development and implementation of a new soa scheme in ...menut/pp/200806-carb-chimere.pdf · 1...
TRANSCRIPT

1
Development and implementation of a new SOA scheme in CHIMERE -Model evaluation against organic and elemental carbon observations
1Bertrand BESSAGNET, 2Laurent MENUT, 3Gabriele CURCI, 4Alma HODZIC, 5Bruno
GUILLAUME, 5Catherine LIOUSSE, 6Sophie MOUKHTAR, 7Betty PUN, 7Christian
SEIGNEUR, 8Michael SCHULZ
(1) INERIS, Institut National de l’Environnement Industriel et des Risques, Parc
technologique ALATA, 60550 Verneuil en Halatte, France.
(2) Institut P.-S. Laplace, Laboratoire de Meteorologie Dynamique, Ecole Polytechnique,
F-91128 Palaiseau, France.
(3) CETEMPS, Universita degli Studi dell’Aquila, via Vetoio, 67010 Coppito - L’Aquila,
Italy.
(4) NCAR, National Center for Atmospheric Research, 3450 Mitchell Lane, 80301, CO, USA.
(5) LA/OMP, Laboratoire d’Aerologie / Observatoire Midi-Pyrenees, 14, avenue Edouard
Belin, 31400 Toulouse, France.
(6) Centre for Atmospheric Chemistry - York University - 4700 Keele Street, Toronto, Canada.
(7) Atmospheric & Environmental Research, 2682 Bishop Drive, Suite 120, San Ramon, CA
94583, USA.
(8) Laboratoire des Sciences du Climat et de l’Environnement, IPSL/CEA-CNRS-UVSQ,
F-91190 Gif-sur-Yvette, France.
Short title: CARBONACEOUS SPECIES IN CHIMERE1

2
Abstract.2
The current state of the art of chemistry-transport modeling shows severe limitations for3
Secondary Organics Aerosols (SOA) concentrations estimation. This often implies difficulties to4
forecast or investigate polluted situations since important sources and their corresponding chemistry5
are not taken into account. In this study, a new and complete SOA chemistry scheme was implemented6
in the CHIMERE chemistry-transport model. Moreover, in order to improve the emission sources, the7
MEGAN biogenic emissions inventory was implemented. Hourly simulations were performed over the8
entire year of 2003 in Western Europe. The model results were clearly improved by implementing the9
new SOA scheme. In this study, a clear underestimation of OC concentrations was diagnosed during10
winter. A possible explanation could be missing (but observed) wood burning emissions in Portugal,11
Italy, Slovakia and Hungary as suggested by other modeling works. In addition, this work suggests that12
during the higher fire emission periods, fires can be the dominant source of primary organic carbon over13
the Mediterranean Basin. The contribution of SOA from fire emissions is low. This study highlights14
more precisely reasons of discrepancies between observations and modeling choices. We show that15
isoprene chemistry has a strong impact on SOA formation when using the current available kinetic16
schemes. Such results could explain large underestimates of OC concentrations in the southern Europe17
when this specific chemistry is not accounted for.18

3
1. Introduction19
Particulate matter (PM) pollution control is one of the main challenge highlighted20
by the Thematic Strategy on Air Pollution (CAFE as Clean Air For Europe), adopted by21
the European Commission in October 2005, under its 6th Environmental Action Program22
(Decision No 1600/2002/EC of the European Parliament and of the Council of July 22, 200223
laying down the Sixth Community Environment Action Programme). The CAFE strategy24
states that particulate matter (and especially fine particles with diameter smaller than 2.5 µm25
- PM2.5) is responsible today for an average reduction of life expectancy of about 8 months26
in Europe: recent epidemiological studies highlight the role of the smallest part of these27
particles on health [Schlesinger et al., 2006; Lee et al., 2007; Heinrich and Slama, 2007]. The28
fine particles are composed of a large fraction of organic (OC) and elemental carbon (EC)29
([Putaud et al., 2004]). According to [Van Dingenen et al., 2004], 70% to 80% of particles30
number consists mainly of carbonaceous material. This carbonaceous material is injected into31
the atmosphere by diffusion and mixing of surface emissions or by direct injection of biomass32
burning. They can also be formed by chemical reactions of Volatile Organic Compounds33
(VOC) in the atmosphere, the so called Secondary Organic Aerosols (SOA) (see [Kroll and34
Seinfeld, 2008] for a complete review of the SOA chemistry in gas and aqueous phases). This35
includes the large contribution of terpenes and isoprene ([Surratt et al., 2006]) emitted by36
the vegetation. While in winter biomass burning emissions are the main source with sizable37
additional contribution from fossil fuel combustion, the SOA originate mainly from non fossil38
sources in summer. Based on the CARBOSOL measurements, [Gelencser et al., 2007] showed39

4
that non fossil sources represent 63-76% of the Total Carbon (hereafter TC). [Pio et al.,40
2007] shows that 50-80% of OC is water soluble and suggest that OC has to be considered41
in discussing the role of clouds on climate over Europe. Humic-like substances (HULIS) are42
contributors of water soluble compounds (3-7%) and are directly emitted by biomass burning43
and probably formed by chemical reactions in the atmosphere ([Mayol-Bracero et al., 2002;44
Graber and Rudich, 2006; Lukacs et al., 2007; Schmidl et al., 2008]). A chemical mechanism45
in aqueous phase through the photo-oxidation of methylglyoxal is proposed by [Altieri et al.,46
2008] for HULIS formation. According [Legrand and Puxbaum, 2007] and references therein,47
it seems that most OC is contained in oligomeric or polymeric matter. OC emitted by the48
decomposition of vegetative debris is an other source of the organic coarse size fraction49
([Puxbaum and Tenze-Kunit, 2003]), [Bauer et al., 2002] found a contribution of 6% in a50
remote site.51
In the framework of the CARBOSOL campaign, and in a model context, the modeled52
EC and OC concentrations fields of the EMEP model were compared to measurements.53
OC is generally underpredicted in most of the sites certainly due to missing wood burning54
contributions. The model underpredicts TC in the southern Europe mainly due to a lack55
of SOA ([Simpson et al., 2007]). For EC, the largest uncertainties probably lie in EC56
emissions from residential wood/fossil combustion possibilly associated with both emission57
factors and spatial and temporal variation ([Tsyro et al., 2007]). During a specific modeling58
intercomparison exercize, a potential large underestimate of EC components has been recently59
reported in [Stern et al., 2008] in Eastern Germany, due to underestimated sources in Eastern60
Europe. Over the United-States, the role of isoprene in secondary organic aerosol formation61

5
([Dommen et al., 2006; Kroll et al., 2006]) has been extensively studied by [van Donkelaar62
et al., 2007] and [Zhang et al., 2007] that suggests a high sensitivity to the values of the63
enthalpy of vaporization used in models.64
The CHIMERE model was extensively validated on PM10, sulfate, nitrate and ammonium65
components in [Bessagnet et al., 2004, 2005; Vautard et al., 2005; Hodzic et al., 2005]. In this66
work, the SOA scheme implemented in CHIMERE is presented, a new emission inventory67
for biogenic VOC emissions has been implemented. A first evaluation of the CHIMERE68
model is proposed for carbonaceous species by using OC and EC observations from the69
CARBOSOL and EMEP available data for the year 2003. This year was characterized by70
huge fire emissions in the southern Europe taken into account in this study.71
2. The chemistry transport model CHIMERE72
2.1. General description73
Given a set of NOx, SOx, NH3, PM, VOC’s and CO emissions, the CHIMERE model74
calculates the concentrations of 44 gas-phase and aerosol species. In this study, a version75
of CHIMERE for a domain covering the western Europe is used [Schmidt et al., 2001;76
Bessagnet et al., 2004]: from 14oW to 28oE in longitude and from 35oN to 58.2oN in77
latitude, with a constant horizontal resolution of 0.4o× 0.4o. The vertical grid contains78
15 layers from surface to 500 hPa. The dynamics and gas-phase parts of the model are79
described in [Schmidt et al., 2001], and improvements have successively been brought80
([Vautard et al., 2003, 2005]). The model documentation can be found on the web server81

