files.shareholder.comfiles.shareholder.com/downloads/amda-mfnla/0x0xs1047469-15-151… · use these...
TRANSCRIPT

KYTHERA BIOPHARMACEUTICALS INC
FORM 10-K(Annual Report)
Filed 03/02/15 for the Period Ending 12/31/14
Address 27200 WEST AGOURA ROADSUITE 200CALABASAS, CA 91301
Telephone 818-587-4500CIK 0001436304
Symbol KYTHSIC Code 2834 - Pharmaceutical Preparations
Industry ConglomeratesSector Conglomerates
Fiscal Year 12/31
http://www.edgar-online.com© Copyright 2015, EDGAR Online, Inc. All Rights Reserved.
Distribution and use of this document restricted under EDGAR Online, Inc. Terms of Use.

Use these links to rapidly review the document TABLE OF CONTENTS PART IV KYTHERA BIOPHARMACEUTICALS, INC.
Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
Commission file number: 001-35663
KYTHERA Biopharmaceuticals, Inc. (Exact Name of Registrant as Specified in its Charter)
(818) 587-4500 (Registrant's Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes � No �
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes � No �
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes � No �
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes � No �
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. �
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act:
Delaware (State or Other Jurisdiction of Incorporation or Organization)
03-0552903 (I.R.S. Employer
Identification No.)
30930 Russell Ranch Road, 3 rd Floor
Westlake Village, California (Address of Principal Executive Offices)
91362
(Zip Code)
(Mark One) �
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2014
OR
�
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to

Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes � No �
The aggregate market value of common stock held by non-affiliates of the Registrant as of June 30, 2014 (the last business day of the Registrant's most recently completed second quarter) was $674,448,828.
As of February 23, 2015, the number of outstanding shares of the registrant's common stock, par value $0.00001 per share, was 22,755,124.
DOCUMENTS INCORPORATED BY REFERENCE
Certain sections of the registrant's definitive Proxy Statement for the 2015 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission pursuant to Rule 14A not later than 120 days after end of this fiscal year covered by this Form 10-K are incorporated by reference into Part III of this Form 10-K.
Large accelerated filer � Accelerated filer � Non-accelerated filer �
(Do not check if a smaller reporting company)
Smaller reporting company �

Table of Contents
TABLE OF CONTENTS
i
Page PART I
Item 1. Business 1 Item 1A. Risk Factors 42 Item 1B. Unresolved Staff Comments 73 Item 2. Properties 73 Item 3. Legal Proceedings 73 Item 4. Mine Safety Disclosures 73
PART II Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of
Equity Securities
74 Item 6. Selected Financial Data 76 Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations 78 Item 7A. Quantitative and Qualitative Disclosures About Market Risk 92 Item 8. Consolidated Financial Statements and Supplementary Data 93 Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure 93 Item 9A. Controls and Procedures 93 Item 9B. Other Information 95
PART III Item 10. Directors, Executive Officers and Corporate Governance 95 Item 11. Executive Compensation 95 Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder
Matters
95 Item 13. Certain Relationships and Related Transactions, and Director Independence 95 Item 14. Principal Accounting Fees and Services 95
PART IV Item 15. Exhibits, Financial Statement Schedules 96
SIGNATURES 97

Table of Contents
Forward-Looking Statements
This Annual Report on Form 10-K, including "Business" in Part I Item I and "Management's Discussion and Analysis of Financial Condition and Results of Operations" in Part II Item 7, contains "forward-looking statements" within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Such forward-looking statements are subject to risks, uncertainties and other factors that could cause the results of KYTHERA Biopharmaceuticals, Inc. (KYTHERA or the Company) and the timing of certain events to differ materially from future results expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be deemed forward-looking statements. These statements are often identified by the use of words such as "may," "will," "expect," "believe," "anticipate," "intend," "could," "should," "estimate," or "continue," and similar expressions or variations. The risks and uncertainties referred to above include, without limitation, risks related to our research and development efforts, need for future capital, timely completion of our clinical trials, uncertainty of clinical trial results or regulatory approvals or clearances, manufacturing of our product candidates at scales and costs appropriate for commercialization, enforcement of our patent and proprietary rights, reliance upon our collaborative partners, potential competition and other risks that are described herein and that are otherwise described from time to time in KYTHERA's Securities and Exchange Commission reports including, but not limited to, the factors described in Item 1A, "Risk Factors," of this Annual Report. KYTHERA assumes no obligation and does not intend to update these forward- looking statements.
PART I
ITEM 1. Business.
Overview
We are a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of novel prescription products for the aesthetic medicine market. Our objective is to develop first-in-class, prescription products using an approach that relies on the scientific rigor of biotechnology to address unmet needs in the rapidly-growing market for aesthetic medicine. Our initial focus is on the facial aesthetics market, which comprises the majority of the aesthetic medicine market. Our product candidate, ATX-101, is a potential first-in-class, injectable drug in late-stage clinical development for the treatment of submental fullness associated with submental fat, which commonly presents as an undesirable "double chin." Based on clinical trials conducted to date, ATX-101 has exhibited significant, meaningful and long-lasting results in the reduction of submental fat. These results correspond with patient satisfaction measures demonstrating meaningful improvement in perceived chin appearance. If approved by applicable regulatory authorities, we believe ATX-101 will be an attractive solution for the treatment of submental fullness, representing a new product category within the rapidly growing facial aesthetics market.
In 2013, consumers spent nearly $12.0 billion on 11.0 million physician-administered aesthetic procedures, according to the American Society of Aesthetic Plastic Surgery, or ASAPS. Additionally, ASAPS estimates that from 1997 to 2013, surgical aesthetic procedures increased by more than 89%, and non-surgical procedures increased by 521%, reflecting increasing demand for aesthetic procedures over more than a decade. The primary driver of this procedural growth is the demand for facial injectables, such as botulinum toxins, which increased 15.6% from the prior year, and dermal fillers, which increased 31.5% from the prior year. Injectables overall saw a 21% increase in 2013. While botulinum toxins and dermal fillers have created a market for non-surgical facial rejuvenation of the upper and mid-face as Food and Drug Administration, or FDA, approved injectables, liposuction and other surgical procedures remain the only proven treatment options for submental fat reduction. Although effective at fat reduction, these invasive procedures often involve significant pain, downtime and expense, as well as risks associated with invasive surgery.
1

Table of Contents
We believe ATX-101 will be an attractive non-surgical solution for the treatment of submental fullness. ATX-101 is a proprietary formulation of a purified synthetic version of deoxycholic acid, a naturally occurring molecule in the body that aids in the breakdown of dietary fat. ATX-101 treatment contours the area under the chin by destroying fat cells while leaving surrounding tissue largely unaffected. Our Phase II and Phase III studies have demonstrated that ATX-101, when injected subcutaneously into the target fat deposit, reduces the localized fat deposit while leaving the surrounding tissue largely unaffected.
In March 2014, we announced the acquisition of rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer Consumer Care AG ("Bayer"), our former collaborator. We originally entered into a collaboration arrangement with Bayer in August 2010, pursuant to which Bayer was to develop and commercialize ATX-101 for all indications outside the United States and Canada. Under the collaboration arrangement, we received an aggregate of $76.8 million from Bayer. This was comprised of an upfront license fee of $21.3 million and $22.2 million for Phase III development activities, followed by $15.8 million contingent event payment and an additional $17.4 million for further development activities in connection with Bayer's decision to pursue continued development and regulatory approval for ATX-101 after receipt of positive results from the European Phase III clinical trials for ATX-101 in 2012. Under the Assignment and Novation Agreement between Bayer and KYTHERA Holdings Ltd. ("KHL"), a wholly-owned Bermuda subsidiary of KYTHERA, Bayer transferred all of its right, title, and interest in the Amended License Agreement, and assigned all of its rights and delegated all of its obligations, liabilities and duties thereunder, to KHL for $84.0 million comprised of $33.0 million in KYTHERA common stock, plus a note for $51.0 million which bears interest at 5%, payable no later than 2024. Bayer is also eligible to receive certain long-term sales milestone payments on annual sales outside of the United States and Canada.
In the United States and Canada, we conducted two pivotal Phase III trials of ATX-101 for the reduction of submental fat. We initiated this pivotal Phase III clinical program in March 2012 and completed enrollment of 1,022 patients in these trials in August 2012. In these multi-center, randomized, double-blind, placebo-controlled pivotal trials, ATX-101 was found to be well tolerated and resulted in a statistically significant reduction in submental fat, as assessed by validated clinician and patient scales. These scales were validated using scientific principles and process recommendations per the FDA's Patient Reported Outcome Guidance (FDA, 2009) in an effort to ensure reliability, construct validity and sensitivity to change over time, and are similar to other rating scales used for approved aesthetic drug products and medical devices, such as botulinum toxins and dermal fillers. ATX-101 also demonstrated statistically significant reduction in submental volume using MRI in a subset of patients. In addition, treatment with ATX-101 resulted in improvements in patients' self-perception related to the appearance of their "chin fat" as measured by a patient-reported treatment impact scale.
We reported positive topline results in September 2013. These trials formed the basis for our ATX-101 New Drug Application, or NDA, which we submitted in May 2014. The Dermatology and Ophthalmic Drugs Advisory Committee of the FDA is scheduled to review KYTHERA's NDA for ATX-101 (deoxycholic acid) for "improvement in the appearance of moderate to severe submental fullness" on March 9, 2015. The NDA will be subject to a standard review and has a Prescription Drug User Fee Act (PDUFA) action date of May 13, 2015. In addition, in August 2014, we filed a New Drug Submission (NDS) to Health Canada seeking approval for ATX-101 (deoxycholic acid), as a potential first-in-class treatment of submental fullness. In October 2014 we submitted a marketing authorization application, or MAA, in Switzerland and in February 2015 we submitted a drug application to Australia's Therapeutic Goods Administration (TGA) for ATX-101 as a potential first-in-class treatment of submental fullness.
KYTHERA expects to make additional ex-United States regulatory submissions in 2015.
2
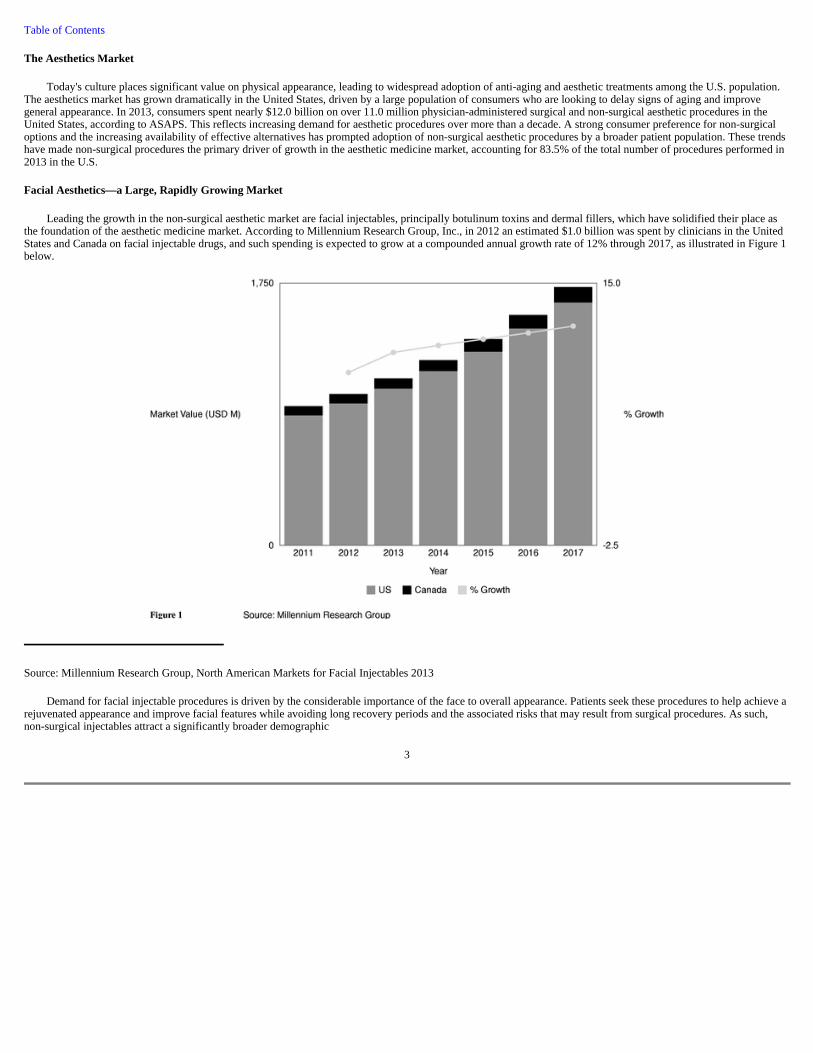
Table of Contents
The Aesthetics Market
Today's culture places significant value on physical appearance, leading to widespread adoption of anti-aging and aesthetic treatments among the U.S. population. The aesthetics market has grown dramatically in the United States, driven by a large population of consumers who are looking to delay signs of aging and improve general appearance. In 2013, consumers spent nearly $12.0 billion on over 11.0 million physician-administered surgical and non-surgical aesthetic procedures in the United States, according to ASAPS. This reflects increasing demand for aesthetic procedures over more than a decade. A strong consumer preference for non-surgical options and the increasing availability of effective alternatives has prompted adoption of non-surgical aesthetic procedures by a broader patient population. These trends have made non-surgical procedures the primary driver of growth in the aesthetic medicine market, accounting for 83.5% of the total number of procedures performed in 2013 in the U.S.
Facial Aesthetics—a Large, Rapidly Growing Market
Leading the growth in the non-surgical aesthetic market are facial injectables, principally botulinum toxins and dermal fillers, which have solidified their place as the foundation of the aesthetic medicine market. According to Millennium Research Group, Inc., in 2012 an estimated $1.0 billion was spent by clinicians in the United States and Canada on facial injectable drugs, and such spending is expected to grow at a compounded annual growth rate of 12% through 2017, as illustrated in Figure 1 below.
Source: Millennium Research Group, North American Markets for Facial Injectables 2013
Demand for facial injectable procedures is driven by the considerable importance of the face to overall appearance. Patients seek these procedures to help achieve a rejuvenated appearance and improve facial features while avoiding long recovery periods and the associated risks that may result from surgical procedures. As such, non-surgical injectables attract a significantly broader demographic
3

Table of Contents
compared to surgical interventions. ASAPS reports that younger patients are increasingly opting for facial injectable treatments to delay the appearance of lines and wrinkles. In addition, older men are increasingly trying to minimize and delay the signs of aging and are seeking facial injectable treatments, a market that has traditionally been dominated by female patients. In the United States, facial injectable procedures represented 51.6% of all physician-administered aesthetic procedures in 2013, and are the fastest growing segment of the aesthetic medicine market, according to ASAPS.
According to ASAPS, approximately 7.2 million facial injectable procedures were performed in the United States in 2013. Based on ASAPS reports and other industry sources, we estimate that approximately 2.1 million patients received botulinum toxin and dermal filler procedures in 2013. The number of facial injectable procedures in North America is expected to grow at a compounded annual growth rate of over 12% through 2017, according to industry sources. We believe this projected growth in the facial injectable market will be driven by a number of trends including:
• high patient demand for non-invasive aesthetic procedures;
• positive media promotion leading to heightened patient awareness and education and expanding patient pool/demographics;
• affordability (relative to invasive surgical aesthetic procedures) driven by competitive pricing pressures;
• more indications for current products, new product approvals and emerging alternative technologies;
• increasing popularity of receiving several treatments in combination; and
• more physician specialties performing facial injectable procedures.
Beyond the United States and Canada, the facial aesthetics market is also growing at a significant rate, with industry sources projecting the European facial injectable market to reach an annual growth rate of nearly 8% by 2017. We believe the long-term aggregate potential of ex-U.S. markets will approximate the North American market.
Patient and Physician Preference for Facial Injectables
We believe several factors contribute to patients' increased preference for facial injectables, including:
• significant and predictable results;
• non-surgical alternative;
• minimal or no downtime; and
• high-degree of patient satisfaction.
In addition to offering features that are desirable to patients, facial injectable treatments also deliver benefits that are attractive to physicians, including:
• ease of injectable administration, with office visits typically lasting 15 to 20 minutes;
• high-degree of patient satisfaction leading to patient trust and loyalty;
• ability to command premium procedure fees to reflect physician expertise and artistry;
• patient self-pay, insulating physicians from medical reimbursement risk;
• no capital equipment requirements and limited upfront costs; and
• demonstrated safety and efficacy supported by FDA approval.
4

Table of Contents
We believe these benefits will drive rapid adoption of new FDA-approved injectable treatments among dermatologists and plastic surgeons, who together perform a significant majority of these procedures.
Unmet Need for Injectable Therapy For Double Chin
Botulinum toxins and dermal fillers have created a market for non-surgical facial rejuvenation of the upper and mid-face. Despite this, undesirable submental fullness, or "double chin," remains an important yet unaddressed aesthetic target for injectable facial treatments. The following graphic depicts the areas of the face in which currently-approved injectable products are used, and highlights the lack of treatments for undesirable submental fullness.
(1) American Society of Aesthetic Plastic Surgery, ASAPS, 2013
5

Table of Contents
According to the 2014 American Society for Dermatologic Surgery (ASDS) Consumer Survey on Cosmetic Dermatologic Procedures, 68% of consumers were somewhat to extremely bothered by excess fat under the chin.
Despite the patient and physician shift in favor of non-surgical and injectable procedures, there are no FDA-approved drugs for the treatment of submental fullness, and liposuction and other surgical procedures remain the only proven treatment options. Although effective at fat reduction, these procedures often involve significant pain, downtime and expense, and involve the risks associated with invasive surgery.
Our Injectable Solution for Submental Fullness
ATX-101 is an injectable, facial treatment that we are initially developing for treatment of submental fullness. Excess submental fat has been found to impact adversely a patient's self-perception. If approved by applicable regulatory authorities, we believe ATX-101 will be an attractive non-surgical solution for the treatment of submental fullness, representing a new product category within the rapidly growing facial aesthetics market, for the following reasons:
• Significant, meaningful and durable results. Our clinical studies demonstrated a marked reduction in submental fat, which corresponded with patient satisfaction measures demonstrating meaningful improvement in perceived chin appearance. As is consistent across our U.S. Phase IIb study, our pivotal U.S. and Canadian Phase III trials, and the pivotal European Phase III trials, ATX-101 demonstrated statistically significant reduction of submental fat in all three efficacy measures: patient assessment, physician assessment and objective measures (magnetic resonance imaging, or MRI, in the U.S. and Canadian Phase III trials and Phase IIb study, and calipers in the European Phase III trials). In addition, the results from our U.S. and Canadian Phase III trials and the European Phase III trials demonstrated that treatment with ATX-101 resulted in a statistically significant improvement in patient happiness with the appearance of their "chin fat" and associated self-perception of youthfulness, as well as a reduction in feeling embarrassed, bothered and self-conscious about the appearance of their "chin fat." Further, an interim analysis of our long-term follow-up study monitoring Phase II patients who received ATX-101 treatment and were responders in their predecessor study indicates that 87% sustained or improved their response (defined as ≥ 1 grade change from baseline on the Clinician-Reported Submental Fat Rating Scale, or CR-SMFRS) at four years (n=33). As is consistent with previous findings, no long-term safety concerns were noted in this interim analysis.
• Ready group of target patients. Our market research surveys indicated that 78% of existing botulinum toxin or dermal filler patients had excess treatable submental fat and that 89% of dermal filler patients were also botulinum toxin patients. Taken together, we believe these results indicate that patients who have undergone, or are currently receiving facial injectable treatments, are highly likely to adopt new, similar procedures. Our market research in 385 patients receiving botulinum toxin or dermal fillers indicated that 61% of such patients were likely to try ATX-101 after reading the product profile. Accordingly, we believe that there is a large, readily addressable market of experienced patients, already in our target physicians' practices, who are likely to adopt an injectable facial treatment for submental fullness to enhance their facial appearance.
• Low barrier to physician adoption. If approved, we expect ATX-101 to be an easily adopted solution for the treatment of submental fullness. We believe the short treatment sessions and ease of administration should facilitate physician adoption of ATX-101 as a natural complementary therapy for physicians already administering botulinum toxins and dermal fillers. Unlike some aesthetic procedures that require a physician practice to acquire and make space
6

Table of Contents
for often expensive and bulky capital equipment, ATX-101 will not require capital equipment. In addition, given the patient self-pay model in aesthetic medicine, physicians receive procedure fees from patients at the time of service and are not exposed to potential reimbursement risks.
• Facilitates physician practice expansion. We believe ATX-101 will provide dermatologists and plastic surgeons an opportunity to expand the scope and reach of their current aesthetic medicine practices. If approved, ATX-101 for the treatment of submental fullness will be positioned as a naturally complementary procedure for experienced toxin and dermal filler patients, while potentially being an attractive point of entry for new patients that have never received an aesthetic procedure. For example, our market research indicates that men have a significantly higher interest in an injectable solution for submental fat reduction versus other facial injectable treatment options, but according to ASAPS, only comprised 9% of the facial injectable market in 2013. We believe ATX-101 provides an opportunity for dermatologists and plastic surgeons to address a broader patient demographic within the facial aesthetics market while also expanding the scope of treatments they can offer to their patient base.
• A favorable safety profile with no or limited downtime. ATX-101 has been well tolerated across all clinical studies completed to date, with the most notable side effects being local to the treatment area and typically mild to moderate in severity. Treatment with ATX-101 also avoids the inherent risks and recovery time associated with general anesthesia and invasive surgical methods. Together, these attributes could make ATX-101 an aesthetic option with limited or no downtime with most patients able to resume normal daily activity immediately after treatment.
We believe ATX-101, if approved, has the potential to reach peak year annual sales of $500 million in the United States.
Our Strategy
In order to achieve our objective of developing first-in-class, non-surgical, prescription products in aesthetic medicine, our near- and long-term strategies include:
Near-term:
• Develop and commercialize ATX-101 in the United States and Canada for the non-surgical treatment of submental fullness. We believe ATX-101 will satisfy a significant unmet need and view it as a beachhead for our entry into the aesthetics market. If we are successful in receiving regulatory approval, we expect to build a sales organization to commercialize ATX-101 in the United States and Canada. We believe this would include establishing a specialty field sales force initially focused on core aesthetic dermatologists and plastic surgeons.
• Maximize the strategic global value of ATX-101. We will develop our global business model and a long-term global strategy to maximize the value of ATX-101 on a territory-by-territory basis.
• Assess and prioritize future indications for ATX-101. While we are initially developing ATX-101 for the treatment of submental fullness, we expect to assess future potential treatment indications for ATX-101. Over time we may develop ATX-101 for the reduction of other small, localized fat deposits with high aesthetic value.
Long-term:
• Build KYTHERA into a leading aesthetics company. We are focused on high-value, self-pay aesthetic products that yield high patient satisfaction. We expect that ATX-101 will serve as a foundation for us to develop a franchise of aesthetic products. We intend to leverage ATX-101 to build KYTHERA into a leading biopharmaceutical company.
7

Table of Contents
• Leverage our biotechnology and aesthetics experience. We strive to attract, retain and incentivize a unique team with extensive biotech and aesthetic medicine capabilities to create an environment of high achievement. Our management team, with significant biotechnology and aesthetics experience, played a leadership role in securing FDA approval of and successfully commercializing marquee aesthetic products, such as BOTOX® and JUVEDERM®, and in successfully commercializing established biotechnology products such as Aranesp®, NEUPOGEN® and Neulasta®.
• Apply rigorous clinical, regulatory and scientific practices to the development of novel, FDA-regulated, aesthetic medicine product candidates such as ATX-101. We were founded on the premise of combining the scientific and clinical rigor of biotechnology with the attractive market potential of aesthetics. We believe applying scientific rigor to our product candidates allows both patients and physicians to be confident that the product is efficacious and safe based on clinical data.
• Be capital efficient and opportunistically expand our product portfolio. Our capital deployment decisions are an integral part of executing our strategy. To date, we have advanced ATX-101 through Phase III clinical development by taking a disciplined approach that utilizes a network of clinical research organizations and contract manufacturers. We are focused on differentiated opportunities in aesthetics, and have been disciplined in our evaluation of opportunities to date. We will continue to search for new opportunities and intend to advance only those product candidates that meet our criteria for innovation, novelty and large addressable markets.
• Continue to build a robust and defensible patent portfolio. Our ATX-101 patent estate consists, on a worldwide basis, of over 100 issued or allowed patents, including foreign counterparts, and over 100 additional pending patent applications. We will continue to aggressively pursue additional patent protection as well as take appropriate measures to obtain and maintain proprietary protection for our innovative technologies.
Our Drug Candidate: ATX-101
If approved, we believe ATX-101 will be a first-in-class submental contouring injectable drug. We have completed Phase III clinical trials and submitted our NDA, and have made regulatory filings outside the United States, in Canada, Switzerland and Australia. ATX-101 (deoxycholic acid [DCA] injection) is a first-in-class, injectable drug that has been developed for the treatment of submental fullness. It has been observed to be well tolerated in Phase I, II and III studies. Deoxycholic acid has a long track record of safe administration to humans and is a common excipient in the formulation of several marketed drug products.
Mechanism of Action
ATX-101 is a proprietary formulation of purified synthetic deoxycholic acid (non-animal derived) available as a sterile, ready-to-use injectable drug. When injected subcutaneously, ATX-101 results in focal adipocytolysis (the destruction of fat cells) and a predicted local tissue response resulting in a reduction in submental fat with unchanged or improved skin laxity in most patients.
Nonclinical Program
Although DCA is a well-characterized endogenous substance, KYTHERA completed a comprehensive panel of nonclinical studies to characterize the toxicity and safety profile of ATX-101, including:
1) In vitro and in vivo pharmacology studies (nonclinical efficacy);
2) Cardiovascular, respiratory and central nervous system safety pharmacology studies;
8

Table of Contents
3) Absorption, distribution and toxicokinetic studies;
4) Single-dose through chronic repeat-dose toxicity, reproductive and genetic toxicology studies;
5) Synthetic DCA and animal-derived sodium deoxycholate (NaDC) bridging toxicity studies; drug product formulation bridging toxicity studies; and impurity evaluations.
Overall, the results of the extensive nonclinical program encompassing pharmacologic, pharmacokinetic (PK), and toxicological evaluations support the safe use of ATX-101 at the intended maximum dosage for the treatment of submental fullness.
Clinical Development Program
To support approval of ATX-101, we are pursuing a rigorous, science-based clinical development program. ATX-101 is the subject of four pivotal Phase III clinical trials for the reduction of submental fat, consisting of two completed trials in the United States and Canada, with total enrollment of 1,022 patients, and two completed trials in Europe that involved 723 total patients. These clinical trials were designed based on promising results observed in three multi-center, double-blind, placebo-controlled Phase II studies that evaluated various dosing regimens for ATX-101 and also served to validate potential efficacy endpoints, including patient and physician submental fat rating scales. Altogether, a total of 681 patients have participated in 12 Phase I and Phase II clinical studies of ATX-101 for the treatment of submental fat reduction including two early studies for the treatment of superficial lipomas, a common type of benign fatty tumor found under the skin. This included a 218-patient cardiac safety (QT/QTc) Phase I study that met the pre-specified FDA agreed primary endpoint, demonstrating that ATX-101, at and above therapeutic levels, did not prolong the rate corrected QT interval in healthy individuals. An additional open-label long-term safety study of ATX-101 for the reduction of submental fat was completed in 165 patients, demonstrating ATX-101 was well tolerated and may be effective in reducing SMF by both clinician- and patient-reported outcome measures. Overall, subcutaneous treatment with ATX-101 has been observed to be well tolerated across all clinical studies to date.
Clinical development of ATX-101 in the United States is being conducted under an Investigational New Drug Application, or IND, that was opened in December 2007 and is currently active with the FDA. In Canada, Europe and other territories, clinical development is being conducted under similar national clinical trial applications.
Phase I Clinical Studies
Seven Phase I studies were conducted in a total of 319 patients and evaluated safety, tolerability, pharmacokinetics and/or histopathology, and cardiac safety study following administration of ATX-101, as described below:
Pharmacokinetic studies (2008-2009, 2011, 2012). Three Phase I studies have been conducted to evaluate the pharmacokinetics of ATX-101. The first study was an open-label, single-center, dose-escalation, safety study and was conducted in 24 patients. Each patient received a single ATX-101 treatment at 0-4mg/cm 2 , given subcutaneously into the submental fat, using a variety of concentrations, volumes and injection grid patterns. Treatment with ATX-101 was well tolerated. The most common adverse effects were mild-to-moderate local injection site reactions. Peak deoxycholate blood levels occurred within about 30 minutes after administration of ATX-101 and were dose-related. At all doses, deoxycholate returned to endogenous levels within 12 to 24 hours. The second study was an open-label, single-center, cross-over study in five patients that compared blood levels of deoxycholate following injection of ATX-101 into the abdomen or submental fat. Comparable overall exposure was observed when ATX-101 was injected into the two locations,
9

Table of Contents
although peak exposure was somewhat higher following injection into submental fat. The third study was an open-label, single-center study conducted in 24 patients to determine the pharmacokinetics of injecting a single dose of ATX-101 in its final proposed commercial formulations. Each patient received a single ATX-101 treatment at 2 mg/cm 2 , given subcutaneously into the submental fat. No serious adverse events were reported and no subjects were discontinued due to adverse events.
Histology study (2008-2009). In this open-label, vehicle-controlled, multi-center study, various doses of ATX-101 were injected into the abdominal fat of 14 patients who were scheduled to undergo abdominoplasty at differing time points. Histological evaluation of the excised fat tissue demonstrated the initial destruction of adipocytes followed by a local immune response and thickening of fibrous septae within the fat tissue consistent with the proposed mechanism of action of ATX-101.
Lipid study (2010-2011). This 10-patient, open-label, single-center study evaluated blood levels of lipids and other biological markers following administration of ATX-101. Results confirmed that adipocytolysis induced by ATX-101 did not result in adverse levels of serum lipids (including cholesterol, triglycerides and free fatty acids) or metabolic and inflammatory markers.
Formulation tolerability study (2010). The comfort and tolerability of two formulations of ATX-101, one with and one without a benzyl alcohol preservative, were compared in 24 patients in this single-dose study. Both formulations were considered acceptable for use in future studies. We intend to use the benzyl alcohol formulation for the U.S. and Canadian markets.
Cardiac safety study (2013). This 218-patient cardiac safety study of ATX-101, known as the QT/QTc study, met the pre-specified FDA agreed primary endpoint, demonstrating that ATX-101, at and above therapeutic levels, did not prolong the QTc interval in healthy individuals. Further, the study found no relationship between QTc interval and plasma ATX-101 concentrations.
Clinical Development in Reduction of Submental Fat
The following key measures were developed, and assessed in Phase II and Phase III clinical trials:
• Clinician-Reported Submental Fat Rating Scale (CR-SMFRS): a physician assessment of the prominence and convexity of submental fat on a 5-point ordinal scale (0-none, 1-mild, 2-moderate, 3-severe, 4-extreme).
• Patient-Reported Submental Fat Rating Scale (PR-SMFRS): a patient self-assessment of the amount of their "chin fat" on a 5-point ordinal scale (0-none, 1-slight, 2-moderate, 3-large, 4-very large).
• Subject-Self Rating Scale (SSRS): a patient self-assessment of satisfaction with the appearance of their face and chin on a 7-point ordinal scale (0-extremely dissatisfied, 1-dissatisfied, 2-somewhat dissatisfied, 3-neither satisfied nor dissatisfied, 4-somewhat satisfied, 5-satisfied, 6-extremely satisfied) when asked the following question:
"...how satisfied do you feel with your appearance at the present time whether or not in your judgment it is due to treatment with ATX-101?"
• Patient-Reported Submental Fat Impact Scale (PR-SMFIS): a patient self-assessment, each on a 10-point ordinal scale, of their perception measuring: • How happy are you with the appearance with your chin fat?
• How bothered are you by the appearance of your chin fat?
• How self-conscious are you about the appearance of your chin fat?
• How embarrassed are you about the appearance of your chin fat?
10

Table of Contents
• How much older do you look because of your chin fat?
• How much overweight do you look because of your chin fat?
• Patient Satisfaction with Treatment: a patient self-assessment of satisfaction with treatment measured on a 7-point ordinal scale (0-extremely dissatisfied, 1-moderately dissatisfied, 2-a little dissatisfied, 3-neither satisfied nor dissatisfied, 4-a little satisfied, 5-moderately satisfied, 6-extremely satisfied).
• Submental Skin Laxity Grade (SMSLG): a physician assessment of submental wrinkling, skin folding and apposition to underlying neck structures on a 4-point photo-numeric grading scale (1-none, 2-mild, 3-moderate, 4-severe).
• Change in Submental Volume via Magnetic Resonance Imaging, or MRI: an objective measure of submental volume within a 1 cm wide sagittal slice acquired through MRI measured in cubic millimeters (mm 3 ).
• Change in Submental Fat Thickness via Calipers: an objective measure of submental fat thickness measured in millimeters (mm).
11
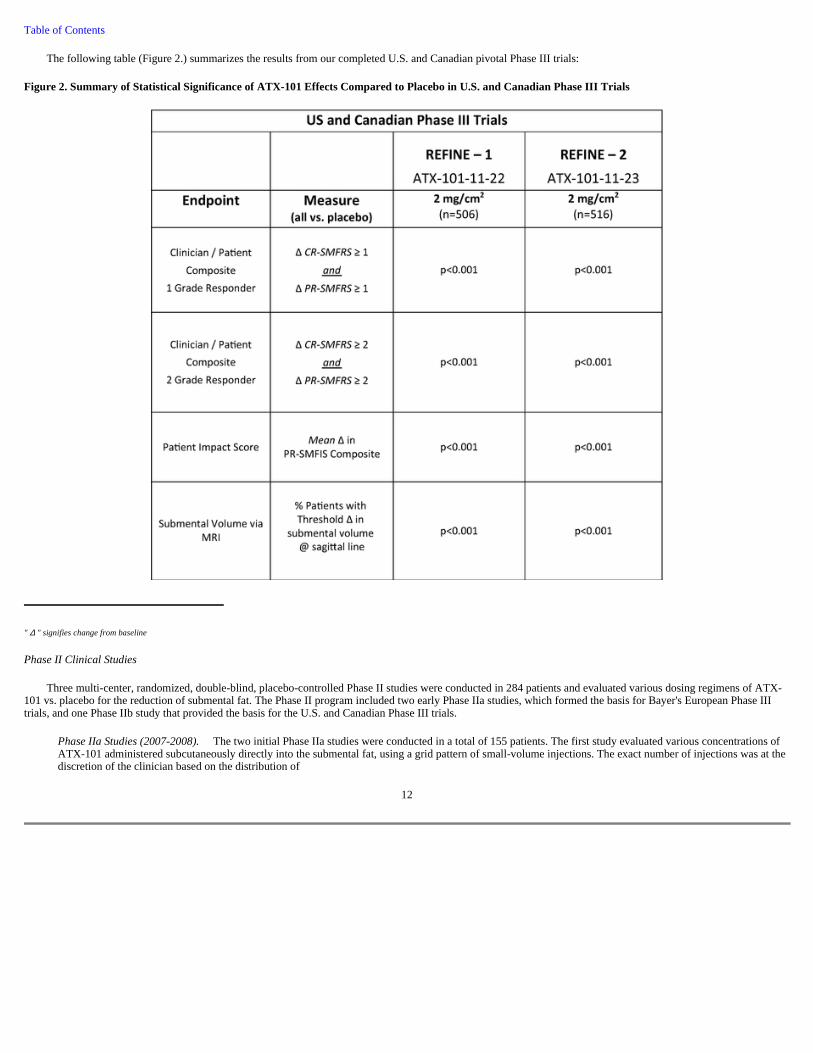
Table of Contents
The following table (Figure 2.) summarizes the results from our completed U.S. and Canadian pivotal Phase III trials:
Figure 2. Summary of Statistical Significance of ATX-101 Effects Compared to Placebo in U.S. and Canadian Phase III Trials
" ∆ " signifies change from baseline
Phase II Clinical Studies
Three multi-center, randomized, double-blind, placebo-controlled Phase II studies were conducted in 284 patients and evaluated various dosing regimens of ATX-101 vs. placebo for the reduction of submental fat. The Phase II program included two early Phase IIa studies, which formed the basis for Bayer's European Phase III trials, and one Phase IIb study that provided the basis for the U.S. and Canadian Phase III trials.
Phase IIa Studies (2007-2008). The two initial Phase IIa studies were conducted in a total of 155 patients. The first study evaluated various concentrations of ATX-101 administered subcutaneously directly into the submental fat, using a grid pattern of small-volume injections. The exact number of injections was at the discretion of the clinician based on the distribution of
12

Table of Contents
submental fat in each particular patient, but could not exceed 10 mL. In a similar manner, the second Phase IIa study utilized a constant concentration of ATX-101 and varied the volume of each injection and the spacing of the grid pattern. In both studies, patients received up to four treatment cycles at 28-day intervals, and agreed to maintain consistent diet and exercise practices throughout the study in order to minimize potential changes in body weight. Principal efficacy measures in these two early studies were the CR-SMFRS and a 7-point patient-reported outcome scale (SSRS), or PRO, assessing satisfaction with appearance of the face and chin. These rating instruments are similar to other rating scales used for approved aesthetic drug products and/or medical devices, such as botulinum toxins and dermal fillers. For both measures, we observed statistically significant improvements in patients treated with ATX-101, particularly patients dosed at 1 or 2 mg/cm 2 and, following discussions with various European regulatory authorities, the Phase IIa data were deemed sufficient to inform the design of Bayer's European pivotal Phase III clinical trials.
Phase IIb Study (2009-2010). Subsequent discussions with the FDA led to the development of additional submental fat assessment tools, including the PR-SMFRS, which parallels the clinician-reported assessment, and the PR-SMFIS, which assesses the visual and psychological impacts of "chin fat." These patient-reported scales, along with the CR-SMFRS, were rigorously validated, including demonstrations of multi-patient and multi-physician inter- and intra-rater reliability. These scales were used along with MRI assessments in a definitive Phase IIb dose-ranging study in 129 patients. This study compared ATX-101 at 1 or 2 mg/cm 2 vs. placebo in patients with moderate to severe submental fat. As in our Phase IIa studies, the exact number of injections of ATX-101 in our Phase IIb study was at the discretion of the clinician based on the distribution of submental fat for each particular patient, with a maximum of 10 mL injected per treatment cycle. Patients were allowed to receive up to six treatments at 28-day intervals and, as in previous studies, were requested to maintain consistent diet and exercise practices. Results from this study indicated that both doses were superior to placebo in most measures; however, the higher 2 mg/cm 2 dose appeared to consistently outperform the lower 1 mg/cm 2 dose. Compared to placebo-treated patients, dose-related improvements were observed in ATX-101-treated patients for mean changes in CR-SMFRS ratings, mean changes in PR-SMFRS ratings and mean changes in submental fat volume as assessed using MRI. In addition, treatment with ATX-101 resulted in improvements in patients' self-perception related to their submental fat as measured by the PR-SMFIS. Treated patients reported statistically significant increases in happiness with the appearance of their "chin fat" and self-perceptions of youthfulness and looking less overweight. The patients also reported feeling significantly less embarrassed, bothered and self-conscious about the appearance of their "chin fat."
Across all three Phase II studies, ATX-101 was well tolerated in all dosing regimens. The observed safety profile of ATX-101 is characterized by transient, local injection site reactions of typically mild to moderate severity. Pain was the most common injection site reaction, followed by numbness, bruising, swelling, induration and redness. Less frequently, itching, nodules and tingling were reported. Most of the injection site reactions resolved within the 28-day treatment interval, with some cases extending beyond the treatment interval gradually diminishing over the treatment period. Rare instances of transient facial nerve or muscle impairment (neuropraxia), which typically manifests as an asymmetric smile, were reported.
Long-Term Phase II Follow-Up Study. In an effort to assess long-term safety and duration of treatment effects, we are conducting an ongoing observational study to capture up to five years of long-term follow-up data on a subset of patients who completed Phase II studies of ATX-101 for submental fat reduction. An interim analysis of patients who received ATX-101 and were responders in their predecessor Phase II study was conducted. At the longest follow-up timepoint with available data (ongoing Study 12), 4 years after treatment in the early Phase 2 studies, 1-grade
13

Table of Contents
CR-SMFRS responses were observed to have been maintained in 29 of the 33 responders (87.9%) who attended the 4-year visit. As is consistent with previous findings at the two-year mark, no long-term safety concerns were noted in this interim analysis.
Summary of Regulatory Endpoints
The table (Figure 3) below summarizes the regulatory endpoints developed during the Phase II studies and used in the Phase III clinical trials to measure ATX-101 performance. These patient and physician scales are similar to other rating scales used for approved aesthetic drug products and medical devices, such as botulinum toxins and dermal fillers, and the CR-SMFRS, PR-SMFRS and PR-SMFIS were validated using scientific principles and process recommendations per the FDA's Patient Reported Outcome Guidance (FDA, 2009) in an effort to ensure reliability, construct validity and sensitivity to change over time.
Figure 3. Summary of Phase III Regulatory Endpoints
European Pivotal Phase III Clinical Trials, (Trial ATX-101-10-16 and Trial ATX-101-17) (2010-2012)
In Europe, Bayer, our former collaborator, conducted two identical pivotal, multi-center, randomized, double-blind, placebo-controlled, Phase III clinical trials designed to assess the efficacy, safety and tolerability of ATX-101, dosed at 1 or 2 mg/cm 2 , vs. placebo for the reduction of moderate to severe submental fat. The design of these trials was based on results from the prior Phase IIa studies and further informed by the then-ongoing development of additional patient-reported endpoints. Patients received up to four treatments at 28-day intervals and, as in previous studies, were requested to maintain consistent diet and exercise practices. Together, the trials enrolled 723 patients in 57 centers in the United Kingdom, France, Germany, Belgium, Spain and Italy.
Both trials met the pre-specified primary endpoints by demonstrating statistically significant reduction of moderate to severe submental fat, for both the 1 and 2 mg/cm 2 doses compared to placebo, as assessed by: percentage of patients with at least a 1-point improvement on the 5-point Clinician-Reported Submental Fat Rating Scale, or CR-SMFRS and percentage of patients expressing satisfaction with their appearance in association with their face and chin as measured by a rating of 4 or higher on the 7-point Subject Self Rating Scale, or SSRS.
14

Table of Contents
In addition, treatment with ATX-101 resulted in dose-dependent, statistically significant improvements in a number of secondary outcomes measured by: the 5-point Patient-Reported Submental Fat Rating Scale, or PR-SMFRS, caliper measurements of the submental area and patients' self-perception related to the appearance of their submental fat as measured by the Patient-Reported Submental Fat Impact Scale, or PR-SMFIS. Treated patients reported statistically significant increases in happiness with the appearance of their "chin fat" and associated self-perceptions of youthfulness and looking less overweight. The patients also reported feeling less embarrassed, bothered and self-conscious about the appearance of their "chin fat." Patients also reported a statistically significant difference in overall satisfaction with treatment as compared to placebo.
In these trials, the observed safety profile of ATX-101 was characterized by transient, local injection site reactions of predominately mild to moderate severity. Pain was the most common injection site reaction, followed by swelling, bruising, numbness, redness and induration. Less frequently, itching, tingling and nodules were reported. Rare instances of transient facial nerve or muscle impairment (neuropraxia), which typically manifests as an asymmetric smile, were reported.
U.S. and Canadian Pivotal Phase III Clinical Trials—"REFINE-1" and "REFINE-2" (2012-2013)
Based on study results to date, as well as substantive discussions with the FDA, our U.S. and Canadian pivotal Phase III trials were designed following the treatment regimen used in our completed Phase IIb study. These trials were designed to test against regulatory approval requirements using rigorous statistical analysis of validated physician and patient scales. These patient and physician scales are similar to other rating scales used for approved aesthetic drug products and medical devices, such as botulinum toxins and dermal fillers. As a substantiating objective measure, MRI was used to measure the change in submental volume following treatment with ATX-101. In addition, secondary and other measures evaluated patient satisfaction, perception of change and the psychological impacts associated with reduction of submental fat.
Our pivotal U.S. and Canadian Phase III trials, known as REFINE-1 and REFINE-2, or R andomized Double-blind E valuation of Submental F at Reduction IN ATX-101 Tr E ated Patients, were initiated in March 2012 and were designed to confirm the efficacy, safety and tolerability of ATX-101 for reduction of moderate to severe submental fat. These trials were identical multi-center, randomized, double-blind, placebo-controlled trials that compared subcutaneous injections of ATX-101 dosed at 2 mg/cm 2 to similar injections of placebo, with the primary efficacy assessments at 12 weeks after final treatment. Patients received up to six treatments at 28-day intervals and agreed to maintain consistent diet and exercise practices throughout the study in order to minimize potential changes in body weight. The trials were conducted in approximately 70 centers across the United States and Canada. We enrolled 1,022 patients across the two trials, randomized 1:1 to ATX-101 or placebo (REFINE-1 enrolled 506 patients and REFINE-2 enrolled 516 patients).
These Phase III trials had as principal efficacy measures both the CR-SMFRS and the PR-SMFRS. The two scales were used together as a composite to define a responder as a patient who achieved a pre-defined threshold of improvement simultaneously on both measures. Such a composite analysis was designed to eliminate variances that may be seen in data from either scale alone and substantially increased the difficulty in achieving any given threshold of improvement. These trials pre-specified two different thresholds as co-primary endpoints: a 1-grade improvement in the CR-SMFRS / PR-SMFRS composite, and a 2-grade improvement in the CR-SMFRS / PR-SMFRS composite. As co-primary endpoints, both had to be achieved for trial success. ATX-101 studies to date indicate that a 1-grade improvement represents a meaningful and positive change to both patients and physicians as indicated by high patient satisfaction and the patient's global impression of change in submental fat. Based on FDA guidance on interpretation of patient reported outcomes, these scores are used as an "anchor-based" method to establish a clinically meaningful threshold of response. The requirement of a 2-grade
15

Table of Contents
composite threshold represents a more difficult statistical standard for both product and scale performance.
To better understand treatment outcomes, secondary endpoints measured patients' self-perception related to the appearance of their submental fat as measured by the Patient-Reported Submental Fat Impact Scale, or PR-SMFIS, including treatment related impacts in patients' happiness with the appearance of their "chin fat" as well as associated self-perceptions of youthfulness, looking overweight, or feeling embarrassed, bothered or self-conscious about the appearance of their "chin fat." In addition, these trials measured the change in submental volume using MRI in a subset of approximately 400 patients at selected centers. We completed enrollment in these trials in August 2012 and reported positive topline results in September 2013. These trials form the basis for our NDA for approval of ATX-101 for the treatment of moderate to severe submental fat.
Trial ATX-101-11-22 (REFINE-1) and Trial ATX-101-11-23 (REFINE-2).
Pivotal Phase III Studies 22 and 23 each met their co-primary efficacy endpoints: the 1-grade composite SMFRS response rate (ie, proportion of patients with at least 1-grade simultaneous improvement on both the CR-SMFRS and the PR-SMFRS) and 2-grade composite SMFRS response rate (ie, proportion of patients with at least 2-grade simultaneous improvement in both the CR-SMFRS and the PR-SMFRS) at 12 weeks after last treatment, as summarized in Figure 4 below.
Figure 4: Co-primary Efficacy Results—Pivotal Studies 22 and 23
ATX-101 = deoxycholic acid injection; SMFRS = Submental Fat Rating Scale.
Note: Results shown are for protocol-specified co-primary analyses based on intent-to-treat (ITT) datasets consisting of all randomized patients. Source: Table 1, Section 2.5 of NDA 206333
These trials met the pre-specified co-primary endpoints by demonstrating statistically significant reduction of moderate to severe submental fat for the 2 mg/cm 2 dose compared to placebo.
Primary Efficacy Endpoints
REFINE-1 (Study ATX-101-11-22)
• 70.0% of ATX-101 (2mg/cm 2 ) REFINE-1 subjects demonstrated a simultaneous improvement of at least one grade from baseline on the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS) vs. 18.6% in placebo (p<0.001) (Figure 25).
16

Table of Contents
• 13.4% of ATX-101 (2mg/cm 2 ) REFINE-1 subjects demonstrated a simultaneous improvement of at least two grades from baseline on the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS) vs. 0% in placebo (p<0.001) (Figure 27).
REFINE-2 (Study ATX-101-11-23)
• 66.5% of ATX-101 (2mg/cm 2 ) REFINE-2 subjects demonstrated a simultaneous improvement of at least one grade from baseline on the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS) vs. 22.2% in placebo (p<0.001) (Figure 26).
• 18.6% of ATX-101 (2mg/cm 2 ) REFINE-2 subjects demonstrated a simultaneous improvement of at least two grades from baseline on the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS) vs. 3.0% in placebo (p<0.001) (Figure 27).
Secondary Efficacy Endpoints
• Additionally, an assessment of the trials' first secondary endpoint showed 46.3% of REFINE-1 and 40.2% of REFINE-2 patients achieved a predefined, statistically significant reduction in the volume of their submental region vs. 5.3% and 5.2% for placebo, respectively (both p<0.001), as measured by MRI (Figure 28).
• Subjects also rated the visual and psychological impacts of submental fat using the Patient-Reported Submental Fat Impact Scale (PR-SMFIS), which assessed whether they perceived themselves to be happier, less bothered, less self-conscious, less embarrassed, younger or less overweight after treatment with ATX-101. Statistical significance was achieved for the change from baseline in PR-SMFIS with 3.61 vs. 1.10 for REFINE-1 (Figure 30), and 3.44 vs. 1.46, for ATX-101 (2mg/cm 2 ) and placebo for REFINE-2 (p<0.001 for both). Each individual component within the PR-SMFIS also demonstrated statistical significance vs. placebo in both trials (p<0.001 for all PR-SMFIS measures).
Additional Efficacy Analyses
• 79.1% of ATX-101 (2mg/cm 2 ) REFINE-1 subjects demonstrated an improvement of at least one grade from baseline based on clinician assessment using the validated clinician-rated scale (CR-SMFRS) vs. 36.2% in placebo (p<0.001) (Figure 25).
• 77.9% of ATX-101 (2mg/cm 2 ) REFINE-2 subjects demonstrated an improvement of at least one grade from baseline based on clinician assessment using the validated clinician-rated scale (CR-SMFRS) vs. 34.5% in placebo (p<0.001) (Figure 26).
• 82.3% of ATX-101 (2mg/cm 2 ) REFINE-1 subjects demonstrated an improvement of at least one grade from baseline using the validated patient-rated scale (PR-SMFRS) vs. 38.5% in placebo (p<0.001) (Figure 25).
• 78.4% of ATX-101 (2mg/cm 2 ) REFINE-2 subjects demonstrated an improvement of at least one grade from baseline using the validated patient-rated scale (PR-SMFRS) vs. 37.8% in placebo (p<0.001) (Figure 26).
• Furthermore, 88.9% of REFINE-1 and 84.2% of REFINE-2 subjects reported satisfaction with treatment received in the trial, compared to 37.7% and 43.6% of placebo subjects, respectively (p<0.001) (Figure 29).
Additionally, there were no treatment-related serious adverse events. The most common adverse events, which were predominantly mild to moderate, were bruising, pain, numbness, edema, and swelling (Figure 11). Most AEs were mild or moderate in severity (98% of events in ATX-101 patients; 99% of events in placebo patients), and typically required no action to be taken for the event to resolve
17

Table of Contents
(88% of events in ATX-101 patients; 85% of events in placebo patients). Rare instances of transient facial nerve or muscle impairment (neuropraxia), which typically manifest as an asymmetric smile, were reported. 1.6% of subjects discontinued the trial due to adverse events.
Analyses of primary efficacy measures shown in the figures below are based on the protocol specified "Intent-to-Treat," or ITT, population which comprises all randomized subjects with baseline efficacy data (CR-SMFRS/PR-SMFRS). For secondary measures of PR-SMFIS and MRI, analysis includes only subjects with available data. Statistically significant results at various levels are denoted by asterisks or p-values in the figures below. The p-value is the probability that the reported result was achieved purely by chance (e.g., a p-value ≤ 0.01 means that there is a 1% or less chance that the difference between the placebo group and the treatment group is purely due to chance). A p-value ≤ 0.05 is a commonly used criterion for statistical significance. The symbol "n" is used to denote sample size per group.
Figure 5. U.S. and Canadian Phase III Trials, ATX-101-11-22 (REFINE-1), Percent of Patients with at least a 1-Grade Improvement in Clinician and Patient Rating Scales, CR- SMFRS/PR-SMFRS, at Week 32 by Treatment Group
All statistical comparisons are ATX-101 vs. placebo, ITT population with multiple imputation
18

Table of Contents
Figure 6. U.S. and Canadian Phase III Trials, ATX-101-11-23 (REFINE-2), Percent of Patients with at least a 1-Grade Improvement in Clinician and Patient Rating Scales, CR- SMFRS/PR-SMFRS, at Week 32 by Treatment Group
All statistical comparisons are ATX-101 vs. placebo, ITT population with multiple imputation
Figure 7. U.S. and Canadian Phase III Trials, ATX-101-11-22 and ATX-101-11-23, Percent of Patients with at least a 2-Grade Improvement in Clinician and Patient Rating Scales Composite, CR-SMFRS/PR-SMFRS, at Week 32 by Treatment Group
All statistical comparisons are ATX-101 vs. placebo, ITT population with multiple imputation
19

Table of Contents
Figure 8. U.S. and Canadian Phase III Trials, ATX-101-11-22 and ATX-101-11-23, Percent of Patients with MRI Volume Threshold Change, at Week 32 by Treatment Group
Per protocol, Magnetic Resonance Imaging ("MRI") were planned for approximately 200 subjects per study
All statistical comparisons are ATX-101 vs. placebo, ITT population with multiple imputation
Figure 9. U.S. and Canadian Phase III Trials, ATX-101-11-22 and ATX-101-11-23, Percent of Patients Treated with ATX-101 Expressing Satisfaction with Treatment, at Week 32 by Treatment Group
20
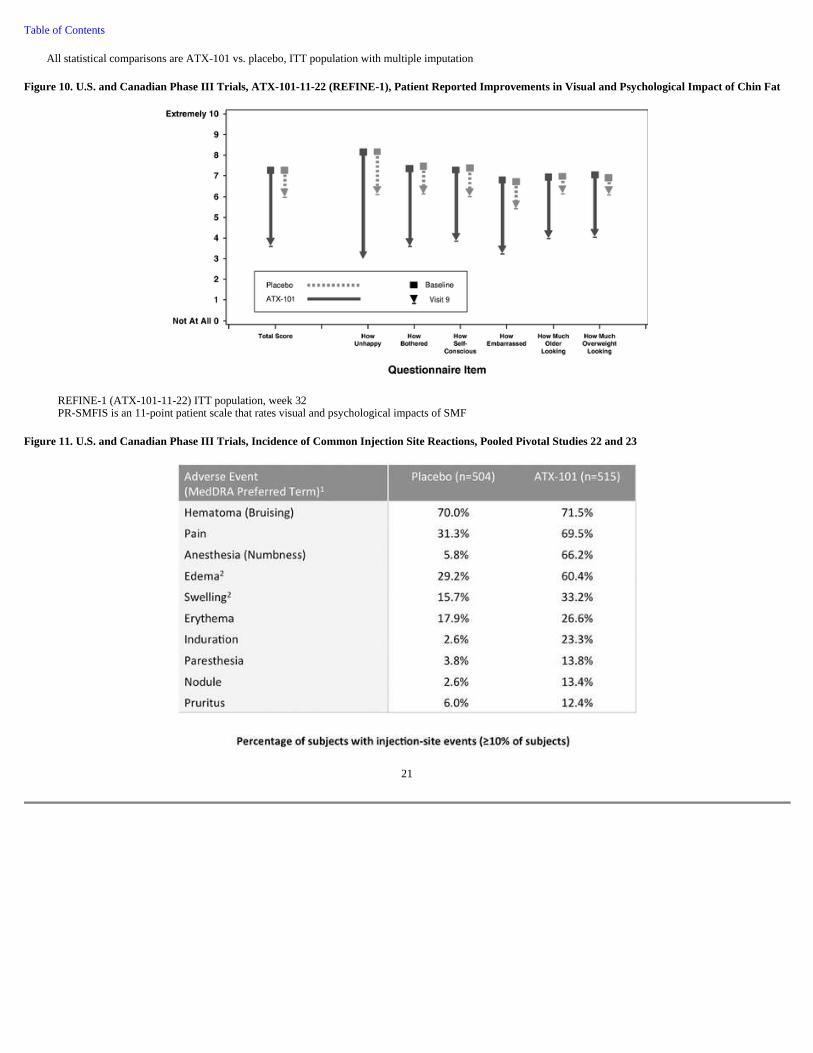
Table of Contents
All statistical comparisons are ATX-101 vs. placebo, ITT population with multiple imputation
Figure 10. U.S. and Canadian Phase III Trials, ATX-101-11-22 (REFINE-1), Patient Reported Improvements in Visual and Psychological Impact of Chin Fat
REFINE-1 (ATX-101-11-22) ITT population, week 32 PR-SMFIS is an 11-point patient scale that rates visual and psychological impacts of SMF
Figure 11. U.S. and Canadian Phase III Trials, Incidence of Common Injection Site Reactions, Pooled Pivotal Studies 22 and 23
21

Table of Contents
Rare instances of transient facial nerve or muscle impairment (neuropraxia), which typically manifests as an asymmetric smile, were reported. In the pivotal studies, neuropraxia occurred in 0.9% of ATX-101 treatments, as shown in figure 12 below. In the broader safety population of all SMF studies in the development program, neuropraxia occurred in 0.7% of ATX-101 treatments. Most events were mild or moderate in severity and events were temporary and recovered/resolved without sequelae.
Figure 12: Rate of Occurrence of Neuropraxia on All SMF Studies and Pivotal Studies by ATX-101 Patients vs. Placebo Patients
Representative before/after photographs of seven Phase III patients are shown in Figures 13, 14, 15, 16, 17, 18 and 19.
Figure 13. U.S. and Canadian Phase III, 26-year-old female
This 26-year-old female entered the Phase III trial with her baseline submental fat rated as grade 2 (moderate) on the clinician-rated scale (CR- SMFRS) and a grade 3 (severe) on the patient-rated scale (PR-SMFRS). By the end of study at week 32, she achieved a 2-grade reduction in submental fat according to the CR-SMFRS/PR-SMFRS composite.
* Satisfaction based on Subject Self-Rating Scale (SSRS); Laxity based on Submental Skin Laxity Grading Scale (SMSLG)
22
Rate Per Treatment Rate Per Patient ATX -101 Placebo ATX -101 Placebo All SMF Studies 0.7 % 0.1 % 2.9 % 0.3 % Pivotal Studies 0.9 % 0.1 % 4.3 % 0.4 %
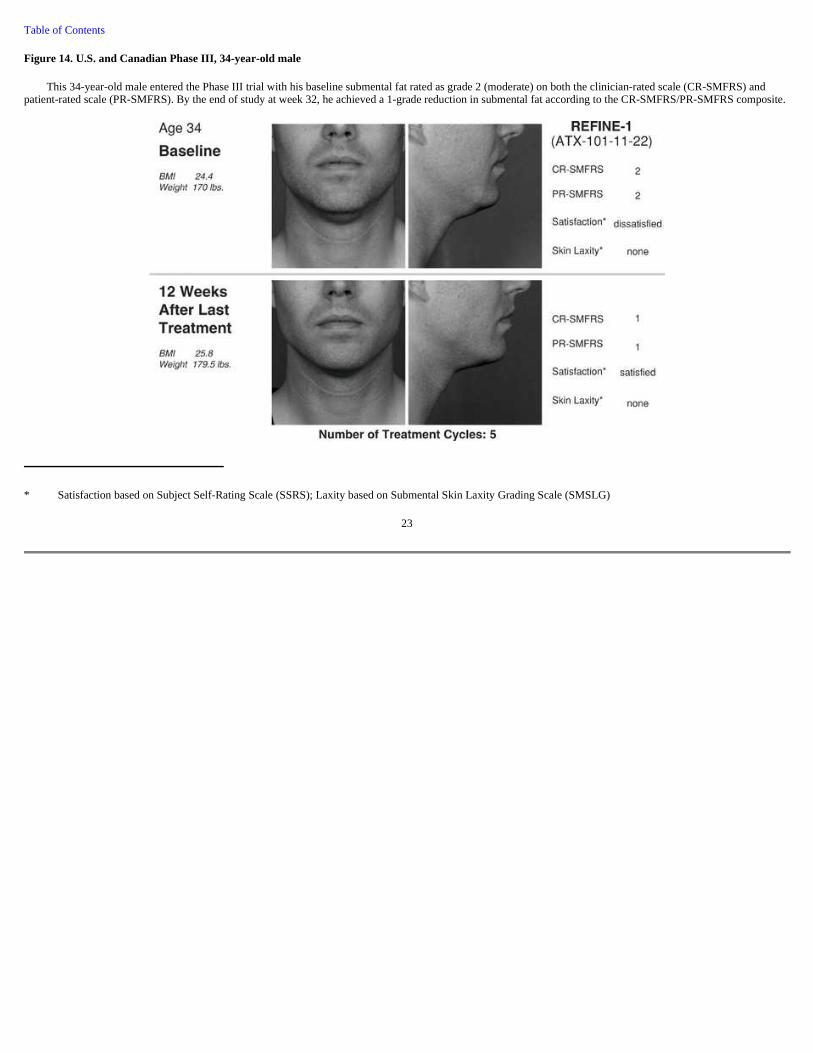
Table of Contents
Figure 14. U.S. and Canadian Phase III, 34-year-old male
This 34-year-old male entered the Phase III trial with his baseline submental fat rated as grade 2 (moderate) on both the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS). By the end of study at week 32, he achieved a 1-grade reduction in submental fat according to the CR-SMFRS/PR-SMFRS composite.
* Satisfaction based on Subject Self-Rating Scale (SSRS); Laxity based on Submental Skin Laxity Grading Scale (SMSLG)
23

Table of Contents
Figure 15. U.S. and Canadian Phase III, 35-year-old female
This 35-year-old female entered the Phase III trial with her baseline submental fat rated as grade 2 (moderate) on both the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS). By the end of study at week 32, she achieved a 1-grade reduction in submental fat according to the CR-SMFRS/PR-SMFRS composite.
* Satisfaction based on Subject Self-Rating Scale (SSRS); Laxity based on Submental Skin Laxity Grading Scale (SMSLG)
24

Table of Contents
Figure 16. U.S. and Canadian Phase III, 45-year-old female
This 45-year-old female entered the Phase III trial with her baseline submental fat rated as grade 3 (severe) on the clinician-rated scale (CR- SMFRS) and a grade 2 (moderate) on the patient-rated scale (PR-SMFRS). By the end of study at week 32, she achieved a 1-grade reduction in submental fat according to the CR-SMFRS/PR-SMFRS composite.
* Satisfaction based on Subject Self-Rating Scale (SSRS); Laxity based on Submental Skin Laxity Grading Scale (SMSLG)
25
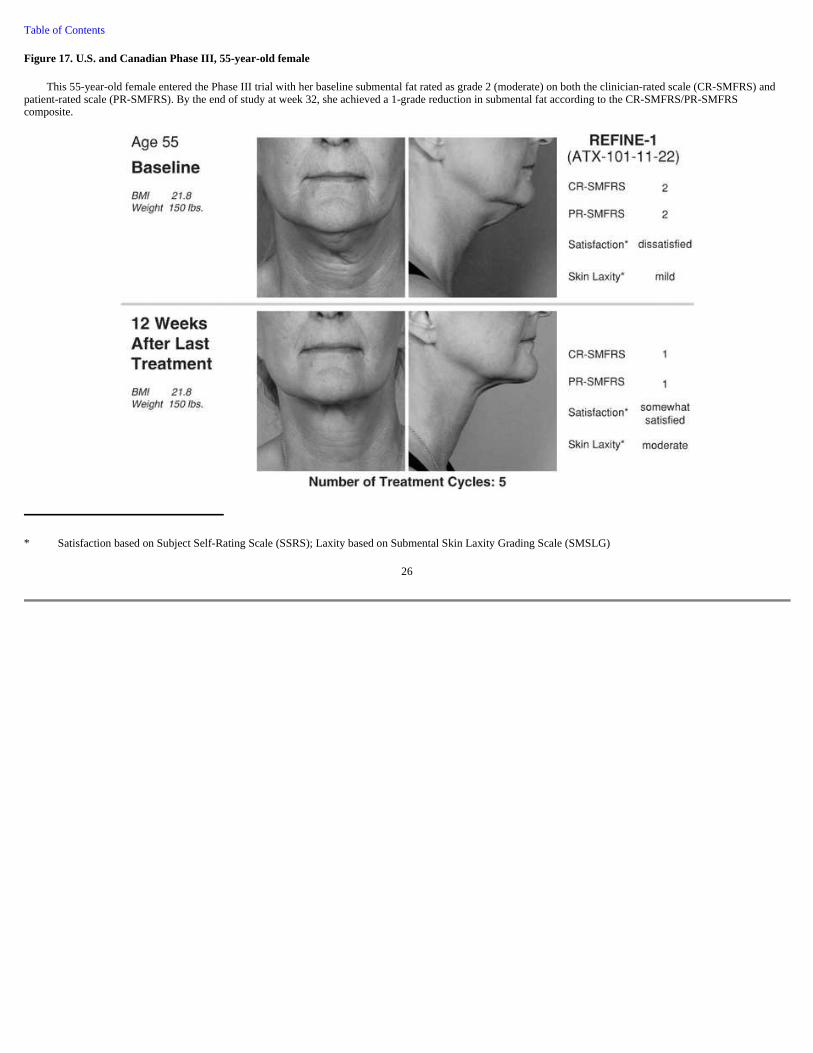
Table of Contents
Figure 17. U.S. and Canadian Phase III, 55-year-old female
This 55-year-old female entered the Phase III trial with her baseline submental fat rated as grade 2 (moderate) on both the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS). By the end of study at week 32, she achieved a 1-grade reduction in submental fat according to the CR-SMFRS/PR-SMFRS composite.
* Satisfaction based on Subject Self-Rating Scale (SSRS); Laxity based on Submental Skin Laxity Grading Scale (SMSLG)
26
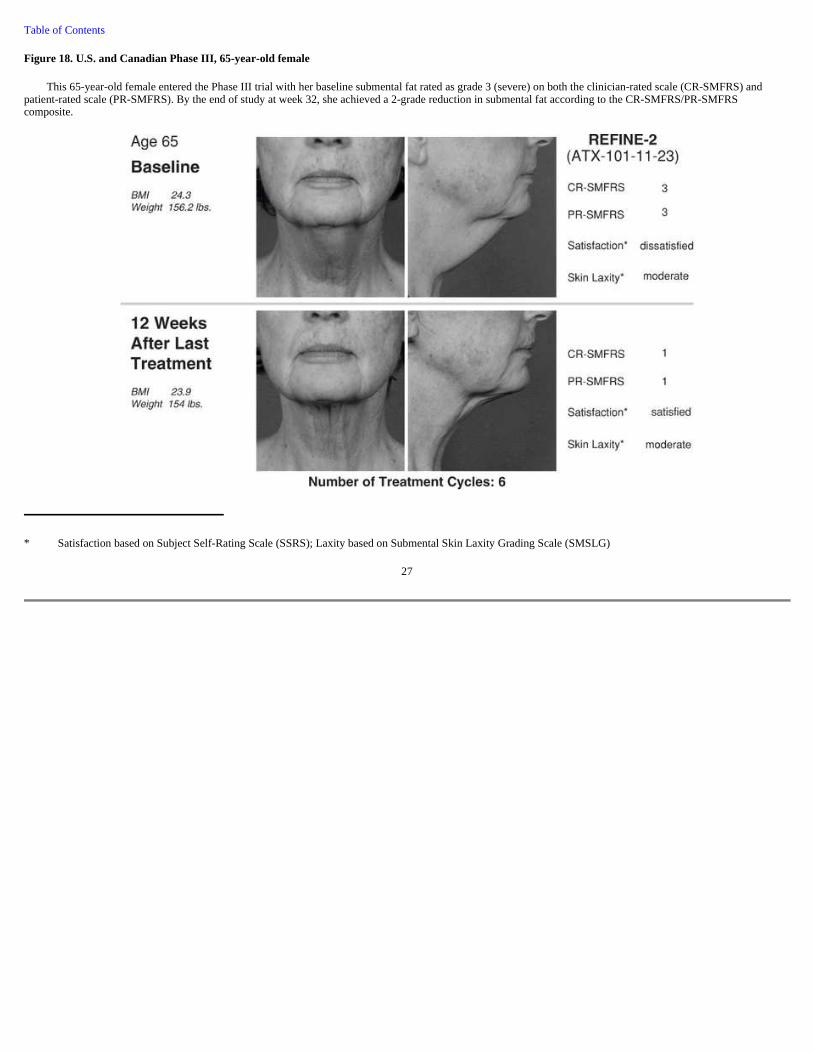
Table of Contents
Figure 18. U.S. and Canadian Phase III, 65-year-old female
This 65-year-old female entered the Phase III trial with her baseline submental fat rated as grade 3 (severe) on both the clinician-rated scale (CR-SMFRS) and patient-rated scale (PR-SMFRS). By the end of study at week 32, she achieved a 2-grade reduction in submental fat according to the CR-SMFRS/PR-SMFRS composite.
* Satisfaction based on Subject Self-Rating Scale (SSRS); Laxity based on Submental Skin Laxity Grading Scale (SMSLG)
27
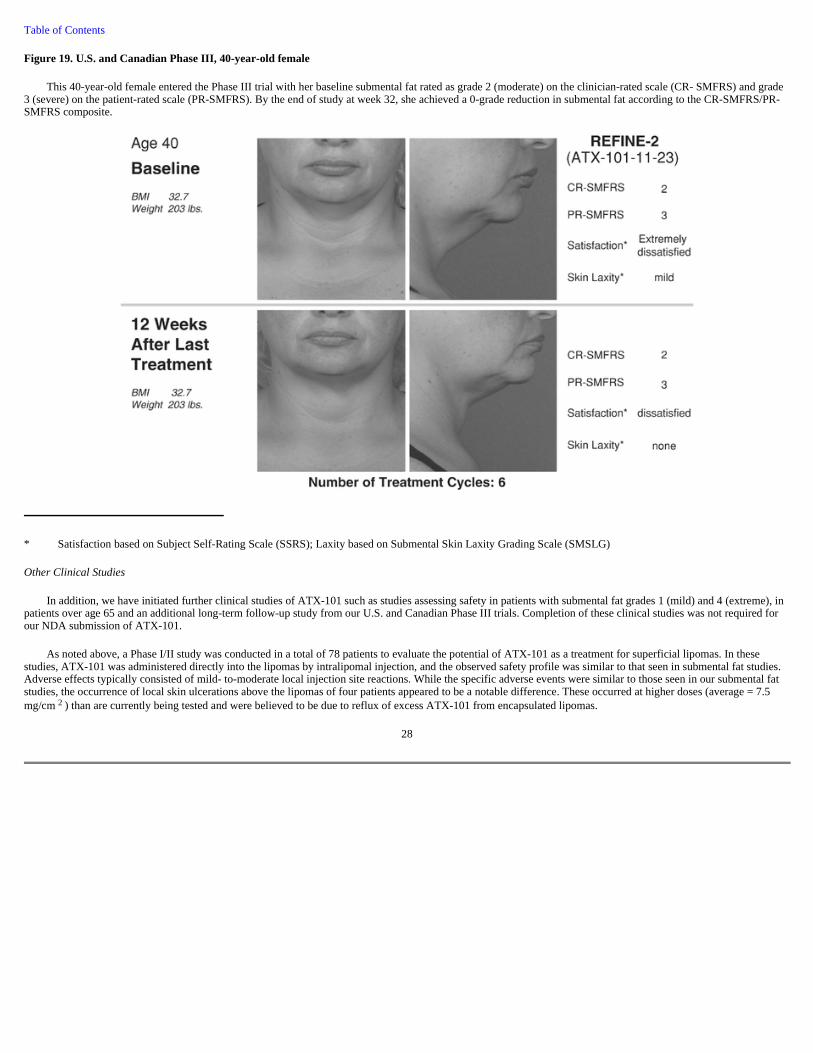
Table of Contents
Figure 19. U.S. and Canadian Phase III, 40-year-old female
This 40-year-old female entered the Phase III trial with her baseline submental fat rated as grade 2 (moderate) on the clinician-rated scale (CR- SMFRS) and grade 3 (severe) on the patient-rated scale (PR-SMFRS). By the end of study at week 32, she achieved a 0-grade reduction in submental fat according to the CR-SMFRS/PR-SMFRS composite.
* Satisfaction based on Subject Self-Rating Scale (SSRS); Laxity based on Submental Skin Laxity Grading Scale (SMSLG)
Other Clinical Studies
In addition, we have initiated further clinical studies of ATX-101 such as studies assessing safety in patients with submental fat grades 1 (mild) and 4 (extreme), in patients over age 65 and an additional long-term follow-up study from our U.S. and Canadian Phase III trials. Completion of these clinical studies was not required for our NDA submission of ATX-101.
As noted above, a Phase I/II study was conducted in a total of 78 patients to evaluate the potential of ATX-101 as a treatment for superficial lipomas. In these studies, ATX-101 was administered directly into the lipomas by intralipomal injection, and the observed safety profile was similar to that seen in submental fat studies. Adverse effects typically consisted of mild- to-moderate local injection site reactions. While the specific adverse events were similar to those seen in our submental fat studies, the occurrence of local skin ulcerations above the lipomas of four patients appeared to be a notable difference. These occurred at higher doses (average = 7.5 mg/cm 2 ) than are currently being tested and were believed to be due to reflux of excess ATX-101 from encapsulated lipomas.
28

