estudo do efeito do tratamento corona...
TRANSCRIPT

VÍTOR CÉSAR DE ALMEIDA LOUZI
ESTUDO DO EFEITO DO TRATAMENTO CORONA
APLICADO A MONOFILAMENTOS DE POLÍMEROS
SINTÉTICOS
CAMPINAS
2015



iv

v

vi

vii
RESUMO
Fibras poliméricas sintéticas vêm sendo muito utilizadas em diversas aplicações (medicina, têxtil,
etc), especialmente na forma de monofilamentos, com diversos diâmetros, devido às suas
características de resistência mecânica. No entanto apresentam baixa hidrofilidade e energia livre
de superfície com propriedades adesivas relativamente fracas, limitando assim suas aplicações.
Com objetivo de ampliar as aplicações deste material, técnicas estão sendo desenvolvidas no
sentido de melhorar estas propriedades, especialmente as de superfície. Dentre as técnicas de
modificação de superfície destaca-se a descarga corona a qual pode operar em condições
ambientes de temperatura e pressão, sem necessidade de agentes químicos, minimizando a
geração de resíduos. Desse modo, o presente trabalho estuda os efeitos promovidos pela descarga
corona em três diferentes fibras poliméricas: polipropileno (PP), poli(tereftalato de etileno) (PET)
e poliamida-6 (PA-6). Amostras desses polímeros foram submetidas à descarga corona sob
condições ambientes controladas. O tratamento corona foi realizado em função dos parâmetros:
distância entre os eletrodos e tempo de tratamento. Após tratadas, as amostras foram submetidas
às técnicas de caracterização: ângulo de contato (Método de Wilhelmy), espectroscopia no
infravermelho (FT-IR/ATR), microscopia eletrônica de varredura (MEV), calorimetria
exploratória diferencial (DSC), ensaios de tração e absorção de água. Os valores de ângulo de
contato dinâmico foram reduzidos em até 29,6%, 27,9% e 18,6% para amostras de PP, PET e PA-
6 tratadas, respectivamente. Os valores de energia de superfície aumentaram em 50,3%, 26% e
15% para PP, PET e PA-6. Amostras de PP, PET e PA-6 tratadas por descarga corona (4mm e
120s) apresentaram grau de absorção de água igual a 9,4%, 8,5% e 9,1%. A partir dos resultados
obtidos com amostras de monofilamentos de PP, PET e PA-6, sem tratamento e após corona,
pode-se concluir que o tratamento corona promove aumento da hidrofilidade e as propriedades
térmicas e mecânicas não são afetadas.
Palavras-chave: corona (eletricidade), fibras têxteis, monofilamentos poliméricos, polipropileno,
poli(tereftalato de etileno), poliamida-6.

viii

ix
ABSTRACT
Synthetic polymer fibers have been widely used in various applications, especially in the form of
monofilament and under various diameters, due to its characteristics of mechanical resistance.
However synthetic polymer fibers exhibit low hydrophilicity and surface free energy with
relatively weak adhesive properties, thus limiting their applications. Aiming to expand the
applications of these materials, techniques are being developed to improve these properties,
especially the surface. Among modification techniques there is the corona discharge which can
operate in ambient temperature and pressure, without the need for chemical agents, minimizing
the generation residue. Thus, this research work studies the effects caused by the corona
discharge in three different polymeric fibers: polypropylene (PP), poly(ethylene terephthalate)
(PET) and polyamide-6 (PA-6). Samples of these polymers were subjected to corona discharge
under controlled environmental conditions. The corona treatment was performed on the basis of
the parameters: distance between the electrodes and time of treatment. After treated, the samples
were subjected to characterization techniques: contact angle (Wilhelmy Method), infrared
spectroscopy (FT-IR/ATR), scanning electron microscopy (SEM), differential scanning
calorimetry (DSC), tensile and water absorption. The dynamic contact angle values were reduced
by up to 29,6%, 27,9% and 18,6% for PP, PET and PA-6 treated samples, respectively. The
surface energy values increased by 50,3%, 26% and 15% for PP, PET and PA-6. PP, PET and
PA-6 samples treated by corona discharge (4mm and 120s), showed the degree of water
absorption equal of 9,4%, 8,5% and 9,1%. From the results obtained with samples of PP, PET
and PA-6, without treatment and after treatment, can be concluded that the corona treatment
enhances the hydrophilicity and the thermal and mechanical properties are not affected.
Keywords: corona (electricity), textile fibers, monofilament polymer, polypropylene,
poly(ethylene terephthalate), polyamide-6.

x

xi
SUMÁRIO
1 INTRODUÇÃO ..................................................................................................................... 1
1.1 OBJETIVOS ......................................................................................................................... 3
1.1.1 Objetivo Geral ................................................................................................................... 3
1.1.2 Objetivos Específicos ........................................................................................................ 3
2 REVISÃO BIBLIOGRÁFICA ............................................................................................ 4
2.1 ADESÃO EM POLÍMEROS ............................................................................................... 4
2.1.1 Molhabilidade e Ângulo de Contato .................................................................................. 5
2.1.2 Energia Livre de Superfície ............................................................................................... 7
2.2 DETERMINAÇÃO DA ENERGIA DE SUPERFÍCIE DE SÓLIDOS ............................... 8
2.2.1 Método de Zisman ............................................................................................................. 8
2.2.2 Teoria de Fowkes ............................................................................................................ 10
2.2.3 Média Geométrica ........................................................................................................... 10
2.2.4 Média Harmônica ............................................................................................................ 11
2.3 FIBRAS TÊXTEIS E SUAS APLICAÇÕES ..................................................................... 12
2.3.1 Classificação de fibras ..................................................................................................... 13
2.3.2 Propriedades de fibras ..................................................................................................... 14
2.3.3 Monofilamentos ............................................................................................................... 15
2.3.4 Polipropileno - PP ............................................................................................................ 15
2.3.5 Poli(tereftalato de etileno) - PET ..................................................................................... 17
2.3.6 Poliamida 6 – PA-6 ......................................................................................................... 18
2.4 MÉTODOS DE MODIFICAÇÃO DE SUPERFÍCIE ....................................................... 20
2.4.1 Tratamento Químico ........................................................................................................ 21

xii
2.4.2 Tratamento por Plasma .................................................................................................... 22
2.4.3 Tratamento por Descarga Corona .................................................................................... 24
2.5 TÉCNICAS DE CARACTERIZAÇÃO ............................................................................. 27
2.5.1 Medidas de Ângulo de Contato Dinâmico ...................................................................... 27
2.5.2 Espectroscopia de Infravermelho com Refletância Total Atenuada ............................... 30
2.5.3 Microscopia Eletrônica Exploratória ............................................................................... 34
2.5.4 Calorimetria Exploratória Diferencial ............................................................................. 35
2.5.5 Ensaio Mecânico de Tração ............................................................................................. 39
3 MATERIAIS E MÉTODOS ............................................................................................... 41
3.1 MATERIAIS ...................................................................................................................... 41
3.2 LIMPEZA DAS AMOSTRAS ........................................................................................... 41
3.3 DESCARGA CORONA ..................................................................................................... 41
3.3.1 Descrição do Sistema Fio-plano ...................................................................................... 41
3.3.2 Tratamento da Superfície das Amostras .......................................................................... 42
3.4 MEDIDAS DE ÂNGULO DE CONTATO DINÂMICO E ESTÁTICO .......................... 43
3.5 ESPECTROSCOPIA NO INFRAVERMELHO/ATR (FT-IR/ATR) ................................ 44
3.6 CALORIMETRIA EXPLORATÓRIA DIFERENCIAL (DSC) ........................................ 44
3.7 MICROSCOPIA ELETRÔNICA EXPLORATÓRIA ....................................................... 46
3.8 ENSAIO MECÂNICO DE TRAÇÃO ............................................................................... 46
3.9 ABSORÇÃO DE ÁGUA.................................................................................................... 46
3.10 CÁLCULO DA ENERGIA LIVRE DE SUPERFÍCIE ................................................... 47
4 RESULTADOS E DISCUSSÕES ...................................................................................... 49
4.1 MEDIDAS DE ÂNGULO DE CONTATO DINÂMICO .................................................. 49
4.1.1 Influência da limpeza das amostras ................................................................................. 49

xiii
4.1.2 Influência do número de ciclos de medidas .................................................................... 50
4.1.3 Influência da velocidade de imersão ............................................................................... 52
4.2 TEMPO E DISTÂNCIA INTER-ELETRODOS NO TRATAMENTO CORONA .......... 53
4.3 COMPOSIÇÃO QUÍMICA DE SUPERFÍCIE (FT-IR/ATR) ........................................... 58
4.4 ANÁLISE TOPOGRÁFICA DE MONOFILAMENTOS (MEV) ..................................... 62
4.5 PROPRIEDADES TÉRMICAS (DSC) .............................................................................. 66
4.6 PROPRIEDADES MECÂNICAS DE TRAÇÃO .............................................................. 71
4.7 ABSORÇÃO DE ÁGUA.................................................................................................... 72
4.8 CÁLCULO DA ENERGIA LIVRE DE SUPERFÍCIE ..................................................... 74
5 CONCLUSÕES E SUGESTÕES PARA TRABALHOS FUTUROS ............................. 88
5.1 CONCLUSÕES .................................................................................................................. 88
5.2 SUGESTÕES PARA TRABALHOS FUTUROS .............................................................. 89
6 REFERÊNCIAS .................................................................................................................. 90

xiv

xv
AGRADECIMENTOS
Agradeço primeiramente e imensamente a Deus, pelo dom da vida e por Ele estar presente
sempre em minha vida me apoiando e realizando obras maravilhosas.
Aos professores da FEQ, Lúcia Mei, Antônio Carlos Lisboa, Ana Rita Morales, Marisa
Beppu e Aline Costa, pelos ensinamentos e transferência de conhecimento.
Ao meu orientador, Prof. Dr. João Sinézio de Carvalho Campos, pelos ensinamentos.
Agradeço à Profa. Dra. Sandra Cristina dos Santos Rocha, responsável pelo Lab. de
Fluidodinâmica e Secagem (LFS)/FEQ/UNICAMP, pela realização de ensaios de molhabilidade
e cálculo de energia de superfície.
Aos meus pais César e Alda e aos meus irmãos Igor e Felipe pelo amor, carinho e
credibilidade dados a mim durante esse período.
Aos meus tios Lúcia e Milton e às minhas primas Carol e Marina pela estrutura e suporte
que me deram desde que cheguei a Campinas e por todo carinho e atenção desde sempre!
Ao Mauro Anastácio por colaborar no desenvolvimento deste projeto, pelo apoio,
conselhos, paciência, colaboração, estímulo, perseverança e amizade.
Ao Lucas, Daniel, Júnior, Fernando, Thalles e Matheus pela amizade e por terem me
proporcionado imensos momentos de alegria e diversão.
Aos técnicos: Adilson, Celso e Lucélia do Laboratório de Recursos Analíticos e de
Calibração (LRAC)/FEQ/UNICAMP, pelas análises das amostras.
À empresa Mazzaferro Ltda. pela disposição em conceder amostras dos monofilamentos
usados nesta pesquisa.
A CAPES e à Unicamp pela bolsa de estudo concedida.
Por fim, agradeço de modo sincero a todos que contribuíram para realização e conclusão
deste trabalho e etapa da minha vida.
Muito Obrigado!

xvi

xvii
LISTA DE FIGURAS
Figura 1: Representação de ângulo de contato. a) Gota de um líquido depositada sobre uma
superfície; b) Material sólido emerso de um líquido (Fonte: Gupta, 2013). .............................. 6
Figura 2: Determinação da tensão superficial crítica ................................................................. 9
Figura 3: Unidade de repetição do polipropileno ..................................................................... 16
Figura 4: Unidade de repetição do PET ................................................................................... 17
Figura 5: Unidade repetitiva do nylon-6................................................................................... 19
Figura 6: Modificação da superfície de polímeros (Fonte: Adaptado de Ma, Mao, Gao, (2007)).
.................................................................................................................................................. 21
Figura 7: Corona em ar (Fonte: Adaptado de Akshev et. al, (2005) e Antao et. al. (2009)). ... 25
Figura 8: Oxidação da superfície de polipropileno (Fonte: Adaptado de Sellin (2002)). ........ 26
Figura 9: Representação de sistema utilizado no Método de WilHelmy (Fonte: Adaptado de
Sheridan et. al., (1994)) ............................................................................................................ 28
Figura 10: Representação da espectroscopia de infravermelho (Fonte: Adaptado de Alves, 2014).
.................................................................................................................................................. 31
Figura 11: Reflexão interna na interface amostra/cristal (Fonte: Adaptado de Alkor Crystal
Optics, 2014) ............................................................................................................................ 32
Figura 12: Interior de um microscópio eletrônico de varredura (Fonte: Lopes, 2012). ........... 35
Figura 13: Representação de um DSC de fluxo de calor (Fonte: Lopes, 2012). ...................... 37
Figura 14: Representação de um DSC de compensação de potência (Fonte: Lopes, 2012). ... 38
Figura 15: Diagrama de DSC em polímeros (Fonte: Lopes, 2012) .......................................... 39
Figura 16: Ilustração de uma curva de tensão em função da deformação (Fonte: Sena, 2011).40
Figura 17: Representação do sistema corona fio-plano ............................................................ 42
Figura 18: Ângulo de contato em função do tempo de tratamento para PP. a) ângulo de avanço; b)
ângulo de recuo; c) ângulo de equilíbrio; d) histerese .............................................................. 54

xviii
Figura 19: Ângulo de contato em função do tempo de tratamento para PET. a) ângulo de avanço;
b) ângulo de recuo; c) ângulo de equilíbrio; d) histerese ......................................................... 55
Figura 20: Ângulo de contato em função do tempo de tratamento para PA-6. a) ângulo de
avanço; b) ângulo de recuo; c) ângulo de equilíbrio; d) histerese ............................................ 56
Figura 21: Espectros FT-IR/ATR dos monofilamentos de PP sem tratamento, e tratados por 10s,
60s e 120s. ................................................................................................................................ 59
Figura 22: Espectros FT-IR/ATR dos monofilamentos de PET sem tratamento, e tratados por 10s,
60s e 120s. ................................................................................................................................ 60
Figura 23: Espectros FT-IR/ATR dos monofilamentos de PA-6 sem tratamento, e tratados por
10s, 60s e 120s. ......................................................................................................................... 61
Figura 24: Topografias de monofilamentos de PP, obtidos por MEV. a) sem tratamento corona e
tratados por: b) 10s; c) 60s; d) 120s. ........................................................................................ 63
Figura 25: Topografias de monofilamentos de PET, obtidos por MEV. a) sem tratamento corona
e tratados por: b) 10s; c) 60s; d) 120s. ...................................................................................... 64
Figura 26: Topografias de monofilamentos de PA-6, obtidos por MEV. a) sem tratamento corona
e tratados por: b) 10s; c) 60s; d) 120s. ...................................................................................... 65
Figura 27: Curvas de DSC de monofilamentos de PP antes a pós tratamento corona (picos de
fusão das amostras) ................................................................................................................... 67
Figura 28: Curvas de DSC de monofilamentos de PP antes e após tratamento corona (picos de
cristalização das amostras) ....................................................................................................... 67
Figura 29: Curvas de DSC de monofilamentos de PET antes a pós tratamento corona (picos de
fusão das amostras) ................................................................................................................... 68
Figura 30: Curvas de DSC de monofilamentos de PET antes a pós tratamento corona (picos de
cristalização das amostras) ....................................................................................................... 69
Figura 31: Curvas de DSC de monofilamentos de PA-6 antes a pós tratamento corona (picos de
fusão das amostras) ................................................................................................................... 70

xix
Figura 32: Curvas de DSC de monofilamentos de PA-6 antes a pós tratamento corona (picos de
cristalização das amostras) ....................................................................................................... 70
Figura 33: Grau de absorção de água de amostras de PP, PET e PA-6 não tratadas e tratadas por
corona em 10s, 60s e 120s. ....................................................................................................... 73
Figura 34: Ângulos de avanço em função do tempo de tratamento para amostras de PP. ....... 75
Figura 35: Ângulos de recuo em função do tempo de tratamento para amostras de PP. ......... 75
Figura 36: Ângulos de equilíbrio em função do tempo de tratamento para amostras de PP. ... 76
Figura 37: Ângulos de avanço em função do tempo de tratamento para amostras de PET. .... 77
Figura 38: Ângulos de recuo em função do tempo de tratamento para amostras de PET. ....... 77
Figura 39: Ângulos de equilíbrio em função do tempo de tratamento para amostras de PET. 78
Figura 40: Ângulos de avanço em função do tempo de tratamento para amostras de PA-6. ... 79
Figura 41:Ângulos de recuo em função do tempo de tratamento para amostras de PA-6. ...... 79
Figura 42: Ângulos de equilíbrio em função do tempo de tratamento para amostras de PA-6.80
Figura 43: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de avanço para amostras de PP. .................................................................................... 80
Figura 44: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de recuo para amostras de PP. ...................................................................................... 81
Figura 45: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de equilíbrio para amostras de PP. ................................................................................ 81
Figura 46: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de avanço para amostras de PET. ................................................................................. 82
Figura 47: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de recúo para amostras de PET. .................................................................................... 83
Figura 48: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de equilíbrio para amostras de PET. ............................................................................. 83

xx
Figura 49: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de avanço para amostras de PA-6. ................................................................................ 84
Figura 50: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de recúo para amostras de PA-6. .................................................................................. 85
Figura 51: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de equilíbrio para amostras de PA-6. ............................................................................ 85

xxi
LISTA DE TABELAS
Tabela 1: Tensão superficial crítica de polímeros (Fonte: Zisman, 1964) ................................. 9
Tabela 2: Grupos funcionais e faixa de absorção no infravermelho ........................................ 34
Tabela 3: Propriedades térmicas do PP, PET e PA-6 (ASTM D3418-12) ............................... 36
Tabela 4: Niveis das variáveis estudadas no tratamento corona .............................................. 43
Tabela 5: Condições de ensaio DSC para PP, PET e PA-6 ...................................................... 45
Tabela 6: Energia de superfície e suas componentes para água e a e diiodo metano ............... 47
Tabela 7: Influência da limpeza dos monofilamentos nos valores de ângulo de contato de avanço
e retrocesso ............................................................................................................................... 49
Tabela 8: Influência do número de ciclos nos valores de ângulo de contato de avanço e retrocesso
para monofilamentos de PP, PET e PA-6. Velocidade de 4,5mm/min .................................... 51
Tabela 9: Influência da velocidade nos valores de ângulo de contato de avanço e retrocesso para
monofilamentos de PP, PET e PA-6. Velocidade de 9,0mm/min ............................................ 52
Tabela 10: Valores de ângulo dinâmicos de avanço e retrocesso adotados como padrão de
controle para monofilamentos limpos e sem tratamento corona .............................................. 53
Tabela 11: Propriedades Térmicas para amostras de PP .......................................................... 68
Tabela 12: Propriedades Térmicas para amostras de PET ....................................................... 69
Tabela 13: Propriedades Térmicas para amostras de PA-6 ...................................................... 71
Tabela 14: Propriedades mecânicas de monofilamento de PP, PET e PA-6 ............................ 72

xxii

xxiii
LISTA DE SIGLAS
ASTM American Society for Testing and Materials;
CGS Centimetro-Grama-Segundo;
DSC Calorimetria Exploratória Diferencial;
FT – IR/ATR Espectroscopia no Infravermelho com Transformadas de Fourier
utilizando dispositivo de Refletância Total Atenuada;
MEV Microscopia Eletrônica de Varredura;
NTPP Não Tecido de Polipropileno;
PA-6 Poliamida-6
PET Poli(tereftalato de etileno)
PP Polipropileno
PPS Poli (p-sulfeto de etileno)
SI Sistema Internacional de Unidades
UV Ultravioleta
XPS Espectroscopia Fotoelétrica de Raio-X

xxiv

xxv
NOMENCLATURA
𝐴 Área da seção transversal;
𝑎 Coeficiente linear;
𝑏 Coeficiente angular;
𝑐𝑝 Calor específico;
𝑑 Diâmetro;
𝐷𝑝 Profundidade de penetração;
𝜎 Tensão;
𝜎𝐸𝑠𝑐𝑜𝑎𝑚𝑒𝑛𝑡𝑜 Limite de escoamento;
𝜎𝑀á𝑥 𝑅𝑢𝑝 Resistência máxima à ruptura;
𝛥𝐻𝑐 Entalpia de cristalização;
𝛥𝐻𝑚 Entalpia de fusão;
𝛥𝐻𝑟 Entalpia de reação;
Xc Grau de cristalinidade;
𝐸 Módulo de elasticidade;
𝜀 Deformação;
𝜀𝑅𝑢𝑝 Deformação na ruptura;
𝐹𝑡𝑜𝑡𝑎𝑙 Força total de equilíbrio;
𝐺𝐴 Grau de absorção de água;
𝛷 Ângulo de incidência da radiação infravermelha;
𝑔 Aceleração da gravidade;
𝛾𝐿𝑉𝑑 Componente dispersiva da energia de superfície do líquido;

xxvi
𝛾𝑆𝑉𝑑 Componente dispersiva da energia de superfície do sólido;
𝛾𝐿𝑉𝑝
Componente polar da energia de superfície do líquido;
𝛾𝑆𝑉𝑝
Componente polar da energia de superfície do sólido;
𝛾𝐿𝑉 Energia livre de superfície na interface líquido-gás;
𝛾𝑆𝐿 Energia livre de superfície na interface sólido-líquído;
𝛾𝑆𝑉 Energia livre de superfície do sólido;
∆𝜃 Diferença entre ângulo de avanço e recuo (Histerese);
𝐼 Intensidade do feixe com amostra;
𝐼𝑜 Intensidade do feixe sem amostra;
𝜆 Comprimento de onda;
𝑚 Massa;
𝑚𝑠 Massa seca;
𝑚𝑢 Massa úmida final;
𝑛1 Índice de refração do elemento de reflexão interna;
𝑛212 Razão entre o índice de refração da amostra e do elemento de reflexão
interna;
𝑃 Perímetro;
𝜌𝐿 Densidade do líquido;
𝑇 Transmitância;
𝑇𝑎 Temperatura da amostra;
𝑇𝑅 Temperatura de referencia;
𝑇𝑐 Temperatura de cristalização;
𝑇𝑔 Temperatura de transição vítrea
𝑇𝑚 Temperatura de fusão;

xxvii
𝜃 Ângulo de contato;
𝜃𝑎 Ângulo de avanço;
𝜃𝑟 Ângulo de recuo;
𝜃𝐸𝑄 Ângulo de equilíbrio;

xxviii

1
1 INTRODUÇÃO
Materiais poliméricos são utilizados em ampla gama de aplicações por apresentarem
durabilidade mecânica, fácil processamento e moldagem, substituindo outros materiais como os
metais e as cerâmicas. No entanto, a aplicação da maioria dos polímeros no campo da adesão é
limitada devido ao baixo valor de energia de superfície que apresentam (Mazzola; Bemporad;
Carassiti, 2010).
Indústrias têxteis têm desenvolvido produtos cada vez mais versáteis através do uso de
fibras de polímeros sintéticos como polipropileno, poli(tereftalato de etileno) e poliamida. Por
conseguinte, as fibras podem ser usadas em várias aplicações ligadas, por exemplo, a fenômenos
de absorção de umidade e adsorção, a produtos biocompatíveis e/ou a adesivos (John e
Anandjiwala, 2009).
Uma grande desvantagem de algumas fibras sintéticas é seu alto caráter hidrofóbico, o
que impede a aplicação de corantes e produtos de acabamento durante a impressão e tingimento.
Muitas dessas aplicações requerem melhorias da interação da superfície das fibras com outros
materiais como, por exemplo, promoção de adesão (John e Anandjiwala, 2009).
Assim, a modificação da superfície de materiais têxteis é realizada com o intuito de
melhorar várias propriedades relacionadas à hidrofilidade como molhabilidade, absorção de
umidade, adesão e energia de superfície (John e Anandjiwala, 2009).
Propriedades de superfície dos biomateriais têxteis como monofilamentos (dispositivos
biomédicos, órgãos artificiais e bionsensores) tais como molhabilidade
(hidrofilidade/hidrofobicidade), energia livre de superfície, composição química e rugosidade são
importantes na avaliação da biocompatibilidade material-organismo (Lee et. al., 1998). Sabe-se
que o aumento da hidrofilidade em materiais implantáveis é necessário para que suportem a
adesão e proliferação celular. Desta forma, a utilização de biomateriais que apresentam
molhabilidade moderada resulta em resposta celular positiva (Ma; Mao; Gao, 2007).
Superfícies de polímeros podem ser modificadas sob variados tipos de tratamento como:
químico, plasma, irradiação ultravioleta ou corona (Sellin, 2002). Como técnica de modificação
superficial a descarga corona tem sido um método útil na melhoria da propriedade hidrófila de

2
superfícies poliméricas. Diante de outros métodos químicos e físicos de modificação superficial,
a descarga corona apresenta vantagens por tratar amostras rapidamente a condições ambientes
(Xu e Liu, 2003).
Durante o tratamento corona, superfícies de poliolefinas sofrem oxidação pela quebra de
cadeias e criação de grupos polares na superfície sem alterar suas propriedades de volume. Desta
maneira, tem-se como resultado uma superfície mais polar, com maior caráter hidrofílico (Sellin
e Campos, 2003). De acordo Sellin, 2002, o tratamento corona propicia alteração na topografia
do material, ocasionando aumento na rugosidade superficial, facilitando o ancoramento de tintas,
corantes ou adesivos na superfície de polímeros.
A descarga corona pode ser usada para melhoria do tingimento de fios têxteis naturais ou
sintéticos e também como etapa de copolimerização produzindo um material com funcionalidade
diferente, tendo em vista que após irradiação, um novo grupo funcional é produzido e assim um
monômero pode ser enxertado na superfície do material tratado (Xu e Liu, 2003).
A modificação da superfície de polímeros por descarga corona é bastante estudada por
vários autores e largamente usada na indústria, no entanto a aplicação deste tratamento de
superfície em fibras é pouco praticada, gerando problemas quanto a sua aplicação por falta de
melhor entendimento de propriedades como: molhabilidade, energia de superfície.
Muitos trabalhos foram realizados com o intuito de se avaliar os efeitos do tratamento por
descarga corona na superfície de filmes e tecidos com pouca atenção em relação a fios
provenientes de polímeros sintéticos. O tratamento com descarga corona já é largamente
empregado em filmes poliméricos a fim de se obter maior adesão à tintas. Dispositivos
biomédicos tem sido modificados superficialmente para melhoria da adesão celular e
biocompatibilidade. Tecidos sintéticos e naturais são tratados com descarga corona com o intuito
de melhorar a interação da superfície com corantes durante o tingimento. Almeja-se, então,
estudar e analisar as características apresentadas por monofilamentos de polímeros sintéticos
frente ao tratamento por descarga corona, contribuindo, assim, para que posteriores estudos e
possíveis aplicações possam ser realizados, caracterizando as amostras sem tratamento e após,
através de técnicas adequadas.

