direct visualization of a vast cortical calcium...
TRANSCRIPT

1895Journal of Cell Science 108, 1895-1909 (1995)Printed in Great Britain © The Company of Biologists Limited 1995
Direct visualization of a vast cortical calcium compartment in Paramecium by
secondary ion mass spectrometry (SIMS) microscopy: possible involvement
in exocytosis
Nicole Stelly1,*, Sylvain Halpern2, Gisèle Nicolas3, Philippe Fragu2 and André Adoutte1
1Laboratoire de Biologie Cellulaire 4 (CNRS, URA 1134), Bâtiment 444, Université Paris-Sud, 91405 Orsay Cedex, France2Equipe de Microscopie Ionique (INSERM U66), Institut Gustave Roussy, 94800 Villejuif, France3Centre Interuniversitaire de Microscopie Electronique (CNRS, URA 1488) et Laboratoire de Cytologie, Université Pierre et MarieCurie, 7 quai St Bernard, Bâtiment A, 75252 Paris Cedex 05, France
*Author for correspondence
The plasma membrane of ciliates is underlaid by a vastcontinuous array of membrane vesicles known as corticalalveoli. Previous work had shown that a purified fractionof these vesicles actively pumps calcium, suggesting thatalveoli may constitute a calcium-storage compartment.Here we provide direct confirmation of this hypothesisusing in situ visualization of total cell calcium on sectionsof cryofixed and cryosubstituted cells analyzed by SIMS(secondary ion mass spectrometry) microscopy a methodnever previously applied to protists. A narrow, continuous,Ca-emitting zone located all along the cell periphery wasobserved on sections including the cortex. In contrast, Naand K were evenly distributed throughout the cell. Variouscontrols confirmed that emission was from the alveoli, in
particular, the emitting zone was still seen in mutantstotally lacking trichocysts, the large exocytotic organellesdocked at the cell surface, indicating that they make nomajor direct contribution to the emission. Calcium con-centration within alveoli was quantified for the first time inSIMS microscopy using an external reference and wasfound to be in the range of 3 to 5 mM, a value similar tothat for sarcoplasmic reticulum. After massive induction oftrichocyst discharge, this concentration was found todecrease by about 50%, suggesting that the alveoli are themain source of the calcium involved in exocytosis.
Key words: calcium, SIMS, Paramecium
SUMMARY
INTRODUCTION
The question of the subcellular location of calcium (Ca) storesin eukaryotic cells has attracted considerable attention in recentyears (see Koch, 1990; Meldolesi et al., 1990; Tsien and Tsien,1990; Lytton and Nigam, 1992; Meldolesi and Villa, 1993, forreview). The measurement of free, cytosolic calcium (Ca2+)concentration and its oscillation in single cells has becomepossible with the advent of fluorescent probes (Grynkiewicz etal., 1985; see Williams and Fay, 1990, for review). This con-centration, however, is several orders of magnitude lower thanthat of total cellular calcium; part of this large amount ofcalcium is bound to cytosolic proteins but the majority, by far,appears to be segregated inside membrane-bounded intracellu-lar organelles where it is complexed with low-affinity, high-capacity proteins (Carafoli, 1987; Koch, 1990). Release ofCa2+ from these organelles plays a key role in severalprocesses, especially in the response to various extracellularstimuli.
Identification of these Ca-sequestering organelles hasproven difficult, however. Two major approaches have beenused, a direct one, seeking to visualize the element itself by
electron probe microanalysis of tissue sections (Somlyo, 1985;Andrews et al., 1987), and an indirect one, seeking to identifythe organelles by virtue of the presence of a set of ‘marker’proteins involved in Ca2+ homeostasis: Ca2+-ATPases, Ca2+
channels (ryanodine receptor, inositol-triphosphate (InsP3)receptor) and Ca-binding proteins (calsequestrin, calreticulin,etc.). These proteins were detected either, by physiological andbiochemical methods (such as by measurement of Ca2+ uptakeand release in subcellular fractions; see Pietrobon et al., 1990for review) or by immunocytological approaches (subcellularlocalization, at the EM level, of compartments reacting withantibodies directed against the marker proteins; e.g. see Volpeet al., 1988).
The results of these approaches are summarized in recentreviews (cited above). Except for the striated muscle cell, anextensively studied model system in which the situation is con-siderably clarified because of the amplification of the Castorage compartment achieved in the form of the sarcoplasmicreticulum, the number, location and functional role of Castorage compartments in eukaryotic cells has until recentlybeen unclear. Except for mitochondria, which do not appear tobe involved in Ca2+ storage, the major intracellular Ca store

1896 N. Stelly and others
has, for many years, been assumed to be the endoplasmicreticulum. Currently, at least two and more probably three oreven four types of compartments are implicated (Burgoyne andCheek, 1991; Lytton and Nigam, 1992; Sitia and Meldolesi,1992; Meldolesi and Villa, 1993), depending on the cell typeanalyzed, on the basis of the receptors, channels, pumps andcalcium-binding proteins that they contain. All of these com-partments appear to belong to the general intracellular proteinsorting compartments (RER, Golgi, endosomes, etc.) althoughthey are more or less distantly connected to it.
In this paper, we present an approach complementary tothose just cited, aimed at directly visualizing total cellular Ca.It involves the use of both a new cell type and a differentmethod. The cell type is the ciliated protozoan Paramecium,in which we have recently shown that a vast vesicular networklying just beneath the plasma membrane actively pumps Ca2+
(Stelly et al., 1991). This ‘primitive’ organism thereforeappeared to offer a naturally amplified Ca storage compart-ment (akin to the sarcoplasmic reticulum), facilitating directvisualization. The method is secondary ion mass spectrome-try (SIMS; Castaing and Slodzian, 1962) microscopy, whichhas been extensively used in solid state physics to character-ize surface composition of samples, but much less in biolog-ical applications (see reviews by Chandra and Morrison,1988; Fragu et al., 1992). A primary ion beam is focused ontothe surface of a tissue section, leading to the sputtering of themost superficial atoms, themselves partly in the form of ions.These secondary ions are then collected, analyzed with a massspectrometer and the corresponding image is reconstructed.One therefore obtains an image of the distribution of a specificatom at the surface of the specimen analyzed. This methodoffers three main advantages. First, it allows visualization ofall the elements of Mendeleiev’s table as well as discrimina-tion between many of their stable and radioactive isotopes.By successively eroding the same section one can map severaldifferent ions (for example, Ca2+, Na+, K+, etc.) in the sametissue and cells. It should be stressed that ions are highlyprone to extraction from cytological preparations duringspecimen preparation (see Mentré and Escaig, 1988). A pre-requisite for all the approaches just outlined is to take suitableprecautions to avoid loss and/or redistribution of diffusiblecompounds. This can be achieved by rapid freezing of thecells, avoiding the use of any fixative, then either cryosubsti-tuting the samples (as done in the present work) or usingfreeze-fracturing followed by freeze-drying (Chandra andMorrison, 1992). Second, its sensitivity is at least as good asthat of X-ray microanalysis (EPMA) and probably slightlybetter (see below). This level of sensitivity is comparable orslightly inferior to that of electron energy loss spectrometrymethods (EELS); Third, images of ion distribution can beobtained over large areas of cells in a short time, making themethod especially valuable when there is a need to observewhole tissues or extended portions of large cells. The majordrawback of SIMS microscopy is its limited lateral resolution,especially when compared with EELS, being of the order of0.5 µm for the present instruments, yielding images equiva-lent to those from a light microscope. This drawback is partlycompensated by the sensitivity of SIMS, the very low levelof background noise and the relative ease with which datafrom large areas can be collected (for a detailed comparisonof the merits and limitations of the various microprobe
methods used in biological microanalysis, see Linton andGoldsmith, 1992).
Paramecia have already been studied by microanalysis at theEM level both in the EPMA (Schmitz et al., 1985; Zierold etal., 1989) and the EELS mode (Knoll et al., 1993), usingexcellent techniques of cryofixation followed by either freeze-drying or cryosubstitution. The major result of these studieswas the identification of a peripheral Ca-containing zone in thecortex of Paramecium (not including the exocytotic organellesknown as trichocysts) and preliminary evidence for redistrib-ution of this calcium after induction of exocytosis in the EELShigh-resolution study (Knoll et al., 1993) but not in the EPMAone (Zierold et al., 1989). Here, we extend these studies usingthe different SIMS approach, which we adapted for fast-swimming single cells. We confirm the occurrence of anintensely emitting peripheral rim of calcium and, through theuse of mutants devoid of trichocysts, demonstrate that the exo-cytotic organelle is not the location of the ion. By a variety ofcontrols, we show that this rim cannot be due solely to arte-factual displacement or external adhesion of calcium. Thisprovides definitive confirmation of the existence of a vast sub-membranal calcium compartment in this cell, in which most ofthe cell calcium is stored (at least one order of magnitude morethan in the rest of the cytoplasm). We also provide the firstquantification of the amount of calcium by SIMS microscopyusing an internal standard; this concentration is in the mil-limolar range, equivalent to that found in the sarcoplasmicreticulum. Finally, we provide preliminary evidence that thiscalcium is involved in the exocytotic process, by observing a50% reduction of its amount in cells fixed 15 to 30 secondsafter massive induction of exocytosis.
MATERIALS AND METHODS
Biological material and sample preparation Paramecium
Strains and culture conditionsThe wild-type (WT) cells used in these experiments were from stockd4-2 of Paramecium tetraurelia. Cells were grown at 27°C inphosphate-buffered wheat grass powder infusion, bacterized the daybefore use with Klebsiella pneumoniae and supplemented with 0.5µg/ml β-sitosterol.
