extracellular matrix proteins as drivers of inflammation ...20schi%f8ler%20schultz.pdf · mekanisme...
TRANSCRIPT

U N I V E R S I T Y O F C O P E N H A G E N
F A C U L T Y O F S C I E N C E
Industrial PhD Thesis
Heidi Schiøler Schultz
Extracellular matrix proteins as drivers of
inflammation in rheumatoid arthritis Thesis subtitle
Academic advisor: Martin W. Berchtold
Submitted: 13/03/2015

2
Extracellular matrix proteins as drivers of
inflammation in rheumatoid arthritis
Industrial PhD Thesis
Heidi Schiøler Schultz
Department of Biology, Section for Cell and Developmental Biology, Faculty of Science,
University of Copenhagen.
Copenhagen, Denmark
&
Department of Immuobiology, AID, Novo Nordisk A/S
Måløv, Denmark
2015
”It is good to have an end to journey towards, but it is the journey that matters in the end”
– Ursula K. Le Guin

3
Extracellular matrix proteins as drivers of inflammation in rheumatoid arthritis
The PhD project was financially supported by the Danish Ministry of Science, Technology and
Innovation and R&D Academic Relations, Novo Nordisk A/S
This thesis was submitted to the Graduate School of the Faculty of Science, University of
Copenhagen, March 2015
Principal supervisor
Professor Martin W. Berchtold
Department of Cell and Developmental Biology, Faculty of Science,
University of Copenhagen, Denmark
Co-supervisors
Principal Scientist, PhD, MD, Svetlana Panina
Department of Immunobiology
Novo Nordisk A/S, Måløv, Denmark
Senior Scientist, PhD Christine Brender Read
Department of Autoimmune Disease Research
Novo Nordisk A/S, Måløv, Denmark
Principal Scientist, PhD, Olle Björkdahl
Department of Immunogenicity Assessment
Novo Nordisk A/S, Måløv, Denmark
Assessment committee
Senior Research Fellow, PhD, Kalle Søderstrøm
NDORMS, Botnar Research Center
Oxford University, United Kingdom

4
Research Scientist, PhD, Anders Aspberg
Department of Clinical Sciences
Lund University, Lund, Sweden
Section Head, DSc, Niels Behrendt (chairman)
Finsen Laboratory, Rigshospitalet,
BRIC, University of Copenhagen, Denmark
Cover:
The drawing shows pathways of differentiation of monocyte-derived cells within the joints of
patients with rheumatoid arthritis. The drawings are made by S. Panina and kindly given to me.
PhD Thesis 2015 © Heidi Schiøler Schultz

5
List of publications
Papers included in this PhD Thesis
I. Heidi S. Schultz, Louise M. Nitze,, Louise H. Zeuthen, Pernille Keller, Albrecht Gruhler,
Jesper Pass, Jianhe Chen, Li Guo, Andrew J. Fleetwood, John A. Hamilton, Martin W.
Berchtold and Svetlana Panina. (2015) Collagen Induces Maturation of Human
Monocyte-Derived Dendritic Cells by Signaling through Osteoclast-Associated. J.
Immunology. 194 (epub, doi 10.4049/jimmunol.1402800)
II. Heidi S. Schultz, Li Guo, Pernille Keller, Andrew J. Fleetwood, Wei Guo, John A.
Hamilton, Olle Björkdahl, Martin W. Berchtold and Svetlana Panina. OSCAR-collagen
signaling mediates activation of monocytes. To be submitted to J. Immunology March,
2015.
Poster abstracts
III. Extracellular matrix proteins may control tissue-specific maturation of dendritic
cells: implications for therapy of Rheumatoid Arthritis. Heidi S. Schultz, Louise M.
Nitze, Louise H. Zeuthen, Pernille Keller, Li Guo and Svetlana Panina. Poster presented
at the PhD Day: Building research through dissemination, Department of Biology,
University of Copenhagen, 2013 Nov 1-14th
.
Relevant papers not included in the PhD
IV. Fleetwood, A. J., A. Achuthan, H. Schultz, A. Nansen, K. Almholt, P. Usher, and J. A.
Hamilton. (2014). Urokinase plasminogen activator is a central regulator of
macrophage three-dimensional invasion, matrix degradation, and adhesion. J.
Immunology. 192: 3540-3547.

6
Abstract
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease that primarily affects the
peripheral diarthroidal joints. The joint inflammation in RA is characterized by hyperplasia of the
synovium and pannus formation as well as by the presence of pro-inflammatory cytokines and
chemokines which causes a massive influx of immune cells into the synovial tissue (ST). A
hallmark of RA pathogenesis is the enhanced formation and activation of osteoclasts (OC). The
progressive inflammation leads to remodeling of the extracellular matrix (ECM), and ultimately
results in destruction of cartilage and bone in the joint. RA can to some degree be suppressed by
non-biologics disease-modifying anti-rheumatic drugs (nbDMARDS) or biologics (bDMARDS)
such as e.g. TNF-α inhibitors. However, the efficacy of the current drugs is limited as they are only
suppressing inflammation, but are not able to halt joint destruction completely. Identification of
factors that regulate inflammation and bone/cartilage loss is crucial for development of a new
generation of drugs to treat RA.
The OSteoClast-Associated Receptor (OSCAR) was proposed as a new target for
treatment of RA. OSCAR has been reported to be up-regulated in the synovium of RA patients and
on the circulating monocytes where its expression level has been shown to positively correlate with
the disease activity. In mice, OSCAR expression is limited to OC. It was proposed that OSCAR
provides a co-stimulatory signaling in RANK-dependent osteoclastogenesis. In humans, OSCAR is
expressed in OC, DCs, monocytes, macrophages and neutrophils. We and others have identified the
ligands of OSCAR to be collagen type I (ColI) and –type II (ColII). In RA, excessive degradation of
ECM by enzymes secreted by activated immune cells, fibroblasts and OC leads to collagens
exposure and makes them available for interaction with the immune cells. Thus, we hypothesize
that at such pathological conditions ColI/II serve as naturally occurring activators of the OSCAR
signaling.
In the present study, we have investigated the functional outcome of the OSCAR-
collagen interaction in human myeloid cells. We have observed that OSCAR engagement by ColI/II
induced activation and maturation of DCs characterized by up-regulation of cell surface markers
and secretion of pro-inflammatory cytokines. These collagen-matured DCs (Col-DCs) were
efficient drivers of naïve T cell proliferation. The T cells expanded by Col-DCs secreted cytokines
with no clear T-cell polarization pattern. Global RNA profiling revealed that multiple pro-
inflammatory mediators, including cytokines and cytokine receptors, components of the stable

7
immune synapse (namely, CD40, CD86, CD80, ICAM-1), as well as components of TNF- and TLR
signaling, are transcriptional targets of OSCAR in DCs.
The functional role of the OSCAR-collagen interaction was further addressed in
monocytes and macrophages. We have shown that the OSCAR-ColII signaling supports survival of
monocytes under conditions of growth factor withdrawal. Moreover, ColII stimulated release of
pro-inflammatory cytokines by monocytes from healthy donors, and this release could be
completely blocked by an antagonistic anti-OSCAR mAb. Similarly, mononuclear cells from
synovial fluid of RA patients cultured on ColII secreted TNF-α and IL-8 in an OSCAR-dependent
manner. Global profiling of gene expression in monocytes showed that components of multiple
signaling pathways relevant for the RA pathogenesis are transcriptional targets of OSCAR.
Surprisingly, OSCAR engagement by ColII in macrophages, which were differentiated in vitro from
monocytes of healthy donors, did not lead to secretion of cytokines. Microarray analysis did not
reveal any gene regulation by the OSCAR-ColII signaling in macrophages. However, degradation
of collagen-rich matrix, gelatin, by macrophages was significantly abolished by an antagonistic
anti-OSCAR mAb, indicating that OSCAR modulates the activity of proteolytic enzymes on a post-
transcriptional level.
Taken together, our findings indicate the existence of a novel pathway mediating a
sustained inflammation in the RA joints where collagens become exposed during tissue remodeling
and can thus activate the OSCAR signaling in monocyte-derived cells. We hypothesize that the
OSCAR-collagen pathway can potentially contribute to the RA pathogenesis on multiple levels and
may thus represents a new target for therapeutic intervention.

8
Sammendrag (Danish summary)
Kronisk leddegigt (reumatoid artrit) er en systemisk, autoimmun sygdom, der hovedsageligt
påvirker de perifere led. Karakteristisk for betændelsestilstanden i leddegigt er en øget celledeling
af synovialvævet og tilstedeværelsen af pro-inflammatoriske cytokiner og chemokiner, som øger
tilstrømning af immunceller til synovialleddet. Et af kendetegnene ved leddegigt er den øgede
dannelse og aktivering af de knoglenedbrydende osteoklaster. Den progressive betændelsestilstand
medfører en omstrukturering af den ekstracellulære matrix, som fører til ødelæggelse af knogle og
brusk i leddet. Leddegigt kan til en vis grad hæmmes af sygdomsmodificerende lægemidler, som
methotrexate, eller biologiske lægemidler såsom TNF-α inhibitors. Desværre er effekten af de
nuværende behandlingsformer begrænset, da det i de fleste tilfælde kun er betændelsestilstanden der
mindskes og ikke nedbrydningen af knogler og brusk. Det er derfor vigtigt for udviklingen af ny
behandling til gigtpatienter, at identificere nye faktorer der kan regulere både betændelsestilstanden
og knogle/brusk nedbrydelsen.
Som en ny behandlingsform af leddegigtspatienter er receptoren OSteoClast-
Associated Receptor (OSCAR) blevet foreslået. Studier har vist, at der er et øget niveau af OSCAR
i leddene hos leddegigtpatienter, og at forekomsten af OSCAR på monocytter i blodet korrelerer
positivt med sygdomsaktiviteten. I mus er OSCAR kun udtrykt på osteoklaster, hvor OSCAR ses at
have en co-stimulatorisk rolle i RANK-medieret osteoklast-dannelse. I mennesket er OSCAR
udtrykt på både osteoklaster, dendritiske celler, monocytter, makrofager og neutrofile granulocytter.
Liganden til OSCAR har vi og andre identificeret til at være Collagen type I og -II.
I leddegigt udskiller aktiveret immunceller, fibroblaster og osteoklaster høje mængder
enzym, som nedbryder den ekstracellulære matrix, hvilket blotter collagen og muliggør interaktion
mellem collagenen og immuncellerne i leddet. Under de patologiske forhold i forbindelse med
leddegigt, antages det derfor, at collagen type I og -II vil fungere som naturlige ligander for OSCAR
signalering.
Vi har i dette studie undersøgt den funktionelle betydning af interaktionen mellem
OSCAR og collagen i celler af myeloid oprindelse. Vi observerede at interaktionen mellem OSCAR
og collagen øgede aktiveringen og modningen af dendritiske celler, hvilket var karakteriseret ved
opregulering af overflademarkører og udskillelse af pro-inflammatoriske cytokiner. Endvidere så vi,
at de dendritiske celler, som var modnet med collagen, effektivt stimulerede T celle deling, men
disse T celler udviste ikke nogen klar cytokin polarisering. Genanalyse af de collagen stimulerede

9
dendritiske celler viste at flere betændelsesmarkører blev opreguleret, såsom cytokiner,
cytokinereceptorer, dele af den stabiliserende immunologiske synapse (herunder CD40, CD86,
CD80, ICAM-1) og dele af TNF- og TLR signalering.
Derudover blev den funktionelle rolle af OSCAR-collagen interaktionen også
undersøgt i monocytter og makrofager. I fravær af overlevelsesfaktorer kunne vi i monocytter
konstatere, at OSCAR-collagen interaktionen forlængede deres overlevelse. Derudover stimulerede
collagen udskillelsen af flere pro-inflammatoriske cytokiner i monocytter fra raske kontroller,
hvilket vi kunne hæmme ved at tilsætte et antagonistisk anti-OSCAR antistof. Global genanalyse
viste at flere transkriptionelle mål for OSCAR signaleringen i monocytter er involveret i
patogenesen for leddegigt. Vi noterede ydermere at celler fra synovialvæsken fra patienter med
leddegigt udskilte TNF-α og IL-8 når de blev stimuleret med collagen, hvilket var afhængigt af
OSCAR. Overraskende nok så vi ikke cytokin-udskillelse ved collagen-stimulering af monocyte-
genereret makrofager og tilsvarende så vi ingen effekt på gen niveau. Imidlertid konstaterede vi,
ved at hæmme OSCAR signaleringen i makrofager, at nedbrydelsen af collagen-rig matrix blev
mindsket, hvilket kunne indikere at OSCAR kan modulere enzym aktiviteten post-transkriptionelt.
Tilsammen indikerer vores fund at OSCAR-collagen interaktionen er en vigtig
mekanisme der bidrager til betændelsestilstanden i leddet hos patienter med leddegigt, hvor
collagen bliver blottet på grund af strukturelle ændringer i ledvævet og derfor kan interagere med
OSCAR i monocyt-genererede myeloide celler. Vi mener derfor at OSCAR-collagen interaktionen
kan bidrage til øget forståelse af patogenesen i leddegigt på flere niveauer og at mekanismen
ligeledes kan bruges som nyt, effektivt mål for udvikling af behandling.

10
Preface
This thesis was written in order to obtain the PhD degree from the Faculty of Science, University of
Copenhagen. The thesis is the result of three years experimental work at the Biopharmaceutical
Research Unit, Novo Nordisk A/S, Måløv, Denmark, and Department of Biology, University of
Copenhagen, Denmark. The thesis was carried out in the period between April 2012 and March
2015. Professor Martin W. Berchtold was my internal supervisor at Department of Cell and
Developmental Biology, Faculty of Science, University of Copenhagen. My main external
supervisor was Principal Scientist Svetlana Panina, Department of Immunobiology,
Biopharmaceutical Research Unit, Novo Nordisk A/S. A part of the PhD studies was carried out at
the University of Melbourne, Australia, under the supervision of Professor John Hamilton and
Research Fellow Andrew Fleetwood.
The PhD project was proposed by Novo Nordisk A/S in connection with the drug discovery
program for treatment of rheumatoid arthritis. During my PhD study, several changes took place in
the R&D research focus and the company structure. In December 2013 Novo Nordisk A/S decided
to discontinue the OSCAR project. The OSCAR project was closed due to challenges connected
with restricted expression of OSCAR in rodents, making it difficult to conduct in vivo studies using
animal disease models. I took a decision to continue with the OSCAR project as described in the
PhD proposal with adjustments to keep it in line with company activities. In March 2014, Novo
Nordisk re-organized their whole inflammation area, resulting in closure of Dept. of
Immunobiology. I continued my PhD in Department of Cellular Pharmacology, under the
supervision of Scientist and Team leader Christine Brender Read. In September 2014, the company
took a strategic decision to discontinue all activities within autoimmune inflammation disease
(AID) research and the whole function area was closed down. I therefore continued my PhD in
Immunogenicity Assessment, Non-Clinical development, being supervised by Principal Scientist
Olle Björkdahl.
The PhD thesis consists of an introduction summarizing the current knowledge about the interface
between the bone biology and inflammation. It outlines the pathogenesis of rheumatoid arthritis,
focusing on the contribution of immune cells, cytokines and extracellular matrix, as well as the
immunoreceptor OSCAR. The results are arranged in tree sections, based on two manuscripts

11
(results part I and part II) and a section written in a paper format, called results part III,
summarizing unpublished data. Paper I describes the role of OSCAR in dendritic cells and it was
recently published in Journal of Immunology. Paper II focuses on the OSCAR function in
monocytes and is currently being prepared for submission to Journal of Immunology. The third
section shows preliminary data on the OSCAR biology in macrophages. The methodologies used to
conduct the studies are described in the two papers and in part III of the results. Finally, the results
are discussed in a general Discussion section based on the two papers and my unpublished data on
the OSCAR biology in the context of rheumatoid arthritis.
________________________________
Copenhagen, March 2015
Heidi Schultz

12
Acknowledgements
It has been three educating and challenging years during my PhD studies, which I could not have
done without help from my supervisors, colleagues, collaborators, friends and family. It has been
truly rewarding to be a part of the daily routine at the departments of Immunobiology, Cellular
Pharmacology and Immunogenicity Assessment.
Many colleagues have contributed to the work in this thesis and I would like to direct my gratitude
to several people.
Special thanks to my first supervisor Svetlana Panina. Svetlana was the Biology Coordinator of the
OSCAR project and proposed my PhD project. Svetlana has a deep scientific knowledge and a great
passion for science. I would like to thank Svetlana for her expertise, interesting discussions and
encouragement when needed. Svetlana has my deepest respect for still finding time and making the
effort to guide and help me in the last stretch of my PhD, even though she has not been my official
supervisor since June 2014. For that I am very grateful.
Professor Martin Berchtold has been my internal university supervisor. He has engaged me in his
group and challenged me every time he could, encouraging me in my work. Your advice has always
been of great help.
I would also like to thank my supervisor Christine Brender Read, who, even though it was for a
short period of time, welcomed me into her little but very competent team.
Last but not least, thanks to Olle Björkdahl. Olle was so kind to be my supervisor in the challenging
period after Inflammation research was closed down. He made sure that I had what I needed to
finish my PhD. I would like to thank Olle for engaging in me and my project, asking questions and
guiding me through the last part. Special thanks for always offering your help willingly, and for
rapid and constructive corrections of my writings.

13
Special thanks to Professor John A. Hamilton, Andrew Fleetwood and colleagues at the Arthritis
and Inflammation Research Centre, Department of Medicine, Royal Melbourne Hospital, Australia,
for an amazing stay. It has been rewarding in many ways both scientifically and personally.
I wish to acknowledge everyone who contributed to my scientific work, in particular Pernille Keller
for her help with micro array analysis, as well as Lise W. F. Jensen, Gou Li, Lotte Bentzon and
Sofie Hedlund Møller. Thanks to all my colleagues, especially in Inflammation Biology for
practical help with laboratory techniques and encouraging talks. Thanks to the PhD student Mette
Dandanell Nielsen, Kasper Vadstrup and master student Pernille Damgaard Petersen for daily chats
and problem solving discussions. I thank Suzi Høgh Madsen for letting me borrow some of her own
figures for my introduction (Fig. 2 and 5).
An acknowledgement to the Danish Ministry of Science, Technology and Innovation and R&D
Academic relations at Novo Nordisk A/S for financial support.
I want to express my gratitude to the assessment committee for evaluating this thesis.
Finally, I wish to direct my thankfulness to friends and family. Special thanks to Mie and Palle S.
Schultz, Ninna S. Hansen and Andreas E. Clemmensen for their unconditional love and support.
The strength gained from such love is tremendous. Andreas, you are the best part of our little team.

14
Abbreviations
(m)Ab: (Monoclonal) Antibody
Ag: Antigen
APC: Allophycocyanin
APC: Antigen-presenting cell
CCL: CC Chemokine ligand
CCLR: CC chemokine receptor
CD: Cluster of differentiation
Col I/II: Collagen type 1 or 2
CIA: Collagen-induced arthritis
CXCL: CXC chemokine ligand
CXCR: CXC chemokine receptor
Cy7: Cyanin-7
DAP12: DNAX activation protein of 12 kDa
(i)DC: (immature) Dendritic cell
DDR: Discoidin-domain receptor
DMARDs: Disease-modifying anti-rheumatic drugs
DMSO: Dimethyl sulfoxide
ECM: Extracellular matrix
EDTA: Ethylene diamine tetra acetic acid
FACS: Fluorescence activated cell sorter
FcR: Fc receptor common gamma chain
FCS: Fetal calf serum
FITC: Fluorescein isothiocyanate
FSC: Forward scatter
GM-CSF: Granulocyte-macrophage colony-stimulating factor
GPVI: Glycoprotein VI
HLA: Human leukocyte antigen
HSA: Human serum albumin
HSC: Hematopoietic stem cell
ICAM-1: Intracellular cell adhesion molecule-1
Ig: Immunoglobulin
IL: Interleukin
INF: Interferon gamma
ITAM: Immunoreceptor tyrosine-based activation motif
ITIM: Immunoreceptor tyrosine-based inhibition motif
LAIR-1: Leukocyte-associated Ig-like receptor-1
LPS: Lipopolysaccharide
MAPK: Mitogen-activated protein kinases
MCP-1: Monocyte chemotactic protein-1
15
M-CSF: Macrophage colony-stimulating factor
MHC: Major Histocompatibility complex
MIP: Macrophage inflammatory protein
MMP: Matrix metalloproteinase
MNC: Mononuclear cells
Mo: Monocyte
NFATc1: Nuclear factor of activated T-cells
NFB: Nuclear factor kappa B
NSAID: Non-steroidal anti-inflammatory drug
OB: Osteoblasts
OC: Osteoclast
OPG: Osteoprotegerin
(s)OSCAR: (soluble) Osteoclast-associated receptor
PAI: Plasminogen activator inhibitor
PBMC: Peripheral blood mononuclear cells
PBS: Phosphate buffer saline
PE: Phycoerythrin
PerCP: Peridinin chlorophyll protein
Plg: Plasminogen
P/S: Penicillin/Streptomycin
RA: Rheumatoid arthritis
RANK: Receptor activator of nuclear factor kappa B
RANKL: RANK ligand
RANTES: Regulated and normal T cell expressed and secreted
RBC: Red blood cell
Rh: Recombinant human
RNA: Ribonucleic acid
rpm: Revolutions per minute
SD: Standard deviation
SSC: Side scatter
STAT: Signal transducer and activator of transcription
TCR: T cell receptor
TGF-β: Transforming growth factor beta
Th: T helper
TLR: Toll-like receptors
TNF-: Tumor necrosis factor alpha
TNFR: TNF receptor
TNP: Trinitrophenyl
TRAP: Tartrate-resistant acid phosphatase
TREM: Triggering receptor expressed on myeloid cells
uPA: Urokinase-type plasminogen activator
VCAM-1: Vascular cell adhesion molecule-1

16
Table of contents
1. INTRODUCTION........................................................................................................... 18
1.1 Osteoimmunology ..................................................................................................................................................... 18 1.1.1 Immune cells ....................................................................................................................................................... 18 1.1.2 Cells regulating bone homeostasis ...................................................................................................................... 20
1.2 Rheumatoid arthritis ................................................................................................................................................ 23 1.2.1 Cellular contribution to rheumatoid arthritis ....................................................................................................... 24 1.2.2 Cytokines and chemokines in rheumatoid arthritis ............................................................................................. 26 1.2.3 Rheumatoid arthritis treatment ............................................................................................................................ 27
1.3 Extracellular matrix ................................................................................................................................................. 28 1.3.1 Extracellular matrix of bone and cartilage .......................................................................................................... 28 1.3.2 Extracellular matrix turnover .............................................................................................................................. 30 1.3.3 Collagen receptors .............................................................................................................................................. 32
1.4 Osteoclast-associated receptor (OSCAR) ............................................................................................................... 35 1.4.1 Murine OSCAR .................................................................................................................................................. 35 1.4.2 Human OSCAR .................................................................................................................................................. 36 1.4.3 OSCAR function ................................................................................................................................................. 36 1.4.4 OSCAR ligand .................................................................................................................................................... 37 1.4.5 Regulation of OSCAR ........................................................................................................................................ 38 1.4.6 OSCAR in disease ............................................................................................................................................... 38
2. RATIONALE AND AIM ................................................................................................. 40
3. RESULTS ....................................................................................................................... 41
3.1 OSCAR function in dendritic cells .......................................................................................................................... 42
3.2 OSCAR function in monocytes ................................................................................................................................ 54 3.2.1 Abstract ............................................................................................................................................................... 55 3.2.2 Introduction ......................................................................................................................................................... 56 3.2.3 Materials and methods ........................................................................................................................................ 58 3.2.4 Results ................................................................................................................................................................. 62 3.2.5 Discussion ........................................................................................................................................................... 69
3.3 OSCAR function in macrophages ........................................................................................................................... 73 3.3.1 Introduction ......................................................................................................................................................... 73 3.3.2 Material and methods .......................................................................................................................................... 74 3.3.3 Results ................................................................................................................................................................. 78 3.3.4 Discussion ........................................................................................................................................................... 84
4. DISCUSSION ................................................................................................................. 87
5. CONCLUSION ............................................................................................................... 94
6. PERSPECTIVES ............................................................................................................ 95
6.1 OSCAR project ......................................................................................................................................................... 95

17
6.2 Atherosclerosis .......................................................................................................................................................... 96
6.3 Limitations to the OSCAR project .......................................................................................................................... 96
7. REFERENCES ............................................................................................................... 98
8. APPENDIX ................................................................................................................... 114
8.1 Appendix I ............................................................................................................................................................... 114
8.2 Appendix II ............................................................................................................................................................. 116
8.3 Co-author contributions ......................................................................................................................................... 117

18
1. Introduction
1.1 Osteoimmunology
Osteoimmunology is a new field of research that deals with the role of the immune system in bone
remodeling1. Immune cells and bone cells are closely interconnected sharing signaling molecules
and site of origin, namely the bone marrow. Attention was drawn to this field due to the observation
that bone destruction can be caused by an abnormal activation of the immune system. Osteoclast-
mediated bone loss has been observed in various autoimmune diseases, like rheumatoid arthritis and
diabetes mellitus2. On the contrary, it has also been reported that bone cells influence immune
cells3.
1.1.1 Immune cells
The cellular component of the immune system consists of two main types, the myeloid and the
lymphoid cells. Both lineages derive from the hematopoietic stem cells (HSCs). The myeloid cells
represent an integral part of the natural immune system and participate in the first-line response
against infectious agents. The myeloid cells are phagocytic cells that scavenge debris generated by
physiological or pathological processes4.
Fig. 1. Development of the cells of the immune system. Immune cells originate from the hematopoietic stem cells in
the bone marrow. Hematopoietic stem cells are capable of generating progenitor cells that will later give rise to different
types of immune cells. There are two main types of progenitors, the myeloid progenitors and the lymphoid progenitors.
The figure was produced by Schultz, H. S. using Servier Medical art figures as a template.

19
Monocytes derive from bone marrow progenitors, circulate in the blood and are capable of
differentiating into macrophages, dendritic cells and osteoclasts depending on signals from the
extracellular environment. Monocytes can function as antigen-presenting cells (APCs) and can
secrete various inflammatory factors5.
Macrophages are heterogenic tissue-resident cells. They have high proteolytic and
catabolic activities contributing to their ability to scavenge and digest pathogens, dead cells and
cellular debris. Macrophages have the capability to present antigens, though they do it less efficient
compared to other APCs like dendritic cells (DCs) and B-cells. At a steady-state, they are
considered to be anti-inflammatory, maintaining organ homeostasis5. Under inflammatory
conditions, they become activated and take part in host response to pathogens6.
DCs are specialized APCs which play an important role in processing and presentation
of antigens (Ag) to T cells. DCs secrete cytokines important for both innate and adaptive immunity.
At present, four main DCs types are categorized being: conventional DCs (cDCs), plasmacytoid
DCs (pDCs), Langerhans cells and monocyte-derived DCs. These subsets constitute a
heterogeneous population, classified further by their anatomical location, phenotype, functional
properties and migratory capability7;8
. Most human DCs are differentiated from blood monocytes or
monocyte precursors to immature DCs (iDCs) under the control of granulocyte-macrophage colony-
stimulating factor (GM-CSF) and Interleukin (IL)-49. DCs migrate from the bone marrow to
peripheral tissues, where they reside in an immature state, awaiting antigen encounter. iDCs can
become mature in response to several stimuli, like IL-1, TNF-, LPS, CD40-CD40L interaction or
by exposure to extracellular matrix (ECM) components, such as collagen10
. The activity of DCs can
be regulated by Toll-like receptors (TLR), as well as activating and inhibiting immunoreceptors,
which transduce signals trough immunoreceptor tyrosine-based activating motifs (ITAM) and
through immunoreceptor tyrosine-based inhibiting motifs (ITIM), respectively11;12
. When
encountering an Ag the DCs process the Ag and display the peptides via their MHC complex.
While maturing, the DCs migrate to the T cells areas of secondary lymphoid organs, where they
present Ag to T cells. The outcome - the immune priming or tolerance - depends on the maturation
stage of the DCs13-15
. DCs are important for orchestrating the adaptive immune response and may
also contribute to the development of autoimmunity when dysregulated16
.
Important for the initiation of the adaptive immune response is T cell activation. T
cells are activated by interaction between the T cell receptor (TCR) on the T cell and the major
histocompatibility complex (MHC)-Ag peptide complex on the APCs. The TCR has a low affinity

20
for the MHC molecule, but the interaction is strengthened by the formation of a specialized contact,
an immunological synapse. The immunological synapse is characterized by a specific pattern of
receptor segregation with a central cluster of TCR surrounded by co-stimulatory receptors and a
ring of integrin adhesion molecules17
. For T cells to be activated two signals are needed. The first
signal is the peptide-MHC:TCR complex, stabilized by either CD4 or CD8 co-receptor. The second
signal is provided by the co-stimulatory molecules such as CD80 and CD86 on the APCs that
interacts with CD28 on the T cells. Without co-stimulation T cells become anergic, a mechanism to
protect against autoimmunity. The interaction between the T cell and the APC is enhanced by
adhesion molecules, like lymphocyte function-associated antigen 1 (LFA-1) expressed on T cells
and intercellular adhesion molecule 1 (ICAM-1) present on APC18
. The outcome of T cell
activation is dependent on the state of APC maturation, e.g. number of MHC-peptide complexes as
well as duration of TCR engagement. The activation generally results in T cell proliferation and
polarization, and expression of multiple cytokines including IL-2, IFNγ and TNF-19;20
.
1.1.2 Cells regulating bone homeostasis
Bone contains several types of cells that are important for its development, maintenance and repair.
In adults, bone is constantly being remodeled, enabling the tissue to regenerate damages, adapt to
changing stress and to control calcium homeostasis. The main cell types facilitating bone
remodeling are the specialized bone-resorbing cells, osteoclasts (OCs) and the bone-synthesizing
cells, osteoblasts (OBs)2.
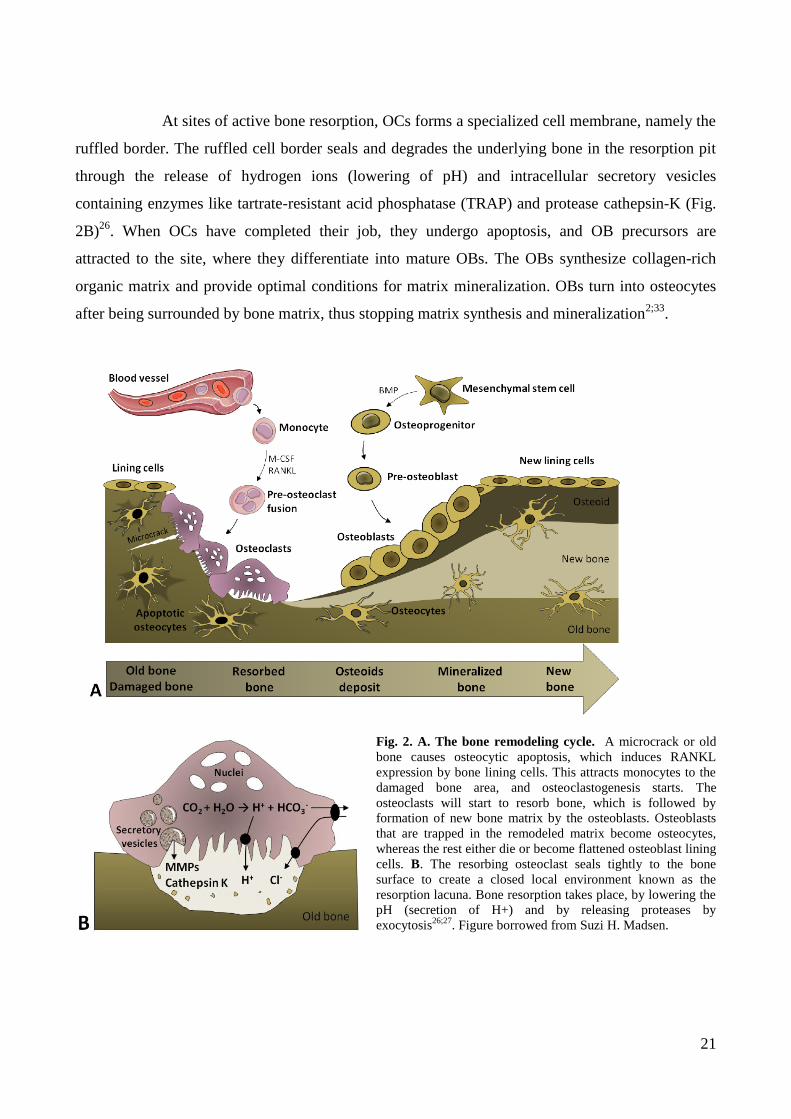
Bone remodeling (Fig. 2A) is initiated by osteocytes21;22
. Osteocytes are DC-like cells
trapped in the bone matrix. Osteocytes act as mechanosensors in bone tissue and activate other
osteocytes and bone lining cells when microdamage occurs. This signaling results in attraction of
OC precursors and initiation of osteoclastogenesis. OCs originates from cells of the myeloid
lineage, which fuse into large multinucleated OCs. Essential for OCs differentiation, activity and
survival are macrophage-colony stimulating factor (M-CSF) and receptor activator of nuclear
factor-kB ligand (RANKL). M-CSF induces early differentiation, promotes proliferation and
survival of OCs precursors. RANKL is a member of the TNF superfamily and signals through its
receptor, RANK, which is expressed on a variety of cells including OCs. RANK-RANKL
interaction induces the final differentiation of OCs and their bone-resorbing activity23-27
. M-CSF
and RANKL are believed to be sufficient for inducing OC differentiation in culture28
. However, co-
stimulatory signals mediated by ITAM-harboring receptors have been proven to be important co-
stimulatory signaling component in osteoclastogenesis and are essential for bone remodeling29-32
.
21
At sites of active bone resorption, OCs forms a specialized cell membrane, namely the
ruffled border. The ruffled cell border seals and degrades the underlying bone in the resorption pit
through the release of hydrogen ions (lowering of pH) and intracellular secretory vesicles
containing enzymes like tartrate-resistant acid phosphatase (TRAP) and protease cathepsin-K (Fig.
2B)26
. When OCs have completed their job, they undergo apoptosis, and OB precursors are
attracted to the site, where they differentiate into mature OBs. The OBs synthesize collagen-rich
organic matrix and provide optimal conditions for matrix mineralization. OBs turn into osteocytes
after being surrounded by bone matrix, thus stopping matrix synthesis and mineralization2;33
.
Fig. 2. A. The bone remodeling cycle. A microcrack or old
bone causes osteocytic apoptosis, which induces RANKL
expression by bone lining cells. This attracts monocytes to the
damaged bone area, and osteoclastogenesis starts. The
osteoclasts will start to resorb bone, which is followed by
formation of new bone matrix by the osteoblasts. Osteoblasts
that are trapped in the remodeled matrix become osteocytes,
whereas the rest either die or become flattened osteoblast lining
cells. B. The resorbing osteoclast seals tightly to the bone
surface to create a closed local environment known as the
resorption lacuna. Bone resorption takes place, by lowering the
pH (secretion of H+) and by releasing proteases by
exocytosis26;27
. Figure borrowed from Suzi H. Madsen.

22
Modulation of osteoclastogenesis
OCs function is mainly regulated by TNF-, TNFR and TNF-like proteins such as osteoprotegerin
(OPG), RANK and RANKL. Additionally, hormones, cytokines and humoral factors regulate bone
density. TNF-, IL-1, IL-6 and IL-17 increase bone-resorption by locally enhancing RANKL
expression34
, which together with IL-1β promotes survival of OCs26
.
The RANK signaling pathway is negatively regulated by OPG. OPG blocks OCs
formation in vitro and bone resorption in vivo35
. OPG functions by blocking RANKL binding to
RANK. Accessibility of RANKL and OPG is coordinated to regulate bone resorption by controlling
the activation state of RANK on OCs26
. Some cytokines like IFN- and IL-4 have an inhibitory
effect on osteoclastogenesis36-38
.
Trans-differentiation
During in vivo inflammatory bone conditions, considerable de novo osteoclastogenesis is observed.
It has been investigated whether cell types other than those from the monocyte/macrophage lineage
can generate multinucleated giant cell with bone resorption capacity. Several recent in vitro studies
have reported that both human (monocyte-derived) and mouse (bone marrow-derived) DCs can
develop into functional OCs when cultured in the presence of M-CSF, RANKL and bone-like
matrix9;39-41
. This process is greatly enhanced by the microenvironment in the joint, involving IL-
1, TNF- and the extracellular matrix hyaluronic acid9;40
. The osteoclastogenic potential is
restricted to iDCs, which upon cell maturation is lost, even after addition of RANKL42
.
Additionally, in mice, it has been observed that CD11c+
DCs upon interaction with CD4+
T cells
develop into functional OCs in response to microbial or protein Ags in a RANKL/RANK-
dependent manner39
. Taken together, these findings indicate a high plasticity of DCs and suggest
that DCs may directly contribute to osteoclastogenesis.

