edacc primary analysis pipelines
DESCRIPTION
EDACC Primary Analysis Pipelines. Cristian Coarfa Bioinformatics Research Laboratory Molecular and Human Genetics. Data Levels. ChIP-Seq Shotgun Bisulfite Sequencing Methyl-C Reduced Representation Bisulfite Sequencing RRBS MRE-Seq MeDIP-Seq Chromatin Accessibility - PowerPoint PPT PresentationTRANSCRIPT

EDACCPrimary Analysis Pipelines
Cristian CoarfaBioinformatics Research Laboratory
Molecular and Human Genetics

Data Levels

Data Types Submitted To EDACC
• ChIP-Seq • Shotgun Bisulfite Sequencing
– Methyl-C • Reduced Representation Bisulfite Sequencing
– RRBS • MRE-Seq • MeDIP-Seq • Chromatin Accessibility • small RNA-Seq • mRNA-Seq

Read Mapping• Common processing step to all pipelines• High throughput
– Sequence space: Illumina– Color space: SOLID
• Quick and accurate anchoring• Reads size varies 36-76 bp• Short read aligners
– 1st generation: Maq, soap• Ungapped alignment
– 2nd generation: bowtie, bwa, soap 2• Tradeoff speed for sensitivity, good enough for many applications
• Mapping tools– Robust to indels– Sensitive to variable number of mismatches

Pash 3.0
• Positional Hashing
• Regular reads mapping• Bisulfite sequencing mapping• Integrate basepair variation with epigenetic variation
• SAM output, easy integration with other analysis tools• Accuracy without sacrificing efficiency

Bisulfite Sequencing• Current tools: BSMAP, RMAP-BS, mrsFast, Zoom
• Pash 3.0– Integrate mutation discovery with basepair-level methylation discovery– Speedup
• General approach– Covert C’s to T’s in reads and/or reference– Use mappings, reads and reference to determine methylated sites
• Pash 3– Generate and hash all possible kmers for reads– CTT: CCC, CCT, CTC, CTT– Map against forward and reverse complement chromosome strands
• Superior sensitivity to other tools, without loss of efficiency

Galaxy/Genboree
• Developed at Penn State University• Benefits
– Rapid deployment tool– Share pipelines w/ others
• Alan Harris, Sriram Raghuram– Deployed Galaxy/Genboree– Integration w/ Genboree
• API for upload/download– Adaptors for LFF file format support– EDACC XML validation tools
• Sriram Raghuram, Andrew Jackson, Cristian Coarfa– Integration with compute clusters
• Arpit Tandon, Sriram Raghuram– Deployed analysis tools
http://genboree.org/galaxy

Primary Analysis Pipelines
• Implemented & exposed via Galaxy/Genboree– Read mapping– Bisulfite Sequencing read mapping– Peak calling (ChIP-Seq, MeDIP-Seq)
• MACS (Harvard), FindPeaks (UBC)– Chromatin accessibility
• HotSpot (UW)– Small RNA-seq
• Coming soon– mRNA seq– Expression, alternative splicing– Gene fusion
• Typical user interaction– Use Galaxy for user input– Submit jobs to a cluster– Upload results to Genboree
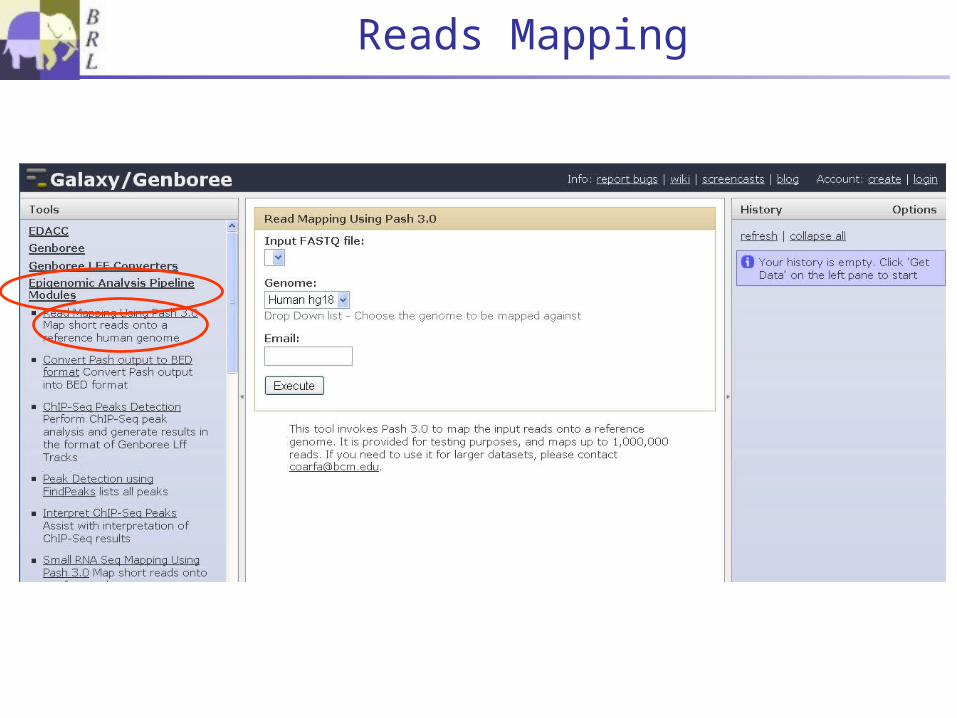
Reads Mapping

ChIP-Seq
• Select uniquely mapping reads • Build read density maps
– Extend each read 200bp along the mapping strand– Remove monoclonal reads– Generate WIG data– Can be visualized in Genboree and UCSC
• Peak calling– FindPeaks, MACS
• Intepret Peaks– Overlap with genomic features of interest: gene promoters, etc

MeDIP-Seq
• Select uniquely mapping reads • Build read density maps• Determine methylated CpGs
– FindPeaks

Finding methylated CpGs

MeDIP-Seq Signal Visualization

MRE-Seq
• Select uniquely mapping reads • Determine unmethylated CpGs

Bisulfite Sequencing
• Shotgun Bisulfite Sequencing– Methyl-C– Genome wide
• Reduced Representation Bisulfite Sequencing– RRBS– Enzyme cocktail
• Map using Pash• Build methylation maps

Bisulfite Sequencing Read Mapping

Methylation MapsPosition Strand CHHStatus Methylation Unmethylated TotalReads50100242 + CG 1 0 150100243 - CG 40 11 5150100250 + CG 1 0 150100251 - CG 37 8 46

Small RNA-Seq
• Trim adapters• Map reads onto target genome
– up to 100 locations per read• Interpret
– Overlap w/ miRNAs, piRNAs, sno/scaRNAs

Exercise
• Download the input MeDIP-Seq file from the workshop wiki
• Analyze it using FindPeaks in Galaxy– Obtain results in Genboree Lff format
• Upload the results to Genboree database• View the results in a tabular view• Find the largest peaks• Explore them in the Genboree browser