3
1.1 OBJETIVOS
1.1.1 Objetivo Geral
O objetivo deste trabalho é analisar os efeitos promovidos por descarga corona em
monofilamentos de polipropileno (PP), poliamida (PA-6) e politereftalato de etileno (PET), com
propósito de avaliar modificações de propriedades físico-químicas de superfície, compreendendo
o tratamento para possíveis aplicações na (indústria têxtil, medicina,etc) onde se requer melhoria
das propriedades de adesão de monofilamentos sintéticos.
1.1.2 Objetivos Específicos
Levantamento bibliográfico através de pesquisa de artigos, teses e livros;
Obter matérias-primas sob a forma de monofilamentos de PP, PET e PA-6 para
realização do tratamento de superfície;
Realizar limpeza das amostras verificando-se os efeitos sobre sua propriedade de
superfície através das estimativas de ângulo de contato dinâmico;
Aplicar a técnica de descarga corona sobre a superfície de amostras de
monofilamentos de PP, PA-6 e PET;
Analisar a influência do tempo de tratamento e distância inter-eletrodos no
tratamento corona sobre a superfície das amostras;
Caracterizar amostras antes a antes e após tratamento de superfície através das
técnicas de medidas de ângulo de contato dinâmico, absorção de água,
espectroscopia no infravermelho (FT-IR/ATR), calorimetria exploratória
diferencial (DSC), microscopia eletrônica de varredura (SEM) e ensaio mecânico
de tração.
Adaptar o método de WilHelmy para determinação de valores de ângulo de
contato estático e cálculo da energia livre de superfície;
Estimar valores de energia livre de superfície de amostras antes e após tratamento
corona através do método da média harmônica;

4
2 REVISÃO BIBLIOGRÁFICA
2.1 ADESÃO EM POLÍMEROS
O fenômeno da adesão é conhecido como uma série de interações interatômicas e
moleculares decorrentes na interface de duas superfícies. Um conjunto de mecanismos baseados
na difusão, fenômenos mecânicos, moleculares, químicos e termodinâmicos são utilizados na
tentativa de descrever a adesão (Awaja et. al., 2009).
Materiais poliméricos estão sendo cada vez mais utilizados por apresentarem excelentes
propriedades físicas e químicas. Estes materiais apresentam como vantagens o seu baixo custo de
processo, baixa toxidade, durabilidade mecânica e são de fácil processamento e moldagem
(Mazzola; Bemporad; Carassiti, 2010).
No entanto, a maioria dos polímeros possuem limitações quanto às suas propriedades de
superfície, ficando muitas vezes impedidos de serem utilizados no campo da adesão, impressão,
metalização e como materiais biocompatíveis (Mazzola; Bemporad; Carassiti, 2010).
Desse modo, variadas técnicas de modificação de superfície tem sido desenvolvidas e
aprimoradas para aumento da molhabilidade, adesão e impressão de superfícies poliméricas. Tais
técnicas induzem a formação de grupos polares, com pouca atenção à especificidade dos grupos
funcionais introduzidos na superfície. Caso o material tenha aplicação pré-determinada, estas
técnicas devem ser adaptadas com a função de introduzir grupos funcionais específicos (Goddard
e Hotchikiss, 2007).
Recentemente, os setores automotivo e aeroespacial estão direcionando seu interesse para
o campo da adesão de polímeros e resinas epóxi, promoção de melhor aderência entre tintas e
superfícies poliméricas através da inserção de grupos químicos na interface. A indústria da
construção utiliza polímeros e selantes para os casos em que o material de vedação apresente
capacidade de resistência a expansão e contração térmica e boa ligação ao substrato. (Awaja et.
al., 2009).
Na biomedicina, vários estudos têm investigado os mecanismos de adesão entre a
superfície de polímeros e as células do tecido no qual é implantado com o intuito de melhorar a
aderência durante a aplicação (Klee et. al., 2003).

5
No setor têxtil, monofilamentos poliméricos tem sido cada vez mais usados em aplicações
que envolvem absorção de umidade como em lenços, absorventes e meios filtrantes, técnicas de
tingimento e impressão. Para tais aplicações é necessário que a superfície do material apresente
hidrofilidade e boa molhabilidade, que pode ser verificada através das interações entre a
superfície e um meio líquido (John e Anandjiwala, 2009).
A adesão é dependente, basicamente, de duas propriedades de superfície: molhabilidade e
energia livre de superfície, sendo os seus valores muito baixos em se tratando de polímeros
(Mazzola; Bemporad; Carassiti, 2010). O conceito para tais propriedades são melhor descritos
nos itens a seguir: 2.1.1 e 2.2.2.
Outras propriedades como rugosidade, polaridade e composição química de superfície
também descrevem e explicam a adesão utilizando determinadas técnicas de caracterização
(Awaja et. al., 2009).
2.1.1 Molhabilidade e Ângulo de Contato
A molhabilidade pode ser definida como a capacidade de um determinado líquido em se
espalhar por uma superfície sólida. Sua compreensão pode ser dada através da medição do ângulo
de contato (θ) formado entre o sólido e o líquido envolvidos durante a adesão (Sellin, 2002;
Zisman, 1964).
O ângulo de contato de um líquido é definido geometricamente como o resultado de um
processo de equilíbrio gerado pela deposição de uma gota na superfície sólida (suave,
homogênea, não deformável e isotrópica), sob a ação de forças de superfície (Awaja, et. al.,
2009), como mostra a Fig. 1a.
Quando um material é imerso ou retirado de um determinado fluido, ângulos de contato
dinâmicos são produzidos, gerando dois ângulos: ângulo de avanço e de retrocesso. O ângulo de
avanço decorre da imersão na amostra no fluido, enquanto que o ângulo de retrocesso, também
conhecido como ângulo de recúo ocorre na emersão da amostra sobre o fluido (John e
Anandjiwala, 2009), como mostra Fig.1b.

6
Na Fig. 1, 𝛾𝑆𝑉, 𝛾𝐿𝑆 e 𝛾𝐿𝑉 representam as tensões, ou energia de superfície, interfaciais
sólido/ar, sólido/líquido e líquido/ar, respectivamente. Young estabeleceu uma relação qualitativa
entres essas tensões que pode ser escrita de forma algébrica (Zisman, 1964; Awaja et. al., 2009).
𝛾𝑆𝑉 − 𝛾𝐿𝑆 = 𝛾𝐿𝑉. cos 𝜃 (1)
Figura 1: Representação de ângulo de contato. a) Gota de um líquido depositada sobre uma
superfície; b) Material sólido emerso de um líquido (Fonte: Gupta, 2013).
A Eq. 1 é denominada Equação de Young. Os valores do ângulo de contato encontram-se
numa faixa que varia de 0° a 180°. Quando θ = 0° toda a superfície sólida é molhada
completamente pelo líquido que se espalha sobre ela a uma taxa que varia com a viscosidade do
líquido e rugosidade da superfície em que se encontra. Caso o ângulo seja maior que zero (θ > 0)
o líquido não molhará a superfície completamente (Sellin, 2002).

7
Experimentos comprovam que cada tipo de líquido se espalha em algum tipo de sólido em
determinada amplitude, mas nunca completamente, ou seja, θ > 180° (Sellin, 2002). Desta forma,
pode-se concluir que o ângulo de contato é uma medida contrária à molhabilidade.
Para que a Equação de Young possa ser aplicada algumas suposições são seguidas
possibilitando que a energia de superfície do sólido possa ser determinada. O líquido utilizado
para medições do ângulo de contato deve ser puro, não sendo empregadas soluções surfactantes
ou misturas de líquidos já que podem apresentar adsorção preferencial ao material. O valor da
tensão de interface líquido/vapor deve ser constante ao se medir o ângulo de contato. Desta
forma, é necessário impedir que reações químicas ou físicas ocorram entre o sólido e o líquido. A
energia interfacial entre a fase líquido/vapor deve ser maior que a energia de interface
sólido/vapor a ser determinada, para que não ocorra completa molhabilidade (Sena, 2011).
2.1.2 Energia Livre de Superfície
A energia livre de superfície é também denominada tensão superficial em líquidos ou
energia de superfície em sólidos, sendo expressa no sistema CGS em dyn/cm ou crg/cm² ou no
sistema SI em mN/m (milinewtons por metro) ou mJ/m² (milijoules por metro quadrado).
Geralmente, utiliza-se o símbolo 𝛾 (letra grega gama) acompanhado por letras subscritas que
indicam quais fases estão presentes na interface (Sena, 2011).
Moléculas que estão predominantemente no interior de uma substância estão sob ação de
forças intermoleculares do tipo Van Der Waals em todas as direções, no entanto o mesmo
fenômeno não pode ser observado para as moléculas que se encontram na superfície (Sellin,
2002).
As moléculas da superfície orientam-se, de forma preferencial, para o interior da
substância, por sofrerem atração simétrica pelas moléculas do volume interior o que gera uma
forma geométrica com menor área como, por exemplo, gotas esféricas em líquidos. No caso dos
líquidos, a energia livre de superfície pode ser definida como uma força que atua em paralelo à
superfície por unidade de comprimento. Entretanto, no caso de materiais sólidos, não ocorre
equivalência entre os átomos da superfície como ocorre nos líquidos e o fenômeno observado em
líquidos não poderá ocorrer da mesma forma (Costa, 1982).

8
A água utilizada na grande maioria dos testes, como líquido de prova, apresenta tensão
superficial de 72 mN/m. Líquidos apresentam em geral energia de superfície menor que 100
mN/m. Materiais sólidos como metais, rubi, diamante e sílica, apresentam tensão superficial entre
500-5000 mN/m, sendo suas superfícies consideradas com elevada energia. Em se tratando dos
polímeros, sua energia livre superficial encontra-se numa faixa de 20 a 30 mN/m, sendo
considerados materiais de baixa energia livre de superfície (Sellin, 2002).
2.2 DETERMINAÇÃO DA ENERGIA DE SUPERFÍCIE DE SÓLIDOS
Uma série de fenômenos relacionados a adesão e engenharia de superfície são
compreendidos pela determinação da energia de superfície dos materiais no estado sólido. Vale
ressaltar que seu valor não é facilmente determinado a partir da Equação de Young devido à
existência de dois parâmetros não determinados: 𝛾𝑆𝑉 e 𝛾𝑆𝐿. Utilizando-se, então, no mínimo um
dois ou três líquidos diferentes, juntamente com o valor de ângulo de contato obtido será possível
fazer uma estimativa de tais parâmetros (Sena, 2011).
A Termodinâmica apresenta como vantagem analisar a aderência apenas como um
processo de equilíbrio decorrente na interface. Muitas teorias foram introduzidas com o intuito de
descrever e medir a energia de superfície de materiais com aplicações a sistemas poliméricos
(Awaja et. al., 2009). Metodologias para determinação da energia de superfície como: Métodos
de Zisman, Teoria de Fowkes, Média Geométrica e Média Harmônica serão explicados a seguir.
2.2.1 Método de Zisman
O Método de Zisman caracteriza um sólido determinando sua tensão superficial crítica.
Mede-se o ângulo de contato entre diferentes líquidos e a superfície, reunindo-se os valores
medidos em um gráfico cos 𝜃 versus tensão superficial dos líquidos de referência. Obtém-se uma
relação linear extrapolada até o ponto em que o líquido molha totalmente a superfície, θ = 0° ou
cos 𝜃 =1. O valor da tensão crítica do sólido será projetado no eixo das abcissas (Zisman, 1964),
como mostra a Fig.2.

9
Figura 2: Determinação da tensão superficial crítica
Os resultados apresentados na Tab. 1 mostram que a tensão superficial crítica dos
polímeros no estado sólido varia com a estrutura química molecular superficial. Dessa forma, a
substituição de átomos de flúor por hidrogênio faz com que haja um aumento da tensão
superficial crítica. Essa adição de energia pode ser verificada também nos polímeros
clorocarbonados, onde há a substituição de átomos de hidrogênio por átomos de cloro (Zisman,
1964).
Tabela 1: Tensão superficial crítica de polímeros (Fonte: Zisman, 1964)
Polímero 𝜸𝒄(mN/m) a 20°C
Poli (hexafluorpropileno) 16,2
Poli (tetrafluoretileno) 18,5
Poli (trifluoretileno) 22
Poli (fluoreto de vinilideno) 25
Poli (fluoreto de vinila) 28
Polietileno 31
Poli (trifluorcloroetileno) 31
Polipropileno 32
Poliestireno 33
Poli (álcool de vinila) 37
Poli (cloreto de vinilideno) 40
Poli (cloreto de vinila) 40
Poli (tereftalato de etileno) 43

10
Através de seus resultados, Zisman (1964) deduziu que a molhabilidade ou tensão
superficial crítica acontece devido aos arranjos atômicos que se encontram na interface
sólido/líquido, sendo independente de arranjos mais distantes.
2.2.2 Teoria de Fowkes
Fowkes dividiu a energia livre de superfície (𝛾𝑆𝑉) de um sólido em: uma componente
dispersiva (𝛾𝑆𝑉𝑑 ) consistindo em forças de London como dispersão, orientação e indução e a
componente polar (𝛾𝑆𝑉𝑝
) atribuída a formação de ligações de hidrogênio (Awaja.et.al.,2009).
Dessa forma, a energia de superfície total de um sólido foi definida de acordo a Eq. 2:
𝛾𝑆𝑉 = 𝛾𝑆𝑉𝑑 + 𝛾𝑆𝑉
𝑝 (2)
A Equação de Fowkes considera apenas forças de dispersão atuando na interface
sólido/líquido, de forma que o cálculo para energia livre de superfície fornece aproximações para
sistemas simples (Awaja et. al., 2009). Assim, nos sistemas em que existem apenas forças de
dispersão atuantes, o modelo para energia interfacial sólido/líquido proposto por Fowkes em
1962 foi:
𝛾𝐿𝑆 = 𝛾𝑆𝑉 + 𝛾𝐿𝑉 − 2(𝛾𝑆𝑉𝑑 . 𝛾𝐿𝑉
𝑑 )1/2 (3)
Em que 𝛾𝑆𝑉𝑑 e 𝛾𝐿𝑉
𝑑 são as componentes de dispersão da energia livre superficial para o
sólido e líquido, respectivamente. Rearranjando a Eq. 3 e combinando-a com a Eq. 1, tem-se:
𝛾𝐿𝑉(1 + cos(𝜃)) = 2(𝛾𝑆𝑉𝑑 . 𝛾𝐿𝑉
𝑑 )1/2 (4)
Nesta equação, o cálculo da energia livre de superfície considera apenas interações
dispersivas do sistema não sendo confiável para estimativas de sistemas complexos. Porém, sua
aplicação a sistemas simples fornece aproximações úteis (Awaja et. al., 2009).
2.2.3 Média Geométrica
Devido a existência de outros modos de interação entre as fases sólidas e líquidas em suas
interfaces, a Eq. 4 foi reescrita apropriando-se a mesma para forças polares. A relação de Fowkes
foi estendida incluindo-se o termo referente às ligações de hidrogênio usando-se a média

11
geométrica para reunir as forças de dispersão com as forças polares, obtendo-se a Eq. 5, onde 𝛾𝑆𝑉𝑝
e 𝛾𝐿𝑉𝑝
são componentes polares relativos à energia livre superficial do sólido. (Awaja et. al.,
2009; Sellin, 2002; Sena, 2011).
𝛾𝐿𝑉(1 + cos θ) = 2(𝛾𝑆𝑉𝑑 . 𝛾𝐿𝑉
𝑑 )1
2 + 2(𝛾𝑆𝑉𝑝 . 𝛾𝐿𝑉
𝑝 )1
2 (5)
Para a obtenção das componentes polar e dispersiva do sólido (𝛾𝑆𝑉𝑑 , 𝛾𝑆𝑉
𝑝 ), é necessário
utilizar dois líquidos conhecidos (um polar e outro apolar). Através da Eq. 6, calcula-se o valor
da componente dispersiva ( 𝛾𝑆𝑉𝑑 ) através de 𝜃 e 𝛾𝐿𝑉 do líquido apolar.
𝛾𝑆𝑉𝑑 =
1
4[𝛾𝐿𝑉(1 + cos θ)2] (6)
O valor da componente polar (𝛾𝑆𝑉𝑝
) é estimado a partir do valor da componente dispersiva
(𝛾𝑆𝑉𝑑 ) calculado usando-se os valores de 𝜃, 𝛾𝐿𝑉 e das componentes do líquido polar (𝛾𝐿𝑉
𝑑 , 𝛾𝐿𝑉𝑝
),
pela Eq. 7.
𝛾𝑆𝑉𝑝 = [𝛾𝐿𝑉(1 + cos θ) − 4(𝛾𝑆𝑉
𝑑 . 𝛾𝐿𝑉𝑑 )] 4𝛾𝐿𝑉
𝑝⁄ (7)
A média geométrica é mais indicada para sistemas de alta energia como adesivos em
metais (Sellin, 2002), por isso não será usada neste trabalho para determinação da energia de
superfície das amostras de monofilamentos.
2.2.4 Média Harmônica
Wu aproximou a Equação de Fowkes ao utilizar o método da média harmônica para
cálculo da energia de superfície (Awaja et. al., 2009; Sena, 2011), introduzindo na Equação 3 a
componente polar, obtendo a Equação 8, através da média harmônica.
𝛾𝐿𝑆 = 𝛾𝑆𝑉 + 𝛾𝐿𝑉 − 4 [𝛾𝑆𝑉
𝑑 .𝛾𝐿𝑉𝑑
𝛾𝑆𝑉𝑑 + 𝛾𝐿𝑉
𝑑 +𝛾𝑆𝑉
𝑝.𝛾𝐿𝑉
𝑝
𝛾𝑆𝑉𝑝
+ 𝛾𝐿𝑉𝑝 ] (8)
Substituindo a Equação 1 na Equação 8, obtém-se a aproximação de Wu, como mostrado
na Equação 9.
𝛾𝐿𝑉(1 + cos 𝜃) = 4 [𝛾𝑆𝑉
𝑑 .𝛾𝐿𝑉𝑑
𝛾𝑆𝑉𝑑 + 𝛾𝐿𝑉
𝑑 +𝛾𝑆𝑉
𝑝.𝛾𝐿𝑉
𝑝
𝛾𝑆𝑉𝑝
+ 𝛾𝐿𝑉𝑝 ] (9)

12
Para obtenção das componentes dispersiva e polar do sólido é necessário o uso de dois
líquidos (polar e apolar). O valor da componente dispersiva é inicialmente determinado através
dos valores de 𝜃, 𝛾𝐿𝑉 do líquido apolar pela Equação 10.
𝛾𝑆𝑉𝑑 = [𝛾𝐿𝑉(1 + cos θ)] (3 − cos 𝜃)⁄ (10)
O valor da componente polar é calculado a partir do valor da componente dispersiva e dos
valores de 𝜃, 𝛾𝐿𝑉 e das componentes do líquido polar (𝛾𝐿𝑉𝑑 , 𝛾𝐿𝑉
𝑝), pela Equação 11.
𝛾𝑆𝑉𝑝 = [𝛾𝐿𝑉
𝑝 (𝐴 − 𝐵)] [4𝛾𝑆𝑉𝑝 − (𝐴 − 𝐵)]⁄ (11)
Em que:
𝐴 = 𝛾𝐿𝑉(1 + cos θ);
𝐵 = (4𝛾𝑆𝑉𝑑 . 𝛾
𝐿𝑉𝑑 𝛾
𝑆𝑉𝑑 + 𝛾
𝐿𝑉𝑑 )⁄ ;
Wu (1971) mostrou que a Eq. 9 é precisamente aplicável a ambos os sistemas polares e
não polares, sendo mais vantajosa em relação à Equação proposta por Fowkes e ao método da
média geométrica proposto por Owens e Wendt.
A média harmônica, de acordo Wu (1971) fornece resultados mais plausíveis para
interações entre sistemas de baixa energia como, por exemplo, líquidos orgânicos, água e
polímeros no estado fundido ou no estado sólido. No entanto, não se espera usar esse método
para fases que apresentam grandes diferenças de polaridade como água e mercúrio.
2.3 FIBRAS TÊXTEIS E SUAS APLICAÇÕES
Fibras são consideradas o alicerce para fabricação de materiais têxteis, havendo uma forte
correlação entre as propriedades da fibra e propriedades dos produtos têxteis resultantes (Kothari,
2008). Fibras têxteis podem ser aplicadas em produtos têxteis e em outras aplicações industriais
(Carvalho, 2011).
O termo fibra é genérico e aplicável a variados tipos de materiais, naturais ou artificais,
compondo os elementos básicos para fins têxteis (Araújo, 1986; Martinez, 1972). Segundo a
ASTM D123-03 (2006), as fibras são definidas como uma unidade de material que apresentam
comprimento pelo menos cem vezes maior que o seu diâmetro.

13
2.3.1 Classificação de fibras
De acordo Araújo (1986) e Martinez (1972) apud Giordano (2007), fibras têxteis podem
ser classificadas como:
Naturais: provenientes e encontradas na natureza sendo de origem vegetal ou
animal ou mineral (algodão, linho, lã, seda e amianto);
Químicas: produzidas a partir de processos industriais e subclassificadas como:
artificiais (raion, viscose e acetato) e sintéticas (poliamida, poliéster,
polipropileno, elastano).
Fibras vegetais são constituídas basicamente por celulose encontradas na natureza
combinada a outras substâncias como a lignina, contendo impurezas como gomas, gorduras, ceras
e pigmentos. Dentre suas propriedades, as fibras vegatais apresentam: alta absorção de umidade,
moderada resistência à tensão e abrasão, baixa resistência a ácidos fortes e boa resistência a
álcalis (Araújo, 1986; Martinez, 1972).
Dentre as fibras animais, a lã pode ser obtida do pelo de ovelhas. Seu principal
componente é a queratina. Outras fibras animais podem ser aplicadas na indústria têxtil como os
de camelho, coelho, alpaca, etc. Fios de seda, por exemplo, são produzidas por uma lagarta
Bombyx mori. A seda é constituída essencialmente por fibroina e sericina (Giordano, 2007).
As fibras artificiais são elaboradas a partir da celulose e também chamadas de fibras
celulósicas. As fibras artificiais mais usadas são a viscose e o acetato. O acetato é produzido pela
reação entre anidro acético com celulose. Em seguida, é feita a dissolução em um solvente,
realizando-se extrusão contra um jato de ar quente para que o solvente evapore. A viscose é
obtida por regeneração da celulose (Araújo, 1986; Martinez, 1972).
Inúmeros polímeros são produzidos como plásticos industriais, no entanto somente alguns
podem ser convertidos em fibras sintéticas. Isso porque polímeros usados na fabricação de fibras
devem ser lineares, simples ou com grupos laterais polares pequenos, possam ser fundíveis,
dissolvidos em solventes adequados, sendo orientáveis (Gupta, 2013).

