electric field effects in the chemisorption of co on bimetallic rhcu surface models
TRANSCRIPT

Surface Science 548 (2004) 209–219
www.elsevier.com/locate/susc
Electric field effects in the chemisorption of CO on bimetallicRhCu surface models
Silvia Gonz�aalez a,b, Carmen Sousa a, Francesc Illas a,*
a Departament de Qu�ıımica F�ıısica i Centre Especial de Recerca en Qu�ıımica Te�oorica, Universitat de Barcelona i Parc Cient�ııfic
de Barcelona, C/ Mart�ıı i Franqu�ees 1, 08028 Barcelona, Spainb Departamento de Qu�ıımica, Universidad Aut�oonoma Metropolitana-Iztapalapa, P.O. Box 55-534, C.P. 09340 M�eexico D.F., Mexico
Received 13 August 2003; accepted for publication 7 November 2003
Abstract
Cluster models and hybrid density functional theory calculations are used to study the effect of an external electric
field on the properties and bonding mechanism of CO on various RhCu bimetallic surfaces. The presence of an external
electric field induces changes in the geometry and vibrational frequency of the chemisorbed molecule. Several methods
of analysis establish that the electric field effects are not just of electrostatic nature but do affect the bonding mecha-
nisms as well. In any case, the electric field effects observed for various RhCu bimetallic surfaces are not at all different
from what is known for monometallic surfaces. The present study permits to conclude that the effect of an external
electric field on the properties of CO adsorbed on bimetallic RhCu surfaces of different composition follows the same
trend exhibited by the single metal surfaces.
� 2003 Elsevier B.V. All rights reserved.
Keywords: Metallic surfaces; Alloys; Surface electronic phenomena (work function, surface potential, surface states, etc.); Density
functional calculations; Carbon monoxide; Chemisorption
1. Introduction
Promotion is one of the fundamental issues in
heterogeneous catalysis [1,2] because it permits one
to improve the catalytic properties of a given
material with a minimal manipulation. Promotion
may be caused by the introduction of a chemical
additive (chemical promotion) or by the applica-
tion of an external electric potential (electro-
chemical promotion). While chemical promotion
* Corresponding author. Tel.: +34-934021229; fax: +34-
934021231.
E-mail address: [email protected] (F. Illas).
0039-6028/$ - see front matter � 2003 Elsevier B.V. All rights reserv
doi:10.1016/j.susc.2003.11.009
has been long known and used, electrochemical
promotion (or non-faradaic electrochemical mod-ification of catalytic activity; NEMCA effect) was
discovered only twenty years ago [3,4]. The interest
in this phenomenon has been continuously grow-
ing because of its unique way to allow modifying
the activity and selectivity of the catalytic system
in a controlled manner [5–8]. Electrochemical
promoted processes do not only exhibit very large
increases in reaction rate with respect to the un-promoted catalytic processes; significant improve-
ments in the selectivity have also been observed [4].
The easiness to control the electric potential in
electrochemically promoted systems contrasts with
the difficulty to control the rather large number of
ed.

210 S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219
variables that play a role in any electrochemical
process. In fact, electrochemical systems are
characterized by the presence of many simulta-
neous effects such as solvent, impurities, adsorbed
species, competing reactions, etc. Therefore, for a
better understanding of electrochemical promo-tion but also of the complex electrochemical
environment, it is desirable to identify and sepa-
rate contributions arising from each different
effect. To this end, many authors have used theo-
retical models to analyze the role played by an
external electric field in the chemisorption prop-
erties of various adsorbates on metallic surfaces
[9–24]. Bagus et al. [9–11] concluded that thechanges in the vibrational frequencies of simple
molecules like CO and CN� adsorbed on Cu(1 0 0)
induced by the presence of an external electric field
were due to a vibrational Stark effect; this is
without any significant change in the bonding
mechanism of these molecules to the surface. This
conclusion was questioned by Head-Gordon and
Tully [12] who suggested that electrostatic andchemical effects could not be rigorously separated.
Later on, several analysis have shown that, in fact,
the presence of an external electric field induces
noticeable chemical changes in the chemisorption
bond [13,14,20,21,23,24]. In particular, it has been
shown that the electric field provokes changes in
the population 2p� molecular orbital of the ad-
sorbed CO molecule and, hence, in the importanceof the metal to CO backdonation bonding mech-
anism [13,14]. Lambert et al. have used this argu-
ment to explain the changes in the selectivity of
some reactions on chemical promoted metallic
surfaces in terms of changes in the electronic
density in the molecular orbital caused by an
internal electric field [25].
Up to now, almost all studies regarding the ef-fect of an external electric field on the properties
of adsorbed molecules on metals concern mono-
metallic surfaces. However, bimetallic systems are
increasingly used and are preferred as catalysts
in many industrial chemical reactions because,
compared to each separate component, the com-
bination of two metals with different catalytic pro-
perties results in a better catalytic performance[26–30]. Bimetallic RhCu constitutes a paradig-
matic example which has been studied as early as
1975 [31] and experienced renewed interest in the
past few years [32–42]. Recently, chemisorption of
CO on RhCu(1 1 1) surface models has been theo-
retically studied [41]. In particular, it has been
shown that from the study of the geometry and
vibrational frequency of the chemisorbed mole-cule it is not possible to obtain information about
the alloy composition. On the other hand, it has
also been shown that the vibrational frequency
of chemisorbed CO permits to assign the chemi-
sorption site in a rather univocal way.
In the present work, we continue the previous
study about bimetallic RhCu by analyzing the ef-
fect of a uniform external electric field on theproperties of CO chemisorbed on various bime-
tallic surfaces of different Rh–Cu composition.
