effects of good s buffers and ph on the structural ...l−1 horseradish peroxidase (hrp) for h 2 o 2...
TRANSCRIPT

Effects of Good’s Buffers and pH on the Structural Transformation ofZero Valent Iron and the Oxidative Degradation of ContaminantsChuanshu He,†,⊥ Di He,‡,§,⊥ Richard N. Collins,∥ Shikha Garg,∥ Yang Mu,† and T. David Waite*,∥
†CAS Key Laboratory of Urban Pollutant Conversion, Collaborative Innovation Centre of Suzhou Nano Science and Technology,Department of Chemistry, University of Science and Technology of China, Hefei 230026, China‡Institute of Environmental Health and Pollution Control, Guangdong University of Technology, Guangzhou 510006, China§Guangzhou Key Laboratory of Environmental Catalysis and Pollution Control, School of Environmental Science and Engineering,Guangzhou University of Technology, Guangzhou 510006, China∥School of Civil and Environmental Engineering, University of New South Wales, Sydney, NSW 2052, Australia
*S Supporting Information
ABSTRACT: The presence of Good’s buffers caused rapidZVI corrosion and a dramatic release of Fe(II) leading to theFe(II)-catalyzed transformation of ferrihydrite to lepidocrociteand/or the direct formation of lepidocrocite from theoxidation of Fe(II) in the pH range 4.0−6.2. In comparison,in the absence of Good’s buffers, elution of Fe(II) wasinsignificant with ferrihydrite being the only Fe(III) oxy-hydroxide detected following the oxidative transformation ofZVI. The rapid ZVI corrosion in the presence of Good’s bufferis possibly due to either (i) disruption of the Fe oxide surfacelayer as a result of attack by Good’s buffers and/or (ii)interaction of Good’s buffer with the outer Fe oxide surfaceand surface-associated Fe(II)/Fe(III) causing the Fe oxidesurface layers to be more porous with both these processesfacilitating continuous O2 access to the Fe(0) core and allowing the diffusion of Fe atoms outward. Our results further show thatthe deprotonated forms of Good’s buffers and the surface charge of the Fe oxides formed at the ZVI surface strongly affect thesorption of the target compound (i.e., formate) and hence the oxidation of these compounds via surface-associated Fe(II)-mediated heterogeneous Fenton processes.
■ INTRODUCTION
Zero valent iron (ZVI) has been widely applied to degrade avariety of pollutants via reductive pathways, including thereductive transformation of halogenated solvents and reducibleinorganic contaminants to less toxic products.1−4 Morerecently, it has been demonstrated that the corrosion of ZVIin aerated systems can lead to the oxidation of inorganiccontaminants and refractory organic pollutants that are difficultto remove through reductive pathways.5−8 The corrosion ofZVI driven by dissolved oxygen (DO) has been reported toproduce reactive oxygen intermediates (O2
•‑ and H2O2) andFe(II) with further reaction between these products generatingstrongly oxidizing hydroxyl radicals (HO•) and/or Fe(IV)O2+
species via the Fenton reaction.6,7,9,10 The Fe(II) speciesformed during Fe(0) corrosion are oxidized, by both O2 andH2O2, with the resulting Fe(III) species readily hydrolyzing andprecipitating to form various Fe(III) (oxyhydr)oxides thatprovide surface adsorption sites for contaminants.11 Theoxidation products of ZVI vary with the oxidants (e.g., H2O2,O2, KMnO4, NaClO)
8,12−14 and targeted contaminants.12,15
Although various types of iron oxyhdroxides, such as goethite,
hematite, magnetite and other amorphous phases of ironoxides/hydroxides can be produced from the intensive surfacereaction between ZVI and strong oxidants such as H2O2,KMnO4, NaOCl,
8 however ferrihydrite and lepidocrocite arethe main products formed during ZVI oxidation in air.12−14
The elution of Fe(II) and structural transformation ofFe(III) (oxyhydr)oxides could play important role in the ZVI-mediated sequestration of contaminants. It is to be expectedthat the nature and properties of the Fe(III) (oxyhydr)oxidesinitially formed and the relevant surface reactions, includingFe(II) desorption, structural transformation of the Fe(III)(oxyhydr)oxides and the sequestration of contaminants, will behighly pH dependent.16,17 In nonbuffered aerated systems, ithas been observed that pH values converge toward neutralvalues during ZVI corrosion, regardless of the initial pH, withthe reactions shown in eqs 1−7 likely to be occurring. As
Received: August 6, 2017Revised: December 17, 2017Accepted: January 7, 2018Published: January 8, 2018
Article
pubs.acs.org/estCite This: Environ. Sci. Technol. 2018, 52, 1393−1403
© 2018 American Chemical Society 1393 DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403

reported by Sun et al.,16 under acidic conditions, protons areinitially consumed by the Fe(0) corrosion process (eq 1)leading to the formation of Fe(II) and an increase in pH giventhat the subsequent proton-generating Fe(II) oxidation step(eq 2) is relatively slow under acidic conditions although thesubsequent precipitation of Fe(OH)3(s) is accompanied by thegeneration of protons (eq 3). At neutral pH, Fe(II) oxidationoccurs more rapidly with the associated proton generation (eq4) balancing the proton consumption of the corrosion process(eq 1) and leads to the precipitation of Fe(III) (oxyhydr)oxides(eq 5). At alkaline pH, the extent of proton generation in therapid Fe2+ oxidation step (eq 6) is greater than the extent ofproton consumption during the corrosion step (eq 1) andprecipitation step (eq 7) with the result that the pH of theseinitially high pH systems decreases.
+ + → ++ +2Fe O 4H 2Fe 2H O02
22 (1)
+ + → ++ + +4Fe O 6H O 4Fe(OH) 4H (acidic)22 2 2 (2)
→ ++ +Fe(OH) FeOOH(s) H (acidic)2 (3)
+ + → ++ +4Fe O 10H O 4Fe(OH) 8H (neutral)22 2 3
0
(4)
→ +Fe(OH) FeOOH(s) H O(neutral)30
2 (5)
+ + → ++ − +4Fe O 14H O 4Fe(OH) 12H (alkaline)22 2 4
(6)
+ → +− +Fe(OH) H FeOOH(s) 2H O(alkaline)4 2 (7)
In order to undertake studies at fixed pH, buffers arecommonly used with the zwitterionic tertiary amine “Good’s”buffers popular because of their supposed minimal interferencewith Fe chemistry,7,18−20 though an increasing number ofstudies have confirmed the formation of metal ion-Good’sbuffer complexes in recent years.21,22 In fact, pronouncedvariations in contaminant removal rates by ZVI and Fe(III)oxides have been observed in recent studies following theaddition of Good’s buffers.18,19,23−25 Reasons for the effects ofGood’s buffers are varied with, on the one hand, the presence ofGood’s buffers possibly accelerating the elution of Fe(II) fromeither ZVI or magnetite (Fe3O4) resulting in enhancement ofthe rate and extent of contaminant reductive degradation.18,25
On the other hand, complexation between surface-boundFe(II) and Good’s buffers may retard the reduction of targetcontaminants.24 Additionally, Good’s buffers could also serve aseffective •OH scavengers and, as a result, strongly inhibit theoxidative degradation of organic contaminants via Fentonprocesses.26−28
The effect of Good’s buffers on ZVI-mediated oxidativedegradation of contaminants at pH values between 4.0 and 9.0are not well understood. Thus, in this study, insights gainedfrom detailed investigations of Fe(II) elution, ZVI structuraltransformations and ZVI-mediated contaminant oxidation,using 14C-labeled formate (H14COO−) as the target compound,in the absence and presence of Good’s buffers are used tosystematically address the effect of Good’s buffers on ZVI-mediated Fenton process at pH 4.0, 6.2, and 9.0. Note thatoxidation of Good’s buffers by the ZVI/O2 system may alsooccur. However, understanding of transformation of Good’sbuffers does not lie within the scope of this work. Furthermore,since the Good’s buffers used were present in excess in ourstudy, their overall concentration did not change significantly
during experiments and hence the portion of the oxidant(produced by ZVI/O2) scavenged by Good’s buffer remainedconstant during the experiments.