14
2.3.2 Propriedades de fibras (Gupta, 2013)
A estrutura física das fibras determina suas propriedades físicas e mecânicas em muitos
casos. Nas regiões cristalinas, as cadeias poliméricas encontram-se firmemente ligadas, dispostas
regularmente, como encontrado num cristal composto de átomos inorgânicos. Nas regiões
amorfas as cadeias encontram-se irregularmente dispostas com distância relativamente maior que
nas estruturas cristalinas, com ligações mais fracas entre as moléculas.
O tamanho, forma e orientação dos cristais e a divisão entre as frações das suas fases
depende das propriedades do polímero e as condições de fabricação. O grau de cristalinidade para
fibras mais úteis situa-se no intervalo de 30-70%, proporcionando equilíbrio de propriedades. As
regiões cristalinas contribuem para estabilidade térmica e mecânica das fibras, enquanto as
regiões amorfas contribuem para boas propriedades de volume, promovendo flexibilidade,
estiramento e recuperação elástica das fibras.
A escolha de um tipo de fibra para determinada aplicação baseia-se na capacidade da
mesma em ser processada e convertida para o dispositivo final desejado e em seu potencial para
realização da função do produto acabado. Dentre as características físicas principais consideradas
importantes para esta finalidade encontram-se: tamanho e forma das fibras; propriedades de
tração; transições térmicas; energia livre de superfície e capacidade de absorção de umidade.
Fibras são caracterizadas em termos de tamanho através de sua densidade linear expressa
em termos de massa por unidade de comprimento. O tamanho de uma fibra afeta sua força,
rigidez, resistência à abrasão, degradabilidade, capilaridade, entre outras propriedades. Se por um
lado, um aumento na densidade linear promove aumento da força e resistência à abrasão, por
outro lado, este aumento provoca diminuição da flexibilidade da fibra.
Uma variedade de fibras com propriedades mecânicas distintas podem ser fabricadas
variando-se o peso molecular e estrutura macromolecuar durante sua produção. As propriedades
de tração incluem os valores de tensão de ruptura (tenacidade), módulo de elasticidade, entre
outras.
A capacidade de uma determinada fibra em resistir a ruptura está diretamente ligada à sua
densidade linear. Propriedades como módulo de elasticidade, e flexibilidade de fibras diminuem à

15
medidade que a temperatura aumenta, em fibras hidrófilas, a capacidade de absorção de umidade
aumenta.
As fibras que dispõem de grupos funcionais em seu interior, e não somente em sua
superfície tais como: celulose, nylon e polipetídeos, absorvem água para sua estrutura interna.
Nesse caso, tais materiais possuem espaços livres ou um grau de cristalinidade baixo no qual as
moléculas de água penetram, deslocando cadeias, formando ligações com grupos funcionais
existentes no meio.
2.3.3 Monofilamentos
Dentre as fibras sintéticas produzidas, os monofilamentos são amplamente usados em
tecidos ou como fios de engenharia devido à sua alta tenacidade (Kamath e Bhat, 2008).
Polímeros típicos são usados na produção de monofilamentos: poliamida, poliéster,
polipropileno, polietileno e polifluoreto de vinilideno (Giles, Wagner, Mount, 2005).
Monofilamentos tem seu uso difundido em diversas aplicações que vão desde a linha de
produtos técnicos e de engenharia à vida cotidiana e biomateriais implantáveis. Desse modo,
monofilamentos podem ser aplicados como fibras ópticas e compósitos com borracha (Spiridinov
Lambrinos e Peng, 2005).
Na área esportiva, eles são usados para linha de pesca e como cordas de raquetes de tênis,
entre outras. Monofilamentos são usados em processos de tecelagem na produção de estufas,
cercas. Também são combinados para produzir fitas decorativas, embalagens e cerdas de escovas
de dentes (Giles, Wagner, Mount, 2005). Na área biomédica, monofilamentos são amplamente
usados como fios de sutura (Gupta, 2013).
2.3.4 Polipropileno - PP
Poliolefinas, como o polipropileno (PP), são empregadas industrialmente por serem
versáteis em suas aplicações, apresentando boa processabilidade, excelente resistência química,
alta flexibilidade, baixa densidade e baixo custo, quando comparadas a outros materiais. Porém,

16
apresentam baixa energia de superfície e fraca adesão, sendo necessária a modificação de suas
propriedades de superfície tornando-as adequadas para impressão e adesão (Sena, 2011).
O polipropileno apresenta superfície hidrofóbica, devido a grupos metílicos presentes em
suas cadeias puramente formadas por hidrocarbonetos, restringindo suas aplicações em que se é
necessário polaridade da superfície (Paul, 2009).
O polipropileno é um termoplástico obtido através da polimerização do monômero
propileno. O polímero formado apresenta-se como homopolímero e copolímeros, sendo o
homopolímero mais rígido e estável termicamente. Pode ser obtido nas formas isotático,
sindiotático e atático, sendo sua forma isotática linear e altamente cristalina, de grande interesse
comercial (Sellin, 2002). Homopolímeros de polipropileno são semi-cristalinos, sendo o grau de
cristalinidade dependente da taticidade da cadeias (Paul, 2009).
Figura 3: Unidade de repetição do polipropileno
Fibras de polipropileno podem ser usadas em carpetes, interiores de automóveis, não-
tecidos, entre outros materiais devido à sua versatilidade, resistência a ataques químicos e leveza,
com boas propriedades de isolantes sendo insensíveis a umidade e sujeira (Maier e Calafut,
1998). Monofilamentos de polipropileno são aplicados em produtos de tecelagem que exigem boa
resistência mecânica à tração, tais como cordas. Também apresentam baixa absorção de umidade
e baixa densidade (Maier e Clafut, 1998).
Como material biomédico, monofilamentos de polipropileno usados em suturas são
produzidos a partir de polipropileno isotático. Após fiação, os monofilamentos são submetidos a
uma série de etapas incluindo recozimento para aumento da cristalinidade. Suturas de PP são
esterilizadas com óxido de etileno, alto-clave ou radiação. Apresentam uma das mais baixas
respostas do tecido, não sendo suscetíveis à hidrólise após implante (He e Benson, 2011) e

17
dificuldade em se manterem amarradas, devido à suavidade e capacidade de lubrificação do fio
(Taylor e Shalaby, 2013).
2.3.5 Poli(tereftalato de etileno) - PET
Considerado como um dos mais importantes polímeros em engenharia devido à sua
grande aplicabilidade, o poli(tereftalato de etileno) (PET) apresenta excelentes propriedades
mecânicas de resistência a tração e impacto, boa resistência química a ácidos, bases e alguns
solventes, óleos e gorduras, claridade, processabilidade e estabilidade térmica moderada (Awaja e
Pavel, 2005).
A cadeia de PET é rígida acima da temperatura de transição vítrea. Sua baixa flexibilidade
decorre da presença dos grupos etileno e p-fenileno, afetando propriedades relacionadas a
estrutura e transições térmicas, apresentando temperatura de transição vítrea numa faixa de 67°C
a 140°C e temperatura de fusão em torno de 255°C a 265°C (Awaja e Pavel, 2005).
Figura 4: Unidade de repetição do PET
Polímeros como o PET com alto peso molecular são usados na produção de filamentos
industriais, podendo ter alta massa molar para o uso em borrachas e correrias transportadoras,
cintos de segurança, mangueiras e cordas e tecidos para revestimento, ou podem ser mais finas
para aplicações em linhas de costura e produção de tecidos leves. (Santhana e Kulthari, 2008).
Na indústria têxtil, fibras de PET são amplamente usadas na produção de tecidos tingidos,
o que requer boas propriedades de adesão. Devido à sua pouca hidrofilidade e baixa absorção de
água, fibras de PET são tingidas por corantes em altas temperaturas e pressões, aumentando-se a
demanda energética do processo para garantir qualidade do produto final (Xu e Liu, 2003).

18
Entre suturas não absorvíveis à base de poliéster as mais comumente usadas são PET e
PBT. Suturas de PET são comercialmente disponíveis como monofilamentos revestidos ou não,
ou como multifilamentos trançados. O PET é biologicamente estável não havendo evidências de
degradação, apresenta força de resistência por longo período de tempo. A resposta do tecido a
suturas de PET é dependente da configuração: alta reatividade a multifilamentos e pouca
reatividade a monofilamentos (He e Benson, 2011).
2.3.6 Poliamida 6 – PA-6
Dentre os polímeros sintéticos, poliamidas são materiais de engenharia importantes,
comumente usados em fitas, cabos isolantes, embalagens alimentícias, materiais cirúrgicos,
roupas e produtos ligados a moda e inovação, e outras diversas aplicações. A palavra nylon é uma
forma genérica de representar a classe dos polímeros de poliamida (Deopura, 2008).
As poliamidas são termoplásticos processáveis, isto é, podem ser fundidos e moldados
novamente a uma nova forma. Sua estrutura química é caracterizada por inúmeras repetições de
ligações amida, com cadeias carbônicas lineares. Sua síntese ocorre a partir de uma reação de
policondensação entre moléculas com grupos amina (NH2) e moléculas com grupos ácidos
(COOH) (Kothari, 2008).
A poliamida resultante de um processo de policondensação é nomeada com base no
número de átomos de carbono que separam os grupos ácidos dos grupos amina. As poliamidas
formadas terão diferentes características químicas que resultam em diferentes propriedades e
aplicações (Khotari, 2008).
Uma variedade de poliamidas são atualmente fabricadas e comercializadas com diversos
nomes comerciais diferentes. Entre eles o nylon 6 e o nylon 66 que são as duas poliamidas mais
fabricadas. Outros tipos de poliamidas como nylon 46, nylon 10 e nylon 12, são usados para
aplicações distintas (Deopura, 2008).
O nylon-6 é um polímero semicristalino com excelentes propriedades físicas e mecânicas
tem sua formação pela polimerização de abertura de anel da caprolactana. Apesar de formar
suturas não-absorvíveis, as ligações entre amidas são suscetíveis a hidrólise, o que diminui sua
resistência mecânica com o tempo (Taylor e Shalaby, 2013).

19
Figura 5: Unidade repetitiva do nylon-6
Essas características únicas se devem a estruturas químicas das cadeias de poliamidas,
devido à mobilidade e flexibilidade de grupos –CH2– e ligações de hidrogênio entre as cadeias
poliméricas e também ao peso molecular do polímero formado. Poliamidas são semicristalinas
por natureza e sua estrutura cristalina é afetada de maneira significativa por interações entre
átomos de hidrogênio e grupos –CO– e –NH– presentes nas cadeias poliméricas (Deopura, 2008).
Além disso, a morfologia do polímero e seu grau de cristalinidade também influenciam e
modificam essas propriedades. A estrutura química e interações moleculares também influenciam
em propriedades térmicas como temperatura de transição vítrea, temperatura de fusão e
cristalização das poliamidas, modificando assim as propriedades finais do produto acabado
(Deopura, 2008).
A resistência à ruptura de filamentos de poliamidas pode ser avaliada com base na
morfologia das fibras. Uma fibra de poliamida é composta por regiões cristalinas e amorfas. Os
domínios amorfos são elementos fracos e caso algum esforço seja feito pelo material, tais
domínios iniciariam a formação de trincas (Deopura, 2008).
Poliamidas sob a forma de fibras tem excelente comportamento de recuperação elástica
com a remoção de estresse. Durante a deformação de nylons, grupos –CH2– em regiões amorfas
são tipicamente móveis. No entanto, as restrições a essa mobilidade são impostas por ligações de
hidrogênio presentes no meio. A quebra de ligações de hidrogênio resulta no comportamento da
fluência das cadeias poliméricas através de uma carga imposta. Na remoção desta carga, o
processo de recuperação elástica volta a ocorrer com a formação das ligações de hidrogênio
quebradas (Deopura, 2008).
Fibras de nylon são usadas tanto para produção de roupas quanto em setores industriais.
Roupas leves para mulheres e roupas íntimas são produzidas a partir de nylon 6 e nylon 66 que
apresentam propriedades de particular importância para esta aplicação com alta elongação e

20
excelente elasticidade. Este polímero está presente também em cintos de segurança nos carros,
mangueiras, lonas e mochilas para bagagens por causa de sua alta resistência à cargas pesadas e
impactos (Deopura, 2008).
Diferentes configurações de suturas de poliamida não absorvíveis são utilizadas, por
exemplo, como monofilamentos e multifilamentos trançados. Suturas multifilamentares
geralmente são revestidas com silicone para redução do arraste de tecido. A reação dos tecidos a
suturas de nylon mostram-se independentes da configuração, provocando pouca reação
inflamatória. Suturas de poliamida são usadas por serem flexíveis e manterem a resistência
necessária a puxamentos e aos nós feitos durante um implante (He e Benson, 2011).
2.4 MÉTODOS DE MODIFICAÇÃO DE SUPERFÍCIE
Muitos polímeros mostram inércia química de superfície. Geralmente, estes materiais
apresentam baixa energia livre superficial, com fraca capacidade de adesão. A modificação da
superfície de polímeros é fundamental para as indústrias que requerem boas propriedades
adesivas. Por esses motivos, inúmeros materiais poliméricos são tratados superficialmente com
técnicas adequadas (Sellin, 2002).
O grande desafio enfrentado na engenharia de superfície é a introdução de grupos
funcionais na superfície dos biomateriais, aumentando o valor da energia de superfície com
promoção da adesão celular ao material implantado. Nesse sentido, ao invés de se desenvolver
novos biomateriais realiza-se técnicas de modificação superficial ou de revestimento superficial
em materiais já existentes. (Klee et. al., 2003).
Geralmente, introduz-se grupos polares como aminas e carbonilas em superfícies de
biomateriais para a formação de sítios de acoplamento que promovem ligações covalentes com
proteínas (Ma; Mao; Gao, 2007).
Diante de diferentes técnicas de modificação de superfície (mecânica, química, térmica,
ozônio, ultravioleta, feixe de íon, plasma, etc.) (Sena, 2011), os tratamentos por plasma e corona
promovem a oxidação da superfície de polímeros, introduzindo grupos polares como hidroxilas,
carboxilas e aminas. Outro método utilizado é enxertar polímeros hidrófilos na superfície dos

21
biomateriais via copolimerização (Ma; Mao; Gao, 2007), como ilustra a Fig. 6. Os tratamentos:
químico, via plasma e por descarga corona serão apresentados com mais detalhes a seguir.
Figura 6: Modificação da superfície de polímeros (Fonte: Adaptado de Ma, Mao, Gao, (2007)).
2.4.1 Tratamento Químico
Industrialmente, o tratamento químico tem sido utilizado com o objetivo de tratar objetos
com grandes dimensões. Assim, superfícies poliméricas hidrofóbicas e lisas são tratadas com
reagentes químicos convertendo-as em superfícies hidrofílicas e rugosas. A seleção do reagente
está sujeita ao tipo de polímero a ser tratado. O método propicia a obtenção de uma superfície
com ancoramentos físicos e químicos pontuais receptíveis aos revestimentos (Carneiro, 2001).
Durante o tratamento, polímeros são imersos em reagentes químicos num determinado
período a uma dada temperatura. Dessa forma, ocorre a oxidação da superfície e o aumento da
rugosidade, proporcionando aumento das propriedades de superfície como molhabilidade e
adesão (Sellin, 2002). A técnica visa a criação de novos grupos funcionais na interface de dois
materiais submetidos a adesão (Awaja et. al., 2009).

22
Solventes como xileno, tolueno, peróxido de hidrogênio, misturas de ácidos nítricos e
sulfúrico, bem como soluções de permanganato e ácido nítrico são amplamente aplicados no pré-
tratamento da modificação superficial de polímeros. Tais solventes induzem mudanças
moleculares com a introdução de grupos funcionais contendo oxigênio, melhorando as
propriedades de adesão (Awaja et. al., 2009; Sena, 2011; Xu et. al., 2010).
A modificação de polímeros, especialmente borrachas (vulcanizadas ou não) por meio de
modificação química com reagentes ácidos e oxidantes mostrou aumento na polaridade
superficial. O aumento da polaridade da superfície é devido ao aumento de forças moleculares,
com aumento da força de aderência (Awaja et. al., 2009).
Modificações da superfície de poli(p-sulfeto de fenileno) (PPS) tem ocorrido através da
inserção de grupos funcionais via oxidação química e tratamento por plasma. Amostras tratadas
foram caracterizadas por espectroscopia de FT-IR, XPS e medidas de ângulo de contato. Os
resultados mostram aumento da adesão do material tratado devido a duas condições: aumento da
polaridade por causa do tratamento superficial e aumento na rugosidade medido por microscopia
de força atômica (Awaja et. al., 2009).
Apesar de o tratamento químico ser uma alternativa para modificação superficial de
polímeros, tendo em vista a melhoria das propriedades superficiais e adesivas desses materiais,
muitas pesquisas têm apontado que esta técnica não é adequada por causar poluição ao meio
ambiente e altos custos associados ao tratamento quando comparado a outros métodos (Awaja et.
al., 2009).
2.4.2 Tratamento por Plasma
A modificação de superfícies poliméricas usando o tratamento por plasma tem-se tornado
cada vez mais popular devido às vantagens apresentadas por esse método. Tal técnica não utiliza
solventes tóxicos ao meio ambiente, sendo de fácil configuração e reprodutibilidade, com rápidos
resultados (Vandencasteele, 2005).
O plasma é definido como um estado diluído da matéria, em que partículas carregadas
negativamente e positivamente (átomos, moléculas, íons, espécies radicalares e excitadas)

23
encontram-se distribuídas em determinadas proporções, sendo semelhante a um gás ionizado. Tal
estado é conhecido como quarto estado da matéria (Sellin, 2002).
Plasmas iônicos são criados numa variedade de gases (He, Ar, Kr, Ne, ar, NH3, N2, CO2,
O2) aplicando-se uma corrente elétrica direta (entre eletrodos), rádio frequência ou microondas,
geralmente formados em câmaras de baixa pressão, da ordem de mTorr (miliTorr). Os parâmetros
para essa técnica são: geometria da amostra, câmara, pressão, frequência, potência e atmosfera do
gás utilizado (Lopes, 2012).
Durante o tratamento, espécies reativas provenientes do plasma chocam-se com a
superfície do polímero, promovendo a quebra de ligações químicas e o surgimento de radicais
livres, permitindo a formação na superfície de grupos funcionais polares contendo oxigênio,
aumentando assim a energia livre de superfície do substrato (Salmoria; Martins; Fucio, 2012).
Excepcionalmente, o plasma de nitrogênio promove a formação de aminas e amidas juntamente
com hidroxilas, aumentando a energia livre de superfície (Awaja et. al., 2009).
Esta forma de tratamento modifica a superfície do polímero sem alteração total das
propriedades de volume, uma vez que as modificações se estendem a uma pequena profundidade
abaixo da superfície. Além disso, o tratamento de superfícies por plasma de oxigênio pode
promover aumento da rugosidade de superfície e alteração na área de contato (Salmoria; Martins;
Fucio, 2012).
Salmoria, Martins e Fúcio (2012) investigaram a influência do processo de tratamento da
superfície de cateteres de poliamida 11 por plasma de oxigênio, avaliando a composição química,
rugosidade e molhabilidade das amostras tratadas. Observou-se aumento da hidrofilicidade e
rugosidade do material indicadas através de medidas de ângulo de contato (gota séssil) e MEV,
respectivamente. Análises de ângulo de contato constataram que mudanças na molhabilidade
foram diretamente dependentes do tempo de exposição dos cateteres ao tratamento. A partir das
análises de MEV os autores perceberam mudanças significativas na superfície com o
aparecimento de rugosidades que se intensificaram devido ao tempo de tratamento, com aumento
dos desníveis de relevo.
Em seus experimentos Lopes (2012) analisou os efeitos das técnicas de descarga corona
(atmosfera ambiente) e plasma por radiofrequência (atmosferas de O2, N2 e Ar) aplicadas ao
material polimérico NTPP (não tecido de polipropileno). Os resultados revelaram aumento na

24
molhabilidade do material, com aumento da energia livre de superfície em função do tempo de
tratamento para as ambas as técnicas. Análises de espectrometria no infravermelho indicaram a
formação de grupos funcionais como hidroxilas (C-OH) e carboxilas (COOH) na superfície do
material. Análises de DSC reveleram que as amostras tratadas não apresentaram mudanças nas
propriedades de volume em relação as amostras sem tratamento.
2.4.3 Tratamento por Descarga Corona
As descargas coronas são produzidas em pontas finas, bordas ou fios finos onde um
campo elétrico é suficientemente largo (Antao et. al., 2009), em meio a atmosfera gasosa
(geralmente ar) a partir da aplicação de alta tensão elétrica entre dois eletrodos, à temperatura e
umidade relativa ambientes. O fenômeno ocorre com formação de espécies ativas (íons, elétrons
e moléculas excitadas) e outros componentes que interagem com o material provocando
mudanças em sua superfície (Lopes, 2012; Sellin, 2002).
A descarga corona tem sido uma técnica útil por aumentar a molhabilidade de polímeros,
tendo como consequência ativação da superfície polimérica, com aumento da energia livre de
superfície e promoção de caráter hidrofílico (Pascual; Balart; Sánchez, 2008).
Esta técnica é utilizada também para precipitação eletrostática, produção de ozônio,
controle de odor, remoção de compostos orgânicos voláteis, eliminação de contaminantes gasosos
e tratamento de água. No entanto, é responsável por transmitir energia como rádio ruídos e perdas
energéticas (Antao et. al., 2009).
Diferentes geometrias do sistema corona, como: sistema fio-plano e ponta-plano, podem
ser utilizadas sob atmosfera ambiente promovendo formas variadas de descarga corona auto-
sustentada. A corona pode ser negativa ou positiva dependendo da tensão (positiva ou negativa)
aplicada (Antao et. al., 2009).
Ao avaliarem as condições de operação de um sistema ponta-plano com fluxo de ar,
Antao et. al. (2009) e Akishev et. al.(2005) observaram a formação de descarga corona negativa
acompanhada por luz difusa e estável seguida de faíscas, como mostrado na Fig. 7. Na Fig. 7, A e
C mostram luz difusa e estável em fluxo de ar; em B, faísca transiente; O sistema apresenta
distância entre eletrodos de 15mm (A e B) e 3mm (C).

25
Sob voltagem crítica em fluxo de ar, faíscas decompõem-se rapidamente promovendo
rápidas transições entre a luz difusa (descarga luminescente). Em ar estático, as faíscas
filamentares são acompanhadas de descargas luminescentes que movem-se de forma desordenada
no espaço inter-eletrodos devido a convecção do ar aquecido através dos filamentos. Neste caso,
a rápida transição de descarga corona para faíscas é devido ao desenvolvimento de instabilidade
durante a ionização no espaço entre os eletrodos (Antao et. al., 2009).
Figura 7: Corona em ar (Fonte: Adaptado de Akshev et. al, (2005) e Antao et. al. (2009)).
As mudanças ocorridas nas propriedades adesivas dos materiais dependem de
determinados fatores estabelecidos durante o processo. Tem-se como exemplo a tensão aplicada,
o tempo de tratamento, a atmosfera utilizada, temperatura, umidade relativa e as características
do sistema gerador incluindo a distância entre os eletrodos (Carneiro, 2001; Lopes, 2012, Sellin,
2002; Sena, 2011).
Alguns estudos mostram que o efeito primário deste tratamento em polímeros é a
oxidação da superfície do material o que melhora suas propriedades como molhabilidade e
adesão, entretanto outro possível resultado para este tipo de tratamento é o aumento da
rugosidade superficial dos materiais que influencia em suas propriedades de superfície (Sellin,
2002).
A descarga corona, em ar atmosférico, é constituída de íons, elétrons e espécies excitadas
ou metaestáveis de oxigênio e nitrogênio. Essas espécies possuem energia suficiente (1-20eV)
para quebrar ligações carbono-carbono (2,54eV) e carbono-hidrogênio (3,79eV) da superfície de
polímeros como PP, gerando radicais livres (Carneiro, 2001;Sellin, 2002; Sena, 2011).

26
Os radicais podem reagir com os átomos de oxigênio e ozônio, formando grupos
funcionais na superfície do polímero. Outros grupos funcionais podem ser formados através de
reações ocorridas na superfície em presença de vapor d’água, ozônio, óxidos de nitrogênio e
derivados, mesmo em pequenas quantidades (Carneiro, 2001; Sena, 2011. Lopes, 2012; Sellin,
2002). A Fig. 8 mostra o esquema do mecanismo de oxidação da superfície de polipropileno,
envolvendo apenas oxigênio como espécie reativa.
Figura 8: Oxidação da superfície de polipropileno (Fonte: Adaptado de Sellin (2002)).
O aumento da molhabilidade produzida na superfície de poliolefinas, como filmes de PP,
após tratamento corona são atribuídos a introdução de grupos polares originando carbonilas
(C=O), ácidos carboxílicos (COOH), éster (COC=O), hidroxilas (C-OH) e peróxidos (C-O-O)
(Carneiro, 2001; Sena, 2011; Sellin, 2002; Lopes, 2012).
Ao estudar os efeitos promovidos em não-tecido de polipropileno utilizando um sistema
com geometria fio-plano, Lopes (2012) observou que o tratamento promove mudanças físico-
químicas significativas na superfície das amostras, com a introdução de grupos polares livre
superficiais. O controle do processo foi feito aplicando-se uma tensão fixa, variando-se o tempo
de tratamento. Dentre as mudanças observadas destacam-se a melhoria da molhabilidade e
aumento da energia livre de superfície do material provocadas pela introdução de grupos
funcionais oxigenados na superfície do não tecido.