23
1.2 Rheumatoid arthritis
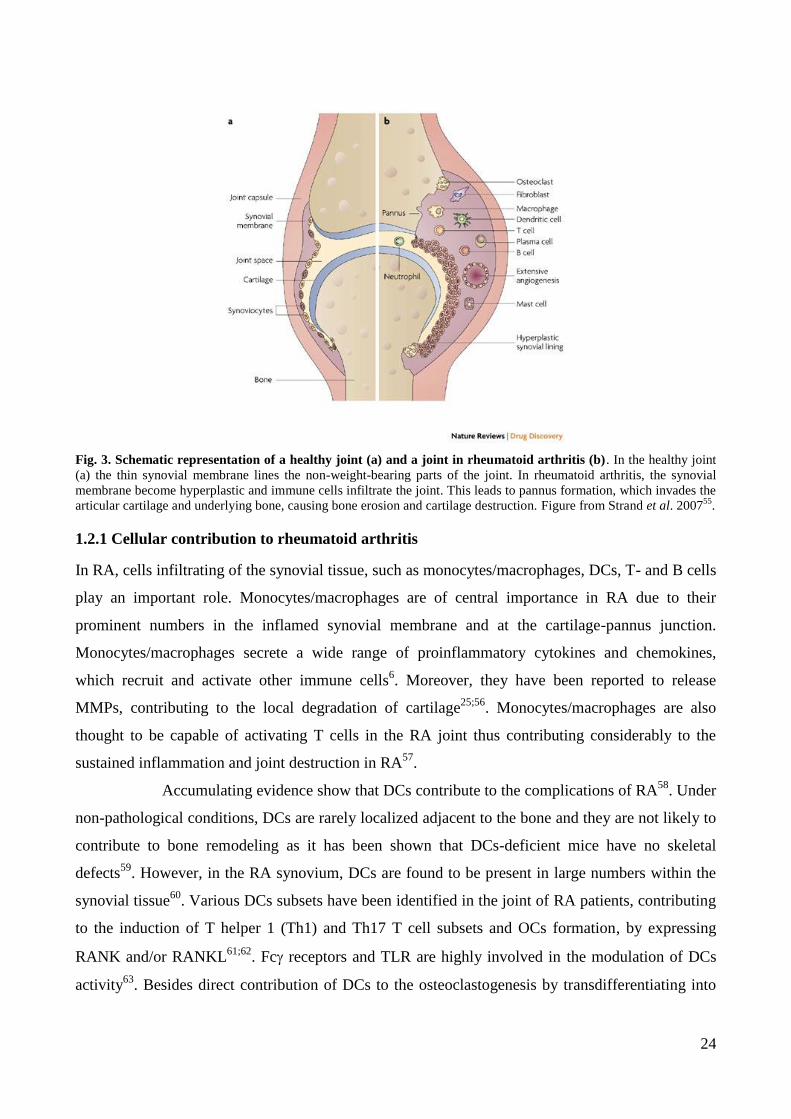
Rheumatoid arthritis (RA) is a chronic systemic autoimmune inflammatory disease that targets the
articular cartilage and bone at the joint margins, as well as periarticular and subchondral bone,
leading to joint destruction43
. It is characterized by the presence of autoantibodies, synovial
inflammation, pannus formation, cartilage damage and bone erosion. RA affects up to 1% of the
population, women more often than men, and is associated with significant morbidity and
mortality44
. The molecular mechanisms involved in RA pathogenesis remain unclear. It has been
proposed that the development of RA can be divided into three stages45
. First, autoimmunity
develops based on a complex interplay of genetic and environmental factors. Genetic studies of RA
have identified at least 46 risk loci46
. Especially the human leukocyte antigen (HLA)-DR, protein
tyrosine phosphatase non-receptor type 22 (PTPN22) and peptidyl arginine deiminase type IV
(PADI4) loci, involved in antigen presentation, TCR regulation and citrullination of peptides
respectively, have been associated with RA46-49
. Environmental factors, such as tobacco smoking
and infectious agents have been shown to alter post-translational protein modifications leading to
recognition of autoantigens and the loss of immune tolerance25;50
. This together with the genetic
predispositions can trigger an unwanted immune-response51;52
. Most of the RA patients at this stage
develop autoantibodies, such as rheumatoid factor, anti-citrullinated protein antibody (ACPA) or
anti-collagen type II antibody, which may predict a more aggressive progression of the disease44
. At
the next stage, the inflammation attacks the joint tissue. Activated immune cells accumulate in the
joint and secrete pro-inflammatory cytokines, chemokines and matrix degrading enzymes, including
MMPs, serine proteinases and aggrecanases25;53
. The inflammation increases the formation and
activity of the bone eroding OCs25;43;54
. In the synovial lining resident synovial fibroblasts
proliferate, which causes hyperplasia and pannus formation. The inflamed synovium invades
adjacent cartilage and bone ultimately leading to articular destruction25;44
. Finally, if the local
inflammation is not down-regulated, it develops into a chronic systemic inflammatory condition via
a positive feedback loop that permanently destructs the joints and affects other organs45
.
24
Fig. 3. Schematic representation of a healthy joint (a) and a joint in rheumatoid arthritis (b). In the healthy joint
(a) the thin synovial membrane lines the non-weight-bearing parts of the joint. In rheumatoid arthritis, the synovial
membrane become hyperplastic and immune cells infiltrate the joint. This leads to pannus formation, which invades the
articular cartilage and underlying bone, causing bone erosion and cartilage destruction. Figure from Strand et al. 200755
.
1.2.1 Cellular contribution to rheumatoid arthritis
In RA, cells infiltrating of the synovial tissue, such as monocytes/macrophages, DCs, T- and B cells
play an important role. Monocytes/macrophages are of central importance in RA due to their
prominent numbers in the inflamed synovial membrane and at the cartilage-pannus junction.
Monocytes/macrophages secrete a wide range of proinflammatory cytokines and chemokines,
which recruit and activate other immune cells6. Moreover, they have been reported to release
MMPs, contributing to the local degradation of cartilage25;56
. Monocytes/macrophages are also
thought to be capable of activating T cells in the RA joint thus contributing considerably to the
sustained inflammation and joint destruction in RA57
.
Accumulating evidence show that DCs contribute to the complications of RA58
. Under
non-pathological conditions, DCs are rarely localized adjacent to the bone and they are not likely to
contribute to bone remodeling as it has been shown that DCs-deficient mice have no skeletal
defects59
. However, in the RA synovium, DCs are found to be present in large numbers within the
synovial tissue60
. Various DCs subsets have been identified in the joint of RA patients, contributing
to the induction of T helper 1 (Th1) and Th17 T cell subsets and OCs formation, by expressing
RANK and/or RANKL61;62
. Fc receptors and TLR are highly involved in the modulation of DCs
activity63
. Besides direct contribution of DCs to the osteoclastogenesis by transdifferentiating into

25
OCs (see 1.1.2), they also play an indirect role in bone degradation by producing cytokines such as
IL-1, IL-6 and TNF-, which increase expression and release of cathepsin K and TRAP by resident
OCs8. It has been shown that ColII-pulsed DCs can induce arthritis in a mouse model after adoptive
transfer, indicating a potential role of DCs in driving RA immunopathology64
.
Traditionally, RA has been considered a Th1 cell mediated disorder, driven by the
cytokines IFN- and TNF-25
. Recently, a new model has been proposed, implicating Th17 cells
and their effector cytokines, IL-17, IL-21 and IL-22 as crucial factors for the RA development53;65
.
Th17 cells support osteoclastogenesis, likely through an IL-17-mediated induction of RANKL on
osteoblastic cells and synovial fibroblasts66
. IL-17 facilitates local inflammation by activating
macrophages, monocytes, fibroblasts and endothelial cells leading to release of TNF- and IL-1,
which further enhance the RANKL expression25
. Th1 and Th2 cells have been proposed to inhibit
osteoclastogenesis by acting on the OCs precursor cells, mainly through IFN- and IL-4,
respectively25
.
Fig. 4. IL-17-mediated regulation of RANKL-induced osteoclastogenesis in the RA synovial tissue. Th17 cells
function as a pro-osteoclastogenic Th cell subset by stimulating local inflammation, inducing RANKL on the cells
supporting osteoclastogenesis , and expressing RANKL themselves. It is notable that RANKL on Th17 cells alone is
not sufficient for the induction of OCs differentiation (dotted line). Op, osteoclast precursor cell. Figure from Sato et al.
200665
.
B cells may contribute to RA in several ways, for example by acting as APCs, cytokine producers
and by expressing RANKL. In RA, B cells secrete autoantibodies to citrullinated proteins or anti-Ig
(rheumatoid factor) which lead to formation of immune complexes and complement deposition in
the joint57
. In some RA patients, ectopic germinal centers are formed in the joint. The formation of
germinal centers contributes to B cell maturation and generation of high affinity Abs25;57
. The
importance of B cells in RA has been emphasized by the fact that B cell depleting mAb Rituximab
has shown efficacy in RA patients67
.

26
1.2.2 Cytokines and chemokines in rheumatoid arthritis
Cytokines are directly implicated in the immune processes associated with the pathogenesis of RA.
The cytokines are crucial for maintenance of chronic inflammation and driving the destruction of
joint tissue in the RA patients25
. Elevated levels of proinflammatory cytokines, such as TNF-, IL-
1, IL-6, IL-15, IL-18 and GM-CSF are found in the RA synovium44
. TNF- is a pleiotropic
cytokine that induces the expression of inflammatory factors like cytokines and prostaglandins as
well as enhances OCs differentiation6;43
. TNF- may also induce activation and survival of
leukocytes, endothelial cells and fibroblasts25
. The IL-1 levels in the synovial fluid have been
shown to correlate with the score of the joint inflammation68
. This cytokine appears to mediate the
articular damage to a high degree69
. IL-1 induces MMP-1 and MMP-3 production by macrophages
and fibroblasts, which enhance collagen degradation6;70
. Both TNF-, IL-1 are shown to up-
regulate RANKL on the bone stromal cells, which enhances OCs generation and bone resorption43
.
Overproduction of IL-6 is closely related to the RA pathology71;72
. The level of IL-6 has been found
to be significantly elevated in the synovial fluid from RA patients, especially during the acute
phase73
. IL-6 is secreted by several cell types in the synovium, especially by monocytes/
macrophages, and it can stimulate hepatocytes to produce acute-phase factors. In synergy with
TNF- or IL-1, IL-6 induces the production of vascular endothelial growth factor (VEGF), a
factor important for angiogenesis74
.
Various regulatory cytokines, such as TGF-, IL-10, IL-11 and IL-1RA, are expressed
in the joint. However, they seem not to be present in sufficient amounts to counteract the activities
of the pro-inflammatory cytokines75
. Similarly, the T cell derived cytokines IL-2 and IL-4 are
absent, which may impair generation of regulatory T cells (Treg), and hence would lead to
preferential Th1 and Th17 differentiation6;75
.
Several chemokines have been shown to be elevated in the RA synovial fluid
contributing to the RA pathogenesis. Macrophages secrete IL-8 and MCP-1, which mediate
leukocyte chemotaxis76-78
. T cells and fibroblasts secrete Regulated on Activation Normal T-cell
Expressed and Secreted (RANTES) that attracts T cells to the RA joints76;79
. Fibroblasts,
macrophages and neutrophils secrete Macrophage Inflammatory Protein-1 (MIP-1) that recruits
macrophages and T cells, activates granulocytes and enhances the production of proinflammatory
cytokines80;81
.

27
1.2.3 Rheumatoid arthritis treatment
Different types of treatments exist for patients with RA. The current treatments can improve
symptoms and modify the progression of the disease, but they do not cure RA. The treatments used
for RA include anti-inflammatory agents, analgesics and disease-modifying anti-rheumatic drugs
(DMARDs), such as Methotrexate. DMARDs hinder disease progression, but have a limited effect
and a complete halt of joint destruction is not achieved in the vast majority of the cases82-84
.
Recently, the focus has been on biological agents as alternative treatment options. Especially
targeting cytokines, in particular TNF-, IL-1 and IL-6, has been proposed44
. Blocking TNF- has
been proved successful for treatment of RA and other autoimmune diseases and has highlighted the
importance of inflammation in joint destruction25;40
. Anti-TNF- treatment has been shown to
target inflammation at multiple levels. For instance, it caused decrease in plasma levels of IL-1, IL-
6 and acute-phase proteins, suppressed OCs and DCs maturation as well as leukocyte migration into
the synovium. It has also been shown to led to the recovery of Treg cell formation and function85-88
.
However, a significant number of RA patients show only partial responses or fail to respond to anti-
TNF- treatment25;89
. Furthermore, a whole new type of treatment, consisting of small molecules
that interrupt intracellular signaling through kinase inhibition, is being examined as RA
therapeutics90
. An example of this is Tofacitinib, a JAK inhibitor, which showed promising results
in a phase II study, where it decreased structural damage and improved disease activity in RA
patients91
.
The major challenge in RA management still remains, namely to halt ongoing bone
and cartilage destruction. A promising treatment for halting bone erosion is an anti-RANKL mAb
(Denosumab). Denosumab has been shown to have an anti-resorptive effect in osteoporosis92;93
and
to inhibit structural damage in RA94
. Anti-RANKL therapy might, in combination with anti-
inflammatory drugs, block the disease manifestations.

28
1.3 Extracellular matrix
Extracellular matrix (ECM) is a 3-D structure that surrounds cells and defines their
microenvironment95
. It consists of multiple components of which collagen is the most abundant96
.
There are two main types of ECM, the basement membrane, mainly consisting of collagen type IV
(ColIV), laminins, entactin and proteoglycans, and the interstitial matrix, primarily consisting of
collagen type I (ColI), -type II (ColII), –type III (ColIII) and fibronectins97
. Besides supporting
structural integrity, ECM is important for controlling various cellular functions. Cells use ECM for
homing, migration and invasion. ECM also regulates cell differentiation, function,
activation/maturation, and seems to enhance complement receptor- and Fc receptor mediated
phagocytosis, as well as to stimulate cytokine production98-100
.
1.3.1 Extracellular matrix of bone and cartilage
The human skeleton consists of specialized tissues, such as bone and cartilage. Bone is a dynamic
organ that is continuously being formed, shaped and repaired, involving break down (resorption)
and build-up (synthesis), thus providing maximal strength with minimal mass26
. Bone is composed
of cells and ECM, the latter being further subdivided into an inorganic and organic part. The
inorganic part primarily contains crystals of calcium and phosphorus, whereas the organic part
mainly consists of ColI (approximately 95%), as well as other collagens, non-collagenous proteins
and proteoglycans2. Bone homeostasis depends on the balance between the bone-forming OBs and
the bone-resorbing OCs. The balance is tightly regulated by a number of osteogenic cytokines,
growth factors and hormones101
. Most adult skeletal diseases are due to excess osteoclastic activity,
leading to imbalance in bone remodeling favoring resorption. These include diseases such as
osteoporosis, periodontal disease, RA, multiple myeloma and metastatic cancer26
.
In adults, articular cartilage is an avascular tissue composed of collagens,
proteoglycans and non-collagenous proteins (Fig. 5). The collagen network in cartilage consists of
approximately 60% ColII102;103
. The most prominent proteoglycan in cartilage is aggrecan that
attracts water and gives cartilage protective resiliency. The cellular compartment of cartilage is
constituted of chondrocytes, which regulate cartilage homeostasis104
. The chondrocytes synthesize
new ECM and are also the source of proteinases for the ECM degradation. The turnover of cartilage
is limited and the repair capacity is restricted upon damage104
. Chondrocytes’ function is affected
by the inflammatory environment in RA, where the articular cartilage is damaged mainly by

29
cytokine-induced proteolytic enzymes. Cartilage degradation can be examined by assessing
degradation fragments, such as telopeptides of ColII97
.
Fig. 5. Cartilage components. The articular cartilage is mainly composed of collagen type II, chondrocytes and
aggrecan attached to hyaluronic acid102;103
. Figure borrowed from Suzi H. Madsen.
Collagen
Collagen is the key component of the ECM. It provides essential structural support for connective
tissue and is involved in a spectrum of cellular functions98;99
. All collagens consist of three
polypeptide chains termed chains that are characterized by repeating glycine-X-X’ sequences.
Amino acids in position X and X’ are often proline and 4-hydroxyproline (O), respectively105
. The
three chains are twisted around each other into a right-handed super helix. In vertebrates, 28
different types of collagen have been identified, composed of at least 46 distinct collagen
polypeptide chains106
. Several collagens carry glycosaminoglycans chains and are also considered
proteoglycans107
. Collagens can assemble into supramolecular structures, such as fibrils, which are
stabilized by the formation of covalent cross-links. ColI and ColII belong to the group of classical
fibril forming collagens107
. Collagen interacts with cell surface receptors. Some binding sites are
only available for interaction after denaturation and these motifs may be important in the removal of
degraded and denatured collagen or in autoimmune responses99
.

30
Fig. 6. Schematic representation of the structure of collagen. (a) Surface representation of a collagen triple helix, as
a model for the collagen-like peptide (PPG)9. The three α-chains wind around one another with a one-residue stagger.
(b) Schematic representation of how individual collagen triple helices assemble into a quarter-staggered array to form a
collagen fibril. (c) Electron micrograph of a heterotypic collagen fibril isolated from human articular cartilage. Figure
from Leitinger, B. 201199
.
1.3.2 Extracellular matrix turnover
The pleiotropic ECM functions are defined by a highly dynamic structure of ECM and its
remodeling capabilities to control cellular behaviour108
. Four classes of enzymes can degrade matrix
proteins: cysteine, aspartate- and serine-dependent proteases, and metalloproteinases. The cysteine
and aspartate-dependent proteases act primarily intracellularly, while serine-dependent proteases
and metalloproteinases act extracellularly109
. Connective tissue destruction is majorly mediated by
metalloproteinases. Within the metalloproteinases classes, two main families exist: the matrix
metalloproteinase (MMP) and a disintegrin and metalloproteinase with trombospondin motifs
(ADAMTS)108
. Native fibrillar collagens are resistant to proteases such as pepsin, trypsin or
chrymotrypsin98
.
Matrix metalloproteinases
MMPs are zinc-dependent endopeptidases which currently comprise 23 related, but distinct
proteins110;111
. MMPs are classified into five subtypes: 1) the gelatinases, which degrade ColIV and
other basement membrane proteins; 2) the collagenases, which degrade the fibrillar collagens (ColI,
ColII and ColIII); 3) the stromelysins, which degrade non-collagen matrix-proteins; 4) membrane-
type MMPs and 5) a diverse subgroup110;112
. Fibril-forming collagens (I-III) are cleaved by MMP-1,
-8 and 13. Furthermore, MMP-2 and 14 are capable of cleaving ColI107
.

31
In steady-state tissue MMP activity is very low. However, the expression of the
MMPs can be regulated at the transcription level by several factors, such as cytokines, hormones,
cell-cell interactions and cell interactions with ECM113;114
. The MMPs are secreted as inactive pro-
MMPs that need to be activated. They are converted into the active form by cleavage of their pro-
domain. The mode of MMP activation is not fully elucidated, but they can be activated by other
active MMPs, oxidants and by plasmin110;115;116
. The active MMPs can be inhibited by
internalization or by inhibitors, like tissue inhibitors of metalloproteinases (TIMPs)117
.
Plasmin
Additional mediators of ECM degradation are the serine proteases, like plasmin, elastase and
cathepsin G109
. The zymogen plasminogen (plg) is mainly produced by the liver and circulates in
the plasma at high concentrations (2μM). Plg is converted to its active form, plasmin, by tissue Plg
activator (tPA) or urokinase Plg activator (uPA). When converted, plasmin binds to the ECM and
degrades multiple ECM proteins118;119
. Plasmin can also activate other ECM-degrading proteinases,
such as MMP-3, MMP-9, MMP-12, and MMP-13120
. Plg-dependent activation of MMP-9 has been
shown to be required for inflammatory macrophage migration across ECM121
. The Plg system is
regulated at several levels and potent inhibitors are Plg activator inhibitor–1 (PAI-1), PAI-2, α2-
antiplasmin and aprotinin118;119
. Urokinase-type Plasminogen Activator Receptor Associated Protein
(uPARAP) has been shown to be important for degradation of collagen by macrophages122
.

32
Fig. 7. Plasminogen activating system. The uPA receptor (uPAR) binds both pro-uPA and active uPA, thus restricting
the activation of plasminogen to the cell membrane. When pro-uPA is bound to the uPAR it facilitates plasmin-
mediated activation of uPA. Activated uPA then cleaves plasminogen; generating active plasmin, creating a positive
feed-back loop. Plasmin cleaves and activates MMPs and together they can degrade ECM. Plasminogen can also be
converted to its active form by tissue plasminogen activator (tPA). The major inhibitors of the Plg system are Plg
activator inhibitor–1 (PAI-1), PAI-2, aprotinin and α2-antiplasmin. Figure from Rao, J. 2003121;123
.
1.3.3 Collagen receptors
A structurally and functionally diverse group of surface receptors mediates recognition of collagen,
such as integrins, discoidin domain receptors (DDRs), glycoprotein VI (GPVI), and leukocyte-
associated immunoglobulin-like receptor-1 (LAIR-1) (Fig. 8). These receptors regulate cell
adhesion and migration, homeostasis and immune functions99
. The importance of proper
collagen:collagen-receptor interaction is emphasized by their involvement in pathogenesis of
various diseases, where dysregulation of expression of collagens or collagen-receptors contribute to
disease pathology, such as fibrosis, wound healing, inflammatory disorders and tumor
angiogenesis99
.
Integrins are the main family of collagen receptors and primarily mediate cell
adhesion124
. Integrins are heterodimers composed of non-covalently associated and subunits,
forming 24 distinct integrins. The α1β1 and α2β1 integrins are the most broadly expressed collagen-
binding integrins. The discoidin domain receptors, DDR1 and DDR2, constitute a subfamily of
receptor tyrosine kinases that are characterized by the presence of a discoidin domain in their

33
extracellular region. DDRs form constitutive dimers in the cell membrane99
. DDRs regulate
important cellular processes such as proliferation, migration and differentiation125
. In smooth
muscle cells and fibroblasts DDRs are found to influence remodeling of ECM through the induction
of MMPs126;127
. Both integrins and DDRs display exquisite specificity in their interaction with
collagen by binding specific amino acid side chains in the collagen triple-helix. Their binding
motifs are distinct and non-overlapping99
. The structurally related receptors GPVI and LAIR-1 have
similar collagen-binding mode but mediate opposing functions128
. GPVI is an activating receptor on
platelets and LAIR-1 is an inhibitory receptor on immune cells99
. GPVI is central to haemostasis
where it activates platelets upon binding to collagen129
. LAIR-1 contributes to the regulation of the
immune system by delivering inhibitory signals via its ITIM signaling motif130
. Besides LAIR-1,
LAIR-2 is a part of the subfamily and is a soluble homolog of LAIR-1. It is hypothesized that
LAIR-2 may function as an inhibitor of LAIR-1 signaling in vivo99
.
Recently, a potential crosstalk between different collagen receptors have been
studied99
. Xu and colleagues have reported that the DDRs positively modulate integrin-mediated
cell adhesion131
. Mocsai and co-authors found that activation of integrins was decreased when
ITAM signaling was defective31
. Moreover, ITIM-associated receptors are potent inhibitors of
ITAM-signaling. It has been observed that LAIR-1 inhibits signals by ITAM-bearing receptors
leading to suppression of T cell activity, down-regulation of B cell function and blocking of
cytokine-induced signals99
.
Fig. 8. Schematic domain structures of the collagen receptors. Four classes of collagen binding transmembrane
receptors, the collagen-binding integrins, discoidin domain receptors (DDRs), glycoprotein VI (GPVI) and leukocyte-
associated immunoglobulin-like receptor-1 (LAIR-1) Figure from Leitinger, B. 201199
.

34
Extracellular matrix receptors on myeloid cells
The cells of the myeloid lineage have a large number of receptors regulating their activity. Several
studies have addressed the involvement of the ECM proteins in the control of the immune cell
functions. It has been observed that collagens stimulate maturation of DCs10
as well as induce
cytokine production by PBMCs100
and RA synovial fluid cells132
.
In recent years, a growing number of receptors of the lectin and immunoglobulin
superfamilies (IgSFs), linked to ITAMs/ITIMs signaling, have been described133
. ITAMs are mainly
found in the cytoplasmic tail of transmembrane adaptors, such as CD3, FcR or DAP12, and play a
role of signaling subunits when associated with cell surface receptors. ITAM signaling induces
cellular activation via a phosphorylation cascade triggered by protein tyrosine kinases134
. The
transmembrane adaptors DAP12 and FcR, associated with TREM2 and OSCAR receptors,
respectively, have both been identified to be involved in OCs and DCs differentiation and
function29;32;135;136
. Combined with the increasing evidence that differentiation of OCs occurs
locally in the inflamed synovial membrane, where OCs can form from monocytic precursor cells, a
growing number of studies was focused on the monocyte/OCs receptors TREM2 and OSCAR. My
project is focused on the biology of human OSCAR in various immune cells present in the RA
joint.

35
1.4 Osteoclast-associated receptor (OSCAR)
1.4.1 Murine OSCAR
Osteoclast associated receptor (OSCAR), was initially identified in mice by Kim and colleagues,
and characterized as a novel member of the immunoglobulin (Ig)-like surface receptor family29
. In
mice, OSCAR has been found to be specifically expressed on osteoclast precursors and mature
osteoclasts29
. OSCAR is a monomeric N-glycosylated cell-surface protein of approximately 45 kDa.
The protein exhibits two C2-type immunoglobulin extracellular domains, a transmembrane domain
that contains a positively charged arginine residue, and a short intracellular domain. OSCAR is
associated with the ITAM harboring Fc receptor -chain (FcR) that serves as a signaling subunit of
the receptor complex135
. OSCAR has been recognized as a co-stimulatory receptor, contributing to
osteoclastogenesis by activating NFATc1, an important transcription factor of OCs
differentiation29;30;135
.
The role of OSCAR and its ITAM-harboring adaptor have been examined in murine
models. FcR-/-
mice show no significant difference compared to wild-type mice in respect to bone
disorders. Additionally, the OCs in the knock-out mice were well developed in the presence of M-
CSF and RANKL, and when cultured in the presence of OBs, suggesting that FcR is not essential
for the differentiation of OCs135
. However, several groups have reported that mice deficient in both
DAP12 and FcR exhibit severe osteopetrosis and show defective differentiation of osteoclasts
31;32.
This cannot simply be attributed to DAP12 deficiency, since single knock out DAP12-/-
mice only
exhibit mild osteopetrosis and normal numbers of OCs in bone tissue. Co-culturing DAP12-/-
OC
precursors with OBs eradicated the effect of knocking out DAP1231
. These results suggest that
OSCAR may co-stimulate osteoclastogenesis independent of TREM2-DAP12 signaling137
and that
DAP12 and FcR functionally compensate for the lack of each other32
.

36
Fig. 9. A schematic model of ITAM-mediated co-stimulatory signal in RANK-stimulated induction of OC
differentiation. ITAM-stimulation by immunoreceptors and RANKL–RANK interaction results in NFATc1 induction.
RANKL may also contribute to efficient ITAM signaling through the induction of immunoreceptors. Figure from Koga
et al.32
1.4.2 Human OSCAR
Human OSCAR (hOSCAR) was identified by Merck et al.136
. The murine and human OSCAR
share approximately 73% amino acid identity and the genes can be regarded as orthologs29
. In
humans, OSCAR is more widely expressed than murine OSCAR and is present on all cells of the
myeloid cell lineage136
. Like in mice, hOSCAR is associated with FcR and can signal to activate
myeloid cells by triggering calcium influx and cytokine release136
. The calcium influx induces the
master transcription factor for osteoclastogenesis, NFATc132
, shown in Fig. 9. The gene coding for
OSCAR, known as PIGR3, has been mapped to chromosome 19q13136
. OSCAR exists as both
membrane-bound and soluble isoforms138-140
presumably resulting from alternative splicing event29
and/or being proteolytically shed from the cell membrane141
.
1.4.3 OSCAR function
OSCAR has diverse functions, playing a role in both bone homeostasis and activation of the
immune response. OSCAR serves as a co-stimulatory molecule for OCs differentiation, acting in an
additive manner with primary stimulation for RANKL, enabling a fine-tuning of OCs
differentiation29;137;141
. Moreover, OSCAR has been suggested to be implicated in the development
and maturation of cells of the myeloid lineage11;134;136
. Most studies on the hOSCAR function in
myeloid cells have been performed using stimulation of the OSCAR signaling by crosslinking of
OSCAR with specific antibodies, since a naturally occurring ligand of OSCAR was not identified
until recently137;142
. Merck and colleagues reported that crosslinking OSCAR on iDCs led to
activation and maturation. The cells were up-regulating the phenotypic markers CD40, CD54,

37
CD80, CD86, HLA-DR and partially CD8311
. Up-regulation of activation and maturation markers
was also observed for monocytes, and to a lesser extent on neutrophils134
.
Crosslinking of hOSCAR in iDCs has been shown to induce high expression levels of
the chemokines TARC/CCL17 and MDC/CCL22. These chemokines are known to be able to attract
Th2 effectors and regulatory T cells143;144
. Moreover, cross-linking of OSCAR on DCs, monocytes
and neutrophils induced secretion of IL-8 and MCP-1, which are involved in the recruitment of
leukocytes to sites of inflammation11
. Furthermore, ligation of OSCAR on neutrophils caused
degranulation, respiratory burst activity and release of antibacterial molecules, namely
myeloperoxidase, MMP-9 and lactoferrin134
.
OSCAR stimulation has further been suggested to promote survival of monocyte-DCs
in the absence of survival factors by maintaining expression of anti-apoptotic molecules of the Bcl-
2 family through activation of the PI3K and ERK signaling pathways11
. The same was observed for
monocytes, but not for neutrophils134
. Merck and colleagues have suggested a role of OSCAR in
antigen endocytosis and presentation in DCs. They demonstrated that upon cross-linking of OSCAR
with an anti-OSCAR mAb the whole receptor/mAb complex was endocytosed. The internalized
mAb peptides reached vesicles characteristic of the lysosomal/late endosomal MHC II-loading
compartment. The peptides derived from the anti-OSCAR mIgG1 mAb were presented to T cells
and stimulated proliferation of the mIgG1 specific T cell clone136
. Merck and colleagues also
observed that OSCAR signaling in DCs could modulate the response of the cells to the Toll-like
receptors (TLR) engagement11
. Co-stimulation of DCs with the OSCAR surrogate ligand
(crosslinking mAb) and TLR ligands, but not by OSCAR cross-linking alone, synergistically
enhanced pro-inflammatory cytokine release and maturation of DCs as well as the ability of the
DCs to induce proliferation of naïve T cells 11
. Synergistic action of OSCAR and TLR ligands was
also observed in monocytes and neutrophils134
.
1.4.4 OSCAR ligand
Kim and colleagues found that an OSCAR-IgFc fusion protein bound to the surface of osteoblasts
indicating that these cells express the ligand of OSCAR29
. The group of Trowsdale then reasoned,
based on the structural homology of OSCAR and collagen receptors GPVI and LAIR1, that the
OSCAR ligands could be ECM proteins and hence screened a library of the triple-helical ColII
peptides (ToolKit II). OSCAR was then identified as a receptor for ColII and was further shown to
bind strongly to collagen I, II and III, weakly to collagen IV, and not to collagen V137
. OSCAR did
not bind to the triple-helical peptide ligands for integrin 21, the GPVI ligand, (GPP)10 control

38
peptides or to the ECM proteins vitronectin or fibronectin137
. Specific binding of collagen to
OSCAR was shown by adding an anti-hOSCAR-blocking mAb that inhibited the interaction137
.
Functionally, the authors were capable of inhibiting the collagen peptide-induced osteoclastogenesis
by adding anti-hOSCAR blocking mAb to the monocytes cultured in the presence of RANKL,
supporting that the co-stimulatory signaling effect of collagen peptides are hOSCAR specific137
.
1.4.5 Regulation of OSCAR
Regulation of OSCAR has not been fully elucidated. The OSCAR gene expression was shown to be
induced by NFATc1 as well as by the microphtalmia-inducing transcription factor (Mitf) that works
synergistically with the transcription factor PU.1145
. The expression of murine OSCAR was shown
to be induced during RANKL-induced osteoclastogenesis29
through stimulation of NFATc1, which
initiates transcription of OCs-specific genes, indicating a positive feedback loop between OSCAR
and NFATc130
. It has also been reported that OSCAR expression is elevated on monocytes from
RA patients stimulated with TNF- but not with RANKL or M-CSF141
.
OSCAR signaling is negatively regulated by ITIM receptors such as CD85j (leukocyte
Ig-like receptor-1/Ig-like transcript 2)12
and LAIR-1 (Leukocyte-Associated Immunoglobulin-Like
Receptor 1)99
. For example, Tenca and colleagues12
showed that numerous DCs functions triggered
by hOSCAR stimulation were negatively regulated by CD85j. In DCs, CD85j has been shown to
inhibit intracellular calcium mobilization and strongly reduce OSCAR-mediated secretion of IL-8
and IL-12(p40). Moreover, CD85j was able to counteract the anti-apoptotic effect of OSCAR, by
reducing the Bcl-2 expression enhanced by OSCAR stimulation12
. These observations are in line
with the findings that osteoclastogenesis is enhanced in mice lacking SHP-1 or SHIP-1
phosphatases, which counterbalance the ITAM-signal in the immune system32
.
1.4.6 OSCAR in disease
OSCAR has been linked to bone disorders as well as inflammatory diseases. A single nucleotide
polymorphism within the OSCAR gene has been associated with low bone mass and osteoporosis in
postmenopausal women146
. Herman and colleagues were the first to address the role of OSCAR in
RA, looking into the OSCAR expression in the synovial tissue of RA patients. OSCAR was
expressed by OCs at the bone erosion front and by mononuclear cells (MNCs) surrounding synovial
micro vessels141
. The OSCAR expression was also associated with microvasculature in patients
with RA and OA, which is not seen in healthy tissue139
. Additionally, it was observed that the
OSCAR expression was enhanced in peripheral blood monocytes of RA patients as compared to

39
healthy subjects141
. The expression level of OSCAR positively correlated with disease activity as
well as with the potency of monocytes to differentiate into OCs in vitro, specifying the importance
of OSCAR as an enhancer of osteoclastogenesis.
Contradictory results have been shown for soluble OSCAR (sOSCAR) in synovial
fluid and plasma from RA patients138-140
. One study by Zhao et al. showed that serum levels of
sOSCAR were significantly lower in RA patients than in healthy controls138
. Opposite data was
seen in a study including 136 RA patients, who had significantly higher plasma levels of sOSCAR
compared to the healthy controls. The level of sOSCAR was also shown to correlate with the
disease progression, as the patients with destructive RA had significantly higher amounts of
sOSCAR compared to non-destructive RA patients140
.
OSCAR was also reported to be expressed on endothelial cells147;148
. In endothelial
cells, OSCAR expression is increased by oxidized low-density lipoproteins147
. Further analysis of
the OSCAR signaling in the endothelial cells revealed its involvement in proliferation,
inflammatory response and cell-cell signaling148
. Moreover, OSCAR expression in monocytic cells
was shown to be induced in atherosclerotic mice and was suggested to be regulated by pro-
atherogenic stimuli, indicating that OSCAR may play a role in the pathogenesis of
atherosclerosis149
.

40
2. Rationale and aim
As a part of the NN project “OSCAR intervention for treatment of RA”, the PhD project aimed to
gain further insights into the pathogenesis of RA. One of the characteristic features of rheumatoid
arthritis is the presence of immune cells in the synovium. Immune cells, such as DCs, monocytes
and macrophages are shown to express OSCAR136
. OSCAR has been shown to serve as a co-
stimulatory molecule for OCs differentiation29;137
and a functional role of OSCAR in human
myeloid cells have been proposed by Merck and colleagues11;134;136
. Collagen was recently
identified as naturally occurring OSCAR ligand137;142
and the functional implication of OSCAR-
collagen interaction in myeloid cells was not addressed so far. In the current project, we aimed to
assess the role of OSCAR-collagen signaling in DCs, monocytes and macrophages, using an in
vitro approach with human primary cells. Since collagen is the major component of the bone and
cartilage, it was hypothesize that OSCAR engagement by collagen on myeloid cells could
contribute to the RA pathogenesis as a driver of sustained inflammation, cartilage damage and
inflammatory bone resorption. The project therefore also intended to evaluate the effect of blocking
OSCAR-collagen interaction in DCs, monocytes and macrophages.
The outcome of the work was anticipated to provide new knowledge of the mechanisms controlling
synovial inflammation, inflammatory bone erosion and cartilage degradation, with the perspective
of identifying new therapeutic targets.

41
3. Results
The aim for this PhD thesis is to examine the functional role of the OSCAR-collagen interaction in
human immune cells of the myeloid origin.
The results are presented in the following three parts:
Results part 3.1: exploring the role of OSCAR in DCs
Results part 3.2: studies on the OSCAR-collagen interaction in monocytes from healthy donors
and synovial fluid cells from RA patients
Results part 3.3: combines preliminary data on the role of OSCAR in macrophages

42
3.1 OSCAR function in dendritic cells
This part of my studies has been published in:
Heidi S. Schultz, Louise M. Nitze, Louise H. Zeuthen, Pernille Keller, Albrecht Gruhler, Jesper
Pass, Jianhe Chen, Li Guo, Andrew J. Fleetwood, John A. Hamilton, Martin W. Berchtold and
Svetlana Panina. (2015) Collagen Induces Maturation of Human Monocyte-Derived Dendritic
Cells by Signaling through Osteoclast-Associated. J. Immunology. 194 (epub, doi 10.4049/
jimmunol.1402800)
For supplemental data see Appendix I

The Journal of Immunology
Collagen Induces Maturation of Human Monocyte-DerivedDendritic Cells by Signaling through Osteoclast-AssociatedReceptor
Heidi S. Schultz,*,† Louise M. Nitze,*,† Louise H. Zeuthen,* Pernille Keller,*
Albrecht Gruhler,* Jesper Pass,* Jianhe Chen,‡ Li Guo,‡ Andrew J. Fleetwood,x
John A. Hamilton,x Martin W. Berchtold,† and Svetlana Panina*
Osteoclast-associated receptor (OSCAR) is widely expressed on human myeloid cells. Collagen types (Col)I, II, and III have been
described as OSCAR ligands, and ColII peptides can induce costimulatory signaling in receptor activator for NF-kB–dependent
osteoclastogenesis. In this study, we isolated collagen as an OSCAR-interacting protein from the membranes of murine osteo-
blasts. We have investigated a functional outcome of the OSCAR–collagen interaction in human monocyte-derived dendritic cells
(DCs). OSCAR engagement by ColI/II-induced activation/maturation of DCs is characterized by upregulation of cell surface
markers and secretion of cytokines. These collagen-matured DCs (Col-DCs) were efficient drivers of allogeneic and autologous
naive T cell proliferation. The T cells expanded by Col-DCs secreted cytokines with no clear T cell polarization pattern. Global
RNA profiling revealed that multiple proinflammatory mediators, including cytokines and cytokine receptors, components of the
stable immune synapse (namely CD40, CD86, CD80, and ICAM-1), as well as components of TNF and TLR signaling, are
transcriptional targets of OSCAR in DCs. Our findings indicate the existence of a novel pathway by which extracellular matrix
proteins locally drive maturation of DCs during inflammatory conditions, for example, within synovial tissue of rheumatoid
arthritis patients, where collagens become exposed during tissue remodeling and are thus accessible for interaction with infil-
trating precursors of DCs. The Journal of Immunology, 2015, 194: 000–000.
Rheumatoid arthritis (RA) is a systemic autoimmune dis-ease that primarily targets the peripheral diarthrodialjoints. It is characterized by the presence of autoanti-
bodies, synovial inflammation, pannus formation, cartilage dam-age, and bone erosion. The molecular mechanisms involved in RApathogenesis remain obscure. A multistep mechanism of RA de-velopment has been proposed (1, 2). First, unknown environmentalfactors cause alterations in posttranslational protein modifications,leading to recognition of autoantigens and the loss of immune
tolerance. Next, a tissue-specific inflammatory response is elicitedin the joints. Activated immune cells accumulate in the synovialtissue and secrete cytokines, chemokines, and matrix-degradingenzymes, causing tissue damage and remodeling. By way ofa positive feedback loop, the local inflammation is sustained andfinally progresses into a systemic disorder.The cellular infiltrates in the synovial tissue are mainly com-
posed of blood-derived cells, including T and B cells, as wellas macrophages and dendritic cells (DCs) differentiated frommonocytes in the RA synovial microenvironment. Cells of themyeloid lineage are regarded as pivotal regulators of RA. Bothimmature and mature DC subsets are present in large numberswithin the RA joint, and strong evidence supports the importance ofDCs in synovial inflammation (3, 4). DCs are professional APCsthat are highly capable of inducing T cell responses. In RA, theDCs are essential for initiation and perpetuation by Ag presenta-tion as well as for cytokine and chemokine secretion (5, 6). Fur-thermore, DCs are capable of transdifferentiating into osteoclasts,contributing directly to bone erosion (7, 8). The maturation stateof the DCs is crucial for determining the balance between toler-ance and immunity. Under homeostatic conditions, DCs arelargely immature and are thought to induce tolerance. Changes inthe microenvironment stimulate maturation of DCs, which canpromote immunogenicity.A human ortholog of murine OSCAR was identified by Merck
et al. (9). OSCAR is expressed on cells of myeloid origin, and itsignals through the ITAM-harboring adaptor protein FcRg (9, 10).In DCs, cross-linking of OSCAR with specific mAbs enhancedAg presentation and promoted a semimature phenotype, causingchemokine but not proinflammatory cytokine secretion. In synergywith TLR signaling, but not alone, OSCAR cross-linking en-hanced the ability of DCs to induce naive CD4+ T cell prolifer-
*Biopharmaceutical Research Unit, Novo Nordisk A/S, 2760 Maløv, Denmark;†Department of Biology, Copenhagen University, Copenhagen 2200, Denmark;‡Novo Nordisk Research Center China, Beijing 102206, China; and xDepartmentof Medicine, University of Melbourne, Royal Melbourne Hospital, Parkville, Victoria3050, Australia
Received for publication November 5, 2014. Accepted for publication January 22,2015.
H.S.S. and L.M.N. received Ph.D. fellowships from the Danish Agency for Science,Technology, Innovation and Research.
The microarray dataset presented in this article has been submitted to the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2904/)under accession number E-MTAB-2904.
Address correspondence and reprint requests to Dr. Svetlana Panina at the currentaddress: Novo Nordisk A/S, Hvidkildevej 111, 2400 Copenhagen, Denmark. E-mailaddress: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: Col, collagen type; Col-DC, collagen-matured DC;DC, dendritic cell; ECD, extracellular domain; ECM, extracellular matrix; FDR,false discovery rate; iDC, immature DC; LC-MS, liquid chromatography–mass spec-trometry; MS, mass spectrometry; NP-40, Nonidet P-40; OSCAR, osteoclast-associated receptor; RA, rheumatoid arthritis; SPR, surface plasmon resonance.
This article is distributed under The American Association of Immunologists, Inc.,Reuse Terms and Conditions for Author Choice articles.
Copyright� 2015 by The American Association of Immunologists, Inc. 0022-1767/15/$25.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1402800

