generic substitution matrix based sequence comparison q: m a t w l i. a: m a - w t v. scr: 45 -?11 3...
TRANSCRIPT

Generic substitution matrix based sequence comparison
Q: M A T W L I .
A: M A - W T V .
Scr: 45 -? 11 -1 3Scr: 45 -2 -2 1
Q: M A T W L I .
A: M A W T V A .
Total: 5
-1
Total = 22 - ?
Blosum 62:
Gap openning: -6 ~ -15
Gap Extension: -2 ~ -6
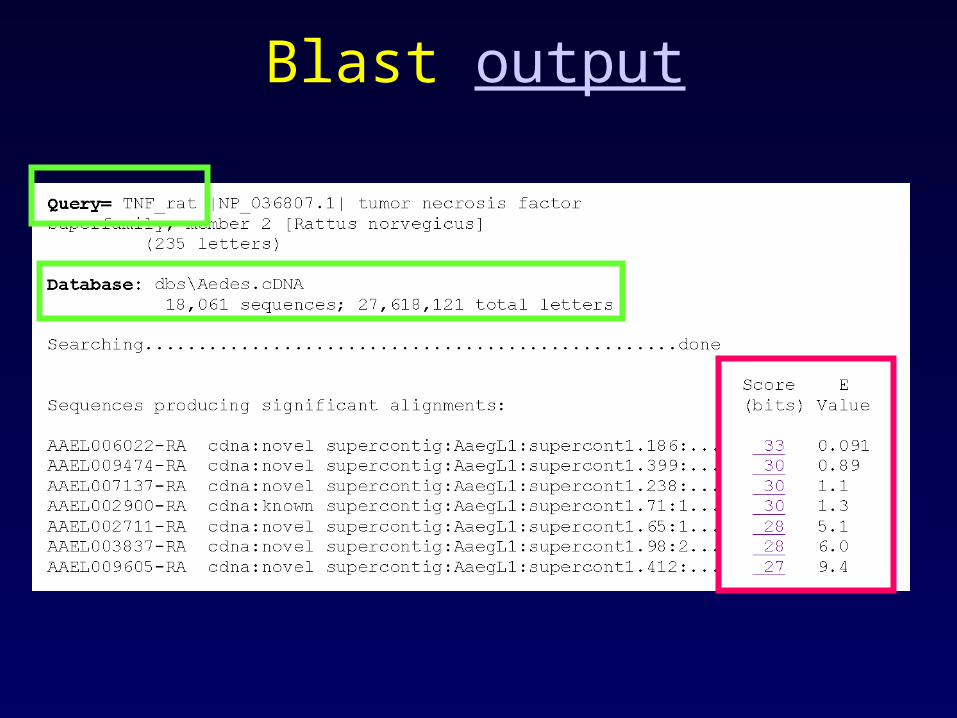
Blast output
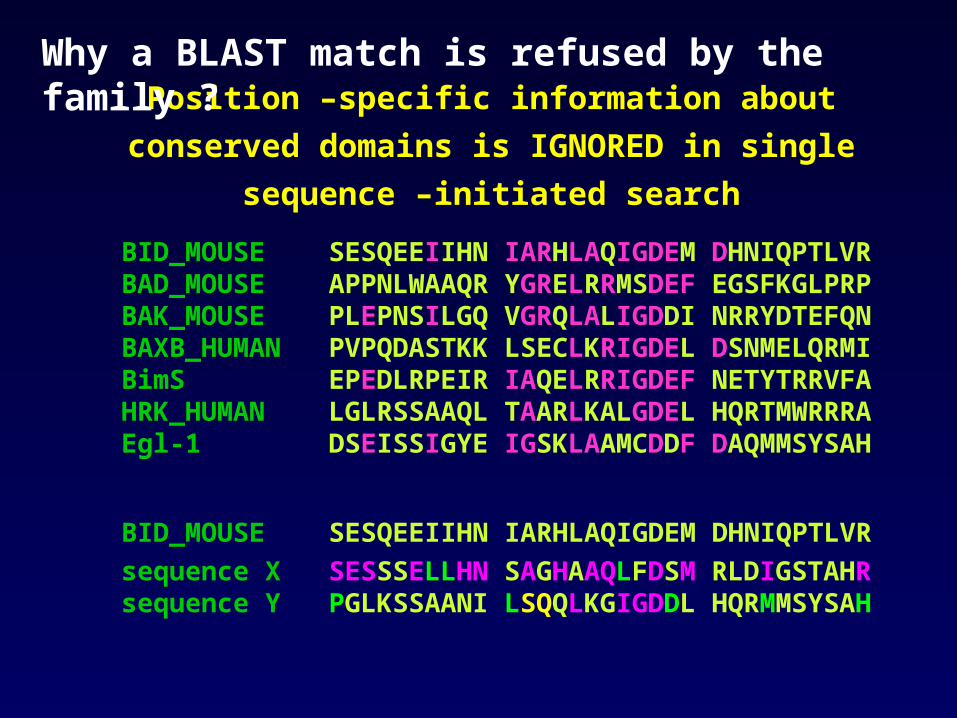
Position –specific information about conserved domains is
IGNORED in single sequence –initiated search
BID_MOUSE SESQEEIIHN IARHLAQIGDEM DHNIQPTLVRBAD_MOUSE APPNLWAAQR YGRELRRMSDEF EGSFKGLPRPBAK_MOUSE PLEPNSILGQ VGRQLALIGDDI NRRYDTEFQNBAXB_HUMAN PVPQDASTKK LSECLKRIGDEL DSNMELQRMIBimS EPEDLRPEIR IAQELRRIGDEF NETYTRRVFAHRK_HUMAN LGLRSSAAQL TAARLKALGDEL HQRTMWRRRAEgl-1 DSEISSIGYE IGSKLAAMCDDF DAQMMSYSAH
BID_MOUSE SESQEEIIHN IARHLAQIGDEM DHNIQPTLVR
sequence X SESSSELLHN SAGHAAQLFDSM RLDIGSTAHRsequence Y PGLKSSAANI LSQQLKGIGDDL HQRMMSYSAH
Why a BLAST match is refused by the family ?