27
A cinética de introdução de grupos polares na superfície de filmes de PP tratados por
descarga corona pode ser verificada através da quantificação do índice de carbonila (C=O),
calculado a partir de análises dos espectros gerados por FT-IR/ATR (Sellin, 2002).
Sellin (2002) observou que a concentração de carbonila na superfície do filme de PP
aumenta com o aumento do tempo de exposição à corona. A presença desse grupo (C=O)
aumenta a propriedade hidrofílica, via ângulo de contato, e adesiva, através do Pell Test, e seu
índice pode ser associado ao controle da taxa de inserção desses grupos polares na superfície.
Amostras de filmes de polipropileno tratadas durante 120s, num sistema em que a distância inter-
eletrodos era de 3mm apresentaram índice de carbonila igual a 1,3. Amostras sem tratamento
apresentam índice de carbonila de aproximadamente 0,2, confirmando a introdução de carbonila
na superfície de PP, devido ao tratamento corona.
Ao tratar tecidos de poliéster com corona sob diferentes tensões aplicadas (6kV a 12kV),
Xu e Liu (2003) constataram que amostras tratadas aumentam a capacidade em absorver água,
preservando suas propriedades hidrófilas por longo período de tempo. Os autores relataram que
boas propriedades hidrófilas foram observadas aumentando-se a tensão aplicada, verificadas
através de testes de absorção, aumento da afinidade da superfície do tecido tratado por cola de
amido e corantes usados durante o tingimento.
A descarga corona em meio atmosférico é atóxica, de fácil operação de baixo custo,
simplicidade, apresentando rapidez, baixa produção de resíduos sendo amplamente utilizada para
a modificação da superfície de polímeros, melhorando suas propriedades de superfície e adesão.
Mudanças nas propriedades de superfície e de volume dos materiais tratados por descarga corona
podem ser verificadas através de técnicas de caracterização adequadas. Algumas técnicas
utilizadas neste trabalho serão descritas no item 2.5.
2.5 TÉCNICAS DE CARACTERIZAÇÃO
2.5.1 Medidas de Ângulo de Contato Dinâmico
Mudanças na hidrofilidade dos materiais podem ser verificadas através de medidas de
ângulo de contato (método de Wilhelmy), a condições ambientes. A base dessa técnica é o

28
equilíbrio de forças estabelecido no ponto de contato da interface sólido/líquido/vapor. As
medidas de ângulo de contato podem ser realizadas pelo método estático ou dinâmico.
No método estático não há movimento da interface sólido/líquido durante a medida. No
método dinâmico os valores de ângulos de contato dinâmicos são medidos em duas direções,
através do avanço e retrocesso do líquido sobre a superfície da amostra, sendo chamados de
ângulo de avanço (𝜃𝑎) e ângulo de retrocesso (𝜃𝑟), (Alves, 1999).
No método de Wilhelmy, a amostra é ligada a um feixe de uma eletrobalança e mantida na
vertical, numa posição fixa durante o teste, enquanto um recipiente contendo o líquido de teste é
levantado ou abaixado através de uma plataforma de condução (Tran et. al., 2011).
Quando o líquido toca a amostra, uma força é detectada pela eletro-balança. Mantendo-se
o contato entre líquido e amostra, forças de avanços e retrocessos são obtidas a velocidade
constante (Huang et. al., 2006; Saihi et. al., 2002; Tran et. al., 2011).
Diferentes medidas de velocidade de avanço e retrocesso podem ser utilizadas como, por
exemplo, 2µm/s a 8µm/s (Tran et. al., 2011), 20,2µm/s (Saihi et. al., 2002) e 100µm/s (Carneiro,
2001). A Fig. 9 apresenta o sistema descrito e usado na determinação de ângulo de contato.
Figura 9: Representação de sistema utilizado no Método de WilHelmy (Fonte: Adaptado de
Sheridan et. al., (1994))
A força total de equilíbrio estabelecido na balança é a soma da força de molhamento, o
peso da amostra e a força de flutuação (empuxo), dada pela Equação 12:
𝐹𝑡𝑜𝑡𝑎𝑙 = 𝑃𝛾𝐿𝑉 cos 𝜃 + 𝑚𝑔 + 𝜌𝐿𝑔𝐴𝑑 (12)

29
Em que 𝐹𝑡𝑜𝑡𝑎𝑙 é a força total, 𝑃 é o perímetro da amostra, 𝛾𝐿𝑉 é a energia livre de
superfície do líquido de teste, 𝜃 é o ângulo de contato formado na interface sólido-líquido, 𝑚 é a
massa da amostra, 𝑔 é a acelaração da gravidade, 𝜌𝐿 é a densidade do líquido de teste, 𝐴 é a área
da superfície da amostra e 𝑑 é a distância de imersão da placa.
Antes de realizar qualquer medição, a balança deve ser ajustada para zero, medindo-se o
peso da amostra antes, ajustando-o a zero no equilíbrio. A extrapolação para profundidade zero
elimina o efeito da força de flutuação (Huang et. al., 2006; Tran et. al., 2011). Dessa forma, os
ângulos de avanço (𝜃𝑎) e retrocesso (𝜃𝑟) serão calculados através de perfis de força versus
distância de imersão da amostra.
A partir desses perfis é realizada uma regressão linear para cada um dos ciclos de avanço
e retrocesso, dando origem aos coeficientes angular (𝑏) e linear (𝑎) (Alves, 1999), sendo a força
medida uma função da distância de imersão do material no líquido:
𝐹𝑡𝑜𝑡𝑎𝑙 = 𝑏. 𝑑 + 𝑎 (13)
Sendo os coeficientes 𝑎 e 𝑏, definidos como:
𝑎 = 𝑃𝛾𝐿𝑉 cos 𝜃 (14)
𝑏 = 𝜌𝐿𝑔𝐴 (15)
A diferença entre os ângulos de contato, Eq. 16, de avanço e retrocesso é ocasionada pela
diferença de forças medidas durante imersão e emersão da amostra (Alves, 1999) e denominada,
neste trabalho, como histerese. O termo histerese, neste caso, é amplamente utilizado na
literatura, sendo designado como a diferença entre o ângulo de avanço e recúo, como em Alves
(1999), Huang et. al. (2006), Lam et. al. (2001), Saihi et. al. (2002) e Tran et. al. (2011). Apesar
de ser amplamente estudada, e suas causas não são tão bem esclarecidas. Geralmente, sua
existência é atribuída a características de superfície como heterogeneidade, rugosidade,
metaestabilidade, mobilidade molecular de superfície e adsorção de líquidos pelo material (Long
et. al., 2005; Lam et. al., 2001).
∆𝜃 = 𝜃𝑎 − 𝜃𝑟 (16)
De acordo Alves (1999), a diferença entre os ângulo de avanço e recuo tem apresenta
explicações: termodinâmicas e cinéticas. Deformações na superfície podem resultar em

30
diferenças de ângulo de avanço e recúo termodinâmica e cinética. Rugosidade e heterogeneidade,
associada a grandes variações de energia de superfície, com algumas regiões hidrofílicas e outras
hidrofóbicas, promovem diferenças do tipo termodinâmica. Interações da superfície do sólido
com o líquido, como hidratação do sólido, extração do material na fase sólida para a fase líquida
e penetração do líquido na superfície sólida, causam diferenças do tipo cinética. Reorientação e
movimento das moléculas de superfície em resposta a mudanças no ambiente provocam
diferenças atribuídas como cinética (Alves, 1999).
2.5.2 Espectroscopia de Infravermelho com Refletância Total Atenuada
A energia denominada infravermelha corresponde à região do espectro eletromagnético
situada na faixa de número de ondas entre 14290 e 200 cm-1
. A região que apresenta número de
ondas entre 4000 a 400 cm-1
é denominada de infravermelho médio, enquanto que a região com
números de onda entre 14290 a 4000 cm-1
é chamada de infravermelho próximo (Barbosa, 2008).
Espectrocopia na região do infravermelho é uma técnica de caracterização que pode ser
aplicada a materiais poliméricos, servindo como ferramenta na identificação e determinação de
grupos funcionais, estudos da conformação de macromoléculas e obtenção do espectro
vibracional molecular completo do material analisado (Colthup; Daly; Wiberley 1964; Brame e
Grasseli, 1977 apud Lopes, 2013).
A espectrometria do infravermelho com Transformada de Fourier, como ilustra a Fig. 10,
utiliza um interferômetro de Michelson. Durante a técnica, um feixe de radiação infravermelha,
proveniente de uma fonte, é produzido e enviado a um divisor de feixe que o divide em dois raios
de intensidades aproximadamente iguais. Um dos feixes é enviado a um espelho fixo enquanto o
outro é enviado a um espelho móvel (Lopes, 2012; Canevarolo, 2003).
Após reflexões nos espelhos fixo e móvel, os feixes são recombinados no divisor de feixe.
A radiação combinada é enviada ao compartimento da amostra e referência, através de um
conjunto de espelhos. Logo após atravessar o compartimento, a radiação é transmitida a um
detector. Os dados dão origem a um interferograma que corresponde às variações no sinal medido
pelo detector, em função do tempo (Silverstein; Webster; Kiemle, 2007; Holler; Skoog; Crouch,
2009 apud Lopes, 2012).

31
Figura 10: Representação da espectroscopia de infravermelho (Fonte: Adaptado de Alves, 2014).
Os espectros de feixe simples são obtidos por cálculos da Transformada de Fourier a
partir de um interferograma. O espectro em transmitância é obtido pela razão entre o espectro de
feixe simples pelo espectro de feixe obtido sem amostra (Lopes, 2012). Dessa forma, a
transmitância é determinada conforme Eq. 16:
T =𝐼
𝐼0 (16)
Em que:
T : transmitância
𝐼: intensidade do feixe com amostra;
𝐼0: intensidade do feixe sem amostra;
Caso a resposta da intensidade do feixe for a mesma com e sem amostra a transmitância
terá valor igual a um. Se a amostra absorver luz, o valor da transmitância será menor que um,
pois menor quantidade de energia chegará ao detector. O valor da transmitância será igual a zero
quando a amostra absorver totalmente a luz. Os valores de transmitância podem ser multiplicados
por 100, obtendo-se assim valores de transmitância percentual (Cuca, 2010).

32
O espectro é um gráfico em que, na ordenada, é geralmente apresentada a porcentagem de
transmitância. Na abscissa, a posição no ponto de mínima transmitância (máxima absorbância) da
banda é apresentada em número de onda (em unidade cm-1
). O espectro no infravermelho
apresenta grande número de bandas, porém sua interpretação não requer a atribuição de todas elas
(Barbosa, 2008).
A radiação infravermelha causa alteração nos modos rotacionais e vibracionais das
moléculas. O espectro na região de 4000 a 400cm-1
, que é a região mais importante do ponto de
vista da caracterização de grupos funcionais orgânicos, geralmente apresenta bandas de absorção
em vez de linhas, já que para cada mudança de nível vibracional está associada uma série de
transições rotacionais (Barbosa, 2008).
Na técnica de FTIR/ATR, a amostra com menor índice de refração é colocada em contato
com um material com alto índice de refração, tal como um cristal, como ilustra a Fig. 11. O feixe
de infravermelho é incidido na superfície do cristal, atravessando a amostra até a profundidade de
alguns micrômetros, sofrendo múltiplas reflexões no interior da interface cristal/amostra (Sellin,
2002; Carneiro, 2001).
Figura 11: Reflexão interna na interface amostra/cristal (Fonte: Adaptado de Alkor Crystal
Optics, 2014)
O uso da técnica ATR baseia-se no fato de que, embora ocorra reflexão interna total na
interface da amostra/cristal, a radiação na verdade penetra determinada distância no meio com
menor índice de refração (menos denso). Essa radiação denominada onda evanescente pode ser

33
parcialmente absorvida, pondo-se a amostra em contato com o meio mais denso. Esse meio mais
denso é denominado prisma ou elemento de reflexão interna (Barbosa, 2008).
Segundo Barbosa (2008), a profundidade de penetração da radiação é da ordem do
comprimento de onda (µm) e pode ser calculada pela Eq.17:
𝐷𝑝 = 𝜆
2𝜋𝑛1√(𝑠𝑒𝑛2𝜙− 𝑛212
(17)
Em que:
𝐷𝑝: profundidade de penetração (µm);
𝜆: comprimento de onda (µm);
𝑛1: índice de refração do elemento de reflexão interna;
𝑛212 : razão entre o índice de refração da amostra e do elemento de reflexão interna;
𝜙: ângulo de incidência da radiação.
Os espectros de absorção podem ser obtidos para diferentes materiais como gases,
líquidos, soluções de sólidos e líquidos viscosos, emulsão, pastilhas e filmes finos. No caso de
amostras líquidas, a técnica deve ser empregada utilizando células especiais (Barbosa, 2008;
Carneiro, 2001).
As vantagens do uso de ATR incluem a possibilidade de estudar amostras que absorvem
fortemente na região do infravermelho, como amostras dissolvidas em água, bem como realizar
estudos de superfícies de amostras como polímeros, papéis, fibras, entre outros (Barbosa, 2008).
A Tab. 2 apresenta os principais grupos funcionais, formados após tratamento de
superfície de poliolefinas, observados por vários autores através de análise de FT-IR/ATR
(Sellin, 2002; Barbosa, 2010; Sena, 2011; Lopes, 2012).

34
Tabela 2: Grupos funcionais e faixa de absorção no infravermelho
Grupo Funcional Faixa de absorção (cm-1
)
O – H 3600 – 3200
C – O 1300 – 1000
C = O 1700 – 1720
C – O – C 1300 - 1050
C – H 3300 - 2800
2.5.3 Microscopia Eletrônica Exploratória
De acordo Lopes (2012), a microscopia eletrônica exploratória (siga em inglês SEM)
utiliza um microscópio que permite a observação e caracterização de materiais através de um
feixe de elétrons, produzindo imagens com alta ampliação (30 a 300.000x) e resolução (cerca de
10nm). Seu princípio de funcionamento baseia-se na leitura da superfície de um material por um
feixe de elétrons.
O microscópio eletrônico de varredura tem grande capacidade em gerar imagens com
resolução espacial de poucos nanômetros com profundidade em alguns casos superior a de um
microscópio óptico (Sena, 2011). É um dos instrumentos mais versáteis à observação de
características microestruturais de materiais, pois, possibilita o uso de amostras de grandes
dimensões (volume na ordem de cm³) e permite a observação de amostras com superfícies
irregulares e topografia complexa (Magalhães, 2009).
As amostras requerem certas condições para que possam ser analisadas por MEV,
possuindo boa condutividade elétrica de superfície, necessitando a aplicação de um revestimento
condutor como: ouro (Au), ouro-paládio (Au-Pd), platina (Pt), alumínio (Al) ou carbono. Durante
a interação com o feixe de elétrons, propriedades físico-químicas da amostra não podem sofrer
alteração (Canevarolo, 2003; Magalhães, 2009; Lopes, 2012).
A Fig. 12 apresenta o interior de um microscópio para análise de MEV. Nesta técnica, um
canhão de elétrons produz um feixe, com elétrons acelarados a energia cinética na faixa de
centenas de eV a dezenas de keV, com correntes elétricas na ordem de 10-6
a 10-12
A, causando
aquecimento. Os elétrons formados são atraídos fortemente para um anodo (Canevarolo, 2003;
Lopes, 2012).

35
A correção do percurso do feixe de elétrons é realizada por lentes magnéticas
condensadoras, que alinham o feixe em direção à abertura da objetiva. Assim, a objetiva ajusta o
foco do feixe eletrônico antes de atingirem a amostra (Canevarolo, 2003; Magalhães, 2009,
Lopes, 2012).
Figura 12: Interior de um microscópio eletrônico de varredura (Fonte: Lopes, 2012).
A amostra é fixada em uma placa de platina que suporta e possibilita o seu deslocamento.
O feixe de elétrons atinge a amostra e deve ter corrente suficiente para formar uma imagem
definida. Dois conjuntos de bobinas de varredura permitem a deflecção do feixe de elétrons de
forma a explorar a região de interesse. Condições de vácuo são feitas por um conjunto de bombas
(Magalhães, 2009).
2.5.4 Calorimetria Exploratória Diferencial
Mudanças físicas ou químicas sofridas pelos materiais são acompanhadas por uma
variação correspondente em sua entalpia. A técnica conhecida como calorimetria exploratória
diferencial (DSC) compreende esse processo através de medidas da variação de entalpia no

36
material em estudo, usando-se uma amostra inerte como referência (Lucas; Soares; Monteiro,
2001).
Nesta técnica, a análise pode ser realizada por um programa de aquecimento ou
resfriamento, com taxas de aquecimento programáveis (5 a 20°C/min). O sistema pode também
ser mantido a uma temperatura constante, operando no modo isotérmico, a qualquer temperatura
dentro da faixa de operação do equipamento, durante um tempo determinado (Lucas; Soares;
Monteiro, 2001).
A partir das análises de DSC, pode-se determinar temperaturas de transição em polímeros:
temperatura de transição vítrea (Tg), temperatura de fusão (Tm), temperatura de cristalização
(Tc), bem como realizar medidas quantitativas do calor específico (Cp), entalpia de fusão (ΔHm),
de cristalização (ΔHc) e de reação (ΔHr), além de possibilitar outros estudos na determinação do
grau de cristalinidade, perda de água, vulcanização, degradação, entre outros (Lucas; Soares;
Monteiro, 2001; Canevarolo, 2003).
As propriedades térmicas dos polímeros PP, PET e PA-6 segundo a ASTM D3418-12 são
apresentados na Tab. 3.
Tabela 3: Propriedades térmicas do PP, PET e PA-6 (ASTM D3418-12)
Polímero Tg (°C) Tc (°C) Tm (°C) Hc (J/g) Hm (J/g)
PP - 111,7 160,6 95,6 89,6
PET 76,2 194,9 249,2 45,9 41,1
PA-6 - 183,4 220,3 63,8 62,0
Há dois tipos de equipamentos que promovem a DSC denominados, DCS de fluxo de
calor e DSC de compensação de potência. A faixa de temperatura utilizada pode variar de um
equipamento a outro, variando de um modo geral de -180°C a 700°C (Lopes, 2012; Wendlandt,
1986).
No DSC de fluxo de calor, como mostra a Fig. 13, amostra e referência são colocadas em
cápsulas idênticas, posicionadas sobre um disco termoelétrico e aquecidas por uma única fonte de
calor. O calor é transferido para as cápsulas por meio do disco, com fluxo de calor diferencial

37
entre ambas as cápsulas sendo controlado por meio de termopares conectados ao disco (Matos e
Machado, 2003; Canevarolo, 2003; Lopes, 2012).
Figura 13: Representação de um DSC de fluxo de calor (Fonte: Lopes, 2012).
A temperatura da amostra, monitorada por um termopar, é comparada com a temperatura
de referência. À medida que a temperatura do local onde estão as cápsulas é elevada, a uma taxa
de aquecimento constante, a temperatura da amostra (TA) e da referência (TR) irão se manter
igualadas até que ocorra alguma mudança física ou química na amostra (Höne; Hemminger;
Flammersheim, 1996; Lucas; Soares; Monteiro, 2001).
Se a variação for exotérmica, a amostra irá liberar calor e TA será maior que TR por um
determinado período de tempo. Caso a variação seja endotérmica, TA será temporariamente
menor que TR, sendo a diferença de temperatura, ΔT, (Eq.18) medida e registrada (Lucas; Soares;
Monteiro, 2001; Canevarolo, 2003). Segundo Höne, Hemminger e Flammersheim (1996), a
magnitude de ΔT, em um dado instante, é proporcional á variação de entalpia medida.
ΔT = TA – TR (18)
No DSC de compensação de potência, amostra e referência são dispostas em fornos
separados idênticos, com fontes de aquecimento individuais, em que temperatura e energia são
monitoradas e geradas por filamentos de platina idênticos, atuando como termômetros resistivos e
aquecedores, como ilustra a Fig. 14 (Höne; Hemminger; Flammersheim, 1996; Lucas; Soares;
Monteiro, 2001; Canevarolo, 2003).

38
Figura 14: Representação de um DSC de compensação de potência (Fonte: Lopes, 2012).
A técnica pressupõe que amostra e referência sejam mantidas em condições isotérmicas.
Desse modo, quando a amostra sofre alguma variação de temperatura através de um evento
exotérmico ou endotérmico, um sistema de controle modifica a energia fornecida para um dos
fornos (amostra ou referência), igualando a temperatura de ambos (Lucas; Soares; Monteiro,
2001; Canevarolo, 2003).
Os parâmetros instrumentais podem influenciar no valor da temperatura, na qual ocorre
alguma transição, obtida pela curva de análise térmica. Desse modo, a resolução da análise é
dependente de fatores instrumentais e de fatores associados à amostra e à referência, tais como a
taxa de aquecimento, a atmosfera do forno e tipo de gás fluente (Höne; Hemminger;
Flammershein, 1996; Lopes, 2012; Lucas; Soares; Monteiro, 2001).
Os registros das curvas de DSC, ilustrado na Fig. 15, são expressos em termos de fluxo de
calor versus temperatura ou tempo. De posse de tal curva, as transições de interesse podem ser
determinadas (Lucas; Soares; Monteiro, 2001).
A temperatura de transição vítrea (Tg) é observada por um desnível na curva de
aquecimento, a temperatura de cristalização (Tc) é registrada na transição endotérmica enquanto
que a temperatura de (Tm) na transição exotérmica. A área entre os picos e a linha base da curva
equivale a entalpia de cristalização (∆𝐻𝑐) e entalpia fusão (∆𝐻𝑚) da amostra analisada.

39
O grau de cristalinidade dos materiais pode ser calculado através da Eq. (19). Na Eq. (19),
Xc representa o grau de cristalinidade do polímero em %, ∆𝐻𝑚 representa a entalpia de fusão da
amostras e ∆𝐻𝑚𝑜 representa a entalpia de fusão de um polímero com grau de cristalinidade igual a
100%.
𝑋𝑐 =∆𝐻𝑚
∆𝐻𝑚𝑜 𝑥 100 (19)
Figura 15: Diagrama de DSC em polímeros (Fonte: Lopes, 2012)
2.5.5 Ensaio Mecânico de Tração
Neste ensaio, corpos de prova são presos em uma máquina de ensaios, onde sofrem
alongamento a uma velocidade predeterminada até a ruptura (ASTM D2256/D2256M).
Propriedades mecânicas podem ser determinadas, realizando-se os ensaios em quintuplicatas.

40
A partir do ensaio mecânico de resistência à tração, propriedades como módulo de
elasticidade (𝐸), limite de escoamento (𝜎𝐸𝑠𝑐𝑜𝑎𝑚𝑒𝑛𝑡𝑜), deformação na ruptura (𝜀𝑅𝑢𝑝) e resistência
máxima à ruptura (𝜎𝑀á𝑥 𝑅𝑢𝑝) são determinados, como mostra a Fig. 16.
Figura 16: Ilustração de uma curva de tensão em função da deformação (Fonte: Sena, 2011).

41
3 MATERIAIS E MÉTODOS
3.1 MATERIAIS
Foram usados monofilamentos de polipropileno (PP), politereftalato de etileno (PET) e
poliamida-6 (PA-6), produzidos por Mazzaferro Ltda., com diâmetros iguais a 200µm, 170µm e
300µm, respectivamente. Destes monofilamentos, cortaram-se fios com aproximadamente 50mm
de comprimento.
3.2 LIMPEZA DAS AMOSTRAS
A limpeza é necessária para remover impurezas e particulados depositados na superfície
de materiais durante seu armazenamento (Sellin, 2002; Sena, 2011). Usou-se etanol comercial
(98% Ecibra) para limpeza das amostras. Desse modo, as amostras foram previamente imersas
em um béquer contendo etanol, durante um tempo aproximado de 1minuto, sendo posteriormente
postas em superfícies de papel toalha e secas a condições controladas de temperatura e umidade
(22°C e 38%), por aproximadamente 1 hora para remoção do etanol residual. Foram avaliados os
efeitos da limpeza das amostras de monofilamentos medindo-se o ângulo de contato antes e após
a limpeza.
3.3 DESCARGA CORONA
3.3.1 Descrição do Sistema Fio-plano
Como ilustra a Fig. 17, o equipamento gerador de corona do tipo fio-plano é constituído
por um fio (aço inox, diâmetro: 200 µm) acoplado entre duas barras isolantes, um plano (placa de
alumínio retangular de 12 x 10 x 2 cm), uma fonte de tensão fixa em 35 kV e medidor de
corrente. A distância fio-plano pode ser previamente ajustada (2 a 15 mm). Durante os ensaios
manteve-se o ar condicionado ligado no sentido de estabilizar a umidade relativa e temperatura
do ambiente (22°C e 38%).
O equipamento foi construído pelo Prof. Dr. João Sinézio de Carvalho Campos,
coordenador do grupo de FisPol/FEQ/UNICAMP, com fonte de alta tensão adaptadas de
monitores de computadores. O equipamento gerador de corona já foi utilizado em diversos

42
trabalhos do grupo FisPol: Sellin, 2002; Carneiro, 2001; Sena, 2011 e Lopes, 2012. Durante o
tratamento corona, uma amostra de monofilamento era posicionada no plano, paralela ao fio de
aço (eletrodo móvel).
Figura 17: Representação do sistema corona fio-plano
3.3.2 Tratamento da Superfície das Amostras
Amostras limpas foram submetidas à descarga corona com tensão de 35 kV, a condições
ambientes estabilizadas, utilizando o equipamento descrito em 3.3.1.
Estudou-se a influência das variáveis: tempo de tratamento e distância entre os eletrodos
sobre a molhabilidade das amostras apresentadas na Tab 4. Amostras higienizadas e sem
tratamento serviram como padrão de controle na verificação de alterações das propriedades de
superfície observadas após tratamento.

43
Tabela 4: Niveis das variáveis estudadas no tratamento corona
Variável Nível inferior Nível médio Nível superior
Distância inter-eletrodos (mm) 4 7 10
Tempo (s) 10 60 120
Como respostas, estimou-se valores do ângulo de contato dinâmico, ângulo de contato
estático e energia livre de superfície, em relação às superfícies de PP, PET e PA-6. Ressalta-se
que as amostras, após limpeza, eram armazenadas em ambiente limpo com condições controladas
de umidade relativa e temperatura.
3.4 MEDIDAS DE ÂNGULO DE CONTATO DINÂMICO E ESTÁTICO
Medidas de ângulo de contato, com água deionizada, sobre as superfícies dos
monofilamentos limpos e após tratamento corona podem fornecer uma indicação de modificação
na hidrofilidade da superfície analisada. Devido à dificuldade de se medir ângulos de contato de
materiais na forma de fios, optou-se em utilizar o Método de Wilhelmy.
Para cada monofilamento, realizou-se 3 ciclos de medidas de força em condições
ambientes controladas de temperatura e umidade relativa (22°C e 38%).. Foram usadas duas
velocidades de imersão/emersão: 4,5 mm/min. (velocidade mínima da plataforma) e 9,0 mm/min.
(dobro da velocidade mínima), com o intuito de se verificar sua influência nos valores de ângulo
de contato dinâmico. Os valores de força foram obtidos com profundidade mínima de imersão de
1 mm e máxima de 9 mm da amostra na fase líquida.
Valores de ângulo de contato dinâmico fornecem estimativas de mudanças na
molhabilidade das amostras, frente ao tratamento de superfície. No entanto, os valores de ângulo
de contato de sistemas estáticos são mais estudados e usados para o cálculo da energia livre de
superfície.
Dessa forma, para estimativas do ângulo de contato estático de amostras de
monofilamentos, usou-se o Método de Wilhelmy adaptado. Durante as medições dos valores de
ângulo de contato, foram utilizados água deionizada e diiodo metano 99% da Sigma-Aldrich.
Durante os movimentos de avanço e retrocesso, o movimento do béquer contendo os líquidos
usados era interrompido a cada 1mm de imersão da amostra durante um período mínimo de 30s

44
para estabelecimento do equilíbrio de forças envolvidas. Os dados de força versus imersão foram
usados para cálculo do ângulo de contato estático de cada amostra, sendo estes usados na
estimativa da energia livre de superfície das amostras.
Como líquidos de medida foram usados água deionizada e diiodo metano. As medidas
foram feitas antes e após tratamento corona. Os valores de ângulos de contato dinâmico foram
calculados a partir dos dados recebidos de força versus profundidade conforme item 2.5.1. Os
valores de força foram obtidos por um tensiômetro automático de Du Noüy, marca Sigma, KSV
Instruments®, Finlândia, pertecente ao Laboratório de Fluidodinâmica e Secagem
(FEQ/UNICAMP).
3.5 ESPECTROSCOPIA NO INFRAVERMELHO/ATR (FT-IR/ATR)
O tratamento corona aplicado em amostras PP, PET e PA-6 pode levar à formação de
grupos polares de superfície contendo oxigênio que podem ser identificados por espectroscopia
no infravermelho com reflexão total atenuada (FT-IR/ATR). Para caracterização da composição
química de superfície das amostras de monofilamentos de PP, PET e PA-6 antes e após
tratamento corona, foram obtidos espectros de FT-IR/ATR, nas condições ambientes.
Análises foram realizadas com um Espectrômetro de Infravermelho com Transformada de
Fourier (FT-IR), marca: Thermo Scientific, modelo: Nicolet 6700 (Madison/USA). As medidas
foram realizadas no modo ATR utilizando o acessório SMART OMNI SAMPLER, em uma faixa
de 4000-675 cm-1
, com resolução de 4 cm-1
, no Laboratório de Recursos Analíticos e de
Calibração (FEQ/UNICAMP).
3.6 CALORIMETRIA EXPLORATÓRIA DIFERENCIAL (DSC)
Análises de DSC permitem avaliar mudanças nas propriedades térmicas dos materiais. Já
que a modificação provocada nos monofilamentos pelo tratamento corona é superficial, espera-se
que os monofilamentos mantenham suas propriedades térmicas após tratamento. Os termogramas
obtidos após análise em forneceram informações sobre a temperatura de transição vítrea (Tg),

45
temperatura de fusão (Tm), temperatura de cristalização (Tc), entalpia de fusão (ΔHm) e de
cristalização (ΔHc) das amostras.
Durante o ensaio, uma massa de aproximadamente 4,5 mg de cada amostra foi pesada em
Balança Microanalítica marca: Mettler Toledo modelo: MX5 (Zürich, Suíça) e acondicionada em
porta amostra de alumínio. Usou-se atmosfera inerte de N2. As condições específicas de ensaio
para cada tipo de polímero usado são descritas na Tab. 5. Os ensaios foram realizados com as
seguintes etapas:
a) Aquecimento da amostra da temperatura ambiente à temperatura máxima de operação,
mantendo-se constante a temperatura máxima com isoterma de 5 minutos para remoção do
histórico térmico do material;
b) Resfriamento da amostra da temperatura máxima à temperatura mínima de trabalho para
identificação de Tc e ΔHc;
c) Aquecimento da amostra da temperatura mínima à temperatura máxima de trabalho para
identificação de Tg, Tm e ΔHm.
Tabela 5: Condições de ensaio DSC para PP, PET e PA-6
Condições de ensaio PP PET PA-6
Vazão de gás inerte (mL/min) 50 40 50
Faixa de temperatura (°C) -50 a 250 0 a 300 10 a 250
Taxa de aquecimento/resfriamento (°C/min) 10 10 10
Os valores do grau de cristalinidade das amostras foram obtidos através do cálculo pela
Eq. (19), usando-se como entalpia de fusão dos polímeros 100% cristalinos iguais a 165J/g
(Sellin, 2002; Nihlstrand et. al., 1996), 140J/g (Gao et. al., 2013) e 190J/g (Yousfi, et. al., 2006),
para o PP, PET e PA-6, respectivamente.
Usou-se o equipamento com fluxo de calor, Calorímetro Exploratório Diferencial (DSC),
marca: Mettler-Toledo modelo: DSC (Zürich, Suíça), para determinação das propriedades
térmicas, do Laboratório de Recursos Analíticos e de Calibração (FEQ/UNICAMP).

