelices have been replaced by a single helix (α4) that packs parallelto helices α1 and α2 to form a three-helix bundle that comprisesmost of domain I (Fig. 1 E and F). This topology subtly resemblesthe L domain of papain (57) and markedly reduces the size ofdomain I and its contribution to Ub binding relative to otherOTU DUBs that have been determined in complex with Ub (38,43–45, 48). The compact fold of arterivirus PLP2 may be re-quired for its function in replicase polyprotein maturation, anactivity that is not shared by cellular OTU DUBs or the OTUprotease of CCHFV (58).A more striking deviation from known OTU domain struc-
tures is the presence of the zinc finger in PLP2 domain II (Fig.1C) and its critical importance for catalytic activity (16). Ub-binding domains that contain zinc fingers exist within a numberof OTU DUBs, but none comprise part of the distal Ub-bindingsite within the OTU fold. Instead, they exist as accessory domainsthat are connected to the OTU domain through flexible linkers(42), where, in the case of A20, they appear to provide additionalpolyubiquitin linkage specificity and possibly target the protein tospecific signaling complexes (59). For PLP2, the zinc finger is anintegral part of the OTU fold, and it plays a central role in bindingand positioning the distal Ub molecule on the protease surface(Fig. 2A). Currently, OTU DUBs are grouped into three sub-classes based on their structural characteristics: the Otubains, theA20-like OTUs, and the OTUs (42). Given the unique featuresof EAV PLP2, we propose that arterivirus PLP2 enzymes rep-resent a fourth subclass of zinc-dependent OTUs.
Fig. 6. EAV lacking PLP2 DUB activity elicits an enhanced innate immune response. ELFs were infected with EAV encoding WT or mutant PLP2 at m.o.i. 0.25and at 20 and 24 hpi RNA was isolated for real-time qRT-PCR measurement of the levels of (A) IFN-β mRNA, (B) MX1 mRNA, (C) IL8 mRNA, or (D) EAV genomicRNA. Results, which were obtained in three independent experiments, were analyzed using the standard curve method and normalized against the relativequantities of GAPDH and β-actin mRNA. Error bars represent SDs, and asterisks indicate a significant difference relative to WT at the same time point. *P <0.05; **P < 0.01. AU, arbitrary units; hpi, hours postinfection.
E844 | www.pnas.org/cgi/doi/10.1073/pnas.1218464110 van Kasteren et al.

We believe that the structure-guided inactivation of the PLP2DUB function may contribute to the engineering of improvedmodified live vaccines (MLVs) for arteriviruses. Although generallyeffective strategies for the prevention and control of equine viralarteritis have been developed, several field strains are poorlyneutralized by antibodies from horses vaccinated with theARVAC vaccine strain (60), and EAV outbreaks continue tocause significant disruptions of the horse breeding industry (61).Although our current work focuses on EAV PLP2, the corre-sponding protease of PRRSV has been shown to have very similarimmune evasive properties in a variety of experimental settings(18, 30, 31). Since its discovery in the late 1980s, PRRSV hasspread around the globe and now ranks among the most impor-tant swine pathogens. PRRSV infection was estimated to causeannual losses in the order of $500 million in the United Statesalone (62), and the emergence of highly virulent strains in Chinais of particular concern (63–65). Moreover, the virus has provendifficult to control, and it has been suggested to counteract innateimmunity, thus undermining the overall immune response andviral clearance in infected animals (66). Consequently, in-activation of the PLP2 DUB function may be an important step inthe design of improved vaccine candidates. Virus lacking PLP2DUB activity should induce a more robust innate response andtherefore also a more potent adaptive immune reaction than thatachieved with the currently available MLVs. Multiple studieshave suggested that additional arterivirus nsps may contribute toinnate immune evasion (67, 68), and consequently, vaccine effi-cacy may be further bolstered by targeting a combination of suchfunctions. As in this study, detailed insight into the molecularinteractions of these other arterivirus proteins and their hostligands will likely be required to achieve this goal without crip-pling the basic replication capacity of the virus. In cell culture, ourmost promising EAV PLP2 mutant, carrying mutations T312A/I313V/I353R, induced an approximately eightfold increase in IFN-βmRNA levels compared withWT virus. Although wewere unableto investigate this effect in vivo due to the lack of a small animalmodel for EAV infection, the disabling of immune evasion mech-anisms can result in significant virus attenuation. One striking ex-ample is the modification of the immune evasive influenza virusNS1 protein, which yielded attenuated viruses that are promisingvaccine candidates (69).In addition to opening possibilities for vaccine development,
