feasibility of metatranscriptome analysis from infant gut...
TRANSCRIPT

Research ArticleFeasibility of Metatranscriptome Analysis fromInfant Gut Microbiota Adaptation to Solid Foods Results inIncreased Activity of Firmicutes at Six Months
Floor Hugenholtz1 Jarmo Ritari2 Lotta Nylund3 Mark Davids4
Reetta Satokari235 andWillemM de Vos125
1Laboratory of Microbiology Wageningen University Dreijenplein 10 6703 HBWageningen Netherlands2Department of Basic Veterinary Medicine University of Helsinki Helsinki Finland3Functional Foods Forum University of Turku Turku Finland4Laboratory of Systems and Synthetic Biology Wageningen University Dreijenplein 10 6703 HBWageningen Netherlands5RPU Immunobiology Department of Bacteriology and Immunology University of Helsinki Helsinki Finland
Correspondence should be addressed to Willem M de Vos willemdevoswurnl
Received 12 April 2017 Revised 17 June 2017 Accepted 4 July 2017 Published 24 August 2017
Academic Editor Barbara H Iglewski
Copyright copy 2017 Floor Hugenholtz et al This is an open access article distributed under the Creative Commons AttributionLicense which permits unrestricted use distribution and reproduction in any medium provided the original work is properlycited
Newborns are rapidly colonized by microbes and their intestinal tracts contain highly dynamic and rapidly developing microbialcommunities in the first months of life In this study we describe the feasibility of isolating mRNA from rapidly processed faecalsamples and applying deep RNA-Seq analysis to provide insight into the active contributors of the microbial community in earlylife Specific attention is given to the impact of removing rRNA from the mRNA on the phylogenetic and transcriptional profilingand its analysis depth A breastfed baby was followed in the first six months of life during adaptation to solid food dairy productsand formula It was found that in the weaning period the total transcriptional activity of Actinobacteria mainly represented byBifidobacterium decreased while that of Firmicutes increased over time Moreover Firmicutes and Actinobacteria including thecanonical Bifidobacteria as well as Collinsella were found to be important contributors to carbohydrate fermentation and vitaminbiosynthesis in the infant intestine Finally the expression of Lactobacillus rhamnosus-like genes was detected likely followingtransfer from the mother who consumed L rhamnosus GG The study indicates that metatranscriptome analysis of the infant gutmicrobiota is feasible on infant stool samples and can be used to provide insight into the core activities of the developing community
1 Background
After birth newborns are rapidly colonized by intestinalmicrobiota originating from the surrounding environmentincluding maternal faecal microbes and vaginal or skin spe-cies depending on the mode of delivery [1 2] It is generallyassumed that the first colonizers of the infant intestine arefacultative anaerobes such as Streptococcus Enterococcus andLactobacillus spp and Escherichia coli [3 4] followed by an-aerobic bacteria such as Bifidobacterium Clostridium andRuminococcus spp [5 6] Defining a normal intestinal micro-biota at early age is challenging since the composition andtemporal patterns of the microbial communities vary widelyamong infants During maturation the variation among
infants becomes smaller when obtaining a more adult-likemicrobiota composition but this has recently been found totake over 5 years of time [7ndash9] Bifidobacterium and to a lesserextent Bacteroides are considered the predominant bacterialgenera colonizing the early infant gut when the infant isbreastfed as these bacteria have the rather unique capacityto degrade human milk oligosaccharides [10] Introductionof solid foods significantly alters the gut microbiota switch-ing the microbial composition to be more dominated byBacteroides and Clostridium spp [7 9 11] The increase inabundance of the phyla Firmicutes and Bacteroidetes afterweaning is an indication of the adaptation to a more complexdiet [6 7]
HindawiInternational Journal of MicrobiologyVolume 2017 Article ID 9547063 9 pageshttpsdoiorg10115520179547063
2 International Journal of Microbiology
Table 1 Overview of the infant diet
Sample code T1 T2 T3Age (days) 131 165 171Breast milk Exclusively Yes YesFormula No No YesPotato amp roots First baby food Yes YesFruits amp berries No Yes YesVegetables No Yes YesGrain products No Yes YesMeatchickenfisheggs No Yes YesMilk amp dairy products No No Yes
Various culturing approaches have shown that there isvertical transmission of specific bacteria frommother to babyThis was shown elegantly for endogenous Bifidobacteria [12]as well as probiotic bacteria consumed by the mother thatin the case of Lactobacillus rhamnosus GG were found inthe baby [13 14] Culture-independent evidence for maternaltransmission of early life microbiota is limited notably as thebaby microbiota differs so much from that of the motherIn some cases signatures of parental microbes have beendescribed to be present in the infant microbiome [15] andrecently it was reported that the microbiota in children at3 years of age shows signatures present similar to their ownmother and were not seen in between children and unrelatedmothers [16]
While considerable attention has been given to the com-positional development during early life only recently highthroughput omics-based approaches have been applied Arecent study analysed the faecal metagenome during the firstyear of life and observed striking differences between vaginaland C-section delivered infants [7] Moreover some earlystudies addressed the transcriptome of Bifidobacteria in theintestinal tract of babies that were breastfed or on a formula-diet and showed differential expression of genes involvedin sugar catabolism exopolysaccharide production or folatebiosynthesis [17] Moreover metatranscriptome analysis ininfants and their mothers showed differences in expressionof higher capacity of mucin utilization higher capacity folatebiosynthesis and decreased starch degradation in infants[18] Globalmetatranscriptome analysis in theGI tractmicro-biota could enable the elucidation of the specific functionalroles microbes have in this complex community Initial meta-transcriptome studies in the human large intestine revealedthat different functions are expressed between individualswhile core functions of the microbiota appeared to beconsistently expressed among individuals [19ndash21]These find-ings imply that metatranscriptomics could provide insightinto the differential activity profiles in the gut microbiotaenabling the reconstruction of the metabolic activity profileof microbial communities
In this study we describe the feasibility of using deepRNA-Seq analysis to get further insight in the active con-tributors of the microbial community in early life Specificattention is given to the rapid sampling and impact ofremoving rRNA from the mRNA on the phylogenetic and
transcriptional profiling and its analysis depth A breastfedbaby was followed in the first six months of life duringadaptation to solid food dairy products and formula Theresults indicate that Bifidobacterium is an active memberof the community and over time various members of theFirmicutes become more active that are involved in vitaminproduction and sugar metabolism at 6 months
2 Methods
21 Subject Dietary Information and Sampling One vagi-nally born breastfed Finnish baby girl was followed duringthe introduction of first solid foods into the diet Faecalsamples were collected at three time points specifically atthe ages of 131 165 and 171 days These samples were givenwith consent of the mother and ethics are within nationaland international regulation The time points and numberof samples were taken for practical reasons as the faecalsamples were taken at home and immediately processed inRNAlater as to preserve the mRNA as good as possible Theinfant did not receive any probiotic supplementation but hermother consumed dairy products containing LactobacillusrhamnosusGG Both infant and her mother were healthy anddid not receive any antibiotics during the study period At thefirst time point (131 days) the infant consumed exclusivelybreast-milk and some mashed potato and roots as the firstbaby food which was extended in the second time point(165 days) to fruits vegetables and grain and meat productsBefore the last time point the infant was also introduced todairy products and formula milk (Table 1)
22 RNA Extraction rRNA Removal cDNA Synthesis LibraryPreparation and Illumina MiSeq 2500 Sequencing Freshfaecal samples were collected and immediately processed asdescribed previously [22] For this purpose one ml RNAlaterwas added to each gram of sample and stored at minus70∘Cuntil later processing The amount of faecal material usedfor the RNA extraction was 681 g for T1 412 g for T2and 442 g for T3 respectively Total RNA was extractedas described before using the Macaloid procedure [23] andfinal RNA concentrations weremeasured with theNanoDrop1000 Here we obtained 3509 3804 and 3182 120583g of totalRNA for T1 T2 and T3 respectively From sample T25 120583g of RNA was also used for rRNA removal by using the
International Journal of Microbiology 3
Ribo-Zero kit (Epicentre) which is a magnetic beads-basedrRNA hybridization technique to remove the 23S 16S and5S rRNA from the sample One sample was taken onlyto see the effect of the rRNA removal in the sequencingresults This portion was further named T2 M while thesample containing total RNA was labelled T2 T 500 ng oftotal RNA from the samples T1 T2 T and T3 and dueto the rRNA removal 244 ng of mRNA of T2 M was usedfor cDNA synthesis and library construction (TrueSeq RNASample Preparation Kit Illumina San Diego CA) Due tothe quality differences slightly different lengths were takenfor the sequencing T1 300ndash700 bp T2 T 300ndash570 bp T2 M300ndash500 bp and T3 300ndash500 bp Sequencing was performedusing the Illumina MiSeq instrument on 2 times 150 bp pairedend mode Between 3 and 4 million reads were obtained persample
23 RNA-Seq Data Processing The raw reads data are avail-able in the MG-RAST server under the following accessioncodes 46217943 46217953 46217963 46217973 4621798346217993 46218003 and 46218013 After fastq quality filter-ing reads were taxonomically assigned by matching againsta human intestinal 16S rRNA database (httpsgithubcommicrobiomeHITdb Ritari et al unpublished) at 97 iden-tity threshold For the mRNA analysis rRNA reads wererapidly filtered from the samples using SortmeRNA (ver-sion 12) after which the remaining reads were assembledusing idba ud using the pipeline described previously [24]Essential single copy genes (ESCG) identified using HMMsearch (httphmmerorg) were taxonomically classifiedusing MEGAN after being aligned against full NR databaseThe remainder of the proteins was taxonomically classified byaligning them against all proteins in the NR database belong-ing tomembers of nine identified bacterial orders Functionalannotation was performed to all predicted protein sequencesby assignment of KEGG orthology identifiers using theKEGG KAAS server Expression levels of the predicted ORFswere determined by aligning the reads against the assemblyusing bowtie2 and counting readsmapped to eachORF usingBEDtools
3 Results and Discussion
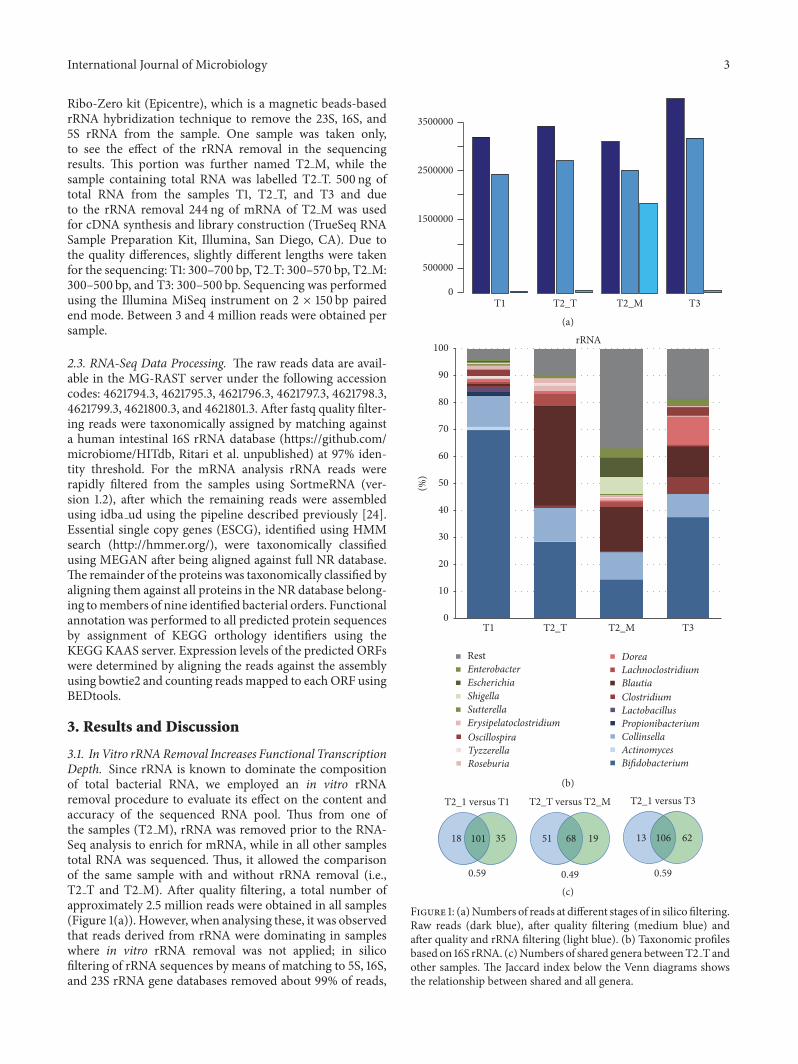
31 InVitro rRNARemoval Increases Functional TranscriptionDepth Since rRNA is known to dominate the compositionof total bacterial RNA we employed an in vitro rRNAremoval procedure to evaluate its effect on the content andaccuracy of the sequenced RNA pool Thus from one ofthe samples (T2 M) rRNA was removed prior to the RNA-Seq analysis to enrich for mRNA while in all other samplestotal RNA was sequenced Thus it allowed the comparisonof the same sample with and without rRNA removal (ieT2 T and T2 M) After quality filtering a total number ofapproximately 25 million reads were obtained in all samples(Figure 1(a)) However when analysing these it was observedthat reads derived from rRNA were dominating in sampleswhere in vitro rRNA removal was not applied in silicofiltering of rRNA sequences by means of matching to 5S 16Sand 23S rRNA gene databases removed about 99 of reads
0T1 T3
500000
1500000
2500000
3500000
T2_T T2_M
(a)
T1 T2_T T2_M T3
rRNA
RestEnterobacterEscherichiaShigellaSutterellaErysipelatoclostridiumOscillospiraTyzzerellaRoseburia
DoreaLachnoclostridiumBlautiaClostridiumLactobacillusPropionibacteriumCollinsellaActinomycesBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
(b)
059059
T2_1 versus T1 T2_T versus T2_M T2_1 versus T3
18 51 68 19 13 106 62101 35
049(c)
Figure 1 (a)Numbers of reads at different stages of in silico filteringRaw reads (dark blue) after quality filtering (medium blue) andafter quality and rRNA filtering (light blue) (b) Taxonomic profilesbased on 16S rRNA (c)Numbers of shared genera betweenT2 T andother samples The Jaccard index below the Venn diagrams showsthe relationship between shared and all genera
4 International Journal of Microbiology
with the exception of sample T2 M where only about 28of reads were removed This showed that T2 M containedmuch less rRNA than untreated samples and confirmed theefficiency of the in vitro rRNA removal procedure As aconsequence for sample T2 M the number of filteredmRNAreads was approximately 250-fold higher than that of theuntreated samples