46
3.7 MICROSCOPIA ELETRÔNICA EXPLORATÓRIA
Empregou-se MEV, a fim de examinar alterações na topografia, com possíveis formações
de trincas e superfície rugosas nos monofilamentos tratados por corona. As amostras foram
fixadas em fita de carbono, submetidas a recobrimento metálico com ouro, para que a superfície
se tornasse condutora. Foram obtidas micrografias de monofilamentos de PP, PET e PA-6
tratados com corona e sem tratamento para comparação, usando MEV a condições ambientes e
ampliação de 3000 X. Para obtenção das micrografias, usou-se um microscópio eletrônico marca
LEO Electron Microscopy, modelo Leo-440i com resolução de 10μm, tensão de aceleração de
15kV e corrente de feixe igual a 100pA, do Laboratório de Recursos Analíticos e de Calibração
(FEQ/UNICAMP).
3.8 ENSAIO MECÂNICO DE TRAÇÃO
Propriedades mecânicas dos monofilamentos de PP, PET e PA-6 tratados com corona
foram determinadas usando um equipamento servo-hidráulico para ensaios mecânicos, modelo
810-Flex Text 40, fabricante: MTS, com capacidade de 100kN, usando-se célula de carga de
1,5kN. Amostras sem tratamento serviram como padrão de controle para avaliação de possíveis
modificações nos valores das propriedades. Os corpos de prova foram preparados com 800 mm
de comprimento, fixados e tensionados verticalmente em duas garras da máquina de ensaio
distantes em 100 mm, a uma velocidade de 120 mm/min sob condições ambientes controladas.
Cada valor relacionado às propriedades obtidas equivalem a média de 5 amostras testadas. Os
testes foram realizados no Laboratório de Ensaios Mecânicos do (DEMM/FEM/UNICAMP).
3.9 ABSORÇÃO DE ÁGUA
A capacidade de absorção de fluidos pelos monofilamentos tratados com corona foi
testada usando água a fim de se analisar o efeito deste solvente no comportamento dos
monofilamentos. Pesaram-se as amostras secas, em uma Balança analítica da marca: Unibloc,

47
modelo: AUY 220, pertencente ao Laboratório de Física de Polímeros (FEQ/UNICAMP),
determinando-se a massa seca de cada amostra (𝑚𝑠). As amostras pesadas forma imersas em um
Becker contendo aproximadamente 40mL de água destilada, por 24 horas, a condições ambientes.
A metodologia utilizada, neste trabalho, é semelhante à utilizada por Giordano (2007), que
determinou o grau de absorção de água de têxteis .
Após este período, o excesso de água foi levemente retido em papel toalha, pesando-se
novamente as amostras, determinando-se a massa úmida final (𝑚𝑢). O grau de absorção (𝐴) das
amostras é dado em porcentagem e calculado pela seguinte Eq. (20):
𝐺𝐴 = 𝑚𝑢− 𝑚𝑠
𝑚𝑠 𝑥 100 (20)
3.10 CÁLCULO DA ENERGIA LIVRE DE SUPERFÍCIE
Valores da energia de superfície das amostras de monofilamentos de PP, PET e PA-6 foram
estimados através de valores de ângulo de contato estático utilizando-se água deionizada e diiodo
metano como líquidos de medida. De acordo Chibowski e Terpilowski (2009) os valores de energia
livre de superfície e suas componentes polar e dispersiva para água e diiodo metano são mostrados na
Tab. 6.
Tabela 6: Energia de superfície e suas componentes para água e a e diiodo metano
Líquido 𝜸𝑳𝑽 𝜸𝑳𝑽𝒑
𝜸𝑳𝑽𝒅
Água (H2O) 72,8 51,0 21,8
Diiodo metano (CH2I2) 50,8 50,8 0,0
Os valores das componentes polar e dispersiva e energia de superfície total das amostras foram
estimados através da média harmônica, pois fornece resultados mais consistentes para interações entre
sistemas de baixa energia como, por exemplo, líquidos e polímeros (Wu, 1971). A media harmônica
foi utilizada neste trabalho pelo fato de se ter trabalhado com grandezas inversamente proporcionais.
Neste caso, quanto maior for o valor da energia livre de superfície, menores serão os valores de

48
ângulo de contato.
Para obtenção das componentes dispersiva e polar dos materiais sem tratamento e tratados por
corona realizou-se as seguintes etapas: calculou-se inicialmente a componente dispersiva da energia
livre de superfície através da Eq. 10, utilizando os valores de ângulo de contato e a energia de
superfície para o diiodo metano. Em seguida, o valor da componente polar foi calculado a partir dos
valores da componente dispersiva e dos valores de ângulo de contato, energia de superfície da água,
pela Eq. 11. A soma da componente dispersiva e componente polar pela Eq. 2 resultou nos valores da
energia livre de superfície total para as amostras de PP, PET e PA-6 sem tratamento e tratadas por
descarga corona.

49
4 RESULTADOS E DISCUSSÕES
4.1 MEDIDAS DE ÂNGULO DE CONTATO DINÂMICO
4.1.1 Influência da limpeza das amostras
A remoção de impurezas da superfície de polímeros pode ser feita através do uso de
solventes sob diferentes formas de aplicação: imersão, pulverização sob pressão ou em fase
vapor. Solventes possuem alto poder de dissolução de compostos que contaminam a superfície do
material alterando ou encobrindo suas propriedades de superfície. Neste estudo, optou-se em
trabalhar com etanol para limpeza das amostras dos monofilamentos de PP, PET e PA-6, devido
ao seu baixo custo e fácil acesso.
A Tab.7 apresenta os resultados das determinações dos ângulos de contato dinâmico para
as superfícies dos monofilamentos de PP, PET e PA-6 antes e após limpeza com etanol. Durante
a imersão dos monofilamentos em água para obtenção dos perfis de força versus profundidade,
foi utilizada velocidade mínima de imersão igual a 4,5mm/min. Foi utilizado apenas 1 ciclo de
medida de avanço e retrocesso para verificar a influência da limpeza das amostras uma vez que
pode haver diferenças nos valores de ângulo de contato entre o 1° ciclo e os demais ciclos de
medida. As medidas foram realizadas em triplicata.
Tabela 7: Influência da limpeza dos monofilamentos nos valores de ângulo de contato de avanço
e retrocesso
Ângulo de avanço (°) Ângulo de retrocesso (°) ∆𝜽 (°)
Amostra Não limpa Limpa Não limpa Limpa Não limpa Limpa
PP 86,1° ±3,3° 104,0° ±3,3° 75,0° ±2,6° 73,8° ±0,7° 11,1° 30,2°
PET 77,7° ±2,7'° 79,0° ±2,9° 53,2° ±1,4° 33,3° ±0,6° 24,5° 45,7°
PA-6 60,0° ±0,3° 73,7° ±0,6° 30,7° ±0,7° 24,0° ±2,2° 29,3° 49,7°
Observa-se da Tab.7 que amostras limpas com etanol apresentam maiores ângulo de
avanço indicando remoção de possíveis impurezas presentes na superfície, que tendem a diminuir
este parâmetro. Houve um aumento de 17,9°, 1,3° e 13,7° nos valores de ângulo de avanço da

50
superfície de monofilamentos de PP, PET e PA-6 respectivamente após limpeza com etanol.
Nota-se que os valores de ângulo de retrocesso para a superfície de amostras limpas são
relativamente menores que ângulos de retrocessos de amostra não limpas, com diminuição de
1,2°, 19,9° e 6,7° nos valores de ângulo de retrocesso de amostras de PP, PET e PA-6 após
limpeza, sugerindo maiores interações entre as superfícies limpas e a água, após mergulhadas e
retiradas do líquido. Amostras limpas apresentaram maior ∆θ que amostra não limpas,
Impurezas na superfície dos monofilamentos podem ocorrer devido a condições de
armazenamento inadequado, condições de processamento ou até mesmo do manuseio impróprio.
Pode haver acúmulo de poeira sobre a superfície dos monofilamentos durante armazenamento
sendo importante acomodá-los em local adequado.
O manuseio inapropriado pode ocorrer através da impregnação de gordura das mãos na
superfície dos monofilamentos. Durante o processamento de polímeros, aditivos como
plastificantes e lubrificantes são usados para melhorar as propriedades desses materiais podendo
migrar do volume para a superfície do polímero.
A existência desses contaminantes influencia os valores de ângulo de contato dos
monofilamentos, alterando valores reais de propriedades de superfície como molhabilidade,
energia livre de superfície e adesão. Desta forma, a limpeza da superfície das amostras torna-se
importante. Os resultados obtidos indicam que o etanol usado como solvente promove remoção
dos contaminantes de superfície.
Em seu trabalho, Sena (2011) constatou que o uso de etanol como solvente para limpeza
de filmes de PP apresenta eficiência por remover impurezas da superfície das amostras, elevando-
se o valor de ângulo de contato para os filmes limpos. O autor verificou ainda que não há
diferenças significativas quanto ao valor de ângulo de contato, quando amostras de PP eram
submetidas à limpeza com acetona.
4.1.2 Influência do número de ciclos de medidas
A Tab. 8 apresenta os valores dos ângulos de avanço e retrocesso e seus respectivos
valores para amostras de PP, PET e PA-6 limpas sem tratamento corona, registrados durante 3

51
ciclos de medida por amostra a uma velocidade de 4,5mm/min. Os valores correspondem à média
para 3 amostras.
Nota-se pela Tab. 8 diferenças nos valores médios de ângulo de avanço e retrocesso para
os monofilamentos de PP, PET e PA-6 devido ao número de ciclos em que uma amostra foi
submetida. Diferenças entre o primeiro e os próximos ciclos podem ocorrer, por exemplo, devido
a hidratação dos monofilamentos.
Tabela 8: Influência do número de ciclos nos valores de ângulo de contato de avanço e retrocesso
para monofilamentos de PP, PET e PA-6. Velocidade de 4,5mm/min
Ângulo de avanço (°) Ângulo de retrocesso (°)
Ciclos PP PET PA-6 PP PET PA-6
1 104,0° ±3,3° 79,0° ±2,9° 73,7° ±0,6° 73,8° ±0,7° 33,3° ±0,6° 25,0° ±2,6°
2 107,0° ±3,0° 80,9° ±0,6° 76,2° ±0,9° 73,4° ±4,0° 32,0° ±4,0° 32,7° ±0,4°
3 107,2° ±1,3° 79,6° ±1,4° 76,0° ±0,3° 73,6° ±4,0° 33,9° ±2,3° 33,6° ±0,7°
Nota-se pela Tab. 8 diferenças nos valores médios de ângulo de avanço e retrocesso para
os monofilamentos de PP, PET e PA-6 devido ao número de ciclos em que uma amostra foi
submetida. Diferenças entre o primeiro e os próximos ciclos podem ocorrer, por exemplo, devido
a hidratação dos monofilamentos.
Observa-se através da Tab. 8 molhabilidade mais acentuada em monofilamentos de PA-6,
dentre os 3 polímeros sintéticos estudados, tendo em vista menores valores de ângulo de contato
apresentados por este material. Monofilamentos de PET apresentaram menores valores de ângulo
de contato que amostras de PP.
Possíveis explicações para a molhabilidade das amostras testadas estão relacionadas à
estrutura química dos polímeros usados. O polipropileno apresenta superfície hidrofóbica, devido
a grupos metílicos presentes em suas cadeias puramente formadas por átomos de carbono e
hidrogênio (Sellin, 2002). A alta hidrofilidade de poliamidas diante dos outros materiais é afetada
de maneira significativa por interações entre átomos de hidrogênio e grupos polares (–CO– e –
NH–) presentes nas cadeias poliméricas (Deopura, 2008). Por conter alguns grupos oxigenados
em sua estrutura, cadeias de PET são mais hidrofílicas que cadeias de PP. No entanto, a

52
hidrofobicidade de poliésteres é justificada pela falta de grupos polares em suas cadeias (Zaman
et. al.,2013).
Fatores como irregularidades e deformações da superfície dos monofilamentos, aliadas a
possível hidratação dos materiais usados também podem ser responsáveis pelo comportamento
apresentado nos ciclos de avanço e retrocesso, promovendo a diferenças entre os ângulos de
avanço e recuo (histerese), Alves (1999).
4.1.3 Influência da velocidade de imersão
Outro fator avaliado durante as medidas de ângulo de avanço e recúo das amostras foi a
velocidade de imersão. Estimou-se, então, ângulos de contato de amostras limpas de
monofilamentos de PP, PET e PA-6 limpas e sem tratamento de superfície a uma velocidade de
imersão igual a 9,0mm/min. Os valores podem ser observados na Tab.9.
Nestas condições, a mudança de velocidade não interferiu em alterações relevantes nos
valores de ângulo de contato de avanço e retrocesso estimados, além de fornecer valores em
menor intervalo de tempo. Desse modo, estimativas de ângulo de contato de amostras tratadas
por descarga corona foram realizadas usando-se velocidade de imersão igual a 8,92mm/min.
Tabela 9: Influência da velocidade nos valores de ângulo de contato de avanço e retrocesso para
monofilamentos de PP, PET e PA-6. Velocidade de 9,0mm/min
Ângulo de avanço (°) Ângulo de retrocesso (°)
Ciclos PP PET PA-6 PP PET PA-6
1 104,0° ±2,1° 77,1° ±0,9° 73,1° ±0,6° 73,4° ±3,1° 33,8° ±1,6° 25,4° ±2,3°
2 108,6° ±1,6° 80,4° ±0,3° 76,8° ±1,1° 76,8° ±1,6° 32,9° ±0,9° 33,7° ±2,0°
3 107,2° ±1,3° 80,6° ±0,8° 76,7° ±0,7° 77,9° ±2,9° 33,2° ±1,4° 33,8° ±2,1°
Nota-se que para os segundo e terceiro ciclos, os ângulos de avanço mantiveram-se
aproximadamente constantes. Por essa razão, o primeiro ciclo não foi usado no cálculo dos
valores médios dos ângulos de contato de avanço e retrocesso para amostras limpas sem
tratamento e amostras tratadas por descarga corona.

53
É muito comum por pesquisadores estimar apenas os valores de ângulo de avanço, no
entanto, ambos os ângulos de avanço e retrocesso são importantes para se descrever bem o
sistema. A Tab. 10 apresenta a estimativa dos valores de ângulo de contato dinâmico de avanço e
retrocesso e a diferença entre estes, das amostras de monofilamentos limpas e sem tratamento de
superfície.
Os valores da Tab. 10 serão usados como padrão de controle para análises posteriores das
amostras de monofilamentos limpas submetidas ao tratamento corona sob diferentes condições. O
ângulo de equilíbrio (𝜽𝑬𝑸), calculado neste trabalho, é apenas uma estimativa da média entre o
ângulo de avanço e retrocesso obtidos experimentalmente. Ressalta-se que os valores obtidos
para ângulo de contato de avanço e recúo para amostras de PP, PET e PA-6 situam-se numa faixa
de valores encontrados por diversos autores como: Della Volpe (1998), Wu (1971), Tran et. al
(2011) e Gotoh, 2004.
Tabela 10: Valores de ângulo dinâmicos de avanço e retrocesso adotados como padrão de
controle para monofilamentos limpos e sem tratamento corona
Polímero 𝜽𝒂 (°) 𝜽𝒓 (°) 𝜽𝑬𝑸 (°) ∆𝜽(°)
PP 107,5 77,4 92,5 30,1
PET 80,5 33,1 56,8 47,4
PA-6 76,8 33,8 55,3 43,0
4.2 TEMPO E DISTÂNCIA INTER-ELETRODOS NO TRATAMENTO CORONA
Em um sistema corona, a distância inter-eletrodos influencia nas propriedades físicas do
plasma, ocasionando mudanças na superfície de polímeros. A partir das Fig. 18, 19 e 20,
referentes às respectivas amostras de PP, PET e PA-6, observa-se o comportamento dos valores
de 𝜃 em função do tempo de tratamento, a diferentes distâncias inter-eletrodos.
Nota-se em todos os casos que os ângulos de avanço, retrocesso e equilíbrio tendem a
diminuir com menores distâncias entre os eletrodos. Ressalta-se que, neste estudo, menores níveis
de molhabilidade foram obtidos na distância de 10mm. Níveis intermediários de molhabidade
foram atingidos na distância de 7mm. Maiores níveis de molhabilidade foram obtidos
diminuindo-se a distância inter-eletrodos a 4mm.

54
A partir da Fig. 18, nota-se o comportamento dos valores de 𝜃 para amostras de PP
tratadas por corona. Valores de ângulo de avanço não sofreram mudanças bruscas após 10 s de
tratamento com diminuição de 5% do valor do ângulo de uma amostra não tratada. Valores de
ângulo de recuo tiveram mudanças significativas após 10s de tratamento com uma diminuição de
29,6%, quando amostras de monofilamentos de PP foram submetidas ao tratamento corona com
distância inter-eletrodos igual a 4 mm, durante 120 s. Nestas mesmas condições de operação,
houve uma redução de 15,4% do valor inicial do ângulo de equilíbrio. Menores distâncias inter-
eletrodos e o aumento do tempo de tratamento promoveram aumento de 58,1% dos valores de ∆𝜃
inicial, denominados, neste trabalho, como histerese.
Figura 18: Ângulo de contato em função do tempo de tratamento para PP. a) ângulo de avanço; b)
ângulo de recuo; c) ângulo de equilíbrio; d) histerese
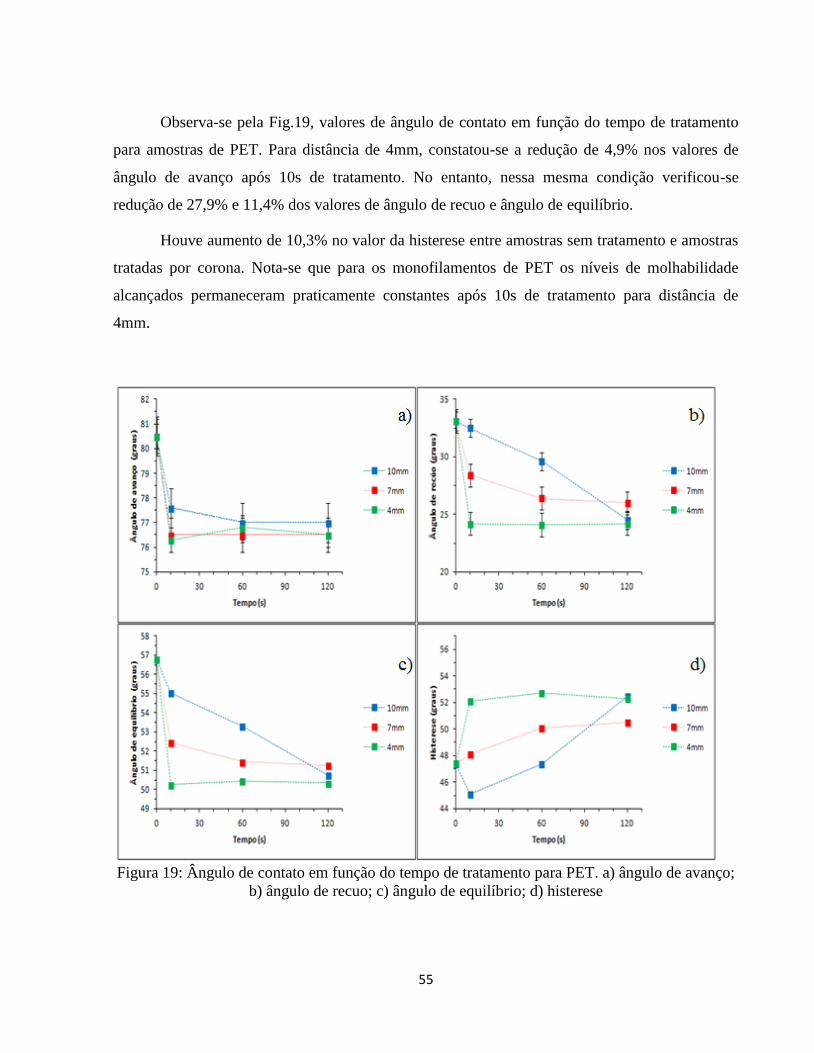
55
Observa-se pela Fig.19, valores de ângulo de contato em função do tempo de tratamento
para amostras de PET. Para distância de 4mm, constatou-se a redução de 4,9% nos valores de
ângulo de avanço após 10s de tratamento. No entanto, nessa mesma condição verificou-se
redução de 27,9% e 11,4% dos valores de ângulo de recuo e ângulo de equilíbrio.
Houve aumento de 10,3% no valor da histerese entre amostras sem tratamento e amostras
tratadas por corona. Nota-se que para os monofilamentos de PET os níveis de molhabilidade
alcançados permaneceram praticamente constantes após 10s de tratamento para distância de
4mm.
Figura 19: Ângulo de contato em função do tempo de tratamento para PET. a) ângulo de avanço;
b) ângulo de recuo; c) ângulo de equilíbrio; d) histerese
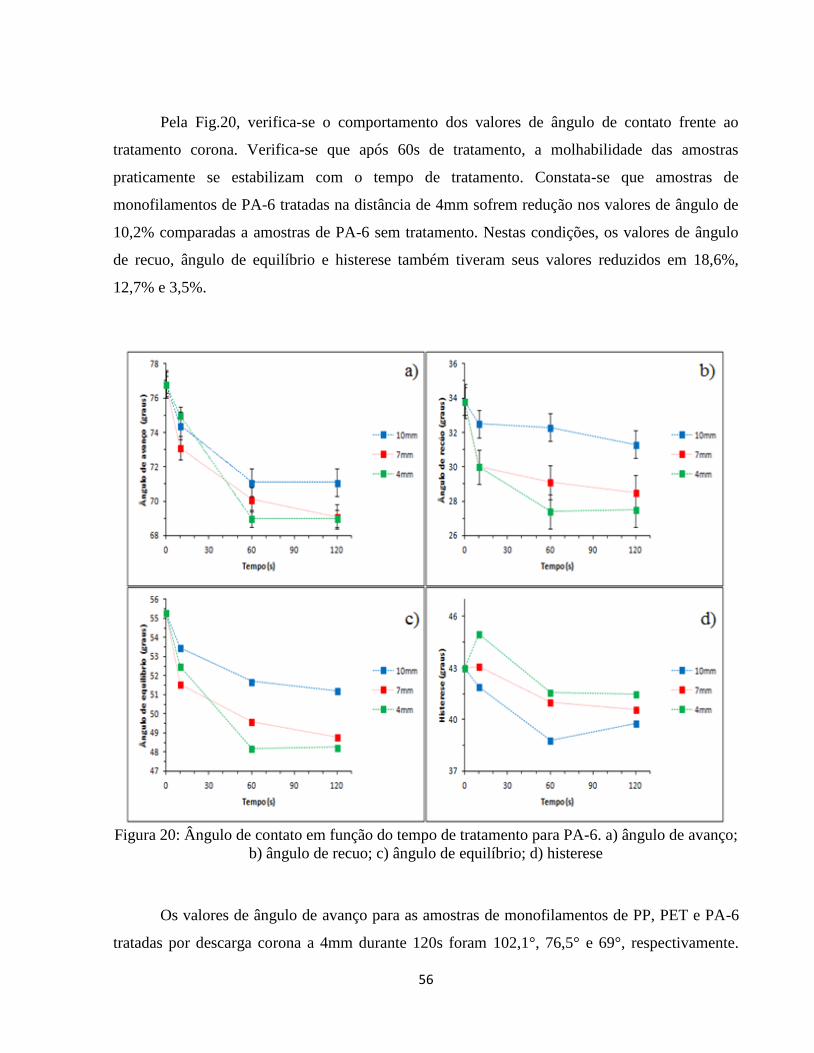
56
Pela Fig.20, verifica-se o comportamento dos valores de ângulo de contato frente ao
tratamento corona. Verifica-se que após 60s de tratamento, a molhabilidade das amostras
praticamente se estabilizam com o tempo de tratamento. Constata-se que amostras de
monofilamentos de PA-6 tratadas na distância de 4mm sofrem redução nos valores de ângulo de
10,2% comparadas a amostras de PA-6 sem tratamento. Nestas condições, os valores de ângulo
de recuo, ângulo de equilíbrio e histerese também tiveram seus valores reduzidos em 18,6%,
12,7% e 3,5%.
Figura 20: Ângulo de contato em função do tempo de tratamento para PA-6. a) ângulo de avanço;
b) ângulo de recuo; c) ângulo de equilíbrio; d) histerese
Os valores de ângulo de avanço para as amostras de monofilamentos de PP, PET e PA-6
tratadas por descarga corona a 4mm durante 120s foram 102,1°, 76,5° e 69°, respectivamente.