ation (9, 11, 12). It was concluded that OSCAR can inducea semimatured phenotype in DCs, which is characterized by ex-pression of costimulatory molecules but an inability to secreteproinflammatory cytokines. Such semimatured DCs are implicatedin the induction of tolerance.Recent studies have assessed the role of OSCAR in RA. OSCAR
is expressed on mononuclear cells surrounding synovial micro-vessels and on multinucleated giant cells at the bone resorptionareas. OSCARwas also found to be upregulated in peripheral bloodmonocytes from RA patients compared with healthy subjects, andits expression correlated to disease activity (13).In the present study we aimed to examine OSCAR function in
DCs with relevance to RA pathogenesis. We identified collagentypes (Col)I/II as OSCAR ligands and investigated the func-tional outcomes of OSCAR–collagen interaction in DCs. Duringthe preparation of this manuscript, Trowsdale and colleagues (14)identified ColI, -II and -III to be ligands of OSCAR (14). Theauthors showed that within joints collagens are exposed toOSCAR-expressing osteoclasts and osteoclast precursors. In vitro,OSCAR-binding collagen peptides promoted receptor activator forNF-kB ligand–induced osteoclast differentiation from blood-derived monocytes, indicating that OSCAR is an activating re-ceptor on osteoclast precursor cells. In the present study, we showthat OSCAR–collagen interaction provides an activating signalalso in monocyte-derived DCs. This indicates an important, yetpoorly recognized, role of the extracellular matrix (ECM) in thearthritic joint, where ColI/II become exposed due to ongoing tis-sue remodeling, thus providing activating signals to myeloid cellsto drive a sustained inflammation. Blocking the OSCAR signaling,for example, with a mAb, may be considered as a novel thera-peutic approach for RA.
Materials and MethodsProteins and Abs for functional assays
The extracellular domain (ECD) of human OSCAR and OSCAR–Fc fusionprotein were produced by recombinant technique as follows. The fragmentencoding ECD (amino acid residues 1–227; sequence ID NP_573399) wasamplified by RT-PCR using human universal cDNA (Clontech) as tem-plate, cloned into the pJSV002 expression vector, and sequenced. Therecombinant protein was expressed in HEK293-6E cells. The purifiedrecombinant protein is an N-glycosylated monomer of ∼25 kDa. For ex-pression of OSCAR–Fc protein, the ECD-encoding cDNA was cloned in-frame with a sequence encoding the Fc fragment of human IgG4. Thesequence was verified and the protein was expressed in HEK293-6E cells.The purified protein is an N-glycosylated dimer of ∼100 kDa. For cellbinding studies, OSCAR-ECD was labeled using an Alexa Fluor 488protein staining kit (Life Technologies) according to the manufacturer’sprotocol.
A panel of murine mAbs against OSCAR-ECD and isotype controlswere generated using hybridoma technology and then humanized usingrecombinant technology. The CDR of the generated mAb was grafted ona germline-encoded, human-mutated IgG1 Fc part, Fc-hIgG1.1 (15, 16).Five point mutations introduced in the Fc-hIgG1.1 hinder its binding tohigh-affinity Fc receptors. As isotype controls, an anti-OSCAR non-blocking high-affinity mAb as well as anti-TNP hIgG1.1 were used. Mostblocking experiments were performed with the use of several differenthigh-affinity anti-OSCAR–blocking mAbs and the two above-mentionedisotype controls with identical results. For simplicity, data are shownonly for 1 mg/ml mAb unless specified otherwise. For functional assays,the following commercially available mAbs were used: anti-CD3 mAb(clone OKT3; eBioscience), anti-CD3/CD28 expander beads (Dynal),and anti–integrin a2b1 mAb (clone BHA2.1; Millipore). ColI purifiedfrom human skin was purchased from Sigma-Aldrich. ColII purifiedfrom human cartilage was purchased from Millipore. ColIII purified fromhuman placenta was purchased from Millipore. ColIV from humanplacenta was purchased from Sigma-Aldrich. Endotoxin level in theprotein solutions was measured using the turbidimetric kinetic Limulusamebocyte lysate assay (reagents supplied by Charles River Laborato-ries). All proteins used for functional assays in this study were essen-tially endotoxin-free.
Identification of OSCAR ligand
OSCAR-binding proteins were purified from membranes of MC3T3-E1cells by affinity chromatography on an OSCAR–Fc matrix followed byidentification of hits using a liquid chromatography–mass spectrometry(LC-MS) approach. In brief, for extraction of membrane proteins, the cellswere washed in PBS, scraped with a plastic policeman, and homogenizedmechanically using a glass pastel homogenizer in the buffer containing 10mM imidazole and 1 mM EDTA (pH 7.4) on ice. The homogenate wasspun at 20,000 3 g for 10 min, and the pellet was resuspended in thehomogenization buffer. This washing cycle was repeated three times withone last time in a buffer containing 2 mM imidazole/1 mM EDTA (pH7.4). The pellet was then washed twice in 50 mM Tris-HCl/150 mM NaCl(pH 8.2), centrifuged, and resuspended in a RIPA buffer (150 mM NaCl,50 mM Tris-HCl [pH 8.2], 1 mM EDTA, 1% Nonidet P-40 [NP-40], 1 mg/mlBSA, protease inhibitor [cOmplete, Roche] and phosphatase inhibitormixture [Sigma-Aldrich]) for 30 min on ice. The protein extracts wereclarified by centrifugation at 20,000 3 g. Protein concentration wasmeasured using a bicinchoninic acid protein assay (Pierce).
Preparation of an affinity resin was performed as follows. HumanOSCAR–Fc IgG4 or Fc IgG4 (control) recombinant proteins were bound toa protein G–Sepharose 4B Fast Flow affinity matrix (Sigma-Aldrich)according to the manufacturer’s protocol and then chemically cross-linked as described in Schneider et al. (16). This technique allows opti-mal orientation of OSCAR-Fc on the beads and thus efficient binding ofinteracting proteins with minimal/no leakage of the bait during the elutionprocess. After cross-linking, the beads were thoroughly washed in 0.1 Mborate buffer, equilibrated in 5 ml 0.5 M Tris-HCl (pH 8.2), and sedi-mented by centrifugation. Cell membrane extracts in RIPA buffer wereadded to the affinity beads. NaCl concentration was adjusted to final 0.5 Mand the beads were incubated for 3 h at 4˚C with gentle agitation. Thebeads were then washed sequentially with three buffers containing 1) 0.5 MNaCl, 50 mM Tris-HCl, 1 mM EDTA, and 0.5% NP-40 (pH 8.2); 2) 150mM NaCl, 50 mM Tris-HCl, 0.5% NP-40, and 0.1% SDS (pH 8.2); and 3)150 mM NaCl and 0.5% CHAPS. The latter wash was repeated twice. Theproteins bound to the affinity matrix were eluted in 50 mM diethylamine/0.5% CHAPS (pH 11.5) and pH was immediately adjusted to neutral usingPBS and borate buffer. The samples were subjected to a 4–12% SDS-PAGE. The gels were stained using a ProteoSilver plus silver stain kit(Sigma-Aldrich). The bands differentially bound to the OSCAR–Fc matrixwere subjected to LC-MS analysis as follows. Bands were excised from theSDS-PAGE gel, destained with the SilverQuest silver staining kit (Invi-trogen), reduced with DTT, alkylated with iodoacetamide, and digestedwith trypsin with a slightly modified version of the method described inShevchenko et al. (17).
LC-MS analysis
Peptide mixtures were analyzed by nanoscale liquid chromatography–electrospray ionization–tandem MS on an Easy-nLC (Proxeon Biosystems)connected to an LTQ Orbitrap mass spectrometer (Thermo Fisher Scien-tific). Reversed phase chromatography was performed with 0.1% formicacid (buffer A) and acetonitrile/0.1% formic acid (buffer B). Peptides weretrapped on a precolumn (Proxeon Biosystems) and separated on a 10-cmfused-silica column (internal diameter 75 mm) packed with 3-mmReproSil-Pur C18-AQ (Proxeon Biosystems). MS analysis was performedon the LTQ Orbitrap in data-dependent mode, where one MS survey scanwas followed by MS fragmentation spectra of the four most intense ions.
Analysis of the MS data were performed with Proteome Discoverer 1.4(Thermo Fisher Scientific). Peak lists of the MS fragmentation spectra weregenerated and searched in MASCOT against the mammalian part of theSwissProt database with oxidation (P), oxidation (M), and deamidated (NQ)as dynamic modifications and carbamidomethyl (C) as fixed modification.Results were filtered for a false discovery rate (FDR) of 0.01 and aminimumMASCOT peptide score of 25.
Binding of OSCAR to collagen
Binding of the soluble extracellular domain of OSCAR to human collagenswas tested by surface plasmon resonance (SPR) on a ProteOn instrument(Bio-Rad). Human collagens were immobilized on a GLC sensor chip usingthe amine coupling kit (Bio-Rad). The experiment was performed at 15˚Cusing HBS-EP+ (GE Healthcare) as running buffer for the immobilizationand HBS-EP+/0.1% BSA for binding analysis. The chip was activated with1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride/normalhuman serum (diluted 2:1 with H2O) according to instructions. Collagenswere diluted in 10 mM NaAc buffer (pH 4.5) to 25 mg/ml and injected for200 s followed by quenching with ethanolamine. OSCAR-ECD wasinjected at 2 mM, 1 mM, 500 nM, 250 nM, and 125 nM for 200 s at a flow
2 OSCAR-DEPENDENT MATURATION OF DCs

of 60 ml/min followed by regeneration with 10 mM glycine (pH 2.0) for30 s. Binding curves were double referenced by subtraction of the signalsfrom reference cells and a buffer injection using the ProteOn Managersoftware.
Competition of OSCAR binding to collagen
Competition of anti-OSCAR mAb with OSCAR binding to collagen wasmeasured by SPR on a Biacore T100 instrument using the same buffers asdescribed above.
Human ColI was diluted with 10 mM NaAc (pH 4.5) to a final con-centration of 2.5 mg/ml and immobilized on a CM5 sensor chip (GEHealthcare) using the amine coupling kit (GE Healthcare). Immobilizationwas carried out at 4˚C with HBS-EP+ as buffer using 200 resonance unitsas a target level for the immobilized ligand. OSCAR was diluted to 25 nMin HBS-EP+ buffer containing 0.1% serum albumin and increasing con-centrations of the anti-OSCAR Abs from 0 to 20 mg/ml. The OSCAR/mAbsolutions were injected for 240 s followed by 200 s dissociation at a flowrate of 50 ml/min at 4˚C followed by regeneration with 10 mM glycine (pH2.0). Binding curves were double referenced by subtraction of the signalsfrom reference cells and a buffer injection and further analyzed using theBiacore T100 evaluation software.
Cells
Buffy coats from healthy individuals were obtained anonymously from theBlood Bank, Rigshospitalet, Copenhagen, Denmark. All donors gave in-formed consent according to the protocol approved by the Ethics Committeefor Copenhagen, Denmark, for research use (approval no. H-D-2008-113).PBMCs were freshly purified by Ficoll-Plaque density centrifugation.Monocytes were purified from PBMCs using the CD14MicroBeads positiveselection kit (Miltenyi Biotec). To induce DC differentiation, monocyteswere cultured in RPMI 1640/10% FCS in the presence of 50 ng/mlrecombinant human GM-CSF and 50 ng/ml recombinant human IL-4(Novo Nordisk) for 6 d. These cells are referred to as immature DCs(iDCs). Naive CD4+RA+ cells were purified from PBMCs using a humannaive CD4+ T cell enrichment kit (StemCell Technologies). For freezing,T cells were resuspended at a density of 7 3 106/ml in RPMI 1640/50%FBS/10% DMSO, and cryovials were placed in a Mr. Frosty container(Thermo Fisher Scientific) at 280˚C overnight and then transferred intoa 2140˚C freezer. Viability of the thawed cells was ∼95% as measuredusing a NucleoCounter (ChemoMetec).
DC culture on collagen
Untreated virgin polystyrene 24-well plates were coated with ColI or ColIIin PBS at ∼3 mg/cm2 overnight at 4˚C. iDCs (13 106) were added per wellafter being preincubated for 20 min with indicated mAb. The cells werecultured for 20 h unless otherwise specified. ColI and ColII had similareffects in all experiments. ColII was always slightly more potent than ColI.Functional data in the manuscript are shown for ColII only unless other-wise specified.
Allogeneic and autologous MLR
For allogeneic MLRs freshly purified cells were used. For autologousreactions, T cells were kept frozen in RPMI 1640/50% FBS/10% DMSOuntil the autologous DCs became matured. T cells (1 3 105) werecocultured in triplicate wells with 1 3 103 DCs in RPMI 1640/10% FCSgrowth medium. No live DCs were detected in the coculture at the time oftermination of the experiment. To assess T cell proliferation pulse labelingof cells with [3H]thymidine was used to monitor DNA synthesis. Briefly, atday 4 of coculture the cells were pulsed with 0.5 mCi [3H]thymidine(PerkinElmer) for 18 h. [3H]thymidine incorporation was measured byliquid scintillation counting using a TopCount NXT (PerkinElmer).
Cytokine secretion
Multiplex analysis of cytokines was performed using the BioPlex 200cytokine assay platform (Bio-Rad). IL-23 (p19/p40) was detected using thePlatinum ELISA kit (eBioscience).
Expression of cell surface markers
For analysis of cell surface marker expression, the following Abs were used:BD multicolor PE-conjugated mouse monoclonal anti-human CD86,PerCP-Cy5.5–conjugated mouse anti-human CD209, allophycocyanin-conjugated mouse anti-human CD83 (BD Biosciences), FITC-conju-gated mouse anti-human HLA-DR (BD Pharmingen), allophycocyanin-conjugated mouse anti-human CD14 (BD Pharmingen), PE-conjugatedmouse anti-human OSCAR (clone 11.1CN5; Beckman Coulter), and Pa-
cific Blue–conjugated anti-human CD3 (BD Pharmingen). Cells werewashed twice in PBS with 2% FCS and then stained for 30 min at 37˚C inPBS with 2% FCS in the presence of 1% human serum albumin to blockunspecific binding. Cells were rinsed three times in PBS with 2% FCSprior to flow cytometry analysis. Data acquisition was done usinga FACSFortessa (BD Biosciences). Only live cells, that is, negative forallophycocyanin-H7 dead cell stain (Invitrogen/Life Technologies) wereanalyzed. For compensation single stain was used with one drop of neg-ative control beads and anti-mouse IgG beads (BD Biosciences). For thedead cell marker compensation, amine reactive compensation beads wereused (Invitrogen/Life Technologies). Data analysis was performed usingKaluza software version 1.2 (Beckman Coulter).
Microarray analysis
Total RNAwas obtained 4 and 20 h after stimulating DCs with ColI/II in theabsence or presence of anti-OSCARmAb or isotype control mAb. RNAwasextracted using TRIzol (Invitrogen/Life Technologies) followed by puri-fication with an RNeasy MinElute cleanup kit (Qiagen). The RNA integritywas evaluated on an Agilent 2100 bioanalyzer using Agilent RNA 6000Nano Kit chips (Agilent Technologies), with RNA integrity number scoresof $9.9. Microarray experiments were performed according to the pro-tocols supplied by Affymetrix and run on the HT HG-U133+ PM chipfrom Affymetrix using an Affymetrix GeneTitan machine in a 96-welllayout. Array data were normalized using the robust multiarray averagealgorithm with a custom chip definition file obtained from BrainArray(University of Michigan) based on Ensembl gene annotations (ENSG).Microarray data were analyzed for significant differences using QlucoreOmics Explorer (v2.2) software. A paired two-group comparison wasperformed with Benjamini–Hochberg correction for multiple testing ap-plying a FDR of 5% and a biological threshold of .2-fold regulation.Differentially expressed genes were subjected to gene ontology analysisfor biological processes using Ingenuity Pathway Analysis. The datasetand technical information compliant with minimum information abouta microarray experiment can be found at the EMBL-EBI ArrayExpressdepository, accession no. E-MTAB-2904 (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2904/).
Statistical analysis
Statistical analyses were performed in GraphPad Prism 5 using Student two-tailed t tests. A p value ,0.05 was considered statistically significant.Unless otherwise indicated, means and SD are shown.
ResultsIdentification of OSCAR ligand
It was previously shown that a putative ligand for murine OSCAR isexpressed on the cell surface of osteoblasts (18). In the presentstudy, we screened a panel of cell lines for expression of a putativeligand for human OSCAR by monitoring binding of fluorescentlylabeled OSCAR-ECD to the surface of live cells. A murineMC3T3-E1 preosteoblast cell line was selected for purification ofthe OSCAR ligand. Using affinity chromatography followed byliquid chromatography–tandem MS analysis of eluted proteins, wehave identified a1 ColI as OSCAR-interacting protein. Directinteraction of OSCAR with native human ColI an ColII wasconfirmed by SPR, whereas no binding was detected to ColIII orColIV (Supplemental Fig. 1). Thus, ColI and ColII serve as nat-urally occurring OSCAR ligands.Anti-OSCAR mAb capable of blocking OSCAR–collagen in-
teraction as well as nonblocking mAb were selected for furtherfunctional assays using SPR (Supplemental Fig. 2).
OSCAR–collagen signaling promotes DC survival
It has previously been found that cross-linking of OSCAR with anmAb promoted survival of monocyte-derived DCs (11). In thepresent study, we tested whether the naturally occurring OSCARligands, namely ColI/II, could promote DC survival. We differ-entiated iDCs from peripheral blood monocytes in the presence ofGM-CSF and IL-4. This method allows generation of cells phe-notypically resembling “inflammatory” DCs, which differentiatein vivo from blood-derived precursors during inflammation (19).
The Journal of Immunology 3
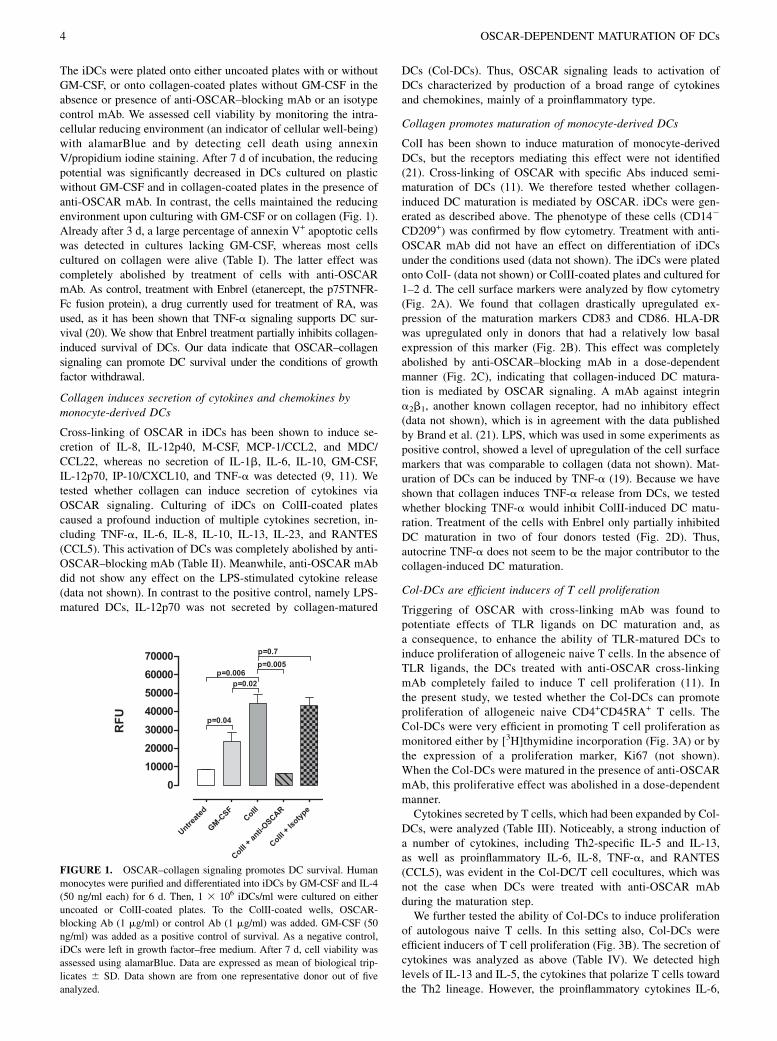
The iDCs were plated onto either uncoated plates with or withoutGM-CSF, or onto collagen-coated plates without GM-CSF in theabsence or presence of anti-OSCAR–blocking mAb or an isotypecontrol mAb. We assessed cell viability by monitoring the intra-cellular reducing environment (an indicator of cellular well-being)with alamarBlue and by detecting cell death using annexinV/propidium iodine staining. After 7 d of incubation, the reducingpotential was significantly decreased in DCs cultured on plasticwithout GM-CSF and in collagen-coated plates in the presence ofanti-OSCAR mAb. In contrast, the cells maintained the reducingenvironment upon culturing with GM-CSF or on collagen (Fig. 1).Already after 3 d, a large percentage of annexin V+ apoptotic cellswas detected in cultures lacking GM-CSF, whereas most cellscultured on collagen were alive (Table I). The latter effect wascompletely abolished by treatment of cells with anti-OSCARmAb. As control, treatment with Enbrel (etanercept, the p75TNFR-Fc fusion protein), a drug currently used for treatment of RA, wasused, as it has been shown that TNF-a signaling supports DC sur-vival (20). We show that Enbrel treatment partially inhibits collagen-induced survival of DCs. Our data indicate that OSCAR–collagensignaling can promote DC survival under the conditions of growthfactor withdrawal.
Collagen induces secretion of cytokines and chemokines bymonocyte-derived DCs
Cross-linking of OSCAR in iDCs has been shown to induce se-cretion of IL-8, IL-12p40, M-CSF, MCP-1/CCL2, and MDC/CCL22, whereas no secretion of IL-1b, IL-6, IL-10, GM-CSF,IL-12p70, IP-10/CXCL10, and TNF-a was detected (9, 11). Wetested whether collagen can induce secretion of cytokines viaOSCAR signaling. Culturing of iDCs on ColII-coated platescaused a profound induction of multiple cytokines secretion, in-cluding TNF-a, IL-6, IL-8, IL-10, IL-13, IL-23, and RANTES(CCL5). This activation of DCs was completely abolished by anti-OSCAR–blocking mAb (Table II). Meanwhile, anti-OSCAR mAbdid not show any effect on the LPS-stimulated cytokine release(data not shown). In contrast to the positive control, namely LPS-matured DCs, IL-12p70 was not secreted by collagen-matured
DCs (Col-DCs). Thus, OSCAR signaling leads to activation ofDCs characterized by production of a broad range of cytokinesand chemokines, mainly of a proinflammatory type.
Collagen promotes maturation of monocyte-derived DCs
ColI has been shown to induce maturation of monocyte-derivedDCs, but the receptors mediating this effect were not identified(21). Cross-linking of OSCAR with specific Abs induced semi-maturation of DCs (11). We therefore tested whether collagen-induced DC maturation is mediated by OSCAR. iDCs were gen-erated as described above. The phenotype of these cells (CD142
CD209+) was confirmed by flow cytometry. Treatment with anti-OSCAR mAb did not have an effect on differentiation of iDCsunder the conditions used (data not shown). The iDCs were platedonto ColI- (data not shown) or ColII-coated plates and cultured for1–2 d. The cell surface markers were analyzed by flow cytometry(Fig. 2A). We found that collagen drastically upregulated ex-pression of the maturation markers CD83 and CD86. HLA-DRwas upregulated only in donors that had a relatively low basalexpression of this marker (Fig. 2B). This effect was completelyabolished by anti-OSCAR–blocking mAb in a dose-dependentmanner (Fig. 2C), indicating that collagen-induced DC matura-tion is mediated by OSCAR signaling. A mAb against integrina2b1, another known collagen receptor, had no inhibitory effect(data not shown), which is in agreement with the data publishedby Brand et al. (21). LPS, which was used in some experiments aspositive control, showed a level of upregulation of the cell surfacemarkers that was comparable to collagen (data not shown). Mat-uration of DCs can be induced by TNF-a (19). Because we haveshown that collagen induces TNF-a release from DCs, we testedwhether blocking TNF-a would inhibit ColII-induced DC matu-ration. Treatment of the cells with Enbrel only partially inhibitedDC maturation in two of four donors tested (Fig. 2D). Thus,autocrine TNF-a does not seem to be the major contributor to thecollagen-induced DC maturation.
Col-DCs are efficient inducers of T cell proliferation
Triggering of OSCAR with cross-linking mAb was found topotentiate effects of TLR ligands on DC maturation and, asa consequence, to enhance the ability of TLR-matured DCs toinduce proliferation of allogeneic naive T cells. In the absence ofTLR ligands, the DCs treated with anti-OSCAR cross-linkingmAb completely failed to induce T cell proliferation (11). Inthe present study, we tested whether the Col-DCs can promoteproliferation of allogeneic naive CD4+CD45RA+ T cells. TheCol-DCs were very efficient in promoting T cell proliferation asmonitored either by [3H]thymidine incorporation (Fig. 3A) or bythe expression of a proliferation marker, Ki67 (not shown).When the Col-DCs were matured in the presence of anti-OSCARmAb, this proliferative effect was abolished in a dose-dependentmanner.Cytokines secreted by T cells, which had been expanded by Col-
DCs, were analyzed (Table III). Noticeably, a strong induction ofa number of cytokines, including Th2-specific IL-5 and IL-13,as well as proinflammatory IL-6, IL-8, TNF-a, and RANTES(CCL5), was evident in the Col-DC/T cell cocultures, which wasnot the case when DCs were treated with anti-OSCAR mAbduring the maturation step.We further tested the ability of Col-DCs to induce proliferation
of autologous naive T cells. In this setting also, Col-DCs wereefficient inducers of T cell proliferation (Fig. 3B). The secretion ofcytokines was analyzed as above (Table IV). We detected highlevels of IL-13 and IL-5, the cytokines that polarize T cells towardthe Th2 lineage. However, the proinflammatory cytokines IL-6,
FIGURE 1. OSCAR–collagen signaling promotes DC survival. Human
monocytes were purified and differentiated into iDCs by GM-CSF and IL-4
(50 ng/ml each) for 6 d. Then, 1 3 106 iDCs/ml were cultured on either
uncoated or ColII-coated plates. To the ColII-coated wells, OSCAR-
blocking Ab (1 mg/ml) or control Ab (1 mg/ml) was added. GM-CSF (50
ng/ml) was added as a positive control of survival. As a negative control,
iDCs were left in growth factor–free medium. After 7 d, cell viability was
assessed using alamarBlue. Data are expressed as mean of biological trip-
licates 6 SD. Data shown are from one representative donor out of five
analyzed.
4 OSCAR-DEPENDENT MATURATION OF DCs
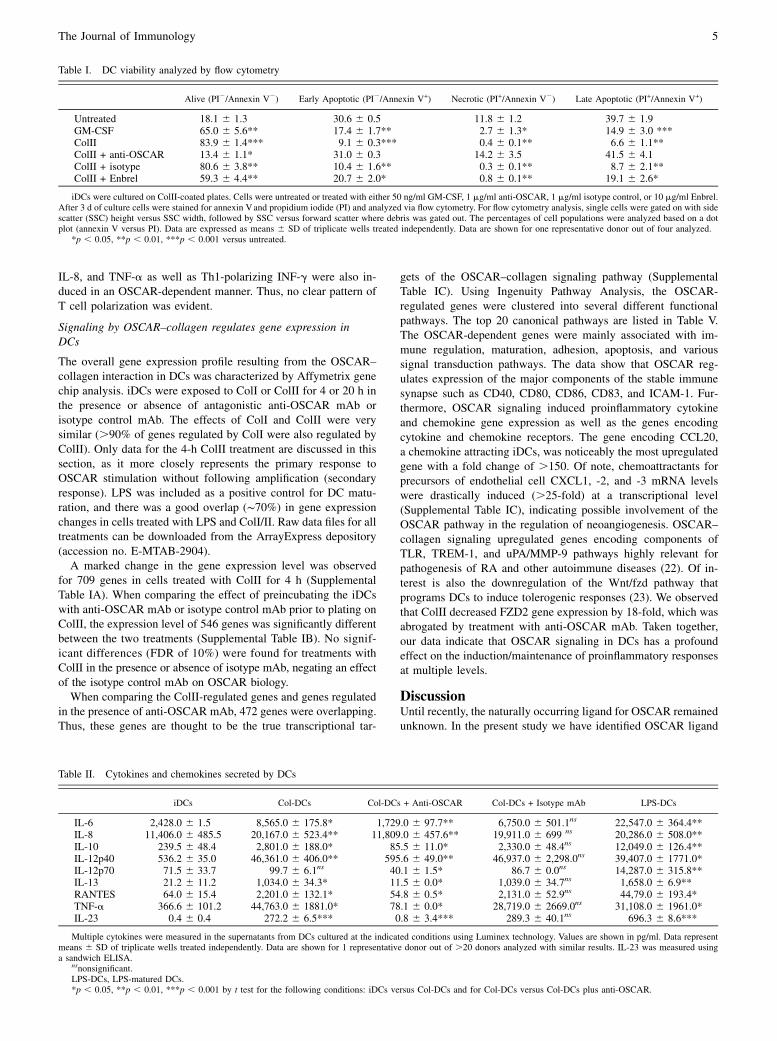
IL-8, and TNF-a as well as Th1-polarizing INF-g were also in-duced in an OSCAR-dependent manner. Thus, no clear pattern ofT cell polarization was evident.
Signaling by OSCAR–collagen regulates gene expression inDCs
The overall gene expression profile resulting from the OSCAR–collagen interaction in DCs was characterized by Affymetrix genechip analysis. iDCs were exposed to ColI or ColII for 4 or 20 h inthe presence or absence of antagonistic anti-OSCAR mAb orisotype control mAb. The effects of ColI and ColII were verysimilar (.90% of genes regulated by ColI were also regulated byColII). Only data for the 4-h ColII treatment are discussed in thissection, as it more closely represents the primary response toOSCAR stimulation without following amplification (secondaryresponse). LPS was included as a positive control for DC matu-ration, and there was a good overlap (∼70%) in gene expressionchanges in cells treated with LPS and ColI/II. Raw data files for alltreatments can be downloaded from the ArrayExpress depository(accession no. E-MTAB-2904).A marked change in the gene expression level was observed
for 709 genes in cells treated with ColII for 4 h (SupplementalTable IA). When comparing the effect of preincubating the iDCswith anti-OSCAR mAb or isotype control mAb prior to plating onColII, the expression level of 546 genes was significantly differentbetween the two treatments (Supplemental Table IB). No signif-icant differences (FDR of 10%) were found for treatments withColII in the presence or absence of isotype mAb, negating an effectof the isotype control mAb on OSCAR biology.When comparing the ColII-regulated genes and genes regulated
in the presence of anti-OSCAR mAb, 472 genes were overlapping.Thus, these genes are thought to be the true transcriptional tar-
gets of the OSCAR–collagen signaling pathway (SupplementalTable IC). Using Ingenuity Pathway Analysis, the OSCAR-regulated genes were clustered into several different functionalpathways. The top 20 canonical pathways are listed in Table V.The OSCAR-dependent genes were mainly associated with im-mune regulation, maturation, adhesion, apoptosis, and varioussignal transduction pathways. The data show that OSCAR reg-ulates expression of the major components of the stable immunesynapse such as CD40, CD80, CD86, CD83, and ICAM-1. Fur-thermore, OSCAR signaling induced proinflammatory cytokineand chemokine gene expression as well as the genes encodingcytokine and chemokine receptors. The gene encoding CCL20,a chemokine attracting iDCs, was noticeably the most upregulatedgene with a fold change of .150. Of note, chemoattractants forprecursors of endothelial cell CXCL1, -2, and -3 mRNA levelswere drastically induced (.25-fold) at a transcriptional level(Supplemental Table IC), indicating possible involvement of theOSCAR pathway in the regulation of neoangiogenesis. OSCAR–collagen signaling upregulated genes encoding components ofTLR, TREM-1, and uPA/MMP-9 pathways highly relevant forpathogenesis of RA and other autoimmune diseases (22). Of in-terest is also the downregulation of the Wnt/fzd pathway thatprograms DCs to induce tolerogenic responses (23). We observedthat ColII decreased FZD2 gene expression by 18-fold, which wasabrogated by treatment with anti-OSCAR mAb. Taken together,our data indicate that OSCAR signaling in DCs has a profoundeffect on the induction/maintenance of proinflammatory responsesat multiple levels.
DiscussionUntil recently, the naturally occurring ligand for OSCAR remainedunknown. In the present study we have identified OSCAR ligand
Table I. DC viability analyzed by flow cytometry
Alive (PI2/Annexin V2) Early Apoptotic (PI2/Annexin V+) Necrotic (PI+/Annexin V2) Late Apoptotic (PI+/Annexin V+)
Untreated 18.1 6 1.3 30.6 6 0.5 11.8 6 1.2 39.7 6 1.9GM-CSF 65.0 6 5.6** 17.4 6 1.7** 2.7 6 1.3* 14.9 6 3.0 ***ColII 83.9 6 1.4*** 9.1 6 0.3*** 0.4 6 0.1** 6.6 6 1.1**ColII + anti-OSCAR 13.4 6 1.1* 31.0 6 0.3 14.2 6 3.5 41.5 6 4.1ColII + isotype 80.6 6 3.8** 10.4 6 1.6** 0.3 6 0.1** 8.7 6 2.1**ColII + Enbrel 59.3 6 4.4** 20.7 6 2.0* 0.8 6 0.1** 19.1 6 2.6*
iDCs were cultured on ColII-coated plates. Cells were untreated or treated with either 50 ng/ml GM-CSF, 1 mg/ml anti-OSCAR, 1 mg/ml isotype control, or 10 mg/ml Enbrel.After 3 d of culture cells were stained for annexin Vand propidium iodide (PI) and analyzed via flow cytometry. For flow cytometry analysis, single cells were gated on with sidescatter (SSC) height versus SSC width, followed by SSC versus forward scatter where debris was gated out. The percentages of cell populations were analyzed based on a dotplot (annexin V versus PI). Data are expressed as means 6 SD of triplicate wells treated independently. Data are shown for one representative donor out of four analyzed.
*p , 0.05, **p , 0.01, ***p , 0.001 versus untreated.
Table II. Cytokines and chemokines secreted by DCs
iDCs Col-DCs Col-DCs + Anti-OSCAR Col-DCs + Isotype mAb LPS-DCs
IL-6 2,428.0 6 1.5 8,565.0 6 175.8* 1,729.0 6 97.7** 6,750.0 6 501.1ns 22,547.0 6 364.4**IL-8 11,406.0 6 485.5 20,167.0 6 523.4** 11,809.0 6 457.6** 19,911.0 6 699 ns 20,286.0 6 508.0**IL-10 239.5 6 48.4 2,801.0 6 188.0* 85.5 6 11.0* 2,330.0 6 48.4ns 12,049.0 6 126.4**IL-12p40 536.2 6 35.0 46,361.0 6 406.0** 595.6 6 49.0** 46,937.0 6 2,298.0ns 39,407.0 6 1771.0*IL-12p70 71.5 6 33.7 99.7 6 6.1ns 40.1 6 1.5* 86.7 6 0.0ns 14,287.0 6 315.8**IL-13 21.2 6 11.2 1,034.0 6 34.3* 11.5 6 0.0* 1,039.0 6 34.7ns 1,658.0 6 6.9**RANTES 64.0 6 15.4 2,201.0 6 132.1* 54.8 6 0.5* 2,131.0 6 52.9ns 44,79.0 6 193.4*TNF-a 366.6 6 101.2 44,763.0 6 1881.0* 78.1 6 0.0* 28,719.0 6 2669.0ns 31,108.0 6 1961.0*IL-23 0.4 6 0.4 272.2 6 6.5*** 0.8 6 3.4*** 289.3 6 40.1ns 696.3 6 8.6***
Multiple cytokines were measured in the supernatants from DCs cultured at the indicated conditions using Luminex technology. Values are shown in pg/ml. Data representmeans 6 SD of triplicate wells treated independently. Data are shown for 1 representative donor out of .20 donors analyzed with similar results. IL-23 was measured usinga sandwich ELISA.
nsnonsignificant.LPS-DCs, LPS-matured DCs.*p , 0.05, **p , 0.01, ***p , 0.001 by t test for the following conditions: iDCs versus Col-DCs and for Col-DCs versus Col-DCs plus anti-OSCAR.
The Journal of Immunology 5
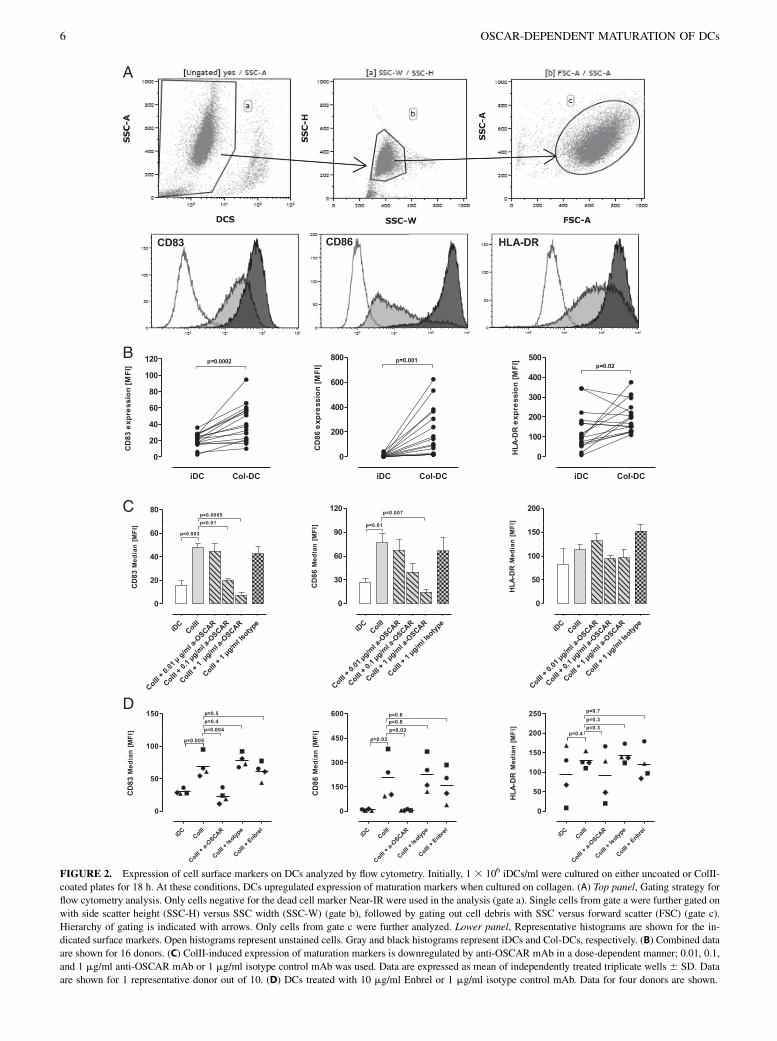
FIGURE 2. Expression of cell surface markers on DCs analyzed by flow cytometry. Initially, 1 3 106 iDCs/ml were cultured on either uncoated or ColII-
coated plates for 18 h. At these conditions, DCs upregulated expression of maturation markers when cultured on collagen. (A) Top panel, Gating strategy for
flow cytometry analysis. Only cells negative for the dead cell marker Near-IR were used in the analysis (gate a). Single cells from gate a were further gated on
with side scatter height (SSC-H) versus SSC width (SSC-W) (gate b), followed by gating out cell debris with SSC versus forward scatter (FSC) (gate c).
Hierarchy of gating is indicated with arrows. Only cells from gate c were further analyzed. Lower panel, Representative histograms are shown for the in-
dicated surface markers. Open histograms represent unstained cells. Gray and black histograms represent iDCs and Col-DCs, respectively. (B) Combined data
are shown for 16 donors. (C) ColII-induced expression of maturation markers is downregulated by anti-OSCAR mAb in a dose-dependent manner; 0.01, 0.1,
and 1 mg/ml anti-OSCAR mAb or 1 mg/ml isotype control mAb was used. Data are expressed as mean of independently treated triplicate wells 6 SD. Data
are shown for 1 representative donor out of 10. (D) DCs treated with 10 mg/ml Enbrel or 1 mg/ml isotype control mAb. Data for four donors are shown.
6 OSCAR-DEPENDENT MATURATION OF DCs