32 Impact of rRNA Removal on 16S rRNA Based TaxonomicProfiles Comparison of taxonomical profiles of sampleswith and without in vitro rRNA removal (T2 T versusT2 M) showed a significant difference between the sam-ples (Figure 1(b)) Indeed the difference between T2 T andT2 M was more pronounced than the difference betweensamples T2 T and T3 representing different time pointsThe difference was especially evident in the abundance ofBifidobacterium spp that showed an approximately twofoldreduction in sample T2 M as compared to the identicalbut not treated sample T2 T (Figure 1(b)) Furthermorethe amount of 16S rRNA sequences of Blautia was alsoapproximately 2-fold reduced while that of Proteobacteriaincreased over 15-fold in T2 M altogether indicating a strongeffect of the rRNA removal by the kit In addition the numberof common genera between the samples T2 T and T2 Mwas lower than that between other samples T2 T had morein common with T1 and T3 than with T2 M (Figure 1(c))Moreover comparing the log relative abundances betweenT2 T and T2 M indicated that there were several noncorre-lating genera which were abundant in T2 M but rare in T2 T(Supplementary Figure 1 in SupplementaryMaterial availableonline at httpsdoiorg10115520179547063)
Altogether the results indicate that the used in vitrorRNA removal procedure causes bias to the taxonomic com-position most likely because the procedure does not targetall taxonomic groups equally Thus it generates taxonomicprofileswith different bacterial content and abundanceswhencompared to untreated samples This has also been observedfor other rRNA removal methods in a study with a syntheticcommunity consisting of 5 genera that are not commoninhabitants of the intestinal tract [25] Here we confirmand extend this analysis and show that the rRNA removalusing the Ribo-Zero kit (Epicentre) specifically reduces therRNA fraction of the Firmicutes and Actinobacteria fromthe intestinal microbiome of the infant while increasing thatof the Proteobacteria Hence taxonomic analysis based on16S rRNA sequences from in vitro treated material should betakenwith caution even though there would be enough readsleft after the removal
33 Metatranscriptome For further analysis at mRNA-levelthe data was filtered to remove rRNA sequences adaptersequences and poor quality reads SortMeRNA [26] was usedto rapidly filter out rRNA sequences using the precompileddatabases for eukaryotes bacteria and archaea To determinethe function and taxonomy of the mRNA reads they weremerged and de novo assembled into larger contigs creatinga single contig reference set for all samples A total of 11558contigs larger than 300 bp could be assembled with an overalllength of 9843629 bases These contigs encoded a total of
Table 2 Sequencing assembly and mapping information of thesamples
Total reads(pairs)
mRNA reads(pairs)
Mapped(single) mapped
T1 1591215 6229 3801 305T2 T 1710009 13749 17570 639T2 M 1550435 1084270 1480261 683T3 1986769 15661 15657 500
T1 T2_T T2_M T3
mRNA
RestSutterellaVeillonellaNA_ErysipelotrichaceaeErysipelatoclostridiumNA_FirmicutesTyzzerellaRoseburiaCoprococcus
NA_LachnospiraceaeLachnoclostridiumBlautiaClostridiumStreptococcusNA_CoriobacteriaceaeCollinsella NA_BifidobacteriaceaeBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Figure 2 Taxonomic profiles based on mRNA
16422 predicted open reading frames Altogether 30 to 68of themRNA reads could bemapped and assigned to protein-encoding gene regions (Table 2)
The protein assigned mRNA reads were used for adetailed taxonomic analysis (Figure 2) As the mapping isnot without bias and quite variable (Table 2) we observedvarious differences in the phylogenetic composition of thebaby samples based the 16S rRNA (Figure 2) and mRNAdata (Figure 2) These differences in abundances may alsobe explained by varying gene expression levels that do notnecessarily reflect the real abundances of underlying bacterialgroups Remarkably the phylogenetic composition based on
International Journal of Microbiology 5
T1 T2_T T2_M T3
NANA_FirmicutesTyzzerellaNA_LachnospiraceaeBifidobacterium
0102030405060708090
100
()
-galactosidase
Figure 3 Taxonomic profiles based on expression of the 120573-galactosidase gene
the mapped mRNA appeared to be not affected by the invitro rRNA removal as indicated by a Pearson correlationcoefficient of T2 T andT2 Msamples of 099 (SupplementaryFigure 2) This suggests that the rRNA removal is likely topreserve mRNA levels with high fidelity
Themain taxonomic groups expressing protein-encodingtranscripts belonged to Bifidobacterium Collinsella BlautiaLachnoclostridium and unidentified Firmicutes (Figure 2)The expression of mRNA derived from Bifidobacterium andCollinsella decreased from T1 to T3 most likely due to theintroduction of new solid foods into the infant diet afterT1 As the weaning process proceeds a wider variety ofcarbon sources becomes available in the infant intestinethus supporting the establishment of more diverse microbialcommunity This was demonstrated by the high expressionof bifidobacterial 120573-galactosidase while the infant receivedexclusively breast-milk (T1) whereas unidentified Firmicuteswere the main producer of this enzyme at T3 when theinfant diet included also a variety of solid foods and dairyproducts (Figure 3) Transcripts derived from Bacteroideteswere hardly found in the dataset While Bacteroidetes havebeen reported to be present during weaning [6 7] theirabsence in 3-month healthy and breastfed babies is notunusual [18 27] As the rRNA-derived composition alsoindicated a low level of Bacteroidetes (Figure 1(b)) thisindicated that the infantrsquos gut microbiota during weaning isstill adapting towards more adult-like composition Howeverwe must point out here that we have not obtained specificinformation on the sampling time during the day and theexact amount and frequency of the food intake this couldchange activity patterns considerably and should be takeninto consideration in future experiments
34 In-Depth Functional and Taxonomic Distributions ofMetabolic Pathways Metagenome analyses have indicatedthat the production of vitamins may be one of the benefits of
the early life intestinal microbiota [7 9] However evidencebased on expression studies is lacking apart from indica-tions of folate biosynthesis by Bifidobacteria in 8-month-old infants [17] The sequencing depth in sample T2 M wassufficient for determining the taxonomic distribution of thevitamin pathways (Supplementary Figure 3) as well as thatof sugar transport and the taxonomic distribution of theglycosylases expression Remarkably the expressed genesinvolved in biosynthesis of the vitamin folate (B9) reflectsimilar taxonomic distribution as the 120573-galactosidase and theoverall mRNA expression profiles to some extent with Bifi-dobacterium Collinsella andBlautia as themain contributorsto folate metabolism (Figure 4) Other vitamins such as B6B7 and B12 were each produced by quite different organismsincluding Blautia Lachnoclostridium and Lachnospiraceaewhich mainly belonged to phylum Firmicutes This indicatesthat other bacteria than the canonical Bifidobacterium sppcontribute to important functions such as vitamin productionat 6 months of age
Wewere also able to detect in sampleT2 M the expressionof a large variety of genes for sugar transporters belongingto PEP-dependent phosphotransferase (PTS) or ATP bind-ing cassette (ABC) systems that are characterized by theinvolvement of multiple proteins (Table 3) Not all transcriptsof the transporter module proteins functions were equallyexpressed but this may be due to differential gene expres-sion or transcript stability Metagenomic analysis had alsorevealed the presence of these transporters in infants of 4andor 12 months [7] Altogether the expression of thesetransporter genes indicates that a large range of sugars istransported by the gut microbiota in an infant of approxi-mately 6 months old Moreover the expression of glycosidasegenes specific for the hydrolysis of the transported saccha-rides was also detected indicating the functional use of thesesugars including alpha- and beta-glycosidases (altogetherdenoted as glycosylase genes) (Figure 5 Supplementary Table1) These glycosidase genes had been previously found to bepresent before the introduction of solid food [6] While Acti-nobacteria expressed the largest amount of sugar transportand degradation genes around 25 of the transcripts derivedfrom Firmicutes indicating that members of both phyla areimportant contributors to the carbohydrate degradation
35 Expression Levels of Genes from Lactobacillus rhamnosusGG While breastfeeding the mother consumed dairy prod-ucts containing Lactobacillus rhamnosusGG (LGG) which isvery common in many dairy products and drinks in Finlandand has the capacity to strongly interact with human mucusvia its protruding pili [28] Previously it has been shown thatmother-infant transfer of LGG may occur frequently [14]To study whether we could deduce the presence of LGG orrelated bacteria in the transcriptome of the studied infantmRNA reads were mapped to LGG genome at 95 similaritythreshold This analysis confirmed the expression of LGG-like genes including these present in variable regions presentin LGG but absent in many other L rhamnosus strains [29]Comparative analysis revealed significantly more LGG-likegenes ( of total mRNA reads) in sample T1 when comparedto samples T2 and T3 (Figure 6) The same result was seen
6 International Journal of Microbiology
B9 Folate
BifidobacteriumCollinsellaBlautia
B6
CollinsellaBlautiaLachnoclostridium
B7
BlautiaNA-LachnospiraceaeVeillonellaNA
B12
NA-Clostridiales
B9 B6 B7 B12Expression level 55 67 15 29
Figure 4 Relative taxonomic profiles based on the final enzyme for the corresponding vitaminmetabolism of sample T2 M KEGG numbersfor the final step in the corresponding vitamin pathways for this figure are K00287 (B9 Folate) K00868 (B6) K01012 (B7 Biotin) K02233(B12)
Table 3 Overview of the expressed PTS and ABC transport modules involved in sugar transport in sample T2 M 119901 values and adjusted 119901values indicate the likelihood of a KEGG module to be enriched on a background of amongst all detected KEGG orthologs
(a) PTS transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedproteins-encodinggenes
Description
M00266 0 0006 3 2 Maltose and glucose-specific II componentM00269 0 0006 3 2 Sucrose-specific II componentM00268 0 0006 3 2 Arbutin-like II componentM00273 0 0006 3 2 Fructose-specific II componentM00270 0001 0011 4 2 Trehalose-specific II componentM00277 0001 0011 4 2 N-Acetylgalactosamine-specific II componentM00267 0031 0555 3 1 N-Acetylglucosamine-specific II componentM00282 0031 0555 3 1 D-Glucosamine-specific II componentM00272 0031 0555 3 1 Arbutin- cellobiose- and salicin-specific II componentM00303 0031 0555 3 1 N-Acetylmuramic acid-specific II componentM00279 0031 0555 3 1 Galactitol-specific II componentM00283 0031 0555 3 1 Ascorbate-specific II component
(b) ABC sugar transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedprotein-encodinggenes
Description
M00601 0 0009 3 2 Putative chitobiose transport systemM00219 0001 0018 4 2 AI-2 transport systemM00211 0025 048 2 1 Putative ABC transport systemM00217 0038 0716 3 1 D-Allose transport systemM00210 0038 0716 3 1 Putative ABC transport systemM00605 005 0949 4 1 Glucosemannose transport systemM00206 005 0949 4 1 Cellobiose transport systemM00606 005 0949 4 1 NN1015840-Diacetylchitobiose transport systemM00197 005 0949 4 1 Putative fructooligosaccharide transport system
International Journal of Microbiology 7
T2_M
Taxonomic groupsexpressing glycosylases
NA
NA_FirmicutesTyzzerellaRoseburiaLachnoclostridiumBlautiaNA_ActinobacteriaCollinsellaBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Erysipelatoclostridium
Figure 5 Taxonomic profiles based on expression of the glycosy-lases genes in sample T2 M
when mapping rRNA reads from faecal samples to LGG 16SrRNA sequence at 97 similarity While only a few differentLGG-like functions could be detected in the transcriptome ofsample T1 in that of sample T2 M the expression of the LGG-like genes related to glycolysis is clearly visible indicative ofits high activity (Supplementary Figure 4) Since the sampleT1 represents the microbiota of an infant with breast-milk asthe main source of nutrients and whose mother consumedLGG containing products it may be speculated that LGGwas acquired during or soon after the birth from the motherAt later time points the abundance of LGG-like transcriptsdropped suggesting that LGG would then not permanentlycolonize the infant intestinal tract ecosystem However asthis coincides with the introduction of formula and solidfood one may speculate that these factors also contribute tothe clearance of LGG
4 Conclusions
We demonstrate here the feasibility of metatranscriptomeanalysis using high throughput RNA-Seq on the infant gutto get in-depth insight in the active community structurein the weaning period A relevant technical observation is
Relative number of rRNA reads mapping to LGG 16S
T1 T3T2_T T2_M0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped 0004
0005
(a)
0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped
0004
0005
T1
Relative number of mRNA reads mapping to LGG genes
T3T2_T T2_M
(b)
Figure 6 Relative numbers of mRNA (b) and rRNA (a) readmapping to LGG Error bars represent standard deviations based onbinomial sampling
that rRNA removal with Ribo-Zero kit (Epicentre) has apronounced effect on the rRNA distribution but not on thatof mRNA It was found that in the weaning period of thebreastfed infant a number of major activity changes occurredwith a decrease in activity of the Actinobacteria mainlyrepresented by Bifidobacterium and an increase in activity ofmembers of the Firmicutes increased in activity over timeMoreover the Firmicutes and Actinobacteria including thecanonical Bifidobacteria and Collinsella were found to beimportant contributors to vitamin production in the infantintestine including that of vitamin B6 B7 B9 and B12Finally the mother consumed the probiotic LGG and wedetected LGG-like transcripts in the infant transcriptomeindicating that LGG has been transferred is an active mem-ber at the earliest time point and is decreasing in abundanceand activity during weaning showing that LGG is not likelyto remain a permanent member of the intestinal microbiota