The motivation to carry out such study comes
from the fact that, upon alloying, large effects on
the electronic structure of the components are
observed by photoemission and X-ray Absorption
Near Edge Structure [43,44]. Changes in the elec-
tronic structure of the alloy have also been ob-served by Rodriguez and Goodman [29] who
found a linear relationship between the core level
shift of the metals and the adsorption energy of
CO. The formation of the heterometallic bond and
the concomitant changes in the electronic structure
may result or not in a different response of the
bimetallic surface to the presence of an external
electric field. Therefore, the main goal of thepresent paper is to investigate the effect of an
external electric field on a bimetallic system and to
see whether the changes induced by such electric
field are similar to those encountered for mono-
metallic surfaces or if, on the contrary, there are
significant differences in the properties of the ad-
sorbed molecule. To this end, cluster models are
employed to study the effect of an external electricfield in the interaction of CO with RhCu and we
make use of density functional theory (DFT) and
of the DFT implementation [45] of the constrained
space orbital variation (CSOV) technique [46–48]
to compute the interaction energy and to dissect it
into its various physically meaningful contribu-
tions. An important conclusion of this work is that
the effect of an external electric field on thechemisorption properties does not depend on the
composition of the alloy since, for each compo-

S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219 211
sition, the electric field modifies chemisorption
properties in a similar way.
2. Surface cluster models and computational details
In this paper different cluster models have been
used to simulate mono (Rh and Cu) and RhCu
bimetallic (1 1 1) surfaces. The employed models
are the same used in previous studies about
molecular hydrogen dissociation [40] and CO
chemisorption on these bimetallic systems [41].
The Rh10, Rh9Cu, Rh4Cu6, RhCu9 and Cu10
cluster models contain seven atoms in the firstlayer and three in the second one in such a way
that the central atom in the upper layer has the
coordination of a surface atom in a (1 1 1) surface.
Rh10 and Cu10 model the corresponding (1 1 1)
surfaces and have been used as reference to com-
pare the activity of the bimetallic systems towards
CO adsorption. Bimetallic Rh9Cu and RhCu9
models represent the Rh- or Cu-rich phases pre-dicted by the RhCu phase diagram [49] whereas
the Rh4Cu6 cluster represents a mixed composition
and has been included mainly for comparison
purposes. The inter-atomic distances in the clusters
considered have been kept fixed. For the mono-
metallic models, the cluster geometries have been
fixed as in Rh or Cu bulk [50] with dRhRh ¼ 2:69 �AAand dCuCu ¼ 2:56 �AA, whereas in the bimetalliccompounds dRhCu ¼ 2:59 �AA is a weighted interpo-
lated value between the two first distances. Notice
that these distances are similar to those experi-
mentally measured by extended X-ray absorption
fine structure (EXAFS) for RhCu clusters of �14�AA (dRhRh ¼ 2:68 �AA, dCuCu ¼ 2:62 �AA and dRhCu ¼2:64 �AA) [51]. Additional details about these cluster
models can be found in previous works [40,41]. Inall cases, the CO molecule adsorbs perpendicular
to the surface with the C atom interacting directly
with the surface metal atom located at the centre
of the cluster model. Hence, for Rh10, Rh4Cu6 and
RhCu9 the CO is adsorbed on top of a Rh atom
while for Cu10 and Rh9Cu the CO molecule
interacts directly with a Cu atom.
To study the effect of an external electric fieldeffect in the CO chemisorption on the above de-
scribed surface cluster models a uniform electric
field has been included in the Hamiltonian on the
system. Calculations including the presence of this
uniform electric field have been carried out using a
first principles density functional theory based
method. In particular, we use the B3LYP hybrid
functional, which combines the gradient correctedBecke�s three parameters hybrid exchange func-
tional [52] with the correlation functional of Lee,
Yang and Parr [53], as implemented in the HON-
DO99 [54] version of HONDO95.3 [55] and in the
Gaussian98 [56] computational packages. The
cluster metal atoms were described by relativistic
small-core effective core potentials and double-fbasis sets reported by Hay and Wadt [57]. Forthe CO molecule we use the Alhrichs TZV basis
augmented with d functions [58]. Calculations
including explicitly a uniform electric field have
been carried out to obtain the equilibrium geo-
metry of the adsorbed molecule, the internal CO
vibrational frequency, the interaction energy with
respect to the CO and M10 separated systems,
and the occupation of the CO(2p�) and CO(5r)molecular orbitals as function of the intensity of a
uniform external electric field. For each electric
field intensity, the interaction energy is decom-
posed into Pauli repulsion, CO or metal cluster
intra-unit polarization and inter-unit donation,
the usual various contributions to chemisorption
bond as defined by Bagus et al. [46–48]. The inter-
unit donation arises almost exclusively from theCO(5r) molecular orbital to the metal surface and
the backdonation from the d atomic orbitals of the
cluster metal atom directly interacting with CO to
the CO(2p�) molecular orbital.
Chemisorption geometry and internal CO
stretching frequency have been obtained by means
of the Gaussian98 computational package; the
interaction energy and CSOV analysis was carriedout using HONDO99; finally, the extent of back-
donation from metal d orbitals to the CO(2p�)
orbital has been calculated by means of the orbital
projection technique introduced by Nelin et al. [59]
using Kohn–Sham orbitals. In all cases, the lowest
singlet closed-shell electronic state has been con-
sidered; this neglects possible low-lying open shell
states which are likely to arise from cluster arti-facts. On the other hand, it has been shown that
low-lying electronic states of small metal clusters

212 S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219
are very close in energy and exhibit a similar
chemistry [60]. Electric field intensities of F ¼�0:01, )0.005, +0.005 and +0.01 a.u. (0.01 a.u.¼5.14 · 10�7 V/cm) have been considered because
these are of the order of the electric fields which
are present in the electrochemical environment.The use of larger values for the intensity of the
electric field does not bring any additional effect in
the stretching frequency of free and adsorbed CO
[19]. The electric field is oriented perpendicular to
the surface in the C–O molecular axis in such a
way that negative values pull electrons out of the
surface towards the vacuum.
