devenir du médicament a.d.m.e. dans...
TRANSCRIPT

41
Devenir du médicament dans l’organisme
AbsorptionDistributionMétabolismeExcrétion
Clairances
Pharmacocinétique (PK)Aire sous la courbe (ASC)Biodisponibilité (F)Demi vie (T1/2 )Volume de distribution (Vd)
PLAN :
A.D.M.E.
3)Distribution
Cible (récepteurs)
2) Métabolisme (FOIE, intestin)
1) Absorption (intestin)
4) Elimination (BILE-REINS)
Absorption intestinale des médicaments
C’est la forme non ionisée des médicaments qui est principalementabsorbée.
Pour une Base, c’est la forme B B + H+ ⇔ BH +En milieu acide (estomac), essentiellement sous forme BH +
⇒ mauvaise absorption; devient meilleure après duodénumEn milieu basique (succs pancréatiques), essentiellement sous forme B
⇒ bonne absorption
Pour un Acide, c’est la forme AH A- + H+ ⇔ AHEn milieu acide (estomac), essentiellement sous forme AH
⇒ bonne absorption; diminue après duodénumEn milieu basique (succs pancréat), essentiellement sous forme A-
⇒ mauvaise absorption
Absorption intestinale des médicaments
Facteurs modulant l’absorption intestinale médicaments:
pH gastrique : (pansements - antiulcéreux - maladies -repas)
Vidange gastrique : (vomissements - diabète - repas)
Temps de contact : accélérateurs ou ralentisseurs transit
Interaction Atazanavir - Omeprazole
Agarwala S, et al. CROI 2005. Abstract 658.
ATV/r 300/100 ATV/r 300/100 + 40 mg OMP
100
1000
10,000
0 2 4 6 8 10 12 14 16 18 20 22 24
Time (hr)
Ata
zana
vir T
roug
h (n
g/m
L)
Distribution des médicaments
Les médicaments (IV-PO) se distribuent en premier lieu dans lesecteur vasculaire
-Les lipophiles (bien absorbés +++) sont fortement fixés auxprotéines plasmatiques (80 à 99% fixés aux protéines)
Leur fraction libre (non liée aux protéines plasmatiques =active ) est faible
- Les hydrophiles (pas très bien absorbés+++) sont faiblementfixés aux protéines plasmatiques (inf 50% fixation). Leur fractionlibre active est donc plus élevée

41
Distribution des médicaments
Les médicaments (IV-PO) se distribuent ensuite dans différentscompartiments en fonction de leurs propriétés physicochimiqueset des phénomènes de transport (qui limitent ou facilitent lespassages du sang vers l’organe)
Certains se concentrent plus spécifiquement dans certains organes(poumon, foie, reins, os, graisses), souvent médicamentslipophiles
Certains se « diluent » dans l’eau corporelle totale, d’autres dansle milieu extracellulaire, souvent médicaments hydrophiles
