guia analisisevolmolec
TRANSCRIPT

Documento
Rodrigo A. Moreno y Marco A. Méndez. 2009. Métodos de reconstrucción filogenética para caracteres morfológicos y moleculares. Guía de pasos metodológicos para principiantes en sistemática filogenética. Versión 1.1. Laboratorio de Genética y Evolución, Facultad de Ciencias, Universidad de Chile. 33 pp.
Este documento es propiedad intelectual del Laboratorio de Genética y Evolución, Facultad de Ciencias, Universidad de Chile.
-2009-

RA Moreno & MA Méndez
2
Introducción
La presente guía que ponemos a disposición como material docente para el curso de
postgrado “Procesos Evolutivos: Métodos de Reconstrucción Filogenética” de la
Universidad de Chile, tiene como objetivo brindar apoyo metodológico a los estudiantes
que recién se inician en el estudio de la sistemática filogenética. Hemos querido
ejemplificar de forma simple los métodos rutinarios que se aplican en las reconstrucciones
filogenéticas para evaluar hipótesis en ecología y evolución. En esta guía se presentan
detalladamente los pasos metodológicos y los programas computacionales informáticos ad
hoc para reconstruir hipótesis filogenéticas basadas en caracteres o rasgos morfológicos y
moleculares (secuencias nucleotídicas).
Esperamos que este material contribuya a desarrollar en términos pedagógicos la habilidad
de los estudiantes de comprender el uso adecuado de cada herramienta y su potencial
utilidad y restricción metodológica e interpretativa desde el punto de vista estadístico y
biológico. Finalmente, entregamos las directrices como realizar análisis filogenéticos
utilizando métodos como Neighbor Joining, Máxima Parsimonia, Máxima Verosimilitud
(Maximum Likelihood) e Inferencia Estadística Bayesiana en distintas plataformas
computacionales en su mayoría de libre distribución.
A continuación se procede a mostrar con ejemplos tipos la forma de iniciar el análisis de los
datos en una sección dedicada a caracteres morfológicos y una segunda a caracteres
moleculares. Cabe destacar que esta guía esta desarrollada exclusivamente para efectuar
análisis en ambiente Windows XP.

RA Moreno & MA Méndez
3
1.- Reconstrucción filogenética para caracteres morfológicos
Procedimiento
1) Abrir bloc de notas
2) Proceda a crear un archivo (matriz) en lenguaje #NEXUS
Ejemplo tipo: Vertebrados
3) Defina el grupo externo “outgroup” [por convención se puede rellenar con ceros]
4) guarde su archivo como Vertebrados. nex
5) Este archivo con extensión nexus podrá ser utilizado en la plataforma del software PAUP
[Phylogenetic Analysis using Parsimony *and other methods]

RA Moreno & MA Méndez
4
Análisis Filogenético de Máxima Parsimonia en Software PAUP
Citas
Swofford, D.L. 2002. PAUP*: Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer, Sunderland, Massachusetts. Page, R. 2001. TREEVIEW 1.6.6. Tree drawing software for Apple Macintosh and Windows. Disponible en http://taxonomy.zoology.gla.ac.uk/rod/treeview.html.
Procedimiento
1) Abrir PAUP 2) Abra el archivo tipo Vertebrados. nex 3) Ejecute los siguientes comandos _ = un espacio
3.1. Defina su tipo de Búsqueda alltree Búsqueda Exhaustiva: <12 taxa hsearch Búsqueda Heurística bandb Búsqueda Heurística: [Branch and Bound]