Table of Contents
Sales and Marketing
We currently are building our sales and marketing capabilities. We have retained all commercial rights to ATX-101. If ATX-101 is approved by the FDA, we expect to access the market through a focused, specialized sales force in the United States and Canada. We currently plan to hire 8 regional managers and 73 sales reps in 2015.
Core Physicians
If approved for sale, we will focus our initial marketing of ATX-101 on those core dermatologists, plastic surgeons and facial plastic surgeons we identify as having substantial experience in performing facial injectable procedures. While plastic surgery specialties currently have tools, such as liposuction, for submental fat reduction, ATX-101, if approved as of today, would represent a significant opportunity to expand the number of potential patients. For the dermatologist community, ATX-101, if approved, will provide the only approved non-surgical tool for the treatment of submental fullness. In the United States, we plan to initially market to the top 3,000 high-value facial injectable accounts. We plan to engage and train physicians through a training-led launch, which will include on-demand learning, educational congresses, small group trainings and live webinars.
Patient Marketing
Our research indicates that the potential for ATX-101 to provide meaningful changes to lower facial appearance will interest both injectable "experienced" and injectable "naive" patients ( i.e ., patients that have not received injectable toxins or fillers). According to ASAPS, in 2013 there were an estimated 4.1 million cosmetic botulinum toxin procedures and 1.8 million dermal filler procedures performed in the United States and Canada, all of which were for aesthetic purposes. The likely early adopters of ATX-101 will be injectable experienced patients, however, we also plan to market to injectable-naive patients, as we believe ATX-101 provides a solution for a commonly desired aesthetic improvement. In 2013, women comprised approximately 90% of the injectable procedures market; however, our internal market research indicates that men have a significantly higher interest in an injectable solution for reducing submental fat than other facial injectable treatments.
Competition
The aesthetic product market, and the facial aesthetic market in particular, is highly competitive and dynamic, and is characterized by rapid and substantial technological development and product innovations. Demand for ATX-101 could be limited by the treatments, products and technologies offered now or in the future. We are seeking regulatory approval of ATX-101 for the treatment of submental fullness. While we do not believe there are any other injectable treatments approved as drugs by the FDA or equivalent foreign regulatory agencies designed specifically for this indication, we anticipate that ATX-101, if approved, will face significant competition from surgical alternatives for submental fat reduction and other medical device technologies designed for the reduction of fat.
Surgical Submental Fat Reduction Market
Liposuction and neck surgical procedures remain the primary treatment options for submental fat reduction. While we believe that most patients seeking facial aesthetics prefer non-surgical modalities, we do expect that ATX-101 may compete indirectly with liposuction for physician preference and resources.
FDA Cleared Devices for Fat Reduction
The FDA has cleared several medical devices for fat reduction. Although at this time none of these devices have been approved for the treatment of submental fat, there is the possibility that these
29

Table of Contents
devices could develop alternative application methods to accommodate use on the submental region. In addition, we may in the future face competition from new and emerging technologies.
Aesthetic Medicine Market Competition
Injectable botulinum toxins and dermal fillers dominate the aesthetic medicine market for facial rejuvenation. We believe ATX-101 will be a complementary procedure to these existing facial injectables.
For a description of the risks we face related to competition, please see "Risk Factors—Risks Related to Our Business—ATX-101, if approved, will face significant competition and our failure to effectively compete may prevent us from achieving significant market penetration" and "—The commercial success of ATX-101, if approved, will depend significantly on broad physician adoption and use of ATX-101."
We believe that ATX-101 will compete largely on the basis of the following competitive factors: significant, meaningful and durable results; a ready group of target patients; favorable safety profile with limited or no downtime as compared to surgical alternatives; low barrier to physician adoption and the ability to facilitate physician practice expansion; and the effectiveness of sales and marketing programs and initiatives.
Strategic Relationships
Prior ATX-101 Collaboration with Bayer Outside of the United States and Canada
In August 2010, we entered into a License Agreement with Bayer Consumer Care AG that provided for an exclusive license to develop, manufacture and commercialize ATX-101 outside of the United States and Canada. In connection with this license agreement, we entered into a related Services, Research, Development and Collaboration Agreement with Bayer's affiliate, Intendis GmbH, or collaboration agreement. We refer to these agreements jointly as our collaboration arrangement with Bayer, and we refer to Bayer Consumer Care AG and Intendis GmbH jointly as Bayer. In connection with the entry into the collaboration arrangement, Bayer paid us an upfront license fee of approximately $21.3 million and approximately $22.2 million for research and development to fund certain further global development activities of ATX-101. In May 2012, we received a $15.8 million payment from Bayer triggered by Bayer's decision to pursue continued development and regulatory approval for ATX-101 after receipt of positive results from the European Phase III clinical trials for ATX-101. In addition, we also received $17.4 million from Bayer to fund certain further global development activities of ATX-101 under the terms of the collaboration arrangement.
Acquisition of Rights to ATX-101 Outside of the United States and Canada
In March 2014, we announced the acquisition of rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer Consumer Care AG ("Bayer"), our former collaborator. Pursuant to an Assignment and Novation Agreement between Bayer and KYTHERA Holdings Ltd. ("KHL"), a wholly-owned Bermuda subsidiary of KYTHERA, Bayer transferred all of its right, title, and interest in the Amended License Agreement, and assigned all of its rights and delegated all of its obligations, liabilities and duties thereunder, to KHL for $84.0 million comprised of $33.0 million in KYTHERA common stock, plus a note for $51.0 million which bears interest at 5%, payable no later than 2024. Bayer is also eligible to receive certain long-term sales milestone payments on annual sales outside of the United States and Canada.
Los Angeles Biomedical Research Institute
In August 2005, we entered into an exclusive license agreement with Los Angeles Biomedical Research Institute at Harbor/UCLA Medical Center, or LA Biomed, pursuant to which we obtained a
30

Table of Contents
worldwide exclusive license to practice, enforce and otherwise exploit certain patent rights related to ATX-101. Our exclusive license requires us to pay LA Biomed a milestone payment of $0.5 million upon receipt of marketing approval of ATX-101, as well as non-royalty sublicense fees equal to 10% of any sublicense income, up to an aggregate of $5.0 million. In August 2010 and May 2012, we incurred non-royalty sublicense fees of $2.0 million and $1.6 million, respectively, in connection with the license fee paid by Bayer upon execution of the license agreement, and receipt of the $15.8 million contingent event- based payment triggered by Bayer's decision to pursue continued development and regulatory approval for ATX-101 after receipt of positive results from the European Phase III clinical trials for ATX-101. Upon receipt of these non- royalty sublicense fees, we initially paid a total of $0.7 million, with the remaining amount of $2.8 million satisfied subsequent to the initial public offering of which 50% was satisfied with shares of our common stock, and 50% in cash.
Additionally, upon commercialization of a licensed product or service, we are obligated to pay low- to mid-single digit royalties on net product sales of ATX-101. We may terminate this license without penalty upon 90 days' notice to LA Biomed and LA Biomed may terminate the license in certain circumstances if we fail to perform, or violate any term of the agreement, subject to applicable cure provisions. Subject to default by us or earlier termination, the license remains in effect until the last patent or patent application in the licensed patent rights has expired or been revoked, invalidated or abandoned.
Manufacturing
We contract with third parties for the manufacture of ATX-101 and intend to do so in the future. We do not own or operate and we do not expect to own or operate, facilities for product manufacturing, storage and distribution, or testing. Because we rely on contract manufacturers, we have personnel with extensive technical, manufacturing, analytical and quality experience and strong project management discipline to oversee contract manufacturing and testing activities, and to compile manufacturing and quality information for our regulatory submissions.
Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, and which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Our systems and our contractors are required to be in compliance with these regulations, and this is assessed regularly through monitoring of performance and a formal audit program.
Drug Substance
Our current drug substance supply chain involves contractors used for manufacturing and testing of key raw materials, regulatory starting material, and synthetic deoxycholate, which is the drug substance in ATX-101.
We currently have an exclusive supply agreement with Pfizer, Inc. as the single-source supplier of a key raw material used in manufacture of the regulatory starting material. We are not required to purchase any minimum or specific quantities from Pfizer and Pfizer cannot sell the key raw material to any other party for the manufacturing of bile acids and bile acid derivatives. However, under current terms, any purchase of this key raw starting material must be made from Pfizer. Our agreement also contains customary commercial terms for forecasting, payment, pricing, ordering, compliance, quality and indemnification. Our agreement with Pfizer expires in December 2016.
We currently have a non-exclusive supply agreement with Sai Life Sciences, Inc. for the supply of our regulatory starting material. We are not required to purchase any minimum or specific quantities from Sai Life Sciences, Inc. and our agreement contains customary commercial terms for forecasting, payment, pricing, ordering, compliance, quality and indemnification. Our agreement with SAI expires in June 2018.
31

Table of Contents
We currently have a non-exclusive supply agreement with Cambridge Major Laboratories, Inc., or CML for supply of synthetic deoxycholate. The agreement with CML contains customary commercial terms for drug substance supply regarding forecasting, payment, pricing, ordering, current good manufacturing practices, compliance, quality and indemnification. The term of the agreement runs through the fifth anniversary of final FDA approval of ATX-101. Our agreement with CML expires in May 2020.
We believe our current ATX-101 drug substance contractors have the scale, the systems and the experience to supply all clinical and commercial supply requirements. To ensure continuity in our supply chain, we plan to hold adequate inventory at each stage of manufacturing and establish supply arrangements with alternative suppliers as needed.
The process to manufacture the drug substance is being validated at a scale we believe will be sufficient for commercial launch in all territories. Our process uses common synthetic chemistry and readily available materials. We have established an ongoing program to identify possible process changes to improve purity, yield, manufacturability, etc., and process changes will be implemented as warranted and appropriate. The drug substance has historically shown stability adequate to support commercial operations.
Drug substance used to manufacture ATX-101 for nonclinical studies and for early clinical studies was purified from bovine bile. Drug substance used to manufacture ATX-101 for the Phase III trials in Europe, the United States and Canada was chemically synthesized and contains no ingredients of animal origin. The active ingredient in both animal-derived and synthetic drug substance is structurally identical.
Drug Product
ATX-101 drug product is a terminally sterilized liquid parenteral formulation for injection. Drug product manufacturing uses common processes and readily available materials. Changes to the drug product over the course of development have been relatively minor and have included changes in buffer and salt concentration. The formulation used in U.S. Phase III trials is the formulation anticipated for commercial launch in the United States and Canada. In countries outside the United States and Canada and depending on the local regulations, we may choose to commercialize the formulation used for U.S. and Canadian Phase III trials, or we may choose to commercialize the formulation used in the European Phase III trials.
The formulation used in the U.S. and Canadian Phase III trials differs slightly from the formulation used in European Phase III trials; the U.S. Phase III trials tested a single strength that contained benzyl alcohol as a preservative. The European trials tested two strengths without benzyl alcohol. Both formulations have historically shown product stability at room temperature adequate to support commercial operations and stability studies are ongoing.
In November 2010, we entered into a long-term agreement with Hospira, Inc. as our drug product fill/finish supplier for the United States and Canada. In December 2014, the agreement was amended to expand the scope to include Europe, Switzerland, Australia, and New Zealand. Our drug product is manufactured at the Hospira facility located in McPherson, Kansas. The initial term of our agreement expires five years after the first day of the month after our first bona fide sale of ATX-101 to a non-affiliate customer, and may be extended for additional and successive two-year terms. The agreement may be terminated prior to expiration by either party upon 24 months' written notice. The agreement may also be terminated by either party upon 60 days' written notice of an uncured material breach, the bankruptcy or insolvency of the other party or upon notice if the other party is unable to perform for 180 days due to a force majeure. If ATX-101 is approved, Hospira has the right to be our sole provider of drug product for the United States and Canada for a period of three years following commercialization, and, following the completion of the third year, the right to provide no less than
32

Table of Contents
seventy-five percent of our total annual requirement of drug product for the remaining term of the contract. We believe Hospira has the scale, systems and experience to supply ATX-101 drug product for clinical needs and long-term commercial demand.
Distribution
In January 2015, we entered into a three year distribution services agreement with Besse Medical for the distribution of ATX-101 in the United States, with consecutive automatic extensions of one year unless either party provides written notice of intent not to renew the agreement within 180 days. We expect that Besse Medical will be the exclusive reseller of the product in the United States, subject to our ability to sell directly to healthcare providers. We intend to sell ATX-101 to Besse at a fixed price, subject to certain adjustments and cost-sharing provisions.
Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain proprietary protection for our drug candidates, novel biological discoveries, and drug development technology and other know-how, to operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development and implementation of our business. We also rely on trade secrets, know-how, continuing technological innovation and potential in-licensing opportunities to develop and maintain our proprietary position.
As for the pharmaceutical products we develop and commercialize, as a normal course of business, we intend to pursue dosage and formulation patents, as well as method of use patents on novel indications for known compounds or for new compounds and, where possible, composition-of-matter patents. We also seek patent protection with respect to novel biological discoveries, including new targets and aesthetic applications. We have also pursued patents with respect to our proprietary manufacturing processes and drug development processes and technology. We have sought and plan to continue to seek patent protection on an ongoing basis.
Our patent estate, on a worldwide basis, includes over 100 issued or allowed patents and over 100 pending patent applications with claims relating to our current clinical stage drug candidate, ATX-101, which contains synthetic deoxycholate. The issued patents include 14 patents issued by the U.S. Patent and Trademark Office, or U.S. PTO:
33
Coverage Patent
number Expiration Method of Use 7,622,130 12/10/2027 Method of Use 7,754,230 12/10/2027 Method of Use 8,298,556 08/03/2025 Method of Use 8,846,066 12/10/2027 Synthetic DCA 8,242,294 05/16/2028 Synthetic DCA 8,883,770 05/16/2028 Synthetic DCA (Method of Manufacturing) 7,902,387 12/21/2028 Synthetic DCA (Method of Manufacturing) 7,994,351 05/16/2028 Synthetic DCA (Method of Manufacturing) 8,362,285 05/16/2028 Synthetic DCA (Method of Manufacturing) 8,367,852 05/16/2028 Formulation 8,546,367 05/16/2028 Formulation 8,461,140 05/16/2028 Formulation 8,101,593 03/02/2030 Formulation 8,653,058 03/02/2030 Formulation 8,367,649 03/02/2030

Table of Contents
European Patent Coverage
There are two European patents that were granted by the European Patent Office, or EPO, in 2011, both of which relate to Method of Use, or MOU, (EP1758590, which expires on February 8, 2025, and EP1748780, which expires on May 19, 2025), which resulted in counterparts in the major markets of Europe. A European patent was granted by the European Patent Office, or EPO, in 2014 (EP2561876), which relates to the formulation of ATX-101, counterparts are in force throughout Europe.
Rest of World (ROW) Patent Coverage
We also have patents issued in a number of other jurisdictions related to the patents described above. The chart below describes patents related to the various categories (MOU, Synthetic DCA, Methods of Manufacture (MOM), and Formulation) as summarized below:
In the rest of the world:
• Method of Use patents will expire in the 2025-2027 timeframe;
• Synthetic DCA patents will expire in about 2030;
• Methods of manufacture patents will expire in the 2028-2030 timeframe; and
• Formulation patents will expire in 2028-2031.
Some patents listed above may be eligible for term extension (or the equivalent) in some jurisdictions.
Deoxycholate is a naturally occurring substance and in its natural form may not be not eligible for composition-of-matter patent protection in certain jurisdictions. While we believe that Method of Use, or MOU, manufacturing and formulation patents are more relevant to protecting our competitive position in the aesthetics market in particular, we have developed a synthetic form of deoxycholate, which we believe is eligible for composition-of-matter patent protection and we have
34
Country Method of
use Method of
manufacture Synthetic
DCA Formulation Armenia X X Australia X X X Azerbaijan X X Belarus X X Canada X X X Hong Kong X X Israel X Japan X Korea, Republic Of X Kazakhstan X X Kyrgyzstan X X Mexico X Moldova, Republic Of X X New Zealand X X Philippines Russian Federation X X South Africa X X Tajikistan X X Turkmenistan X X Uzbekistan X X

Table of Contents
composition-of-matter patent applications with respect to this synthetic form currently pending with the in major markets.
Individual patents extend for varying periods depending on the date of filing of the patent application or the date of patent issuance and the legal term of patents in the countries in which they are obtained. Generally, patents issued for regularly filed applications in the United States are effective for 20 years from the earliest effective filing date. In addition, in certain instances, a patent term can be extended to recapture a portion of the U.S. PTO delay in issuing the patent as well as a portion of the term effectively lost as a result of the FDA regulatory review period. However, as to the FDA component, the restoration period cannot be longer than five years and the total patent term including the restoration period must not exceed 14 years following FDA approval. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective filing date. In addition to the patents described in the preceding paragraph, our pending patent applications, if issued, will expire on dates ranging from 2025 to 2032. However, the actual protection afforded by a patent varies on a product by product basis, from country to country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
Furthermore, in the United States the patent positions of biotechnology and pharmaceutical products and processes like ATX-101 can be uncertain and involve complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in such patents has emerged. The patent situation outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries can diminish our ability to protect our inventions and enforce our intellectual property rights and more generally could affect the value of intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights. Our ability to maintain and solidify our proprietary position for ATX-101 and its manufacturing will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of the patent applications that we may file or license from third parties will result in the issuance of any patents. The issued patents that we own or may receive in the future, may be challenged, invalidated or circumvented, and the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may be able to independently develop and commercialize similar drugs or duplicate our technology, business model or strategy without infringing our patents. Because of the extensive time required for clinical development and regulatory review of a drug we may develop, it is possible that, before any of our drugs can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of any such patent.
In addition to patents, we rely upon trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, using confidentiality agreements with our commercial partners, collaborators, employees and consultants and invention assignment agreements with our employees. We also have confidentiality agreements or invention assignment agreements with our commercial partners and selected consultants. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our commercial partners, collaborators,
35

Table of Contents
employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Our commercial success will also depend in part on not infringing upon the proprietary rights of third parties. It is uncertain whether the issuance of any third-party patent would require us to alter our development or commercial strategies, or our drugs or processes, obtain licenses or cease certain activities. Our breach of any license agreements or failure to obtain a license to proprietary rights that we may require to develop or commercialize our future drugs may have a material adverse impact on us. If third parties prepare and file patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference proceedings in the U.S. PTO, to determine priority of invention.
In addition, substantial scientific and commercial research has been conducted for many years in the areas in which we have focused our development efforts, which has resulted in third parties having a number of pending patent applications. Patent applications in the United States which are foreign filed or for which publication is requested are published after 18 months from the priority date as are any foreign applications. Certain applications in the United States which are not foreign filed and for which a request for non- publication has been made will not be published until granted. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made. Therefore, patent applications relating to drugs similar to ATX-101 and any future drugs, discoveries or technologies we might develop may have already been filed by others without our knowledge.
Other Research and Development Programs
KYTH-105 (Setipiprant)
In February 2015, we entered into separate license agreements with Actelion Pharmaceuticals Ltd. ("Actelion") and the Trustees of the University of Pennsylvania ("University of Pennsylvania") for a novel approach for the treatment of hair loss. Setipiprant is a selective oral antagonist to the prostaglandin D2 (PGD 2 ) receptor. The elevation of PGD 2 levels in certain regions of the male scalp is associated with hair loss in those regions. In early research studies, PGD 2 inhibitors were found to extend the anagen (growth) phase of the hair cycle, thereby promoting the growth of hair.
We plan to conduct a human proof-of-concept study to establish the efficacy of setipiprant in male subjects with androgenic alopecia (AGA).
To date, our research and development has focused predominantly on the development of ATX-101 for the treatment of submental fullness. However, we have initiated and terminated other development programs in the aesthetic field. Currently, we are working on several additional research programs, from which additional drug candidates may be identified in the future. The most advanced program centers around therapeutic intervention for hair loss. We maintain an active research interest in hair and fat biology, pigmentation modulation and facial contouring. In addition, we expect to assess future potential treatment indications for ATX-101 with high aesthetic value.
Please see "Management's Discussion and Analysis of Financial Condition and Results of Operations—Financial Overview—Research and Development" for the amounts spent on company-sponsored research and development for the past three fiscal years.
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of drugs. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality
36

Table of Contents
control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling, and export and import of our product candidates.
In the United States, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act, or FFDCA, and the FDA's implementing regulations. If we fail to comply with applicable FDA or other requirements at any time during the drug development process, clinical testing, the approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA's refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any FDA enforcement action could have a material adverse effect on us. FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug, can be marketed in the United States.
The process required by the FDA before a drug may be marketed in the United States generally involves:
• completion of extensive preclinical laboratory tests, preclinical animal studies and formulation studies all performed in accordance with the FDA's current good laboratory practice, or GLP regulations;
• submission to the FDA of an investigational new drug, or IND application which must become effective before human clinical trials in the United States may begin;
• approval by an independent institutional review board, or IRB, at each clinical trial site before each trial may be initiated;
• performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug candidate for each proposed indication;
• satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current Good Manufacturing Practice, or CGMP regulations;
• submission to the FDA of an NDA;
• satisfactory completion of a potential review by an FDA advisory committee, if applicable; and
• FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug.
The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all. Preclinical tests include laboratory evaluation of product chemistry, formulation, stability and toxicity, as well as animal studies to assess the characteristics and potential safety and efficacy of the product. The results of pre-clinical tests, together with manufacturing information, analytical data and a proposed clinical trial protocol and other information, are submitted as part of an IND to the FDA. Some pre-clinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions relating to one or more proposed clinical trials and places the clinical trial on a clinical hold, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, our submission of an IND may not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development.
37

Table of Contents
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be used. Each protocol must be submitted to the FDA as part of the IND. An independent institutional review board, or IRB, for each medical center proposing to conduct a clinical trial must also review and approve a plan for any clinical trial before it can begin at that center and the IRB must monitor the clinical trial until it is completed. The FDA, the IRB, or the sponsor may suspend or discontinue a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice, or GCP requirements, including the requirements for informed consent.
All clinical research performed in the United States in support of an NDA must be authorized in advance by the FDA under the IND regulations and procedures described above. However, a sponsor who wishes to conduct a clinical trial outside the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to FDA in support of an NDA so long as the clinical trial is conducted in compliance with an international guideline for the ethical conduct of clinical research known as the Declaration of Helsinki and/or the laws and regulations of the country or countries in which the clinical trial is performed, whichever provides the greater protection to the participants in the clinical trial.
Clinical Trials
For purposes of NDA submission and approval, clinical trials are typically conducted in three or four sequential phases, which may overlap or be combined.
• Phase I: Clinical trials are initially conducted in a limited population of subjects to test the drug candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans or, on occasion, in patients with severe problems or life-threatening diseases to gain an early indication of its effectiveness.
• Phase II: Clinical trials are generally conducted in a limited patient population to evaluate dosage tolerance and appropriate dosage, identify possible adverse effects and safety risks, and evaluate preliminarily the efficacy of the drug for specific targeted indications in patients with the disease or condition under study.
• Phase III: Clinical trials are typically conducted when Phase II clinical trials demonstrate that a dose range of the product candidate is effective and has an acceptable safety profile. Phase III clinical trials are commonly referred to as "pivotal" studies, which typically denotes a study which presents the data that the FDA or other relevant regulatory agency will use to determine whether or not to approve a drug. Phase III clinical trials are generally undertaken with large numbers of patients, such as groups of several hundred to several thousand, to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically-dispersed clinical trial sites.
• Phase IV: In some cases, FDA may condition approval of an NDA for a product candidate on the sponsor's agreement to conduct additional clinical trials after NDA approval. In other cases, a sponsor may voluntarily conduct additional clinical trials post approval to gain more information about the drug. Such post approval trials are typically referred to as Phase IV clinical trials.
Concurrent with clinical trials, companies usually complete additional animal trials and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the drug in commercial quantities in accordance with CGMP requirements.
38

Table of Contents
The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
New Drug Applications
The results of preclinical studies and of the clinical trials, together with other detailed information, including extensive manufacturing information and information on the composition of the drug, are submitted to the FDA in the form of an NDA requesting approval to market the drug for one or more specified indications. The FDA reviews an NDA to determine, among other things, whether a drug is safe and effective for its intended use.
Once an NDA has been accepted for filing, by law the FDA has 180 days to review the application and respond to the applicant. However, the review process is often significantly extended by FDA requests for additional information or clarification. Under the Prescription Drug User Fee Act, the FDA has a goal of responding to NDAs within ten months of submission for standard review, but this timeframe is also often extended. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The FDA may deny approval of an NDA if the applicable statutory and regulatory criteria are not satisfied, or it may require additional clinical data or an additional Phase III clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret data. Once the FDA approves an NDA, or supplement thereto, the FDA may withdraw the approval if ongoing regulatory requirements are not met or if safety problems are identified after the drug reaches the market. Where a withdrawal may not be appropriate, the FDA still may seize existing inventory of such drug or require a recall of any drug already on the market. In addition, the FDA may require testing, including Phase IV clinical trials and surveillance programs to monitor the effect of approved drugs which have been commercialized. The FDA has the authority to prevent or limit further marketing of a drug based on the results of these post-marketing programs.
Drugs may be marketed only for the FDA approved indications and in accordance with the provisions of the approved labeling. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require us to develop additional data or conduct additional preclinical studies and clinical trials.
Before approving an application, the FDA will inspect the facility or the facilities at which the finished drug product, and sometimes the active drug ingredient, is manufactured, and will not approve the drug unless CGMP compliance is satisfactory. The FDA may also inspect the sites at which the clinical trials were conducted to assess their compliance, and will not approve the drug unless compliance with GCP requirements is satisfactory.
The testing and approval processes require substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all. Even if we believe a clinical trial has demonstrated safety and efficacy of one of our drug candidates for the treatment of a disease, the results may not be satisfactory to the FDA. Preclinical and clinical data may be interpreted by the FDA in different ways, which could delay, limit or prevent regulatory approval. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals which could delay or preclude us from marketing drugs. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial
39

Table of Contents
application of the drugs. After approval, certain changes to the approved drug, such as adding new indications, manufacturing changes, or additional labeling claims are subject to further FDA review and approval. Depending on the nature of the change proposed, an NDA supplement must be filed and approved before the change may be implemented. For many proposed post-approval changes to an NDA, the FDA has up to 180 days to review the application. As with new NDAs, the review process is often significantly extended by the FDA requests for additional information or clarification.
Other Regulatory Requirements
Any drugs manufactured or distributed by us pursuant to FDA approvals would be subject to continuing regulation by the FDA, including recordkeeping requirements and reporting of adverse experiences associated with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including CGMPs, which impose certain procedural and documentation requirements upon us and our third party manufacturers. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the CGMP regulations and other ongoing FDA regulatory requirements. If we or our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution or withdraw approval of the NDA for that drug.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available drugs for uses that are not described in the product's labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers' communications regarding off-label use.
Other Healthcare Laws and Regulations
In the future, we may also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we conduct our business. The laws that may affect our ability to operate include:
• the federal healthcare programs' Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs;
• federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent;
• federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; the federal Health Insurance
40

Table of Contents
Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; and
• state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers.
If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and impact our financial results.
International Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing manufacturing, clinical trials, commercial sales and distribution of our future drugs. Whether or not we obtain FDA approval for a drug, we must obtain approval of a drug by the comparable regulatory authorities of foreign countries before commencing clinical trials or marketing of the drug in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized, decentralized or mutual recognition procedure. The centralized procedure provides for the grant of a single marking authorization that is valid for all European Union member states. The decentralized procedure includes selecting one "reference member state," or RMS, and submitting to more than one member state at the same time. The RMS National Competing Authority conducts a detailed review and prepares an assessment report, to which concerned member states provide comment. The mutual recognition procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marking authorization may submit an application to the remaining member states post-initial approval. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval.
In addition to regulations in Europe and the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial distribution of our future drugs.
Environmental Regulation
We are subject to numerous foreign, federal, state, and local environmental, health and safety laws and regulations relating to, among other matters, safe working conditions, product stewardship and end-of-life handling or disposition of products, and environmental protection, including those governing the generation, storage, handling, use, transportation and disposal of hazardous or potentially hazardous materials. Some of these laws and regulations require us to obtain licenses or permits to conduct our operations. Environmental laws and regulations are complex, change frequently and have tended to become more stringent over time. Although the costs to comply with applicable laws and regulations, including requirements in the European Union relating to the restriction of use of hazardous substances in products, have not been material, we cannot predict the impact on our business of new or amended laws or regulations or any changes in the way existing and future laws and regulations are interpreted or enforced, nor can we ensure we will be able to obtain or maintain any required licenses or permits.
41

Table of Contents
Employees
As of December 31, 2014, we had 106 full-time employees, including a total of 23 employees with M.D. or Ph.D. degrees. Within our workforce, 70 employees are engaged in research and development and 36 in marketing, business development, finance, legal, human resources, facilities, information technology and general management and administration. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Additional Information
We view our operations and measure our business as one reportable segment operating primarily in the United States. Additional information required by this item is incorporated herein by reference to Part I Item 6, "Selected Financial Data."
We were originally incorporated in Delaware in June 2004 under the name Dermion, Inc. We commenced operations in August 2005 and later changed our name to AESTHERx, Inc. In July 2006, we changed our name to KYTHERA Biopharmaceuticals, Inc. We completed our initial public offering of our common stock in October 2012. Our mailing address and executive offices are located at 30930 Russell Ranch Road, 3 rd Floor, Westlake Village, California, 91362 and our telephone number at that address is (818) 587-4500. We maintain an Internet website at the following address: www.kytherabiopharma.com. The information on our website is not incorporated by reference in this annual report on Form 10-K or in any other filings we make with the Securities and Exchange Commission, or SEC.
We make available on or through our website certain reports and amendments to those reports that we file with or furnish to the SEC in accordance with the Securities Exchange Act of 1934, as amended. These include our annual reports on Form 10-K, our quarterly reports on Form 10-Q, and our current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act. We make this information available on or through our website free of charge as soon as reasonably practicable after we electronically file the information with, or furnish it to, the SEC.
ITEM 1A. Risk Factors.
Our operations and financial results are subject to various risks and uncertainties, including those described below, which could adversely affect our business, financial condition, results of operations, cash flows, and the trading price of our common stock. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations. You should carefully consider the risks described below and the other information in this Annual Report on Form 10-K.
Risks Related to Our Limited Operating History, Business and Capital Requirements
We have a limited operating history and have incurred significant losses since our inception and we anticipate that we will continue to incur losses for the foreseeable future. We have only one product candidate and no commercial sales, which, together with our limited operating history, makes it difficult to assess our future viability.
We are a clinical-stage biopharmaceutical company with a limited operating history. Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. To date, we have focused principally on developing our only product candidate, ATX-101, for which we completed our U.S. and Canadian Phase III clinical trials and submitted an NDA with the FDA in May 2014. We are not profitable and have incurred losses in each year since our inception in June 2004. We have only a limited operating history upon which one can evaluate our business and prospects. In addition, we have limited experience and have not yet demonstrated an
42

Table of Contents
ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical industry. We have not generated any revenue from product sales to date. We continue to incur significant research and development and other expenses related to our ongoing operations. Our net loss for the years ended December 31, 2014 and 2013 was approximately $135.6 million and $51.9 million, respectively. As of December 31, 2014, we had an accumulated deficit of $307.8 million. We expect to continue to incur losses for the foreseeable future, as we continue our development of, and seek regulatory approvals for, ATX-101, and begin to commercialize ATX-101. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders' equity and working capital.
We are substantially dependent on the success of our only product candidate, ATX-101.
To date, we have invested nearly all of our efforts and financial resources in the research and development of ATX-101, which is currently our only product candidate. In particular, we have completed seven Phase I, three Phase II clinical studies, two European Phase III clinical trials, and two U.S. and Canadian Phase III clinical trials and two Phase IIIb studies.
Our near-term prospects, including our ability to finance our company and to enter into strategic collaborations and generate revenue, will depend heavily on the successful development, regulatory approval and commercialization of ATX-101 and the development of any future product candidates. The clinical and commercial success of ATX-101 will depend on a number of factors, including the following:
• our ability to demonstrate the safety and efficacy of ATX-101 to the satisfaction of the FDA and similar foreign regulatory authorities;
• our ability to obtain, and the timing of, regulatory approval of ATX-101 from the FDA and similar foreign regulatory authorities and the timely receipt of such necessary marketing approvals, where applicable;
• any unexpected results from further analysis of clinical data of our completed clinical trials;
• timely completion of our ongoing or upcoming clinical trials, which may be slower than we currently anticipate and will depend substantially upon the satisfactory performance of third-party contractors;
• whether we are required by the FDA or other similar foreign regulatory agencies to conduct additional clinical trials;
• our ability to successfully commercialize ATX-101, if approved for marketing and sale by applicable regulatory agencies, whether alone or in collaboration with others;
• our success in educating physicians and patients about the benefits, administration and use of ATX-101;
• the incidence, duration and severity of adverse side effects;
• achieving and maintaining compliance with all regulatory requirements applicable to ATX-101;
• the availability, perceived advantages, relative cost, relative safety and relative efficacy of alternative and competing treatments;
• the effectiveness of our own or our potential strategic collaborators' marketing, sales and distribution strategy and operations;
43

Table of Contents
• the ability of our third-party manufacturers to manufacture clinical trial and commercial supplies of ATX-101 or any future product candidates, to remain in good standing with regulatory agencies, and to develop, validate and maintain commercially viable manufacturing processes that are compliant with Current Good Manufacturing Practice, or CGMP regulations;
• our ability to successfully commercialize ATX-101 in the United States and Canada, and in other international territories, if approved for marketing and sale in such countries and territories, whether alone or in collaboration with others;
• our ability to enforce our intellectual property rights in and to ATX-101;
• our ability to avoid third-party patent interference or patent infringement claims;
• acceptance of ATX-101 as safe and effective by patients and the medical community; and
• a continued acceptable safety profile of ATX-101 following approval.
Many of these factors are beyond our control. Accordingly, we cannot assure you that we will ever be able to generate revenue through the sale of ATX-101. If we are not successful in obtaining approval of and commercializing ATX-101, or are significantly delayed in doing so, our business will be materially harmed.
We may be unable to obtain regulatory approval for ATX-101 under applicable regulatory requirements. The denial or delay of any such approval would delay commercialization of ATX-101 and adversely impact our potential to generate revenue, our business and our results of operations.
To gain approval to market a drug product, we must provide the FDA and foreign regulatory authorities with clinical data that adequately demonstrates the safety, efficacy and compliant manufacturing of the product for the intended indication applied for in the NDA or other respective regulatory filing. Drug development is a long, expensive and uncertain process, and delay or failure can occur at any stage of any of our clinical trials. A number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in clinical trials and manufacturing, even after promising results in earlier preclinical or clinical studies. These setbacks have been caused by, among other things, preclinical findings made while clinical studies were underway and safety or efficacy observations made in clinical studies, including previously unreported adverse events. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and the results of clinical trials by other parties, including Bayer, our former collaborator, may not be indicative of the results in trials we may conduct.
In May 2014, we filed an NDA with the FDA for ATX-101 on the basis of the results from our pivotal U.S. and Canadian Phase III clinical trials, and our business currently depends entirely on its successful regulatory approval and commercialization in the United States. We submitted a New Drug Application ("NDS") with Health Canada in August 2014, a Marketing Authorization Application ("MAA") to Swissmedic in October 2014, and a drug application with Australia's Therapeutic Goods Administration ("TGA"), seeking approval for ATX -101 as an injectable treatment for the treatment of submental fullness. We currently have no drug products approved for sale, and we may never obtain regulatory approval to commercialize ATX-101. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, and such regulations differ from country to country. We are not permitted to market ATX-101 in the United States unless and until we receive approval of an NDA from the FDA, and we are not permitted to market ATX-101 in any foreign countries unless and until we receive the requisite approval from the regulatory authorities of such countries.
44

Table of Contents
The FDA or any foreign regulatory bodies can delay, limit or deny approval of ATX-101 for many reasons, including:
• inability to demonstrate to the satisfaction of the FDA or the applicable foreign regulatory body that ATX-101 is safe and effective for the requested indication;
• the FDA's or the applicable foreign regulatory agency's disagreement with the trial protocol or the interpretation of data from preclinical studies or clinical trials;
• inability to demonstrate that the clinical and other benefits of ATX-101 outweigh any safety or other perceived risks;
• the FDA's or the applicable foreign regulatory agency's requirement for additional preclinical or clinical studies;
• the FDA's or the applicable foreign regulatory agency's non-approval of ATX-101's chemistry, manufacturing or controls or labeling;
• the FDA's or the applicable foreign regulatory agency's failure to approve the manufacturing processes or facilities of third-party manufacturers and testing labs with whom we contract; or
• the potential for approval policies or regulations of the FDA or the applicable foreign regulatory agencies to significantly change in a manner rendering our clinical data or regulatory filings insufficient for approval.
Of the large number of drugs in development, only a small percentage successfully complete the FDA or other regulatory approval processes and are commercialized. Further, we did not conduct our U.S. and Canadian Phase III clinical trials under a Special Protocol Assessment, or SPA. In the absence of an agreed SPA there can be no assurance that the FDA will agree with our Phase III clinical trial protocol. In addition, ATX-101 may not be approved by the FDA or applicable foreign regulatory agencies even though it has met its specified endpoints in our U.S. and Canadian Phase III clinical trials and, our former collaborator, Bayer's pivotal European Phase III clinical trials. The FDA and/or applicable foreign regulatory agencies may disagree with the trial design and/or the interpretation of data from clinical trials, and may ask us to conduct additional costly and time consuming clinical trials in order to obtain marketing approval or approval to enter into an advanced phase of development, or may change the requirements for approval even after it has reviewed and commented on the design for the clinical trials.
Even if we eventually complete clinical testing and receive approval of the NDA or foreign regulatory filing for ATX-101, the FDA or the applicable foreign regulatory agency may grant approval contingent on the performance of costly additional clinical trials which may be required after approval. The FDA or the applicable foreign regulatory agency also may approve ATX-101 for a more limited indication or a narrower patient population than we originally requested, and the FDA, or applicable foreign regulatory agency, may not approve the labeling that we believe is necessary or desirable for the successful commercialization of ATX-101. Any delay in obtaining, or inability to obtain, applicable regulatory approval would delay or prevent commercialization of ATX-101 and would materially adversely impact our business and prospects.
Even if ATX-101 or any future product candidates obtain regulatory approval, they may never achieve market acceptance or commercial success.
Even if we obtain FDA or other regulatory approvals, ATX-101 or any future product candidates may not achieve market acceptance among physicians and patients, and may not be commercially
45

Table of Contents
successful. Market acceptance of ATX-101 or any future product candidates for which we receive approval depends on a number of factors, including:
• the safety and efficacy of the product as demonstrated in clinical trials;
• the clinical indications for which the product is approved and whether our desired labeling is approved;
• acceptance by physicians, major operators of clinics and patients of the product as a safe and effective treatment;
• proper training and administration of our products by physicians;
• the potential and perceived advantages of our product candidates over alternative treatments;
• the cost of treatment in relation to alternative treatments and willingness to pay for our products, if approved, on the part of physicians and patients;
• the willingness of patients to pay for ATX-101 and other aesthetic treatments in general, relative to other discretionary items;
• relative convenience and ease of administration;
• the incidence, duration and severity of adverse side effects;
• the effectiveness of our sales and marketing efforts; and
• the degree to which the approved labeling supports promotional initiatives for commercial success.
Any failure by our product candidates that obtain regulatory approval to achieve market acceptance or commercial success would adversely affect our results of operations.
We have only recently begun building an infrastructure to support a commercial organization and we do not currently have a sales organization sufficient to support a commercial launch of ATX-101, if approved. If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell ATX-101 or any future product candidates effectively, if approved, or generate product revenue.
We have only recently begun building an infrastructure to support a commercial organization and we do not currently have a sales organization sufficient to support a commercial launch of ATX-101, if approved. In order to commercialize ATX-101, we must build our marketing, sales, distribution, management and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If ATX-101 receives regulatory approval, we expect to establish a sales organization with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive, require substantial additional capital and be time consuming. We have no prior experience in the marketing, sale and distribution of pharmaceutical products and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain, and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of these products. We may choose to collaborate with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize ATX-101. If we are not successful in commercializing ATX-101 or any future product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we would incur significant additional losses.
46

Table of Contents
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers or are in breach of non-competition or non-solicitation agreements.
Many of our employees were previously employed at other pharmaceutical or medical device companies, including other companies in the aesthetic medicine space, in some cases until recently. Similarly, we may hire additional employees from these companies. In the future, we could be subject to claims that we or our current or future employees have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of these former employers. In addition, we may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. If our defense to those claims fails, in addition to paying monetary damages, we may be enjoined from selling our product candidates (if approved by FDA), lose valuable intellectual property rights or personnel. Any litigation or the threat thereof may adversely affect our ability to hire employees. A loss of key personnel or their work product could hamper or prevent our ability to commercialize product candidates, which could have an adverse effect on our business, results of operations and financial condition.
We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development, other operations or commercialization efforts.
Since our inception, most of our resources have been dedicated to the preclinical and clinical development of ATX-101. As of December 31, 2014, we had working capital of $80.5 million and capital resources consisting of cash and cash equivalents and marketable securities of $99.7 million. We have drawn down the full $15.0 million available under our credit facility and no longer have any established future borrowing capacity under that credit facility. We believe that we will continue to expend substantial resources for the foreseeable future on preparations for a potential commercial launch of ATX-101, the completion of clinical and regulatory development of ATX-101, preparing and filing foreign regulatory filings and development of any other product candidates we may choose to pursue. These expenditures will include costs associated with research and development, conducting preclinical studies and clinical trials, obtaining regulatory approvals, and manufacturing and supply as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of any drug development process is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of ATX-101 or any future product candidates. We expect to seek additional financing in the future to fund our operations, to expand our commercial organization in anticipation of potential commercial launch of ATX-101, and to develop additional product candidates, by selling additional equity, issuing debt or convertible debt securities that may result in dilution to our stockholders.
We believe our existing cash and cash equivalents will allow us to fund our operations through at least the next 12 months. However, our operating plan may change as a result of factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations. Such financing may result in dilution to stockholders, imposition of debt covenants and repayment obligations, or other restrictions that may adversely affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
47

Table of Contents
Our future capital requirements depend on many factors, including:
• the timing of, and costs involved in, obtaining regulatory approval for ATX-101 in the U.S. and other international territories including the cost of any studies or additional activities that the FDA or other regulatory agencies may require us to complete;
• the timing and scope of the investment we make in building our commercial infrastructure and sales force in anticipation of the potential commercial launch of ATX-101 in the U.S.;
• any unexpected results from further analysis of clinical data from our completed clinical trials;
• the timing of, and the costs involved in, obtaining regulatory approvals for any future product candidates;
• the number and characteristics of any additional product candidates we may develop or acquire;
• the scope, progress, results and costs of researching and developing ATX-101 or any future product candidates, including pursuant to our license agreement with Actelion and the University of Pennsylvania, and conducting preclinical and clinical trials;
• the cost of commercialization activities if ATX-101 or any future product candidates are approved for sale, including marketing, sales and distribution costs and preparedness of our corporate infrastructure;
• the cost of manufacturing ATX-101 or any future product candidates and any products we successfully commercialize;
• the timing of the payment of our debt obligations, including whether or not we determine to prepay the $51.0 million note held by Bayer;
• our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements;
• any product liability or other lawsuits related to our products or commenced against us;
• the expenses needed to attract and retain skilled personnel;
• the costs associated with being a public company;
• the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent or other claims, including litigation costs and the outcome of such litigation; and
• the timing, receipt and amount of sales of, or royalties on, future approved products, if any.
Additional funds may not be available when we need them, on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to:
• delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for ATX-101 or any future product candidates;
• delay, limit, reduce or terminate our research and development activities; or
• delay, limit, reduce or terminate our establishment of sales and marketing capabilities or other activities that may be necessary to commercialize ATX-101 or any future product candidates.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical testing is expensive, and can take many years to complete, and its outcome is inherently uncertain. Furthermore, we rely on contract research organizations, or CROs, and clinical trial sites to ensure the proper and timely conduct of our clinical trials and while we have agreements governing
48

Table of Contents
their committed activities, we have limited influence over their actual performance. Failure can occur at any time during the clinical trial process. For example, we have in the past terminated early-stage development and clinical programs for other potential drug candidates due to a lack of sufficient efficacy or the potential for unacceptable adverse reactions to a particular drug candidate, as well as our desire to concentrate our efforts on the development of ATX-101. The results of preclinical and clinical studies of our product candidates may not be predictive of the results of later-stage clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier studies, and we cannot be certain that we will not face similar setbacks. Even if our ongoing or future clinical trials are completed, the results may not be sufficient to obtain regulatory approval for our product candidates.
We may experience delays in our ongoing clinical trials, and we do not know whether future clinical trials, if any, will begin on time, need to be redesigned, enroll an adequate number of patients on time or be completed on schedule, if at all. Clinical trials can be delayed or aborted for a variety of reasons, including delay or failure to:
• obtain regulatory approval to commence a trial;
• reach agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
• obtain institutional review board, or IRB, approval at each site;
• recruit suitable patients to participate in a trial;
• have patients complete a trial or return for post-treatment follow-up;
• have clinical sites observe trial protocol or continue to participate in a trial;
• address any patient safety concerns that arise during the course of a trial;
• address any conflicts with new or existing laws or regulations;
• add a sufficient number of clinical trial sites; or
• manufacture sufficient quantities of product candidate or placebo for use in clinical trials.
Patient enrollment is a significant factor in the timing of clinical trials and is affected by many factors, including the size and nature of the patient population, the proximity of eligible patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials and clinicians' and patients' perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs or treatments that may be approved for the indications we are investigating.
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted, by the Data Safety Monitoring Board, or DSMB, for such trial or by the FDA or other regulatory authorities. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with the requirements of the relevant regulatory filing (including clinical protocol and manufacturing), inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
49

Table of Contents
If we experience delays in the completion of, or terminate, any clinical trial of our future product candidates, if any, the commercial prospects of these product candidates may be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may significantly harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of a clinical trial may also ultimately lead to the denial of regulatory approval of a product candidate.
ATX-101, if approved, will face significant competition and our failure to effectively compete may prevent us from achieving significant market penetration.
The aesthetic product market, and the facial aesthetics market in particular, is highly competitive and dynamic, and is characterized by rapid and substantial technological development and product innovations. We are seeking regulatory approval of ATX-101 for the treatment of submental fullness. A substantial portion of our target physician market is comprised of plastic surgeons and some dermatologists who utilize liposuction and other procedures for fat reduction. Such physicians may find it more advantageous to utilize these surgical and non-surgical procedures to remove localized fat deposits rather than an injectable biopharmaceutical therapy such as ATX-101. In addition, we expect that ATX-101, if approved, will compete with new and existing therapies for the treatment of localized fat, including, liposuction and other procedures, as well as other technologies aimed at fat reduction, including laser energy-based and ultrasound energy-based products. We believe that some of these products have been or may be marketed and used for the reduction of submental fat even though they have not been approved for that purpose.
If approved, ATX-101 may also compete with unregulated, unapproved and off-label fat reduction treatments. For example, we are aware that there are entities such as compounding pharmacies and device manufacturers that have manufactured quantities of deoxycholic acid-based formulations, which have been sold as fat reduction treatments without drug approval from the FDA or other foreign governmental regulatory body equivalents. In order to compete successfully in the aesthetics market, we will have to demonstrate that the treatment of submental fullness with ATX-101 is a worthwhile aesthetic treatment and is a superior alternative to existing therapies for the reduction of submental fat. There may be other drug or device products currently under development or being considered for development for the treatment of submental or other fat of which we are not currently aware, but which upon approval could compete directly with ATX-101. As an example, we are aware that Neothetics, Inc. (formerly known as Lithera, Inc.), has announced Phase 2b study results for LIPO-202, which is reported to be a physician administered injectable pharmaceutical product being developed for reduction of central abdominal bulging in non-obese patients through the non-ablative, non-surgical fat tissue reduction in specific locations.
In addition, a substantial portion of our target physician market is comprised of plastic surgeons who utilize surgical methods for fat reduction. Such physicians may find it more advantageous to utilize surgical techniques to remove localized fat deposits rather than an injectable biopharmaceutical therapy such as ATX-101. Additionally, multiple non-invasive technologies for the reduction of fat or "body-contouring" have received marketing clearance from the FDA. For example, Zeltiq Aesthetics, Inc. has a body-contouring system, CoolSculpting, which utilizes controlled cooling to reduce the temperature of fat cells in the treated area for the selective reduction of fat around the flanks and abdomen. Zeltiq has announced an intention to pursue FDA clearance in the submentum. Zerona, a laser energy-based product marketed by Erchonia Corporation, and Liposonix, an ultrasound energy-based product marketed by Solta Medical, Inc., have also received FDA marketing clearance. These products may be considered as alternative treatments or therapies.
50

Table of Contents
Due to less stringent regulatory requirements for devices outside of the United States, there are many more aesthetic products and procedures available for use in international markets than are approved for use in the United States. There are also fewer limitations on the claims that competitors in international markets can make about the effectiveness of their products and the manner in which they can market them. For example, Aqualyx has been issued a CE mark in Italy as a medical device that appears to be comprised of an injectable pharmacologic agent combined with ultrasound activity for the non-surgical treatment of localized adiposity. As a result, ATX-101 will face more competition in these markets than in the United States.
Many of these potential competitors are large, experienced companies that have substantially greater resources and brand recognition than we do. Competing in the aesthetic market could result in price-cutting, reduced profit margins, and limited market share, any of which would harm our business, financial condition, and results of operations.
The commercial success of ATX-101, if approved, will depend significantly on broad physician adoption and use of ATX-101.
The commercial success of ATX-101, if approved, will depend significantly on the broad adoption and use of ATX-101 by physicians for the treatment of submental fullness. Physician adoption of ATX-101 for the treatment of submental fullness, if approved, will depend on a number of factors, including:
• the safety and effectiveness of ATX-101 for the treatment of submental fullness as compared to alternative treatments or procedures;
• physician willingness to adopt a new therapy to treat submental fullness;
• patient compliance with the treatment regimen;
• overcoming any biases plastic surgeons may have toward surgical procedures for submental fat reduction;
• patient satisfaction with the results and administration of ATX-101;
• patient demand for treatment of submental fullness;
• the revenue and profitability that ATX-101 will offer a physician as compared to alternative treatments or procedures; and
• the difficulty of administering ATX-101 and any potential side effects of the administration and/or use of ATX-101.
If ATX-101 is approved for use and physicians do not broadly adopt it for the treatment of submental fullness, our financial performance will be adversely affected.
We rely completely on third-party suppliers to manufacture and distribute our clinical drug supplies for ATX-101, we intend to rely on third parties for commercial manufacturing and distribution of ATX-101 and we expect to rely on third parties for manufacturing and distribution of preclinical, clinical and commercial supplies of any future product candidates.
We do not currently have, nor do we plan to acquire, the infrastructure or capability to manufacture or distribute preclinical, clinical or commercial quantities of drug substance or drug product, including ATX-101. Facilities used by our contract manufacturers to manufacture drug substance and drug product for commercial sale must be approved by the FDA or other relevant foreign regulatory agencies pursuant to inspections that will be conducted in connection with the review of our NDA or relevant foreign regulatory submission to the applicable regulatory agency.
51

Table of Contents
We do not have direct control over the ability of our contract manufacturers to maintain adequate manufacturing capacity and capabilities to serve our needs, including quality control, quality assurance and qualified personnel. We are dependent on our contract manufacturers for compliance with all national, state and local regulatory requirements, including Current Good Manufacturing Practices, or CGMPs, for manufacture of both active drug substances and finished drug products. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and/or the strict regulatory requirements of the FDA or foreign regulatory agencies, or are in violation of other regulations governing their business, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. Furthermore, these contract manufacturers are engaged with other companies to supply and/or manufacture materials or products for such companies, which also exposes our manufacturers to regulatory risks for the production of such materials and products. As a result, failure to meet the regulatory requirements for the production of those materials and products may also affect the regulatory clearance of a contract manufacturers' facility. If the FDA or a comparable foreign regulatory agency does not approve these facilities for the manufacture of our product candidates, or if it withdraws its approval in the future, we may need to find alternative manufacturing facilities, which would negatively impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We and our contract manufacturers continue to characterize and improve manufacturing processes, certain aspects of which are complex and unique, and we may encounter difficulties with new or existing processes. For example, as we transitioned to our commercial scale fill/finish supplier, we encountered an issue with respect to the compatibility of ATX-101 with certain types of glass vials, which required identification of compatible vials, manufacturing processes, and storage conditions for ATX-101. Depending on the extent of any difficulties encountered, a similar problem in the future could lead to an interruption in clinical or commercial supply.
Our failure to be able to manufacture adequate drug substance or drug product could have an adverse effect on supply of clinical and/or finished drug in our commercial territories, and, as a result, on our operating results.
We expect to continue to depend on third-party contract manufacturers for the foreseeable future. We have entered into an exclusive agreement with Pfizer, Inc., or Pfizer, for the supply of a key raw material to manufacture synthetic deoxycholate. We currently obtain our supply of synthetic deoxycholate from Albany Molecular Research, Inc., or AMRI, and Cambridge Major Laboratories, Inc., or CML. We currently have a long-term agreement with Hospira, Inc., or Hospira, our fill/finish supplier; we have plans to qualify an additional fill/finish supplier, but failure by Hospira to supply drug product will have an adverse effect on market supply and operating results. Our supply agreements, if any, cannot however guarantee that a contract manufacturer or supplier will provide services adequate for our needs. For example, a supplier or manufacturer may go bankrupt or be acquired, and consequently be unable or unwilling to provide us with manufacturing or supply. In February 2015 Pfizer announced that it would acquire Hospira. If a contract manufacturer/supplier becomes financially distressed or insolvent, is acquired or merged, or discontinues manufacturing supply for us beyond the term of the existing agreement, if any, or for any other reason, this could result in substantial management time and expense to identify and qualify alternative manufacturers or suppliers, and could lead to an interruption in clinical or commercial supply.
In addition, we expect to depend on third-party distributors for the marketing and selling of any future products. For example, we have entered into a distribution agreement with Besse Medical, or Besse, for the distribution of ATX-101. We may depend on distributors' efforts to market our products, yet we are unable to control their efforts completely. Distributors typically sell a variety of other, non-competing products that may limit the resources they dedicate to selling our future products, if any. In addition, we may be unable to ensure that our distributors comply with all applicable laws regarding the sale of our future products. If our distributors fail to effectively market and sell our
52

Table of Contents
products, in full compliance with applicable laws, our operating results and business may suffer. Recruiting and retaining qualified third-party distributors and training them in any future product offering requires significant time and resources. Further, if our relationship with a successful distributor terminates, we may be unable to replace that distributor without disruption to our business. If we fail to maintain positive relationships with our distributors, fail to develop new relationships with other distributors, including in new markets, fail to manage, train or incentivize existing distributors effectively, or fail to provide distributors with competitive products on attractive terms, or if these distributors are not successful in their sales efforts, our revenue may decrease and our operating results, reputation and business may be harmed.
Our reliance on contract manufacturers and distributors further exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may misappropriate our trade secrets or other proprietary information.
We currently rely on a few contract manufacturers and testing labs. Failure of these vendors to supply or otherwise perform adequately can materially and adversely affect our business.
There are a limited number of providers for manufacture, testing and distribution of ATX-101, and we do not have direct control over our contract manufacturers and testing labs. Nor do we have direct control over the processes or timing for the acquisition of the raw materials necessary to manufacture, test or distribute our product candidates. If these raw materials are not available at the volumes and quantity levels required, it could have a material and adverse impact on the supply of drug substance, drug product, and finished drug product. We work closely with our contract manufacturers and testing labs to enable timely delivery of required drug substance and drug product, but these efforts may be insufficient and we may experience delays or our contract manufacturers and testing labs may be unable to provide adequate drug substance or drug product. Although we generally do not begin a clinical trial unless we believe we have a sufficient supply of a product candidate to complete the study, a delay in providing the drug supply required could delay completion of a clinical trial and could result in delays to the clinical program, regulatory approval, and the generation of revenue.
We rely on third parties such as Pfizer, the supplier of a key raw material for manufacture of the regulatory starting material, Sai Life Science, Inc. the supplier of our regulatory starting material, AMRI and CML, the suppliers of the drug substance, synthetic deoxycholate, and Hospira, the supplier of drug product, at key stages in our ATX-101 supply chain. To manufacture, test and distribute ATX-101 in the quantities that we believe will be required to meet anticipated market demand, our third-party manufacturers may need to increase capacity, which could involve significant challenges and will require additional regulatory approvals. In addition, the development of commercial-scale manufacturing capabilities may require us and our third-party manufacturers to invest substantial additional funds and hire and retain the technical personnel who have the necessary manufacturing experience. Neither we nor our third-party manufacturers may successfully complete any required increase to existing manufacturing capacity in a timely manner, or at all.
If there is a disruption to our or our third-party manufacturers' or suppliers' relevant operations, we will have no other means of producing ATX-101 until the affected facilities are restored or we or they procure and qualify alternative facilities. Additionally, any damage to or destruction of our or our third-party manufacturers' or suppliers' facilities or equipment may significantly impair our ability to manufacture ATX-101 on a timely basis.
53

Table of Contents
Manufacturing and supply of drug substance, drug product and finished drug product is a complex and technically challenging undertaking, and there is potential for failure at many points in the manufacturing, testing, quality assurance and distribution supply chain, as well as the potential for latent defects after product has been manufactured and distributed.
Manufacturing and supply of drug substance, drug product and finished drug product is technically challenging. Changes that may be made outside the purview of our direct control can have an impact on the success of our processes, on quality, and successful delivery of product to physicians. Mistakes and mishandling are not uncommon and can affect successful production and supply. Some of these risks include:
• failure of our manufacturers to follow CGMP requirements or mishandling of our product while in production or in preparation for transit;
• transportation and import/export risk, particularly given the global nature of our supply chain;
• delays in analytical results or failure of analytical techniques that we depend on for quality control and release of product;
• natural disasters, labor disputes, financial distress, lack of raw material supply, issues with facilities and equipment or other forms of disruption to business operations at our contract manufacturers/suppliers; and
• latent defects that may become apparent after product has been released and which may result in recall and destruction of drug.
We rely on third parties to conduct all our preclinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize ATX-101 or any future product candidates.
We do not have the ability to independently conduct preclinical studies or clinical trials. We rely on medical institutions, clinical investigators, contract laboratories, collaborative partners and other third parties, such as CROs, to conduct clinical trials on our drug candidates. The third parties with whom we contract for execution of our clinical trials play a significant role in the conduct of these trials and the subsequent collection and analysis of data. However, these third parties are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount, quality or timing of resources that they devote to our programs. For example, following the completion of a Phase I pharmacokinetic study of ATX-101, we discovered that the CRO utilized an incorrect isomer in a testing assay, which resulted in erroneous initial trial results. As a result, we were required to complete a new Phase I pharmacokinetic study of ATX-101. Although we rely on these third parties to conduct our preclinical studies and clinical trials, we remain responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, referred to as Good Clinical Practice, or GCP, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial subjects are adequately informed of the potential risks of participating in clinical trials.
In addition, the execution of preclinical studies and clinical trials, and the subsequent compilation and analysis of the data produced, requires coordination among various parties. In order for these functions to be carried out effectively and efficiently, it is imperative that these parties communicate and coordinate with one another. Moreover, these third parties may also have relationships with other commercial entities, some of which may compete with us. These third parties may terminate their agreements with us upon as little as 30 days prior written notice of a material breach by us that is not cured within 30 days. Many of these agreements may also be terminated by such third parties under
54

Table of Contents
certain other circumstances, including our insolvency or our failure to comply with applicable laws. In general, these agreements require such third parties to reasonably cooperate with us at our expense for an orderly winding down of services of such third parties under the agreements. If the third parties conducting our clinical trials do not perform their contractual duties or obligations, experience work stoppages, become insolvent or undergo restructuring, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical trial protocols or GCPs, or for any other reason, we may need to enter into new arrangements with alternative third parties, which could be difficult, costly, time consuming, or impossible, and our clinical trials may be extended, delayed or terminated or may need to be repeated. If any of the foregoing were to occur, we may not be able to obtain regulatory approval for or commercialize the product candidate being tested in such trials.
Our ability to market ATX-101, if approved, will be limited to use for treatment of submental fullness, and if we want to expand the indications for which we may market ATX-101, we will need to obtain additional regulatory approvals, which may not be granted.
We are currently seeking regulatory approval for ATX-101 in the United States, Canada and other international territories for the treatment of submental fullness. If ATX-101 is approved, the applicable regulatory agency will restrict our ability to market or advertise ATX-101 for other specific body areas, which could limit physician and patient adoption. We may attempt to develop, seek regulatory approval for, promote and commercialize new treatment indications and protocols for ATX-101 in the future, but we cannot predict when or if we will receive the clearances required to do so. In addition, we would be required to conduct additional clinical trials or studies to support our applications, which would be time-consuming and expensive, and may produce results that do not result in regulatory approvals. If we do not obtain additional regulatory approvals, our ability to expand our business in the United States, Canada and other international territories will be limited.
Even if ATX-101 is approved for commercialization, if there is not sufficient patient demand for ATX-101 procedures, our financial results and future prospects will be harmed.
Treatment of submental fullness with ATX-101 is an elective procedure, the cost of which must be borne by the patient, and we do not expect it to be reimbursable through government or private health insurance. The decision by a patient to elect to undergo treatment with ATX-101 may be influenced by a number of factors, such as:
• the success of any sales and marketing programs that we, or any third parties we engage, undertake, and as to which we have limited experience;
• the extent to which physicians recommend ATX-101 to their patients;
• the extent to which ATX-101 satisfies patient expectations;
• our ability to properly train physicians in the use of ATX-101 such that their patients do not experience excessive discomfort during treatment or adverse side effects;
• the cost, safety, and effectiveness of ATX-101;
• consumer sentiment about the benefits and risks of aesthetic procedures generally and ATX-101 in particular;
• the success of any direct-to-consumer marketing efforts we may initiate; and
• general consumer confidence, which may be impacted by economic and political conditions.
Our financial performance will be materially harmed if we cannot generate significant patient demand for ATX-101.
55

Table of Contents
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
As of December 31, 2014, we had 106 full-time employees. We will need to continue to expand our managerial, operational, finance and other resources in order to manage our operations and clinical trials, continue our development activities and commercialize ATX-101 in the U.S., Canada and other territories acquired in the March 2014 transaction with Bayer, or any future product candidates. Our management and personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively execute our growth strategy requires that we:
• manage our clinical trials effectively;
• identify, recruit, retain, incentivize and integrate additional employees;
• manage our internal development efforts effectively while carrying out our contractual obligations to third parties; and
• continue to improve our operational, financial and management controls, reporting systems and procedures.
Successfully transitioning the ATX-101 program we acquired from Bayer in March 2014 could require significant management attention and disrupt our business, and if we are unable to successfully transition this program the anticipated benefits of the program's acquisition may not be fully realized.
In March 2014, we announced the acquisition of rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer. We believe that the acquisition of these rights to ATX-101 will allow us to maximize the global long-term value of ATX-101. However, our ability to realize the anticipated additional benefits depends on successfully transitioning and expanding our existing ATX-101 development program into a global development program. The development of this program by us is subject to numerous risks, including operating in international markets, which will require significant resources and management attention and will subject us to regulatory, economic and political risks that are different from those in the United States. We may also encounter unexpected and more restrictive laws and regulations, including those laws governing the enforcement and ownership of intellectual property and other activities important to our business.
In addition, Bayer has been leading the clinical and regulatory development of ATX-101 in Europe and we will now be transitioning the existing clinical and regulatory data into our organization and assuming the responsibility for future development and commercialization. As a company, we do not have any prior experience securing regulatory approval of a drug product in the European Union, and we may encounter unexpected delays in the process. If we are not able to successfully transition and integrate the acquired ATX-101 program into our organization, or successfully secure regulatory approval and subsequently successfully commercialize ATX-101 outside the U.S. and Canada, the anticipated benefits of the acquisition of these rights may not be realized fully, or at all, or may take longer to realize than expected.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of ATX-101 or any future product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, and a breach of warranties, among others. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of
56

Table of Contents
our product candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
• decreased demand for ATX-101 or any future product candidates;
• injury to our reputation;
• withdrawal of clinical trial participants;
• costs to defend the related litigation;
• a diversion of management's time and our resources;
• substantial monetary awards to trial participants or patients;
• regulatory investigations, product recalls, withdrawals or labeling, marketing or promotional restrictions;
• loss of revenue; and
• the inability to commercialize ATX-101 or any future product candidates.
Our inability to obtain and maintain sufficient product liability insurance at an acceptable cost and scope of coverage to protect against potential product liability claims could prevent or inhibit the commercialization of ATX-101 or any future products we develop. We currently carry product liability insurance covering our clinical trials in the amount of $10.0 million in the aggregate. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions and deductibles, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. If and when we obtain approval for marketing ATX-101, we intend to expand our insurance coverage to include the sale of ATX-101; however, we may be unable to obtain this liability insurance on commercially reasonable terms.
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop ATX-101 or any future product candidates, conduct our clinical trials and commercialize ATX-101 or any future product candidates.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel. We are highly dependent upon our senior management, particularly our President and Chief Executive Officer, as well as our senior scientists and other members of our senior management team. The loss of services of any of these individuals could delay or prevent the successful development of our product pipeline, completion of our planned clinical trials or the commercialization of ATX-101 or any future product candidates. Although we have entered into employment agreements with our senior management team, these agreements do not provide for a fixed term of service.
Although we have not historically experienced unique difficulties attracting and retaining qualified employees, we could experience such problems in the future. For example, competition for qualified personnel in the biotechnology and pharmaceuticals field is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to hire additional personnel as we expand our clinical development and commercial activities. We may not be able to attract and retain quality personnel on acceptable terms, or at all. In addition, to the extent we
57