expressed on murine preosteoblasts as being a1 ColI. OSCARinteraction with native human ColI and ColII was confirmed bythe SPR technique. While we were conducting our experiments,the Trowsdale et al. (14) published their discovery of collagens asligands of OSCAR. In this study, the authors used a syntheticColII peptide library and identified a number of peptides showingdirect binding to human OSCAR. Some of these peptides wereefficient stimulators of receptor activator for NF-kB ligand–de-pendent osteoclastogenesis. The authors confirmed binding ofOSCAR to native human ColI, ColII, and ColIII. We neither sawa binding to native ColIII using SPR analysis nor a biologicaleffect on DCs, which might be attributed to differences in qualitiesof the native protein preparations used in the two studies.Collagens are broadly distributed in the human and, in addition
to their structural function, they play an important role in celladhesion, migration, proliferation, differentiation, and survival(24). These multiple and diverse effects are highly dependent onthe ECM structure and remodeling (25). ColI is the main proteinconstituent of bone, whereas ColII is the major protein componentof cartilage. Under pathological conditions, such as, for example,RA, the ECM undergoes excessive remodeling with drastic up-regulation of the ColI–III turnover within the joints (26). Wesuggest that under such conditions, collagens of bone and cartilagemay become available for interaction with infiltrating immunecells and may considerably influence their activation status andviability within RA joints. Indeed, it has been shown that OSCAR-expressing mononuclear cells are in contact with collagens in thejoints and the OSCAR-binding sites are expected to be on thesurface of collagens exposed to the infiltrating cells (14).Among the cells expressing the OSCAR protein and potentially
capable of entering synovial tissue upon inflammation aremonocytes, monocyte-derived DCs, and lineage-negative CD11c+
DC (9). Whereas the latter are thought to represent a rare het-erogeneous population of cells found in health and disease,monocytes may serve as precursors of inflammatory DCs, whichare only generated in pathological conditions, such as, for ex-ample, RA (27). Development of such inflammatory, fully func-tional DCs is commonly modeled in vitro by treatment ofperipheral blood monocytes with GM-CSF and IL-4 (19). Themonocyte-derived DCs are potent APCs, which secrete multipleproinflammatory cytokines and express CD1a, a surface marker
that was shown to be specific for this cell type (28). CD1a+ cellswere found in RA synovial tissue, where they are thought to befunctional (29). In the present study, we chose to focus on thispopulation of inflammatory DCs to mechanistically address theoutcome of OSCAR–collagen signaling with relevance to the RApathogenesis, although we cannot exclude an impact of theOSCAR–collagen interaction on the Ag-presenting function insubpopulations of the CD11c+ cells, which should further beaddressed in clinical studies.Merck et al. (9, 11) have shown that cross-linking of OSCAR
with the receptor-specific Abs can provide survival, activation, andmaturation signals to human monocyte-derived DCs. The use ofcross-linking mAbs to stimulate the ITAM-bearing receptors viaaggregation is an efficient way to address the signaling potential ofa receptor when its natural ligand is unknown. However, an en-dogenous ligand may induce a signal of different magnitude andquality, depending on the affinity of interaction and cross-talkingsignaling pathways modulating the ITAM signal. Therefore, it isessential to investigate the outcome of the OSCAR signaling in thepresence of the native ligand in the cells where other collagenreceptors are coexpressed.OSCAR expression in human monocyte-derived DCs (11) and
monocytes (30) was suggested to support cell survival in the ab-sence of growth factors via induction of antiapoptotic moleculesBcl-2 and Bcl-xL through PI3K and ERK signaling. In our study,the DCs plated on ColI/II showed prolonged viability in the ab-sence of the essential growth factor, GM-CSF, indicating thatOSCAR could play an important role in cell survival. Expressionof genes encoding multiple components of the ERK pathway(including MAP3K1 and MAP3K8), as well as BCL2A1 encodinga Bcl-2 related protein, was strongly upregulated by the OSCAR–Col signaling. Autocrine TNF-a was previously shown to providesurvival signals to DCs (20). Blocking of TNF-a with eithersoluble p55TNFR-Fc or anti–TNF-a mAb (infliximab) signifi-cantly reduced survival of LPS-matured DCs (31). We show in thepresent study that survival of Col-DCs was impaired by treatmentwith Enbrel, although to a much lower degree than by treatmentwith anti-OSCAR mAb. These data indicate that collagen supportssurvival of DCs at least in part via an autocrine TNF-a signaling.DCs plated on ColI/II secreted a number of cytokines with
a proinflammatory function. In contrast to the OSCAR cross-
FIGURE 3. ColII-matured DCs induce proliferation of T cells. (A) Allogeneic MLR. Proliferation of CD3+CD4+CD45+RA+ T cells was estimated by
[3H]thymidine incorporation. T cells were incubated for 4 d with the indicated allogeneic DCs: iDCs, ColII-matured DCs, or DCs that were maintained on
collagen in the presence of anti-OSCAR mAb (data shown for 0.01, 0.1, and 1 mg/ml mAb) or isotype control mAb (1 mg/ml), as well as T cells alone (no
DCs) as negative control and T cells stimulated with CD3/CD28 beads as positive control. (B) Proliferation of autologous CD3+CD4+CD45+RA+ T cells in
the presence of the indicated DCs; costimulation of TCR was achieved by treatment with an agonistic OKT-3 mAb; all treatments and controls are as in (A).
Data are mean 6 SD of triplicate wells from one representative experiment out of seven (allogeneic) or three (autologous) performed with similar results.
The Journal of Immunology 7
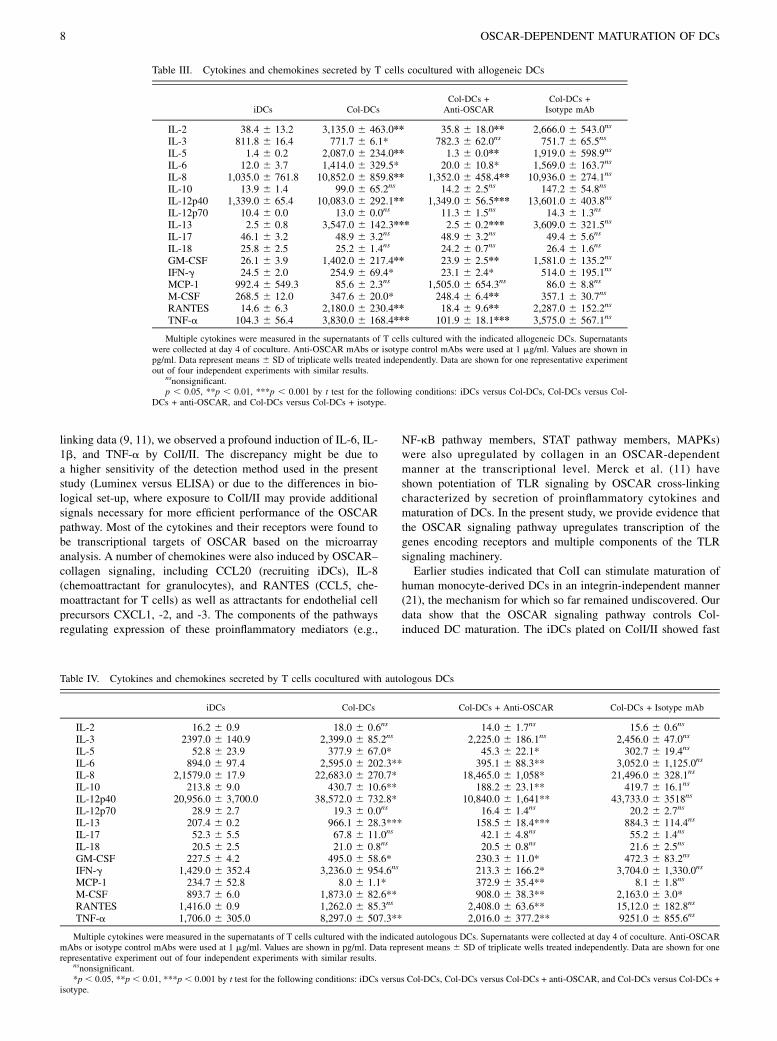
linking data (9, 11), we observed a profound induction of IL-6, IL-1b, and TNF-a by ColI/II. The discrepancy might be due toa higher sensitivity of the detection method used in the presentstudy (Luminex versus ELISA) or due to the differences in bio-logical set-up, where exposure to ColI/II may provide additionalsignals necessary for more efficient performance of the OSCARpathway. Most of the cytokines and their receptors were found tobe transcriptional targets of OSCAR based on the microarrayanalysis. A number of chemokines were also induced by OSCAR–collagen signaling, including CCL20 (recruiting iDCs), IL-8(chemoattractant for granulocytes), and RANTES (CCL5, che-moattractant for T cells) as well as attractants for endothelial cellprecursors CXCL1, -2, and -3. The components of the pathwaysregulating expression of these proinflammatory mediators (e.g.,
NF-kB pathway members, STAT pathway members, MAPKs)were also upregulated by collagen in an OSCAR-dependentmanner at the transcriptional level. Merck et al. (11) haveshown potentiation of TLR signaling by OSCAR cross-linkingcharacterized by secretion of proinflammatory cytokines andmaturation of DCs. In the present study, we provide evidence thatthe OSCAR signaling pathway upregulates transcription of thegenes encoding receptors and multiple components of the TLRsignaling machinery.Earlier studies indicated that ColI can stimulate maturation of
human monocyte-derived DCs in an integrin-independent manner(21), the mechanism for which so far remained undiscovered. Ourdata show that the OSCAR signaling pathway controls Col-induced DC maturation. The iDCs plated on ColI/II showed fast
Table III. Cytokines and chemokines secreted by T cells cocultured with allogeneic DCs
iDCs Col-DCsCol-DCs +Anti-OSCAR
Col-DCs +Isotype mAb
IL-2 38.4 6 13.2 3,135.0 6 463.0** 35.8 6 18.0** 2,666.0 6 543.0ns
IL-3 811.8 6 16.4 771.7 6 6.1* 782.3 6 62.0ns 751.7 6 65.5ns
IL-5 1.4 6 0.2 2,087.0 6 234.0** 1.3 6 0.0** 1,919.0 6 598.9ns
IL-6 12.0 6 3.7 1,414.0 6 329.5* 20.0 6 10.8* 1,569.0 6 163.7ns
IL-8 1,035.0 6 761.8 10,852.0 6 859.8** 1,352.0 6 458.4** 10,936.0 6 274.1ns
IL-10 13.9 6 1.4 99.0 6 65.2ns 14.2 6 2.5ns 147.2 6 54.8ns
IL-12p40 1,339.0 6 65.4 10,083.0 6 292.1** 1,349.0 6 56.5*** 13,601.0 6 403.8ns
IL-12p70 10.4 6 0.0 13.0 6 0.0ns 11.3 6 1.5ns 14.3 6 1.3ns
IL-13 2.5 6 0.8 3,547.0 6 142.3*** 2.5 6 0.2*** 3,609.0 6 321.5ns
IL-17 46.1 6 3.2 48.9 6 3.2ns 48.9 6 3.2ns 49.4 6 5.6ns
IL-18 25.8 6 2.5 25.2 6 1.4ns 24.2 6 0.7ns 26.4 6 1.6ns
GM-CSF 26.1 6 3.9 1,402.0 6 217.4** 23.9 6 2.5** 1,581.0 6 135.2ns
IFN-g 24.5 6 2.0 254.9 6 69.4* 23.1 6 2.4* 514.0 6 195.1ns
MCP-1 992.4 6 549.3 85.6 6 2.3ns 1,505.0 6 654.3ns 86.0 6 8.8ns
M-CSF 268.5 6 12.0 347.6 6 20.0* 248.4 6 6.4** 357.1 6 30.7ns
RANTES 14.6 6 6.3 2,180.0 6 230.4** 18.4 6 9.6** 2,287.0 6 152.2ns
TNF-a 104.3 6 56.4 3,830.0 6 168.4*** 101.9 6 18.1*** 3,575.0 6 567.1ns
Multiple cytokines were measured in the supernatants of T cells cultured with the indicated allogeneic DCs. Supernatantswere collected at day 4 of coculture. Anti-OSCAR mAbs or isotype control mAbs were used at 1 mg/ml. Values are shown inpg/ml. Data represent means 6 SD of triplicate wells treated independently. Data are shown for one representative experimentout of four independent experiments with similar results.
nsnonsignificant.p , 0.05, **p , 0.01, ***p , 0.001 by t test for the following conditions: iDCs versus Col-DCs, Col-DCs versus Col-
DCs + anti-OSCAR, and Col-DCs versus Col-DCs + isotype.
Table IV. Cytokines and chemokines secreted by T cells cocultured with autologous DCs
iDCs Col-DCs Col-DCs + Anti-OSCAR Col-DCs + Isotype mAb
IL-2 16.2 6 0.9 18.0 6 0.6ns 14.0 6 1.7ns 15.6 6 0.6ns
IL-3 2397.0 6 140.9 2,399.0 6 85.2ns 2,225.0 6 186.1ns 2,456.0 6 47.0ns
IL-5 52.8 6 23.9 377.9 6 67.0* 45.3 6 22.1* 302.7 6 19.4ns
IL-6 894.0 6 97.4 2,595.0 6 202.3** 395.1 6 88.3** 3,052.0 6 1,125.0ns
IL-8 2,1579.0 6 17.9 22,683.0 6 270.7* 18,465.0 6 1,058* 21,496.0 6 328.1ns
IL-10 213.8 6 9.0 430.7 6 10.6** 188.2 6 23.1** 419.7 6 16.1ns
IL-12p40 20,956.0 6 3,700.0 38,572.0 6 732.8* 10,840.0 6 1,641** 43,733.0 6 3518ns
IL-12p70 28.9 6 2.7 19.3 6 0.0ns 16.4 6 1.4ns 20.2 6 2.7ns
IL-13 207.4 6 0.2 966.1 6 28.3*** 158.5 6 18.4*** 884.3 6 114.4ns
IL-17 52.3 6 5.5 67.8 6 11.0ns 42.1 6 4.8ns 55.2 6 1.4ns
IL-18 20.5 6 2.5 21.0 6 0.8ns 20.5 6 0.8ns 21.6 6 2.5ns
GM-CSF 227.5 6 4.2 495.0 6 58.6* 230.3 6 11.0* 472.3 6 83.2ns
IFN-g 1,429.0 6 352.4 3,236.0 6 954.6ns 213.3 6 166.2* 3,704.0 6 1,330.0ns
MCP-1 234.7 6 52.8 8.0 6 1.1* 372.9 6 35.4** 8.1 6 1.8ns
M-CSF 893.7 6 6.0 1,873.0 6 82.6** 908.0 6 38.3** 2,163.0 6 3.0*RANTES 1,416.0 6 0.9 1,262.0 6 85.3ns 2,408.0 6 63.6** 15,12.0 6 182.8ns
TNF-a 1,706.0 6 305.0 8,297.0 6 507.3** 2,016.0 6 377.2** 9251.0 6 855.6ns
Multiple cytokines were measured in the supernatants of T cells cultured with the indicated autologous DCs. Supernatants were collected at day 4 of coculture. Anti-OSCARmAbs or isotype control mAbs were used at 1 mg/ml. Values are shown in pg/ml. Data represent means 6 SD of triplicate wells treated independently. Data are shown for onerepresentative experiment out of four independent experiments with similar results.
nsnonsignificant.*p , 0.05, **p , 0.01, ***p , 0.001 by t test for the following conditions: iDCs versus Col-DCs, Col-DCs versus Col-DCs + anti-OSCAR, and Col-DCs versus Col-DCs +
isotype.
8 OSCAR-DEPENDENT MATURATION OF DCs

and drastic upregulation of maturation markers. Microarrayanalysis has shown that most of the major components of thestable immune synapse, including CD40, CD80, CD86, andICAM-1, are transcriptional targets of the OSCAR–collagen sig-naling. Treatment of cells with anti-OSCAR mAb completelyblocked this collagen-induced maturation process. The cellsurface expression of HLA-DR was upregulated in an OSCAR-dependent manner. However, expression of the MHC class IIgenes was not influenced by collagen exposure, indicating reg-ulation at a posttranscriptional level. These results are in linewith the previously published studies showing posttranscrip-tional regulation of the surface expression of MHC class II (32),particularly via protein recycling/trafficking to and from theplasma membrane (33, 34).
In contrast to the OSCAR cross-linking data (11), in our studytriggering of the OSCAR signaling in DCs with a native ligand ledto formation of matured cells, which efficiently stimulated pro-liferation of allogeneic and autologous naive T cells. Thus, even inthe absence of microbial or inflammatory stimulation, OSCARengagement by collagen was sufficient to induce a full DC mat-uration program. The T cells expanded by Col-DCs producedmultiple cytokines mainly of the proinflammatory type, but withno clear signs of T cell polarization. Additional studies on phe-notyping of these cells (e.g., microarray and transcription factorprofiling) are needed to fully understand the T cell polarizingeffect of the Col-DCs.Upregulation of OSCAR expression on the surface of blood
monocytes was found in RA patients with active disease, and
Table V. Top 20 canonical pathways regulated by ColII and reversed by anti-OSCAR
Ingenuity Canonical Pathways Molecules 2Log (p Value)
TLR signaling MAP3K14, IL1A, MAP3K1, TLR8, TNFAIP3, MAPK13, NFKB1,TLR2, NFKBIA, IL1RN, IL1B, MAP2K3, TNF, TRAF1, IRAK2
1.05E+01
Hepatic fibrosis/hepatic stellate cell activation IL4R, IL1A, ICA, VEGFA, CXCL3, CCL2, CD40, EDN1, CSF1,TNFSF8, IL1B, SERPINE1, IL1RAP, TNF, MMP9, TNFSF14
1.04E+01
Atherosclerosis signaling IL1A, ICAM1, PDGFA, NFKB1, F3, TNFRSF12A, SELPLG,CD40, CCL2, CSF1, IL1RN, LPL, IL1B, SERPINA1, TNF, MMP9,
TNFSF14
9.06E+00
Type I diabetes mellitus signaling SOCS3, MAP3K14, IFNGR1, MAPK13, MAP3K5, JAK2, NFKB1,NFKBIA, CD80, IL1B, CD86, BID, MAP2K3, CASP8, IL1RAP,
TNF
8.89E+00
Role of macrophages, fibroblasts, andendothelial cells in RA
SOCS3, IL1A, ICAM1, PDGFA, TLR8, JAK2, NFKB1, VEGFA,NFAT5, NFKBIA, CCL2, OSM, MAPKAPK2, IL1RAP, FZD2,
TRAF1, MAP3K14, C5AR1, CREB3, TLR2, CSF1, IL1RN, IL1B,MAP2K3, TNF, IRAK2
8.86E+00
Acute phase response signaling SOCS3, MAP3K14, IL1A, C3, MAP3K1, JAK2, MAPK13,MAP3K5, NFKB1, NFKBIA, SOD2, IL1RN, OSM, IL1B,
SERPINA1, MAP2K3, SERPINE1, IL1RAP, TNF
8.48E+00
Granulocyte adhesion and diapedesis IL1A, C5AR1, ICAM1, CCL20, CCL22, SDC4, SELPLG, CXCL3,CLDN12, CCL2, CLDN1, IL1RN, IL1B, CXCL1, CXCL2,
IL1RAP, TNF, MMP9, CCL1
8.14E+00
IL-6 signaling SOCS3, MAP3K14, IL1A, MAPK13, JAK2, NFKB1, VEGFA,NFKBIA, IL1RN, IL1B, MAP2K3, MAPKAPK2, IL1RAP, TNF,
MCL1
7.71E+00
IL-10 signaling SOCS3, MAP3K14, IL1A, IL4R, NFKBIA, IL1RN, IL1B,MAP2K3, MAPK13, NFKB1, TNF, IL1RAP
7.54E+00
Altered T cell and B cell signaling in RA MAP3K14, IL1A, SLAMF1, TLR8, NFKB1, TLR2, CD80, CD40,CSF1, IL1RN, CD86, IL1B, TNF
7.46E+00
TREM1 signaling TLR2, CXCL3, TREM1, ICAM1, CCL2, CD40, TLR8, CD86,IL1B, JAK2, NFKB1, TNF
7.27E+00
CD40 signaling MAP3K14, ICAM1, NFKBIA, CD40, TNFAIP3, MAP2K3,MAPK13, PTGS2, MAPKAPK2, NFKB1, TRAF1
7.03E+00
CD27 signaling in lymphocytes MAP3K14, NFKBIA, MAP3K1, BID, MAP3K8, MAP2K3,MAP3K5, CASP8, NFKB1, MAP3K2
6.99E+00
Agranulocyte adhesion and diapedesis IL1A, C5AR1, ICAM1, CCL20, CCL22, SDC4, SELPLG, CXCL3,CLDN12, CCL2, CLDN1, IL1RN, IL1B, CXCL1, CXCL2, TNF,
MMP9, CCL1
6.98E+00
NF-kB signaling MAP3K14, IL1A, FLT1, MAP3K1, TLR8, TNFAIP3, MALT1,NFKB1, TGFBR2, TLR2, NFKBIA, CD40, IL1RN, IL1B,
MAP3K8, CASP8, TNF
6.86E+00
Role of IL-17A in arthritis CXCL3, NFKBIA, CCL2, CCL20, MAP2K3, CXCL1, MAPK13,PTGS2, MAPKAPK2, NFKB1
6.67E+00
DC maturation MAP3K14, IL1A, ICAM1, CREB3, MAPK13, JAK2, NFKB1,STAT4, TLR2, NFKBIA, CD40, CD80, IL1RN, CD86, IL1B, IRF8,
TNF
6.64E+00
BCR signaling BLNK, MAP3K14, CREB3, MAP3K1, MAPK13, MALT1,MAP3K5, NFKB1, INPP5D, NFKBIA, NFAT5, SYK, MAP2K3,
MAP3K8, RASSF5, BCL2A1, MAP3K2
6.57E+00
TWEAK signaling CASP6, MAP3K14, NFKBIA, BID, CASP8, NFKB1,TNFRSF12A, TRAF1
6.42E+00
TNFR1 signaling CASP6, MAP3K14, NFKBIA, MAP3K1, TNFAIP3, BID, CASP8,NFKB1, TNF
6.17E+00
iDCs were cultured on ColII for 4 h in the presence or absence of GM-CSF (50 ng/ml), anti-OSCAR mAb (1 mg/ml), or isotype control (1 mg/ml). Total cellular RNA wasisolated, and genome-wide mRNA levels were determined using Affymetrix GeneChip microarray analysis. The table shows canonical pathways for OSCAR-ColII–regulatedgenes at an FDR of 5% and a biological criterion of minimum 2-fold change. A complete list of all genes regulated by the OSCAR-ColII signaling at 4 h time point is provided inSupplemental Table IC. Samples from six independent experiments were analyzed.
The Journal of Immunology 9

OSCAR-expressing mononuclear cells were detected in peri-vascular areas of the synovium (13). The RA synovium is con-sidered an ectopic lymphoid organ (35). Both immature andmature DCs are present in the RA synovium in about a 1:1 ratio.The immature cells are primarily located in the sublining or lininglayer of the synovium and in the perivascular infiltrates, whereasmature DCs are associated with perivascular lymphocytic infil-trates (36, 37). These perivascular DCs were suggested to origi-nate from blood-derived precursors that migrated through theactivated endothelium and received differentiation signals, such asGM-CSF and IL-13, within the joint (reviewed in Ref. 38). It isthought that functional DC/T cell interaction can take place lo-cally (39, 40), and the perivascular region may act as a site for cellactivation (41). Intriguingly, surgical removal of cartilage in RApatients led to reduced inflammation and suppression of functionaldifferentiation of DCs in the synovium (42). Because ColII makesup to 60% of dry weight of articular cartilage (43), we speculatethat this observed effect might at least partially be attributed tointerference with the OSCAR signaling in DCs.Taken together, our data indicate that the principal role of
OSCAR in DCs is proinflammatory. Although OSCAR is expressedin resting myeloid cells (9, 13), it may not be functional becausethe ligand accessibility is limited. We hypothesize that theOSCAR–collagen signaling plays a pivotal role in pathology, such asin RA, where ligands become available due to ongoing tissueremodeling. OSCAR may act at multiple levels contributing toperpetuation of local synovial inflammation and may impactsystemic inflammation in RA via expansion of proinflammatoryT cells.
AcknowledgmentsWe thank Lise F.W. Jensen, JetteW. Platou, JingWang, Bing Zhang, Helene
Bregnbak, Jian Zhou, Wenjuan Xia, Xiaoai Wu, Li Yang, and Chunyan Ma
for excellent technical assistance.
DisclosuresThe authors have no financial conflicts of interest.
References1. McInnes, I. B., and G. Schett. 2011. The pathogenesis of rheumatoid arthritis. N.
Engl. J. Med. 365: 2205–2219.2. Holmdahl, R., V. Malmstrom, and H. Burkhardt. 2014. Autoimmune priming,
tissue attack and chronic inflammation—the three stages of rheumatoid arthritis.Eur. J. Immunol. 44: 1593–1599.
3. Thomas, R., L. S. Davis, and P. E. Lipsky. 1994. Rheumatoid synovium is enrichedin mature antigen-presenting dendritic cells. J. Immunol. 152: 2613–2623.
4. Radstake, T. R., A. W. van Lieshout, P. L. van Riel, W. B. van den Berg, andG. J. Adema. 2005. Dendritic cells, Fcg receptors, and Toll-like receptors: po-tential allies in the battle against rheumatoid arthritis. Ann. Rheum. Dis. 64:1532–1538.
5. Lebre, M. C., and P. P. Tak. 2008. Dendritic cell subsets: their roles in rheu-matoid arthritis. Acta Reumatol. Port. 33: 35–45.
6. Radstake, T. R., R. van der Voort, M. ten Brummelhuis, M. de Waal Malefijt,M. Looman, C. G. Figdor, W. B. van den Berg, P. Barrera, and G. J. Adema.2005. Increased expression of CCL18, CCL19, and CCL17 by dendritic cellsfrom patients with rheumatoid arthritis, and regulation by Fcg receptors. Ann.Rheum. Dis. 64: 359–367.
7. Rivollier, A., M. Mazzorana, J. Tebib, M. Piperno, T. Aitsiselmi, C. Rabourdin-Combe, P. Jurdic, and C. Servet-Delprat. 2004. Immature dendritic cell trans-differentiation into osteoclasts: a novel pathway sustained by the rheumatoidarthritis microenvironment. Blood 104: 4029–4037.
8. Alnaeeli, M., J. M. Penninger, and Y. T. Teng. 2006. Immune interactions withCD4+ T cells promote the development of functional osteoclasts from murineCD11c+ dendritic cells. J. Immunol. 177: 3314–3326.
9. Merck, E., C. Gaillard, D. M. Gorman, F. Montero-Julian, I. Durand,S. M. Zurawski, C. Menetrier-Caux, G. Carra, S. Lebecque, G. Trinchieri, andE. E. Bates. 2004. OSCAR is an FcRg-associated receptor that is expressed bymyeloid cells and is involved in antigen presentation and activation of humandendritic cells. Blood 104: 1386–1395.
10. Ishikawa, S., N. Arase, T. Suenaga, Y. Saita, M. Noda, T. Kuriyama, H. Arase,and T. Saito. 2004. Involvement of FcRg in signal transduction of osteoclast-associated receptor (OSCAR). Int. Immunol. 16: 1019–1025.
11. Merck, E., B. de Saint-Vis, M. Scuiller, C. Gaillard, C. Caux, G. Trinchieri, andE. E. Bates. 2005. Fc receptor g-chain activation via hOSCAR induces survivaland maturation of dendritic cells and modulates Toll-like receptor responses.Blood 105: 3623–3632.
12. Tenca, C., A. Merlo, E. Merck, E. E. Bates, D. Saverino, R. Simone, D. Zarcone,G. Trinchieri, C. E. Grossi, and E. Ciccone. 2005. CD85j (leukocyte Ig-likereceptor-1/Ig-like transcript 2) inhibits human osteoclast-associated receptor-mediated activation of human dendritic cells. J. Immunol. 174: 6757–6763.
13. Herman, S., R. B. M€uller, G. Kronke, J. Zwerina, K. Redlich, A. J. Hueber,H. Gelse, E. Neumann, U. M€uller-Ladner, and G. Schett. 2008. Induction ofosteoclast-associated receptor, a key osteoclast costimulation molecule, inrheumatoid arthritis. Arthritis Rheum. 58: 3041–3050.
14. Barrow, A. D., N. Raynal, T. L. Andersen, D. A. Slatter, D. Bihan, N. Pugh,M. Cella, T. Kim, J. Rho, T. Negishi-Koga, et al. 2011. OSCAR is a collagenreceptor that costimulates osteoclastogenesis in DAP12-deficient humans andmice. J. Clin. Invest. 121: 3505–3516.
15. Canfield, S. M., and S. L. Morrison. 1991. The binding affinity of human IgG forits high affinity Fc receptor is determined by multiple amino acids in the CH2domain and is modulated by the hinge region. J. Exp. Med. 173: 1483–1491.
16. Schneider, C., R. A. Newman, D. R. Sutherland, U. Asser, and M. F. Greaves.1982. A one-step purification of membrane proteins using a high efficiencyimmunomatrix. J. Biol. Chem. 257: 10766–10769.
17. Shevchenko, A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric se-quencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68: 850–858.
18. Kim, N., M. Takami, J. Rho, R. Josien, and Y. Choi. 2002. A novel member ofthe leukocyte receptor complex regulates osteoclast differentiation. J. Exp. Med.195: 201–209.
19. Sallusto, F., and A. Lanzavecchia. 1994. Efficient presentation of soluble antigenby cultured human dendritic cells is maintained by granulocyte/macrophagecolony-stimulating factor plus interleukin 4 and downregulated by tumor ne-crosis factor alpha. J. Exp. Med. 179: 1109–1118.
20. Ludewig, B., D. Graf, H. R. Gelderblom, Y. Becker, R. A. Kroczek, and G. Pauli.1995. Spontaneous apoptosis of dendritic cells is efficiently inhibited by TRAP(CD40-ligand) and TNF-a, but strongly enhanced by interleukin-10. Eur. J.Immunol. 25: 1943–1950.
21. Brand, U., I. Bellinghausen, A. H. Enk, H. Jonuleit, D. Becker, J. Knop, andJ. Saloga. 1998. Influence of extracellular matrix proteins on the development ofcultured human dendritic cells. Eur. J. Immunol. 28: 1673–1680.
22. Liu, Z., N. Li, L. A. Diaz, M. Shipley, R. M. Senior, and Z. Werb. 2005. Synergybetween a plasminogen cascade and MMP-9 in autoimmune disease. J. Clin.Invest. 115: 879–887.
23. Pulendran, B., H. Tang, and S. Manicassamy. 2010. Programming dendritic cellsto induce TH2 and tolerogenic responses. Nat. Immunol. 11: 647–655.
24. Hynes, R. O. 2009. The extracellular matrix: not just pretty fibrils. Science 326:1216–1219.
25. Daley, W. P., S. B. Peters, and M. Larsen. 2008. Extracellular matrix dynamics indevelopment and regenerative medicine. J. Cell Sci. 121: 255–264.
26. Karsdal, M. A., T. Woodworth, K. Henriksen, W. P. Maksymowych, H. Genant,P. Vergnaud, C. Christiansen, T. Schubert, P. Qvist, G. Schett, et al. 2011. Bio-chemical markers of ongoing joint damage in rheumatoid arthritis—current andfuture applications, limitations and opportunities. Arthritis Res. Ther. 13: 215.
27. Shortman, K., and S. H. Naik. 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7: 19–30.
28. Leon, B., M. Lopez-Bravo, and C. Ardavın. 2005. Monocyte-derived dendriticcells. Semin. Immunol. 17: 313–318.
29. Steenbakkers, P. G., D. Baeten, E. Rovers, E. M. Veys, A. W. Rijnders, J. Meijerink,F. De Keyser, and A. M. Boots. 2003. Localization of MHC class II/human cartilageglycoprotein-39 complexes in synovia of rheumatoid arthritis patients usingcomplex-specific monoclonal antibodies. J. Immunol. 170: 5719–5727.
30. Merck, E., C. Gaillard, M. Scuiller, P. Scapini, M. A. Cassatella, G. Trinchieri,and E. E. Bates. 2006. Ligation of the FcRg chain-associated human osteoclast-associated receptor enhances the proinflammatory responses of human mono-cytes and neutrophils. J. Immunol. 176: 3149–3156.
31. Baldwin, H. M., T. Ito-Ihara, J. D. Isaacs, and C. M. Hilkens. 2010. Tumournecrosis factor alpha blockade impairs dendritic cell survival and function inrheumatoid arthritis. Ann. Rheum. Dis. 69: 1200–1207.
32. Pai, R. K., D. Askew, W. H. Boom, and C. V. Harding. 2002. Regulation of classII MHC expression in APCs: roles of types I, III, and IV class II transactivator. J.Immunol. 169: 1326–1333.
33. Shin, J. S., M. Ebersold, M. Pypaert, L. Delamarre, A. Hartley, and I. Mellman.2006. Surface expression of MHC class II in dendritic cells is controlled byregulated ubiquitination. Nature 444: 115–118.
34. Chow, A., D. Toomre, W. Garrett, and I. Mellman. 2002. Dendritic cell matu-ration triggers retrograde MHC class II transport from lysosomes to the plasmamembrane. Nature 418: 988–994.
35. Kratz, A., A. Campos-Neto, M. S. Hanson, and N. H. Ruddle. 1996. Chronic inflam-mation caused by lymphotoxin is lymphoid neogenesis. J. Exp. Med. 183: 1461–1472.
36. Page, G., S. Lebecque, and P. Miossec. 2002. Anatomic localization of immatureand mature dendritic cells in an ectopic lymphoid organ: correlation with selectivechemokine expression in rheumatoid synovium. J. Immunol. 168: 5333–5341.
37. Manzo, A., S. Bugatti, R. Caporali, R. Prevo, D. G. Jackson, M. Uguccioni,C. D. Buckley, C. Montecucco, and C. Pitzalis. 2007. CCL21 expression patternof human secondary lymphoid organ stroma is conserved in inflammatorylesions with lymphoid neogenesis. Am. J. Pathol. 171: 1549–1562.
38. Thomas, R., K. P. MacDonald, A. R. Pettit, L. L. Cavanagh, J. Padmanabha, andS. Zehntner. 1999. Dendritic cells and the pathogenesis of rheumatoid arthritis.J. Leukoc. Biol. 66: 286–292.
10 OSCAR-DEPENDENT MATURATION OF DCs

39. Baeten, D., P. G. Steenbakkers, A. M. Rijnders, A. M. Boots, E. M. Veys, andF. De Keyser. 2004. Detection of major histocompatibility complex/humancartilage gp-39 complexes in rheumatoid arthritis synovitis as a specific andindependent histologic marker. Arthritis Rheum. 50: 444–451.
40. Timmer, T. C., B. Baltus, M. Vondenhoff, T. W. Huizinga, P. P. Tak,C. L. Verweij, R. E. Mebius, and T. C. van der Pouw Kraan. 2007. Inflammationand ectopic lymphoid structures in rheumatoid arthritis synovial tissues dissectedby genomics technology: identification of the interleukin-7 signaling pathway intissues with lymphoid neogenesis. Arthritis Rheum. 56: 2492–2502.
41. Krenn, V., N. Schalhorn, A. Greiner, R. Molitoris, A. Konig, F. Gohlke, andH. K. M€uller-Hermelink. 1996. Immunohistochemical analysis of proliferating andantigen-presenting cells in rheumatoid synovial tissue. Rheumatol. Int. 15: 239–247.
42. Li, T. F., J. Mandelin, M. Hukkanen, J. Lassus, J. Sandelin, S. Santavirta,I. Virtanen, and Y. T. Konttinen. 2002. Dendritic cells in rheumatoid synovialmembrane after total removal of the hyaline articular cartilage. Rheumatology(Oxford) 41: 319–323.
43. Sophia Fox, A. J., A. Bedi, and S. A. Rodeo. 2009. The basic science of articularcartilage: structure, composition, and function. Sports Health 1: 461–468.
The Journal of Immunology 11

54
3.2 OSCAR function in monocytes
This part of my studies is being submitted for publication in J. Immunology:
OSCAR-collagen signaling mediates activation of monocytes
Heidi S. Schultz*†1
, Li Guo‡, Pernille Keller
*, Andrew J. Fleetwood
§, Wei Guo
‡, John A. Hamilton
§,
Olle Bjørkdahl*, Martin W. Berchtold
† and Svetlana Panina
*¶
*Biopharmaceutical Research Unit, Novo Nordisk A/S, Novo Nordisk Park 1, 2760 Måløv,
Denmark. †Department of Biology, Copenhagen University, Copenhagen 2200, Denmark.
‡Novo
Nordisk Research Centre China, 20 Life Science Park Rd, Changping District, CA 102206 Beijing,
China. §Department of Medicine, University of Melbourne, The Royal Melbourne Hospital,
Parkville, Victoria 3050, Australia.
¶Corresponding author, Svetlana Panina, [email protected], Tel: (+45) 53834930
For supplemental data see Appendix II
1 HSS PhD fellowship was supported by the Danish Agency for Science, Technology, Innovation and Research.

55
3.2.1 Abstract
Osteoclast Associated Receptor (OSCAR) is an activating receptor expressed in human myeloid
cells. Collagen type I (ColI) and collagen type II (ColII) serve as ligands for OSCAR. OSCAR-
collagen interaction stimulates RANK-dependent osteoclastogenesis. We have recently reported
that in monocyte-derived dendritic cells (DC), OSCAR promotes activation and functional
maturation. OSCAR is up-regulated in monocytes from rheumatoid arthritis (RA) patients with
active disease and these monocytes show an increased pro-osteoclastogenic potential. In the current
study, we have addressed a functional role of OSCAR-collagen interaction in monocytes. We show
that the OSCAR-ColII signaling supports survival of monocytes under conditions of growth factor
withdrawal. Moreover, ColII stimulated the release of pro-inflammatory cytokines by monocytes
from healthy donors, which can be completely blocked by an anti-OSCAR monoclonal antibody
(mAb). Mononuclear cells from synovial fluid of RA patients plated on ColII secreted TNF-α and
IL-8 in an OSCAR-dependent manner. Global RNA profiling showed that components of multiple
signaling pathways relevant for the RA pathogenesis are transcriptional targets of OSCAR in
monocytes. Thus, OSCAR plays a pro-inflammatory role in monocyte-derived cells and may
crucially contribute to the RA pathogenesis on multiple levels.

56
3.2.2 Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by a dysregulated
immune response and profound cartilage and bone destruction43
. Infiltration of the synovium by
pro-inflammatory immune cells is the hallmark of RA pathogenesis44;150
. These cells secrete
multiple cytokines and matrix degrading enzymes causing tissue damage. Monocytes are believed
to crucially contribute to the pathophysiology of RA at multiple levels5. The circulating monocytes
of RA patients were shown to have an activated phenotype characterized by production of pro-
inflammatory cytokines IL-1 and IL-6 as well as by up-regulation of the cell surface marker,
integrin CD11b151;152
. Monocytes massively infiltrate the progressively inflamed RA synovium and
produce large amounts of the master pro-inflammatory cytokine, TNF-, thus critically contributing
to the sustained joint inflammation. In the course of inflammation, monocytes serve as precursors
for macrophages, dendritic cells and osteoclasts5. In a number of cross-sectional studies, synovial
sublining monocyte/macrophage infiltration was shown to correlate with scores of disease activity
in RA patients153
. Therapeutic targeting of monocytes/macrophages has been suggested for
treatment of RA154-156
.
The remarkable plasticity of monocytes is influenced by the RA joint
microenvironment, resulting in an altered differentiation, activation and maturation status157
.
Among microenvironmental factors influencing the immune cell activation in RA patients the
extracellular matrix (ECM) proteins has been suggested to be of importance. It has been speculated
that in the joints of RA patients collagens become exposed due to profound synovial architectural
reorganization, thus enabling interaction with ECM receptors137
. Fragments of ColII, the major
collagen of the articular cartilage102
, have been detected in synovial fluid (SF) of patients with joint
diseases158-160
. Several studies have addressed ECM interaction with immune cells. ColI has been
shown to stimulate cytokine secretion by PBMCs100
and by synovial fluid mononuclear cells from
RA patients132
. However, the molecular mechanisms of the ECM-mediated cell activation still
remain obscure. We have recently shown that ColI and ColII stimulate cytokine secretion and
functional maturation of monocyte-derived DCs via the OSCAR signaling pathway142
. OSCAR has
been described as an important player in “osteoimmunology”101
. OSCAR is associated with the
FcRγ136
and functions as a stimulatory co-receptor in osteoclastogenesis signaling via the nuclear
factor of activated T cells c1 (NFATc1) 30
. OSCAR is expressed on all human myeloid cells and is
capable of modulating the immune response11;134;136;142
. The ECM proteins ColI, ColII137;142
and

57
collagen type III137
serve as naturally occurring ligands of OSCAR. It has been proposed that
OSCAR plays a role in skeletal146
and joint disorders139;141
. Monocytes isolated from RA patients
express high levels of OSCAR and show an elevated activation status141
. Furthermore, high levels
of soluble OSCAR, sOSCAR, (presumably, membrane-shed or a secreted splice form) have been
found in plasma, synovial fluid and vasculature of RA patients139-141
. The plasma level of sOSCAR
has been reported to be positively correlating with bone destruction and cardiovascular risk in
patients with RA140
. In contrast, a reverse correlation of sOSCAR present in the serum of RA
patients with the disease activity was reported by the group of Schett141
. Both studies were done on
small patients populations and with a use of different methodologies, thus the prognostic value of
sOSCAR and its function remains unclear.
The function of OSCAR in monocytes has so far been addressed with the use of cross-
linking mAb134
. The authors have shown that triggering of OSCAR induces the release of multiple
chemokines and promotes survival of the cells. Cross-linking of OSCAR in the presence of TLR
ligands, but not alone, potentiated TLR-induced release of pro-inflammatory cytokines134
. In the
current study we addressed the OSCAR-mediated response of monocytes in the presence of the
natural ligand, collagen. We show here that peripheral blood monocytes exposed to ColII increased
their viability in an OSCAR dependent manner. In contrast to the published cross-linking data,
OSCAR engagement by its natural ligand induced profound secretion of pro-inflammatory
cytokines, most of which turned out to be transcriptional targets of the OSCAR-ColII signaling. Our
data suggest that OSCAR-ColII interaction in monocytes may directly contribute to the sustained
inflammation and the joint damage in the RA patients.