■ MATERIALS AND METHODSReagents. All chemicals were purchased from Sigma-
Aldrich Co. in analytical reagent grade and were used asreceived unless otherwise stated. All solutions were preparedusing 18 MΩ·cm Milli-Q water (Millipore). Glassware wassoaked in a 5% v/v HCl container for at least 72 h and rinsedbefore use. For experiments conducted under anaerobicconditions, Milli-Q water, methanol and stock solutions werepurged with high purity argon gas for at least 3 h before beingtransferred into the anaerobic chamber. Microsized ZVI (Fe0(s),powder, ∼70 mesh, 99%) was obtained from Acros Organics(product no. 197815000) and stored in an anaerobic chamber(855 Series; Plas-Lab Inc.) after receipt. Previous experimentshave indicated that this ZVI has an average particle size of ∼167μm and is principally Fe(0) with traces of FeO (wustite).29
Commercial ZVI is usually covered with discontinuous passivelayers of iron oxides.30 To avoid interference from thisdiscontinuous iron oxide layer, depassivation of ZVI wasexecuted in the anaerobic chamber before any experiments inthe following manner: 10 g of ZVI was preweighed in a 250 mLconical flask containing 100 mL of 0.5 M HCl. The mixture wasthen placed on an orbital shaker at 210 rpm keeping particles insuspension for 1 h with the particles subsequently collected bydiscarding the supernatant. The freshly prepared ZVI particleswere further washed with methanol thrice, dried overnight in anoven at 50 °C and subsequently ground into a fine powder inthe anaerobic chamber prior to use. Unless specifically stated,ZVI in the following sections represents acid-washed ZVI. Notethat acid washing does not affect or confound the effects seen inthe presence of Good’s buffer since the same acid-washed ZVIwas used in nonbuffered and buffered systems.Three commonly applied buffers: 1,4-piperazinedipropane-
sulfonic acid (PIPPS; pH 3.2−4.4), 3-(N-morpholino)propanesulfonic acid (MOPS; pH 6.5−7.9) and N, N, N′,N′-tetrathlethylenediamine (TEEN; pH 9.2−10.4) were usedin the fixed pH experiments at pH 4.0, 6.2, and 9.0, respectively.In order to minimize the variation in pH, a constant Good’sbuffer (PIPPS, MOPS and TEEN) concentration of 100 mMwas used in this study unless otherwise noted. A 20 mM H2O2stock solution was prepared weekly from a 30% w/v H2O2solution and was standardized by UV−Vis spectrometry (usinga molar extinction coefficient of 22.7 M−1cm−1 at 250 nm).31 A0.2 mM radiolabeled formic acid (H14COONa) stock solutionwas prepared from a concentrated H14COONa solution.Lepidocrocite (Lpd) and ferrihydrite (Fhy) were synthesizedaccording to procedures outlined previously.32 Stock solutionsof 200 μM Amplex Red (AR, Invitrogen) mixed with 100 kUL−1 horseradish peroxidase (HRP) for H2O2 measurementswere prepared as reported earlier14 and stored at −80 °C whennot in use. For determination of aqueous and total Fe(II)concentrations, a 3.0 mM 1,10-phenanthroline stock solutionwas prepared in 0.1 M acetic acid solution followed by theadjustment of pH to 6.0 in order to prevent further dissolutionof ZVI during the measurement of total Fe(II). The solutionpH was adjusted when necessary using 1 M NaOH or 1 MHCl.
Experimental Procedures. All experiments evaluating theeffects of Good’s buffers on the oxygenation of ZVI werecarried out at controlled room temperature (22 ± 2 °C) in
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1394

gastight suites placed on an orbital shaker table at 210 rpm asillustrated in Supporting Information (SI) Figure S1. The suiteconsisted of a 500 mL reaction vessel equipped with a spargerinput port and an outflow line to a CO2 trapping vesselcontaining 100 mL of 1.0 M CO2-free NaOH solution. Toinitiate reaction, 2 g·L−1 ZVI or an aliquot of Fe(II) was addedinto 100 mL air-saturated (or deoxygenated) solutionscontaining 1 μM H14COO− as the probe compound. VariablepH experiments were performed at initial pH values of 4.0, 6.2and 9.0, respectively, while fixed pH experiments at pH 4.0, 6.2,and 9.0 were performed in 100 mM corresponding Good’sbuffer systems. Simultaneous measurement of adsorptive andoxidative capacities of ZVI was achieved by continuous air-sparging (or nitrogen-sparging in the anaerobic chamber) inorder to separate 14CO2 from H14COO−. Formation of oxidizedH14COO− (i.e., 14CO2) resulted in an increase in theconcentration of 14CO3
2− in the downstream trapping vesselwhere any 14CO2 driven from the reaction vessel was rapidlyconverted to 14CO3
2− as it passed through the 1.0 M NaOHtrap solution. At predetermined time intervals, the samplestaken from both reaction and trapping vessels were filteredthrough 0.22 μm PVDF syringe filters (Millipore) forsubsequent analysis unless otherwise stated. Following thecompletion of the oxidation process after 24 h, 20 mL of anacidic quench solution (∼12 M HCl and 1 mM formic acid)was introduced into the reaction vessel for dissolution of theZVI and any iron oxides formed on the surface of the ZVIparticles. Note that addition of the quenching solution does notfacilitate oxidation of H14COO− via Fenton processes since anyadditional oxidant generated will be quenched by the largeexcess (1000 times) of nonradiolabeled formic acid present inthe quenching solution. The release of H14COO− into thesolution of the reaction vessel after complete dissolution of thesolids represents “adsorbed” H14COO− while an increase in theconcentration of 14CO3
2− in the trapping vessel enablesquantification of “precipitated” carbonate (in the form of 14C-labeled siderite (FeCO3(s)) resulting from the oxidation ofH14COO−.All experiments were conducted (at least) intriplicate with the data averaged. Given that any sorbedH14COO− is continuously undergoing oxidation, the totalconcentration of sorbed H14COO− at any time point is given bythe sum of oxidized and sorbed H14COO− concentrations.To explore the HO• scavenging ability of Good’s buffer, 1
mL 3% (w/w) H2O2 was added to 100 mL of 0.1 mM Fe(II) inthe absence or presence of 100 mM MOPS (pH 4.0) to initiatethe Fenton reaction. Hydroxyl radicals generated during theFenton reaction were trapped using DMPO (5,5-dimethyl-1-pyrroline N-oxide) (Sigma-Aldrich) and analyzed using anelectron spin (paramagnetic) resonance (EPR) spectrometerJES-FA200 (JEOL Ltd., Tokyo, Japan). To measure the impactof Good’s buffer on HO• generation rate in the ZVI/O2 system,2g·L−1 ZVI was added to the nonbuffered and MOPS bufferedpH 6.2 solutions to initiate generation of HO•. The HO•