Representation of positional information in specific motif
M-C-N-S-S-C-[MV]-G-G-M-N-R-R.
Binary patterns:
Positional matrix:
-2.499 -2.269 -5.001 -4.568 -2.418 -4.589 -3.879 1.971 -4.330 1.477 -1.241 -4.221 -4.590 -4.097 -4.293 -3.808 0.495 2.545 -3.648 -3.265
1.627 -2.453 -1.804 -1.746 -3.528 2.539 1.544 -3.362 -1.440 -3.391 -2.490 -1.435 -3.076 -1.571 0.501 0.201 -1.930 -2.707 -3.473 -3.024
-1.346 -2.872 -1.367 0.699 -2.938 -2.427 -0.936 -2.632 -0.095 1.147 -1.684 -1.111 -2.531 1.174 2.105 1.057 -1.400 -2.255 -2.899 -2.260
1.045 1.754 -1.169 -0.576 -2.756 -2.212 1.686 -2.576 0.951 -2.438 -1.544 0.857 -2.301 1.891 1.556 -1.097 -1.180 -2.155 -2.751 -2.060
-3.385 -2.965 -5.039 -4.313 -1.529 -5.006 -3.577 0.429 -4.094 3.154 -0.121 -4.440 -4.199 -3.292 -3.662 -4.198 -3.281 -1.505 -3.043 -3.120
2.368 -3.197 -2.285 -1.533 -3.721 -2.945 1.815 -3.235 0.067 -3.061 -2.259 -1.680 -3.231 1.195 2.287 -2.009 -2.044 -2.825 -3.324 -2.844
1.046 1.742 0.576 -0.734 -2.072 -2.234 -0.851 0.436 -0.548 -0.129 -0.974 -1.039 -2.318 2.368 0.667 -1.135 -1.076 -1.304 -2.398 -1.821
0.715 -1.778 -3.820 -3.359 -1.535 -3.463 -2.571 3.060 -3.008 0.262 1.566 -2.996 -3.575 0.450 -3.001 -2.571 -1.765 0.193 -2.389 -2.068
-2.053 0.965 -2.767 -3.509 -4.520 3.654 -3.242 -4.548 -3.338 -4.968 -3.789 -2.462 -4.048 -3.738 -3.266 -2.596 -3.573 -3.990
A C D E F G H I K L M N ….
Pos. 123456789