8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
2 International Journal of Microbiology
Table 1 Overview of the infant diet
Sample code T1 T2 T3Age (days) 131 165 171Breast milk Exclusively Yes YesFormula No No YesPotato amp roots First baby food Yes YesFruits amp berries No Yes YesVegetables No Yes YesGrain products No Yes YesMeatchickenfisheggs No Yes YesMilk amp dairy products No No Yes
Various culturing approaches have shown that there isvertical transmission of specific bacteria frommother to babyThis was shown elegantly for endogenous Bifidobacteria [12]as well as probiotic bacteria consumed by the mother thatin the case of Lactobacillus rhamnosus GG were found inthe baby [13 14] Culture-independent evidence for maternaltransmission of early life microbiota is limited notably as thebaby microbiota differs so much from that of the motherIn some cases signatures of parental microbes have beendescribed to be present in the infant microbiome [15] andrecently it was reported that the microbiota in children at3 years of age shows signatures present similar to their ownmother and were not seen in between children and unrelatedmothers [16]
While considerable attention has been given to the com-positional development during early life only recently highthroughput omics-based approaches have been applied Arecent study analysed the faecal metagenome during the firstyear of life and observed striking differences between vaginaland C-section delivered infants [7] Moreover some earlystudies addressed the transcriptome of Bifidobacteria in theintestinal tract of babies that were breastfed or on a formula-diet and showed differential expression of genes involvedin sugar catabolism exopolysaccharide production or folatebiosynthesis [17] Moreover metatranscriptome analysis ininfants and their mothers showed differences in expressionof higher capacity of mucin utilization higher capacity folatebiosynthesis and decreased starch degradation in infants[18] Globalmetatranscriptome analysis in theGI tractmicro-biota could enable the elucidation of the specific functionalroles microbes have in this complex community Initial meta-transcriptome studies in the human large intestine revealedthat different functions are expressed between individualswhile core functions of the microbiota appeared to beconsistently expressed among individuals [19ndash21]These find-ings imply that metatranscriptomics could provide insightinto the differential activity profiles in the gut microbiotaenabling the reconstruction of the metabolic activity profileof microbial communities
In this study we describe the feasibility of using deepRNA-Seq analysis to get further insight in the active con-tributors of the microbial community in early life Specificattention is given to the rapid sampling and impact ofremoving rRNA from the mRNA on the phylogenetic and
transcriptional profiling and its analysis depth A breastfedbaby was followed in the first six months of life duringadaptation to solid food dairy products and formula Theresults indicate that Bifidobacterium is an active memberof the community and over time various members of theFirmicutes become more active that are involved in vitaminproduction and sugar metabolism at 6 months
2 Methods
21 Subject Dietary Information and Sampling One vagi-nally born breastfed Finnish baby girl was followed duringthe introduction of first solid foods into the diet Faecalsamples were collected at three time points specifically atthe ages of 131 165 and 171 days These samples were givenwith consent of the mother and ethics are within nationaland international regulation The time points and numberof samples were taken for practical reasons as the faecalsamples were taken at home and immediately processed inRNAlater as to preserve the mRNA as good as possible Theinfant did not receive any probiotic supplementation but hermother consumed dairy products containing LactobacillusrhamnosusGG Both infant and her mother were healthy anddid not receive any antibiotics during the study period At thefirst time point (131 days) the infant consumed exclusivelybreast-milk and some mashed potato and roots as the firstbaby food which was extended in the second time point(165 days) to fruits vegetables and grain and meat productsBefore the last time point the infant was also introduced todairy products and formula milk (Table 1)
22 RNA Extraction rRNA Removal cDNA Synthesis LibraryPreparation and Illumina MiSeq 2500 Sequencing Freshfaecal samples were collected and immediately processed asdescribed previously [22] For this purpose one ml RNAlaterwas added to each gram of sample and stored at minus70∘Cuntil later processing The amount of faecal material usedfor the RNA extraction was 681 g for T1 412 g for T2and 442 g for T3 respectively Total RNA was extractedas described before using the Macaloid procedure [23] andfinal RNA concentrations weremeasured with theNanoDrop1000 Here we obtained 3509 3804 and 3182 120583g of totalRNA for T1 T2 and T3 respectively From sample T25 120583g of RNA was also used for rRNA removal by using the
International Journal of Microbiology 3
Ribo-Zero kit (Epicentre) which is a magnetic beads-basedrRNA hybridization technique to remove the 23S 16S and5S rRNA from the sample One sample was taken onlyto see the effect of the rRNA removal in the sequencingresults This portion was further named T2 M while thesample containing total RNA was labelled T2 T 500 ng oftotal RNA from the samples T1 T2 T and T3 and dueto the rRNA removal 244 ng of mRNA of T2 M was usedfor cDNA synthesis and library construction (TrueSeq RNASample Preparation Kit Illumina San Diego CA) Due tothe quality differences slightly different lengths were takenfor the sequencing T1 300ndash700 bp T2 T 300ndash570 bp T2 M300ndash500 bp and T3 300ndash500 bp Sequencing was performedusing the Illumina MiSeq instrument on 2 times 150 bp pairedend mode Between 3 and 4 million reads were obtained persample
23 RNA-Seq Data Processing The raw reads data are avail-able in the MG-RAST server under the following accessioncodes 46217943 46217953 46217963 46217973 4621798346217993 46218003 and 46218013 After fastq quality filter-ing reads were taxonomically assigned by matching againsta human intestinal 16S rRNA database (httpsgithubcommicrobiomeHITdb Ritari et al unpublished) at 97 iden-tity threshold For the mRNA analysis rRNA reads wererapidly filtered from the samples using SortmeRNA (ver-sion 12) after which the remaining reads were assembledusing idba ud using the pipeline described previously [24]Essential single copy genes (ESCG) identified using HMMsearch (httphmmerorg) were taxonomically classifiedusing MEGAN after being aligned against full NR databaseThe remainder of the proteins was taxonomically classified byaligning them against all proteins in the NR database belong-ing tomembers of nine identified bacterial orders Functionalannotation was performed to all predicted protein sequencesby assignment of KEGG orthology identifiers using theKEGG KAAS server Expression levels of the predicted ORFswere determined by aligning the reads against the assemblyusing bowtie2 and counting readsmapped to eachORF usingBEDtools
3 Results and Discussion
31 InVitro rRNARemoval Increases Functional TranscriptionDepth Since rRNA is known to dominate the compositionof total bacterial RNA we employed an in vitro rRNAremoval procedure to evaluate its effect on the content andaccuracy of the sequenced RNA pool Thus from one ofthe samples (T2 M) rRNA was removed prior to the RNA-Seq analysis to enrich for mRNA while in all other samplestotal RNA was sequenced Thus it allowed the comparisonof the same sample with and without rRNA removal (ieT2 T and T2 M) After quality filtering a total number ofapproximately 25 million reads were obtained in all samples(Figure 1(a)) However when analysing these it was observedthat reads derived from rRNA were dominating in sampleswhere in vitro rRNA removal was not applied in silicofiltering of rRNA sequences by means of matching to 5S 16Sand 23S rRNA gene databases removed about 99 of reads
0T1 T3
500000
1500000
2500000
3500000
T2_T T2_M
(a)
T1 T2_T T2_M T3
rRNA
RestEnterobacterEscherichiaShigellaSutterellaErysipelatoclostridiumOscillospiraTyzzerellaRoseburia
DoreaLachnoclostridiumBlautiaClostridiumLactobacillusPropionibacteriumCollinsellaActinomycesBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
(b)
059059
T2_1 versus T1 T2_T versus T2_M T2_1 versus T3
18 51 68 19 13 106 62101 35
049(c)
Figure 1 (a)Numbers of reads at different stages of in silico filteringRaw reads (dark blue) after quality filtering (medium blue) andafter quality and rRNA filtering (light blue) (b) Taxonomic profilesbased on 16S rRNA (c)Numbers of shared genera betweenT2 T andother samples The Jaccard index below the Venn diagrams showsthe relationship between shared and all genera
4 International Journal of Microbiology
with the exception of sample T2 M where only about 28of reads were removed This showed that T2 M containedmuch less rRNA than untreated samples and confirmed theefficiency of the in vitro rRNA removal procedure As aconsequence for sample T2 M the number of filteredmRNAreads was approximately 250-fold higher than that of theuntreated samples
32 Impact of rRNA Removal on 16S rRNA Based TaxonomicProfiles Comparison of taxonomical profiles of sampleswith and without in vitro rRNA removal (T2 T versusT2 M) showed a significant difference between the sam-ples (Figure 1(b)) Indeed the difference between T2 T andT2 M was more pronounced than the difference betweensamples T2 T and T3 representing different time pointsThe difference was especially evident in the abundance ofBifidobacterium spp that showed an approximately twofoldreduction in sample T2 M as compared to the identicalbut not treated sample T2 T (Figure 1(b)) Furthermorethe amount of 16S rRNA sequences of Blautia was alsoapproximately 2-fold reduced while that of Proteobacteriaincreased over 15-fold in T2 M altogether indicating a strongeffect of the rRNA removal by the kit In addition the numberof common genera between the samples T2 T and T2 Mwas lower than that between other samples T2 T had morein common with T1 and T3 than with T2 M (Figure 1(c))Moreover comparing the log relative abundances betweenT2 T and T2 M indicated that there were several noncorre-lating genera which were abundant in T2 M but rare in T2 T(Supplementary Figure 1 in SupplementaryMaterial availableonline at httpsdoiorg10115520179547063)
Altogether the results indicate that the used in vitrorRNA removal procedure causes bias to the taxonomic com-position most likely because the procedure does not targetall taxonomic groups equally Thus it generates taxonomicprofileswith different bacterial content and abundanceswhencompared to untreated samples This has also been observedfor other rRNA removal methods in a study with a syntheticcommunity consisting of 5 genera that are not commoninhabitants of the intestinal tract [25] Here we confirmand extend this analysis and show that the rRNA removalusing the Ribo-Zero kit (Epicentre) specifically reduces therRNA fraction of the Firmicutes and Actinobacteria fromthe intestinal microbiome of the infant while increasing thatof the Proteobacteria Hence taxonomic analysis based on16S rRNA sequences from in vitro treated material should betakenwith caution even though there would be enough readsleft after the removal
33 Metatranscriptome For further analysis at mRNA-levelthe data was filtered to remove rRNA sequences adaptersequences and poor quality reads SortMeRNA [26] was usedto rapidly filter out rRNA sequences using the precompileddatabases for eukaryotes bacteria and archaea To determinethe function and taxonomy of the mRNA reads they weremerged and de novo assembled into larger contigs creatinga single contig reference set for all samples A total of 11558contigs larger than 300 bp could be assembled with an overalllength of 9843629 bases These contigs encoded a total of
Table 2 Sequencing assembly and mapping information of thesamples
Total reads(pairs)
mRNA reads(pairs)
Mapped(single) mapped
T1 1591215 6229 3801 305T2 T 1710009 13749 17570 639T2 M 1550435 1084270 1480261 683T3 1986769 15661 15657 500
T1 T2_T T2_M T3
mRNA
RestSutterellaVeillonellaNA_ErysipelotrichaceaeErysipelatoclostridiumNA_FirmicutesTyzzerellaRoseburiaCoprococcus
NA_LachnospiraceaeLachnoclostridiumBlautiaClostridiumStreptococcusNA_CoriobacteriaceaeCollinsella NA_BifidobacteriaceaeBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Figure 2 Taxonomic profiles based on mRNA
16422 predicted open reading frames Altogether 30 to 68of themRNA reads could bemapped and assigned to protein-encoding gene regions (Table 2)
The protein assigned mRNA reads were used for adetailed taxonomic analysis (Figure 2) As the mapping isnot without bias and quite variable (Table 2) we observedvarious differences in the phylogenetic composition of thebaby samples based the 16S rRNA (Figure 2) and mRNAdata (Figure 2) These differences in abundances may alsobe explained by varying gene expression levels that do notnecessarily reflect the real abundances of underlying bacterialgroups Remarkably the phylogenetic composition based on
International Journal of Microbiology 5
T1 T2_T T2_M T3
NANA_FirmicutesTyzzerellaNA_LachnospiraceaeBifidobacterium
0102030405060708090
100
()
-galactosidase
Figure 3 Taxonomic profiles based on expression of the 120573-galactosidase gene
the mapped mRNA appeared to be not affected by the invitro rRNA removal as indicated by a Pearson correlationcoefficient of T2 T andT2 Msamples of 099 (SupplementaryFigure 2) This suggests that the rRNA removal is likely topreserve mRNA levels with high fidelity