6
http://euler.lmd.polytechnique.fr/chimere. For both ozone and PM10, the model has undergone82
extensive modeled aerosols intercomparisons at European and city scales ([Vautard et al.,83
2007; Van Loon et al., 2007; Schaap et al., 2007]).84
The CHIMERE model is hourly driven by the meteorological model MM5 for the85
dynamical parameters (wind, temperature, humidity, pressure). In order to be consistent86
with the PREVAIR operational forecast system using MM5 and CHIMERE, the current87
MM5 configuration ([Dudhia, 1993]) was chosen to be the same than the one used and88
validated during the last three years for the daily forecast, [Honore et al., 2008]. The MM589
vertical grid contains 32 levels ranging from surface to 10hPa. The horizontal resolution is90
54km over a domain encompassing the european CHIMERE domain. The meteorological91
boundary conditions as well as the nudging is performed with the six-hourly ECMWF analysis92
meteorological fields.93
The aerosol module is that described in [Bessagnet et al., 2004]. Anthropogenic gas94
emissions are taken from the Co-operative Programme for Monitoring and Evaluation of the95
Long-range Transmission of Air pollutants in Europe - EMEP - ([Vestreng, 2003]). Three96
particulate model species are considered: PPM (primary particle material) that contains only97
mineral dust from anthropogenic sources (EMEP inventory) assumed to be coarse particles,98
primary OC and EC assumed to be in the fine mode. OC and EC emissions are from a specific99
inventory discussed in the next section. Calculation of model species emissions that can be100
used by the CHIMERE CTM, is made in several steps. The spatial emission distribution from101
the original grid to the CHIMERE grid is performed using an intermediate fine grid with a102
1km resolution (GLCF dataset, [Hansen et al., 2000]). Soil types being known on the fine103

7
grid allows for a better apportionment of the emissions according to urban, rural, maritime104
and continental areas. Standard time variation profiles are applied to get hourly emission105
from annual data, as required by the model. The other modeled species are sulfates, nitrates,106
ammonium, secondary organic aerosols, sea-salt (considered as inert here) and dust. The107
particle size distribution ranges from about 40 nm to 10 µm and are distributed into 8 bins.108
The 8 bins used are defined between the following intervals: 0.039, 0.078, 0.156, 0.312, 0.625,109
1.25, 2.5, 5, 10 µm. The gas - particle partitioning of the ensemble Sulfate/Nitrate/Ammonium110
is treated by the code ISORROPIA ([Nenes et al., 1998]) implemented in CHIMERE. For the111
main gas and aerosols, boundary conditions are issued from a 5 years climatology (2001-2005)112
of the global model LMDzT-INCA. For aerosol boundary conditions, only elemental and113
organic carbon, desert dust and sulfate are taken into account. The LMDzT-INCA model114
contains a multi-modal aerosol distribution ([Textor et al., 2006; Schulz et al., 2006]). Organic115
and elemental carbon are described as belonging to a soluble and insoluble accumulation116
mode, where ageing processes transfer constantly insoluble matter into the soluble aerosol117
mode. The emissions are described by [Dentener et al., 2006] and have been used for the118
AeroCom model intercomparison experiment B.119
2.2. OC and EC emissions over Europe120
2.2.1. Primary OC and EC Emission inventories over Europe A particular effort has121
been put on primary OC (OCp) and EC emission inventories emitted by fossil fuel and biofuel122
combustion (traffic, industry, residential sources), which are key elements in EC and OC123
modeling. These emissions, which are traditionally obtained by bottom-up approaches are still124

8
hampered by severe uncertainties resulting from large differences in the choice of emission125
factors. Two major different approaches for deriving global fossil fuel and biofuel EC and126
OCp emission inventories are currently available, [Junker and Liousse, 2008] and [Bond et al.,127
2004], the main difference being in technology differentiations. Thus, [Bond et al., 2004]128
consider for each fossil fuel a detailed list of combustion technologies and emission controls129
with associated emission factors (EF), while in [Junker and Liousse, 2008], for each fuel, two130
parameters characterize the emissions. First, the activity sector (domestic, industrial, traffic)131
and second, the national level of development (with three levels: developed, semi-developed132
and developing countries), based on gross national incomes taken as a proxy reflecting133
technology and emission control levels. In each of these methods, the part of uncontrolled134
emissions is differently handled which is the origin of their largest differences. Higher135
estimated emissions may be found for the major fuels (coal, diesel, peat, lignite, coke) in136
[Junker and Liousse, 2008] than in [Bond et al., 2004] with more controlled emissions in [Bond137
et al., 2004] than in [Junker and Liousse, 2008]. Harmonization between these two inventory138
types is in progress at a global scale. At the European scale, detailed inventories are given139
for EC and OCp emissions in [Schaap et al., 2004] (after [Bond et al., 2004] methodology140
and emission factors) and in [Guillaume et al., 2007, 2008]. A technology-splitting approach141
has been adopted with the same considerations of emission controls and details as in [Schaap142
et al., 2004] and in [Bond et al., 2004], while keeping EF values estimated from [Junker and143
Liousse, 2008]. With such a configuration, both newer combustor types and ”large emitters”144
that still exist in Europe, are taken into account. The IIASA fuel dataset ([Klimont et al.,145
2002]) are used, covering 35 European countries excluding Russia and Ukraine and including146

9
informations on controlled and uncontrolled fractions of fuel use which depend on emission147
control set up. Let us note that waste burning is not included. Emission factors (EF) for EC148
and OCp are obtained from [Junker and Liousse, 2008], using a proxy when EF values are not149
welknown. Variation of EC/total particulate matter ratio is the usual proxy used to derive150
unknown EF values. This proxy is based on relationships between EC/OCp and CO/CO2151
ratios. It is also interesting to note that an improved spatial distribution of EC and OCp152
emissions than in previous inventories is used with traffic and domestic emissions scaled on153
rural and urban population densities, but industrial emissions spatialized according to their real154
geographical positions and magnitudes. A comparison between this inventory and [Schaap155
et al., 2004] is proposed in [Guillaume et al., 2007]. In both inventories, EC from brown156
coal and hard coal fuels dominates while slowly replaced by diesel; wood relative importance157
is comparable. Fuel consumption is generally in agreement. Main differences occur in the158
relative importance of controlled and uncontrolled fractions of fuel use and in brown and159
hard coal emission factors selected for industrial and domestic sectors. Emission factors by160
[Schaap et al., 2004], based on [Bond et al., 2004] values are smaller than in [Guillaume161
et al., 2007]. Industrial emissions (especially power plant emissions) are more controlled in162
[Schaap et al., 2004] than in the inventory used in this work. These differences are particularly163
important for Poland emissions, much higher in this inventory than in [Schaap et al., 2004].164
Same differences exist for domestic emissions while traffic emissions are comparable. Finally,165
this European EC inventory is about 1.5 times higher than in [Schaap et al., 2004], while in166
agreement with the European zoom of [Junker and Liousse, 2008] inventory. Both inventories167
have been tested in ORISAM-TM4 global transport model [Guillaume et al., 2007, 2008].168

10
Following these results, EC and OCp European emission inventories for the year 2000 and169
built with a 25kmx25km resolution, have been selected in our study. These emissions are170
extrapolated to the CHIMERE resolution.171
2.2.2. Fire emissions In the past decade, wildfires have devastated vast areas of forest172
and agricultural lands across Europe. In 2003 alone, more than 650,000 hectares of forest area173
and about 45,000 ha of agricultural land have been destroyed in Europe, which released into174
the atmosphere large amount of smoke particles and trace gases (such as CO, CO2, VOC’s,175
NO, NO2, etc.). The VOC speciation of [EPA, 1993] was used. The 2003 wildfire emissions176
contributed significantly to the enhancement of carbonaceous aerosol concentrations and177
perturbations of the surface radiative balance. In order to accurately assess the effects of178
wildfires on the atmospheric chemistry and radiative budget, the amount of emitted species179
needs to be quantified. In this study, and in the same way than in [Hodzic et al., 2007],180
daily wildfires emissions of particulate matter and trace gases together with their geographic181
location were estimated based on satellite information including (i) the location and date of182
the fire event, (ii) the area burned, (iii) the fuel loading factors (mass of biomass per area),183
(iv) the fraction of biomass fuel burned, and (v) the emission factors. These parameters have184
been determined by combining data available from several satellite products according to the185
methodology described in [Wiedinmyer et al., 2006]. The VOC speciation of [EPA, 1993] was186
used. A detailed description of the emission dataset is given by [Hodzic et al., 2007].187

11
2.3. SOA modeling in CHIMERE188
The complete chemical scheme implemented in CHIMERE includes biogenic and189
anthropogenic precursors (Table 1). Biogenic precursors include API (α-pinene and190
sabinene), BPI (β-pinene and δ3-carene), LIM (limonene), TPO (myrcene and ocimene)191
and ISO (isoprene). Anthropogenic precursors include TOL (benzene, toluene and other192
mono-substituted aromatics), TMB (Trimethylbenzene and other poly-substituted aromatics),193
and NC4H10 (higher alkanes). SOA formation is represented according to a single-step194
oxidation of the relevant precursors and gas-particle partitioning of the condensable oxidation195
products. The gas-particle partitioning formulation has been described in detail by [Pun et al.,196
2006]. The overall approach consists in differentiating between hydrophilic SOA that are most197
likely to dissolve into aqueous inorganic particles and hydrophobic SOA that are most likely198
to absorb into organic particles. The dissolution of hydrophilic SOA is governed by Henry’s199
law whereas the absorption of hydrophobic particles is governed by Raoult’s law. The large200
number of condensable organic compounds is represented by a set of surrogate compounds201
that cover the range of physico-chemical properties relevant for aerosol formation, i.e., water202
solubility and acid dissociation for hydrophilic compounds and saturation vapor pressure for203
hydrophobic compounds. These surrogate compounds were selected by grouping identified204
particulate-phase molecular products with similar properties. The molecular weight of each205
surrogate compound is determined based on its structure and functional groups. The Henry’s206
law constant or the saturation vapor pressure of the surrogate species is derived from the207
average properties of the group. Other properties are estimated using the structure of each208