3. Results and discussion
The calculation of the vibrational frequency of
a free CO molecule in absence or presence of an
external electric field permits a direct comparison
with experiment and hence provides a check on the
consistency of the present computational model.The computed value of the harmonic vibrational
frequency for free CO is 2219 and 51 cm�1 larger
than the experimental one which is 2170 cm�1 [61].
This difference can be corrected including points in
the anharmonic region of the potential energy
curve which is next fitted to a third degree poly-
nomial to extract the harmonic frequency. The
stretching frequency computed in this way, 2195
Table 1
Calculated values for the equilibrium perpendicular distance of CO to
vibrational frequency (mCO) and the adsorption energy (Eint) correspon
models. The adsorption energy is defined in a way that a positive val
Adsorption on top of Rh
Rh10 Rh4Cu6
dM–C (�AA) 1.841 (1.84a) 1.880
dCO (�AA) 1.147 (1.15a) 1.153
mCO (cm�1) 2059 (2015c) 2009
Eint (kcalmol�1) 28.1 (39e) 42.48
Experimental data, only available for CO on Rh(1 1 1) and on Cu(1 0aRef. [64].bRef. [65].cRef. [66].dRef. [67].eRef. [70].f Ref. [71].
cm�1 is closer to the experimental one and the
anharmonic constant is 11.02 cm�1 is also in good
agreement with the experimental value of 13.46
cm�1 [61]. As it is well known, the presence of an
external electric field induces changes in the
stretching frequency and this change is usuallyreferred to as vibrational Stark effect. It is char-
acterized by the so-called Stark tuning rate value
(STR), the first derivate of the molecular stretch-
ing frequency with respect to the intensity of the
external electric field (dm=dF ). The present calcu-
lated STR is 4.95 · 10�7 cm�1/V/cm, which is in a
rather good agreement with the value for the free
CO molecule reported by Lambert [62] obtainedusing a semiclassical model and some spectro-
scopic data (4.29 · 10�7 cm�1/V/cm) and is also
close to the more direct experimental measured
value reported later also by Lambert which is of
5.27 ± 0.27 · 10�7 cm�1/V/cm [63].
Before starting the discussion about the electric
field effects on CO chemisorption, let us briefly
consider the geometrical parameters, CO vibra-tional frequency and adsorption energy for ad-
sorbed CO without external electric field reported
in Table 1. These are very close to those previously
reported and the only difference comes from the
basis set of CO; Ahlrichs TZV plus polarization
in the present work compared to the standard
6-31G** basis used in Ref. [41]. Therefore, the
discussion corresponding to the values in absence
the surface (dM–C), the C–O inter-atomic distance (dCO); the COding to the interaction of CO with mono and bimetallic surface
ue indicates an exothermic process
Adsorption on top of Cu
RhCu9 Rh9Cu Cu10
1.946 2.037 2.201 (1.9b)
1.148 1.136 1.131 (1.13b)
2032 2117 2154 (2078d)
11.8 3.1 )3.3 (10f )
0), are given in parenthesis.

S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219 213
of electric field will be omitted and we would like
just to point out that geometries and vibrational
frequencies are in good agreement with available
experimental data [64–67] which have been in-
cluded in Table 1 for comparison. Calculated
chemisorption energies for CO on Rh(1 1 1) sur-faces are comparable to the values reported from
temperature programmed desorption (TPD)
[68,69] and time-resolved electron energy loss
spectroscopy (TREELS) [70] although somewhat
smaller. Interestingly enough, TPD measurements
for CO on Cu(1 1 1) suggest that the adsorption
energy is �30 kcalmol�1 smaller than the one
corresponding to the Rh(1 1 1) surface [71]. Thistrend is also properly predicted by the present
cluster B3LYP calculations. Finally, notice that a
discussion concerning the variation of the CO
adsorption energy with respect to the composition
of the bimetallic RhCu alloy has been reported
also in [41]. In particular, the non-monotonic
trend in this quantity has been explained by the
difference on charge transfer mechanisms and onthe Pauli repulsion due to changes in electronic
charge density caused by heterometallic bond.
The changes induced by the external electric
field will be studied by looking at the effect pro-
duced on the C–O distance (dCO), stretching fre-
quency CO (mCO) and occupation of the 2p�
molecular orbital of the adsorbed molecule. The
adsorption energy on each metallic or bimetallicsurface will also be discussed although we can
anticipate that, compared to the properties men-
tioned above; the changes induced in the adsorp-
tion energies are much smaller. Before starting the
discussion it is convenient to remember that a
negative sign of electric field is such that the elec-
tric field pulls electrons out of the surface toward
the vacuum. In case the electric field effects forbimetallic surfaces follow the trend observed for
single metal surfaces, one would expect that in
going from negative to positive electric fields the
inter-atomic distance of CO decreases because the
backdonation contribution will be favored for
negative fields. In the same way, one expects that
the larger the contribution from backdonation the
larger the red shift of the vibrational frequencyand, consequently, the smaller the absolute value
of the vibrational frequency of adsorbed CO.