Le secteur de diffusion est apprécié en partie par la notion devolume de distribution.
Métabolisme des médicaments
Environ 50 % des médicaments sont métabolisés
Les processus de métabolisme siègent principalement dans le foie(un peu dans l’intestin)
1) Médicament actif Métabolite inactif
2) Médicament inactif (pro drogue) Métabolite actif
Métabolisme des médicaments,Métabolisme des médicaments,ClairanceClairance
Clearance
DrugMetabolite
Enzymes de Phase ICytochromes P450
Metaboliteconjugué
Enzymes Phase II
Métabolisme des médicaments
Médicament ( R ) (R-CH3)
Métabolite (R-OH) (R-H)
Métabolite conjugué (R-O-glucuronide) (R-glucuronide)
Enzyme Phase ICytochromes P450(hydroxylation-déméthylation)
Enzyme Phase IIEnz conjuguaison (UGT)(glucuroconjuguaison)
Bile - Urine
Hyd
roph
ilie
CYP hépatiques humains
CY P 2 E 1CY P2 D 6
CY P 2C1 8
C Y P 2 C 1 9
CYP3ACYP1A2
CYP non quantifiésCYP2C9
CYP2C8
Benzodiazépines Antiarythmiques inhibi HMGCoA-Ralprazolam amiodarone simvastatinemidazolam quinidine lovastatinediazepam lidocaine atorvastatinetriazolam cerivastatine
Autres Antibiotiques inhibiteurs calciquesEthymorphine Erythromycine diltiazemCocaine clarythromycine vérapamil
rifampicine bépridil troleandomycine amlodipine
nifédipine
Anticancéreux Antiprotéases antihistaminiques isofosfamide saquinavir terfenadineimatinib indinavir
nelfinaviramprénavir
analogues non nucléosi Hormones immunosuppresseursdelavirdine estradiol ciclosporineefavirenz éthynyl estradiol tacrolimus
dexaméthasone rapamycineprednisolone
Substrats CYP3A4 (50% med. métabolisés)

41
CYP3A4
CYP3A4
CYP3A4
DrugDrug+ metabolites
Drug+ metabolites
Drug+ metabolites
Drug
Drug
Drug
Effet de premier passage
Intestinal + hepatic CYP3A4+ P-glycoprotein
Effet de premier passage
Barrière à la pénétration de substances étrangères dansl’organisme
• Limite la quantité de médicament inchangé pénétrant dans lecompartiment sanguin
• Limite la biodisponibilité des médicaments oraux
• Associe transport (MDR1 / P-glycoprotéine) intestinal etmétabolisme intestinal et hépatique
Médicamenthydrophile
Médicament inchangé
Bile U
rine
Excrétion
Médicamenthydrophobe
Métabolites hydrophiles
Métabolisme hépatique
Excrétion
Excrétion
Majorité des médicaments excrétés voie rénale (filtration passive ± sécrétion)
médicaments inchangés et/ou métabolites
voie biliaire (sécrétion ± métabolisme) médicaments inchangés et/ou métabolites