generated were trapped using DMPO and analyzed asdescribed above. Note that in the buffered solution, a portionof the HO• generated is scavenged by Good’s buffer and hencethe EPR signal in this case represents an apparent generationrate accounting for the formation as well as consumption ofHO•.Analytical Methods. Determination of aqueous and total
Fe(II) concentrations was carried out using a 1,10-phenanthro-line colorimetric method.29,33 The concentration of aqueousFe(II) (i.e., [Fe(II)]aq) was quantified by addition of 3 mL of 3
mM phenanthroline to 1 mL of diluted (from 1 to 100 times)samples following their filtration through 0.22 μm PVDF filters(Millipore). The total Fe(II) concentration (i.e., [Fe(II)]T,including aqueous and surface associated Fe(II)) was quantifiedby addition of 3 mM phenanthroline prior to filtration. Controlexperiments were performed to ensure that (i) complexation ofFe(II) by phenanthroline outcompetes the oxidation of Fe(II)and (ii) phenanthroline traps both aqueous and surfaceassociated Fe(II) in the measurement system.14
The total H2O2 concentrations (including free and surface-associated H2O2) were measured using the Amplex Redmethod.13,14,34 H2O2 oxidizes Amplex Red (AR) to producehighly fluorescent resorufin (λex = 563 nm, λem = 587 nm), theconcentration of which was quantified using a fluorometer(Varian Cary Eclipse). For measurement, 1 mL of sample wasdiluted with 2 mL of 10.0 mM MOPS buffer (∼pH 7.0)containing 60 μL of AR/HRP solution in a 1 cm quartz cuvette.Calibration was performed daily over the concentration rangeof 0−800 nM by the standard addition of H2O2 to ourexperimental matrix.The concentrations of H14COO− in the reaction vessel and
14CO32− in the trapping vessel were measured by mixing 1 mL
of the sample solution with 5 mL of liquid scintillation fluid(Ultima GOLD, PACKARD) for subsequent analysis in aPackard Tri-Carb 2100TR scintillation counter.35
All pH measurements were made using a Hanna HI9025 pHmeter combined with a glass electrode and Ag/AgCl reference.Dissolved oxygen concentrations in the reaction vessel weredetermined through the course of the reaction by use of aEZDO-7031 microprocessor-based waterproof dissolved oxy-gen meter.The relative proportions of the different Fe-containing solid
phases in the reaction vessel were determined by Fe K-edgeextended X-ray absorption fine structure (EXAFS) spectro-scopic analysis of samples collected through the course of thereaction. EXAFS measurements were conducted at the wigglerXAS beamline (ID 12) at the Australian Synchrotron (Clayton,Australia) which operates a third-generation 3 GeV storagering.13,36 Spectra were recorded in transmission mode using aliquid nitrogen-cooled double crystal fixed-exit monochroma-tors with the beam focused (fully tuned) both vertically andhorizontally with a Rh-coated mirror. Approximately 20−30 mgof freeze-dried powdered reference iron oxides (ferrihydrite andlepidocrocite) and samples were carefully mixed with boronnitride and packed into aluminum slides for EXAFS trans-mission analysis. Samples were cooled to ∼7 K using a closedHe cryostat. Spectra were energy calibrated with the softwareAverage and normalized and background corrected using thestandard features of ATHENA. Linear combination fits (LCF)of oxygenated ZVI samples, using ferrihydrite, lepidocrocite andacid-washed ZVI samples as standards, were also carried out inATHENA over k = 2−12 Å−1 (k3-weighted). The R-factors forthese fits ranged from 0.004 to 0.089 (average 0.05, n = 48)indicating that these three minerals comprised the majority, ifnot all, of the solid phases present during ZVI oxidation inthese experiments. Ferrihydrite and lepidocrocite are quiteoften reported to be the main iron (oxyhydr)oxides formed onZVI oxidation in aerated environments.12−14 Furthermore, theinclusion of magnetite, hematite, goethite and wustite did notlead to improved fits, thereby confirming that the proportion ofthese minerals present was relatively negligible.Transformation kinetic models were fitted to the exper-
imental data using KinTek explorer with this software package
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1395
enabling multiple data sets to be fit simultaneously to a singlemodel based on numerical integration of the rate equationsdescribing the reaction mechanism.37
The morphological characterization of ZVI and surfaceelemental mapping of Fe, C, and O on the ZVI particles after 1h reaction in both nonbuffered suspensions and bufferedsystems was performed using scanning electron microscopy-energy dispersive X-ray (SEM−EDX) analyses (JSM-7001F,JEOL). The samples were collected from the reactionsuspensions by centrifugation. To avoid postreaction ofGood’s buffer on the surface of the particles, the ZVI sampleswere washed two times using ethanol before loading ontosilicon holders and drying overnight in the anaerobic chamber.
■ RESULTS
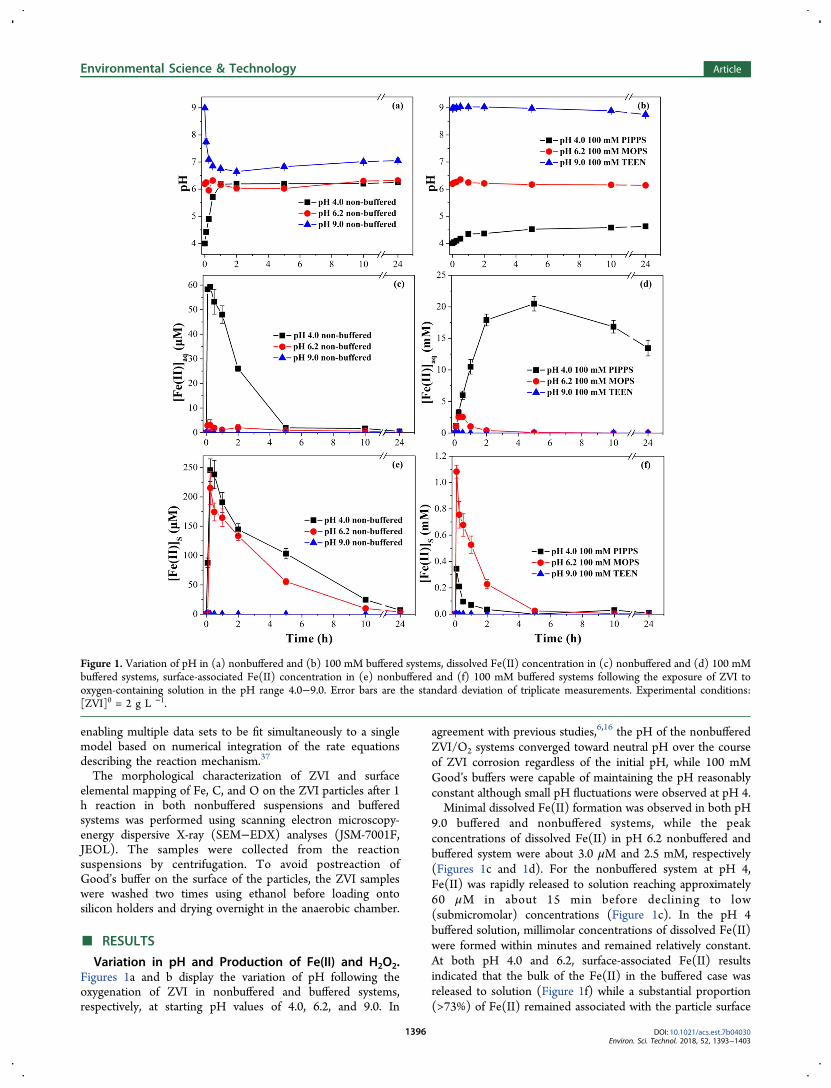
Variation in pH and Production of Fe(II) and H2O2.Figures 1a and b display the variation of pH following theoxygenation of ZVI in nonbuffered and buffered systems,respectively, at starting pH values of 4.0, 6.2, and 9.0. In
agreement with previous studies,6,16 the pH of the nonbufferedZVI/O2 systems converged toward neutral pH over the courseof ZVI corrosion regardless of the initial pH, while 100 mMGood’s buffers were capable of maintaining the pH reasonablyconstant although small pH fluctuations were observed at pH 4.Minimal dissolved Fe(II) formation was observed in both pH
9.0 buffered and nonbuffered systems, while the peakconcentrations of dissolved Fe(II) in pH 6.2 nonbuffered andbuffered system were about 3.0 μM and 2.5 mM, respectively(Figures 1c and 1d). For the nonbuffered system at pH 4,Fe(II) was rapidly released to solution reaching approximately60 μM in about 15 min before declining to low(submicromolar) concentrations (Figure 1c). In the pH 4buffered solution, millimolar concentrations of dissolved Fe(II)were formed within minutes and remained relatively constant.At both pH 4.0 and 6.2, surface-associated Fe(II) resultsindicated that the bulk of the Fe(II) in the buffered case wasreleased to solution (Figure 1f) while a substantial proportion(>73%) of Fe(II) remained associated with the particle surface
Figure 1. Variation of pH in (a) nonbuffered and (b) 100 mM buffered systems, dissolved Fe(II) concentration in (c) nonbuffered and (d) 100 mMbuffered systems, surface-associated Fe(II) concentration in (e) nonbuffered and (f) 100 mM buffered systems following the exposure of ZVI tooxygen-containing solution in the pH range 4.0−9.0. Error bars are the standard deviation of triplicate measurements. Experimental conditions:[ZVI]0 = 2 g L −1.