12
surrogate compound.209
The absorption process in CHIMERE is implemented as in [Bowman et al., 1997]. A210
dynamical approach is adopted to decribe the gas particle conversion.211
Ji =1
τi
(Gi −Geqi ) (1)
Ji (µg.m−3.s−1) is the absorption or desorption flux of species i, τi (s) is a characteristic212
time of the mass transfer that depends on particle size and the chemical properties of species i,213
Gi is the bulk gas-phase concentration of species i and Geqi is the gas-phase concentration of214
species i at thermodynamic equilibrium (i.e., at the surface of the particle). The equilibrium215
gas-phase concentrations are functions of the particle chemical composition, temperature and,216
for hydrophilic species, relative humidity, as described by [Pun et al., 2006].217
The base SOA module was tested against the smog chamber data of [Odum et al., 1997]218
for anthropogenic compounds and those of [Griffin et al., 1999] for biogenic compounds219
and was shown to satisfactorily reproduce SOA formation for those compounds [Pun et al.,220
2006]. Higher alkanes and isoprene were added to the original chemical mechanism of [Pun221
et al., 2006]. The formation of SOA from higher alkanes follows the formulation of [Zhang222
et al., 2007] for the stoichiometric SOA yield and it is assumed that the SOA species can be223
represented by a hydrophobic surrogate compound with a moderate saturation vapor pressure.224
The formation of SOA from the oxidation of isoprene by hydroxyl radicals is represented with225
two surrogate products and follows the formulation of [Kroll et al., 2006; Zhang et al., 2007].226Table 1.

13
2.4. Implementation of the MEGAN biogenic inventory227
The previous biogenic inventory used in CHIMERE was based on Simpson’s algorithms228
([Simpson et al., 1995; Moukhtar et al., 2005]). In CHIMERE, a strong and questionable229
assumption was the use of a unique forest composition for a given country. In order to230
bypass this problem, the Model of Emissions of Gases and Aerosols from Nature (MEGAN,231
[Guenther et al., 2006], v. 2.04) was implemented for this study in the CHIMERE model. It232
estimates emissions of Volatile Organic Compounds and NO from vegetation as :233
ERi = EFi × γi(T, PPFD, LAI)× ρi (2)
where ERi (µg.m−2.h−1) is the emissions rate of species i, EFi (µg.m−2.h−1) is an234
emission factor at canopy standard conditions, γi (unitless) is an emission activity factor that235
accounts for deviations from canopy standard conditions, and ρi is a factor that accounts236
for production/loss within canopy. The canopy standard conditions relevant for this study237
are defined as: air temperature (T) of 303 K, photosynthetic photon flux density (PPFD) of238
1500 µmol.m−2.s−1 at top of the canopy, leaf area index (LAI) of 5 m2.m−2 and a canopy239
with 80% mature, 10% growing and 10% old foliage. The MEGAN model parameterizes the240
bulk effect of changing environmental conditions using three time-dependent input variables241
specified at top of the canopy: temperature (T), radiation (PPFD), and foliage density (LAI).242
The production/loss term within canopy is assumed to be unity (ρ = 1). The equation can then243
be expanded as :244

14
ERi = EFi × γT,i × γPPFD × γLAI (3)
The MEGAN model provides input EF and LAI data over a global grid, herefater245
projected on the CHIMERE model grid. The current available choice for EF’s is restricted246
to following species: isoprene, α-pinene, β-pinene, myrcene, sabinene, limonene, δ3-carene,247
ocimene, and nitrogen oxide. EF’s are static and refer to years 2000-2001. They are obtained248
summing up over several plant functional types (e.g. broadleaf and needle trees, shrubs, etc...).249
LAI database is given as a monthly mean product derived from MODIS observations, referred250
to base year 2000. Hourly emissions are calculated using 2-m temperature and short-wave251
radiation from MM5 model. The optimal choice for this work is the 150 seconds resolution252
(≈5 Km) products proposed in the MEGAN inventory.253Figure 1.
Figure 1 shows the differences for July 2003 between the new inventory MEGAN254
implemented in CHIMERE and the former approach based on [Simpson et al., 1995]. Large255
differences everywhere in Europe are observed for Terpene emissions with lower emissions256
using the MEGAN algorithms. Isoprene emissions are higher with MEGAN in Poland, Spain,257
Italy and Portugal and lower in Greece, United-Kingdom and North Africa.258
3. Comparisons between model and observational data259
3.1. Observational data260
Two databases of measurements are used in this study:261
1. The CARBOSOL data as described in [Pio et al., 2007; Legrand and Puxbaum, 2007]).262

15
EC, OC and diacids C2 to C5 (glutaric, malic, succinic, oxalic, malonic and tartaric263
acids) chemical analyses are available on a weekly basis. These low molecular weight264
dicarboxylic acids are mainly formed in the atmosphere by oxidation of VOC’s, but they265
also have a primary origin ([Limbeck and Puxbaum, 1999; Plewka et al., 2006; Kleefeld266
et al., 2002; Kawamura and Yasui, 2005]). Analyses are carried out for particles smaller267
than 2.5 µm.268
2. The EMEP data issued from the EMEP 2002-2003 campaign ([Yttri et al., 2007]). A269
daily sample per week with only EC and OC analyses is available. Analyses were270
performed using the thermal-optical transmission (TOT) instrument from Sunset Lab271
Inc., operating according to a NIOSH derived temperature program, more details can be272
found in [Yttri et al., 2007]. The routine measurements at the EMEP sites provide daily273
average PM10 concentrations (particle diameter smaller than 10 µm)274
The coordinates and locations in Europe of all stations used in this study are described in275
Table 2 and Figure 2. For the comparisons to modeled concentrations, values are taken in276
the first model layer. This may induce a problem when the topography varies a lot or in277
mountainous regions: the measurements are not really representative of the entire modeled278
cell. This is the case for the Puy de Dome (PDD) and Schauinsland (SIL) stations that are279
the most elevated sites. In order to convert the Organic Material (OM) concentrations to the280
modeled Organic Carbon (OC), the relation proposed by [Turpin and Lim, 2001] was used281
with OM = 1.6×OC.282 Figure 2.
Table 2.

16
3.2. OC comparisons283
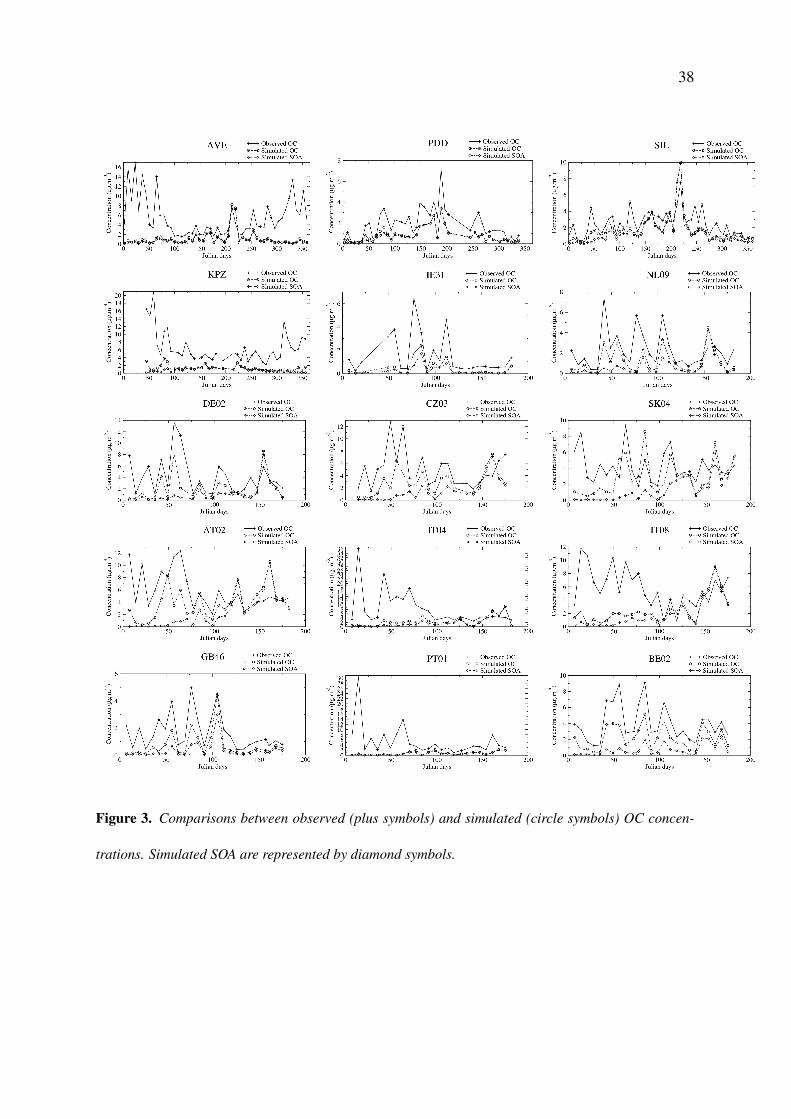
In Figure 3 and Table 3, the comparison of CHIMERE with OC measurements shows a284
systematic underestimation for each site. Temporal correlations for PDD, SIL, BE02, GB46285
and IE31 sites are in the range 0.75 - 0.91. For IT04, IT08, AVE, PT01, SK04 and KPZ286
sites a clear underestimate by the CHIMERE model is observed in wintertime. The same287
underestimate by the EMEP model highlighted by [Simpson et al., 2007] was attributed to288
missing wood burning sources in these countries. In summertime, SOA largely dominates289
the organic fraction in simulated results, in the range 90-95% for the two elevated sites PDD290
(France) and SIL (Germany). These findings are in line with [Gelencser et al., 2007] who291
find that at least 86% of OC could be attributed to SOA. CHIMERE gives surprisingly high292
SOA contributions to total OC in winter for PDD, 71% and SIL, 52% for January-March and293
October-December periods. This is consistent with [Gelencser et al., 2007] findings that show294
a high fraction of biogenic secondary organic carbon in winter, 72% at SIL and 69% at PDD295
for the same periods.296Figure 3.
Table 3. The yearly averaged SOA/OM ratio simulated by CHIMERE are mostly in the range297
30-80 % in Europe (Figure 4). The highest SOA/OM ratio are found over the Pyrenees and298
Massif Central in France and in Spain. Poland displays lower SOA contribution in Europe299
because of higher primary organic emissions from industries in that country as previously300
discussed. In the southern Europe, secondary organic aerosols produced by isoprene chemistry301
dominates the total SOA formation (Figure 5). [Henze and Seinfeld, 2006] found a large302
increase of the SOA global budget by a factor of two by accounting for isoprene chemistry in303