Two mutants were used in this study: mutant tam8, whose tri-chocysts are never attached to the cell surface (Beisson and Rossignol,1975; Lefort-Tran et al., 1981); and a thermosensitive mutant, nd9,whose trichocysts are attached at the cell surface but cannot be dis-charged at 27°C (Beisson et al., 1976).
MicroscopyCells were harvested from early stationary phase cultures and thepellet was fixed either: (1) in 0.5% glutaraldehyde plus 2%paraformaldehyde, 50 mM sodium cacodylate buffer, pH 7.4, for 20minutes at 4°C, or in the same fixative followed by postfixation in 2%OsO4 in the same buffer. Cells were then pre-embedded in fibrinogenpellets, dehydrated and embedded in LR White or Epon-Araldite; or(2) by fast-freeze fixation by slamming the specimen against a coldcopper block cooled by liquid helium (Escaig, 1983) followed byfreeze substitution at −86°C for 72 hours in acetone in the presenceof 20 mM oxalic acid, then warmed to −30°C, maintained for twohours at −30°C, and finally warmed to room temperature andembedded in Epon-Araldite.
Wild-type cells were also cryofixed and cryodehydrated without

1897Calcium stores visualized by SIMS in Paramecium
oxalic acid as controls and embedded in Epon-Araldite or cryoem-bedded in Lowicryl K4M.
ExocytosisSynchronous exocytosis can be achieved with AED (amino ethyldextran), which causes instantaneous release of most of the trichocysts(Plattner et al., 1984, 1985; Kerboeuf and Cohen, 1990). AED, kindlyprovided by J. Cohen and D. Kerboeuf, was used at 6 µM on a pelletof cells. About 30 seconds after stimulation, the cells were slammed onthe cryobloc and cryodehydrated in the same way as the untreated cells.Aliquots of cells were further treated with picric acid and observed witha dark-field microscope to check the extend of exocytosis.
MuscleThe cutaneous muscle of frog was taken up in Ringer’s solution. Verysmall pieces were cryofixed and cryodehydrated exactly the same wayas for Paramecium.
To achieve a good preservation of cells (Paramecium and muscle),ultrathin sections were obtained and stained with uranyl acetatefollowed by lead citrate. They were observed in Siemens Elmiskop102 electron microscope.
SIMS microscopy studySerial semi-thin sections were deposited on glass slides for opticalexamination and on ultrapure gold holders for ion analysis. Sections (1µm) on glass slides were stained with Toluidine Blue and sections (3µm) for ion analysis were deposited over a microdrop of water on thegold holder, and heated to 60°C. In some control experiments sectionswere deposited over the gold holder without any contact with water.
The instrument used in this study was an IMS 3F (CAMECA,Courbevoie, France), fitted with two primary ion sources andconnected to an image processing system. This system (Olivo et al.,1989) allows digitalization of images (512×512 pixels), high-speedsignal integration of the ionic images (to improve signal/noise ratio),histogram equalization (to increase the grey-scale contrast and todecrease background noise) and, finally, image superposition.
The ion microscope was operated with the O2+ primary source witha 15 keV primary beam current of 200 nA. The image field diameterwas 150 µm. A mass resolution (M/∆M) of 2000 was used in orderto eliminate interferences between cluster ions and the specificelements under study. Under these conditions, elemental mapping ofCa, K, Na and Mg is easily achieved.
For quantification and calibration, internal reference elements wereused. In SIMS, the secondary ion beam current intensity (Ia) is afunction of the concentration of the analyzed element (Ca), the areato be analyzed (S), the useful ion yield (Ya) and the primary ion beamcurrent intensity (Ip). Thus, Ia = Ca.S.Ya.Ip. However, when dealingwith insulating specimens such as embedded biological samples, partof the positive primary ion beam is repelled by charge effects whennegative secondary ions are extracted. The real intensity of theprimary ion beam current (Ip) is therefore variable, and the relationbetween Ia and Ca is not directly applicable.
Nevertheless, a calibration is possible by measuring the intensityof the secondary ion beam using an internal reference element (Ir),which is present at a large homogeneous and constant concentrationin the specimen. Then Ia/Ir = K.Ca, where K is a proportionalityconstant which can be determined using a standard with increasingconcentration of the tested element to generate a calibration curve. Asthe carbon content of biological specimens and embedding resins arevirtually similar, this element can be used as an internal reference. Inorder to quantify Ca, calcium octoate (Calcium-Norol bySICCANOR, 59282 Douchy-les-Mines, France) was used as areference. Since calcium octoate is soluble in Epon-Araldite, samplescontaining varying Ca concentrations, from 0.05 mM to 5 mM, wereprepared. Sections of 3 µm thickness were obtained and depositedwithout any water onto the gold holders. Emission was measured overfields of 150 µm in diameter.
In our IMS 3F, the secondary ion beam intensity is measureddirectly with the electron multiplier; the measurements are performedon selected areas, which are limited by adapted apertures. In thepresent work, the measured areas were always of 8 µm diameter. Inorder to obtain statistically significant results, sets of at least 10 mea-surements were carried out on each holder for each of the domainsunder analysis (Ca rim, cytoplasm, etc.). The beam was centered onthe area to be measured and the location of the area was recorded overthe image. Concerning the Ca rim located at the cell periphery, whichis the main subject of this paper, the diameter of the measured circleis larger than that of the rim. The rim was therefore positioned in thecenter of the measured field. We checked that a slight move of therim toward the borders of the field did not significantly modify therecorded values.
Usually, several ions were recorded from the same section (mostfrequently Ca2+, Na+, K+ and Mg2+) and, in most cases, a histologi-cal section immediately following or preceding those analyzed bySIMS was stained with Toluidine Blue on a glass slide, to be observedat the light microscope to provide a reference pattern.
RESULTS
Fast-freezing of wild-type Paramecium allows goodultrastructural preservationIn order to prevent fixation-induced ion loss and redistribution,we adapted the cryoblock method of Escaig (1983) to use witha continuously fast-swimming, fragile cell such as Para-mecium. We used small pieces of filter paper to trap the cellsin a thin layer while keeping them alive and healthy. Under theconditions used, only those cells that happen to be in the mostsuperficial portion of the filter facing the copper bloc werecooled rapidly, a condition essential for preventing formationof ice crystals. After rapid freezing, there are two main alter-natives for preserving the intracellular distribution of ions,freeze-substitution or even better, freeze-drying. The problemof freeze-drying and freeze-sectioning in SIMS is that thesections obtained in that way adhere very poorly to the goldholders used in the following step. Sod et al. (1990) havedeveloped indium holders to overcome this difficulty foranimal cell cultures, but this approach is difficult to apply tosingle cells. In addition, in order to keep the paramecia in astate as close as possible to the physiological one, we avoidedusing high concentrations of centrifuged cells. Under the con-ditions used, the density of cells found in the filter paper is low.This condition in addition to the previous one precluded theuse of the freeze-fracture and freeze-drying techniquedeveloped by Chandra et al. (1986). Therefore we resorted tofreeze-substitution and inclusion in resin, realizing that someredistribution of ions may occur at the final step of inclusionin the resin when cells are brought back to room temperature.Controls embedded in Lowicryl at low temperature were thusincluded to check for major redistribution (see below). Thefrozen pieces of filter paper were then taken through thevarious steps of cryosubstitution in acetone, in the presence (orabsence) of oxalic acid, inclusion in epon resin (or in Lowicryl)and sectioning (see Materials and Methods). Sections were firstobserved by both light and conventional transmission electronmicroscopy, to ascertain the quality of structural preservation.
By conventional light microscopy (after Toluidine Bluestaining) the cell contours and many cellular organelles werereadily recognized. In Fig. 1, for example, we can recognize,

1898 N. Stelly and others
Fig. 1. Rapidly frozen WT Paramecium: semi-thin section stainedwith Toluidine Blue and observed with an optical microscope. N,macronucleus; V, food vacuole; T, trichocysts attached to the cortexC. Bar, 10 µm.
from the periphery to the inside of the cell: cilia in the form ofpatches in some portions of the section, the cortex consistingof adjacent typical cup-shaped cortical units, the carrot-shapeddark trichocysts perpendicular to the surface when cut longi-tudinally, the oral depression or oral apparatus, and finally thedense cytoplasm with food vacuoles containing bacteria invarious stages of digestion and, occasionally, a portion of themacronucleus. The only abnormality observed is that the cellslocated at the front are slightly deformed by the compressiongenerated during slamming on the copper block.
Fig. 2 illustrates the ultrastructural characteristics of stainedsections from cryofixed wild-type cells (b,c) as compared tochemically fixed ones (a). Two other variables were alsoanalyzed (not shown): presence or absence of oxalic acid in thecryosubstitution medium and inclusion in Lowicryl instead ofEpon. Oxalic acid was added in order to promote in situ pre-cipitation of calcium (see Nicaise et al., 1989); Lowicryl wasused in order to check the quality of ultrastructural preserva-tion it provided compared with traditional resins, since its usecould prove useful both for ion distribution studies (byallowing polymerization at low temperature after cryosubsti-tution, thus further preventing ion diffusion) and for immuno-cytochemical studies. In cells located close to the frozen front,good ultrastructural preservation was observed: the cortex withits typical organelles was easily recognized, and even some ofthe cytoskeletal networks which underly the cortex such as theepiplasm and the filamentous infraciliary lattice were seen(Allen, 1971) (Fig. 2b,c). The major differences noted withrespect to conventional chemical fixation are: first, in theappearance of membranes within the cytoplasm and; second,in that of the alveolar lumen. Although the cytoplasmdisplayed its classical ribosome- and glycogen-studded appear-ance, membranes of the rough endoplasmic reticulum, of small
vesicles and even those of mitochondria were difficult torecognize. Thus, mitochondria were revealed more by theiroverall shape, distribution, compactness and ghosts of cristaethan through the conventional architecture of outer and innermembranes. Similarly, the membrane which normallysurrounds the trichocysts was not visible. Treatment of thesections with osmic acid did not modify these patterns. Insummary, cytoplasmic membranes appeared to be extracted.