the mutants described in this paper should provide excellenttools for identifying the cellular targets of PLP2, which to dateremain unknown. Although Western blot analysis suggests thatPLP2 acts in a very promiscuous fashion, causing a generaldecrease in the levels of ubiquitinated host proteins (Fig. 5D),our qRT-PCR results support the idea that there is at leastsome degree of specificity in the inhibition of innate immunesignaling, as evidenced by the PLP2-mediated inhibition ofIFN-β mRNA transcription but not IL8 mRNA transcription(Fig. 6 A and C). Because the expression of IFN-β dependslargely on the activation of the transcription factor IFN regu-latory factor 3 (IRF3) and the expression of proinflammatorycytokines like IL8 does not (27), our findings suggest that PLP2-mediated inhibition of innate immunity is primarily directed atIRF3-dependent signaling. Future experiments will aim at eluci-dating the target specificity of arterivirus PLP2.Arteriviruses are not the only virus family harboring proteases
with multiple substrate specificities. Especially interesting in thisrespect are the distantly related coronaviruses (CoVs), whichinfect a wide variety of species, including livestock, companionanimals, bats, and humans. Six CoV species have now been foundto infect humans, causing symptoms ranging from mild respiratoryillness to acute respiratory syndromes, as in the case of severe acuterespiratory syndrome (SARS) CoV (70) and the recently emergedhuman CoV EMC/2012 (71, 72). The replicase polyproteins ofcoronaviruses harbor one or two papain-like proteases (PLpros)
that participate in polyprotein maturation and presumably pro-mote evasion of innate immunity by means of their DUB activity(32, 34, 37, 73). Although the CoV PLpros belong to the Ub-spe-cific protease (USP) (74, 75) rather than the OTU superfamily ofDUBs, it is likely that coronavirus PLpro substrate specificities canbe decoupled using a similar structure-guided approach. Here wehave illustrated how structure-guided mutagenesis of such a viralprotease may be used to enhance the innate immune response toinfection, a strategy that may be applied in the design of MLVstargeting arteriviruses and other virus families encoding similardual-specificity proteases.
Materials and MethodsPLP2 Plasmids. For bacterial expression of EAV PLP2, a cDNA fragmentencoding residues 261–392 of EAV pp1a and an in-frame C-terminal His6purification tag were inserted downstream of a Ub fusion partner in thepASK3 vector (76), yielding plasmid pASK3-ePLP2. A mammalian expressionconstruct encoding an EAV nsp2-3 polyprotein was made by cloning residues261–1,064 of EAV pp1a in-frame with an N-terminal HA tag in the pcDNA3.1vector (Invitrogen). All mutants were engineered by site-directed mutagenesisusing Pfu DNA polymerase (Fermentas). Primer sequences are available onrequest. All constructs were verified by sequence analysis.
Purification and Crystallization of EAV PLP2 Bound to Ub. E. coli BL21-Gold(DE3) cells were transformed with pASK3-ePLP2 and cultured to an opticaldensity (OD600) of 0.7 in lysogeny broth (LB) medium at 37 °C. The culturewas then supplemented with 200 ng/mL of anhydrous tetracycline and in-cubated for 3 h at 28 °C with shaking to induce expression of the Ub-PLP2-His6 fusion protein. The cells were pelleted and resuspended in ice-cold lysisbuffer [20 mM 2-(N-Morpholino)ethanesulfonic acid (MES), pH 7, 500 mMNaCl, 10% (vol/vol) glycerol, 5 mM imidazole, pH 7.4, 0.5 mM Tris(2-carboxyethyl)phosphine (TCEP)] and lysed using a French pressure cell[American Instrument Company (AMINCO)]. The lysate was clarified bycentrifugation and loaded onto a Ni-NTA column (Qiagen) preequilibratedwith lysis buffer. After washing with lysis buffer supplemented with 15 mMimidazole, recombinant PLP2 was eluted from the column using an equili-bration buffer supplemented with 150 mM imidazole and exchanged into50 mM Tris, pH 8.0, 300 mM NaCl and 5 mM DTT before storing at 4 °C. Theendogenous DUB activity of PLP2 resulted in the efficient removal of theN-terminal Ub-tag from the fusion protein during expression in E. coli;therefore, affinity chromatography yielded highly pure PLP2 carrying aC-terminal His6 purification tag only.