57
Valores de ângulo de recuo, para os respectivos materiais, foram: 54,5°, 24,2° e 27,5°. Nestas
condições, constatou-se que os respectivos valores de ângulo de equilíbrio para o PP, PET e PA-6
foram: 78,3°, 50,4° e 48,3°. É notável que todas as amostras demonstraram maior caráter
hidrofílico após tratamento. No entanto, nota-se que amostras tratadas de PET e PA-6 durante
120s a distância inter-eletrodos de 4mm apresentaram maior molhabilidade, devido a menores
valores de ângulo de contato como observado, frente às amostras de PP.
A redução dos valores de ângulo de contato após descarga corona com consequente
modificação da superfície dos monofilamentos ocorre possivelmente pela formação de grupos
polares contendo átomos de O e/ou N na superfície dos polímeros (Sellin, 2002; Sena 2011;
Lopes, 2012).
Estudos mostraram que o tratamento por descarga corona conduz a formação de grupos
funcionais na superfície de PP como hidroxilas, carbonilas, carboxilas, álcoois, aldeídos, cetonas,
peróxidos, hidroperóxidos, carbonatos e ésteres (Sellin, 2002; Sena, 2011). Estudos em fibras de
poliamida revelam resultados semelhantes como em Labay et. al, 2014. Dessa forma, menores
valores de ângulo de contato, indicam melhoria nas propriedades de molhabilidade, adesão e
energia livre de superfície desse material (Sena, 2011).
Briggs et. al. (1980), estudaram os efeitos da descarga corona em filmes de PET usando
sistema de placas paralelas, mantendo-se uma distância de 2mm entre placas e 15,9kV durante o
tratamento. Os autores constataram que o aumento do tempo de tratamento promove aumento de
propriedades hidrófilas das amostras, com diminuição do ângulo de contato formado entre
superfície e água, aumentando-se a energia livre de superfície das mesmas. Os autores atribuem
tal característica à incorporação de grupos fenólicos e ácidos carboxílicos na superfície das
amostras.
O método de Wilhelmy tem sido amplamente aplicado para análise de ângulo de contato
dinâmico. Ao avaliar os efeitos da técnica de plasma aplicada sobre a superfície de fibras de PP
Huang et. al. (2006) verificaram que fibras sem tratamento apresentaram ângulo de contato de
avanço de aproximadamente 95° e ângulo de recúo de 78°, exibindo histerese de 17°, justificada
pela rugosidade da superfície das fibras. O efeito do tratamento com plasma sobre os valores de
ângulo de contato promoveu diminuição do ângulo de avanço para 83° e ângulo de retrocesso

58
para 52°, após tratamento com plasma, durante 30 segundos. A histerese aumentou de 17° (fibras
sem tratamento) para 31° após tratamento com plasma.
Verificou-se que em menores distâncias inter-eletrodos a molhabilidade das amostras
estudadas foi maior, contatada pela diminuição dos valores de ângulo de contato. Dessa forma,
diante dos estudos realizados neste trabalho, adotou-se 4 mm como distância padrão para
tratamentos dos monofilamentos estudados dando-se continuidade ao trabalho.
Distâncias menores que 3mm foram testadas para realização do tratamento das amostras,
observando-se que nestas condições, o tratamento corona conduzia a uma maior quantidade de
amostras perfuradas, enrugadas ou escurecidas, devido a maior geração de faíscas filamentares,
observadas durante o tratamento. Além disso, menores distâncias induzem a diminuição da área a
ser tratada tendo como consequência, menor criação de grupos polares na superfície (Sena, 2009;
Sellin, 2002).
4.3 COMPOSIÇÃO QUÍMICA DE SUPERFÍCIE (FT-IR/ATR)
Mudanças químicas promovidas na superfície dos monofilamentos tratados por descarga
corona foram caracterizadas por espectros de transmissão por refletância total atenuada (FT-
IR/ATR) na região de 4000-650cm-1
. Tal região foi escolhida, tendo em vista a verificação do
aparecimento de grupos funcionais polares após o tratamento de superfície nos espectros gerados
que ocorrem nessa região para amostras de PP (Sena, 2011; Lopes, 2012; Sellin, 2002), de PET
(Briggs, 1980; Liu et. al., 2013) e PA-6 (Coelho, 2012; Evora et. al., 2002). Monofilamentos não
tratados serviram como padrão comparativo.
A Fig. 21 mostra espectros de FT-IR/ATR de monofilamentos de PP em diferentes
tempos de tratamento por descarga corona. O pico em 2365 cm-1
corresponde ao gás carbônico
presente no meio atmosférico e não interfere na região de análise.

59
Figura 21: Espectros FT-IR/ATR dos monofilamentos de PP sem tratamento, e tratados por 10s,
60s e 120s.
A análise dos espectros mostram que picos na região de 2700-3000 cm-1
correspondem a
deformações axiais de C-H, com vibrações assimétricas e simétricas do grupo metila,
confirmadas na região de 1340-1480cm-1
. Verifica-se nos espectros dos monofilamentos tratados
a presença de novos picos, se comparados a monofilamentos não tratados. Picos na região de
3200-3600 cm-1
referem-se à possíveis estiramentos do grupo hidroxila (-OH).
A região próxima a 1700 cm-1
mostram picos de pequena intensidade que pode ser
atribuídos possivelmente a deformação axial da carbonila (C=O), no grupo ácido carboxílico (-
COOH). Picos na região 1000-1260 cm-1
podem corresponder ao grupo éster (-COO), formados
durante o tratamento corona na superfície dos monofilamentos de PP (Sellin, 2002), indicando
aumento da molhabilidade das amostras de acordo os resultados obtidos pelas estimativas de
ângulo de contato dinâmico.
A partir da Fig. 22, observa-se os espectros de FT-IR/ATR de amostras de
monofilamentos de PET, tratados com corona e sem tratamento. As bandas situadas em 1740 e
730 cm-1
são atribuídas a vibrações de deformações do grupo éster (-COO).

60
Bandas de absorção na região de 2872 cm-1
e 2951 cm-1
correspondem a deformação
simétrica e assimétrica do grupo -CH2 do etileno, respectivamente. Picos em 3049 cm-1
, 1503 cm-
1 e 1598 cm
-1 representam a deformação de grupos =C–H, indicando a presença de anel
benzênico . Bandas nítidas são observadas na região 1400-1600 cm-1
e 950-1250 cm-1
, sendo
atribuídas a estiramento de ligações C-C e C-H no plano para o benzeno (Liu et. al., 2013).
Verifica-se a formação suave de picos na região de 3100-3700 cm-1
indicando a possível
presença de grupos OH, após tratamento por descarga corona em amostras tratadas de PET.
Observa-se ligeira intensificação dos picos e alargamento das bandas de C=O, indicando que tais
grupos são proveninetes de reações químicas ocorridas na superfície do PET, provacadas por
descarga corona (Briggs, 1980).
Figura 22: Espectros FT-IR/ATR dos monofilamentos de PET sem tratamento, e tratados por 10s,
60s e 120s.

61
Os espectros da Fig. 23 correspondem às análise de FTIR/ATR realizadas sobre a
superfície de amostras de monofilamentos de PA-6 sem tratamento e tratadas por corona. Picos
em 1545 cm-1
são atribuídos a presença de N-H no plano e a estiramentos de ligações C-N e CC.
Picos em 3310 cm-1
indicam absorção devido ao estiramento da ligação N-H. Amostras
tratadas por corona apresentam picos levemente mais intensos que amostras não tratadas. Na
região de 3080 cm-1
verifica-se a presença de grupos amida. Picos em 2930 cm-1
e em 2850 cm-1
correspondem a respectivos estiramentos assimétricos e simétricos de –CH2.
Picos na região de 1640 cm-1
correspondem a estiramento das ligações C=O e C-N e
deformação do grupo C-C-N. Percebe-se que nesta região, os picos se tornam levemente mais
intensos em amostras tratadas por corona, indicando possíveis formações de grupos oxigenados e
nitrogenandos na superfície dos materiais.
Figura 23: Espectros FT-IR/ATR dos monofilamentos de PA-6 sem tratamento, e tratados por
10s, 60s e 120s.
Os resultados das análises de FT-IR/ATR realizados para as amostras de PP, PET
e PA-6 sem tratamento e tratadas por descarga corona não apresentaram grandes diferenças entre

62
os espectros de amostras tratadas e sem tratamento. Dessa forma, uma outra técnica de
caracterização, como por exemplo a Espectroscopia de fotoelétrons excitados por raio-X, poderia
ser usada para confirmação da incorporação de átomos de oxigênio e nitrogênio na superfície das
amostras tratadas.
4.4 ANÁLISE TOPOGRÁFICA DE MONOFILAMENTOS (MEV)
Usou-se MEV com o intuito de identificar mudanças topográficas com possíveis
formações de trincas nos monofilamentos de PP, PET e PA-6 estudados sem tratamento e
tratados por corona a 10s; 60s e 120s a uma distância de 4mm. Os resultados obtidos podem ser
observados nas Fig. 24, Fig. 25 e Fig. 26 para o PP, PET e PA-6 respectivamente.
Nota-se que o monofilamento de PP sem tratamento (Fig.24a) apresenta uma superfície
íntegra, isto é, sem danos devido ao tratamento. Após 10s de tratamento, a superfície do
monofilamento de PP defeitos, com pequenas fissuras entre as fibras da superfície.
Percebe-se também um ligeiro aumento da rugosidade desse monofilamento comparado
ao polímero não tratado. Verifica-se que após 60s e 120s o tratamento corona intensifica os
defeitos na superfície do PP, fazendo com que ocorra pequenas cavidades ao longo das fibras
com a propagação de aberturas e erosão na superfície do material.

63
Figura 24: Topografias de monofilamentos de PP, obtidos por MEV. a) sem tratamento corona e
tratados por: b) 10s; c) 60s; d) 120s.
A Fig.25 apresenta a topografia de monofilamentos de PET. Nota-se que a superfície do
monofilamennto não tratado é lisa com a presença de algumas linhas mono-orientadas devido a
extrusão. Nota-se a formação de pequenos grânulos na superfície deste material após 10s de
tratamento com aumento da depressão das linhas unidirecionais.
Com o aumento do tempo de tratamento percebe-se elevação do tamanho dos grânulos em
60s, com a formação de cavidades e protuberâncias na superfície do monofilamento de PET
tratado por 120s.

64
Figura 25: Topografias de monofilamentos de PET, obtidos por MEV. a) sem tratamento corona
e tratados por: b) 10s; c) 60s; d) 120s.
A partir da Fig. 26, nota-se as mudanças ocorridas nas superfícies dos monofilamentos de
PA-6, com o aumento do tempo de tratamento das amostras. Amostra de monofilamento de PA-6
não tratada apresenta em sua superfície ligeira rugosidade, característico do processo de extrusão.
O aumento da rugosidade na superfície dos monofilamentos apresenta a formação de eriçamentos
e protuberâncias. Após 60 segundos, a modificação topográfica ocorrida torna-se mais ampla,
com a formação de grânulos e pequenas fissuras e grânulos.
Observa-se a partir das micrografias que as estruturas dos monofilamentos apresentam
forma fibrilar unidirecional. Tal comportamento está relacionado ao processo de extrusão dos
monofilamentos.

65
Figura 26: Topografias de monofilamentos de PA-6, obtidos por MEV. a) sem tratamento corona
e tratados por: b) 10s; c) 60s; d) 120s.
Nota-se que danos promovidos na superfície dos monofilamentos de PP, PET e PA-6 por
descarga corona se intensificaram devido ao aumento do tempo de tratamento. No entanto, os
danos ocorridos foram pontuais, isto é, não ocorriam em toda a área da superfície dos
monofilamentos, ocasionando a formação de superfícies com topografia heterogênea justificada
pela formação de estrutura granulada, erosões e depressões numerosas e visível distribuição
irregular na superfície das amostras.
A formação de defeitos na superfície dos monofilamentos pode ser explicada através da
modificação local do nível de cristalinidade da superfície das amostras tratadas. Nesse caso, o
pressuposto se baseia em especulações de que quando a descarga atinge a superfície do polímero,
neste ponto, a superfície irá fundir devido à alta energia da descarga, propiciando a criação de
regiões amorfas (Leroux et. al. 2008).

66
É possível que o aumento da rugosidade, visualizada através da formação de trincas,
eriçamentos, protuberâncias e erupções na superfície das amostras tratadas promova aumento na
hidrofilidade (molhabilidade) e energia de superfície dos polímeros (Sellin, 2002; Sena, 2011;
Lopes, 2012), confirmando os resultados de ângulo de contato dinâmico e FT-IR/ATR.
Sugere-se que o aumento da histerese na superfície dos monofilamentos de PP, PET e PA-
6 tratados por corona seja uma consequência da formação de protuberâncias, grânulos e elevada
rugosidade intensificadas com o aumento do tempo de tratamento das amostras. Rugosidade e
heterogeneidade, associada a grandes variações de energia de superfície, com algumas regiões
hidrofílicas e outras hidrofóbicas, promovem histerese (Alves, 1999).
4.5 PROPRIEDADES TÉRMICAS (DSC)
A Fig. 27 e Fig. 28 mostram os resultados obtidos por DSC das amostras de PP sem
tratamento e tratadas por corona durante 10, 60 e 120s. Observa-se que todas as amostras de
monofilamentos apresentaram picos de fusão na temperatura em torno de 164°C (Fig.27) e picos
de cristalização em temperaturas de aproximadamente 119°C (Fig.28), característico do
polipropileno. Não foi possível observar transição vítrea em nenhuma das amostras, que em geral
ocorre a -8°C. Amostras de polipropileno apresentaram grau de cristalinidade em torno de 54,9%.
A Tab.11 apresenta os resultados obtidos por DSC dos monofilamentos de PP.

67
Figura 27: Curvas de DSC de monofilamentos de PP antes a pós tratamento corona (picos de
fusão das amostras)
Figura 28: Curvas de DSC de monofilamentos de PP antes e após tratamento corona (picos de
cristalização das amostras)
-2
-1,8
-1,6
-1,4
-1,2
-1
-0,8
-0,6
-0,4
-0,2
-50 0 50 100 150 200 250
Flu
xo d
e C
alo
r (W
/g)
Temperatura (°C)
Sem tratamento
10s
60s
120s
-0,4-0,2
00,20,40,60,8
11,21,41,61,8
22,22,42,62,8
-50 0 50 100 150 200 250
Flu
xo d
e C
alo
r (W
/g)
Temperatura (°C)
Sem tratamento
10s
60s
120s

68
Tabela 11: Propriedades Térmicas para amostras de PP
Amostra Tg (°C) Tc (°C) Tm (°C) Hc (J/g) Hm (J/g) Xc (%)
PP sem tratamento - 119,1 164,1 92,4 92,4 56,0
PP tratado por 10s - 118,3 163,3 92,9 91,2 55,3
PP tratado por 60s - 119,2 164,2 89,7 91,3 55,3
PP tratado por 120s - 118,9 164,2 90,7 87,2 52,8
A Fig. 29 e Fig. 30 apresenta os resultados obtidos por DSC das amostras de
monofilamentos de PET. Observa-se que as amostras apresentam transições vítreas (Tg) em
77°C, picos de fusão e cristalização em torno de 247°C e 180°C, respectivamente, com grau de
cristalinidade de aproximadamente 26,4%. A Tab.12 mostra os resultados obtidos por DSC das
amostras de PET.
Figura 29: Curvas de DSC de monofilamentos de PET antes a pós tratamento corona (picos de
fusão das amostras)
-1
-0,9
-0,8
-0,7
-0,6
-0,5
-0,4
-0,3
-0,2
-0,1
0
0 50 100 150 200 250 300
Flu
xo d
e C
alo
r (W
/g)
Temperatura (°C)
Sem tratamento
10s
60s
120s
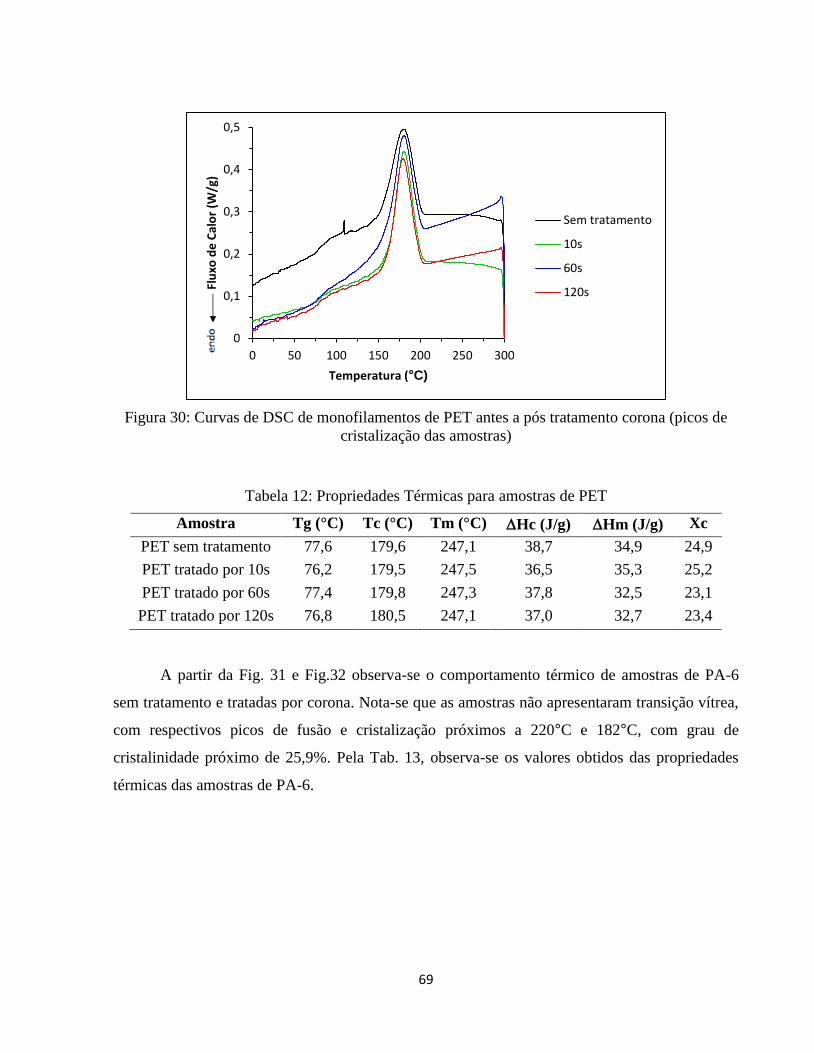
69
Figura 30: Curvas de DSC de monofilamentos de PET antes a pós tratamento corona (picos de
cristalização das amostras)
Tabela 12: Propriedades Térmicas para amostras de PET
Amostra Tg (°C) Tc (°C) Tm (°C) Hc (J/g) Hm (J/g) Xc
PET sem tratamento 77,6 179,6 247,1 38,7 34,9 24,9
PET tratado por 10s 76,2 179,5 247,5 36,5 35,3 25,2
PET tratado por 60s 77,4 179,8 247,3 37,8 32,5 23,1
PET tratado por 120s 76,8 180,5 247,1 37,0 32,7 23,4
A partir da Fig. 31 e Fig.32 observa-se o comportamento térmico de amostras de PA-6
sem tratamento e tratadas por corona. Nota-se que as amostras não apresentaram transição vítrea,
com respectivos picos de fusão e cristalização próximos a 220°C e 182°C, com grau de
cristalinidade próximo de 25,9%. Pela Tab. 13, observa-se os valores obtidos das propriedades
térmicas das amostras de PA-6.
0
0,1
0,2
0,3
0,4
0,5
0 50 100 150 200 250 300
Flu
xo d
e C
alo
r (W
/g)
Temperatura (°C)
Sem tratamento
10s
60s
120s

70
Figura 31: Curvas de DSC de monofilamentos de PA-6 antes a pós tratamento corona (picos de
fusão das amostras)
Figura 32: Curvas de DSC de monofilamentos de PA-6 antes a pós tratamento corona (picos de
cristalização das amostras)
-1,5
-1,2
-0,9
-0,6
-0,3
0
10 50 90 130 170 210 250
Flu
xo d
e C
alo
r (W
/g)
Temperatura (°C)
Sem tratamento
10s
60s
120s
-0,1
0,4
0,9
1,4
1,9
10 50 90 130 170 210 250
Flu
xo d
e C
alo
r (W
/g)
Temperatura (°C)
Sem tratamento
10s
60s
120s

71
Tabela 13: Propriedades Térmicas para amostras de PA-6
Amostra Tg (°C) Tc (°C) Tm (°C) Hc (J/g) Hm (J/g) Xc
PA-6 sem tratamento - 183,0 221,6 61,5 49,8 26,2
PA-6 tratado por 10s - 181,9 220,2 61,2 49,4 26,0
PA-6 tratado por 60s - 182,4 219,5 62,0 50,8 26,7
PA-6 tratado por 120s - 182,1 219,9 58,7 46,7 24,6
Não foi observado, deste modo, influencia do tratamento corona sobre a modificação das
propriedades térmicas dos materiais estudados devido ao aumento do tempo de exposição,
indicando que propriedades de volume dos monofilamentos foram preservadas. Por ser um
tratamento de superfície, mudanças nas propriedades térmicas dos materiais podem não ter sido
detectadas. Os resultados obtidos neste trabalho são similares aos de Sellin, 2002; Sena, 2011;
Lopes, 2012. Os valores das propriedades térmicas para monofilamentos de PP, PET e PA-6 são
semelhantes a valores da ASTM D3418-12 encontrados para estes polímeros.
4.6 PROPRIEDADES MECÂNICAS DE TRAÇÃO
Na Tab.14 são apresentados os resultados de resistência à tração de amostras de
monofilamentos de PP, PET e PA-6 sem tratamento corona e tratados durante 120s, com
distância entre os eletrodos de 4mm.
Dentre os monofilamentos testados, amostras de PET apresentaram maiores módulo de
elasticidade, limite de escoamento e resistência a ruptura. Amostras de PA-6 apresentaram
maiores deformações. Amostras de polipropileno apresentaram propriedades mecânicas de tração
menores que as de PET e PA-6. Observa-se que não há grandes diferenças entre propriedades
mecânicas das amostras associadas ao tratamento por descarga corona.
Verifica-se variações de aproximadamente de 21,41 MPa, 35,93 MPa e 67,94 MPa entre
amostras não tratadas e tratadas por descarga corona durante 120s para PP, PET e PA-6. Nota-se
que todas as amostras, sem tratamento e tratadas por corona, apresentaram alto módulo de
elasticidade, o que as caracteriza como sendo rígidas. Verifica-se pouca influência do tratamento

72
nesta propriedade mecânica, indicando que o tratamento corona não altera a rigidez dos
monofilamentos de PP, PET e PA-6 após o tratamento.
Percebe-se que após o tratamento, amostras de PP, PET e PA-6 tem seus valores de
ruptura ligeiramente modificados, sofrendo uma variação de 2,47%, 8,04% e 4,25%,
respectivamente. Houve pouca modificação nos valores de limite de escoamento de 24,7MPa,
26,7MPa e 7,82MPa, sugerindo que as propriedades mecânicas dos materiais analisados não
sofrem grandes alterações devido ao tratamento de superfície.
Tabela 14: Propriedades mecânicas de monofilamento de PP, PET e PA-6
Amostra 𝑬 (MPa) 𝝈𝑬𝒔𝒄𝒐𝒂𝒎𝒆𝒏𝒕𝒐 (MPa) 𝝈𝑴á𝒙 𝑹𝒖𝒑(MPa) 𝜺𝑹𝒖𝒑 (%)
PP sem tratamento 1013,97 ± 58,73 454,49 ± 9,88 449,70 ± 9,02 50,12 ± 1,15
PP tratado por 120s 1035, 38 ± 71,90 429,82 ± 4,31 418,72 ± 20,15 47,65 ± 1,63
PET sem tratamento 2503,49 ± 48,38 779,37 ± 7,79 776,18 ± 7,96 49,55 ± 1,53
PET tratado por 120s 2339,42 ± 113,25 752,66 ± 22,19 739,64 ± 31,99 57,59 ± 4,02
PA-6 sem tratamento 930,11 ± 53,65 719, 29 ± 24,73 719, 29 ± 25,00 83,66 ± 5,28
PA-6 tratado por 120s 862,17 ± 148,85 727,12 ± 5,78 727,12 ± 5,78 79,41 ± 5,32
4.7 ABSORÇÃO DE ÁGUA
A Fig. 33 apresenta os resultados obtidos do teste de absorção de água para os
monofilamentos de PP, PET e PA-6 tratados por corona com distância inter-eletrodo de 4mm e
tempos de tratamento de 10, 60 e 120s. Amostras de PP, PET e PA-6 sem tratamento também
foram testadas e usadas como padrão de comparação.