Themain taxonomic groups expressing protein-encodingtranscripts belonged to Bifidobacterium Collinsella BlautiaLachnoclostridium and unidentified Firmicutes (Figure 2)The expression of mRNA derived from Bifidobacterium andCollinsella decreased from T1 to T3 most likely due to theintroduction of new solid foods into the infant diet afterT1 As the weaning process proceeds a wider variety ofcarbon sources becomes available in the infant intestinethus supporting the establishment of more diverse microbialcommunity This was demonstrated by the high expressionof bifidobacterial 120573-galactosidase while the infant receivedexclusively breast-milk (T1) whereas unidentified Firmicuteswere the main producer of this enzyme at T3 when theinfant diet included also a variety of solid foods and dairyproducts (Figure 3) Transcripts derived from Bacteroideteswere hardly found in the dataset While Bacteroidetes havebeen reported to be present during weaning [6 7] theirabsence in 3-month healthy and breastfed babies is notunusual [18 27] As the rRNA-derived composition alsoindicated a low level of Bacteroidetes (Figure 1(b)) thisindicated that the infantrsquos gut microbiota during weaning isstill adapting towards more adult-like composition Howeverwe must point out here that we have not obtained specificinformation on the sampling time during the day and theexact amount and frequency of the food intake this couldchange activity patterns considerably and should be takeninto consideration in future experiments
34 In-Depth Functional and Taxonomic Distributions ofMetabolic Pathways Metagenome analyses have indicatedthat the production of vitamins may be one of the benefits of
the early life intestinal microbiota [7 9] However evidencebased on expression studies is lacking apart from indica-tions of folate biosynthesis by Bifidobacteria in 8-month-old infants [17] The sequencing depth in sample T2 M wassufficient for determining the taxonomic distribution of thevitamin pathways (Supplementary Figure 3) as well as thatof sugar transport and the taxonomic distribution of theglycosylases expression Remarkably the expressed genesinvolved in biosynthesis of the vitamin folate (B9) reflectsimilar taxonomic distribution as the 120573-galactosidase and theoverall mRNA expression profiles to some extent with Bifi-dobacterium Collinsella andBlautia as themain contributorsto folate metabolism (Figure 4) Other vitamins such as B6B7 and B12 were each produced by quite different organismsincluding Blautia Lachnoclostridium and Lachnospiraceaewhich mainly belonged to phylum Firmicutes This indicatesthat other bacteria than the canonical Bifidobacterium sppcontribute to important functions such as vitamin productionat 6 months of age
Wewere also able to detect in sampleT2 M the expressionof a large variety of genes for sugar transporters belongingto PEP-dependent phosphotransferase (PTS) or ATP bind-ing cassette (ABC) systems that are characterized by theinvolvement of multiple proteins (Table 3) Not all transcriptsof the transporter module proteins functions were equallyexpressed but this may be due to differential gene expres-sion or transcript stability Metagenomic analysis had alsorevealed the presence of these transporters in infants of 4andor 12 months [7] Altogether the expression of thesetransporter genes indicates that a large range of sugars istransported by the gut microbiota in an infant of approxi-mately 6 months old Moreover the expression of glycosidasegenes specific for the hydrolysis of the transported saccha-rides was also detected indicating the functional use of thesesugars including alpha- and beta-glycosidases (altogetherdenoted as glycosylase genes) (Figure 5 Supplementary Table1) These glycosidase genes had been previously found to bepresent before the introduction of solid food [6] While Acti-nobacteria expressed the largest amount of sugar transportand degradation genes around 25 of the transcripts derivedfrom Firmicutes indicating that members of both phyla areimportant contributors to the carbohydrate degradation
35 Expression Levels of Genes from Lactobacillus rhamnosusGG While breastfeeding the mother consumed dairy prod-ucts containing Lactobacillus rhamnosusGG (LGG) which isvery common in many dairy products and drinks in Finlandand has the capacity to strongly interact with human mucusvia its protruding pili [28] Previously it has been shown thatmother-infant transfer of LGG may occur frequently [14]To study whether we could deduce the presence of LGG orrelated bacteria in the transcriptome of the studied infantmRNA reads were mapped to LGG genome at 95 similaritythreshold This analysis confirmed the expression of LGG-like genes including these present in variable regions presentin LGG but absent in many other L rhamnosus strains [29]Comparative analysis revealed significantly more LGG-likegenes ( of total mRNA reads) in sample T1 when comparedto samples T2 and T3 (Figure 6) The same result was seen
6 International Journal of Microbiology
B9 Folate
BifidobacteriumCollinsellaBlautia
B6
CollinsellaBlautiaLachnoclostridium
B7
BlautiaNA-LachnospiraceaeVeillonellaNA
B12
NA-Clostridiales
B9 B6 B7 B12Expression level 55 67 15 29
Figure 4 Relative taxonomic profiles based on the final enzyme for the corresponding vitaminmetabolism of sample T2 M KEGG numbersfor the final step in the corresponding vitamin pathways for this figure are K00287 (B9 Folate) K00868 (B6) K01012 (B7 Biotin) K02233(B12)
Table 3 Overview of the expressed PTS and ABC transport modules involved in sugar transport in sample T2 M 119901 values and adjusted 119901values indicate the likelihood of a KEGG module to be enriched on a background of amongst all detected KEGG orthologs
(a) PTS transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedproteins-encodinggenes
Description
M00266 0 0006 3 2 Maltose and glucose-specific II componentM00269 0 0006 3 2 Sucrose-specific II componentM00268 0 0006 3 2 Arbutin-like II componentM00273 0 0006 3 2 Fructose-specific II componentM00270 0001 0011 4 2 Trehalose-specific II componentM00277 0001 0011 4 2 N-Acetylgalactosamine-specific II componentM00267 0031 0555 3 1 N-Acetylglucosamine-specific II componentM00282 0031 0555 3 1 D-Glucosamine-specific II componentM00272 0031 0555 3 1 Arbutin- cellobiose- and salicin-specific II componentM00303 0031 0555 3 1 N-Acetylmuramic acid-specific II componentM00279 0031 0555 3 1 Galactitol-specific II componentM00283 0031 0555 3 1 Ascorbate-specific II component
(b) ABC sugar transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedprotein-encodinggenes
Description
M00601 0 0009 3 2 Putative chitobiose transport systemM00219 0001 0018 4 2 AI-2 transport systemM00211 0025 048 2 1 Putative ABC transport systemM00217 0038 0716 3 1 D-Allose transport systemM00210 0038 0716 3 1 Putative ABC transport systemM00605 005 0949 4 1 Glucosemannose transport systemM00206 005 0949 4 1 Cellobiose transport systemM00606 005 0949 4 1 NN1015840-Diacetylchitobiose transport systemM00197 005 0949 4 1 Putative fructooligosaccharide transport system
International Journal of Microbiology 7
T2_M
Taxonomic groupsexpressing glycosylases
NA
NA_FirmicutesTyzzerellaRoseburiaLachnoclostridiumBlautiaNA_ActinobacteriaCollinsellaBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Erysipelatoclostridium
Figure 5 Taxonomic profiles based on expression of the glycosy-lases genes in sample T2 M
when mapping rRNA reads from faecal samples to LGG 16SrRNA sequence at 97 similarity While only a few differentLGG-like functions could be detected in the transcriptome ofsample T1 in that of sample T2 M the expression of the LGG-like genes related to glycolysis is clearly visible indicative ofits high activity (Supplementary Figure 4) Since the sampleT1 represents the microbiota of an infant with breast-milk asthe main source of nutrients and whose mother consumedLGG containing products it may be speculated that LGGwas acquired during or soon after the birth from the motherAt later time points the abundance of LGG-like transcriptsdropped suggesting that LGG would then not permanentlycolonize the infant intestinal tract ecosystem However asthis coincides with the introduction of formula and solidfood one may speculate that these factors also contribute tothe clearance of LGG
4 Conclusions
We demonstrate here the feasibility of metatranscriptomeanalysis using high throughput RNA-Seq on the infant gutto get in-depth insight in the active community structurein the weaning period A relevant technical observation is
Relative number of rRNA reads mapping to LGG 16S
T1 T3T2_T T2_M0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped 0004
0005
(a)
0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped
0004
0005
T1
Relative number of mRNA reads mapping to LGG genes
T3T2_T T2_M
(b)
Figure 6 Relative numbers of mRNA (b) and rRNA (a) readmapping to LGG Error bars represent standard deviations based onbinomial sampling
that rRNA removal with Ribo-Zero kit (Epicentre) has apronounced effect on the rRNA distribution but not on thatof mRNA It was found that in the weaning period of thebreastfed infant a number of major activity changes occurredwith a decrease in activity of the Actinobacteria mainlyrepresented by Bifidobacterium and an increase in activity ofmembers of the Firmicutes increased in activity over timeMoreover the Firmicutes and Actinobacteria including thecanonical Bifidobacteria and Collinsella were found to beimportant contributors to vitamin production in the infantintestine including that of vitamin B6 B7 B9 and B12Finally the mother consumed the probiotic LGG and wedetected LGG-like transcripts in the infant transcriptomeindicating that LGG has been transferred is an active mem-ber at the earliest time point and is decreasing in abundanceand activity during weaning showing that LGG is not likelyto remain a permanent member of the intestinal microbiota
8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
International Journal of Microbiology 3
Ribo-Zero kit (Epicentre) which is a magnetic beads-basedrRNA hybridization technique to remove the 23S 16S and5S rRNA from the sample One sample was taken onlyto see the effect of the rRNA removal in the sequencingresults This portion was further named T2 M while thesample containing total RNA was labelled T2 T 500 ng oftotal RNA from the samples T1 T2 T and T3 and dueto the rRNA removal 244 ng of mRNA of T2 M was usedfor cDNA synthesis and library construction (TrueSeq RNASample Preparation Kit Illumina San Diego CA) Due tothe quality differences slightly different lengths were takenfor the sequencing T1 300ndash700 bp T2 T 300ndash570 bp T2 M300ndash500 bp and T3 300ndash500 bp Sequencing was performedusing the Illumina MiSeq instrument on 2 times 150 bp pairedend mode Between 3 and 4 million reads were obtained persample
23 RNA-Seq Data Processing The raw reads data are avail-able in the MG-RAST server under the following accessioncodes 46217943 46217953 46217963 46217973 4621798346217993 46218003 and 46218013 After fastq quality filter-ing reads were taxonomically assigned by matching againsta human intestinal 16S rRNA database (httpsgithubcommicrobiomeHITdb Ritari et al unpublished) at 97 iden-tity threshold For the mRNA analysis rRNA reads wererapidly filtered from the samples using SortmeRNA (ver-sion 12) after which the remaining reads were assembledusing idba ud using the pipeline described previously [24]Essential single copy genes (ESCG) identified using HMMsearch (httphmmerorg) were taxonomically classifiedusing MEGAN after being aligned against full NR databaseThe remainder of the proteins was taxonomically classified byaligning them against all proteins in the NR database belong-ing tomembers of nine identified bacterial orders Functionalannotation was performed to all predicted protein sequencesby assignment of KEGG orthology identifiers using theKEGG KAAS server Expression levels of the predicted ORFswere determined by aligning the reads against the assemblyusing bowtie2 and counting readsmapped to eachORF usingBEDtools
3 Results and Discussion
31 InVitro rRNARemoval Increases Functional TranscriptionDepth Since rRNA is known to dominate the compositionof total bacterial RNA we employed an in vitro rRNAremoval procedure to evaluate its effect on the content andaccuracy of the sequenced RNA pool Thus from one ofthe samples (T2 M) rRNA was removed prior to the RNA-Seq analysis to enrich for mRNA while in all other samplestotal RNA was sequenced Thus it allowed the comparisonof the same sample with and without rRNA removal (ieT2 T and T2 M) After quality filtering a total number ofapproximately 25 million reads were obtained in all samples(Figure 1(a)) However when analysing these it was observedthat reads derived from rRNA were dominating in sampleswhere in vitro rRNA removal was not applied in silicofiltering of rRNA sequences by means of matching to 5S 16Sand 23S rRNA gene databases removed about 99 of reads
0T1 T3
500000
1500000
2500000
3500000
T2_T T2_M
(a)
T1 T2_T T2_M T3
rRNA
RestEnterobacterEscherichiaShigellaSutterellaErysipelatoclostridiumOscillospiraTyzzerellaRoseburia
DoreaLachnoclostridiumBlautiaClostridiumLactobacillusPropionibacteriumCollinsellaActinomycesBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
(b)
059059
T2_1 versus T1 T2_T versus T2_M T2_1 versus T3
18 51 68 19 13 106 62101 35
049(c)
Figure 1 (a)Numbers of reads at different stages of in silico filteringRaw reads (dark blue) after quality filtering (medium blue) andafter quality and rRNA filtering (light blue) (b) Taxonomic profilesbased on 16S rRNA (c)Numbers of shared genera betweenT2 T andother samples The Jaccard index below the Venn diagrams showsthe relationship between shared and all genera
4 International Journal of Microbiology
with the exception of sample T2 M where only about 28of reads were removed This showed that T2 M containedmuch less rRNA than untreated samples and confirmed theefficiency of the in vitro rRNA removal procedure As aconsequence for sample T2 M the number of filteredmRNAreads was approximately 250-fold higher than that of theuntreated samples