The mechanism-based suicide inhibitor Ub–3Br was prepared according toMessick et al. (38) and Borodovsky et al. (39), as described by James et al.(43). Ub-3Br was covalently bound to purified PLP2 by gently mixing theproteins in a 3:2 molar ratio for 1 h at 37 °C. The resulting PLP2-Ub complexwas purified by gel filtration (Superdex 75) followed by anion exchange(Source 15Q) chromatography and then exchanged into 20 mM Tris, pH 8.0,50 mM NaCl before concentrating to 10 mg/mL and storing at 4 °C.
The PLP2-Ub complex was crystallized by hanging-drop vapor diffusion at10 mg/mL in mother liquor consisting of 100 mM MES, pH 6.2, 18% (wt/vol)polyethylene glycol (PEG) 20,000. Crystals were flash-cooled and stored inliquid nitrogen after sweeping them through mother liquor supplementedwith 20% (vol/vol) glycerol.
X-Ray Data Collection and Crystal Structure Determination. X-ray diffractiondata for a multiwavelength anomalous dispersion (MAD) experiment werecollected at the Canadian Light Source (beam line 08ID-1). Data were col-lected at three different wavelengths over the absorption edge of zinc froma single crystal of the PLP2-Ub complex held at 100 K in an N2 (g) stream. Thedata were processed using MOSFLM and SCALA (77), and structure factorphases were determined using phenix.autosol (78). Initial phases generatedby SOLVE were improved by density modification using RESOLVE within thePHENIX package. After reserving a random subset of reflections for cross-validation using the free R-factor (79), a model was built using phenix.autobuild (78) and manually completed and refined using COOT (80) andphenix.refine (78). Crystallographic data and model refinement statistics aresummarized in Table S1.
In Vitro Enzymatic Assays. To ensure complete removal of the N-terminalUb-tag from recombinant PLP2, E. coli C2523 containing (WT or mutant)pASK3-ePLP2 were cotransformed with plasmid pCG1, which encodes theDUB ubiquitin-specific processing protease 1 (Ubp1) (76). The DUB activity of
van Kasteren et al. PNAS | Published online February 11, 2013 | E845
MICRO
BIOLO
GY
PNASPL
US

PLP2 (WT and mutants) was assayed using 7-amino-4-methylcoumarin(AMC)–labeled versions of Ub (Ub-AMC) (Boston Biochem) and the C-ter-minal peptide motif of Ub: RLRGG-AMC (Enzo Life Sciences). The enzymescleave the AMC label causing a significant increase in the fluorescencequantum yield of the dye. All reactions were performed in 50 mM Tris-Clbuffer at pH 8 and 100 mM NaCl. Time-dependent fluorescence traces werecollected by a Fluorolog-3 Horiba Jobin Yvon fluorimeter. The mono-chromators were set to 360 nm (excitation) and 460 nm (emission). The slitswere set between 1 and 3 nm bandpass depending on substrate concentra-tion. Enzyme activities in all mutants were characterized by the specificityconstant kcat=Km. At substrate concentrations significantly smaller than Km,the formation of product follows pseudo–first-order kinetics; therefore, thetemporal evolution of product fluorescence F follows the equation
F ¼ F∞
�1− exp
�−kcat½E�Km
t��
þ F0; [1]
where F∞is the fluorescence when all AMC is liberated, F0 is the backgroundfluorescence, [E] is the total enzyme concentration, and t is the time elapsed.From fitting the fluorescence time traces to Eq. 1, all parameters includingthe specificity constant kcat=Km are obtained. Our assays indicate that PLP2exhibits pseudo–first-order kinetics for Ub-AMC at concentrations less than0.2 μM and for RLRGG-AMC at concentrations less than 150 μM.