58
3.2.3 Materials and methods
Antibodies and ECM proteins
The following monoclonal antibodies (mAb) were used for flow cytometry analysis: Alexa Fluor
647-conjugated anti-OSCAR mAb (Novo Nordisk A/S), APC-conjugated anti-CD14 (BD
Pharmigen), FITC-conjugated anti-CD14 mAb (eBioscience), FITC-conjugated anti-CD68 mAb
(eBioscience), PerCP-Cy5.5–conjugated anti-CD3 mAb (eBioscience), FITC-conjugated anti-HLA-
DR mAb (BD Pharmigen), PE-Cy5-conjugated anti-CD11b mAb (BD Pharmigen), APC-
conjugated anti-TREM1 mAb (BioSite), PE-Cy5-conjugated anti-ICAM1/CD54 mAb (BD
Pharmigen) and PE-conjugated anti-CD86 mAb (IOTest). The isotype control mAb used were anti-
TNP conjugated with Alexa Fluor 647 (Novo Nordisk A/S), IgG1-FITC (eBioscience), IgG2a-
PerCP Cy5.5 (eBioscience), IgG2B-PE (R&D systems), IgG1-APC (BD Bioscience), IgG2b-
FITC (BD Pharmigen), IgG1-PE-Cy5 (BD Bioscience) and IgG1-PE (BD Bioscience).
For functional assays, the OSCAR blocking humanized IgG1.1 mAb (described in Schultz et al.,
2015) was used142
. As an isotype control, an anti-TNP humanized IgG1.1 was used. In some
experiments, an anti-OSCAR non-blocking high affinity humanized IgG1.1 mAb was used as an
isotype control, which produced identical results with the anti-TNP control. In the manuscript, only
anti-TNP control is shown. IgG1.1 antibodies harbor a human Fc fragment with five point
mutations, which prevents activation of the Fc receptors161
.
ColII purified from human cartilage was purchased from Millipore.
Endotoxin level in all protein solutions used for functional studies was measured using the
turbidimetric kinetic LAL assay (Charles River Laboratories). All proteins were essentially
endotoxin-free (below detection limit, <0.01 EU/ml).
Cells
Peripheral blood mononuclear cells (PBMCs) were isolated from leukocyte-enriched buffy coats by
Ficoll-Plague PLUS (GE Healthcare) density centrifugation. Buffy coats from healthy individuals
were obtained anonymously from the Clinical Immunology Blood Bank, The State University
Hospital (Rigshospitalet), Copenhagen, Denmark. All donors gave informed consent according to
the protocol approved by The Ethics Committee for Copenhagen, Denmark, for research use
(ethical approval number H-D-2008-113). Red blood cells (RBC) were removed using a lysis buffer
(eBioscience). Monocytes were purified from PBMCs using CD14 Microbeads human positive

59
selection kit (MACS; Miltenyi Biotec) according to manufactures’ protocol using an AutoMACS
Pro (Miltenyi Biotec). The purity of the CD14+ preparations was assessed by flow cytometry.
Synovial fluid (SF) from RA patients was obtained by needle aspiration anonymously from Peking
University People’s Hospital (PKUPH), Beijing, China. All donors gave informed consent
according to the protocol approved by the Ethics Committee of PKUPH for research use (approval
#20110513-LZ-PKUPH). Synovial fluid cells were isolated by centrifugation at 300xg for 10 min.
Synovial tissue from RA patients was obtained during joint replacement surgery at PKUPH. The
tissue was placed in a buffer (1g tissue per 3 ml buffer) containing 4 mg/ml Collagenase A (Roche)
and 0.1 mg/ml of DNase I (Roche) in RPMI 1640 medium and then homogenized using a MACS
dissociator. The samples were incubated on a MACS mix tube rotator at 37oC for 30 min. The
reaction was stopped by adding an ice-cold growth medium (RPMI 1640 with 10% FCS). The cell
suspension was filtered using a 70μm strainer and centrifuged at 300xg for 10 min. The cells were
re-suspended in a growth medium and processed for flow cytometry analysis.
Activation of cells by plate-bound collagen
Most experiments were performed with both ColI and ColII. Similar to the results obtained with
DCs142
, ColI and ColII had similar effect with ColII being more potent. We therefore show data for
ColII only. For monocytes, 24-well plates were coated with ColII in PBS at ~3µg/cm2
overnight at
4°C. The plate was washed three times in PBS before cells were added to the wells. 1x106
cells
were seeded per well in growth medium (RPMI1640 with GlutaMAX) (Invitrogen, Life
Technologies) supplemented with 10% heat-inactivated FCS and 1% penicillin-streptomycin
(GIBCO, Life Technologies), pre-incubated 30-60 min with anti-OSCAR or isotype control mAb
(1µg/ml) as indicated. The cells were cultured on ColII for various time points as indicated. In some
of the experiments, 100ng/ml LPS (Sigma) and 60ng/ml PMA (Calbiochem) were included as
controls.
RA SF cells (5x105 cells /well) were cultured on high binding 96-well cell culture plates coated
with ColII as described above.
Analysis of cytokine secretion
Supernatants of monocyte cultures were collected after 24 hours of ColII exposure, frozen at -80oC
and shipped to Myriad RBM (Austin, TX) for a 45-Biomarker Multi-Analyte Profiling.

60
Supernatants from 24 hours cultures of SF cells were collected and analyzed using ELISA. The
TNF- ELISA kit was from eBioscience and the IL-8 ELISA kit was from BD Bioscience.
Monocyte viability
Monocytes were washed and re-plated in fresh growth medium without cytokines at 5x105cells/ml.
The monocytes were left untreated or plated onto ColII-coated plates for 2 days in the absence or
presence of anti-OSCAR mAb (1µg/ml) or isotype control (1µg/ml). As a positive control, M-CSF
(10ng/ml) was added. At day two, the cells were washed in PBS and stained with Annexin V-FITC
(BD-Pharmigen) for 15 min at room temperature followed by flow cytometry analysis using a
FACSFortessaTM
(BD Bioscience).
Analysis of cell surface markers by flow cytometry
Expression of cell surface markers was assessed by flow cytometry. In short, cells were washed
twice in PBS with 2% FCS and then stained for 30 min at 37°C in PBS containing 2% FCS in the
presence of 1% human serum albumin (HSA) to block unspecific binding. Cells were rinsed twice
in PBS with 2% FCS prior to flow cytometry analysis. Data acquisition was done using a
FACSFortessaTM
(BD Bioscience). Data analysis was performed using Kaluza software version 1.2
(Beckman coulter).
Microarray analysis
Total RNA was obtained four hours after stimulating the cells with ColII from six donors in the
absence or presence of anti-OSCAR mAb or isotype control mAb. RNA was extracted using TRIzol
(Invitrogen, Life Technologies) followed by purification with an RNeasy MinElute Cleanup Kit
(Qiagen). The RNA integrity was evaluated on an Agilent 2100 Bioanalyser using Agilent RNA
6000 Nano Kit chips (Agilent Technologies), with RNA Integrity Number (RIN) scores of 9.9 or
above. Microarray experiments and data treatment were performed as described in Schultz et al142
.
A paired two-group comparison was done with Benjamini-Hochberg correction for multiple testing
applying a false discovery rate (FDR) of 5% and a biological threshold of more than 2-fold
regulation. The effect of ColII treatment was evaluated using a Principal component analysis
(PCA). Differentially expressed genes were subjected to Gene Ontology (GO) analysis for
biological processes using Ingenuity Pathway Analysis (IPA). The dataset and technical information

61
compliant with MIAME can be found at the EMBL-EBI ArrayExpress depository, accession
number E-MTAB-3281 (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-3281/).
Statistical analysis
Statistical analysis was performed using a Student 2-tailed t tests in a GraphPad Prism 5.0 software.
A P value of less than 0.05 was considered statistically significant. Unless otherwise indicated,
means and standard deviations are shown.
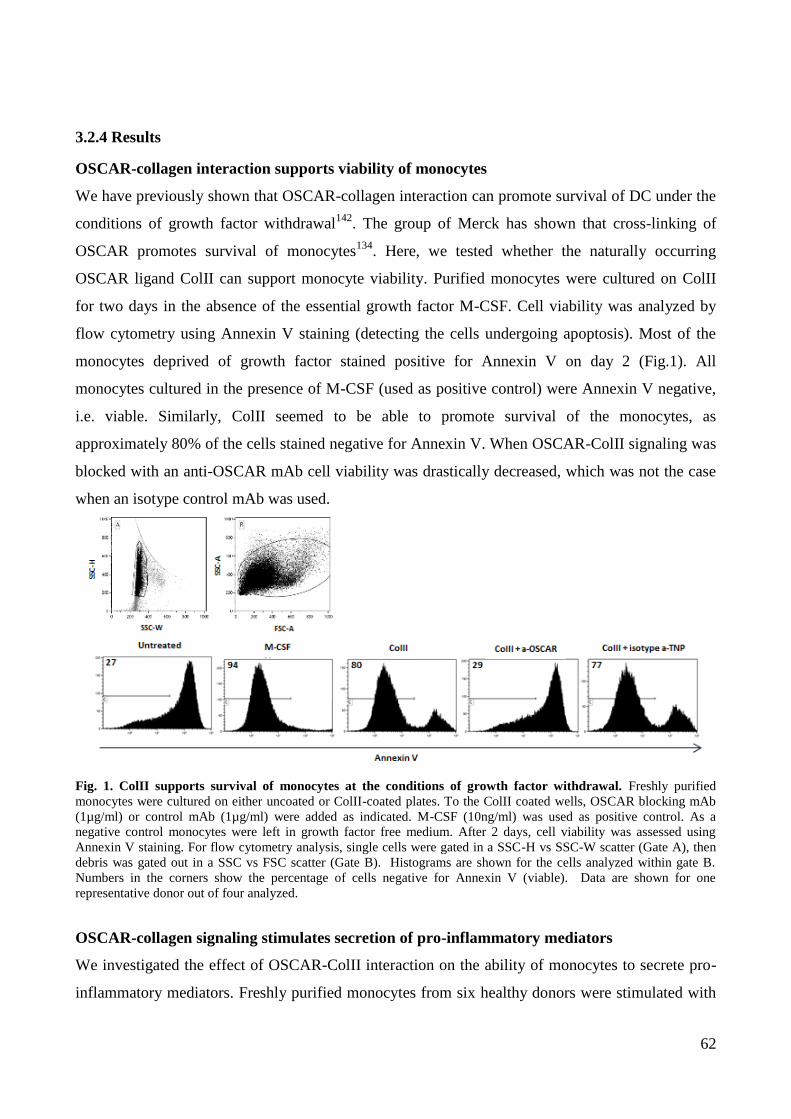
62
3.2.4 Results
OSCAR-collagen interaction supports viability of monocytes
We have previously shown that OSCAR-collagen interaction can promote survival of DC under the
conditions of growth factor withdrawal142
. The group of Merck has shown that cross-linking of
OSCAR promotes survival of monocytes134
. Here, we tested whether the naturally occurring
OSCAR ligand ColII can support monocyte viability. Purified monocytes were cultured on ColII
for two days in the absence of the essential growth factor M-CSF. Cell viability was analyzed by
flow cytometry using Annexin V staining (detecting the cells undergoing apoptosis). Most of the
monocytes deprived of growth factor stained positive for Annexin V on day 2 (Fig.1). All
monocytes cultured in the presence of M-CSF (used as positive control) were Annexin V negative,
i.e. viable. Similarly, ColII seemed to be able to promote survival of the monocytes, as
approximately 80% of the cells stained negative for Annexin V. When OSCAR-ColII signaling was
blocked with an anti-OSCAR mAb cell viability was drastically decreased, which was not the case
when an isotype control mAb was used.
Fig. 1. ColII supports survival of monocytes at the conditions of growth factor withdrawal. Freshly purified
monocytes were cultured on either uncoated or ColII-coated plates. To the ColII coated wells, OSCAR blocking mAb
(1µg/ml) or control mAb (1µg/ml) were added as indicated. M-CSF (10ng/ml) was used as positive control. As a
negative control monocytes were left in growth factor free medium. After 2 days, cell viability was assessed using
Annexin V staining. For flow cytometry analysis, single cells were gated in a SSC-H vs SSC-W scatter (Gate A), then
debris was gated out in a SSC vs FSC scatter (Gate B). Histograms are shown for the cells analyzed within gate B.
Numbers in the corners show the percentage of cells negative for Annexin V (viable). Data are shown for one
representative donor out of four analyzed.
OSCAR-collagen signaling stimulates secretion of pro-inflammatory mediators
We investigated the effect of OSCAR-ColII interaction on the ability of monocytes to secrete pro-
inflammatory mediators. Freshly purified monocytes from six healthy donors were stimulated with

63
ColII for 20 hours and the cell culture supernatants were analyzed for the presence of inflammation
markers. An array of 45 different factors, such as cytokines, chemokines and acute-phase reactants,
was analyzed in the cell culture supernatants using the Multi-Analyte Profiling (MAP) technology
platform (service provided by Myriad RBM Inc.). In the supernatants, 19 inflammation markers
were detected, of which 14 were significantly up-regulated upon ColII exposure as compared to the
untreated control and five proteins showed a tendency to be up-regulated. Stimulation with ColII
triggered production of Alpha-1-Antitrypsin (AAT), Beta-2-Microglobulin (B2M), Fibrinogen, IL-
1, IL-1ra, IL-8, MIP-1, MIP-1, Vascular endothelial growth factor (VEGF), RANTES, tissue
inhibitor or metalloproteinases (TIMP)-1, TNF-, TNFR2 and vitamin D binding protein (VDBP)
in monocytes (Table I), which were down-regulated to the baseline level when OSCAR-collagen
interaction was blocked by an anti-OSCAR mAb, but not by the isotype control. The following
markers, ICAM1, IL-6, IL-10, IL-18 and MCP-1, showed a tendency to be up-regulated when
stimulated with ColII, but the data did not reach a statistical significance.
Table I. Inflammatory mediators secreted by monocytes exposed to ColII
monocytes ColII-monocytes
ColII-monocytes + anti-
OSCAR
ColII-monocytes +
isotype
AAT (ng/ml) 79.0±4.8 92.0±4.9* 67.0±3.0** 96.0±7.1*
B2M (µg/ml) 0.12±0.013 0.16±0.014** 0.10±0.011* 0.16±0.017**
Fribrinogen (ng/ml) 6.9±0.94 10±2.1* 7.2±1.0 10±2.3
ICAM-1 (ng/ml) 0.49±0.018 0.67±0.10 0.53±0.027 0.59±0.048*
IL-1b (pg/ml) 6.3±2.7 15±5.3* 2.9±0.55 14±4.4**
IL-1ra (pg/ml) 147±32 797±213* 90±14* 807±198*
IL-6 (pg/ml) 19±11 51±24 17±6.3 41±18
IL-8 (ng/ml) 0.39±0.18 1.4±0.57* 0.21±0.048 1.2±0.39*
IL-10 (pg/ml) 3.4±0.82 4.8±1.2 3.3±0.58 4.9±1.3
IL-18 (pg/ml) 6.3±1.2 7.8±1.3 5.4±1.0 7.1±1.3
MIP-1a (pg/ml) 24±8.3 58±11*** 14±3.7 60±9.6***
MIP-1b (pg/ml)) 153±50 331±52** 108±28 334±54***
MCP-1 (pg/ml) 17±5.9 29±9.1 15±3.2 30±8.4
RANTES (pg/ml) 3.2±0.39 4.2±0.59* 2.6±0.32*** 3.9±0.42**
TIMP-1 (ng/ml) 22±6.0 54±14* 13±2.5 55±15*
TNF-a ((pg/ml) 22±7.4 89±23** 11±2.0 85±19**
TNFR2 (ng/ml) 2.1±0.23 3.2±0.26*** 1.4±0.13** 3.2±0.2**
VEGF (pg/ml) 307±83 419±83** 212±31 367±61
VDBP (ng/ml) 1.2±0.13 1.4±0.16* 1.1±0.11 1.4±0.14*
Ferritin (ng/ml) 101±16 84±11 92±16* 88±10
IL-1a (pg/ml) 0.63±0.11 0.66±0.11 0.43±0.036 0.7±0.083
CD14+ monocytes were either pre-incubated with 1µg/ml anti-OSCAR mAb or an isotype control for 30 min before plating onto
ColII-coated plates for 20 hours. Supernatant was collected, frozen and shipped to Myriad RBM in Austin, TX for a 45-biomarker
Multi-Analyte profiling. AAT: Alpha-1-Antitrypsin; B2M: Beta-2-Microglobulin; MIP, Macrophage Inflammatory Protein; TNFR2:
Tumor Necrosis Factor Receptor 2; TIMP-1: Tissue Inhibitor of Metalloproteinases 1; VEGF, Vascular Endothelial Growth Factor;
VDBP: Vitamin D-Binding Protein. p<0.05=*; p<0.01=**; p<0.001=*** indicate significant difference from untreated.
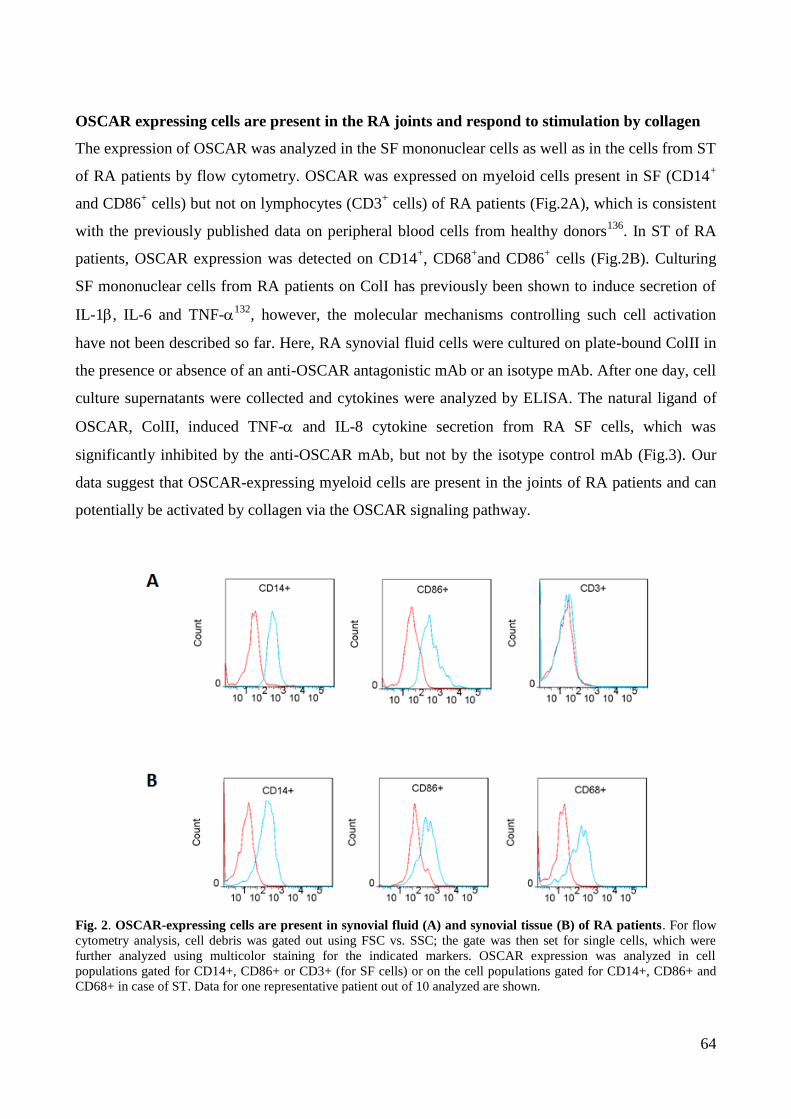
64
OSCAR expressing cells are present in the RA joints and respond to stimulation by collagen
The expression of OSCAR was analyzed in the SF mononuclear cells as well as in the cells from ST
of RA patients by flow cytometry. OSCAR was expressed on myeloid cells present in SF (CD14+
and CD86+ cells) but not on lymphocytes (CD3
+ cells) of RA patients (Fig.2A), which is consistent
with the previously published data on peripheral blood cells from healthy donors136
. In ST of RA
patients, OSCAR expression was detected on CD14+, CD68
+and CD86
+ cells (Fig.2B). Culturing
SF mononuclear cells from RA patients on ColI has previously been shown to induce secretion of
IL-1, IL-6 and TNF-132
, however, the molecular mechanisms controlling such cell activation
have not been described so far. Here, RA synovial fluid cells were cultured on plate-bound ColII in
the presence or absence of an anti-OSCAR antagonistic mAb or an isotype mAb. After one day, cell
culture supernatants were collected and cytokines were analyzed by ELISA. The natural ligand of
OSCAR, ColII, induced TNF- and IL-8 cytokine secretion from RA SF cells, which was
significantly inhibited by the anti-OSCAR mAb, but not by the isotype control mAb (Fig.3). Our
data suggest that OSCAR-expressing myeloid cells are present in the joints of RA patients and can
potentially be activated by collagen via the OSCAR signaling pathway.
Fig. 2. OSCAR-expressing cells are present in synovial fluid (A) and synovial tissue (B) of RA patients. For flow
cytometry analysis, cell debris was gated out using FSC vs. SSC; the gate was then set for single cells, which were
further analyzed using multicolor staining for the indicated markers. OSCAR expression was analyzed in cell
populations gated for CD14+, CD86+ or CD3+ (for SF cells) or on the cell populations gated for CD14+, CD86+ and
CD68+ in case of ST. Data for one representative patient out of 10 analyzed are shown.
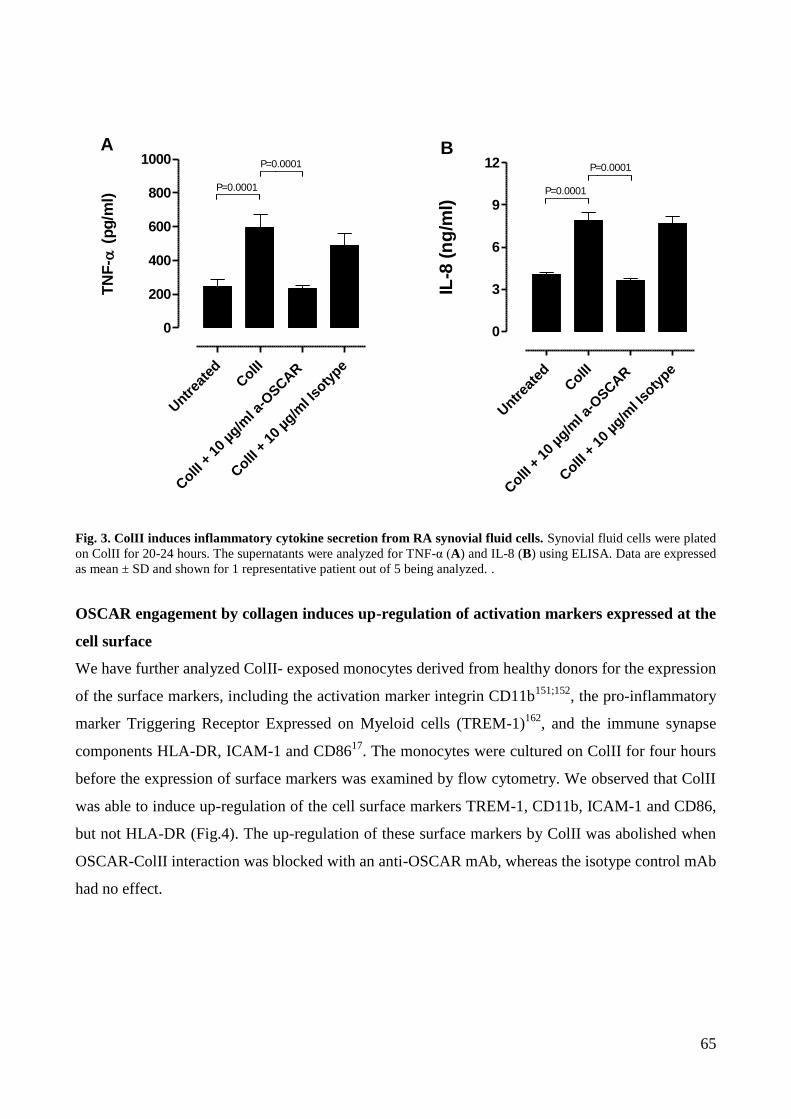
65
Untr
eate
dColII
ColII
+ 1
0 µg
/ml a
-OSCAR
ColII
+ 1
0 µg
/ml I
soty
pe
0
200
400
600
800
1000A
P=0.0001
P=0.0001
TN
F-
(p
g/m
l)
Untr
eate
dColII
ColII
+ 1
0 µg
/ml a
-OSCAR
ColII
+ 1
0 µg
/ml I
soty
pe
0
3
6
9
12B
P=0.0001
P=0.0001
IL-8
(n
g/m
l)
Fig. 3. ColII induces inflammatory cytokine secretion from RA synovial fluid cells. Synovial fluid cells were plated
on ColII for 20-24 hours. The supernatants were analyzed for TNF-α (A) and IL-8 (B) using ELISA. Data are expressed
as mean ± SD and shown for 1 representative patient out of 5 being analyzed. .
OSCAR engagement by collagen induces up-regulation of activation markers expressed at the
cell surface
We have further analyzed ColII- exposed monocytes derived from healthy donors for the expression
of the surface markers, including the activation marker integrin CD11b151;152
, the pro-inflammatory
marker Triggering Receptor Expressed on Myeloid cells (TREM-1)162
, and the immune synapse
components HLA-DR, ICAM-1 and CD8617
. The monocytes were cultured on ColII for four hours
before the expression of surface markers was examined by flow cytometry. We observed that ColII
was able to induce up-regulation of the cell surface markers TREM-1, CD11b, ICAM-1 and CD86,
but not HLA-DR (Fig.4). The up-regulation of these surface markers by ColII was abolished when
OSCAR-ColII interaction was blocked with an anti-OSCAR mAb, whereas the isotype control mAb
had no effect.
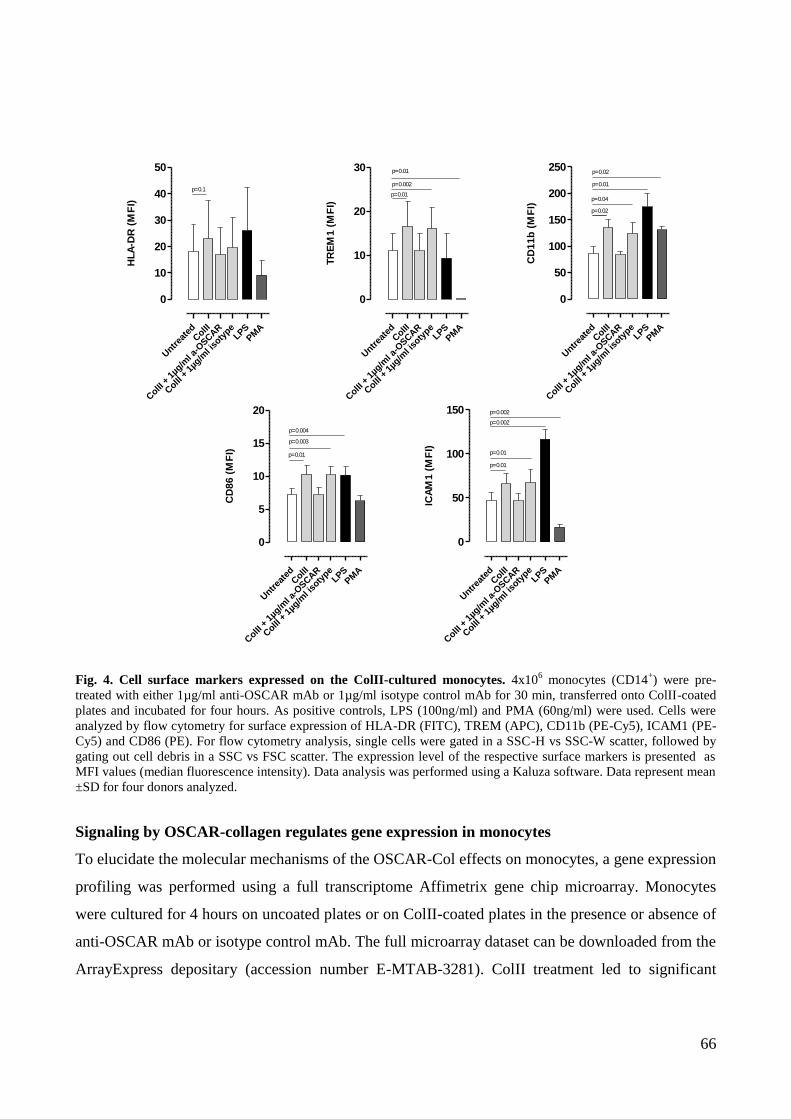
66
Untr
eate
dColII
ColII
+ 1
µg/m
l a-O
SCAR
ColII
+ 1
µg/m
l iso
type
LPSPM
A
0
10
20
30
40
50
p=0.1
HL
A-D
R (
MF
I)
Untr
eate
dColII
ColII
+ 1
µg/m
l a-O
SCAR
ColII
+ 1
µg/m
l iso
type
LPSPM
A
0
10
20
30
p=0.01
p=0.002
p=0.01
TR
EM
1 (
MF
I)
Untr
eate
dColII
ColII
+ 1
µg/m
l a-O
SCAR
ColII
+ 1
µg/m
l iso
type
LPSPM
A
0
50
100
150
200
250
p=0.02
p=0.04
p=0.01
p=0.02
CD
11
b (
MF
I)
Untr
eate
dColII
ColII
+ 1
µg/m
l a-O
SCAR
ColII
+ 1
µg/m
l iso
type
LPSPM
A
0
5
10
15
20
p=0.01
p=0.003
p=0.004
CD
86
(M
FI)
Untr
eate
dColII
ColII
+ 1
µg/m
l a-O
SCAR
ColII
+ 1
µg/m
l iso
type
LPSPM
A
0
50
100
150
p=0.01
p=0.01
p=0.002
p=0.002
ICA
M1
(M
FI)
Fig. 4. Cell surface markers expressed on the ColII-cultured monocytes. 4x106 monocytes (CD14
+) were pre-
treated with either 1µg/ml anti-OSCAR mAb or 1µg/ml isotype control mAb for 30 min, transferred onto ColII-coated
plates and incubated for four hours. As positive controls, LPS (100ng/ml) and PMA (60ng/ml) were used. Cells were
analyzed by flow cytometry for surface expression of HLA-DR (FITC), TREM (APC), CD11b (PE-Cy5), ICAM1 (PE-
Cy5) and CD86 (PE). For flow cytometry analysis, single cells were gated in a SSC-H vs SSC-W scatter, followed by
gating out cell debris in a SSC vs FSC scatter. The expression level of the respective surface markers is presented as
MFI values (median fluorescence intensity). Data analysis was performed using a Kaluza software. Data represent mean
±SD for four donors analyzed.
Signaling by OSCAR-collagen regulates gene expression in monocytes
To elucidate the molecular mechanisms of the OSCAR-Col effects on monocytes, a gene expression
profiling was performed using a full transcriptome Affimetrix gene chip microarray. Monocytes
were cultured for 4 hours on uncoated plates or on ColII-coated plates in the presence or absence of
anti-OSCAR mAb or isotype control mAb. The full microarray dataset can be downloaded from the
ArrayExpress depositary (accession number E-MTAB-3281). ColII treatment led to significant

67
changes in the expression level of 226 genes (>2 fold change; 5% false discovery rate;
Supplemental Table SIA). Treatment of the ColII exposed cells with an anti-OSCAR mAb had an
effect on the expression of 528 genes when compared to the treatment with an isotype mAb
(Supplemental Table SIB). Of the 226 genes regulated by ColII, 218 were clearly regulated via the
OSCAR signaling pathway as the ColII effect was fully abolished by the treatment with an anti-
OSCAR mAb and not by the isotype mAb (“true OSCAR-collagen target genes”; Supplemental
Table S1C). The OSCAR-ColII target genes were clustered into functional categories and pathways
using the Ingenuity Pathway Analysis. The top 20 canonical pathways are listed in Table II. We
found that the OSCAR-ColII signaling regulates the expression of genes involved in
adhesion/diapedesis, cell activation, immune regulation and a number of genes implicated in
various signal transduction pathways implicated in inflammation. OSCAR seems to be involved in
the transcriptional regulation of pro-inflammatory cytokines, their receptors and components of
their signaling pathways. Among those, expression of the gene for IL-1 (IL1A), its receptor IL1R1
as well as the receptor antagonist IL1RN (coding for the IL-1Ra protein) was up-regulated.
OSCAR-ColII up-regulated expression of the gene coding for the key pro-inflammatory cytokine
TNF-α as well as the TNF family member TNFSF14 (coding the LIGHT protein), while expression
of another TNF-related gene, TNFSF10 (coding TRAIL) was down-regulated. A number of genes
encoding chemokines (CCL7, CXCL1, -2 and -3, CXCL5, CCL20 and CCL24) were found to be
transcriptional targets of the OSCAR-ColII signaling. The most pronounced up-regulation was seen
for the gene encoding CCL24, a chemoattratctant for eosinophils, neutrophils and macrophages 163
.
Other OSCAR-ColII regulated molecules and pathways with relevance to inflammation included
components of the NF-κB, TREM-1, Jak2/STAT and TLR signaling pathways as well as ECM
modifying enzymes such as MMP7 and MMP19. A number of genes were down-regulated by the
OSCAR-ColII signaling, including FZD2 (encoding frizzled 2, a receptor for WNT proteins) as
well as chemokine receptors, CCR2 and CX3CR1.

68
Table II. Top 20 canonical pathways regulated by the OSCAR-ColII signaling
Ingenuity Canonical Pathways Molecules -Log (p-value)
Granulocyte Adhesion and Diapedesis MMP7,IL1A,CCL20,CCL24,IL1R1,CXCL5,SDC4,CXCL3,I
L1RN,SDC2,CXCL1,CXCL2,TNF,MMP19,CCL7
9,72E+00
Agranulocyte Adhesion and Diapedesis MMP7,IL1A,CCL20,CCL24,IL1R1,CXCL5,SDC4,CXCL3,I
L1RN,CXCL1,CXCL2,TNF,MMP19,CCL7
8,38E+00
Atherosclerosis Signaling IL1A,IL1RN,LPL,APOC1,CCR2,F3,TNF,TNFSF14,TNFRS
F12A
5,52E+00
Role of IL-17A in Psoriasis CXCL3,CCL20,CXCL1,CXCL5 5,27E+00
TREM1 Signaling CXCL3,NLRP12,TLR8,TLR7,JAK2,TNF,CCL7 5,09E+00
LXR/RXR Activation IL1A,IL1RN,LPL,SERPINF1,APOC1,IL1R1,TNF,CCL7 4,50E+00
Role of Macrophages, Fibroblasts and
Endothelial Cells in Rheumatoid Arthritis
IL1A,F2RL1,RRAS,IL1RN,TLR8,TLR7,CEBPD,IL1R1,JA
K2,TNF,FZD2
3,77E+00
Role of IL-17A in Arthritis CXCL3,CCL20,CXCL1,CXCL5,CCL7 3,71E+00
Altered T Cell and B Cell Signaling in
Rheumatoid Arthritis
IL1A,SPP1,IL1RN,TLR8,TLR7,TNF 3,70E+00
Acute Phase Response Signaling IL1A,RRAS,IL1RN,SERPINF1,IL1R1,JAK2,SERPINE1,TN
F
3,62E+00
NF-B Signaling IL1A,RRAS,IL1RN,TLR8,TLR7,MAP3K8,IL1R1,TNF 3,58E+00
PPAR Signaling PPARG,IL1A,RRAS,IL1RN,IL1R1,TNF 3,55E+00
FXR/RXR Activation PPARG,IL1A,IL1RN,LPL,SERPINF1,APOC1,TNF 3,44E+00
IL-17A Signaling in Airway Cells CXCL3,CCL20,CXCL1,CXCL5,JAK2 3,38E+00
Colorectal Cancer Metastasis Signaling MMP7,RRAS,TLR8,TLR7,RHOU,JAK2,TNF,FZD2,MMP1
9
3,28E+00
Cholecystokinin/Gastrin-mediated
Signaling
IL1A,RRAS,IL1RN,CREM,RHOU,TNF 3,27E+00
Toll-like Receptor Signaling IL1A,IL1RN,TLR8,TLR7,TNF 3,15E+00
Role of Tissue Factor in Cancer F2RL1,RRAS,CXCL1,JAK2,F3,EIF4E 3,14E+00
IL-6 Signaling IL1A,RRAS,IL1RN,IL1R1,JAK2,TNF 3,06E+00
HMGB1 Signaling IL1A,RRAS,RHOU,IL1R1,SERPINE1,TNF 2,99E+00
Monocytes were cultured on ColII for 4 hours in the presence or absence of anti-OSCAR mAb (1µg/ml) or isotype control
(1µg/ml). Total cellular RNA was isolated and genome-wide mRNA expression analysis was done using Affymetrix GeneChip
microarray. The table shows canonical pathways for the OSCAR-ColII regulated genes at a false discovery rate at 5% and a
biological criterion of minimum two-fold change. A complete list of all genes regulated by OSCAR-ColII signaling is provided in
Supplemental Table SIC. Samples from six independent donors were analyzed.

69
3.2.5 Discussion
Differentiation, polarization and activation of leukocytes have been shown to be influenced by their
interaction with ECM proteins10;100;164
, however it still remains unclear which receptors mediate
these effects. We have recently shown that ColI and ColII induce functional maturation and
activation of human monocyte-derived DCs through the OSCAR signaling142
. Besides providing
structural support, collagens exert an important role in various cellular functions, including
proliferation, migration and apoptosis165
. The structure of ECM is pivotal for defining the cellular
response166
. In RA, joint destruction is a hallmark of the disease. The structural damage leads to an
increased turnover of collagens within the joint167
. This makes collagens accessible for interaction
with the infiltrating immune cells, which can alter the cell differentiation and activation status.
Monocytes, the major drivers of synovial inflammation, infiltrate the RA synovial tissue where they
encounter collagen in their environment. OSCAR-expressing mononuclear cells have been detected
in the inflamed synovium in contact with the exposed collagens137;141
. This let us hypothesize that
the activation status of monocytes exiting the circulation might be modulated by the exposed
collagens in the RA joint microenvironment via OSCAR signaling.
Merck and colleagues have shown that cross-linking of OSCAR promotes survival of
monocytes134
. Here, we examined the functional role of OSCAR stimulation with ColII in
monocytes. We show that peripheral blood monocytes exposed to ColII exhibited increased their
viability in the absence of growth factors, and that this effect was mediated by the OSCAR
pathway. Though the molecular mechanisms of this effect were not addressed in the current
manuscript, we speculate based on the microarray data that the OSCAR-collagen signaling
regulates the expression of genes involved in the control of cell death and survival. Among
transcriptional targets of OSCAR are genes encoding components of the NFB- and ERK/MAPK
signaling machinery (both can work in pro-survival pathways), as well as members of the death
receptor signaling like TNF-Related Apoptosis Inducing Ligand (TNFSF10 or TRAIL) and TNF-.
TRAIL triggers programmed cell death and is down-regulated in monocytes stimulated with
collagen. TRAIL has been linked to the pathogenesis of RA where it has been suggested to inhibit
the development of the disease by affecting the viability of the cells in the synovium168
. TNF- is
known to induce survival of myeloid cells169-171
. We observed an up-regulation of TNF- at both
gene and protein levels, possibly contributing to the increased viability of monocytes in an
autocrine and paracrine manner.