Finally, the larger backdonation contribution for
negative fields must be accompanied by a higher
occupation of the CO(2p�) molecular orbital and
hence this value must decrease in going from neg-
ative to positive values of the electric field. Fig. 1
reports the variation of dCO, mCO and the occupa-tion of the CO(2p�) molecular orbital with respect
to the electric field. From this figure one does not
see any differential behavior between monometal-
lic and bimetallic model surfaces considered in
this work, or between the different composition of
the RhCu alloys. Moreover, the trend followed
by the three properties is precisely as anticipated in
the previous discussion and the variation of dC–O,mC–O and the occupation of the CO(2p�) molecular
orbital with respect to the electric field intensity is
almost linear. In fact, a linear relationship for dCOhas also been reported by other authors in study-
ing the electric field effects on single metal surfaces
[13,14,21,23]; the linear trend of mCO with respect
to the electric field intensity agrees with reported
results for CO on several metal surfaces [9,10,13,14,21,23,72] and, finally, the occupation of the
CO(2p�) molecular orbital follows the trend pre-
viously found for CO on Cu(1 0 0) [9] and Pt(1 1 1)
[13,14]. Notice that the change in the CO(2p�)
occupation indicates that a external uniform elec-
tric field produces chemical changes. This is in
agreement with the previous work of several au-
thors for CO on single metal surfaces [13,14,21–24] and is in contrast with the original interpre-
tation of Bagus et al. [9] who suggested that
the electric field effect did not affect the bonding
mechanism. Another important point concerns the
slope associated to this linear dependence of dCO,mCO and the occupation of the CO(2p�) molecular
orbital which is similar for all cases implying that
the differences with composition found in absenceof electric field [41] are also preserved, at least for
the electric field intensity range studied in the
present work. The slope of mCO versus the external
electric field intensity merits an additional com-
ment. The values obtained for the Rh10, Rh4Cu6,
RhCu9, Rh9Cu and Cu10 are 15.00, 14.23, 14.62,
15.39 and 15.39 in units of 10�7 cm�1/V/cm. The
close similarity of these values seems to suggestthat the effect of the electric field on the interac-
tion of CO with these metal surfaces is mainly of
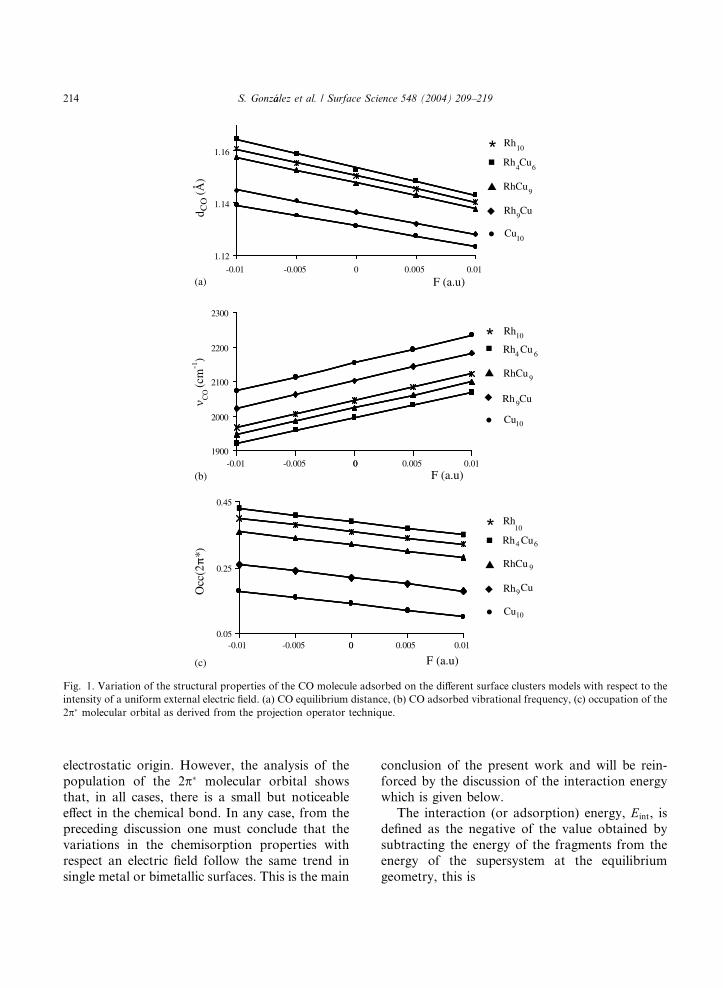
Fig. 1. Variation of the structural properties of the CO molecule adsorbed on the different surface clusters models with respect to the
intensity of a uniform external electric field. (a) CO equilibrium distance, (b) CO adsorbed vibrational frequency, (c) occupation of the
2p� molecular orbital as derived from the projection operator technique.
214 S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219
electrostatic origin. However, the analysis of the
population of the 2p� molecular orbital showsthat, in all cases, there is a small but noticeable
effect in the chemical bond. In any case, from the
preceding discussion one must conclude that the
variations in the chemisorption properties with
respect an electric field follow the same trend in
single metal or bimetallic surfaces. This is the main
conclusion of the present work and will be rein-
forced by the discussion of the interaction energywhich is given below.