17
SOA formation.304 Figure 4.
Figure 5.The evaluation of a SOA scheme is actually impossible because total SOA measurements305
do not exist yet in Europe. However, we can evaluate the impact of adding a SOA scheme on306
PM10 simulations. Some EMEP background sites in Europe have been selected.307 Table 4.
As shown in Table 4, the implementation of the SOA scheme improves the temporal308
correlation at many background sites. Large improvements are observed in Spain and Slovenia309
where biogenic VOC precursor emissions are very large. In Italy, in IT04 (Ispra) temporal310
correlations are impaired, but largely improved in IT01 (Montelibretti). In the forest region311
where the station DE08 is located in Germany, a large improvement is also observed from312
0.58 to 0.70. In Spain, a clear improvement is obtained by adding the isoprene chemistry for313
SOA formation. In Slovakia the temporal correlations are not globally improved. The urban314
site of Perigueux (PERI), a small city on the western part of the Massif Central, is located in a315
strong biogenic precursors emission area in France. In this region the CHIMERE simulation316
exhibits a high contribution of secondary organic species (up to 80%) on the total organic317
carbon as shown in Figure 4. The temporal correlation is largely improved at this site when318
implementing the SOA scheme from 0.39 to 0.51. Considering only the summer period from319
May 1st to September 30, the correlation is improved from 0.24 to 0.63 at Perigueux.320 Table 5.
For the sites AVE, PDD and SIL, the temporal correlation between observed diacid321
(C2-C5) concentrations and modeled SOA is fairly good (Table 5). Such a result could explain322
the good agreement between OC measurements and the simulated OC for PDD and SIL as323
reported in Table 3. For AVE, a good correlation is observed between diacids measurements324
and model SOA and a poor correlation between OC measurements and simulated OC, that325

18
confirms a problem with primary organic carbon in the model certainly due to missing326
emissions.327
3.3. EC comparisons328
Figure 6 and Table 6 display the comparison of EC model results against observations at329
each site. As for OC concentrations, the model underpredict EC concentrations at AVE, PT01,330
IT01, IT04 and KPZ particularly in winter. The assumption made by [Simpson et al., 2007]331
about missing wood burning sources in the model could again explain this behavior. The332
model reproduces well the temporal evolution of EC concentrations at BE02, NL09, IE31 and333
GB46, these sites are very close to well documented sources and are located over flat areas.334
For Mace Head (IE31), EC concentrations given by the model are globally underestimated335
by a factor of two: 0.24 µg.m−3 vs. 0.13 µg.m−3 respectively for the observations and the336
model. The low temporal correlation at the two elevated sites (SIL and PDD) suggest that337
these sites could be influenced by long range transport of anthropogenic pollution. By using338
monthly climatologies, the model is not able to capture daily intercontinental fluxes that can339
influence EC concentrations at elevated sites. That could be the reason why correlations340
for OC concentrations are higher than for EC concentrations at elevated sites because OC341
has a more local secondary origin (from the oxidation of VOC precursors) than EC. EC342
concentrations at remote places are affected by long range transport from anthropic emission343
areas ([Kasper and Puxbaum, 1998; Hitzenberger et al., 1999; Guillaume et al., 2008; Kaiser344
et al., 2007; Jaffe et al., 2003]) and discrepancies in meteorological calculation can largely345
impair model concentration results.346Figure 6.
Table 6.

19
3.4. Potential impact of forest fires347
Fire emissions can be an important contributor to OC concentrations as shown by348
[Langmann et al., 2008]. However, wild fire emissions do not contribute much to the349
atmospheric EC concentrations ([Tsyro et al., 2007]) on a yearly average. The impact of350
fire emissions depends on fire buoyancy and in models, sensitivity to fires depends on the351
altitude of their release on the vertical grid ([Hodzic et al., 2006]). Figure 7 shows the impact352
of adding forest fire respectively on primary and secondary organic material estimated by353
CHIMERE on August 1st - 15, 2003. During this period, intense fires were recorded around354
the Mediterranean basin and in Portugal. CHIMERE estimates give a large contribution of355
these fires to the primary organic concentrations in the South West of Europe with more than356
90 % in Portugal and often more than 50 % over the Mediterranean Sea. Moreover, fires357
emit volatile organic precursors that can be oxidized and form SOA. In Figure 7[bottom], the358
model gives a limited impact of secondary organic material originating from biomass burning359
related VOC’s, less than 10 % during the more intense fire period of the year 2003. The360
modeling results provide insight into a key question in [Gelencser et al., 2007] concerning361
the contribution of secondary organic carbon from VOC emitted by vegetation and biomass362
burning, because analysis methods cannot separate the two contributions.363 Figure 7.
4. Sources of uncertainties in SOA modeling364
From the measurements to the modeled scheme definition, the modeling of SOA includes365
a lot of uncertainties. Recently, the gap between laboratory studies (mandatory to develop366

20
more realistic chemical schemes) and realistic schemes in models was highlighted by [Pun367
and Seigneur, 2007]. One important weakness was identified to be the biogenic precursor368
emissions estimation. A first step towards improving our SOA knowledge is improvements to369
the biogenic emission inventories [Simpson et al., 2007]. Next steps concern more directly the370
representation of atmospheric processes by modeling schemes, several gaps are identified and371
may be described as follows, among others:372
1. Photochemistry of Semi-volatile organic species373
Recent studies pointed out the possible role of semi-volatile organic precursors in374
SOA formation ([Robinson et al., 2007; Donahue et al., 2006; Schauer et al., 2002]).375
These precursors evaporate during the emission dilution process and could be converted376
into the particulate phase after oxidation. The work of [Shrivastava et al., 2006] also377
suggests that aerosol emission factors could be underestimated since they are calculated378
at given temperature and dilution ratio so that a non negligible fraction could be not379
taken into account in current gas and particle inventories.380
2. Gas to particle conversion381
The exact physical and chemical pathways to secondary organic aerosol for most parent382
hydrocarbons are still uncertain and during the last ten years the condensation/sorption383
process described in [Bowman et al., 1997] has been adopted in models. However,384
nucleation burst was observed in rather clean environments such as boreal forest385
([Kavouras et al., 1998]) and in urban areas assuming co-nucleation effects with sulfuric386
acid ([Fan et al., 2006]). If in urban area absorption certainly dominates the issue is387