32 Impact of rRNA Removal on 16S rRNA Based TaxonomicProfiles Comparison of taxonomical profiles of sampleswith and without in vitro rRNA removal (T2 T versusT2 M) showed a significant difference between the sam-ples (Figure 1(b)) Indeed the difference between T2 T andT2 M was more pronounced than the difference betweensamples T2 T and T3 representing different time pointsThe difference was especially evident in the abundance ofBifidobacterium spp that showed an approximately twofoldreduction in sample T2 M as compared to the identicalbut not treated sample T2 T (Figure 1(b)) Furthermorethe amount of 16S rRNA sequences of Blautia was alsoapproximately 2-fold reduced while that of Proteobacteriaincreased over 15-fold in T2 M altogether indicating a strongeffect of the rRNA removal by the kit In addition the numberof common genera between the samples T2 T and T2 Mwas lower than that between other samples T2 T had morein common with T1 and T3 than with T2 M (Figure 1(c))Moreover comparing the log relative abundances betweenT2 T and T2 M indicated that there were several noncorre-lating genera which were abundant in T2 M but rare in T2 T(Supplementary Figure 1 in SupplementaryMaterial availableonline at httpsdoiorg10115520179547063)
Altogether the results indicate that the used in vitrorRNA removal procedure causes bias to the taxonomic com-position most likely because the procedure does not targetall taxonomic groups equally Thus it generates taxonomicprofileswith different bacterial content and abundanceswhencompared to untreated samples This has also been observedfor other rRNA removal methods in a study with a syntheticcommunity consisting of 5 genera that are not commoninhabitants of the intestinal tract [25] Here we confirmand extend this analysis and show that the rRNA removalusing the Ribo-Zero kit (Epicentre) specifically reduces therRNA fraction of the Firmicutes and Actinobacteria fromthe intestinal microbiome of the infant while increasing thatof the Proteobacteria Hence taxonomic analysis based on16S rRNA sequences from in vitro treated material should betakenwith caution even though there would be enough readsleft after the removal
33 Metatranscriptome For further analysis at mRNA-levelthe data was filtered to remove rRNA sequences adaptersequences and poor quality reads SortMeRNA [26] was usedto rapidly filter out rRNA sequences using the precompileddatabases for eukaryotes bacteria and archaea To determinethe function and taxonomy of the mRNA reads they weremerged and de novo assembled into larger contigs creatinga single contig reference set for all samples A total of 11558contigs larger than 300 bp could be assembled with an overalllength of 9843629 bases These contigs encoded a total of
Table 2 Sequencing assembly and mapping information of thesamples
Total reads(pairs)
mRNA reads(pairs)
Mapped(single) mapped
T1 1591215 6229 3801 305T2 T 1710009 13749 17570 639T2 M 1550435 1084270 1480261 683T3 1986769 15661 15657 500
T1 T2_T T2_M T3
mRNA
RestSutterellaVeillonellaNA_ErysipelotrichaceaeErysipelatoclostridiumNA_FirmicutesTyzzerellaRoseburiaCoprococcus
NA_LachnospiraceaeLachnoclostridiumBlautiaClostridiumStreptococcusNA_CoriobacteriaceaeCollinsella NA_BifidobacteriaceaeBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Figure 2 Taxonomic profiles based on mRNA
16422 predicted open reading frames Altogether 30 to 68of themRNA reads could bemapped and assigned to protein-encoding gene regions (Table 2)
The protein assigned mRNA reads were used for adetailed taxonomic analysis (Figure 2) As the mapping isnot without bias and quite variable (Table 2) we observedvarious differences in the phylogenetic composition of thebaby samples based the 16S rRNA (Figure 2) and mRNAdata (Figure 2) These differences in abundances may alsobe explained by varying gene expression levels that do notnecessarily reflect the real abundances of underlying bacterialgroups Remarkably the phylogenetic composition based on
International Journal of Microbiology 5
T1 T2_T T2_M T3
NANA_FirmicutesTyzzerellaNA_LachnospiraceaeBifidobacterium
0102030405060708090
100
()
-galactosidase
Figure 3 Taxonomic profiles based on expression of the 120573-galactosidase gene
the mapped mRNA appeared to be not affected by the invitro rRNA removal as indicated by a Pearson correlationcoefficient of T2 T andT2 Msamples of 099 (SupplementaryFigure 2) This suggests that the rRNA removal is likely topreserve mRNA levels with high fidelity
Themain taxonomic groups expressing protein-encodingtranscripts belonged to Bifidobacterium Collinsella BlautiaLachnoclostridium and unidentified Firmicutes (Figure 2)The expression of mRNA derived from Bifidobacterium andCollinsella decreased from T1 to T3 most likely due to theintroduction of new solid foods into the infant diet afterT1 As the weaning process proceeds a wider variety ofcarbon sources becomes available in the infant intestinethus supporting the establishment of more diverse microbialcommunity This was demonstrated by the high expressionof bifidobacterial 120573-galactosidase while the infant receivedexclusively breast-milk (T1) whereas unidentified Firmicuteswere the main producer of this enzyme at T3 when theinfant diet included also a variety of solid foods and dairyproducts (Figure 3) Transcripts derived from Bacteroideteswere hardly found in the dataset While Bacteroidetes havebeen reported to be present during weaning [6 7] theirabsence in 3-month healthy and breastfed babies is notunusual [18 27] As the rRNA-derived composition alsoindicated a low level of Bacteroidetes (Figure 1(b)) thisindicated that the infantrsquos gut microbiota during weaning isstill adapting towards more adult-like composition Howeverwe must point out here that we have not obtained specificinformation on the sampling time during the day and theexact amount and frequency of the food intake this couldchange activity patterns considerably and should be takeninto consideration in future experiments
34 In-Depth Functional and Taxonomic Distributions ofMetabolic Pathways Metagenome analyses have indicatedthat the production of vitamins may be one of the benefits of
the early life intestinal microbiota [7 9] However evidencebased on expression studies is lacking apart from indica-tions of folate biosynthesis by Bifidobacteria in 8-month-old infants [17] The sequencing depth in sample T2 M wassufficient for determining the taxonomic distribution of thevitamin pathways (Supplementary Figure 3) as well as thatof sugar transport and the taxonomic distribution of theglycosylases expression Remarkably the expressed genesinvolved in biosynthesis of the vitamin folate (B9) reflectsimilar taxonomic distribution as the 120573-galactosidase and theoverall mRNA expression profiles to some extent with Bifi-dobacterium Collinsella andBlautia as themain contributorsto folate metabolism (Figure 4) Other vitamins such as B6B7 and B12 were each produced by quite different organismsincluding Blautia Lachnoclostridium and Lachnospiraceaewhich mainly belonged to phylum Firmicutes This indicatesthat other bacteria than the canonical Bifidobacterium sppcontribute to important functions such as vitamin productionat 6 months of age
Wewere also able to detect in sampleT2 M the expressionof a large variety of genes for sugar transporters belongingto PEP-dependent phosphotransferase (PTS) or ATP bind-ing cassette (ABC) systems that are characterized by theinvolvement of multiple proteins (Table 3) Not all transcriptsof the transporter module proteins functions were equallyexpressed but this may be due to differential gene expres-sion or transcript stability Metagenomic analysis had alsorevealed the presence of these transporters in infants of 4andor 12 months [7] Altogether the expression of thesetransporter genes indicates that a large range of sugars istransported by the gut microbiota in an infant of approxi-mately 6 months old Moreover the expression of glycosidasegenes specific for the hydrolysis of the transported saccha-rides was also detected indicating the functional use of thesesugars including alpha- and beta-glycosidases (altogetherdenoted as glycosylase genes) (Figure 5 Supplementary Table1) These glycosidase genes had been previously found to bepresent before the introduction of solid food [6] While Acti-nobacteria expressed the largest amount of sugar transportand degradation genes around 25 of the transcripts derivedfrom Firmicutes indicating that members of both phyla areimportant contributors to the carbohydrate degradation
35 Expression Levels of Genes from Lactobacillus rhamnosusGG While breastfeeding the mother consumed dairy prod-ucts containing Lactobacillus rhamnosusGG (LGG) which isvery common in many dairy products and drinks in Finlandand has the capacity to strongly interact with human mucusvia its protruding pili [28] Previously it has been shown thatmother-infant transfer of LGG may occur frequently [14]To study whether we could deduce the presence of LGG orrelated bacteria in the transcriptome of the studied infantmRNA reads were mapped to LGG genome at 95 similaritythreshold This analysis confirmed the expression of LGG-like genes including these present in variable regions presentin LGG but absent in many other L rhamnosus strains [29]Comparative analysis revealed significantly more LGG-likegenes ( of total mRNA reads) in sample T1 when comparedto samples T2 and T3 (Figure 6) The same result was seen
6 International Journal of Microbiology
B9 Folate
BifidobacteriumCollinsellaBlautia
B6
CollinsellaBlautiaLachnoclostridium
B7
BlautiaNA-LachnospiraceaeVeillonellaNA
B12
NA-Clostridiales
B9 B6 B7 B12Expression level 55 67 15 29
Figure 4 Relative taxonomic profiles based on the final enzyme for the corresponding vitaminmetabolism of sample T2 M KEGG numbersfor the final step in the corresponding vitamin pathways for this figure are K00287 (B9 Folate) K00868 (B6) K01012 (B7 Biotin) K02233(B12)
Table 3 Overview of the expressed PTS and ABC transport modules involved in sugar transport in sample T2 M 119901 values and adjusted 119901values indicate the likelihood of a KEGG module to be enriched on a background of amongst all detected KEGG orthologs
(a) PTS transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedproteins-encodinggenes
Description
M00266 0 0006 3 2 Maltose and glucose-specific II componentM00269 0 0006 3 2 Sucrose-specific II componentM00268 0 0006 3 2 Arbutin-like II componentM00273 0 0006 3 2 Fructose-specific II componentM00270 0001 0011 4 2 Trehalose-specific II componentM00277 0001 0011 4 2 N-Acetylgalactosamine-specific II componentM00267 0031 0555 3 1 N-Acetylglucosamine-specific II componentM00282 0031 0555 3 1 D-Glucosamine-specific II componentM00272 0031 0555 3 1 Arbutin- cellobiose- and salicin-specific II componentM00303 0031 0555 3 1 N-Acetylmuramic acid-specific II componentM00279 0031 0555 3 1 Galactitol-specific II componentM00283 0031 0555 3 1 Ascorbate-specific II component
(b) ABC sugar transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedprotein-encodinggenes
Description
M00601 0 0009 3 2 Putative chitobiose transport systemM00219 0001 0018 4 2 AI-2 transport systemM00211 0025 048 2 1 Putative ABC transport systemM00217 0038 0716 3 1 D-Allose transport systemM00210 0038 0716 3 1 Putative ABC transport systemM00605 005 0949 4 1 Glucosemannose transport systemM00206 005 0949 4 1 Cellobiose transport systemM00606 005 0949 4 1 NN1015840-Diacetylchitobiose transport systemM00197 005 0949 4 1 Putative fructooligosaccharide transport system
International Journal of Microbiology 7
T2_M
Taxonomic groupsexpressing glycosylases
NA
NA_FirmicutesTyzzerellaRoseburiaLachnoclostridiumBlautiaNA_ActinobacteriaCollinsellaBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Erysipelatoclostridium
Figure 5 Taxonomic profiles based on expression of the glycosy-lases genes in sample T2 M
when mapping rRNA reads from faecal samples to LGG 16SrRNA sequence at 97 similarity While only a few differentLGG-like functions could be detected in the transcriptome ofsample T1 in that of sample T2 M the expression of the LGG-like genes related to glycolysis is clearly visible indicative ofits high activity (Supplementary Figure 4) Since the sampleT1 represents the microbiota of an infant with breast-milk asthe main source of nutrients and whose mother consumedLGG containing products it may be speculated that LGGwas acquired during or soon after the birth from the motherAt later time points the abundance of LGG-like transcriptsdropped suggesting that LGG would then not permanentlycolonize the infant intestinal tract ecosystem However asthis coincides with the introduction of formula and solidfood one may speculate that these factors also contribute tothe clearance of LGG
4 Conclusions
We demonstrate here the feasibility of metatranscriptomeanalysis using high throughput RNA-Seq on the infant gutto get in-depth insight in the active community structurein the weaning period A relevant technical observation is
Relative number of rRNA reads mapping to LGG 16S
T1 T3T2_T T2_M0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped 0004
0005
(a)
0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped
0004
0005
T1
Relative number of mRNA reads mapping to LGG genes
T3T2_T T2_M
(b)
Figure 6 Relative numbers of mRNA (b) and rRNA (a) readmapping to LGG Error bars represent standard deviations based onbinomial sampling