RA Moreno & MA Méndez
5
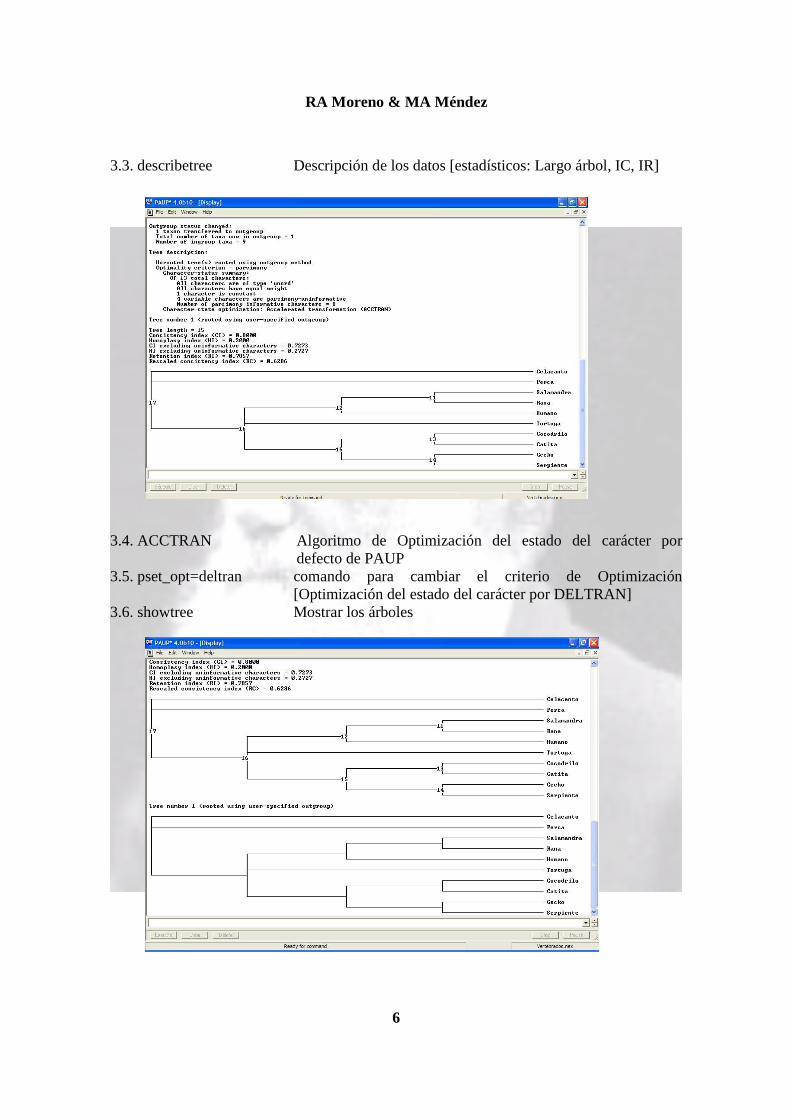
3.2. outgroup _celacanto [definir Outgroup - ver archivo Nexus]
RA Moreno & MA Méndez
6
3.3. describetree Descripción de los datos [estadísticos: Largo árbol, IC, IR] 3.4. ACCTRAN Algoritmo de Optimización del estado del carácter por
defecto de PAUP 3.5. pset_opt=deltran comando para cambiar el criterio de Optimización
[Optimización del estado del carácter por DELTRAN] 3.6. showtree Mostrar los árboles

RA Moreno & MA Méndez
7
3.7. contree Árbol de Consenso 3.8. savetree Guardar árbol [archivo queda con la extensión. tree] 3.9. bootstrap_nrep=10000 Soporte de los nodos (Árbol de consenso del 50% de la regla
de la mayoría)

RA Moreno & MA Méndez
8
3.10. savetree_file= Vertebrados.tree_from=1_to=1_brlens=yes Salvar el árbol como
archivo con largo de ramas (filograma)

RA Moreno & MA Méndez
9
En su carpeta aparecerá Vertebrados con extensión tree para ser desplegado en el software Treeview. Recuerde ir al menú Tree y seleccione show internal Edge labels para que despliegue los valores de soporte de los nodos obtenidos en su análisis de Bootstrap. Guarde la figura como Save as Graphics y seleccione la extensión Enhaced metafile (emf).

RA Moreno & MA Méndez
10
Otros comandos de utilidad 3.11. set_criterion=dist Cambio de criterio a método de distancia nj Neighbor-Joining [vecino más cercano] set_criterion=parsimony Cambio de criterio a método de Máxima Parsimonia 3.12. exclude_uninf Excluir caracteres no informativos 3.13. includeall Incluir todos los caracteres 3.14. showmat Mostrar matriz de datos 3.15. describetree/apolist Muestra la lista de apomorfías de la matriz
4) utilize el software TREEVIEW para desplegar gráficamente el(los) árbol(es) recuperados. 5) Cierre PAUP. Nota: El procedimiento es similar cuando se utiliza una matriz con secuencias nucleotídicas.

RA Moreno & MA Méndez
11
2.- Reconstrucción filogenética para caracteres moleculares
Procedimiento
1) Para obtener secuencias nucleotídicas utilize la plataforma de Genbank http://www.ncbi.nlm.nih.gov/Genbank/index.html 2) Seleccione en el menú Search “Nucleotide” 3) Posteriormente en el buscador “For” coloque el taxón de interés y el tipo de marcador(es) molecular(es).
4) Realice click en el n° de acceso de Genbank y baje “download” la secuencia en formato
FASTA

RA Moreno & MA Méndez
12
5) Genere una carpeta con las secuencias recuperadas en GenBank 6) Cierre la página. 7) Una vez obtenida las secuencias nucleotídicas se procede a generar una base de secuencias en las plataformas CLUSTALX y BIOEDIT para realizar los alineamientos de secuencias nucleotídicas.
Procedimiento CLUSTALX
Extensión .fas (formato FASTA)
Cita Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., Higgins, D.G. 1997. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research 25: 4876-4882. Para cada archivo realizar el siguiente procedimiento:
1) Abrir el Programa CLUSTALX y dentro de éste, abra el archivo .fas. 2) Proceder a alinear las secuencias utilizando la opción que viene por defecto
Multiple Aligment Mode y vaya al menú alignment y ejecute “Do complete alignment” .
3) En su carpeta aparecerá el alineamiento efectuado con la extensión .aln 4) Cierrar el programa CLUSTALX