The interaction (or adsorption) energy, Eint, is
defined as the negative of the value obtained by
subtracting the energy of the fragments from the
energy of the supersystem at the equilibrium
geometry, this is
S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219 215
EintðF Þ ¼ �fEF ðCO–RhmCunÞ� ½EðCOÞ þ EðRhmCunÞ�g; ð1Þ
where EF indicates that the energy of the super-
system has been computed in presence of an
external field of intensity F . The changes in the
total interaction energy, Eint, induced by the elec-
tric field will be given by
DEintðF Þ ¼ EintðF Þ � Eintð0Þ: ð2Þ
These changes are similar for the cluster where
CO interacts directly with Rh or Cu. Accordingly,
a detailed discussion will be given for the interac-
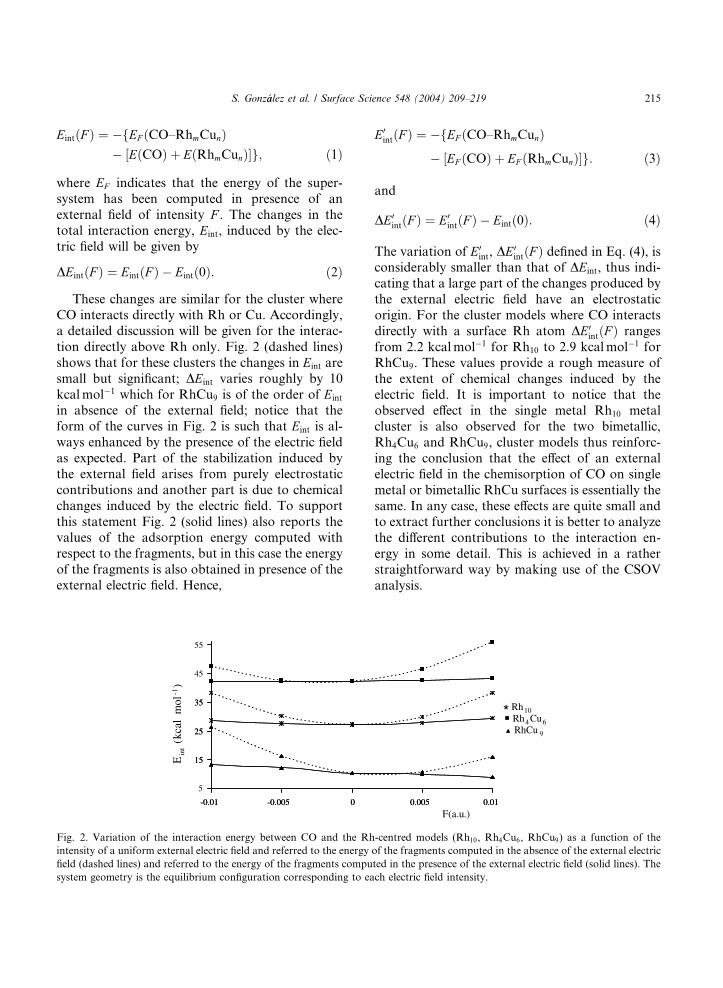
tion directly above Rh only. Fig. 2 (dashed lines)
shows that for these clusters the changes in Eint are
small but significant; DEint varies roughly by 10
kcalmol�1 which for RhCu9 is of the order of Eint
in absence of the external field; notice that the
form of the curves in Fig. 2 is such that Eint is al-
ways enhanced by the presence of the electric field
as expected. Part of the stabilization induced by
the external field arises from purely electrostatic
contributions and another part is due to chemical
changes induced by the electric field. To support
this statement Fig. 2 (solid lines) also reports thevalues of the adsorption energy computed with
respect to the fragments, but in this case the energy
of the fragments is also obtained in presence of the
external electric field. Hence,
Ein
t(k
cal
mol
-1)
15
25
35
45
55
-0.01 -0.005 0
5
15
25
35
-0.01 -0.005 0
Fig. 2. Variation of the interaction energy between CO and the Rh
intensity of a uniform external electric field and referred to the energy
field (dashed lines) and referred to the energy of the fragments compu
system geometry is the equilibrium configuration corresponding to ea
E0intðF Þ ¼ �fEF ðCO–RhmCunÞ
� ½EF ðCOÞ þ EF ðRhmCunÞ�g: ð3Þ
and
DE0intðF Þ ¼ E0
intðF Þ � Eintð0Þ: ð4Þ
The variation of E0int, DE
0intðF Þ defined in Eq. (4), is
considerably smaller than that of DEint, thus indi-
cating that a large part of the changes produced by
the external electric field have an electrostatic
origin. For the cluster models where CO interacts
directly with a surface Rh atom DE0intðF Þ ranges
from 2.2 kcalmol�1 for Rh10 to 2.9 kcalmol�1 for
RhCu9. These values provide a rough measure of
the extent of chemical changes induced by theelectric field. It is important to notice that the
observed effect in the single metal Rh10 metal
cluster is also observed for the two bimetallic,
Rh4Cu6 and RhCu9, cluster models thus reinforc-
ing the conclusion that the effect of an external
electric field in the chemisorption of CO on single
metal or bimetallic RhCu surfaces is essentially the
same. In any case, these effects are quite small andto extract further conclusions it is better to analyze
the different contributions to the interaction en-
ergy in some detail. This is achieved in a rather
straightforward way by making use of the CSOV
analysis.
* Rh10Rh4Cu6RhCu 9
0.005 0.01
F(a.u.)0.005 0.01
-centred models (Rh10, Rh4Cu6, RhCu9) as a function of the
of the fragments computed in the absence of the external electric
ted in the presence of the external electric field (solid lines). The
ch electric field intensity.