70
We detected secretion of several inflammatory markers when monocytes were
exposed to ColII, indicating that collagen activates monocytes in an OSCAR-dependent manner. In
contrast to the data published by Merck et al. reporting that OSCAR cross-linking cannot induce
secretion of pro-inflammatory cytokines, but chemokines134
, we detected a profound secretion of
several pro-inflammatory cytokines including IL-1β, IL-6 and TNF-α. These cytokines are all the
key players in RA pathogenesis and have been found in high concentrations in the synovial fluid of
RA patients25
. IL-1, IL-6 and TNF- are able to increase cellular infiltration of the synovium,
potentiate osteoclastogenesis and induce MMP production within the RA joint25;110;172
. TNF- is
also seen to enhance OSCAR expression on monocytes141
, which might amplify the effect via a
positive feedback mechanism.
Several chemokines were induced by the OSCAR-ColII interaction. Among the
secreted chemokines, IL-8 (recruiting granulocytes), RANTES (recruiting T cells) and MIP-
1α/βchemoattractant for monocytes/macrophages, NK cells and neutrophils) were significantly
up-regulated. Moreover, MIP-1/ has been shown to stimulate the production of reactive oxygen
species and to induce the synthesis of pro-inflammatory cytokines in macrophages and
fibroblasts173-175
. At the transcriptional level, several chemokine genes were up-regulated including
CCL24 (recruiting resting T cells and, to a lesser degree, granulocytes), CXCL3 and CXCL5 (both
attracting neutrophils and acting on the same receptor, CXCR2) and CCL7 (recruiting
monocytes/macrophages). Altogether, these data indicate the potential of OSCAR signaling to
contribute to the amplification of the inflammatory response in RA.
The monocyte chemokine receptors, CCR2 (receptor for MCP1) and CX3CR1
(fractalkine receptor) were down-regulated at the transcriptional level upon exposure of the cells to
ColII in an OSCAR-dependent manner. These receptors, which are critical for the initial
recruitment of monocytes, have been reported to be down-regulated upon extravasation of
monocytes into the inflamed tissue176;177
and their sequential differentiation to macrophages178
,
which might be essential for cell retention within the inflamed tissue. It is likely OSCAR-ColII
signaling is involved, at least in part, in these previously described phenomena.
The expression of several cell surface markers including CD11b, TREM-1, ICAM1
and CD86 was found to be up-regulated in ColII-exposed monocytes in an OSCAR-dependent
manner. CD11b has been shown to be an activation marker in monocytes, which expression was
enhanced in RA patients. It has been suggested that CD11b is involved in monocyte infiltration of
the synovium151
. We have previously shown that OSCAR-collagen signaling is controlling the

71
expression of the major immune synapse components (HLA-DR, CD40, CD80, CD86 and ICAM1)
in monocyte-derived DCs142
. Similar to DCs, monocytes may act as APC, though being less potent.
Here, we show that, unlike DCs, ColII-exposed monocytes did not up-regulate expression of HLA-
DR, though the expression of the adhesion molecule ICAM1 and the co-stimulatory molecule CD86
was increased upon OSCAR engagement by ColII. Thus, OSCAR might potentially contribute to
the monocyte-T cell interaction via stabilization of cell-cell interaction and by enhancing co-
stimulatory signaling.
Another surface marker that was induced by the OSCAR-ColII interaction on the
transcript and protein expression level was TREM-1. The Ingenuity Pathway Analysis (Table II)
has revealed that multiple downstream components of the TREM-1 pathway were up-regulated by
the OSCAR-ColII signaling in monocytes. TREM-1 is a member of the immunoglobulin protein
superfamily that activates myeloid cells through the DAP12 signaling. TREM-1 has been shown to
amplify TLR and NOD-mediated signaling, where it synergistically enhances cytokine
secretion162;179-182
. TREM-1 expression is controlled by the PI3K- and NFB pathways180;183
which
seem to be regulated by the OSCAR signaling. TREM-1 has mainly been implicated in bacterial
infections180
. Besides, TREM-1 expression was also shown to be up-regulated in the RA
synovium184
and blocking TREM-1 signaling in CIA mice models significantly improved the
disease outcome185
indicating that TREM-1 is associated with the RA pathogenesis. We suggest that
the OSCAR pro-inflammatory effect can, at least in part, be mediated by activation of the TREM-1
signaling cascade (Supplemental Figure S1).
We have identified an OSCAR-driven up-regulation of the genes encoding MMP7 and MMP19 in
monocytes cultured on ColII. MMP19 has been detected at the surface of activated peripheral blood
mononuclear cells186
and in blood vessels of the inflamed synovium of RA patients187
. MMP-19 is
able to hydrolyze basement membrane components188
and constituents of cartilage189
. This suggests
that OSCAR-ColII interaction may potentially enhance extravasation and diapedesis of monocytes
during arthritic diseases, leading to an increased invasion of the pannus into the joint, thus
contributing to joint destruction.
In RA, the level of OSCAR expression on monocytes has been shown to positively
correlate with the disease activity and it was suggested that OSCAR functions as a co-stimulatory
receptor enhancing osteoclastogenesis from monocyte-derived precursors 141
. In the synovial tissue
of RA patients, OSCAR expressing mononuclear cells were found to be localized adjacent to the

72
microvasculature139;141
. Here, we tested whether cells present in the synovial fluid of RA patients
can be activated by collagen via OSCAR. The mononuclear SF cells stimulated with ColII secreted
TNF- and IL-8, which was abrogated by blocking the OSCAR-ColII interaction with an anti-
OSCAR antagonistic mAb. Our results support previously published findings on the activation of
mononuclear cells from RA patients cultured on collagen132
and identify OSCAR as being the
receptor mediating this effect. Thus, OSCAR is functional in mononuclear cells present in the SF of
RA patients and these cells - via the OSCAR-ColII interaction - can contribute to the RA
pathogenesis by secreting pro-inflammatory cytokines and chemokines. Collagen fragments have
been identified in the synovial fluid of RA patients158;159;190;191
. Though we did not test whether they
can serve as functional OSCAR ligands, we speculate that they may potentially contribute to
activation of the myeloid cells present in the synovial fluid.
The present study further elucidates the role of OSCAR in myeloid cells. Similar to its
function in DCs142
, the primary function of OSCAR in monocytes seems to be pro-inflammatory.
Our data suggest that the OSCAR-ColII interaction in monocytes may be important in perpetuating
the course of the disease in RA patients. Targeting of OSCAR-ColII signaling may thus represent a
novel approach for therapy of RA.

73
3.3 OSCAR function in macrophages
3.3.1 Introduction
Macrophages are one of the most abundant cell types in the inflamed synovial membrane. The
number of macrophages correlates with damage, pain and inflammation of the joint, indicating an
important role in RA pathogenesis154;192;193
. Macrophages are essential for homeostasis of the body
where they scavenge debris made by physiological or pathological processes. Activation of
macrophages has been observed in RA tissue6. When activated, macrophages secrete a broad
repertoire of proteases, reactive oxygen species, pro-inflammatory cytokines and chemokines. That
in turn recruit and activate other immune cells, thus contributing considerably to the sustained
inflammation and joint destruction in RA6;194
. One of the characteristic features of RA is bone and
cartilage destruction. Macrophages can contribute to the joint destruction by releasing cytokines
such as IL-1 and TNF-, and matrix metalloproteinases (MMPs) or by activating fibroblasts to
produce MMPs43
. Macrophages can differentiate into bone degrading osteoclasts within the RA
synovium under the influence of factors like RANKL and M-CSF26
.
OSCAR is expressed on all myeloid cells in humans, including macrophages135;136
. In
DCs and monocytes OSCAR has been described to have an immunomodulating
role11;134;136;137;141;142
. However, no study has described the function of OSCAR in macrophages. In
house data indicated that cross-linking OSCAR on macrophages induces secretion of MMP-1, -3, -
7, -8 and 12 (preliminary data by S. Panina, Novo Nordisk A/S). In DCs, we have shown by
microarray analysis that MMP-9, MMP-10 and the uPA receptor (uPAR) are transcriptional targets
of the OSCAR signaling142
. Therefore, in this study, we investigated the hypothesis that OSCAR
might be involved in the control of ECM degradation by macrophages and that blocking of OSCAR
signaling could hinder joint damage.

74
3.3.2 Material and methods
Antibodies and ECM proteins
For functional assays several OSCAR-blocking mAb were generated by recombinant technology at
Novo Nordisk A/S (described in Schultz et al., 2015)142
. As an isotype control, anti-TNP human
IgG1.1 was used. IgG1.1 antibodies harbor a Fc fragment with five point mutations that abolish
binding to Fc receptors, as described in161;195
.
ColII purified from human cartilage was purchased from Millipore. Endotoxin level in the protein
solutions was measured using the turbidimetric kinetic LAL assay (reagents supplied by Charles
River Laboratories). All proteins used for functional assays in this study were essentially endotoxin-
free (below detection limit of 0.01 EU/ml).
Macrophage generation and culturing on collagen
Buffy coats from healthy individuals were obtained anonymously from Rigshospitalet Blood Bank,
Copenhagen. All donors gave informed consent according to the protocol approved by The Ethics
Committee for Copenhagen, Denmark, for research use (approval #H-D-2008-113). PBMCs were
purified by Ficoll-Plaque density centrifugation. Monocytes were purified from PBMCs using the
CD14+ Microbeads positive selection kit (Miltenyi Biotec). To induce macrophage differentiation,
purified monocytes were seeded at 3x105 cells/ml in RPMI1640 (Gibco, Life technologies)
supplemented with 10% (v/v) fetal bovine serum, Glutamax (Gibco, Life technologies), 1%
penicillin/streptomycin (Gibco, Life technologies) (referred to as medium), and recombinant human
macrophage colony stimulating factor (CSF-1; Chiron, 2000U/ml or M-CSF, RD systems, 40ng/ml)
(Science Products Dept. or Nunclon) for six days in a 5% CO2 tissue culture incubator at 37°C. At
day three, the medium was replenished. To loosen macrophages from the dish the media was
removed, the cells were quickly flushed with 10ml ice-cold PBS, before adding 10ml PBS
containing 5mM EDTA to the macrophages. The cells were left for 30 min in a 5% CO2 tissue
culture incubator at 37°C. 1x106
cells were thoroughly washed in medium followed by a 20 min
pre-incubation with 1µg/ml OSCAR-blocking, control mAb or “vehicle” (medium without Ab).
Untreated virgin polysterene 24-well plates (Science Products Dept.) were coated with ColII in PBS
at (~3µg/cm2) overnight at 4°C. The plate was washed three times in PBS before cells were added to
the wells. The cells were cultured on ColII for 4 hours or 20 hours as indicated.

75
Analysis of cell surface markers by flow cytometry
Expression of cell surface markers was assessed by flow cytometry. Cells were washed twice in
PBS with 2% FCS and stained with anti-OSCAR-Phycoerythrin (PE)-conjugated mouse mAb
(clone 11.1CN5, Beckman Coulter) or murine IgG2B-PE isotype control (R&D systems) for 30 min
at 37°C in PBS with 2% FCS in the presence of 1% HSA to block unspecific binding. Cells were
washed three times in PBS with 2% FCS prior to flow cytometry analysis. Data acquisition was
performed using a FACSFortessaTM
(BD Bioscience). APC-H7 Dead Cell Stain (Invitrogen, Life
technologies) was used for dead cell exclusion. For compensation, individual Ab stain was used
with one drop of negative control beads and anti-mouse IgG beads (BD Biosciences). For the dead
cell marker compensation arC reactive beads were used (Invitrogen, Life technologies). Data
analysis was performed using Kaluza software version 1.2 (Beckman coulter).
Degradation assay
Macrophage matrix degradation was analyzed using a QCMTM
Gelatin Invadopodia assay (Merck-
Millipore) according to the manufacturer’s protocol. Briefly, poly-L-lysine was absorbed to the
glass coverslip and treated with glutaraldehyde before a layer of FITC-conjugated gelatin was
added to the surface. Subsequently, the fluorescently-coated glass was disinfected with 70% ethanol
and left with medium for 30 min before seeding the cells. Macrophages (3x104 cells) were cultured
on the gelatin surface in the presence of M-CSF (1000U/ml), for 24 hours in a 5% CO2 tissue
culture incubator at 37°C. Following incubation, the cells were fixed with 3.7% formaldehyde and
stained for F-actin (TRICT-Phalloidin) and nuclei (DAPI). Prior to imaging, the coverslips were
transferred to glass slides, containing 1 drop of mounting gel (ProSciTec), sealed and left in dark at
4oC to settle overnight. Cells were imaged using a fluorescence microscope (Zeiss Axioskop 2; Carl
Zeiss; at original magnificationx20). Images were captured by a Zeiss AxioCam MRm (Carl Zeiss)
and visualized with AxioVision 4.4 software. Quantification of the degradation area in percentages
was performed in ImageJ, version 1.46r (NIH, Rasband). The Images were changes to 8 bit images
and the threshold was defined specifically for each donor. The percentage of degradation was
normalized to number of untreated cells. In some experiments, matrix degradation was measured in
the presence of 250nM Plg (Enzyme Research Lab), 10nM aprotinin (Sigma-Aldrich), and 1µg/ml
anti-OSCAR mAb or isotype control.

76
Note, that the manufacturer has disclosed that gelatin used in the assay was extracted from porcine
skin using an acid based method and is known as Type A. Though, they denied disclosing the
composition of the gelatin.
Assessment of MMP secretion
Human MMP base kit Luminex® Performance Assay was used to analyze the quantity of selected
MMPs in supernatants from macrophages, according to manufacturer’s recommendations. In short,
MMP Standard cocktail was reconstituted and diluted in 500μl of Calibrator Diluent RD5-37. After
15 minutes a 3x serial dilution was performed in Calibrator Diluent RD5-37. Standards were ran in
duplicates. 50µl of the microparticle mixture was added to the pre-wetted filter-bottomed
microplate, followed by addition of 50µl of sample or standard. It was left to incubate for 2 hours at
RT on a shaker (500rpm) protected from light. Using a vacuum manifold device the plate was
washed three times in the supplied wash buffer (100µl). Then 50µl Biotin Antibody Cocktail was
added and incubated at the same conditions for one hour. The wash step was repeated. Diluted
streptavidin was added to the wells and incubated at the same conditions for 30min. 100μl Wash
buffer was added per well and incubated on the shaker for 2 min before reading the plate using the
BioPlexTM
200 System (Luminex). Data were analyzed using GraphPad Prism 5.0.
Assessment of cytokine secretion
Multiplex analysis of cytokines and chemokines in the cell culture supernatants was performed
using the BioPlexTM
200 Cytokine Assay platform (Bio-Rad), and a Luminex 200 instrument
(Luminex), according to manufacturer’s protocols. Data was analyzed using GraphPad Prism 5.0.
MMP activity
To measure MMP activity a fluorogenic substrate for several matrix metalloproteinases (MMPs)
(Enzo Life Sciences) was used according to manufacturer’s protocol. Briefly, the peptide was first
dissolved in DMSO to a concentration of 20mM and then diluted in assay buffer (500mM HEPES,
100mM CaCl2 and 0.5% Brij-35 – pH 7) to a 10µM concentration. 100µl of the substrate was added
to a Black microwell 96 well (NUNC) together with 100µl of sample and left to incubate for 1h at
37oC. The Fluorescence intensity was measured using a Paradigm microplate reader (Beckman
Coulter) at the excitation/emission values of 360/450nm set to 37oC every 10 minutes for 10 cycles.

77
Microarray analysis
Total RNA was obtained four hours after culturing macrophages on ColII in the absence or
presence of anti-OSCAR mAb or isotype control mAb. Cells were lysed in 400l TRIzol
(Invitrogen, Life technologies). After adding 80µl chloroform, samples were shaken vigorously for
15 seconds and incubated at room temperature for 3 minutes. The lysate was then centrifuged at
12,000xg for 15 min at 4oC and RNA was purified from the aqueous phase using a RNeasy
MinElute Cleanup Kit (Qiagen, Hilden) according to the manufacturers’ protocol. Subsequent,
RNA was eluted in 14l water. RNA integrity was evaluated on an Agilent 2100 Bioanalyser using
Agilent RNA 6000 Nano Kit chips (Agilent Technologies); RNA Integrity Number (RIN) scores
were of 9.9 or above. The RNA concentration was measured using a NanoDrop® ND-1000
(Saveen&Werner). Microarray experiments were performed according to the protocols supplied by
Affymetrix as described previously in Schultz et al142
.
Statistical analysis
Statistical analyses were performed in GraphPad Prism 5 using Student t tests. A P value less than
0.05 were considered statistically significant. Unless otherwise indicated, means and standard
deviation are shown.
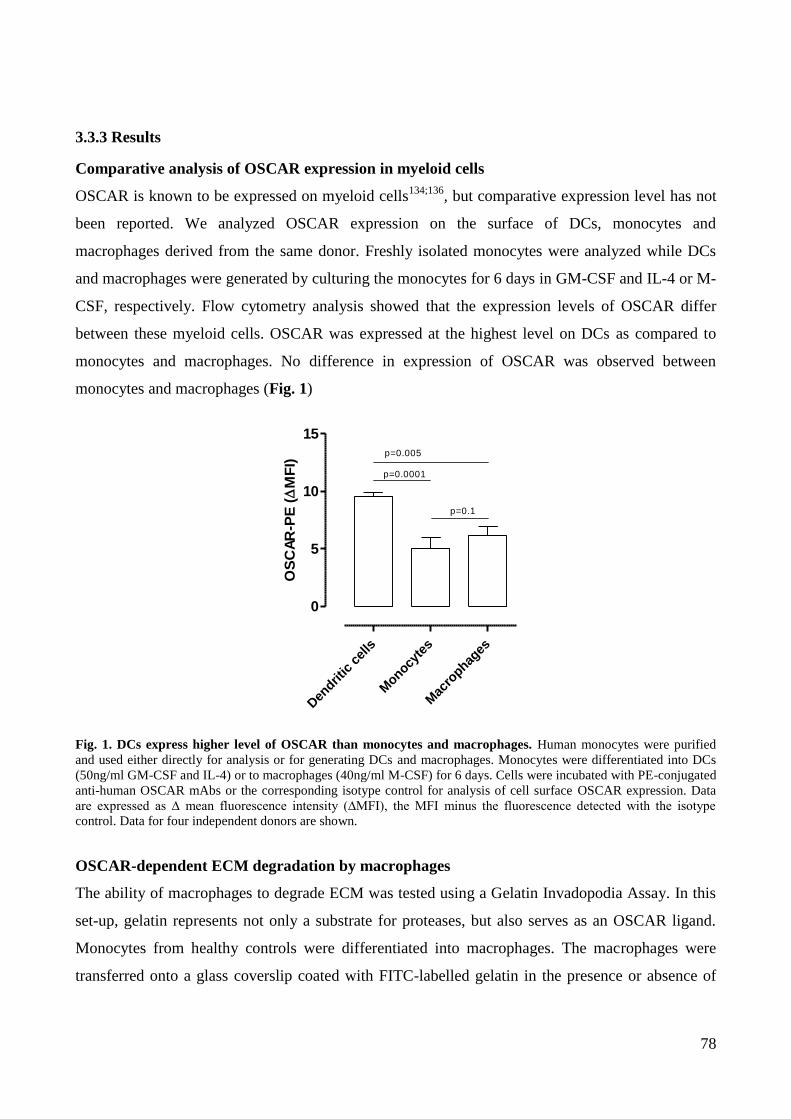
78
3.3.3 Results
Comparative analysis of OSCAR expression in myeloid cells
OSCAR is known to be expressed on myeloid cells134;136
, but comparative expression level has not
been reported. We analyzed OSCAR expression on the surface of DCs, monocytes and
macrophages derived from the same donor. Freshly isolated monocytes were analyzed while DCs
and macrophages were generated by culturing the monocytes for 6 days in GM-CSF and IL-4 or M-
CSF, respectively. Flow cytometry analysis showed that the expression levels of OSCAR differ
between these myeloid cells. OSCAR was expressed at the highest level on DCs as compared to
monocytes and macrophages. No difference in expression of OSCAR was observed between
monocytes and macrophages (Fig. 1)
Den
dritic
cel
ls
Monocy
tes
Mac
rophag
es
0
5
10
15
p=0.0001
p=0.1
p=0.005
OS
CA
R-P
E (
MF
I)
Fig. 1. DCs express higher level of OSCAR than monocytes and macrophages. Human monocytes were purified
and used either directly for analysis or for generating DCs and macrophages. Monocytes were differentiated into DCs
(50ng/ml GM-CSF and IL-4) or to macrophages (40ng/ml M-CSF) for 6 days. Cells were incubated with PE-conjugated
anti-human OSCAR mAbs or the corresponding isotype control for analysis of cell surface OSCAR expression. Data
are expressed as ∆ mean fluorescence intensity (∆MFI), the MFI minus the fluorescence detected with the isotype
control. Data for four independent donors are shown.
OSCAR-dependent ECM degradation by macrophages
The ability of macrophages to degrade ECM was tested using a Gelatin Invadopodia Assay. In this
set-up, gelatin represents not only a substrate for proteases, but also serves as an OSCAR ligand.
Monocytes from healthy controls were differentiated into macrophages. The macrophages were
transferred onto a glass coverslip coated with FITC-labelled gelatin in the presence or absence of
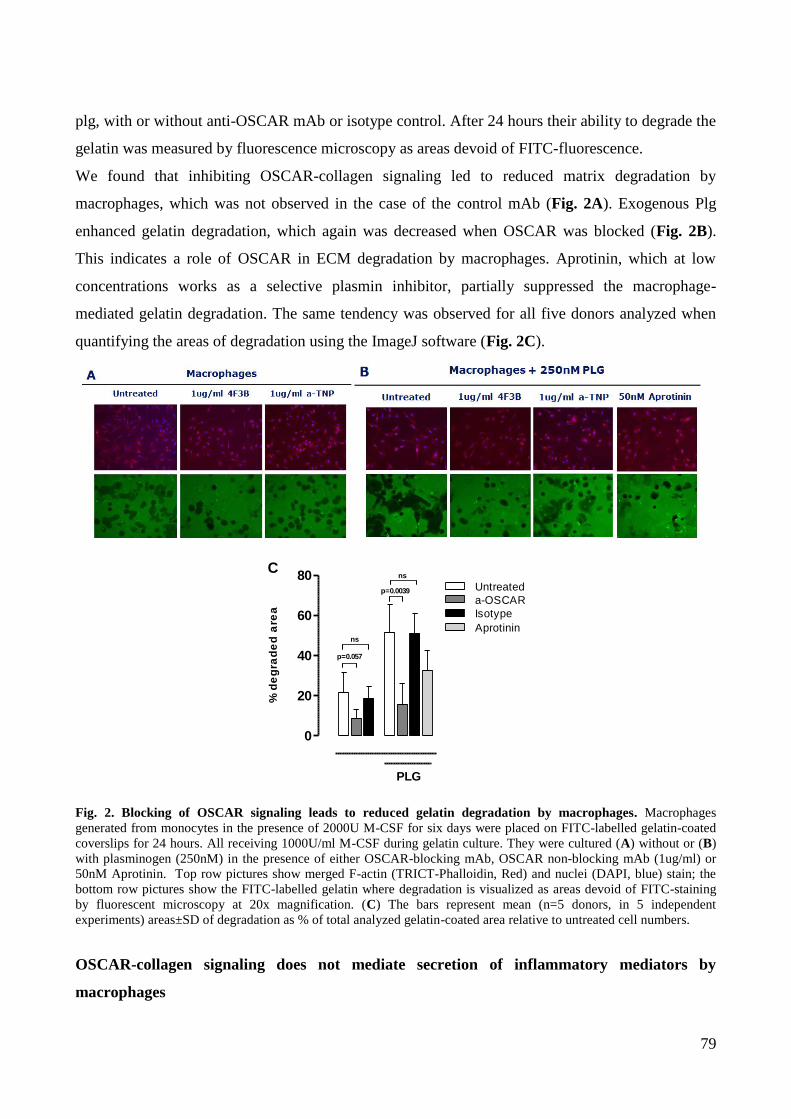
79
plg, with or without anti-OSCAR mAb or isotype control. After 24 hours their ability to degrade the
gelatin was measured by fluorescence microscopy as areas devoid of FITC-fluorescence.
We found that inhibiting OSCAR-collagen signaling led to reduced matrix degradation by
macrophages, which was not observed in the case of the control mAb (Fig. 2A). Exogenous Plg
enhanced gelatin degradation, which again was decreased when OSCAR was blocked (Fig. 2B).
This indicates a role of OSCAR in ECM degradation by macrophages. Aprotinin, which at low
concentrations works as a selective plasmin inhibitor, partially suppressed the macrophage-
mediated gelatin degradation. The same tendency was observed for all five donors analyzed when
quantifying the areas of degradation using the ImageJ software (Fig. 2C).
0
20
40
60
80
PLG
p=0.057
p=0.0039
ns
ns
Untreated
Isotype
Aprotinin
a-OSCAR
C
% d
eg
rad
ed
are
a
Fig. 2. Blocking of OSCAR signaling leads to reduced gelatin degradation by macrophages. Macrophages
generated from monocytes in the presence of 2000U M-CSF for six days were placed on FITC-labelled gelatin-coated
coverslips for 24 hours. All receiving 1000U/ml M-CSF during gelatin culture. They were cultured (A) without or (B)
with plasminogen (250nM) in the presence of either OSCAR-blocking mAb, OSCAR non-blocking mAb (1ug/ml) or
50nM Aprotinin. Top row pictures show merged F-actin (TRICT-Phalloidin, Red) and nuclei (DAPI, blue) stain; the
bottom row pictures show the FITC-labelled gelatin where degradation is visualized as areas devoid of FITC-staining
by fluorescent microscopy at 20x magnification. (C) The bars represent mean (n=5 donors, in 5 independent
experiments) areas±SD of degradation as % of total analyzed gelatin-coated area relative to untreated cell numbers.
OSCAR-collagen signaling does not mediate secretion of inflammatory mediators by
macrophages
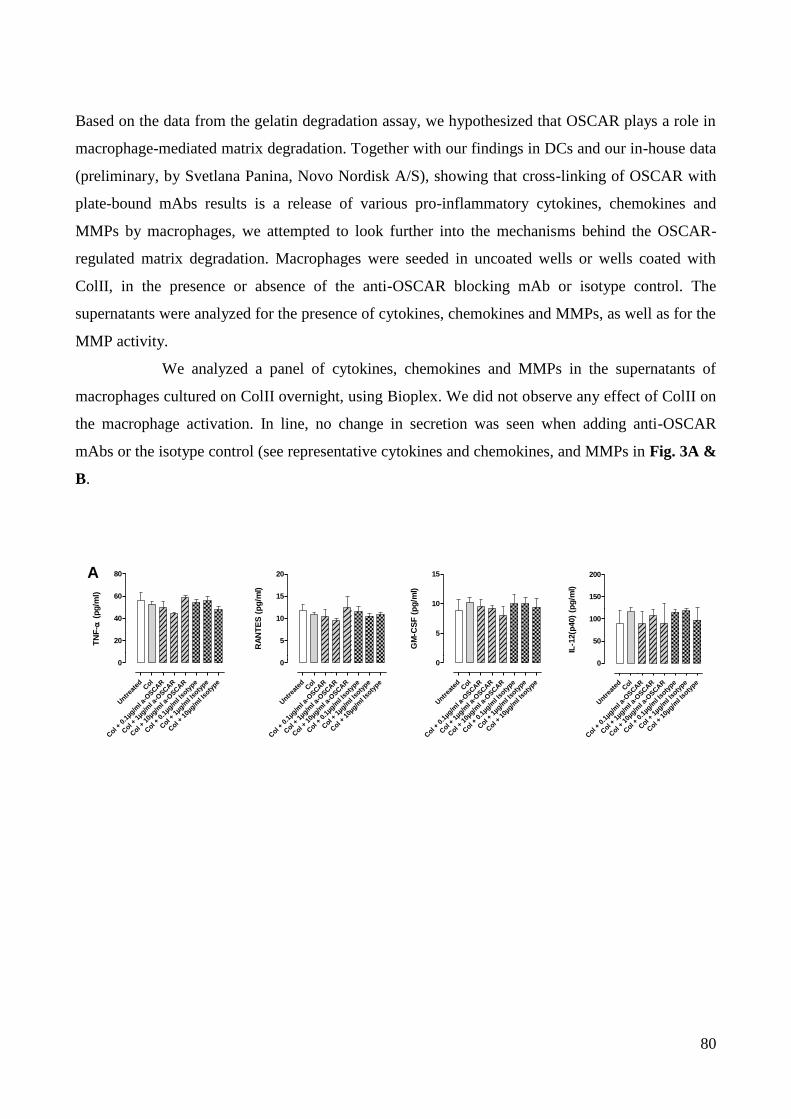
80
Based on the data from the gelatin degradation assay, we hypothesized that OSCAR plays a role in
macrophage-mediated matrix degradation. Together with our findings in DCs and our in-house data
(preliminary, by Svetlana Panina, Novo Nordisk A/S), showing that cross-linking of OSCAR with
plate-bound mAbs results is a release of various pro-inflammatory cytokines, chemokines and
MMPs by macrophages, we attempted to look further into the mechanisms behind the OSCAR-
regulated matrix degradation. Macrophages were seeded in uncoated wells or wells coated with
ColII, in the presence or absence of the anti-OSCAR blocking mAb or isotype control. The
supernatants were analyzed for the presence of cytokines, chemokines and MMPs, as well as for the
MMP activity.
We analyzed a panel of cytokines, chemokines and MMPs in the supernatants of
macrophages cultured on ColII overnight, using Bioplex. We did not observe any effect of ColII on
the macrophage activation. In line, no change in secretion was seen when adding anti-OSCAR
mAbs or the isotype control (see representative cytokines and chemokines, and MMPs in Fig. 3A &
B.
Untr
eate
dCol
Col +
0.1
µg/m
l a-O
SCAR
Col +
1µg
/ml a
-OSC
AR
Col +
10µ
g/ml a
-OSC
AR
Col +
0.1
µg/m
l Iso
type
CoI +
1µg
/ml I
soty
pe
Col +
10µ
g/ml I
soty
pe
0
20
40
60
80
TN
F-
(p
g/m
l)
Untr
eate
dCol
Col +
0.1
µg/m
l a-O
SCAR
Col +
1µg
/ml a
-OSC
AR
Col +
10µ
g/ml a
-OSC
AR
Col +
0.1
µg/m
l Iso
type
CoI +
1µg
/ml I
soty
pe
Col +
10µ
g/ml I
soty
pe
0
5
10
15
20
RA
NT
ES
(p
g/m
l)
Untr
eate
dCol
Col +
0.1
µg/m
l a-O
SCAR
Col +
1µg
/ml a
-OSC
AR
Col +
10µ
g/ml a
-OSC
AR
Col +
0.1
µg/m
l Iso
type
CoI +
1µg
/ml I
soty
pe
Col +
10µ
g/ml I
soty
pe
0
5
10
15
GM
-CS
F (
pg
/ml)
Untr
eate
dCol
Col +
0.1
µg/m
l a-O
SCAR
Col +
1µg
/ml a
-OSC
AR
Col +
10µ
g/ml a
-OSC
AR
Col +
0.1
µg/m
l Iso
type
CoI +
1µg
/ml I
soty
pe
Col +
10µ
g/ml I
soty
pe
0
50
100
150
200IL
-12(p
40)
(pg
/ml)
A
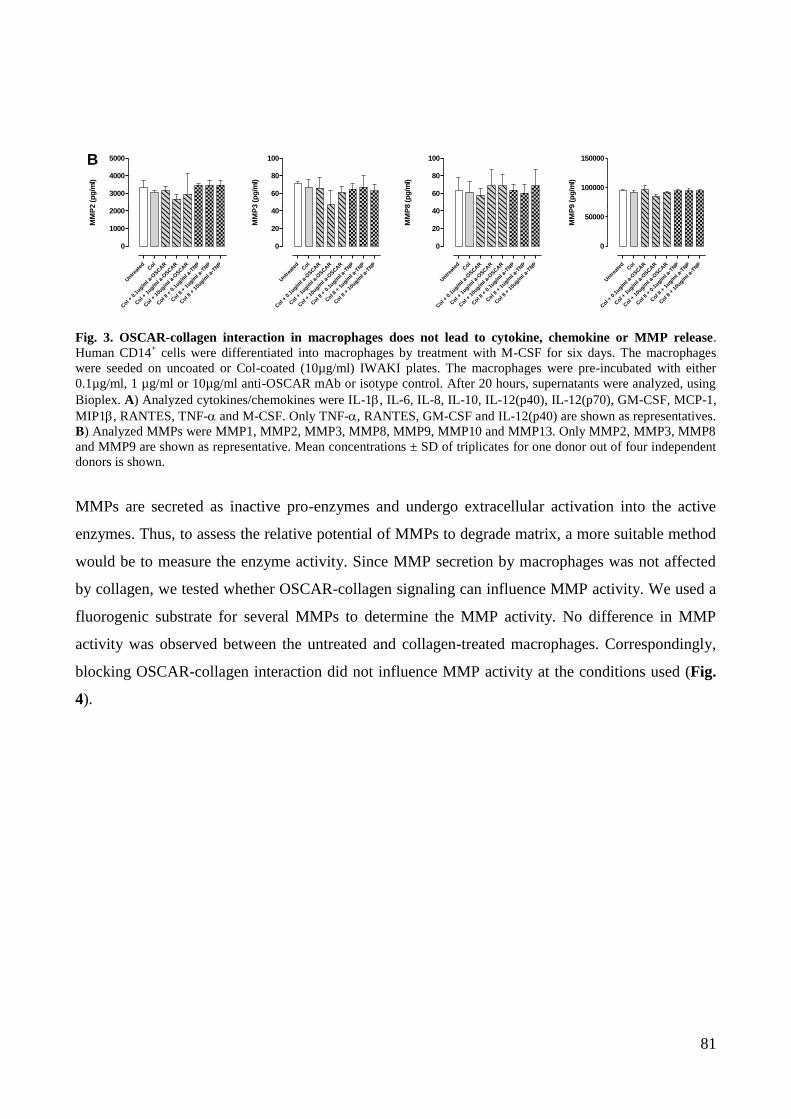
81
Untr
eate
dCol
Col +
0.1
ug/ml a
-OSCAR
Col +
1ug/m
l a-O
SCAR
Col +
10u
g/ml a
-OSCAR
Col I
I + 0
.1ug/m
l a-T
NP
Col I
I + 1
ug/ml a
-TNP
Col I
I + 1
0ug/m
l a-T
NP
0
1000
2000
3000
4000
5000
MM
P2 (
pg
/ml)
Untr
eate
dCol
Col +
0.1
ug/ml a
-OSCAR
Col +
1ug/m
l a-O
SCAR
Col +
10u
g/ml a
-OSCAR
Col I
I + 0
.1ug/m
l a-T
NP
Col I
I + 1
ug/ml a
-TNP
Col I
I + 1
0ug/m
l a-T
NP
0
20
40
60
80
100
MM
P3 (
pg
/ml)
Untr
eate
dCol
Col +
0.1
ug/ml a
-OSCAR
Col +
1ug/m
l a-O
SCAR
Col +
10u
g/ml a
-OSCAR
Col I
I + 0
.1ug/m
l a-T
NP
Col I
I + 1
ug/ml a
-TNP
Col I
I + 1
0ug/m
l a-T
NP
0
20
40
60
80
100
MM
P8 (
pg
/ml)
Untr
eate
dCol
Col +
0.1
ug/ml a
-OSCAR
Col +
1ug/m
l a-O
SCAR
Col +
10u
g/ml a
-OSCAR
Col I
I + 0
.1ug/m
l a-T
NP
Col I
I + 1
ug/ml a
-TNP
Col I
I + 1
0ug/m
l a-T
NP
0
50000
100000
150000
MM
P9 (
pg
/ml)
B
Fig. 3. OSCAR-collagen interaction in macrophages does not lead to cytokine, chemokine or MMP release.
Human CD14+ cells were differentiated into macrophages by treatment with M-CSF for six days. The macrophages
were seeded on uncoated or Col-coated (10µg/ml) IWAKI plates. The macrophages were pre-incubated with either
0.1µg/ml, 1 µg/ml or 10µg/ml anti-OSCAR mAb or isotype control. After 20 hours, supernatants were analyzed, using
Bioplex. A) Analyzed cytokines/chemokines were IL-1, IL-6, IL-8, IL-10, IL-12(p40), IL-12(p70), GM-CSF, MCP-1,
MIP1, RANTES, TNF- and M-CSF. Only TNF-, RANTES, GM-CSF and IL-12(p40) are shown as representatives.
B) Analyzed MMPs were MMP1, MMP2, MMP3, MMP8, MMP9, MMP10 and MMP13. Only MMP2, MMP3, MMP8
and MMP9 are shown as representative. Mean concentrations ± SD of triplicates for one donor out of four independent
donors is shown.
MMPs are secreted as inactive pro-enzymes and undergo extracellular activation into the active
enzymes. Thus, to assess the relative potential of MMPs to degrade matrix, a more suitable method
would be to measure the enzyme activity. Since MMP secretion by macrophages was not affected
by collagen, we tested whether OSCAR-collagen signaling can influence MMP activity. We used a
fluorogenic substrate for several MMPs to determine the MMP activity. No difference in MMP
activity was observed between the untreated and collagen-treated macrophages. Correspondingly,
blocking OSCAR-collagen interaction did not influence MMP activity at the conditions used (Fig.
4).
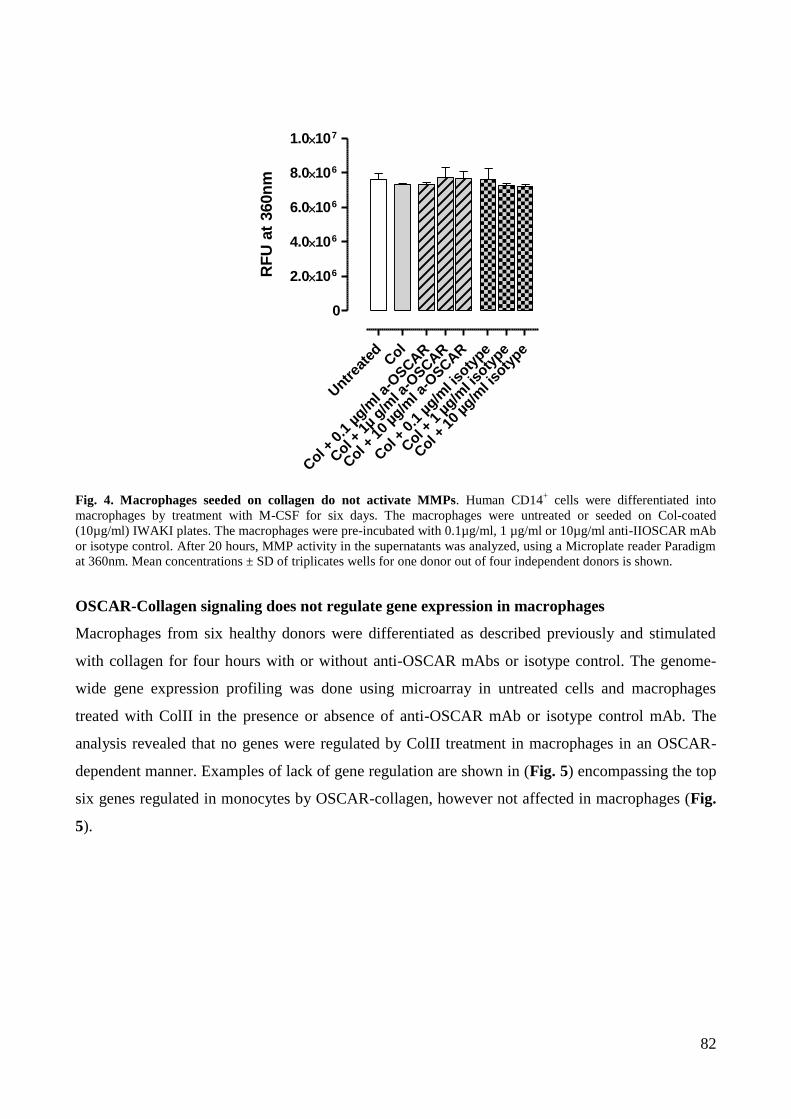
82
Untr
eate
dCol
Col +
0.1
µg/m
l a-O
SCAR
Col +
1µ
g/ml a
-OSCAR
Col +
10
µg/m
l a-O
SCAR
Col +
0.1
µg/m
l iso
type
Col +
1 µ
g/ml i
soty
pe
Col +
10
µg/m
l iso
type
0
2.0106
4.0106
6.0106
8.0106
1.0107
RF
U a
t 360n
m
Fig. 4. Macrophages seeded on collagen do not activate MMPs. Human CD14+ cells were differentiated into
macrophages by treatment with M-CSF for six days. The macrophages were untreated or seeded on Col-coated
(10µg/ml) IWAKI plates. The macrophages were pre-incubated with 0.1µg/ml, 1 µg/ml or 10µg/ml anti-IIOSCAR mAb
or isotype control. After 20 hours, MMP activity in the supernatants was analyzed, using a Microplate reader Paradigm
at 360nm. Mean concentrations ± SD of triplicates wells for one donor out of four independent donors is shown.
OSCAR-Collagen signaling does not regulate gene expression in macrophages
Macrophages from six healthy donors were differentiated as described previously and stimulated
with collagen for four hours with or without anti-OSCAR mAbs or isotype control. The genome-
wide gene expression profiling was done using microarray in untreated cells and macrophages
treated with ColII in the presence or absence of anti-OSCAR mAb or isotype control mAb. The
analysis revealed that no genes were regulated by ColII treatment in macrophages in an OSCAR-
dependent manner. Examples of lack of gene regulation are shown in (Fig. 5) encompassing the top
six genes regulated in monocytes by OSCAR-collagen, however not affected in macrophages (Fig.
5).
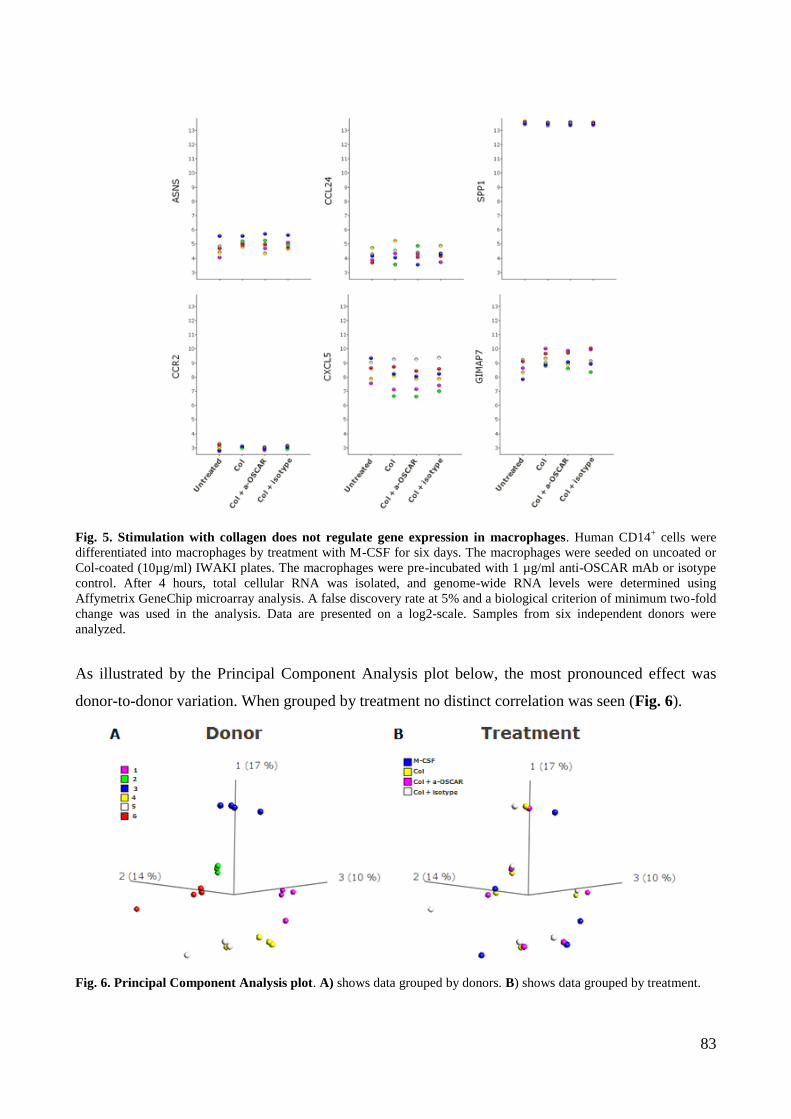
83
Fig. 5. Stimulation with collagen does not regulate gene expression in macrophages. Human CD14+ cells were
differentiated into macrophages by treatment with M-CSF for six days. The macrophages were seeded on uncoated or
Col-coated (10µg/ml) IWAKI plates. The macrophages were pre-incubated with 1 µg/ml anti-OSCAR mAb or isotype
control. After 4 hours, total cellular RNA was isolated, and genome-wide RNA levels were determined using
Affymetrix GeneChip microarray analysis. A false discovery rate at 5% and a biological criterion of minimum two-fold
change was used in the analysis. Data are presented on a log2-scale. Samples from six independent donors were
analyzed.
As illustrated by the Principal Component Analysis plot below, the most pronounced effect was
donor-to-donor variation. When grouped by treatment no distinct correlation was seen (Fig. 6).
Fig. 6. Principal Component Analysis plot. A) shows data grouped by donors. B) shows data grouped by treatment.