A converse result was repeatedely observed for the alveolarlumen: while in chemically fixed cells the lumen of the alveoliis usually swollen and essentially electron transparent(‘empty’), as in Fig. 2a, in all types of cryofixed cells the lumenis more flattened and appears to be filled with a meshwork ofelectron-dense material distributed evenly throughout all of theluminal volume (Fig. 2b,c). Note (Fig. 2c) that this fluffymaterial appears to be more abundant along the inner alveolarmembrane, i.e. on the surface facing the epiplasm. A similarappearance can be seen in pictures published by Glas-Albrechtet al. (1991) of Paramecium cells which were rapidly fixed andfreeze-substituted by a somewhat different method. Thepresence of the meshwork does not depend on the presence ofoxalic acid in the cryosubstitution method, is not affected bythe type of embedding resin used and is seen even on unstainedsections, thus indicating that the meshwork is not an artefactdue to oxalate precipitation or staining. Cryofixation thereforereveals the presence of a genuine meshwork that had appar-ently collapsed or was extracted in all previous studies usingchemical fixation.
The overall ultrastructural conservation at first sightappeared to be slightly less good in Lowicryl than in Epon-Araldite. However, the cytoplasm and, especially, thereticulum membranes appeared to be less extracted, and morematerial seemed to be preserved in various complex organellessuch as the axonemes. All further studies presented in thispaper, except for the Lowicryl control, were carried out withEpon-Araldite-embedded material because: the ultrastructuralpreservation appeared to be quite acceptable, these sectionsadhered more easily to the gold substratum required for SIMSmicroscopy and the loss of ions was more limited during thelate stages of processing of Epon sections than Lowicryl ones(especially during recovery of sections on water).
SIMS reveals a Ca compartment at the periphery ofcryofixed Paramecium cellsCell sections of both chemically fixed and cryofixed cells wereanalyzed by SIMS microscopy as described in Materials andMethods. The distribution of a large number of ions wasexamined; Fig. 3 shows an example of the results observed oncryofixed cells with two physiologically important ones, Na+
and Ca2+, as compared to the light microscopic appearance ofan adjacent section. Fig. 3a corresponds to the Toluidine Blue-stained section and shows portions of seven cells, three ofwhich, in the upper part of the picture, lie along the edge ofthe frozen front. The various organelles pointed out in Fig. 1can be recognized.
The most striking differences in spatial distribution of ionsdepend on the fixation method concerned calcium: in chemi-cally fixed cells, a weak Ca signal was uniformly distributedthroughout the cells (not shown; see Fragu et al., 1992), whilein cryofixed ones (Fig. 3b), the signal was restricted to a rela-tively narrow, bright band located around the cell periphery.

1899Calcium stores visualized by SIMS in Paramecium
Fig. 2. Electron microscopyof ultrathin sections of WTParamecium. alv, corticalalveola; ci, cilium; T,trichocyst; M,mitochondrion; pm, plasmamembrane; oam, outeralveolar membrane; iam,inner alveolar membrane;ep, epiplasm; kf,kinetodesmal fibers. Allspecimens were embeddedin Epon-Araldite. Thesections were stained withuranyl acetate followed bylead citrate. (a) Chemicalfixation with 0.5%glutaraldehyde in 50 mMcacodylate buffer, followedby 2% OsO4 in the samebuffer: cortical alveoli areswollen and empty. Bar, 0.5µm. (b) Cryofixation andcryodehydratation inacetone in the presence of20 mM oxalic acid: corticalalveoli are more flattenedand are filled with ameshwork of electron-densematerial. Trichocysts havebarely decondensed. Bar,0.5 µm. (c) Detail of acortical alveola of acryofixed cell: note the goodpreservation of plasma andcortical membranes and thepresence of dense materialthroughout the alveolarlumen and especially facingthe inner alveolarmembrane. Bar, 0.1 µm.
Thus, as suspected, chemical fixation induced a drastic redis-tribution of ions, most clearly seen with Ca2+. Redistributionwas less striking with Na+, K+ and Mg2+ because theseelements were uniformly distributed in cryofixed cells: Na+
yielded a homogeneously and intensely emitting cytoplasm inwhich only some large circular areas, most probably corre-sponding to food vacuoles, were not labelled (Fig. 3c). Thesame was true for Mg2+; K+ yielded a slightly more granular

1900 N. Stelly and others
Fig. 3. SIMS and light microscopy of adjacent cell sections. Thesections traverse a large number of cells located at the edge of thesample (i.e. facing the frozen copper block). (a) Toluidine Blue-stained section; (b) and (c) Ca and Na SIMS observed sectionadjacent to that in (a). Note the regularity of the Ca peripheral signalin (b), its independence from the presence of trichocysts and itsabsence over the macronucleus. Bar, 30 µm.
Fig. 4. SIMS image of Ca in cryofixed WT Paramecium: the semi-thin section was placed over the gold holder without water. Calciumis located around the cell periphery as in Fig. 3f. Image field is 150µm diameter.
or patchy labelling of the cytoplasm, again excluding vacuoles(not shown). In chemically fixed cells, Na+ and K+ were lessuniformly distributed, with K+ concentrated into large precip-itates both inside and outside the cells. The general intensityof emission also appeared to be reduced (not shown; see Fraguet al., 1992). In the remainder of this paper we will thereforeonly be describing cryofixed cells. The presence or absence ofoxalic acid in the cryosubstitution medium did not modify theresults. Because some loss of elements can occur during thefinal stages of specimen preparation, when the sections arebriefly deposited over a water drop on the gold holder and
immediately heated, we prepared some specimens that had nocontact with water at any stage. These are difficult to preparebecause of the tendency of sections to roll over themselves onthe gold holder in the total absence of water. SIMS of such a‘dry’ specimen is shown in Fig. 4; Ca emission, identical tothat seen on sections deposited on water, is evident. As an addi-tional control, cryofixed cells were included in Lowicryl aftercryosubstitution in order to carry out the whole procedure atlow temperature. Although the appearance of the cells in SIMSmicroscopy was slightly more hazy than in the case of Eponembedding, the Ca rim was clearly visible, indicating that therim is not due to ion redistribution occurring during inclusionin Epon.
In fact, when using low levels of integration, a punctateperipheral Ca pattern with a periodicity identical to that of theadjacent cortical alveoli was often observed (see for example,the three cell sections to the right of Fig. 3b). Three pointsrequired evaluation, however, before a definitive acceptance ofthis hypothesis. First, although the difference observedbetween cryo- and chemically fixed cells was encouraging andsuggested a specific Ca localization in cryofixed cells, couldwe be totally confident of our method? One way of answeringthis question would be to show that our methods reveal thewell-known specific Ca localizations in striated muscle.Second, since the large exocytotic vesicles known as tri-chocysts are docked beneath the cell surface in Paramecium inan highly regular arrangement, could they be contributing tothe Ca signal? Indeed, many types of exocytotic vesiclescontain large amounts of Ca (reviewed by Nicaise et al., 1992).This second question could be explored by using cells devoidof trichocysts. Third, did careful observation of a large numberof sections indeed confirm that both the size of the Ca-emittingzone and its precise shape and distribution agree with what isknown for cortical alveoli at the EM level? In particular, does

1901Calcium stores visualized by SIMS in Paramecium
Fig. 5. Frog cutaneousmuscle. (a) A semi-thinsection of a portion of musclefiber cryofixed and observedfor Ca by SIMS. The brightbands correspond to thesarcoplasmic terminalcisternae within the I band.Bar, 25 µm. (b) ToluidineBlue-stained sections of thesame fiber. Bar, 25 µm. (c) Ultrastructure of the samecryofixed fiber where theterminal cisternae (TC) arealigned along the Z diskwithin the I band. M,mitochondrion. Bar, 1 µm.
the width of the compartment agree with what is known of thesize of alveoli in electron microscopy? This last question couldbe analyzed by carefully comparing successive sections ofmaterial, some being observed by conventional lightmicroscopy to locate the major organelles and consecutiveones by SIMS microscopy, and also by comparing the distrib-ution of Ca2+ with that of another ion such as Na+ to checkwhether extracellular adhesion of Ca occurred.These threepoints are addressed below.
SIMS identifies the expected Ca compartment inmuscle cellsSamples of frog cutaneous muscle were prepared using exactlythe same methods as for Paramecium, i.e. by rapid freezing,cryosubstitution in the presence of oxalic acid and embeddingin Epon-Araldite. Conventional light microscopy shows thetypical striated appearance of sarcomeres (Fig. 5b) and electronmicroscopy indicates a reasonably good ultrastructural preser-vation (Fig. 5c). I and A bands are readily identified and theterminal cisternae are clearly seen in the I bands. In this tissue,EM electron-probe analysis of ultrathin cryosections hasshown that 60 to 70% of total fiber Ca is localized in theterminal cisternae, within the I bands (Somlyo et al., 1981).SIMS microscopy (Fig. 5a) shows a regular alternation ofemitting and non-emitting bands for Ca, corresponding,respectively, to the light and dark striations seen in lightmicroscopy, and therefore to I and A bands, respectively. Asexpected, Ca is thus restricted to the I bands in which theterminal cisternae of the sarcoplasmic reticulum are located,and no major loss or redistribution of calcium seem to haveoccurred during sample preparation.
The periphery of the fiber also strongly emitted Ca,probably because of the presence of a dense array of vesicleslying immediately beneath the sarcolemna, referred to ascaveolae by Franzini-Armstrong (1970) and which possiblycontain high amounts of calcium originating from the exta-cellular medium.