Reverse Genetics. Mutations in the EAV PLP2 coding sequence were engi-neered in an appropriate shuttle vector and subsequently transferred topEAN551/AB, a derivative of EAV full-length cDNA clone pEAN551 carryingadditional (translationally silent) AflII and BspEI restriction sites (17, 81). Thevirus derived from pEAN551/AB was used as a WT control in all experiments.All constructs were verified by sequence analysis.
In vitro RNA transcription from XhoI-linearized WT or mutant EAV full-length cDNA clones was performed using the mMESSAGE mMACHINE T7 Kit(Ambion). Five micrograms of full-length EAV RNA was electroporated into5.0 × 106 BHK-21 cells using the Amaxa Cell Line Nucleofector Kit T and theprogram T-020 of the Amaxa Nucleofector (Lonza) according to the manu-facturer’s instructions. Cells were incubated at 39.5 °C, and virus-containingsupernatants were harvested at 24 h after transfection. Titers were deter-mined by plaque assay on primary ELFs essentially as described before (82).
To verify the presence of the correct mutations, RNA was isolated fromvirus-containing supernatants using the QIAamp Viral RNA Mini Kit (Qiagen)and converted to cDNA using RevertAid H Minus reverse transcriptase(Fermentas) and random hexameric primers. The region of PLP2 encoding themutations was subsequently PCR amplified using Pfu DNA polymerase(Fermentas) and sequenced.
Real-Time qRT-PCR. Confluent ELFs were infected with WT or mutant EAV atm.o.i. 5, 0.5, or 0.25 and incubated at 37 °C. At the indicated time points, cell
lysates were harvested in TriPure Isolation Reagent (Roche). After the ad-dition of chloroform, the aqueous phase was mixed in a 1:1 ratio with bufferRA1 of the Nucleospin RNA II kit (Macherey-Nagel). RNA was isolated as perthe manufacturer’s instructions and reverse transcribed using RevertAid HMinus RT (Fermentas) and the oligo(dT)20 primer. Finally, samples wereassayed by real-time qRT-PCR on a CFX384 Touch Real-Time PCR detectionsystem (BioRad) using iTaq SYBR Green Supermix with ROX (BioRad). Primers(Table S2) targeting mRNAs encoding equine GAPDH, β-actin, IFN-β, MX1,and the EAV genome were designed using Primer3 (83) or sequences werekindly provided by Udeni Balasuriya (University of Kentucky, Lexington, KY)in the case of IL8. The real-time PCR was followed by a melting-curve analysisto verify the specificity of the reaction. Results were quantified using thestandard curve method and normalized against the geometric mean of therelative quantities of GAPDH and β-actin mRNA. Data from three in-dependent experiments were analyzed with SPSS Statistics software usinga one-sample t test or unpaired Student t test, where appropriate. P < 0.05was considered statistically significant.
Deubiquitination During Infection. Confluent ELFs were infected with WTor mutant EAV at m.o.i. 5 and incubated for 10 h at 37 °C. Cells were thenlysed in 500 μL 2× Laemmli sample buffer [250 mM Tris, 2% (wt/vol) SDS,20% (vol/vol) glycerol, 0.01% bromophenol blue, 2 mM DTT, pH 6.8], andtotal ubiquitination was assessed by Western blot analysis as described in SIMaterials and Methods.
Ectopic Expression Experiments. Ectopic expression experiments were per-formed essentially as described before (31) but are described in detail in SIMaterials and Methods, together with a description of the additional plas-mids, cells, and antibodies used.