voie rénale et biliaire médicaments inchangés et/ou métabolites
voies mineures (respiratoire-sudorale)
Insuffisance rénale et insuffisance hépatique
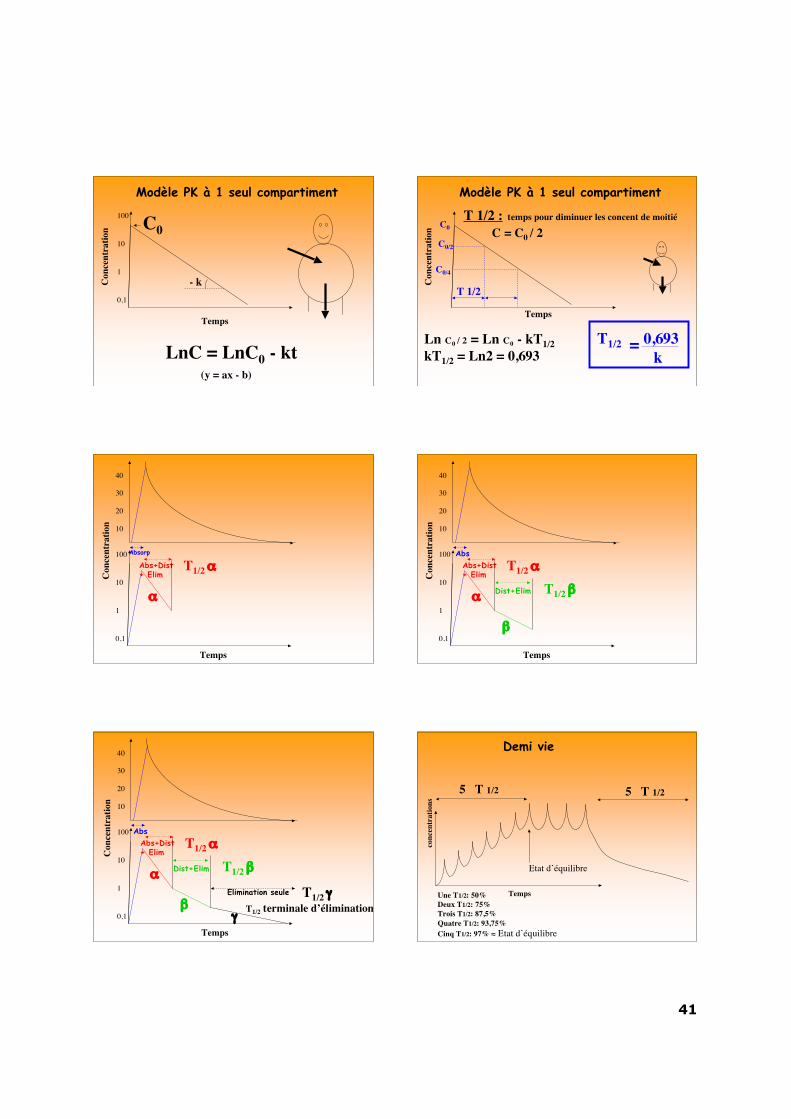
Modèle PK à 1 seul compartiment
Con
cent
ratio
n
Temps
C = C0 e-kt
10987654321
41
Modèle PK à 1 seul compartiment
Con
cent
ratio
n
Temps
LnC = LnC0 - kt (y = ax - b)
100
10
1
0,1
C0
- k
Modèle PK à 1 seul compartiment
Con
cent
ratio
n
Temps
Ln C0 / 2 = Ln C0 - kT1/2kT1/2 = Ln2 = 0,693
T 1/2 : temps pour diminuer les concent de moitié
C = C0 / 2
T1/2 = 0,693k
C0
C0/2
T 1/2
C0/4
Con
cent
ratio
n
Temps
100
10
1
0,1
40
30
20
10
Absorp
Abs+Dist+ Elim
α
T1/2 α Con
cent
ratio
n
Temps
100
10
1
0,1
40
30
20
10
AbsAbs+Dist+ Elim
Dist+Elimα
β
T1/2 α
T1/2 β
Con
cent
ratio
n
Temps
100
10
1
0,1
40
30
20
10
AbsAbs+Dist+ Elim
Dist+Elim
Elimination seule
α
βγ
T1/2 α
T1/2 β
T1/2 γT1/2 terminale d’élimination
5 T 1/2
Etat d’équilibre
conc
entr
atio
ns
Temps
Demi vie
5 T 1/2
Une T1/2: 50%Deux T1/2: 75%Trois T1/2: 87,5%Quatre T1/2: 93,75%Cinq T1/2: 97% ≈ Etat d’équilibre

41
5 T 1/2
Etat d’équilibreconc
entr
atio
ns
Temps
IV
Demi vie
5 T 1/2
Etat d’équilibre
conc
entr
atio
ns
Temps
5 T 1/2
Etat d’équilibre
conc
entr
atio
ns
Etat d’équilibre
Dose de Charge IV
Temps
conc
entr
atio
ns
Temps
T1/2 T1/2 T1/2 T1/2 T1/2
Demi vie: limites
5 T 1/2
Etat d’équilibre
conc
entr
atio
ns
5 T 1/2
conc
entr
atio
ns
Temps
Médicament ne s’accumulant pas dans l’organismeIntervalle d’administration >5 T 1/2
Médicament s’accumulant dans l’organisme
Conc
entr
ations
(PK
)
Effe
t (P
D)
La PD ne suit pas toujours la PK du médicament

41
Biodisponibilité : FPourcentage de médicament arrivant dans la circulation sanguine
Si voie IV : F = 100%
Biodisponibilité absolue : Rapport ASC0-∞ per os / ASC0-∞ IV
Biodisponibilité relative :RapportASC0-∞ per os nouvelle galénique / ASC0-∞ per os ancienne galénique
Pour les génériques Biodisponibilité relative avec la forme originaleASC0-∞ générique + ou - 20 % ASC0-∞ forme originale
Forme originale ASC0-∞ = 100 %;ASC générique entre 80 et 120 % ASC0-∞ forme originale
conc
entr
atio
ns
Temps
IV
PO
conc
entr
atio
ns
Temps
IV
PO
Biodisponibilité : F
Faible biodisponibilité ≈ 25%
Bonne biodisponibilité ≈ 75%
Avant A l’état d’équilibre
Dose
Volume de distribution VdVolume théorique
Ceq
Vd = Dose Ceq
Si Vd > poids corporel:Concentration dans les tissus
Si Vd = 4 à 5 litres:Volume sanguin