84
3.3.4 Discussion
Macrophages are known to be crucial for synovial inflammation. They exert a variety of immuno-
regulatory, phagocytic and matrix degrading functions. In the joint of RA patients, macrophages are
found to secrete multiple proinflammatory cytokines, as well as proteases that are involved in the
degradation of ECM5;6
. In a variety of proof-of-principle clinical studies, it has been shown that the
number of macrophages infiltrating synovial tissue correlates with disease severity and inversely –
with therapeutic benefit, independently of the type of therapy153
. It has been proposed that reduction
of the macrophage numbers after successful treatment does not result from cell death, but rather
from the egress of macrophages from synovial tissue and/or prevention of the influx from
circulation196;197
. Therefore, it has been suggested that targeting monocyte/macrophage
migration/invasion, survival and activation/differentiation can offer a therapeutic potential.
Here, we have observed that gelatin degradation by macrophages was OSCAR
dependent. In our initial experiments, cross-linking of OSCAR in macrophages led to the secretion
of cytokines, chemokines and MMPs (unpublished, S. Panina, Novo Nordisk A/S), indicating that
OSCAR signaling is functional in this cell type. Hence, OSCAR engagement by its native ligand
collagen was thought to be able to activate macrophages. However, we have not been able to detect
any effect on the regulation of gene expression or on the cytokine, chemokine and MMP secretion,
as well as MMP activity when culturing macrophages on collagen. Since activation of other cell
types (i.e. DCs and monocytes) derived from the same donor was drastic, the absence of response in
macrophages to collagen is difficult to explain based on our observations, and would require
additional experiments. The observed discrepancies between responses of the macrophages and
DCs/monocytes to collagen may, at least in part, be attributed to the different expression levels of
various immunoreceptors, including OSCAR expression, or the components of their signaling
machinery. We have analyzed the expression level of OSCAR in monocytes, macrophages and DCs
derived from the same donors. The cell surface expression of OSCAR in monocytes and
macrophages was comparable and significantly lower than that in DCs. We have previously shown
that OSCAR-collagen interaction activates monocytes142
, so the lower expression level in
macrophages compared to DCs may not account for the entire absence of collagen responsiveness
in macrophages.
Collagens are known to interact with multiple immune cell receptors, including
adhesion molecules (integrins) and non-integrin receptors99
. Integrins may synergize with ITAM
receptors providing activating signals to the cells99
. The non-integrin group includes inhibitory

85
ITIM-type receptors, which are known to suppress intracellular signaling by ITAM receptors, hence
regulating the immune response by balancing activating and inhibitory signals198
. The ITIM
receptor LAIR-1 was recently shown to interact with ColI, -II and -III128;199;200
. The LAIR-1 binding
sites on collagen were found not to overlap with OSCAR binding sites on collagen99
. In-house data
on LAIR-1 expression showed that LAIR-1 was expressed at moderate level on monocytes, at low
level in DCs and was highly expressed in differentiated macrophages (unpublished data by S.
Panina and L. Bentzon, Novo Nordisk A/S). LAIR-1 may potentially inhibit OSCAR activation
when both receptors are engaged by collagen. It is possible that the ratio of inhibitory and activating
signals may define the functional outcome of cell stimulation by collagen. LAIR-1 may therefore
contribute to the lack of the effect of collagen seen in macrophages. One may further speculate that
additional ITIM-receptors could interfere with OSCAR-signaling. The inhibitory receptor leucocyte
Ig-like receptor-1/Ig-like Transcript 2 (CD85j) has been shown to negatively regulate OSCAR
induced survival and cytokine production in DCs12
. A better mechanistic understanding of OSCAR-
collagen signaling in macrophages could be explored by inhibiting ITIM-signaling. This could be
performed using pharmacological inhibitors of ITIM signaling components or by knock-down of
individual ITIM receptors, or by using specific receptor-blocking antibodies. Moreover, the
interfering signals from other collagen receptors could also be avoided by using specific OSCAR-
binding collagen peptides instead of full-length collagen. If collagen still cannot induce activation in
macrophages when ITIM-signaling is blocked, another possibility is that OSCAR requires a co-
receptor for full activation. Such putative co-receptor might be expressed on monocytes and DCs,
but not on macrophages. Merck and colleagues have shown that the effect of OSCAR ligation in
neutrophils is weaker compared to the effect observed in monocytes134
. They speculated that it
could be explained by a limited availability of FcRγ in neutrophils, which serves as signaling
subunit for multiple receptors expressed and functioning in immune cells.
Despite the lack of effect of collagen on the activation of macrophages, we have shown that anti-
OSCAR mAb inhibits degradation of gelatin by macrophages. The effect was reproducible and seen
in at least five independent experiments. Gelatin has a high content of collagen that, besides serving
as a substrate for macrophage degradation, functions as a ligand for OSCAR. Gelatin degradation is
primarily mediated by MMPs and various proteases, like plasmin201
. We found that macrophage
degradation of gelatin was greatly potentiated by the addition of plasminogen. When OSCAR was
blocked at these conditions, the degradation was inhibited to the level observed when no

86
plasminogen was added. This suggests that OSCAR-signaling induce factors important for plasmin
activation. We have previously identified several genes involved in ECM turnover being regulated
by OSCAR-signaling in DCs and monocytes. These genes include several MMPs and urokinase-
type plasminogen activator receptor (uPAR)142
. We speculate that OSCAR signaling may
potentially control ECM degradation via regulation of MMP and conversion of plasminogen to
plasmin. Unfortunately, I did not study the mechanisms of OSCAR involvement in macrophage
mediated degradation of gelatin further during my stay at the University of Melbourne.
We have tried to clarify the mechanisms of macrophage-mediated gelatin breakdown
by using a more simple system, namely a purified collagen, as the natural ligand of OSCAR. Even
though gelatin contains collagen in its polymerized form, the composition of the two substrates used
in our study is very different and this fact may be the major reason why collagen did not promote
activation of macrophages while gelatin did. Besides collagen, other ECM components are present
in gelatin, which may interact with ECM receptors and potentiate the effect of the OSCAR
signaling. Alternatively, binding sites for inhibitory (e.g. ITIM) receptors might not be accessible in
gelatin. Studies have shown that various matrix proteins are crucial for macrophage MMP
secretion202-204
. It has been shown that macrophages cultured on ColI and ColII but not ColIV,
laminin, fibronectin and elastin can induce collagenase (MMP-1, -8 and -13) production202
. Lepidi
et al. reported that MMP synthesis by macrophages was decreased on polymerized ColI and slightly
increased on monomeric collagen203
. Moreover, in a study searching for immune-modulating
biomaterial to regulate macrophage activity, Franz et al. found that ECM composed of ColI and
high-sulfated hyaluronan dampened the activity of pro-inflammatory macrophages204
. Taken
together, these reported data indicate that the nature of ECM may drastically influences macrophage
activity. Further experiments should be performed to mechanistically assess activation of the
OSCAR signaling in macrophages using gelatin as a surrogate ligand.
In summary:
1. OSCAR is expressed on macrophages and can potentially be functional, as shown by cross-
linking studies
2. Purified ColII does not stimulate macrophage activation at the conditions used
3. OSCAR signaling may control macrophage-mediated degradation of collagen-rich ECM,
gelatin, possibly by regulating MMP secretion and/or activation and converting
plasminogen into plasmin

87
4. Discussion
It is well established that OSCAR plays a role in osteoclastogenesis, as it has been shown in in vivo
and in vitro studies29;32;137
. In humans, OSCAR is more widely expressed than in mice, where the
expression is restricted to osteoclasts29
. Before collagen was discovered as the ligand of
OSCAR137;142
, the functions of OSCAR in immune cells were studied by cross-linking OSCAR
with mAbs11;134;136
. Therefore, the main goal of this PhD project was to investigate the function of
OSCAR following interaction with its natural ligand, collagen, in human immune cells.
OSCAR is constitutively expressed on human myeloid cells. Consequently, the
availability of the OSCAR ligand will most likely determine the activation of OSCAR. OSCAR
plays an important role in OC function and in the maintenance of bone205
. However, in immune
cells the function of OSCAR is still not yet elucidated. The group of Trowsdale speculated that
collagen could be regarded as a “damage-associated molecular pattern” that could be sensed by
OSCAR on immune cells to detect alterations in the ECM137
. This would be beneficial at the
homeostatic conditions, but could potentially be harmful in chronic inflammatory situations.
Collagens are recognized to play a major role in RA. From animal models we know
that collagen can induce arthritis (collagen-induced arthritis, CIA, models). Moreover, collagen
fragments and antibodies against collagens have been identified in the sera and synovial fluid of
some RA patients 158;159;206;207
. It has further been suggested that the severity of RA may correlate
with an increase in collagen degradation190
. In RA, it is likely that collagen, which is normally
embedded in the ECM or under the layer of endothelial cells, chondrocytes and fibroblasts,
becomes exposed due to cellular reorganization and ECM remodeling within the RA joint43;104;137
.
OSCAR has been demonstrated to be expressed on OCs and mononuclear cells in the joint tissue of
RA patients and being up-regulated on the circulating monocytes141
. We have speculated that the
collagen exposure upon ECM remodeling in the RA joints and the concurrent influx of
inflammatory OSCAR-expressing immune cells like monocytes, macrophages and DCs, may
significantly contribute to the RA disease pathology. This was supported by the findings by
Trowsdale and colleagues who have identified OSCAR-expressing mononuclear cells in contact
with collagen type I and II at the bone remodeling sites137
. It is therefore crucial to gain further
knowledge about the functional outcome of the OSCAR-collagen interaction in immune cells, as a
way to discover new therapeutic interventions.

88
During the course of inflammation, monocyte-derived DCs (mo-DCs) may develop in the inflamed
tissue from blood monocytes208;209
. Due to the relevance in RA, we chose this DCs type to study the
effect of OSCAR-collagen interaction. In paper I we describe the identification of ColI and ColII
as the ligands of OSCAR and investigate the functional outcome of the OSCAR-collagen
interaction in DCs142
. Monocytes and macrophages are central to the inflammation observed in
RA5. They massively infiltrate the joint, have an activated phenotype and produce high levels of
TNF-6;151;152;210
. When entering the joint, monocytes become further activated upon adherence to
ECM and exposure to the complex inflammatory environment encountered211
. The functional
outcome of monocyte activation by ECM would provide useful information about the pathogenesis
of RA. Paper II primarily describes the activation of monocytes by the OSCAR-collagen
interaction142
. Macrophages are a major source of cytokines and chemokines, and degrading
enzymes which drive inflammation and joint destruction6. Treatments which have been shown to
improve the disease activity led to a decrease in macrophage numbers in the joint153;194
. Based on
the findings in papers I and II, part III of the result section assess the role of OSCAR in
macrophages using two different substrates: ColII and collagen-rich gelatin. Have
A thorough discussion of the results is provided in the Discussion section of the included paper I,
paper II and part III. Hence, the section below covers a combined discussion with a focus on the
mechanisms and biology of OSCAR-collagen interaction in DCs, monocytes and macrophages,
related to the RA pathology.
The identification of collagen as a ligand for OSCAR was done prior to the start of my PhD project
by the group of Dr. Panina (Novo Nordisk A/S) in parallel with the group of Trowsdale, with the
latter being the first to publish the data137
. Both ColI and ColII were found to bind OSCAR (Paper
I) and initially both collagens were used to stimulate the DCs. Our results showed that ColI and
ColII elicited the same effect in DCs. However, ColII was more potent than ColI, as ColII generated
a greater cytokine response and induced higher expression of surface activation markers than ColI.
When comparing the gene profiles of ColI- and ColII-stimulated DCs, we saw that over 90% of the
genes overlapped. We therefore chose to continue the studies using ColII as our primary OSCAR
ligand.

89
To understand the extent of the involvement of OSCAR in DC biology, we investigated the effect
of adding anti-OSCAR-blocking mAb while generating iDCs from blood monocytes. We found that
adding anti-OSCAR mAb did not have an effect on the differentiation of monocytes to iDCs under
the conditions used in these studies. This suggests that iDC differentiation does not require
OSCAR-collagen signaling. Further, to exclude any indiscriminate inhibitory effect on DC
activation by anti-OSCAR mAb, we evaluated the effect of anti-OSCAR mAb on LPS activated
DCs. We found that anti-OSCAR mAb did not have an effect on the LPS-driven cytokine release.
Hence, we believe that adding OSCAR mAb has no effect on DCs when collagen is not present.
As demonstrated in paper I and II, we had several main findings when exploring the outcome of
the OSCAR engagement by collagen in DCs and monocytes. Culturing DCs and monocytes on
collagen led to an enhanced secretion of a variety of inflammatory markers, including chemokines
and cytokines, in an OSCAR-dependent manner. Chemokine and cytokines are important in the
orientation and amplification of the adaptive immune response212
. Among the chemokines detected,
IL-8 and RANTES were found at high levels. These chemokines recruit immune cells, including T
cells, neutrophils, granulocytes and monocytes to the joints. These cells further mediate the
generation of reactive oxygen species, promote angiogenesis, and modulate the function of other
immune cells213;214
. Merck and colleagues suggested that the role of OSCAR in DCs was not
proinflammatory since they could not detect any or low amounts of cytokines when cross-linking
OSCAR on DCs or monocytes11;134
. In contrast, we observed a drastic induction of proinflammatory
cytokines, like TNF-, IL-1 and IL-6, by DCs and monocytes when cultured on collagen,
suggesting that OSCAR on DCs and monocytes have a proinflammatory function. Additionally, we
found that collagen, in an OSCAR dependent manner, induced secretion of TNF- and IL-8 by
synovial fluid cells from RA patients, showing that the OSCAR pathway is active in immune cells
present in the RA synovium. TNF-, IL-1, IL-6 and IL-8 may all be found at high levels in the RA
joints and seem to play a crucial role in the course of the disease25
. The pro-inflammatory cytokines
contribute to the RA pathology in several ways. They stimulate osteoclastogenesis, increase the
bone-resorbing capacity of osteoclasts, stimulate secretion of MMPs and nitric oxide and prevent
new synthesis of collagens and proteoglycans, hence contributing to bone erosion and cartilage
degradation43;104;113
.

90
To evaluate the underlying molecular mechanisms of the OSCAR-collagen mediated
cell activation, we analyzed the global gene expression profile upon OSCAR engagement in DCs
and monocytes. OSCAR engagement by collagen up-regulated expression of genes involved in the
mitogen-activated protein kinases (MAPK) pathways, like p38 and ERK, as well as PI3K and the
NFB family of transcription factors. Presumably as a result of the activation of NFB and MAPK
signaling, several pro-inflammatory genes, which are known to be transcriptional targets of these
pathways, were strongly up-regulated. Of importance in DCs, OSCAR-collagen interaction
regulated the expression of genes involved in the immune regulation and activation. Among these,
the most up-regulated genes were TNF-, IL-1 and IL-1, together with several chemokines
including CCL20, CCL5/RANTES, CXCL8/IL-8 as well as chemoattractants for endothelial
precursor cells CXCL1, -2 and -3. In monocytes, expression of genes which products are involved
in the immune regulation and cell activation were also induced by OSCAR-collagen interaction. Of
interest was the up-regulation of genes coding for TNF-, IL-1, CCL24, CCL20, CCL7, CXCL2
and -3, which are essential for amplification of the immune response and attracting lymphocytes,
monocytes, macrophages and endothelial cells25;44
. For both DCs and monocytes, genes involved in
cell-matrix interaction, such as osteopontin, syndecans and MMPs were found to be up-regulated.
The gene profile analysis supports our findings on the protein level that OSCAR signaling can
activate monocytes and DCs to secrete proinflammatory mediators.
In the absence of growth factors, most cells would undergo apoptosis. Paper I and II make it
evident that OSCAR-collagen interaction protects monocytes and DCs from apoptosis induced by
the absence of growth factors. Our results are in line with the findings made by other groups,
showing the pro-survival effects of OSCAR11;134;147
. Based on our microarray analysis one may
speculate about the mechanism by which OSCAR-collagen interaction promotes survival of DCs
and monocytes. The gene profile analysis of DCs and monocytes revealed that several pro- and anti-
apoptotic pathways were affected, like the MAPK and the TNF related weak inducer of apoptosis
(TWEAK) signaling. However, the specific mechanisms behind the increased viability appear
different for the DCs and monocytes. OSCAR-collagen interaction in DCs seems to protect them
from apoptosis by up-regulating anti-apoptotic genes, such as BCL2A1 and multiple components of
the MAPK/ERK pathway, and by down-regulating members of the caspase family, such as caspase
6 and 8. Fewer genes controlling cell survival were found to be regulated in monocytes by OSCAR.
Nevertheless, the death receptor signaling pathway promoting apoptosis was seen to be down-

91
regulated (TNFSF10/TRAIL), while TNFRSF4/CD134 which is involved in suppressing apoptosis
was up-regulated by Col in an OSCAR-dependent manner.
Additionally, we have shown that both DCs and monocytes produce TNF- when
stimulated with collagen, which in an autocrine manner could contribute to the increased
viability169;215;216
. The increased viability of DCs and monocytes could ensure a long-term activity,
sustaining the inflammation seen in the joints of RA patients.
DCs are potent APC cells that are crucial for the communication between the innate and adaptive
immune system. After Ag capture, iDCs migrate to the lymph nodes and the spleen, where they
undergo maturation and present their antigen-peptide complexes to T cells. In RA, DCs are found in
the synovial tissue and synovial fluid, where they may play a role of powerful immune stimulators.
In the RA synovial tissue, DCs have been found within lymphoid aggregates expressing a variety of
co-stimulatory receptors and being capable of activating T cells57;217
. Paper I shows that upon
OSCAR-collagen interaction the DCs increase the expression of numerous phenotypic markers for
cellular activation and maturation, such as CD83, CD86 and HLA-DR. Moreover, several
components of the immune synapse including CD40, CD80, CD83, CD86 and ICAM-1, were found
to be transcriptional targets of the OSCAR-collagen signaling. The interaction of DCs with T cells
is central for the inflammation seen in RA57
. When DCs mature they become capable of inducing
clonal expansion of T cells and the cytokines secreted by the DCs can skew the nature of the T cell
responses217
. The Col-DCs showed a significant difference in their ability to induce T cell
proliferation in the DC-T cell co-culture studies compared to the control iDCs. When adding an
anti-OSCAR blocking mAb during DC maturation, these DCs had a reduced T cell stimulatory
capacity. The T cells that were co-cultured with the Col-DCs produced high amounts of
proinflammatory cytokines and chemokines. We found both Th1 and Th2 cytokines in the
supernatants of the Col-DCs-T cell co-cultures. We did not see any clear polarization of the T cells,
as the cytokine levels did not seems to be skewed. The generated T cells may further contribute to
bone remodeling by expressing RANKL hence increasing osteoclastogenesis more.
Part III of my PhD work describes the role of OSCAR-collagen interaction in macrophages. We
and others136
have found OSCAR to be expressed on human DCs, monocytes and macrophages.
When analyzing the OSCAR expression levels on these cells, we identified a higher expression of
OSCAR on DCs compared to monocytes and macrophages. In-house data by the group of Dr.

92
Panina have shown that OSCAR may potentially be functional in macrophages as cross-linking of
OSCAR enhanced the secretion of cytokines and MMPs (preliminary data by Dr. Panina, Novo
Nordisk A/S). However, in macrophages the activity of OSCAR seems to be different from DCs
and monocytes, as we did not observe any collagen-mediated effects in the analyzed macrophages.
We speculated that the unresponsiveness could partly be due to the lower OSCAR expression than
that in DCs in combination with a high expression of the inhibitory collagen receptor LAIR1, or
maybe a defective capacity of the OSCAR-signaling machinery. However, when assessing
degradation of gelatin by macrophages, we found that blocking OSCAR effectively decreased the
degradation. Hence, indicating that OSCAR plays a role in enhancing macrophage-mediated gelatin
degradation. Gelatin degradation is primarily mediated by MMPs and other proteases such as
plasmin. Through gene analysis in DCs and monocytes we have found that OSCAR-signaling
affects genes involved in the ECM remodeling, including MMPs and the uPA receptor142
. When
plasminogen was present, matrix degradation was more efficient; however blocking OSCAR-
signaling inhibited the degradation to the level observed when no plasminogen was added. This
indicates that OSCAR signaling induces factors important for plasmin activation. Hence, the
mechanisms of OSCAR-regulated gelatin degradation might be MMP and plasmin dependent.
Altogether, OSCAR engagement in macrophages might contribute to the invasion of macrophages
into the synovium and to joint destruction in RA.
We have shown that gelatin but not collagen had an OSCAR-dependent effect serving
as a ligand for OSCAR. Although the substrates are related there is a clear difference in structural
composition and preparation of ColII and gelatin. Gelatin is a mixture of several collagens, mainly
ColI and III, and other ECM components like elastin, fibrillin and fibronectin are present98;106;218-220
.
The ColII used in my experiments is relatively pure (90%), with only little contribution of other
collagens. The OSCAR and LAIR1 binding sites may therefore be different between ColII and
gelatin. The gelatin experiments were conducted during my stay in Australia and unfortunately due
to a time limitation I did not study the mechanisms behind my observations. It remains to be
investigated what is causing the difference in the OSCAR-collagen effects between macrophages
and DCs/monocytes. One strategy could be to evaluate the contribution of ITIM-receptors, which
could be investigated by using specific receptor-blocking antibodies, inhibitors of ITIM signaling
components or knocking-down individual ITIM receptors. The interfering signals from other
collagen receptors could also be avoided by using specific OSCAR-binding collagen peptides
instead of full-length collagen. Finally, there is a possibility that OSCAR requires a putative co-
93
receptor for full activation, which might be expressed on monocytes and DCs, but not on
macrophages.
Figure. 10. Role of OSCAR in rheumatoid arthritis. Potential outcome of OSCAR-collagen interaction in the RA
joint is indicated in red boxes. OSCAR is a co-stimulator of osteoclastogenesis. In monocytes, DCs and macrophages
OSCAR shows immune-modulatory effects, including increased viability, promotion of activation, maturation (leading
to T cell expansion and polarization) and ECM degradation. The secreted cytokines and chemokines recruit more
immune cells, enhance cell stimulation and stimulate the release of proteases. Altogether, inflammation, and increased
osteoclastogenesis, mediates bone and cartilage degradation. Blocking of OSCAR signaling in the synovial tissue may
inhibit macrophage invasion, dampen DC-driven local T cell expansion and hence cytokine level and inhibit cartilage
and bone damage.

94
5. Conclusion
This PhD thesis demonstrates that collagen is a functional ligand of OSCAR in DCs and
monocytes. Our data emphasizes the importance of ECM remodeling in chronic inflammatory
conditions, as the exposed ECM interacts with immune cells contributing to the inflammation seen
in the joints of RA patients. We have shown that OSCAR-collagen interaction in DCs leads to an
increased viability, enhanced secretion of pro-inflammatory cytokines and chemokines, augmenting
the recruitment of immune cells to the joint. OSCAR promotes maturation of DCs, which leads to
expansion and activation/polarization of T cell characterized by cytokine secretion, thus further
enhancing the inflammatory response. We found that OSCAR promotes cell survival and activation
of monocytes. Monocytes were shown to secrete inflammatory cytokines and chemokines and to
up-regulate surface markers when stimulated with collagen in an OSCAR-dependent manner.
Furthermore, OSCAR was expressed and shown to be functional on synovial fluid cells from RA
patients. Surprisingly, the OSCAR-collagen pathway was not capable of activating macrophages.
Though, preliminary data indicate that OSCAR can be involved in the control of degradation of
collagen-rich gelatin by macrophages, the mechanisms controlling this effect need further
investigation. Altogether, our studies indicate that OSCAR is not only involved in
osteoclastogenesis, but may activate the immune response, hence contributing to the pathogenesis
of RA on multiple levels. This underlines the possibility that inhibition of OSCAR signaling may
halt both, the inflammatory response and the bone erosion associated with RA.

95
6. Perspectives
Current treatment options for RA are to reduce inflammation and delay joint destruction. The
simplified RA treatment paradigm relies on steroids and Non-steroidal anti-inflammatory drug
(NSAIDS) for mild RA, DMARDS for moderate RA and biologics, like TNF- inhibitors, for
severe RA, which all primarily target inflammation. Unfortunately, a large number of the patients
fail to respond to the therapeutics and the therapies can cause undesired side effects221;222
. Recently,
myeloid cells have come into focus as potential targets for RA treatment. Anti-RANKL mAbs have
been developed and demonstrated a favorable bone-protecting effect, but no effect on inflammation
in RA222
. So, even though new therapies were developed in the recent years, there is still a strong
need for better treatment options. Novo Nordisk A/S therefore explored the option of targeting
OSCAR for treatment of RA. The expectations were that targeting OSCAR would inhibit OCs
formation as well as myeloid cell activation to limit both bone erosion and inflammation.
6.1 OSCAR project
The purpose of this PhD was to investigate the function of OSCAR on immune cells when engaged
by its natural ligand collagen. A thorough examination and understanding of OSCAR biology have
been achieved in DCs and monocytes. Certainly, more experiments could be conducted. It could be
interesting to assess the contribution of collagen signaling through OSCAR in the presence of TLR
ligands or other inflammatory mediators, to evaluate any synergistic effects.
A better understanding of OSCAR-collagen signaling in macrophages could be
achieved. The interplay between ITAM and ITIM receptors would need to be further examined.
This could be accomplished by working at conditions were ITAM signaling is absent, for example
by inhibiting ITIM-signaling or by using specific OSCAR-binding peptides. Additional mechanistic
studies using gelatin as a ligand for OSCAR could be conducted to assess macrophage activation.
If a putative OSCAR co-receptor exists, it could be identified by co-
immunoprecipitation using anti-OSCAR Ab followed by mass-spectrometry.
Several studies have shown high plasticity of myeloid cells. Mo-DCs are seen to be
capable of transdifferentiating into bone-resorbing OCs9. OSCAR might play a role in this
transdifferentiation of iDCs to OCs. Long term culture of DCs on collagen could be conducted and
examined for the presence of multinucleated OCs expressing cathepsin K and TRAP (cytostaining)

96
and if such cells are found in the culture – continue further with functional assays, where the cells
will be examined for the ability to degrade bone slices.
In addition, it would be interesting to assess the ability of the Col-DCs activated T
cells to promote osteoclastogenesis from OCs-precursor cells. This could be tested in an
osteoclastogenesis assay using the T cells in co-culture with CD14+ cells in the presence or absence
of RANKL/M-CSF and/or RA synovial fluid.
6.2 Atherosclerosis
Another relevant indication for the use of anti-OSCAR mAbs could be in the inflammatory disease
atherosclerosis. The atherosclerotic lesions (especially, non-occlusive vulnerable plaques) contain
large numbers of activated immune cells, including monocytes, macrophages and DCs223-225
. The
immune cells are found to be located mainly at the rupture prone areas, increasing the risk of
thrombus formation, heart attacks and strokes226
. High levels of MMPs and oxidants are found
within the lesions causing ECM remodeling and protein modifications, which would make
collagens accessible for interaction with OSCAR expressed on the immune cells. When analyzing
target genes and pathways regulated by OSCAR-collagen signaling in DCs and monocytes, one of
the main regulated pathways was predicted to be “atherosclerosis signaling”. Of interest, one of the
most up-regulated genes in both DCs and monocytes was the LPL gene, coding for Lipoprotein
Lipase. LPL mediates lipoprotein uptake and dysregulation has been linked to disorders of
lipoprotein metabolism. Altogether, suggesting that OSCAR could play a role in the pathogenesis of
atherosclerosis.
6.3 Limitations to the OSCAR project
OSCAR is an immune receptor broadly expressed on myeloid cells in human, while expression is
restricted to OCs in mice and several other animals except higher primates (In-house data from
Novo Nordisk A/S)29;136
. In vitro data indicate that targeting of OSCAR may be efficient for
dampening inflammation and bone erosion. However, the difference in the expression pattern
makes it difficult to conduct in vivo proof-of-principle studies using animal disease models. Novo
Nordisk A/S has addressed the function of OSCAR on bone erosion and in osteoclastogenesis in
mouse models of inflammatory bone destruction (Collagen-Induced Arthritis (CIA) model and
Delayed-Type Hypersensitivity (DTH) model). Unfortunately, blocking OSCAR did not show any

97
significant effect on bone erosion, and OC formation and activity in the animal models examined.
The lack of efficacy may be explained by the fact that OSCAR is not expressed on peripheral
monocytes which seem to serve as precursors of OCs in the CIA and DTH models. The murine
bone marrow derived OCs precursors, even though they express OSCAR, may not contribute
significantly to the inflammatory bone loss. Thus, neither CIA nor DTH murine models are optimal
for testing OSCAR contribution to bone destruction. The OSCAR project group came up with
several proposals for addressing OSCAR function in other in vivo setups. The only species
expressing OSCAR in a similar to human pattern are monkeys. However, for ethical reasons Novo
Nordisk A/S does not conduct intervention studies in monkeys. It has been proposed to generate a
humanized mouse model (knock-in), where OSCAR would have the same expression pattern as in
humans, and/or mechanistically address OSCAR mode of action in a small clinical study.
Nevertheless, the management board decided to discontinue the OSCAR project due to
unpredictability of the outcome and high costs.

98
7. References
Reference List
1. Arron JR, Choi Y. Bone versus immune system. Nature 2000;408:535-536.
2. Rauner M, Sipos W, Pietschmann P. Osteoimmunology. Int.Arch.Allergy Immunol.
2007;143:31-48.
3. Takayanagi H. New developments in osteoimmunology. Nat.Rev.Rheumatol. 2012;8:684-
689.
4. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and
relationship with dendritic cells. Annu.Rev.Immunol. 2009;27:669-692.
5. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat.Rev.Immunol.
2005;5:953-964.
6. Kinne RW, Stuhlmuller B, Burmester GR. Cells of the synovium in rheumatoid arthritis.
Macrophages. Arthritis Res.Ther. 2007;9:224.
7. Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage.
Nat.Immunol. 2012;13:1145-1154.
8. Alnaeeli M, Park J, Mahamed D, Penninger JM, Teng YT. Dendritic cells at the osteo-
immune interface: implications for inflammation-induced bone loss. J.Bone Miner.Res.
2007;22:775-780.
9. Rivollier A, Mazzorana M, Tebib J et al. Immature dendritic cell transdifferentiation into
osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood
2004;104:4029-4037.
10. Brand U, Bellinghausen I, Enk AH et al. Influence of extracellular matrix proteins on the
development of cultured human dendritic cells. Eur.J.Immunol. 1998;28:1673-1680.
11. Merck E, de Saint-Vis B, Scuiller M et al. Fc receptor gamma-chain activation via hOSCAR
induces survival and maturation of dendritic cells and modulates Toll-like receptor
responses. Blood 2005;105:3623-3632.
12. Tenca C, Merlo A, Merck E et al. CD85j (leukocyte Ig-like receptor-1/Ig-like transcript 2)
inhibits human osteoclast-associated receptor-mediated activation of human dendritic cells.
J.Immunol. 2005;174:6757-6763.
13. Sallusto F, Schaerli P, Loetscher P et al. Rapid and coordinated switch in chemokine
receptor expression during dendritic cell maturation. Eur.J.Immunol. 1998;28:2760-2769.
14. Cyster JG. Leukocyte migration: scent of the T zone. Curr.Biol. 2000;10:R30-R33.

99
15. Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway
regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells.
J.Exp.Med. 2001;194:1111-1122.
16. Gallo PM, Gallucci S. The dendritic cell response to classic, emerging, and homeostatic
danger signals. Implications for autoimmunity. Front Immunol. 2013;4:138.
17. Grakoui A, Bromley SK, Sumen C et al. The immunological synapse: a molecular machine
controlling T cell activation. Science 1999;285:221-227.
18. Bromley SK, Burack WR, Johnson KG et al. The immunological synapse.
Annu.Rev.Immunol. 2001;19:375-396.
19. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and
bounded. Nat.Rev.Immunol. 2013;13:257-269.
20. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell
differentiation. Nat.Rev.Immunol. 2012;12:749-761.
21. Noble BS, Stevens H, Loveridge N, Reeve J. Identification of apoptotic changes in
osteocytes in normal and pathological human bone. Bone 1997;20:273-282.
22. Gu G, Mulari M, Peng Z, Hentunen TA, Vaananen HK. Death of osteocytes turns off the
inhibition of osteoclasts and triggers local bone resorption. Biochem.Biophys.Res.Commun.
2005;335:1095-1101.
23. Gori F, Hofbauer LC, Dunstan CR et al. The expression of osteoprotegerin and RANK
ligand and the support of osteoclast formation by stromal-osteoblast lineage cells is
developmentally regulated. Endocrinology 2000;141:4768-4776.
24. Teitelbaum SL. Bone resorption by osteoclasts. Science 2000;289:1504-1508.
25. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis.
Nat.Rev.Immunol. 2007;7:429-442.
26. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature
2003;423:337-342.
27. Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function.
Nat.Rev.Genet. 2003;4:638-649.
28. Lacey DL, Timms E, Tan HL et al. Osteoprotegerin ligand is a cytokine that regulates
osteoclast differentiation and activation. Cell 1998;93:165-176.
29. Kim N, Takami M, Rho J, Josien R, Choi Y. A novel member of the leukocyte receptor
complex regulates osteoclast differentiation. J.Exp.Med. 2002;195:201-209.
30. Kim K, Kim JH, Lee J et al. Nuclear factor of activated T cells c1 induces osteoclast-
associated receptor gene expression during tumor necrosis factor-related activation-induced
cytokine-mediated osteoclastogenesis. J.Biol.Chem. 2005;280:35209-35216.

100
31. Mocsai A, Abram CL, Jakus Z et al. Integrin signaling in neutrophils and macrophages uses
adaptors containing immunoreceptor tyrosine-based activation motifs. Nat.Immunol.
2006;7:1326-1333.
32. Koga T, Inui M, Inoue K et al. Costimulatory signals mediated by the ITAM motif
cooperate with RANKL for bone homeostasis. Nature 2004;428:758-763.
33. Takahashi N, Akatsu T, Udagawa N et al. Osteoblastic cells are involved in osteoclast
formation. Endocrinology 1988;123:2600-2602.
34. Nakashima T, Takayanagi H. The dynamic interplay between osteoclasts and the immune
system. Arch.Biochem.Biophys. 2008;473:166-171.
35. Simonet WS, Lacey DL, Dunstan CR et al. Osteoprotegerin: a novel secreted protein
involved in the regulation of bone density. Cell 1997;89:309-319.
36. Zupan J, Jeras M, Marc J. Osteoimmunology and the influence of pro-inflammatory
cytokines on osteoclasts. Biochem.Med.(Zagreb.) 2013;23:43-63.
37. Cheng J, Liu J, Shi Z et al. Interleukin-4 inhibits RANKL-induced NFATc1 expression via
STAT6: a novel mechanism mediating its blockade of osteoclastogenesis. J.Cell Biochem.
2011;112:3385-3392.
38. Takayanagi H, Ogasawara K, Hida S et al. T-cell-mediated regulation of osteoclastogenesis
by signalling cross-talk between RANKL and IFN-gamma. Nature 2000;408:600-605.
39. Alnaeeli M, Penninger JM, Teng YT. Immune interactions with CD4+ T cells promote the
development of functional osteoclasts from murine CD11c+ dendritic cells. J.Immunol.
2006;177:3314-3326.
40. Lebre MC, Tak PP. Dendritic cell subsets: their roles in rheumatoid arthritis. Acta
Reumatol.Port. 2008;33:35-45.
41. Maitra R, Follenzi A, Yaghoobian A et al. Dendritic cell-mediated in vivo bone resorption.
J.Immunol. 2010;185:1485-1491.
42. Alnaeeli M, Teng YT. Dendritic cells differentiate into osteoclasts in bone marrow
microenvironment in vivo. Blood 2009;113:264-265.
43. Goldring SR. Pathogenesis of bone and cartilage destruction in rheumatoid arthritis.
Rheumatology.(Oxford) 2003;42 Suppl 2:ii11-ii16.
44. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature 2003;423:356-361.
45. Holmdahl R, Malmstrom V, Burkhardt H. Autoimmune priming tissue attack and chronic
inflammation - The three stages of rheumatoid arthritis. Eur.J.Immunol. 2014
46. Eyre S, Bowes J, Diogo D et al. High-density genetic mapping identifies new susceptibility
loci for rheumatoid arthritis. Nat.Genet. 2012;44:1336-1340.

101
47. Stahl EA, Raychaudhuri S, Remmers EF et al. Genome-wide association study meta-
analysis identifies seven new rheumatoid arthritis risk loci. Nat.Genet. 2010;42:508-514.
48. Zhernakova A, Stahl EA, Trynka G et al. Meta-analysis of genome-wide association studies
in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci.
PLoS.Genet. 2011;7:e1002004.
49. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to
understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis
Rheum. 1987;30:1205-1213.
50. Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease.
Nature 2005;435:584-589.
51. Karlson EW, Chang SC, Cui J et al. Gene-environment interaction between HLA-DRB1
shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis.
Ann.Rheum.Dis. 2010;69:54-60.
52. Scally SW, Petersen J, Law SC et al. A molecular basis for the association of the HLA-
DRB1 locus, citrullination, and rheumatoid arthritis. J.Exp.Med. 2013;210:2569-2582.
53. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N.Engl.J.Med.
2011;365:2205-2219.
54. Schett G. Cells of the synovium in rheumatoid arthritis. Osteoclasts. Arthritis Res.Ther.
2007;9:203.
55. Strand V, Kimberly R, Isaacs JD. Biologic therapies in rheumatology: lessons learned,
future directions. Nat.Rev.Drug Discov. 2007;6:75-92.
56. Zhou M, Qin S, Chu Y et al. Immunolocalization of MMP-2 and MMP-9 in human
rheumatoid synovium. Int.J.Clin.Exp.Pathol. 2014;7:3048-3056.
57. Tran CN, Lundy SK, Fox DA. Synovial biology and T cells in rheumatoid arthritis.
Pathophysiology. 2005;12:183-189.
58. Lutzky V, Hannawi S, Thomas R. Cells of the synovium in rheumatoid arthritis. Dendritic
cells. Arthritis Res.Ther. 2007;9:219.
59. McKenna HJ, Stocking KL, Miller RE et al. Mice lacking flt3 ligand have deficient
hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer
cells. Blood 2000;95:3489-3497.
60. Page G, Lebecque S, Miossec P. Anatomic localization of immature and mature dendritic
cells in an ectopic lymphoid organ: correlation with selective chemokine expression in
rheumatoid synovium. J.Immunol. 2002;168:5333-5341.
61. Page G, Miossec P. RANK and RANKL expression as markers of dendritic cell-T cell
interactions in paired samples of rheumatoid synovium and lymph nodes. Arthritis Rheum.
2005;52:2307-2312.