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1396
in the nonbuffered solution (Figure 1e). The concentration ofsurface-associated Fe(II) (at least at pH 4.0 and 6.2) in bothbuffered and nonbuffered systems increased rapidly initially butgradually decreased to low levels thereafter.In comparison to the noteworthy differences in the extent of
Fe(II) generated in nonbuffered and buffered systems, thesteady-state concentrations of H2O2 in both nonbuffered andbuffered cases displayed relatively similar profiles (SI Figure S2,SI). Total H2O2 concentrations reached maximum valueswithin 0.5 h but decreased slowly thereafter in bothnonbuffered and buffered systems at all pH values investigatedhere. The reactions potentially contributing to this decrease inH2O2 concentrations include (i) depletion of Fe(0) (eqs 8 and9), (ii) interplay among Fe(II), O2 and H2O2 (eqs 11−13), and(iii) scavenging interaction between Fe(0) and H2O2 (eq14)13,38
+ ⎯ →⎯⎯⎯⎯ − ++
−Fe O 2H Fe Fe H On n0
2 10 II
2 2 (8)
+ → − + +−−Fe 2H O Fe Fe H (g) 2OHn n
02 1
0 II2 (9)
− ⇌ +− −Fe Fe Fe Fe (aq)n n10 II
10 II
(10)
− + → − + +− −• −Fe Fe H O Fe Fe HO OHn n1
0 II2 2 1
0 III
(11)
− + → − +− −•−Fe Fe O Fe Fe On n1
0 II2 1
0 III2 (12)
+ ⎯ →⎯⎯⎯⎯ +•− •−+
O O 2H H O O2 2 2 2 2 (13)
+ ⎯ →⎯⎯⎯⎯ − ++
−Fe H O 2H Fe Fe 2H On n0
2 2 10 II
2 (14)
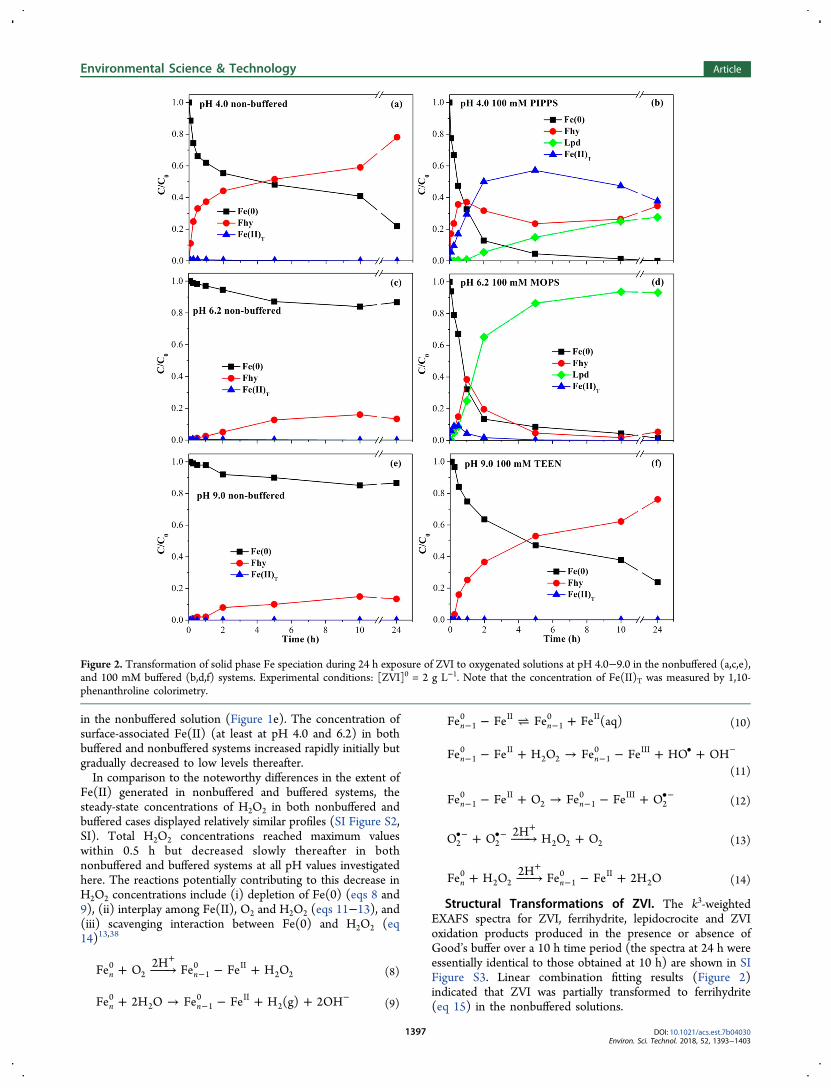
Structural Transformations of ZVI. The k3-weightedEXAFS spectra for ZVI, ferrihydrite, lepidocrocite and ZVIoxidation products produced in the presence or absence ofGood’s buffer over a 10 h time period (the spectra at 24 h wereessentially identical to those obtained at 10 h) are shown in SIFigure S3. Linear combination fitting results (Figure 2)indicated that ZVI was partially transformed to ferrihydrite(eq 15) in the nonbuffered solutions.
Figure 2. Transformation of solid phase Fe speciation during 24 h exposure of ZVI to oxygenated solutions at pH 4.0−9.0 in the nonbuffered (a,c,e),and 100 mM buffered (b,d,f) systems. Experimental conditions: [ZVI]0 = 2 g L−1. Note that the concentration of Fe(II)T was measured by 1,10-phenanthroline colorimetry.