ACKNOWLEDGMENTS. We thank Adolfo García-Sastre, John-Paul Bacik,John Hiscott, Udeni B. Balasuriya, Alexander E. Gorbalenya, Aartjan J. W. teVelthuis, Adriaan H. de Wilde, Kathleen C. Lehman, and Diede Oudshoornfor helpful discussions. We thank V. Larmour for technical assistance andS. Labiuk and the staff of the Canadian Light Source (CLS) beamline 08ID-1for assistance with data collection. We kindly thank the following peoplefor providing us with reagents: Erwin van den Born, Craig E. Cameron, Nata-lia Frias-Staheli, Michaela U. Gack, Paul N. Moynagh, Adolfo García-Sastre,and Gijs A. Versteeg. The CLS is supported by Natural Sciences and Engineer-ing Research Council of Canada (NSERC), the National Research Council, theCanadian Institutes of Health Research, and the University of Saskatchewan.This research was supported in part by the Division of Chemical Sciences ofthe Netherlands Organization for Scientific Research (NWO-CW) throughECHO Grant 700.59.008 (to M. Kikkert and E.J.S.) and by NSERC Grant311775-2010 (to B.L.M.). B.L.M. holds a Manitoba Research Chair award.The research was also supported in part by the European Union SeventhFramework Programme (FP7/2007-2013) under SILVER Grant 260644.
1. Firth AE, Brierley I (2012) Non-canonical translation in RNA viruses. J Gen Virol
93(Pt 7):1385–1409.2. Dougherty WG, Semler BL (1993) Expression of virus-encoded proteinases: Functional
and structural similarities with cellular enzymes. Microbiol Rev 57(4):781–822.3. Gorbalenya AE, Donchenko AP, Blinov VM, Koonin EV (1989) Cysteine proteases of
positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein
superfamily with a common structural fold. FEBS Lett 243(2):103–114.4. Gorbalenya AE, Koonin EV, Lai MM (1991) Putative papain-related thiol proteases of
positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and
delineation of a novel conserved domain associated with proteases of rubi-, alpha-
and coronaviruses. FEBS Lett 288(1-2):201–205.5. Hellen CU, Kräusslich HG, Wimmer E (1989) Proteolytic processing of polyproteins in
the replication of RNA viruses. Biochemistry 28(26):9881–9890.6. Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW (1982) Inhibition of HeLa
cell protein synthesis following poliovirus infection correlates with the proteolysis of
a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap
binding protein complex. J Biol Chem 257(24):14806–14810.7. Kräusslich HG, Nicklin MJ, Toyoda H, Etchison D, Wimmer E (1987) Poliovirus
proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220.
J Virol 61(9):2711–2718.8. Li XD, Sun L, Seth RB, Pineda G, Chen ZJ (2005) Hepatitis C virus protease NS3/4A
cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate
immunity. Proc Natl Acad Sci USA 102(49):17717–17722.9. Meylan E, et al. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and
is targeted by hepatitis C virus. Nature 437(7062):1167–1172.10. Ventoso I, MacMillan SE, Hershey JW, Carrasco L (1998) Poliovirus 2A proteinase
cleaves directly the eIF-4G subunit of eIF-4F complex. FEBS Lett 435(1):79–83.11. Balasuriya UB, MacLachlan NJ (2004) The immune response to equine arteritis virus:
Potential lessons for other arteriviruses. Vet Immunol Immunopathol 102(3):107–129.
12. Huang YW, Meng XJ (2010) Novel strategies and approaches to develop the next
generation of vaccines against porcine reproductive and respiratory syndrome virus
(PRRSV). Virus Res 154(1-2):141–149.13. Fang Y, Snijder EJ (2010) The PRRSV replicase: Exploring the multifunctionality of an
intriguing set of nonstructural proteins. Virus Res 154(1–2):61–76.14. Ziebuhr J, Snijder EJ, Gorbalenya AE (2000) Virus-encoded proteinases and proteolytic
processing in the Nidovirales. J Gen Virol 81(Pt 4):853–879.15. Han J, Rutherford MS, Faaberg KS (2009) The porcine reproductive and respiratory
syndrome virus nsp2 cysteine protease domain possesses both trans- and cis-cleavage
activities. J Virol 83(18):9449–9463.16. Snijder EJ, Wassenaar AL, Spaan WJ, Gorbalenya AE (1995) The arterivirus Nsp2
protease. An unusual cysteine protease with primary structure similarities to both
papain-like and chymotrypsin-like proteases. J Biol Chem 270(28):16671–16676.17. Posthuma CC, et al. (2008) Formation of the arterivirus replication/transcription complex:
A key role for nonstructural protein 3 in the remodeling of intracellular membranes.