21
still open over forested and remote areas ([Svendby et al., 2008; Wexler and Johnston,388
2008; Bonn and Moortgat, 2002; Holmes, 2007]). [Kerminen et al., 1999] showed389
in clean environment (Arctic) a high amount of dicarboxylic acids such as glutaric,390
malonic, succinic and glutaric acid, in the coarse particles. These acids were observed391
in coarse urban and suburban aerosols corroborating the possible condensation/sorption392
of semi-volatile species onto pre-existing coarse particles in summer ([Jaffrezo et al.,393
2005]) or the possible role of in-cloud processes ([Hsieh et al., 2007; Oliveira et al.,394
2007]). Other missing processes could be mentioned such as oligomerization, oxidation395
of semi-volatile primary emissions, high/low NOx regimes.396
3. Deposition397
Particle formation by nucleation or condensation of semi- and non volatile compounds398
onto pre-existing particles involve different behaviors due to coagulation, growth and399
deposition processes. The way of modeling gas to particle conversion will have a strong400
impact on particle concentrations and compositions. Due to very high kinetic rates,401
secondary organic aerosols are rapidly produced over forested area and as reported402
by [Noll and Aluko, 2006; Petroff et al., 2008a, b] particle deposition over vegetation403
canopies is still uncertain and could be a source of model discrepancies. Moreover,404
secondary organic aerosols are partitioned between the gas phase and particles, in this405
work deposition velocities of semi-volatile organic species in the gas phase are set to406
zero. Accounting for a non zero velocity deposition for these species could largely407
affect the results.408

22
5. Conclusion409
In order to improve atmospheric composition modeling, an improved Secondary Organic410
Aerosols (SOA) scheme was implemented in the CHIMERE chemistry-transport model. This411
addition was done in parallel with the implementation of the MEGAN biogenic emissions412
inventory. Accounting for these processes allowed a clear improvement of the model results413
over the whole year of 2003 (in average and in comparison to PM10 surface data with an414
increase of 5 to 10% of temporal correlation).415
The insertion of more detailed schemes is a natural source of more model variability.416
That reveals problems not known before. In this study, a clear underestimation of OC417
concentrations was diagnosed during winter. A possible explanation is that wood burning418
emissions in Portugal, Italy, Slovakia and Hungary are missing in the model emission419
inventory as suggested by a recent modeling work ([Simpson et al., 2007]) carried out with420
different model and input data (meteorology and emissions). In addition, this work suggests421
that during the higher fire emission periods, OC concentrations from fires can be the major422
part of primary organic carbon. The contribution of SOA from fire emissions is low.423
Surprisingly, good correlations are observed between model and measurements for424
elevated sites such as Puy de Dome (France) and Schauinsland (Germany). In this specific425
case, the contribution of SOA to the total OC even in winter is quite high, showing that adding426
SOA is beneficial towards model performance.427
Finally, this work suggests that isoprene chemistry has a strong contribution to SOA428
concentrations and could explain large underestimate of OC concentrations in the southern429

23
Europe when this specific chemistry is not accounted for. The formulation of VOC oxidation430
to SOA remains too simple in the model and needs to be improved by adding aqueous and431
heterogeneous pathways, as well as taking into account the multi-step oxidation processes and432
their dependence on the NOx regime.433
Acknowledgments. This work was funded by the French Ministry in charge of Ecology434
(MEEDDAT). We thank David Simpson (EMEP, Norway) for providing us the CARBOSOL data,435
CARBOSOL was financed by European Commission (EVK2-2001-113). Yann Martinet (CITEPA,436
France) is aknowledged for his contribution in emission speciation. We also thank Alex Guenther437
(NCAR, USA) for providing us the MEGAN biogenic inventory. G. Curci has been supported by438
EU/FP6 CIRCE project.439

24
References440
Altieri, K. E., S. P. Seitzinger, A. G. Carlton, B. J. Turpin, G. C. Klein, and A. G. Marshall441
(2008), Oligomers formed through in-cloud methylglyoxal reactions: Chemical composition,442
properties, and mechanisms investigated by ultra-high resolution FT-ICR mass spectrometry,443
Atmos. Environ., 42, 1476–1490.444
Bauer, H., A. Kasper-Giebl, M. Loflund, H. Giebl, R. Hitzenberger, F. Zibuschka, and H. Puxbaum445
(2002), The contribution of bacteria and fungal spores to the organic carbon content of cloud446
water, precipitation and aerosols, Atmospheric Research, 64, 109–119.447
Bessagnet, B., A. Hodzic, R. Vautard, M. Beekmann, S. Cheinet, C. Honore, C. Liousse, and L. Rouıl448
(2004), Aerosol modeling with CHIMERE: preliminary evaluation at the continental scale,449
Atmos. Environ., 38, 2803–2817.450
Bessagnet, B., A. Hodzic, O. Blanchard, M. Lattuati, O. Le Bihan, H. Marfaing, and L. Rouıl451
(2005), Origin of particulate matter pollution episodes in wintertime over the Paris Basin,452
Atmos. Environ., 39, 6159–6174.453
Bond, T., D. Streets, K. Yarber, S. Nelson, J.-H. Woo, and Z. Klimont (2004), A technology-based454
global inventory of black and organic carbon emissions from combustion., Journal of455
Geophysical Research, 109, D14,203, doi:10.1029/2003JD003697.456
Bonn, B., and G. K. Moortgat (2002), New particle formation during α and β-pinene oxidation by457
O3, OH and NO3, and the influence of water vapour: particle size distribution studies, Atmos.458
Chem. Phys., 2, 183–196.459
Bowman, F. m., J. R. Odum, J. H. Seinfeld, and S. N. Pandis (1997), Mathematical model for460
gas-particle parttioning of secondary organic aerosols, Atmos. Environ., 31, 3921–3931.461

25
Dentener, F., et al. (2006), Emissions of primary aerosol and precursor gases in the years 2000 and462
1750 prescribed data-sets for AeroCom, Atmos. Chem. Phys., 6, 4321–4344.463
Dommen, J., et al. (2006), Laboratory observation of oligomers in the aerosol from isoprene/NOx464
photooxidation, Geophysical Research Letters, 33, doi:10.1029/2006GL026523.465
Donahue, N. M., A. L. Robinson, C. O. Stanier, and S. N. Pandis (2006), Coupled partitioning,466
dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643,467
doi:10.1021/es052297.468
Dudhia, J. (1993), A nonhydrostatic version of the Penn State/NCAR mesoscale model: validation469
tests and simulation of an Atlantic cyclone and cold front., Monthly Weather Review, 121,470
1493–1513.471
EPA (1993), Volatile Organic Compounds (VOC)/Particulate Matter (PM) Speciation Data System472
(SPECIATE), Version 1.5, Emission Inventory Branch (MD-14), Tech. rep., Office of Air473
Quality Planning and Standards, U.S. Environmental Protection Agency, Research Triangle474
Park, NC.475
Fan, J., R. Zhang, D. Collins, and G. Li (2006), Contribution of secondary condensable organics to476
new particle formation : A case study in Houston, Texas, Geophysical Research Letters, 33,477
doi:10.1029/2006GL026295.478
Gelencser, A., B. May, D. Simpson, A. Sanchez-Ochoa, A. Kasper-Giebl, H. Puxbaum, A. Caseiro,479
C. Pio, and M. Legrand (2007), Source apportionment of PM2.5 organic aerosol over480
Europe:primary/secondary, natural/anthropogenic, and fossil/biogenic origin, J. Geophys. Res.,481
112, doi:10.1029/2006JD008094.482

26
Graber, E. R., and Y. Rudich (2006), Atmospheric HULIS: How humic-like are they? A comprehensive483
and critical review, Atmos. Chem. Phys., 6, 729–753.484
Griffin, R. J., E. R. Cocker, R. C. Flagan, and J. H. Seinfeld (1999), Organic aerosol formation from the485
oxidation of biogenic hydrocarbons, J. Geophys. Res., 104, 3555–3567.486
Guenther, A., T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer, and C. Geron (2006), Estimates of global487
terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from488
Nature), Atmos. Chem. Phys., 6, 3181–3210.489
Guillaume, B., C. Liousse, R. Rosset, H. Cachier, B. Bessagnet, P. V. Velthoven, and N. Poisson (2007),490
Global modelling of internally mixed size-resolved inorganic/organic aerosols with a focus on491
carbonaceous components, Tellus B, 9, 283–302.492
Guillaume, B., C. Liousse, C. Galy-Lacaux, R. Rosset, E. Gardrat, H. Cachier, B. Bessagnet, and493
N. Poisson (2008), Modeling exceptional high concentrations of carbonaceous aerosols494
observed at Pic du Midi in spring-summer 2003:Comparison with Sonnblick and Puy de Dome,495
Atmos. Environ., in press.496
Hansen, M., R. DeFries, J. Townshend, and R. Sohlberg (2000), Global land cover classification at497
1km resolution using a decision tree classifier, International Journal of Remote Sensing, 21,498
1331–1365.499
Heinrich, J., and R. Slama (2007), Fine particles, a major threat to children, International Journal of500
Hygiene and Environmental Health, 210, 617–622.501
Henze, D. K., and J. H. Seinfeld (2006), Global secondary organic aerosol from isoprene oxidation,502
Geophysical Research Letters, 33, doi:10.1029/2006GL025976.503
Hitzenberger, R., A. Berner, H. Giebl, R. Kromp, S. Larson, A. Rouc, K. A., S. Marischka, and504