DNA Pattern – Transcription factor binding site
Practice: identify potential transcription factor binding sites on a promoter
sequence.
Using TESS : Transcription Element Search System
http://www.cbil.upenn.edu/cgi-bin/tess/tess33?RQ=WELCOME
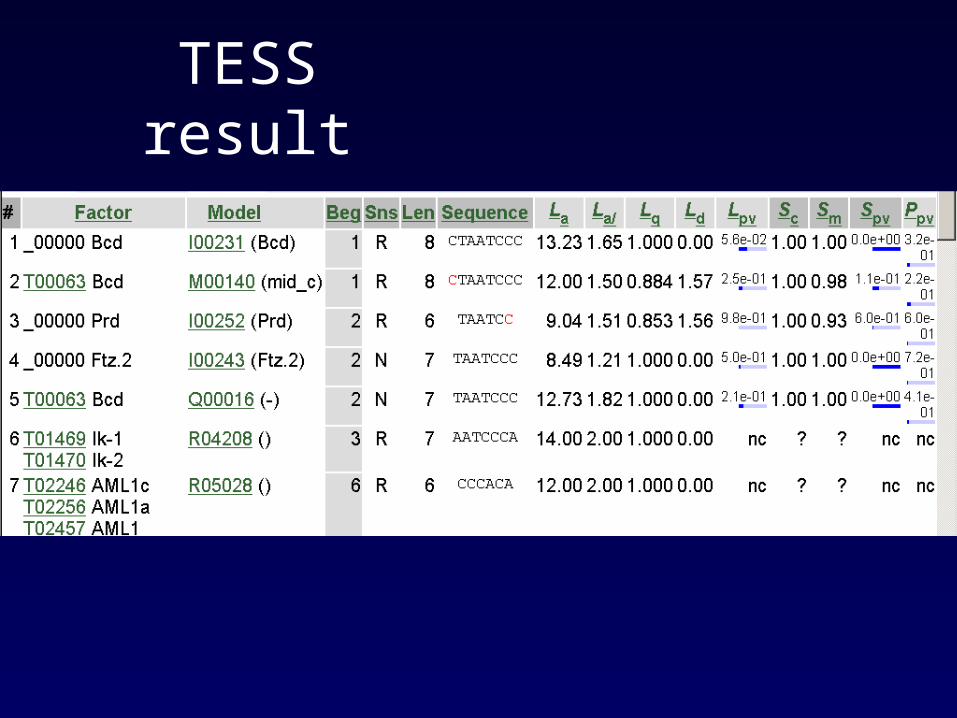
TESS result

Why are there many false positives for TF binding site scan?
Contextual dependency is not considered.
Stringency of the matrices.