Trichocyst-deprived cells still display the peripheralCa compartmentTwo approaches were used to obtain cells devoid of tri-chocysts at the cortex; massive induction of trichocystsdischarge by AED from wild-type cells (Plattner et al., 1984),or use of a mutant (tam8) lacking attached trichocysts at thecortex (Beisson and Rossignol, 1975; Lefort-Tran et al.,1981). The wild-type cells were frozen within 30 seconds afterdischarge; the extent of discharge was monitored byobserving, in a dark-field microscope, aliquots of the AED-treated cell suspension to which picric acid had been added.Although sometimes irregular, AED-induced discharge wasusually quite effective as shown by light and EM microscopycontrols. In these cells rapidly frozen after discharge, somemodifications in the ultrastructural aspect of the alveoli areapparent: they appear to be somewhat collapsed, with theouter membrane disjointed from the plasma membrane, and tocontain less fluffy material than the controls. The mutant cellswere processed as wild-type cells. tam8, clearly, lackedattached trichocysts at the cortex; its trichocysts lay randomlywithin the cytoplasm. One additional mutant strain was used,nd9. This is a conditional mutant, defective in exocytosis at27°C, but displaying a normal complement of trichocystsattached at the cortex. It is therefore defective in only the veryfinal steps of exocytosis (Beisson et al., 1980). This straintherefore allows discrimination between effects due to theabsence of trichocysts at the surface from effects only due tolack of exocytosis potential.
In all cases (AED-treated WT, tam8, nd9), the peripheral Cacompartment was still observed by SIMS microscopy (Fig.6a,b,c). The Ca image of nd9 cells appeared to be identical tothat of WT ones. While in AED-treated WT cells and in tam8ones, the width and intensity of the Ca zone appeared to beslightly reduced. This led us to a more quantitative study asdescribed below. In any case, it became clear that trichocystscannot be the major contributors to the peripheral Ca signal,since this signal was always present in tam8 cells, where con-
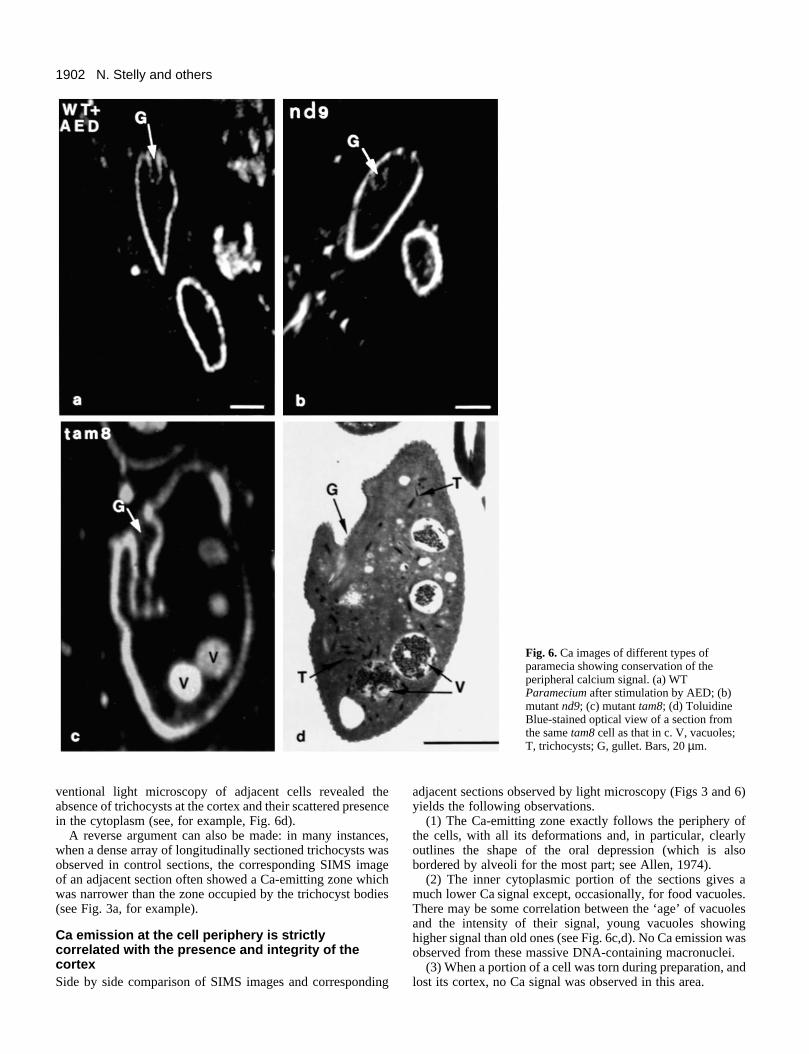
1902 N. Stelly and others
Fig. 6. Ca images of different types ofparamecia showing conservation of theperipheral calcium signal. (a) WTParamecium after stimulation by AED; (b)mutant nd9; (c) mutant tam8; (d) ToluidineBlue-stained optical view of a section fromthe same tam8 cell as that in c. V, vacuoles;T, trichocysts; G, gullet. Bars, 20 µm.
ventional light microscopy of adjacent cells revealed theabsence of trichocysts at the cortex and their scattered presencein the cytoplasm (see, for example, Fig. 6d).
A reverse argument can also be made: in many instances,when a dense array of longitudinally sectioned trichocysts wasobserved in control sections, the corresponding SIMS imageof an adjacent section often showed a Ca-emitting zone whichwas narrower than the zone occupied by the trichocyst bodies(see Fig. 3a, for example).
Ca emission at the cell periphery is strictlycorrelated with the presence and integrity of thecortexSide by side comparison of SIMS images and corresponding
adjacent sections observed by light microscopy (Figs 3 and 6)yields the following observations.
(1) The Ca-emitting zone exactly follows the periphery ofthe cells, with all its deformations and, in particular, clearlyoutlines the shape of the oral depression (which is alsobordered by alveoli for the most part; see Allen, 1974).
(2) The inner cytoplasmic portion of the sections gives amuch lower Ca signal except, occasionally, for food vacuoles.There may be some correlation between the ‘age’ of vacuolesand the intensity of their signal, young vacuoles showinghigher signal than old ones (see Fig. 6c,d). No Ca emission wasobserved from these massive DNA-containing macronuclei.
(3) When a portion of a cell was torn during preparation, andlost its cortex, no Ca signal was observed in this area.
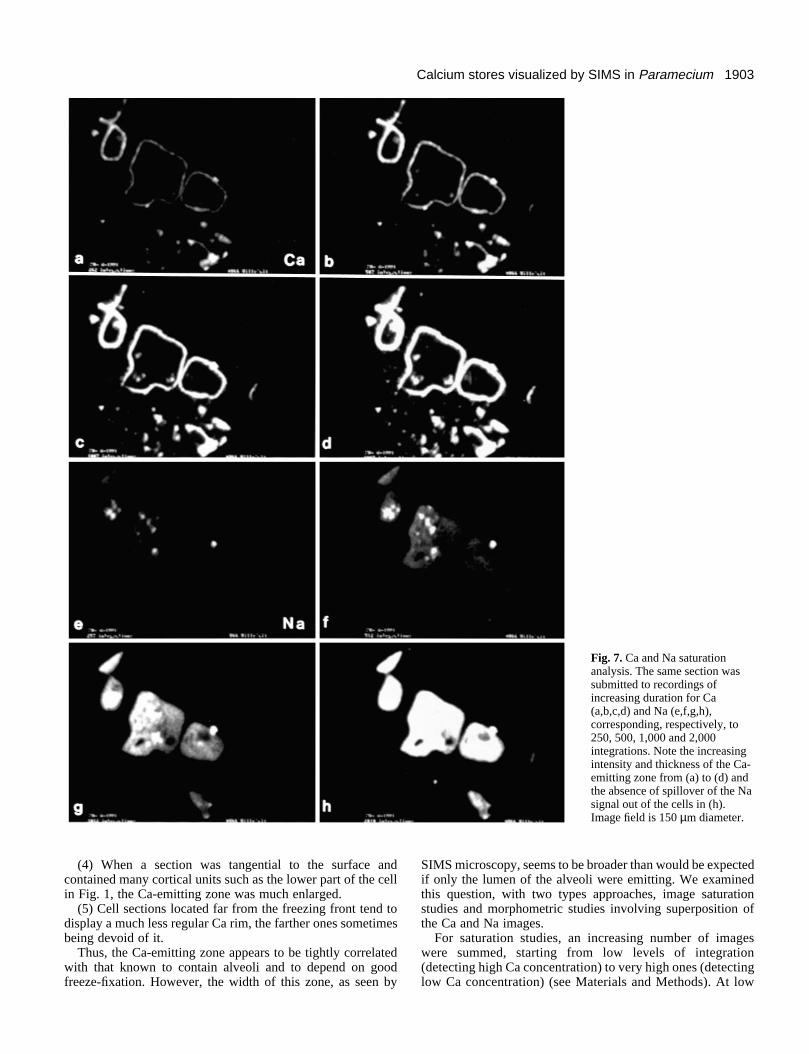
1903Calcium stores visualized by SIMS in Paramecium
Fig. 7. Ca and Na saturationanalysis. The same section wassubmitted to recordings ofincreasing duration for Ca(a,b,c,d) and Na (e,f,g,h),corresponding, respectively, to250, 500, 1,000 and 2,000integrations. Note the increasingintensity and thickness of the Ca-emitting zone from (a) to (d) andthe absence of spillover of the Nasignal out of the cells in (h).Image field is 150 µm diameter.
(4) When a section was tangential to the surface andcontained many cortical units such as the lower part of the cellin Fig. 1, the Ca-emitting zone was much enlarged.