that rRNA removal with Ribo-Zero kit (Epicentre) has apronounced effect on the rRNA distribution but not on thatof mRNA It was found that in the weaning period of thebreastfed infant a number of major activity changes occurredwith a decrease in activity of the Actinobacteria mainlyrepresented by Bifidobacterium and an increase in activity ofmembers of the Firmicutes increased in activity over timeMoreover the Firmicutes and Actinobacteria including thecanonical Bifidobacteria and Collinsella were found to beimportant contributors to vitamin production in the infantintestine including that of vitamin B6 B7 B9 and B12Finally the mother consumed the probiotic LGG and wedetected LGG-like transcripts in the infant transcriptomeindicating that LGG has been transferred is an active mem-ber at the earliest time point and is decreasing in abundanceand activity during weaning showing that LGG is not likelyto remain a permanent member of the intestinal microbiota
8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
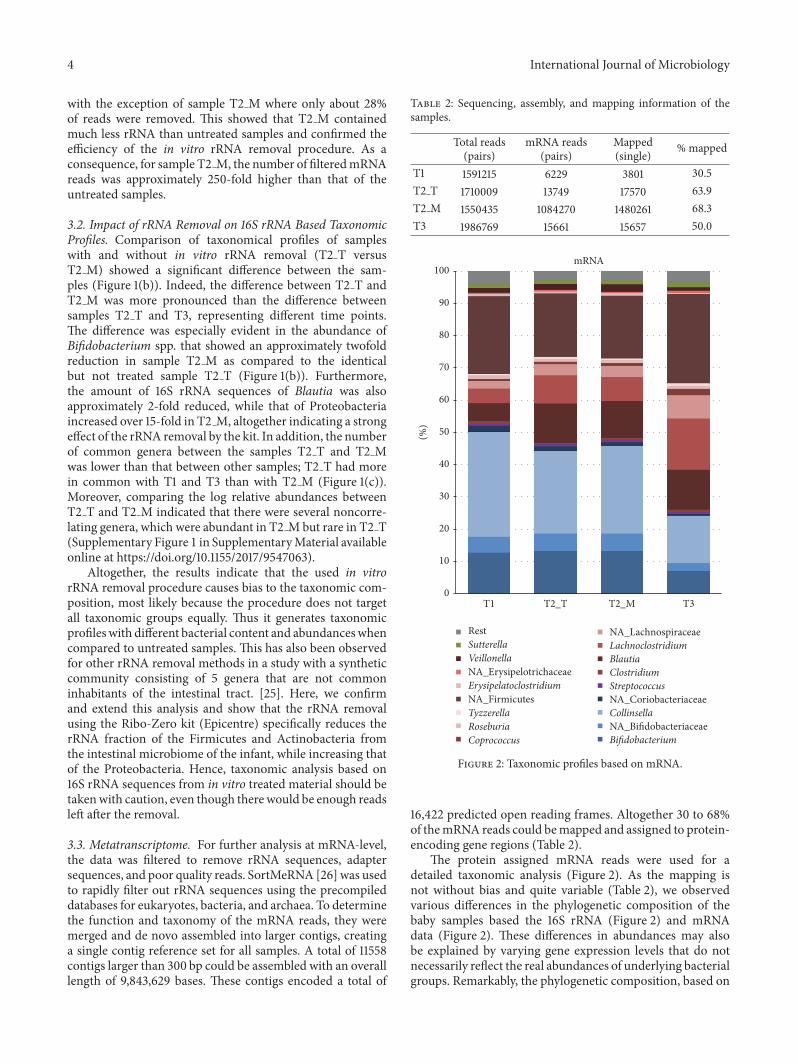
4 International Journal of Microbiology
with the exception of sample T2 M where only about 28of reads were removed This showed that T2 M containedmuch less rRNA than untreated samples and confirmed theefficiency of the in vitro rRNA removal procedure As aconsequence for sample T2 M the number of filteredmRNAreads was approximately 250-fold higher than that of theuntreated samples
32 Impact of rRNA Removal on 16S rRNA Based TaxonomicProfiles Comparison of taxonomical profiles of sampleswith and without in vitro rRNA removal (T2 T versusT2 M) showed a significant difference between the sam-ples (Figure 1(b)) Indeed the difference between T2 T andT2 M was more pronounced than the difference betweensamples T2 T and T3 representing different time pointsThe difference was especially evident in the abundance ofBifidobacterium spp that showed an approximately twofoldreduction in sample T2 M as compared to the identicalbut not treated sample T2 T (Figure 1(b)) Furthermorethe amount of 16S rRNA sequences of Blautia was alsoapproximately 2-fold reduced while that of Proteobacteriaincreased over 15-fold in T2 M altogether indicating a strongeffect of the rRNA removal by the kit In addition the numberof common genera between the samples T2 T and T2 Mwas lower than that between other samples T2 T had morein common with T1 and T3 than with T2 M (Figure 1(c))Moreover comparing the log relative abundances betweenT2 T and T2 M indicated that there were several noncorre-lating genera which were abundant in T2 M but rare in T2 T(Supplementary Figure 1 in SupplementaryMaterial availableonline at httpsdoiorg10115520179547063)
Altogether the results indicate that the used in vitrorRNA removal procedure causes bias to the taxonomic com-position most likely because the procedure does not targetall taxonomic groups equally Thus it generates taxonomicprofileswith different bacterial content and abundanceswhencompared to untreated samples This has also been observedfor other rRNA removal methods in a study with a syntheticcommunity consisting of 5 genera that are not commoninhabitants of the intestinal tract [25] Here we confirmand extend this analysis and show that the rRNA removalusing the Ribo-Zero kit (Epicentre) specifically reduces therRNA fraction of the Firmicutes and Actinobacteria fromthe intestinal microbiome of the infant while increasing thatof the Proteobacteria Hence taxonomic analysis based on16S rRNA sequences from in vitro treated material should betakenwith caution even though there would be enough readsleft after the removal
33 Metatranscriptome For further analysis at mRNA-levelthe data was filtered to remove rRNA sequences adaptersequences and poor quality reads SortMeRNA [26] was usedto rapidly filter out rRNA sequences using the precompileddatabases for eukaryotes bacteria and archaea To determinethe function and taxonomy of the mRNA reads they weremerged and de novo assembled into larger contigs creatinga single contig reference set for all samples A total of 11558contigs larger than 300 bp could be assembled with an overalllength of 9843629 bases These contigs encoded a total of
Table 2 Sequencing assembly and mapping information of thesamples
Total reads(pairs)
mRNA reads(pairs)
Mapped(single) mapped
T1 1591215 6229 3801 305T2 T 1710009 13749 17570 639T2 M 1550435 1084270 1480261 683T3 1986769 15661 15657 500
T1 T2_T T2_M T3
mRNA
RestSutterellaVeillonellaNA_ErysipelotrichaceaeErysipelatoclostridiumNA_FirmicutesTyzzerellaRoseburiaCoprococcus
NA_LachnospiraceaeLachnoclostridiumBlautiaClostridiumStreptococcusNA_CoriobacteriaceaeCollinsella NA_BifidobacteriaceaeBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Figure 2 Taxonomic profiles based on mRNA
16422 predicted open reading frames Altogether 30 to 68of themRNA reads could bemapped and assigned to protein-encoding gene regions (Table 2)
The protein assigned mRNA reads were used for adetailed taxonomic analysis (Figure 2) As the mapping isnot without bias and quite variable (Table 2) we observedvarious differences in the phylogenetic composition of thebaby samples based the 16S rRNA (Figure 2) and mRNAdata (Figure 2) These differences in abundances may alsobe explained by varying gene expression levels that do notnecessarily reflect the real abundances of underlying bacterialgroups Remarkably the phylogenetic composition based on
International Journal of Microbiology 5
T1 T2_T T2_M T3
NANA_FirmicutesTyzzerellaNA_LachnospiraceaeBifidobacterium
0102030405060708090
100
()
-galactosidase
Figure 3 Taxonomic profiles based on expression of the 120573-galactosidase gene
the mapped mRNA appeared to be not affected by the invitro rRNA removal as indicated by a Pearson correlationcoefficient of T2 T andT2 Msamples of 099 (SupplementaryFigure 2) This suggests that the rRNA removal is likely topreserve mRNA levels with high fidelity
Themain taxonomic groups expressing protein-encodingtranscripts belonged to Bifidobacterium Collinsella BlautiaLachnoclostridium and unidentified Firmicutes (Figure 2)The expression of mRNA derived from Bifidobacterium andCollinsella decreased from T1 to T3 most likely due to theintroduction of new solid foods into the infant diet afterT1 As the weaning process proceeds a wider variety ofcarbon sources becomes available in the infant intestinethus supporting the establishment of more diverse microbialcommunity This was demonstrated by the high expressionof bifidobacterial 120573-galactosidase while the infant receivedexclusively breast-milk (T1) whereas unidentified Firmicuteswere the main producer of this enzyme at T3 when theinfant diet included also a variety of solid foods and dairyproducts (Figure 3) Transcripts derived from Bacteroideteswere hardly found in the dataset While Bacteroidetes havebeen reported to be present during weaning [6 7] theirabsence in 3-month healthy and breastfed babies is notunusual [18 27] As the rRNA-derived composition alsoindicated a low level of Bacteroidetes (Figure 1(b)) thisindicated that the infantrsquos gut microbiota during weaning isstill adapting towards more adult-like composition Howeverwe must point out here that we have not obtained specificinformation on the sampling time during the day and theexact amount and frequency of the food intake this couldchange activity patterns considerably and should be takeninto consideration in future experiments
34 In-Depth Functional and Taxonomic Distributions ofMetabolic Pathways Metagenome analyses have indicatedthat the production of vitamins may be one of the benefits of
the early life intestinal microbiota [7 9] However evidencebased on expression studies is lacking apart from indica-tions of folate biosynthesis by Bifidobacteria in 8-month-old infants [17] The sequencing depth in sample T2 M wassufficient for determining the taxonomic distribution of thevitamin pathways (Supplementary Figure 3) as well as thatof sugar transport and the taxonomic distribution of theglycosylases expression Remarkably the expressed genesinvolved in biosynthesis of the vitamin folate (B9) reflectsimilar taxonomic distribution as the 120573-galactosidase and theoverall mRNA expression profiles to some extent with Bifi-dobacterium Collinsella andBlautia as themain contributorsto folate metabolism (Figure 4) Other vitamins such as B6B7 and B12 were each produced by quite different organismsincluding Blautia Lachnoclostridium and Lachnospiraceaewhich mainly belonged to phylum Firmicutes This indicatesthat other bacteria than the canonical Bifidobacterium sppcontribute to important functions such as vitamin productionat 6 months of age
Wewere also able to detect in sampleT2 M the expressionof a large variety of genes for sugar transporters belongingto PEP-dependent phosphotransferase (PTS) or ATP bind-ing cassette (ABC) systems that are characterized by theinvolvement of multiple proteins (Table 3) Not all transcriptsof the transporter module proteins functions were equallyexpressed but this may be due to differential gene expres-sion or transcript stability Metagenomic analysis had alsorevealed the presence of these transporters in infants of 4andor 12 months [7] Altogether the expression of thesetransporter genes indicates that a large range of sugars istransported by the gut microbiota in an infant of approxi-mately 6 months old Moreover the expression of glycosidasegenes specific for the hydrolysis of the transported saccha-rides was also detected indicating the functional use of thesesugars including alpha- and beta-glycosidases (altogetherdenoted as glycosylase genes) (Figure 5 Supplementary Table1) These glycosidase genes had been previously found to bepresent before the introduction of solid food [6] While Acti-nobacteria expressed the largest amount of sugar transportand degradation genes around 25 of the transcripts derivedfrom Firmicutes indicating that members of both phyla areimportant contributors to the carbohydrate degradation
35 Expression Levels of Genes from Lactobacillus rhamnosusGG While breastfeeding the mother consumed dairy prod-ucts containing Lactobacillus rhamnosusGG (LGG) which isvery common in many dairy products and drinks in Finlandand has the capacity to strongly interact with human mucusvia its protruding pili [28] Previously it has been shown thatmother-infant transfer of LGG may occur frequently [14]To study whether we could deduce the presence of LGG orrelated bacteria in the transcriptome of the studied infantmRNA reads were mapped to LGG genome at 95 similaritythreshold This analysis confirmed the expression of LGG-like genes including these present in variable regions presentin LGG but absent in many other L rhamnosus strains [29]Comparative analysis revealed significantly more LGG-likegenes ( of total mRNA reads) in sample T1 when comparedto samples T2 and T3 (Figure 6) The same result was seen
6 International Journal of Microbiology
B9 Folate
BifidobacteriumCollinsellaBlautia
B6
CollinsellaBlautiaLachnoclostridium
B7
BlautiaNA-LachnospiraceaeVeillonellaNA
B12
NA-Clostridiales
B9 B6 B7 B12Expression level 55 67 15 29
Figure 4 Relative taxonomic profiles based on the final enzyme for the corresponding vitaminmetabolism of sample T2 M KEGG numbersfor the final step in the corresponding vitamin pathways for this figure are K00287 (B9 Folate) K00868 (B6) K01012 (B7 Biotin) K02233(B12)
Table 3 Overview of the expressed PTS and ABC transport modules involved in sugar transport in sample T2 M 119901 values and adjusted 119901values indicate the likelihood of a KEGG module to be enriched on a background of amongst all detected KEGG orthologs
(a) PTS transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedproteins-encodinggenes
Description
M00266 0 0006 3 2 Maltose and glucose-specific II componentM00269 0 0006 3 2 Sucrose-specific II componentM00268 0 0006 3 2 Arbutin-like II componentM00273 0 0006 3 2 Fructose-specific II componentM00270 0001 0011 4 2 Trehalose-specific II componentM00277 0001 0011 4 2 N-Acetylgalactosamine-specific II componentM00267 0031 0555 3 1 N-Acetylglucosamine-specific II componentM00282 0031 0555 3 1 D-Glucosamine-specific II componentM00272 0031 0555 3 1 Arbutin- cellobiose- and salicin-specific II componentM00303 0031 0555 3 1 N-Acetylmuramic acid-specific II componentM00279 0031 0555 3 1 Galactitol-specific II componentM00283 0031 0555 3 1 Ascorbate-specific II component
(b) ABC sugar transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedprotein-encodinggenes
Description
M00601 0 0009 3 2 Putative chitobiose transport systemM00219 0001 0018 4 2 AI-2 transport systemM00211 0025 048 2 1 Putative ABC transport systemM00217 0038 0716 3 1 D-Allose transport systemM00210 0038 0716 3 1 Putative ABC transport systemM00605 005 0949 4 1 Glucosemannose transport systemM00206 005 0949 4 1 Cellobiose transport systemM00606 005 0949 4 1 NN1015840-Diacetylchitobiose transport systemM00197 005 0949 4 1 Putative fructooligosaccharide transport system
International Journal of Microbiology 7
T2_M
Taxonomic groupsexpressing glycosylases
NA
NA_FirmicutesTyzzerellaRoseburiaLachnoclostridiumBlautiaNA_ActinobacteriaCollinsellaBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Erysipelatoclostridium
Figure 5 Taxonomic profiles based on expression of the glycosy-lases genes in sample T2 M
when mapping rRNA reads from faecal samples to LGG 16SrRNA sequence at 97 similarity While only a few differentLGG-like functions could be detected in the transcriptome ofsample T1 in that of sample T2 M the expression of the LGG-like genes related to glycolysis is clearly visible indicative ofits high activity (Supplementary Figure 4) Since the sampleT1 represents the microbiota of an infant with breast-milk asthe main source of nutrients and whose mother consumedLGG containing products it may be speculated that LGGwas acquired during or soon after the birth from the motherAt later time points the abundance of LGG-like transcriptsdropped suggesting that LGG would then not permanentlycolonize the infant intestinal tract ecosystem However asthis coincides with the introduction of formula and solidfood one may speculate that these factors also contribute tothe clearance of LGG
4 Conclusions