DNA Pattern – Transcription factor binding site
• Pattern strings / Matrixes are extracted from known binding sequence.
• Core vs whole.
• Some short and/or ambiguous patterns will have many hits.
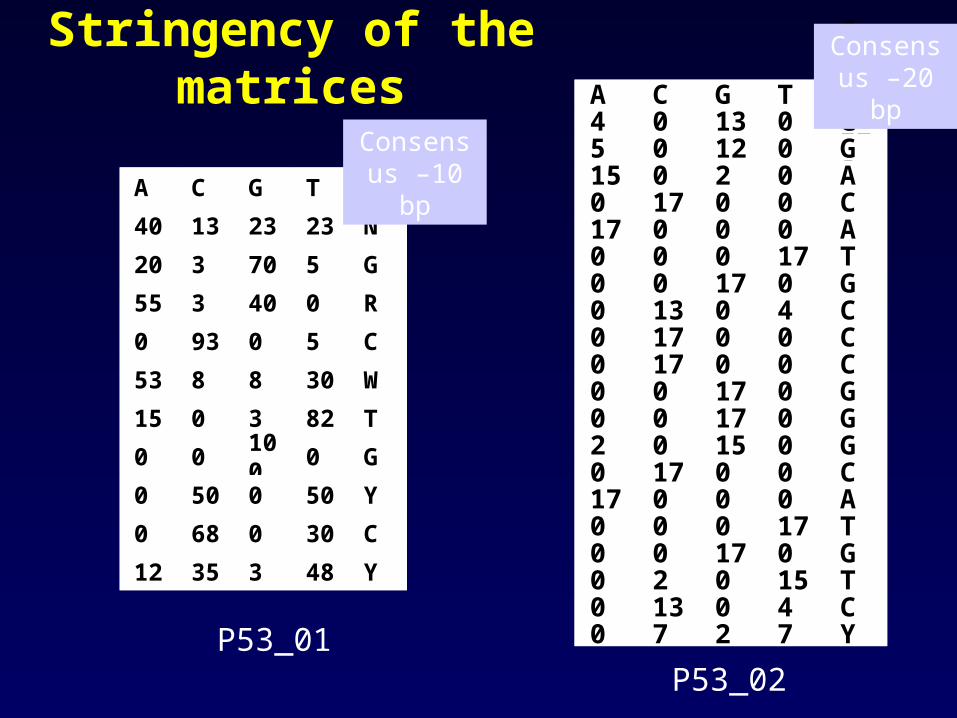
Stringency of the matrices
A C G T Consens
us 40 13 23 23 N
20 3 70 5 G
55 3 40 0 R
0 93 0 5 C
53 8 8 30 W
15 0 3 82 T
0 0 100 0 G
0 50 0 50 Y
0 68 0 30 C
12 35 3 48 Y
A C G T
Consensus
4 0 13 0 G 5 0 12 0 G
15 0 2 0 A 0 17 0 0 C
17 0 0 0 A 0 0 0 17 T 0 0 17 0 G 0 13 0 4 C 0 17 0 0 C 0 17 0 0 C 0 0 17 0 G 0 0 17 0 G 2 0 15 0 G 0 17 0 0 C
17 0 0 0 A 0 0 0 17 T 0 0 17 0 G 0 2 0 15 T 0 13 0 4 C 0 7 2 7 Y P53_01
P53_02
Consensus –10 bp
Consensus –20 bp