J Virol 82(9):4480–4491.18. Frias-Staheli N, et al. (2007) Ovarian tumor domain-containing viral proteases evade
ubiquitin- and ISG15-dependent innate immune responses. Cell HostMicrobe 2(6):404–416.19. Behrends C, Harper JW (2011) Constructing and decoding unconventional ubiquitin
chains. Nat Struct Mol Biol 18(5):520–528.20. Komander D (2009) The emerging complexity of protein ubiquitination. Biochem Soc
Trans 37(Pt 5):937–953.21. Enesa K, et al. (2008) NF-kappaB suppression by the deubiquitinating enzyme
Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem
283(11):7036–7045.22. Kayagaki N, et al. (2007) DUBA: A deubiquitinase that regulates type I interferon
production. Science 318(5856):1628–1632.23. Li S, et al. (2010) Regulation of virus-triggered signaling by OTUB1- and OTUB2-
mediated deubiquitination of TRAF3 and TRAF6. J Biol Chem 285(7):4291–4297.
E846 | www.pnas.org/cgi/doi/10.1073/pnas.1218464110 van Kasteren et al.

24. Wertz IE, et al. (2004) De-ubiquitination and ubiquitin ligase domains of A20downregulate NF-kappaB signalling. Nature 430(7000):694–699.
25. Jiang X, Chen ZJ (2012) The role of ubiquitylation in immune defence and pathogenevasion. Nat Rev Immunol 12(1):35–48.
26. Oudshoorn D, Versteeg GA, Kikkert M (2012) Regulation of the innate immunesystem by ubiquitin and ubiquitin-like modifiers. Cytokine Growth Factor Rev 23(6):273–282.
27. Jensen S, Thomsen AR (2012) Sensing of RNA viruses: A review of innate immunereceptors involved in recognizing RNA virus invasion. J Virol 86(6):2900–2910.
28. O’Neill LA, Bowie AG (2010) Sensing and signaling in antiviral innate immunity. CurrBiol 20(7):R328–R333.
29. Makarova KS, Aravind L, Koonin EV (2000) A novel superfamily of predicted cysteineproteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem Sci25(2):50–52.
30. Sun Z, Chen Z, Lawson SR, Fang Y (2010) The cysteine protease domain of porcinereproductive and respiratory syndrome virus nonstructural protein 2 possessesdeubiquitinating and interferon antagonism functions. J Virol 84(15):7832–7846.
31. van Kasteren PB, et al. (2012) Arterivirus and nairovirus ovarian tumor domain-containing Deubiquitinases target activated RIG-I to control innate immune signaling.J Virol 86(2):773–785.
32. Devaraj SG, et al. (2007) Regulation of IRF-3-dependent innate immunity by thepapain-like protease domain of the severe acute respiratory syndrome coronavirus.J Biol Chem 282(44):32208–32221.
33. Inn KS, et al. (2011) Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J Virol 85(20):10899–10904.
34. FriemanM, Ratia K, Johnston RE, Mesecar AD, Baric RS (2009) Severe acute respiratorysyndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domainregulate antagonism of IRF3 and NF-kappaB signaling. J Virol 83(13):6689–6705.
35. Jiang J, Tang H (2010) Mechanism of inhibiting type I interferon induction byhepatitis B virus X protein. Protein Cell 1(12):1106–1117.
36. Wang D, et al. (2011) The leader proteinase of foot-and-mouth disease virusnegatively regulates the type I interferon pathway by acting as a viral deubiquitinase.J Virol 85(8):3758–3766.
37. Zheng D, Chen G, Guo B, Cheng G, Tang H (2008) PLP2, a potent deubiquitinase frommurine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res18(11):1105–1113.
38. Messick TE, et al. (2008) Structural basis for ubiquitin recognition by the Otu1 ovariantumor domain protein. J Biol Chem 283(16):11038–11049.
39. Borodovsky A, et al. (2002) Chemistry-based functional proteomics reveals novelmembers of the deubiquitinating enzyme family. Chem Biol 9(10):1149–1159.
40. Andreini C, Bertini I, Cavallaro G (2011) Minimal functional sites allow a classificationof zinc sites in proteins. PLoS ONE 6(10):e26325.