Table of Contents
hire personnel from other pharmaceutical or medical device companies, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output.
If we are not successful in discovering, developing, acquiring and commercializing additional product candidates, our ability to expand our business and achieve our strategic objectives would be impaired.
Although a substantial amount of our effort will focus on the continued clinical testing and potential approval of ATX-101, a key element of our strategy is to discover, develop and commercialize a portfolio of products to serve the aesthetic market. We are seeking to do so through our internal research programs and may explore strategic collaborations for the development or acquisition of new products. For example, in February 2015 we announced the acquisition of setipiprant, a potentially novel treatment for hair loss. Research programs to identify product candidates require substantial technical, financial and human resources, whether or not any product candidates are ultimately identified. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for many reasons, including the following:
• the research methodology used may not be successful in identifying potential product candidates;
• competitors may develop alternatives that render our product candidates obsolete or less attractive;
• product candidates we develop may nevertheless be covered by third parties' patents or other exclusive rights;
• a product candidate may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria;
• a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all;
• a product candidate may not be accepted as safe and effective by patients, the medical community or third-party payors, if applicable; and
• FDA or other regulatory authorities may not approve or agree with the intended use of a new product candidate.
If we fail to develop and successfully commercialize other product candidates, our business and future prospects may be harmed and our business will be more vulnerable to any problems that we encounter in developing and commercializing ATX-101.
We incur significant costs as a result of operating as a public company, and our management devotes substantial time to new compliance initiatives. We may fail to comply with the rules that apply to public companies, including Section 404 of the Sarbanes-Oxley Act of 2002, which could result in sanctions or other penalties that would harm our business.
We have incurred and will continue to incur significant legal, accounting and other expenses as a public company, including costs resulting from public company reporting obligations under the Securities Exchange Act of 1934, as amended, and regulations regarding corporate governance practices. The listing requirements of The NASDAQ Global Select Market require that we satisfy certain corporate governance requirements relating to director independence, distributing annual and interim reports, stockholder meetings, approvals and voting, soliciting proxies, conflicts of interest and a code of conduct. Our management and other personnel devote a substantial amount of time to ensure that we comply with all of these requirements. Moreover, the reporting requirements, rules and
58

Table of Contents
regulations have increased our legal and financial compliance costs and have made some activities more time-consuming and costly. Any changes made to comply with these obligations may not be sufficient to allow us to satisfy our obligations as a public company on a timely basis, or at all. These reporting requirements, rules and regulations, coupled with the increase in potential litigation exposure associated with being a public company, could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors or board committees or to serve as executive officers, or to obtain certain types of insurance, including directors' and officers' insurance, on acceptable terms.
Section 404 of The Sarbanes-Oxley Act of 2002, or Section 404, and the related rules of the Securities and Exchange Commission, or SEC, require our management and independent registered public accounting firm to report on the effectiveness of our internal control over financial reporting. However, for so long as we remain an emerging growth company as defined in the JOBS Act, we intend to continue to take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not emerging growth companies, including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404, as well as reduced reporting disclosure on executive compensation and no requirement for non-binding advisory votes on executive compensation. Once we are no longer an emerging growth company or, if prior to such date, we opt to no longer take advantage of the applicable exemption, we will be required to include an opinion from our independent registered public accounting firm on the effectiveness of our internal controls over financial reporting. We will remain an emerging growth company until the earlier of (1) December 31, 2017 (the last day of the fiscal year following the fifth anniversary of the completion of our initial public offering), (2) the last day of the fiscal year in which we have total annual gross revenue of at least $1.0 billion, (3) the last day of the fiscal year in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th, and (4) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
In addition, we are required to file accurate and timely quarterly and annual reports with the SEC under the Securities Exchange Act of 1934, as amended. In order to report our results of operations and consolidated financial statements on an accurate and timely basis, we depend on CROs to provide timely and accurate notice of their costs to us.
If we fail to maintain proper and effective internal controls over financial reporting, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, our ability to operate our business and investors' views of us and have a materially adverse effect on our stock price.
During the course of our review and testing of our internal controls, we may identify deficiencies and be unable to remediate them before we must provide the required reports. Furthermore, if we have a material weakness in our internal controls over financial reporting, we may not detect errors on a timely basis and our consolidated financial statements may be materially misstated. We or our independent registered public accounting firm, when required, may not be able to conclude on an ongoing basis that we have effective internal control over financial reporting, which could harm our operating results, cause investors to lose confidence in our reported financial information and cause the trading price of our stock to fall.
We cannot provide assurance that a material weakness will not occur in the future, or that we will be able to conclude on an ongoing basis that we have effective internal controls over financial reporting in accordance with Section 404 and the related rules and regulations of the SEC when required. A material weakness in internal control over financial reporting is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company's annual or interim consolidated financial statements will not be prevented or detected on a timely basis by the company's internal controls. If we cannot in the future
59

Table of Contents
favorably assess, or our independent registered public accounting firm, when required, is unable to provide an unqualified attestation report on, the effectiveness of our internal controls over financial reporting, investor confidence in the reliability of our financial reports may be adversely affected, which could have a material adverse effect on our stock price. In addition, any failure to report our financial results on an accurate and timely basis could result in sanctions, lawsuits, delisting of our shares from The NASDAQ Global Select Market or other adverse consequences that would materially harm our business.
Under the related agreements for the acquisition of the rights to develop and commercialize ATX-101 outside of the United States and Canada, Bayer has the right to seek indemnification from us for certain damages it suffers as a result of a material breach of the representations and warranties we made or if we fail to perform any of our other contractual obligations under these agreements, as well as for certain damages arising out of the ATX-101 program, which amounts could be significant.
We have agreed to indemnify Bayer and its affiliates against losses it suffers as a result of our material breach of representations and warranties and our other obligations in the agreements related to the acquisition of rights from Bayer to ATX-101 outside of the United States and Canada. If one or more of our representations and warranties were not true at the time we made them to Bayer, or if we fail to perform any of our other contractual obligations under these agreements, we would be in breach of the applicable agreement. In the event of a breach or failure by us to perform, Bayer has the right to seek indemnification from us for damages suffered by them as a result of such breach or failure to perform. In addition, we have agreed, subject to certain exceptions, to indemnify Bayer and its affiliates for damages it suffers as a result of our development and commercialization of ATX-101, including as a result of a personal injury or death caused by a manufacturing defect in the product produced by us or our suppliers, or a violation of any regulatory authorization related to the marketing and sale of ATX-101. The amounts for which we could become liable may be significant.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. Furthermore, the market for aesthetic medical procedures may be particularly vulnerable to unfavorable economic conditions. In particular, we do not expect ATX-101 to be reimbursed by any government or third-party payor and, as a result, demand for this product will be tied to discretionary spending levels of our targeted patient population. A severe or prolonged economic downturn, such as the most recent global financial crisis, could result in a variety of risks to our business, including, weakened demand for ATX-101, if approved, and our ability to raise additional capital when needed on acceptable terms, if at all. This is particularly true in Europe, which is undergoing a continued severe economic crisis. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our services. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
We or the third parties upon whom we depend may be adversely affected by earthquakes or other natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Our corporate headquarters and other facilities are located in the Northern Los Angeles Area, which in the past has experienced both severe earthquakes and wildfires. We do not carry earthquake insurance. Earthquakes, wildfires or other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects.
60

Table of Contents
If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as our enterprise financial systems or manufacturing resource planning and enterprise quality systems, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place currently are limited and are unlikely to prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, particularly when taken together with our lack of earthquake insurance, could have a material adverse effect on our business.
Furthermore, integral parties in our supply chain are geographically concentrated and operating from single sites, increasing their vulnerability to natural disasters or other sudden, unforeseen and severe adverse events. If such an event were to affect our supply chain, it could have a material adverse effect on our business.
Our business involves the use of hazardous materials and we and our third-party manufacturers and suppliers must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our research and development activities and our third-party manufacturers' and suppliers' activities involve the controlled storage, use and disposal of hazardous materials owned by us, including the components of our product and product candidates and other hazardous compounds. We and our manufacturers and suppliers are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. In some cases, these hazardous materials and various wastes resulting from their use are stored at our and our manufacturers' facilities pending their use and disposal. We cannot eliminate the risk of contamination, which could cause an interruption of our commercialization efforts, research and development efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we believe that the safety procedures utilized by our third-party manufacturers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance. We do not currently carry biological or hazardous waste insurance coverage.
Risks Related to Intellectual Property
We may become subject to third parties' claims alleging infringement of patents and proprietary rights or seeking to invalidate our patents or proprietary rights, which would be costly, time consuming and, if successfully asserted against us, delay or prevent the development and commercialization of ATX-101 or any future product candidates.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. We cannot assure that ATX-101 or any future product candidates will not infringe existing or future patents. Because patent applications can take many years to issue and may be confidential for 18 months or more after filing, there may be applications now pending of which we are unaware and which may later result in issued patents that we may infringe by commercializing ATX-101 or any future product candidates. Moreover, we may face claims from non-practicing entities, which have no relevant product revenue and against whom our own patent portfolio may thus have no deterrent effect.
61

Table of Contents
We may be subject to third-party claims that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages, including treble damages and attorney's fees if we are found to be willfully infringing a third party's patents. We have agreed to indemnify our former collaborator Bayer against such claims brought against it, and may undertake similar obligations on behalf of other future collaborators in the future. If a patent infringement suit were brought against us or our future collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit. As a result of patent infringement claims, or in order to avoid potential claims, we or our future collaborators may choose to seek, or be required to seek, a license from the third-party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our future collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or forced to redesign it, or to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our future collaborators are unable to enter into licenses on acceptable terms. Even if we are successful in defending such claims, infringement and other intellectual property litigation can be expensive and time-consuming to litigate and would divert management's attention from our core business. Any of these events could harm our business significantly.
In addition to infringement claims against us, if third parties prepare and file patent applications in the United States that also claim technology to which we have rights, we may have to participate in proceedings in the United States Patent and Trademark Office, or the U.S. PTO, to determine the rights to invention. We may also become involved in similar opposition proceedings in the European Patent Office or similar offices in other jurisdictions regarding our intellectual property rights with respect to our products and technology.
If our efforts to protect the proprietary nature of the intellectual property related to ATX-101 or any future product candidates are not adequate, we may not be able to compete effectively in our market.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to ATX-101 and our research and development programs. Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be uncertain. The patent applications that we own or license may fail to result in issued patents in the United States or in foreign countries. Even if the patents do successfully issue, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. For example, patents granted by the European Patent Office may be opposed by any person within nine months from the publication of the grant. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. For example, although ATX-101 contains synthetic deoxycholate, the most common form of deoxycholate is a naturally occurring substance (as are certain other aesthetic pharmaceuticals, including botulinum toxin), and is therefore not eligible for composition-of-matter patent protection in certain jurisdictions, including the United States. To the extent that naturally-occurring deoxycholate products (or other synthetic deoxycholate products) do not infringe claims in our method-of-use, formulation or other patents and patent applications, competitors may be able to offer and sell such products and compete with ATX-101. If the breadth or strength of protection provided by the patents and patent applications we hold or pursue with respect to ATX-101 or any future product candidates is challenged, then it
62

Table of Contents
could threaten our ability to commercialize ATX-101 or any future product candidates. Further, if we encounter delays in our clinical trials, the period of time during which we could market ATX-101 or any future product candidates under patent protection would be reduced. Since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to file any patent application related to our product candidates. Furthermore, an interference proceeding can be provoked by a third-party, or instituted by the U.S. PTO, to determine who was first to invent any of the subject matter covered by the patent claims of our applications.
Even where laws provide protection, costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and some of our competitors have substantially greater intellectual property portfolios than we have.
We also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that may not be patentable, processes for which patents may be difficult to obtain and/or enforce and any other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees to assign their inventions to us, and endeavor to execute confidentiality agreements with all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology, we cannot be certain that we have executed such agreements with all parties who may have helped to develop our intellectual property or who had access to our proprietary information, nor can we be certain that our agreements will not be breached. We cannot guarantee that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
Our issued patents could be found invalid or unenforceable if challenged in court.
If we or one of our future collaborators were to initiate legal proceedings against a third party to enforce a patent covering ATX-101, or one of our future products, the defendant could counterclaim that our patent is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before the U.S. PTO, even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on ATX-101 or a future product. Such a loss of patent protection would have a material adverse impact on our business.
63

Table of Contents
If we fail to comply with our obligations under our intellectual property licenses we could lose license rights that are important to our business.
We are a party to a license agreement with Los Angeles Biomedical Research Institute, or LA Biomed, pursuant to which we license key intellectual property relating to the active ingredient in ATX-101. This existing license imposes various diligence, royalty, insurance and other obligations on us. If we fail to comply with these obligations, LA Biomed may have the right to terminate the license, in which event we would not be able to develop or market ATX-101. If we lose such license rights, our business, results of operations, financial condition and prospects would be materially adversely affected. In addition, we have entered into a license agreement with Actelion and the University of Pennsylvania pursuant to which we license intellectual property relating to the use of prostaglandin D 2 receptor for the treatment of hair loss. If we fail to comply with obligations under these agreements, we may not be able to develop our potential hair loss treatment product.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involve both technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the United States has enacted and is currently implementing wide- ranging patent reform legislation. The United States Supreme Court has ruled on several patent cases relatively recently, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. PTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The U.S. PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
We do not yet have registered trademarks for commercial trade names for ATX-101 and failure to secure such registrations could adversely affect our business.
We do not yet have registered trademarks for commercial trade names for ATX-101. During trademark registration proceedings, we may receive rejections. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the U.S. PTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. Moreover, any name we propose to use with our product candidates in the United States must be approved by the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA objects to any of our proposed
64

Table of Contents
proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Risks Related to Government Regulation
The regulatory approval process is highly uncertain and we may not obtain regulatory approval for the commercialization of ATX-101 or any future product candidates.
The research, testing, manufacturing, labeling, approval, selling, import, export, marketing and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, which regulations differ from country to country. We are not permitted to market ATX-101 or any future product candidate in the United States and other countries until we receive approval of an NDA from the FDA or foreign equivalents. We have not obtained marketing approval for ATX-101 anywhere in the world. Obtaining regulatory approval of an NDA or foreign equivalent can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable United States and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions or other actions, including:
• warning letters;
• civil and criminal penalties;
• injunctions;
• withdrawal of approved products;
65

Table of Contents
• product seizure or detention;
• product recalls;
• total or partial suspension of production; and
• refusal to approve pending NDAs or supplements to approved NDAs.
Prior to obtaining approval to commercialize a drug candidate in the United States or abroad, we must demonstrate with substantial evidence from well controlled clinical trials, and to the satisfaction of the FDA or other foreign regulatory agencies, that such drug candidates are safe and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. Even if we believe the preclinical or clinical data for our drug candidates are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities. Administering drug candidates to humans may produce undesirable side effects, which could interrupt, delay or halt clinical trials and result in the FDA or other regulatory authorities denying approval of a drug candidate for any or all targeted indications.
Regulatory approval of an NDA or NDA supplement, or foreign equivalents, is not guaranteed, and the approval process is expensive and may take several years. The FDA and other foreign regulatory authorities also have substantial discretion in the approval process. Despite the time and expense exerted, failure can occur at any stage, and we or our future collaborators could encounter problems that cause us to abandon or repeat clinical trials, or perform additional preclinical studies and clinical trials. The number of preclinical studies and clinical trials that will be required for approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to address, and the regulations applicable to any particular drug candidate. The FDA and other foreign regulatory authorities can delay, limit or deny approval of a drug candidate for many reasons, including, but not limited to, the following:
• a drug candidate may not be deemed safe or effective;
• they may not find the data from preclinical studies and clinical trials sufficient;
• they might not approve our third party manufacturers' processes or facilities; or
• they may change their approval policies or adopt new regulations.
If ATX-101 or any future product candidate fails to demonstrate safety and efficacy in clinical trials or does not gain regulatory approval, our business and results of operations will be materially and adversely harmed.
Even if we receive regulatory approval for ATX-101 or any future product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense. Additionally, any product candidates, if approved, could be subject to labeling and other restrictions and market withdrawal and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
Any regulatory approvals that we receive for ATX-101 or any future product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase IV clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. In addition, if the applicable regulatory agency approves ATX-101 or any future product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, and continued compliance with CGMP and GCP,
66

Table of Contents
for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with ATX-101 or any future product candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls;
• fines, warning letters or holds on clinical trials;
• refusal by the FDA or other regulatory agencies to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals;
• product seizure or detention, or refusal to permit the import or export of products; and
• injunctions or the imposition of civil or criminal penalties.
Regulatory agency policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business.
Our failure to obtain regulatory approvals in foreign jurisdictions for ATX-101 would prevent us from marketing ATX-101 internationally.
In order to market any product in the EEA (which is composed of the 28 Member States of the European Union plus Norway, Iceland and Liechtenstein) and many other foreign jurisdictions, separate regulatory approvals are required. In the EEA, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. Before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
The approval procedures vary among countries and can involve additional clinical testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other foreign countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not be able to file for regulatory approvals or to do so on a timely basis, and even if we do file we may not receive necessary approvals to commercialize our products in any market.
If approved, ATX-101 or any future products may cause or contribute to adverse medical events that we are required to report to regulatory agencies and if we fail to do so, we could be subject to sanctions that would materially harm our business.
Some participants in our clinical studies have reported adverse effects after being treated with ATX-101. If we are successful in commercializing ATX-101 or any other products, FDA and foreign regulatory agency regulations require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of
67

Table of Contents
the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA or a foreign regulatory agency could take action including criminal prosecution, the imposition of civil monetary penalties, seizure of our products, or delay in approval or clearance of future products.
We may in the future be subject to various federal, state and foreign laws pertaining to health care fraud and abuse, including anti-kickback, self-referral, false claims and fraud laws, and any violations by us of such laws could result in fines or other penalties.
While we do not expect that ATX-101, if approved, will be covered for patients in whole or in part by Medicare, Medicaid or other federal and foreign healthcare programs, we may still be subject to the various U.S. federal, state and foreign laws intended to prevent health care fraud and abuse. The federal anti-kickback statute prohibits the offer, receipt, or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or in part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. Many states have similar laws that apply to their state health care programs as well as private payors. Violations of the anti-kickback laws can result in exclusion from federal health care programs and substantial civil and criminal penalties.
The federal False Claims Act, or FCA, imposes liability on persons who, among other things, present or cause to be presented false or fraudulent claims for payment by a federal health care program. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. The FCA includes a whistleblower provision that allows individuals to bring actions on behalf of the federal government and share a portion of the recovery of successful claims. If our marketing or other arrangements were determined to violate anti-kickback or related laws, including the FCA, then our revenues could be adversely affected, which would likely harm our business, financial condition, and results of operations.
State and federal authorities have aggressively targeted pharmaceutical and medical technology companies for alleged violations of these anti-fraud statutes, based on improper research or consulting contracts with doctors, certain marketing arrangements that rely on volume-based pricing, off-label marketing schemes, and other improper promotional practices. Companies targeted in such prosecutions have paid substantial fines in the hundreds of millions of dollars or more, have been forced to implement extensive corrective action plans, and have often become subject to consent decrees severely restricting the manner in which they conduct their business. If we become the target of such an investigation or prosecution based on our contractual relationships with providers or institutions, or our marketing and promotional practices, we could face similar sanctions, which would materially harm our business.
Also, the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business. We cannot assure you that our internal control policies and procedures will protect us from reckless or negligent acts committed by our employees, contractors, future distributors, partners, future or former collaborators or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties, or prosecution and have a negative impact on our business, results of operations and reputation.
We will also be subject to similar laws in foreign countries where we may conduct business. For example, within the EU, the control of unlawful marketing and promotional activities is a matter of
68

Table of Contents
national law in each of the member states. The member states of the EEA closely monitor perceived unlawful marketing and promotional activities by companies. We could face civil, criminal and administrative sanctions if any member state determines that we have breached our obligations under its national laws. Industry associations also closely monitor the activities of member companies. If these organizations or authorities name us as having breached our obligations under their regulations, rules or standards, our reputation would suffer and our business and financial condition could be adversely affected.
Legislative or regulatory healthcare reforms in the United States may make it more difficult and costly for us to obtain regulatory clearance or approval of ATX-101 or any future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory clearance or approval, manufacture, and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of ATX-101 or any future product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
• changes to manufacturing, testing or distribution methods;
• recall, replacement, or discontinuance of one or more of our products; and
• additional record keeping.
Each of these would likely entail substantial time and cost and could materially harm our business and our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition, and results of operations.
Risks Related to Our Common Stock
Our stock price may be volatile and investors in our common stock could incur substantial losses.
From October 11, 2012, the first day of trading of our common stock, to December 31, 2014, our stock has had low and high closing sales prices in the range of $19.05 to $55.98 per share. The trading price of our common stock is highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include those discussed in this "Risk Factors" section of this Annual Report on Form 10-K and others such as:
• announcements of regulatory approval or disapproval of ATX-101 or any future product candidates;
• delays in the commercialization of ATX-101 or any future product candidates;
• negative perceptions with regard to the acquisition of rights to ATX-101 from Bayer;
• results from, and any delays in, our trials for ATX-101, or any other future clinical development programs;
• changes in revenue and earnings estimates or recommendations by securities analysts;
• changes in financial estimates or guidance, including our ability to meet our future revenue and operating profit or loss estimates or guidance;
69

Table of Contents
• failure or discontinuation of any of our research and development programs;
• announcements relating to future licensing, collaboration or development agreements;
• acquisitions and sales of new products, technologies or businesses;
• manufacturing and supply issues related to our product candidates for clinical trials or future product candidates for commercialization;
• quarterly variations in our results of operations or those of our future competitors;
• announcements by us or our competitors of new products, significant contracts, commercial relationships, acquisitions or capital commitments;
• developments with respect to intellectual property rights;
• our commencement of, or involvement in, litigation;
• any major changes in our board of directors or management;
• new legislation in the United States or other foreign territories relating to the sale or pricing of pharmaceuticals;
• FDA or other U.S. or foreign regulatory actions affecting us or our industry;
• product liability claims or other litigation or public concern about the safety of our drug candidates or future drugs;
• market conditions in the pharmaceutical, biopharmaceutical and biotechnology sectors; and
• general economic conditions in the United States and abroad.
In addition, the stock markets in general, and the markets for pharmaceutical, biopharmaceutical and biotechnology stocks in particular, have experienced extreme volatility that may have been unrelated to the operating performance of the issuer. These broad market fluctuations may adversely affect the trading price or liquidity of our common stock. In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business.
We are an "emerging growth company" and as a result of the reduced disclosure and governance requirements applicable to emerging growth companies, our common stock may be less attractive to investors.
We are an "emerging growth company," as defined in the JOBS Act, and we are taking advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we are relying on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earlier of (1) December 31, 2017 (the last day of the fiscal year following the fifth anniversary of our initial public offering), (2) the last day of the fiscal year in which we have total annual gross revenue of at least $1.0 billion, (3) the last day of the fiscal year in which we are deemed
70

Table of Contents
to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th, and (4) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
If we sell or issue shares of our common stock in future financings or transactions, stockholders may experience immediate dilution and, as a result, our stock price may decline.
We may from time to time issue additional shares of common stock at a discount from the current trading price of our common stock. As a result, our stockholders would experience immediate dilution upon the purchase or issuance of any shares of our common stock sold at such discount. In addition, as opportunities present themselves, we may enter into financing or similar arrangements in the future, including the issuance of debt securities, preferred stock or common stock. If we issue common stock or securities convertible into common stock, our common stockholders would experience additional dilution and, as a result, our stock price may decline.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
As of February 23, 2015, our executive officers, directors, and their respective affiliates beneficially owned approximately 9.5% of our outstanding voting stock. These stockholders have the ability to influence us through this ownership position and may be able to determine all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that existing stockholders may feel are in their best interest.
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline. As of February 23, 2015, we have a total of approximately 22.8 million shares of common stock outstanding. In addition, as of February 23, 2015, approximately 5.7 million shares of common stock were either subject to outstanding stock awards or reserved for future issuance under our equity incentive plans are eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 and Rule 701 under the Securities Act. If these additional shares of common stock are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could delay or prevent changes in control or changes in our management without the consent of our board of directors. These provisions include the following:
• a classified board of directors with three-year staggered terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors;
• no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates;
• the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors;
71

Table of Contents
• the ability of our board of directors to authorize the issuance of shares of preferred stock and to determine the price and other terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquiror;
• the ability of our board of directors to alter our bylaws without obtaining stockholder approval;
• the required approval of at least 66 2 / 3 % of the shares entitled to vote at an election of directors to adopt, amend or repeal our bylaws or repeal the provisions of our amended and restated certificate of incorporation regarding the election and removal of directors;
• a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders;
• the requirement that a special meeting of stockholders may be called only by the chairman of the board of directors, the chief executive officer, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and
• advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders' meeting, which may discourage or deter a potential acquiror from conducting a solicitation of proxies to elect the acquiror's own slate of directors or otherwise attempting to obtain control of us.
In addition, these provisions apply even if we were to receive an offer that some stockholders may consider beneficial.
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation may not, in general, engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the board of directors has approved the transaction.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful third-party claims against us and may reduce the amount of money available to us.
Our amended and restated certificate of incorporation and amended and restated bylaws provide that we will indemnify our directors and officers, in each case to the fullest extent permitted by Delaware law.
In addition, as permitted by Section 145 of the Delaware General Corporation Law, our amended and restated bylaws and our indemnification agreements that we have entered into with our directors and officers provide that:
• We will indemnify our directors and officers for serving us in those capacities or for serving other business enterprises at our request, to the fullest extent permitted by Delaware law. Delaware law provides that a corporation may indemnify such person if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the registrant and, with respect to any criminal proceeding, had no reasonable cause to believe such person's conduct was unlawful.
• We may, in our discretion, indemnify employees and agents in those circumstances where indemnification is permitted by applicable law.
• We are required to advance expenses, as incurred, to our directors and officers in connection with defending a proceeding, except that such directors or officers shall undertake to repay such advances if it is ultimately determined that such person is not entitled to indemnification.
72

Table of Contents
• We will not be obligated pursuant to our amended and restated bylaws to indemnify a person with respect to proceedings initiated by that person against us or our other indemnitees, except with respect to proceedings authorized by our board of directors or brought to enforce a right to indemnification.
• The rights conferred in our amended and restated bylaws are not exclusive, and we are authorized to enter into indemnification agreements with our directors, officers, employees and agents and to obtain insurance to indemnify such persons.
• We may not retroactively amend our amended and restated bylaw provisions to reduce our indemnification obligations to directors, officers, employees and agents.
Our employment agreements with our executive officers may require us to pay severance benefits to any of those persons who are terminated in connection with a change in control of us, which could harm our financial condition or results.
Certain of our executive officers are parties to employment agreements that contain change in control and severance provisions providing for cash payments for severance and other benefits and acceleration of vesting of stock options in the event of a termination of employment in connection with a change in control. The accelerated vesting of options could result in dilution to our existing stockholders and harm the market price of our common stock. The payment of these severance benefits could harm our financial condition and results. In addition, these potential severance payments may discourage or prevent third parties from seeking a business combination with us.
We do not currently intend to pay dividends on our common stock, and, consequently, our stockholders ability to achieve a return on their investment will depend on appreciation in the price of our common stock.
We do not currently intend to pay any cash dividends on our common stock for the foreseeable future. We currently intend to invest our future earnings, if any, to fund our growth. Additionally, the terms of our credit facility restrict our ability to pay dividends. Therefore, our stockholders are not likely to receive any dividends on our common stock for the foreseeable future. Since we do not intend to pay dividends, our stockholders ability to receive a return on their investment will depend on any future appreciation in the market value of our common stock. There is no guarantee that our common stock will appreciate or even maintain the price at which our holders have purchased it.
ITEM 1B. Unresolved Staff Comments.
Not applicable.
ITEM 2. Properties.
Our corporate headquarters are located in Westlake Village, California, where we lease and occupy 33,198 square feet of office space. The current terms of our lease expire July 31, 2017, with an option to renew for an additional 36 months.
ITEM 3. Legal Proceedings.
We may from time to time be involved in various legal proceedings of a character normally incident to the ordinary course of our business. We are not currently a party to any material litigation or other material legal proceedings.
ITEM 4. Mine Safety Disclosures.
Not applicable.
73

Table of Contents
PART II
ITEM 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Our common stock has been publicly traded on The NASDAQ Global Select Market under the symbol "KYTH" since our initial public offering on October 11, 2012. Prior to that time, there was no public market for our common stock. The following table sets forth on a per share basis, for the periods indicated, the low and high sale intraday prices of our common stock as reported by The NASDAQ Global Select Market.
Holders
As of February 23, 2015, there were approximately 36 holders of record of our common stock. This number does not include beneficial owners whose shares are held by nominees in street name.
Dividend Policy
We have never declared or paid cash dividends on our capital stock. We intend to retain all available funds and any future earnings, if any, to fund the development and expansion of our business and we do not anticipate paying any cash dividends in the foreseeable future. In addition, unless waived, the terms of our credit facility with Lighthouse Capital Partners VI, L.P. prohibit us from paying any cash dividends. Any future determination related to dividend policy will be made at the discretion of our board of directors.
Performance Graph
This graph is not "soliciting material," is not deemed "filed" with the SEC and is not to be incorporated by reference into any filing of KYTHERA Biopharmaceuticals, Inc. under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
The following graph shows the total stockholder return of an investment of $100 in cash on October 11, 2012 (the first day of trading of our common stock), through December 31, 2014 for (i) our common stock, (ii) the NASDAQ Composite Index (U.S.) and (iii) the NASDAQ Biotechnology Index. Pursuant to applicable Securities and Exchange Commission rules, all values assume reinvestment of the full amount of all dividends, however no dividends have been declared on our common stock to date. The stockholder return shown on the graph below is not necessarily indicative
74
High Low Year ended December 31, 2013 First Quarter $ 31.49 $ 14.07 Second Quarter $ 27.33 $ 18.56 Third Quarter $ 47.50 $ 23.03 Fourth Quarter $ 47.85 $ 35.25 Year ended December 31, 2014 First Quarter $ 56.36 $ 33.67 Second Quarter $ 40.22 $ 29.50 Third Quarter $ 40.98 $ 31.39 Fourth Quarter $ 39.78 $ 29.86

Table of Contents
of future performance, and we do not make or endorse any predictions as to future stockholder returns.
Comparison of Cumulative Total Return*
Among Kythera Biopharmaceuticals, Inc., the NASDAQ Composite Index, and the NASDAQ Biotechnology Index
* Assume $100 invested in stock or index on October 11, 2012 including reinvestment of dividends
Recent Sales of Unregistered Securities
From January 1, 2014 through December 31, 2014, we did not issue any securities in a transaction not registered under the Securities Act that was not been previously disclosed in a Quarterly Report on Form 10-Q or Current Report on Form 8-K.
Use of Proceeds
On October 10, 2012, the Securities and Exchange Commission declared effective our registration statement on Form S-1 (File No. 333-181476), as amended, filed in connection with the initial public offering of our common stock. As of December 31, 2014, all of the proceeds from our initial public offering have been applied.
75
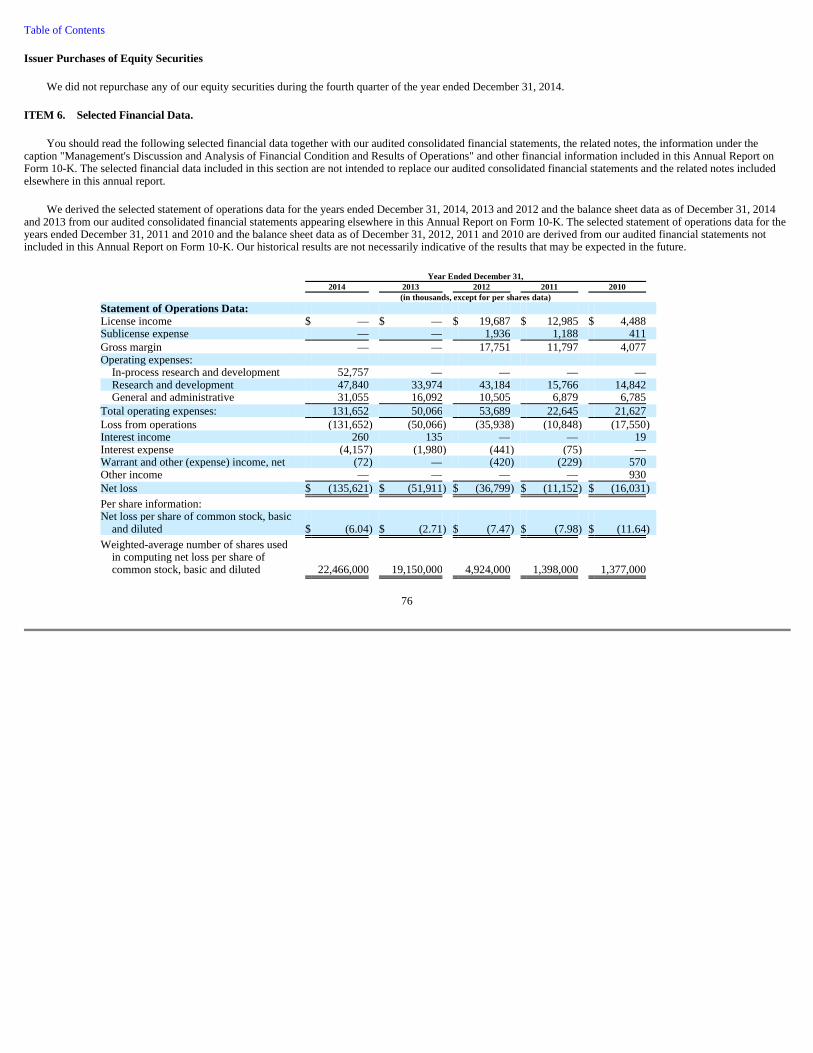
Table of Contents
Issuer Purchases of Equity Securities
We did not repurchase any of our equity securities during the fourth quarter of the year ended December 31, 2014.
ITEM 6. Selected Financial Data.
You should read the following selected financial data together with our audited consolidated financial statements, the related notes, the information under the caption "Management's Discussion and Analysis of Financial Condition and Results of Operations" and other financial information included in this Annual Report on Form 10-K. The selected financial data included in this section are not intended to replace our audited consolidated financial statements and the related notes included elsewhere in this annual report.
We derived the selected statement of operations data for the years ended December 31, 2014, 2013 and 2012 and the balance sheet data as of December 31, 2014 and 2013 from our audited consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K. The selected statement of operations data for the years ended December 31, 2011 and 2010 and the balance sheet data as of December 31, 2012, 2011 and 2010 are derived from our audited financial statements not included in this Annual Report on Form 10-K. Our historical results are not necessarily indicative of the results that may be expected in the future.
76
Year Ended December 31, 2014 2013 2012 2011 2010 (in thousands, except for per shares data) Statement of Operations Data: License income $ — $ — $ 19,687 $ 12,985 $ 4,488 Sublicense expense — — 1,936 1,188 411 Gross margin — — 17,751 11,797 4,077 Operating expenses:
In-process research and development 52,757 — — — — Research and development 47,840 33,974 43,184 15,766 14,842 General and administrative 31,055 16,092 10,505 6,879 6,785
Total operating expenses: 131,652 50,066 53,689 22,645 21,627 Loss from operations (131,652 ) (50,066 ) (35,938 ) (10,848 ) (17,550 ) Interest income 260 135 — — 19 Interest expense (4,157 ) (1,980 ) (441 ) (75 ) — Warrant and other (expense) income, net (72 ) — (420 ) (229 ) 570 Other income — — — — 930 Net loss $ (135,621 ) $ (51,911 ) $ (36,799 ) $ (11,152 ) $ (16,031 )
Per share information: Net loss per share of common stock, basic
and diluted $ (6.04 ) $ (2.71 ) $ (7.47 ) $ (7.98 ) $ (11.64 )
Weighted-average number of shares used in computing net loss per share of common stock, basic and diluted 22,466,000 19,150,000 4,924,000 1,398,000 1,377,000
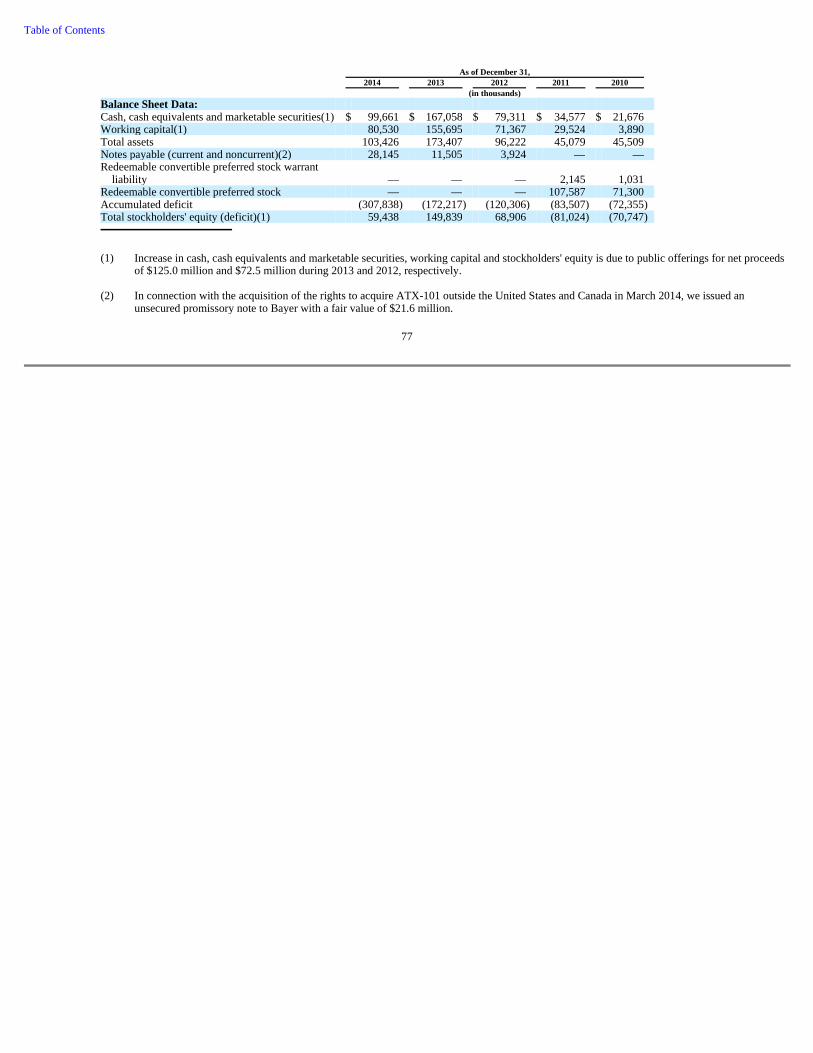
Table of Contents
77
As of December 31, 2014 2013 2012 2011 2010 (in thousands) Balance Sheet Data: Cash, cash equivalents and marketable securities(1) $ 99,661 $ 167,058 $ 79,311 $ 34,577 $ 21,676 Working capital(1) 80,530 155,695 71,367 29,524 3,890 Total assets 103,426 173,407 96,222 45,079 45,509 Notes payable (current and noncurrent)(2) 28,145 11,505 3,924 — — Redeemable convertible preferred stock warrant
liability — — — 2,145 1,031 Redeemable convertible preferred stock — — — 107,587 71,300 Accumulated deficit (307,838 ) (172,217 ) (120,306 ) (83,507 ) (72,355 ) Total stockholders' equity (deficit)(1) 59,438 149,839 68,906 (81,024 ) (70,747 )
(1) Increase in cash, cash equivalents and marketable securities, working capital and stockholders' equity is due to public offerings for net proceeds of $125.0 million and $72.5 million during 2013 and 2012, respectively.
(2) In connection with the acquisition of the rights to acquire ATX-101 outside the United States and Canada in March 2014, we issued an unsecured promissory note to Bayer with a fair value of $21.6 million.

Table of Contents
ITEM 7. Management's Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of financial condition and results of operations together with the section entitled "Selected Financial Data" and our consolidated financial statements and related notes included elsewhere in this Annual Report on Form 10-K. This discussion and other parts of this Annual Report contain forward-looking statements that involve risk and uncertainties, such as statements of our plans, objectives, expectations and intentions. Our actual results could differ materially from those discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in the "Risk Factors" section in Part I Item IA.
Overview
We are a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of novel prescription products for the aesthetic medicine market. Our objective is to develop first-in-class, prescription products using an approach that relies on the scientific rigor of biotechnology to address unmet needs in the rapidly-growing market for aesthetic medicine. Our initial focus is on the facial aesthetics market, which comprises the majority of the aesthetic medicine market. Our product candidate, ATX-101, is a potential first-in-class, injectable drug in late-stage development for the treatment of submental fullness, which commonly presents as an undesirable "double chin." Based on clinical trials conducted to date, ATX-101 has exhibited significant, meaningful and long-lasting results in the reduction of submental fat. These results correspond with patient satisfaction measures demonstrating meaningful improvement in perceived chin appearance. If approved by applicable regulatory authorities, we believe ATX-101 will be an attractive solution for the treatment of submental fullness, representing a new product category within the rapidly growing facial aesthetics market. ATX-101 is our only product candidate and we are substantially dependent on its regulatory approval and successful commercialization.
Since commencing operations in August 2005, we have devoted substantially all our efforts to identify and develop products for the aesthetics market, recruiting personnel and raising capital. We have devoted predominantly all of our resources to the preclinical and clinical development of ATX-101 and we have not generated any revenue from product sales. On May 12, 2014, we submitted our New Drug Application ("NDA") with the U.S. Food and Drug Administration ("FDA"), which was accepted for filing by the FDA on July 10, 2014. The Company was notified by the Dermatology and Ophthalmic Advisory Committee of the FDA that it is scheduled to review the NDA in a half-day meeting on the morning of March 9, 2015. The NDA will be subject to a standard review with a Prescription Drug User Fee Act (PDUFA) action date of May 13, 2015. The PDUFA date is the goal date for the FDA to complete its review of the NDA. We submitted a New Drug Submission ("NDS") with Health Canada in August 2014, a Marketing Authorization Application ("MAA") to Swissmedic in October 2014 and a drug application with Australia's Therapeutic Goods Administration ("TGA") in February 2015, seeking approval for ATX-101 as an injectable treatment of submental fullness. Historically we have funded substantially all of our operations through the sale and issuance of our common and preferred stock, convertible debt, amounts received from U.S. Government grants and pursuant to our collaboration arrangement with Bayer and borrowings under our existing credit facility.
In March 2014, we entered into an agreement whereby we acquired the rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer in exchange for 698,103 shares of KYTHERA common stock with a fair value of $31.4 million and a $51.0 million unsecured promissory note with a fair value of $21.6 million. The promissory note, which is subordinated to amounts owed under our existing credit facility, bears interest at 5% per annum and is payable no later than 2024. Additionally, Bayer is eligible to receive up to $123.8 million upon the achievement of certain long-term sales milestone payments on annual sales of ATX-101 outside of the United States and Canada. The transaction was accounted for as an asset acquisition and, as a result, a one-time
78

Table of Contents
charge to in-process research and development of $52.8 million was recognized during the first quarter of 2014.
We believe the acquisition of these rights is a transformative opportunity for KYTHERA and that we have the right leadership and operational team to shape the future path of ATX-101. We continue to evaluate on a territory-by-territory basis where and how to best maximize the value of ATX-101. As a result of acquiring the rights to develop and commercialize ATX-101 outside of the United States and Canada, we experienced modest increases in our operating expense and cash usage for regulatory costs outside the United States since the acquisition.
In February 2015, we entered into licensing arrangements with Actelion and University of Pennsylvania pursuant to which we obtained exclusive worldwide rights to setipiprant, a clinical-stage selective and potent oral antagonist to the prostaglandin D 2 (PGD 2 ) receptor and exclusive worldwide rights to certain patent rights
owned by the University of Pennsylvania covering the use of PGD 2 receptor antagonists for the treatment of hair loss. For additional discussion, refer to Note 13, "Subsequent Events (unaudited)".
We have never been profitable and, as of December 31, 2014, we had an accumulated deficit of $307.8 million. We incurred net losses of $135.6 million, $51.9 million and $36.8 million for the years ended December 31, 2014, 2013 and 2012, respectively. We may continue to incur net operating losses for at least the next several years as we advance ATX-101 through clinical development, seek regulatory approval, prepare for and, if approved, proceed to commercialization, and develop other clinical assets, if any. We have no manufacturing and distribution facilities and all of our manufacturing and distribution activities are contracted out to third parties. Additionally, we currently utilize third-party clinical research organizations, or CROs, to carry out our clinical development and we do not yet have a sales organization. We expect to seek additional funding to support our operating activities, especially as we approach anticipated regulatory approval in the United States and Canada and possibly internationally, and begin to establish our sales capabilities. Adequate funding may not be available to us on acceptable terms, or at all. Our failure to obtain sufficient funds on acceptable terms when needed could have a material adverse effect on our business, results of operations, and financial condition.
Financial Overview
Revenue
We have not generated any revenue from product sales. In the future, if ATX-101 is approved for commercial sale, we may generate revenue from product sales.
To date, all of our revenue has been derived from license fees we received pursuant to our collaboration arrangement with Bayer. In May 2012, we recorded $15.8 million in revenue for a payment received from Bayer triggered by Bayer's decision to pursue continued development and regulatory approval for ATX-101 after receipt of positive results from the European Phase III clinical trials for ATX-101. As of March 2014, we acquired rights to ATX-101 outside of the United States and Canada from Bayer and, as a result, no longer expect future revenues from this license agreement.
Even if ATX-101 is approved for commercial sale, we do not expect to generate revenue from product sales until at least the latter part of 2015, if at all. If ATX-101 is approved by the FDA, we expect to access the market through a focused, specialized sales force in the United States. If approved for sale, we intend to focus our initial marketing of ATX-101 on those core dermatologists, plastic surgeons and facial plastic surgeons we identify as having substantial experience in performing facial injectable procedures. If we fail to complete the development of ATX-101, or other product candidates, in a timely manner or to obtain regulatory approval, our ability to generate future revenue, and our results of operations and financial position, would be materially adversely affected.
79

Table of Contents
In-Process Research and Development
In March 2014, we incurred a one-time charge to in-process research and development of $52.8 million related to obtaining the rights to ATX-101 outside of the United States and Canada from Bayer. See Note 8 to the Consolidated Financial Statements, "Commitments, Collaborations and Contingencies" for a more detailed discussion of this acquisition.
Research and Development Expenses
We expense all research and development costs in the periods in which they are incurred. Major components of our research and development costs are personnel costs, including cash and stock-based compensation expense, pre-clinical studies, clinical trials and related manufacturing, materials and supplies, regulatory affairs activities, medical affairs and fees paid to consultants and other entities that conduct certain research and development activities on our behalf. To date, our research and development expenses have related predominately to the development of ATX- 101. In the years ended December 31, 2014, 2013 and 2012 we incurred $47.8 million, $34.0 million and $43.2 million, respectively, in research and development expenses, excluding the charge for in-process research and development in 2014. Since inception through December 31, 2014, we have incurred approximately $174.6 million in research and development expenses related to the development of ATX-101, excluding cash and stock- based compensation expenses but including the charge for in-process research and development. We do not allocate cash and stock-based compensation expense to individual product candidates, as we are organized and record expense by functional department and our employees may allocate time to more than one development project. We do not utilize a formal time allocation system to capture expenses on a project-by-project basis.
Conducting significant research and development is central to our business and strategy. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and greater duration of late stage clinical trials as compared to earlier clinical and preclinical development. However, if ATX-101 is approved, elements of manufacturing costs attributed to the future production of ATX-101 will be capitalized as inventory and expensed as cost of goods when sold. We expect our research and development expenses, excluding the charge for in-process research and development, will be similar to 2014 as we complete our Advisory Committee and NDA approval process, continue to develop our medical affairs capability, establish commercial manufacturing and supply chain prior to approval, continue with regulatory submissions outside the United States and Canada, complete Phase IIIb trials started in 2014, and incur costs related to our planned hair loss prevention program based on the intellectual property acquired from Actelion and the University of Pennsylvania in 2015.
General and Administrative Expenses
Our general and administrative costs primarily consist of personnel costs, including cash compensation and stock-based compensation expense, associated with our executive, accounting and finance, legal, marketing and human resources departments. Other general and administrative expenses include costs in connection with patent filing, prosecution and defense, facility costs and professional fees for legal, consulting, marketing, audit and tax services. For the years ended December 31, 2014, 2013 and 2012, our general and administrative expenses totaled approximately $31.1 million, $16.1 million and $10.5 million, respectively. We expect our general and administrative costs will rise in 2015 as we increase our headcount and expand our support staffing, realize the full year's effect of occupying larger facility space leased in 2014, hire sales leadership, and, if approved, field based sales representatives, establish and complete commercial infrastructure including information technology systems and personnel support for the commercial organization, and other activities to support our company growth as we prepare for a potential commercial launch of ATX-101, both in the United States and internationally.
80

Table of Contents
Interest Expense
Interest expense relates to the interest on the outstanding balances of the notes payable. Interest expense increased significantly as compared to 2013 due to the additional interest expense on the note payable to Bayer issued in March 2014. Interest expense in 2015 should be similar to 2014.
Former Collaboration Arrangement with Bayer
In August 2010, we entered into a license agreement with Bayer Consumer Care AG and a related collaboration agreement with Bayer's affiliate, Intendis GmbH. We refer to these agreements jointly as our collaboration arrangement with Bayer, and we refer to Bayer Consumer Care AG and Intendis GmbH jointly as Bayer. Pursuant to our collaboration arrangement, we licensed to Bayer all of the development and commercial rights to ATX-101 outside the United States and Canada. In connection with establishing the collaboration arrangement, we received upfront payments of $43.6 million in 2010 comprised of license fees and amounts to fund certain further global development activities of ATX-101. In May 2012, we received a $15.8 million payment from Bayer triggered by Bayer's decision to pursue continued development and regulatory approval for ATX-101 after receipt of positive results from the European Phase III clinical trials for ATX-101. In addition, in May 2012, we also received $17.4 million from Bayer to fund certain further Bayer global development activities of ATX-101 under the terms of the collaboration arrangement.
In March 2014, we acquired the rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer. As a result of this agreement, Bayer was issued 698,103 shares of our common stock with a fair value of $31.4 million and we issued a note payable for $51.0 million, but accounted for it at its fair market value of $21.6 million. The promissory note, which is subordinated to amounts owed under our existing credit facility, bears interest at 5% per annum and is payable no later than 2024. Additionally, Bayer is eligible to receive up to $123.8 million upon the achievement of certain long-term sales milestone payments on annual sales of ATX-101 outside of the United States and Canada. We accounted for this transaction as an asset acquisition. Since the acquired rights represent acquired in-process research and development with no alternative future uses, we recorded a one-time charge to in-process research and development of $52.8 million as a result of expensing the resulting value assigned to such rights.
License Fees
License fees received in 2010 of approximately $21.3 million were deferred and recognized on a straight-line basis over the expected period of substantial involvement in the collaboration activities that were required to be conducted relative to the upfront license fee and development funds received from Bayer and completion of which was a condition to Bayer's decision to pursue continued development and regulatory approval for ATX-101. These activities were completed as of May 31, 2012, and pursuant to the acquisition of rights from Bayer to ATX-101 outside the U.S. and Canada we will not receive future license fees from Bayer.
Collaboration Development Funds
Additionally, we received approximately $39.6 million to fund certain further global development activities of ATX-101, which were recorded as restricted cash and deferred development funds and were an offset to research and development expenses as the restricted cash was utilized to fund development activities. Amounts recognized as offsets to research and development expenses were $3.6 million, $11.7 million, and $10.1 million for the years ended December 31, 2014, 2013 and 2012, respectively.
81

Table of Contents
Los Angeles Biomedical Research Institute
We entered into a license agreement with the Los Angeles Biomedical Research Institute, or LA Biomed, in August 2005, which granted us exclusive worldwide rights to key intellectual property for the active ingredient in ATX-101. As part of this license agreement, we incur sublicense fees equal to 10% of any non-royalty sublicense income, up to a total of an aggregate of $5.0 million. We are obligated to pay LA Biomed low- to mid-single digit royalties on net product sales of ATX-101. Additionally, we will incur a milestone payment of $0.5 million upon initial receipt of marketing approval.
In August 2010, due to the receipt of the license fee income from Bayer, we incurred non-royalty sublicense fees of $2.0 million, which was deferred and recorded as sublicense expense on a straight-line basis over the same period as the license income was recorded. During 2010, we made payments to LA Biomed in cash and stock totaling $0.4 million related to the non-royalty sublicense fee incurred and the remaining $1.6 million was paid upon our IPO in 50% cash and 50% stock. Due to the receipt of the $15.8 million contingent event-based payment from Bayer in May 2012, we incurred an additional non-royalty sublicense fee of $1.6 million due to LA Biomed which was paid in 50% cash and 50% stock.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of our consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of our consolidated financial statements as well as the reported revenues and expenses during the reported periods. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not apparent from other sources. Actual results may differ materially from these estimates.
While our significant accounting policies are described in the notes to our consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K, we believe that the following critical accounting policies are most important to understanding and evaluating our reported financial results.
Revenue Recognition
To date, we have only recognized revenue derived from license fees pursuant to our license agreement with Bayer executed in August 2010. In the future, we may receive revenue from the sale of our products, if approved. We recognize revenue when all of the following four criteria are present: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured. For arrangements that involve the delivery or performance of multiple products, services and/or rights to use assets, the elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has "standalone value" to the collaborator and whether there is objective and reliable evidence of the fair value of the undelivered obligation(s). The estimated selling price of each deliverable is determined using the following hierarchy of values: (i) vendor-specific objective evidence of fair value, (ii) third-party evidence of selling price (TPE) and (iii) best estimate of selling price (BESP). The BESP reflects our best estimate of what the selling price would be if the deliverable was regularly sold by us on a standalone basis. In most cases we expect to use TPE or BESP for allocating consideration to each deliverable. The consideration received is allocated among the separate units either on the basis of each unit's fair value or using the residual method and the applicable revenue recognition criteria is applied to each of the separate units. Analyzing the arrangement to identify deliverables
82

Table of Contents
requires the use of judgment, and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation.
The non-refundable upfront license payment we received upon execution of our license agreement with Bayer had continuing performance obligations under the terms of our collaboration arrangement with Bayer, including development and clinical manufacturing supply obligations. Due to these ongoing performance obligations, we determined that the license did not have standalone value. We also did not have objective and reliable evidence of the fair value of these undelivered obligations. Accordingly, amounts received upfront under the license agreement were recorded as deferred revenue and revenue was recognized on a straight-line basis over the expected period of substantial involvement in the collaboration activities. The period over which these activities were to be performed was based upon management's estimate of the period to complete the development activities required to be conducted relative to the upfront license fee and development funds received from Bayer, and all revenue was recognized as of May 31, 2012.
We recognize revenue from milestone payments when earned; provided that (i) the milestone event is substantive in that it can only be achieved based in whole or in part on either our performance or on the occurrence of a specific outcome resulting from our performance and its achievability was not reasonably assured at the inception of the agreement, (ii) we do not have ongoing performance obligations related to the achievement of the milestone and (iii) it would result in the receipt of additional payments. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment is non-refundable; achievement of the milestone was not reasonably assured at the inception of the arrangement; substantive effort is involved to achieve the milestone; and the amount of the milestone payments appears reasonable in relation to the effort expended, the other milestones in the arrangement and the related risk associated with the achievement of the milestone. Any amounts received under the agreements in advance of performance, if deemed substantive, are recorded as deferred revenue and recognized as revenue as we complete our performance obligations.
See related discussion below in Recent Accounting Pronouncements regarding newly issued accounting standard on revenue recognition.
Clinical Trial Accruals
As part of the process of preparing our consolidated financial statements, we are required to estimate our accrued expenses. Our clinical trial accrual process seeks to account for expenses resulting from our obligations under contract with vendors, consultants and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided to us under such contracts. Our objective is to reflect the appropriate trial expense in our consolidated financial statements by matching the appropriate expenses with the period in which services and efforts are expended. We account for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. We determine accrual estimates through financial models that take into account discussion with applicable personnel and outside services providers as to the progress or state of completion of trials, or the services completed. During the course of a clinical trial, we adjust our rate of clinical expense recognition if actual results differ from our estimates. We make estimates of our accrued expenses as of each balance sheet date in our consolidated financial statements based on the facts and circumstances known to us at that time. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in us reporting amounts that are too high or too low for any particular period. Through December 31, 2014, there have been no material adjustments to our prior period estimates of accrued expenses for clinical trials. Our clinical trial accrual is dependent, in part, upon the receipt of timely and accurate reporting from clinical research organizations and other third-party vendors.
83

Table of Contents
Stock-Based Compensation
We account for all stock-based compensation awards issued to employees and directors based on the estimated fair value of the awards on the date of grant, net of forfeitures.
We estimate the fair value of our stock options issued to employees and directors using the Black-Scholes option pricing model. The Black-Scholes model requires the input of various assumptions, including (a) the expected stock price volatility, (b) the calculation of the expected term of the award, (c) the risk free interest rate and (d) expected dividends. Due to our limited operating history and a lack of company specific historical and implied volatility data, we have based our estimate of expected volatility on the historical volatility of a group of similar companies, which are publicly-traded. When selecting these public companies on which we have based our expected stock price volatility, we selected companies with comparable characteristics to us, including enterprise value, risk profiles, position within the industry, and with historical share price information sufficient to meet the expected life of our stock-based awards. The historical volatility data was computed using the daily closing prices for the selected companies' shares during the equivalent period of the calculated expected term of our stock-based awards. We will continue to apply this process until a sufficient amount of historical information regarding the volatility of our own stock price becomes available. We have estimated the expected life of our employee stock options using the "simplified" method, whereby, the expected life equals the arithmetic average of the vesting term and the original contractual term of the option. The risk-free interest rates for periods within the expected life of the option are based on the U.S. Treasury yield curve in effect during the period the options were granted. We have never paid, and do not expect to pay dividends in the foreseeable future. These inputs, which are complex and subjective, can significantly impact the estimated grant date fair value for our stock-based awards and the resulting compensation expense recognized.
We are also required to estimate forfeitures at the time of grant, and revise those estimates in subsequent periods if actual forfeitures differ from our estimates. We use historical data to estimate pre-vesting option forfeitures and record stock-based compensation expense only for those awards that are expected to vest. To the extent that actual forfeitures differ from our estimates, the difference is recorded as a cumulative adjustment in the period the estimates were revised. For the years ended December 31, 2014, 2013 and 2012 we applied a forfeiture rate, which was determined based on historical forfeitures. No forfeiture rate was applied for any period prior to January 1, 2011 as forfeitures prior to such date had been insignificant.
Total compensation cost recorded in the consolidated statements of operations and comprehensive loss, includes stock-based compensation expense related to stock options and restricted stock units issued to employees and directors, and the value of stock options issued to non-employees for services is as follows for the years ended December 31, 2014, 2013 and 2012, respectively: $12.4 million, $6.5 million and $1.9 million.
As of December 31, 2014, there was $28.6 million of unrecognized compensation expense related to unvested employee stock award agreements expected to vest, which is expected to be recognized over a weighted-average period of approximately 2.74 years.
Net Operating Loss Carryforwards
We recorded net deferred tax assets of approximately $94.7 million as of December 31, 2014, which have been fully offset by a valuation allowance due to uncertainties surrounding our ability to generate future taxable income to realize these assets. The deferred tax assets are primarily composed of federal and state tax net operating losses, or NOL carryforwards, R&D credit carryforwards, stock compensation and start-up expenditures. As of December 31, 2014, we have federal and California NOL carryforwards of approximately $219.8 million and $215.2 million, which will begin to expire in 2025 and 2015, respectively. In general, if we experience a greater than 50 percentage point aggregate change in ownership of certain significant stockholders over a three-year period (a "Section 382
84

Table of Contents
ownership change"), utilization of our pre-change NOL carryforwards are subject to an annual limitation under Section 382 of the Internal Revenue Code (and similar state laws). Such limitations may result in expiration of a portion of the NOL carryforwards before utilization and may be substantial. We have determined that we have experienced ownership changes in the past. If we experience a Section 382 ownership change as a result of future changes in our stock ownership, some of which changes are outside our control, the tax benefits related to the NOL carryforwards may be further limited or lost.
JOBS Act
In April 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other companies.
Results of Operations
Comparison of Years Ended December 31, 2014 and 2013
In-process research and development. In-process research and development of $52.8 million for the year ended December 31, 2014 represents the one-time charge for acquiring the rights to ATX-101 outside of the United States and Canada from Bayer in March 2014.
Research and development expenses. Research and development expenses increased $13.9 million, or 41%, for the year ended December 31, 2014. The increase is primarily attributable to higher costs from an increase in headcount and related personnel costs of $6.1 million, including increased stock compensation expense of $2.5 million resulting from an increase in stock option grants as well as increase in share price and the issuance of restricted stock units, an increase in manufacturing costs of $1.7 million due to establishing commercial manufacturing and supply chain capabilities in preparation for commercial launch as we prepare for the potential approval of ATX-101 as well as drug product manufacturing in support of international regulatory filings, an increase in external costs associated with our regulatory filings and preparation for our expected FDA Advisory Committee meeting of
85
Year Ended
December 31, Change 2014 2013 $ % (in thousands, except percentages) Operating expenses:
In-process research and development $ 52,757 $ — $ 52,757 * % Research and development 47,840 33,974 13,866 41 General and administrative 31,055 16,092 14,963 93
Total operating expenses 131,652 50,066 81,586 163 Loss from operations (131,652 ) (50,066 ) (81,586 ) 163 Interest income 260 135 125 93 Interest expense (4,157 ) (1,980 ) (2,177 ) 110 Other income (expense), net (72 ) — (72 ) * Net loss $ (135,621 ) $ (51,911 ) $ (83,710 ) 161 %
* Percentage not meaningful

Table of Contents
$4.8 million, and an increase in external costs of $3.0 million for the development and execution of our medical affairs program. The increase was partially offset by a decrease in clinical trial costs of approximately $2.8 million due to the completion of the U.S. and Canadian Phase III pivotal trials offset by the costs of previously announced additional trials initiated during the year.
General and administrative expenses. General and administrative expenses increased by $15.0 million, or 93%, for the year ended December 31, 2014. The increase is primarily due to increased personnel costs associated with an increase in headcount in support of the growing organization of approximately $6.1 million, including increased stock compensation expense of $3.5 million resulting from an increase in stock option grants, an increase in share price, and the issuance of restricted stock units, increased external costs of $3.3 million associated with marketing activities in preparation of commercial launch as we prepare for the potential approval of ATX-101, increased external costs of $2.8 million for the expansion of our information technology infrastructure to support our potential commercial launch of ATX-101, and increased corporate development, accounting, legal, director fees and insurance costs of $1.4 million as a result of supporting a growing public company.
Interest income. Interest income represents interest earned on marketable securities held during 2014 offset by the amortization of premiums paid on the purchased securities.
Interest expense. Interest expense increased by $2.2 million from $2.0 million for the year ended December 31, 2013 to $4.2 million for the year ended December 31, 2014. The increase is primarily due to the interest expense incurred on the note payable to Bayer issued in March 2014. The Bayer note bears interest at a rate of 5%, but was recorded at its fair value assuming a borrowing rate of 15%. Included in interest expense is the amortization of the discount on the note being recognized utilizing the effective interest method.
Comparison of Years Ended December 31, 2013 and 2012
License income. License income in the year ended December 31, 2012 represents the amortization of upfront license fees received in 2010 from our license agreement with Bayer, which was entered into in August 2010, and was fully amortized by May 31, 2012.
86
Year Ended
December 31, Change 2013 2012 $ % (in thousands, except percentages) License income $ — $ 19,687 $ (19,687 ) (100 )% Sublicense expense — 1,936 (1,936 ) (100 ) Gross margin — 17,751 (17,751 ) (100 ) Operating expenses:
Research and development 33,974 43,184 (9,210 ) (21 ) General and administrative 16,092 10,505 5,587 53
Total operating expenses 50,066 53,689 (3,623 ) (6 ) Loss from operations (50,066 ) (35,938 ) (14,128 ) 39 Interest income 135 — 135 * Interest expense (1,980 ) (441 ) (1,539 ) 349 Warrant and other income (expense), net — (420 ) 420 (100 ) Net loss $ (51,911 ) $ (36,799 ) $ (15,112 ) 41 %
* Percentage not meaningful

Table of Contents
Sublicense expense. Sublicense expense in the year ended December 31, 2012 represents the amortization of our $2.0 million non-royalty sublicense fee payable to LA Biomed as a result of our receipt of license fee income pursuant to our license agreement with Bayer, and was recognized on a straight-line basis over the same period that the license income was recorded and was fully amortized by May 31, 2012.
Research and development expenses. Research and development expenses decreased $9.2 million, or 21%, from $43.2 million for the year ended December 31, 2012 to $34.0 million for the year ended December 31, 2013. The decrease is primarily attributable to a decrease in the U.S. and Canadian Phase III clinical trial costs of approximately $14.6 million as we transitioned from the enrollment and initiation of patient treatment during the first half of 2012 to completion in late 2013 and a related decrease of approximately $2.0 million in manufacturing expense due to the timing of manufacturing clinical supply for the U.S. and Canadian Phase III clinical trials in 2012, partially offset by higher costs from increases in headcount and related personnel costs of $6.0 million, including increased stock compensation expense of $3.0 million, and increased consulting and other costs of approximately $1.9 million, net for NDA filing preparations.
General and administrative expenses. General and administrative expenses increased by $5.6 million, or 53%, from $10.5 million for the year ended December 31, 2012 to $16.1 million for the year ended December 31, 2013. The increase is primarily due to increased personnel costs associated with an increase in headcount in support of the growing organization of approximately $2.5 million, including increased stock compensation expense of $1.6 million, increased accounting, legal, director fees and insurance costs of $1.8 million as a result of being a public company and increased marketing costs of $1.6 million in connection with ATX-101 brand development and market research.
Interest income. Interest income for the year ended December 31, 2013 represents interest earned on marketable securities purchased during 2013 offset by the amortization of premiums paid on the purchased securities.
Interest expense. Interest expense increased by $1.5 million from $0.4 million for the year ended December 31, 2012 to $2.0 million for the year ended December 31, 2013. The increase is primarily due to the interest expense incurred on the amount of outstanding balances of the notes payable related to the credit facility.
Warrant and other income (expense), net. Warrant and other income (expense), net, of $0.4 million for the year ended December 31, 2012 relates to the fair value of the warrants due to revaluation. All warrants were exercised on a cashless basis in December 2012 and no warrants were outstanding as of December 31, 2013.
Liquidity and Capital Resources
Since our inception, we have incurred net losses and negative cash flows from our operations. We incurred net losses of $135.6 million, $51.9 million and $36.8 million for years ended December 31, 2014, 2013 and 2012, respectively. At December 31, 2014, we had an accumulated deficit of $307.8 million.
As of December 31, 2014, we had working capital of $80.5 million. Historically, we have principally financed our operations through our public offerings, private placements of redeemable convertible preferred stock and convertible debt, and amounts received pursuant to our collaboration arrangement with Bayer for the development of our product candidate, ATX-101. We received net proceeds of $72.5 million from our initial public offering in 2012. In the fourth quarter 2013, we completed a public offering of shares of common stock at an offering price of $45.75 per share and we received aggregate net proceeds of approximately $125.0 million, after deducting the underwriting discount and offering related transaction costs.
87

Table of Contents
Additionally, we have drawn $15.0 million under our credit facility and have no further borrowing capacity under our existing credit facility. In connection with our acquisition of the rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer in March 2014, we issued a $51.0 million unsecured promissory note, recorded at its fair market value of $21.6 million. The promissory note, which is subordinated to amounts owed under our existing credit facility, bears interest at 5% and is payable no later than 2024. The discount on the note payable was calculated assuming a borrowing rate of 15%. Bayer is also eligible to receive certain long-term sales milestone payments on annual sales outside of the United States and Canada. We have the right to repay the outstanding obligation including accrued interest before its due date in 2024. The terms provide for the option to defer up to 80% of the accrued interest, adding any unpaid interest to the principal amount annually.
At December 31, 2014, we had capital resources consisting of cash and cash equivalents and marketable securities of $99.7 million. Our funds are currently invested in money market funds, U.S. government agency securities, corporate debt securities and demand deposit accounts.
Summary Statement of Cash Flows
The following table shows a summary of our cash flows for each of the three years ended December 31, 2014, 2013 and 2012.
Net cash used in operating activities. Net cash used in operating activities was $59.5 million for the year ended December 31, 2014 and consisted primarily of net loss of $135.6 million offset by non-cash in-progress research and development costs of $53.0 million and stock based compensation of $12.4 million. The significant items in the change in operating assets and liabilities include a decrease in restricted cash of $5.1 million and an increase in accounts payable and other accrued liabilities of $4.6 million, offset by a decrease in deferred development funds of $2.1 million. The increase in accounts payable and other accrued liabilities is primarily due to an increase in accrued interest of $2.5 million related to the interest accruing on the Bayer note issued in March 2014 as well as an increase in accrued personnel costs of $1.6 million related to the increase in headcount and related personnel costs. The decrease in the deferred development funds and restricted cash is due to the termination of our obligations under our collaboration agreement with Bayer.
Net cash used in operating activities was $44.9 million for the year ended December 31, 2013 and consisted primarily of a net loss of $51.9 million offset by stock based compensation of $6.5 million. The significant items in the change in operating assets and liabilities include a decrease in deferred development funds of $12.0 million offset by a decrease in restricted cash of $10.9 million. The decrease in the deferred development funds and restricted cash is due to the continuing performance of obligations under our collaboration agreement with Bayer.
Net cash used in operating activities was $32.7 million for the year ended December 31, 2012 and consisted primarily of a net loss of $36.8 million offset by stock based compensation and payment for services in stock of $3.4 million. The significant items in the change in operating assets and liabilities
88
Year Ended December 31, 2014 2013 2012 (in thousands) Net cash (used in) provided by
Operating activities $ (59,529 ) $ (44,869 ) $ (32,688 ) Investing activities (18,296 ) (56,502 ) (190 ) Financing activities (4,467 ) 133,578 77,612
Net (decrease) increase in cash and cash equivalents $ (82,292 ) $ 32,207 $ 44,734

Table of Contents
include an increase in restricted cash of $8.6 million, a decrease in deferred revenue of $3.8 million and a decrease in payable to a licensor of $1.6 million, offset by an increase in deferred development funds of $9.2 million and an increase in accounts payable and other accrued liabilities of $3.2 million. The decrease in deferred revenue is due to the recognition of the remaining unamortized portion of the upfront license payment received as part of our license agreement with Bayer. The increase in the deferred development funds and restricted cash is due to the receipt of $17.4 million under the collaboration agreement with Bayer, offset by funds utilized for the continuing performance of obligations under the collaboration agreement with Bayer.
Net cash used in investing activities. Net cash used in investing activities for the year ended December 31, 2014 was $18.3 million and consisted primarily of purchases of marketable securities of $63.5 million offset by proceeds from the sales of marketable securities of $47.3 million. Net cash used in investing activities for the year ended December 31, 2013 was $56.5 million and was comprised mainly of purchases of marketable securities. Net cash used in investing activities for the year ended December 31, 2012 was $0.2 million and consisted of purchases of fixed assets.
Net cash (used in) provided by financing activities. Net cash used in financing activities was $4.5 million for the year ended December 31, 2014, which was primarily due to repayment of notes payables related to the credit facility. Net cash provided by financing activities was $133.6 million for the year ended December 31, 2013, which was primarily due to proceeds from the public offering of $125.0 million and borrowing of $10.0 million from our credit facility, offset by repayments on the notes of $2.8 million. Net cash provided by financing activities was $77.6 million for the year ended December 31, 2012, which was primarily due to proceeds from our initial public offering and borrowings of $5.0 million from our credit facility.
Operating and Capital Expenditure Requirements
We believe that our existing capital resources will be sufficient to fund our operations for at least the next 12 months. We believe that we will continue to expend substantial resources for the foreseeable future on the preparations for potential commercial launch of ATX-101, completion of clinical and regulatory development of ATX-101, preparing and filing foreign regulatory applications, and development of any other product candidates we may choose to pursue. These expenditures will include costs associated with research and development, conducting preclinical studies and clinical trials, obtaining regulatory approvals, and manufacturing and supply as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise.
We expect to seek additional financing in the future to fund our operations, to expand our commercial organization in anticipation of potential commercial launch of ATX-101, or develop additional product candidates, by selling additional equity or issuing debt or convertible debt securities that may result in dilution to our stockholders. If we raise additional funds through the issuance of convertible debt securities, these securities could have rights senior to those of our common stock and could contain covenants that restrict our operations. There can be no assurance that we will be able to obtain additional equity or debt financing on terms acceptable to us, if at all. Our failure to obtain sufficient funds on acceptable terms when needed could have a material adverse effect on our business, results of operations, and financial condition.
Our future capital requirements depend on many factors, including:
• the timing of, and the costs involved in, obtaining regulatory approvals for ATX-101 in the U.S., and other international territories, including the cost of any studies or additional activities that the FDA or other regulatory agencies may require us to complete;
• the timing and scope of the investment we make in building our commercial infrastructure and sales force in anticipation of the potential commercial launch of ATX-101 in the U.S.;
89

Table of Contents
• any unexpected results from further analysis of clinical data from our completed clinical trials;
• the timing of, and the costs involved in, obtaining regulatory approvals for any future product candidates;
• the number and characteristics of any additional product candidates we may develop or acquire;
• the scope, progress, results and costs of researching and developing ATX-101 or any future product candidates, including pursuant to our license agreements with Actelion and the University of Pennsylvania, and conducting preclinical and clinical trials;
• the cost of commercialization activities if ATX-101 or any future product candidates are approved for sale, including marketing, sales and distribution cost and preparedness of our corporate infrastructure;
• the cost of manufacturing ATX-101 or any future product candidates and any products we successfully commercialize;
• the timing of the payment of our debt obligations, including whether or not we determine to prepay the $51.0 million note held by Bayer;
• our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements;
• any product liability or other lawsuits related to our products or commenced against us;
• the expenses needed to attract and retain skilled personnel;
• the costs associated with being a public company;
• the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent or other claims, including litigation costs and the outcome of such litigation; and
• the timing, receipt and amount of sales of, or royalties on, future approved products, if any.
Please see "Risk Factors" for additional risks associated with our substantial capital requirements.
Contractual Obligations and Commitments
The following is a summary of our long-term contractual cash obligations as of December 31, 2014.
90
Payments Due by Period
Contractual Obligations Total Less than One Year 1 - 3 Years 3 - 5 Years
More than 5 Years
(in thousands) Operating lease obligations(1) $ 2,650 $ 962 $ 1,688 $ — $ — Note payable to Bayer(2)(3) 81,616 510 1,082 1,791 78,233 Notes payable related to credit
facility(2) 7,534 6,381 1,153 — — Total contractual obligations $ 91,800 $ 7,853 $ 3,923 $ 1,791 $ 78,233
(1) We lease our facilities under operating leases
(2) Notes payable payments include both principal and interest
(3) Payments on the note payable to Bayer assume that we will pay 20% of the accrued interest, with the remainder increasing the principal balance on an annual basis. For additional discussion, refer to Note 5, "Notes Payable".