How are the motif matrices derived? - example: Hidden Markov Model
• A specification on “how events will happen” so that statistical assessment can be readily made
• Used for Speech recognition and characterizing sequence patterns
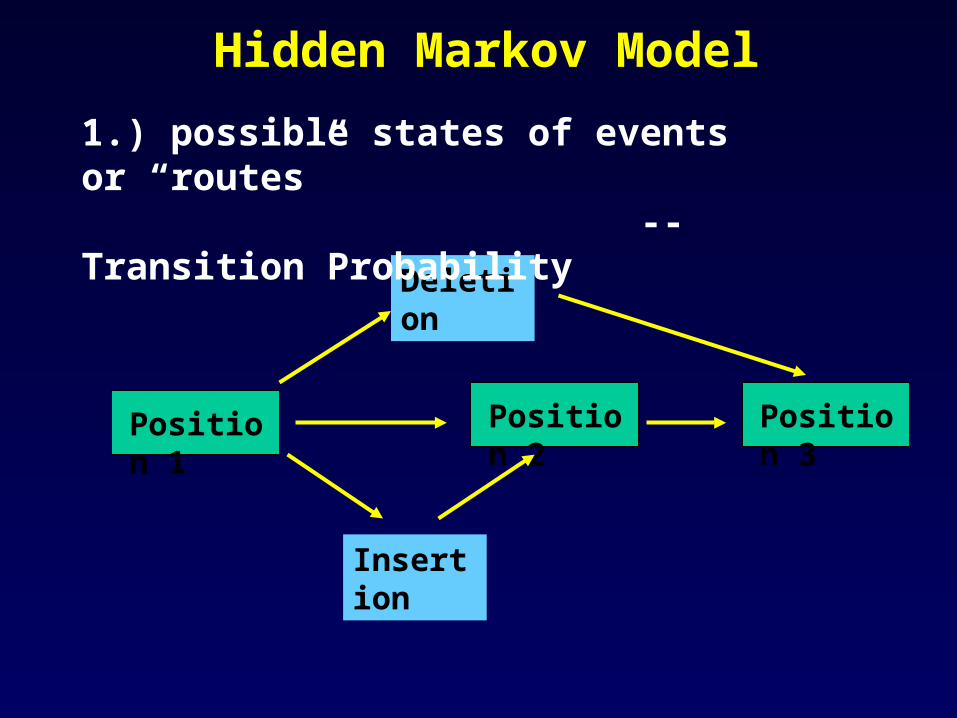
Hidden Markov Model
Position 1 Position 2
Deletion
Insertion
Position 3
1.) possible states of events or “routes” --Transition Probability
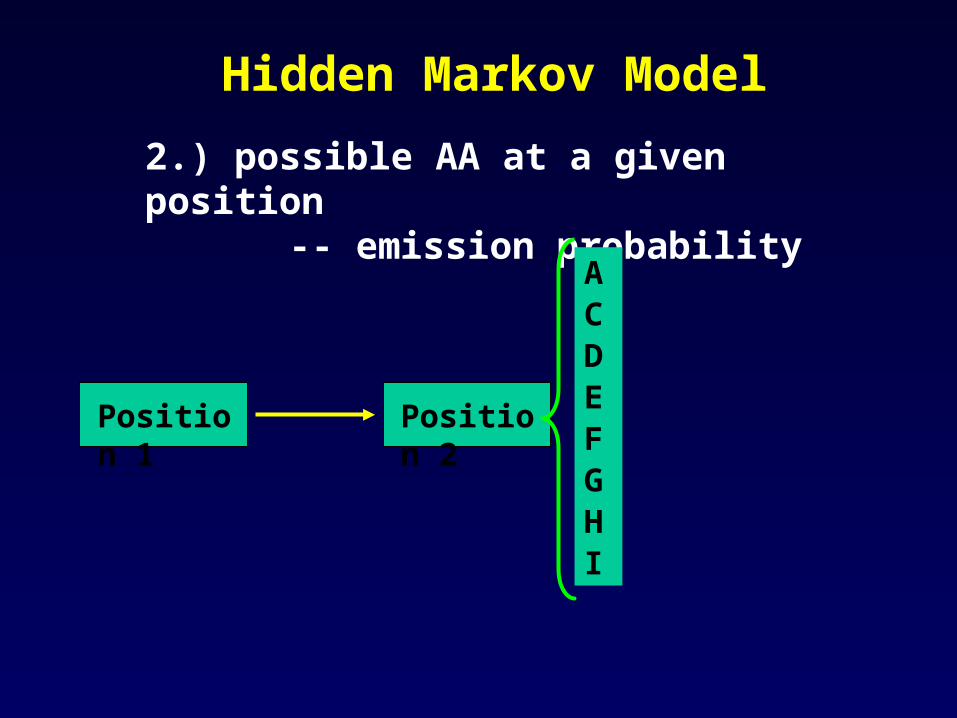
Hidden Markov Model
Position 1 Position 2
2.) possible AA at a given position -- emission probability
ACDEFGHI

Hidden Markov Model3.) That’s all. Let’s give it a try and you will know what it is about

Hidden Markov Model How to make a HMM for my motif ?
• Collect related sequences
• MEME : http://meme.sdsc.edu/meme4_3_0/cgi-bin/meme.cgi
*Selection of sequences determines the model*