102
62. Jongbloed SL, Lebre MC, Fraser AR et al. Enumeration and phenotypical analysis of
distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthritis
Res.Ther. 2006;8:R15.
63. Radstake TR, van Lieshout AW, van Riel PL, van den Berg WB, Adema GJ. Dendritic cells,
Fc{gamma} receptors, and Toll-like receptors: potential allies in the battle against
rheumatoid arthritis. Ann.Rheum.Dis. 2005;64:1532-1538.
64. Leung BP, Conacher M, Hunter D et al. A novel dendritic cell-induced model of erosive
inflammatory arthritis: distinct roles for dendritic cells in T cell activation and induction of
local inflammation. J.Immunol. 2002;169:7071-7077.
65. Sato K, Suematsu A, Okamoto K et al. Th17 functions as an osteoclastogenic helper T cell
subset that links T cell activation and bone destruction. J.Exp.Med. 2006;203:2673-2682.
66. Kotake S, Udagawa N, Takahashi N et al. IL-17 in synovial fluids from patients with
rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J.Clin.Invest
1999;103:1345-1352.
67. Edwards JC, Szczepanski L, Szechinski J et al. Efficacy of B-cell-targeted therapy with
rituximab in patients with rheumatoid arthritis. N.Engl.J.Med. 2004;350:2572-2581.
68. Dayer JM, Bresnihan B. Targeting interleukin-1 in the treatment of rheumatoid arthritis.
Arthritis Rheum. 2002;46:574-578.
69. van den Berg WB, Joosten LA, Kollias G, van de Loo FA. Role of tumour necrosis factor
alpha in experimental arthritis: separate activity of interleukin 1beta in chronicity and
cartilage destruction. Ann.Rheum.Dis. 1999;58 Suppl 1:I40-I48.
70. Yang G, Im HJ, Wang JH. Repetitive mechanical stretching modulates IL-1beta induced
COX-2, MMP-1 expression, and PGE2 production in human patellar tendon fibroblasts.
Gene 2005;363:166-172.
71. Sack U, Kinne RW, Marx T et al. Interleukin-6 in synovial fluid is closely associated with
chronic synovitis in rheumatoid arthritis. Rheumatol.Int. 1993;13:45-51.
72. Madhok R, Crilly A, Watson J, Capell HA. Serum interleukin 6 levels in rheumatoid
arthritis: correlations with clinical and laboratory indices of disease activity.
Ann.Rheum.Dis. 1993;52:232-234.
73. Houssiau FA, Devogelaer JP, Van DJ, de Deuxchaisnes CN, Van SJ. Interleukin-6 in
synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory
arthritides. Arthritis Rheum. 1988;31:784-788.
74. Nakahara H, Song J, Sugimoto M et al. Anti-interleukin-6 receptor antibody therapy reduces
vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum.
2003;48:1521-1529.
75. Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis.
Annu.Rev.Immunol. 1996;14:397-440.

103
76. Robinson E, Keystone EC, Schall TJ, Gillett N, Fish EN. Chemokine expression in
rheumatoid arthritis (RA): evidence of RANTES and macrophage inflammatory protein
(MIP)-1 beta production by synovial T cells. Clin.Exp.Immunol. 1995;101:398-407.
77. Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Monocyte chemotactic
proteins MCP-1, MCP-2, and MCP-3 are major attractants for human CD4+ and CD8+ T
lymphocytes. FASEB J. 1994;8:1055-1060.
78. Koch AE, Kunkel SL, Harlow LA et al. Enhanced production of monocyte chemoattractant
protein-1 in rheumatoid arthritis. J.Clin.Invest 1992;90:772-779.
79. Rathanaswami P, Hachicha M, Sadick M, Schall TJ, McColl SR. Expression of the cytokine
RANTES in human rheumatoid synovial fibroblasts. Differential regulation of RANTES
and interleukin-8 genes by inflammatory cytokines. J.Biol.Chem. 1993;268:5834-5839.
80. Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid
arthritis: chemokines in the joints of patients. FEBS J. 2008;275:4448-4455.
81. Koch AE, Kunkel SL, Harlow LA et al. Macrophage inflammatory protein-1 alpha. A novel
chemotactic cytokine for macrophages in rheumatoid arthritis. J.Clin.Invest 1994;93:921-
928.
82. Toussirot E, Wendling D. The use of TNF-alpha blocking agents in rheumatoid arthritis: an
update. Expert.Opin.Pharmacother. 2007;8:2089-2107.
83. Choi Y, Arron JR, Townsend MJ. Promising bone-related therapeutic targets for rheumatoid
arthritis. Nat.Rev.Rheumatol. 2009;5:543-548.
84. Nell VP, Machold KP, Eberl G et al. Benefit of very early referral and very early therapy
with disease-modifying anti-rheumatic drugs in patients with early rheumatoid arthritis.
Rheumatology.(Oxford) 2004;43:906-914.
85. Maini RN, Taylor PC. Anti-cytokine therapy for rheumatoid arthritis. Annu.Rev.Med.
2000;51:207-229.
86. Lipsky PE, van der Heijde DM, St Clair EW et al. Infliximab and methotrexate in the
treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis
with Concomitant Therapy Study Group. N.Engl.J.Med. 2000;343:1594-1602.
87. Redlich K, Hayer S, Ricci R et al. Osteoclasts are essential for TNF-alpha-mediated joint
destruction. J.Clin.Invest 2002;110:1419-1427.
88. Ehrenstein MR, Evans JG, Singh A et al. Compromised function of regulatory T cells in
rheumatoid arthritis and reversal by anti-TNFalpha therapy. J.Exp.Med. 2004;200:277-285.
89. Feldmann M, Brennan FM, Foxwell BM et al. Anti-TNF therapy: where have we got to in
2005? J.Autoimmun. 2005;25 Suppl:26-28.
90. Kelly V, Genovese M. Novel small molecule therapeutics in rheumatoid arthritis.
Rheumatology.(Oxford) 2013;52:1155-1162.

104
91. van der Heijde D, Tanaka Y, Fleischmann R et al. Tofacitinib (CP-690,550) in patients with
rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month
phase III randomized radiographic study. Arthritis Rheum. 2013;65:559-570.
92. McClung MR, Lewiecki EM, Cohen SB et al. Denosumab in postmenopausal women with
low bone mineral density. N.Engl.J.Med. 2006;354:821-831.
93. McClung MR. Inhibition of RANKL as a treatment for osteoporosis: preclinical and early
clinical studies. Curr.Osteoporos.Rep. 2006;4:28-33.
94. Cohen SB, Dore RK, Lane NE et al. Denosumab treatment effects on structural damage,
bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month,
multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis
Rheum. 2008;58:1299-1309.
95. Aumailley M, Gayraud B. Structure and biological activity of the extracellular matrix.
J.Mol.Med.(Berl) 1998;76:253-265.
96. Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans,
flies and worms. Trends Genet. 2004;20:33-43.
97. Karsdal MA, Nielsen MJ, Sand JM et al. Extracellular matrix remodeling: the common
denominator in connective tissue diseases. Possibilities for evaluation and current
understanding of the matrix as more than a passive architecture, but a key player in tissue
failure. Assay.Drug Dev.Technol. 2013;11:70-92.
98. Gelse K, Poschl E, Aigner T. Collagens--structure, function, and biosynthesis. Adv.Drug
Deliv.Rev. 2003;55:1531-1546.
99. Leitinger B. Transmembrane collagen receptors. Annu.Rev.Cell Dev.Biol. 2011;27:265-290.
100. Pacifici R, Basilico C, Roman J et al. Collagen-induced release of interleukin 1 from human
blood mononuclear cells. Potentiation by fibronectin binding to the alpha 5 beta 1 integrin.
J.Clin.Invest 1992;89:61-67.
101. Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology.
J.Mol.Med.(Berl) 2005;83:170-179.
102. Yasuda T. Cartilage destruction by matrix degradation products. Mod.Rheumatol.
2006;16:197-205.
103. Sophia Fox AJ, Bedi A, Rodeo SA. The basic science of articular cartilage: structure,
composition, and function. Sports Health 2009;1:461-468.
104. Goldring MB, Marcu KB. Cartilage homeostasis in health and rheumatic diseases. Arthritis
Res.Ther. 2009;11:224.
105. Brodsky B, Persikov AV. Molecular structure of the collagen triple helix. Adv.Protein
Chem. 2005;70:301-339.

105
106. Shoulders MD, Raines RT. Collagen structure and stability. Annu.Rev.Biochem.
2009;78:929-958.
107. Ricard-Blum S. The collagen family. Cold Spring Harb.Perspect.Biol. 2011;3:a004978.
108. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in
development and disease. Cold Spring Harb.Perspect.Biol. 2011;3:
109. Cawston TE, Young DA. Proteinases involved in matrix turnover during cartilage and bone
breakdown. Cell Tissue Res. 2010;339:221-235.
110. Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of
inflammation and innate immunity. Nat.Rev.Immunol. 2004;4:617-629.
111. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and
TIMPs. Cardiovasc.Res. 2006;69:562-573.
112. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front
Biosci. 2006;11:529-543.
113. Arend WP, Dayer JM. Inhibition of the production and effects of interleukin-1 and tumor
necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum. 1995;38:151-160.
114. Yasuda T. Cartilage destruction by matrix degradation products. Mod.Rheumatol.
2006;16:197-205.
115. Davis GE, Pintar Allen KA, Salazar R, Maxwell SA. Matrix metalloproteinase-1 and -9
activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel
contraction and capillary tube regression in three-dimensional collagen matrices. J.Cell Sci.
2001;114:917-930.
116. Ramos-DeSimone N, Hahn-Dantona E, Sipley J et al. Activation of matrix
metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances
tumor cell invasion. J.Biol.Chem. 1999;274:13066-13076.
117. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution,
structure and function. Biochim.Biophys.Acta 2000;1477:267-283.
118. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat.Rev.Mol.Cell Biol.
2010;11:23-36.
119. Green KA, Lund LR. ECM degrading proteases and tissue remodelling in the mammary
gland. Bioessays 2005;27:894-903.
120. Carmeliet P, Moons L, Lijnen R et al. Urokinase-generated plasmin activates matrix
metalloproteinases during aneurysm formation. Nat.Genet. 1997;17:439-444.
121. Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires
MMP-9 activation by plasminogen in mice. J.Clin.Invest 2008;118:3012-3024.

106
122. Madsen DH, Leonard D, Masedunskas A et al. M2-like macrophages are responsible for
collagen degradation through a mannose receptor-mediated pathway. J.Cell Biol.
2013;202:951-966.
123. Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases.
Nat.Rev.Cancer 2003;3:489-501.
124. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002;110:673-687.
125. Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: an update on discoidin
domain receptor function. Cell Signal. 2006;18:1108-1116.
126. Ferri N, Carragher NO, Raines EW. Role of discoidin domain receptors 1 and 2 in human
smooth muscle cell-mediated collagen remodeling: potential implications in atherosclerosis
and lymphangioleiomyomatosis. Am.J.Pathol. 2004;164:1575-1585.
127. Olaso E, Labrador JP, Wang L et al. Discoidin domain receptor 2 regulates fibroblast
proliferation and migration through the extracellular matrix in association with
transcriptional activation of matrix metalloproteinase-2. J.Biol.Chem. 2002;277:3606-3613.
128. Brondijk TH, de RT, Ballering J et al. Crystal structure and collagen-binding site of immune
inhibitory receptor LAIR-1: unexpected implications for collagen binding by platelet
receptor GPVI. Blood 2010;115:1364-1373.
129. Farndale RW, Slatter DA, Siljander PR, Jarvis GE. Platelet receptor recognition and cross-
talk in collagen-induced activation of platelets. J.Thromb.Haemost. 2007;5 Suppl 1:220-
229.
130. Meyaard L. LAIR and collagens in immune regulation. Immunol.Lett. 2010;128:26-28.
131. Xu H, Bihan D, Chang F et al. Discoidin domain receptors promote alpha1beta1- and
alpha2beta1-integrin mediated cell adhesion to collagen by enhancing integrin activation.
PLoS.One. 2012;7:e52209.
132. Jeng KC, Liu MT, Lan JL et al. Collagen induces cytokine production by synovial fluid
mononuclear cells in rheumatoid arthritis. Immunol.Lett. 1995;45:13-17.
133. Dietrich J, Nakajima H, Colonna M. Human inhibitory and activating Ig-like receptors
which modulate the function of myeloid cells. Microbes.Infect. 2000;2:323-329.
134. Merck E, Gaillard C, Scuiller M et al. Ligation of the FcR gamma chain-associated human
osteoclast-associated receptor enhances the proinflammatory responses of human monocytes
and neutrophils. J.Immunol. 2006;176:3149-3156.
135. Ishikawa S, Arase N, Suenaga T et al. Involvement of FcRgamma in signal transduction of
osteoclast-associated receptor (OSCAR). Int.Immunol. 2004;16:1019-1025.
136. Merck E, Gaillard C, Gorman DM et al. OSCAR is an FcRgamma-associated receptor that
is expressed by myeloid cells and is involved in antigen presentation and activation of
human dendritic cells. Blood 2004;104:1386-1395.

107
137. Barrow AD, Raynal N, Andersen TL et al. OSCAR is a collagen receptor that costimulates
osteoclastogenesis in DAP12-deficient humans and mice. J.Clin.Invest 2011;121:3505-
3516.
138. Zhao S, Guo YY, Ding N, Yang LL, Zhang N. Changes in serum levels of soluble
osteoclast-associated receptor in human rheumatoid arthritis. Chin Med.J.(Engl.)
2011;124:3058-3060.
139. Crotti TN, Dharmapatni AA, Alias E et al. The immunoreceptor tyrosine-based activation
motif (ITAM) -related factors are increased in synovial tissue and vasculature of rheumatoid
arthritic joints. Arthritis Res.Ther. 2012;14:R245.
140. Ndongo-Thiam N, de SG, Kastrup J, Miossec P. Levels of soluble osteoclast-associated
receptor (sOSCAR) in rheumatoid arthritis: link to disease severity and cardiovascular risk.
Ann.Rheum.Dis. 2014;73:1276-1277.
141. Herman S, Muller RB, Kronke G et al. Induction of osteoclast-associated receptor, a key
osteoclast costimulation molecule, in rheumatoid arthritis. Arthritis Rheum. 2008;58:3041-
3050.
142. Schultz HS, Nitze LM, Zeuthen LH et al. Collagen Induces Maturation of Human
Monocyte-Derived Dendritic Cells by Signaling through Osteoclast-Associated Receptor.
J.Immunol. 2015
143. Bonecchi R, Bianchi G, Bordignon PP et al. Differential expression of chemokine receptors
and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J.Exp.Med.
1998;187:129-134.
144. Iellem A, Mariani M, Lang R et al. Unique chemotactic response profile and specific
expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells.
J.Exp.Med. 2001;194:847-853.
145. So H, Rho J, Jeong D et al. Microphthalmia transcription factor and PU.1 synergistically
induce the leukocyte receptor osteoclast-associated receptor gene expression. J.Biol.Chem.
2003;278:24209-24216.
146. Kim GS, Koh JM, Chang JS et al. Association of the OSCAR promoter polymorphism with
BMD in postmenopausal women. J.Bone Miner.Res. 2005;20:1342-1348.
147. Goettsch C, Rauner M, Sinningen K et al. The osteoclast-associated receptor (OSCAR) is a
novel receptor regulated by oxidized low-density lipoprotein in human endothelial cells.
Endocrinology 2011;152:4915-4926.
148. Goettsch C, Kliemt S, Sinningen K et al. Quantitative proteomics reveals novel functions of
osteoclast-associated receptor in STAT signaling and cell adhesion in human endothelial
cells. J.Mol.Cell Cardiol. 2012;53:829-837.
149. Sinningen K, Rauner M, Goettsch C et al. Monocytic expression of osteoclast-associated
receptor (OSCAR) is induced in atherosclerotic mice and regulated by oxidized low-density
lipoprotein in vitro. Biochem.Biophys.Res.Commun. 2013;437:314-318.

108
150. van den Berg WB. Animal models of arthritis. What have we learned? J.Rheumatol.Suppl
2005;72:7-9.
151. Liote F, Boval-Boizard B, Weill D, Kuntz D, Wautier JL. Blood monocyte activation in
rheumatoid arthritis: increased monocyte adhesiveness, integrin expression, and cytokine
release. Clin.Exp.Immunol. 1996;106:13-19.
152. Torsteinsdottir I, Arvidson NG, Hallgren R, Hakansson L. Monocyte activation in
rheumatoid arthritis (RA): increased integrin, Fc gamma and complement receptor
expression and the effect of glucocorticoids. Clin.Exp.Immunol. 1999;115:554-560.
153. Vieira-Sousa E, Gerlag DM, Tak PP. Synovial tissue response to treatment in rheumatoid
arthritis. Open.Rheumatol.J. 2011;5:115-122.
154. Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and
articular damage in rheumatoid arthritis. Arthritis Rheum. 1996;39:115-124.
155. Haringman JJ, Gerlag DM, Zwinderman AH et al. Synovial tissue macrophages: a sensitive
biomarker for response to treatment in patients with rheumatoid arthritis. Ann.Rheum.Dis.
2005;64:834-838.
156. Davignon JL, Hayder M, Baron M et al. Targeting monocytes/macrophages in the treatment
of rheumatoid arthritis. Rheumatology.(Oxford) 2013;52:590-598.
157. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation.
Nat.Rev.Immunol. 2008;8:958-969.
158. Cheung HS, Ryan LM, Kozin F, McCarty DJ. Identification of collagen subtypes in
synovial fluid sediments from arthritic patients. Am.J.Med. 1980;68:73-79.
159. Wotton SF, Dieppe PA, Duance VC. Type IX collagen immunoreactive peptides in synovial
fluids from arthritis patients. Rheumatology.(Oxford) 1999;38:338-345.
160. Lohmander LS, Atley LM, Pietka TA, Eyre DR. The release of crosslinked peptides from
type II collagen into human synovial fluid is increased soon after joint injury and in
osteoarthritis. Arthritis Rheum. 2003;48:3130-3139.
161. Canfield SM, Morrison SL. The binding affinity of human IgG for its high affinity Fc
receptor is determined by multiple amino acids in the CH2 domain and is modulated by the
hinge region. J.Exp.Med. 1991;173:1483-1491.
162. Bleharski JR, Kiessler V, Buonsanti C et al. A role for triggering receptor expressed on
myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune
response. J.Immunol. 2003;170:3812-3818.
163. Menzies-Gow A, Ying S, Sabroe I et al. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce
recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of
early- and late-phase allergic reactions following cutaneous injection in human atopic and
nonatopic volunteers. J.Immunol. 2002;169:2712-2718.

109
164. Pacifici R, Carano A, Santoro SA et al. Bone matrix constituents stimulate interleukin-1
release from human blood mononuclear cells. J.Clin.Invest 1991;87:221-228.
165. Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326:1216-1219.
166. Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and
regenerative medicine. J.Cell Sci. 2008;121:255-264.
167. Karsdal MA, Woodworth T, Henriksen K et al. Biochemical markers of ongoing joint
damage in rheumatoid arthritis--current and future applications, limitations and
opportunities. Arthritis Res.Ther. 2011;13:215.
168. Neve A, Corrado A, Cantatore FP. TNF-related apoptosis-inducing ligand (TRAIL) in
rheumatoid arthritis: what's new? Clin.Exp.Med. 2014;14:115-120.
169. Baldwin HM, Ito-Ihara T, Isaacs JD, Hilkens CM. Tumour necrosis factor alpha blockade
impairs dendritic cell survival and function in rheumatoid arthritis. Ann.Rheum.Dis.
2010;69:1200-1207.
170. Lo SZ, Steer JH, Joyce DA. Tumor necrosis factor-alpha promotes survival in methotrexate-
exposed macrophages by an NF-kappaB-dependent pathway. Arthritis Res.Ther.
2011;13:R24.
171. Flad HD, Grage-Griebenow E, Petersen F et al. The role of cytokines in monocyte
apoptosis. Pathobiology 1999;67:291-293.
172. Hounoki H, Sugiyama E, Mohamed SG et al. Activation of peroxisome proliferator-
activated receptor gamma inhibits TNF-alpha-mediated osteoclast differentiation in human
peripheral monocytes in part via suppression of monocyte chemoattractant protein-1
expression. Bone 2008;42:765-774.
173. Szekanecz Z, Koch AE. Macrophages and their products in rheumatoid arthritis.
Curr.Opin.Rheumatol. 2007;19:289-295.
174. Fahey TJ, III, Tracey KJ, Tekamp-Olson P et al. Macrophage inflammatory protein 1
modulates macrophage function. J.Immunol. 1992;148:2764-2769.
175. Narni-Mancinelli E, Soudja SM, Crozat K et al. Inflammatory monocytes and neutrophils
are licensed to kill during memory responses in vivo. PLoS.Pathog. 2011;7:e1002457.
176. Fantuzzi L, Borghi P, Ciolli V et al. Loss of CCR2 expression and functional response to
monocyte chemotactic protein (MCP-1) during the differentiation of human monocytes: role
of secreted MCP-1 in the regulation of the chemotactic response. Blood 1999;94:875-883.
177. Katschke KJ, Jr., Rottman JB, Ruth JH et al. Differential expression of chemokine receptors
on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in
rheumatoid arthritis. Arthritis Rheum. 2001;44:1022-1032.
178. Phillips RJ, Lutz M, Premack B. Differential signaling mechanisms regulate expression of
CC chemokine receptor-2 during monocyte maturation. J.Inflamm.(Lond) 2005;2:14.

110
179. Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered
by TREM-1, a novel receptor expressed on neutrophils and monocytes. J.Immunol.
2000;164:4991-4995.
180. Knapp S, Gibot S, de VA et al. Cutting edge: expression patterns of surface and soluble
triggering receptor expressed on myeloid cells-1 in human endotoxemia. J.Immunol.
2004;173:7131-7134.
181. Sharif O, Bolshakov VN, Raines S, Newham P, Perkins ND. Transcriptional profiling of the
LPS induced NF-kappaB response in macrophages. BMC.Immunol. 2007;8:1.
182. Netea MG, Azam T, Ferwerda G et al. Triggering receptor expressed on myeloid cells-1
(TREM-1) amplifies the signals induced by the NACHT-LRR (NLR) pattern recognition
receptors. J.Leukoc.Biol. 2006;80:1454-1461.
183. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function.
Nat.Rev.Mol.Cell Biol. 2007;8:49-62.
184. Kuai J, Gregory B, Hill A et al. TREM-1 expression is increased in the synovium of
rheumatoid arthritis patients and induces the expression of pro-inflammatory cytokines.
Rheumatology.(Oxford) 2009;48:1352-1358.
185. Murakami Y, Akahoshi T, Aoki N et al. Intervention of an inflammation amplifier,
triggering receptor expressed on myeloid cells 1, for treatment of autoimmune arthritis.
Arthritis Rheum. 2009;60:1615-1623.
186. Sedlacek R, Mauch S, Kolb B et al. Matrix metalloproteinase MMP-19 (RASI-1) is
expressed on the surface of activated peripheral blood mononuclear cells and is detected as
an autoantigen in rheumatoid arthritis. Immunobiology 1998;198:408-423.
187. Kolb C, Mauch S, Peter HH, Krawinkel U, Sedlacek R. The matrix metalloproteinase RASI-
1 is expressed in synovial blood vessels of a rheumatoid arthritis patient. Immunol.Lett.
1997;57:83-88.
188. Stracke JO, Hutton M, Stewart M et al. Biochemical characterization of the catalytic domain
of human matrix metalloproteinase 19. Evidence for a role as a potent basement membrane
degrading enzyme. J.Biol.Chem. 2000;275:14809-14816.
189. Stracke JO, Fosang AJ, Last K et al. Matrix metalloproteinases 19 and 20 cleave aggrecan
and cartilage oligomeric matrix protein (COMP). FEBS Lett. 2000;478:52-56.
190. Hakala M, Risteli L, Manelius J, Nieminen P, Risteli J. Increased type I collagen
degradation correlates with disease severity in rheumatoid arthritis. Ann.Rheum.Dis.
1993;52:866-869.
191. Hakala M, Aman S, Luukkainen R et al. Application of markers of collagen metabolism in
serum and synovial fluid for assessment of disease process in patients with rheumatoid
arthritis. Ann.Rheum.Dis. 1995;54:886-890.

111
192. Tak PP, Breedveld FC. Analysis of serial synovial biopsies as a screening method for
predicting the effects of therapeutic interventions. J.Clin.Rheumatol. 1997;3:186.
193. Tak PP, Smeets TJ, Daha MR et al. Analysis of the synovial cell infiltrate in early
rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum.
1997;40:217-225.
194. Hamilton JA, Tak PP. The dynamics of macrophage lineage populations in inflammatory
and autoimmune diseases. Arthritis Rheum. 2009;60:1210-1221.
195. Schneider C, Newman RA, Sutherland DR, Asser U, Greaves MF. A one-step purification
of membrane proteins using a high efficiency immunomatrix. J.Biol.Chem.
1982;257:10766-10769.
196. Herenius MM, Thurlings RM, Wijbrandts CA et al. Monocyte migration to the synovium in
rheumatoid arthritis patients treated with adalimumab. Ann.Rheum.Dis. 2011;70:1160-1162.
197. Wijbrandts CA, Remans PH, Klarenbeek PL et al. Analysis of apoptosis in peripheral blood
and synovial tissue very early after initiation of infliximab treatment in rheumatoid arthritis
patients. Arthritis Rheum. 2008;58:3330-3339.
198. Verbrugge A, Meyaard L. Signaling by ITIM-Bearing Receptors. Current Immunology
Reviews 2015;1:201-212.
199. Lebbink RJ, de RT, Adelmeijer J et al. Collagens are functional, high affinity ligands for the
inhibitory immune receptor LAIR-1. J.Exp.Med. 2006;203:1419-1425.
200. Lebbink RJ, Raynal N, de RT et al. Identification of multiple potent binding sites for human
leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol.
2009;28:202-210.
201. Fleetwood AJ, Achuthan A, Schultz H et al. Urokinase plasminogen activator is a central
regulator of macrophage three-dimensional invasion, matrix degradation, and adhesion.
J.Immunol. 2014;192:3540-3547.
202. Shapiro SD, Kobayashi DK, Pentland AP, Welgus HG. Induction of macrophage
metalloproteinases by extracellular matrix. Evidence for enzyme- and substrate-specific
responses involving prostaglandin-dependent mechanisms. J.Biol.Chem. 1993;268:8170-
8175.
203. Lepidi S, Kenagy RD, Raines EW et al. MMP9 production by human monocyte-derived
macrophages is decreased on polymerized type I collagen. J.Vasc.Surg. 2001;34:1111-1118.
204. Franz S, Allenstein F, Kajahn J et al. Artificial extracellular matrices composed of collagen I
and high-sulfated hyaluronan promote phenotypic and functional modulation of human pro-
inflammatory M1 macrophages. Acta Biomater. 2013;9:5621-5629.
205. Nemeth K, Schoppet M, Al-Fakhri N et al. The role of osteoclast-associated receptor in
osteoimmunology. J.Immunol. 2011;186:13-18.

112
206. Trentham DE, Kammer GM, McCune WJ, David JR. Autoimmunity to collagen: a shared
feature of psoriatic and rheumatoid arthritis. Arthritis Rheum. 1981;24:1363-1369.
207. Stuart JM, Huffstutter EH, Townes AS, Kang AH. Incidence and specificity of antibodies to
types I, II, III, IV, and V collagen in rheumatoid arthritis and other rheumatic diseases as
measured by 125I-radioimmunoassay. Arthritis Rheum. 1983;26:832-840.
208. Dalod M, Chelbi R, Malissen B, Lawrence T. Dendritic cell maturation: functional
specialization through signaling specificity and transcriptional programming. EMBO J.
2014;33:1104-1116.
209. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development.
Nat.Rev.Immunol. 2007;7:19-30.
210. Swirski FK, Nahrendorf M, Etzrodt M et al. Identification of splenic reservoir monocytes
and their deployment to inflammatory sites. Science 2009;325:612-616.
211. Sengupta TK, Chen A, Zhong Z, Darnell JE, Jr., Ivashkiv LB. Activation of monocyte
effector genes and STAT family transcription factors by inflammatory synovial fluid is
independent of interferon gamma. J.Exp.Med. 1995;181:1015-1025.
212. Sallusto F, Lanzavecchia A, Mackay CR. Chemokines and chemokine receptors in T-cell
priming and Th1/Th2-mediated responses. Immunol.Today 1998;19:568-574.
213. Baggiolini M. Chemotactic and inflammatory cytokines--CXC and CC proteins.
Adv.Exp.Med.Biol. 1993;351:1-11.
214. Mukaida N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases.
Am.J.Physiol Lung Cell Mol.Physiol 2003;284:L566-L577.
215. Ludewig B, Graf D, Gelderblom HR et al. Spontaneous apoptosis of dendritic cells is
efficiently inhibited by TRAP (CD40-ligand) and TNF-alpha, but strongly enhanced by
interleukin-10. Eur.J.Immunol. 1995;25:1943-1950.
216. Slobodin G, Kessel A, Peri R et al. Etanercept impairs maturation of human monocyte-
derived dendritic cells by inhibiting the autocrine TNFalpha-mediated signaling.
Inflammation 2009;32:146-150.
217. Sarkar S, Fox DA. Dendritic cells in rheumatoid arthritis. Front Biosci. 2005;10:656-665.
218. Turner NJ, Pezzone D, Badylak SF. Regional Variations in the Histology of Porcine Skin.
Tissue Eng Part C.Methods 2014
219. Watson RE, Gibbs NK, Griffiths CE, Sherratt MJ. Damage to skin extracellular matrix
induced by UV exposure. Antioxid.Redox.Signal. 2014;21:1063-1077.
220. Agarwal P, Zwolanek D, Keene DR et al. Collagen XII and XIV, new partners of cartilage
oligomeric matrix protein in the skin extracellular matrix suprastructure. J.Biol.Chem.
2012;287:22549-22559.

113
221. Seymour HE, Worsley A, Smith JM, Thomas SH. Anti-TNF agents for rheumatoid arthritis.
Br.J.Clin.Pharmacol. 2001;51:201-208.
222. Curtis JR, Singh JA. Use of biologics in rheumatoid arthritis: current and emerging
paradigms of care. Clin.Ther. 2011;33:679-707.
223. Lusis AJ. Atherosclerosis. Nature 2000;407:233-241.
224. Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin.Immunol.
2010;134:25-32.
225. Bobryshev YV, Lord RS. S-100 positive cells in human arterial intima and in atherosclerotic
lesions. Cardiovasc.Res. 1995;29:689-696.
226. Yilmaz A, Lochno M, Traeg F et al. Emergence of dendritic cells in rupture-prone regions
of vulnerable carotid plaques. Atherosclerosis 2004;176:101-110.
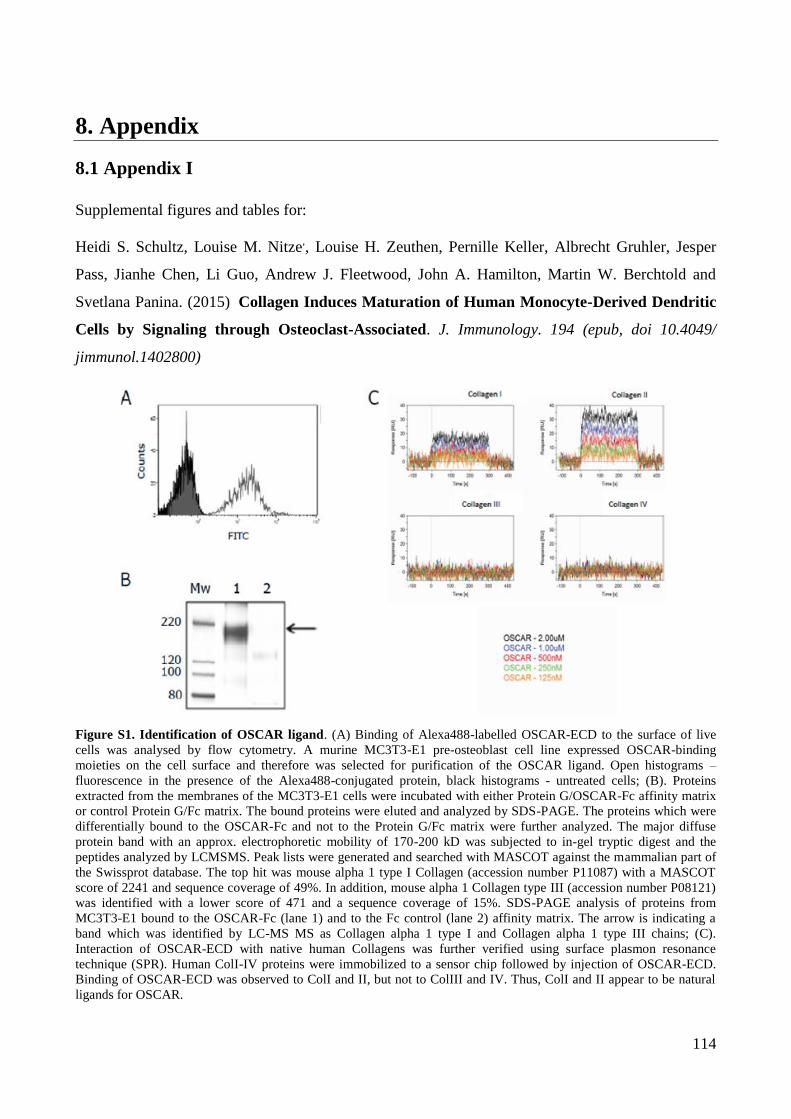
114
8. Appendix
8.1 Appendix I
Supplemental figures and tables for:
Heidi S. Schultz, Louise M. Nitze,, Louise H. Zeuthen, Pernille Keller, Albrecht Gruhler, Jesper
Pass, Jianhe Chen, Li Guo, Andrew J. Fleetwood, John A. Hamilton, Martin W. Berchtold and
Svetlana Panina. (2015) Collagen Induces Maturation of Human Monocyte-Derived Dendritic
Cells by Signaling through Osteoclast-Associated. J. Immunology. 194 (epub, doi 10.4049/
jimmunol.1402800)
Figure S1. Identification of OSCAR ligand. (A) Binding of Alexa488-labelled OSCAR-ECD to the surface of live
cells was analysed by flow cytometry. A murine MC3T3-E1 pre-osteoblast cell line expressed OSCAR-binding
moieties on the cell surface and therefore was selected for purification of the OSCAR ligand. Open histograms –
fluorescence in the presence of the Alexa488-conjugated protein, black histograms - untreated cells; (B). Proteins
extracted from the membranes of the MC3T3-E1 cells were incubated with either Protein G/OSCAR-Fc affinity matrix
or control Protein G/Fc matrix. The bound proteins were eluted and analyzed by SDS-PAGE. The proteins which were
differentially bound to the OSCAR-Fc and not to the Protein G/Fc matrix were further analyzed. The major diffuse
protein band with an approx. electrophoretic mobility of 170-200 kD was subjected to in-gel tryptic digest and the
peptides analyzed by LCMSMS. Peak lists were generated and searched with MASCOT against the mammalian part of
the Swissprot database. The top hit was mouse alpha 1 type I Collagen (accession number P11087) with a MASCOT
score of 2241 and sequence coverage of 49%. In addition, mouse alpha 1 Collagen type III (accession number P08121)
was identified with a lower score of 471 and a sequence coverage of 15%. SDS-PAGE analysis of proteins from
MC3T3-E1 bound to the OSCAR-Fc (lane 1) and to the Fc control (lane 2) affinity matrix. The arrow is indicating a
band which was identified by LC-MS MS as Collagen alpha 1 type I and Collagen alpha 1 type III chains; (C).
Interaction of OSCAR-ECD with native human Collagens was further verified using surface plasmon resonance
technique (SPR). Human ColI-IV proteins were immobilized to a sensor chip followed by injection of OSCAR-ECD.
Binding of OSCAR-ECD was observed to ColI and II, but not to ColIII and IV. Thus, ColI and II appear to be natural
ligands for OSCAR.
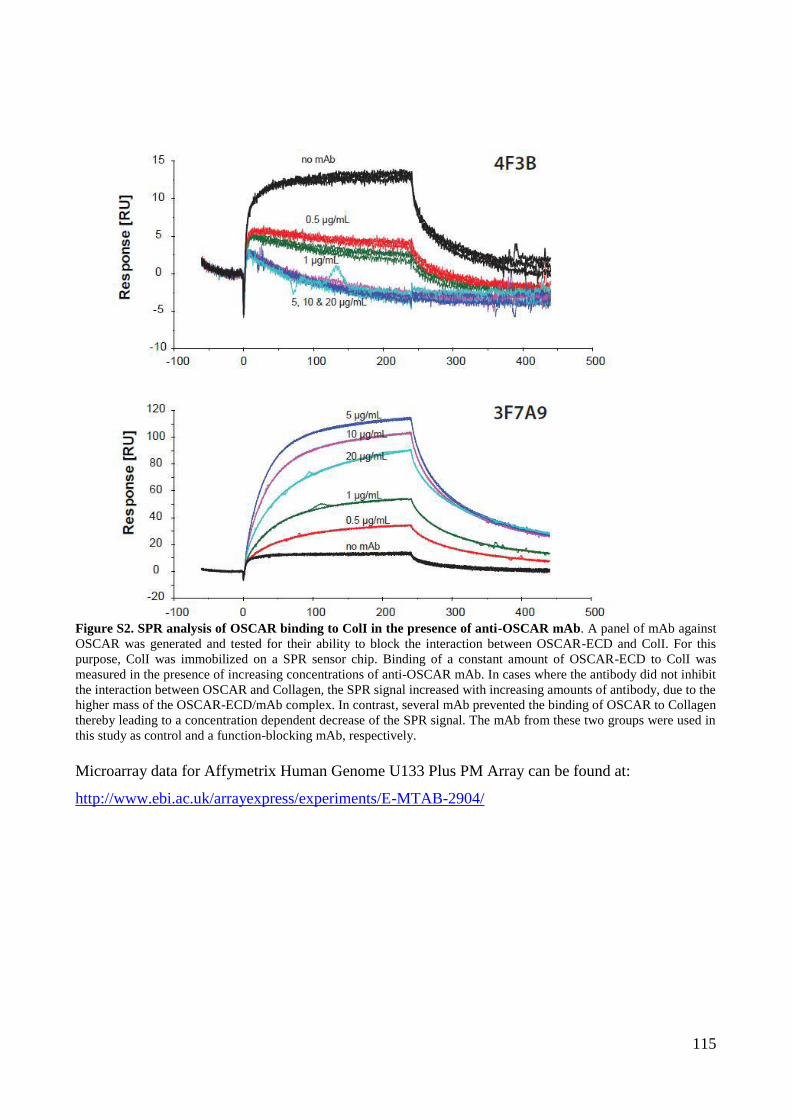
115
Figure S2. SPR analysis of OSCAR binding to ColI in the presence of anti-OSCAR mAb. A panel of mAb against
OSCAR was generated and tested for their ability to block the interaction between OSCAR-ECD and ColI. For this
purpose, ColI was immobilized on a SPR sensor chip. Binding of a constant amount of OSCAR-ECD to ColI was
measured in the presence of increasing concentrations of anti-OSCAR mAb. In cases where the antibody did not inhibit
the interaction between OSCAR and Collagen, the SPR signal increased with increasing amounts of antibody, due to the
higher mass of the OSCAR-ECD/mAb complex. In contrast, several mAb prevented the binding of OSCAR to Collagen
thereby leading to a concentration dependent decrease of the SPR signal. The mAb from these two groups were used in
this study as control and a function-blocking mAb, respectively.
Microarray data for Affymetrix Human Genome U133 Plus PM Array can be found at:
http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2904/

116
8.2 Appendix II
Supplemental figures and tables for:
Heidi S. Schultz, Li Guo, Pernille Keller, Andrew J. Fleetwood, Wei Guo, John A. Hamilton, Olle
Björkdahl, Martin W. Berchtold and Svetlana Panina. OSCAR-collagen signaling mediates
activation of monocytes. To be submitted to J. Immunology March, 2015.
Figure S1. TREM-1 is the top biological upstream regulator of the genes regulated by OSCAR-ColII signaling.
Bright orange fillings indicate predicted activation and pale orange indicate less predicted activation. Pale blue fillings
indicate less predicted inhibition. The lines indicate predicted relationships: orange, leads to activation; blue, leads to
inhibition; Grey, effect not predicted and yellow, findings inconsistent with state of downstream molecule.
Microarray data for Affymetrix Human Genome U133 Plus PM Array can be found at:
http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-3281/

117
8.3 Co-author contributions



120
D E P A R T M E N T O F C E L L A N D D E V E L O P M E N T A L B I O L O G Y
F A C U L T Y O F S C I E N C E
U N I V E R S I T Y O F C O P E N H A G E N
P H D T H E S I S 2 0 1 5
H E I D I S C H I Ø L E R S C H U L T Z