Table of Contents
Purchase Commitments
We have no material non-cancelable purchase commitments with contract manufacturers or service providers as we have generally contracted on a cancelable purchase order basis.
Other Milestone-Based Commitments
We have obligations to make future payments to third parties that become due and payable on the achievement of certain development, regulatory and commercial milestones. Because the achievement and timing of these milestones are not fixed and determinable, such commitments have not been included on our balance sheet or in the Contractual Obligations and Commitments table above. The total aggregate amount of such commitments is approximately $1.9 million. Due to the acquisition of rights to ATX-101, Bayer is eligible to receive up to $123.8 million upon the achievement of certain long-term sales milestone payments on annual sales of ATX-101 outside of the United States and Canada. Additionally, Actelion is eligible to receive up to $25.5 million and University of Pennsylvania is eligible to receive up to $4.1 million in milestone payments if setipiprant is successfully commercialized for the treatment of hair loss.
Notes Payable
Note Payable to Bayer
In March 2014, as part of the agreement to acquire the rights to develop and commercialize ATX-101 outside of the United States and Canada, we entered into a $51.0 million unsecured promissory note with Bayer. The promissory note is subordinated to amounts owed under our existing credit facility. Interest is accrued at 5% per annum and is payable on the anniversary date of the note. We have the option of deferring up to 80% of the accrued interest, adding any unpaid interest to the principal amount. The note is due on the ten year anniversary from issuance, with the option to prepay at any point in time without penalty. As the note payable to Bayer includes a favorable interest rate, which was below the borrowing rate available to us at issuance, it was recorded at its fair value of $21.6 million, which is the present value of the future cash flows assuming a borrowing rate of 15%, which approximates the market rate for debt with similar terms and consideration of default and credit risk. The discount on the note is being amortized to interest expense utilizing the effective interest method.
Notes Payable Related to Credit Facility
On March 21, 2011, we entered into a credit facility with Lighthouse Capital Partners VI, L.P., or Lighthouse, which we refer to as our credit facility, providing access to borrow up to $15.0 million of senior loan financing through January 1, 2012. In December 2011, July 2012 and November 2012, we amended the credit facility to provide for an extension of our drawdown period. As of December 31, 2014 and 2013, we had drawn $15.0 million and no longer have any further borrowing capacity under the credit facility. The credit facility includes various covenants, and we were in compliance with all covenants at December 31, 2014 and 2013.
For each drawdown, we will make six months of interest only payments at a fixed rate of 11.5% followed by 30 months of interest and principal payments at a fixed rate of 8.5%, and a final payment of 6% of the amount drawn. The debt is secured by all of our assets, except for intellectual property, which is subject to a negative pledge agreement. In accordance with the terms of the credit facility, we issued to Lighthouse warrants to purchase 33,700 shares of our Series C redeemable convertible preferred stock at a price per share of $13.3530 and 86,306 shares of our Series D redeemable convertible preferred stock at a price per share of $13.9040. The estimated fair value of the warrants at issuance of approximately $1.4 million was recorded as a deferred financing cost offsetting the notes payable liability and is being amortized to interest expense over the term of the loan.
91

Table of Contents
All of the warrants issued to Lighthouse converted to warrants to purchase 120,006 shares of common stock in connection with our initial public offering and were subsequently net exercised on a cashless basis for 49,435 shares of common stock in December 2012.
Indemnification
In the normal course of business, we enter into contracts and agreements that contain a variety of representations and warranties and provide for general indemnifications. Our exposure under these agreements is unknown because it involves claims that may be made against us in the future, but have not yet been made. To date, we have not paid any claims or been required to defend any action related to our indemnification obligations. However, we may record charges in the future as a result of these indemnification obligations.
In accordance with our certificate of incorporation and bylaws, we have indemnification obligations to our officers and directors for specified events or occurrences, subject to some limits, while they are serving at our request in such capacities. There have been no claims to date, and we have director and officer insurance that may enable us to recover a portion of any amounts paid for future potential claims.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements (as defined by applicable SEC regulations) that are reasonably likely to have a current or future material effect on our financial condition, changes in financial condition, revenues or expenses results of operations, liquidity, capital expenditures or capital resources, except stock options.
Recent Accounting Pronouncements
In May 2014, a new standard was issued related to revenue recognition, which requires an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The new standard will replace most of the existing revenue recognition standards in U.S. GAAP when it becomes effective on January 1, 2017. Early adoption is not permitted. The new standard allows for either "full retrospective" adoption, whereby the new standard is applied to each prior reporting period presented or "modified retrospective" adoption, whereby the new standard is only applied to the most current period presented with the cumulative effect of the change recognized at the date of the initial application. We are assessing the potential impact of the new standard on our consolidated statements of financial position and results of operations and comprehensive income (loss) and have not yet selected a method of adoption.
In August 2014, a new standard was issued which will require management to evaluate if there is substantial doubt about the entity's ability to continue as a going concern and, if so, disclose it in both annual and interim reporting periods. The new standard will become effective for the Company's annual filing for the period ending December 31, 2016 and interim periods thereafter, with early adoption permitted. We do not believe the adoption of this accounting standard will have a material impact on our consolidated financial statements and related disclosures.
ITEM 7A. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to market risks in the ordinary course of our business. These market risks are principally limited to interest rate fluctuations and foreign currency exchange rates fluctuations. Due to the fixed interest rate of our credit facility, we do not currently have any exposure to changes in our interest expense as a result of changes in interest rates.
92

Table of Contents
Cash and Cash Equivalents. Marketable Securities and Restricted Cash
As of December 31, 2014, we had cash and cash equivalents and marketable securities of $99.7 million. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. At December 31, 2014, our cash and cash equivalents and marketable securities were comprised of funds in cash, money market accounts, U.S. government agency securities and highly rated, highly liquid corporate debt securities. Due to the short-term nature of our investment portfolio, we do not believe an immediate 10% increase or decrease in interest rates would have a material effect on the fair market value of our portfolio. Additionally, we do not expect our operating results or cash flows to be materially affected to any degree by a sudden change in market interest rates.
Notes Payable
Our notes payable related to the credit facility are at an effective interest rate which approximates the borrowing rates currently available to the Company for debt with similar terms and consideration of default and credit risk. Our note payable to Bayer is at a rate that is less than market, but the note was recorded at fair value based on an interest rate which approximates the borrowing rates currently available to the Company for debt with similar terms and consideration of default and credit risk. As the interest rates on our notes payable are fixed for specified periods, changes in interest rates do not affect our operating results or cash flows.
Foreign Currency Exchange Rate Fluctuations
With the recent acquisition of rights to ATX-101 outside of the United States and Canada from Bayer, in the future, we may be subject to fluctuations in foreign currency exchange rate risk. We are not currently exposed to any material foreign currency exchange rate risk and, as a result, we do not currently hedge any foreign currency exposure.
ITEM 8. Consolidated Financial Statements and Supplementary Data.
Our consolidated financial statements, together with the independent registered public accounting firm report thereon, are incorporated by reference from the applicable information set forth in Part IV Item 15, "Exhibits, Financial Statement Schedules" of this Annual Report on Form 10-K.
ITEM 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
ITEM 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our chief executive and financial officers, evaluated the effectiveness of our disclosures controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of December 31, 2014. The term "disclosure controls and procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company's management, including its principal
93

Table of Contents
executive and principal financial officers, as appropriate, to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2014, our chief executive officer and chief financial officer concluded that, as of such date, our disclosure controls and procedures were effective at a reasonable assurance level.
Management's Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed by, or under the supervision of, our CEO and CFO, and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
• Pertain to the maintenance of records that accurately and fairly reflect in reasonable detail the transactions and dispositions of the assets of our company;
• Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
• Provide reasonable assurances regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material adverse effect on our consolidated financial statements.
Our management assessed our internal control over financial reporting as of December 31, 2014, the end of our fiscal year. Management based its assessment on criteria established in "Internal Control—Integrated Framework (2013)" issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on management's assessment of our internal control over financial reporting, management concluded that, as of December 31, 2014, our internal control over financial reporting was effective.
Internal control over financial reporting has inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements will not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Attestation Report on Internal Control over Financial Reporting
This Annual Report on Form 10-K does not include an attestation report of our independent registered public accounting firm due to the deferral allowed under the JOBS Act for emerging growth companies.
Changes in Internal Control over Financial Reporting
There were no changes in our internal controls over financial reporting during the quarter ended December 31, 2014 identified in connection with the evaluation required by Rule 13a-15(d) and
94

Table of Contents
15d-15(d) of the Exchange Act that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. Other Information.
None.
PART III
ITEM 10. Directors, Executive Officers and Corporate Governance.
The information required by this item is incorporated by reference from the applicable information set forth in "Executive Officers," "Election of Directors," "Information about the Board of Directors and its Committees," and "Security Ownership of Directors and Executive Officers—Section 16(a) Beneficial Ownership Reporting Requirements" which will be included in our definitive Proxy Statement for our 2015 Annual Meeting of Stockholders to be filed with the SEC.
ITEM 11. Executive Compensation.
The information required by this item is incorporated by reference from the applicable information set forth in "Executive Officers," "Compensation of Executive Officers" and "Compensation of Directors" which will be included in our definitive Proxy Statement for our 2015 Annual Meeting of Stockholders to be filed with the SEC.
ITEM 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item is incorporated by reference from the applicable information set forth in "Executive Officers," "Security Ownership of Principal Stockholders and Management" and "Equity Compensation Plan Information" which will be included in our definitive Proxy Statement for our 2015 Annual Meeting of Stockholders to be filed with the SEC.
ITEM 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item is incorporated by reference from the applicable information set forth in "Other Information—Related Person Transactions" and "Information about the Board of Directors and its Committees" which will be included in our definitive Proxy Statement for our 2015 Annual Meeting of Stockholders to be filed with the SEC.
ITEM 14. Principal Accounting Fees and Services.
The information required by this item is incorporated by reference from the applicable information set forth in "Ratification of Selection of Independent Registered Accounting Firm" which will be included in our definitive Proxy Statement for our 2015 Annual Meeting of Stockholders to be filed with the SEC.
95

Table of Contents
PART IV
ITEM 15. Exhibits, Consolidated Financial Statement Schedules.
(a) Documents filed as part of this report
1. Financial Statements
The following consolidated financial statements are included herein:
2. Consolidated Financial Statement Schedules
None, as all required disclosures have been made in the Consolidated Financial Statements and notes thereto or are not applicable.
(b) Exhibits: The exhibits listed on the accompanying Exhibit Index are filed as part of, or hereby incorporated by reference into, this Annual Report on Form 10-K.
96
Page Report of Independent Registered Public Accounting Firm F-2 Consolidated Balance Sheets at December 31, 2014 and 2013 F-3 Consolidated Statements of Operations and Comprehensive Loss for each of the three years in the period
ended December 31, 2014
F-4 Consolidated Statements of Stockholders' Equity (Deficit) for each of the three years in the period ended
December 31, 2014
F-5 Consolidated Statements of Cash Flows for each of the three years in the period ended December 31, 2014 F-6 Notes to Consolidated Financial Statements F-7

Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Keith R. Leonard, Jr. and John W. Smither his true and lawful attorney-in-fact and agent, with full power of substitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated opposite his/her name.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
97
KYTHERA Biopharmaceuticals, Inc. March 2, 2015
By:
/s/ JOHN W. SMITHER
John W. Smither Chief Financial Officer (Principal Financial and
Accounting Officer)
Signature Title Date
/s/ KEITH R. LEONARD, JR.
Keith R. Leonard, Jr. Director, President and Chief Executive
Officer (Principal Executive Officer) March 2, 2015
/s/ JOHN W. SMITHER
John W. Smither
Chief Financial Officer (Principal Financial and Accounting Officer)
March 2, 2015
/s/ F. MICHAEL BALL
F. Michael Ball
Director
March 2, 2015
/s/ NATHANIEL DAVID
Nathaniel David, Ph.D.
Director
March 2, 2015

Table of Contents
98
Signature Title Date
/s/ DENNIS FENTON
Dennis Fenton, Ph.D. Director March 2, 2015
/s/ FRANÇOIS KRESS
François Kress
Director
March 2, 2015
/s/ CAMILLE SAMUELS
Camille Samuels
Director
March 2, 2015
/s/ HOLLINGS C. RENTON
Hollings C. Renton
Director
March 2, 2015
/s/ JOSEPH L. TURNER
Joseph L. Turner
Director
March 2, 2015

Table of Contents
EXHIBIT INDEX
99
Incorporated by Reference
Exhibit Number
Filed Herewith
Exhibit Description Form Date Number
3.1 Seventh Amended and Restated Certificate of Incorporation of KYTHERA Biopharmaceuticals, Inc.
8-K 10/16/2012 3.1
3.2 Amended and Restated Bylaws of KYTHERA
Biopharmaceuticals, Inc. 8-K 10/16/2012 3.2
4.1 Reference is made to exhibits 3.1 and 3.2. 4.2 Specimen Common Stock Certificate. S-
1/A 08/31/2012 4.2
4.3 Form of Indenture S-3 09/02/2014 4.3 10.1(a ) Third Amended and Restated Investor Rights Agreement,
dated August 30, 2011, between KYTHERA Biopharmaceuticals, Inc. and certain of its stockholders.
S-1/A
05/17/2012 10.1(a)
10.1(b ) Amendment, dated January 27, 2012, to Third Amended
and Restated Investor Rights Agreement, dated August 30, 2011, between KYTHERA Biopharmaceuticals, Inc. and certain of its stockholders.
S-1/A
05/17/2012 10.1(b)
10.2 # Form of Indemnity Agreement for directors and officers. S-
1/A 05/17/2012 10.2
10.3(a ) Loan and Security Agreement No. 1991, dated as of
March 21, 2011, by and between Lighthouse Capital Partners VI, L.P. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.3(a)
10.3(b ) Amendment No. 1, dated December 30, 2011, to that
certain Loan and Security Agreement No. 1991, dated as of March 21, 2011, by and between Lighthouse Capital Partners VI, L.P. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.3(b)
10.3(c ) Amendment No. 2, dated July 23, 2012, to that certain
Loan and Security Agreement No. 1991, dated as of March 21, 2011, by and between Lighthouse Capital Partners VI, L.P. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
08/31/2012 10.3(c)
10.3(d ) Amendment No. 3, dated November 30, 2012, to that
certain Loan and Security Agreement No. 1991, dated as of March 21, 2011, as amended, by and between Lighthouse Capital Partners VI, L.P. and KYTHERA Biopharmaceuticals, Inc.
8-K 12/05/2012 10.1
10.4 Office Lease, dated March 12, 2014, by and between
Russell Ranch Road, LLC and KYTHERA Biopharmaceuticals, Inc.
10-K
03/17/2014 10.20
10.5(a) † License Agreement, dated August 26, 2010, by and
between Bayer Consumer Care AG and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.5(a)
10.5(b) † First Amendment, dated March 21, 2011, to that certain
License Agreement, dated August 26, 2010, by and between Bayer Consumer Care AG and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.5(b)

Table of Contents
100
Incorporated by Reference
Exhibit Number
Filed Herewith
Exhibit Description Form Date Number
10.5(c) † Second Amendment, dated April 2, 2012, to that certain License Agreement, dated August 26, 2010, by and between Bayer Consumer Care AG and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.5(c)
10.6(a) † Services, Research, Development and Collaboration
Agreement, dated August 26, 2010, by and between Intendis GmbH and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.6(a)
10.6(b) † First Amendment, dated November 17, 2010, to that certain
Services, Research, Development and Collaboration Agreement, dated August 26, 2010, by and between Intendis GmbH and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.6(b)
10.6(c) † Second Amendment, dated April 2, 2012, to that certain
Services, Research, Development and Collaboration Agreement, dated August 26, 2010, by and between Intendis GmbH and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.6(c)
10.7(a) † License Agreement, dated August 29, 2005, by and between
KYTHERA Biopharmaceuticals and Los Angeles Biomedical Research Institute at Harbor/UCLA Medical Center.
S-1/A
05/17/2012 10.7(a)
10.7(b ) Amendment, dated January 5, 2010, to that certain License
Agreement, dated August 29, 2005, by and between KYTHERA Biopharmaceuticals and Los Angeles Biomedical Research Institute at Harbor/UCLA Medical Center.
S-1/A
05/17/2012 10.7(b)
10.7(c ) Amendment, dated August 18, 2010, to that certain License
Agreement, dated August 29, 2005, by and between KYTHERA Biopharmaceuticals and Los Angeles Biomedical Research Institute at Harbor/UCLA Medical Center.
S-1/A
05/17/2012 10.7(c)
10.8(a) † Manufacturing and Supply Agreement, dated July 7, 2009,
by and between Pfizer, Inc. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
06/11/2012 10.8(a)
10.8(b) † First Amendment, dated May 21, 2012, to Manufacturing
and Supply Agreement, dated July 7, 2009 by and between Pfizer, Inc. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
06/11/2012 10.8(b)
10.8(c ) Second Amendment, dated October 17, 2014, to
Manufacturing and Supply Agreement, dated July 7, 2009, as amended, by and between Pfizer, Inc. and KYTHERA Biopharmaceuticals, Inc.
X
10.9 † Development and Supply Agreement, dated November 29,
2010, by and between Hospira Worldwide, Inc. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.9
10.10(a) # KYTHERA Biopharmaceuticals, Inc. 2004 Stock Plan, as
amended. S-
1/A 05/17/2012 10.10
(a)
10.10(b) # Form of Stock Option Grant Notice and Stock Option
Agreement under the 2004 Stock Plan, as amended. S-
1/A 05/17/2012 10.10
(b)

Table of Contents
101
Incorporated by Reference
Exhibit Number
Filed Herewith
Exhibit Description Form Date Number
10.11(a) # KYTHERA Biopharmaceuticals, Inc. 2012 Equity Incentive Award Plan.
10-Q
11/13/2012 10.3(a)
10.11(b) # Form of Stock Option Grant Notice and Stock Option
Agreement under the 2012 Equity Incentive Award Plan. S-
1/A 05/17/2012 10.11
(b)
10.11(c) # Form of Restricted Stock Award Agreement and Restricted
Stock Unit Award Grant Notice under the 2012 Equity Incentive Award Plan.
S-1/A
05/17/2012 10.11(c)
10.12 # Amended and Restated Employment Agreement, dated
April 2, 2012, by and between Keith R. Leonard, Jr. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.12
10.13 # Amended and Restated Employment Agreement, dated
April 2, 2012, by and between John W. Smither and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.13
10.14 # Amended and Restated Employment Agreement, dated
April 2, 2012, by and between Patricia S. Walker, M.D., Ph.D., and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.14
10.14(b) # Separation Agreement and Release, dated March 27, 2013,
by and between Patricia S. Walker, M.D., Ph.D., and KYTHERA Biopharmaceuticals, Inc.
8-K 04/02/2013 10.1
10.14(c) # Senior Advisor to the Board Agreement, effective March 28,
2013, by and between Patricia S. Walker, M.D., Ph.D., and KYTHERA Biopharmaceuticals, Inc.
8-K 04/02/2013 10.2
10.15 # Amended and Restated Employment Agreement, dated
April 2, 2012, by and between Keith L. Klein, J.D. and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.15
10.16 # Amended and Restated Employment Agreement, dated
April 2, 2012, by and between Jeffrey D. Webster and KYTHERA Biopharmaceuticals, Inc.
S-1/A
05/17/2012 10.16
10.17(a) # Non-employee Director Compensation Program. S-
1/A 05/17/2012 10.17
10.7(b) # Amendment to Non-Employee Director Compensation
Program 8-K 09/19/2013 *
10.18 # Employment agreement, dated March 23, 2013, by and
between Frederick Beddingfield, III, M.D., Ph.D., and KYTHERA Biopharmaceuticals, Inc.
10-K
03/26/2013 10.18
10.19 † Commercial Development and Supply Agreement by and
between KYTHERA Biopharmaceuticals, Inc., and Cambridge Major Laboratories, Inc.
10-Q
05/13/2013 10.4
10.20 † License Agreement, dated February 11, 2015, by and
between KYTHERA Holdings, Ltd., and Actelion Pharmaceuticals, Ltd.
X
10.21 † License Agreement, dated February 11, 2015 by and
between KYTHERA Holdings, Ltd., and University of Pennsylvania
X
10.22 † Distribution Services Agreement, dated January 16, 2015 by
and between KYTHERA Biopharmaceuticals, Inc., and Besse Medical.
X

Table of Contents
102
Incorporated by Reference
Exhibit Number
Filed Herewith
Exhibit Description Form Date Number
10.23 Securities Purchase Agreement, dated March 7, 2014, by and among KYTHERA Biopharmaceuticals, Inc., KYTHERA Holdings Ltd. and Bayer Consumer Care AG.
8-K 03/10/2014 10.1
10.24 Restructuring Agreement, dated March 7, 2014 by and
between Bayer Consumer Care AG and KYTHERA Biopharmaceuticals, Inc.
10-Q 05/08/2014 10.2
10.25 Assignment and Novation Agreement, dated March 7,
2014, by and between Bayer Consumer Care AG and KYTHERA Holdings Ltd.
10-Q 05/08/2014 10.3
10.26 Note Agreement, dated March 7, 2014, by and between
KYTHERA Biopharmaceuticals, Inc. and KYTHERA Holdings Ltd.
8-K 03/10/14 4.1
10.27(a ) Registration Rights Agreement, dated March 7, 2014, by
and between KYTHERA Biopharmaceuticals, Inc. and Bayer Consumer Care AG.
8-K 03/10/14 4.2
10.27(b ) Termination and Waiver Agreement, dated November 5,
2014, by and among KYTHERA Biopharmaceuticals, Inc., KYTHERA Holdings Ltd., and Bayer Consumer Care AG.
8-K 11/05/14 10.1
10.28(a) # KYTHERA Biopharmaceuticals, Inc. 2014 Employment
Commencement Incentive Plan. S-8 09/02/14 99.1
10.28(b) # Form of Stock Option Grant Notice and Stock Option
Agreement under the 2014 Employment Commencement Incentive Plan.
S-8 09/02/14 99.2
10.28(c) # Form of Restricted Stock Unit Award Grant Notice and
Restricted Stock Unit Award Agreement under the 2014 Employment Commencement Incentive Plan.
S-8 09/02/14 99.3
10.29 # KYTHERA Biopharmaceuticals, Inc. 2015 Employee
Stock Purchase Plan. S-8 02/13/15 99.1
21.1 List of Subsidiaries X 23.1 Consent of Independent Registered Public Accounting
Firm. X
24.1 Power of Attorney (included on signature page to this
Annual Report on Form 10-K). X
31.1 Certification of Chief Executive Officer of KYTHERA
Biopharmaceuticals, Inc., as required by Rule 13a- 14(a) or Rule 15d-14(a).
X
31.2 Certification of Chief Financial Officer of KYTHERA
Biopharmaceuticals, Inc., as required by Rule 13a- 14(a) or Rule 15d-14(a).
X
32.1 ** Certification by the Chief Executive Officer, as required
by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 36 of Title 18 of the United States Code (18 U.S.C. §1350).
X
32.1 ** Certification by the Chief Financial Officer, as required
by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 36 of Title 18 of the United States Code (18 U.S.C. §1350).
X

Table of Contents
103
Incorporated by Reference
Exhibit Number
Filed Herewith
Exhibit Description Form Date Number
101.INS XBRL Instance Document X 101.SCH XBRL Taxonomy Extension Schema Document X 101.CAL XBRL Taxonomy Extension Calculation Linkbase
Document X
101.DEF XBRL Taxonomy Extension Definition Linkbase
Document X
101.LAB XBRL Taxonomy Extension Labels Linkbase Document X 101.PRE XBRL Taxonomy Extension Presentation Linkbase
Document X
† Confidential treatment has been granted for certain information contained in this exhibit. Such information has been omitted and filed separately with the Securities and Exchange Commission.
# Indicates management contract or compensatory plan.
* Reference is made to the text of the Current Report on Form 8-K under Item 5.02.
** The certifications attached as Exhibits 32.1 that accompany this Annual Report on Form 10-K is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of KYTHERA Biopharmaceuticals, Inc. under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Form 10-K, irrespective of any general incorporation language contained in such filing.

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Index to Consolidated Financial Statements
F-1
Page Report of Independent Registered Public Accounting Firm F-2 Consolidated Balance Sheets F-3 Consolidated Statements of Operations and Comprehensive Loss F-4 Consolidated Statements of Stockholders' Equity (Deficit) F-5 Consolidated Statements of Cash Flows F-6 Notes to Consolidated Financial Statements F-7

Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of KYTHERA Biopharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of KYTHERA Biopharmaceuticals, Inc. as of December 31, 2014 and 2013, and the related consolidated statements of operations and comprehensive loss, stockholders' equity (deficit), and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of KYTHERA Biopharmaceuticals, Inc. at December 31, 2014 and 2013, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
Los Angeles, California March 2, 2015
F-2
/s/ Ernst & Young LLP

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Consolidated Balance Sheets
See accompanying notes.
F-3
December 31, 2014 2013 Assets Current assets:
Cash and cash equivalents $ 29,226,000 $ 111,518,000 Restricted cash, current portion — 5,062,000 Marketable securities 70,435,000 55,540,000 Prepaid expenses and other current assets 1,498,000 1,156,000
Total current assets 101,159,000 173,276,000 Property and equipment, net 1,847,000 94,000 Other assets 420,000 37,000 Total assets $ 103,426,000 $ 173,407,000
Liabilities and stockholders' equity Current liabilities:
Accounts payable $ 4,690,000 $ 3,228,000 Accrued personnel costs 4,858,000 3,114,000 Accrued costs for services 2,103,000 3,191,000 Accrued interest 2,857,000 375,000 Notes payable, current portion 5,484,000 5,526,000 Deferred development funds — 2,147,000 Other current liabilities 637,000 —
Total current liabilities 20,629,000 17,581,000 Notes payable—net of current portion 22,661,000 5,979,000 Other liabilities 698,000 8,000 Total liabilities 43,988,000 23,568,000 Commitments and contingencies Stockholders' equity:
Preferred stock, $0.00001 par value, 5,000,000 shares authorized and none issued and outstanding — —
Common stock, $0.00001 par value, 300,000,000 shares authorized, 22,691,000 and 21,745,000 shares issued and outstanding at December 31, 2014 and 2013, respectively — —
Additional paid in capital 367,329,000 322,052,000 Accumulated other comprehensive (loss) income (53,000 ) 4,000 Accumulated deficit (307,838,000 ) (172,217,000 )
Total stockholders' equity 59,438,000 149,839,000 Total liabilities and stockholders' equity $ 103,426,000 $ 173,407,000
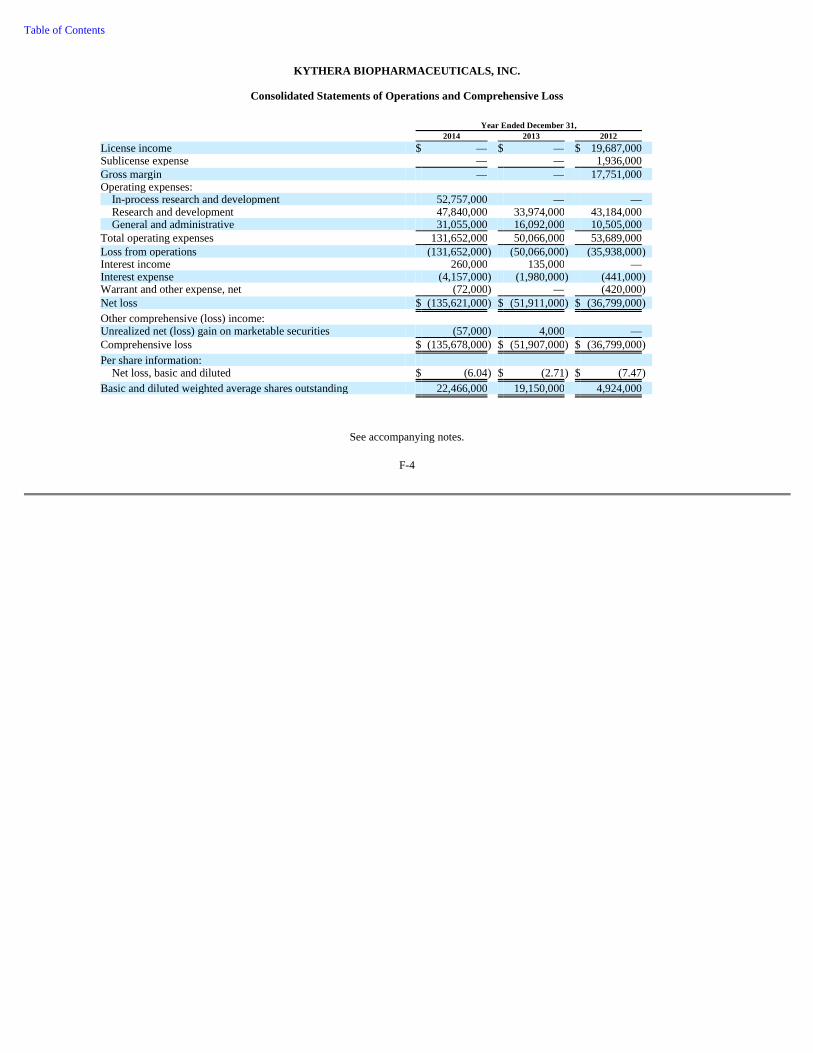
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Consolidated Statements of Operations and Comprehensive Loss
See accompanying notes.
F-4
Year Ended December 31, 2014 2013 2012 License income $ — $ — $ 19,687,000 Sublicense expense — — 1,936,000 Gross margin — — 17,751,000 Operating expenses:
In-process research and development 52,757,000 — — Research and development 47,840,000 33,974,000 43,184,000 General and administrative 31,055,000 16,092,000 10,505,000
Total operating expenses 131,652,000 50,066,000 53,689,000 Loss from operations (131,652,000 ) (50,066,000 ) (35,938,000 ) Interest income 260,000 135,000 — Interest expense (4,157,000 ) (1,980,000 ) (441,000 ) Warrant and other expense, net (72,000 ) — (420,000 ) Net loss $ (135,621,000 ) $ (51,911,000 ) $ (36,799,000 )
Other comprehensive (loss) income: Unrealized net (loss) gain on marketable securities (57,000 ) 4,000 — Comprehensive loss $ (135,678,000 ) $ (51,907,000 ) $ (36,799,000 )
Per share information: Net loss, basic and diluted $ (6.04 ) $ (2.71 ) $ (7.47 )
Basic and diluted weighted average shares outstanding 22,466,000 19,150,000 4,924,000
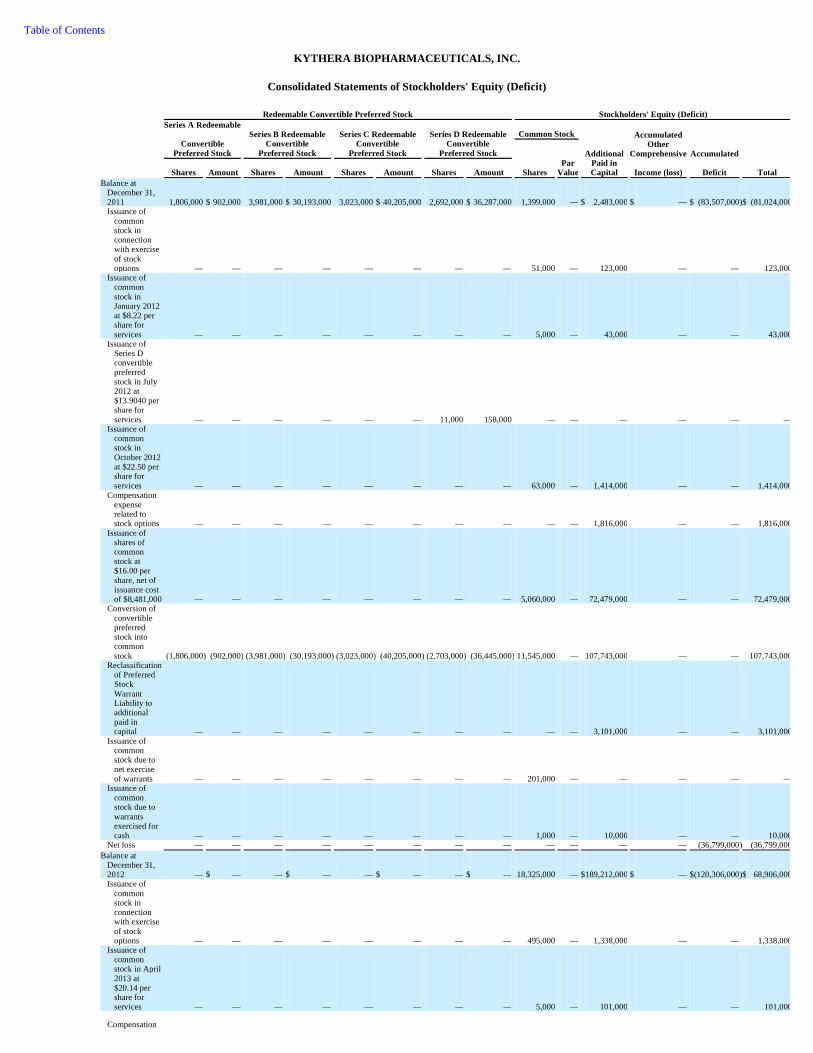
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Consolidated Statements of Stockholders' Equity (Deficit)
Redeemable Convertible Preferred Stock Stockholders' Equity (Deficit)
Series A Redeemable
Convertible Preferred Stock
Series B Redeemable Convertible
Preferred Stock
Series C Redeemable Convertible
Preferred Stock
Series D Redeemable Convertible
Preferred Stock
Common Stock
Accumulated Other
Comprehensive
Income (loss)
Par
Value
Additional Paid in Capital
Accumulated
Deficit
Shares Amount Shares Amount Shares Amount Shares Amount Shares Total Balance at
December 31, 2011 1,806,000 $ 902,000 3,981,000 $ 30,193,000 3,023,000 $ 40,205,000 2,692,000 $ 36,287,000 1,399,000 — $ 2,483,000 $ — $ (83,507,000 ) $ (81,024,000 Issuance of
common stock in connection with exercise of stock options — — — — — — — — 51,000 — 123,000 — — 123,000
Issuance of common stock in January 2012 at $8.22 per share for services — — — — — — — — 5,000 — 43,000 — — 43,000
Issuance of Series D convertible preferred stock in July 2012 at $13.9040 per share for services — — — — — — 11,000 158,000 — — — — — —
Issuance of common stock in October 2012 at $22.50 per share for services — — — — — — — — 63,000 — 1,414,000 — — 1,414,000
Compensation expense related to stock options — — — — — — — — — — 1,816,000 — — 1,816,000
Issuance of shares of common stock at $16.00 per share, net of issuance cost of $8,481,000 — — — — — — — — 5,060,000 — 72,479,000 — — 72,479,000
Conversion of convertible preferred stock into common stock (1,806,000 ) (902,000 ) (3,981,000 ) (30,193,000 ) (3,023,000 ) (40,205,000 ) (2,703,000 ) (36,445,000 ) 11,545,000 — 107,743,000 — — 107,743,000
Reclassification of Preferred Stock Warrant Liability to additional paid in capital — — — — — — — — — — 3,101,000 — — 3,101,000
Issuance of common stock due to net exercise of warrants — — — — — — — — 201,000 — — — — —
Issuance of common stock due to warrants exercised for cash — — — — — — — — 1,000 — 10,000 — — 10,000
Net loss — — — — — — — — — — — — (36,799,000 ) (36,799,000 Balance at
December 31, 2012 — $ — — $ — — $ — — $ — 18,325,000 — $ 189,212,000 $ — $ (120,306,000 ) $ 68,906,000 Issuance of
common stock in connection with exercise of stock options — — — — — — — — 495,000 — 1,338,000 — — 1,338,000
Issuance of common stock in April 2013 at $20.14 per share for services — — — — — — — — 5,000 — 101,000 — — 101,000
Compensation

See accompanying notes.
F-5
expense related to stock options — — — — — — — — — — 6,402,000 — — 6,402,000
Issuance of shares of common stock at $45.75 per share, net of issuance cost of $8,591,000 — — — — — — — — 2,920,000 — 124,999,000 — — 124,999,000
Other comprehensive income — — — — — — — — — — — 4,000 — 4,000
Net loss — — — — — — — — — — — — (51,911,000 ) (51,911,000 Balance at
December 31, 2013 — $ — — $ — — $ — — $ — 21,745,000 — $ 322,052,000 $ 4,000 $ (172,217,000 ) $ 149,839,000 Issuance of
common stock in connection with exercise of stock options — — — — — — — — 231,000 — 1,399,000 — — 1,399,000
Issuance of common stock in connection with vesting of restricted stock units — — — — — — — — 17,000 — — — — —
Compensation expense related to stock awards — — — — — — — — — — 12,449,000 — — 12,449,000
Issuance of shares of common stock in March 2014 at $45.02 per share in connection with asset acquisition — — — — — — — — 698,000 — 31,429,000 — — 31,429,000
Other comprehensive loss — — — — — — — — — — — (57,000 ) — (57,000
Net loss — — — — — — — — — — — — (135,621,000 ) (135,621,000 Balance at
December 31, 2014 — $ — — $ — — $ — — $ — 22,691,000 — $ 367,329,000 $ (53,000 ) $ (307,838,000 ) $ 59,438,000
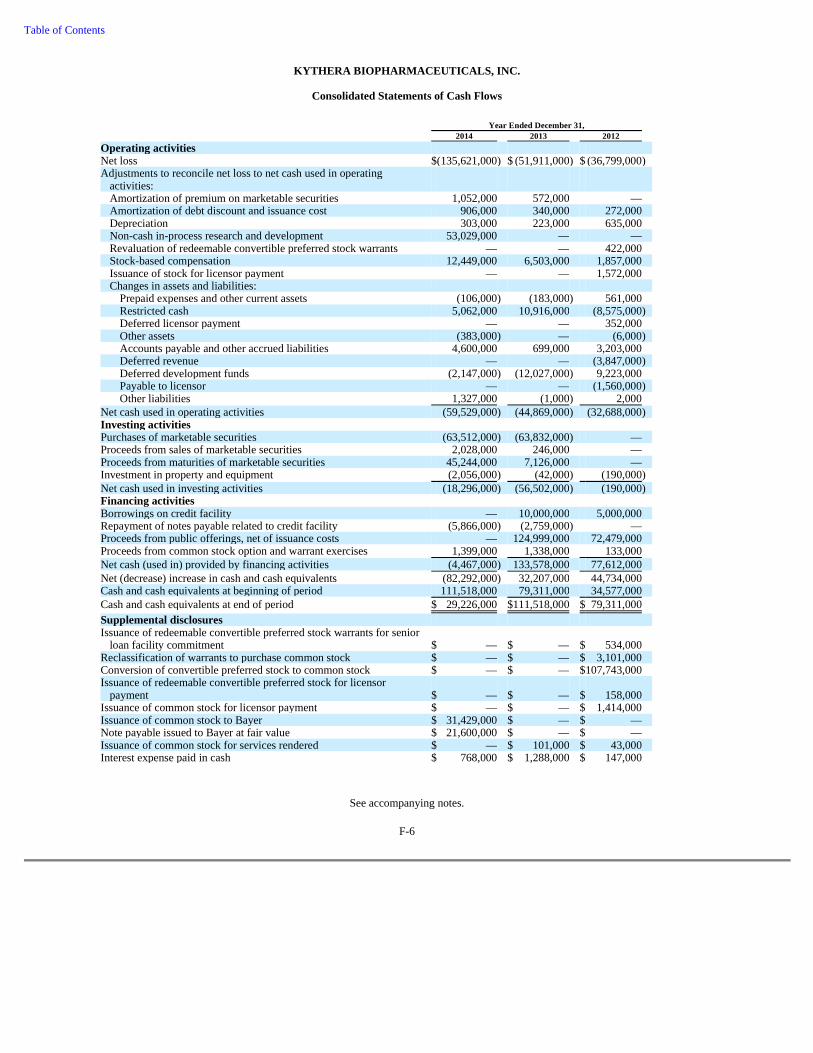
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Consolidated Statements of Cash Flows
See accompanying notes.
F-6
Year Ended December 31, 2014 2013 2012 Operating activities Net loss $ (135,621,000 ) $ (51,911,000 ) $ (36,799,000 ) Adjustments to reconcile net loss to net cash used in operating
activities: Amortization of premium on marketable securities 1,052,000 572,000 — Amortization of debt discount and issuance cost 906,000 340,000 272,000 Depreciation 303,000 223,000 635,000 Non-cash in-process research and development 53,029,000 — — Revaluation of redeemable convertible preferred stock warrants — — 422,000 Stock-based compensation 12,449,000 6,503,000 1,857,000 Issuance of stock for licensor payment — — 1,572,000 Changes in assets and liabilities:
Prepaid expenses and other current assets (106,000 ) (183,000 ) 561,000 Restricted cash 5,062,000 10,916,000 (8,575,000 ) Deferred licensor payment — — 352,000 Other assets (383,000 ) — (6,000 ) Accounts payable and other accrued liabilities 4,600,000 699,000 3,203,000 Deferred revenue — — (3,847,000 ) Deferred development funds (2,147,000 ) (12,027,000 ) 9,223,000 Payable to licensor — — (1,560,000 ) Other liabilities 1,327,000 (1,000 ) 2,000
Net cash used in operating activities (59,529,000 ) (44,869,000 ) (32,688,000 ) Investing activities Purchases of marketable securities (63,512,000 ) (63,832,000 ) — Proceeds from sales of marketable securities 2,028,000 246,000 — Proceeds from maturities of marketable securities 45,244,000 7,126,000 — Investment in property and equipment (2,056,000 ) (42,000 ) (190,000 ) Net cash used in investing activities (18,296,000 ) (56,502,000 ) (190,000 ) Financing activities Borrowings on credit facility — 10,000,000 5,000,000 Repayment of notes payable related to credit facility (5,866,000 ) (2,759,000 ) — Proceeds from public offerings, net of issuance costs — 124,999,000 72,479,000 Proceeds from common stock option and warrant exercises 1,399,000 1,338,000 133,000 Net cash (used in) provided by financing activities (4,467,000 ) 133,578,000 77,612,000 Net (decrease) increase in cash and cash equivalents (82,292,000 ) 32,207,000 44,734,000 Cash and cash equivalents at beginning of period 111,518,000 79,311,000 34,577,000 Cash and cash equivalents at end of period $ 29,226,000 $ 111,518,000 $ 79,311,000
Supplemental disclosures Issuance of redeemable convertible preferred stock warrants for senior
loan facility commitment $ — $ — $ 534,000 Reclassification of warrants to purchase common stock $ — $ — $ 3,101,000 Conversion of convertible preferred stock to common stock $ — $ — $ 107,743,000 Issuance of redeemable convertible preferred stock for licensor
payment $ — $ — $ 158,000 Issuance of common stock for licensor payment $ — $ — $ 1,414,000 Issuance of common stock to Bayer $ 31,429,000 $ — $ — Note payable issued to Bayer at fair value $ 21,600,000 $ — $ — Issuance of common stock for services rendered $ — $ 101,000 $ 43,000 Interest expense paid in cash $ 768,000 $ 1,288,000 $ 147,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements
1. Organization and Basis of Presentation
KYTHERA Biopharmaceuticals, Inc. ("KYTHERA" or the "Company") is a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of novel prescription products for the aesthetic medicine market. The Company's objective is to develop first-in-class, prescription aesthetic products using an approach that relies on the scientific rigor of biotechnology to address unmet needs in the rapidly-growing market for aesthetic medicine. The Company's initial focus is on the facial aesthetics market, which comprises the majority of the aesthetics medicine market. The Company's product candidate, ATX-101, is a potential first-in-class, injectable drug in late stage development for the treatment of submental fullness, which commonly presents as an undesirable "double chin."
KYTHERA was originally incorporated in Delaware in June 2004 under the name Dermion, Inc. It commenced operations in August 2005 and its name was changed to AESTHERx, Inc. In July 2006, the Company changed its name to KYTHERA Biopharmaceuticals, Inc.
Since commencement of operations in August 2005, the Company has devoted substantially all its efforts to the development of ATX-101, recruiting personnel and raising capital. In 2010, the Company entered into a License Agreement with Bayer Consumer Care AG and a Services, Research, Development and Collaboration Agreement with Bayer's Affiliate, Intendis GmbH, collectively referred to as the collaboration arrangement with Bayer. Bayer Consumer Care AG and Intendis GmbH are referred to jointly as Bayer. On March 7, 2014, the Company entered into an agreement whereby it acquired the rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer. In connection with this acquisition, the Company recorded a $52,757,000 charge for acquired in-process research and development during the quarter ended March 31, 2014. See Note 8, "Commitments, Collaborations and Contingencies" for a more detailed discussion of this acquisition.
The accompanying consolidated financial statements for the years ended December 31, 2014, 2013, and 2012 have been prepared on a going-concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business for the foreseeable future. Since inception in 2004, the Company has not been profitable and has incurred operating losses in each year. The Company has a limited operating history upon which you can evaluate its business and prospects. The Company has not generated any revenue from product sales to date and will continue to incur significant research and development and other expenses related to its ongoing operations. The Company has recorded net losses of $135,621,000, $51,911,000 and $36,799,000 for the years ended December 31, 2014, 2013 and 2012, respectively, and had an accumulated deficit of $307,838,000 and $172,217,000 as of December 31, 2014 and 2013, respectively. The Company has funded its operations primarily through the sale and issuance of common and preferred stock, convertible debt, and amounts received pursuant to the collaboration arrangement with Bayer and borrowings under its credit facility. Net working capital at December 31, 2014 and 2013, was $80,530,000 and $155,695,000, respectively. The Company may continue to incur losses for the next several years. At December 31, 2014, the Company had capital resources consisting of cash and cash equivalents and marketable securities of $99,661,000. At December 31, 2014, the Company had no further borrowing capacity under its existing credit facility.
The Company currently estimates that it has sufficient capital resources to meet its anticipated cash needs to fund its operations through at least the next twelve months. The Company expects to seek additional financing in the future to fund its operations, to expand its commercial organization in anticipation of potential commercial launch of ATX-101, and to develop additional product candidates, by selling additional equity, issuing debt or convertible debt securities. Additional funds may not be
F-7

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
1. Organization and Basis of Presentation (Continued)
available when the Company needs them, on terms that are acceptable to the Company, or at all. If adequate funds are not available to the Company on a timely basis, it could have a negative impact on its current business plans, including timing of launch of ATX-101.
On September 11, 2012, the Board of Directors approved a 1-for-2.6443 reverse stock split of the Company's capital stock. All share and per share information included in the accompanying consolidated financial statements has been adjusted to reflect this reverse stock split.
On October 16, 2012, the Company completed its initial public offering ("IPO") of 5,060,000 shares of common stock, which includes the exercise in full by the underwriters of their option to purchase up to 660,000 additional shares of common stock, at an offering price of $16.00 per share. The Company received net proceeds of $72,479,000, after deducting underwriting discounts, commissions and offering related transaction costs. In connection with the IPO, the Company's outstanding shares of redeemable convertible preferred stock were automatically converted into 11,545,000 shares of common stock and warrants exercisable for redeemable convertible preferred stock were automatically converted into warrants exercisable for 365,000 shares of common stock, resulting in the reclassification of the related redeemable convertible preferred stock warrant liability of $3,101,000 to additional paid-in capital.
In connection with the completion of its IPO, on October 16, 2012, the Company filed an amended and restated certificate of incorporation and bylaws, which, among other things, changed the number of authorized shares of common stock to 300,000,000 shares and preferred stock to 5,000,000 shares.
On October 15, 2013, the Company completed its secondary offering of 2,623,000 shares of common stock at an offering price of $45.75 per share. On November 12, 2013, the underwriters purchased an additional 297,000 shares of common stock pursuant to their option to purchase additional shares. The Company received aggregate net proceeds of $124,999,000, after deducting the underwriting discounts, commissions and offering related transaction costs.
2. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements include the consolidated accounts of the Company and its wholly-owned subsidiary, KYTHERA Holdings Ltd. ("KHL"), a Bermuda corporation. Intercompany accounts and transactions have been eliminated in consolidation. The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, or GAAP.
Reclassifications
Certain prior year amounts have been reclassified to conform to the current year presentation.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts in the financial statements and accompanying notes. Actual results could materially differ from those estimates. Management considers many factors in selecting appropriate financial accounting policies and controls, and in developing the estimates and assumptions that are used in the preparation of these consolidated financial statements.
F-8

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Management must apply significant judgment in this process. In addition, other factors may affect estimates, including: expected business and operational changes, sensitivity and volatility associated with the assumptions used in developing estimates, and whether historical trends are expected to be representative of future trends. The estimation process often may yield a range of potentially reasonable estimates of the ultimate future outcomes and management must select an amount that falls within that range of reasonable estimates. This process may result in actual results differing materially from those estimated amounts used in the preparation of the financial statements.
Segment Reporting
To date, the Company has viewed its operations and manages its business as one segment operating primarily in the United States.
Cash and Cash Equivalents and Marketable Securities
All highly liquid securities with maturities of 90 days or less from the date of purchase are considered to be cash equivalents. As of December 31, 2014 and 2013, cash and cash equivalents are comprised of funds invested in cash and money market accounts. The Company maintains cash balances in excess of amounts insured by the Federal Deposit Insurance Corporation, or FDIC. The accounts are monitored by management to mitigate the risk.
Investments with maturities of more than 90 days from the date of purchase are classified as marketable securities. Marketable securities are stated at fair value. The Company has classified its entire marketable securities portfolio as available-for-sale securities. Accordingly, any unrealized gain or loss on the investments is reported as a component of accumulated other comprehensive income (loss). The amortized cost of these securities is adjusted for amortization of premiums and accretion of discounts to maturity, as applicable. Such amortizations and accretions are included as a component of interest income. The entire marketable securities portfolio is considered available for use in current operations and, accordingly, all such investments are considered current assets although the stated maturity of individual investments may be one year or more beyond the balance sheet date.
Realized gains and losses on sales of marketable securities are determined on a specific identification basis and declines in value, if any, judged to be other-than-temporary on available-for-sale securities are reported in interest income. When securities are sold, any associated unrealized gain or loss previously reported in accumulated other comprehensive income (loss) is reclassified out of stockholders' equity and recorded in the statement of operations and comprehensive loss for the period. Accrued interest and dividends are included in interest income.
On a quarterly basis, the Company reviews its available-for-sale securities for other-than temporary declines in fair value below the amortized cost basis. This evaluation is based on a number of factors including the length of time and extent to which the fair value has been below the cost basis and any adverse conditions related specifically to the security, including any changes to the credit rating of the security. If the Company concludes that an other-than-temporary impairment exists, it recognizes an impairment charge to reduce the investment to fair value and records the related charge as a reduction of interest income. No impairment charges were recognized during the years ended December 31, 2014, 2013 and 2012.
Realized gains or losses from the sale of marketable securities for the years ended December 31, 2014 and 2013 were immaterial and there were no realized gains or losses from sales for the year
F-9

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
ended December 31, 2012. As of December 31, 2012 the Company did not have any marketable securities.
Restricted Cash
There was no restricted cash at December 31, 2014. Restricted cash of $5,062,000 at December 31, 2013 represents restricted cash received through the collaboration arrangement with Bayer that will be used to fund certain further global development activities of ATX-101. See Note 8, "Commitments, Collaborations and Contingencies."
Payments the Company received to fund collaboration efforts under the terms of the collaboration agreement with Bayer were recorded as restricted cash and deferred development funds, and were recognized as an offset to development expenses as the restricted cash was utilized to fund such development activities.
Property and Equipment
Property and equipment are recorded at historical cost and consisted of the following:
With the exception of leasehold improvements, discussed below, depreciation is recorded using the straight-line method over the estimated useful lives of the assets (one to five years). The Company reviews its property and equipment assets for impairment in value whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. There were no impairments recognized during the years ended December 31, 2014, 2013 and 2012. During the year ended December 31, 2014, the Company disposed of certain assets totaling $254,000, consisting primarily of leasehold improvements and furniture and fixtures from the Company's previously leased facilities. These assets were fully depreciated at the date of disposal. The Company had no asset disposals during the years ended December 31, 2013 and 2012.
The $1,465,000 in leasehold improvements as of December 31, 2014 relates to the leasehold improvements constructed at the Company's newly leased facility. These leasehold improvements were placed into service in August 2014 when the Company began to occupy the newly leased facility and are being depreciated on a straight-line basis over the three year lease term, which is the shorter of the improvements expected useful lives and the lease term.
Cameras were used in the Company's clinical trials, are fully depreciated as of December 31, 2014 and are being held for disposal.
F-10
December 31, 2014 2013 Cameras $ 923,000 $ 923,000 Computer hardware and electronics 420,000 182,000 Leasehold improvements 1,465,000 126,000 Furniture & fixtures 345,000 108,000 Software 9,000 21,000
3,162,000 1,360,000 Less accumulated depreciation (1,315,000 ) (1,266,000 )
$ 1,847,000 $ 94,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Concentration of Credit Risk
Financial instruments, which potentially subject the Company to significant concentrations of credit risk, consist primarily of cash and cash equivalents and marketable securities. The primary objectives for the Company's investment portfolio are the preservation of capital and the maintenance of liquidity. The Company does not enter into any investment transactions for trading or speculative purposes.
The Company's investment policy limits investments to certain types of instruments such as certificates of deposit, money market instruments, obligations issued by U.S. government and U.S. government agencies and corporate debt securities and places restrictions on maturities and concentration by type and issuer. The Company maintains cash balances in excess of amounts insured by the FDIC. The accounts are monitored by management to mitigate the risk.
Stock-Based Compensation
The Company accounts for stock options issued to employees and directors using an option pricing model for estimating fair value at the date of grant. The Company accounts for restricted stock units issued to employees at fair value based on the market price of the Company's stock on the date of grant. Compensation expense is recognized for the portion of the award that is ultimately expected to vest over the period during which the recipient renders the required services to the Company generally using the straight-line single option method.
In accordance with authoritative guidance, the fair value of non-employee stock based awards is remeasured as the awards vest, and the resulting increase in fair value, if any, is recognized as expense in the period the related services are rendered.
Redeemable Convertible Preferred Stock Warrants
Prior to the Company's IPO, freestanding warrants that related to the purchase of redeemable convertible preferred stock were classified as liabilities and recorded at fair value each reporting period regardless of the timing of the redemption feature or the redemption price or the likelihood of redemption. The warrants were subject to remeasurement at each balance sheet date and any change in fair value was recognized as a component of warrant and other income (expense), net. Pursuant to the terms of these warrants, upon the conversion of the class of preferred stock underlying the warrant, the warrants automatically became exercisable for shares of the Company's common stock based upon the conversion ratio of the underlying class of preferred stock. The consummation of the Company's IPO resulted in the conversion of all classes of the Company's preferred stock into common stock. Upon such conversion of the underlying classes of preferred stock, the warrants were reclassified as a component of equity and were no longer subject to remeasurement.
Fair Value of Financial Instruments
The carrying amounts reported in the accompanying consolidated financial statements for accounts payable and accrued liabilities approximate their fair value because of the short-term nature of these accounts. The fair value of cash equivalents, marketable securities and the notes payable are discussed in Note 9, "Fair Value Measurements."
F-11

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Revenue Recognition
The Company recognizes revenue when all of the following four criteria are present: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured.
For arrangements that involve the delivery of more than one element, each product, service and/or right to use assets is evaluated to determine whether it qualifies as a separate unit of accounting. This determination is based on whether the deliverable has "stand-alone value" to the customer. The consideration that is fixed or determinable is then allocated to each separate unit of accounting based on the relative selling price of each deliverable. The estimated selling price of each deliverable is determined using the following hierarchy of values: (i) vendor-specific objective evidence of fair value, (ii) third-party evidence of selling price (TPE) and (iii) best estimate of selling price (BESP). The BESP reflects the Company's best estimate of what the selling price would be if the deliverable was regularly sold by us on a stand-alone basis. In most cases the Company expects to use TPE or BESP for allocating consideration to each deliverable. The consideration allocated to each unit of accounting is recognized as the related goods or services are delivered, limited to the consideration that is not contingent upon future deliverables. Analyzing the arrangement to identify deliverables requires the use of judgment, and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation.
The Company's revenue earned during the year ended December 31, 2012 is related to the license agreement with Bayer executed in 2010. This agreement provided for various types of payments, including non-refundable upfront license fees, milestone payments, and future royalties on Bayer's net product sales of ATX-101.
The Company received a non-refundable upfront license payment of approximately $21,319,000 from Bayer upon execution of the license agreement. The terms of the collaboration arrangement with Bayer included continuing performance obligations and development and clinical manufacturing supply obligations that were conditions to Bayer's decision to pursue continued development and regulatory approval for ATX-101. Due to these ongoing performance obligations, the Company determined that the license did not have stand-alone value. The Company also did not have objective and reliable evidence of the fair value of these undelivered obligations. Accordingly, amounts received upfront under the license agreement were recorded as deferred revenue and were recognized on a straight-line basis over the expected period of substantial involvement in these collaboration activities, which were completed as of May 31, 2012. The period over which these activities were to be performed was based upon management's estimate of the period to complete the development activities required to be conducted relative to the upfront license fee and development funds received from Bayer.
The Company recognizes revenue from milestone payments when earned, provided that (i) the milestone event is substantive in that it can only be achieved based in whole or in part on either the entity's performance or on the occurrence of a specific outcome resulting from the entity's performance and its achievability was not reasonably assured at the inception of the collaboration arrangement and (ii) the Company does not have ongoing performance obligations related to the achievement of the milestone earned and (iii) it would result in additional payments being due to the Company. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment is non-refundable; achievement of the milestone was not reasonably assured at the inception of the arrangement; substantive effort is involved to achieve the milestone; and the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement and the
F-12

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
related risk associated with the achievement of the milestone. Any amounts received under the collaboration arrangement in advance of performance, if deemed substantive, are recorded as deferred revenue and recognized as revenue as the Company completes its performance obligations.
Research and Development Costs
Major components of research and development (R&D) costs include cash compensation, stock-based compensation, pre-clinical studies, clinical trials and related clinical manufacturing, materials and supplies, and fees paid to consultants and other entities that conduct certain research and development activities on the Company's behalf. R&D costs, including upfront fees and milestones paid to collaborators, are expensed as goods are received or services rendered. Costs to acquire technologies to be used in research and development that have not reached technological feasibility and have no alternative future use are also expensed as incurred. Costs incurred in connection with clinical trial activities for which the underlying nature of the activities themselves do not directly relate to active research and development, such as costs incurred for market research and focus groups linked to clinical strategy as well as costs to build the Company's brand, are not included in R&D costs but are reflected as general and administrative expenses.
The Company enters into agreements with various research institutions, contract laboratories, contract manufacturers and consultants. These agreements are generally on a fee-for-service basis and are cancelable.
Clinical Trial Accruals
As part of the process of preparing its consolidated financial statements, the Company is required to estimate its expenses resulting from its obligations under contracts with vendors and consultants and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided to us under such contracts. The Company's objective is to reflect the appropriate trial expenses in its consolidated financial statements by matching those expenses with the period in which services and efforts are expended. The Company accounts for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. The Company determines accrual estimates through financial models taking into account discussion with applicable personnel and outside service providers as to the progress or state of consummation of trials, or the services completed. During the course of a clinical trial, the Company adjusts its rate of clinical expense recognition if actual results differ from its estimates. The Company makes estimates of its accrued expenses as of each balance sheet date in its consolidated financial statements based on the facts and circumstances known to us at that time. Although the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in us reporting amounts that are too high or too low for any particular period. Through December 31, 2014, there have been no material adjustments to the Company's prior period estimates of accrued expenses for clinical trials. The Company's clinical trial accrual is dependent in part upon the timely and accurate reporting of contract research organizations and other third-party vendors.
F-13

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Basic and Diluted Net Loss Per Common Share
Basic net loss per common share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period, excluding the effects of converting redeemable preferred stock, warrants to purchase redeemable convertible preferred stock or common stock and stock options and restricted stock units outstanding. Diluted net loss per common share is computed by dividing the net loss by the sum of the weighted average number of common shares outstanding during the period plus the potential dilutive effects of redeemable convertible preferred stock, warrants to purchase redeemable convertible preferred stock or common stock and stock options and restricted stock units outstanding during the period calculated in accordance with the treasury stock method. Because the impact of these items is anti-dilutive during periods of net loss, there was no difference between the weighted average number of shares used to calculate basic and diluted net loss per common share for the years ended December 31, 2014, 2013 and 2012. The calculation of weighted average diluted shares outstanding excludes the dilutive effect of redeemable convertible preferred stock, warrants to purchase redeemable convertible preferred stock or common stock and stock options and restricted stock units, as the impact of such items are anti-dilutive during periods of net loss. Shares excluded from the calculation, prior to the use of the treasury stock method, unweighted, were 3,113,000, 2,335,000 and 1,956,000 at December 31, 2014, 2013 and 2012, respectively.
Income Taxes
The Company accounts for income taxes under the asset and liability method, which requires, among other things, that deferred income taxes be provided for temporary differences between the tax bases of the Company's assets and liabilities and their financial statement reported amounts. A valuation allowance is recorded when it is more likely than not that some portion or all of a deferred tax asset will not be realized. Management has considered estimated future taxable income and ongoing prudent and feasible tax planning strategies in assessing the amount of the valuation allowance. When the Company establishes or reduces the valuation allowance against its deferred tax assets, its provision for income taxes will increase or decrease, respectively, in the period such determination is made.
Additionally, the Company recognizes the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities based on the technical merits of the position. The tax benefit recognized in the consolidated financial statements for a particular tax position is based on the largest benefit that is more likely than not to be realized upon settlement. Accordingly, the Company establishes reserves for uncertain tax positions.
Comprehensive Income (Loss)
Comprehensive income (loss) is comprised of net loss and other comprehensive income (loss). For the years ended December 31, 2014 and 2013, the only component of other comprehensive income (loss) relates to net unrealized gains and losses on marketable securities. The Company had no other comprehensive income (loss) for the year ended December 31, 2012.
There were no material reclassifications out of accumulated other comprehensive income during the years ended December 31, 2014 and 2013.
F-14