27
H. Puxbaum (1999), Contribution of carbonaceous material to cloud condensation nuclei505
concentrations in European background (Mt. Sonnblick) and urban (Vienna) aerosols, Atmos.506
Environ., 33, 2647–2659.507
Hodzic, A., R. Vautard, B. Bessagnet, M. Lattuati, and F. Moreto (2005), Long-term urban aerosol508
simulation versus routine particulate matter observations, Atmos. Environ., 39, 5851–5864.509
Hodzic, A., R. Vautard, H. Chepfer, P. Goloub, L. Menut, P. Chazette, J. Deuze, A. Apituley, and510
P. Couvert (2006), Evolution of aerosol optical thickness over Europe during the August511
2003 heat wave as seen from CHIMERE model simulations and POLDER data, Atmospheric512
Chemistry and Physics, 6, 1853–1864.513
Hodzic, A., S. Madronich, B. Bohn, S. Massie, L. Menut, and C. Wiedinmyer (2007), Wildfire514
particulate matter in Europe during summer 2003: Meso-scale modeling of smoke emissions,515
transport and radiative effects, Atmospheric Chemistry and Physics, 7, 4043–4064.516
Holmes, N. S. (2007), A review of particle formation events and growth in the atmosphere in the various517
environments and discussion of mechanistic implications, Atmos. Environ., 41, 2183–2201.518
Honore, C., et al. (2008), Predictability of European air quality: The assessment of three years519
of operational forecasts and analyses by the PREV’AIR system, J. Geophys. Res., 113,520
doi:10.1029/2007JD008761.521
Hsieh, L.-Y., S.-C. Kuo, C.-L. Chen, and Y. I. Tsai (2007), Origin of low-molecular-weight522
dicarboxylic acids and their concentration and size distribution variation in suburban aerosol,523
Atmos. Environ., 41, 6648–6661.524
Jaffe, D., I. McKendry, T. Anderson, and H. Price (2003), Six new episodes of trans-Pacific transport525
of air pollutants, Atmos. Environ., 37, 391–404.526

28
Jaffrezo, J.-L., G. Aymoz, and J. Cozic (2005), Size distribution of EC and OC in the aerosol of Alpine527
valleys during summer and winter, Atmos. Chem. Phys., 5, 2915–2925.528
Junker, C., and C. Liousse (2008), A global emission inventory of carbonaceous aerosol from historic529
records of fossil fuel and biofuel consumption for the period 1860-1997, Atmos. Chem. Phys.,530
8, 1195–1207.531
Kaiser, A., H. Scheifinger, W. Spangl, A. Weiss, S. Gilge, W. Fricke, L. Ries, D. Cemas, and532
B. Jesenovec (2007), Transport of nitrogen oxides, carbon monoxide and ozone to the533
Alpine Global Atmosphere Watch stations Jungfraujoch (Switzerland), Zugspitze and534
Hohenpeissenberg (Germany), Sonnblick (Austria) and Mt. Krvavec (Slovenia), Atmos.535
Environ., 41, 9273–9287.536
Kasper, A., and H. Puxbaum (1998), Seasonal variation of SO2, HNO3, NH3 and selected aerosol537
components at Sonnblick (3106 m a.s.l.), Atmos. Environ., 32(23), 3925–3939.538
Kavouras, I., N. Mihalopoulos, and E. Stephanou (1998), Formation of atmospheric particles from539
organic acids produced by forests, Nature, 395, 683–686.540
Kawamura, K., and O. Yasui (2005), Diurnal changes in the distribution of dicarboxylic acids,541
ketocarboxylic acids and dicarbonyls in the urban Tokyo atmosphere, Atmos. Environ., 39,542
1945–1960.543
Kerminen, V.-M., K. Teinila, R. Hillamo, and T. Makela (1999), Size-segregated chemistry of544
particulate dicarboxylic acids in the Arctic atmosphere, Atmos. Environ., 33, 2089–2100.545
Kleefeld, S., A. Hoffer, Z. Krivacsy, and S. G. Jennings (2002), Importance of organic and blackcarbon546
in atmospheric aerosols at Mace Head, on the West Coast of ireland (53’19’N, 9’54’W), Atmos.547
Environ., 36, 4479–4490.548

29
Klimont, Z., J. Cofala, I. Bertok, M. Amann, C. Heyes, and F. Gyarfas (2002), Estimating costs for549
controlling emissions of volatile organic compounds (VOC) from stationary sources in Europe.,550
Tech. rep., IIASA Interim Report IR-02-76., Laxenburg, Austria.551
Kroll, J. H., and J. H. Seinfeld (2008), Review : Chemistry of secondary organic aerosol: Formation552
and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624.553
Kroll, J. H., N. L. Ng, S. M. Murphy, R. C. Flagan, and J. H. Seinfeld (2006), Secondary organic554
aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877,555
doi:10.1021/es0524301.556
Langmann, B., S. Varghese, E. Marmer, E. Vignati, J. Wilson, P. Stier, and C. O’Dowd (2008), Aerosol557
distribution over Europe: a model evaluation study with detailed aerosol microphysics, Atmos.558
Chem. Phys., 8, 1591–1607.559
Lee, J.-T., J.-Y. Son, and C. Y.-S. (2007), The adverse effects of fine particle air pollution on respiratory560
function in the elderly, The Science of the Total Environment, 385, 28–36.561
Legrand, M., and H. Puxbaum (2007), Summary of the CARBOSOL project: Present and562
retrospective state of organic versus inorganic aerosol over Europe, J. Geophys. Res., 112,563
doi:10.1029/2006JD008271.564
Limbeck, A., and H. Puxbaum (1999), Organic acids in continental background aerosols, Atmos.565
Environ., 33, 1847–1852.566
Lukacs, H., A. Gelencser, H. Puxbaum, C. Pio, M. Legrand, A. Caseiro, A. Limbeck, and S. Preunkert567
(2007), Seasonal trends and possible sources of brown carbon based on two-years aerosol568
measurements at six sites in Europe, J. Geophys. Res., doi:10.1029/2006JD008151.569
Mayol-Bracero, O. L., P. Guyon, B. Graham, G. Roberts, M. O. Andreae, S. Decesari, M. C. Facchini,570

30
S. Fuzzi, and P. Artaxo (2002), Water-soluble organic compounds in biomass burning aerosols571
over amazonia: 2. apportionment of the chemical composition and importance of the polyacidic572
fraction, J. Geophys. Res., 107, doi:10.1029/2001JD000522.573
Moukhtar, S., B. Bessagnet, L. Rouıl, and V. Simon (2005), Monoterpene emissions from beech (fagus574
sylvatica) in a french forest and impact on secondary pollutants formation at regional scale,575
Atmos. Environ., 39, 3535–3547.576
Nenes, A., C. Pilinis, and S. N. Pandis (1998), ISORROPIA : a new thermodynamic equilibrium model577
for multiphase multicomponent marine aerosols, Aquatic Geochemistry, 4, 123–152.578
Noll, K. E., and O. Aluko (2006), Changes in large particle size distribution due to dry deposition579
processes, Journal of Aerosol Science, 37, 1797–1808.580
Odum, J. R., T. P. W. Jungkamp, R. J. Griffin, J. L. Forstner, R. C. Flagan, and J. H. Seinfeld (1997),581
Aromatics, reformulated gasoline and atmospheric organic aerosol formation, Environ. Sci.582
Technol., 31, 1890–1897.583
Oliveira, C., et al. (2007), Seasonal distribution of polar organic compounds in the urban atmosphere of584
two large cities from the north and south of Europe, Atmos. Environ., 41, 5555–5570.585
Petroff, A., A. Mailliat, M. Amielh, and F. Anselmet (2008a), Aerosol dry deposition on vegetative586
canopies. Part I: Review of present knowledge, Atmos. Environ., 42, 3625–3653.587
Petroff, A., A. Mailliat, M. Amielh, and F. Anselmet (2008b), Aerosol dry deposition on vegetative588
canopies. Part II: A new modelling approach and applications, Atmos. Environ., 42, 3654–3683.589
Pio, C. A., et al. (2007), Climatology of aerosol composition (organic versus inorganic) at nonurban590
sites on a west-east transect across Europe, J. Geophys. Res., 112, doi:10.1029/2006JD008038.591
Plewka, A., T. Gnauk, E. Bruggemann, and H. Herrmann (2006), Biogenic contributions to the592