41. Holm L, Rosenström P (2010) Dali server: Conservation mapping in 3D. Nucleic AcidsRes 38(Web Server issue, Suppl 2):W545-9.
42. Komander D, Clague MJ, Urbé S (2009) Breaking the chains: Structure and function ofthe deubiquitinases. Nat Rev Mol Cell Biol 10(8):550–563.
43. James TW, et al. (2011) Structural basis for the removal of ubiquitin and interferon-stimulated gene 15 by a viral ovarian tumor domain-containing protease. Proc NatlAcad Sci USA 108(6):2222–2227.
44. Akutsu M, Ye Y, Virdee S, Chin JW, Komander D (2011) Molecular basis for ubiquitinand ISG15 cross-reactivity in viral ovarian tumor domains. Proc Natl Acad Sci USA 108(6):2228–2233.
45. Capodagli GC, et al. (2011) Structural analysis of a viral ovarian tumor domainprotease from the Crimean-Congo hemorrhagic fever virus in complex with covalentlybonded ubiquitin. J Virol 85(7):3621–3630.
46. Juang Y-C, et al. (2012) OTUB1 co-opts Lys48-linked ubiquitin recognition to suppressE2 enzyme function. Mol Cell 45(3):384–397.
47. Wiener R, Zhang X, Wang T, Wolberger C (2012) The mechanism of OTUB1-mediatedinhibition of ubiquitination. Nature 483(7391):618–622.
48. Huang OW, et al. (2012) Phosphorylation-dependent activity of the deubiquitinaseDUBA. Nat Struct Mol Biol 19(2):171–175.
49. Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y (2012) Nonstructural protein 2 of porcinereproductive and respiratory syndrome virus inhibits the antiviral function ofinterferon-stimulated gene 15. J Virol 86(7):3839–3850.
50. Durfee LA, Lyon N, Seo K, Huibregtse JM (2010) The ISG15 conjugation system broadlytargets newly synthesized proteins: Implications for the antiviral function of ISG15.Mol Cell 38(5):722–732.
51. Dikic I, Wakatsuki S, Walters KJ (2009) Ubiquitin-binding domains - from structures tofunctions. Nat Rev Mol Cell Biol 10(10):659–671.
52. Gack MU, et al. (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446(7138):916–920.
53. Morrison JM, Racaniello VR (2009) Proteinase 2Apro is essential for enterovirusreplication in type I interferon-treated cells. J Virol 83(9):4412–4422.
54. Ventoso I, Carrasco L (1995) A poliovirus 2A(pro) mutant unable to cleave 3CD showsinefficient viral protein synthesis and transactivation defects. J Virol 69(10):6280–6288.
55. Yu SF, Lloyd RE (1991) Identification of essential amino acid residues in the functionalactivity of poliovirus 2A protease. Virology 182(2):615–625.
56. Yu SF, Benton P, Bovee M, Sessions J, Lloyd RE (1995) Defective RNA replication bypoliovirus mutants deficient in 2A protease cleavage activity. J Virol 69(1):247–252.
57. Kamphuis IG, Kalk KH, Swarte MBA, Drenth J (1984) Structure of papain refined at1.65 A resolution. J Mol Biol 179(2):233–256.
58. Bergeron E, Albariño CG, Khristova ML, Nichol ST (2010) Crimean-Congo hemorrhagicfever virus-encoded ovarian tumor protease activity is dispensable for virus RNApolymerase function. J Virol 84(1):216–226.
59. Bosanac I, et al. (2010) Ubiquitin binding to A20 ZnF4 is required for modulation ofNF-κB signaling. Mol Cell 40(4):548–557.
60. Zhang J, et al. (2010) Molecular epidemiology and genetic characterization of equinearteritis virus isolates associated with the 2006-2007 multi-state disease occurrence inthe USA. J Gen Virol 91(Pt 9):2286–2301.
61. Holyoak GR, Balasuriya UB, Broaddus CC, Timoney PJ (2008) Equine viral arteritis:Current status and prevention. Theriogenology 70(3):403–414.
62. Neumann EJ, et al. (2005) Assessment of the economic impact of porcine reproductiveand respiratory syndrome on swine production in the United States. J Am Vet Med Assoc227(3):385–392.