73
Figura 33: Grau de absorção de água de amostras de PP, PET e PA-6 não tratadas e tratadas por
corona em 10s, 60s e 120s.
Nota-se que amostras de PP sem tratamento apresentaram o menor grau de absorção (0,92
± 0,14%) enquanto que amostras de PET e PA-6 não tratadas apresentaram respectivos valores de
absorção iguais a 2,90 ± 0,53% e 2,19 ± 0,41%. Observa-se aumento do grau de absorção de água
das amostras devido ao aumento do tempo de tratamento.
Amostras tratadas em 120s, apresentaram maiores graus de absorção que amostras
tratadas em 10s e 60s. Em 10s, as amostras de PP, PET e PA-6 mostraram graus de absorção
iguais a: 1,92%; 3,57% e 3,89%. Em 60s, os respectivos valores foram de: 2,1%; 7,7% e 7,6%. O
grau de absorção de água para amostras de PP, PET e PA-6 tratadas por corona durante 120s foi
de, respectivamente, 9,4%; 8,5% e 9,1%, respectivamente. Tais resultados mostram que amostras
de PP e PA-6 após tratamento por descarga corona apresentaram maior capacidade em absorver
água que amostras de PET.
Tais resultados estão de acordo as estimativas de ângulo de contato dinâmico
apresentadas, que mostram que a molhabilidade dos monofilamentos aumentam com o tempo de
tratamento. Os resultados mostram que amostras de PP apresentaram maior aumento da
capacidade de absorção de água, em seguida PA-6 e PET. A criação de zonas na superfície com
menor cristalinidade devido à fusão das amostras pela aplicação de descarga corona pode ser um
dos fatores que contribuem para que o grau de absorção de água das amostras após tratamento
0
2
4
6
8
10
0 40 80 120
Gra
u d
e a
bso
rção
(
%)
Tempo de tratamento (s)
PP - 4mm
PET - 4mm
PA-6 - 4mm

74
aumente consideravelmente (Leroux et. al. 2008). Devido ao tratamento corona, a formação de
trincas, rugosidade, protuberâncias e pequenas cavidades podem facilitar a difusão de água para o
interior dos monofilamentos. Tais defeitos podem contribuir também para que ocorra maior
ancoragem de moléculas de água na superfície, favorecendo a absorção.
4.8 CÁLCULO DA ENERGIA LIVRE DE SUPERFÍCIE
As Fig. 34, Fig. 35 e Fig. 36 apresentam os valores de ângulo de contato estático de
avanço, recuo e equilíbrio para amostras de PP tratadas por corona com 4mm de distância inter-
eletrodos, em função do tempo de tratamento. Os valores de ângulo de contato foram estimados
com água deionizada (líquido polar) e diiodo metano (líquido apolar). A fim de se verificar a
intensidade do tratamento devido ao tempo de exposição das amostras, foram estimados valores
de ângulo de contato para 180s e 240s.
Observa-se que os valores de ângulo de contato apresentam a mesma tendência
(diminuição do valor inicial) para os dois líquidos usados. Percebe-se que valores de ângulo de
contato para água são maiores que para o diiodo metano, indicando maior afinidade da superfície
das amostras de PP com o diiodo metano.
A partir da Fig. 34, verifica-se que os valores de ângulo de avanço sofrem ligeira queda
devido ao aumento do tempo de exposição das amostras ao tratamento corona. Os valores
medidos com água sofrem redução de 100,3° para 91,5° após 240s de tratamento. Valores
medidos com diiodo metano são reduzidos de 60,7° para 56,7°.
Pela Fig. 35, nota-se expressivas diminuições dos valores de ângulo de recuo quando
comparados aos ângulos de avanço medidos sob as mesmas condições. Em 240s, o valor de
ângulo de recúo medido com água diminui bruscamente de 78,4° (sem tratamento) para 34,2°
(em 240s), mostrando que o tempo de tratamento aumentou a hidrofilidade das amostras de PP.
Medidas de ângulo de recuo com diiodo metano mostram redução de 44,1° para 38,7°.

75
Figura 34: Ângulos de avanço em função do tempo de tratamento para amostras de PP.
Figura 35: Ângulos de recuo em função do tempo de tratamento para amostras de PP.
A Fig. 36 apresenta os valores de ângulo de equilíbrio em função do tempo de tratamento.
Percebe-se que os valores de ângulo de equilíbrio para água diminuem de 89,4° para 62,9° após
240s de tratamento. Usando diiodo metano, os valores de ângulo de equilíbrio se alteram de 52,4°
para 47,7°.
50
60
70
80
90
100
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PP - DiiodoMetano/Av
PP - Agua/Av
30
40
50
60
70
80
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PP - DiiodoMetano/Ret
PP - Agua/Ret
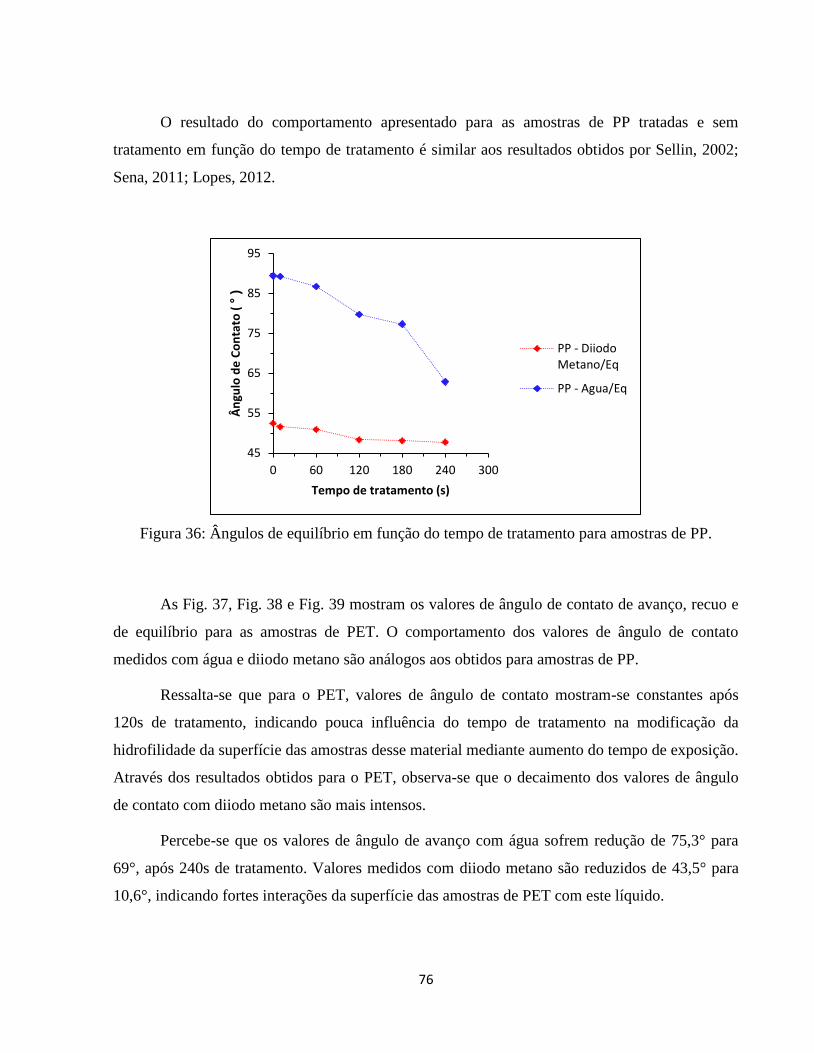
76
O resultado do comportamento apresentado para as amostras de PP tratadas e sem
tratamento em função do tempo de tratamento é similar aos resultados obtidos por Sellin, 2002;
Sena, 2011; Lopes, 2012.
Figura 36: Ângulos de equilíbrio em função do tempo de tratamento para amostras de PP.
As Fig. 37, Fig. 38 e Fig. 39 mostram os valores de ângulo de contato de avanço, recuo e
de equilíbrio para as amostras de PET. O comportamento dos valores de ângulo de contato
medidos com água e diiodo metano são análogos aos obtidos para amostras de PP.
Ressalta-se que para o PET, valores de ângulo de contato mostram-se constantes após
120s de tratamento, indicando pouca influência do tempo de tratamento na modificação da
hidrofilidade da superfície das amostras desse material mediante aumento do tempo de exposição.
Através dos resultados obtidos para o PET, observa-se que o decaimento dos valores de ângulo
de contato com diiodo metano são mais intensos.
Percebe-se que os valores de ângulo de avanço com água sofrem redução de 75,3° para
69°, após 240s de tratamento. Valores medidos com diiodo metano são reduzidos de 43,5° para
10,6°, indicando fortes interações da superfície das amostras de PET com este líquido.
45
55
65
75
85
95
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PP - DiiodoMetano/Eq
PP - Agua/Eq
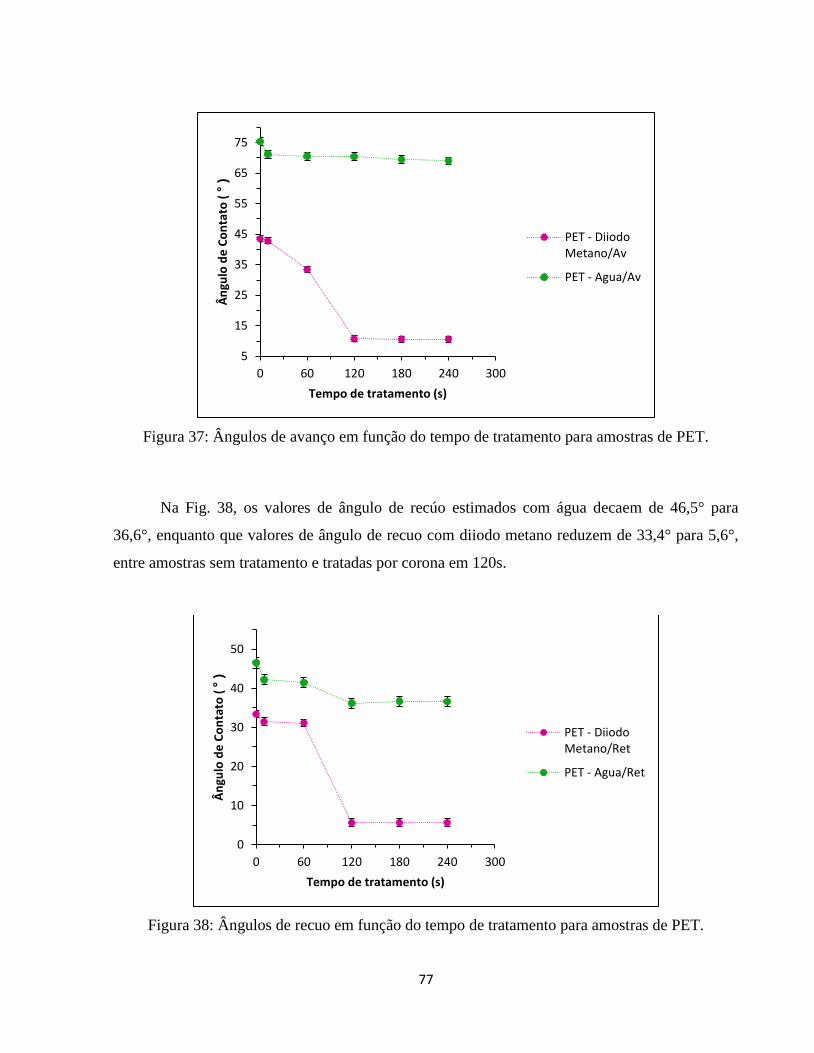
77
Figura 37: Ângulos de avanço em função do tempo de tratamento para amostras de PET.
Na Fig. 38, os valores de ângulo de recúo estimados com água decaem de 46,5° para
36,6°, enquanto que valores de ângulo de recuo com diiodo metano reduzem de 33,4° para 5,6°,
entre amostras sem tratamento e tratadas por corona em 120s.
Figura 38: Ângulos de recuo em função do tempo de tratamento para amostras de PET.
5
15
25
35
45
55
65
75
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PET - DiiodoMetano/Av
PET - Agua/Av
0
10
20
30
40
50
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PET - DiiodoMetano/Ret
PET - Agua/Ret

78
A partir da Fig. 39 verifica-se que os valores de ângulo de equilíbrio em função do tempo
de tratamento para água diminuem de 89,4° para 62,9° após 240s de tratamento. Usando-se
diiodo metano, os valores de ângulo de equilíbrio se alteram de 52,4° para 47,7°. O
comportamento apresentado pelos valores de ângulo de contato devido à exposição dos
monofilamentos à descarga corona é similar aos resultados obtidos por Briggs et. al., 1980.
Figura 39: Ângulos de equilíbrio em função do tempo de tratamento para amostras de PET.
As Fig. 40, Fig. 41 e Fig. 42 revelam o comportamento dos valores de ângulo de contato
de avanço, recuo e de equilíbrio para as amostras de PA-6. O comportamento dos valores de
ângulo de contato medidos com água e diiodo metano são análogos aos obtidos para amostras de
PP e PET. Não se nota resultados expressivos na diminuição dos valores de ângulo de contato
medidos devido ao aumento do tempo de tratamento das amostras de PA-6.
Nesse caso, apesar de amostras de PA-6 sem tratamento apresentarem caráter hidrofílico,
verificou-se que após 10s os valores sofrem uma leve queda e que após 120s há pouco
decréscimo nos valores de ângulo com água e diiodo metano.
5
15
25
35
45
55
65
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PET - DiiodoMetano/Eq
PET - Agua/Eq
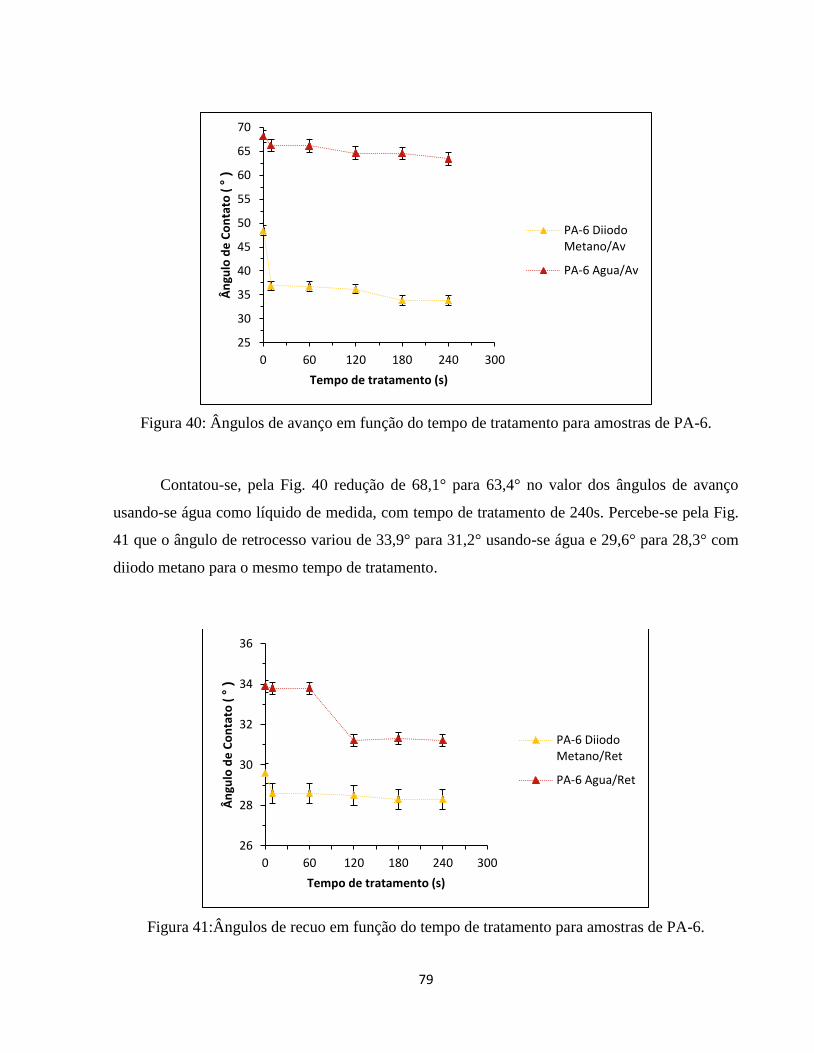
79
Figura 40: Ângulos de avanço em função do tempo de tratamento para amostras de PA-6.
Contatou-se, pela Fig. 40 redução de 68,1° para 63,4° no valor dos ângulos de avanço
usando-se água como líquido de medida, com tempo de tratamento de 240s. Percebe-se pela Fig.
41 que o ângulo de retrocesso variou de 33,9° para 31,2° usando-se água e 29,6° para 28,3° com
diiodo metano para o mesmo tempo de tratamento.
Figura 41:Ângulos de recuo em função do tempo de tratamento para amostras de PA-6.
25
30
35
40
45
50
55
60
65
70
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PA-6 DiiodoMetano/Av
PA-6 Agua/Av
26
28
30
32
34
36
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PA-6 DiiodoMetano/Ret
PA-6 Agua/Ret
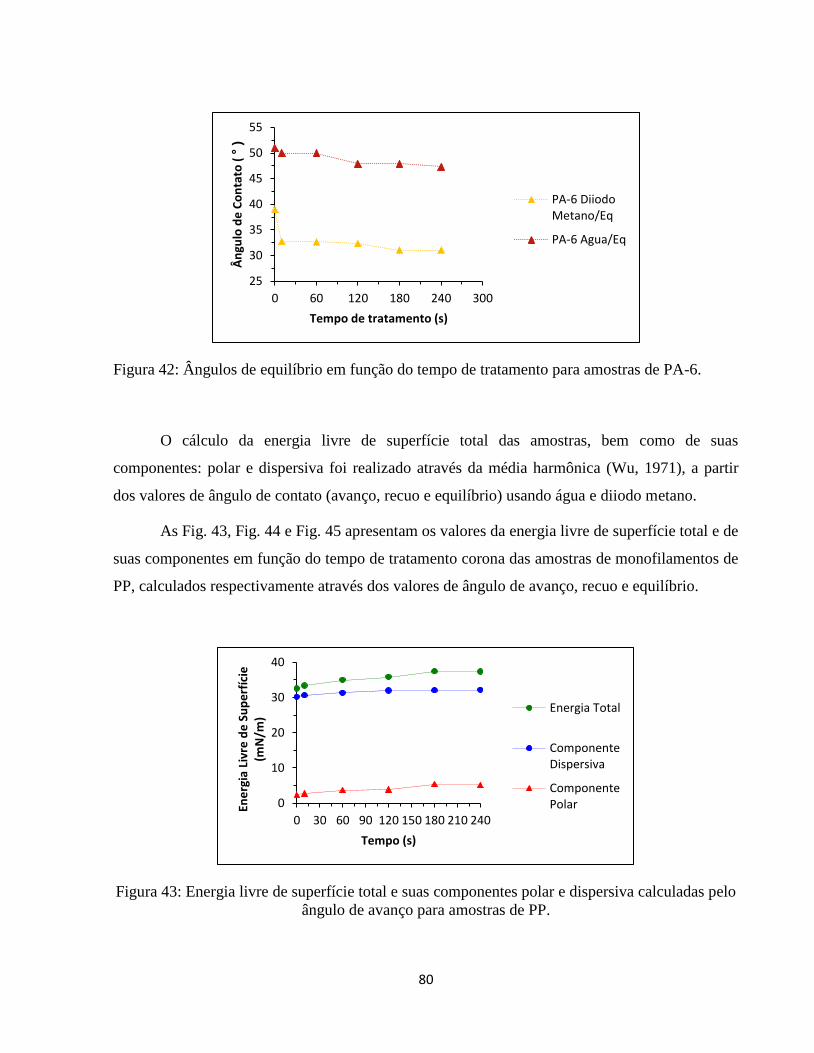
80
Figura 42: Ângulos de equilíbrio em função do tempo de tratamento para amostras de PA-6.
O cálculo da energia livre de superfície total das amostras, bem como de suas
componentes: polar e dispersiva foi realizado através da média harmônica (Wu, 1971), a partir
dos valores de ângulo de contato (avanço, recuo e equilíbrio) usando água e diiodo metano.
As Fig. 43, Fig. 44 e Fig. 45 apresentam os valores da energia livre de superfície total e de
suas componentes em função do tempo de tratamento corona das amostras de monofilamentos de
PP, calculados respectivamente através dos valores de ângulo de avanço, recuo e equilíbrio.
Figura 43: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de avanço para amostras de PP.
25
30
35
40
45
50
55
0 60 120 180 240 300
Ân
gulo
de
Co
nta
to (
° )
Tempo de tratamento (s)
PA-6 DiiodoMetano/Eq
PA-6 Agua/Eq
0
10
20
30
40
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
ComponentePolar

81
Nota-se que o aumento da energia livre de superfície devido ao tempo de tratamento
corona corresponde às estimativas de ângulo de contato. Verifica-se que há um claro aumento na
componente polar das amostras PP, com razoável acréscimo da componente dispersiva, devido ao
aumento do tempo de tratamento das amostras.
Figura 44: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de recuo para amostras de PP.
Figura 45: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de equilíbrio para amostras de PP.
0
10
20
30
40
50
60
70
80
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
ComponentePolar
0
10
20
30
40
50
60
70
0 30 60 90 120 150 180 210 240Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
Componente Polar

82
Observa-se respectivos aumentos da energia de superfície das amostras de PP calculadas
pelos ângulos de avanço, recuo e equilíbrio de 32,5mN/m para 37,3mN/m, 47,7mN/m para
71,1mN/m e 39,9mN/m para 53,7mN/m após 240s de tratamento. Tais valores obtidos após
tratamento por descarga corona encontram-se de acordo com trabalhos realizados por Sellin,
2002; Sena, 2011 e Lopes, 2012, em que encontraram valores de energia de superfície total ou
superior a 40mN/m para superfície de filmes de PP.
A partir das Fig.46, Fig. 47 e Fig. 48 observa-se o comportamento da energia livre de
superfície para o PET, calculados pelos valores de ângulo de avanço, recuo e equilíbrio. Percebe-
se que devido ao tempo de tratamento corona, a energia livre de superfície total se eleva.
Verifica-se que após 120s, os valores da energia de superfície e de suas componentes tornam-se
constantes, indicando possível estabilidade adquirida na superfície nestas condições (4 mm;
120s).
Figura 46: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de avanço para amostras de PET.
No caso das amostras de PET, houve um aumento expressivo da componente dispersiva
devido o aumento do tempo de tratamento das amostras, indicando maior afinidade da superfície
do PET pelo diiodo metano. Percebe-se um leve aumento da componente polar indicando
aumento da hidrofilidade da superfície das amostras de PET.
0
10
20
30
40
50
60
70
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
Componente Polar
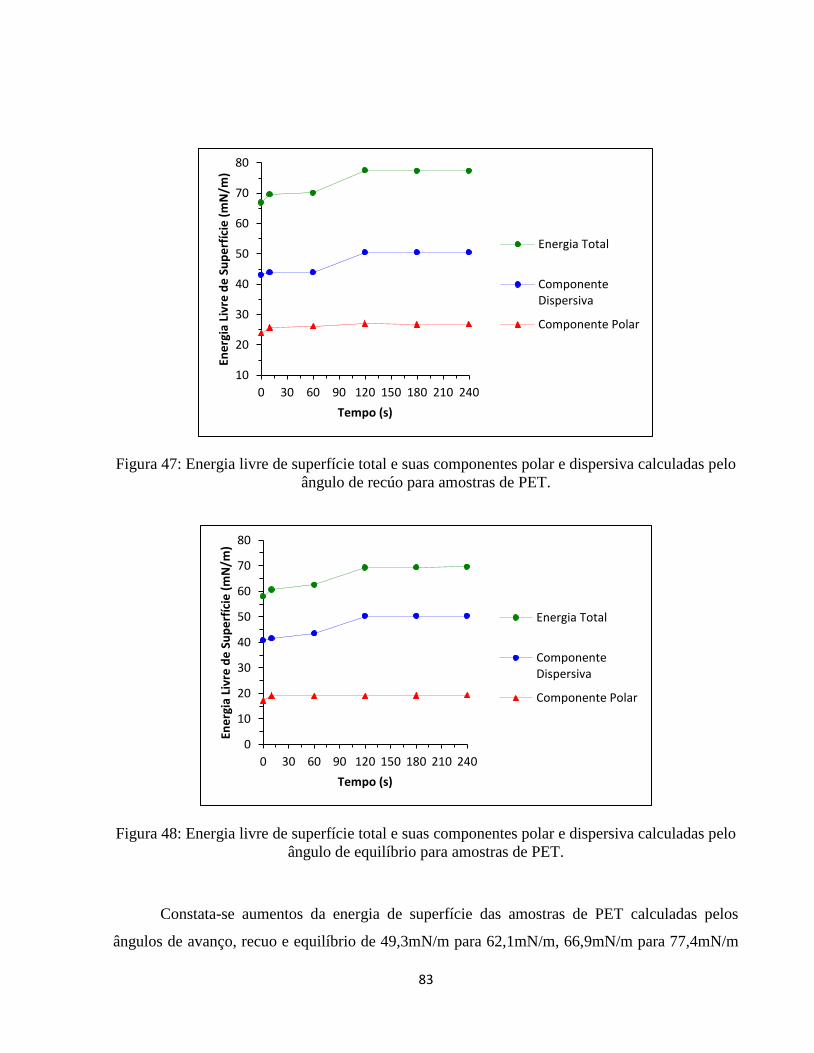
83
Figura 47: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de recúo para amostras de PET.
Figura 48: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de equilíbrio para amostras de PET.
Constata-se aumentos da energia de superfície das amostras de PET calculadas pelos
ângulos de avanço, recuo e equilíbrio de 49,3mN/m para 62,1mN/m, 66,9mN/m para 77,4mN/m
10
20
30
40
50
60
70
80
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
Componente Polar
0
10
20
30
40
50
60
70
80
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
Componente Polar