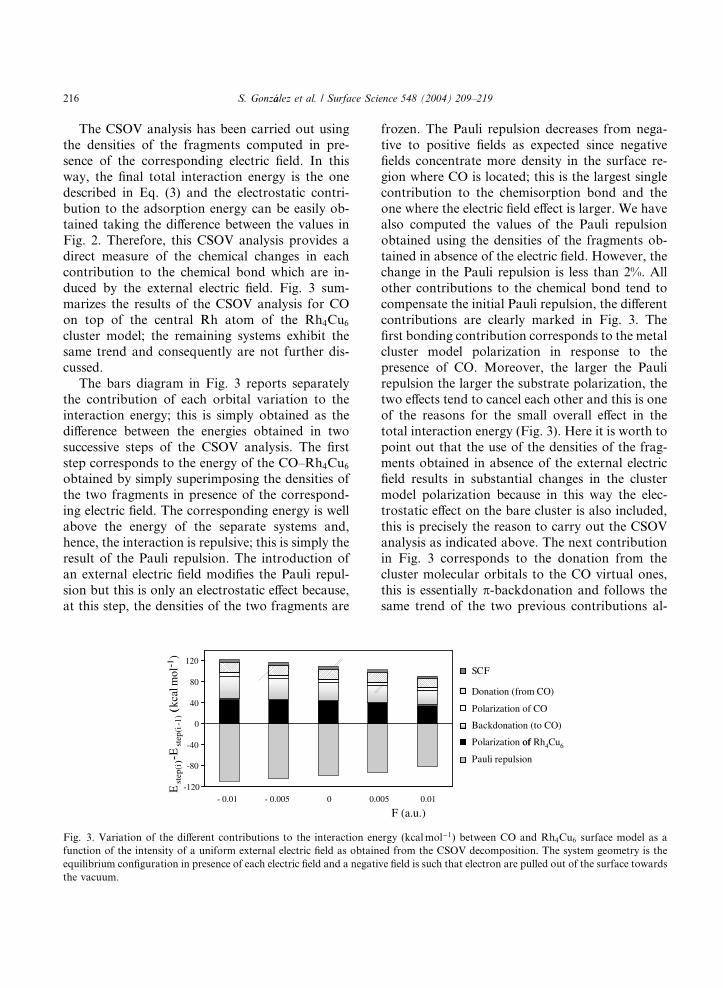
216 S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219
The CSOV analysis has been carried out using
the densities of the fragments computed in pre-
sence of the corresponding electric field. In this
way, the final total interaction energy is the one
described in Eq. (3) and the electrostatic contri-
bution to the adsorption energy can be easily ob-tained taking the difference between the values in
Fig. 2. Therefore, this CSOV analysis provides a
direct measure of the chemical changes in each
contribution to the chemical bond which are in-
duced by the external electric field. Fig. 3 sum-
marizes the results of the CSOV analysis for CO
on top of the central Rh atom of the Rh4Cu6
cluster model; the remaining systems exhibit thesame trend and consequently are not further dis-
cussed.
The bars diagram in Fig. 3 reports separately
the contribution of each orbital variation to the
interaction energy; this is simply obtained as the
difference between the energies obtained in two
successive steps of the CSOV analysis. The first
step corresponds to the energy of the CO–Rh4Cu6
obtained by simply superimposing the densities of
the two fragments in presence of the correspond-
ing electric field. The corresponding energy is well
above the energy of the separate systems and,
hence, the interaction is repulsive; this is simply the
result of the Pauli repulsion. The introduction of
an external electric field modifies the Pauli repul-
sion but this is only an electrostatic effect because,at this step, the densities of the two fragments are
-120
-80
-40
0
40
80
120
- 0.01 - 0.005 0 0.0
Est
ep(i
)-E
step
(i-1
) (k
cal m
ol-1
))-
(-
Fig. 3. Variation of the different contributions to the interaction en
function of the intensity of a uniform external electric field as obtain
equilibrium configuration in presence of each electric field and a negati
the vacuum.
frozen. The Pauli repulsion decreases from nega-
tive to positive fields as expected since negative
fields concentrate more density in the surface re-
gion where CO is located; this is the largest single
contribution to the chemisorption bond and the
one where the electric field effect is larger. We havealso computed the values of the Pauli repulsion
obtained using the densities of the fragments ob-
tained in absence of the electric field. However, the
change in the Pauli repulsion is less than 2%. All
other contributions to the chemical bond tend to
compensate the initial Pauli repulsion, the different
contributions are clearly marked in Fig. 3. The
first bonding contribution corresponds to the metalcluster model polarization in response to the
presence of CO. Moreover, the larger the Pauli
repulsion the larger the substrate polarization, the
two effects tend to cancel each other and this is one
of the reasons for the small overall effect in the
total interaction energy (Fig. 3). Here it is worth to
point out that the use of the densities of the frag-
ments obtained in absence of the external electricfield results in substantial changes in the cluster
model polarization because in this way the elec-
trostatic effect on the bare cluster is also included,
this is precisely the reason to carry out the CSOV
analysis as indicated above. The next contribution
in Fig. 3 corresponds to the donation from the
cluster molecular orbitals to the CO virtual ones,
this is essentially p-backdonation and follows thesame trend of the two previous contributions al-
SCF
Donation (from CO)
Polarization of CO
Backdonation (to CO)
Polarization of Rh4Cu6
Pauli repulsion
of
F (a.u.)05 0.01
ergy (kcalmol�1) between CO and Rh4Cu6 surface model as a
ed from the CSOV decomposition. The system geometry is the
ve field is such that electron are pulled out of the surface towards

S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219 217
though to a lesser extent. This is because although
backdonation is expected to be increased when, as
a result of the external field, the electron density is
pulled towards CO, the extent of this effect does
also depend on the capability of the CO 2p�
molecular orbital to accept electronic population.Consequently, backdonation is larger for negative
fields and in going from a field intensity of )0.01to one of +0.01 a.u. varies from 50.1 to 37.2
kcalmol�1, this is a strong indication that chemi-
cal changes are important in agreement with the
results of the occupation of the CO(2p�) orbital
as obtained from the projection operator tech-
nique. It is worth comparing these values withthose corresponding to the interaction of CO
with Rh10 (48.1 kcalmol�1 for F ¼ �0:01 and 34.0
kcalmol�1 for F ¼ þ0:01) and with RhCu9 (42.0
kcalmol�1 for F ¼ �0:01 and 27.9 kcalmol�1 for
F ¼ þ0:01), again providing strong evidence that
single metal and bimetallic surfaces exhibit the
same behavior. We close this discussion of the
CSOV analysis by pointing out that the COpolarization contributions is only little affected by
the presence of the external electric field and that
the CO donation to the metal surface follows the
opposite trend of the p-backdonation with respect
to the electric field intensity as expected from the
reversal sense of this donation.