63. Tong GZ, et al. (2007) Highly pathogenic porcine reproductive and respiratorysyndrome, China. Emerg Infect Dis 13(9):1434–1436.
64. Li Y, et al. (2007) Emergence of a highly pathogenic porcine reproductive andrespiratory syndrome virus in the Mid-Eastern region of China. Vet J 174(3):577–584.
65. Tian K, et al. (2007) Emergence of fatal PRRSV variants: Unparalleled outbreaksof atypical PRRS in China and molecular dissection of the unique hallmark. PLoS ONE2(6):e526.
66. Kimman TG, Cornelissen LA, Moormann RJ, Rebel JM, Stockhofe-Zurwieden N (2009)Challenges for porcine reproductive and respiratory syndrome virus (PRRSV) vaccinology.Vaccine 27(28):3704–3718.
67. Chen Z, et al. (2010) Identification of two auto-cleavage products of nonstructuralprotein 1 (nsp1) in porcine reproductive and respiratory syndrome virus infected cells:nsp1 function as interferon antagonist. Virology 398(1):87–97.
68. Beura LK, et al. (2010) Porcine reproductive and respiratory syndrome virusnonstructural protein 1beta modulates host innate immune response by antagonizingIRF3 activation. J Virol 84(3):1574–1584.
69. Richt JA, García-Sastre A (2009) Attenuated influenza virus vaccines with modifiedNS1 proteins. Curr Top Microbiol Immunol 333:177–195.
70. Perlman S, Netland J (2009) Coronaviruses post-SARS: Update on replication andpathogenesis. Nat Rev Microbiol 7(6):439–450.
71. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolationof a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med367(19):1814–1820.
72. van Boheemen S, et al. (2012) Genomic characterization of a newly discoveredcoronavirus associated with acute respiratory distress syndrome in humans.mBio 3(6).
73. Clementz MA, et al. (2010) Deubiquitinating and interferon antagonism activities ofcoronavirus papain-like proteases. J Virol 84(9):4619–4629.
74. Wojdyla JA, et al. (2010) Papain-like protease 1 from transmissible gastroenteritisvirus: Crystal structure and enzymatic activity toward viral and cellular substrates.J Virol 84(19):10063–10073.
75. Ratia K, et al. (2006) Severe acute respiratory syndrome coronavirus papain-likeprotease: Structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA103(15):5717–5722.
76. Gohara DW, et al. (1999) Production of “authentic” poliovirus RNA-dependent RNApolymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli.Protein Expr Purif 17(1):128–138.
77. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: Programs forprotein crystallography. Acta Crystallogr D Biol Crystallogr 50(Pt 5):760–763.
78. Adams PD, et al. (2010) PHENIX: A comprehensive Python-based system formacromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66(Pt 2):213–221.
79. Brünger AT (1992) Free R value: A novel statistical quantity for assessing the accuracyof crystal structures. Nature 355(6359):472–475.
80. Emsley P, Cowtan K (2004) Coot: Model-building tools for molecular graphics. ActaCrystallogr D Biol Crystallogr 60(Pt 12 Pt 1):2126–2132.
81. van Dinten LC, den Boon JA, Wassenaar AL, Spaan WJ, Snijder EJ (1997) An infectiousarterivirus cDNA clone: Identification of a replicase point mutation that abolishesdiscontinuous mRNA transcription. Proc Natl Acad Sci USA 94(3):991–996.
82. Nedialkova DD, Gorbalenya AE, Snijder EJ (2010) Arterivirus Nsp1 modulates theaccumulation of minus-strand templates to control the relative abundance of viralmRNAs. PLoS Pathog 6(2):e1000772.
83. Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologistprogrammers. Bioinformatics Methods and Protocols: Methods in Molecular Biology,eds Krawetz S, Misener S (Humana Press, Totowa, NJ), pp 365–386.
84. DeLano WL (2002) The PyMOL Molecular Graphics System (DeLano Scientific, PaloAlto, CA).
van Kasteren et al. PNAS | Published online February 11, 2013 | E847
MICRO
BIOLO
GY
PNASPL
US