We demonstrate here the feasibility of metatranscriptomeanalysis using high throughput RNA-Seq on the infant gutto get in-depth insight in the active community structurein the weaning period A relevant technical observation is
Relative number of rRNA reads mapping to LGG 16S
T1 T3T2_T T2_M0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped 0004
0005
(a)
0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped
0004
0005
T1
Relative number of mRNA reads mapping to LGG genes
T3T2_T T2_M
(b)
Figure 6 Relative numbers of mRNA (b) and rRNA (a) readmapping to LGG Error bars represent standard deviations based onbinomial sampling
that rRNA removal with Ribo-Zero kit (Epicentre) has apronounced effect on the rRNA distribution but not on thatof mRNA It was found that in the weaning period of thebreastfed infant a number of major activity changes occurredwith a decrease in activity of the Actinobacteria mainlyrepresented by Bifidobacterium and an increase in activity ofmembers of the Firmicutes increased in activity over timeMoreover the Firmicutes and Actinobacteria including thecanonical Bifidobacteria and Collinsella were found to beimportant contributors to vitamin production in the infantintestine including that of vitamin B6 B7 B9 and B12Finally the mother consumed the probiotic LGG and wedetected LGG-like transcripts in the infant transcriptomeindicating that LGG has been transferred is an active mem-ber at the earliest time point and is decreasing in abundanceand activity during weaning showing that LGG is not likelyto remain a permanent member of the intestinal microbiota
8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
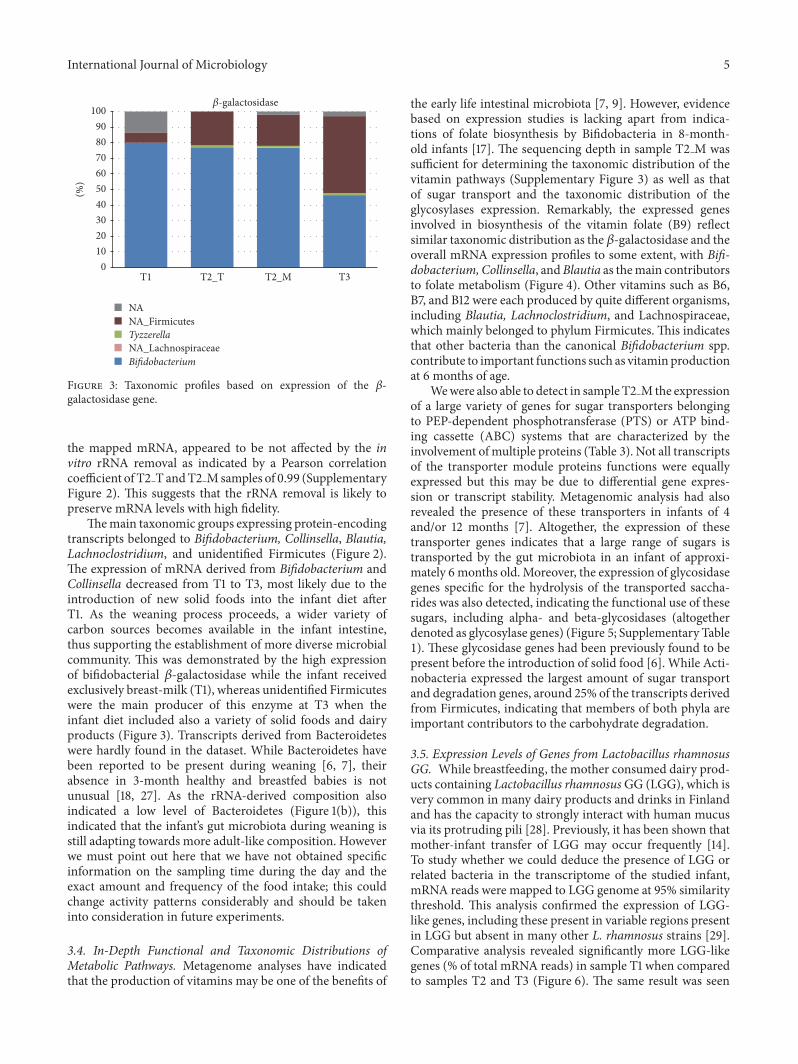
International Journal of Microbiology 5
T1 T2_T T2_M T3
NANA_FirmicutesTyzzerellaNA_LachnospiraceaeBifidobacterium
0102030405060708090
100
()
-galactosidase
Figure 3 Taxonomic profiles based on expression of the 120573-galactosidase gene
the mapped mRNA appeared to be not affected by the invitro rRNA removal as indicated by a Pearson correlationcoefficient of T2 T andT2 Msamples of 099 (SupplementaryFigure 2) This suggests that the rRNA removal is likely topreserve mRNA levels with high fidelity
Themain taxonomic groups expressing protein-encodingtranscripts belonged to Bifidobacterium Collinsella BlautiaLachnoclostridium and unidentified Firmicutes (Figure 2)The expression of mRNA derived from Bifidobacterium andCollinsella decreased from T1 to T3 most likely due to theintroduction of new solid foods into the infant diet afterT1 As the weaning process proceeds a wider variety ofcarbon sources becomes available in the infant intestinethus supporting the establishment of more diverse microbialcommunity This was demonstrated by the high expressionof bifidobacterial 120573-galactosidase while the infant receivedexclusively breast-milk (T1) whereas unidentified Firmicuteswere the main producer of this enzyme at T3 when theinfant diet included also a variety of solid foods and dairyproducts (Figure 3) Transcripts derived from Bacteroideteswere hardly found in the dataset While Bacteroidetes havebeen reported to be present during weaning [6 7] theirabsence in 3-month healthy and breastfed babies is notunusual [18 27] As the rRNA-derived composition alsoindicated a low level of Bacteroidetes (Figure 1(b)) thisindicated that the infantrsquos gut microbiota during weaning isstill adapting towards more adult-like composition Howeverwe must point out here that we have not obtained specificinformation on the sampling time during the day and theexact amount and frequency of the food intake this couldchange activity patterns considerably and should be takeninto consideration in future experiments
34 In-Depth Functional and Taxonomic Distributions ofMetabolic Pathways Metagenome analyses have indicatedthat the production of vitamins may be one of the benefits of
the early life intestinal microbiota [7 9] However evidencebased on expression studies is lacking apart from indica-tions of folate biosynthesis by Bifidobacteria in 8-month-old infants [17] The sequencing depth in sample T2 M wassufficient for determining the taxonomic distribution of thevitamin pathways (Supplementary Figure 3) as well as thatof sugar transport and the taxonomic distribution of theglycosylases expression Remarkably the expressed genesinvolved in biosynthesis of the vitamin folate (B9) reflectsimilar taxonomic distribution as the 120573-galactosidase and theoverall mRNA expression profiles to some extent with Bifi-dobacterium Collinsella andBlautia as themain contributorsto folate metabolism (Figure 4) Other vitamins such as B6B7 and B12 were each produced by quite different organismsincluding Blautia Lachnoclostridium and Lachnospiraceaewhich mainly belonged to phylum Firmicutes This indicatesthat other bacteria than the canonical Bifidobacterium sppcontribute to important functions such as vitamin productionat 6 months of age
Wewere also able to detect in sampleT2 M the expressionof a large variety of genes for sugar transporters belongingto PEP-dependent phosphotransferase (PTS) or ATP bind-ing cassette (ABC) systems that are characterized by theinvolvement of multiple proteins (Table 3) Not all transcriptsof the transporter module proteins functions were equallyexpressed but this may be due to differential gene expres-sion or transcript stability Metagenomic analysis had alsorevealed the presence of these transporters in infants of 4andor 12 months [7] Altogether the expression of thesetransporter genes indicates that a large range of sugars istransported by the gut microbiota in an infant of approxi-mately 6 months old Moreover the expression of glycosidasegenes specific for the hydrolysis of the transported saccha-rides was also detected indicating the functional use of thesesugars including alpha- and beta-glycosidases (altogetherdenoted as glycosylase genes) (Figure 5 Supplementary Table1) These glycosidase genes had been previously found to bepresent before the introduction of solid food [6] While Acti-nobacteria expressed the largest amount of sugar transportand degradation genes around 25 of the transcripts derivedfrom Firmicutes indicating that members of both phyla areimportant contributors to the carbohydrate degradation
35 Expression Levels of Genes from Lactobacillus rhamnosusGG While breastfeeding the mother consumed dairy prod-ucts containing Lactobacillus rhamnosusGG (LGG) which isvery common in many dairy products and drinks in Finlandand has the capacity to strongly interact with human mucusvia its protruding pili [28] Previously it has been shown thatmother-infant transfer of LGG may occur frequently [14]To study whether we could deduce the presence of LGG orrelated bacteria in the transcriptome of the studied infantmRNA reads were mapped to LGG genome at 95 similaritythreshold This analysis confirmed the expression of LGG-like genes including these present in variable regions presentin LGG but absent in many other L rhamnosus strains [29]Comparative analysis revealed significantly more LGG-likegenes ( of total mRNA reads) in sample T1 when comparedto samples T2 and T3 (Figure 6) The same result was seen
6 International Journal of Microbiology
B9 Folate
BifidobacteriumCollinsellaBlautia
B6
CollinsellaBlautiaLachnoclostridium
B7
BlautiaNA-LachnospiraceaeVeillonellaNA
B12
NA-Clostridiales
B9 B6 B7 B12Expression level 55 67 15 29
Figure 4 Relative taxonomic profiles based on the final enzyme for the corresponding vitaminmetabolism of sample T2 M KEGG numbersfor the final step in the corresponding vitamin pathways for this figure are K00287 (B9 Folate) K00868 (B6) K01012 (B7 Biotin) K02233(B12)
Table 3 Overview of the expressed PTS and ABC transport modules involved in sugar transport in sample T2 M 119901 values and adjusted 119901values indicate the likelihood of a KEGG module to be enriched on a background of amongst all detected KEGG orthologs
(a) PTS transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedproteins-encodinggenes
Description
M00266 0 0006 3 2 Maltose and glucose-specific II componentM00269 0 0006 3 2 Sucrose-specific II componentM00268 0 0006 3 2 Arbutin-like II componentM00273 0 0006 3 2 Fructose-specific II componentM00270 0001 0011 4 2 Trehalose-specific II componentM00277 0001 0011 4 2 N-Acetylgalactosamine-specific II componentM00267 0031 0555 3 1 N-Acetylglucosamine-specific II componentM00282 0031 0555 3 1 D-Glucosamine-specific II componentM00272 0031 0555 3 1 Arbutin- cellobiose- and salicin-specific II componentM00303 0031 0555 3 1 N-Acetylmuramic acid-specific II componentM00279 0031 0555 3 1 Galactitol-specific II componentM00283 0031 0555 3 1 Ascorbate-specific II component
(b) ABC sugar transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedprotein-encodinggenes
Description
M00601 0 0009 3 2 Putative chitobiose transport systemM00219 0001 0018 4 2 AI-2 transport systemM00211 0025 048 2 1 Putative ABC transport systemM00217 0038 0716 3 1 D-Allose transport systemM00210 0038 0716 3 1 Putative ABC transport systemM00605 005 0949 4 1 Glucosemannose transport systemM00206 005 0949 4 1 Cellobiose transport systemM00606 005 0949 4 1 NN1015840-Diacetylchitobiose transport systemM00197 005 0949 4 1 Putative fructooligosaccharide transport system
International Journal of Microbiology 7
T2_M
Taxonomic groupsexpressing glycosylases
NA
NA_FirmicutesTyzzerellaRoseburiaLachnoclostridiumBlautiaNA_ActinobacteriaCollinsellaBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Erysipelatoclostridium
Figure 5 Taxonomic profiles based on expression of the glycosy-lases genes in sample T2 M
when mapping rRNA reads from faecal samples to LGG 16SrRNA sequence at 97 similarity While only a few differentLGG-like functions could be detected in the transcriptome ofsample T1 in that of sample T2 M the expression of the LGG-like genes related to glycolysis is clearly visible indicative ofits high activity (Supplementary Figure 4) Since the sampleT1 represents the microbiota of an infant with breast-milk asthe main source of nutrients and whose mother consumedLGG containing products it may be speculated that LGGwas acquired during or soon after the birth from the motherAt later time points the abundance of LGG-like transcriptsdropped suggesting that LGG would then not permanentlycolonize the infant intestinal tract ecosystem However asthis coincides with the introduction of formula and solidfood one may speculate that these factors also contribute tothe clearance of LGG
4 Conclusions
We demonstrate here the feasibility of metatranscriptomeanalysis using high throughput RNA-Seq on the infant gutto get in-depth insight in the active community structurein the weaning period A relevant technical observation is
Relative number of rRNA reads mapping to LGG 16S
T1 T3T2_T T2_M0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped 0004
0005
(a)
0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped
0004
0005
T1
Relative number of mRNA reads mapping to LGG genes
T3T2_T T2_M
(b)
Figure 6 Relative numbers of mRNA (b) and rRNA (a) readmapping to LGG Error bars represent standard deviations based onbinomial sampling
that rRNA removal with Ribo-Zero kit (Epicentre) has apronounced effect on the rRNA distribution but not on thatof mRNA It was found that in the weaning period of thebreastfed infant a number of major activity changes occurredwith a decrease in activity of the Actinobacteria mainlyrepresented by Bifidobacterium and an increase in activity ofmembers of the Firmicutes increased in activity over timeMoreover the Firmicutes and Actinobacteria including thecanonical Bifidobacteria and Collinsella were found to beimportant contributors to vitamin production in the infantintestine including that of vitamin B6 B7 B9 and B12Finally the mother consumed the probiotic LGG and wedetected LGG-like transcripts in the infant transcriptomeindicating that LGG has been transferred is an active mem-ber at the earliest time point and is decreasing in abundanceand activity during weaning showing that LGG is not likelyto remain a permanent member of the intestinal microbiota
8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

6 International Journal of Microbiology
B9 Folate
BifidobacteriumCollinsellaBlautia
B6
CollinsellaBlautiaLachnoclostridium
B7
BlautiaNA-LachnospiraceaeVeillonellaNA
B12
NA-Clostridiales
B9 B6 B7 B12Expression level 55 67 15 29
Figure 4 Relative taxonomic profiles based on the final enzyme for the corresponding vitaminmetabolism of sample T2 M KEGG numbersfor the final step in the corresponding vitamin pathways for this figure are K00287 (B9 Folate) K00868 (B6) K01012 (B7 Biotin) K02233(B12)
Table 3 Overview of the expressed PTS and ABC transport modules involved in sugar transport in sample T2 M 119901 values and adjusted 119901values indicate the likelihood of a KEGG module to be enriched on a background of amongst all detected KEGG orthologs
(a) PTS transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedproteins-encodinggenes
Description
M00266 0 0006 3 2 Maltose and glucose-specific II componentM00269 0 0006 3 2 Sucrose-specific II componentM00268 0 0006 3 2 Arbutin-like II componentM00273 0 0006 3 2 Fructose-specific II componentM00270 0001 0011 4 2 Trehalose-specific II componentM00277 0001 0011 4 2 N-Acetylgalactosamine-specific II componentM00267 0031 0555 3 1 N-Acetylglucosamine-specific II componentM00282 0031 0555 3 1 D-Glucosamine-specific II componentM00272 0031 0555 3 1 Arbutin- cellobiose- and salicin-specific II componentM00303 0031 0555 3 1 N-Acetylmuramic acid-specific II componentM00279 0031 0555 3 1 Galactitol-specific II componentM00283 0031 0555 3 1 Ascorbate-specific II component
(b) ABC sugar transporter modules
Module number 119901 value Adjusted 119901 valueNumber ofproteins inmodules
Number oftran-scribedprotein-encodinggenes
Description