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1397

− → −− −Fe Fe(III) Fe Fhyn n10
m0
(15)
As observed from Figure 2a, biphasic transformation of ZVIto Fhy occurred with the transformation (or “corrosion”) ratein stage I (0−2 h) corresponding to the change of pH (Figure1a) with lower pH coincident with faster corrosion while thecorrosion rate at stage II (2−10 h) remained similar in thethree cases in accord with the shift of pH to the neutral pHrange.Due to a large amount of Fe(II) (∼1 mM) associated with
the surface of ZVI in the buffered system at pH 6.2, Fe(II)-catalyzed transformation of ferrihydrite to lepidocrocite (eq 16)occurred rapidly and extensively, in a similar manner asreported for nano-ZVI.13 Furthermore, direct oxidation ofdissolved Fe(II) will also contribute to lepidocrocite formation(eq 17). At pH 4.0−4.5, a significant decrease in the maximumconcentration of surface-associated Fe(II) (e.g., ∼ 0.3 mM) was
observed due to increased proton competition and the morepositively charged nature of the oxidized ZVI surface and, assuch, the Fe(II)-catalyzed transformation of ferrihydrite tolepidocrocite is not expected to be as favorable as at pH 6.2. Itshould be recognized, however, that the presence of a highconcentration of dissolved Fe(II) (∼20 mM) at pH 4.0−4.5may well contribute to the formation of lepidocrocite throughdirect oxidation of Fe(II) (eq 17). Nevertheless, this direct andthe indirect Fe(II)-catalyzed pathways lead, cumulatively, tosubstantially less lepidocrocite formation at pH 4.0 thanobserved in the pH 6.2 buffered system.39
− ⎯ →⎯⎯⎯⎯⎯⎯⎯⎯⎯>
−− −Fe FhyFe(II)
Fe Lpdn m n m0 0
(16)
⎯→⎯Fe(II)O
Lpd(aq)2
(17)
Figure 3. Time course results showing proportion of H14COO− remaining in the reaction vessel and proportion converted to 14CO2 in the collectionvessel following addition of ZVI to air-saturated water containing radio-labeled H14COO− at pH 4.0−9.0 in (a,c,e) nonbuffered system and (b,d,f)100 mM buffered systems. Proportion of H14COO− converted to various products after 24 h reaction time is shown in the column graph withproportions of adsorbed H14COO− and H14COO− converted to siderite determined by the increase in 14C signal in reaction and collection vesselsfollowing acid digestion of solids in reaction vessel, respectively (as depicted by arrow). Error bars are the standard deviation of triplicatemeasurements. Experimental conditions: [ZVI]0 = 2 g L−1. Note that the concentration of total 14C represents the sum of 14C concentration in thesolution phases of reaction and collection vessels. The concentration of total 14C coincides with the concentration of H14COO− in the reaction vesselas there is no formation of 14CO2 in TEEN buffered system at pH 9.0.
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1398

As neither aqueous nor surface-associated Fe(II) wasobserved in the pH 9.0 buffered system, it is perhaps notsurprising that there was no apparent conversion of ferrihydriteto lepidocrocite at this pH. The exact reason for more rapidtransformation of Fe(0) to ferrihydrite in the buffered system(pH 9.0) compared to the nonbuffered system (pH 9.0) isunclear but could be associated with the formation of moreporous ZVI oxide layer due to deposition of TEEN buffer and/or disruption of ZVI surface oxide layer by TEEN buffer withboth these processes increasing the accessibility of H2O/O2 tothe underlying Fe(0) with this process contributing to morerapid formation of Fe(II) which quickly transforms toferrihydrite at pH 9.0 due to the fast Fe(II) oxidation kineticsunder alkaline conditions.Oxidation of H14COO−. To understand the effect of
Good’s buffers on the oxidizing capacity of ZVI, the conversionof H14COO− to 14CO2 was measured. Results shown in Figure3 reveal that substantial removal of H14COO−from solution(∼68.0−76.6%) was achieved in the nonbuffered systems andwithin 2 h ∼ 27.5%, 12.5%, and 7.1% of H14COO− wasconverted to14CO2 at pH 4.0, 6.2, and 9.0, respectively. Thehighly pH dependent oxidation of H14COO− is likely related to
decrease in the extent of generation of the oxidants (within thefirst hour) with increase in pH. The decrease in oxidantgeneration could occur as a result of either a decrease in Fe(II)concentration produced with increase in pH (Figure 1) and/ora stronger ability of O2 to outcompete H2O2 for Fe(II) speciesat higher pH as reported earlier10 with both effects leading to areduction in the generation of oxidizing radicals. Surprisingly,only 20.0%, 29.2% and 1.6% of H14COO−was removed fromthe aqueous phase after 24 h reaction in the buffered system atpH 4.0, 6.2, and 9.0, respectively (Figures 3b, d, f), whereas theconversion of H14COO− to 14CO2 was ∼10.9%, 24.9% ,and 0%at pH 4.0, 6.2, and 9.0 respectively.To elucidate the role of homogeneous Fenton reaction in the
ZVI/O2 systems, Fe(II) and H2O2 concentrations similar to themeasured steady-state concentration of these species after 0.5 hfollowing ZVI oxidation at pH 6.2 (i.e., 6 μM Fe(II) and 1.6μM H2O2 and 3 mM Fe(II) and 2.5 μM H2O2 for thenonbuffered and MOPS buffered solutions, respectively) wereadded to pH 6.2 solutions containing 1 μM H14COO−. Asdepicted in Figure S4, ∼ 0% and 2.8% H14COO− were oxidizedin the nonbuffered and MOPS-buffered systems respectively,following 24 h of reaction. Minimal oxidation of H14COO− via
Figure 4. From left to right: SEM images and EDS elemental maps (Fe, O, and C and combined Fe, O, C) of ZVI following 1 h exposure to pH 4.0(a) nonbuffered and (b) 100 mM PIPPS buffered systems, pH 6.2 (c) nonbuffered and (d) 100 mM MOPS buffered systems, and pH 9.0 (e)nonbuffered system and (f) 100 mM TEEN buffered system.
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1399

homogeneous Fenton reaction strongly suggests that the ZVI-mediated oxidation of H14COO−occurs at the surface of theseparticles in the presence as well as the absence of Good’sbuffers.
■ DISCUSSIONCorrosion of ZVI. The present study demonstrates that the
use of Good’s buffers in ZVI/O2 systems induces severecorrosion of ZVI with a marked release of Fe(II) resulting inFe(II)-catalyzed transformation of ferrihydrite to lepidocrociteand/or the formation of lepidocrocite from the direct oxidationof Fe(II) in the pH range 4.0−6.2. Since the pH in bothnonbuffered and buffered systems was maintained at ∼6.2 inthe pH 6.2 system, the difference in the ZVI corrosion behaviorwere mainly induced by the presence of the Good’s buffersrather than any variation in pH. Even in the pH 4 system, whencomparing the buffered system with the nonbuffered system inwhich pH was manually maintained at 4 by HCl addition (SIFigure S5b), the concentrations of Fe(II) released aresignificantly different further confirming the dramatic con-tribution of Good’s buffers in enhancing the rate of ZVIcorrosion. The addition of 100 mM Good’s buffer introduces100 mM Na+ in the solution however the increase in the saltconcentration does not contribute to ZVI corrosion. As shownin SI Figure S5, even though addition of 100 mM NaCl (whichis close to the Na+ concentration in the buffered system) in thenonbuffered pH 6.2 solution results in an increase in theconcentration of Fe(II) released (SI Figure S5a), however theimpact of NaCl addition is not significant when compared tothe overall effect of Good’s buffers on Fe(II) release furthersupporting the finding that Good’s buffers induce ZVIcorrosion. Our results further show that the increased ZVIcorrosion rate is not due to the effect of Good’s buffers on thedegree of Fe(II) sorption on Fe(III) oxides, with the resultspresented in SI Figure S6 revealing that the presence of Good’sbuffers had no significant effect on the extent of Fe(II) sorptionto the Fe(III) oxides present under anoxic conditions.Since Fe dissolution and Fe oxide formation both occur on
the ZVI surface,40 surface analyses can facilitate to elucidate themechanism of ZVI corrosion caused by Good’s buffers. SEMimages of ZVI following 1 h exposure to oxygenated water(Figure 4) revealed that the Fe oxides covering the surfaceappear even more loose and porous in the presence of PIPPS(Figure 4b), MOPS (Figure 4d) and TEEN buffer (Figure 4f).Elemental mapping using SEM-EDX (Figure 4) and relativecontents of Fe, C, O calculated via repeated EDX measure-ments (at least three samples of each group) (SI Table S1)showed the presence of significant amounts of carbon at theparticle surfaces in the buffered systems supporting thatdeposition of the Good’s buffers on the particle surface occurin accord with the findings by previous investigators.24 It shouldbe noted that the ZVI suspensions were thoroughly washedbefore being transferred to the silicon holder thereby avoidingthe deposition of Good’s buffers onto the surface of ZVIparticles during drying. Furthermore, even though ethanol wasused for washing the ZVI suspension, most of the ethanol usedevaporated during drying as confirmed by the low carboncontent of the ZVI suspension prepared in the nonbufferedsolution (Figure 4). As such, the high abundance of C on theZVI surface in suspensions containing Good’s buffers can beattributed to the association of the Good’s buffers with theFe(III) oxyhydroxides that are formed at the surface of the ZVIparticles as they corrode. Generally, the surface oxide layers
formed on the ZVI surface by the hydrolysis of Fe(III) wouldprotect the remaining Fe(0) from attack by oxygen11 and limitfurther corrosion of Fe(0). However, the presence of Good’sbuffers may attack and disrupt the surface oxide layers during itsformation, as has been reported to occur in the presence ofinorganic anions like Ca2+, Mg2+, Fe2+,41,42 allowing continuousaccess of H2O/O2 to the Fe(0) core. Alternatively, it is alsopossible that the incorporation of Good’s buffer into thegrowing iron oxide coatings results in the formation of porousassemblages due to electrostatic and/or steric effects with theseassemblages more prone to H2O and/or O2 penetration.In accord with above hypotheses, the consumption of
dissolved oxygen (DO) in the Good’s buffer systems was fasterthan that observed in the nonbuffered systems during the initial2 h of reaction (Figure S7). At the initial stage (0−0.5 h), DOwould most likely be consumed by Fe(0) with the generation ofH2O2 and Fe(II) according to eq 831 with observation of thedramatic release of Fe(II) within the first 0.5 h (Figure 1)consistent with such a process. The so formed Fe(II) canundergo oxygenation via the reaction shown in eq 12 therebyresulting in further consumption of DO as confirmed by theresults in Figure S8 which show DO consumption in solutioncontaining dissolved Fe(II) at pH 6.2. In the nonbufferedsystem, the formation of compact surface Fe oxide layers couldprevent the Fe(0) core from continuous attack by O2 while themuch more significant variation of DO over the course of ZVIcorrosion in suspensions containing Good’s buffers providesconfirmation that the presence of Good’s buffers may indeeddisrupt/transform the protective oxide layer facilitating thecontinuous access of O2 to the Fe(0) core. The impact ofGood’s buffer on the reactivity of iron oxide layers is furthersupported by the difference in the dissolution rates of ironoxides (i.e., ferrihydrite) formed in the presence and absence ofMOPS buffer (see Section S2 for a detailed description of theseresults). As shown in Figure S9, the measured dissolution rateconstant of ferrihydrite formed in the presence of MOPS bufferwas higher than that observed in the absence of MOPS buffer.This observation is in agreement with the increased corrosionof ZVI observed in the presence of Good’s buffer.
Oxidation of H14COO−. Given that a surface associatedFenton process is responsible for oxidation of H14COO− underthe experimental conditions investigated here, the presence ofGood’s buffer may impact the oxidation of H14COO− by ZVI asa result of
(1) modification of surface oxide layers;(2) decrease in the extent of H14COO− sorption on the ZVI
surface; and/or(3) competition between H14COO− and Good’s buffer
molecules for the HO• generated by ZVI/O2 system.
The results shown in Figure 1 and 2 support the conclusionthat the extent and the nature of the surface oxide layer formedin the buffered and nonbuffered systems varies significantly andhence will have implications to oxidant generation and HCOO−
sorption. Since a higher surface associated Fe(II) concentrationis observed in the buffered-solution, the overall oxidantgeneration via Fenton reaction is expected to be higher underthese conditions. This is supported by the results in SI FigureS11 that show higher HO• yield on oxidation of ZVI in thebuffered solution compared to that measured in the non-buffered solution at pH 6.2.Our results support the conclusion that the presence of
Good’s buffers diminished the extent of HCOO− sorption on
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1400

the ZVI surface (SI Table S2). Even though rapid trans-formation of ZVI to ferrihydrite occurred in the bufferedsolution, limited HCOO− sorption occurred in these solutionsat all pH values investigated here with this result indicating thatGood’s buffers interfere with the sorption of HCOO− on theZVI surface, presumably due to steric or electrostatic repulsionby the Good’s buffer. The interference of Good’s buffer onsorption of HCOO− is further supported by the observationthat less H14COO− is sorbed on the ferrihydrite surface in thepresence of 100 MOPS or 100 mM PIPPS compared to thatsorbed in the absence of buffer under neutral pH conditions (SIFigure S12). The difference in the extent of HCOO− sorptionin buffered and nonbuffered ZVI systems is more pronouncedin the systems containing more negatively charged Good’sbuffers (i.e., PIPPS and TEEN in view of the pKa values ofPIPPS (pKa1 = 3.79, pKa2 = 7.97), MOPS (pKa = 7.09) andTEEN (pKa1 = 6.58, pKa2 = 9.88)) due to increasedelectrostatic repulsion.The scavenging of HO• by Good’s buffer has been reported
to occur in previous studies43,44 and is also supported by our
EPR analysis (SI Figure S10). However, the observation of ahigher HO• yield on oxidation of ZVI in the buffered systemcompared to that in nonbuffered system (SI Figure S11)suggests that the impact of Good’s buffer on scavenging of HO•
plays a minor role compared to the impact of Good’s buffer onthe rate of oxidant generation via Fenton processes as a resultof the increased concentration of surface associated Fe(II)under these conditions.We thus conclude that (i) the rapid transformation of ZVI,
(ii) higher surface associated Fe(II) concentration and (iii)lower HCOO− sorption in the buffered system compared tothe nonbuffered system control the overall extent of oxidationof HCOO−. Even though the overall oxidation of H14COO− islower in the buffered system as result of lower sorption, thefraction of sorbed H14COO− that is oxidized to 14CO3
2 isexpected to be higher under these conditions as a result of thehigher HO• generation rate in the buffered-system (SI FigureS11). To probe this we calculated the fraction of sorbedH14COO− that is oxidized to 14CO3
2− using the followingequation:
=+
+ +− −
− −
− − −%oxidized HC OO of sorbed HC OO(oxidized HC OO preciptated HC OO )
(oxidized HC OO preciptated HC OO sorbed HC OO )14 14
14 14
14 14 14(18)
Note that the denominator in eq 18 represents the totalsorbed H14COO− and includes both the adsorbed and oxidizedfractions of H14COO− since any adsorbed H14COO− will beundergoing continuous oxidation.As shown in SI Table S2, 69.4% and 31.8% of the adsorbed
H14COO− was oxidized in the nonbuffered system at pH 4.0and 6.2 respectively while 84.1% and 94.9% of the adsorbedH14COO− was converted to 14CO3
2− in the correspondingbuffered systems confirming that a much higher degree ofsorbed H14COO− is mineralized to 14CO3
2− in the PIPPS andMOPS-buffered systems than in the nonbuffered solution. Notethat this comparison cannot be performed for pH 9 since noH14COO− is sorbed under this pH condition in the bufferedsolution. The higher degree of conversion of sorbed H14COO−
in buffered solution compared to nonbuffered solution is inagreement with the observation of a higher concentration ofsurface-associated Fe(II) (produced from rapid ZVI corrosion)in buffered solution which will result in increased oxidantgeneration (and hence more efficient H14COO− oxidation)through a heterogeneous Fenton process.The pH dependence of the conversion rate of H14COO− in
the buffered solution further confirms that the extent ofoxidation of sorbed H14COO− is related to the surfaceassociated Fe(II) concentration. As shown in SI Table S2,less sorbed H14COO− was converted to 14CO3
2− in the pH 4buffered system (84.1%) compared to the pH 6.2 bufferedsystem (94.9%) due to lower concentration of surface-associated Fe(II) at pH 4 than at pH 6.2 (Figure 1f).Environmental Implications. The results presented above
indicate that caution must be exercised when assessing theviability of ZVI-based technologies for oxidation of targetcontaminants, especially if Good’s buffers have been used inlaboratory investigations. The current study was performed at aconstant Good’s buffer concentration of 100 mM with furtherwork required to more clearly elucidate the impact of bufferconcentration on ZVI transformation and contaminantoxidation. Our results further suggest that there is a strongpossibility that organic compounds present in waters and/or
wastewaters may induce similar effects to those observed forGood’s buffers in this study. Indeed, as shown in SI Figure S13,it has been confirmed that the adsorption of natural organicmatter (humic acid in this instance) on Fe oxides contributes tothe enhanced corrosion of ZVI, leading to the rapid release ofdissolved Fe(II), with porous Fe oxide assemblages formed in amanner similar to that observed for Good’s buffers in this study.Additionally, the presence of abundant surface-associatedFe(II) formed during the organically enhanced ZVI corrosionprocess could further facilitate the structural transformation ofthe Fe (oxyhydr)oxides formed. As the adsorption affinities ofFe(III) (oxyhydr)oxides toward negatively charged contami-nants under circumneutral pH conditions is highly dependenton their crystallinity (with the adsorption capacity following theorder: magnetite ≈ hematite < goethite < lepidocrocite <ferrihydrite), the rate and extent of transformation from highlyamorphous iron (oxyhydr)oxides to more crystalline forms mayinfluence both the association of organic contaminants withiron (oxyhydr)oxide-coated ZVI particles and any subsequentoxidative degradation of these contaminants.45 Further studies,however, are required to evaluate the effects of natural/wastewater organic matters on the oxidative capacity of ZVI/O2systems as well as the corrosion behavior of ZVI in realwastewaters.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.est.7b04030.
Sketch of the experimental setup and additionalexperimental data on relative contents of Fe, C, Oelements in the nonbuffered and buffered samples, totalH2O2 concentrations following the exposure of ZVI tooxygen-containing water in the presence and absence ofGood’s buffers; the k3-weighted EXAFS spectra for ZVI,ferrihydrite, lepidocrocite and oxidized ZVI over 10 h inthe pH range 4.0−9.0; oxidation of H14COO− in the
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1401

homogeneous nonbuffered and MOPS buffered systemsat pH 6.2; dissolved Fe(II) concentration in nonbufferedsolution containing 100 mM NaCl and 100 mM MOPSsolution at pH 6.2, dissolved Fe(II) concentration innonbuffered solution in which pH was manuallymaintained at 4.0 by HCl addition; effect of MOPS onthe adsorption of Fe(II) by ferrihydrite and lepidocrociteat pH 6.2; and consumption of DO by ZVI or Fe(II) inthe presence or absence of Good’s buffers; variation ofdissolved oxygen following addition of Fe(II) in 100 mMMOPS solution; dissolution rate constant of 1 μMferrihydrite suspensions following their formation in theabsence and presence of MOPS buffer; EPR spectra ofthe hydroxyl radical generated on Fenton reaction in theabsence and presence of MOPS; EPR spectra of thehydroxyl radical generated 708 on oxidation of ZVI at 0.5h in the absence and presence of MOPS; sorption ofH14COO− on the surface of ferrihydrite in buffered andnonbuffered solution; Fraction of sorbed H14COO− andfraction of sorbed H14COO− oxidized to 14CO3
2− at 24 hin buffered and nonbuffered solution; variation indissolved Fe(II) concentration formed in the absenceand presence of 100 mg·L−1 humic acid at pH 3.0 andpH 6.5, SEM images of ZVI following 24 h exposure topH 3.0 control and 100 mg·L−1 humic acid solution(PDF)
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] N. Collins: 0000-0001-8895-7031Yang Mu: 0000-0001-7338-7398T. David Waite: 0000-0002-5411-3233Author Contributions⊥The authors contributed equally to this work.NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe gratefully acknowledge the funding provided by theAustralian Synchrotron through Grant No. XAS10764. YangMu and Chuan-Shu He wish to thank the Natural ScienceFoundation of China (51538012) for partial financial support.
■ REFERENCES(1) Gillham, R. W.; O’Hannesin, S. F. Enhanced degradation ofhalogenated aliphatics by zero-valent iron. Groundwater 1994, 32 (6),958−967.(2) Matheson, L. J.; Tratnyek, P. G. Reductive Dehalogenation ofChlorinated Methanes by Iron Metal. Environ. Sci. Technol. 1994, 28(12), 2045−2053.(3) Agrawal, A.; Tratnyek, P. G. Reduction of nitro aromaticcompounds by zero-valent iron metal. Environ. Sci. Technol. 1996, 30(1), 153.(4) Farrell, J.; Kason, M.; Melitas, N.; Li, T. Investigation of the long-term performance of zero-valent iron for reductive dechlorination oftrichloroethylene. Environ. Sci. Technol. 2000, 34 (3), 514−521.(5) Guan, X. H.; Sun, Y. K.; Qin, H. J.; Li, J. X.; Lo, I. M.; He, D.;Dong, H. R. The limitations of applying zero-valent iron technology incontaminants sequestration and the corresponding countermeasures:the development in zero-valent iron technology in the last two decades(1994−2014). Water Res. 2015, 75, 224−248.
(6) Katsoyiannis, I. A.; Ruettimann, T.; Hug, S. J. pH dependence offenton reagent generation and As(III) oxidation and removal bycorrosion of zero valent iron in aerated water. Environ. Sci. Technol.2008, 42 (19), 7424−7430.(7) Keenan, C. R.; Sedlak, D. L. Factors affecting the yield of oxidantsfrom the reaction of nanoparticulate zero-valent iron and oxygen.Environ. Sci. Technol. 2008, 42 (4), 1262−1267.(8) Guo, X. J.; Yang, Z.; Dong, H. Y.; Guan, X. H.; Ren, Q. D.; Lv, X.F.; Jin, X. Simple combination of oxidants with zero-valent-iron (ZVI)achieved very rapid and highly efficient removal of heavy metals fromwater. Water Res. 2016, 88, 671−680.(9) Joo, S. H.; Feitz, A. J.; Waite, T. D. Oxidative degradation of thecarbothioate herbicide, molinate, using nanoscale zero-valent iron.Environ. Sci. Technol. 2004, 38 (7), 2242−2247.(10) Joo, S. H.; Feitz, A. J.; Sedlak, D. L.; Waite, T. D. Quantificationof the oxidizing capacity of nanoparticulate zero-valent iron. Environ.Sci. Technol. 2005, 39 (5), 1263−1268.(11) Nakatsuji, Y.; Salehi, Z.; Kawase, Y. Mechanisms for removal ofp-nitrophenol from aqueous solution using zero-valent iron. J. Environ.Manage. 2015, 152, 183−191.(12) Qin, H. J.; Li, J. X.; Bao, Q. Q.; Li, L. N.; Guan, X. H. Role ofdissolved oxygen in metal (loid) removal by zerovalent iron atdifferent pH: its dependence on the removal mechanisms. RSC Adv.2016, 6 (55), 50144−50152.(13) He, D.; Ma, J. X.; Collins, R. N.; Waite, T. D. Effect of structuraltransformation of nanoparticulate zero-valent iron on generation ofreactive oxygen species. Environ. Sci. Technol. 2016, 50 (7), 3820−3828.(14) Ma, J. X.; He, D.; Collins, R. N.; He, C. S.; Waite, T. D. Thetortoise versus the hare-possible advantages of microparticulatezerovalent iron (mZVI) over nanoparticulate zerovalent iron (nZVI)in aerobic degradation of contaminants. Water Res. 2016, 105, 331−340.(15) Zhang, T. C.; Huang, Y. H. Profiling iron corrosion coating oniron grains in a zerovalent iron system under the influence of dissolvedoxygen. Water Res. 2006, 40 (12), 2311−2320.(16) Sun, Y. K.; Guan, X. H.; Wang, J. M.; Meng, X. G.; Xu, C. H.;Zhou, G. M. Effect of weak magnetic field on arsenate and arseniteremoval from water by zerovalent iron: an XAFS investigation. Environ.Sci. Technol. 2014, 48 (12), 6850−6858.(17) Boland, D. D.; Collins, R. N.; Miller, C. J.; Glover, C. J.; Waite,T. D. Effect of solution and solid-phase conditions on the Fe(II)-accelerated transformation of ferrihydrite to lepidocrocite and goethite.Environ. Sci. Technol. 