(5) Cell sections located far from the freezing front tend todisplay a much less regular Ca rim, the farther ones sometimesbeing devoid of it.
Thus, the Ca-emitting zone appears to be tightly correlatedwith that known to contain alveoli and to depend on goodfreeze-fixation. However, the width of this zone, as seen by
SIMS microscopy, seems to be broader than would be expectedif only the lumen of the alveoli were emitting. We examinedthis question, with two types approaches, image saturationstudies and morphometric studies involving superposition ofthe Ca and Na images.
For saturation studies, an increasing number of imageswere summed, starting from low levels of integration(detecting high Ca concentration) to very high ones (detectinglow Ca concentration) (see Materials and Methods). At low

1904 N. Stelly and others
Fig. 8. Na and Ca image superposition. The same section was submitted to Ca analysis (a) followed by Na analysis (b) and the two resultingimages were superposed in the computer (c). Note that a peripheral yellow to orange rim is obtained in c, reflecting the good superposition ofthe Ca rim within the limits of the Na border. Also note that the most rapidly frozen front is to the right of the image, as can be seen by theslight deformation of the cells due to their slamming over the copper block and by the presence of extracellular calcium-rich deposists. Incontrast, note that cells deeper in the sample (to the left of the image), although they are detected in the Na image (b), lack the red Ca rim (a),probably because of poor fixation. Bar, 10 µm.
levels, a fine Ca line first appears all along the cell periphery,only very slightly spilling over (as based on the Na and Kimages; see below), but it thickens with increasing integra-tions, reaching a width of approximately 5 µm (Fig. 7a to d).This is broader than the largest width of alveoli as measuredon EM images (approx. 3 µm). Thus, the width of the Ca-emitting zone is broader than what strictly corresponds to thealveoli. The same phenomenon was observed using ‘dry’sections; it is therefore not due to diffusion induced by thewater drop used on the gold holder. Under the same satura-tion conditions, the Na, K and Mg emissions did not spill overacross the cell boundary but remained confined within theirinitial area of distribution (Fig. 7g and h), again indicatingthat the situation observed for Ca reflects a real in situ dis-tribution and not nonspecific diffusion. It should be stressed,however, that the level of integration required to see thewidening of the calcium rim is at least an order of magnitudehigher than that needed to see the initial peripheral signal.Thus, the amount of the excess calcium is much lower thanthat seen at low integration levels.
Widening of the Ca-emitting zone occurred mainly inward.This was established by measurements on the micrographs,superposition of hand-drawn tracings and, most directly, bycomputer superposition of the calcium and sodium images(Fig. 8), using the programs of Olivo et al. (1989). On suchimages, it can be seen that at low integration levels, the red rim(Ca) superposes well over the green background (Na) giving ayellow-orange border, with no indication of a red externalmargin; this remains the case even at higher levels of integra-tion. This shows: first, that the calcium rim detected at lowlevels of integration is located inside the cell; and, second, thatthe outer boundary of calcium distribution does not extendmuch beyond that of Na.
At extremely high integration levels (10 to 20-fold higherthan necessary to see the rim), a weak Ca signal was eventu-ally seen throughout the cytoplasm, most probably reflecting
the quasi-uniform distribution of the much smaller amount oftotal cytoplasmic Ca than that located in the alveoli.
Taken together, these saturation studies indicate thepresence of a thin intracellular peripheral compartment with avery high Ca concentration, surrounded, on the outside, by anarrow Ca-containing zone and, on the inside, by a broader Ca-containing domain where Ca concentration decreases in agraded manner towards the cytoplasm of the cell.
Quantification of alveolar Ca reveals a two-folddecrease after exocytosisAs briefly indicated above, SIMS analysis revealed, in bothtam8 cells and the wild type cells in which a massive releaseof trichocysts was induced, a Ca emission lower than that ofcontrol cells. This had to be quantified rigorously becausesection to section comparison in SIMS microscopy may not bereliable. A proportionality coefficient (Ca/C) was thereforeestablished for each of our measurements by measuring simul-taneously the Ca and C beam intensities within the samevolume of sample. Each measure provided corresponds to themean of 10 measurements carried out over a surface of 8 µmdiameter. This allows a sample to sample comparison (seeMaterials and Methods). In addition, absolute quantification ofCa was achieved using an internal standard consisting ofcalcium octoate included in Epon-Araldite (see Materials andMethods).
Table 1 summarizes the quantitative observations indifferent types of cell sections. As expected, Ca concentrationis significantly higher (about 7-fold) at the cell periphery thanwithin the cytoplasm in all types of cells and conditions (2 to3 mM vs 0.3 to 0.4 mM). It is interesting to compare the Caconcentration within the cortex of Paramecium with that inmuscle, using SIMS. We found the muscle values to oscillatearound 10 mM. This is quite comparable to the value found bySomlyo et al. (1981) using EM microanalysis: their valuesrange from 10 to 117 mmol/kg dry weight, the highest value

1905Calcium stores visualized by SIMS in Paramecium
Table 1. Quantitative evaluation of Ca amountsCortex n1† n2 Cytoplasm n1 n2
Wild type 17.9±8.4.10−1* 35 22 3.0±1.2.10−1 34 30(3 exp.) 3.4±1.8 mM 0.4±0.2 mM
Wild type + 10.5±5.10−1 36 20 3.7±1.5.10−1 15 11AED 1.8±1.2 mM 0.6±0.4 mM
(2 exp.)
tam8 12.4±6.2.10−1 41 14 4.4±3.10−1 11 11(1 exp.) 2.3±1.2 mM 0.7±0.6 mM
tam8 + AED 10.6±4.5.10−1 19 9 4.8±1.7.10−1 7 6(1 exp.) 1.8±1.0 mM 0.8±0.4 mM
nd9 17.9±5.1.10−1 25 12 3.3±1.3.10−1 6 6(1 exp.) 3.4±1.2 mM 0.5±0.2 mM
*The upper line represents the mean of Ca count/C count ± s.d. The secondline is the Ca concentration in mM, calculated using Calcium reference.
†The numbers in the n1 columns refer to the number of surface spots overwhich measurements were carried out; the numbers in the n2 columns refer tothe number of different cell sections examined.
corresponding to a position of their narrow (20 µm) probewithin the terminal cisternae. When converted to mM, the unitsused in the present work, by taking into account the wateramount, their values are 3 to 30 mM, i.e. with an average veryclose to our 10 mM value obtained by SIMS with a much largerprobe diameter.
Thus, the total Ca concentration in the cortex is in the samerange as that of the sarcoplasmic reticulum.
After induction of massive exocytosis by AED, a markeddecrease in Ca concentration in the cortex was repeatedlyobserved (from 3.4±1.8 mM to 1.8±1.2 mM). This decrease isstatistically highly significant using the t-test (P<0.0001).Interestingly, this decrease in the cortex appears to be corre-lated with an increase in Ca concentration within thecytoplasm, but this is barely significant statistically. Thekinetics of these changes was not studied. All the datapresented are from experiments with cells that were cryofixedabout 30 seconds after exocytosis.
Concerning trichocyst mutant strains, nd9 displayed acortical Ca concentration identical to that of wild type, indi-cating that, when trichocysts are attached at the cortex, inca-pacity to carry out exocytosis does not lead per se to a modi-fication of Ca concentration.
In contrast, tam8 displayed a significantly lower amount,placing it at an intermediate level between normal wild typeand wild type after exocytosis. Treatment of tam8 cells withAED further decreased the Ca concentration in the cortex butthis was only marginally significant statistically (P<0.02).
Trichocyst exocytosis or lack of attached trichocysts at thecortex therefore lead to a significant decrease in cortical Caconcentration.
DISCUSSION
The major result presented in this paper is the visualization ofa subcortical Ca compartment in Paramecium using a newmethod, SIMS microscopy, applied to sections of ultrarapidlyfrozen cells. In addition, the amount of Ca in this compartmentwas found to decrease substantially after massive induction ofexocytosis.
The idea that the cortical alveoli, a network of large, inter-connected membrane vesicles directly underlying the plasmamembrane in Paramecium and other ciliates might correspondto a Ca-sequestering compartment akin to the sarcoplasmicreticulum of muscle cells is old (Allen and Eckert, 1969; Satirand Wissig, 1982). It received strong support when wesucceeded in purifying these vesicles and showed that theyactively pump Ca2+ in an ATP- and Mg2+-dependent process(Stelly et al., 1991). Additional evidence was provided by thework of Schmitz et al. (1985), Zierold (1991) and Knoll et al.(1993) using EM microanalysis methods. Here, we have soughtto provide direct visualization of this compartment over largeareas of many cells using a new approach, that of SIMSmicroscopy. The main advantage of this method lies in the factthat it provides images of total Ca distribution over individualcell sections and is thus especially suited for identifying Castorage sites (as compared to methods detecting only freeCa2+). Since the lateral resolution of the method is limited,however, the compartment must be of sufficient size to be iden-tifiable. Because cortical alveoli are typically about 0.2 to 2 µm× 1 to 3 µm in size, we hoped that, if they indeed containedlarge amounts of Ca, SIMS microscopy would allow their visu-alization. In addition, there was a clear prediction as to theexpected intracellular location of the signal, namely through-out the cell periphery.
These predictions were fulfilled remarkably: a continuous,peripheral, Ca-emitting zone was immediately seen, providedthat cells were fixed by rapid freezing.