31
chemical composition of airborne particles in a coniferous forest in Germany, Atmos. Environ.,593
40, S103–S115.594
Pun, B., and C. Seigneur (2007), Investigative modeling of new pathways for secondary organic aerosol595
formation, Atmos. Chem. Phys., 7, 2199–2216.596
Pun, B., C. Seigneur, and K. Lohman (2006), Modeling secondary organic aerosol via multiphase597
partitioning with molecular data, Environ. Sci. Technol., 40, 4722–4731.598
Putaud, J.-P., et al. (2004), A European aerosol phenomenology–2: chemical characteristics of599
particulate matter at kerbside, urban, rural and background sites in Europe, Atmos. Environ., 38,600
2579–2595.601
Puxbaum, H., and M. Tenze-Kunit (2003), Size distribution and seasonal variation of atmospheric602
cellulose, Atmos. Environ., 37, 3693–3699.603
Robinson, A. L., N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop,604
T. E. Lane, J. R. Pierce, and S. N. Pandis (2007), Rethinking organic aerosols: Semivolatile605
emissions and photochemical aging, Science, 315, doi:10.1126/science.1133061.606
Schaap, M., et al. (2004), Anthropogenic black carbon and fine aerosol distribution over europe, J.607
Geophys. Res., 109, D18,207, doi:10.1029/2003JD004330.608
Schaap, M., et al. (2007), Evaluation of long-term aerosol simulations from seven air quality models609
and their ensemble in the eurodelta study, Atmos. Environ., p. submitted.610
Schauer, J. J., M. J. Kleeman, G. R. Cass, and B. R. T. Simoneit (2002), Measurement of emissions611
from air pollution sources. 5. C1-C32 organic compounds from gasoline-powered motor612
vehicles, Environ. Sci. Technol., 36, 1169–1180.613
Schlesinger, R. B., N. Kunzli, G. M. Hidy, T. Gotschi, and M. Jerrett (2006), The health relevance614

32
of ambient particulate matter characteristics: coherence of toxicological and epidemiological615
inferences, Inhal. Toxicol., 18, 96–125.616
Schmidl, C., I. L. Marr, A. Caseiro, P. Kotianova, A. Berner, H. Bauer, A. Kasper-Giebl, and617
H. Puxbaum (2008), Chemical characterisation of fine particle emissions from wood stove618
combustion of common woods growing in mid-European Alpine regions, Atmos. Environ., 42,619
126–141.620
Schmidt, H., C. Derognat, R. Vautard, and M. Beekmann (2001), A comparison of simulated and621
observed ozone mixing ratios for the summer of 1998 in western Europe, Atmos. Environ., 35,622
6277–6297.623
Schulz, M., et al. (2006), Radiative forcing by aerosols as derived from the AeroCom present-day and624
pre-industrial simulations, Atmos. Chem. Phys., 6, 5225–5246.625
Shrivastava, M. K., E. M. Lipsky, C. O. Stanier, and A. L. Robinson (2006), Modeling semivolatile626
organic aerosol mass emissions from combustion systems, Environ. Sci. Technol., 40,627
2671–2677, doi:10.1021/es0522231.628
Simpson, D., A. Guenther, C. N. Hewitt, and R. Steinbrecher (1995), Biogenic emissions in Europe. 1.629
estimates and uncertainties, J. Geophys. Res., 100D, 22,875–22,890.630
Simpson, D., K. E. Yttri, Z. Klimont, K. Kupiainen, A. Caseiro, A. Gelencser, C. Pio, H. Puxbaum, and631
M. Legrand (2007), Modeling carbonaceous aerosol over Europe: Analysis of the CARBOSOL632
and EMEP EC/OC campaigns, J. Geophys. Res., 112, doi:10.1029/2006JD008158.633
Stern, R., et al. (2008), A model inter-comparison study focussing on episodes with elevated PM10634
concentrations, Atmos. Environ., doi:10.1016/j.atmosenv.2008.01.068.635

33
Surratt, J. D., et al. (2006), Chemical composition of secondary organic aerosol formed from the636
photooxidation of isoprene, J. Phys. Chem., 110, 9665–9690.637
Svendby, T. M., M. Lazaridis, and K. Tørseth (2008), Temperature dependent secondary organic aerosol638
formation from terpenes and aromatics, J. Atmos. Chem., 59, 25–46, doi:10.1007/s10874-007-639
9093-7.640
Textor, C., et al. (2006), Analysis and quantification of the diversities of aerosol life cycles within641
AeroCom, Atmos. Chem. Phys., 6, 1777–1813.642
Tsyro, S., D. Simpson, L. Tarrason, Z. Klimont, K. Kupiainen, C. Pio, and K. E. Yttri (2007), Modeling643
of elemental carbon over Europe, J. Geophys. Res., 112, doi:10.1029/2006JD008164.644
Turpin, B. J., and H.-J. Lim (2001), Species contributions to PM2.5 mass concentrations : Revisiting645
common assumptions for estimating organic mass, Aerosol Sci. Tech., 35, 602–610.646
Van Dingenen, R., et al. (2004), A European aerosol phenomenology–1: physical characteristics of647
particulate matter at kerbside, urban, rural and background sites in Europe, Atmos. Environ., 38,648
2561–2577.649
van Donkelaar, A., R. V. Martin, R. J. Park, C. L. Heald, T.-M. Fu, H. Liao, and A. Guenther (2007),650
Model evidence for a significant source of secondary organic aerosol from isoprene, Atmos.651
Environ., 41, 1267–1274.652
Van Loon, M., et al. (2007), Evaluation of long-term ozone simulations from seven regional air quality653
models and their ensemble average, Atmos. Environ., 41, 2083–2097.654
Vautard, R., B. Bessagnet, M. Chin, and L. Menut (2005), On the contribution of natural aeolian655
sources to particulate matter concentrations in Europe: Testing hypotheses with a modelling656
approach, Atmos. Environ., 39, 3291–3303.657

34
Vautard, R., et al. (2003), Paris emission inventory diagnostics from ESQUIF airborne measurements658
and a chemistry transport model, Journal of Geophysical Research, 108(D17), 8564,659
doi:10.1029/2002JD002797.660
Vautard, R., et al. (2007), Evaluation and intercomparison of ozone and PM10 simulations by several661
chemistry-transport models over 4 european cities within the city-delta project, Atmos. Environ.,662
41, 173–188.663
Vestreng, V. (2003), Review and Revision. Emission data reported to CLRTAP, Tech. rep., EMEP664
MSC-W, http://www.emep.int/mscw/mscw publications.html.665
Wexler, A. S., and M. V. Johnston (2008), What have we learned from highly time-resolved666
measurements during EPA’s Supersites program and related studies?, Journal of the Air &667
Waste Management Association, 58, 303–319.668
Wiedinmyer, C., B. Quayle, C. Geron, A. Belote, D. McKenzie, X. Y. Zhang, S. O’Neill, and K. K.669
Wynne (2006), Estimating emissions from fires in North America for air quality modeling,670
Atmos. Environ., 40(19), 3419–3432.671
Yttri, K. E., et al. (2007), Elemental and organic carbon in PM10: a one year measurement campaign672
within the European Monitoring and Evaluation Programme EMEP, Atmos. Chem. Phys., 7,673
5711–5725.674
Zhang, Y., J.-P. Huang, D. K. Henze, and J. H. Seinfeld (2007), Role of isoprene in secondary organic675
aerosol formation on a regional scale, J. Geophys. Res., doi:10.1029/2007JD008675.676
Bertrand BESSAGNET, INERIS, Institut National de l’Environnement Industriel677

35
et des Risques, Parc technologique ALATA, 60550 Verneuil en Halatte, France.678
[[email protected]]679
Received680
This manuscript was prepared with AGU’s LATEX macros v5, with the extension package681
‘AGU++’ by P. W. Daly, version 1.6 from 1999/02/24.682

36
Figure Captions683
(a) Total terpenes (former algorithm) (b) Total terpenes (MEGAN)
(c) Isoprene (former algorithm) (d) Isoprene (MEGAN)
Figure 1. Biogenic emissions (terpenes and isoprene) for July 2003 (in Mg/cell) calculated with the
former algorithm in CHIMERE and the new MEGAN inventory.

37
Figure 2. Location of stations, red triangles : OC and EC measurements available; green circles :
PM10 measurements. For IT04, SK04, AT02 sites both PM10 and OC/EC measurements are available.
38
Figure 3. Comparisons between observed (plus symbols) and simulated (circle symbols) OC concen-
trations. Simulated SOA are represented by diamond symbols.

39
Figure 4. Average contribution (%) in 2003 of secondary organic carbon to the total organic carbon
calculated by CHIMERE.
Figure 5. Average contribution (%) in 2003 of isoprene secondary organic carbon to the total secondary
organic carbon calculated by CHIMERE.