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Collaboration Arrangements
From time to time, the Company enters into certain collaboration arrangements with third parties regarding the research and development, manufacture and/or commercialization of products and product candidates. These collaborations generally provide for non-refundable, upfront license fees, research and development and commercial performance milestone payments, cost sharing and royalty payments. The collaboration agreements with third parties are performed on a "best efforts" basis with no guarantee of either technological or commercial success. The Company evaluates whether an arrangement is a collaboration arrangement at its inception based on the facts and circumstances specific to the arrangement. The Company reevaluates whether an arrangement qualifies or continues to qualify as a collaboration arrangement whenever there is a change in the anticipated or actual ultimate commercial success of the endeavor. See Note 8, "Commitments, Collaborations and Contingencies."
Recent Accounting Pronouncements
In May 2014, a new standard was issued related to revenue recognition, which requires an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The new standard will replace most of the existing revenue recognition standards in U.S. GAAP when it becomes effective on January 1, 2017. Early adoption is not permitted. The new standard allows for either "full retrospective" adoption, whereby the new standard is applied to each prior reporting period presented or "modified retrospective" adoption, whereby the new standard is only applied to the most current period presented with the cumulative effect of the change recognized at the date of the initial application. The Company is assessing the potential impact, including method of adoption, of the new standard on its consolidated statements of financial position and results of operations and, comprehensive income (loss) and has not yet selected a method of adoption.
In August 2014, a new standard was issued which will require management to evaluate if there is substantial doubt about the entity's ability to continue as a going concern and, if so, disclose it in both annual and interim reporting periods. The new standard will become effective for the Company's annual filing for the period ending December 31, 2016 and interim periods thereafter, with early adoption permitted. The Company does not believe that the eventual adoption of this accounting standard will have a material impact on the Company's consolidated financial statements and related disclosures.
F-15
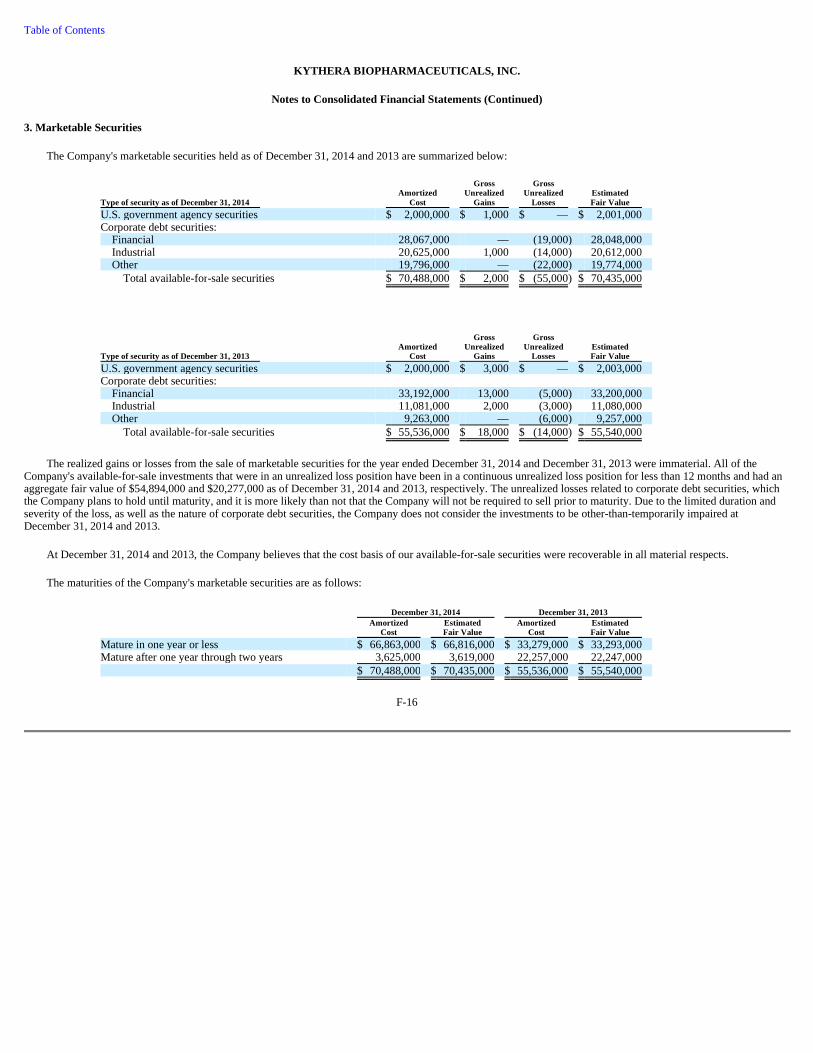
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
3. Marketable Securities
The Company's marketable securities held as of December 31, 2014 and 2013 are summarized below:
The realized gains or losses from the sale of marketable securities for the year ended December 31, 2014 and December 31, 2013 were immaterial. All of the Company's available-for-sale investments that were in an unrealized loss position have been in a continuous unrealized loss position for less than 12 months and had an aggregate fair value of $54,894,000 and $20,277,000 as of December 31, 2014 and 2013, respectively. The unrealized losses related to corporate debt securities, which the Company plans to hold until maturity, and it is more likely than not that the Company will not be required to sell prior to maturity. Due to the limited duration and severity of the loss, as well as the nature of corporate debt securities, the Company does not consider the investments to be other-than-temporarily impaired at December 31, 2014 and 2013.
At December 31, 2014 and 2013, the Company believes that the cost basis of our available-for-sale securities were recoverable in all material respects.
The maturities of the Company's marketable securities are as follows:
F-16
Type of security as of December 31, 2014 Amortized
Cost
Gross Unrealized
Gains
Gross Unrealized
Losses Estimated Fair Value
U.S. government agency securities $ 2,000,000 $ 1,000 $ — $ 2,001,000 Corporate debt securities:
Financial 28,067,000 — (19,000 ) 28,048,000 Industrial 20,625,000 1,000 (14,000 ) 20,612,000 Other 19,796,000 — (22,000 ) 19,774,000
Total available-for-sale securities $ 70,488,000 $ 2,000 $ (55,000 ) $ 70,435,000
Type of security as of December 31, 2013 Amortized
Cost
Gross Unrealized
Gains
Gross Unrealized
Losses Estimated Fair Value
U.S. government agency securities $ 2,000,000 $ 3,000 $ — $ 2,003,000 Corporate debt securities:
Financial 33,192,000 13,000 (5,000 ) 33,200,000 Industrial 11,081,000 2,000 (3,000 ) 11,080,000 Other 9,263,000 — (6,000 ) 9,257,000
Total available-for-sale securities $ 55,536,000 $ 18,000 $ (14,000 ) $ 55,540,000
December 31, 2014 December 31, 2013
Amortized
Cost Estimated Fair Value
Amortized Cost
Estimated Fair Value
Mature in one year or less $ 66,863,000 $ 66,816,000 $ 33,279,000 $ 33,293,000 Mature after one year through two years 3,625,000 3,619,000 22,257,000 22,247,000
$ 70,488,000 $ 70,435,000 $ 55,536,000 $ 55,540,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
4. Bridge Financing
In January 2006, the Company entered into bridge financing transactions by issuing convertible promissory notes and warrants, for aggregate proceeds of $8,051,000 from certain existing and new investors. The convertible notes bore interest at 8% and were automatically convertible into shares of the Company's preferred stock upon the occurrence of a qualified preferred stock financing as described in the convertible promissory note agreement. The estimated fair value of the warrants, totaling approximately $440,000, was treated as a deduction from the note proceeds (debt discount) and amortized as interest expense over the period the notes were outstanding.
The increase in the fair value of the warrants from issuance through IPO in October of 2012, totaled $1,205,000 and has been recorded in warrant and other income (expense), net, and included $349,000 of interest expense in the statements of operations and comprehensive loss for the year ended December 31, 2012. Effective immediately prior to the closing of the Company's IPO in October of 2012, the warrants for convertible preferred stock became warrants for common stock and were reclassified to equity. All but 178 warrants were exercised during 2012, with the remaining warrants being exercised in January 2013, resulting in the net issuance of 153,000 shares of common stock.
5. Notes Payable
Notes payable consisted of the following:
Future maturities of the Notes payable as of December 31, 2014 are as follows:
Note Payable to Bayer
In March 2014, as part of the agreement to acquire the rights to develop and commercialize ATX-101 outside of the United States and Canada, the Company issued a $51,000,000 unsecured promissory note to Bayer. The promissory note is subordinated to amounts owed under the Company's existing credit facility. Interest is accrued at 5% per annum and is payable on the anniversary date of
F-17
December 31,
2014 December 31,
2013 Note payable to Bayer $ 22,166,000 $ — Notes payable related to credit facility 5,979,000 11,505,000
28,145,000 11,505,000 Less current portion (5,484,000 ) (5,526,000 ) Notes payable-net of current portion $ 22,661,000 $ 5,979,000
2015 $ 5,823,000 2016 552,000 2024 51,000,000
57,375,000 Unamortized debt issuance cost (29,230,000 )
28,145,000 Current portion (5,484,000 ) Notes payable—net of current portion $ 22,661,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
5. Notes Payable (Continued)
the note. The Company has the option of deferring up to 80% of the accrued interest, adding any unpaid interest to the principal amount. The note is due on the ten year anniversary from issuance, with the option to prepay at any point in time without penalty.
As the note payable to Bayer bears interest at 5%, which was below the borrowing rate available to the Company at issuance, it was recorded at its fair value of $21,600,000, which is the present value of the future cash flows assuming a borrowing rate of 15%. The difference between face value and fair value of the note was recorded as a discount and is being amortized over the life of the note utilizing the effective interest method. The balance of the note payable at December 31, 2014 is as follows:
Included in interest expense for the year ended December 31, 2014 was amortization of the debt discount of $566,000 and interest expense of $2,089,000 related to the note payable to Bayer. The balance of accrued interest expense at December, 2014 was $2,089,000 and the balance of the remaining unamortized discount was $28,834,000 at December, 2014.
Notes Payable Related to Credit Facility
On March 21, 2011, the Company executed a debt financing agreement whereby the Company had access to borrow up to $15,000,000 of senior loan financing through January 1, 2012. Subsequently, the credit facility was amended to provide for an extension of the drawdown period through December 1, 2012, provided that it had drawn down at least $5,000,000 as of August 1, 2012. On July 23, 2012, the Company obtained an additional 60 day extension to the initial drawdown date from August 1, 2012 to October 1, 2012. For each draw, the Company shall make six months of interest only payments at a fixed rate of 11.5% followed by 30 months of interest and principal payments at a fixed rate of 8.5% and a final payment of 6% of the amount drawn due with the last principal payment. The debt is secured by all the Company's assets, except for the Company's intellectual property, which is subject to a negative pledge agreement.
On October 1, 2012 the Company drew $5,000,000 and in November 2012, obtained an extension allowing for the draw of the remaining $10,000,000 in $5,000,000 increments by January 31, 2013 and February 28, 2013. On January 31, 2013 the Company drew down an additional $5,000,000 of available funds under its credit facility and on February 28, 2013, the Company drew the remaining $5,000,000. As of December 31, 2014 and December 31, 2013, there was no further borrowing capacity under this facility.
Upon entry into the agreement, the Company issued warrants to purchase 34,000 shares of Series C redeemable convertible preferred stock ("Series C Preferred") at an exercise price of $13.3530. The estimated fair value of the warrants at issuance of approximately $345,000 was recorded as a deferred financing cost offsetting the notes payable liability and is being amortized to interest expense over the expected term of the loan.
F-18
December 31,
2014 Face value of note payable to Bayer $ 51,000,000 Debt discount at date of issuance (29,400,000 ) Amortization of debt discount 566,000 Note payable to Bayer $ 22,166,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
5. Notes Payable (Continued)
Upon extension of the credit facility in December 2011, the Company issued a warrant to purchase up to an additional 86,000 shares of its Series D redeemable convertible preferred stock ("Series D Preferred") at an exercise price of $13.9040. The estimated fair value of the warrant at issuance of approximately $1,074,000 was recorded as a deferred financing cost offsetting the notes payable liability and is being amortized to interest expense over the expected term of the loan.
The increase in the fair value of the Series C Preferred warrants from issuance in March 2011 through the IPO in October of 2012, totaled $55,000 and was recorded in warrant and other income (expense), net, in the statement of operations and comprehensive loss, and included expense of $91,000 for the year ended December 31, 2012. The decrease in value of the Series D Preferred warrants from issuance in December 2011 through the IPO in October of 2012 totaled $18,000 and was recorded in warrant and other income (expense), net, in the statement of operations and comprehensive loss for the year ended December 31, 2012. There was no change in the fair value of the Series D Preferred warrants during the year ended December 31, 2011 due to the timing of issuance.
All warrants were exercised on a cashless basis in December of 2012 resulting in the net issuance of 49,000 shares of common stock and there were no warrants outstanding as of December 31, 2012.
Included in interest expense for the years ended December 31, 2014, 2013 and 2012 was amortization of debt issuance costs of $340,000, $340,000 and $272,000 and interest expense of $1,162,000, $1,640,000 and $169,000 related to the notes payable, respectively. The balance of accrued interest expense at December 31, 2014 and 2013 was $768,000 and $375,000, respectively. The remaining unamortized debt issuance cost at December 31, 2014 and 2013 was $396,000 and $736,000, respectively.
The credit facility includes various covenants. As of December 31, 2014 and 2013, the Company was in compliance with all covenants associated with the credit facility.
6. Redeemable Convertible Preferred Stock
Upon the Company's IPO in October of 2012, all shares of convertible preferred stock converted into 11,545,000 shares of common stock. The Series A Preferred, Series C Preferred and Series D Preferred were converted into common stock on a 1-for-1 basis. The Series B Preferred was converted into shares of common stock on a 1-for-1.00785 basis.
Dividends
The holders of the Preferred Stock were entitled to receive dividends, when and if declared by the board of directors. No dividends had been declared through the date of conversion.
F-19

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
7. Stockholders' Equity
Shares Reserved for Future Issuance
Shares of the Company's common stock reserved for future issuance are as follows:
Stock Awards
In September 2012, the Company's board of directors approved the 2012 Equity Incentive Award Plan ("the 2012 Plan"), which became effective upon completion of the Company's IPO. The 2012 Plan provides for the granting of incentive and nonstatutory stock options, restricted stock, restricted stock units, stock appreciation rights, deferred stock, dividend equivalents, performance awards, stock payments and other stock-based and cash-based awards to its employees, directors, and consultants at the discretion of the compensation committee of the board of directors. The 2012 Plan is the successor to the Amended and Restated 2004 Stock Plan. Any remaining shares available for future issuance under the 2004 Stock Plan are available for issuance under the 2012 Plan. In addition, the plan reserve will automatically increase annually from January 1, 2013 through January 1, 2022 by an amount equal to the smaller of: (a) 1,512,687 shares, (b) four percent of the number of shares of common stock issued and outstanding on the immediately preceding December 31, or (c) an amount determined by the Company's board of directors. As of December 31, 2014, 2013 and 2012, a total of 4,094,000, 3,224,000 and 2,491,000 shares, respectively, were authorized for grant under the 2012 Plan.
In September 2014, the Company's board of directors approved the 2014 Employment Commencement Incentive Plan ("the 2014 Plan"), under which up to 900,000 shares are available for grant. The 2014 Plan provides for the granting of nonstatutory stock options, restricted stock, restricted stock units, performance awards, dividend equivalents, stock payments, stock appreciation rights or other incentive awards or performance share awards to new employees as inducement to enter employment.
Stock options may be granted with exercise prices not less than the estimated fair market value of the Company's common stock. Under the 2012 Plan, incentive stock options granted to individuals owning more than 10% of the total combined voting power of all classes of stock ("a 10% stockholder") are exercisable up to five years from the date of grant. The 2012 Plan provides that the exercise price of any incentive stock option granted to a 10% stockholder cannot be less than 110% of the estimated fair value of the common stock on the date of grant. Except as set forth above, options granted under the Company's plans expire no later than ten years from the date of grant. Options granted to employees under the Company's plans vest over periods determined by the board of directors, which generally vest over four years. Additionally, the Company has granted certain performance-based stock option awards and restricted stock units that vest based on the achievement of certain predetermined milestones. Prior to the Company's IPO, the board of directors determined the estimated fair value of its common stock based on assistance from an independent third-party valuation expert. Since the Company's IPO, the fair value of the Company's common stock is the closing share
F-20
December 31, 2014 2013 Common stock awards outstanding 3,113,000 2,335,000 Common stock awards available for grant 1,040,000 295,000 Total common shares reserved for future issuance 4,153,000 2,630,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
7. Stockholders' Equity (Continued)
price as reported on The NASDAQ Global Select Market on the date of grant. The fair value of the Company's common stock was determined to be $34.68, $37.35 and $30.34 at December 31, 2014, 2013 and 2012, respectively.
The Company estimates the fair value of its share-based awards to employees and directors using the Black-Scholes option pricing model. The Black-Scholes model requires the input of complex and subjective assumptions, including (a) the expected stock price volatility, (b) the expected term of the award, (c) the risk free interest rate and (d) expected dividends. Due to the Company's limited operating history and a lack of company specific historical and implied volatility data, the Company has based its estimate of expected volatility on the historical volatility of a group of similar companies that are publicly traded. When selecting these public companies on which it has based its expected stock price volatility, the Company selected companies with comparable characteristics to it, including enterprise value, risk profiles, position within the industry, and with historical share price information sufficient to meet the expected life of the stock-based awards. The historical volatility data was computed using the daily closing prices for the selected companies' shares during the equivalent period of the calculated expected term of the stock-based awards. The Company will continue to apply this process until a sufficient amount of historical information regarding the volatility of its own stock price becomes available. The Company has estimated the expected life of its employee stock options using the "simplified" method, whereby, the expected life equals the arithmetic average of the vesting term and the original contractual term of the option due to its lack of sufficient historical data. The risk-free interest rates for periods within the expected life of the option are based on the U.S. Treasury yield curve in effect during the period the options were granted. The Company has never paid, and does not expect to pay dividends in the foreseeable future. The Company also grants restricted stock units and the grant date fair value per share of those awards is equal to the closing stock price on the date of grant.
The weighted-average assumptions used to estimate the fair value of stock options using the Black-Scholes option pricing model were as follows:
The Company is also required to estimate forfeitures at the time of grant, and revise those estimates in subsequent periods if actual forfeitures differ from its estimates. The Company uses historical data to estimate pre-vesting option forfeitures and record stock-based compensation expense only for those awards that are expected to vest. To the extent that actual forfeitures differ from the Company's estimates, the difference is recorded as a cumulative adjustment in the period the estimates were revised. For the years ended December 31, 2014, 2013 and 2012, the Company applied a forfeiture rate based on the Company's historical forfeitures. No forfeiture rate was applied prior to January 1, 2011 as forfeitures prior to such date had been insignificant.
F-21
Year Ended December 31, 2014 2013 2012 Weighted-average exercise price of options granted $ 39.32 $ 25.90 $ 10.19 Expected volatility 70 % 67 % 75 % Expected term (in years) 6.1 5.9 6.1 Weighted-average risk free interest rate 1.89 % 1.28 % 1.05 % Expected dividends 0 % 0 % 0 % Weight-average grant date fair value 24.99 17.85 7.09
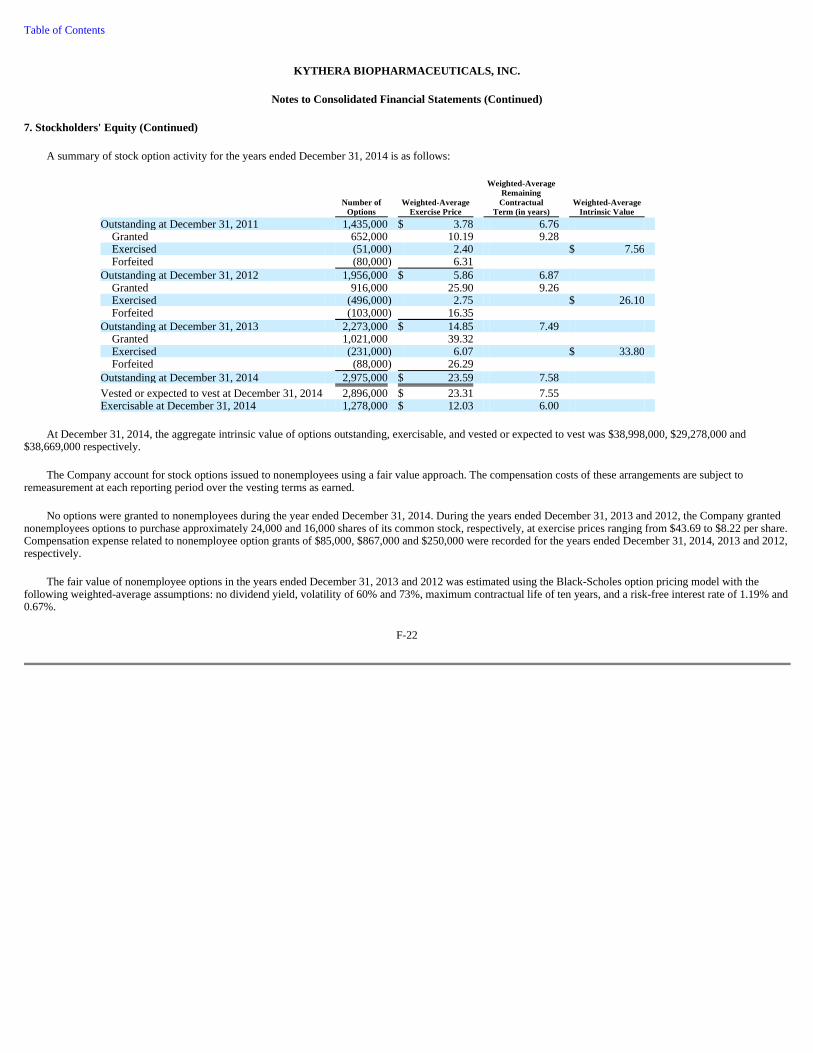
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
7. Stockholders' Equity (Continued)
A summary of stock option activity for the years ended December 31, 2014 is as follows:
At December 31, 2014, the aggregate intrinsic value of options outstanding, exercisable, and vested or expected to vest was $38,998,000, $29,278,000 and $38,669,000 respectively.
The Company account for stock options issued to nonemployees using a fair value approach. The compensation costs of these arrangements are subject to remeasurement at each reporting period over the vesting terms as earned.
No options were granted to nonemployees during the year ended December 31, 2014. During the years ended December 31, 2013 and 2012, the Company granted nonemployees options to purchase approximately 24,000 and 16,000 shares of its common stock, respectively, at exercise prices ranging from $43.69 to $8.22 per share. Compensation expense related to nonemployee option grants of $85,000, $867,000 and $250,000 were recorded for the years ended December 31, 2014, 2013 and 2012, respectively.
The fair value of nonemployee options in the years ended December 31, 2013 and 2012 was estimated using the Black-Scholes option pricing model with the following weighted-average assumptions: no dividend yield, volatility of 60% and 73%, maximum contractual life of ten years, and a risk-free interest rate of 1.19% and 0.67%.
F-22
Number of
Options Weighted-Average
Exercise Price
Weighted-Average Remaining Contractual
Term (in years) Weighted-Average
Intrinsic Value Outstanding at December 31, 2011 1,435,000 $ 3.78 6.76
Granted 652,000 10.19 9.28 Exercised (51,000 ) 2.40 $ 7.56 Forfeited (80,000 ) 6.31
Outstanding at December 31, 2012 1,956,000 $ 5.86 6.87 Granted 916,000 25.90 9.26 Exercised (496,000 ) 2.75 $ 26.10 Forfeited (103,000 ) 16.35
Outstanding at December 31, 2013 2,273,000 $ 14.85 7.49 Granted 1,021,000 39.32 Exercised (231,000 ) 6.07 $ 33.80 Forfeited (88,000 ) 26.29
Outstanding at December 31, 2014 2,975,000 $ 23.59 7.58
Vested or expected to vest at December 31, 2014 2,896,000 $ 23.31 7.55 Exercisable at December 31, 2014 1,278,000 $ 12.03 6.00
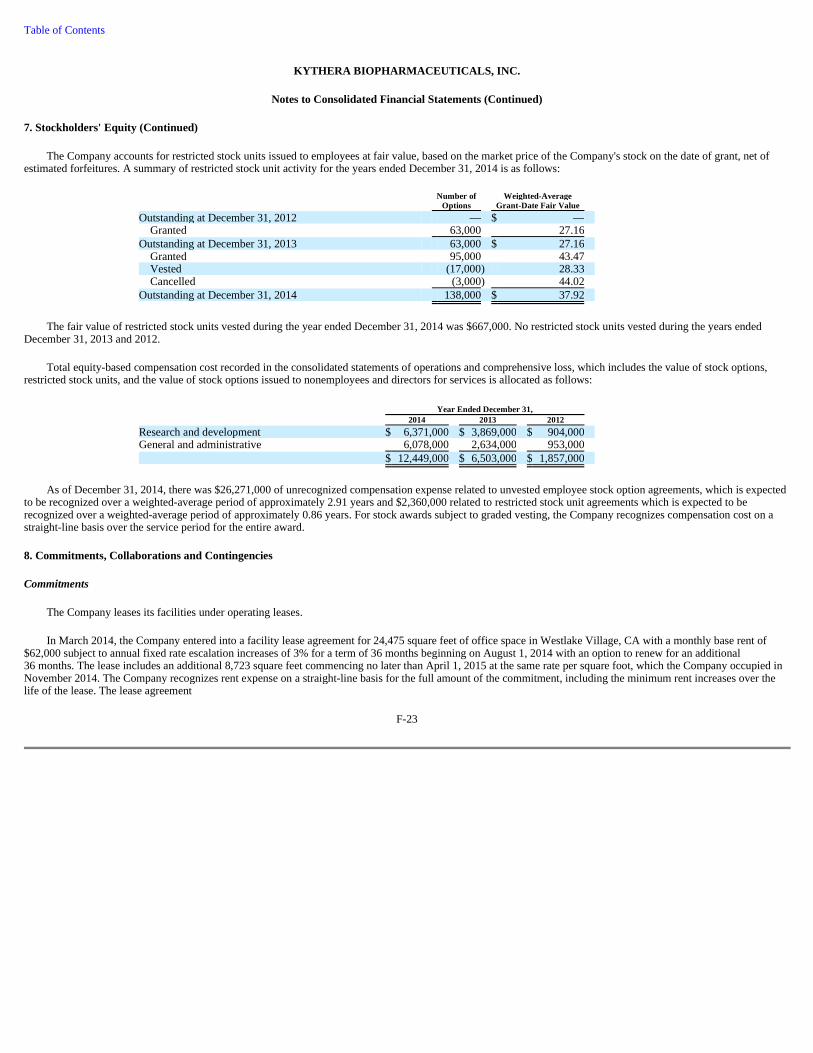
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
7. Stockholders' Equity (Continued)
The Company accounts for restricted stock units issued to employees at fair value, based on the market price of the Company's stock on the date of grant, net of estimated forfeitures. A summary of restricted stock unit activity for the years ended December 31, 2014 is as follows:
The fair value of restricted stock units vested during the year ended December 31, 2014 was $667,000. No restricted stock units vested during the years ended December 31, 2013 and 2012.
Total equity-based compensation cost recorded in the consolidated statements of operations and comprehensive loss, which includes the value of stock options, restricted stock units, and the value of stock options issued to nonemployees and directors for services is allocated as follows:
As of December 31, 2014, there was $26,271,000 of unrecognized compensation expense related to unvested employee stock option agreements, which is expected to be recognized over a weighted-average period of approximately 2.91 years and $2,360,000 related to restricted stock unit agreements which is expected to be recognized over a weighted-average period of approximately 0.86 years. For stock awards subject to graded vesting, the Company recognizes compensation cost on a straight-line basis over the service period for the entire award.
8. Commitments, Collaborations and Contingencies
Commitments
The Company leases its facilities under operating leases.
In March 2014, the Company entered into a facility lease agreement for 24,475 square feet of office space in Westlake Village, CA with a monthly base rent of $62,000 subject to annual fixed rate escalation increases of 3% for a term of 36 months beginning on August 1, 2014 with an option to renew for an additional 36 months. The lease includes an additional 8,723 square feet commencing no later than April 1, 2015 at the same rate per square foot, which the Company occupied in November 2014. The Company recognizes rent expense on a straight-line basis for the full amount of the commitment, including the minimum rent increases over the life of the lease. The lease agreement
F-23
Number of
Options Weighted-Average
Grant -Date Fair Value Outstanding at December 31, 2012 — $ —
Granted 63,000 27.16 Outstanding at December 31, 2013 63,000 $ 27.16
Granted 95,000 43.47 Vested (17,000 ) 28.33 Cancelled (3,000 ) 44.02
Outstanding at December 31, 2014 138,000 $ 37.92
Year Ended December 31, 2014 2013 2012 Research and development $ 6,371,000 $ 3,869,000 $ 904,000 General and administrative 6,078,000 2,634,000 953,000
$ 12,449,000 $ 6,503,000 $ 1,857,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
8. Commitments, Collaborations and Contingencies (Continued)
provides for free rent for three months for the initial lease space and until April 1, 2015 on the additional space, as well as leasehold improvement allowances of $319,000. These lease incentives were accounted for as deferred rent and are amortized to rent expense over the life of the lease. Under the terms of the lease agreement, the Company expects to incur net rent expense associated with this lease of $854,000 per year.
Rent expense under the Company's operating leases for the years ended December 31, 2014, 2013 and 2012, was $741,000, $409,000 and $379,000, respectively.
Future minimum lease payments are as follows at December 31, 2014:
Collaboration Arrangements
From time to time, the Company enters into collaborative arrangements for its research and development and manufacturing activities. Each collaboration is unique in nature and the Company's significant agreements are discussed below.
Bayer
In August 2010, the Company entered into a license agreement with Bayer Consumer Care AG that provides Bayer with an exclusive license to develop, manufacture and commercialize ATX-101 outside of the United States and Canada. In connection with the License Agreement the Company also entered into a related Services, Research, Development and Collaboration Agreement with Bayer's affiliate, Intendis GmbH. These agreements are referred to jointly as the collaboration arrangement with Bayer, and Bayer Consumer Care AG and Intendis GmbH are referred to jointly as Bayer. In connection with the entry into the collaboration arrangement, Bayer paid the Company an upfront license fee of approximately $21,319,000 and approximately $22,247,000 for research and development to fund a portion of Bayer's European Phase III clinical trials. The Company received a contingent event-based payments of $15,800,000 on May 31, 2012, triggered by the completion of Bayer's European Phase III clinical trial for ATX-101, the receipt of two consecutive validated manufacturing lots and Bayer's subsequent decision to pursue continued development and seek regulatory approval of ATX-101. This amount was recorded as revenue upon receipt as all revenue recognition criteria had been met. At this time, Bayer also elected to pursue additional research and development activities related to ATX-101, which are distinct from the original development activities, and subsequently funded $17,400,000 for these additional Bayer development activities. In March 2014, the Company acquired the rights to ATX-101 outside the United States and Canada from Bayer and no longer expects to recognize revenue from Bayer.
License fees of $21,319,000 were deferred and recognized on a straight-line basis over period of substantial involvement in collaboration activities that were required to be conducted relative to the
F-24
Operating
Leases Years ended December 31:
2015 $ 962,000 2016 1,059,000 2017 629,000
Total future minimum lease payments $ 2,650,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
8. Commitments, Collaborations and Contingencies (Continued)
upfront license fee and development funds received from Bayer. These activities were completed as of May 31, 2012.
Amounts to fund collaboration efforts of $22,247,000 and $17,400,000 were recorded as restricted cash and deferred development funds and recognized as an offset to research and development expenses as the restricted cash was utilized to fund development activities. Amounts recognized as offsets to research and development expense were $3,570,000, $11,739,000 and $10,075,000 for the years ended December 31, 2014, 2013 and 2012, respectively.
Deferred development funds remaining at the time that the Company purchased the rights to ATX-101 outside of the United States and Canada in March 2014 of $967,000 were no longer considered restricted cash and were deducted from the one-time charge to in-process research and development, reducing the in-process research and development expense recorded in March 2014.
In March 2014, the Company entered into an agreement whereby it acquired the rights to develop and commercialize ATX-101 outside of the United States and Canada from Bayer. Under the new agreement, KYTHERA Holdings Ltd., a wholly-owned Bermuda subsidiary of the Company, purchased these rights. As a result of this agreement, Bayer was issued 698,103 shares of the Company's common stock valued at $33,000,000 based on an average closing stock price of $47.27 over a period set forth in the agreement. Additionally, the Company issued an unsecured promissory note for $51,000,000, which is subordinated to amounts owed under the Company's existing credit facility, bears interest at 5% per annum and is payable no later than 2024. Additionally, Bayer is eligible to receive up to $123,800,000 upon the achievement of certain long-term sales milestone payments on annual sales of ATX-101 outside of the United States and Canada. As of December 31, 2014, the achievement of the related sales levels is not considered to be probable.
The acquisition date costs allocated to the acquired rights consists of the following:
The fair value of the common stock issued is based on the closing price of the Company's common stock on the acquisition date of $45.02 per share. As the note payable to Bayer includes an interest rate which was below the borrowing rate available to the Company at issuance, it was recorded at its fair value of $21,600,000, which is the present value of the future cash flows assuming a borrowing rate of 15%, which approximates the market rate for debt with similar terms and consideration of default and credit risk. Other represents the net impact of the reduction in costs resulting from the residual restricted cash of $967,000 that reverted to the Company, partially offset by $695,000 of transaction costs related to the acquisition.
This transaction was accounted for as an acquisition of a group of assets. As the acquired rights to ATX-101, represent acquired-in-process research and development, with no alternative future uses, the costs allocated to such rights, $52,757,000, were immediately expensed upon acquisition and reflected as in-process research and development in the consolidated statement of operations and comprehensive loss. This charge is considered to be a component of research and development expense.
See Note 5, "Notes Payable" for a more detailed discussion of the promissory note.
F-25
Fair value of common stock issued $ 31,429,000 Fair value of note payable to Bayer 21,600,000 Other (272,000 ) Total in-process research and development $ 52,757,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
8. Commitments, Collaborations and Contingencies (Continued)
Los Angeles Biomedical Research
In August 2005, the Company entered into an exclusive license agreement with Los Angeles Biomedical Research Institute at Harbor/UCLA Medical Center, or LA Biomed, pursuant to which it obtained a worldwide exclusive license to practice, enforce and otherwise exploit certain patent rights related to the use of the active ingredient in ATX-101. The exclusive license requires the Company to pay LA Biomed a milestone payment of $500,000 upon receipt of initial marketing approval of ATX-101, as well as non-royalty sublicense fees equal to 10% of any sublicense income, up to a total of $5,000,000, 50% of which the Company may elect to satisfy through the issuance of capital stock. Additionally, upon commercialization of a licensed product or service, the Company is obligated to pay low- to mid- single digit royalties on net product sales of ATX- 101. The Company may terminate this license without penalty upon 90 days' notice to LA Biomed and LA Biomed may terminate the license in certain circumstances if the Company fails to perform, or violates any term of the agreement, subject to applicable cure provisions. Subject to default by the Company or termination, the license remains in effect until the last patent or patent application in the licensed patent rights has expired or been revoked, invalidated or abandoned.
In August 2010, due to the receipt of the upfront license fee from Bayer, the Company incurred non-royalty sublicense fees of $1,950,000, which was deferred and recognized as sublicense fee expense on a straight-line basis over the same period as the license income. During 2010, the Company made payments of cash and stock to LA Biomed totaling $390,000 related to the non-royalty sublicense fee incurred. The remainder of $1,560,000 was paid upon our IPO in 50% cash and 50% stock.
Additionally, due to the receipt of the contingent event-based payment of $15,800,000 payable from Bayer on May 31, 2012, the Company incurred an additional non-royalty sublicense fee of $1,580,000 payable to LA Biomed, which was paid in 50% cash and 50% stock.
Supply Arrangements
Pfizer, Inc.
The Company currently has an exclusive supply agreement with Pfizer, Inc., or Pfizer, as the single-source supplier of a key raw material for ATX-101. The agreement with Pfizer expires in December 2016. The agreement can also be terminated prior to expiration by either party (i) upon 60 days' written notice of an uncured material breach, (ii) upon 24 months' written notice, (ii) the bankruptcy or insolvency of the other party or (iv) by Pfizer if the Company abandons development of ATX-101. The Company is not required to purchase any minimum or specific quantities from Pfizer, however, any purchase of the key starting material must be made from Pfizer. Payments are due to Pfizer 30 days after receipt of an invoice following shipment of the material.
Hospira, Inc .
In November 2010, the Company entered into a long-term agreement with Hospira, Inc., or Hospira, as its drug product fill/finish supplier. The initial term of the agreement expires five years after the first day of the month after our first bona fide sale of ATX-101 to a non-affiliate customer after ATX-101 receives regulatory approval, and may be extended for additional and successive two year terms. The agreement may be terminated prior to expiration by either party upon 24 months' written notice. The agreement may also be terminated by either party upon 60 days' written notice of an uncured material breach, the bankruptcy or insolvency of the other party or upon notice that the
F-26

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
8. Commitments, Collaborations and Contingencies (Continued)
other party is unable to perform for 180 days due to a force majeure. The Company is not required to purchase any minimum or specific quantities from Hospira; however, if ATX-101 is approved, Hospira has the right to be the Company's sole provider of drug product for a period of three year following commercialization, and, following the completion of the third year, the right to provide no less than seventy-five percent of the Company's total annual requirement of drug product. Payments are due to Hospira 30 days after receipt of an invoice or, with respect to certain pre- specified events, upon completion of such event.
Contingencies
The Company is subject to certain legal proceedings, complaints and claims arising out of the conduct of its business. In accordance with ASC 450, Contingencies ("ASC 450"), the Company records a reserve for estimated losses when the loss is probable and the amount can be reasonably estimated. Since the Company believes that it has provided adequate reserves, when necessary, it anticipates that the ultimate outcome of any matters currently pending against the Company will not materially affect the consolidated financial position, results of operations or consolidated cash flows of the Company.
9. Fair Value Measurements
The Company applies various valuation approaches in determining the fair value of its financial assets and liabilities within a hierarchy that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company's assumptions about the inputs that market participants would use in pricing the asset or liability and are developed based on the best information available in the circumstances. The fair value hierarchy is broken down into three levels based on the source of inputs as follows:
Level 1—Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access.
Level 2—Valuations based on quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active and models for which all significant inputs are observable, either directly or indirectly.
Level 3—Valuations based on inputs that are unobservable and significant to the overall fair value measurement.
The availability of observable inputs can vary among the various types of financial assets and liabilities. To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, for financial statement disclosure purposes, the level in the fair value hierarchy within which the fair value measurement is categorized is based on the lowest level input that is significant to the overall fair value measurement.
F-27

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
9. Fair Value Measurements (Continued)
At December 31, 2014 and 2013, the Company had cash equivalents comprised of U.S. Treasury securities money market accounts whose value is based using quoted market prices with no adjustments applied. Accordingly, these securities are classified as Level 1. U.S. Government agency securities and corporate debt securities are measured at fair value using Level 2 inputs. These securities are investment grade with maturity dates of two years or less from the balance sheet date. The Company reviews trading activity and pricing for these investments as of the measurement date. When sufficient quoted pricing for identical securities is not available, the Company uses market pricing and other observable market inputs for similar securities obtained from various third party data providers. These inputs represent quoted prices for similar assets in active markets or these inputs have been derived from observable market data. This approach results in the classification of these securities as Level 2 of the fair value hierarchy. There were no transfers of assets or liabilities between the fair value measurement levels during the years ended December 31, 2014 and 2013.
The following fair value hierarchy table presents information about each major category of the Company's financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2014 and 2013.
The Company has notes payable, which are liabilities for which fair value is disclosed as of December 31, 2014 and 2013. Based on the borrowing rates currently available to the Company for debt with similar terms and consideration of default and credit risk, the Company believes that the carrying value of the notes payable related to the credit facility (Level 2) approximates its fair value at December 31, 2014 and 2013. At December 31, 2014, the Company had a note payable with Bayer at a
F-28
Fair Value Measurement at December 31, 2014 Using:
Quoted Prices in Active
Markets for Identical
Assets (Level 1)
Significant Other
Observable Inputs
(Level 2)
Significant Unobservable
Inputs (Level 3)
Balance as of December 31, 2014
Assets Cash equivalents $ 22,691,000 $ — $ — $ 22,691,000 U.S. government agency securities — 2,001,000 — 2,001,000 Corporate debt securities — 68,434,000 — 68,434,000
Total $ 22,691,000 $ 70,435,000 $ — $ 93,126,000
Fair Value Measurement at December 31, 2013 Using:
Quoted Prices in Active
Markets for Identical
Assets (Level 1)
Significant Other
Observable Inputs
(Level 2)
Significant Unobservable
Inputs (Level 3)
Balance as of December 31, 2013
Assets Cash equivalents $ 95,537,000 $ — $ — $ 95,537,000 U.S. government agency securities — 2,003,000 — 2,003,000 Corporate debt securities — 53,537,000 — 53,537,000
Total $ 95,537,000 $ 55,540,000 $ — $ 151,077,000

Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
9. Fair Value Measurements (Continued)
below market interest rate of 5%. The carrying value of the note was calculated assuming a discount rate of 15% based on an analysis to determine the market rate available to the Company for similar debt at issuance. Based on an analysis of various factors at December 31, 2014, including the Company's cost of equity, the effective interest rate on the credit facility notes payable, the rate of public company structured debt arrangements for companies at a similar stage, its subordinated nature, other debt issuance by investment firms to similar companies and management's judgment, the Company believes that the carrying value of this note payable approximates its fair value. Since several of the factors analyzed are considered to be unobservable inputs, this is considered to be a Level 3 valuation.
The fair values of cash equivalents, accounts payable and accrued liabilities approximate their carrying values due to the short-term nature of these accounts.
10. 401(k) Savings Plan
In 2006, the Company sponsored a defined contribution savings plan under Section 401(k) of the Internal Revenue Code ("the 401(k) Plan"). The 401(k) Plan covers all employees who meet defined minimum age and service requirements, and allows participants to defer a portion of their annual compensation on a pretax basis. The Company matches a percentage of its eligible employees' contributions. For the years ended December 31, 2014, 2013 and 2012, the total amounts included in expense for its contributions were $362,000, $341,000 and $208,000, respectively.
11. Income Taxes
There is no provision for income taxes because the Company has incurred operating losses since inception. At December 31, 2014, the Company has concluded that it is more likely than not that the Company may not realize the benefit of its deferred tax assets due to its history of losses. Accordingly, the net deferred tax assets have been fully reserved.
The Company recorded net deferred tax assets of approximately $94,691,000 and $73,687,000 as of December 31, 2014 and 2013, respectively, which have been fully offset by a valuation allowance due to uncertainties surrounding its ability to generate future taxable income to realize these assets. The deferred tax assets are primarily composed of federal and state tax net operating losses, or NOL carryforwards, R&D credit carryforwards, stock compensation and start-up expenditures. As of December 31, 2014, the Company has federal and California NOL carryforwards of approximately $219,792,000 and $215,159,000, which will begin to expire in 2025 and 2015, respectively. Of these deferred tax assets, approximately $9,655,000 will be credited directly to additional paid in capital. At December 31, 2014, the Company has federal and state tax credit carryforwards of approximately $8,773,000 and $5,957,000 available to reduce future federal and state tax liability, respectively. The federal tax credits begin to expire in 2026. Under California law, California tax credits do not have an expiration date.
In general, if the Company experiences a greater than 50 percentage point aggregate change in ownership of certain significant stockholders over a three-year period (a "Section 382 ownership change"), utilization of its pre-change NOL carryforwards are subject to an annual limitation under Section 382 of the Internal Revenue Code (and similar state laws). The annual limitation generally is determined by multiplying the value of the Company's stock at the time of such ownership change (subject to certain adjustments) by the applicable long-term tax-exempt rate. Such limitations may
F-29
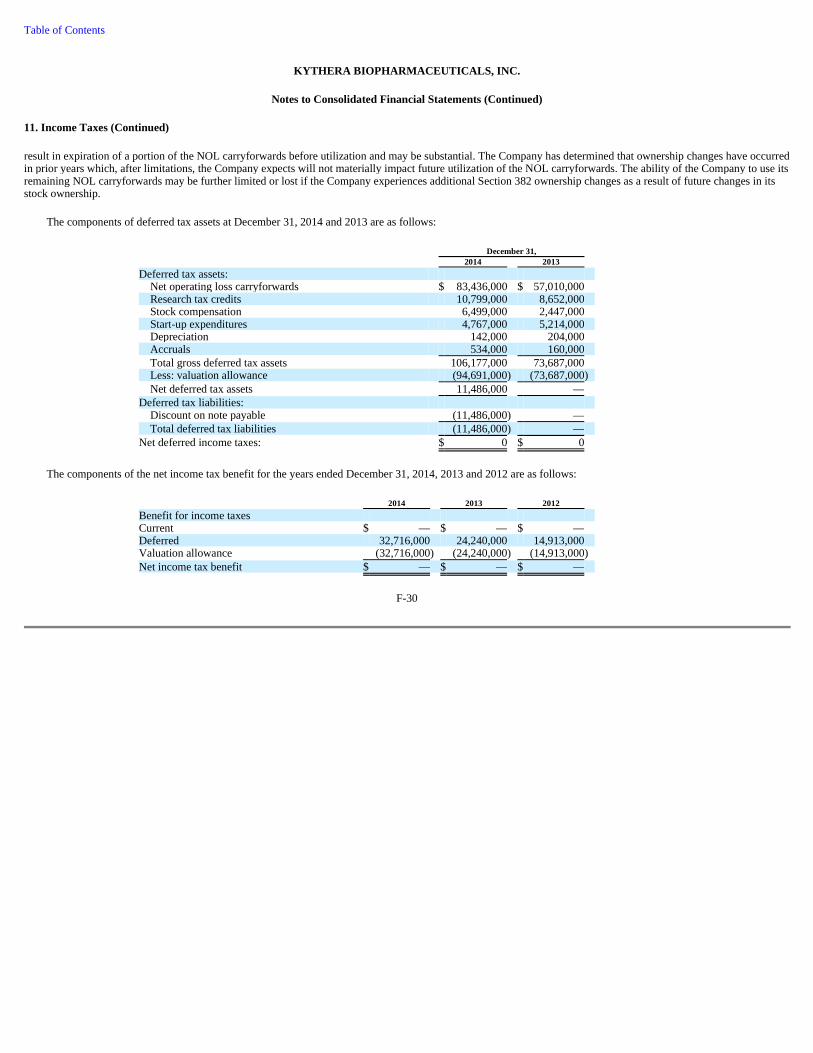
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
11. Income Taxes (Continued)
result in expiration of a portion of the NOL carryforwards before utilization and may be substantial. The Company has determined that ownership changes have occurred in prior years which, after limitations, the Company expects will not materially impact future utilization of the NOL carryforwards. The ability of the Company to use its remaining NOL carryforwards may be further limited or lost if the Company experiences additional Section 382 ownership changes as a result of future changes in its stock ownership.
The components of deferred tax assets at December 31, 2014 and 2013 are as follows:
The components of the net income tax benefit for the years ended December 31, 2014, 2013 and 2012 are as follows:
F-30
December 31, 2014 2013 Deferred tax assets:
Net operating loss carryforwards $ 83,436,000 $ 57,010,000 Research tax credits 10,799,000 8,652,000 Stock compensation 6,499,000 2,447,000 Start-up expenditures 4,767,000 5,214,000 Depreciation 142,000 204,000 Accruals 534,000 160,000 Total gross deferred tax assets 106,177,000 73,687,000 Less: valuation allowance (94,691,000 ) (73,687,000 ) Net deferred tax assets 11,486,000 —
Deferred tax liabilities: Discount on note payable (11,486,000 ) — Total deferred tax liabilities (11,486,000 ) —
Net deferred income taxes: $ 0 $ 0
2014 2013 2012 Benefit for income taxes Current $ — $ — $ — Deferred 32,716,000 24,240,000 14,913,000 Valuation allowance (32,716,000 ) (24,240,000 ) (14,913,000 ) Net income tax benefit $ — $ — $ —
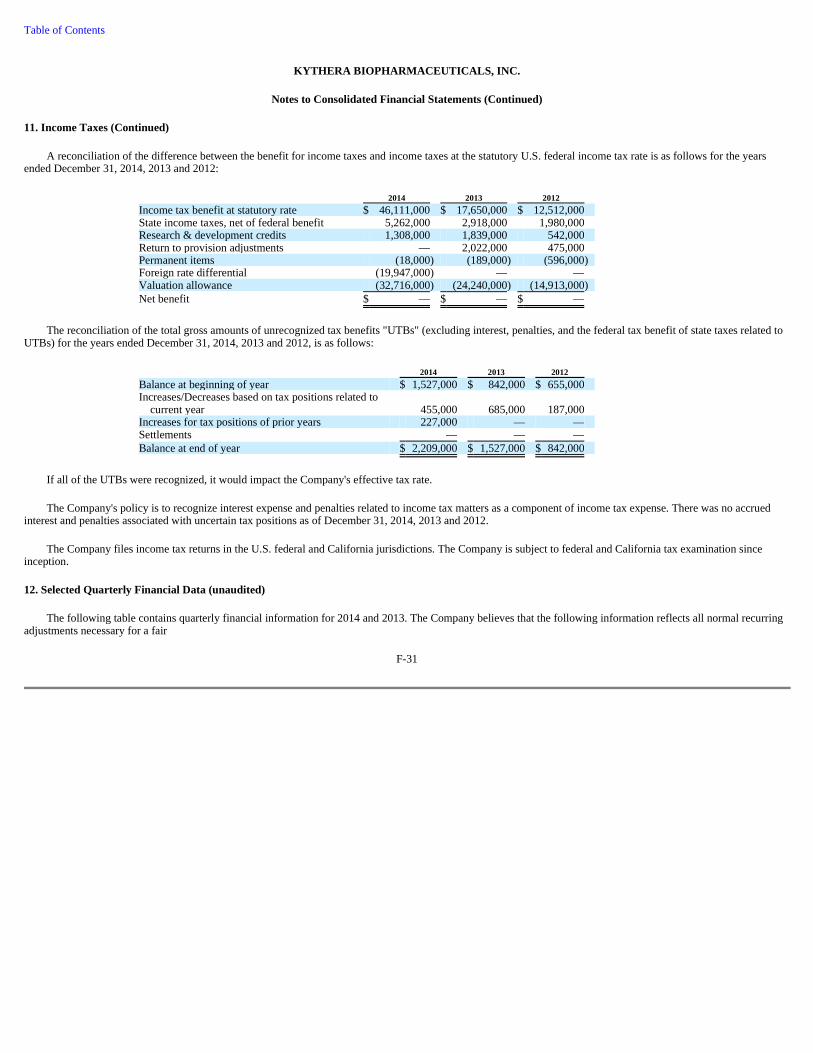
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
11. Income Taxes (Continued)
A reconciliation of the difference between the benefit for income taxes and income taxes at the statutory U.S. federal income tax rate is as follows for the years ended December 31, 2014, 2013 and 2012:
The reconciliation of the total gross amounts of unrecognized tax benefits "UTBs" (excluding interest, penalties, and the federal tax benefit of state taxes related to UTBs) for the years ended December 31, 2014, 2013 and 2012, is as follows:
If all of the UTBs were recognized, it would impact the Company's effective tax rate.
The Company's policy is to recognize interest expense and penalties related to income tax matters as a component of income tax expense. There was no accrued interest and penalties associated with uncertain tax positions as of December 31, 2014, 2013 and 2012.
The Company files income tax returns in the U.S. federal and California jurisdictions. The Company is subject to federal and California tax examination since inception.
12. Selected Quarterly Financial Data (unaudited)
The following table contains quarterly financial information for 2014 and 2013. The Company believes that the following information reflects all normal recurring adjustments necessary for a fair
F-31
2014 2013 2012 Income tax benefit at statutory rate $ 46,111,000 $ 17,650,000 $ 12,512,000 State income taxes, net of federal benefit 5,262,000 2,918,000 1,980,000 Research & development credits 1,308,000 1,839,000 542,000 Return to provision adjustments — 2,022,000 475,000 Permanent items (18,000 ) (189,000 ) (596,000 ) Foreign rate differential (19,947,000 ) — — Valuation allowance (32,716,000 ) (24,240,000 ) (14,913,000 ) Net benefit $ — $ — $ —
2014 2013 2012 Balance at beginning of year $ 1,527,000 $ 842,000 $ 655,000 Increases/Decreases based on tax positions related to
current year 455,000 685,000 187,000 Increases for tax positions of prior years 227,000 — — Settlements — — — Balance at end of year $ 2,209,000 $ 1,527,000 $ 842,000
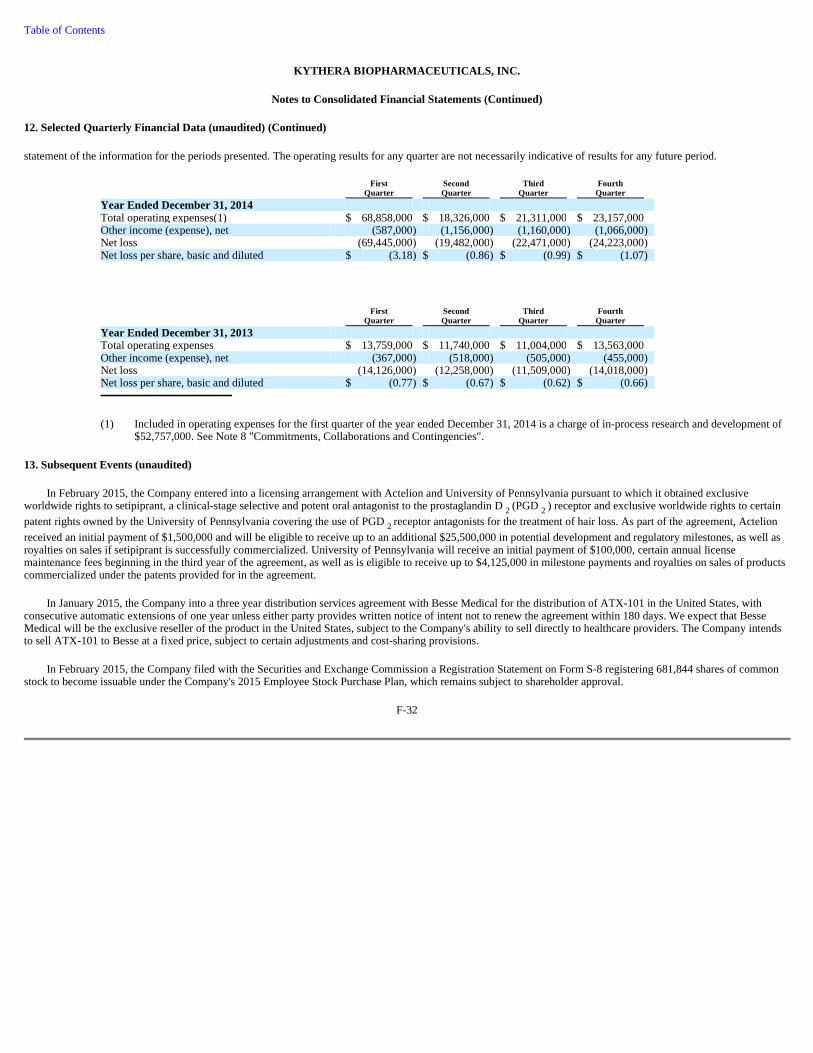
Table of Contents
KYTHERA BIOPHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
12. Selected Quarterly Financial Data (unaudited) (Continued)
statement of the information for the periods presented. The operating results for any quarter are not necessarily indicative of results for any future period.
13. Subsequent Events (unaudited)
In February 2015, the Company entered into a licensing arrangement with Actelion and University of Pennsylvania pursuant to which it obtained exclusive worldwide rights to setipiprant, a clinical-stage selective and potent oral antagonist to the prostaglandin D 2 (PGD 2 ) receptor and exclusive worldwide rights to certain patent rights owned by the University of Pennsylvania covering the use of PGD 2 receptor antagonists for the treatment of hair loss. As part of the agreement, Actelion received an initial payment of $1,500,000 and will be eligible to receive up to an additional $25,500,000 in potential development and regulatory milestones, as well as royalties on sales if setipiprant is successfully commercialized. University of Pennsylvania will receive an initial payment of $100,000, certain annual license maintenance fees beginning in the third year of the agreement, as well as is eligible to receive up to $4,125,000 in milestone payments and royalties on sales of products commercialized under the patents provided for in the agreement.
In January 2015, the Company into a three year distribution services agreement with Besse Medical for the distribution of ATX-101 in the United States, with consecutive automatic extensions of one year unless either party provides written notice of intent not to renew the agreement within 180 days. We expect that Besse Medical will be the exclusive reseller of the product in the United States, subject to the Company's ability to sell directly to healthcare providers. The Company intends to sell ATX-101 to Besse at a fixed price, subject to certain adjustments and cost-sharing provisions.
In February 2015, the Company filed with the Securities and Exchange Commission a Registration Statement on Form S-8 registering 681,844 shares of common stock to become issuable under the Company's 2015 Employee Stock Purchase Plan, which remains subject to shareholder approval.
F-32
First
Quarter Second Quarter
Third Quarter
Fourth Quarter
Year Ended December 31, 2014 Total operating expenses(1) $ 68,858,000 $ 18,326,000 $ 21,311,000 $ 23,157,000 Other income (expense), net (587,000 ) (1,156,000 ) (1,160,000 ) (1,066,000 ) Net loss (69,445,000 ) (19,482,000 ) (22,471,000 ) (24,223,000 ) Net loss per share, basic and diluted $ (3.18 ) $ (0.86 ) $ (0.99 ) $ (1.07 )
First
Quarter Second Quarter
Third Quarter
Fourth Quarter
Year Ended December 31, 2013 Total operating expenses $ 13,759,000 $ 11,740,000 $ 11,004,000 $ 13,563,000 Other income (expense), net (367,000 ) (518,000 ) (505,000 ) (455,000 ) Net loss (14,126,000 ) (12,258,000 ) (11,509,000 ) (14,018,000 ) Net loss per share, basic and diluted $ (0.77 ) $ (0.67 ) $ (0.62 ) $ (0.66 )
(1) Included in operating expenses for the first quarter of the year ended December 31, 2014 is a charge of in-process research and development of $52,757,000. See Note 8 "Commitments, Collaborations and Contingencies".


Exhibit 10.8(c)
Second Amendment to Manufacturing and Supply Agreement
This Second Amendment to the Manufacturing and Supply Agreement (this “Second Amendment”) is made as of October 17, 2014 (the “Effective Date of this Second Amendment”) by and between Pfizer, Inc., a corporation organized and existing under the laws of the State of Delaware, with offices at 235 E. 42 Street, New York, NY 10017 (“Pfizer”) and Kythera Biopharmaceuticals, Inc., a corporation organized and existing under the laws of the State of Delaware, having an address of 30930 Russell Ranch Road, Third Floor, Westlake Village, CA 91362 (hereinafter “Customer”).
WHEREAS, Pfizer and Customer have entered into that certain Manufacturing and Supply Agreement by and between Pfizer Inc., and Kythera Biopharmaceuticals, Inc., dated as of July 7, 2009, (the “Agreement”) and amended by a First Amendment dated May 21, 2012;
WHEREAS, Pfizer and Customer entered into a Product Addendum to the Supply Agreement (the “Product Addendum”) also dated July 7, 2009
as amended by a First Amendment dated May 21, 2012.
WHEREAS, Pfizer and Customer desire to extend the term of the Agreement. NOW, THEREFORE, in consideration of the mutual covenants, conditions and agreements set forth herein and for other good and valuable
consideration, the receipt and sufficiency of which are hereby acknowledged, Pfizer and Customer agree as follows: 1. Section 9.1 Term of Agreement is hereby DELETED in its entirety and REPLACED with the following:
“Section 9.1 Term of Agreement . This Agreement shall commence on the Effective Date and shall remain in effect until December 31, 2016 (the “Term” of this Agreement) unless terminated pursuant to the Article or extended upon the mutual written agreement of the Parties.”
2. Section 10. Notices is hereby DELETED in its entirety and REPLACED with the following:
Section 10 Notices. Any notice required to be given hereunder shall be in writing in the English language and shall be deemed to have been sufficiently given: (i) when delivered in person, (ii) on the fifth Business Day after mailing by registered or certified mail, postage prepaid, return receipt requested, (iii) on the next Business Day after mailing by overnight courier service, or (iv) when delivered via facsimile with original delivered via one of the preceding methods; to the address specified below:
1
If to Company: Pfizer Inc.
7000 Portage Road
Kalamazoo, MI 49001
nd

Either Party may, by notice to the other Party, change the addresses and names given above.
3. Except as specifically amended above, all terms and conditions of the Product Addendum, as amended, and Agreement, as amended, shall
remain in full force and effect and are hereby ratified and confirmed.
4. In the event that there are any conflicts between the terms of this Second Amendment and the terms of the Agreement, the terms of this Second Amendment shall control. The terms of this Second Amendment shall be controlling over any terms of any purchase order, sales acknowledgement, invoice or other such documents issued by either Party.
5. This Second Amendment may be executed in two or more counterparts, each of which shall be deemed to be an original, and all of which
shall together constitute one and the same agreement, and shall become effective when signed by each of the parties hereto and delivered to the other party in person or by facsimile or other reliable electronic means.
IN WITNESS WHEREOF, the parties hereto have caused their authorized representatives to execute this Second Amendment as of the date first
written above. 2
Attn: President, Pfizer CentreSource
Fax. No.: 269-833-4229
With copy to: Pfizer Inc.
7000 Portage Road
Kalamazoo, MI 49001
Attn: Senior Corporate Counsel
Fax. No.: 269-833-3661 If to Purchaser: Kythera Biopharmaceuticals, Inc.
30930 Russell Ranch Road, Third Floor
Westlake Village, CA 91362
Telephone: 818-587-4500
Facsimile: 818-587-4563
Attn: Majed Kheir, ED Supply Chain and Planning With a copy to: Kythera Biopharmaceuticals, Inc.
30930 Russell Ranch Road, Third Floor
Westlake Village, CA 91362
Telephone: 818-587-4500
Facsimile: 818-587-4591
Attn: Jeff Webster, SVP Operations

3
Pfizer, Inc.
Kythera Biopharmaceuticals, Inc. By: /s/ Michael J. Kosko
By: /s/ Jeff Webster
Name: Michael J. Kosko
Name: Jeff Webster Title: President, Pfizer CenterSource
Title: SVP, OPS
Signed on the 30th day of October 2014
Signed on the 17 day of October 2014

Exhibit 10.20
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission.
Confidential treatment has been requested with respect to the omitted portions. CONFIDENTIAL INFORMATION
EXECUTION COPY
LICENSE AGREEMENT
This license agreement (the “ Agreement ”) is effective as of February __, 2015 (the “ Effective Date ”) by and among ACTELION PHARMACEUTICALS, LTD., a Swiss company located at Gewerbestrasse 16, CH-4123 Allschwil, Switzerland (“ Actelion ”), KYTHERA HOLDINGS, LTD. , a Bermuda company located at Clarendon House, 2 Church Street, Hamilton HM 11, Bermuda (“ Kythera ”), and KYTHERA BIOPHARMACEUTICALS, INC. , a Delaware company located at 30930 Russell Ranch Road, 3rd Floor, Westlake Village, CA 91362, United States of America (“ KBI ”). Actelion, Kythera and KBI (but only with respect to section 12.5) are referred to individually as a “ Party ” and collectively as the “ Parties ”.
RECITALS
A. Actelion is a multinational biopharmaceutical company that has discovered and developed small molecule antagonists to the target known as CRTH2, including the compound known as setipiprant. B. Kythera is a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of novel prescription products for the aesthetic medicine market. C. Kythera and Actelion desire to enter into an agreement under which Actelion would grant to Kythera a license under certain technology and intellectual property rights controlled by Actelion covering Actelion’s CRTH2 antagonists for the development and commercialization of products including such compounds. NOW, THEREFORE, the Parties agree as follows: 1. DEFINITIONS Capitalized terms used in this Agreement (other than the headings of the Sections or Articles ) have the following meanings set forth in this Article 1 , or, if not listed in this Article 1 , the meanings as designated in the text of this Agreement.
1.1 “Actelion Licensed Know-How” means all Information (other than Patents) that is Controlled by Actelion or its Affiliates, as of the Effective Date, and that: (a) relates to compositions of matter, methods of use, formulations of, methods of manufacture of, or companion diagnostics for, Compounds; (b) is not generally known; and (c) is necessary or reasonably useful for Kythera to develop setipiprant for hair growth or the prevention of hair loss.
1.2 “Actelion Licensed Patents” means all Patents that are listed on Exhibit 1.2 . 1.3 “Actelion Related Patents” means all Patents that: (a) are conceived or developed by (or on behalf of) Actelion prior to the
Effective Date; (b) are not listed on Exhibit 1.2 , (c) have been filed before, or will be filed after, the Effective Date; and (d) claim inventions that relate to compositions of matter, methods of use, formulations of, methods of manufacture of, or companion diagnostics for, Compounds.
1

CONFIDENTIAL INFORMATION
EXECUTION COPY 1.4 “Affiliate” means, with respect to a Party, a person, corporation, partnership, or other entity that now or hereinafter controls,
is controlled by or is under common control with such Party. For the purposes of the definition in this Section 1.4 , the word “ control ” (including, with correlative meaning, the terms “ controlled by ” or “ under the common control with ”) means the actual power, either directly or indirectly through one (1) or more intermediaries, to direct or cause the direction of the management and policies of such entity, whether by the ownership of at least fifty percent (50%) of the voting stock of such entity, by contract or otherwise.
1.5 “ANDA” means an abbreviated new drug application submitted to the FDA in conformance with applicable laws and
regulations, or the foreign equivalent of any such application in any other country. 1.6 “Commercialize” means to promote, market, import, distribute, sell (and offer for sale or contract to sell) or provide product
support for a Licensed Product, including by way of example: (a) detailing and other promotional activities in support of a Licensed Product; (b) advertising and public relations in support of a Licensed Product, including market research, development and distribution of selling, advertising and promotional materials, field literature, direct-to-consumer advertising campaigns, media or journal advertising, and exhibiting at seminars and conventions; (c) developing reimbursement programs and information and data specifically intended for national accounts, managed care organizations, governmental agencies (e.g., federal, state and local), and other group purchasing organizations, including pull-through activities; (d) co-promotion activities not included in the above; and (e) conducting Phase IV Clinical Trials.
1.7 “Commercially Reasonable Efforts” means the effort customarily exerted by a Party with respect to its own products of
similar nature and commercial potential, taking into account, by example and without limitation, such factors as intellectual property position, the cost of development, regulatory risk, safety and efficacy, reimbursement factors, parallel importation considerations, pricing, product life cycle and cost of sales and marketing, and also including the timing and promptness with which such efforts and resources would be applied.
1.8 “Compendia Listing” means a listing for an indication in the United States for a Licensed Product that is supported by a
citation in at least one of the following authoritative drug reference books: (a) the American Society of Health-System Pharmacists’ American Hospital Formulary Service (AHFS); or (b) the U.S. Pharmacopoeia Drug Information, or in another similar authoritative drug reference book that is relied on by Third Party payors in authorizing reimbursement for such Licensed Product for such indication.
1.9 “Compounds” means: (a) Actelion’s compound known either as setipiprant or ACT-129968; (b) all other compounds [***] ;
and (c) the [***] of the compounds described in (a) and/or (b). 1.10 “Controlled” means, with respect to any physical material, Information or intellectual property right, that the Party owns or
has a license to such physical material, Information or intellectual property right and has the ability to grant to the other Party access, a license or a sublicense (as applicable) to such physical material, Information or intellectual property right as provided for herein without violating the terms of any agreement or other arrangements
2
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY with any Third Party existing at the time such Party would be first required hereunder to grant the other Party such access, license or sublicense.
1.11 “CRTH2” means: (a) the gene encoding the chemoattractant receptor-homologous molecule expressed on Th2 cells for any species (which molecule is also known as GPR44 or DP2); (b) the protein encoded by such gene; and (c) all [***] thereof.
1.12 “CRTH2 Antagonists” means any molecule with a [***] antagonizing CRTH2. 1.13 “Development” means, with respect to a Licensed Product, those activities, including clinical trials and related regulatory
activities, that are necessary or useful to: (a) obtain the approval by the applicable Regulatory Authorities of the Regulatory Approval Application with respect to such Licensed Product in the applicable regulatory jurisdiction, whether alone or for use together, or in combination, with another active agent or pharmaceutical product; (b) maintain such approvals; or (c) obtain or maintain Compendia Listings with respect to such Licensed Product. To avoid confusion, Development does not include the conduct of Research or Phase IV Clinical Trials.
1.14 “Dollars” or “$” means the legal tender of the United States. 1.15 “Executive Officers” means: (a) in the case of Actelion, the EVP and Chief Business Development Officer, or that officer’s
designee; and (b) in the case of Kythera, the Vice President for Strategy and Corporate Development, or that officer’s designee. 1.16 “FDA” means the U.S. Food and Drug Administration, and any successor thereto. 1.17 “Field” means any and all uses (including therapeutic, diagnostic or prophylactic uses) in humans and other vertebrates. 1.18 “First Commercial Sale” means, for each Licensed Product in each country, the first arm’s-length sale to a Third Party for
use or consumption by the public of such Licensed Product in such country after Regulatory Approval of such Licensed Product in such country. A First Commercial Sale will not include any Licensed Product supplied for use in clinical trials, for research or for other non-commercial uses, or as part of a compassionate use program (or similar program for providing Licensed Product before it has received Regulatory Approval in a country).
1.19 “Generic Licensed Product” means, with respect to a particular Licensed Product in a country, a pharmaceutical product
that: (a) contains the same Compound as such Licensed Product; and (b) is approved for use in such country pursuant to 21 U.S.C. 355(b)(2), an ANDA, a separate NDA, Compendia Listing, or equivalent drug approval application.
1.20 “IND” means an investigational new drug application submitted to the FDA in conformance with applicable laws and
regulations, or the foreign equivalent of any such application in any other country. 1.21 “Information” means information, materials, results and data of any type, in any tangible or intangible form, including,
preclinical data, clinical trial data, databases, practices, methods, techniques, specifications, formulations, formulae, knowledge, know-how, skill,
3
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY experience, test data including pharmacological, biological, chemical, biochemical, toxicological and clinical materials, test data, analytical and quality control data, stability data, studies and procedures. For clarity, Information does not include any Patents.
1.22 “Invention” means any and all inventions or improvements that are conceived or made by or on behalf of a Party (or its Affiliates) in the performance of its obligations, or the exercise of its rights, under this Agreement.
1.23 “Joint Invention” means any Invention conceived or made jointly by or on behalf of the employee(s), contractor(s) or agent
(s) of both Parties (or their Affiliates). 1.24 “Licensed Product” means any product preparation in final form that contains one or more Compounds 1.25 “Major European Countries” means [***] . 1.26 “Manufacturing” means all activities related to the production, manufacture, processing, filling, finishing, packaging,
labeling, inspection, receiving, holding and shipping of Compounds, Licensed Products, or any raw materials or packaging materials with respect thereto, or any intermediate of any of the foregoing, including process and cost optimization, process qualification and validation, clinical manufacture, commercial manufacture, stability and release testing, quality assurance and quality control.
1.27 “Manufacturing Information” means all Information that is Controlled by Actelion or its Affiliates as of the Effective Date
and/or during the Term, that is related to the Manufacturing of Compounds and Licensed Products, and that is reasonably necessary or useful to enable Kythera or a Third Party manufacturer selected by Kythera (as appropriate) to Manufacture Compounds.
1.28 “Materials” means Compounds in the possession of Actelion. 1.29 “NDA” means a new drug application submitted to the FDA in conformance with applicable laws and regulations, or the
foreign equivalent of such application in any other country. 1.30 “Net Sales” means the gross amount received by Kythera (or its Affiliate, licensee or sublicensee) for sales of a Licensed
Product to a Third Party purchaser, less the following to the extent included in such invoice or otherwise actually allowed or incurred with respect to such sales: (a) discounts, including cash, trade and quantity discounts, price reduction programs, retroactive price adjustments with respect to sales of a Licensed Product, charge-back payments and rebates granted to managed health care organizations or to federal, state and local governments (or their respective agencies, purchasers and reimbursers) or to trade customers, including but not limited to, wholesalers and chain and pharmacy buying groups; (b) credits or allowances for rejections or returns of Licensed Products, including for recalls or damaged goods; (c) freight, postage, shipping and insurance charges for delivery of Licensed Products, to the extent billed; and (d) taxes, duties or other governmental charges levied on, absorbed or otherwise imposed on sale of Licensed Products, including value-added taxes, or other governmental charges otherwise measured by the billing amount, when included in billing, as adjusted for rebates and refunds, but specifically excluding taxes based on net income of the seller; provided that all of the foregoing deductions are calculated in accordance with US GAAP.
4
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY For sake of clarity and avoidance of doubt, sales by Kythera (or its Affiliate, licensee or sublicensee) of a Licensed Product to a Third Party distributor of such Licensed Product in a given country will be considered a sale to a Third Party purchaser. Any Licensed Products used (but not sold for consideration) for promotional or advertising purposes (including free samples), or used for clinical or other research purposes, will not be considered in determining Net Sales hereunder. If a Licensed Product is sold as a combined product or service, Net Sales, for purposes of determining royalty payments on such Licensed Product, will be calculated by multiplying the Net Sales of the combined product or service by the fraction A/A+B, in which A is the gross selling price (in the applicable country) of the Licensed Product portion of combined product or service when such Licensed Product is sold separately during the applicable accounting period in which the sales of the combined product were made, and B is the gross selling price (in the applicable country) of the other products or services, as the case may be, of the combined product and/or service sold separately during the accounting period in question. All gross selling prices of the components of the combined product or service will be calculated as the average gross selling price of the components during the applicable accounting period for which the Net Sales are being calculated. In any country, if no separate sale of either such above-designated Licensed Product or such above designated elements of the end-user product and/or service are made during the accounting period in which the sale was made, or if gross retail selling price for an active functional element, component or service, as the case may be, cannot be determined for an accounting period, Net Sales allocable to the Licensed Product in each such country will be determined by mutual agreement reached in good faith by the Parties prior to the end of the accounting period in question based on an equitable method of determining same that takes into account, on a country-by-country basis, variations in potency, the relative contribution of each active agent, component or service, as the case may be, in the combination, and relative value to the end user of each active agent, component or service, as the case may be.
1.31 “Patent” means all: (a) unexpired patents (including inventor’s certificates and utility models), including any substitution, extension, registration, confirmation, reissue, re-examination, restoration, supplementary protection certificates, confirmation patents, patent of additions, renewal or any like filing thereof; (b) pending applications for letters patent, including any continuation, division or continuation-in-part thereof and any provisional or other priority applications; and (c) any international counterparts, and counterparts in any country, to clauses (a) and (b) above.
1.32 “Phase I Clinical Trial” means a human clinical trial in any country that would satisfy the requirements of 21 CFR 312.21
(a). 1.33 “Phase II Clinical Trial” means a human clinical trial in any country that would satisfy the requirements of 21 CFR 312.21
(b). 1.34 “Phase IIb Clinical Trial” means a human clinical trial that is conducted on sufficient numbers of patients and that is
designed to provide a preliminary determination of safety and efficacy of a Licensed Product in the target patient population over a range of doses and dose regimens. For clarification, the Proof of Concept Trial planned by Kythera to study the use of the Licensed Product to treat alopecia is not a Phase IIb Clinical Trial.
5
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY 1.35 “Phase III Clinical Trial” means a human clinical trial in any country that would satisfy the requirements of 21 CFR 312.21
(c). 1.36 “Phase IV Clinical Trial” means a product support clinical trial of a Licensed Product commenced after receipt of
Regulatory Approval in the country where such trial is conducted. A Phase IV Clinical Trial may include epidemiological studies, modeling and pharmacoeconomic studies, postmarketing commitments required by health authorities (preclinical, CMC, clinical or other) and investigator-sponsored clinical trials studying Licensed Product that are approved by Kythera and that otherwise fit the foregoing definition.
1.37 “Proof of Concept Trial” means a Phase II Clinical Trial of a Licensed Product demonstrating that such Licensed Product
is: (a) [***] ; and (b) [***] to progress to a Phase IIb Clinical Trial or a Phase III Clinical Trial. 1.38 “Regulatory Approval” means any and all approvals (including Regulatory Approval Applications, supplements,
amendments, pre- and post-approvals, pricing and reimbursement approvals, where applicable), licenses, registrations or authorizations of any Regulatory Authority that are necessary for the manufacture, distribution, use or sale of a Licensed Product in a regulatory jurisdiction.
1.39 “ Regulatory Approval Application ” means: (a) in the United States, an NDA (or a supplemental NDA); and (b) in any
other country or regulatory jurisdiction, an equivalent application for regulatory approval required before commercial sale or use of a Licensed Product (or with respect to a subsequent indication) in such country or regulatory jurisdiction.
1.40 “Regulatory Authority” means the applicable national (e.g., the FDA), supra-national (e.g., the European Commission),
regional, state or local regulatory agency, department, bureau, commission, council or other governmental entity that, in each case, governs the approval of a Licensed Product in such applicable regulatory jurisdiction.
1.41 “Research” means any non-clinical research, including the following activities: (a) identifying molecules as potential
CRTH2 Antagonists; (b) conducting a lead optimization program to optimize such potential CRTH2 Antagonists (including the conduct of medicinal chemistry; biochemical and cellular assays; in vitro profiling of absorption, distribution, metabolism, and excretion; structural biology; preliminary pharmacokinetic studies; and preliminary pharmacodynamic studies); and (c) conducting preclinical development on such CRTH2 Antagonists to prepare them for IND submission (including the conduct of toxicological studies, and related bioanalytical and pharmacokinetic activities). To avoid confusion, Research excludes the conduct of Development.
1.42 “Sole Invention” means any Invention conceived or made solely by or on behalf of a Party (or its Affiliate) and its
employees, contractors and/or agents. 1.43 “Territory” means the world. 1.44 “Third Party” means any entity other than: (a) Actelion; (b) Kythera; or (c) an Affiliate of either Party.
6
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY 1.45 “United States” or “U.S.” means the United States of America, and its territories, districts and possessions. 1.46 “US GAAP” means U.S. generally accepted accounting principles, consistently applied. 1.47 “Valid Claim” means a claim in an issued Actelion Licensed Patent (including a claim under patent term extensions,
supplementary protection certificates or the like) that has not: (a) expired or been canceled; (b) been declared invalid by an unreversed and unappealable or unappealed decision of a court or other appropriate body of competent jurisdiction; (c) been admitted to be invalid or unenforceable through reissue, disclaimer or otherwise; or (d) been abandoned in accordance with or as permitted by the terms of this Agreement or by mutual written agreement of the Parties.
Additional Definitions The following table identifies the location of definitions set forth in various Sections of the Agreement.
2. TRANSFER OF TECHNOLOGY
2.1 Technology Transfer. Beginning on the Effective Date, Actelion will use Commercially Reasonable Efforts to transfer to Kythera all items of Actelion Licensed Know-How (including Manufacturing know-how specified in Exhibit 2.1, but excluding the technology transfer required to transfer drug substance and drug product Manufacturing Information to Kythera or its designated contract manufacturing organization pursuant to Section 5.2 and as specified in Exhibit 5.2 ) and Materials (“ Actelion Technology ”). Notwithstanding the foregoing Actelion will not be
7
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL INFORMATION
EXECUTION COPY required to transfer any Compounds other than setipiprant. The process, timelines and content of the transfer will be described in Exhibit 2.1 to ensure that the scope of the transfer, the format of the documents and the transfer itself are clearly documented to ensure that the chain of custody is preserved and documented. Additionally, Actelion will provide a reasonable amount of advice or support in connection with the foregoing transfer, as requested in writing by Kythera. Kythera will reimburse Actelion for the time spent, at a rate of $ [***] /hour, and any out of pocket costs in relation to the transfer of Actelion Technology as per Exhibits 2.1, 2.2(a)(i) and 5.2. Kythera will use the Actelion Technology for the purposes of exercising its rights, and performing its obligations, under the Agreement. On termination of this Agreement, any Actelion Technology supplied by Actelion will be returned to Actelion or destroyed as agreed upon in writing by the Parties.
2.2 Transfer of Actelion Regulatory Filings and Safety Information for Compounds.
(a) In accordance with the process and timelines described Exhibit 2.1, Actelion will: (i) allow timely access, use and
Definition
Location (Section)
Actelion FTO Patents 6.1(d) Actelion Sole Invention Patents 8.1(b) Actelion Technology 2.1 CMC 11.2(i) Confidential Information 9.1 Disputed Matter 13.1(a) force majeure 13.6 Indemnitee 12.3 Insolvent Party 10.2(b) Joint Invention Patents 8.1(b) Joint Research Agreement 8.1(d) Kythera Prosecuted Patents 8.3(a)(i) Kythera Sole Invention Patents 8.1(b) Liaison 2.3(a) Losses 12.1 Prior CDA 9.4 Rules 13.1(b) Term 10.1

support of their existing regulatory and technical documents related to Actelion’s setipiprant program for Kythera’s submission of an IND for a Licensed Product, including the transfer of copies of the then-current FDA regulatory filings on setipiprant to Kythera (in eCTD format, or for the documents listed in Exhibit 2.2(a)(i) , in the format listed in such Exhibit) for incorporation into an IND to be produced by Kythera and filed for a Licensed Product; and (ii) transfer to Kythera all safety-related Information regarding setipiprant, including all adverse event and significant adverse event reporting from pharmacovigilance databases maintained by or on behalf of Actelion in its clinical development of setipiprant, in a format that is agreed to by the Parties for incorporation into a new Kythera pharmacovigilance database for a Licensed Product. Such access, use and support would also include Actelion providing reasonable access to Actelion staff to assist with the knowledge transfer to Kythera and the preparation of Kythera’s initial IND for a Licensed Product, pursuant to Section 2.2(b) . Actelion will provide, at a cost of $ [***] /hour, a quality check of the investigator brochure on Compound after Kythera has made the update. Kythera acknowledges that Actelion has made it aware that i) the [***] of the setipiprant carcinogenicity studies is completed (ii) the samples from the aforementioned carcinogenicity studies are archived but not yet analyzed; iii) the full evaluation of the study may cost [***] ( [***] ) CHF or more and may take [***] ( [***] ) months or more and iv) will not be performed nor funded by Actelion. Actelion will maintain, or cause the maintenance of, the archived samples and instruct the CRO who performed the study to transfer it to Kythera, and confirm to the CRO that, upon written request from Kythera, it can proceed to the testing of the stored samples at Kythera’s expense.
(b) During the [***] ([***]) month period after completion of the transfer of the documents and Information required
under Section 2.2(a) , Kythera may, via the Liaison, consult with applicable Actelion employees with experience with Licensed Products, on a reasonable basis, to advise Kythera regarding specific issues and assist Kythera in its use and understanding of all such documents and Information. All such consultations will occur at mutually agreeable times and places. Kythera will reimburse Actelion for the time spent in connection therewith at a rate of $ [***] /hour as well as for any reasonable out-of-pocket costs or expenses (such as needed transportation and lodging costs, if requested by Kythera) that Actelion incurs in connection with such consultations and that are pre-approved by Kythera in writing.
8
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY 2.3 Liaisons.
(a) Appointment. Within thirty (30) days after the Effective Date, each of the Parties will appoint a single individual to act as a single point of contact between the Parties to assure a successful transfer of Actelion Technology and to communicate with respect to matters under the Agreement (each, a “ Liaison ”). Each Party will notify the other Party promptly in writing of the identity of, and contact information for, the notifying Party’s Liaison, and each Party may change its designated Liaison from time to time upon written notice to the other Party. Any Liaison may designate a substitute to temporarily perform the functions of that Liaison by written notice to the other Party.
(b) Responsibilities . The Liaisons will: (i) coordinate the transfer of Actelion Technology; (ii) be the point of first
referral in all matters of conflict resolution; (iii) identify and bring disputes to the attention of the applicable Party in a timely manner; (iv) plan and coordinate cooperative efforts and internal and external communications; and (v) otherwise take responsibility for ensuring that relevant action items resulting from such Party interactions are appropriately carried out or otherwise addressed.
2.4 Independence . Subject to the terms of this Agreement, the activities and resources of each Party will be managed by such
Party, acting independently and in its individual capacity. The relationship between Actelion and Kythera is that of independent contractors and neither Party will have the power to bind or obligate the other Party in any manner.
3. RESEARCH, DEVELOPMENT, MANUFACTURING AND COMMERCIALIZATION OF PRODUCTS
3.1 Research, Development, and Manufacturing of Licensed Products
(a) Scope & Diligence . Kythera will have sole control and responsibility for the Research, Development, Manufacture (except as described in Section 5.1 ) and Commercialization of all Licensed Products. As between the Parties, Kythera will bear all costs and expenses associated with such activities. Kythera will use Commercially Reasonable Efforts to Develop and Commercialize a Licensed Product in the Territory. As part of such Commercially Reasonable Efforts, Kythera will initiate (i.e., treat its first patient for) a Proof of Concept Trial within [***] ( [***] ) months of the Effective Date. If Kythera is unable to initiate a Proof of Concept Trial within such initial [***] ( [***] )-month period, and the inability is due to factors not within Kythera’s reasonable control (including a delay in obtaining required quantities of Licensed Product, delays in obtaining approvals from an ethics committee or a Regulatory Authority, or a force majeure), then upon receipt of Kythera’s written request, Actelion will extend such initial [***] ( [***] )-month period for initiation of the Proof of Concept Trial by an additional [***] ( [***] ) months. If Kythera is unable to initiate a Proof of Concept Trial within such initial [***] ( [***] )-month period, and the inability is due to factors within Kythera’s reasonable control, then Kythera may seek a [***] ( [***] ) month extension to initiate the Proof of Concept Trial by paying Actelion [***] Dollars ($ [***] ) [***] . Such payment will be creditable against the milestone owed under Section 7.2(a)(i) . If Kythera is unable to initiate a Proof of Concept Trial within such additional [***] ( [***] )-month or [***] -month period (as applicable), then the Parties will meet to agree on whether a further extension may be agreed upon or whether the agreement is terminated. In case the Parties, acting in good faith, cannot agree on a
9
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY further extension within one (1) month from the date of the first meeting, this Agreement will be terminated with immediate effect.
(b) Reports. Beginning forty-five (45) days after the Effective Date, and every six (6) months thereafter during the term of the Agreement, Kythera will submit to Actelion a written progress report (substantially in the form of Exhibit 3.1(b) ) summarizing the Development and Commercialization performed by or on behalf of Kythera on Licensed Products. All such reports will be considered Confidential Information of Kythera.
3.2 Standards of Conduct. Kythera will perform, or will ensure that its Affiliates, licensees, sublicensees and Third Party
contractors perform, all Research, Development, Manufacturing and Commercialization activities in a good scientific and ethical business manner and in compliance with the terms of this Agreement and all applicable laws, rules and regulations.
4. REGULATORY
4.1 Kythera’s Role . Kythera will have sole responsibility for (and bear all costs and expenses associated with) all regulatory activities regarding Licensed Products. Kythera will also have sole responsibility for (and bear all costs and expenses associated with) worldwide pharmacovigilance for such Licensed Product. Kythera will have the sole right and responsibility to participate in all pricing and reimbursement approval proceedings relating to each Licensed Product in the Territory.
4.2 Ownership of Regulatory Dossier . Kythera will own all regulatory filings for Licensed Products made after the Effective
Date. Kythera will prepare and draft all filings (including any supplements or modifications thereto and including the preparation of any electronic submission of a Regulatory Approval Application) to Regulatory Authorities in each such country in the Territory for such Licensed Product.
4.3 Recalls in the Territory . Any decision to initiate a recall or withdrawal of a Licensed Product in the Territory will be
made by Kythera. In the event of any recall or withdrawal, Kythera will take any and all necessary action to implement such recall or withdrawal in accordance with applicable law, with assistance from Actelion as reasonably requested by Kythera to the extent that Actelion manufactured or had manufactured concerned Licensed Product. The costs of any such recall or withdrawal in the Territory will be borne solely by Kythera.
5. ACTELION MANUFACTURING
5.1 Manufacture of Licensed Product. Actelion will Manufacture, or arrange with Third Parties for the Manufacture of Kythera’s requirements of the Licensed Product as bulk packaged drug product, i.e., bulk tablets of [***] dose and matching placebo(s), excluding finished packaging for the Proof of Concept Trial. As part of such Manufacture, Actelion and Kythera and/or Third Parties agree to enter into a Quality Agreement, and Actelion will provide the documentation to enable Kythera’s regulatory and quality function to test and release all supplies of such Licensed Product for such Proof of Concept Trial (if applicable). The cost of such Manufacturing will be at a price equal to [***] for such Licensed Product. [***] shall mean the [***] and [***] (including [***]
10
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY [***] ) and indirect costs (including [***] ) specifically [***] of the Licensed Product.
5.2 Transfer of Manufacturing Information. The transfer of the drug product Manufacturing Information will be done during the manufacturing campaign of Licensed Product for the Proof of Concept Trial, or any other such date as the Parties agree. Actelion will introduce Kythera to and facilitate Kythera’s interaction with Actelion’s manufacturers to enable the transfer to occur directly between Actelion’s manufacturer and Kythera, given that most of the know-how related to manufacturing is located at Actelion’s manufacturers. If not done so sooner as per the previous sentence, within [***] ( [***] ) days after Kythera’s receipt of final results from Kythera’s Proof of Concept Trial, or sooner if so requested by Kythera or reflected in Exhibit 5.2, Kythera will notify Actelion in writing if Kythera wants the Manufacturing Information transferred to: (a) Kythera; or (b) a Third Party manufacturer reasonably acceptable to Kythera. As soon as is practicable after receiving Kythera’s notice, Actelion will cause its third party manufacturer to transfer to Kythera or such Third Party manufacturer, as the case may be, the Manufacturing Information listed in Exhibit 5.2 . The reasonable costs and expenses incurred by Actelion in carrying out such transfer and approved in advance by Kythera will be reimbursed [***] percent ( [***] %) by Kythera.
6. LICENSES
6.1 Licenses to Kythera.
(a) Subject to the terms and conditions of this Agreement, Actelion hereby grants to Kythera an exclusive (even as to Actelion except to the extent necessary to fulfill its obligations under this Agreement), sublicensable (pursuant to Section 6.3 ), transferable (solely pursuant to Section 13.9 ), royalty-bearing license under the Actelion Licensed Patents to Research, Develop, make, have made, use, sell, offer for sale, import and otherwise Commercialize Compounds and/or Licensed Products in the Field in the Territory.
(b) Subject to the terms and conditions of this Agreement, Actelion hereby grants to Kythera a non-exclusive,
sublicensable (pursuant to Section 6.3 ), transferable (solely pursuant to Section 13.9 ), royalty-free license under the Actelion Licensed Know-How to Research, Develop, make, have made, use, sell, offer for sale, import and otherwise Commercialize Compounds and/or Licensed Products in the Field in the Territory. Kythera shall support Actelion or other non-exclusive licensees regarding any request for information or documentation from health authorities relating to such non-exclusive licenses on the condition that Actelion and other non-exclusive licensees support Kythera regarding any request for information or documentation from health authorities relating to such non-exclusive licenses.
(c) Subject to the terms and conditions of this Agreement, Actelion hereby grants to Kythera a non-exclusive,
sublicensable (pursuant to Section 6.3 ), transferable (solely pursuant to Section 13.9 ), royalty-free license under the Actelion Related Patents to Research, Develop, make, have made, use, sell, offer for sale, import and otherwise Commercialize Compounds and/or Licensed Products in the Field in the Territory.
11
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY (d) Subject to the terms and conditions of this Agreement, Actelion hereby grants to Kythera a non-exclusive,
sublicensable (pursuant to Section 6.3 ), transferable (solely pursuant to Section 13.9 ), royalty-free license under the Actelion FTO Patents to Research, Develop, make, have made, use, sell, offer for sale, import and otherwise Commercialize Compounds and/or Licensed Products in the Territory. The “ Actelion FTO Patents ” mean any Patents that: (i) are not Actelion Licensed Patents or Actelion Related Patents; (ii) cover (A) manufacturing processes (including intermediates) or formulations of Compounds or Licensed Products, or (B) uses of Compounds or Licensed Products for hair growth or the prevention of hair loss; and (iii) Actelion or its Affiliates Controls as of the Effective Date or during the Term.
6.2 No Additional Licenses. Except as expressly provided in Section 6.1, nothing in this Agreement grants either Party any
right, title or interest in and to the intellectual property rights of the other Party (expressly, by implication or by estoppel). For clarity, Actelion does not grant Kythera the right to use in any Licensed Product any compound, which compound or use thereof is covered by any intellectual property right (including any kind of regulatory exclusivity) Controlled by Actelion, other than one or more Compounds.
6.3 Sublicensing. The licenses granted to Kythera in Section 6.1 will be freely sublicensable by Kythera as follows: (a) Kythera
has the right to sublicense [***] ; (b) Kythera has the right to sublicense [***] ; and (c) Kythera has the right to sublicense [***] . Kythera will remain responsible for each permitted sublicensee’s compliance with the applicable terms and conditions of this Agreement. Any agreement with a sublicensee must include provisions substantially the same as those contained herein to ensure compliance with this Agreement.
6.4 Retained Rights . Actelion retains the right for itself and its Affiliates to Manufacture, or arrange with Third Parties for the
Manufacture of, Licensed Products solely to the extent required under Section 5.1 . 6.5 Actelion Exclusivity . Actelion will not (and Actelion will ensure that its Affiliates will not) Research, Develop,
Manufacture (outside the scope of this Agreement) or Commercialize the Compounds during the Term.
7. COMPENSATION
7.1 License Fee. Within [***] ( [***] ) days after the Effective Date, Kythera will pay Actelion a license fee of One Million Five Hundred Thousand US Dollars ($1,500,000).
7.2 Milestone Payments to Actelion.
(a) Development and Regulatory Milestones. Subject to Section 7.2(b) , Kythera will make the following, one-time milestone payments set forth below to Actelion after the first achievement of each indicated event for a Licensed Product by Kythera (or any of its Affiliates, licensees or sublicensees).
12
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY
(b) Milestone Payment Limitations . The milestone events described in Sections 7.2(a)(i) – (vi) will be paid once
each, [***] for a Licensed Product. Milestone events described in Sections 7.2(a)(vii) – (ix) will be paid once each, solely with respect to a Licensed Product [***] , only after [***] . No other milestone payments will be owed by Kythera (or its Affiliates, licensees and sublicensees) with respect to a Licensed Product.
(c) Milestone Payment Process . Kythera will notify Actelion within [***] ( [***] ) days after the achievement of each
milestone event described in Sections 7.2(a)(i) – (ix) . Actelion will invoice Kythera for the amount associated with the applicable milestone event, and Kythera will pay Actelion the applicable amount within [***] ( [***] ) days of receipt of the invoice.
7.3 Royalty Payments to Actelion for Net Sales of Licensed Products. Kythera will pay to Actelion royalties on Net Sales by
Kythera (or its Affiliates, licensees or sublicensees) of Licensed Products. Royalties for Net Sales of a given unit of a Licensed Product will be owed under either Section 7.3(a) or under Section 7.3(b) , and, if both an oral and a topical formulation are being sold royalties will be owed under Section 7.3(a) for units that are oral formulations and Section 7.3(b) for units that are topical formulations.
(a) For orally-dosed formulations of Licensed Products:
13
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Event Milestone Payment
(i) [***] $ [***] (ii) [***] $ [***] (iii) [***] $ [***] (iv) [***] $ [***] (v) [***] $ [***] (vi) [***] $ [***] (vii) [***] $ [***] (viii) [***] $ [***] (ix) [***] $ [***]
Portion of Net Sales of Licensed Products in the Territory in a calendar year Royalty Rate
[***] %

CONFIDENTIAL INFORMATION
EXECUTION COPY
(b) For topical formulations of Licensed Products:
7.4 Third Party Royalties for Licensed Products in the Territory. Kythera may deduct from the royalties it would otherwise
owe to Actelion pursuant to Section 7.3 for a particular Licensed Product, [***] percent ( [***] %) of all royalties payable to one or more Third Party(ies) in consideration for rights [***] to Develop, Manufacture or Commercialize such Licensed Product (where such rights are identified by Kythera after the Effective Date), up to a maximum deduction of [***] percent ( [***] %) of the royalties otherwise due to Actelion for such Licensed Product pursuant to Section 7.3 .
7.5 Quarterly Payments and Reports. All royalties due under Section 7.3 will be paid quarterly, on a country-by-country
basis, within [***] ( [***] ) days after the end of the relevant quarter for which royalties are due. Kythera will provide to Actelion within [***] ( [***] ) days after the end of each quarter a report that summarizes the Net Sales of a Licensed Product during such quarter. Such reports will also include detailed information regarding the calculation of royalties due pursuant to Section 7.3 , including allowable deductions in the calculation of Net Sales of Licensed Products on which royalties are paid.
7.6 Term of Royalties. Actelion’s right to receive royalties under Section 7.3 will begin on the First Commercial Sale of a
Licensed Product in a country and expire, on a country-by-country and Licensed Product-by-Licensed Product basis, upon the later to occur of: (a) seven (7) years from First Commercial Sale of such Licensed Product in such country, and (b) expiration in such country of the last Valid Claim of such Licensed Product (the “Royalty Term”); provided, however, that if such last Valid Claim expires prior to seven (7) years after such First Commercial Sale, then, as of the date of the expiry of such last Valid Claim, royalties on Net Sales of Licensed Products in such country will be reduced by [***] % for the remainder of the Royalty Term. Notwithstanding the foregoing, the Royalty Term for Net Sales in a country in the Territory will
14
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL INFORMATION
EXECUTION COPY expire when reported sales revenues for a Generic Licensed Product over any [***] ( [***] ) consecutive calendar quarters in such country have achieved [***] percent ( [***] %) or more of the average Net Sales of Licensed Products in such country during the [***] ( [***] ) consecutive calendar quarters [***] in which such Generic Licensed Product is first sold commercially in such country.
7.7 Payment Method. All payments due under this Agreement to Actelion will be made by bank wire transfer in immediately available funds to an account designated by Actelion. All payments hereunder will be made in Dollars.
7.8 Taxes. Except for taxes on Actelion’s income, Kythera shall be liable for and pay all taxes imposed in connection with any
and all payments under this Agreement. 7.9 Sublicenses. In the event Kythera grants any permitted licenses or sublicenses to Third Parties to sell Licensed Products that
are subject to royalty payments under Section 7.3 , Kythera will have the responsibility to account for and report sales of any Licensed Product by a licensee or a sublicensee on the same basis as if such sales were Net Sales by Kythera. Kythera will pay to Actelion: (a) royalties on such sales as if such sales of the licensee or sublicensee were Net Sales of Kythera or any of its Affiliates; and (b) milestones payments pursuant to Section 7.2 based on the achievement by such licensee or sublicensee of any milestone event contemplated in such Section 7.2 as if such milestone event had been achieved by Kythera or any of its Affiliates hereunder. Any sales by Kythera’s Affiliates and sublicensees of Kythera or such sublicensee’s Affiliates, in each case to Third Parties, will be aggregated with sales by Kythera for the purpose of calculating the aggregate Net Sales in Section 7.3 .
Portion of Net Sales of Licensed Products in the Territory in a calendar year Royalty Rate
Less than or equal to $ [***]
Greater than $ [***] and less than or equal to $ [***]
[***] % Greater than $ [***]
[***] %
Portion of Net Sales of Licensed Products in the Territory in a calendar year
Royalty Rate Less than or equal to $ [***]
[***] %
Greater than $ [***] and less than or equal to $ [***]
[***] %
Greater than $ [***]
[***] %

7.10 Foreign Exchange. Conversion of sales recorded in local currencies to Dollars will be performed in a manner consistent
with Kythera’s normal practices used to prepare its audited financial statements for internal and external reporting purposes, which uses a widely accepted source of published exchange rates, and such methodology will be included in the Net Sales Report provided to Actelion.
7.11 Records. Kythera will keep (and will ensure that its Affiliates, licensees and sublicensees will keep) such records as are
required to determine, in a manner consistent with GAAP and this Agreement, the sums or credits due under this Agreement. All such books, records and accounts will be retained by Kythera until the later of: (a) three (3) years after the end of the period to which such books, records and accounts pertain; and (b) the expiration of the applicable tax statute of limitations (or any extensions thereof), or for such longer period as may be required by applicable law, rule or regulation. Kythera will require its licensees and sublicensees to provide to Kythera a report detailing the Net Sales calculations made by such licensee or sublicensee, which report will be made available to Actelion in connection with any audit conducted by Actelion pursuant to Section 7.12 .
7.12 Audits. Actelion will have the right to have an independent certified public accountant, reasonably acceptable to Kythera, to
have access during normal business hours, and upon reasonable prior written notice, to examine only those records of Kythera (and its Affiliates, licensees and sublicensees) as may be reasonably necessary to determine, with respect to any calendar year ending not more than three (3) years prior to Actelion’s request, the correctness or completeness of any report or payment made under this Agreement. The foregoing right of review
15
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY may be exercised only once per year and only once with respect to each such periodic report and payment. Results of any such examination will be: (a) limited to information relating to the Licensed Products; (b) made available to both Parties; and (c) subject to Article 9. Actelion will bear the full cost of the performance of any such audit, unless such audit discloses a variance to the detriment of Actelion of more than five percent (5%) from the amount of the original report, royalty or payment calculation, in which case Kythera will bear the full cost of the performance of such audit. The results of such audit will be final, absent manifest error.
7.13 Interest. Any undisputed payments or portions thereof due hereunder that are not paid on the date such payments are due
under this Agreement will bear interest at a rate equal to the lesser of: (a) [***] ( [***] ) percentage points above the Prime Rate as published by Citibank, N.A., New York, New York, or any successor thereto, at 12:01 a.m. on the first day of each quarter in which such payments are overdue; or (b) the maximum rate permitted by law, in each case calculated on the number of days such payment is delinquent, compounded monthly.
7.14 Payments to or Reports by Affiliates. Any payment required under any provision of this Agreement to be made to either
Party or any report required to be made by any Party will be made to or by an Affiliate of that Party if designated in writing by that Party as the appropriate recipient or reporting entity.
8. PATENT PROSECUTION AND ENFORCEMENT
8.1 Ownership.
(a) The inventorship of all Sole Inventions and Joint Inventions will be determined in accordance with the U.S. patent law.
(b) Actelion will own the entire right, title and interest in and to any and all of its Actelion Related Patents and of its Sole
Inventions, and Patents claiming such Sole Inventions (“ Actelion Sole Invention Patents ”). Kythera will own the entire right, title and interest in and to any and all of its Sole Inventions, and Patents claiming only such Sole Inventions (and no Joint Inventions) (“ Kythera Sole Invention Patents ”). Kythera and Actelion will be joint owners in and to any and all Joint Inventions and Patents claiming such Joint Inventions (“ Joint Invention Patents ”). Kythera and Actelion as joint owners will each have the right to exploit, to grant licenses under, to assign and to otherwise dispose of such Joint Inventions, without an accounting or obligation to, or consent required from, the other Party, but subject to the licenses hereunder and the other terms and conditions of this Agreement. Each Party grants to the other Party a nonexclusive, fully-paid, royalty-free, irrevocable, perpetual and sublicensable license under the Joint Invention Patents to make, use, sell, offer for sale and import inventions claimed in the Joint Invention Patents
(c) All employees, agents and contractors of each Party will be under written obligation to assign any inventions and
related intellectual property to the Party for whom they are employed or are providing services. (d) Solely for the purposes of this Section 8, the Parties acknowledge and agree that this Agreement will be deemed to be
a “Joint Research Agreement” as defined under 35 U.S.C. 103(c).
16
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY 8.2 Disclosure. Each Party will submit a written report to the other Party, no less frequently than within [***] ( [***] ) days
after the end of each quarter, describing any Joint Invention arising during the prior quarter in the course of the Agreement which it believes may be patentable or at such earlier time as may be necessary to preserve patentability of such invention. For the avoidance of doubt, if a Party believes that there was no patentable Joint Invention during the prior quarter, no report will be submitted. Actelion will provide (at its expense) to Kythera such assistance and execute such documents as are reasonably necessary to permit the filing and prosecution of such patent application to be filed on such Joint Invention, or the issuance, maintenance, extension or assignment of any resulting Patent.
8.3 Patent Prosecution and Maintenance; Abandonment.
(a) Prosecution and Maintenance.
(i) Filing, Prosecution and Maintenance by Kythera. Within [***] ( [***] ) days after the Effective Date, Actelion will provide Kythera all files related to the preparation, filing, prosecution and maintenance (including any interferences, reissues, reexaminations and oppositions) of the Actelion Licensed Patents. Kythera will be responsible for the preparation, filing, prosecution and maintenance (including any interferences, reissues, reexaminations and oppositions) of all Actelion Licensed Patents that are exclusively licensed to Kythera under Section 6.1 and the Joint Invention Patents (collectively, the “ Kythera Prosecuted Patents ”), provided that such responsibilities may be carried out by external patent counsel or vendor selected by Kythera, or by Kythera internal patent counsel in conjunction with external patent counsel or vendor selected by Kythera (and, regarding payment of annuities and maintenance fees, Kythera may seek to maintain the current vendor relationship with the existing annuity service provider for the Actelion Licensed Patents); and, provided further , that: (1) Kythera will begin prosecuting the Actelion Licensed Patents (as indicated on Exhibit 1.2 ) within [***] ( [***] )] days of receiving the last of the files for the Actelion Licensed Patents; and (2) Kythera will begin maintenance of the Actelion Licensed Patents (as indicated on Exhibit 1.2 ) within [***] ( [***] ) days of receiving the last of the files for the Actelion Licensed Patents. Kythera, or its external counsel or vendor, will provide Actelion with a written update of the filing, prosecution and maintenance status (including payments of annuities and maintenance fees) for each of the Kythera Prosecuted Patents on a quarterly basis, and Kythera (or its external counsel) will provide Actelion with drafts of proposed filings and prosecution documents to permit Actelion a reasonable opportunity for review and comment before such filings or documents are due. Actelion will provide any comments at least [***] ( [***] ) days prior to the original response deadlines that are required for such filing (without regard to applicable extensions). In the absence of a response from Actelion, Kythera may submit such proposed filings or prosecution documents in keeping with the terms of this Agreement.
(ii) Patent Term Extension, Abandonment or Inadequate Maintenance by Kythera .
(1) Patent Term Extension. If available, patent term extension (e.g., an extension in the United States under 35 USC 156 or equivalent extension in other countries) shall be requested and filed on Actelion Licensed Patents in any country in accordance with the respective regulations and requirements of that country.
17
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY (2) Abandonment. If Kythera decides not to continue the prosecution or maintenance of a patent
application or patent within the Kythera Prosecuted Patents in any country, Kythera will provide Actelion with notice of this decision at least [***] ( [***] ) days prior to any pending lapse or abandonment thereof (without regard to applicable extensions), and Actelion will thereafter have the right to assume responsibility for the filing, prosecution and maintenance of such patent or patent application by so notifying Kythera in writing. If Actelion assumes such responsibility for such filing, prosecution and maintenance, then: (A) Kythera will cooperate as reasonably requested by Actelion to facilitate control of such filing, prosecution and maintenance by Actelion (especially, Kythera will immediately provide the respective files related to the preparation, filing, prosecution and maintenance (including any interferences, reissues, reexaminations and oppositions) of the patent application or patent); and (B) such patent application or patent will no longer be an Actelion Licensed Patent subject to the license in Section 6.1(a) and all rights will revert to Actelion.
(3) [***] by Kythera. If Kythera [***] within the Kythera Prosecuted Patents in a given country, and
such patent is [***] in that country solely because of [***] , then the royalty obligation under Section 7.3 associated with Licensed Products covered by such abandoned/not-extended patent in such country will continue in such country until the [***] , had the patent [***] .
(4) [***]. Should Kythera [***] in a particular country, then for that country, the royalty obligation under
Section 7.3 associated with Licensed Products in such country will [***] , or not [***] . In the event that Kythera [***] and such [***] , the [***] will [***] this Section 8.3(a)(ii)(4) .
(5) Royalty Term in case of Section 8.3 (a)(ii)(3) or Section 8.3 (a)(ii)(4). For purposes of Section 7.6,
a Valid Claim is [***] , and any available extension thereof (as described in Section 8.3(a)(ii)(1)), had the [***] . The royalty reduction of [***] % within the [***] and the expiry of the Royalty Term [***] will only apply after the [***] , and any [***] , had the [***] .
(iii) Kythera Sole Invention Patents . Kythera will be responsible for the filing, prosecution (including any
interferences, reissues, reexaminations and oppositions) and maintenance of all Sole Invention Patents Controlled by Kythera.
18
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY (iv) Copies of Files. Kythera will provide copies of files of any Actelion Licensed Patent if requested by Actelion
with good cause (e.g. in case needed for requesting patent term extensions and the like).
(b) Payment of Prosecution Costs. Kythera will pay the expenses associated with the filing, prosecution (including any interferences, reissue proceedings, reexaminations and oppositions) and maintenance of: (i) its Sole Inventions Patents; and (ii) any Kythera Prosecuted Patents prosecuted by Kythera under Section 8.3(a)(i) . Actelion will pay the expenses associated with the filing, prosecution (including any interferences, reissue proceedings, reexaminations and oppositions) and maintenance of any Actelion Related Patents and any Actelion FTO Patents.
8.4 Enforcement of Patent Rights. If either Party becomes aware of a suspected infringement of any Actelion Licensed Patents
or Joint Invention Patents through the development, manufacture or sale of a Licensed Product by a Third Party, such Party will notify the other Party promptly, and following such notification, the Parties will confer. Kythera will have the first right, but will not be obligated, to bring an infringement action against such Third Party at its own expense and by counsel of its own choice, and Actelion will have the right to participate in such action, at its own expense and by counsel of its own choice. If Kythera fails to bring such an action or proceeding prior to the earlier of: (a) [***] ( [***] ) days following Kythera’s receipt of notice of alleged infringement; or (b) [***] ( [***] ) days before the time limit, if any, set forth in the appropriate laws and regulations for the filing of such actions, Actelion will have the right to bring and control any such action, at its own expense and by counsel of its own choice, and Kythera will have the right to be represented in any such action, at its own expense and by counsel of its own choice. If a Party brings an infringement action pursuant to this Section 8.4 , the other Party will reasonably assist the enforcing Party (at the enforcing Party’s expense) in such actions or proceedings if so requested, and will lend its name to such actions or proceedings if required by law in order for the enforcing Party to bring such action. Neither Party will have the right to settle any patent infringement litigation under this Section 8.4 in a manner that diminishes the rights or interests of the other Party without the prior written consent of such other Party, such consent not to be unreasonably withheld or delayed. In case Kythera brings such an action or proceeding, any recovery realized as a result of such litigation, after [***] , will be [***] . In case Actelion brings such an action or proceeding, any recovery realized as a result of such litigation will be [***] .
8.5 Data Exclusivity and Orange Book Listings . With respect to data exclusivity periods (such as those periods listed in the
FDA’s Orange Book (including any available pediatric extensions) or periods under national implementations of Directive 2001/EC/83, and all international equivalents), Kythera will use Commercially Reasonable Efforts consistent with its obligations under applicable law (including any applicable consent order) to seek, maintain and enforce all such data exclusivity periods available for the Licensed Products. With respect to filings in the FDA Orange Book (and foreign equivalents) for issued patents for a Licensed Product, upon request by Kythera (and at Kythera’s expense), Actelion will provide reasonable cooperation to Kythera in filing and maintaining such Orange Book (and foreign equivalent) listings.
8.6 Patent Term Extension . Actelion will cooperate with any of Kythera’s efforts to obtain patent term extension (including
any pediatric exclusivity extensions as may be available) or
19
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY supplemental protection certificates or their equivalents in any country with respect to patent rights covering the Licensed Products.
8.7 Notification of Patent Certification . Actelion will notify and provide Kythera with copies of any allegations of alleged patent invalidity, unenforceability or non-infringement of an Actelion Licensed Patent hereunder, including allegations pursuant to a Paragraph IV Patent Certification by a third party filing an Abbreviated New Drug Application, an application under §505(b)(2) of the Federal Food, Drug, and Cosmetic Act or other similar patent certification by a third party, and any foreign equivalent thereof. Such notification and copies will be sent to Kythera by Actelion as soon as practicable and at least within [***] ( [***] ) days after Actelion receives such certification.
8.8 Defense of Third Party Claims. If a claim is brought by a Third Party that any Compound under the Agreement infringes
the intellectual property rights of such Third Party, each Party will give prompt written notice to the other Party of such claim, and following such notification, the Parties will confer on how to respond.
8.9 Kythera may not limit the scope of protection of any Actelion Licensed Patent, for instance in prosecution, opposition,
litigation and the like, without prior written consent of Actelion, such consent not to be unreasonably withheld or delayed.
9. CONFIDENTIALITY
9.1 Nondisclosure of Confidential Information. Subject to Section 9.2 , all Information and Materials disclosed by one Party to the other Party pursuant to this Agreement, and, subject to Section 9.7 , Information that is generated pursuant to this Agreement with respect to Compounds or Licensed Products (for so long as such Compound or Licensed Product is not removed from the Agreement as a result of a Licensed Product specific termination pursuant to Section 10.2 ), will be “ Confidential Information ” for all purposes hereunder. The terms of this Agreement will be considered Confidential Information of both Parties. The Parties agree that during the period from the Effective Date through the Term, and for a period of [***] ( [***] ) years thereafter, a Party receiving Confidential Information of the other Party will: (a) use Commercially Reasonable Efforts to maintain in confidence such Confidential Information (but not less than those efforts as such Party uses to maintain in confidence its own proprietary industrial information of similar kind and value) and not to disclose such Confidential Information to any Third Party without prior written consent of the other Party (such consent not to be unreasonably withheld, delayed or conditioned), except for disclosures made in confidence to any Third Party under terms consistent with this Agreement; and (b) not use such other Party’s Confidential Information for any purpose except those permitted by this Agreement (it being understood that this Section 9.1 will not create or imply any rights or licenses not expressly granted under Article 6 , and it is also understood that, upon termination of this Agreement, a Party’s right to use the Confidential Information of the other Party will cease).
9.2 Exceptions. The obligations in Section 9.1 will not apply with respect to any portion of the Confidential Information that
the receiving Party can show by competent written proof:
(a) is publicly disclosed by the disclosing Party, either before or after it is disclosed to the receiving Party hereunder;
20
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY (b) was known to the receiving Party or any of its Affiliates, without obligation to keep it confidential, prior to disclosure
by the disclosing Party; (c) is subsequently disclosed to the receiving Party or any of its Affiliates by a Third Party lawfully in possession thereof
and without obligation to keep it confidential; (d) is published by a Third Party or otherwise becomes publicly available or enters the public domain, either before or
after it is disclosed to the receiving Party, and is not directly or indirectly supplied by the receiving Party in violation of this Agreement; or (e) has been independently developed by employees or contractors of the receiving Party or any of its Affiliates without
the aid, application or use of the disclosing Party’s Confidential Information.
9.3 Authorized Disclosure. A Party may disclose the Confidential Information disclosed by the other Party to the extent such disclosure is reasonably necessary in the following instances; provided that notice of any such disclosure will be sent as soon as practicable to the other Party:
(a) during the Term, when filing or prosecuting Patents covering Sole Inventions, Joint Inventions or Licensed Products,
in each case pursuant to activities under this Agreement; (b) during the Term, when making regulatory filings; (c) when prosecuting or defending litigation; (d) when complying with applicable governmental laws and regulations or complying with the requirements of the
national securities exchanges or other stock markets on which such Party’s securities are traded; and (e) when disclosing to a receiving Party’s: Affiliates; potential or actual collaborators, partners, and licensees (including
potential co-marketing and co-promotion contractors); potential or actual investment bankers, acquirers, lenders or investors; employees; consultants; and agents, each of whom, prior to disclosure, must be bound by similar obligations of confidentiality and non-use as set forth in this Article 9 ; provided that a confidentiality and non-use period of [***] ( [***] ) years will be sufficient.
9.4 Termination of Prior Agreements . As of the Effective Date, this Agreement terminates the confidential disclosure
agreement that is between Actelion and Kythera and that is effective as of December 13, 2013 (such confidential disclosure agreement, as amended, the “ Prior CDA ”). All Information exchanged between the Parties with respect to Licensed Products under the Prior CDA will be deemed Confidential Information and will be subject to the terms of this Article 9.
9.5 Publicity. The Parties agree that the public announcement by Kythera of the execution of this Agreement will be
substantially in the form of the press release attached as Exhibit 9.5 . Any other publication, news release or other public announcement relating to the execution of this Agreement will first be reviewed and approved by both Parties; provided,
21
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY however , that any securities filing that is required by law, rule or regulation, including disclosures required by the U.S. Securities and Exchange Commission, or made pursuant to the requirements of the national securities exchange or other stock market on which such Party’s securities are traded, as advised by the disclosing Party’s counsel, will be made pursuant to Section 9.6 .
9.6 Securities Filings . If a Party is required by applicable law, rule or regulation to make a securities filing relating to the
execution of this Agreement with the appropriate governmental authorities (including the U.S. Securities and Exchange Commission, and any securities exchange on which securities of such Party are listed), then the Party under such requirement will prepare a draft of such securities filing for review and comment by the other Party. If such securities filing includes the disclosure of this Agreement and its terms, the Party under such disclosure obligation will include a confidential treatment request and a proposed redacted version of this Agreement as part of such draft. Such draft securities filing will be provided to the other Party reasonably in advance of the deadline for such securities filing, and the other Party agrees to promptly (and in any event, no less than [***] ( [***] ) days (or such shorter time to meet any filing deadline where it was not practical to provide the other Party with [***] ( [***] )-days’ notice) after receipt of such confidential treatment request and proposed redactions) give its input in a reasonable manner in order to allow the Party seeking disclosure to file its request within the timelines proscribed by the regulations of applicable governmental authorities or securities exchange. The Party seeking such disclosure will exercise Commercially Reasonable Efforts to obtain confidential treatment of this Agreement from the applicable governmental authority or securities exchange as represented by the redacted version reviewed by the other Party.
9.7 Publications. Subject to Section 9.3 and Section 9.6 (for securities filings), each Party agrees to provide the other Party the
opportunity to review any proposed disclosure that contains Confidential Information of the other Party and that would or may constitute an oral, written or electronic public disclosure if made (including the full content of proposed abstracts, manuscripts or presentations relating to Compound), or which otherwise may contain Confidential Information, at least [***] ( [***] ) days prior to its intended submission for publication and agrees, upon request, not to submit any such abstract or manuscript for publication until the other Party is given a reasonable period of time to secure patent protection for any material in such publication which it believes to be patentable, such reasonable period not to exceed [***] ( [***] ) months. Both Parties understand that a reasonable commercial strategy may require delay of publication of information or filing of patent applications. The Parties agree to review and consider delay of publication and filing of patent applications under certain circumstances. The Liaisons (or the Parties), as appropriate, will review such requests and recommend subsequent action. Subject to Section 9.3 and Section 9.6 (for securities filings), neither Party will have the right to publish or present Confidential Information of the other Party which is subject to Section 9.1 . Nothing contained in this Section 9.7 will prohibit the inclusion of Confidential Information of the non-filing Party necessary for a patent application, provided the non-filing Party is given a reasonable opportunity to review the extent and necessity for its Confidential Information to be included prior to submission of such patent application related to the Agreement. Any disputes between the Parties regarding delaying a publication or presentation to permit the filing of a patent application will be referred to the Liaisons (or the Parties), as appropriate.
22
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL INFORMATION
EXECUTION COPY 10. TERM AND TERMINATION
10.1 Term. This Agreement will become effective on the Effective Date and will remain in effect until the expiration of all payment obligations under Article 7 , unless earlier terminated in accordance with Section 10.2 or by mutual written agreement of the Parties (the “Term” ). Upon the expiration of the Term with respect to a Licensed Product in a country, Kythera will have a fully-paid, perpetual license under Section 6.1 for the making, using, selling, offering for sale and importing of such Licensed Product in such country.
10.2 Termination Provisions.
(a) Termination for Uncured Material Breach . Subject only to Section 12.5, if either Party believes that the other is in material breach of this Agreement (including any material breach of a representation or warranty made in this Agreement), then the non-breaching Party may deliver notice of such breach to the other Party. In such notice the non-breaching Party will identify the actions or conduct that such Party would consider as an acceptable cure of such breach. The allegedly breaching Party will have thirty (30) days to cure such breach; provided, however , if the allegedly breaching Party disputes in good faith the existence of a material breach, the thirty (30) day cure period will be tolled until such time as the dispute is resolved by the Parties pursuant to Section 13.1(a) . If the Party receiving notice of breach fails to cure such breach within the 30-day period (as may be tolled by the foregoing sentence), then the Party originally delivering the notice may terminate this Agreement by providing at least thirty (30) days advance written notice to the allegedly breaching Party.
(b) Termination in case of insolvency . If either Party (the “ Insolvent Party ”): (i) becomes insolvent, or institutes or
has instituted against it a petition for bankruptcy or is adjudicated bankrupt; or (ii) executes a bill of sale, deed of trust, or a general assignment for the benefit of creditors; or (iii) is dissolved or subject to Section 13.9, transfers a substantial portion of its assets to a Third Party; or (iv) a receiver is appointed for the benefit of its creditors, or a receiver is appointed on account of insolvency; then the Insolvent

Party will immediately notify the other Party in writing of such event, and such other Party will be entitled to terminate this Agreement immediately upon written notice to the Insolvent Party.
(c) Termination by Kythera At-will . Kythera will have the right to terminate this Agreement at-will, in whole or on a
Licensed Product-by-Licensed Product basis, upon providing at least ninety (90) days prior written notice to Actelion, at the end of which the termination will be effective. Notwithstanding the foregoing, Kythera may not terminate at-will during an on-going clinical trial of a Licensed Product, except for reasons of patient safety.
10.3 Survival; Effect of Termination.
(a) In the event of termination of this Agreement, the following provisions of this Agreement will survive: Articles 1 (Definitions) and 12 (Indemnification); and Sections 7.11 (Records) , 8.1 (Ownership of IP) , 8.3 (solely with respect to Joint Invention Patents) , 8.4 (solely with respect to Joint Invention Patents) , 9.1, 9.2, 9.3, 9.4, 10.3, 10.4, 10.5 (but only in case of termination by Kythera under 10.2 (b)) , 13.1, 13.2, 13.3, 13.7, 13.8, 13.10 and 13.13 .
(b) In any event, termination of this Agreement will not: (i) relieve the Parties of any liability which accrued hereunder
prior to the effective date of such termination; (ii) preclude
23 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY either Party from pursuing all rights and remedies it may have hereunder or at law or in equity with respect to any breach of this Agreement; or (iii) prejudice either Party’s right to obtain performance of any obligation.
10.4 Licenses and Rights on Termination.
(a) Termination under Section 3.1. or Section 10.2(a) or Section 10.2(b) by Actelion or under Section 10.2(c) by Kythera . If Actelion terminates this Agreement pursuant to Section 3.1, Section 10.2(a) or Section 10.2(b) , or if Kythera terminates this Agreement pursuant to Section 10.2(c) , then the license granted to Kythera under Section 6.1 will automatically terminate and revert to Actelion. All [***] Compound and/or Licensed Product [***] in a [***] . Kythera shall provide Actelion with a report regarding the status, including, but not limited to possible outstanding issues, of any filings with Regulatory Authorities. Furthermore, the Parties shall notify relevant Regulatory Authorities of the transfer as required by local rules or regulations.
(b) Kythera’s Alternative Right to Termination. If Kythera has the right to terminate this Agreement pursuant to
Section 10.2(a) because Actelion is the breaching Party, then Kythera will have the right, in lieu of its right to terminate the entire Agreement, to have all licenses granted by Actelion under this Agreement survive, subject to Kythera’s fulfillment of fifty percent (50%) of its payment obligations under Article 7 after what would be the effective date of such termination.
10.5 Assignment of Actelion Licensed Patent s. Actelion hereby irrevocably assigns to Kythera the Actelion Licensed Patents;
provided, however, that such assignment will be under the suspensive condition of a termination of this Agreement by Kythera pursuant to Section 10.2(b) . Kythera hereby accepts such assignment. In consideration for such assignment, Kythera will pay to Actelion an amount equal to [***] , where the Parties agree that such [***] will be equal to the [***] . Furthermore, the Parties agree that such [***] will be paid by Kythera to Actelion [***] . Once Kythera has paid Actelion [***] , Kythera will automatically owe no further amounts to Actelion for the assignment of the Actelion Licensed Patents.
11. REPRESENTATIONS AND WARRANTIES AND COVENANTS
11.1 Mutual Authority. Actelion and Kythera each represents and warrants to the other Party as of the Effective Date that: (a) it has the authority and right to enter into and perform this Agreement; (b) this Agreement is a legal and valid obligation binding upon it and is enforceable in accordance with its terms, subject to applicable limitations on such enforcement based on bankruptcy laws and other debtors’ rights; and (c) the execution, delivery and performance of this Agreement will not conflict in any material fashion with the terms of any other agreement or instrument to which it is or becomes a party or by which it is or becomes bound, nor violate any law
24
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY or regulation of any court, governmental body or administrative or other agency having authority over it.
11.2 Actelion Representations and Warranties. Actelion represents and warrants to Kythera that, as of the Effective Date:
(a) Actelion owns the Actelion Licensed Patents and has made all filings and payments, and taken all other actions required to be made or taken, to maintain the Actelion Licensed Patents so that the Actelion Licensed Patents are not abandoned;
(b) To the best of Actelion’s knowledge, the composition of matter or method of manufacture of the Compounds do not
infringe any Patents owned by a Third Party or misappropriate any Third Party’s trade secrets; (c) Actelion and its Affiliates do not own, or have a license to, any Patents directed to or claiming any Compounds, other
than the Actelion Licensed Patents; (d) no Compound is subject either to a license with a Third Party, or an option for a license, pursuant to an agreement
between Actelion and any Third Party (other than material transfer agreements between Actelion and academic institutions); (e) there are no rights that Actelion or its Affiliates currently own, or to which Actelion or its Affiliates currently has a
license, that are within the Actelion Licensed Patents or Actelion Licensed Know-How (or that would be but for the terms of any agreement pursuant to which it has given up Control thereof, or pursuant to which it has rights to such Patents or Information but lacks Control thereof), to which it cannot grant a license of the scope in this Agreement;
(f) Actelion has unencumbered rights to grant licenses of the scope in this Agreement under Patents that are either owned
by Actelion or its Affiliates or are the subject of a license from a Third Party to Actelion that includes the right to sublicense, in each case to the extent such Patents claim any Compound;
(g) the scientific Information relating to setipiprant that Actelion or its Affiliates delivered or made available to Kythera
(whether directly or through its Third Party advisors) prior to the Effective Date (including any Information regarding the safety, efficacy, toxicology, potential side effects, pharmacokinetics, and pharmacodynamics of setipiprant) is representative of the complete set of scientific Information in the Control of Actelion and its Affiliates as of the Effective Date that relates to setipiprant, and Actelion and its Affiliates have not withheld from Kythera any unfavorable data or results of any pre-clinical or clinical studies of setipiprant, including unfavorable data and results of any pharmacokinetic, absorption, distribution, metabolism or excretion studies undertaken with respect to setipiprant;
(h) Actelion’s IND for setipiprant is accurate, true and complete up to the date of inactivation, which is [***] , except for
any data provided by Actelion to Kythera pursuant to Section 2.2(a)(iii) ;
25
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY (i) except for the [***] tablet form of setipiprant, the Chemistry, Manufacturing and Controls (“ CMC ”) portion of
Actelion’s IND for setipiprant is accurate, true and complete up to the date of inactivation, which is [***] ; (j) the scientific Information given to Kythera purporting to be associated with the Compounds actually is so associated,
and any physical samples given to Kythera under this Agreement are Compounds; and (k) To the best of Actelion’s knowledge, Actelion and its Affiliates Control, with respect to the license set forth in
Section 6.1 for the Compounds and Licensed Products containing or comprising such Compounds, all reasonably relevant intellectual property relating to Compounds that was generated by any Third Party in the course of such Third Party’s testing or other use of the Compounds pursuant to an agreement between Actelion (or its Affiliate) and such Third Party entered into prior to the Effective Date (other than material transfer agreements between Actelion and academic institutions).
11.3 Performance by Affiliates. The Parties recognize that each Party may perform some or all of its obligations under this
Agreement through Affiliates; provided, however , that each Party will remain responsible and guarantee performance by its Affiliates and will cause its Affiliates to comply with the provisions of this Agreement in connection with such performance. In particular, if any Affiliate of a Party participates under this Agreement with respect to Compounds: (a) the restrictions of this Agreement which apply to the activities of a Party with respect to Compounds will apply equally to the activities of such Affiliate; and (b) the Party affiliated with such Affiliate will assure, and hereby guarantees, that any intellectual property developed by such Affiliate will be governed by the provisions of this Agreement (and subject to the licenses set forth in Article 6 ) as if such intellectual property had been developed by the Party.
12. INDEMNIFICATION AND LIMITATION OF LIABILITY
12.1 Mutual Indemnification. Subject to Section 12.3 , each Party hereby agrees to indemnify, defend and hold harmless the other Party, its Affiliates, and their respective directors, employees and agents from and against any and all Third Party suits, claims, actions, demands, liabilities, expenses and/or losses, including reasonable legal expenses and reasonable attorneys’ fees (“ Loss(es) ”) to the extent such Losses result from any: (a) breach of any representation or warranty contained in the Agreement by the indemnifying Party; (b) breach of the Agreement or applicable law by such indemnifying Party; or (c) negligence or willful misconduct of the indemnifying Party, its Affiliates or (sub)licensees, or their respective directors, employees and agents in the performance of the Agreement.
12.2 Indemnification by Kythera. Subject to Section 12.3 , Kythera hereby agrees to indemnify, defend and hold harmless
Actelion and its directors, employees and agents from and against any and all Losses to the extent such Losses result from the manufacture, use, handling, storage, sale or other disposition of Licensed Products by Kythera or its Affiliates, agents or sublicensees, except to the extent such Losses result from any negligence, gross negligence or willful misconduct by Actelion, its Affiliates or (sub)licensees, or their respective directors, employees and agents in the performance of the Agreement.
12.3 Conditions to Indemnification. As used herein, “ Indemnitee ” will mean a Party entitled to indemnification under the
terms of Sections 12.1 or 12.2 . A condition precedent to each
26
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY Indemnitee’s right to seek indemnification under such Sections 12.1 or 12.2 is that such Indemnitee will:
(a) inform the indemnifying Party under such applicable Section 12.1 or Section 12.2 of a Loss as soon as reasonably practicable after it receives notice of the Loss;
(b) permit the indemnifying Party to assume direction and control of the defense, litigation, settlement, appeal or other
disposition of the Loss (including the right to settle the claim solely for monetary consideration); provided , that the indemnifying Party will seek the prior written consent (such consent not to be unreasonably withheld, delayed or conditioned) of any such Indemnitee as to any settlement which would materially diminish or materially adversely affect the scope, exclusivity or duration of any Patents licensed under this Agreement, would require any payment by such Indemnitee, would require an admission of legal wrongdoing in any way on the part of an Indemnitee, or would effect an amendment of this Agreement; and
(c) fully cooperate (including providing access to and copies of pertinent records and making available for testimony
relevant individuals subject to its control) as reasonably requested by, and at the expense of, the indemnifying Party in the defense of the Loss.
If an Indemnitee has complied with all of the conditions described in Sections 12.3(a) – (c) , as applicable, the indemnifying Party will supply attorneys reasonably acceptable to the Indemnitee to defend against any such Loss. Subject to the foregoing, an Indemnitee may participate in any proceedings involving such Loss using attorneys of the Indemnitee’s choice and at the Indemnitee’s expense. In no event may an Indemnitee settle or compromise any Loss for which the Indemnitee intends to seek indemnification from the indemnifying Party hereunder without the prior written consent of the indemnifying Party (such consent not to be unreasonably withheld, delayed or conditioned), or the indemnification under such Section 12.1 or 12.2 as to such Loss will be null and void.
12.4 Limitation of Liability. EXCEPT FOR INDEMNIFICATION PROTECTION FROM THE OTHER PARTY PURSUANT TO SECTIONS 12.1 AND 12.2 , IN NO EVENT WILL EITHER PARTY, ITS DIRECTORS, OFFICERS, EMPLOYEES, AGENTS OR AFFILIATES BE LIABLE TO THE OTHER PARTY FOR ANY INDIRECT, INCIDENTAL, SPECIAL, PUNITIVE, EXEMPLARY OR CONSEQUENTIAL DAMAGES, WHETHER BASED UPON A CLAIM OR ACTION OF CONTRACT, WARRANTY, NEGLIGENCE, STRICT LIABILITY OR OTHER TORT, OR OTHERWISE, ARISING OUT OF THE AGREEMENT, UNLESS SUCH DAMAGES ARE DUE TO THE GROSS NEGLIGENCE OR WILLFUL MISCONDUCT OF THE LIABLE PARTY (INCLUDING GROSS NEGLIGENCE OR WILLFUL BREACH WITH RESPECT TO A PARTY’S REPRESENTATIONS AND WARRANTIES IN ARTICLE 11 ).
12.5 KBI Guarantee . KBI hereby irrevocably guarantees to Actelion the due and punctual performance by Kythera of [***] and
undertakes, upon the occurrence and continuance of [***] , that KBI will duly and properly [***] . In the event of Kythera’s failure to fulfill its obligations as described in the previous sentence, within [***] ( [***] ) days after the date that Actelion has notified KBI in
27
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY writing of such failure, if Kythera has not [***] , KBI shall [***] . In case such failure at the same time constitutes a material breach of this Agreement according to Section 10.2 (a), Actelion will have the right to terminate this Agreement by providing [***] ( [***] ) days advance written notice only in case KBI should also [***] within [***] ( [***] ) days after the date that Actelion has notified KBI in writing of such failure. 13. MISCELLANEOUS
13.1 Dispute Resolution.
(a) Except for any dispute described in Section 13.3 (which will be handled exclusively in accordance with Section 13.3 ), if any dispute, controversy or claim arises out of, relates to or connects with any provision of the Agreement (each, a “ Disputed Matter ”), the Parties will try to settle the Disputed Matter using informal dispute resolution and by referring the Disputed Matter to the Party’s respective Executive Officers. Either Party may initiate such informal dispute resolution by sending written notice of the dispute to the other Party, and, within [***] ( [***] ) days after such notice, such Executive Officers will meet for attempted resolution by good faith negotiations. If such Executive Officers are unable to resolve such Disputed Matter within [***] ( [***] ) days after their first meeting for such negotiations, the Parties will resolve their dispute using binding arbitration under Section 13.1(b) .
(b) If the Parties are unable to resolve a Disputed Matter using the process described in Section 13.1(a) , then a Party
seeking further resolution of the Disputed Matter will submit the Disputed Matter to resolution by final and binding arbitration administered by the International Chamber of Commerce, under the Rules of Arbitration of the International Chamber of Commerce (the “ Rules ”), except as otherwise provided herein and applying the substantive law specified in Section 13.2 . Moreover, the Parties agree on the applicability of the “IBA Rules on the Taking of Evidence in International Commercial Arbitration”. Whenever a Party will decide to institute arbitration proceedings, it will give written notice to that effect to the other Party, and the place of arbitration will be in Zurich, Switzerland. The arbitration will be conducted by a panel of three (3) arbitrators, who will be appointed as follows: each Party will appoint a single arbitrator, and the two (2) arbitrators will agree on a third (3 ) arbitrator who will act as the chair of the arbitral tribunal, within ten (10) days after their appointment. If the two (2) arbitrators are unable to agree on the third (3 ) arbitrator within such ten (10) days, then the third (3 ) arbitrator will be appointed by the International Chamber of Commerce in accordance with the Rules. Each arbitrator must have business or legal experience in the biotechnology or pharmaceutical industry and, in addition, be knowledgeable in the specific field of dispute in question. The arbitrators will not have the power to award damages excluded pursuant to Section 12.4, and any arbitral award that purports to award such damages is expressly prohibited and void ab initio . Decisions of the arbitrators that conform to the terms of this Section 13.1(b) will be final and binding on the Parties, and enforcement of the award so rendered may be entered in any court of competent jurisdiction. Either Party may apply to the arbitrators for interim injunctive relief until the arbitration award is rendered or the controversy is otherwise resolved. The losing Party, as determined by the arbitrators, will pay all of the administrative costs and fees of the arbitration and the fees and costs of the arbitrators, and the arbitrators will be directed to provide for payment or reimbursement of such fees and costs by the losing Party. If the arbitrators determine that there is no losing Party, the Parties will each bear
28
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
rd
rd
rd

CONFIDENTIAL INFORMATION
EXECUTION COPY or pay one-half (1/2) of those costs and fees and the arbitrator’s award will so provide. Notwithstanding the foregoing, each Party is to bear or pay its own attorneys’ fees, expert or witness fees, and any other fees and costs, and no such fees or costs will be shifted to the other Party. Except to the extent necessary to confirm an award, enforce an award, or as may be required by law, neither a Party nor the arbitrators may disclose the existence, content, or results of an arbitration without the prior written consent of the Parties. In no event shall an arbitration be initiated after the date when commencement of a legal or equitable proceeding based on the dispute, controversy or claim would be barred by the laws of Switzerland.
13.2 Governing Law. Resolution of all disputes, controversies or claims arising out of, relating to or in connection with the Agreement or the performance, enforcement, breach or termination of the Agreement and any remedies relating thereto, will be governed by and construed under the substantive laws of Switzerland, to the exclusion of the UN Convention on Contracts for the International Sale of Goods, except as described in Section 8.1(a) where the inventorship of all Sole Inventions and Joint Inventions will be determined in accordance with the U.S. patent law.
13.3 Patents and Trademarks; Equitable Relief.
(a) Any dispute, controversy or claim arising out of, relating to or in connection with: (i) the scope, validity, enforceability or infringement of any Patent rights covering the Research, Development, Manufacture or Commercialization any Licensed Product; or (ii) any trademark rights related to any Licensed Product, will in each case be submitted to a court of competent jurisdiction in the territory in which such Patent or trademark rights were granted or arose.
(b) Any dispute, controversy or claim arising out of, relating to or in connection with the need to seek preliminary or
injunctive measures or other equitable relief (e.g., in the event of a potential or actual breach of the confidentiality and non-use provisions in Article 9 ) need not be resolved through the procedure described in Section 13.1 but may be immediately brought in a court of competent jurisdiction.
13.4 Entire Agreement; Amendments. This Agreement sets forth the complete, final and exclusive agreement and all the
covenants, promises, agreements, warranties, representations, conditions and understandings between the Parties hereto and supersedes and terminates all prior agreements and understandings between the Parties. There are no covenants, promises, agreements, warranties, representations, conditions or understandings, either oral or written, between the Parties other than as are set forth herein and therein. No subsequent alteration or amendment of this Agreement will be binding upon the Parties unless it is covered in a document that identifies the specific provision that will be altered or amended, and such document is signed by an authorized officer of each Party.
13.5 Export Control. This Agreement is made subject to any restrictions concerning the export of products or technical
information from the U.S. or other countries which may be imposed upon or related to Actelion or Kythera from time to time. Each Party agrees that it will not export, directly or indirectly, any technical information acquired from the other Party under this Agreement or any products using such technical information to a location or in a manner that at the time of export requires an export license or other governmental approval, without first obtaining the written consent to do so from the appropriate agency or other governmental entity.
29
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY 3.6 Force Majeure. Each Party will be excused from the performance of its obligations under this Agreement to the extent that
such performance is prevented by force majeure (defined below) and the nonperforming Party promptly provides notice of the prevention to the other Party. Such excuse will be continued so long as the condition constituting force majeure continues and the nonperforming Party takes reasonable efforts to remove the condition. For purposes of this Agreement, “ force majeure ” will include conditions beyond the control of the Parties, including an act of God, acts of terrorism, compliance with any regulation, law or order of any government, war, civil commotion, labor strike or lock-out, epidemic, failure or default of public utilities or common carriers, destruction of production facilities or materials by fire, earthquake, storm or like catastrophe.
13.7 Notices. Any notices given under this Agreement will be in writing, addressed to the Parties at the following addresses, and
delivered by person, by facsimile (with receipt confirmation), by email (with receipt confirmation), or by FedEx or other reputable courier service. Any such notice will be deemed to have been given: (a) as of the day of personal delivery; (b) one (1) day after the date sent by confirmed facsimile or confirmed email; or (c) on the day of successful delivery to the other Party confirmed by the courier service. Unless otherwise specified in writing, the mailing addresses of the Parties will be as described below.
For Actelion: Actelion Pharmaceuticals, Ltd.
Gewerbestrasse 16 CH-4123 Allschwil Switzerland Facsimile No./Email: [***] Attention: EVP and Chief Business Development Officer
with a copy to (which will not constitute notice):
Facsimile No./Email: [***] Attention: General Counsel
For Kythera: Kythera Holdings, Ltd.
Clarendon House 2 Church Street, Hamilton HM 11, Bermuda Email: [email protected] Attention: Vice President for Strategy and Corporate Development
with a copy to (which will not constitute notice):
Latham & Watkins LLP 140 Scott Drive Menlo Park, California 94025 Facsimile No.: (650) 463-2600 Attention: Alan Mendelson, Esq. and Judith Hasko, Esq.
30
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL INFORMATION
EXECUTION COPY 13.8 Maintenance of Records Required by Law or Regulation. Each Party will keep and maintain all records required by
applicable law, rule or regulation with respect to Licensed Products and will make copies of such records available to the other Party upon request. Each Party retains the ownership of the documents generated by such Party during the Term. Records shall be retained in the format they were generated in and archived for a period of at least five (5) years or according to applicable rules and regulations if longer. Any agreement with a sublicensee must include provisions substantially the same as those contained herein. Kythera will ensure compliance by such sublicensee with the above record retention obligation. Kythera will keep (and will ensure that its Affiliates, licensees and sublicensees will keep) such records as are required by applicable law, rule or regulation.
13.9 Assignment. Neither Party may assign or transfer this Agreement without the prior written consent of the other Party (such
consent not to be unreasonably withheld, delayed or conditioned), except a Party may make such an assignment without the other Party’s consent to an Affiliate or to a Third Party successor to all or substantially all of the business of such Party to which this Agreement relates, whether in a merger, sale of stock, sale of assets or other transaction; provided that any such permitted successor or assignee of rights and/or obligations hereunder has the financial, technical and business resources reasonably necessary to perform the obligations of the assignor (or, in the case of Kythera assigning to an Affiliate, if such Affiliates performance is guaranteed by KBI in accordance with the terms set forth in Section 12.5. hereof); provided further that any such permitted successor or assignee of rights and/or obligations hereunder is obligated, by reason of operation of law or pursuant to a written agreement with the other Party, to assume performance of this Agreement or such rights

and/or obligations; and provided, further , that if assigned to an Affiliate, the assigning Party will remain jointly and severally responsible for the performance of this Agreement by such Affiliate (unless in the case of Kythera assigning to an Affiliate, if such Affiliates performance is guaranteed by KBI in accordance with the terms set forth in Section 12.5 hereof). Any permitted assignment will be binding on the successors of the assigning Party. Any assignment or attempted assignment by either Party in violation of the terms of this Section 13.9 will be null and void.
13.10 Further Actions. Each Party agrees to execute, acknowledge and deliver such further instruments, and to do all such other
acts, as may be necessary or reasonably appropriate to carry out the purposes and intent of this Agreement. 13.11 Severability. If any of the provisions of this Agreement are held to be invalid or unenforceable, the provision will be
considered severed from this Agreement and will not serve to invalidate any remaining provisions hereof. The Parties will make a good faith effort to replace any invalid or unenforceable provision with a valid and enforceable one such that the objectives contemplated by the Parties when entering this Agreement may be realized.
13.12 No Waiver. Any delay in enforcing a Party’s rights under this Agreement or any waiver as to a particular default or other
matter will not constitute a waiver of such Party’s rights to the future enforcement of its rights under this Agreement, excepting only as to a waiver contained in a document that identifies the provision that is waived, identifies the period of time for the waiver, and is signed by the Parties.
13.13 Construction of this Agreement. Except where the context otherwise requires, wherever used, the use of any gender will
be applicable to all genders, and the word “ or ” are used
31
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY in the inclusive sense. When used in this Agreement, “ including ” means “ including without limitation ”. References to either Party include the successors and permitted assigns of that Party. The headings of this Agreement are for convenience of reference only and in no way define, describe, extend or limit the scope or intent of this Agreement or the intent of any provision contained in this Agreement. The Parties have each consulted counsel of their choice regarding this Agreement, and, accordingly, no provisions of this Agreement will be construed against either Party on the basis that the Party drafted this Agreement or any provision thereof. If the terms of this Agreement conflict with the terms of any Exhibit, then the terms of this Agreement will govern. The official text of this Agreement and any Exhibits hereto, any notice given or accounts or statements required by this Agreement, and any dispute proceeding related to or arising hereunder, will be in English. In the event of any dispute concerning the construction or meaning of this Agreement, reference will be made only to this Agreement as written in English and not to any translation into any other language.
13.14 Counterparts. This Agreement may be executed in two (2) or more counterparts, each of which will be an original and all of which will constitute together the same document. Counterparts may be signed and delivered by facsimile, or electronically in PDF format, each of which will be binding when sent.
Signature page follows.
32
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONFIDENTIAL INFORMATION
EXECUTION COPY IN WITNESS WHEREOF , the Parties have executed this Agreement by their proper officers. The date that this Agreement is
signed will not be construed to imply that the document was made effective on that date.
33
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
ACTELION PHARMACEUTICALS, LTD.,
KYTHERA HOLDINGS, LTD.
/s/ Nicholas Franco
/s/ Keith Leonard By: Nicholas Franco
By: Keith Leonard
Title: EVP & Chief Business Dev. Officer
Title: Director
Date: February 10, 2015
Date: February 10, 2015
ACTELION PHARMACEUTICALS, LTD.,
Agreed to be bound by the terms and conditions of this Agreement pursuant to Section 12.5:
By: /s/ Douglas B. Snyder
KYTHERA BIOPHARMACEUTICALS, INC.
Title: General Counsel
Date: February 10, 2015
By: Keith Leonard
Title:_ President and CEO
Date: February 10, 2015

CONFIDENTIAL INFORMATION
EXECUTION COPY Exhibit 1.2
Actelion Licensed Patents
ACT-[***] Patent Portfolio (Status: October 2014)
1
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Case Act [***] ACT- [***]
Publ. [***] [***]
Title [***]
Country Application No Appl. Date Grant No Grant Date Status [***] [***] [***] [***] [***] [***]

CONFIDENTIAL INFORMATION
EXECUTION COPY
2
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL INFORMATION
EXECUTION COPY Exhibit 2.1
Process of Technology Transfer of
Actelion Licensed Know-How (including specified Manufacturing know-how) and Materials
1
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
[***] [***] [***] [***] [***] [***]
Phase Key Activities and Outputs 1) [***] Estimated Timing: [***]
The purpose of Phase 1 and the activities described below is to [***] . To this end, within [***] days of License Agreement execution: [***]
2) [***] Estimated Timing: [***]
The purpose of Phase 2 and the activities described below is for Actelion to [***] . To this end: [***]
3) [***] Estimated Timing: [***]
The purpose of Phase 3 is to [***] .
4) [***] Estimated Timing: [***]
The purpose of Phase 4 is for [***] .
5) [***] Timing: [***]
The purpose of Phase 5 is for [***] .

CONFIDENTIAL INFORMATION
EXECUTION COPY
Know-How and Materials to be Transferred within 30 days or as otherwise determined hereinafter*
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Function Know-How / Materials to be Transferred In Data Room?
General • [***]
N/A
• [***] No Research & Nonclinical
• [***] No
Regulatory • [***] No
• [***]
No
Clinical • [***]
Incomplete
CMC • [***]
No
[***] No
CONFIDENTIAL INFORMATION
EXECUTION COPY
Know-How and Materials to be discussed for transfer under phase 2: [***]
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Function Know-How / Materials to be Transferred In Data Room?
General • [***]
No
• [***]
N/A
Research & Nonclinical
• [***] o [***]
No
o [***] N/A
o [***] N/A
Regulatory • [***] Incomplete
Clinical • [***] Incomplete
• [***] No CMC [***]
• [***] Incomplete
• [***] No
• [***] Incomplete
• [***]
o [***] No

CONFIDENTIAL INFORMATION
EXECUTION COPY
* [***] [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL INFORMATION
EXECUTION COPY Know-How and Materials to be discussed for transfer under phases 3, 4 and 5 (may in some points be redundant with previous
section (transfer under phase 2)*
* [***] [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
o [***] Incomplete
o [***] Incomplete
o [***] Incomplete
o [***] No
• [***]
N/A
• [***]
N/A
Function Know-How / Materials to be Transferred In Data Room?
General • [***]
No
Nonclinical • [***] No Clinical • [***]
• [***] • [***]
No
• [***] No
CMC [***] No
• [***]
• [***] No
• [***] o [***]
Incomplete
o [***] Incomplete
o [***] No
o [***] No
o [***] Incomplete
o [***] No
o [***] Incomplete
o [***] Incomplete
o [***] No
o [***] No
o [***] No
o [***] No
o [***]
Incomplete

CONFIDENTIAL INFORMATION
EXECUTION COPY Exhibit 2.2 (a)(i)
Regulatory Documents to be provided in format other than eCTD
Tables, Listings and Figures for all clinical studies will be provided in rich text format (rtf). The following setipiprant documents will be provided in Microsoft Word format:
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
IND Section Number Document Title eCTD Sequence Number [***] [***] [***]

CONFIDENTIAL INFORMATION
EXECUTION COPY
N/A = Not Applicable [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
[***] [***] [***]

CONFIDENTIAL INFORMATION
EXECUTION COPY
Exhibit 3.1(b)
Form of Semi - Annual Development Report
Period: MM/YY – MM/YY 1. Planned Development and Regulatory Timelines:
2. Development Updates:
3. Regulatory Updates:
4. Explanation for any deviation from Planned Development or Regulatory Timelines
5. Publication Updates:
6. Commercialization Update:
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL INFORMATION
EXECUTION COPY Exhibit 5.2
Items of Manufacturing Information transferred
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Licensed Compound
Planned Proof of Concept Trial Initiation (MM/YY)
Planned Phase III Initiation (MM/YY)
Planned 1 Regulatory Filing (MM/YY)
Licensed Compound
Indication Phase New / Ongoing Trial? Results
Licensed Compound
Indication Regulatory Status (Filed, Approved)
Major changes in Regulatory Status during Period
US Europe Japan
Licensed Compound
Indication Comments
Licensed Compound
Indication Publication Reference
Licensed Compound
Comments
Activity Description Analytical Method Transfer
[***]
API Process Transfer [***] Drug Product (capsules, tablets, active and placebo) Process Transfer
[***]
Transfer of material [***] Container closure systems
[***]
st


CONFIDENTIAL INFORMATION
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Project Management [***]

CONFIDENTIAL INFORMATION
EXECUTION COPY Exhibit 9.5
Press Release
KYTHERA Acquires Worldwide Rights to Clinical Compound and Key Intellectual Property for Potential Novel
Treatment of Hair Loss
Westlake Village, Calif., XX, 2015 – KYTHERA Biopharmaceuticals, Inc. (NASDAQ: KYTH) today announced that it has entered into license agreements with Actelion Pharmaceuticals Ltd. and the University of Pennsylvania for a novel approach for the treatment of hair loss, which together could allow KYTHERA to bring a new treatment to the very large and still highly unsatisfied hair loss market. “These two licenses build on KYTHERA’s focus on developing and commercializing high-value, self-pay aesthetic products that have the promise to yield high patient satisfaction and enhanced self image,” said Keith Leonard, KYTHERA’s president and CEO. “Based on several years of research and clinical work already accomplished, we believe we have the possibility of reaching initial proof-of-concept data in a very capital efficient manner.” Under the terms of the agreement with Actelion, KYTHERA Holdings Ltd., a wholly-owned Bermuda subsidiary of KYTHERA Biopharmaceuticals, Inc., will license the worldwide rights to setipiprant, a clinical-stage selective and potent oral antagonist to the prostaglandin D (PGD ) receptor. As part of the agreement, Actelion will be eligible to receive up to $27 million in potential development and regulatory milestones, as well as royalties on sales if setipiprant is successfully commercialized. Under the terms of the agreement with the University of Pennsylvania, KYTHERA Holdings Ltd., will acquire exclusive worldwide rights to the University of Pennsylvania’s patent estate covering the use of PGD receptor antagonists for the treatment of hair loss (often presenting as male pattern baldness, or androgenic alopecia). The discovery of the relationship between PGD and hair loss was made in the lab of Dr. George Cotsarelis and published in late 2011 . KYTHERA has been working with Dr. Cotsarelis’ labs at the University of Pennsylvania over the last two years under Sponsored Research and Option Agreements to further develop and elucidate the initial reported observations. “The University of Pennsylvania’s discovery and patent estate combined with access to the PGD antagonist setipiprant provide KYTHERA a strong foundation for this novel approach to hair loss. These observations are potentially the most innovative new thinking in hair loss over the last two decades,” said Frederick Beddingfield, III, M.D., Ph.D., KYTHERA’s Chief Medical Officer. “Setipiprant is believed to directly affect this hair loss pathway, and our own preclinical and human hair models confirmed this effect. It is a well-characterized molecule with a large safety database and we believe we can quickly initiate a development program to study it in hair loss. Putting these two pieces of the puzzle together is ideal and represents an example of our approach to scientifically sound and efficient drug development.”
Science Translational Medicine, Med 21 March 2012:Vol. 4, Issue 126, p. 126ra34 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
2 2
2
2 1
2
1

CONFIDENTIAL INFORMATION
EXECUTION COPY KYTHERA plans to conduct a human proof-of-concept study to establish the efficacy of setipiprant in male subjects with androgenic alopecia (AGA). About Setipiprant Setipiprant is a selective oral antagonist to the prostaglandin D (PGD ) receptor. The elevation of PGD levels in certain regions of the male scalp is associated with hair loss in those regions. In early research studies, PGD inhibitors were found to extend the anagen (growth) phase of the hair cycle, thereby promoting the growth of hair. Setipiprant had previously been studied as a potential allergic inflammation treatment and had undergone eight clinical trials, including a Phase III study in seasonal allergic rhinitis patients and a Phase II proof of concept study in asthma patients, resulting in a safety database of more than 1,000 patients. Actelion suspended the development of setipiprant due to a lack of efficacy seen in clinical trials for inflammatory disorders. Treatment in all studies was well tolerated across all treatment groups and no serious adverse events were reported. About KYTHERA Biopharmaceuticals, Inc. KYTHERA Biopharmaceuticals, Inc. is a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of novel prescription products for the aesthetic medicine market. KYTHERA’s lead product candidate, ATX-101, is currently in late-stage clinical development for the reduction of submental fat, which commonly presents as a double chin, and is a potential first-in-class submental contouring injectable drug. KYTHERA submitted its New Drug Application for ATX-101 in May 2014, and will have a Prescription Drug User Fee Act (PDUFA) action date of May 13, 2015. KYTHERA also maintains an active research interest in hair and fat biology, pigmentation modulation and facial contouring. Find more information at www.kytherabiopharma.com . [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
2 2 2
2

Exhibit 10.21
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
LICENSE AGREEMENT
DATED AS OF FEBRUARY , 2015
BY AND BETWEEN
THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
AND
KYTHERA HOLDINGS, LTD.
AND
KYTHERA BIOPHARMACEUTICALS, INC.
CONFIDENTIAL
EXECUTION COPY

TABLE OF CONTENTS
EXHIBIT A PENN PATENT RIGHTS EXHIBIT B CERTAIN FINANCIAL TERMS APPENDIX I FORM OF FINANCIAL REPORT APPENDIX II DEVELOPMENT PLAN APPENDIX III SUBLICENSE DEVELOPMENT REPORT
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Page
ARTICLE 1 DEFINITIONS 1
ARTICLE 2 LICENSES AND OTHER RIGHTS 7
ARTICLE 3 DILIGENCE 9
ARTICLE 4 FINANCIAL PROVISIONS 10
ARTICLE 5 INTELLECTUAL PROPERTY 14
ARTICLE 6 REPRESENTATIONS, WARRANTIES AND COVENANTS 19
ARTICLE 7 INDEMNIFICATION; INSURANCE AND LIMITATION OF LIABIL ITY 21
ARTICLE 8 TERM AND TERMINATION 23
ARTICLE 9 ADDITIONAL PROVISIONS 25
CONFIDENTIAL -i-
EXECUTION COPY

LICENSE AGREEMENT
This License Agreement (this “ Agreement ”) is dated as of February , 2015 (the “ Effective Date ”) by and among The Trustees of the
University of Pennsylvania, a Pennsylvania nonprofit corporation (“ Penn ”), Kythera Holdings, Ltd., a Bermuda company (“ Licensee ”), and Kythera Biopharmaceuticals, Inc., a Delaware Corporation (“KBI”). Penn, Licensee and KBI (for purposes of Section 6.4.1) may be referred to herein as a “Party ” or, collectively, as “ Parties ”.
RECITALS:
WHEREAS, Penn owns and controls certain innovative technology for regrowth of hair as further defined herein that was developed in the
course of research at Penn by Dr. George Cotsarelis (the “ Inventor(s) ”); WHEREAS, Penn desires to license Penn’s intellectual property rights in such technology, in a manner that will benefit the public and best
facilitate the distribution of useful products and the utilization of new technology, consistent with Penn’s educational and research missions and goals; and
WHEREAS, Licensee desires to license from Penn, Penn’s intellectual property rights in such technology, to develop, manufacture and
commercialize such technology, all on the terms and conditions of this Agreement. NOW, THEREFORE, in consideration of the various promises and undertakings set forth herein, the Parties agree as follows:
ARTICLE 1 DEFINITIONS
Unless otherwise specifically provided herein, the following terms shall have the following meanings:
1.1 “ Achievement Date ” means, with respect to a Diligence Event, the corresponding date such Diligence Event is to be achieved as provided in Exhibit B attached hereto subject to modification pursuant to Section 3.3 below.
1.2 “ Affiliate ” means a Person that controls, is controlled by or is under common control with a Party, but only for so long as such control exists.
For the purposes of this Section 1.2, the word “control” (including, with correlative meaning, the terms “controlled by” or “under the common control with”) means the actual power, either directly or indirectly through one or more intermediaries, to direct the management and policies of such Person or entity, whether by the ownership of more than fifty percent (50%) of the voting stock of such entity, or by contract or otherwise.
1.3 “ Bundled Sublicense” means a sublicense entered into by Licensee: 1) that includes a conveyance of rights to both the Penn Patent Rights as
well as [***] and 2) where Licensee is required to economically compensate the non-Affiliated Third Party for sublicensing of such Third Party’s [***] (e.g., by paying Third Party Royalties to such non-Affiliated Third Party).
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL 1
EXECUTION COPY

1.4 “ Commercially Reasonable Efforts ” means the efforts and resources that a similarly situated biotechnology company would use for its own
internally discovered technology of similar commercial potential and similar stage of development, considering factors such as the likely timing of the technology’s entry into the market and, any patent and other proprietary position of the Product. Without limiting the foregoing, Commercially Reasonable Efforts requires, with respect to such obligations, that the Party (a) promptly assign responsibility for such obligation to specific employee(s) who are accountable for progress and monitor such progress on an on-going basis, (b) set annual objectives for carrying out such obligations, and (c) allocate resources designed to advance progress with respect to such objectives. For clarity, Commercially Reasonable Efforts will not mean that a Party guarantees that it will actually accomplish the applicable task or objective.
1.5 “ Compulsory License ” means a compulsory license under Penn Patent Rights obtained by a Third Party through the order, decree, or grant of
a competent Governmental Body or court, authorizing such Third Party to develop, make, have made, use, sell, offer to sell or import a Product in any country in the Territory.
1.6 “ Confidential Information ” of a Party, means (i) information relating to the business, operations or products of a Party or any of its Affiliates,
including any know-how, that such Party discloses to the other Party under this Agreement, or otherwise becomes known to the other Party by virtue of this Agreement, and (ii) the terms of this Agreement; provided that Confidential Information shall not include information that:
(a) is or becomes generally available to the public other than as a result of disclosure by the recipient; (b) is already known by or in the possession of the recipient at the time of disclosure by the disclosing Party; (c) is independently developed by recipient without use of or reference to the disclosing Party’s Confidential Information; or (d) is obtained by recipient from a Third Party that has not breached any obligations of confidentiality.
1.7 “ Controlled ” means, with respect to intellectual property rights, that a Party or one of its Affiliates owns or has a license or sublicense to such intellectual property rights and has the ability to provide to, grant a license or sublicense to, or assign its right, title and interest in and to, such intellectual property rights as provided for in this Agreement without violating the terms of any agreement or other arrangement with any Third Party.
1.8 “ Development Plan ” means the development plan provided by Licensee to Penn that provides the activities, and the associated timelines of
when such activities shall be conducted (including in detail the activities that shall be conducted in the calendar year following the submission of such Development Plan to Penn), in order to develop a Product for commercialization. The initial Development Plan is attached hereto as Appendix II .
1.9 “ Diligence Event ” means each of the events that Licensee is expected to accomplish in the development of a Product as provided in Exhibit B
attached hereto. 1.10 “ Essential Milestone ” means the event as provided in Exhibit B .
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL 2
EXECUTION COPY

1.11 “ Field of Use ” means hair growth and hair removal in humans. 1.12 “ First Commercial Sale ” means, on a country-by-country basis, the first Sale of Product in such country to a Third Party by Licensee, or any
of its Affiliates or Sublicensees, in each case, after marketing approvals have been obtained for such country, if applicable. 1.13 “ First Patient First Dose” or “FPFD” means the first dosing of the first patient in a clinical trial. 1.14 “ GAAP ” means United States generally accepted accounting principles applied on a consistent basis. 1.15 “ Governmental Approval ” means, with respect to a Product in a country or region, all approvals, licenses, registrations and authorizations of
the relevant Governmental Body, if applicable, required for the commercialization of such Product in such country. 1.16 “ Governmental Body ” means any: (a) nation, principality, state, commonwealth, province, territory, county, municipality, district or other
jurisdiction of any nature; (b) federal, provincial, state, local, municipal, foreign or other government; (c) governmental or quasi-governmental authority of any nature (including any governmental division, subdivision, department, agency, bureau, branch, office, commission, council, board, instrumentality, officer, official, representative, organization, unit, body or entity and any court or other tribunal); (d) multi-national or supranational organization or body; or (e) individual, entity, or body exercising, or entitled to exercise, any executive, legislative, judicial, administrative, regulatory, police, military or taxing authority or power of any nature.
1.17 “Intellectual Property” means the Penn Patent Rights. 1.18 “ Law ” or “ Laws ” means all applicable laws, statutes, rules, regulations, ordinances and other pronouncements having the binding effect of
law of any Governmental Body. 1.19 “ Net Sales ” means the gross consideration invoiced by Licensee or any of its Affiliates or Sublicensees for Sales of Product (including any cash
amounts plus the fair market value of any other forms of consideration), less the following deductions (to the extent included in and not already deducted from the gross amounts invoiced or otherwise charged to the extent reasonable and customary):
1.19.1 trade discounts, including trade, cash and quantity discounts or rebates, credits or refunds; 1.19.2 allowances or credits actually granted upon claims, returns or rejections of products, including recalls, regardless of the party requesting
such recall; 1.19.3 charges included in the gross sales price for freight, insurance, transportation, postage, handling and any other charges relating to the
sale, transportation, delivery or return of such Product; 1.19.4 amounts deemed to be uncollectible due to non-payment and actually written off in accordance with GAAP (“ bad debts ”) relating to
Sales of Products during the applicable calculation period, net of subsequent recoveries;
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL 3
EXECUTION COPY

1.19.5 rebates and chargebacks or retroactive price reductions made to federal, state or local governments (or their agencies), or any Third Party
payor, administrator or contractor, including managed health organizations; and 1.19.6 payments required by law to be made under Medicaid, Medicare or other government special medical assistance programs (including,
but not limited to, payments made under the new “Medicare Part D Coverage Gap Discount Program” and the “Annual Fee on Branded Pharmaceutical Manufacturers”, specific to Product or allocable to Product).
Even if there is overlap between any of deductions described above, each individual item shall only be deducted once in the overall Net Sales calculation.
1.20 “ Patent Rights ” means any of the following, whether existing now or in the future anywhere in the world: issued patent, including inventor’s certificates, substitutions, extensions, confirmations, reissues, re-examination, renewal or any like governmental grant for protection of inventions, and any pending application for any of the foregoing.
1.21 “ Penn Patent Rights ” means (a) the Patent Rights listed in Exhibit A , (b) Patent Rights on inventions or improvements conceived under the
Penn SRA to the extent added to Exhibit A by amendment to this Agreement pursuant to Section 2.6, (c) any continuations, provisionals, continued prosecution applications, substitutions, extensions and term restorations, registrations, confirmations, reexaminations, renewals or reissues thereof, including divisions, but excluding continuations-in-part except to the extent of claims entirely supported in the specification and entitled to the priority date of the parent application, and (d) any corresponding foreign Patent Rights to the foregoing (a)-(c). Notwithstanding the above, Penn Patent Rights does not include the Carve-Out Patent Rights.
1.22 “Penn SRA ” means the sponsored research agreement that is between Penn and Licensee’s Affiliate (Kythera Biopharmaceuticals, Inc.) and
that is signed on February 27, 2013 and March 13, 2013, as amended. 1.23 “ Person ” means any natural person, corporation, firm, business trust, joint venture, association, organization, company, partnership or other
business entity, or any government or agency or political subdivision thereof. 1.24 “ Product ” means any (a) process, service or method covered by a Valid Claim or whose use or practice would, absent the License, constitute
an infringement, inducement of infringement or contributory infringement of any Valid Claim (“ Method ”), (b) article, composition, apparatus, substance, chemical or any other material covered by a Valid Claim or whose manufacture, import, use offer for sale or sale would, absent the License, constitute an infringement, inducement of infringement or contributory infringement of any Valid Claim; or (c) service, article, composition, apparatus, chemical, substance or any other material made, used or sold by or utilizing or practicing a Method.
1.25 “ Sale ” means any transaction for which consideration is received or expected by Licensee, its Affiliates or Sublicensees for sale, use, lease,
transfer or other disposition of a Product to or for the benefit of a Third Party. For clarity, sale, use, lease, transfer or other disposition of a Product by Licensee or any of its Affiliates or Sublicensees to another of these entities for resale by such entity to a Third Party shall not be deemed a Sale. For further clarity, a Sale excludes any Product supplied at cost: (a) for use in clinical trials; (b) for research or for other non-commercial uses (other than Product sold for profit as a research reagent, which will be deemed a Sale); or (c) as part of a compassionate use program (or similar program for providing Product before it has
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
CONFIDENTIAL 4
EXECUTION COPY

received marketing approval in a country), which, with respect to (b) and (c), in no event shall exceed ten percent (10%) of Sales in any four consecutive Quarters after the First Commercial Sale.
1.26 “ Sublicensee ” means a Person (including any Affiliate) to which a Sublicense is granted pursuant to the terms of Section 2.4. 1.27 “ Sublicense Documents ” means any and all agreements, amendments or written understandings entered into with a Sublicensee (including any
of its Affiliates) that are directly or indirectly related to a Sublicense, Penn Patent Rights or Product. For clarity, a development agreement or distribution agreement for a Product is a Sublicense Document.
1.28 “ Sublicense Income ” means income received by Licensee or its Affiliates in consideration for a Sublicense or other agreement providing the
right to negotiate or obtain a Sublicense. Sublicense Income includes income received from a Sublicensee in the form of license issue fees, milestone payments and the like but specifically excludes (a) royalties on the Sale or distribution of Product, (b) consideration received for purchase of equity in Licensee or its Affiliates up to the fair market value of such equity, and (c) payments made in connection with research and development services in accordance with [***] .
1.29 “ Tax ” means all taxes, duties, fees, premiums, assessments, imposts, levies, rates, withholdings, dues, government contributions and other
charges of any kind whatsoever, whether direct or indirect, together with all interest, penalties, fines, additions to tax or other additional amounts, imposed by any Governmental Body.
1.30 “ Third Party ” means any Person other than Penn, Licensee or any of their respective Affiliates. 1.31 “ Third Party Royalties ” means any consideration Licensee owes to one or more Third Parties pursuant to one or more licenses to issued
Patent Rights entered into by Licensee [***] with respect to the manufacture, use or sale of any Product. 1.32 “ United States ” or “ US ” means the United States of America, its territories and possessions. 1.33 “ USD ” or “ $ ” means the lawful currency of the United States of America. 1.34 “ Valid Claim ” means a claim of (a) an issued and unexpired patent in Penn Patent Rights which claim has not been revoked or held
unenforceable or invalid by a decision of a court of governmental agency of competent jurisdiction from which no further appeal can be taken or has been taken within the time allowed for appeal, and has not been abandoned, disclaimed, denied or admitted to be invalid or unenforceable through reissue or disclaimer; or (b) a pending patent application that is included in Penn Patent Rights which was filed and is being prosecuted in good faith, and has not been abandoned or finally disallowed without the possibility of appeal or re-filing of the application.
1.35 Other Terms . The definition of each of the following terms is set forth in the section of the Agreement indicated below:
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Defined Term Section Advance Payment 5.2.2 Agreement Preamble
CONFIDENTIAL 5
EXECUTION COPY

[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
ARTICLE 2
LICENSES AND OTHER RIGHTS
2.1 Grant of License . Subject to the terms and conditions of this Agreement, Penn hereby grants to Licensee an exclusive, worldwide, royalty-
bearing right and license (with the right to sublicense as provided in, and subject to, the provisions of Section 2.4) under Penn Patent Rights to make, have made, use, sell, offer for sale and import Product in the Field of Use during the Term (the “ License ”).
2.2 Retained Rights . Notwithstanding the License, Penn retains the right under Penn Patent Rights in the Field of Use to conduct educational and
non-commercial research activities, and to authorize non-commercial Third Parties to conduct educational and non-commercial research activities. Penn may request permission from Licensee to conduct clinical activities under the Penn Patent Rights in the Field of Use, provided, however, that Penn may not conduct any such clinical activities in the Field of Use without Licensee’s written consent, which may be withheld in Licensee’s sole discretion. For clarity, Penn retains the right to use, and to authorize others to use, the Penn Patent Rights outside the Field of Use for any purpose.
2.3 U.S. Government Rights . The License is expressly subject to all applicable provisions of any license to the United States Government
executed by Penn and is subject to any overriding obligations to the United States Federal Government under 35 U.S.C. §§200-212, applicable governmental implementing regulations, and the U.S. Government sponsored research agreement or other guidelines, including that products that result from intellectual property funded by the United States Federal Government that are sold in the United States be substantially manufactured in the United States. In the event that Licensee or its Sublicensee believes in good faith that substantial manufacture of such product is not commercially feasible in the United States, and Licensee makes a request to Penn in writing to assist in obtaining a waiver of such requirement from the United States Government, then Penn shall, at the expense of Licensee, use reasonable efforts to assist Licensee or Sublicensee in obtaining such waiver.
2.4 Grant of Sublicense by Licensee .
2.4.1 Penn grants to Licensee the right to grant sublicenses, in whole or in part, under the License (each, a “ Sublicense ”) subject to the terms and conditions of this Agreement and specifically this Section 2.4. The term Sublicense shall include any grant of rights under the License by a Sublicensee to any downstream Third Party, such downstream Third Party shall also be considered a Sublicensee for purposes of this Agreement.
Bankruptcy Action 8.3.6 Carve-Out Patent Rights 5.1.2 Disclosure Recitals Effective Date Preamble Equity Issuance Agreement 4.1 Financial Report 4.7 Historic Patent Cost 5.2.1 Infringement Notice 5.4.1 Inventor(s) Recitals Issue Fee 4.1 License 2.1 Licensee Preamble Maintenance Fee 4.2 Milestone 4.3.1 Milestone Payment 4.3.1 Ongoing Patent Costs 5.2.2 Option Agreement 4.1 Parties Preamble Party Preamble Patent Costs 5.2.1 Patent Counsel 5.1.1 Patent Termination Notice 5.3 Penn Preamble Penn Indemnitees 7.1.1 Penn Sublicense Income 4.5 Progress Reports 3.4.1 Prosecution Request 5.1.2 Royalty 4.4.1 Sublicense 2.4.1 Term 8.1
CONFIDENTIAL 6
EXECUTION COPY

2.4.2 All Sublicenses will be (a) issued in writing, (b) to the extent applicable, include all of the rights of Penn and require the performance of
obligations due to Penn (and, if applicable, the U.S. Government under 35 U.S.C. §§200-212) contained in this Agreement and (c) shall include no less than the following terms and conditions:
(a) Reasonable record keeping, audit and reporting obligations sufficient to enable Licensee and Penn to reasonably verify the
payments due to Licensee and Penn under such Sublicense and to reasonably monitor such Sublicensee’s progress in developing and/or commercializing Product, provided that such obligations shall be no less stringent that those provided in this Agreement for Licensee.
CONFIDENTIAL 7
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(b) Infringement and enforcement provisions that do not conflict with the restrictions and procedural requirements imposed on
Licensee and do not provide greater rights to Sublicensee than as provided in Section 5.4. (c) Confidentiality provisions with respect to Confidential Information of Penn consistent with the restrictions on Licensee in
Section 5.6 of this Agreement. (d) Covenants by Sublicensee that are equivalent to those made by Licensee in Section 6.3. (e) A requirement of indemnification of Penn by Sublicensee that is equivalent to the indemnification of Penn by Licensee under
Section 7.1 of this Agreement. (f) A requirement of obtaining and maintaining insurance by Sublicensee that is equivalent to the insurance requirements of
Licensee under Section 7.2 of this Agreement, including coverage under such insurance of Penn as provided in Section 7.2. (g) Restriction on use of Penn’s names etc. consistent with Section 9.4 of this Agreement. (h) A requirement of antidiscrimination by Sublicensee no less stringent then that provided in Section 9.5 of this Agreement. (i) A requirement that Penn is a third party beneficiary of such Sublicense. Any Sublicense that does not include all of the terms and conditions set forth in this Section 2.4.2 or which is not issued in accordance with the terms and conditions set forth in this Section 2.4, shall be considered null and void with no further notice from Penn.
2.4.3 Within thirty (30) days after of the execution of a Sublicense Document, Licensee shall provide a complete and accurate copy of such Sublicense Document to Penn, in the English Language. Penn’s receipt of a Sublicense Document, however, will constitute neither an approval nor disapproval of the Sublicense Document nor a waiver of any right of Penn or obligation of Licensee under this Agreement.
2.4.4 Licensee shall provide an annual Sublicense Development Report on or before December 1 of each year during the Term (“ SDR Report
”) a form of which is attached hereto as Appendix III .
2.5 No Implied License . Each Party acknowledges that the rights and licenses granted in this Agreement are limited to the scope expressly granted. Accordingly, except for the rights expressly granted under this Agreement, no right, title, or interest of any nature whatsoever is granted whether by implication, estoppel, reliance, or otherwise, by either Party to the other Party. All rights with respect to any know-how, patent or other intellectual property right rights that are not specifically granted herein are reserved to the owner thereof.
2.6 License Amendment . Pursuant to the Penn SRA, the License may be amended to include in the Penn Patent Rights listed in Exhibit A certain
inventions or improvements conceived under the Penn SRA and related to the Penn Patent Rights in the Field of Use consistent with the terms of this Agreement upon execution of an amendment to this Agreement by the Parties.
CONFIDENTIAL 8
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

ARTICLE 3 DILIGENCE
3.1 Development Plan . No later than December 1 of each year during the Term, Licensee shall submit an updated Development Plan, which shall
include amendments and revisions to any long term development activities and detailed activities to be conducted in the following calendar year. 3.2 General Diligence . Licensee shall use Commercially Reasonable Efforts to develop and commercialize at least one Product. 3.3 Diligence Events .
3.3.1 Licensee shall achieve each Diligence Event by the corresponding Achievement Date. Licensee may extend any Achievement Date for a Diligence Event by [***] increments, but not more than [***] ( [***] ) per Diligence Event, by making a [***] dollar ($ [***] ) payment to Penn for each requested extension prior to the expiration of the Achievement Date for such Diligence Event.
3.3.2 Penn acknowledges that the timelines for achievement of certain Diligence Events are based on the assumption that development and
commercialization of Product does not encounter material regulatory or other delays for reasons outside of Licensee’s reasonable control. Where such circumstances exist, in addition to the extension permitted due to additional payments under Section 3.3.1, Penn agrees to negotiate in good faith with Licensee, upon Licensee’s written request and provided such request is made at least [***] prior to the Achievement Date for a Diligence Event, an extension of the Achievement Date for a Diligence Event.
3.3.3 Penn’s sole and exclusive remedy with respect to Licensee’s failure to achieve a Diligence Event by the corresponding Achievement
Date shall be its right to terminate this Agreement.
3.4 Progress Reports .
3.4.1 So long as Licensee continues to develop Products, Licensee on an annual basis, but in no event later than December 1 of each calendar year, shall submit to Penn a progress report (each, a “ Progress Report ”) covering Licensee’s (and any Affiliates’ and Sublicensees’) activities related to the development of all Products and the obtaining of Governmental Approvals necessary for commercialization of Products.
3.4.2 Each Progress Report must include all of the following for each annual period:
(a) Summary of work completed, including against the Development Plan for such period; (b) Summary of work in progress; (c) Current schedule of anticipated events or milestones, including anticipated timeline for achievement of Diligence Events; (d) Market plans for introduction of Product; (e) An updated SDR report listing of any and all Sublicenses granted by Licensee; and
CONFIDENTIAL 9
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
st

(f) The names and addresses of all Sublicensees, and a current and valid phone number and e-mail address for a principal point of
contact at each such Sublicensee who is responsible for administering the Sublicensee.
ARTICLE 4 FINANCIAL PROVISIONS
4.1 Issue Fee . In partial consideration of the License, Licensee will pay to Penn a license issue fee of one hundred thousand US Dollars ($100,000)
in two installments (“ Issue Fee ”). Licensee will pay the first installment of fifty thousand US Dollars ($50,000) to Penn within [***] ( [***] ) days of the date that Licensee achieves the Essential Milestone, and, provided that the Essential Milestone was achieved, shall pay the second installment of fifty thousand US Dollars ($50,000) to Penn on the [***] of the Effective Date. For clarity, if the Essential Milestone was not achieved, and the Agreement is terminated prior to the payment of the second installment of the Issue Fee, such second installment is not payable to Penn on the termination date. The Issue Fee is non-refundable and non-creditable against any other amounts, including any royalties due by Licensee, other than any Historical Patent Costs as outlined under Section 5.2.1 of this Agreement, which Historical Patent costs totaling [***] ($ [***] ) will be automatically credited against the Issue Fee. Payment of each installment may be made in [***] and [***] (at Licensee’s sole discretion and [***] ).
4.2 License Maintenance Fee . As further consideration for the License, Licensee will pay an annual maintenance fee (“ Maintenance Fee ”) as
set forth on Exhibit B attached hereto beginning on the third anniversary of the Effective Date. The Maintenance Fee will not be due and payable on any anniversary of the Effective Date if on that date Licensee is commercially selling Product and paying an earned royalty to Penn on the Sales of that Product. For clarity, the Maintenance Fee is non-refundable, is not an advance against royalties due to Penn or any other amounts due to Penn. Provided that Licensee is [***] , Licensee may reduce such Maintenance Fee payment by [***] percent ( [***] %) of such [***] , provided that in no case shall such Maintenance Fee payment be reduced by more than [***] percent ( [***] %) of the amount otherwise owed to Penn.
4.3 Milestone Payments .
4.3.1 As additional consideration for the License, Licensee will pay Penn the milestone payments (each, a “ Milestone Payment ”) provided in Exhibit B attached hereto upon each Product to achieve the corresponding milestone (each, a “ Milestone ”), whether achieved by Licensee or an Affiliate or Sublicensee. Licensee shall promptly notify Penn in writing of the achievement of any such Milestone and Licensee shall pay Penn in full the corresponding Milestone Payment within [***] ( [***] ) days of such achievement. For clarity, each Milestone Payment is non-refundable, is not an advance against royalties due to Penn or any other amounts due to Penn.
4.3.2 Each time a Milestone is achieved, then any other Milestone Payments with respect to earlier Milestones that have not yet been paid will
be due and payable together with the Milestone Payment for the Milestone that is actually achieved.
CONFIDENTIAL 10
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

4.3.3 For clarity, milestones are due and payable on Products and on products that, upon FDA approval, would become Products.
4.4 Royalties .
4.4.1 As further consideration for the License, Licensee shall pay to Penn a non-refundable, non-creditable (except to the extent described in Section 4.11) royalty in accordance with the table below on all Net Sales of Product in a given calendar year (“ Royalty ”).
4.4.2 If Licensee is obligated to pay aggregate royalties for a Product (i.e., royalties to Penn and to all Third Parties) that are greater than or
equal to [***] percent ( [***] %) of Net Sales, then Licensee may deduct [***] percent ( [***] %) of the portion of Third Party Royalties paid to a Third Party in excess of [***] percent ( [***] %) of Net Sales of a Product for a license under such Patent Rights from any Royalties due under this Agreement, provided that:
(a) On an ongoing basis and prior to reduction of any Royalty for a given calendar quarter, Licensee first provides written evidence
to Penn of Licensee’s obligation to pay a Third Party such Third Party Royalties; and (b) In no event shall Royalties due to Penn in any reporting period be reduced to less than [***] : 1) [***] percent ( [***] %) of
[***] under this Agreement or 2) [***] percent ( [***] %).
4.4.3 Notwithstanding anything in this Section 4.4, in the event that Penn or Licensee receives a request for a Compulsory License anywhere in the world, it shall promptly notify the other Party. If any Third Party obtains a Compulsory License in any country, then Licensor or Licensee (whoever has first notice) shall promptly notify the other Party. Thereafter, as of the date the Third Party obtained such Compulsory License in such country, the royalty rate payable under this Section 4.4 to Penn for Net Sales in such country will be adjusted to [***] .
4.4.4 Licensee must pay Royalties owed to Penn on a calendar quarter basis on or before the following dates:
(a) [***] for any Sales that took place on or before the last day of the calendar quarter ending [***] , of the prior year;
Aggregate Annual Net Sales
Royalty Percent
Portion less than $ [***] [***] %
Portion between $ [***] - $ [***] [***] %
Portion between $ [***] – $ [***] [***] %
Portion between $ [***] - $ [***] [***] %
Portion greater than $ [***] [***] %
CONFIDENTIAL 11
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(b) [***] for any Sales that took place on or before the last day of the calendar quarter ending [***] of such calendar year; (c) [***] for any Sales that took place on or before the last day of the calendar quarter ending [***] of such calendar year; and (d) [***] for any Sales that took place on or before the last day of the calendar quarter ending [***] of such calendar year.
4.5 Penn Sublicense Income . Licensee will pay to Penn a percentage of Sublicense Income as provided in Exhibit B attached hereto (“ Penn Sublicense Income ”). Payment of up to [***] percent ( [***] %) of the Penn Sublicense Income may be [***] (at Licensee’s sole discretion and [***] ). Licensee will make such payment to Penn on or before the following dates:
4.5.1 [***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending [***] , of the prior year; 4.5.2 [***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending [***] of such calendar
year; 4.5.3 [***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending [***] of such calendar
year; and 4.5.4 [***] for any Sublicense Income received by Licensee on or before the last day of the calendar quarter ending [***] of such calendar
year.
4.6 Mode of Payment and Currency . Except as permitted in Section 4.1 and Section 4.5, all cash payments to Penn hereunder shall be made by deposit of USD in the requisite amount to the “The Trustees of the University of Pennsylvania” and will be made by delivery to any one of the following:
Payments under this Agreement shall be made in USD. All Royalties payable shall be calculated first in the currency of the jurisdiction in which payment was made, and if not in the United States, then converted into USD. The exchange rate for such conversion shall be the average of the rate quoted in The Wall Street Journal for the last business day of each month in the calendar quarter for such Royalty payment made.
4.7 Royalty and Penn Sublicense Income Reports . Within [***] ( [***] ) days after the end of each calendar quarter (i.e., March 31, June 30, September 30 and December 31), Licensee shall deliver to Penn a report (“ Financial Report ”) setting out all details necessary to calculate the
Royalty and Penn Sublicense Income due under this Article 4 for such calendar quarter, including:
By ACH/Wire: By Check (direct mail): By Check (lockbox): [***] The Trustees of the The Trustees of the ABA # [***] University of Pennsylvania University of Pennsylvania SWIFT CODE: [***] c/o Penn Center for Innovation c/o Penn Center for Innovation
Attention: Financial PO Box 785546 Account Number: [***] Coordinator Philadelphia, PA 19178-5546 Payment should include the necessary amount to cover any bank charges incurred
3160 Chestnut Street, Suite 200 Philadelphia, PA 19104-6283
CONFIDENTIAL 12
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

4.7.1 Number of each Product Sold by Licensee, its Affiliates and Sublicensees in each country, the corresponding name of each such Product; 4.7.2 Gross sales, Net Sales of each Product made by Licensee, its Affiliates and Sublicensees; 4.7.3 Royalties; 4.7.4 Sublicense Income and the calculation of Penn Sublicense Income including documentation clearly providing rationale for a Bundled
Sublicense (if relevant); 4.7.5 The method and currency exchange rates (if any) used to calculate the Royalties and Penn Sublicense Income; 4.7.6 A specification of all deductions and their dollar value that were taken to calculate Net Sales; 4.7.7 A list of all countries in which Product is being manufactured (on a Product-by-Product basis); and 4.7.8 Date of First Commercial Sale in the United States (this need only be reported in the first royalty report following such First Commercial
Sale in the United States).
4.8 Late Payments . In addition to any other remedies available to Penn, including the right to terminate this Agreement, any failure by Licensee to make a payment within [***] ( [***] ) days after the date when due shall obligate Licensee to pay computed interest, the interest period commencing on the due date and ending on the actual payment date, to Penn at a rate per annum equal to [***] percent ( [***] %) per month, or the highest rate allowed by Law, whichever is lower.
4.9 Accounting . Each Party shall calculate all amounts, and perform other accounting procedures required, under this Agreement and applicable to
it in accordance with GAAP. 4.10 Books and Records . Licensee will keep accurate books and records of all Products developed, manufactured, used or sold and all Sublicenses,
collaboration agreements and joint venture agreements entered into by Licensee that involved Penn Patent Rights. Licensee will preserve these books and records for at least [***] ( [***] ) years from the date of the Financial Report to which they pertain. Upon reasonable notice, key personnel, books and records will be made reasonably available and will be open to examination by representatives or agents of Penn during regular office hours to determine their accuracy and assess Licensee’s compliance with the terms of this Agreement, provided that Licensee shall not have an obligation to provide access more than [***] in any given [***] ( [***] ) month period.
4.11 Audits . In addition to the right of Penn to examine the books and records and interview key personnel as provided in Section 4.10 above, Penn,
at its own cost, through an independent auditor reasonably acceptable to Licensee (and who has executed an appropriate confidentiality agreement reasonably acceptable to Licensee that requires the auditor to keep any information learned by it confidential except as needed to report its audit conclusions to Penn), may inspect and audit the relevant records of Licensee pertaining to the calculation of any Milestones, Royalties and Penn Sublicense Income due to Penn under this Agreement. Licensee shall provide such auditors with access to the records during reasonable business hours. Such access need not be given to any such set of records more often than [***] each year or more than [***] ( [***] ) years after the date of any report to be audited. Penn shall provide Licensee with written notice
CONFIDENTIAL 13
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

of its election to inspect and audit the records related to the Royalty due hereunder not less than [***] ( [***] ) days prior to the proposed date of review of Licensee’s records by Penn’s auditors. Should the auditor find any underpayment of Milestones, Royalties or Penn Sublicense Income by Licensee, Licensee shall (a) promptly pay Penn the amount of such underpayment; (b) shall reimburse Penn for the cost of the audit, if such underpayment equals or exceeds the higher of (i) [***] United States dollars ($ [***] ) or (ii) [***] percent ( [***] %) of Royalties or Penn Sublicense Income paid during the time period audited; and (c) provide such auditors with an audit right exercisable within [***] ( [***] ) months after Licensor receives the audit report. If the auditor finds overpayment by Licensee, then Licensee shall have the right to deduct the overpayment from any future royalties due to Penn by Licensee or, if no such future royalties are payable, then Penn shall refund the overpayment to Licensee within [***] ( [***] ) days after Licensor receives the audit report. Licensee may designate competitively sensitive information which such auditor may see and review but which it may not disclose to Licensor; provided, however, that such designation shall not restrict the auditor’s investigation or conclusions.
4.12 Taxes .
4.12.1 All payments made by Licensee to Penn under the Agreement shall be made free and clear of and without any deduction for or on account of any Taxes on or with respect to such payments.
4.12.2 Penn is a tax exempt entity in the U.S. and qualifies under treaties for tax exempt treatment in many jurisdictions. All payments by
Licensee to Penn are made without deduction for or net of Taxes. To the extent that Taxes are required to be withheld in any jurisdiction on account of any payments hereunder, Licensee shall pay such Taxes on behalf of Penn to the appropriate governmental authority, without reducing amounts payable to Penn hereunder. The Parties will cooperate with one another with respect to all documentation required by any taxing authority or reasonably requested by Licensee or Penn to secure a reduction in the rate of applicable withholding Taxes.
ARTICLE 5
INTELLECTUAL PROPERTY
5.1 Patent Filing Prosecution and Maintenance .
5.1.1 Penn Patent Rights will be held in the name of Penn and obtained with counsel selected by Penn and reasonably acceptable to Licensee (“ Patent Counsel ”). Penn shall control all actions and decisions with respect to the filing, prosecution and maintenance of Penn Patent Rights and will consider any reasonable comments, suggestions, drafts or directions by Licensee with respect to same. Penn will instruct Patent Counsel to copy Licensee on all correspondence related to Penn Patent Rights (including copies of correspondence to and from patent offices, correspondence related to Patent Rights, reporting letters to Penn, each patent application, office action, response to office action, request for terminal disclaimer, and request for reissue or reexamination of any patent or patent application) and to interact with Licensee with respect to the preparation, filing, prosecution and maintenance of Penn Patent Rights. The Parties have the right to take action to preserve rights or minimize cost. Penn has the right to act where Licensee has not commented. Penn will use reasonable efforts to not allow any Penn Patent Rights for which Licensee is licensed and is underwriting the costs to lapse or become abandoned without Licensee’s written authorization under this Agreement. Penn shall have no [***] as set forth in this Agreement. Within
[***] ( [***] ) days of the Effective Date, Penn will cause Patent Counsel to [***] . Licensee will bear and shall reimburse Penn for all Patent Costs associated with such [***] . Licensee may assist in preparation of [***] .
5.1.2 Licensee has the right to request a country filing via a written request to Penn within a reasonable time but not less than [***] ( [***] ) days prior to the deadline set by the patent office in the territory in which filing is to take place (“ Prosecution Request ”). The absence of a given Prosecution Request by such deadline will be considered an election not to secure the Patent Rights associated with the specific phase of patent prosecution in such territory, and such patent application(s) and patent(s) (“ Carve-Out Patent Rights ”) will not be part of Penn Patent Rights and therefore not subject to this Agreement, including the License, and Licensee will have no further rights or license to them.
5.2 Patent Costs .
5.2.1 Within [***] ( [***] ) days of the date that Licensee achieves the Essential Milestone, Penn will invoice Licensee for all out-of pocket costs for the filing, prosecution and maintenance of Penn Patent Rights, including all accrued attorney fees, expenses, official and filing fees (“ Patent Costs ”), incurred prior to the Effective Date and not already paid or payable pursuant to the Option Agreement or reimbursed to Penn by any party for past filing, prosecution and maintenance (“ Historic Patent Costs ” ). Such Historical Patent Costs total [***] ($ [***] ).
5.2.2 Licensee will bear all Patent Costs in the Field of Use incurred during the Term (“ Ongoing Patent Costs ”). Penn will invoice Licensee
for all Ongoing Patent Costs promptly after receipt and docketing in Penn’s invoicing system of such invoices for such Ongoing Patent Costs from Patent Counsel. If Penn enters into a license agreement with a Third Party for any of the Penn Patent Rights outside the Field of Use, then Penn will [***] . For the sake of clarity, Licensee will not [***] ; whether [***] .
5.2.3 At any time, at Penn’s request, Licensee shall [***] for regional validations (including, European validation and associated costs), new
original patent applications (including significant new matter continuations-in-part) reexaminations and national phase entry filings requested by Licensee [***] (“ [***] ” ). If the Patent Counsel’s [***] , the portion of such [***] shall be [***] .
CONFIDENTIAL 14
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

5.2.4 Notwithstanding [***] , Licensee shall bear all Patent Costs incurred during the Term and shall pay such amounts within [***] ( [***] )
days of receipt of invoice for such patent actions. For clarity, the term “Patent Costs” means and includes Historic Patent Costs and Ongoing Patent Costs.
5.3 Termination of Rights in, and Obligations with respect to, Certain Penn Patent Rights . Licensee may terminate its rights in, and
obligations with respect to any or all of Penn Patent Rights by providing written notice to Penn of Licensee’s decision to terminate such Penn Patent
CONFIDENTIAL 15
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

Rights (“ Patent Termination Notice ”). Termination of Licensee’s rights in and obligation with respect to such Patent Right will be effective [***] ( [***] ) months after receipt of such Patent Termination Notice by Penn. Penn will immediately curtail Patent Costs chargeable to Licensee under this Agreement after the receipt of the Patent Termination Notice is received. Penn may continue prosecution and maintenance of such Patent Rights at its sole discretion and expense, and such Patent Rights will then be Carve-Out Patent Rights and therefore not subject to this Agreement, including the License, and Licensee will have no further rights or license to them.
5.4 Infringement . 5.4.1 If either Party believes that an infringement by a Third Party with respect to any Penn Patent Right is occurring or may potentially occur,
the knowledgeable Party will provide the other Party with (a) written notice of such infringement or potential infringement and (b) evidence of such infringement or potential infringement (the “ Infringement Notice ”). During the period in which, and in the jurisdiction where, Licensee has exclusive rights under this Agreement, neither Penn or Licensee will notify such a Third Party (including the infringer) of infringement or put such Third Party on notice of the existence of Penn Patent Rights without first obtaining the written consent of the other Party (such consent not to be unreasonably withheld, delayed or conditioned). Both Penn and Licensee will use their diligent efforts to cooperate with each other to terminate such infringement without litigation.
5.4.2 If infringing activity of potential commercial significance has not been abated within [***] ( [***] ) days following the date the
Infringement Notice for such activity was provided, then during the period in which, and in the jurisdiction where, Licensee has exclusive rights under this Agreement, Licensee may institute suit for patent infringement against the infringer after providing Penn (a) a written estimate of the expenses that would be reasonably incurred in connection with such action, including an estimate from an outside law firm, acceptable to Penn, regarding the legal costs associated with such suit and (b) financial records reasonably sufficient to reasonably demonstrate that Licensee has the financial wherewithal to pay such expenses as they fall due through the conclusion of such suit by means of judgment or other final non-appealable decision. Penn may voluntarily join such suit at Licensee’s reasonable expense, but may not thereafter commence suit against the infringer for the acts of infringement that are the subject of Licensee’s suit or any judgment rendered in such suit. Licensee may not join Penn in a suit initiated by Licensee without Penn’s prior written consent, such consent not to be unreasonably withheld, delayed or conditioned. If in a suit initiated by Licensee, Penn is involuntarily joined other than by Licensee, then Licensee will pay any documented costs incurred by Penn arising out of such suit, including any documented legal fees of counsel that Penn selects and retains to represent it in the suit. Licensee shall be free to enter into a settlement, consent judgment or other voluntary disposition, provided that any settlement, consent judgment or other voluntary disposition that (i) limits the scope, validity or enforcement of Penn Patents or (ii) admits fault or wrongdoing on the part of Licensee or Penn must be approved in advance by Penn in
CONFIDENTIAL 16
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

5.4.3 writing, such approval not to be unreasonably withheld, delayed or conditioned. Licensee’s request for such approval shall include
complete copies of final settlement documents, a detailed summary of such settlement, and any other information material to such settlement. Penn shall provide Licensee notice of its approval or denial within [***] ( [***] ) days of any request for such approval by Licensee, provided that (x) in the event Penn wishes to deny such approval, such notice shall include a detailed written description of Penn’s reasonable objections to the proposed settlement, consent judgment, or other voluntary disposition and (y) Penn shall be deemed to have approved of such proposed settlement, consent judgment, or other voluntary disposition in the event it fails to provide such notice within such [***] ( [***] ) day period in accordance herewith.
5.4.4 If, within [***] ( [***] ) days following the date the Infringement Notice was provided, infringing activity of potential commercial
significance has not been abated and if Licensee has not brought suit against the infringer, then Penn may institute suit for patent infringement against the infringer. If Penn institutes such suit, then Licensee may not join such suit without the prior written consent of Penn and may not thereafter commence suit against the infringer for the acts of infringement that are the subject of Penn’s suit or any judgment rendered in such suit.
5.4.5 Notwithstanding Sections 5.4.2 and 5.4.3, in the event that any Penn Patent Rights are infringed by a Third Party if any of the infringed
Penn Patent Rights are also licensed by Penn to a Third Party, prior to any enforcement action being taken by either Party regarding such infringement, the Parties shall discuss, and will mutually agree, in writing, as to how to handle such infringement by such Third Party.
5.4.6 Any recovery or settlement received in connection with any suit will first be shared by Penn and Licensee equally to cover any litigation
costs each incurred and next shall be paid to Penn or Licensee to cover any litigation costs it incurred in excess of the litigation costs of the other. Any remaining recoveries shall be allocated as follows:
For any portion of the recovery or settlement related to the infringement of the Penn Patent Rights, other than for amounts attributable and paid as enhanced damages for willful infringement: (a) for any suit that is initiated by Licensee and in which Penn was not a party in the litigation, Penn shall receive [***] percent
( [***] %) of the recovery, and the Licensee shall receive the remainder; and (b) for any suit that is initiated by the Licensee or Penn and that the other Party joins voluntarily (but only to the extent such
voluntary joining is allowed under this Agreement or expressly by the other Party in a separate agreement) or involuntarily, the non-initiating party’s percentage of such recovery shall be [***] percent ( [***] %) .
For any portion of the recovery or settlement related to the infringement of the Penn Patent Rights paid as enhanced damages for willful infringement: (c) for any suit that is initiated by Licensee or Penn and the other Party voluntarily but only to the extent such voluntary joining is
allowed under this Agreement or expressly by the other Party in a separate agreement) or involuntarily, the non-initiating party’s percentage of such recovery shall be [***] percent ( [***] %) and Licensee shall receive the remainder; and
CONFIDENTIAL 17
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(d) for any suit that is initiated by Licensee and in which Penn was not a party in the litigation, Penn shall receive [***] percent
( [***] %) and the Licensee shall receive the remainder. For any portion of the recovery or settlement received in connection with any suit that is initiated by Penn and in which Licensee was not a party in the litigation, any recovery in excess of litigation costs will [***] .
5.4.7 Each Party will reasonably cooperate and assist with the other in litigation proceedings instituted hereunder but at the expense of the Party who initiated the suit (unless such suit is being jointly prosecuted by the Parties). For clarity, such requirement does not require a Party to join a suit unless otherwise specifically required under this Agreement. If Penn is [***] related to the Penn Patent Rights or Products licensed to Licensee hereunder, [***] with respect to same.
5.5 Patent Marking . Licensee shall place in a conspicuous location on any Product (or its packaging where appropriate and practicable) made or
sold under this Agreement a patent notice in accordance with the Laws concerning the marking of patented articles where such Product is made or sold, as applicable.
5.6 Confidentiality .
5.6.1 Each Party agrees that, for the Term and for [***] ( [***] ) years thereafter, such Party shall (a) use the same degree of care to maintain the secrecy of the Confidential Information of the other Party that it uses to maintain the secrecy of its Confidential Information of like kind, (b) use the Confidential Information only to accomplish the purpose of this Agreement or for audit or management purposes and (c) ensure that any employees, customers, and distributors are bound to it by similar obligations of confidence and to make sure such disclosure only as required to accomplish the purposes of this Agreement. Licensee may disclose for the purposes set forth above Penn’s Confidential Information on a need to know basis to Licensee’s: Affiliates; potential or actual collaborators, partners, and Sublicensees (including potential co-marketing and co-promotion contractors); potential or actual investment bankers, acquirers, lenders or investors; employees; consultants; and agents, provided that (i) prior to disclosure each such recipient, is bound by written obligations of confidentiality and non-use at least as restrictive as those as set forth in this Agreement (but, in any event, not less than reasonable measures to protect the secrecy of, and avoid the unauthorized disclosure or use of, Penn Confidential Information). and (ii) such recipient may not further disclose such Confidential Information to any third party. Licensee shall be primarily liable to Penn for any breach of this Agreement or such obligations of non-use and non-disclosure by Licensee’s Affiliates, Sublicensees or any such other recipient(s) of Penn’s Confidential Information.
5.6.2 A Party may disclose the Confidential Information of the other Party to the extent required by Law or court order; provided, however,
that the recipient promptly provides to the disclosing Party prior written notice of such disclosure and provides reasonable assistance in obtaining an order or other remedy protecting the Confidential Information from public disclosure.
5.6.3 If Licensee is required by applicable law, rule or regulation to make a securities filing relating to the execution of this Agreement with
the appropriate governmental authorities
CONFIDENTIAL 18
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(including the U.S. Securities and Exchange Commission, and any securities exchange on which securities of Licensee are listed), then Licensee will prepare a draft of such securities filing for review and comment by Penn. If such securities filing includes the disclosure of this Agreement and its terms, Licensee will include a confidential treatment request and a proposed redacted version reasonably in advance of the deadline for such securities filing, and Penn agrees to promptly (and in any event, no less than [***] ( [***] ) days (or such shorter time to meet any filing deadline where it was not practical to provide the other Party with [***] ( [***] )-days’ notice after receipt of such confidential treatment request and proposed redactions) give its input in a reasonable manner in order to allow Licensee to file its request within the timelines proscribed by the regulations of applicable governmental authority or securities exchanged as represented by the redacted version reviewed by Penn.
ARTICLE 6 REPRESENTATIONS, WARRANTIES AND COVENANTS
6.1 Mutual Representations and Warranties . Each Party represents and warrants to the other Party that, as of the Effective Date:
6.1.1 such Party is duly organized and validly existing under the Laws of the jurisdiction of its incorporation or organization; 6.1.2 such Party has taken all action necessary to authorize the execution and delivery of this Agreement and the performance of its
obligations under this Agreement; 6.1.3 this Agreement is a legal and valid obligation of such Party, binding upon such Party and enforceable against such Party in accordance
with the terms of this Agreement, except as enforcement may be limited by applicable bankruptcy, fraudulent conveyance, insolvency, reorganization, moratorium and other laws relating to or affecting creditors’ rights generally and by general equitable principles; and
6.1.4 such Party has all right, power and authority to enter into this Agreement, to perform its obligations under this Agreement, and, with
respect to Penn, to the knowledge of the current staff of the Penn Center for Innovation, as of the Effective Date, Penn is an owner of the Penn Patent Rights.
6.2 Disclaimer of Representations and Warranties .
6.2.1 Other than the representations and warranties provided in Section 6.1 above, PENN MAKES NO REPRESENTATIONS AND WARRANTIES, WHETHER EXPRESS OR IMPLIED, AND EXPLICI TLY DISCLAIMS ANY REPRESENTATION AND WARRANTY, INCLUDING WITH RESPECT TO ANY ACCURACY, C OMPLETENESS, MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, COMMERCIAL UTILITY, NON- INFRINGEMENT OR TITLE FOR THE INTELLECTUAL PROPERTY, PATENT RIGHTS, LICENSE AND A NY PRODUCT .
6.2.2 Furthermore, nothing in this Agreement will be construed as:
(a) A representation or warranty by Penn as to the validity or scope of any Penn Patent Right;
CONFIDENTIAL 19
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(b) A representation or warranty that anything made, used, sold or otherwise disposed of under the License is or will be free from
infringement of patents, copyrights, trademarks or any other forms of intellectual property rights or tangible property rights of Third Parties;
(c) Obligating Penn to bring or prosecute actions or suits against Third Parties for patent, copyright or trademark infringement; (d) Conferring by implication, estoppel or otherwise any license or rights under any Patent Rights of Penn other than Penn Patent
Rights as defined herein, regardless of whether such Patent Rights are dominant or subordinate to Penn Patent Rights; and (e) Obligating Penn to furnish any know-how.
6.3 Covenants of Licensee .
6.3.1 Licensee and its Affiliates will not, directly or indirectly (including where such is done by a Third Party on behalf of Licensee or its Affiliates, at the urging of Licensee or its Affiliates or with the assistance of the Licensee or its Affiliates) challenge the validity, scope, or enforceability of or otherwise oppose any Penn Patent Right, provided that if any Penn Patent Right is asserted against Licensee or its Affiliate for activities authorized under this Agreement, then such Licensee or its Affiliates is entitled to all and any defenses available to it including challenging the validity or enforceability of such Patent Right. Licensee will comply with all Laws that apply to its activities or obligations under this Agreement. For example, Licensee will comply with applicable United States export laws and regulations. The transfer of certain technical data and commodities may require a license from the applicable agency of the United States government and/or written assurances by Licensee that Licensee will not export data or commodities to certain foreign countries without prior approval of the agency.
6.3.2 Licensee will not grant a security interest in the License or this Agreement.
6.4 Kythera Biopharmaceuticals, Inc. Guarantee
6.4.1 Kythera Biopharmaceuticals, Inc. (“ KBI ”) hereby irrevocably guarantees to Penn due and punctual performance by Licensee of [***] and undertakes, upon the occurrence and continuance of [***] , that KBI will duly and properly [***] . The liability of KBI as guarantor hereunder shall not be released or diminished by (i) any [***] , unless agreed to by all Parties to the Agreement, (ii) by any [***] , (iii) any [***] , (iv) the [***] or (v) any other act, event or omission, which [***] .
CONFIDENTIAL 20
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

ARTICLE 7
INDEMNIFICATION; INSURANCE AND LIMITATION OF LIABIL ITY
7.1 Indemnification by Licensee .
7.1.1 Licensee shall defend, indemnify and hold Penn and its respective trustees, officers, faculty, students, employees, contractors and agents (the “ Penn Indemnitees ”) harmless from and against any and all liability, damage, loss, cost or expense (including reasonable attorneys’ fees), including, without limitation, bodily injury, risk of bodily injury, death and property damage to the extent arising out of Third Party claims or suits related to (a) this Agreement or any Sublicense, including (i) the development, testing, use, manufacture, promotion, sale or other disposition of any Product (including any product liability claim), (ii) any enforcement action or suit brought by Licensee against a Third Party for infringement of Penn Patent Rights, (iii) any claim by a Third Party that the practice of Penn Patent Rights or the design, composition, manufacture, use, sale or other disposition of any Product infringes or violates any patent, copyright, trade secret, trademark or other intellectual property right of such Third Party, (iv) any breach of this Agreement or Laws by Licensee, its Affiliates or Sublicensees and (b) Licensee’s negligence, omissions or willful misconduct, provided that Licensee’s obligations pursuant to this Section 7.1 shall not apply to the extent such claims or suits result from the negligence, gross negligence or willful misconduct of any of Penn Indemnitees as determined by a court of law.
7.1.2 As a condition to a Penn Indemnitee’s right to receive indemnification under this Section 7.1, Penn shall: (a) promptly notify Licensee as
soon as it becomes aware of a claim or suit for which indemnification may be sought pursuant hereto; (b) reasonably cooperate, and cause the individual Penn Indemnitees to reasonably cooperate, with Licensee in the defense, settlement or compromise of such claim or suit; and (c) permit the Licensee to control the defense, settlement or compromise of such claim or suit, including the right to select defense counsel. In no event, however, may Licensee compromise or settle any claim or suit in a manner which (a) admits fault or negligence on the part of Penn or any other Penn Indemnitee; (b) commits Penn or any other Penn Indemnitee to take, or forbear to take, any action, without the prior written consent of Penn (which consent shall not be unreasonably withheld, delayed or conditioned), or (c) grant any rights under the Penn Patent Rights except for Sublicenses permitted under Article 2. Penn shall reasonably cooperate with Licensee and its counsel in the course of the defense of any such suit, claim or demand, such cooperation to include without limitation using reasonable efforts to provide or make available documents, information and witnesses.
7.1.3 Notwithstanding Section 7.1.2 above, in the event that Penn believes in good faith that a bonafide conflict exists between Licensee and
Penn or any other Penn Indemnitee with respect to a claim or suit subject to indemnification hereunder, then Penn or any other Penn Indemnitee shall have the right to defend against any such claim or suit itself, including by selecting its own counsel, with any [***] . Licensee will [***] .
7.2 Insurance .
7.2.1 Licensee, at its sole cost and expense, must insure its activities in connection with the exercise of its rights under this Agreement and obtain, and keep in force and maintain
CONFIDENTIAL 21
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

7.2.2 Commercial Form General Liability Insurance (contractual liability included) with limits as follows:
Prior to the commencement of clinical trials, if applicable, involving Product:
Prior to the First Commercial Sale of a Product:
Penn may review periodically the adequacy of the minimum amounts of insurance for each coverage required by this Section 7.2.1, and has the right to require Licensee to adjust the limits in Penn’s reasonable discretion to keep in line with then-applicable industry practices.
7.2.3 If the above insurance is written on a claims-made form, it shall continue for [***] ( [***] ) years following termination or expiration of this Agreement. The insurance shall have a retroactive date of placement prior to or coinciding with the Effective Date of this Agreement.
7.2.4 Licensee expressly understands, however, that the coverages and limits in Section 7.2.1 do not in any way limit Licensee’s liability or
indemnification obligations. Licensee’s insurance will:
(a) Be issued by an insurance carrier with an A.M. Best rating of “A” or better; (b) State that Penn is endorsed as an additional insured with respect to the coverages in Section 7.2.1; and (c) Include a provision that the coverages will be primary and will not participate with nor will be excess over any valid and
collective insurance or program of self insurance carried or maintained by Penn. Licensee will provide thirty (30) day advance written notice to Penn of any modification in coverage amount of Licensee’s insurance.
7.2.5 Licensee must furnish to Penn with (a) valid certificate of insurance evidencing compliance with all requirements of this Agreement and (b) additional insured endorsements for Licensee’s applicable policies naming “The Trustees of the University of Pennsylvania” as an additional insured. Licensee must furnish both documents within thirty (30) days of the Effective Date, once per year thereafter and at any time there is a modification in the coverage amount of such insurance.
7.3 LIMITATION OF LIABILITY . IN NO EVENT SHALL EITHER PARTY OR ANY OF ITS AFFILIATES BE LIABLE TO THE OTHER
PARTY OR ANY OF ITS AFFILIATES FOR SPECIAL, INDIRECT, INCIDENTAL, CONSEQUENTIAL OR PUNITIVE DAMAGES, INCLUDING LOSS OF PROFITS, WHETHER IN CONTRACT, WARRANTY, TORT, NEGLIGENCE, STRICT LIABILITY OR OTHERWISE ARISING OUT OF OR RELATING
TO THIS AGREEMENT, THE TRANSACTIONS CONTEMPLATED HEREIN OR ANY BREACH HEREOF. NOTWITHSTANDING THE FOREGOING, NOTHING IN THIS AGREEMENT SHALL LIMIT LICENSEE’S INDEMNIFICATION OBLIGATIONS UNDER SECTION 7.1 ABOVE.
ARTICLE 8
TERM AND TERMINATION
8.1 Term . The term of this Agreement (the “ Term ”) shall commence on the Effective Date and, unless terminated sooner, shall continue in full force and effect until the expiration or abandonment of the last Valid Claim of the last Penn Patent Right. Notwithstanding the foregoing, this Agreement shall automatically terminate in its entirety upon the failure of Licensee to achieve the Essential Milestone as set forth in Exhibit B . For the avoidance of doubt, the expiration of the Term due to Licensee’s failure to achieve the Essential Milestone will not require written or verbal notice that such milestone has not been achieved as required or that the Agreement has expired due to such failure.
8.2 Termination of the Agreement for Convenience . At any time during the Term, Licensee may, at its convenience, terminate this Agreement
upon providing at least sixty (60) days prior written notice to Penn of such intention to terminate, provided that Licensee ceases using the License or making, using, or selling Products.
8.3 Termination For Cause .
8.3.1 If Licensee fails to fulfill its obligations under Section 3.2 (i.e. use Commercially Reasonable Efforts to develop and commercialize a Product), Penn may provide written notice to Licensee of such failure. If Licensee fails to address such failure to the reasonable satisfaction of Penn within three (3) months of receiving such written notice, Penn may terminate this Agreement upon written notice to
(a) Each occurrence $ [***] ; (b) General aggregate $ [***]
(c) Clinical trials liability insurance $ [***]
(d) Products liability insurance $ [***]
CONFIDENTIAL 22
EXECUTION COPY
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

Licensee. 8.3.2 If, after [***] as set forth in the Essential Milestone, Licensee fails at any time during the Term to maintain such [***] , and Licensee
does not (within [***] ( [***] ) months of the termination of the [***] ) obtain [***] and provide Penn with written documentation thereof, then Penn may terminate this Agreement effective upon thirty (30) days written notice to Licensee.
8.3.3 In the event Licensee fails to achieve any Diligence Event by the corresponding Achievement Date (subject to any extensions under
Section 3.3), Penn has the right and option to terminate this Agreement, upon written notice, with immediate effect. 8.3.4 If Licensee materially breaches any of its material obligations under this Agreement, Penn may give to Licensee a written notice
specifying the nature of the default, requiring it to cure such breach, and stating its intention to terminate this Agreement. If such breach is not cured within thirty (30) days of such notice, such termination shall become effective upon a notice of termination by Penn thereafter. For clarity, a breach of a material obligation includes:
CONFIDENTIAL EXECUTION COPY
23
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(a) failure to deliver to Penn any payment at the time or times that such payment is due to Penn under this Agreement; (b) failure to provide reports as set forth in Sections 3.4.1 and 4.7; (c) failure to possess and maintain insurance as set forth in Section 7.2; (d) grant of a sublicense under the Penn Patent Rights that is not in accordance with the terms of this Agreement; (e) failure to terminate a Sublicense where Sublicensee is in breach of the obligations on it pursuant to (i) Sections 2.4.2(a), 2.4.2
(c), 2.4.2(f) and 2.4.2(g) and fails to cure such breach within thirty (30) days of being notified of such breach and (ii) Section 2.4.2(d).
8.3.5 In addition to all other remedies available to it, Penn may terminate this Agreement, upon written notice, with immediate effect, upon a
breach of Section 6.3, Covenants of Licensee. 8.3.6 Penn may terminate this Agreement, upon written notice, with immediate effect if, at any time, Licensee is unable to pay its debts,
including any debts related to exclusive Sublicensees, when they come due, or files in any court or agency pursuant to any statute or regulation of any state, country or jurisdiction, a petition in bankruptcy or insolvency or for reorganization or for an arrangement or for the appointment of a receiver or trustee of Licensee or of its assets, or if Licensee proposes a written agreement of composition or extension of its debts, or if Licensee is served with an involuntary petition against it, filed in any insolvency proceeding, and such petition is not dismissed within ninety (90) days after the filing thereof, or if Licensee proposes or is a party to any dissolution or liquidation, or if Licensee makes an assignment for the benefit of its creditors of all or substantially all its assets (in each case, “Bankruptcy Action ”).
8.4 Effects of Termination .
8.4.1 Notwithstanding the termination of this Agreement, the following provisions shall survive: Sections 4.8-4.12, inclusive, 5.6 and 8.4 and Articles 6, 7 and 9.
8.4.2 Termination of this Agreement shall not relieve the Parties of any obligation or liability that, at the time of termination, has already
accrued hereunder, or which is attributable to a period prior to the effective date of such termination. Termination of this Agreement shall not preclude either Party from pursuing all rights and remedies it may have hereunder or at Law or in equity with respect to any breach of this Agreement nor prejudice either Party’s right to obtain performance of any obligation.
8.4.3 If this Agreement is terminated for any reason, all outstanding Sublicenses (including all Sublicense Documents for each Sublicense) not
in default will be assigned by Licensee to Penn, and such assignment will be accepted by Penn. Each assigned Sublicense will remain in full force and effect with Penn as the licensor or sublicensor instead of Licensee, but the duties and obligations of Penn under the assigned Sublicenses will not be greater than the duties of Penn under this Agreement, and the rights of Penn under the assigned Sublicenses will not be less than the rights of Penn under this Agreement, including all financial consideration and other rights of Penn. Penn may, at its sole
CONFIDENTIAL EXECUTION COPY
24
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

discretion, amend such outstanding Sublicenses to contain the terms and conditions found in this Agreement.
ARTICLE 9
ADDITIONAL PROVISIONS
9.1 Relationship of the Parties . Nothing in this Agreement is intended or shall be deemed, for financial, tax, legal or other purposes, to constitute a partnership, agency, joint venture or employer-employee relationship between the Parties. The Parties are independent contractors and at no time will either Party make commitments or incur any charges or expenses for or on behalf of the other Party.
9.2 Expenses . Except as otherwise provided in this Agreement, each Party shall pay its own expenses and costs incidental to the preparation of this
Agreement and to the consummation of the transactions contemplated hereby. 9.3 Third Party Beneficiary . The Parties agree that each Sublicensee is a third party beneficiary of this Agreement with respect to Section 8.4.3. 9.4 Use of Names . Licensee, its Affiliates and Sublicensees may not use the name, logo, seal, trademark, or service mark (including any adaptation
of them) of Penn or any Penn school, organization, employee, student or representative, without the prior written consent of Penn. Notwithstanding the foregoing, Licensee may use the name of Penn in a non-misleading and factual manner solely in (a) executive summaries, business plans, offering memoranda and other similar documents used by Licensee for the purpose of raising financing for the operations of Licensee as related to Product, or entering into commercial contracts with Third Parties, but in such case only to the extent necessary to inform a reader that the Penn Patent Rights has been licensed by Licensee from Penn, and to inform a reader of the identity and published credentials of Inventors of the Intellectual Property, and (b) any securities reports required to be filed with the Securities and Exchange Commission.
9.5 No Discrimination . To the extent required by applicable law, neither Penn nor Licensee will discriminate against any employee or applicant
for employment because of race, color, sex, sexual or affectional preference, age, religion, national or ethnic origin, handicap, or veteran status. 9.6 Successors and Assignment .
9.6.1 The terms and provisions hereof shall inure to the benefit of, and be binding upon, the Parties and their respective successors and permitted assigns.
9.6.2 Licensee may not assign or transfer this Agreement or any of Licensee’s rights or obligations created hereunder, by operation of law or
otherwise, without the prior written consent of Penn, provided that Penn shall not unreasonably withhold, condition or delay its consent, and provided further that Licensee, without Penn’s consent, may assign this Agreement in its entirety to: (i) an Affiliate of Licensee, so long as such Affiliate’s performance and payment obligations under the License are guaranteed by KBI in accordance with the terms set forth in Section 6.4.1 of this Agreement, or (ii) a Third Party in connection with a sale of all or substantially all of the business or assets of both Licensee and KBI, provided that: (a) the each such assignee agrees in writing to be legally bound by this Agreement and to deliver to Penn an updated Development Plan within ninety (90) days after the closing of the proposed transaction; (b) Licensee
CONFIDENTIAL EXECUTION COPY
25
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

provides Penn with a copy of assignee’s undertaking; and (c) Licensee is not in breach under the terms and conditions of, and has paid all amounts owed to Penn under, this Agreement as of the date of such assignment. Any permitted assignment will not relieve Licensee of responsibility for performance of any obligation of Licensee that has accrued at the time of the assignment.
9.6.3 Any assignment not in accordance with this Section 9.5 shall be void.
9.7 Further Actions . Each Party agrees to execute, acknowledge and deliver such further instruments and to do all such other acts as may be
necessary or appropriate in order to carry out the purposes and intent of this Agreement. 9.8 Entire Agreement of the Parties; Amendments . This Agreement, the Exhibits and Appendices or Schedules hereto, and any Equity Issuance
Agreement constitute and contain the entire understanding and agreement of the Parties respecting the subject matter hereof and cancel and supersede any and all prior negotiations, correspondence, understandings and agreements between the Parties, whether oral or written, regarding such subject matter, including the Confidential Disclosure Agreement by and between Penn and Licensee’s Affiliate (Kythera Biopharmaceuticals, Inc.) dated June 14, 2012 (the “ Prior CDA ”) and the Option Agreement, and any confidential information disclosed under the Prior CDA will be governed by the terms of this Agreement. No waiver, modification or amendment of any provision of this Agreement shall be valid or effective unless made in a writing referencing this Agreement and signed by a duly authorized officer of each Party.
9.9 Governing Law . This Agreement shall be governed by and interpreted in accordance with the laws of the Commonwealth of Pennsylvania,
excluding application of any conflict of laws principles that would require application of the law of a jurisdiction outside of the Commonwealth of Pennsylvania.
9.10 Dispute Resolution . If a dispute arises between the Parties concerning this Agreement, then the Parties will confer, as soon as practicable, in an
attempt to resolve the dispute. If the Parties are unable to resolve such dispute amicably, then the Parties will submit to the exclusive jurisdiction of, and venue in, the state and Federal courts located in the Eastern District of Pennsylvania.
9.11 Notices and Deliveries . Any notice, request, approval or consent required or permitted to be given under this Agreement shall be in writing and
directed to a Party at its address or facsimile number shown below or such other address or facsimile number as such Party shall have last given by notice to the other Party. A notice will be deemed received: if delivered personally, on the date of delivery; if mailed, five (5) days after deposit in the United States mail; if sent via courier, one (1) business day after deposit with the courier service; or if sent via facsimile or email, upon receipt of confirmation of transmission provided that a confirming copy of such notice is sent by certified mail, postage prepaid, return receipt requested.
For Penn with a copy to :
Penn Center for Innovation University of Pennsylvania 3160 Chestnut Street, Suite 200 Philadelphia, PA 19104-6283 Attention: Executive Director
University of Pennsylvania Office of General Counsel 133 South 36th Street, Suite 300 Philadelphia, PA 19104-3246 Attention: General Counsel
CONFIDENTIAL EXECUTION COPY
26
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

9.12 Waiver . A waiver by either Party of any of the terms and conditions of this Agreement in any instance shall not be deemed or construed to be a
waiver of such term or condition for the future, or of any other term or condition hereof. All rights, remedies, undertakings, obligations and agreements contained in this Agreement shall be cumulative and none of them shall be in limitation of any other remedy, right, undertaking, obligation or agreement of either Party.
9.13 Severability . When possible, each provision of this Agreement will be interpreted in such manner as to be effective and valid under law, but if
any provision of this Agreement is held to be prohibited by or invalid under law, such provision will be ineffective only to the extent of such prohibition or invalidity, without invalidating the remainder of this Agreement. The Parties shall make a good faith effort to replace the invalid or unenforceable provision with a valid one which in its economic effect is most consistent with the invalid or unenforceable provision.
9.14 Interpretation . The words “include,” “includes” and “including” shall be deemed to be followed by the phrase “without limitation.” All
references herein to Articles, Sections, Schedules and Exhibits shall be deemed references to Articles and Sections of, Schedules and Exhibits to, this Agreement unless the context shall otherwise require. Except as otherwise expressly provided herein, all terms of an accounting or financial nature shall be construed in accordance with GAAP, as in effect from time to time. Unless the context otherwise requires, countries shall include territories. References to any specific Law or article, section or other division thereof, shall be deemed to include the then-current amendments or any replacement Law thereto.
9.15 Counterparts . This Agreement may be executed in counterparts, each of which will be deemed an original, and all of which together will be
deemed to be one and the same instrument. A facsimile or a portable document format (PDF) copy of this Agreement, including the signature pages, will be deemed an original.
[SIGNATURE PAGE FOLLOWS]
For Licensee: with a copy to:
Kythera Holdings, Ltd.
Latham & Watkins LLP Clarendon House, 2 Church Street
140 Scott Drive Hamilton HM 11, Bermuda
Menlo Park, CA 94025 Email: [email protected]
Fax: (650) 463-2600 Attention: Vice President for Strategy
Attention: Alan Mendelson, Esq. and and Corporate Development
Judith Hasko, Esq.
For KBI: with a copy to:
Kythera Biopharmaceuticals, Inc.
Latham & Watkins LLP 30930 Russell Ranch Road, 3 Flr.
140 Scott Drive Westlake Village, CA 91362
Menlo Park, CA 94025 Attention: Vice President for Strategy and Corporate Development
Fax: (650) 463-2600
Attention: Alan Mendelson, Esq. and Judith Hasko, Esq.
CONFIDENTIAL EXECUTION COPY
27
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
rd

IN WITNESS WHEREOF, duly authorized representatives of the Parties have executed this Agreement as of the Effective Date.
THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA
KYTHERA HOLDINGS, LTD. By: /s/ John S. Swartley
By: /s/ Keith Leonard
Name: John S. Swartley, PhD
Name: Keith Leonard Title: Associate Vice Provost for Research and Executive Director,
Penn Center for Innovation
Title: Director
Agreed to be bound by the terms and conditions of this Agreement pursuant to Section 6.4:
KYTHERA BIOPHARMACEUTICALS, INC.
By: /s/ Keith Leonard
Name: Keith Leonard
Title: President and CEO
CONFIDENTIAL EXECUTION COPY
28
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Exhibit A
Penn Patent Rights
Exhibit B
Certain Financial Terms
Docket [***] [***] Serial No Patent No App Type File Date Status Country Issue Date
[***] [***] [***] [***] [***] [***] [***] Docket [***] [***]
Serial No Patent No App Type File Date Status Country Issue Date [***] [***] [***] [***] [***] [***] [***]
CONFIDENTIAL EXECUTION COPY
29
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
DEVELOPMENT & COMMERCIALIZATION
Diligence Events
Diligence Event Achievement Date
[***]
[***]
[***] [***]
Essential Milestone • Licensee shall secure a [***] within [***] ( [***] ) months after the Effective Date of this Agreement in a manner that
will permit [***] (the “ Essential Milestone ”). Licensee shall provide written documentation to Penn of such Essential Milestone achievement promptly upon securing such [***] , and shall provide Penn with prompt written documentation within [***] ( [***] ) business days of any change in status of [***] .
CONFIDENTIAL EXECUTION COPY
30
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CERTAIN FINANCIAL
TERMS
• License Maintenance Fee .
Anniversary: 3 , [***] [***]and thereafter
License Maintenance Fee:
$ [***] per year $ [***] per year
• Milestone Payments . Payable to Penn on each Product
Milestone Milestone Payment
[***] $ [***]
[***] $ [***]
[***] $ [***]
[***] $ [***]
[***] $ [***]
Total Milestone Payments Due per Product $4,125,000
• Penn Sublicense Income . Licensee shall pay Penn a percent of any Sublicense Income in accordance with the table
below. For clarity, a Bundled Sublicense must meet all conditions as set forth in Section 1.3 of this Agreement to qualify as such.
Anniversary of Effective Date Percent of Sublicense Income Percent of Sublicense Income if a Bundled Sublicense
Prior to [***] [***] % [***] %
Prior to [***] [***] % [***] %
After [***] [***] % [***] %
CONFIDENTIAL EXECUTION COPY
31
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
rd

Appendix II
Development Plan
[***]
CONFIDENTIAL EXECUTION COPY
32
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Appendix II I Sublicense Development Report Unive rsity of Pennsylvania Center for Technology Transfer Annual Sublicense Reporting and Tracking * L icensee Report Date Prepared by: Period Covered Certif ied by: License Inception Date Listing of Sublicensees and Specific Te rms (all on-going as well as those new and terminated in the past year) Sublicensee Name Agreement Inception Date Amendment Date , if any Date complete executed copy of agreement and amendment, if any, sent to Penn (note terminations, if any) Technologies licensed (1) Field(s) of Use Jur isdiction / Terr itory Current Agreement Year History to Date Information Fees, Royalties and Other Payments (2) Due to Penn (3) Fees, Royalties & Other Payments (2) Due to Penn Sublicensee Name Da te Due Amount Due Date Received Amount Received Percentage Amount Date Paid Amount Due Amount Received Percentage Amount* All references to Sublicensee shall inc lude all permitted sub-sublicensees and all references to sublicense shall include all permitted sub-sublicensee. Sublicensee Compliance Section Sublicensee Name Agreement Inception Date Technologies licensed Indicate across from each sublicensee whether the sublicensee complies w ith all mater ial te rms of the sublicense agreement AND the L icense Agreement Notes: (1) include Penn Docket Number and relevant issued patent or patent applica tion number (2) All amounts in US dollars; specify conversion ra te. (3) Provide detailed support for any apportionment percentages as well as detailed information on deductions to arr ive at amounts due to Penn CONFIDENTIAL EXECUTION COPY 33 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidentia l treatment has been requested with respec t to the omitted portions.


Exhibit 10.22
DISTRIBUTION SERVICES AGREEMENT
This Distribution Services Agreement (“Agreement”) dated as of January 16, 2015 (“Effective Date”), is made by and between KYTHERA
Biopharmaceuticals, Inc. (“Company”) and Besse Medical, a division of ASD Specialty Healthcare, Inc. (“Distributor”).
RECITALS
A. The Company is a manufacturer and supplier of biological therapeutic products including the products listed on Exhibit A (“Products” ) . B. The Distributor is a national distributor of vaccines, biologicals and injectables and other specialty pharmaceutical products. C. The Distributor desires to purchase Product(s) from the Company and become an authorized distributor of the Product. D. The Company and the Distributor mutually desire to enter into an exclusive supply and distribution agreement in accordance with and pursuant to
the terms and conditions set forth in this Agreement.
AGREEMENT
NOW THEREFORE , in consideration of the mutual promises and obligations contained in this Agreement, the parties agree as follows:
1. Appointment as Authorized Distributor . The Company will sell the Product(s) to the Distributor on the terms and conditions set forth in this Agreement. The Company hereby appoints the Distributor as an authorized distributor of record for the Product during the term of this Agreement, and with respect to any Products in Distributor’s possession after the termination or expiration of this Agreement for a period of [***] days thereafter. The Company will comply with all applicable federal and state laws, requiring it to publicly identify all of its authorized distributors.
2. Exclusivity . During the term of this Agreement:
(a) Sole Distributor and Service Provider . Distributor will be the sole and exclusive reseller of the Product, and the sole provider of distribution and warehousing functions with respect to the Product, in the United States, its territories and possessions (the “Territory”); and
(b) No Other Distributors or Service Providers . The Company will not sell the Product to any person or entity, other than Distributor, in the
Territory, and will not authorize or permit any person or entity, other than Distributor, to distribute or resell the Product (other than to patients or payors), or to provide distribution and warehousing functions with respect to the Product, in the Territory.
(c) Direct Sales . Notwithstanding the above, the Company reserves the right to sell the Product directly to healthcare service providers (“HCPs”),
in which case the Company will utilize a third party logistics provider (“3PL”) to ship Product directly to HCPs; provided that, if the Company elects to sell the Product directly to HCPs through a 3PL, the Company will [***] under this Agreement to [***] , including any [***] .

3. Product Pricing and Payment Terms to Distributor .
(a) Pricing . The Company will establish a List Price (“List Price”). The List Price is defined as the price at which the Company will sell the Product to the Distributor, and the Distributor will not [***] , provided that nothing herein shall prevent the Distributor from [***] . The List Price is set forth in Exhibit A. The List Price is subject to change [***].
(b) Customers . “Customer” shall be defined as a HCP who is duly licensed, who has fulfilled the Company’s training requirement, and who has
established a customer account with the Distributor. Unless otherwise specified, all Customers are able to purchase the Product at the List Price. (c) Terms of Payment . The Company will invoice the Distributor for Product purchased. The Distributor will pay all of the Company invoices for
undisputed orders for Products in accordance with the due dates specified; provided that (i) terms for the purchased Product are net [***] days, from the date the Distributor receives the Product plus [***] additional float days for payment by electronic funds transfer, and (ii) terms for purchased Product will always be [***] days longer than the standard terms that are offered by the Company to Customers under Section 11 of this Agreement. The standard terms the Company will offer to Customers will be net [***] days.
The Company may ask the Distributor to offer longer than standard payment terms to select customers. In such cases the Distributor will continue to pay the Company’s invoice under the standard terms of [***] days, which represents the standard Customer terms of [***] days plus Distributor terms of [***] days. The Distributor will [***] on a monthly basis consistent with the methodology described in Exhibit E. If the [***] in a month, then the Distributor may [***] for that month. If the [***] , the parties agree to [***] . The Distributor will not be deemed in default or lose any cash discount by reason of any delay in receipt or non-receipt by the Company of funds transferred by electronic funds transfer if the transfer was timely initiated by the Distributor, unless the delay or non-receipt is the result of the negligence or willful misconduct of the Distributor . Without the Distributor’s prior written consent, the Company will not have the right to debit the Distributor’s account electronically.
4. Shipping, Delivery, Title and Risk of Loss . All shipments from the Company under this Agreement are FOB Distributor’s destination at its distribution centers located at:
2
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
345 International Boulevard 5360 Capital Court
Suite 400 Suite 102
Brooks, KY 40109 Reno, NV 89502

The Company will prepay freight and insurance on all shipments to the Distributor. Risk of loss for, and title to, the Products ordered by the Distributor will pass to the Distributor upon receipt of the Product by the Distributor.
5. The Company’s Obligations .
(a) Electronic Orders . The Company will accept Distributor orders electronically in a manner reasonably acceptable to the Company’s 3PL. (b) Minimum Orders . During the first 12 months after the Product launch the minimum order shall be [***] case ( [***] packs). The Company
will provide weekly replenishment shipments from the 3PL to the Distributor and, on an “as-needed” basis, the Company will provide [***] . Thereafter, if weekly sales volumes are [***] , the minimum order quantity shall [***] . As actual and forecasted sales change, the Company and the Distributor will work together to set mutually agreeable minimum purchase order amounts which may differ from those set forth above.
(c) Delivery Times . The Company will make commercially reasonable efforts to ship all Distributor orders completely and to have Product from
these orders shipped to the Distributor within a mutually agreeable schedule of up to [***] of order placement. Notification of Product identified as backordered or unavailable must be provided to Distributor, in writing, within at least [***] . This notification will include the reason for the delay and the expected availability date. The Company will honor the Distributor’s order for [***] days from the date of the order in the event that Product ordered is “backordered” and will not require the Distributor to re-order the Product.
(d) Shipping Labels . The 3PL will clearly label all cartons and pallets of Products with the following shipping information:
Distributor Purchase Order # Ship-From Address Ship-To Address Product Description Item Number Case Quantity NB: Distributor Item Number will be included on the packing list inside the carton Lot Number(s)
(e) Invoicing . The Company will not invoice orders until Product has been shipped to the Distributor. (f) Product Dating . The Company will ship Product to the Distributor on a first expired, first out (“FEFO”) basis; [***] . FEFO is a method of
inventory management by which Product with the shortest expiration dating is shipped first. (g) Price Protection . If at any time the Company reduces the List Price of the Product, the Company will provide an appropriate credit to the
Distributor in an amount equal to the difference between (i) the List Price and (ii) the List Price in effect before the price reduction, for each unit of Product on hand at or in transit to the Distributor’s distribution center on the date of the price
3
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

reduction. The Company may increase the List Price from time to time, in which case the Distributor agrees not to materially change the order pattern for the Product ahead of the price change for the purpose of taking advantage of the price differential.
(h) Accounts Receivable Statement . Upon the Distributor’s request, the Company will provide the Distributor with a monthly accounts receivable
statement of all open transactions. (i) Date of Price . The Company will accept purchase orders at the List Prices in effect on the day the order is transmitted. The Company will
notify the Distributor on the day the order is placed of any Product adjustments or held purchase order delays. (j) Credits . The Company will pay the Distributor all compensation due (including without limitation, payments, credits, product allocations,
and/or bill-back program amounts) within [***] days of the date the amount due is determined. Exceptions must be resolved with Distributor’s Accounts Payable Department. All billing disputes shall be resolved within the terms of this Agreement.
(k) Product Recalls . If the Company conducts a recall, market withdrawal or field correction of any Products, the Company will abide by all
applicable laws and regulations. (l) Supporting Information . The Company will provide any documentation or instructions to the Distributor reasonably necessary for full
compliance with federal, state and local laws with respect to the handling, storage and distribution of the Product. The Company must maintain federal, state and local registrations necessary for the lawful handling of Product and immediately notify the Distributor of any denial, revocation or suspension of any registration or any changes in the Product that the Distributor is authorized to distribute. The Company must report any administrative, civil or criminal action currently pending or arising after the Effective Date of this Agreement by local, state or federal authorities against the Company, its officers or employees, regarding alleged violations of the Controlled Substances Act of 1970, as amended, or other comparable legislation, and provide the Distributor with complete information concerning the disposition of the action .
(m) Product Quality . All Product must meet all applicable requirements and regulations of the Federal Food, Drug and Cosmetic Act and/or the
Federal Food and Drug Administration. No generic Product will be sold to the Distributor.
6. Distributor ’s Services .
(a) Services . The Distributor will provide the services listed on Exhibit C (“Services”). The Services will be of good quality and performed consistent with industry standards. Where appropriate, the parties may agree on metrics, which will be specified in Exhibit C , used to measure the performance of services. Performance of Services will be formally reviewed quarterly by the Company and the Distributor in an operating review.
(b) Launch Date and Readiness Date . The expected launch date (“Launch Date”) is [***] . The Company is not liable for any fees other than the
program launch fee in Exhibit B prior to the Launch Date. The Launch Date is dependent upon approval by the FDA of the Company’s New Drug Application (“NDA”). If the Launch Date is delayed for any reason, the Company agrees to pay the Distributor stand-by fees consisting of the Customer Service Management Fee and the System Access and Support Fee as outlined in Exhibit B.
4
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

The Company agrees to pay the Distributor a one-time program readiness fee in the amount of $ [***] payable in [***] $[***] installments on [***] and [***] . The program readiness date shall be [***] , or as mutually agreed to by both parties. On that date the Distributor will be ready to deliver the Services in Exhibit C, subject to Product availability from the Company.
(c) Fees . The fees for the Services will be calculated on a monthly basis, and will be equal to the applicable service fees as stated on Exhibit B , as adjusted in accordance with the terms of this Agreement.
The parties agree that the fees for Services earned under this Agreement represent fair market value for bona fide services that the Distributor provides to and on behalf of the Company.
Except as specifically provided in this Agreement, the Distributor shall be responsible for all of its own expenses for the performance of the Services. The Distributor may not incur any expenses chargeable to the Company, except as specifically authorized in advance in writing by the Company or as specifically provided for herein.
(d) Credit Card Processing . The Distributor agrees to accept credit card payments from customers for Product purchased from the Distributor. The
Distributor will pass through, on a direct cost basis [***] , all processing charges incurred by the Distributor from credit card sales to the Company on a monthly basis. These fees will be invoiced by the Distributor and paid by the Company in the same manner as other fees for Services under this Agreement. The Distributor will accept payments utilizing American Express, Visa Card, Master Card, and any other credit cards that the Distributor deems acceptable.
(e) Terms of Payment . All fees for Services will be invoiced monthly. All fees for Services are due in full [***] days upon receipt of invoice. The
Company will notify Distributor of any disputed charges in writing within [***] days of receipt of the invoice covering these charges. In the absence of any notice of dispute, all invoices will be deemed to be correct and due in full per the payment terms above. A late fee of [***] % per month (or any portion thereof) will be charged as of the due date on all amounts not paid after [***] days of receipt of the invoice, except any amount disputed by the Company in good faith.
(f) [***] . If the Distributor can reasonably demonstrate to the Company that the [***] as a result of any [***] , then Distributor may [***] (“ [***]
”). However, Distributor agrees that it shall not have the right to [***] unless all of the following conditions are met:
(i) [***]; and (ii) [***]. The Distributor will notify the Company of any [***] at least [***] ( [***] ) days prior to [***] .
5 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

The Company anticipates increasing its List Price from time to time. In such cases the Distributor will [***] . Any [***] shall be [***] from List Price increases. The parties also agree that there shall be no [***] in the [***] period following [***] or the product launch date, whichever date shall occur last.
(g) CPI Adjustment . In addition, the fees for the Services in Exhibit B that are expressed in U.S. dollars will be reviewed and adjusted annually
to reflect increases in the Consumer Price Index for All Urban Consumers, U.S. City Average, for all items, 1982-84=100 (the “CPI-U”), published by the United States Department of Labor on its website at http://www.bls.gov/cpi. The adjustment will be effective [***] after each one year anniversary of the Effective Date. By way of example only, if the Effective Date is January 1, 2011, the adjustment would be effective on [***] . All fees for Services on Exhibit A that are expressed in U.S. dollars will be multiplied by the percent increase in the CPI-U during each twelve month period. The CPI Adjustment shall be limited to [***] % per year.
An example of the calculation of the increase is set forth on Exhibit D . If publication of the CPI-U ceases the parties will mutually agree upon the use of an appropriate substitute index published by the Bureau or any successor agency.
(h) Determination . All Cost Adjustments and CPI-U adjustments will be determined under generally accepted accounting principles (“GAAP”) and
cost allocation methods applied on a consistent basis. If the Company objects to any Cost Adjustment or CPI-U adjustment and the parties are unable in good faith to resolve the objection to the reasonable satisfaction of both parties, then either party may terminate this Agreement upon 180 days’ prior written notice to the other party.
(i) Retention of Services Fees . The Distributor will not pass on any portion of the service fees paid under this Agreement to any of its Customers.
7. Other Distributor Obligations .
(a) Orders . The Distributor will ship Product to its customers, freight pre-paid, on all orders in accordance with the terms set forth in Exhibit C .
Expedited shipments made at the request of a customer will be at the customer’s expense. Additional charges for orders outside of standard delivery, emergency and/or overnight deliveries will be the responsibility of the customer and will be added to the Customer’s invoice.
(b) Storage Condition/Product Handling . The Distributor will ensure that the Product supplied by the Company is handled, maintained, stored,
transported, delivered and/or otherwise managed and distributed in accordance with the handling and storage requirements applicable to each Product as contained in the package insert for the Product approved by the Food and Drug Administration and any other specific instructions provided by the Company in writing to the Distributor, and in accordance with federal and state laws, regulations, rules and practices applicable to the distribution of pharmaceuticals in the jurisdictions in which the Distributor operates. The Distributor will keep and maintain records thereof (including without limitation complete records as to the amount and type of Product sold, lot number(s), customer address, corresponding pre-
6 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
sale storage and tracking information, product codes and invoice dates). These records will be made available for inspection by the Company during normal business hours, on reasonable advance written notice, and in a manner that does not unreasonably interfere with the normal business operations of Distributor.
(c) Licenses . The Distributor must maintain all necessary licenses, permits, certificates, and other requisite documents, including all necessary
governmental approvals and registrations, and pay all applicable customs duties and taxes required for and/or in connection with its marketing, sale and distribution of Product under this Agreement.
(d) Recalls . If requested to do so in writing by the Company, the Distributor will cooperate fully with the Company in recalling or returning any
Product that the Company identifies to the Distributor as being the subject of a recall or withdrawal. The recall or withdrawal will be at the Company’s expense and the Company will credit the Distributor for the full purchase price of all Product recalled or withdrawn, and reimburse the Distributor for all actual costs and expenses incurred as a result of the recall or withdrawal in accordance with the Healthcare Distribution Management Association’s (“HDMA”) published guidelines ; except that the Distributor will be responsible for the costs of the recall or withdrawal to the extent that the recall or withdrawal is attributable to the negligence or intentional misconduct of the Distributor.
The Distributor will maintain complete and accurate records of all Products sold to facilitate compliance with this Section. The Distributor will use commercially reasonable efforts to comply with the Company’s written instructions concerning communications with the public and the procedures to be observed during a recall or return of Products. I f the Company needs to distribute ‘Dear HCP’ notifications, the Distributor will provide a list of Customer’s affected by the recall in an expedited manner within one business day of the Company’s request.
(e) Adverse Events, Medical Device Reports, or Product Complaints . If a Customer notifies the Distributor of an adverse event, medical device
report, or Product complaint concerning a Product, the Distributor will [***] , provide the Customer with the Company’s Drug Safety Hotline information [***] , advise the Customer to call to the Company’s Drug Safety Hotline directly.
(f) Discounts . To the extent required by applicable law and regulations, including, but not limited to, 42 U.S.C. 1320a-7b(b) and 42 C.F.R.
1001.952(h), the Distributor will advise and inform each of its customers to fully report, as required by law or contract, any discounts, rebates, or reductions in prices on Product and provide the discount information supplied by the Distributor to the Department of Health and Human Services or a state agency upon request, consistent with the requirements of 42 U.S.C. 1320a-7b(b) and 42 C.F.R. 1001.952(h).

(g) Theft and Diversion. The Distributor will comply with all applicable laws, regulations and prudent business practices to prevent theft and/or diversion of Product supplied by the Company. The Distributor will conduct regular inventory control procedures to ensure all inventory of Product is fully accounted for. Should the Distributor become aware of any theft or diversion of the Product, suspected or confirmed, the Distributor will immediately notify the Company. In such an event, the Company and the Distributor shall work together in good faith to determine the root cause of the security failure, and to make any necessary adjustments to their business practices to prevent such occurrence in the future.
7 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

8. Damaged Products . The Distributor will visually inspect each shipment of Product for external damage or loss in transit and will notify the
Company in writing ( or by e-mail or fax) of any shortage or other non-conformity in any order delivery within [***] ( [***] ) business days of the date of delivery. With respect to non-conformities of Product that by their nature are not discoverable upon a reasonable visual inspection (“Hidden Defects”), (i) the Distributor will notify the Company promptly upon learning of or discovering a Hidden Defect; and (ii) the parties will meet promptly to discuss the situation and agree on a reasonable and appropriate resolution under the circumstances, subject to the terms of the Continuing Guaranty (defined below).
9. Shipment Errors . In the event of an incomplete shipment, a shortage in shipment, the misdirection of any delivery, or any overshipment, the
Company (or its designated agent) will immediately contact the Distributor purchasing department and will comply with any reasonable directions provided by the Distributor. The Company will be responsible for any related freight or accessorial charges caused by the error.
10. Returns . The Distributor shall have the right to request replacement Product from the Company under the following conditions:
(a) The Distributor has the right to exchange damaged Product as set forth in paragraph 8 of this Agreement. (b) The Distributor will administer the Company’s customer return policy. Product returned to the Distributor under the terms of that policy shall be
inspected by the Distributor and restocked if deemed salable. If Product is non-salable, the Company will replace that Product at no charge. (c) The Distributor will rely on the Company’s sales forecast to determine inventory levels. In a Distributor [***] , the Company will [***] . The Company will develop a written customer return policy to define the conditions under which the Distributor will accept returns from the Customer. The customer return policy shall be signed by the Company and made effective 60 days prior to the Launch Date. The Company shall have the right to update the customer return policy from time to time with at least 30 days prior written notice to the Distributor. In conjunction with the written customer return policy, the Company and Distributor will jointly establish a protocol for accepting Customer returns which will include the following elements: receiving the Customer’s request to return Product, notifying the Company’s sales representative, authorizing the request and documenting reason codes, facilitating and receiving the return shipment, inspecting the returned Product and determining salability of the Product, confirming the Product is not counterfeit, confirming return is being received from a legitimate customer, restocking salable Product and/or destroying non-salable Product. Any suspected counterfeit Product should be immediately segregated from other Product and not moved to any other location without notice and approval from the Company. The Distributor shall notify the Company immediately in the event suspected counterfeit Product is received. In addition, at least 60 days prior to the Product launch date, the Company and Distributor shall establish a mutually acceptable process for replacing damaged or unsalable Product in the possession of the Distributor as a result of the Distributor’s administration of the Company’s Customer return policy.
8 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

11. Special Pricing Programs .
(a) Customer Pricing. The Company may institute [***] . Such [***] . In such cases the Company will inform the Distributor of the [***] , and will submit all updates and changes immediately to the Distributor’s Contracts Department for prompt application. In no event will the Company make any representation to the Participating Customer regarding Distributor’s delivery performance that is inconsistent with the terms and conditions of Exhibit C .
(b) Contract and Chargeback Administration Policy . All customer contract and chargeback matters not set forth in this Agreement are governed by
the Distributor’s Contract and Chargeback Administration Policy in effect as of August 7, 2014, or as may be amended from time to time upon at least 30 days prior written notice to Company.
12. Confidential Information .
(a) Definition . As used in this Agreement, “Confidential Information” means any confidential or proprietary information that is disclosed by one party (“Disclosing Party”) to the other party (“Recipient”), whether in writing or other tangible form, orally or otherwise. Confidential Information includes (I) information about processes, systems, strategic plans, business plans, operating data, financial information and other information and (II) any analysis, compilation, study or other material prepared by the Recipient (regardless of the form in which it is maintained) that contains or otherwise reflects any information disclosed or made available by the Disclosing Party to the Recipient. Confidential Information does not include information that:
i. at the time of disclosure to the Recipient, is generally available to the public; ii. after disclosure to the Recipient, becomes generally available to the public other than as a result of a breach of this Agreement by the
Recipient (including any of its affiliates); iii. the Recipient can establish was already in its possession at the time the information was received from the Disclosing Party if its source was
not known by the Recipient to be bound to an obligation of confidentiality with respect to this information; iv. the Recipient receives from a third party if its source was not known by the Recipient to be bound to an obligation of confidentiality with
respect to the information; or v. the Recipient can establish was developed independently by the Recipient without use, directly or indirectly, of any Confidential
Information.
(b) Limitations on Disclosure and Use . Confidential Information must be kept strictly confidential and may not be disclosed or used by the Recipient except that the Recipient may use the Confidential Information of the Disclosing Party in the performance of its obligations or exercise of its rights under this Agreement or as specifically authorized in advance in writing by the Disclosing Party. The Recipient may not take any action that causes the Confidential Information to lose its confidential and proprietary nature or fail to take any reasonable action necessary to prevent any Confidential Information from losing its confidential and proprietary nature. The Recipient will limit access to the Confidential Information to its employees, officers, directors or
9 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

other authorized representatives (or those of its affiliates) and to those who (a) need to know the Confidential Information in connection with this Agreement and (b) are obligated to the Recipient to maintain the Confidential Information under terms and conditions at least as stringent as those under this Agreement. The Recipient will inform all these persons of the confidential and proprietary nature of the Confidential Information and will take all reasonable steps to ensure they do not breach their confidentiality obligations, including taking any steps the Recipient would take to protect its own similarly confidential information. The Recipient will be responsible for any breach of confidentiality obligations by these persons.
(c) Equitable Relief . Each party acknowledges that, when it is the Recipient, money damages would not be a sufficient remedy for the Disclosing
Party in the event of any breach of this Agreement and that the Disclosing Party is entitled to seek specific performance and injunctive or other equitable relief as a remedy for any breach. The Recipient further waives any requirement for the posting of any bond in connection with any remedy. This remedy will be in addition to any other available remedies at law or in equity.
(d) Disclosures Required by Law . If the Recipient is required by law to disclose any Confidential Information, the Recipient will give the
Disclosing Party prompt notice and will use all reasonable means to obtain confidential treatment for any Confidential Information that it is required to disclose before making any disclosure. If the Recipient cannot assure confidential treatment and it has exhausted all reasonable efforts to do so, the Recipient may disclose the Confidential Information if it is required by law to disclose the information it discloses. Notwithstanding the foregoing, the Disclosing Party may request the Recipient to take additional steps to seek confidential treatment before the Recipient discloses the Confidential Information even though the Recipient has otherwise exhausted all reasonable efforts to do so. In that event, the Recipient will undertake additional steps at the Disclosing Party’s expense.
(e) Effect of Termination . Promptly after the termination or expiration of this Agreement, each party will return to the other any Confidential
Information of the other party and provide a written verification of the return or, at that disclosing party’s request, destroy the Confidential Information and provide written notification of the destroyed Confidential Information. Notwithstanding the foregoing, each party may retain a copy of the Confidential Information in its confidential legal files, and the obligation to destroy or return will not apply to Confidential Information that is stored on back-up tapes and similar media that are not readily accessible to the Recipient. Each party’s obligation to maintain the confidentiality of the Confidential Information will survive for a period of [***] ( [***] ) years following termination of this Agreement.
13. Term and Termination .
(a) Term . This Agreement is effective as of the Effective Date and will continue for three years (“Initial Term”), unless sooner terminated under the terms of this Agreement. Thereafter, this Agreement will automatically renew for subsequent terms of one additional year (“Renewal Term”), unless either party provides the other party with written notice of its intent not to renew this Agreement at least 180 days before expiration of the current term.
(b) Other Termination, Including Termination for Breach . Either party may terminate this Agreement for cause, upon 30 days’ written notice of a
material default to the other party with a reason for termination, and failure of that party to cure the default within the 30 day period after receiving written notice of the material default.
10 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(c) Termination for Specific Events . Either party may immediately terminate this Agreement upon written notice to the other party upon the other
party’s: (i) filing an application for or consenting to appointment of a trustee, receiver or custodian of its assets; (ii) having an order for relief entered in Bankruptcy Code proceedings; (iii) making a general assignment for the benefit of creditors; (iv) having a trustee, receiver, or custodian of its assets appointed unless proceedings and the person appointed are dismissed within 30 days; or (v) insolvency within the meaning of Uniform Commercial Code Section 1-201 or failing generally to pay its debts as they become due within the meaning of Bankruptcy Code Section 303(h)(1), as amended (each, a “Bankruptcy Event”). Each party will provide immediate notice to the other party upon a Bankruptcy Event.
(d) Effect of Termination . The rights and obligations of the parties contained in Sections 12(e), 13(d), (e) and (f), and 16 through 20 of this
Agreement, any payment obligations, and any other provision if its context shows that the parties intend it to survive, will survive expiration or termination of this Agreement and, except as expressly provided, expiration or termination will not affect any obligations arising prior to the expiration or termination date.
(e) Inventory upon Termination . If this Agreement is terminated (i) by the Distributor following a breach by the Company or (ii) by the Company
without cause, at the Distributor’s option the Company will promptly repurchase from the Distributor all Product in its possession or control, at the List Price paid by the Distributor, [***] .
(f) Data upon Termination . In the event that this Agreement is terminated or expires, the Distributor will provide the Company any data reasonably
requested by the Company that has been previously exchanged between the parties and is readily available to Distributor. Such data will be provided for a period of [***] unless otherwise agreed to by an officer of the Company. Any such data shared by the Distributor with the Company in the course of its business relationship will remain property of [***] .
14. Use of Marks . For the purposes of this Agreement, the Company hereby grants to the Distributor a non-exclusive, non-transferable, revocable
license to use the Company’s trademarks, trade names and service marks used and/or owned by the Company with respect to the Product (collectively, the “Marks”). The Marks will be used by the Distributor solely in connection with its marketing, packaging, sale, distribution and/or delivery of the Product purchased from and supplied by the Company, and the Services being performed by the Distributor, under this Agreement. The Company shall have the right to review and approve the Distributor’s use of Marks. The ownership of and goodwill in all Marks will remain the sole and exclusive property of the Company and inure exclusively to the Company’s sole benefit, both during the Initial Term and Renewal Term(s), if any, of this Agreement and thereafter. Nothing in this Agreement will give the Distributor any right, title or interest in or to the Marks other than the right to use the same in the manner contemplated by this Agreement and only for so long as this Agreement is in force. To the extent the Distributor may accumulate or otherwise benefit from any goodwill deriving from or in connection with the Distributor’s use of any of the Marks under this Agreement, the goodwill will upon termination or expiration of this Agreement be assigned and/or otherwise transferred to the Company without any further action by either party.
15. Representations and Warranties .
(a) By Distributor . The Distributor represents and warrants to the Company that:
11 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

i. The Distributor has and will maintain, in full force and effect, all licenses and permits required under applicable federal, state or local law
for the Distributor to sell and distribute Products under this Agreement; and ii. The Distributor will comply with all applicable federal, state and local laws governing the purchase, handling, sale and distribution of
Products purchased under this Agreement. iii. All distribution services will be performed in accordance with the specifications set forth in Exhibit C , as may be amended from time to
time.
(b) By the Company . The Company represents and warrants to the Distributor that:
i. The Company owns or holds the duly approved NDA or Abbreviated New Drug Application or Biologics License Application, as these terms are defined in the Federal Food, Drug and Cosmetic Act, Title 21, United States Code, as amended, and the rules and regulations promulgated thereunder, for the Products, or is otherwise considered the “manufacturer” of the Products within the meaning of any applicable federal, state or local law relating to pedigrees;
ii. The Company has and will maintain, in full force and effect, all licenses and permits required under applicable federal, state or local law for
the Company to sell and distribute Products under this Agreement; iii. The Company will comply with all applicable federal, state and local laws governing the purchase, handling, sale, distribution, and price
reporting of Products purchased under this Agreement; and iv. The Company has good and marketable title to the Products sold to Distributor under this Agreement, and all Products will be sold to the
Distributor free and clear of all liens, claims, security interests or other encumbrances.
(c) Compliance with IAT Rules . To enable the Distributor to comply with the International ACH Transactions rules and the U.S. Department of Treasury’s Office of Foreign Asset Controls’ requirements, the Company represents and warrants to the Distributor that, except as provided below, with respect to electronic payments that the Distributor may remit to a financial institution for credit to an account designated by the Company, the entire payment amount is being sent to a bank within the territorial jurisdiction of the United States and is not subject to standing instructions to be transferred or forwarded to a foreign bank account or financial institution:
____________________________________________________________________ The Company agrees to provide written notice to the Distributor if in the future the Company decides, as part of a single payment transaction, to transfer or forward the entire amount of any electronic payment that the Distributor makes to the Company to a bank account or financial institution located outside the territorial jurisdiction of the United States.
(d) OFCCP/EO Compliance . The Distributor is an equal opportunity employer and federal contractor or subcontractor. Accordingly, the parties will comply with all applicable requirements of 41 CFR 60-1.4(a), 41 CFR 60-300.5(a) and 41 CFR 60-741.5(a), which are incorporated by reference and prohibit discrimination against qualified individuals based on his or her status as protected veterans or individuals with disabilities, and prohibit discrimination against all individuals based on their race, color, religion, sex, or national origin. These regulations require that covered prime contractors and subcontractors take affirmative action to employ and advance in employment individuals without regard to race, color, religion, sex,
12 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
national origin, protected veteran status or disability. The parties will also comply with all applicable requirements of Executive Order 13496 (29 CFR Part 471, Appendix A to Subpart A), relating to the notice of employee rights under federal labor laws.
16. Indemnification .
(a) By the Company . The Company will defend, indemnify, and hold harmless the Distributor and its affiliates, directors, officers, employees and representatives from any claims, demands, costs, expenses (including reasonable attorneys’ fees) and liabilities or losses (“Claims”) that may be asserted against the Distributor or that person or entity by or on behalf of a third party to the extent that the Claims result from or arise out of the negligence or willful misconduct of the Company in connection with its manufacture or sale of the Product or the Company’s breach of this Agreement.
(b) By Distributor . The Distributor will defend indemnify and hold harmless the Company and its affiliates, directors, officers, employees and
representatives from any Claims that may be asserted against the Company or that person or entity by or on behalf of a third party to the extent that the Claims result from or arise out of the negligence or willful misconduct of the Distributor in connection with its sale and distribution of the Products or the Distributor’s breach of this Agreement.
(c) Indemnification Procedures . The obligations and liabilities of the parties with respect to claims subject to indemnification under this Agreement
or the Continuing Guaranty (“Indemnified Claims”) are subject to the following terms and conditions:
i. The party claiming a right to indemnification under this Agreement (“Indemnified Person”) will give prompt written notice to the indemnifying party (“Indemnifying Person”) of any Indemnified Claim, stating its nature, basis and amount, to the extent known. Each notice will be accompanied by copies of all relevant documentation, including any summons, complaint or other pleading that may have been served or any written demand or other document.

ii. With respect to any Indemnified Claim: (A) the Indemnifying Person will defend or settle the Indemnified Claim, subject to provisions of this subsection, (B) the Indemnified Person will, at the Indemnifying Person’s sole cost and expense, cooperate in the defense by providing access to witnesses and evidence available to it, (C) the Indemnified Person will have the right to participate in any defense at its own cost and expense to the extent that, in its judgment, the Indemnified Person may otherwise be prejudiced thereby, (D) the Indemnified Person will not settle, offer to settle or admit liability as to any Indemnified Claim without the written consent of the Indemnifying Person, and (E) the Indemnifying Person will not settle, offer to settle or admit liability as to any Indemnified Claim in which it controls the defense if the settlement, offer or admission contains any admission of fault or guilt on the part of the Indemnified Person, or would impose any liability or other restriction or encumbrance on the Indemnified Person, without the written consent of the Indemnified Person.
iii. Each party will cooperate with, and comply with all reasonable requests of, each other party and act in a reasonable and good faith manner
to minimize the scope of any Indemnified Claim.
13 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

17. Continuing Guaranty and Indemnification Agreement .
Contemporaneously with the execution of this Agreement, the Company will execute and deliver to the Distributor the Continuing Guaranty and Indemnification Agreement (the “Continuing Guaranty”) in substantially the form attached hereto as Exhibit F . The representations, warranties and indemnification provisions contained in the Continuing Guaranty are in addition to those contained in this Agreement. The Company acknowledges that all purchases of Product by Distributor are subject to the Continuing Guaranty, and the Company will perform its obligations, including its obligations to maintain insurance, set forth in the Continuing Guaranty.
18. Liability; Force Majeure .
(a) NO CONSEQUENTIAL DAMAGES . EXCEPT WITH RESPECT TO CLAIMS SUBJECT TO INDEMNIFICATION UNDER THIS AGREEMENT OR THE CONTINUING GUARANTY , NO PARTY WILL BE LIABLE TO ANY OTHER PARTY FOR ANY CONSEQUENTIAL, INCIDENTAL, INDIRECT, SPECIAL, OR OTHER SIMILAR DAMAGES ARISING OUT OF OR IN CONNECTION WITH A BREACH OF THIS AGREEMENT.
(b) Force Majeure . If the performance of any part of this Agreement by any party will be affected for any length of time by fire or other casualty,
government restrictions, war, terrorism, riots, strikes or labor disputes, lock out, transportation delays, electronic disruptions, internet, telecommunication or electrical system failures or interruptions, and acts of God, or any other cause which is beyond control of a party (financial inability excepted), the party will not be responsible for delay or failure of performance of this Agreement for this length of time. The Distributor and the Company will put in place prudent risk mitigation steps, some of which are outlined in Exhibit C , to mitigate against the impact of Force Majeure events.
19. Notices . Any notice, request or other document to be given under this Agreement to a party will be effective when received and must be given in
writing and delivered in person or sent by overnight courier or registered or certified mail, return receipt requested, as follows:
14
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
If to Distributor: Besse Medical
9075 Centre Pointe Drive, Suite 140
West Chester, OH 45069
Attn: President
With a copy to:
AmerisourceBergen Specialty Group, Inc.
3101 Gaylord Parkway, 1N-E186
Frisco, TX 75034
Attn: Group General Counsel If to the Company:
Kythera Biopharmaceuticals, Inc.
30930 Russell Ranch Road, Suite 300
Westlake Village, CA 91362
Attn: Senior Vice President, Operations With a copy to:
Kythera Biopharmaceuticals, Inc.
30930 Russell Ranch Road, Suite 300
Westlake Village, CA 91362
Attn: General Counsel

20. Other Provisions .
(a) Other Rights . No waiver of any breach of any one or more of the conditions or covenants of this Agreement by a party will be deemed to imply or constitute a waiver of a breach of the same condition or covenant in the future, or a waiver of a breach of any other condition or covenant of this Agreement.
(b) Severability . If any provision or the scope of any provision of this Agreement is found to be unenforceable or too broad by judicial decree, the
parties agree that the provisions will be curtailed only to the extent necessary to conform to law to permit enforcement of this Agreement to its full extent.
(c) Entire Agreement; No Reliance . Each of the parties agrees and acknowledges that this Agreement, including the attachments referred to in this
Agreement, (i) constitutes the entire agreement and supersedes all prior and contemporaneous agreements, understandings, negotiations and discussions, whether oral or written, among the parties with respect to the subject matter of this Agreement, and (ii) is not intended to confer any rights or remedies, or impose any obligations, on any person other than the parties. Each of the parties expressly agrees and acknowledges that, other than those statements expressly set forth in this Agreement, it is not relying on any statement, whether oral or written, of any person or entity with respect to its entry into this Agreement or to the consummation of the transactions contemplated by this Agreement, and each of the parties further waives any claim against the other party that the other party has failed to disclose any fact, occurrence or other matter that relates in any way to its entry into this Agreement.
(d) Amendments and Modifications . This Agreement may be modified only by a written agreement signed by both parties. (e) Assignment . Except as otherwise provided in this Agreement, Distributor may not sell, assign, sub-contract or otherwise delegate any rights or
obligations under this Agreement. The Company may assign all of its rights and interests hereunder to a successor to all or substantially all of its business or assets whether by sale, merger, operation of law or otherwise, if (i) the assignee or transferee has agreed to be bound by the terms and conditions of this Agreement and (ii) the assignee or transferee has the financial and operating capability to perform its obligations to at least the same extent as the Company as of the date of the assignment or transfer.
(f) Applicable Law . This Agreement will be construed and enforced in accordance with the laws of the State of Delaware (excluding the choice of
law provisions thereof). (g) Independent Contractor . Distributor’s relationship with the Company under this Agreement will be that of independent contractor, and neither
party will be considered the agent of, partner of, employee or other member of the workforce of, or participant in a joint venture with, the other party, in its performance of all duties under this Agreement. Neither party will have authority to bind the other party unless otherwise agreed to in writing by the parties.
(h) Publicity . Neither party will have the right to issue a press release, statement or publication regarding the terms and conditions of or the
existence of this Agreement without the prior written consent of the other party.
15 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

(i) Joint Preparation . Each party to this Agreement (i) has participated in the preparation of this Agreement, (ii) has read and understands this
Agreement, and (iii) has been represented by counsel of its own choice in the negotiation and preparation of this Agreement. Each party represents that this Agreement is executed voluntarily and should not be construed against a party solely because it drafted all or a portion of this Agreement.
(j) Counterparts . This Agreement may be executed in multiple counterparts, each of which will be deemed an original but all of which together
will constitute one and the same instrument. Facsimile execution and delivery of this Agreement are legal, valid and binding execution and delivery for all purposes.
21. ASD Specialty Healthcare, Inc. The Distributor has advised the Company that ASD Specialty Healthcare, Inc. operates four divisions – ASD
Healthcare, Besse Medical, Chapin Specialty Healthcare and Oncology Supply. The Company agrees and acknowledges that the obligation to perform the Services under this Agreement is solely that of the Besse Medical division and not the ASD Healthcare, Chapin Specialty Healthcare or Oncology Supply divisions of ASD Specialty Healthcare, Inc.
IN WITNESS WHEREOF, the parties execute this Agreement as of the Effective Date.
16 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
KYTHERA Biopharmaceuticals, Inc. Besse Medical, a division of ASD Specialty Healthcare, Inc.
By: /s/ Keith R. Leonard By: /s/ Rob Besse Name: Keith R. Leonard Name: Rob Besse Title: President & CEO Title: VP – Operations
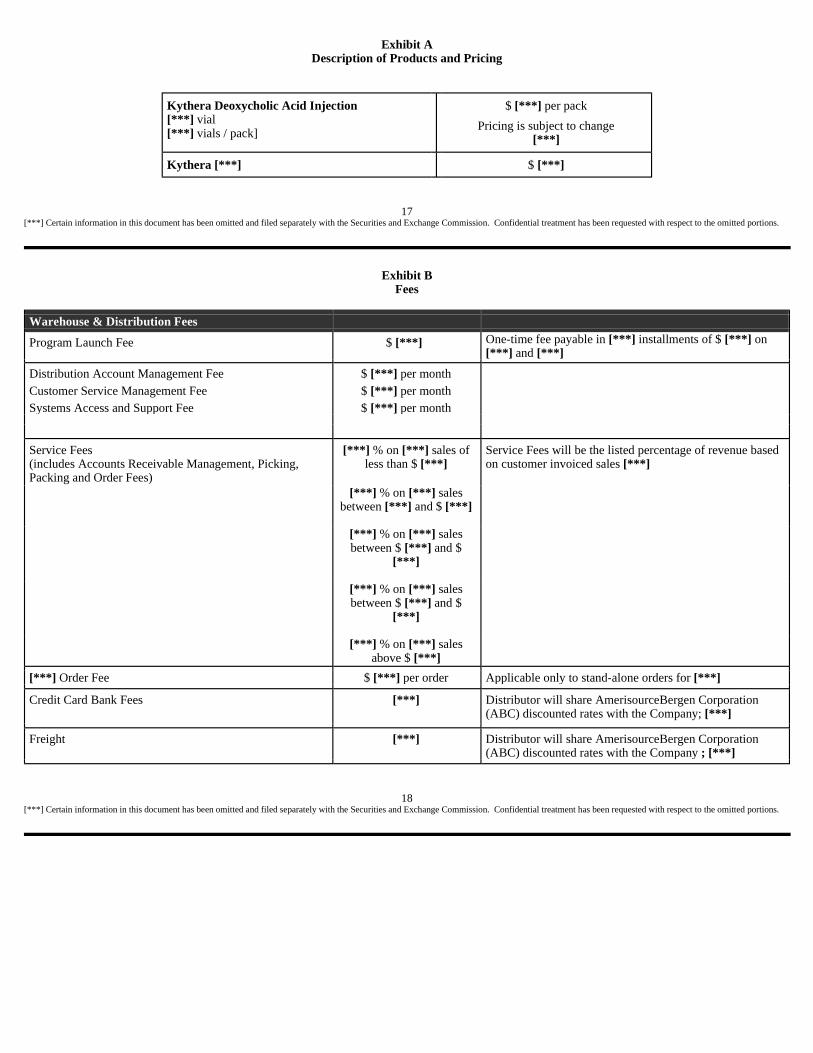
Exhibit A
Description of Products and Pricing
17 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Exhibit B
Fees
18 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Kythera Deoxycholic Acid Injection [***] vial [***] vials / pack]
$ [***] per pack
Pricing is subject to change
[***]
Kythera [***]
$ [***]
Warehouse & Distribution Fees
Program Launch Fee $ [***] One-time fee payable in [***] installments of $ [***] on
[***] and [***] Distribution Account Management Fee Customer Service Management Fee Systems Access and Support Fee
$ [***] per month $ [***] per month $ [***] per month
Service Fees (includes Accounts Receivable Management, Picking, Packing and Order Fees)
[***] % on [***] sales of less than $ [***]
Service Fees will be the listed percentage of revenue based on customer invoiced sales [***]
[***] % on [***] sales
between [***] and $ [***]
[***] % on [***] sales between $ [***] and $
[***]
[***] % on [***] sales between $ [***] and $
[***]
[***] % on [***] sales
above $ [***]
[***] Order Fee $ [***] per order Applicable only to stand-alone orders for [***] Credit Card Bank Fees
[***] Distributor will share AmerisourceBergen Corporation (ABC) discounted rates with the Company; [***]
Freight [***] Distributor will share AmerisourceBergen Corporation
(ABC) discounted rates with the Company ; [***]

19 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Returns Management (if Returns made to Distributor as a result of Company action) RGA Initiation $ [***] per RGA order
created RGA: Returned Goods Authorization. Fee for processing return request from customer and sending the customer an RGA. For clarification, any applicable RGA costs from carrier would also be passed thru. Return freight billed thru freight calculation above.
Return Processing $ [***] per RGA unit
returned. One unit is [***] pack as described
in Exhibit A.
Receipt of physical return at the distribution center. [***]
Credit/Rebill Transactions (if as a result of Kythera action) Credit/Rebill Transactions $ [***] per each Any Company-requested credit or rebill transactions keyed
in the system. IT System Maintenance Fees
Ad Hoc Reports [***] Custom Development Services [***]
$ [***]
$ [***]
Distributor reserves the right to [***]

Exhibit C
Distributor Services
The following list of Services may not be comprehensive. The Company may find it necessary to add, change, or delete certain Services as necessitated by Customer requirements. The Company and the Distributor will mutually agree on changes to these Services as necessary. The Distributor will provide the following Services to the Company: 1. Distribution Services
[***] 2. Management of Customer Agreements
[***]
20 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

3. Data Management
[***]
21 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
4. Specialty Sales Support, Customer Service, Order Management
[***] 5. Receivables Management, Special Pricing Program Management
[***] 6. Risk Mitigation Services
[***]
22 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

23 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
[***]

Exhibit D
Example of Price Adjustment Calculation
All applicable fees would be increased by[***]% effective on [***].
24 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Exhibit E Methodology for Determining [***]
Step 1: The [***] is calculated for the prior month’s sales.
(a) [***] (b) [***]
[***] Step 2: [***] are compared to the table below to determine the [***]. The prior month’s sales are then [***].
[***]
25 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Effective Date: [***] CPI-U for [***]: [***] CPI-U for [***]: [***] (published on or about [***])
Change in CPI-U: [***] Percentage change in CPI-U: [***] = [***]%
Customer Sales (Packs)
Sales ($)
Customer Payment Terms (Days)
[***] (a)
[***] (b)
[***] [***] [***] [***] [***] [***] Total [***] [***]
[***] [***]
[***] [***] [***] [***] [***] [***]

The following conceptual graphic explains how [***] . Both parties have reviewed this illustration and have agreed that it accurately depicts the intent of this agreement.
[***]
26 [***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

Exhibit F
Continuing Guaranty and Indemnification Agreement
(attached)
27
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.

CONTINUING GUARANTY AND INDEMNIFICATION AGREEMENT
The undersigned guarantees to AmerisourceBergen Corporation and each of its subsidiary companies and their successors that (i) any food, drugs, devices, cosmetics, or other merchandise (“Products”) now or hereafter shipped or delivered by or on behalf of the undersigned and its affiliates (“Guarantors”) to or on the order of AmerisourceBergen Corporation or any of its subsidiaries will not be, at the time of such shipment or delivery, adulterated, misbranded, or otherwise prohibited under applicable federal, state and local laws, including applicable provisions of the Federal Food, Drug and Cosmetic Act, 21 U.S.C. §301 et seq. (“FDCA”), and Sections 351 and 361 of the Federal Public Health Service Act, 42 U.S.C. §§ 262 and 264, and their implementing regulations (“Applicable Laws”), each as amended and in effect at the time of shipment or delivery of such Products; (ii) Products are not, at the time of such shipment or delivery, merchandise that may not otherwise be introduced or delivered for introduction into interstate commerce under Applicable Laws, including FDCA section 301 (21 U.S.C. §331); and (iii) Products are merchandise that may be legally transported or sold under the provisions of any other applicable federal, state or local law. Guarantors guarantee further that, in the case of food shipments, only those chemicals or sprays approved by federal, state or local authorities have been used, and any residue in excess of the amount allowed by any such authorities has been removed from Products. Guarantors shall promptly defend, indemnify and hold AmerisourceBergen Corporation and each of its subsidiaries harmless against any and all claims, losses, damages, costs, liabilities and expenses, including attorneys’ fees and expenses, arising as a result of (a) any actual or asserted violation of Applicable Laws or by virtue of which Products made, sold, supplied, or delivered by or on behalf of Guarantors may be alleged or determined to be adulterated, misbranded or otherwise not in full compliance with or in contravention of Applicable Laws, (b) the possession, distribution, sale and/or use of, or by reason of the seizure of, any Products of Guarantors, including any prosecution or action whatsoever by any governmental body or agency or by any private party, including claims of bodily injury, death or property damage, (c) any actual claim that Guarantors’ Products infringe any proprietary or intellectual property rights of any person, including infringement of any trademarks or service names, trade names, trade secrets, inventions, patents or violation of any copyright laws or any other applicable federal, state or local laws, and (d) any actual claim of negligence, willful misconduct or breach of contract by any of Guarantors; except that the indemnity obligations of (a), (b), (c), and (d) shall be limited to the extent arising from the negligence, willful misconduct or breach of contract of AmerisourceBergen or its affiliates. Guarantors shall maintain primary, noncontributory product liability insurance of not less than $ [***] per occurrence for claims relating to Products. This insurance must include AmerisourceBergen Corporation, its subsidiaries and their successors as additional insureds for claims arising out of Products, and provide for at least thirty days’ advance written notice to AmerisourceBergen Corporation of cancellation or material reduction of the required insurance. If the required insurance is underwritten on a “claims made” basis, the insurance must include a provision for an extended reporting period (“ERP”) of not less than [***] months; Guarantors further agree to purchase the ERP if continuous claims made insurance, with a retroactive date not later than the date of this Agreement, is not continually maintained or is otherwise unavailable. This insurance shall be with an insurer and in a form acceptable to AmerisourceBergen Corporation, and any deductible or retained risk must be commercially and financially reasonable and acceptable to AmerisourceBergen Corporation, at its sole discretion. Guarantors warrant that they have sufficient assets to cover any self-insurance or retained risk. Upon request, Guarantors will promptly provide satisfactory evidence of the required insurance. Provisions in this Continuing Guaranty and Indemnification Agreement are in addition to, and not in lieu of, any terms set forth in any purchase orders accepted by Guarantors or any separate agreement entered into between AmerisourceBergen Corporation or any of its subsidiaries and Guarantors. If the language in this Agreement conflicts with the language in any other document, the language in this Agreement controls.
[***] Certain information in this document has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions.
Kythera Biopharmaceuticals, Inc.
By
Name
Title
Date

QuickLinks -- Click here to rapidly navigate through this document
Exhibit 21.1
List of Subsidiaries of KYTHERA Biopharmaceuticals, Inc.
Jurisdiction of Incorporation or Organization
Kythera Biopharmaceuticals (Europe) Ltd. United Kingdom Kythera Biopharmaceuticals Australia Pty. Ltd. Australia Kythera Holdings Ltd. Bermuda

QuickLinks
Exhibit 21.1
List of Subsidiaries of KYTHERA Biopharmaceuticals, Inc.

QuickLinks -- Click here to rapidly navigate through this document
Exhibit 23.1
Consent of Independent Registered Public Accounting Firm
We consent to the incorporation by reference in the following Registration Statements:
• Registration Statement (Form S-8 No. 333-184439) pertaining to the KYTHERA Biopharmaceuticals, Inc. 2004 Stock Plan, as amended, and the KYTHERA Biopharmaceuticals, Inc. 2012 Equity Incentive Award Plan;
• Registration Statements (Form S-8 Nos. 333-187538 and 333-194735) pertaining to the KYTHERA Biopharmaceuticals, Inc. 2012 Equity Incentive Award Plan;
• Registration Statement (Form S-8 No. 333-198512) pertaining to the KYTHERA Biopharmaceuticals, Inc. 2014 Employment Commencement Incentive Plan;
• Registration Statement (Form S-8 No. 333-202104) pertaining to the KYTHERA Biopharmaceuticals, Inc. 2012 Equity Incentive Award Plan and the KYTHERA Biopharmaceuticals, Inc. 2015 Employee Stock Purchase Plan; and
• Registration Statements (Form S-3 No. 333-198511 and Amendment No. 1 thereto) relating to Common Stock; Preferred Stock; Debt Securities; Warrants; Purchase Contracts; and Units of KYTHERA Biopharmaceuticals, Inc. and in the related Prospectus;
of our report dated March 2, 2015, with respect to the consolidated financial statements of KYTHERA Biopharmaceuticals, Inc. included in this Annual Report (Form 10-K) for the year ended December 31, 2014.
Los Angeles, California March 2, 2015
/s/ Ernst & Young LLP

QuickLinks
Exhibit 23.1
Consent of Independent Registered Public Accounting Firm

QuickLinks -- Click here to rapidly navigate through this document
Exhibit 31.1
CERTIFICATION
I, Keith R. Leonard, Jr., certify that:
1. I have reviewed this Annual Report on Form 10-K of KYTHERA Biopharmaceuticals, Inc.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant's other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: (a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that
material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
5. The registrant's other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions): (a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to
adversely affect the registrant's ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: March 2, 2015
/s/ KEITH R. LEONARD, JR.
President and Chief Executive Officer

QuickLinks
Exhibit 31.1
CERTIFICATION

QuickLinks -- Click here to rapidly navigate through this document
Exhibit 31.2
CERTIFICATION
I, John W. Smither, certify that:
1. I have reviewed this Annual Report on Form 10-K of KYTHERA Biopharmaceuticals, Inc.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant's other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: (a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that
material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
5. The registrant's other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions): (a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to
adversely affect the registrant's ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: March 2, 2015
/s/ JOHN W. SMITHER
Chief Financial Officer

QuickLinks
Exhibit 31.2
CERTIFICATION

QuickLinks -- Click here to rapidly navigate through this document
Exhibit 32.1
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of KYTHERA Biopharmaceuticals, Inc. (the "Company") on Form 10-K for the year ended December 31, 2014, as filed with the Securities and Exchange Commission (the "Report"), Keith R. Leonard, Jr., President and Chief Executive Officer of the Company and John W. Smither, Chief Financial Officer of the Company, respectively, do each hereby certify, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that
• The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
• The information in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: March 2, 2015
/s/ KEITH R. LEONARD, JR.
President and Chief Executive Officer
/s/ JOHN W. SMITHER
Chief Financial Officer

QuickLinks
Exhibit 32.1
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002