The good ultrastructural conservation of cells after cryofix-ation, the fact that the peripheral location of Ca strictlydepended on avoiding chemical fixation, and the excellent cor-relation observed between the occurrence of the Ca signal andthe presence of a strip of well frozen cortex in the corre-sponding section, all argue against artifacts. In addition, thefact that it is found also in cortices devoid of trichocystsdemonstrates that the Ca does not emanate predominantly fromthese exocytotic organelles. The most likely Ca-containingcompartment therefore remaining the alveolar one. Additionalevidence on these two points was recently provided by electronprobe microscopy. Using conventional X-ray microanalysis,we found amounts of Ca in a number of alveoli ranging from5 to 10 mM (Stelly, Halpern and Nicaise, unpublished). No Casignal was observed on trichocysts and this is all the more sig-nificant, since these organelles are easily identified in theunstained sections used for microanalysis. Confirmation ofthese two points can be found in a recent study of Knoll et al.(1993) in which one electron energy loss spectrum image ofultrarapidly fixed Paramecium is provided, showing thepresence of Ca in alveoli (not in trichocysts) and its redistrib-ution after exocytosis. It should be pointed out that previousX-ray microanalysis studies had already clearly indicated thepresence of Ca below the cell surface but not in the trichocysts(Schmitz et al., 1985). In summary, all the available evidenceconverges to indicate that trichocysts make little or no contri-bution to the peripheral calcium signal and that the signalemanates predominantly from alveoli.
The width of the Paramecium band, as seen in SIMSmicroscopy, can be estimated as 3 to 5 µm, while the EMobservations indicate that the alveoli do not exceed 2 µm. Inaddition, saturation studies show that this width can reach 10to 20 µm with a narrow Ca zone observed on the outside of the

1906 N. Stelly and others
cell, a phenomenon not seen with Na or K and Mg. Severalexplanations can account for this observation. These fall intotwo broad categories: artefactual diffusion of Ca from thealveoli during sample preparation, or normal presence of Ca inthe proximity of the alveolar compartment. The first hypothe-sis cannot be totally excluded and, in fact, some diffusionwould not be surprising in view of all the steps involved insample preparation. We have excluded the possibility,however, that rapid flotation of sections has a major effect,since sections obtained by completely avoiding any contactwith water showed an identical distribution of all the majorions analyzed. In addition, inclusion in Lowicryl at low tem-perature yielded exactly the same Ca rim as that in Epon-Araldite, indicating that no major diffusion occurs at the timewhen the cryosubstituted samples are warmed up. Finally, thisextracellular emitting zone is Ca-specific and was not observedfor several other ions, indicating that it does not reflect a gen-eralized diffusion from the inside of the cell. The secondhypothesis is therefore much more likely and at least twopossible explanations for a genuine in situ broader Ca zone canbe suggested. First, the cell is covered by cilia and both theplasma and ciliary membranes may be expected to bind a sub-stantial amount of Ca by means of the negatively charged phos-pholipids and the glycosylated cell coat, thus spreading thesignal towards the outside of the cell. Second, concerning thespread of the signal towards the cell’s interior, it should berecalled that a vast filamentous network, the infraciliarynetwork, recently shown to be made up of Ca-binding proteins(Garreau de Loubresse et al., 1991), makes up the deepest ofthe cortical cytoskeletal layers several micrometers below thealveoli. This network may well yield a signal beneath thealveoli on the sections. In addition in Paramecium, especiallyin stationary phase cells, mitochondria tend to be concentratedjust below the cortex, providing a further possible weakercalcium-emitting zone (see Girard et al., 1991, and Rizzuto etal., 1993, for recent evidence of calcium pumping of ER-released calcium by mitochondria). It appears, therefore, thatthree Ca domains can be identified at the cell periphery inParamecium: (i) a narrow zone of very high Ca concentrationcorresponding to alveoli, surrounded by: (ii) a small extracel-lular zone probably corresponding to membrane- and cilium-bound Ca; and (iii) a larger intracellular zone displaying a Caconcentration decreasing towards the inside of the cell.
Through calibration of the absolute amount of Ca using acalcium octoate derivative embedded and analyzed in the sameconditions, the absolute amount of Ca contained in variousareas of the sections was approximated. Within the peripheralband, the mean value was 3.4±1.8 mM. This concentration ishigher by several orders of magnitude than that of freecytosolic Ca2+ (10−4 mM; Eckert, 1972). There are severalimplications of this considerable difference. First, it is mostlikely that Ca is associated with a Ca-sequestering proteinwithin the lumen of the alveoli. In fact, we wonder whether thefluffy material identified in the alveoli only after ultra-rapidfreezing corresponds to such hypothetical sequesteringproteins. A tempting cytological analogy with calsequestrincan indeed be made: only when rapid fixation was used was adiffuse, fluffy material clearly seen in terminal cisternae, whichwas later identified as calsequestrin (Jorgensen and Campbell,1984; Jorgensen et al., 1985). Previously, conventionalchemical fixation had failed to reveal it, as is the case with the
newly discovered material in Paramecium alveoli. Wesearched for a homologue of calsequestrin in the corticalfraction through immunoblotting, Stains-all decoration andradioactive Ca-overlay of gel blots but did not observed asignal in the calsequestrin Mr zone.
The second corollary of the high intra-alveolar Ca concen-tration is the likelihood of a tight control over its release. Pre-viously, we have focused on the analysis of Ca2+ uptake intoalveoli in vitro (Stelly et al., 1991). We are currently charac-terizing the release system kinetically and pharmacologically.There are indirect indications for the operation of an InsP3system in Paramecium (Beisson and Ruiz, 1992) and variouscomponents of the inositide cascade are present (Freund et al.,1992) although InsP3 itself has proven elusive. Similarly, theoccurrence of a Ca2+-induced/Ca2+-release cascade has beensuggested to underly morphogenetic waves during cell division(Le Guyader and Hyver, 1991), but remains to be demon-strated.
As for the function of alveoli in Ca2+ regulation, at leastthree roles might be considered (Stelly et al., 1991), especiallywhen it is noted that the alveoli are in close proximity to threeCa2+-controlled organelles, the cilia, the trichocysts andcytoskeletal networks: (1) general homeostasis of intracellularCa2+ by active sequestration above a given cytosolic level; this,for example, might be the case when a surge of Ca2+ occursthrough depolarization of the ciliary membrane. The alveolibeing immediately adjacent to basal bodies might pump theCa2+ that has entered the ciliary lumen; (2) release of at leastpart of the Ca2+ required for trichocyst exocytosis. In thisrespect, it must be stressed that alveolar membranes are tightlyapposed to the membranes of the tip of trichocysts; (3) releaseof the Ca2+ required for the disassembly of the severalcytoskeletal networks adjacent to the cortex when they undergoreorganization during division (Iftode et al., 1989). Of these,one of the thickest is the infraciliary lattice, which is made upof Ca-binding contractile proteins (Garreau de Loubresse et al.,1991). In the case of this network, the alveoli may also provideCa2+ to regulate contractility.
The present results clearly support the idea that the alveoliprovide at least some of the Ca2+ required during exocytosis,since after massive exocytosis the amount of Ca2+ inside thealveoli dropped by 50%. It should be stressed that such amassive drop in Ca2+ is quite similar to what occurs in the sar-coplasmic reticulum during muscle contraction (Somlyo et al.,1981). In fact, we may not have captured the point of lowestCa2+ concentration, since the cryofixation device used imposesa delay of about 30 seconds between the application of the sec-retagogue (AED) and the fixation of cells. Using faster fixationmethods, Knoll et al. (1993) recently observed a profoundredistribution of alveolar Ca2+ within 80 milliseconds afterAED-induced exocytosis in Paramecium. This observationagrees very well with our results.
The question of the origin of the Ca2+ required for exocyto-sis has been extensively discussed both for Paramecium(Plattner et al., 1991; Knoll et al., 1992, 1993; Cohen andKerboeuf, 1993) and other systems. Our data indicate that acontribution of alveoli to the process is very likely. Interest-ingly, Cohen and Kerboeuf (1993), on the basis of a completelydifferent approach, also concluded that at least part of the Ca2+
involved in trichocyst exocytosis originates from intracellularstores, which, they suggest, might correspond to the alveoli.

1907Calcium stores visualized by SIMS in Paramecium
The exact path followed by alveolar Ca2+ during exocytosis isstill unknown but the tight apposition of alveolar and trichocystmembranes, at the tip of trichocysts, suggests a direct flow,through specific transmembrane proteins.
Assuming that trichocysts themselves contain very littlecalcium, as we concluded above, the fact that exocytoticmutants lacking attached trichocysts show a much decreasedamount of Ca2+ in alveoli, while those having attached tri-chocysts have normal amounts, provides a further hint as to theregulation of alveolar amounts. It suggests that trichocystattachment per se, independently of exocytotic capability,provides a regulatory loop inducing normal Ca2+ pumping andsequestration in alveoli. It should be recalled in this contextthat mutant tam8, which lacks cortex-attached trichocysts, wasshown in our previous work to pump Ca2+ in vitro as efficientlyas the wild type (Stelly et al., 1991). Thus, the in vivo decreasein alveolar Ca described in the present work did not result froman intrinsic defect of the Ca2+-pumping machinery but was duerather to an indirect, presumably regulatory, process.
How wide is the occurrence of such vesicular Ca compart-ments closely apposed to the plasma membrane? The obser-vation made in Paramecium appears to be valid for otherciliates: preliminary SIMS observations carried out in Tetrahy-mena, a genus relatively close to Paramecium in molecularevolutionary terms, but also in Euplotes, a very distant hypotri-chous ciliate (Baroin-Tourancheau et al., 1992), show thepresence of the peripheral Ca band (Stelly, unpublished).Cortical alveoli are in fact a shared ultrastructural character ofthree protist Phyla, ciliates, dinoflagellates and apicomplexa(the Phylum comprising Plasmodium, Toxoplasma, gregarinesand other parasitic protists); these three Phyla have been shownby molecular phylogenetic analysis to form a monophyleticgroup (the ‘alveolata’) (Gajadhar et al., 1991). It is temptingto suggest that in these three groups the alveoli play the roleof Ca-sequestering organelles. In fact, in these three types oforganisms there are exocytotic functions that may correlatewith the presence of alveoli.
Going to much more distant biological groups, the occur-rence of plasma membrane-linked Ca compartments in varioustypes of mammalian cells has been proposed (Putney, 1986).In some highly differentiated metazoan cells, the relation of theendoplasmic reticulum to the plasma membrane becomes quitespecialized, the paradigm being skeletal muscle cells with thetight apposition of a specialized domain of the sarcoplasmicreticulum to the T-tubule membrane, at the level of whichsignal transduction operates (Caswell and Brandt, 1989).
However, the closest analogy at the moment is with eggs ofsea urchins (Luttmer and Longo, 1985) and of ascidians(Gualtieri and Sardet, 1989). There are indeed a number ofstriking similarities between the cortical alveoli of Parame-cium and a vesicular ER network in the cortex of the ova ofvarious sea urchin species (Gardiner and Grey, 1983; Sardet,1984; Terasaki et al., 1991); both actively pump Ca2+ in vitro,both appear to release at least some of the Ca2+ required forexocytosis (trichocysts in Paramecium, cortical granules in seaurchin; Gillot et al., 1991) and both constitute the majorcalcium store of the cell.
The sea urchin egg network has several additional proper-ties which have not been detected in Paramecium: it containscalsequestrin (Henson et al., 1989), releases Ca2+ in responseto InsP3 (Terasaki and Sardet, 1991) and also appears to
contain a ryanodine receptor (McPherson et al., 1992, Sardetet al., 1992). The ciliate Ca stores may therefore be evolution-ary homologues of this ER-derived network and, indeed, ofother Ca storage compartments of ‘higher’ eukaryotes, but thisis an unproven generalization. The search for proteins homol-ogous to those of the compartments of higher eukaryotes inalveolar sacs has been hampered by the great evolutionarydistance separating ciliates from metazoa, which usually leadsto failure of cross-reaction when most antibodies to metazoanproteins are tested in Paramecium. In fact, the exact relation-ship of alveoli to the endoplasmic reticulum in ciliates and theirmode of biogenesis are still unclear. It is being studied atpresent using EM immunocytochemistry by our group, withplasma membrane markers (Charret et al., unpublished;Capdeville et al., 1993).
This work was supported by a grant from the Université Paris-Sud(Action Interdisciplinaire 8922). We thank Dr J. Cohen and D.Kerboeuf for mutant strains and gift of AED, Drs J. P. Mauger and J.Cohen for critical reading of the manuscript, Dr M. Müller for his helpin improving the manuscript and Prof. G. Nicaise for sharing the X-ray results and for comments on the manuscript. We are grateful toMrs N. Narradon for expert photographic assistance and to Mrs C.Couanon for her careful preparation of the manuscript.
REFERENCES
Allen, R. D. and Eckert, R. (1969). A morphological system in ciliatescomparable to the sarcoplasmic reticulum-transverse tubular system instriated muscle. J. Cell. Biol. 43 (2, Pt. 2), 4a (Abstr.)
Allen, R. D. (1971). Fine structure of membranous and microfibrillar systemsin the cortex of Paramecium caudatum. J. Cell Biol. 49, 1-20.
Allen, R. D. (1974). Food vacuole membrane growth with microtubule-associated membrane transport in Paramecium. J. Cell Biol. 63, 904-922.
Andrews, S. B., Leapman, R. D., Landis, D. M. D. and Reese, T. S. (1987).Distribution of calcium and potassium in presynaptic nerve terminals fromcerebellar cortex. Proc. Nat. Acad. Sci. USA 84, 1713-1717.
Baroin-Tourancheau, A., Delgado, P., Perasso, R. and Adoutte, A. (1992).A broad molecular phylogeny of ciliates: Identification of major evolutionarytrends and radiations within the phylum. Proc. Nat. Acad. Sci. USA 89, 9764-9768.
Beisson, J. and Rossignol, M. (1975). Movements and positioning oforganelles in Paramecium aurelia. In Molecular Biology ofNucleocytoplasmic Relationships (ed. S. Puiseux-Dao), pp. 291-294.Elsevier Scientific Publishing Company, Amsterdam, The Netherlands.
Beisson, J., Lefort-Tran, M., Pouphile, M., Rossignol, M. and Satir, B.(1976). Genetic analysis of membrane differentiation in Paramecium.Freeze-fracture study of the trichocyst cycle in wild-type and mutant strains.J. Cell Biol. 69, 126-143.
Beisson, J., Cohen, J., Lefort-Tran, M., Pouphile, M. and Rossignol, M.(1980). Control of membrane fusion in exocytosis. Physiological studies on aParamecium mutant blocked in the final step of the trichocyst extrusionprocess. J. Cell Biol. 85, 213-227.
Beisson, J. and Ruiz, F. (1992). Lithium-induced respecification of pattern inParamecium. Dev. Genet. 13, 194-202.
Burgoyne, R. D. and Cheek, T. R. (1991). Locating intracellular calciumstores. Trends Biochem. Sci. 16, 319-320.
Capdeville, Y., Charret, R., Antony, C., Delorme, J., Nahon, P. andAdoutte, A. (1993). Ciliary and plasma membrane proteins in Paramecium:Description, localization and intracellular transit. In Advances in Cell andMolecular Biology of Membranes, vol. 2A: Membrane Traffic in Protozoa(ed. H. Plattner), pp. 181-226. Jai Press Inc., Greenwich, USA.
Carafoli, E. (1987). Intracellular calcium homeostasis. Annu. Rev. Biochem.56, 395-433.
Castaing, R. and Slodzian, G. (1962). Microanalyse par émission ioniquesecondaire. J. Microsc. 1, 31-38.
Caswell, A. H. and Brandt, N. R. (1989). Open question, Does muscleactivation occur by direct mechanical coupling of transverse tubules tosarcoplasmic reticulum? Trends Biochem. Sci. 14, 161-165.
Chandra, S., Bernius, M. T. and Morrison, G.H. (1986). Intracellular

1908 N. Stelly and others
localization of diffusible elements in frozen-hydrated biological specimenswith ion microscopy. Anal. Chem. 58, 493-496.
Chandra, S. and Morrison, G. H. (1988). Ion microscopy in biology andmedicine. Meth. Enzymol. 158, 157-179.
Chandra, S. and Morrison, G. H. (1992). Sample preparation of animaltissues and cell cultures for secondary ion mass spectrometry (SIMS)microscopy. Biol. Cell 74, 31-42.
Cohen, J. and Kerboeuf, D. (1993). Calcium and trichocyst exocytosis inParamecium: Genetics and physiological studies. In Advances in Cell andMolecular Biology of Membranes, vol. 2A: Membrane Traffic in Protozoa(ed. H. Plattner), pp. 61-81. Jai Press Inc., Greenwich, USA.
Eckert, R. (1972). Bioelectric control of ciliary activity. Locomotion in theciliated protozoa is regulated by membrane-limited calcium fluxes. Science176, 473-481.
Escaig, J. (1983). Techniques de congélation ultra-rapide de specimensbiologiques sans cryo-protecteur. In Microanalyse en Biologie (ed. C.Quintana, and S. Halpern), pp. 105-120. Société Française de MicroscopieElectronique, Paris.
Fragu, P., Briançon, C., Fourré, C., Clerc, J., Casiraghi, O., Jeusset, J.,Omri, F. and Halpern, S. (1992). SIMS microscopy in the biomedical field.Biol. Cell 74, 5-18.
Franzini-Armstrong, C. (1970). Studies of the triad. I. Structure of thejunction in frog twitch fibers. J. Cell Biol. 47, 488-499.
Freund, W. D., Mayr, G. W., Tietz, C. and Schultz, J. E. (1992). Metabolismof inositol phosphates in the protozoan Paramecium. Characterization of anovel inositol-hexakisphosphate-dephosphorylating enzyme. Eur. J.Biochem. 207, 359-367.
Gajadhar, A. A., Marquardt, W. C., Hall, R., Gunderson, J., Ariztia-Carmona, E.V and Sogin, M. L. (1991). Ribosomal RNA sequences ofSarcocystis muris, Theileria annulata and Crypthecodinium cohnii revealevolutionary relationships among apicomplexans, dinoflagellates, andciliates. Mol. Biochem. Parasitol. 45, 147-154.
Gardiner, D. M. and Grey, R. D. (1983). Membrane junctions in Xenopuseggs: Their distribution suggests a role in calcium regulation. J. Cell Biol. 96,1159-1163.
Garreau de Loubresse, N., Klotz, C., Vigues, B., Rutin, J. and Beisson, J.(1991). Ca2+-binding proteins and contractility of the infraciliary lattice inParamecium. Biol. Cell 71, 217-225.
Gillot, I., Ciapa, B., Payan, P. and Sardet, C. (1991). The calcium content ofcortical granules and the loss of calcium from sea urchin eggs at fertilization.Dev. Biol. 146, 396-405.
Girard, J. P., Gillot, I., de Renzis, G. and Payan, P. (1991). Calcium pools insea urchin eggs: Roles of endoplasmic reticulum and mitochondria in relationto fertilization. Cell Calcium 12, 289-299.
Glas-Albrecht, R., Kaesberg, B., Knoll, G., Allmann, K., Pape, R. andPlattner, H. (1991). Synchronised secretory organelle docking inParamecium. Saltatory movement along microtubules transiently formedfrom ciliary basal bodies and selective exclusion of microinjectedheterologous organelles. J. Cell Sci. 100, 45-54.