Identifying motifs using MEME -Multiple EM for Motif Elicitation
• EM: Expectation maximization (P173-177).
• Identifies statistically significant motif(s) in a set of sequences.
• Outline the occurrence of the motifs at the end of the report
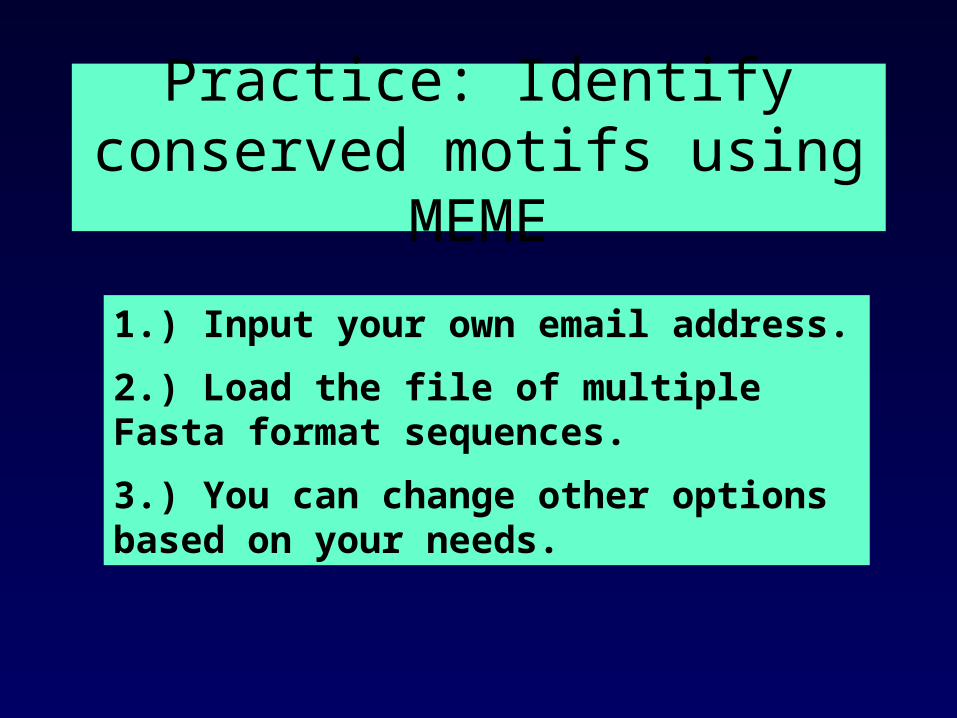
Practice: Identify conserved motifs using MEME
1.) Input your own email address.
2.) Load the file of multiple Fasta format sequences.
3.) You can change other options based on your needs.

Two search examples
The outcome of the search is dependent on the inputting set of sequences.
Compose the inputting set based on your research needs.
Set1: Mammalian P53 plus mosquito hits
Set2: Diverse set of P53 plus mosquito hits

Two search examplesSet1: Mammalian P53 plus mosquito hits
Set2: Diverse set of P53 plus mosquito hits

Secondary structure prediction
Predict the likelihood of amino acid x to be in each of the three (four) types of secondary structure configuration
• Helix• Sheet• Turn• Coil
Coiled-coil is two helices tangled together

Secondary structure prediction- different strategies and algorithms
• Chou-Fasman / Garnier Method
-- based on AA composition
• Nearest Neighbor / Levin Method
-- based on sequence similarity
• Neural Network / PHD
• SOPM, DPM, DSC, etc.
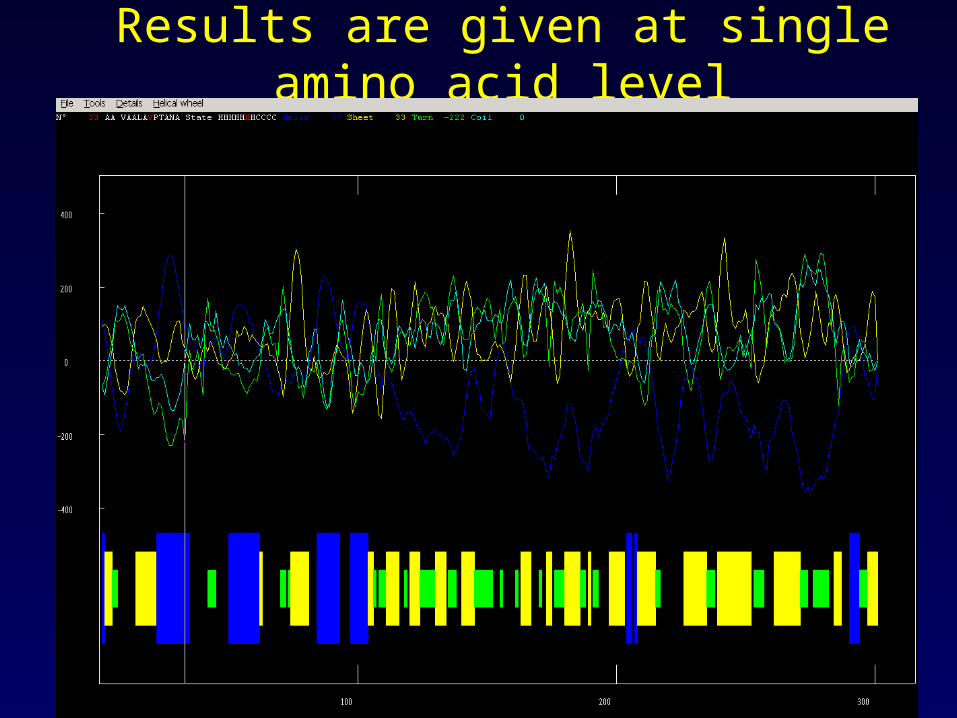
Results are given at single amino acid level