84
e 57,9mN/m para 69,6mN/m após 120s de tratamento. Nestas condições de tratamento, a
descarga corona possibilitou aumento da energia de superfície das amostras de PET,
possibilitando a melhoria de suas propriedades hidrofílicas e adesivas.
Nas Fig. 49, Fig. 50 e Fig. 51 as variações de energia de superfície das amostras de
monofilamentos de PA-6 com o tempo de tratamento corona. Amostras não tratadas de PA-6
apresentam alta energia livre de superfície (50mN/m a 74mN/m). Entretanto, o tratamento corona
não ofereceu variações significativas destes valores mesmo em tempos de tratamento mais
prolongados. Percebe-se que após 10s de tratamento os valores de energia de superfície
aumentam e tornam-se constantes, na faixa de tempo de tratamento estudada.
Figura 49: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de avanço para amostras de PA-6.
0
10
20
30
40
50
60
70
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
ComponentePolar
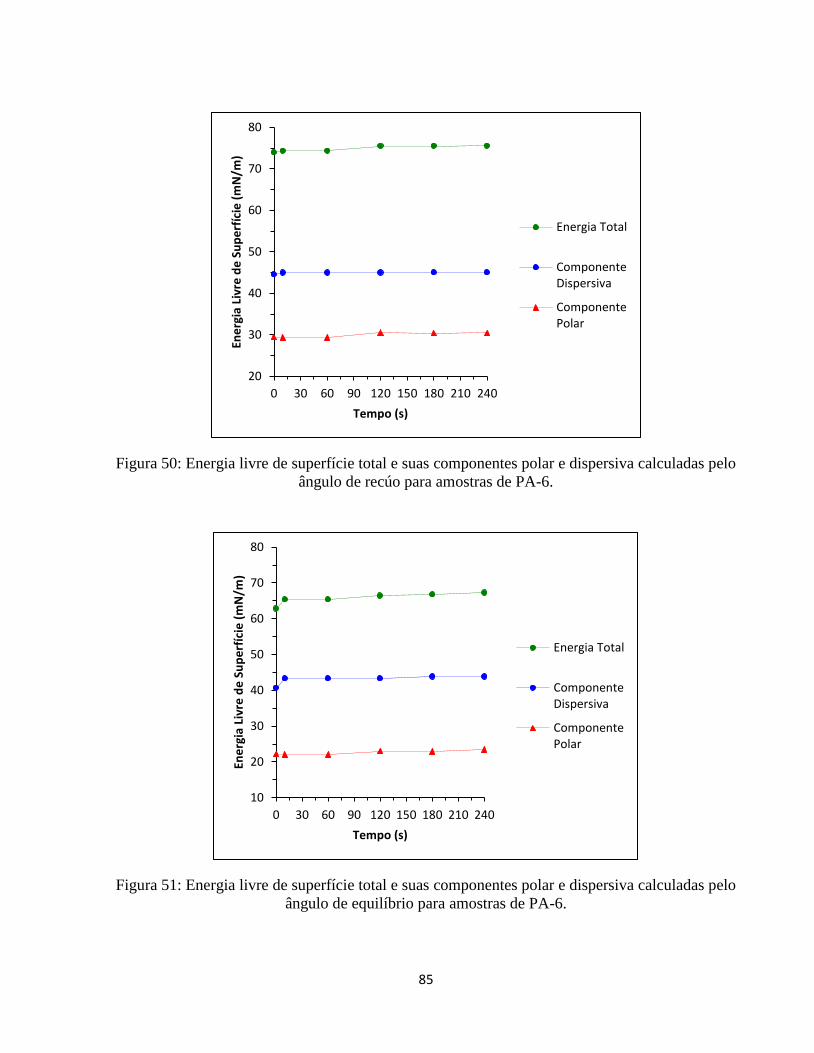
85
Figura 50: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de recúo para amostras de PA-6.
Figura 51: Energia livre de superfície total e suas componentes polar e dispersiva calculadas pelo
ângulo de equilíbrio para amostras de PA-6.
20
30
40
50
60
70
80
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
ComponentePolar
10
20
30
40
50
60
70
80
0 30 60 90 120 150 180 210 240
Ene
rgia
Liv
re d
e S
up
erf
ície
(m
N/m
)
Tempo (s)
Energia Total
ComponenteDispersiva
ComponentePolar

86
Pelos resultados verificou-se que o tratamento por descarga corona aumentou a energia
livre de superfície total das amostras de monofilamentos de PP, PET e PA-6. Dentre os polímeros
estudados, PP e PET apresentaram altos valores da energia de superfície, com aumento de até
50,3% e 26% de seus valores iniciais respectivamente. Amostras de PA-6 obtiveram aumento de
até 15% do valor de energia de superfície. Isto sugere que grupos polares foram introduzidos na
superfície das amostras estudadas, concordando com os resultados obtidos por FT-IR/ATR
mostrados anteriormente.
Sellin (2002) mostrou que propriedades de adesão da superfície de filmes de PP
aumentam com o aumento do tempo de tratamento corona, tendo em vista a formação de grupos
polares contendo oxigênio como hidroxilas (-OH) e, principalmente, na forma de carbonila
(C=O), promovendo adesão, aumentando a molhabilidade e energia livre de superfície total das
amostras estudadas e de suas componentes polar e dispersiva. A autora concluiu também que o
tratamento por descarga corona aumenta a rugosidade dos polímeros devido a formação de novas
estruturas de superfície, com aumento da força de adesão.
Briggs (1980) sugerem que a descarga corona aplicadas em filmes de PET induz a
formação de grupos fenólicos (OH) e ácidos carboxílicos (COOH) na superfície. Os resultados
obtidos mostraram aumento da molhabilidade, via medidas de ângulo de contato de amostras,
com aumento do tempo de tratamento. Além disso, os autores verificaram que a desão ocorre via
ligação de hidrogênio dos grupos fenólicos de filmes tratados com grupos carbonilas (C=O) dos
ácidos carboxílicos introduzidos na superfície.
As análises de FT-IR/ATR para o PA-6 obtidas neste trabalho mostraram a intensificação
de picos para grupos N-H, C=O, sugerindo a introdução de grupos oxigenados e nitrogenados na
superfície desses materiais, o que justifica o aumento da molhabilidade e energia livre na
superfície das amostras.
A baixa energia livre de superfície encontrada para os monofilamentos estudados não
tratados por corona é resultado de forças intermoleculares de coesão/ dispersão (apolar) atuantes
na maioria dos polímeros, fazendo com que sejam pouco aderentes. O aumento da energia livre
de superfície a partir de tratamentos de superfície está relacionado ao aumento da componente
polar devido à introdução de grupos polares na superfície dos materiais (Lopes, 2012).

87
No entanto, sabe-se que o aumento da energia livre de superfície em polímeros, devido a
tratamentos de superfície, não se relaciona somente com o aumento da componente polar, mas
também com um aumento na componente de dispersão (Sellin, 2002).
Dos resultados obtidos anteriormente: ângulo de contato, histerese, FT-IR/ATR e energia
livre de superfície, observa-se que o aumento da componente polar da energia livre de superfície
total das amostras de monofilamentos de PP, PET e PA-6 estão provavelmente relacionados à
introdução de grupos químicos (oxigenados e nitrogenados) na superfície após tratamento por
descarga corona. A componente de dispersão pode estar associada à caracterísiticas topográficas
(Sellin, 2002), tendo em vista seu aumento em função do tempo de tratamento, o que contribuiu
para o aumento da energia de superfície total dos monofilamentos estudados.

88
5 CONCLUSÕES E SUGESTÕES PARA TRABALHOS FUTUROS
5.1 CONCLUSÕES
De acordo com os resultados obtidos neste trabalho referentes ao estudo do efeito do
tratamento corona aplicado na superfície de monofilamentos de polipropileno (PP), politereftalato
de etileno (PET) e poliamida-6 (PA-6) conclui-se que o sistema corona empregado apresenta
configuração e qualidade funcional adequados à realização do tratamento de superfície das
amostras.
Durante o tratamento das amostras, observou-se que as distâncias entre os eletrodos e o
tempo de tratamento influenciam na modificação da superfície das amostras, tendo em vista a
diminuição dos valores de ângulo de contato em função do aumento do tempo de tratamento (10
a 120s) e da diminuição da distância entre os eletrodos fio-plano (10 a 4mm) no sistema corona.
Os valores de ângulo de contato dinâmico foram reduzidos em até 29,6%, 27,9% e 18,6% para
amostras de PP, PET e PA-6 tratadas com corona, durante 120s a uma distância inter-eletrodos
igual a 4mm, respectivamente.
Dentre os polímeros estudados, PP e PET apresentaram altos valores da energia de
superfície, com aumento de aproximadamente 50% e 26% de seus valores iniciais
respectivamente. Amostras de PA-6 obtiveram aumento de até 15% do valor de energia de
superfície, devido à introdução de grupos polares e mudanças na topografia na superfície das
amostras que elevam a componente polar e dispersiva da energia total de superfície. Amostras de
PP, PET e PA-6, tratadas por corona, durante 240s (4mm), alcançaram valores de energia livre de
superfície de 71,1mN/m, 77,4mN/m e 75mN/m, indicando melhoria das propriedades adesivas
desses materiais.
O tratamento por descarga corona não altera propriedades térmicas e mecânicas dos
polímeros. Entretanto, o tratamento promoveu aumento no grau de absorção de água para
amostras de PP, PET e PA-6 tratadas por corona em 120s, de 9,4%, em seguida PA-6, com 9,1%,
e PET, igual a 8,5%.

89
Portanto, a técnica de modificação da superfície por descarga corona usada neste trabalho
enquadra-se como uma ferramenta capaz de aumentar propriedades hidrofílicas e de adesão de
monofilamentos, sem alterar as propriedades de volume destes polímeros, tornando-os aptos a
aplicações para impressão, tingimento e união com adesivos.
Além disso, os monofilamentos podem vir a ser aplicados na área da saúde como
materiais biomédicos implantáveis, como fios de sutura, pois é conhecido que materiais
implantáveis precisam ser resistentes a tração, terem boa adesão e boa molhabilidade.
5.2 SUGESTÕES PARA TRABALHOS FUTUROS
Estudar a influência de outros parâmetros de processo por descarga corona como: tensão
aplicada, umidade relativa do ar e outras configurações operacionais para encontrar
condições adequadas de operação que promovam mudanças na superfície evitando a
geração de danos dos materiais a serem tratados;
Estudar a duração do efeito do tratamento corona e condições de armazenamento dos
monofilamentos;
Realizar testes de impressão, tingimento, revestimento e até mesmo metalização dos
monofilamentos de PP, PET e PA-6, antes e após o tratamento por descarga corona;
Realizar medidas de ângulo de contato e testes de absorção de água à temperatura do
corpo humano, usando também outros líquidos, por exemplo, soro fisiológico uma vez
que os monofilamentos possam vir a ser implantados como fios de sutura;
Vários autores demonstraram que os materiais usados neste trabalho já foram testados in
vitro e in vivo, obtendo-se bons resultados quanto ao implante. No entanto, para garantia
de sua aplicabilidade como biomaterial, os monofilamentos tratados por corona devem ser
testados para analisar o comportamento do material e aceitação do corpo no meio
fisiológico.
Utilizar outras técnicas para identificação de grupos químicos na superfície, bem como
sua quantificação.

90
6 REFERÊNCIAS
AMERICAN SOCIETY FOR TESTING AND MATERIALS. D123 – 03: Standard
Terminology Relating to Textiles. West Conshohocken, 2006. 10p.
AMERICAN SOCIETY FOR TESTING AND MATERIALS. D2256/D2256M - 10ε1
: Standard
test method for tensile properties of yarns by the single method. West Conshohocken, 2010.
13p.
AMERICAN SOCIETY FOR TESTING AND MATERIALS. D3418 - 12 ε1
: Standard test
method for transition temperatures and enthalpies of fusion and crystallization of polymers by
differential scanning calorimetry. West Conshohocken, 2012. 7p.
ARAUJO, MÁRIO de CASTRO, E. M. de Melo. Manual de Engenharia Têxtil. V. 2. Fundação
Cauloste Gulbenkia. Lisboa. Portugal, 1986. 948 P.
AKISHEV, Y.; GRUSHIN, M.; KOCHETOV, I.; KARAL’NIK, V.; NAPARTTOVICH, A.;
TRUSHKIN, N. Negative corona, glow and spark discharges in ambient air and trasitions
between therm. Plasma Sources Science and Technology. v.14, p.S18 – S25, 2005.
ALKOR CRYSTAL OPTICS. Disponível em: < http:// http://www.alkor.net/
ZnSe_ATR_Prisms.html>. Acesso em 08 de março de 2014.
ALVES, G. P. Recobrimento de superíficie de PVC com filmes de Lagmuir-Blodgett usando
fosfatidiletanolamina. 1999. 114p. Dissertação (Mestrado em Engenharia Química). Faculdade
de Engenharia Química, Universidade Estadual de Campinas, Campinas, 1999.
ALVES, O. L. Disponível em: < http: // www.lqes.iqm.unicamp.br/images/vivencia_lqes_
meprotec_espec_fourier.pdf.html >. Acesso em: 02 de março de 2014.

91
ANTAO, D. S.; STAACK, D.A.; FRIDMAN, A.; FAROUK, B. Atmospheric pressure dc corona
discharges: operating regimes and potential applications. Plasma Sources Science and
Technology. v.18, p. 035016 (11 p.), 2009.
AWAJA, F.; GILBERT, M.; KELLY, G.; FOX, B.; PIGRAM, P. J. Adhesion of polymers.
Progress in Polymer Science. v.34, p.948 – 968, 2009.
AWAJA F.; PAVEL, D. Recycling of PET. European Polymer Journal. v.41, p.1453–1477,
2005
BARBOSA, L. C. A. Espectroscopia no infravermelho na caracterização de compostos
orgânicos. Viçosa: Editora UFV, 2007, 189p.
BRAME, JR, E. G.; GRASSELI, J. G. Infrared and Raman Spectroscopy: Part C, New York:
Ed. Marcel Dekker, Inc. p. 718 – 1039, 1977.
BRIGGS, D.; RANCE, D. G.; KENDALL, C. R.; BLYTHE A. R. Surface modification of
poly(ethylene terephthalate) by electrical discharge treatment. Polymer. v.21, issue 8, p. 895 –
900, 1980.
CANEVAROLO, S. V. Técnicas de caracterização de polímeros. São Paulo: Artliber, 2003,
448p.
CARNEIRO, M. P. Caracterização das propriedades de superfície de filmes de polipropileno
tratados com descarga corona. 2001, 76p. Dissertação (Mestrado em Engenharia Química).
Faculdade de Engenharia Química, Universidade Estadual de Campinas, 2001.
CHIBOWSKI, E.; TERPILOWSKI, K.. Surface free energy of polypropylene and polycarbonate
solidifying at different solid surfaces. Applied Surface Science., v. 256, p. 1573-1581, 2009.

92
COELHO,A. F. P. Desenvolvimento de nanocompósitos de nylon 6em um reator de bancada
em batelada: polimerização in situ em unidade experimental. 2012. 176p. Dissertação
(Mestrado em Engenharia Química). Faculdade de Engenharia Química, Universidade Estadual
de Campinas, Campinas, 2012.
COLTHUP, N. B.; DALY, L. H.; WIBERLEY, S. E. Introduction to infrared and raman
spectroscopy. 4°ed. Ed. Academic Press. 1964, 511p.
COSTA, A. C. Modificação de superfície de polietileno de alta e de baixa densidade. 1982, 93p.
Dissertação (Mestrado em Química). Instituto de Química, Universidade Estadual de Campinas,
1982.
CUCA, J. Espectroscopia de absorção de IV por transformada de Fourier. Revista Química e
Derivados. E. QD. Edição 496, abril, 2010.
DELLA VOLPE, C.; DEIMICHEI A.; RICCÓ T. A multiliquid approach to the surfasse free
energy determination of flame-treated surfaces of rubber-toughened polypropylene. Journal of
adhesion science and technology. v.12, p.1141-1180, 1998.
DEOPURA, B. L. Polyamide fibers. In: DEOPURA, B. L.; ALAGIRUSAMY R.; JOSHO M.;
GUPTA B. Poliesters and Polyamides. Ed.: Woodhead Publishing Limited, 2008. p. 41 – 60.
EVORA M.; GONÇALEZ O. L.; DUTRA R. C. L.; DINIZ M. F.; WIEBECK H.; SILVA, L. G.
A. Comparação de técnicas FTIR de transmissão, reflexão e fotoacústica na análise de poliamida
6, reciclada e irradiada. Polímeros: Ciência e Tecnologia. v.12, p.50-68, 2002
GAO, W.; MA X.; LIU Y.; WANG Z.; ZHU Y. Effect of calcium carbonate on PET physical
properties and termal stability. Powder Technology. v.244, p.45-51, 2013.
GODDARD J. M.; HOTCHIKISS, J. H. Polymer surface modification for the attachment of
bioactive compounds. Progress in polymer science. v.32, p.698 – 725, 2007.

93
GILES, H. F.; WAGNER, J. R.; MOUNT, E. M. Monofilament. In: GILES, H. F. Extrusion: the
definitive processing guide and handbook. Ed:William Andrew Publishing, 2005, p. 497 – 504.
GIORDANO, J. B. Tratamento corona sobre superfícies têxteis. 2007, 138p. Tese (Doutorado
em Engenharia Química). Faculdade de Engenharia Química, Universidade Estadual de
Campinas, 2007.
GOTOH, K. Wettability and surfasse free energies of polymeric materials exposed to excimer
ultraviolet light and particle deposition onto their surfaces in water. In: MITTAL, K. L. Polymer
surface modification: relevance to adhesion. Ed: VSP, 2004, p. 125-137.
GUPTA, B. S. Manufacture, types and properties of biotextiles for medical applications. In:
KING, M. W; GUPTA B. S; GUIDOIN R. Biotextiles as medical implants. Ed: Woodhead
Publishin Limited, 2013, p. 3 – 47.
HE, W.; BENSON, R. Polymeric Biomaterials. In: EBNESAJJAD S. Handbook of biopolymers
and biodegradable plastics: properties, processing and applications. Ed. Elsevier, 2011, p. 87 –
107.
HOLLER, F. J.; SKOOG, D. A.; CROUCH, S.R. Princípios de análise instrumental. 6° edição,
2009. 1055 p.
HÖNE, G. W. H.; HEMMINGER W.; FLAMMERSHEIM H. –J. Differential scanning
calorimetry: an introduction for practitioners. New York: Springer, 1996
HUANG F.; WEI Q.; WANG X.; XU W. Dynamic contact angles and morphology of PP fibres
treated with plasma. Polymer Testing. v.25, p.22 – 27, 2006.
JOHN, M. J.; ANANDJIWALA R. D. Surface modification and preparation techniques for textile
materials. In: WEI,Q. Surface modification of textiles. Ed: Woodhead Publishing Limited,
2009. p. 1 – 25.

94
KAMATH, M. G.; BATH G. S. Specialty fibers from polyester and polyamides. In: DEOPURA,
B. L.; ALAGIRUSAMY R.; JOSHO M.; GUPTA B. Poliesters and Polyamides. Ed.: Woodhead
Publishing Limited, 2008. p. 203 – 218.
KLEE, D.; ADEMOVIC, Z.; BOSSERHOFF, A.; HOECKER, H.; MAZIOLIS, G.; ERLI, H.
Surface modification of poly(vinylidenefluoride) to improve the osteoblast adhesion.
Biomaterials. v.24, p. 3663 – 3670, 2003.
KOTHARI, V. K. Polyester and polyamide fibres – appareal applications. In: DEOPURA, B. L.;
ALAGIRUSAMY R.; JOSHO M.; GUPTA B. Poliesters and Polyamides. Ed.: Woodhead
Publishing Limited, 2008. p. 419 – 440.
LABAY, C.; CANAL, J.M.; NAVARRO, A.; CANAL, C. Corona plasma modification of
poliamide 66 for the desing of textile delivery systems for cosmetic therapy. Applied Surface
Science. v. 316, p.251 – 258, 2014.
LAM, C. N. C.; KO, R. H. Y.; YU, L. M. Y.; LI, NG, A.; LI D.; HAIR, M. L.; NEUMANN, A.
W. Dynamic contact angle measurements: study of advancing and receding contact angles.
Journal of Colloid and Interface Science. v.243, p. 208 – 218, 2001.
LEE J. H.; KHANG, G.; LEE, J. W.; LEE H. B. Interaction of different types of cells on polymer
surfaces with wettability gradient. Journal of Colloid and Interface Science. v.205, p. 323 –
330, 1998.
LEROUX, F.; CAMPAGNE, C.; PERWUELZ, A.; GENGEMBRE, L. Polypropylene film
chemical and physical modifications by dielectric barrier discharge plasma treatment at
atmospheric pressure. Journal of Colloid and Interface Science., v.328, p.412–420, 2008.
LIU, X.; SHENG, D.; GAO, X.; LI, T.; YANG, Y. UV-assisted surface modification of PET
fiber for adhesion improvement. Applied Surface Science. v.264, p.61-69, 2013.

95
LONG J.; HYDER M. N.; HUANG R. Y. M.; CHEN P. Thermodynamic modeling of contact
angle on rough, heterogeneous surfaces. Advances in Colloid and Interface Surface Science.
v.118, p. 173 – 190, 2005.
LOPES, S. A. Estudo de propriedades de não tecidos de polipropileno tratados por descarga
corona e plasma de rádio frequência. 2012, 115p. Dissertação (Mestrado em Engenharia
Química). Faculdade de Engenharia Química, Universidade Estadual de Campinas, 2012.
LUCAS, E. F.; SOARES, B. G.; MONTEIRO E. E. C. Caracterização de polímeros:
determinação de peso molecular e análise térmica. Rio de Janeiro: E-papers, 2001, 366p.
MA, Z.; MAO, Z.; GAO, C. Surface modification and property analysis of biomedical polymers
used for tissue engineering. Colloids and Surfaces B: Biointerfaces. v.60, p.137 – 157, 2007.
MAGALHÃES, R. M. S. Desenvolvimento de poli(fluoreto de vinilideno) poroso na fase para
aplicações biomédicas. 2009, 75p. Dissertação (Mestrado em Física). Escola de Ciências,
Universidade do Minho, 2009.
MATOS, J. R. MACHADO, L. D. B. Análise Térmica Diferencial e calorimetria exploratória
diferencial. In: CANEVAROLO JR, S. V. Técnicas de Caracterização de Polímeros. São Paulo:
Artiliber, 2003, p.2209 – 261.
MARTINEZ. P. Química y física de los altos polímeros y matérias plásticas, Editorial
Alhambra, 1972, p.160-165
MAZZOLA, L.; BEMPORAD, E.; CARASSITI, F. Flame treatment on plastic: a new surface free
energy statistical prediction model and characterization of treated surfaces. Applied Surface Science,,
v. 257, p. 2148-2158, 2010.

96
MAIER, C.; CALAFUT, T. Fibers. In: MAIER, C.; CALAFUT, T. Polypropylene: the definitive
user’s guide and databook. Ed. William Andrews, 1998. p. 63 – 67.
NIHLSTRAND, A., HJERTBERG, T., JOHANSSON, K. Plasma treatment of polyolefins:
influence of material composition; 1. Bulk and Surface characterization. Polymer. v.38, n.14,
p.3581-3589, 1997.
PASCUAL M.; BALART R.; SÁNCHEZ L. Study of aging process of corona discharge plasma
effects on low density polyethylene film surface. Journal of Material Science. v.43, p. 4901 –
4909, 2008.
PAUL, S. Surface modification of polypropylene nonwowens to improve adhesion to
elastomers. 2009. 173p. PhD Thesis. Raleigh, NC: North Carolina State University, 2009.
SAÏHI, D.; EL-ACHARI A.; GHENAIM A.; CAZÉ C. Wettability of grafted poly(ethylene
terephtalate) fibers. Polymer Testing. v.21, p. 615 – 618, 2002.
SALMORIA, G. V.; MARTINS, W. F. M.; FUCIO, D. M. Tratamento da superfície de cateteres
de poliamida 11 por plasma de oxigênio. Polímeros: Ciência e Tecnologia. v.23, n. 4, p. 565 –
569, 2013.
SANTHANA, G. K.; KULKARNI, S. T. Polyesters resins. In: DEOPURA, B. L.;
ALAGIRUSAMY R.; JOSHO M.; GUPTA B. Poliesters and Polyamides. Ed: Woodhead
Publishing Limited, 2008. p. 3 – 40.
SELLIN, N. Análise da superfície de polímeros pós-tratamento corona. 2002, 112p. Tese
(Doutorado em Engenharia Química). Faculdade de Engenharia Química, Universidade Estadual
de Campinas, 2002.
SELLIN, N., CAMPOS, J. Surface composition analysis of PP films treated by corona discharge.
Material Research, v. 6, p. 163-166, 2003.

97
SENA, H. C. Efeitos da descarga corona em superfície de polipropileno em temperatura
ambiente e acima. 2011, 84p. Dissertação (Mestrado em Engenharia Química). Faculdade de
Engenharia Química, Universidade Estadual de Campinas, 2011.
SILVERSTEIN, R. M.; WEBSTER, F. X, KIEMLE D. Identificação espectrométrica de
compostos orgânicos. 7° edição, New York: LTC, 2007, p. 70 – 122.
SHERIDAN, P. L.; BUCKTON, G.; STOREY, D. E. The extent of errors associated with contact
angles II. Factors affecting data obtained using a Wilhelmy plate technique for powders.
International Journal of Pharmaceutics. v.109, p. 155-171.
SPIRIDINOV, P.; LAMBRINOS E.; PENG Z. Extrusion of monofilaments of thermoplastic
elastomers. Synthetic Metals. v. 152, p. 61-64, 2005.
TAYLOR, M. S.; SHALABY, S. W. Sutures. In: In: RATNER, B. D; HOFFMAN, A. S.;
SCHOEN, F. J.; LEMONS, J. E. Biomaterials Science – An Introduction to Materials in
Medicine. 3° edição, San Diego: Academic Press, 2013. p. 1010 – 1024.
TRAN, L. Q. N.; FUENTES, C. A.; DUPONT-GILLAIN, C.; VAN VURRE, A. W. Wetting
analysis and surface characterization of coir fiber used as reinforcement for composites. Colloids
and Surfaces A: Physicochemical and Engineering Aspects. v.377, p. 251 – 260, 2011.
WENDLANDT, W. W. Thermal analysis. NY: Chichester J. Wiley, 3º ed., 1986, 814 p.
WU, S. Calculation of Interfacial Tension in Polymer Systems. Journal of Polymer Science, v.
34, p. 19-30, 1971.
XU, W., LIU, X. Surface modification of polyester fabric by corona discharge irraditation.
European Polymer Journal, v. 39, p. 199-202, 2003.

98
XU, L.; FANG, Z.; SONG, P.; PENG, M. Effects of corona discharge on the surface structure,
morphology and properties of multi-walled carbon nanotubes. Applied Surface Science. v.256,
p.6447-6453, 2010.
YOUSFI, N.; LIVI S.; DUMAS A.; LE ROUX C.; CRÉPIN-LEBLOND J.; GREENHILL-
HOOPER, M.; DUCHET-RUMEAU, J. Use of new synthetic talc as reinforcing nanofillers for
polypropylene and polyamide 6 systems: thermal and mechanical properties. Journal of Colloid
and Interface Science. v.403, p.29-42, 2013.
ZAMAN M.; LIU, H.; XIAO, H.; CHIBANTE, F.; NI, Y. Hydrophilic modification of polyester
fabric by applying nanocrystalline cellulose containing surface finish. Carbohydrate Polymers. v.
91, p. 560-567, 2013.
ZISMAN, W.A. Relation of the equilibrium contact angle to liquid and solid constitution. In.
Advances in Chemistry Series 43, p. 1-51, 1964.