Grynkiewicz, G., Poenie, M. and Tsien., R. Y. (1985). A new generation ofCa2+ indicators with greatly improved fluorescence properties. J. Biol. Chem.360, 3440-3450.
Gualtieri, R. and Sardet, C. (1989). The endoplasmic reticulum network inthe Ascidian egg: Localization and calcium content. Biol. Cell 65, 301-304.
Henson, J. H., Begg, D. A., Beaulieu, S. M., Fishkind, D. J., Bonder, E. M.,Terasaki, M., Lebeche, D. and Kaminer, B. (1989). A calsequestrin-likeprotein in the endoplasmic reticulum of the sea urchin: Localization anddynamics in the egg and first cell cycle embryo. J. Cell Biol. 109, 149-161.
Iftode, F., Cohen, J., Ruiz, F., Torres Rueda, A., Chen-Shan, L., Adoutte,A. and Beisson, J. (1989). Development of surface pattern during division inParamecium. I. Mapping of duplication and reorganization of corticalcytoskeletal structures in the wild type. Development 105, 191-211.
Jorgensen, A. O. and Campbell, K. P. (1984). Evidence for the presence ofcalsequestrin in two structurally different regions of myocardialsarcoplasmic reticulum. J. Cell Biol. 98, 1597-1602.
Jorgensen, A. O., Shen, A. C. Y. and Campbell, K. P. (1985). Ultrastructurallocalization of calsequestrin in adult rat atrial and ventricular muscle cells. J.Cell Biol. 101, 257-268.
Kerboeuf, D. and Cohen, J. (1990). A Ca2+ influx associated with exocytosisis specifically abolished in a Paramecium exocytotic mutant. J. Cell Biol.111, 2527-2535.
Knoll, G., Kerboeuf, D. and Plattner, H. (1992). A rapid calcium influxduring exocytosis in Paramecium cells is followed by a rise in cyclic GMPwithin 1 s. FEBS Lett. 304, 265-268.
Knoll, G., Grässle, A., Braun, C., Probst, W., Höhne-Zell, B. and Plattner,H. (1993). A calcium influx in neither strictly associated with nor necessaryfor exocytotic membrane fusion in Paramecium cells. Cell Calcium 14, 173-183.
Koch, G. L. E. (1990). The endoplasmic reticulum and calcium storage.BioEssays 12, 527-531.
Lefort-Tran, M, Aufderheide, K., Pouphile, M., Rossignol, M. and Beisson,J. (1981). Control of exocytotic processes: cytological and physiologicalstudies of trichocyst mutants in Paramecium tetraurelia. J. Cell Biol. 88,301-311
Le Guyader, H. and Hyver, C. (1991). Duplication of cortical units on thecortex of Paramecium: A model involving a Ca2+ wave. J. Theor. Biol. 150,261-276.
Linton, R. W. and Goldsmith, J. G. (1992). The role of secondary ion massspectrometry (SIMS) in biological microanalysis: technique comparisonsand prospects. Biol. Cell 74, 147-154.
Luttmer, S. and Longo, F. J. (1985). Ultrastructural and morphometricobservations of cortical endoplasmic reticulum in Arbacia, Spisula andMouse Eggs. Dev. Growth Differ. 27, 349-359.
Lytton, J. and Nigam, S. K. (1992). Intracellular calcium: molecules andpools. Curr. Opin. Cell Biol. 4, 220-226.
McPherson, S. M., McPherson, P. S., Mathews, L., Campbell, K. P. andLongo, F. J. (1992). Cortical localization of a calcium release channel in seaurchin eggs. J. Cell Biol. 116, 1111-1121.
Meldolesi, J., Madeddu, L. and Pozzan, T. (1990). Intracellular Ca2+ storageorganelles in non-muscle cells: heterogeneity and functional assignment.Biochim. Biophys. Acta 1055, 130-140.
Meldolesi, J. and Villa, A. (1993). Endoplasmic reticulum and the control ofCa2+ homeostasis. In Subcellular Biochemistry, vol. 21: Endoplasmicreticulum (ed. N. Borgese and J. R. Harris), pp. 189-207. Plenum Press, NewYork.
Mentré, P. and Escaig, F. (1988). Localization of cations by pyroantimonate.I. Influence of fixation on distribution of calcium and sodium. An approachby analytical ion microscopy. J. Histochem. Cytochem. 36, 49-54.
Nicaise, G., Gillot, I., Julliard, A. K., Keicher, E., Blaineau, S., Amsellem,J., Meyran, J. C., Hernandez-Nicaise, M. L., Ciapa, B. and Gleyzal, C.(1989). X-ray microanalysis of calcium containing organelles in resinembedded tissue. Scanning Microsc. 3, 199-220.
Nicaise, G., Maggio, K., Thirion, S., Horoyan, M. and Keicher, E. (1992).The calcium loading of secretory granules. A possible key event in stimulus-secretion coupling. Biol. Cell 75, 89-99.
Olivo, J. C., Kahn, E., Halpern, S., Briançon, C., Fragu, P. and Di Paola, R.(1989). Microcomputer system for ion microscopy digital imaging andprocessing. J. Microsc. 56, 105-114.
Pietrobon, D., Di Virgilio, F. and Pozzan, T. (1990). Structural and functionalaspects of calcium homeostasis in eukaryotic cells. Eur. J. Biochem. 193,599-622.
Plattner, H., Matt, H., Kersken, H., Haacke, B. and Stürzl, R. (1984).Synchronous exocytosis in Paramecium cells. I. A novel approach. Exp. CellRes. 151, 6-13.
Plattner, H., Stürzl, R. and Matt, H. (1985). Synchronous exocytosis inParamecium cells. IV. Polyamino compounds as potent trigger agents forrepeatable trigger-redocking cycle. Eur. J. Cell Biol. 36, 32-37.
Plattner, H., Lumpert, C. J., Knoll, G., Kissmehl, R., Höhne, B., Momayezi,M. and Glas-Albrecht, R. (1991). Stimulus-secretion coupling inParamecium cells. Eur. J. Cell Biol. 55, 3-16.
Putney, J. W. Jr (1986). A model for receptor-regulated calcium entry. CellCalcium 7, 1-12.
Rizzuto, R., Brini, M., Murgia, M. and Pozzan, T. (1993). Microdomainswith High Ca2+ close to IP3-sensitive channels that are sensed by neighboringmitochondria. Science 262, 744-747.
Sardet, C. (1984). The ultrastructure of the sea urchin cortex isolated beforeand after fertilization. Dev. Biol. 105, 196-210.
Sardet, C., Gillot, I., Ruscher, A., Payan, P., Girard, J. P. and de Renzis, G.(1992). Ryanodine activates sea urchin eggs. Dev. Growth Differ. 34, 37-42
Satir, B. H. and Wissig, S. L. (1982). Alveolar sacs of Tetrahymena:Ultrastructural characteristics and similaritites to subsurface cisterns ofmuscle and nerve. J. Cell Sci. 55, 13-33.
Schmitz, M., Meyer, R. and Zierold, K. (1985). X-ray microanalysis incryosections of natively frozen Paramecium caudatum with regard to iondistribution in ciliates. Scanning Electron Microsc. 1, 433-445.
Sitia, R. and Meldolesi, J. (1992). Endoplasmic reticulum: A dynamicpatchwork of specialized subregions. Mol. Biol. Cell 3, 1067-1072.
Sod, E. W., Crooker, A. R. and Morrison G. H. (1990). Biological

1909Calcium stores visualized by SIMS in Paramecium
cryosection preparation and practical ion yield evaluation for ionmicroscopic analysis. J. Microsc.160, 55-65.
Somlyo, A. V., Gonzalez-Serratos, H., Shuman, H., McClellan, G. andSomlyo, A. P. (1981). Calcium release and ionic changes in the sarcoplasmicreticulum of tetanized muscle: an electron-probe study. J. Cell Biol. 90, 577-594.
Somlyo, A. P. (1985). Cell calcium measurement with electron probe andelectron energy loss analysis. Cell Calcium 6, 197-212.
Stelly, N., Mauger, J. P., Claret, M. and Adoutte, A. (1991). Cortical alveoliof Paramecium: a vast submembranous calcium storage compartment. J.Cell Biol. 113, 103-112.
Terasaki, M. and Sardet, C. (1991). Demonstration of calcium uptake andrelease by sea urchin egg cortical endoplasmic reticulum. J. Cell Biol. 115,1031-1037.
Terasaki, M., Henson, J., Begg, D., Kaminer, B. and Sardet, C. (1991).Characterization of sea urchin endoplasmic reticulum in corticalpreparations. Dev. Biol. 148, 398-401.
Tsien, R. W. and Tsien, R. Y. (1990). Calcium channels, stores, andoscillations. Annu. Rev. Cell Biol. 6, 715-760.
Volpe, P., Krause, K. H., Hashimoto, S., Zorzato, F., Pozzan T., Meldolesi,J. and Lew, D. P. (1988). ‘Calciosome,’ a cytoplasmic organelle: Theinositol 1,4,5-trisphosphate-sensitive Ca2+ store of nonmuscle cells? Proc.Nat. Acad. Sci. USA 85, 1091-1095.
Williams, D. A. and Fay, F. S. (1990). Intracellular calibration of thefluorescent calcium indicator Fura-2. Cell Calcium 11, 75-83.
Zierold, K., Gerke, I. and Schmitz, M. (1989). X-ray microanalysis of fastexocytotic processes. In Electron Probe Microanalysis. Applications inBiology and Medicine. Springer Series in Biophysics, vol. 4, pp. 281-292,Springer, Berlin.
Zierold, K. (1991). Cryofixation methods for ion localization in cells byelectron probe microanalysis: a review. J. Microsc. 161, 357-366.
(Received 12 August 1994 - Accepted 20 February 1995)