2014, 48 (10), 5477−5485.(18) Zhang, T. C.; Huang, Y. H. Effects of selected Good’s pHbuffers on nitrate reduction by iron powder. J. Environ. Eng. 2005, 131(3), 461−470.(19) Alowitz, M. J.; Scherer, M. M. Kinetics of nitrate, nitrite, and Cr(VI) reduction by iron metal. Environ. Sci. Technol. 2002, 36 (3), 299−306.(20) Liang, L. P.; Yang, W. J.; Guan, X. H.; Li, J. L.; Xu, Z. J.; Wu, J.;Huang, Y. Y.; Zhang, X. Z. Kinetics and mechanisms of pH-dependentselenite removal by zero valent iron. Water Res. 2013, 47 (15), 5846−5855.(21) Azab, H. A.; Orabi, A. S.; El-Salam, E. T. A. Role of biologicallyimportant zwitterionic buffer secondary ligands on the stability of themixed-ligand complexes of divalent metal ions and adenosine 5′-mono-, 5′-di-, and 5′-triphosphate. J. Chem. Eng. Data 2001, 46 (2),346−354.(22) Taha, M.; Gupta, B. S.; Lee, M. J. Complex equilibria in aqueoussolutions of chromium (III) with some biological pH buffers. J. Chem.Eng. Data 2011, 56 (9), 3541−3551.(23) Siantar, D. P.; Schreier, C. G.; Chou, C. S.; Reinhard, M.Treatment of 1, 2-dibromo-3-chloropropane and nitrate-contaminatedwater with zero-valent iron or hydrogen/palladium catalysts. WaterRes. 1996, 30 (10), 2315−2322.(24) Buchholz, A.; Laskov, C.; Haderlein, S. B. Effects of zwitterionicbuffers on sorption of ferrous iron at goethite and its oxidation byCCl4. Environ. Sci. Technol. 2011, 45 (8), 3355−3360.
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1402

(25) Danielsen, K. M.; Gland, J. L.; Hayes, K. F. Influence of aminebuffers on carbon tetrachloride reductive dechlorination by the ironoxide magnetite. Environ. Sci. Technol. 2005, 39 (3), 756−763.(26) Grady, J. K.; Chasteen, N. D.; Harris, D. C. Radicals from“Good’s” buffers. Anal. Biochem. 1988, 173 (1), 111−115.(27) Hicks, M.; Gebicki, J. M. Rate constants for reaction of hydroxylradicals with Tris, Tricine and Hepes buffers. FEBS Lett. 1986, 199(1), 92−94.(28) Tadolini, B. Iron autoxidation in MOPS and HEPES buffers.Free Radical Res. Commun. 1987, 4 (3), 149−160.(29) Stipp, S. L. S.; Hansen, M.; Kristensen, R.; Hochella, M. F., Jr;Bennedsen, L.; Dideriksen, K.; Balic-Zunic, T.; Leonard, D.; Mathieu,H. J. Behaviour of Fe-oxides relevant to contaminant uptake in theenvironment. Chem. Geol. 2002, 190 (1−4), 321−337.(30) Sun, Y. K.; Li, J. X.; Huang, T. L.; Guan, X. H. The influences ofiron characteristics, operating conditions and solution chemistry oncontaminants removal by zero-valent iron: A review. Water Res. 2016,100, 277−295.(31) Qiu, S.; He, D.; Ma, J. X.; Liu, T. X.; Waite, T. D. Kineticmodeling of the electro-Fenton process: quantification of reactiveoxygen species generation. Electrochim. Acta 2015, 176, 51−58.(32) Ai, Z. H.; Gao, Z. T.; Zhang, L. Z.; He, W. W.; Yin, J. J. Core−shell structure dependent reactivity of Fe@Fe2O3 nanowires onaerobic degradation of 4-chlorophenol. Environ. Sci. Technol. 2013, 47(10), 5344−5352.(33) Liu, T. X.; Li, X. M.; Waite, T. D. Depassivation of aged Fe0 bydivalent cations: correlation between contaminant degradation andsurface complexation constants. Environ. Sci. Technol. 2014, 48 (24),14564−14571.(34) Zhou, M. J.; Diwu, Z. J.; Panchuk-Voloshina, N.; Haugland, R.P. A stable nonfluorescent derivative of resorufin for the fluorometricdetermination of trace hydrogen peroxide: applications in detectingthe activity of phagocyte NADPH oxidase and other oxidases. Anal.Biochem. 1997, 253 (2), 162−168.(35) Pham, A. N.; Xing, G. W.; Miller, C. J.; Waite, T. D. Fenton-likecopper redox chemistry revisited: Hydrogen peroxide and superoxidemediation of copper-catalyzed oxidant production. J. Catal. 2013, 301,54−64.(36) Kappen, P.; Webb, J. An EXAFS study of arsenic bonding onamorphous aluminium hydroxide. Appl. Geochem. 2013, 31, 79−83.(37) Johnson, K. A.; Simpson, Z. B.; Blom, T. Global kineticexplorer: a new computer program for dynamic simulation and fittingof kinetic data. Anal. Biochem. 2009, 387 (1), 20−29.(38) Liu, T. X.; Li, X. M.; Waite, T. D. Depassivation of aged Fe0 byferrous ions: implications to contaminant degradation. Environ. Sci.Technol. 2013, 47 (23), 13712−13720.(39) Comarmond, M. J.; Payne, T. E.; Collins, R. N.; Palmer, G.;Lumpkin, G. R.; Angove, M. J. Inhibition of Uranium(VI) Sorption onTitanium Dioxide by Surface Iron(III) Species in Ferric Oxide/Titanium Dioxide Systems. Environ. Sci. Technol. 2012, 46 (20),11128−11134.(40) Zhou, T.; Li, Y. Z.; Ji, J.; Wong, F. S.; Lu, X. H. Oxidation of 4-chlorophenol in a heterogeneous zero valent iron/H2O2 Fenton-likesystem: kinetic, pathway and effect factors. Sep. Purif. Technol. 2008, 62(3), 551−558.(41) Liu, T. X.; Li, X. M.; Waite, T. D. Depassivation of aged Fe0 byferrous ions: Implications to contaminant degradation. Environ. Sci.Technol. 2013, 47 (23), 13712−13720.(42) Liu, T. X.; Li, X. M.; Waite, T. D. Depassivation of aged Fe0 bydivalent cations: correlation between contaminant degradation andsurface complexation constants. Environ. Sci. Technol. 2014, 48 (24),14564−14571.(43) Shiraishi, H.; Kataoka, M.; Morita, Y.; Umemoto, J. Interactionsof hydroxyl radicals with tris (hydroxymethyl) aminomethane andGood’s buffers containing hydroxymethyl or hydroxyethyl residuesproduce formaldehyde. Free Radical Res. Commun. 1993, 19 (5), 315−321.(44) Tadolini, B. Iron autoxidation in Mops and Hepes buffers. FreeRadical Res. Commun. 1987, 4 (3), 149−160.
(45) Hanna, K. Adsorption of aromatic carboxylate compounds onthe surface of synthesized iron oxide-coated sands. Appl. Geochem.2007, 22 (9), 2045−2053.
Environmental Science & Technology Article
DOI: 10.1021/acs.est.7b04030Environ. Sci. Technol. 2018, 52, 1393−1403
1403