RA Moreno & MA Méndez
13
Procedimiento BIOEDIT Cita Hall, T.A. 1999. BIOEDIT: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41: 95-98. 1) En su carpeta aparecerá el alineamiento efectuado con la extensión .aln 2) Abrir Bioedit 3) Seleccionar Edit para cortar secuencias y elimine en el menú edit “delete sequence” la secuencia “Clustal Consensus”. 4) Guardar y cerrar Bioedit

RA Moreno & MA Méndez
14
Procedimiento MEGA4
Cita
Tamura K, Dudley J, Nei M & Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution 24: 1596-1599.
1) Abrir el Programa MEGA4 2) En el menú File elegir la opción Convert To MEGA Format. Esta opción permite convertir el archivo .aln en un archivo .meg

RA Moreno & MA Méndez
15

RA Moreno & MA Méndez
16
3) Abrir el archivo .meg (Open Data en el menú File); ocupar la opción Neighbor-joining en el menú Phylogeny

RA Moreno & MA Méndez
17

RA Moreno & MA Méndez
18

RA Moreno & MA Méndez
19

RA Moreno & MA Méndez
20
4) Volver a realizar este análisis pero utilizando adicionalmente la opción Bootstrap
5) Guardar los árboles correspondientes

RA Moreno & MA Méndez
21

RA Moreno & MA Méndez
22
Software PAUP (Phylogenetic Analysis using Parsimony *and other methods) 1.- Análisis Filogenético de Máxima Parsimonia en Software PAUP (idem a página 4)
2.- Análisis Filogenético de Máxima Verosimilitud (Maximum Likelihood)
Archivo Tipo : Grupo 16s [secuencias nucleotídicas de especies de anfibios del género Bufo] Procedimiento
1) Buscar el mejor modelo de evolución molecular que se ajusta a los datos para ello utilizar el software MODELTEST (incluye 56 modelos). Para el uso en ambiente Windows, utilizar WINMODELTEST . 2) Abrir matriz de datos originales en formato nexus en PAUP.

RA Moreno & MA Méndez
23
3) Abrir además en PAUP el archivo “modelblock4.txt” que se encuentra en la carpeta “System files” de WINMODELTEST ; esto permite calcular los parámetros de los modelos evolutivos en PAUP creando el archivo “model.scores” en la carpeta “System files” de WINMODELTEST .

RA Moreno & MA Méndez
24
4) Abrir WINMODELTEST, hacer click en “New Outfile” y luego en “Run Modeltest”; esto creará un archivo “outfile” en la carpeta “System files” en que aparecerán los parámetros de los modelos escogidos con los criterios de hLRT (Likelihood Radio Test- Prueba de Razón de Verosimilitud) y AIC (Criterio Informativo de Akaike).

RA Moreno & MA Méndez
25
5) Agregar al final del archivo Nexus original las líneas de comandos que aparecen en el archivo “outfile” bajo el modelo escogido (Seleccione el Criterio de Akaike)

RA Moreno & MA Méndez
26
6) Ejecutar el archivo Nexus modificado en PAUP; cambie el criterio a likelihood y realizar una búsqueda exhaustiva o heurística y un bootstrap no-paramétrico. Guarde las salidas (outputs) y cierre PAUP. set_criterion=likelihood. [Cambio de criterio a método de Máxima Verosimilitud].

RA Moreno & MA Méndez
27
7) Recuerde de seguir los mismos pasos que se utilizaron para la reconstrucción por Máxima Parsimonia y grabe su árbol el cuál aparecerá en su carpeta como grupo16s con extensión tree para ser desplegado en el software Treeview. Recuerde ir al menú Tree y seleccione show internal Edge labels para que despliegue los valores de soporte de los nodos obtenidos en su análisis de Bootstrap. Outgroup Barenarum

RA Moreno & MA Méndez
28

RA Moreno & MA Méndez
29
Análisis Filogenético de Inferencia Estadística Bayesiana Plataforma: MRBAYES Cita Huelsenbeck, J.P. & F. Ronquist. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754-755. Procedimiento 1) Copie el archivo Nexus (original) y trasladelo a la carpeta de MRBAYES. Ahora ejecute MRBAYES con los comandos que se indican más abajo. Ejemplo archivo Tipo: Rag1CCQ [secuencias nucleotídicas del gen nuclear Rag 1 -recombination activating gene (RAG) - de géneros de anfibios] exe Rag1CCQ.nex 2) Defina el outgroup Outgroup Leiopelma

RA Moreno & MA Méndez
30
3) Especificar la estructura del modelo lset_nst=6_rates=propinv

RA Moreno & MA Méndez
31
4) Especifique algunos parámetros del análisis mcmcp_ngen=500000_samplefreq=1000_savebrlens=yes 5) Iniciar el análisis: mcmc Para detener el análisis: n

RA Moreno & MA Méndez
32
6) Una vez terminado el análisis realizar un burnin sump_burnin=10 sumt_burnin=10

RA Moreno & MA Méndez
33
7) Ir a la carpeta de MRBAYES y seleccionar el archivo. CON 8) Visualizar el árbol de consenso del 50% de la regla de la mayoría desplegado en Treeview donde se observan las probabilidades a posteriori.