40
Figure 6. Comparisons between observed (plus symbols) and simulated (circle symbols) EC concentra-
tions.

41
Figure 7. Impact of forest fires (in %) estimated by CHIMERE on August 1st - 15, 2003. Figures
displayed the ratio between [top] the primary OC concentrations from fires and total primary OC, and
the ratio between [bottom] SOA concentrations from fires and total SOA

42
Tables684

43
Reactions kinetic rates (molec.cm−3.s−1)685
TOL+OH→ 0.004×AnA0D + 0.001×AnA1D 1.81×10−12exp(355/T)686
+ 0.084×AnBmP + 0.013×AnBlP687
TMB+OH→ 0.002×AnA0D + 0.002× AnA1D + 0.001×AnA2D 9.80×10−9/T688
+ 0.088×AnBmP + 0.006×AnBlP689
NC4H10+OH→ 0.07×AnBmP 1.36×10−12exp(190/T)−2690
API+OH→ 0.30×BiA0D + 0.17×BiA1D + 0.10×BiA2D 1.21×10−11exp(444/T)691
API+O3→ 0.18×BiA0D + 0.16×BiA1D + 0.05×BiA2D 1.01×10−15exp (-732/T)692
API+NO3→ 0.80×BiBmP 1.19×10−12exp(490/T)693
BPI+OH→ 0.07×BiA0D + 0.08×BiA1D + 0.06×BiA2D 2.38×10−11exp(357/T)694
BPI+O3→ 0.09×BiA0D + 0.13×BiA1D + 0.04×BiA2D 1.50×10−17695
BPI+NO3→ 0.80×BiBmP 2.51×10−12696
LIM+OH→ 0.20×BiA0D + 0.25×BiA1D + 0.005×BiA2D 1.71×10−10697
LIM+O3→ 0.09×BiA0D + 0.10×BiA1D 2×10−16698
TPO+OH→ 0.70×BiA0D + 0.075×BiA1D 5.10×−8/T699
TPO+O3→ 0.50×BiA0D + 0.055×BiA1D 7.50×10−14/T700
TPO+NO3→ 0.70×BiA0D + 0.075×BiA1D 4.30×10−9/T701
ISO+OH→ 0.232×ISOPA1 + 0.0288×ISOPA2 2.55×10−11exp(410/T)702

44
Table 1. Gas phase chemical scheme for SOA formation in CHIMERE. The surrogate SOA
compounds consist of six hydrophilic species that include an anthropogenic
nondissociative species (AnA0D), an anthropogenic once-dissociative species
(AnA1D), an anthropogenic twice-dissociative species (AnA2D), a biogenic non
dissociative species (BiA0D), a biogenic once-dissociative species (BiA1D) and
a biogenic twice-dissociative species (BiA2D), three hydrophobic species that
include an anthropogenic species with moderate saturation vapor pressure (AnBmP),
an anthropogenic species with low saturation vapor pressure (AnBlP) and a biogenic
species with moderate saturation vapor pressure (BiBmP), and two surrogate compounds
for the isoprene oxidation products.

45
Station Country Lat. (oN) Long. (oE) Altitude(m)703
Puy de Dome (PDDa) France 45.45 3.00 1450705
Perigueux (PERId) France 45.19 0.73 n.a.706
K-Puszta (KPZa) Hungary 46.97 19.58 125707
Mace Head (IE31b,c) Irland 53.33 -9.90 25708
Kollumerwaard (NL09b,c) The Netherlands 53.33 6.28 0709
Payerne (CH02c) Switzerland 46.82 6.95 489710
Tanikon (CH03c) Switzerland 47.48 8.90 539711
Langenbrugge (DE02b,c) Germany 52.80 10.76 74712
Neuglobsow (DE07c) Germany 53.17 13.03 62713
Schmucke (DE08c) Germany 50.65 10.77 937714
Zingst (DE09c) Germany 54.43 12.73 1715
Schauinsland (SILa) Germany 47.92 7.90 1205716
Kosetice (CZ03b,c) The Czech Republic 49.58 15.08 534717
Stara Lesna (SK04b,c) Slovakia 49.15 20.28 808718
Liesek (SK05c) Slovakia 49.37 19.68 892719
Starina (SK06c) Slovakia 49.05 22.27 345720
Illmitz (AT02b,c) Austria 47.77 16.77 117721
St. Koloman (AT04b,c) Austria 47.65 13.20 851722
Montelibretti (IT01c) Italy 42.10 12.63 48723
Ispra (IT04b,c) Italy 45.80 8.63 209724
San Pietro Capofiume (IT08b) Italy 44.48 11.33 n.a.725

46
Station Country Lat. (oN) Long. (oE) Altitude(m)704
Braganca (PT01b,c) Portugal 41.82 -6.77 691726
Aveiro(AVEa) Portugal 40.57 -8.63 48727
Penicuick (GB46b) Great Brittain 55.95 -3.22 n.a.728
Ghent (BE02b) Belgium 51.05 3.72 n.a.729
Keldsnor (DK05b,c) Denmark 54.73 10.73 9730
Niembro (ES08b,c) Spain 43.44 -4.85 134731
Cabo de Creus (ES10b,c) Spain 42.32 3.32 23732
Barcarrota (ES11b,c) Spain 38.47 -6.92 393733
Zarra (ES12b,c) Spain 39.09 -1.10 885734
Penausende (ES13b,c) Spain 41.28 -5.87 985735
Els Torms (ES14b,c) Spain 41.40 0.72 470736
O Savinao (ES16b,c) Spain 43.23 -7.70 506737
Iskrba (SI08b,c) Slovenia 45.57 14.87 520738
Table 2. Names and coordinates of stations. (a) CARBOSOL sites (EC/OC,
diacids data), (b) EMEP sites of the EC/OC EMEP campaign, (c)
routine EMEP sites (PM10 data), (d) station from the french
monitoring network. Most of the stations are rural background sites,
except IT08, BE02 and PERI that are urban background sites.

47
Station Model Obs. RMSE Corr.
AT02a 3.86 5.82 4.16 0.107
AVEb 1.16 5.43 5.96 0.014
BE02a 2.16 3.97 2.13 0.904
CZ03a 2.84 4.98 3.59 0.328
DE02a 1.99 4.05 3.32 0.594
GB46a 0.70 1.67 1.21 0.823
IE31a 0.42 1.48 1.58 0.831
IT04a 2.49 8.30 10.22 -0.111
IT08a 2.48 5.76 4.76 -0.001
KPZb 1.33 6.46 6.38 0.254
NL09a 1.16 2.25 1.74 0.620
PDDb 0.82 1.84 1.28 0.914
PT01a 0.80 5.30 7.48 -0.081
SILb 1.74 2.47 1.34 0.750
SK04a 2.89 4.16 2.76 0.285
Table 3. Error statistics, mean model values, mean observations, Root Mean Square Errors (RMSE)
and Correlation factor for OC comparisons, (a) daily average values for the EMEP campaign and (b)
weekly average values for the CARBOSOL campaign.

48
Station Corr. without Corr. with SOA Corr. with SOA739
SOA (without isoprene SOAa) (with isoprene SOAb)740
AT02 0.626 0.623 0.600743
AT04 0.504 0.562 0.633744
CH02 0.437 0.434 0.405745
CH03 0.602 0.620 0.612746
DE07 0.803 0.806 0.808747
DE08 0.580 0.651 0.703748
DE09 0.840 0.838 0.837749
DK05 0.846 0.847 0.848750
ES08 0.583 0.664 0.704751
ES10 0.177 0.221 0.275752
ES11 0.778 0.807 0.835753
ES12 0.645 0.660 0.703754
ES13 0.730 0.754 0.781755
ES14 0.526 0.543 0.587756
ES16 0.660 0.716 0.747757
IT01 0.413 0.450 0.486758
IT04 0.607 0.572 0.506759
PERI 0.391 0.488 0.512760
SI08 0.427 0.469 0.503761
SK04 0.440 0.454 0.458762

49
Station Corr. without Corr. with SOA Corr. with SOA741
SOA (without isoprene SOAa) (with isoprene SOAb)742
SK05 0.441 0.416 0.365763
SK06 0.435 0.412 0.353764
Table 4. Impact of the SOA scheme implementation on the temporal correlation coefficients
for PM10 concentrations observed and simulated. In bold, correlation coefficients
improved by implementing the complete SOA scheme. (a) Only terpene and anthropic VOC’s
chemistry, (b) Complete scheme with isoprene chemistry.
Station Correlation
AVE 0.721
KPZ -0.136
PDD 0.646
SIL 0.512
Table 5. Temporal correlation coefficient between modeled SOA and total diacids concentrations during
the CARBOSOL campaign.

50
Station Model Obs. RMSE Corr.
AT02a 1.12 1.04 0.88 0.718
AVEb 0.23 1.09 1.03 0.303
BE02a 1.70 1.65 0.74 0.768
CZ03a 0.90 1.05 0.70 0.522
DE02a 0.73 0.55 0.82 0.598
GB46a 0.31 0.52 0.42 0.746
IE31a 0.13 0.24 0.20 0.894
IT04a 0.72 1.83 1.54 0.582
IT08a 0.58 1.29 0.94 0.467
KPZb 0.33 1.14 1.03 0.395
NL09a 0.54 0.47 0.38 0.732
PDDb 0.09 0.26 0.24 0.226
PT01a 0.11 1.03 1.44 0.295
SILb 0.34 0.30 0.27 0.171
SK04a 0.92 0.85 0.90 0.438
Table 6. Error statistics, mean model values, mean observations, Root Mean Square Errors (RMSE)
and Correlation factor for EC comparisons, (a) daily average values for the EMEP campaign and (b)
weekly average values for the CARBOSOL campaign.