4. Conclusions
The effect of an external electric field on the
properties and bonding mechanism of CO on
various RhCu bimetallic surfaces has been stud-
ied using cluster models and hybrid density
functional theory calculations. The presence of
the external electric field induces changes in thegeometry and vibrational frequency of the chemi-
sorbed molecule. The analysis of the occupation
of the CO(2p�) antibonding molecular orbital of
the adsorbed molecule permits to firmly establish
that the electric field effects are not just of elec-
trostatic nature but do affect the bonding mecha-
nisms as well. The Constrained Space Orbital
Variations method of analysis has also beenapplied to the metallic and bimetallic systems
studied in the present work. This permits to
distinguish between electrostatic and chemical
changes induced by the electric field. Overall, the
electric field effects observed for the present bi-
metallic surfaces are not at all different from what
has been reported for monometallic surfaces.
Therefore, the main conclusion of the presentwork is that the effect of an external electric field
on the properties of CO adsorbed on bimetal-
lic RhCu surfaces of different composition is the
same as in the single metal surfaces. While the
present conclusions have been obtained for a
given bimetallic alloy it is likely that they will
apply to other bimetallic systems as well.
Acknowledgements
This research has been supported by the Span-
ish DGICYT grant BQU2002-04029-CO2-01 and,
in part, by Generalitat de Catalunya grants
2001SGR-00043 and Distinci�oo de la Generalitat de
Catalunya per a la Promoci�oo de la Recerca Uni-
versit�aaria (F.I.). Computer time was provided by
the Centre de Supercomputaci�oo de Catalunya,
CESCA, and Centre Europeu de Paral.lelisme de
Barcelona, CEPBA, through generous grants from
Universitat de Barcelona, Fundaci�oo Catalana per a
la Recerca. S.G. is grateful to the University of
Barcelona and to CONACyT (M�eexico) for sup-
porting her predoctoral research.
References
[1] L.L. Hagedus, R. Aris, A.T. Bell, M. Boudart, N.Y. Chen,
B.C. Gates, W.O. Haag, G.A. Somorjai, J. Wei, Catalyst
Design: Progress and Perspectives, Wiley, New York, 1987.
[2] G. Ertl, H. Kn€oozinger, J. Weitkamp, in: Hanbook of
Heterogeneous Catalysis, vols. 3–5, Wiley-VCH, Weiheim,
1997.
[3] C.G. Vayenas, S. Bebelis, S. Neophytides, J. Phys. Chem.
92 (1988) 5083.
[4] C.G. Vayenas, S. Bebelis, S. Ladas, Nature 343 (1990) 625.
[5] J. Pritchard, Nature 343 (1990) 592.
[6] C.G. Vayenas, S. Brosda, C. Pliangos, J. Catal. 203 (2001)
329.
[7] B. Grzybowska-Swierkosz, J. Haber, Annual Reports on
the Progress of Chemistry No. 91, Royal Soc. of Chem.,
Cambridge, 1994.
[8] C.G. Vayenas, M.M. Jaksic, S. Bebelis, S. Neophytides,
in: J.O.M. Bockris, B.E. Conway, R.E. White (Eds.),

218 S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219
The Electrochemical Activation of Catalysis, Kluwer
Academic/Plenum, New York, 2001.
[9] P.S. Bagus, C.J. Nelin, W. M€uuller, M.R. Philpott, H. Seki,
Phys. Rev. Lett. 58 (1987) 559.
[10] P.S. Bagus, C.J. Nelin, K. Hermann, M.R. Philpott, Phys.
Rev. B 36 (1987) 8169.
[11] P.S. Bagus, G. Pacchioni, Surf. Sci. 236 (1990) 233.
[12] M. Head-Gordon, J.C. Tully, Chem. Phys. 175 (1993)
37.
[13] F. Illas, F. Mele, D. Curulla, A. Clotet, J.M. Ricart,
Electrochem. Acta 44 (1998) 1213.
[14] D. Curulla, A. Clotet, J.M. Ricart, F. Illas, Electrochem.
Acta 45 (1999) 639.
[15] G. Pacchioni, R.J. Lomas, F. Illas, J. Mol. Catal. A 119
(1997) 263.
[16] A. Markovits, M. Garc�ııa-Hern�aandez, J.M. Ricart, F. Illas,
J. Phys. Chem. 103 (1999) 509.
[17] M. Garc�ııa-Hern�aandez, A. Markovits, A. Clotet, J.M.
Ricart, F. Illas, Kluwer series on theoretical chemistry and
physics, Kluwer, Progr. Theor. Chem. Phys. B 7 (2001)
211.
[18] M. Garc�ııa–Hern�aandez, U. Birkenheuer, A. Hu, F. Illas,
N. R€oosch, Surf. Sci. 471 (2001) 151.
[19] M. Garc�ııa-Hern�aandez, D. Curulla, A. Clotet, F. Illas, J.
Chem. Phys. 113 (2000) 364.
[20] D. Curulla, A. Clotet, J.M. Ricart, Surf. Sci. 460 (2000)
101.
[21] M.T.M. Koper, R.A. Van Santen, S.A. Wasileski, M.J.
Weaver, J. Chem. Phys. 113 (2000) 4392.
[22] M.T.M. Koper, R.A. Santen, J. Electroanal. Chem. 476
(1999) 64.
[23] S.A. Wasileski, M.T.M. Koper, M.J. Weaver, J. Phys.
Chem. B 105 (2001) 3518.
[24] S.A. Wasileski, M.J. Weaver, Faraday Discuss. 121 (2002)
285.
[25] R.M. Lambert, A. Palermo, F.J. Williams, M.S. Tikhov,
Solid State Ionics 136 (2000) 677.
[26] J.A. Rodriguez, Surf. Sci. Rep. 24 (1996) 223.
[27] V. Ponec, G.C. Bond, Catalysis by Metals and Alloys,
Elsevier, Amsterdam, 1995.