M00601 0 0009 3 2 Putative chitobiose transport systemM00219 0001 0018 4 2 AI-2 transport systemM00211 0025 048 2 1 Putative ABC transport systemM00217 0038 0716 3 1 D-Allose transport systemM00210 0038 0716 3 1 Putative ABC transport systemM00605 005 0949 4 1 Glucosemannose transport systemM00206 005 0949 4 1 Cellobiose transport systemM00606 005 0949 4 1 NN1015840-Diacetylchitobiose transport systemM00197 005 0949 4 1 Putative fructooligosaccharide transport system
International Journal of Microbiology 7
T2_M
Taxonomic groupsexpressing glycosylases
NA
NA_FirmicutesTyzzerellaRoseburiaLachnoclostridiumBlautiaNA_ActinobacteriaCollinsellaBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Erysipelatoclostridium
Figure 5 Taxonomic profiles based on expression of the glycosy-lases genes in sample T2 M
when mapping rRNA reads from faecal samples to LGG 16SrRNA sequence at 97 similarity While only a few differentLGG-like functions could be detected in the transcriptome ofsample T1 in that of sample T2 M the expression of the LGG-like genes related to glycolysis is clearly visible indicative ofits high activity (Supplementary Figure 4) Since the sampleT1 represents the microbiota of an infant with breast-milk asthe main source of nutrients and whose mother consumedLGG containing products it may be speculated that LGGwas acquired during or soon after the birth from the motherAt later time points the abundance of LGG-like transcriptsdropped suggesting that LGG would then not permanentlycolonize the infant intestinal tract ecosystem However asthis coincides with the introduction of formula and solidfood one may speculate that these factors also contribute tothe clearance of LGG
4 Conclusions
We demonstrate here the feasibility of metatranscriptomeanalysis using high throughput RNA-Seq on the infant gutto get in-depth insight in the active community structurein the weaning period A relevant technical observation is
Relative number of rRNA reads mapping to LGG 16S
T1 T3T2_T T2_M0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped 0004
0005
(a)
0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped
0004
0005
T1
Relative number of mRNA reads mapping to LGG genes
T3T2_T T2_M
(b)
Figure 6 Relative numbers of mRNA (b) and rRNA (a) readmapping to LGG Error bars represent standard deviations based onbinomial sampling
that rRNA removal with Ribo-Zero kit (Epicentre) has apronounced effect on the rRNA distribution but not on thatof mRNA It was found that in the weaning period of thebreastfed infant a number of major activity changes occurredwith a decrease in activity of the Actinobacteria mainlyrepresented by Bifidobacterium and an increase in activity ofmembers of the Firmicutes increased in activity over timeMoreover the Firmicutes and Actinobacteria including thecanonical Bifidobacteria and Collinsella were found to beimportant contributors to vitamin production in the infantintestine including that of vitamin B6 B7 B9 and B12Finally the mother consumed the probiotic LGG and wedetected LGG-like transcripts in the infant transcriptomeindicating that LGG has been transferred is an active mem-ber at the earliest time point and is decreasing in abundanceand activity during weaning showing that LGG is not likelyto remain a permanent member of the intestinal microbiota
8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
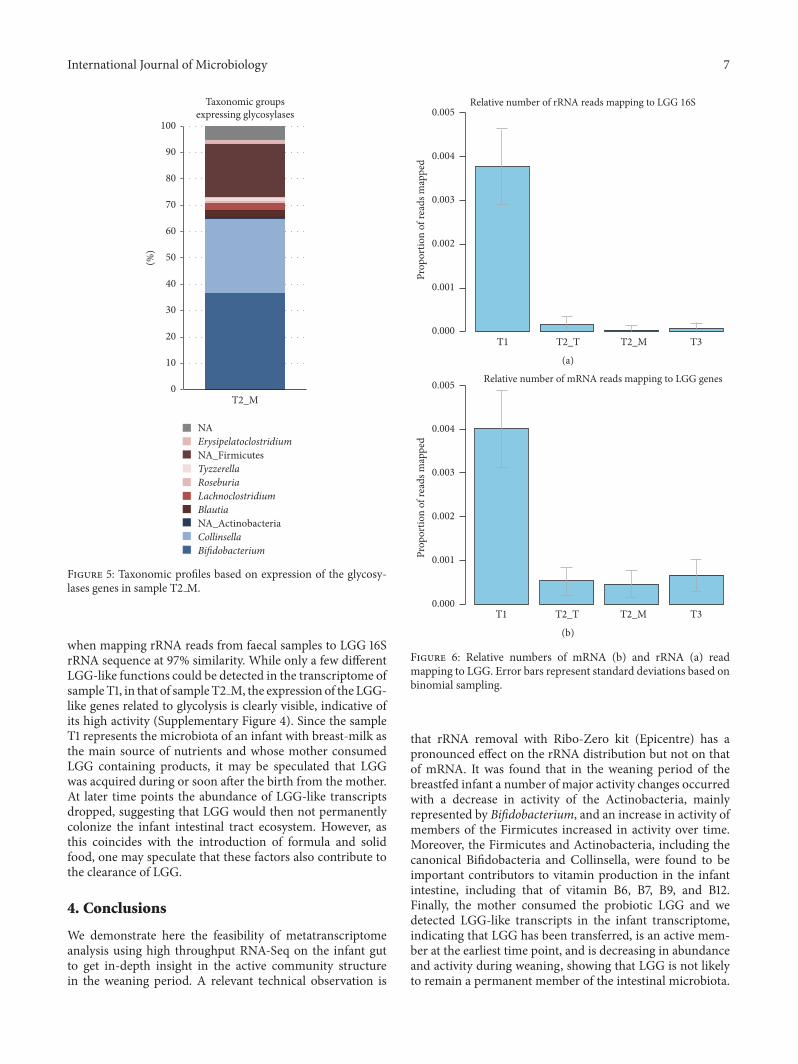
International Journal of Microbiology 7
T2_M
Taxonomic groupsexpressing glycosylases
NA
NA_FirmicutesTyzzerellaRoseburiaLachnoclostridiumBlautiaNA_ActinobacteriaCollinsellaBifidobacterium
0
10
20
30
40
50
60
70
80
90
100
()
Erysipelatoclostridium
Figure 5 Taxonomic profiles based on expression of the glycosy-lases genes in sample T2 M
when mapping rRNA reads from faecal samples to LGG 16SrRNA sequence at 97 similarity While only a few differentLGG-like functions could be detected in the transcriptome ofsample T1 in that of sample T2 M the expression of the LGG-like genes related to glycolysis is clearly visible indicative ofits high activity (Supplementary Figure 4) Since the sampleT1 represents the microbiota of an infant with breast-milk asthe main source of nutrients and whose mother consumedLGG containing products it may be speculated that LGGwas acquired during or soon after the birth from the motherAt later time points the abundance of LGG-like transcriptsdropped suggesting that LGG would then not permanentlycolonize the infant intestinal tract ecosystem However asthis coincides with the introduction of formula and solidfood one may speculate that these factors also contribute tothe clearance of LGG
4 Conclusions
We demonstrate here the feasibility of metatranscriptomeanalysis using high throughput RNA-Seq on the infant gutto get in-depth insight in the active community structurein the weaning period A relevant technical observation is
Relative number of rRNA reads mapping to LGG 16S
T1 T3T2_T T2_M0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped 0004
0005
(a)
0000
0001
0002
0003
Prop
ortio
n of
read
s map
ped
0004
0005
T1
Relative number of mRNA reads mapping to LGG genes
T3T2_T T2_M
(b)
Figure 6 Relative numbers of mRNA (b) and rRNA (a) readmapping to LGG Error bars represent standard deviations based onbinomial sampling
that rRNA removal with Ribo-Zero kit (Epicentre) has apronounced effect on the rRNA distribution but not on thatof mRNA It was found that in the weaning period of thebreastfed infant a number of major activity changes occurredwith a decrease in activity of the Actinobacteria mainlyrepresented by Bifidobacterium and an increase in activity ofmembers of the Firmicutes increased in activity over timeMoreover the Firmicutes and Actinobacteria including thecanonical Bifidobacteria and Collinsella were found to beimportant contributors to vitamin production in the infantintestine including that of vitamin B6 B7 B9 and B12Finally the mother consumed the probiotic LGG and wedetected LGG-like transcripts in the infant transcriptomeindicating that LGG has been transferred is an active mem-ber at the earliest time point and is decreasing in abundanceand activity during weaning showing that LGG is not likelyto remain a permanent member of the intestinal microbiota
8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

8 International Journal of Microbiology
The here described approach and pipeline allow for furtherlongitudinal analysis of infant activity to complement infantcohort metagenome studies that are presently developing
Disclosure
The abstract of this paper was presented before on theDarmendag (Gutflora day) 2015 in Rotterdam Netherlands
Conflicts of Interest
The authors declare that they have no conflicts of interest
Authorsrsquo Contributions
Lotta Nylund Reetta Satokari and Willem M de Vosdesigned the study and Lotta Nylund carried out the RNAextractions and preparation of the sequencing Mark Davidsand Jarmo Ritari preprocessed the RNA-Seq reads and JarmoRitari analysed the rRNA-Seq data Floor Hugenholtz anal-ysed the mRNA-Seq data Floor Hugenholtz and Willem Mde Vos drafted themanuscript All authors read and approvedthe final manuscript
Acknowledgments
This research was partly supported by ERC Advanced Grant250172 MicrobesInside from the European Research Coun-cil the Netherlands Organization for Scientific Research(Spinoza Award and SIAM Gravity Grant 024002002) andthe Finland Academy of Sciences (141130)
References
[1] D M Chu J Ma A L Prince K M Antony M D Seferovicand K M Aagaard ldquoMaturation of the infant microbiomecommunity structure and function across multiple body sitesand in relation to mode of deliveryrdquo Nat Med vol 23 no 3 pp314ndash326 2017
[2] M G Dominguez-Bello K M De Jesus-Laboy N Shen et alldquoPartial restoration of the microbiota of cesarean-born infantsvia vaginal microbial transferrdquo Nature Medicine vol 22 pp250ndash253 2016
[3] L Moles M Gomez H Heilig et al ldquoBacterial diversity inmeconium of preterm neonates and evolution of their fecalmicrobiota during the first month of liferdquo PLoS ONE vol 8 no6 Article ID e66986 2013
[4] H Wopereis R Oozeer K Knipping C Belzer and J KnolldquoThe first thousand days - intestinal microbiology of early lifeEstablishing a symbiosisrdquo Pediatric Allergy and Immunologyvol 25 no 5 pp 428ndash438 2014
[5] C F Favier E E VaughanWMDeVos andAD AkkermansldquoMolecular monitoring of succession of bacterial communitiesin human neonatesrdquo Appl Environ Microbiol vol 68 no 1 pp219ndash226 2002
[6] J E Koenig A Spor N Scalfone et al ldquoSuccession of microbialconsortia in the developing infant gutmicrobiomerdquo Proceedingsof the National Academy of Sciences of the United States ofAmerica vol 108 supplement 1 pp 4578ndash4585 2011
[7] F Backhed J Roswall Y Peng Q Feng H Jia P Kovatcheva-Datchary et al ldquoDynamics and stabilization of the human gutmicrobiome during the first year of liferdquo Cell Host and Microbevol 17 no 5 pp 690ndash703 2015
[8] J Cheng T Ringel-Kulka I Heikamp-de Jong Y Ringel ICarroll and W M de Vos ldquoDiscordant temporal developmentof bacterial phyla and the emergence of core in the fecalmicrobiota of young childrenrdquo ISME J 2015
[9] T Yatsunenko F E ReyM J Manary et al ldquoHuman gutmicro-biome viewed across age and geographyrdquo Nature vol 486 no7402 pp 222ndash227 2012
[10] A Marcobal M Barboza E D Sonnenburg et al ldquoBacteroidesin the infant gut consume milk oligosaccharides via mucus-utilization pathwaysrdquo Cell Host and Microbe vol 10 no 5 pp507ndash514 2011
[11] NOttmanH SmidtWM deVos andC Belzer ldquoThe functionof our microbiota who is out there and what do they dordquoFrontiers in Cellular and Infection Microbiology vol 2 p 1042012
[12] H Makino R Martin E Ishikawa et al ldquoMultilocus sequencetyping of bifidobacterial strains from infantrsquos faeces and humanmilk Are bifidobacteria being sustainably shared during breast-feedingrdquo Beneficial Microbes vol 6 no 4 pp 563ndash572 2015
[13] R K Buddington C HWilliams BM Kostek K K Budding-ton and M J Kullen ldquoMaternal-to-infant transmission of pro-biotics concept validation in mice rats and pigsrdquoNeonatologyvol 97 no 3 Article ID 000253756 pp 250ndash256 2010
[14] M Gueimonde S Sakata M Kalliomaki E Isolauri Y Bennoand S Salminen ldquoEffect of maternal consumption of lactobacil-lus GG on transfer and establishment of fecal bifidobacterialmicrobiota in neonatesrdquo J Pediatr Gastroenterol Nutr vol 42no 2 pp 166ndash170 2006
[15] M G Dominguez-Bello E K Costello M Contreras et alldquoDelivery mode shapes the acquisition and structure of theinitial microbiota across multiple body habitats in newbornsrdquoProceedings of the National Academy of Sciences vol 107 no 26pp 11971ndash11975 2010
[16] L Nylund ldquoEarly Life Intestinal Microbiota in Health and inAtopic Eczemardquo Tech Rep 2015
[17] E S Klaassens R J BoestenMHaarman et al ldquoMixed-speciesgenomic microarray analysis of fecal samples reveals differen-tial transcriptional responses of bifidobacteria in breast- andformula-fed infantsrdquo Appl Environ Microbiol vol 75 no 9 pp2668ndash2676 2009
[18] F Asnicar S Manara M Zolfo D T Truong M Scholz andF Armanini ldquoStudying vertical microbiome transmission frommothers to infants by strain-level metagenomic profilingrdquomSystems vol 2 no 1 pp e00164ndash00116 2017
[19] C CGM Booijink J Boekhorst EG ZoetendalH SmidtMKleerebezem andWM deVos ldquoMetatranscriptome analysis ofthe human fecal microbiota reveals subject-specific expressionprofiles with genes encoding proteins involved in carbohydratemetabolism being dominantly expressedrdquoApplied and Environ-mental Microbiology vol 76 no 16 pp 5533ndash5540 2010
[20] E A Franzosa X C Morgan N Segata et al ldquoRelating themetatranscriptome and metagenome of the human gutrdquo Pro-ceedings of the National Academy of Sciences of the United Statesof America vol 111 no 22 pp E2329ndashE2338 2014
[21] M J Gosalbes A Durban M Pignatelli et al ldquoMetatranscrip-tomic approach to analyze the functional human gut micro-biotardquo PLoS ONE vol 6 no 3 Article ID e17447 2011
International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

International Journal of Microbiology 9
[22] E G Zoetendal H G Heilig E S Klaassens et al ldquoIsolationof DNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 870ndash873 2006
[23] E G Zoetendal C C Booijink E S Klaassens et al ldquoIsolationof RNA from bacterial samples of the human gastrointestinaltractrdquo Nature protocols vol 1 no 2 pp 954ndash959 2006
[24] M Davids F Hugenholtz V Martins dos Santos H Smidt MKleerebezem and P J Schaap ldquoFunctional profiling of unfamil-iar microbial communities using a validated de novo assemblymetatranscriptome pipelinerdquo PLoS One vol 11 no 1 Article IDe0146423 2016
[25] S He O Wurtzel K Singh et al ldquoValidation of two ribosomalRNA removal methods for microbial metatranscriptomicsrdquoNature Methods vol 7 no 10 pp 807ndash812 2010
[26] E Kopylova L Noe and H Touzet ldquoSortMeRNA Fast andaccurate filtering of ribosomal RNAs in metatranscriptomicdatardquo Bioinformatics vol 28 no 24 pp 3211ndash3217 2012
[27] C deWeerth S Fuentes P Puylaert andWM de vos ldquoIntesti-nal microbiota of infants with colic development and specificsignaturesrdquo Pediatrics vol 131 no 2 pp e550ndashe558 2013
[28] M Kankainen L Paulin S Tynkkynen et al ldquoComparativegenomic analysis of Lactobacillus rhamnosus GG reveals pilicontaining a human-mucus binding proteinrdquo Proceedings of theNational Academy of Sciences of the United States of Americavol 106 no 40 pp 17193ndash17198 2009
[29] F P Douillard A Ribbera R Kant et al ldquoComparative Ge-nomic and Functional Analysis of 100 Lactobacillus rhamnosusStrains and Their Comparison with Strain GGrdquo PLoS Geneticsvol 9 no 8 Article ID e1003683 2013
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
Submit your manuscripts athttpswwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 201
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology