gbs: genotyping by sequencing
TRANSCRIPT

2014.12.09

Introduction
• Genetic markers – heritable polymorphisms that can be measured in one or
more populations of individuals – heart of modern genetics – enable the study of important questions in population
genetics, ecological genetics and evolution
• Advent of next-generation sequencing (NGS)– whole genome sequencing– re-sequencing : discovering, sequencing and genotyping
thousands of markers across almost any genome• comprehensive genome-wide association studies for any organ-
ism • genome-wide studies on wild populations

NGS marker discovery and genotyping methods
• RRL and CRoPS (reduced-representation libraries and complexity reduction of polymorphic sequences)
• RAD seq (Restriction-site associated DNA sequenc-ing)
• GBS (Genotyping by sequencing)– the digestion of multiple samples of genomic DNA – a selection or reduction of the resulting restriction fragments – NGS of the final set of fragments, which should be less than
1 kb in size
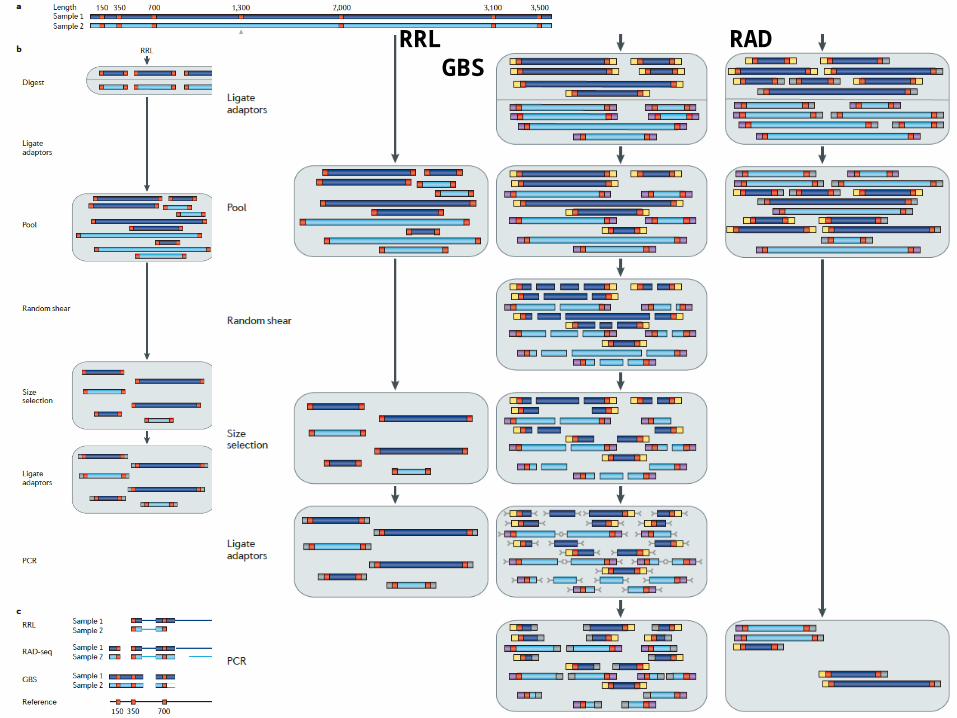
(Davey et al., 2011 Nat Rev Genet )
RRL RAD GBS
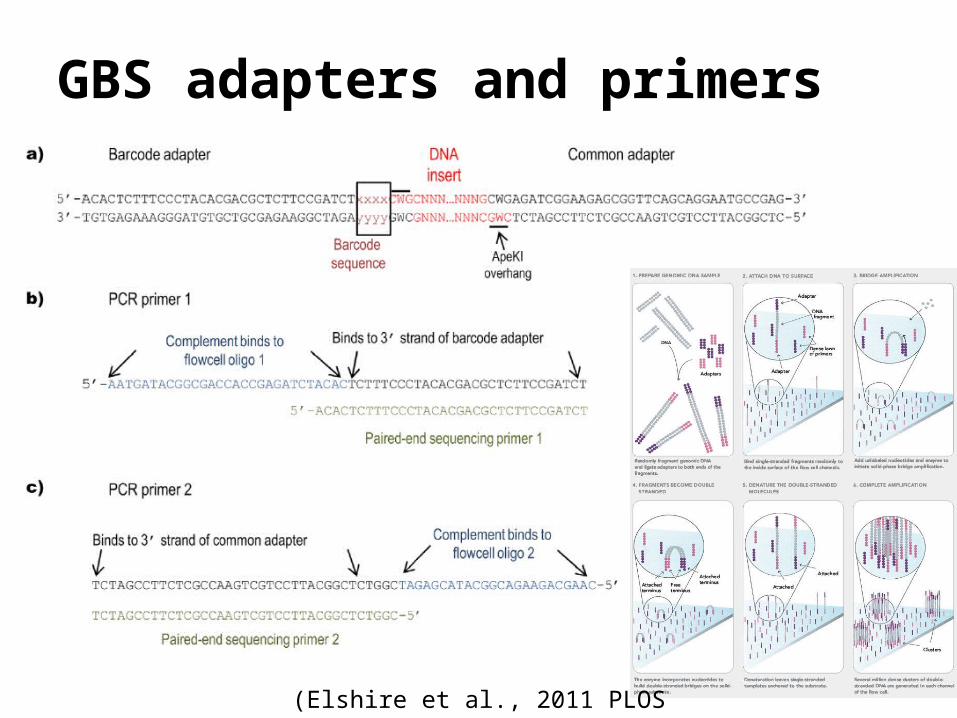
GBS adapters and primers
(Elshire et al., 2011 PLOS One)
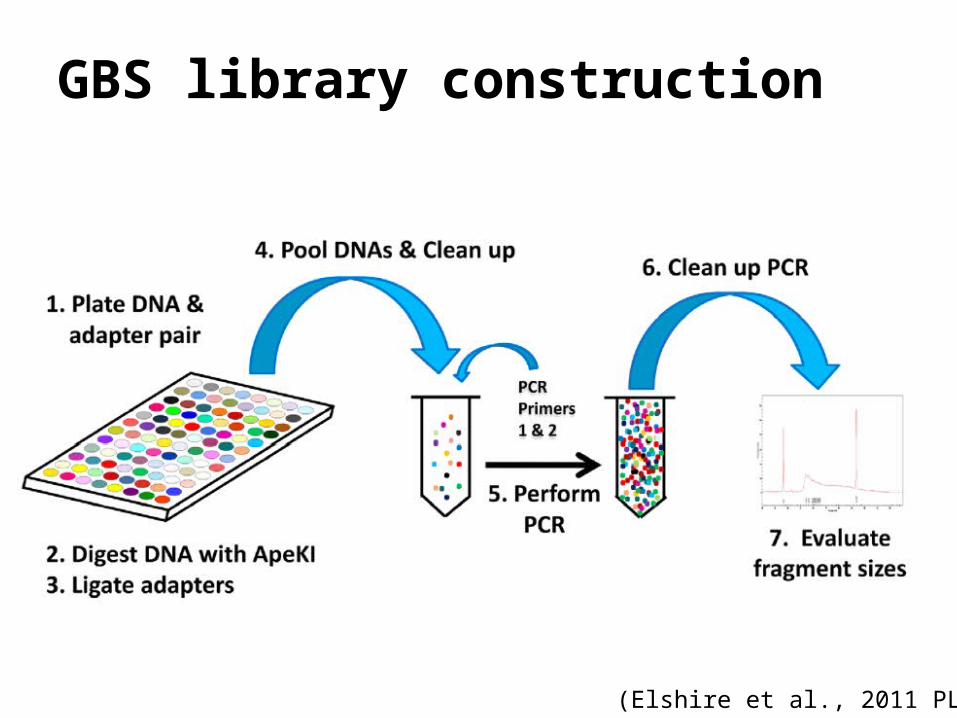
GBS library construction
(Elshire et al., 2011 PLOS One)

GBS results in Maize
• Parental line– 98% of 1,146,449 HQ reads were aligned with maize genome – 868,336 reads that aligned perfectly to the maize genome
• 276 RILs– 6 lanes, 48-plex, 2,090 Mbp per lane on average– From 145,836,644 raw reads, 83% passed filtering process
(120,438,739 GBS reads)– 436,372 reads were produced per DNA sample and 95% of sam-
ples– 809,651 sequence tags covering 51.8 Mbp or 2.3% of the maize
genome– 167,494 of the dominant markers, could be placed upon frame
work map of 25,185 sequence tags.

TASSEL-GBS
• new bottleneck is the efficient bioinformat-ics analysis of the vast and ever-expanding sea of data
• TASSEL-GBS (Trait Analysis by aSSociation, Evolution and Linkage)
– Not limited to the specific restriction enzymes utilized in those protocols:
– work on nearly any restriction enzyme and bar-coding approach specifically
– designed to efficiently handle large quantities of data from large numbers of samples
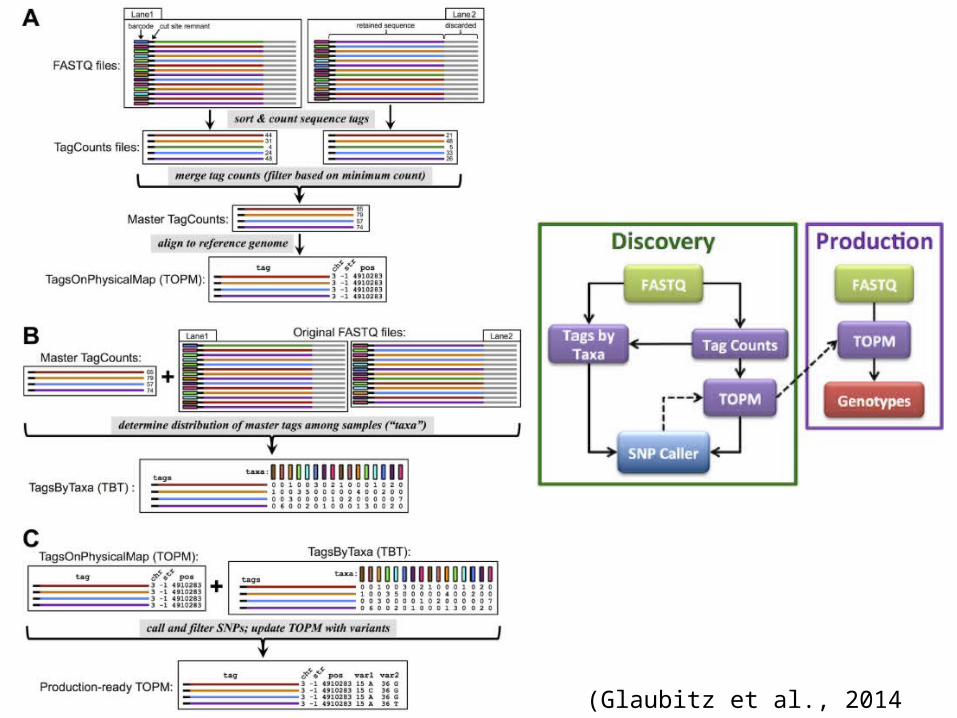
(Glaubitz et al., 2014 PLOS One)
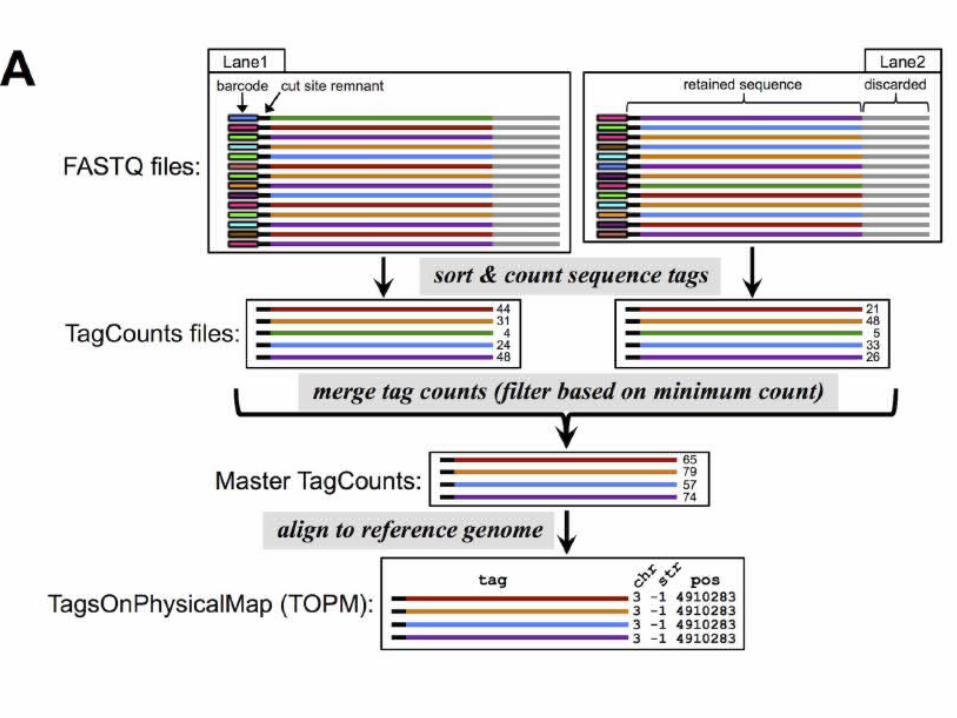
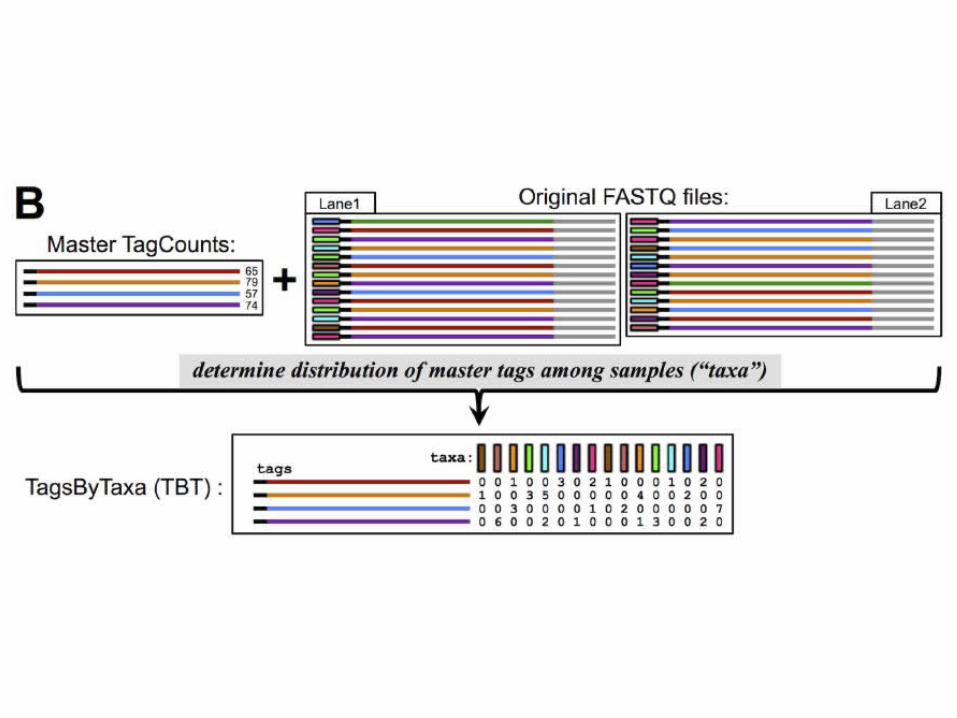
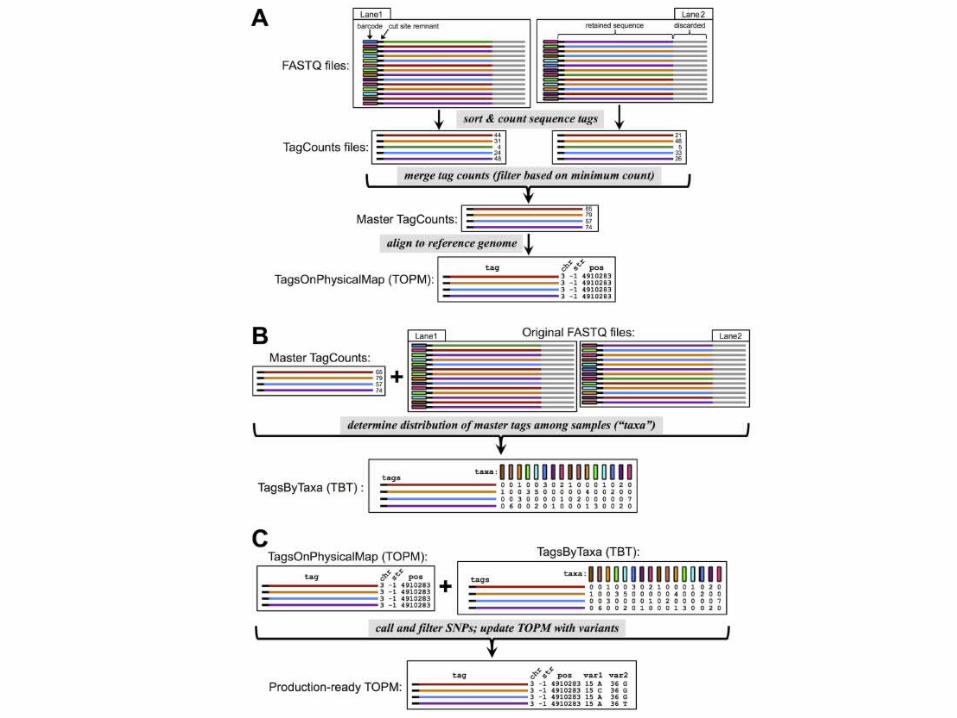

Population genetic-based filtering of putative SNPS
• Putative SNPs from GBS may be of low quality– sequencing error– paralogous sequence tags from different loci
• To detect and filter out error-prone SNPs– minor allele frequency (MAF)– inbreeding coefficient (or ‘‘index of panmixia’’)
𝐹 𝐼𝑇=1−𝐻𝑜𝐻𝑒
𝐻𝑒=2𝑞 (1−𝑞)
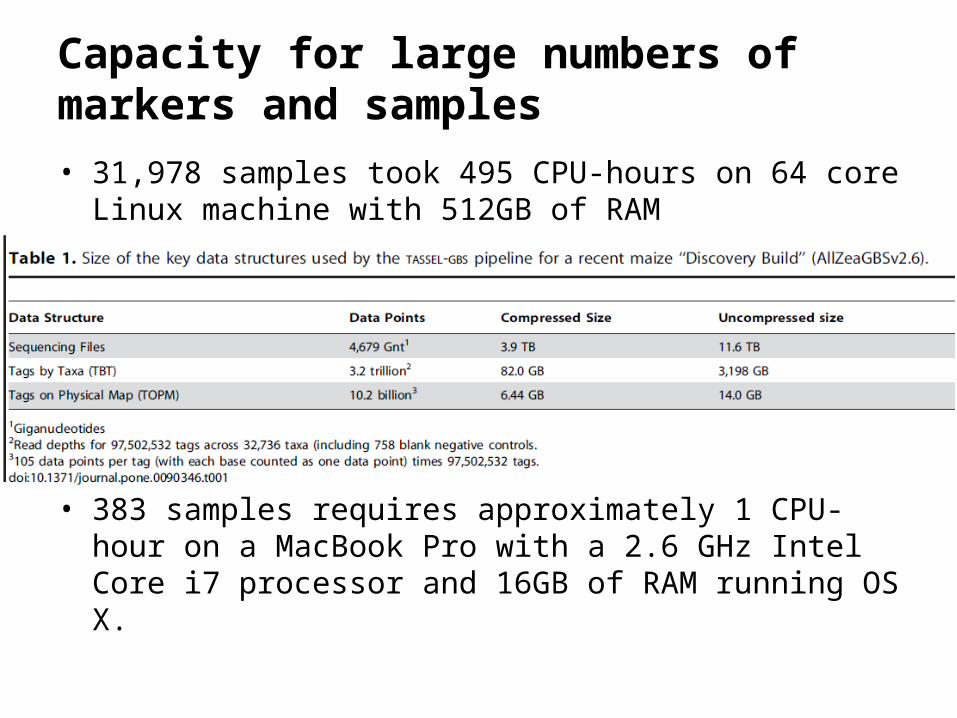
Capacity for large numbers of markers and samples
• 31,978 samples took 495 CPU-hours on 64 core Linux machine with 512GB of RAM
• 383 samples requires approximately 1 CPU-hour on a MacBook Pro with a 2.6 GHz Intel Core i7 processor and 16GB of RAM running OS X.

UNEAK pipeline in TASSEL-GBS• Absence of a reference genome,– SNP calling may be much less accurate with
short-read sequencing technologies,– true SNPs, sequencing errors and SNPs be-
tween paralogs can be difficult to distinguish
• Universal Network-Enabled Analysis Kit (UNEAK)– To enable genome-wide association studies
(GWAS) and genomic selection (GS)
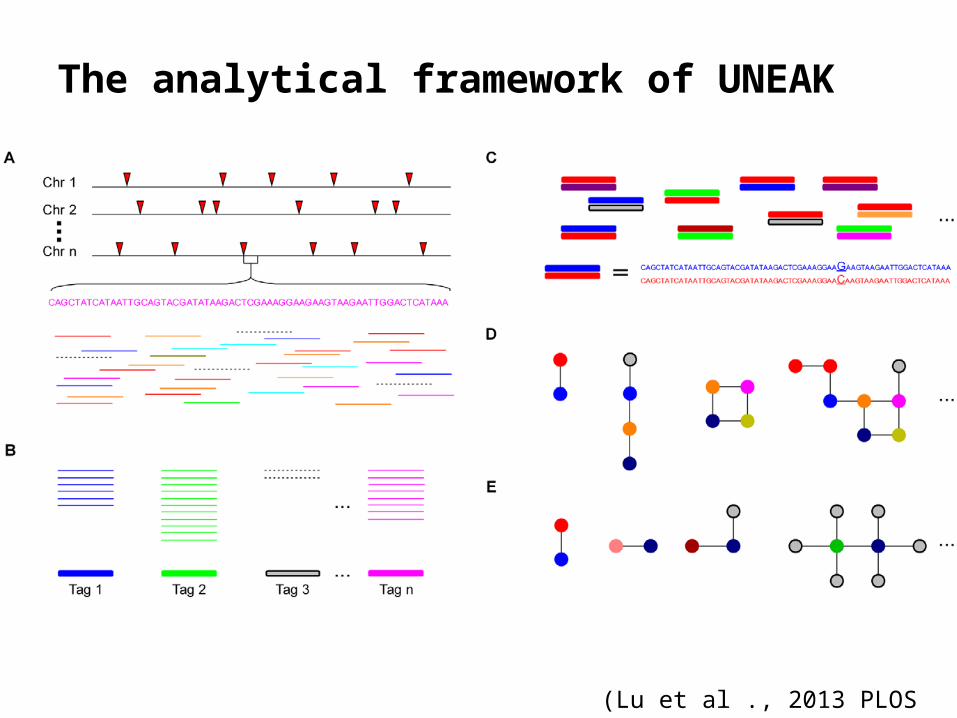
The analytical framework of UNEAK
(Lu et al ., 2013 PLOS Genetics)
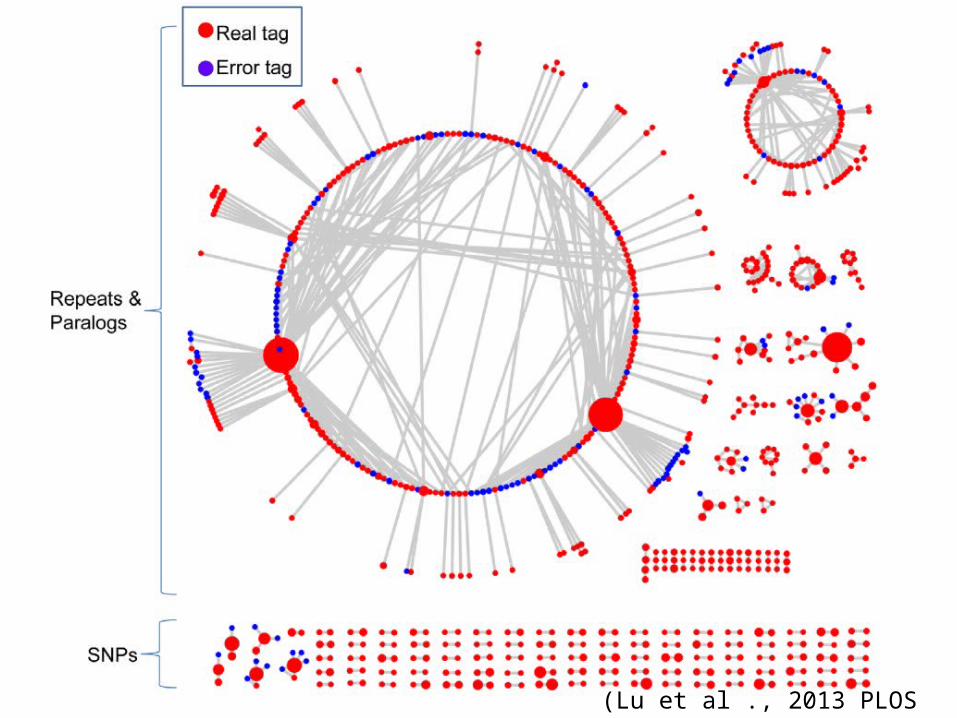
(Lu et al ., 2013 PLOS Genetics)
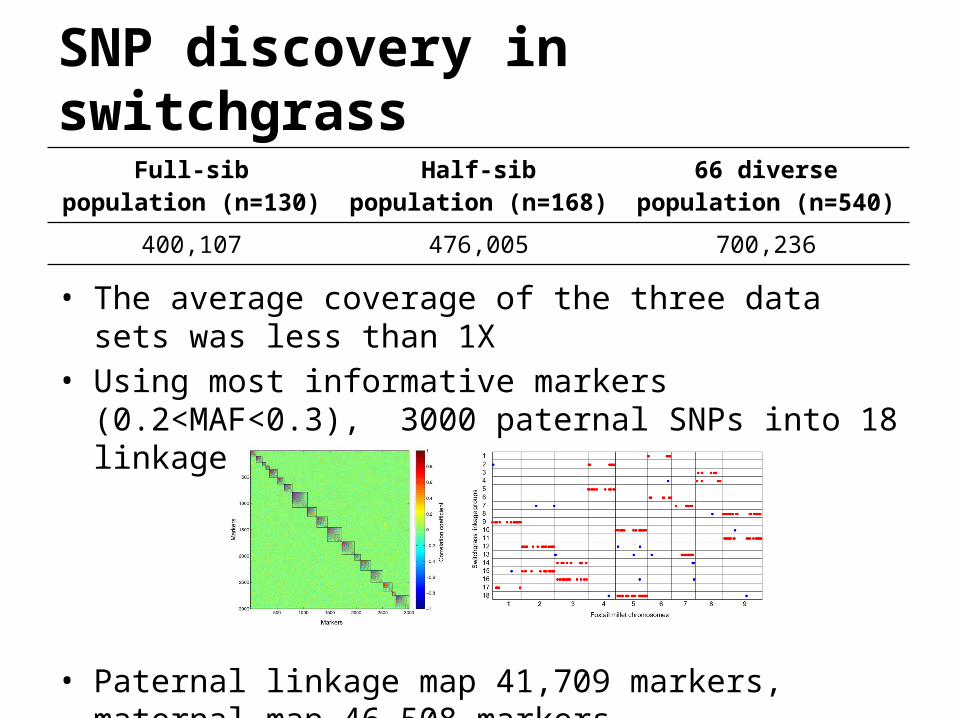
SNP discovery in switch-grassFull-sib population
(n=130)Half-sib population
(n=168)66 diverse popula-
tion (n=540)
400,107 476,005 700,236
• The average coverage of the three data sets was less than 1X
• Using most informative markers (0.2<MAF<0.3), 3000 paternal SNPs into 18 linkage groups
• Paternal linkage map 41,709 markers, maternal map 46,508 markers

Strengths and Weaknesses of GBS
• Strengths of GBS and TASSEL-GBS– The large number of markers potentially pro-
duced– Low cost and minimal startup cost– Integration of SNP discovery with SNP calling
• Weakness– When conducted at low coverage, is the
amount of missing data

Reference
• Elshire R, Glaubitz J, Sun Q, Poland J, Kawamoto K, et al. (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6.
• Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, et al. (2014) TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS ONE 9
• Lu F, Lipka AE, Glaubitz J, Elshire R, Cherney JH, et al. (2013) Switchgrass Genomic Diversity, Ploidy, and Evolution: Novel In-sights from a Network-Based SNP Discovery Protocol. PLoS Genet 9
• Davey J, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, et al. (2011) Genome-wide genetic marker discovery and genotyping using next-genration sequencing. Nat Rev Genet 12:499-510

Thank you for listen-ing !!