Secondary structure prediction
--Interpretation of result
Seq: - D G S L A D E R K
Pre: - B B B H H B B T T
What is the likelihood of helix formation here ?

Secondary structure prediction-Accuracy
• At the amino acid level -- ~ 75% based on testing set
• IF:
Seq: - D G I L A V A S M I V
Pre: - B B H H H H H H H H H Length > 9
> 90% chance a helix formation around this region

Secondary structure prediction-Programs
• There are over a dozen web sites provide 2nd structure predication service – (tools)
• AntheWin has a good sample of different approaches and has other associated tools

Practice: using AnathePro to analyze protein secondary structure
• Open the sequence file.• Chose and run a secondary structure
predication method from the “Methods” menu
• LEFT click the left boundary of an alpha helix and then RIGHT the right boundary, perform “helical Wheel”
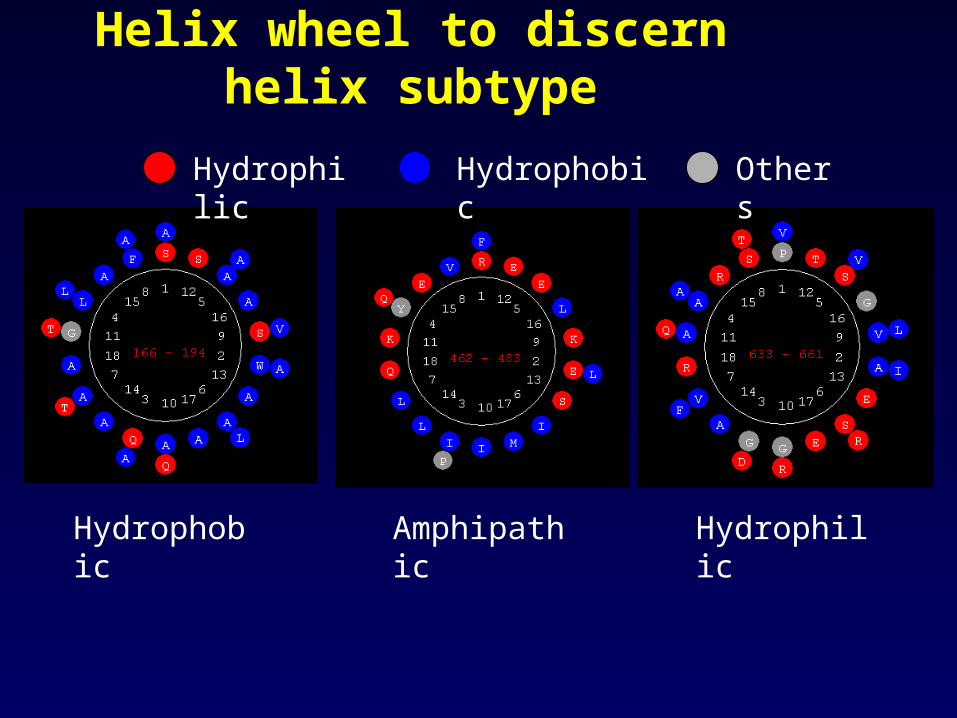
Helix wheel to discern helix subtype
Hydrophilic Hydrophobic Others
AmphipathicHydrophobic Hydrophilic