Volume de distribution VdVolume théorique
Vd modulé par la fixation aux protéines plasmatiquesHypoalbuminémie:(cirrhose -Σ néphrotiques - carences alimentaires)Vd modulé par troubles de l’hydratation extracell ou intracell
Clairance d’un médicament
Volume de plasma épuré d’une substance par unité de temps
Équivalent à un débit, exprimé en ml/min ou litre/h
Clairance médicament IV = DoseASCo-∞
Clairance d’un médicament
Clairance médicament per os = F x DoseASCo-∞
Clpo = Clhep + Clren + Cl diverses
41
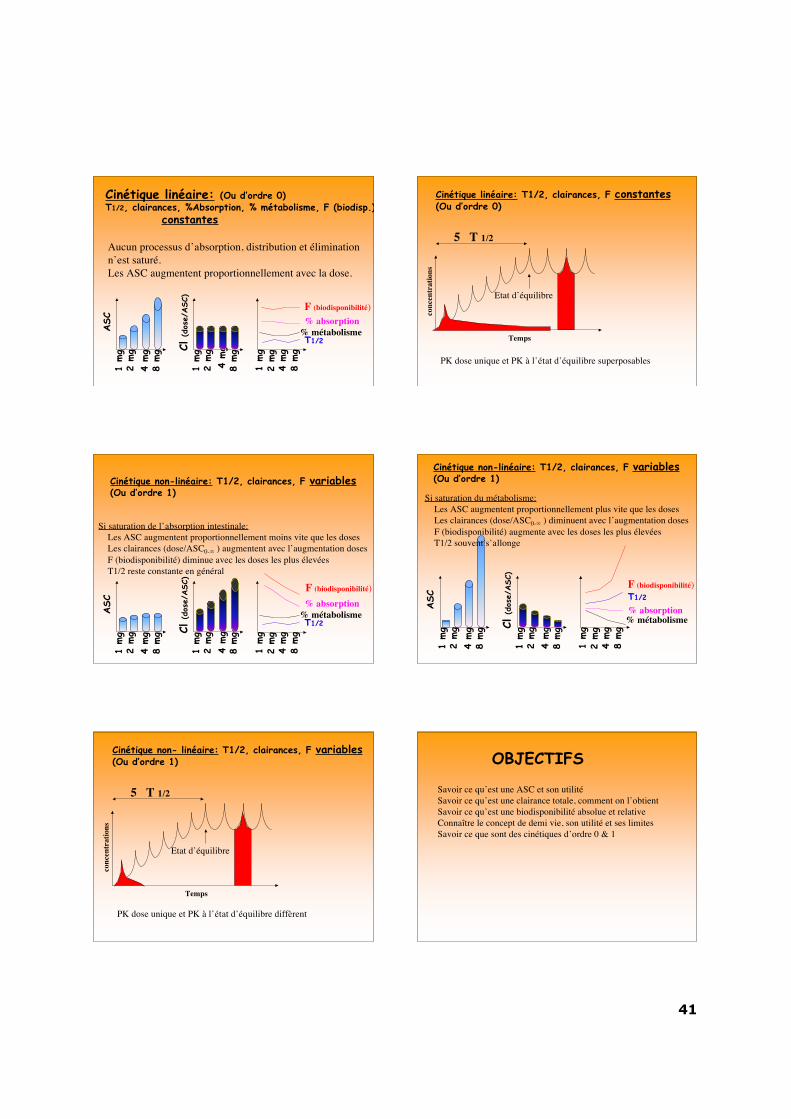
Cinétique linéaire: (Ou d’ordre 0)T1/2, clairances, %Absorption, % métabolisme, F (biodisp.)
constantes
Aucun processus d’absorption, distribution et éliminationn’est saturé.Les ASC augmentent proportionnellement avec la dose.
1 mg
2 mg
4 mg
8 mg
ASC
1 mg
2 mg
4 mg
8 mgCl
(do
se/A
SC)
1 mg
2 mg
4 mg
8 mg
F (biodisponibilité)
T1/2% métabolisme
% absorption
5 T 1/2
Etat d’équilibre
conc
entr
atio
ns
Temps
Cinétique linéaire: T1/2, clairances, F constantes(Ou d’ordre 0)
PK dose unique et PK à l’état d’équilibre superposables
Cinétique non-linéaire: T1/2, clairances, F variables(Ou d’ordre 1)
Si saturation de l’absorption intestinale: Les ASC augmentent proportionnellement moins vite que les doses Les clairances (dose/ASC0-∞ ) augmentent avec l’augmentation doses F (biodisponibilité) diminue avec les doses les plus élevées T1/2 reste constante en général
1 mg
2 mg
4 mg
8 mg
ASC
1 mg
2 mg
4 mg
8 mgCl
(do
se/A
SC)
1 mg
2 mg
4 mg
8 mg
T1/2% métabolisme
% absorptionF (biodisponibilité)
Cinétique non-linéaire: T1/2, clairances, F variables(Ou d’ordre 1)
Si saturation du métabolisme: Les ASC augmentent proportionnellement plus vite que les doses Les clairances (dose/ASC0-∞ ) diminuent avec l’augmentation doses F (biodisponibilité) augmente avec les doses les plus élevées T1/2 souvent s’allonge
1 mg
2 mg
4 mg
8 mg
ASC
1 mg
2 mg
4 mg
8 mgCl
(do
se/A
SC)
1 mg
2 mg
4 mg
8 mg
F (biodisponibilité)T1/2
% métabolisme% absorption
5 T 1/2
Etat d’équilibre
conc
entr
atio
ns
Temps
Cinétique non- linéaire: T1/2, clairances, F variables(Ou d’ordre 1)
PK dose unique et PK à l’état d’équilibre diffèrent
OBJECTIFS
Savoir ce qu’est une ASC et son utilitéSavoir ce qu’est une clairance totale, comment on l’obtientSavoir ce qu’est une biodisponibilité absolue et relativeConnaître le concept de demi vie, son utilité et ses limitesSavoir ce que sont des cinétiques d’ordre 0 & 1