[28] G. Somorjai, Introduction to Surface Chemistry and
Catalysis, John Wiley & Sons Inc., New York, 1994.
[29] J.A. Rodriguez, D.W. Goodman, Science 257 (1992) 897.
[30] J.H. Sinfelt, Bimetallic Catalysts: Discoveries, Concepts
and Applications, Wiley, New York, 1983.
[31] J.K.A. Clarke, A. Peter, J. Chem. Soc. Faraday Trans. 72
(1975) 1817.
[32] B. Coq, R. Dutartre, F. Figueras, A. Rouco, J. Phys.
Chem. 93 (1989) 4904.
[33] J.A. Anderson, C.H. Rochester, Z.J. Wang, J. Mol. Catal.
A 139 (1999) 285.
[34] J. Szanyi, D.W. Goodman, J. Catal. 145 (1994) 508.
[35] R. Khrishnamurthy, S.S.C. Chuang, K. Ghosal, Appl.
Catal. 114 (1994) 109.
[36] F. Solymosi, J. Cser�eenyi, Catal. Lett. 34 (1995) 343.
[37] F.M.T. Mendes, M. Schmal, Appl. Catal. A 151 (1997)
393.
[38] M. Fern�aandez-Garc�ııa, A. Mart�ıınez-Arias, I. Rodriguez-
Ramos, P. Ferreira-Aparicio, A. Guerrero-Ruiz, Langmuir
15 (1999) 5295.
[39] P. Reyes, G. Pecchi, J.L.G. Fierro, Langmuir 17 (2001)
522.
[40] S. Gonzalez, C. Sousa, M. Fern�aandez-Garc�ııa, V. Bertin, F.
Illas, J. Phys. Chem. B 106 (2002) 7839.
[41] S. Gonzalez, C. Sousa, F. Illas, Surf. Sci. 531 (2003)
39.
[42] C. Sousa, V. Bertin, F. Illas, J. Phys. Chem. B 106 (2002)
7839.
[43] M. Fern�aandez-Garc�ııa, J.A. Anderson, G.L. Haller, J.
Phys. Chem. 100 (1996) 16247.
[44] N. Martensson, R. Nyholm, H. Cal�een, J. Hedman, B.
Johansson, Phys. Rev. B 24 (1981) 1725.
[45] A.M. M�aarquez, N. L�oopez, M. Garc�ııa-Hern�aandez, F. Illas,
Surf. Sci. 442 (1999) 463.
[46] P.S. Bagus, K. Hermann, C.W. Bauschlicher Jr., J. Chem.
Phys. 81 (1984) 1966.
[47] P.S. Bagus, K. Hermann, Phys. Rev. B 33 (1986) 2987.
[48] P.S. Bagus, F. Illas, J. Chem. Phys. 96 (1992) 8962.
[49] T.B. Massalsiki, H. Okamoto, P.R. Subramanian, L.
Kacprezak, Binary Alloy Phase Diagrams, second ed.,
ASM, Materials Parks, Ohio, 1992.
[50] See, for instance, Available from <http://www.webele-
ments.com>.
[51] G. Meitzner, G.H. Via, F.W. Lytle, J.H. Sinfelt, J. Chem.
Phys. 78 (1983) 882.
[52] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.
[53] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988)
785.
[54] M. Dupuis, A.M. Marquez, E.R. Davidson, HONDO99,
1999.
[55] M. Dupuis, A.M. Marquez, E.R. Davidson, HONDO
95.3. Quantum Chemistry Program Exchange, QCPE,
Indiana University, Bloomington, IN 47405.
[56] M.J. Frisch et al. Gaussian 98, Revision A.6, Gaussian
Inc., Pittsburgh PA, 1998.
[57] P.J. Hay, W.R. Wadt, J. Chem. Phys. 82 (1985) 299.
[58] A. Schafer, H. Horn, R. Ahlrichs, J. Chem. Phys. 97 (1992)
2571.
[59] C.J. Nelin, P.S. Bagus, M.R. Philpott, J. Chem. Phys. 87
(1987) 2170.
[60] J.M. Ricart, J. Rubio, F. Illas, P.S. Bagus, Surf. Sci. 304
(1994) 335.
[61] K.P. Huber, G. Herzberg, in: Molecular Spectra and
Molecular Structure, Constants of Diatomic Molecules,
vol. 4, Van Nostrand-Reinhold, New York, 1979.
[62] D.K. Lambert, Solid State Commun. 51 (1984) 297.
[63] D.K. Lambert, J. Chem. Phys. 89 (1988) 3847.
[64] M. Gierer, A. Barbieri, M.A. Van Hove, G.A. Somorjai,
Surf. Sci. 391 (1997) 176.
[65] S. Andersson, J.B. Pendry, J. Phys. C 13 (1980) 2547.
[66] R. Linke, D. Curulla, M.J.P. Hoptstaken, J.W. Niemans-
verdriet, J. Chem. Phys. 115 (2001) 8209.
[67] C.J. Hirschmugl, Y.J. Chabal, F.M. Hoffman, G.P.J.
Williams, Vac. Sci. Technol. A 12 (1994) 2229.

S. Gonz�aalez et al. / Surface Science 548 (2004) 209–219 219
[68] D.G. Castner, B.A. Sexton, G.A. Somorjai, Surf. Sci. 71
(1978) 519.
[69] T.W. Root, L.D. Schmidt, Surf. Sci. 150 (1985) 173.
[70] D.H.Wei, D.C. Skelton, S.D.Kevan, Surf. Sci. 381 (1997) 49.
[71] W. Kirstein, B. Kr€uuger, F. Thieme, Surf. Sci. 176 (1986)
505.
[72] P.S. Bagus, G. Pacchioni, Electrochim. Acta 36 (1991)
1669.