inferring phylogenetic trees: maximum likelihood methods prof. william stafford noble department of...
TRANSCRIPT

Inferring phylogenetic trees:Maximum likelihood methods
Prof. William Stafford NobleDepartment of Genome Sciences
Department of Computer Science and EngineeringUniversity of Washington

One-minute responses• First part of class was fine.• I am struggling with Python.• At first it was difficult to complete the program when I get the first half,
but it is getting easier now.• The class lecture is always fine, but the Python problems are getting
tougher. However, they are really interesting and quite informative.• We are learning a lot about programming.• The class is more interesting every day. I enjoy the Python, especially
because I am able to fill in by myself.• Thank you for helping us with sys.stdout.write. It will be very useful for
future work in Python.

Outline
• Parsimony• Distance methods
– Computing distances– Finding the tree
• Maximum likelihood

Revision
Multiple sequencealignment
Pairwisedistancematrix
Phylo-genetic
tree

Revision
• Ideally, distances in a phylogenetic tree would represent time. In practice, however, what do the distance estimate represent?– The expected number of changes per position.
• What is a “back mutation”?– A pair of mutations that reverse one another (e.g.,
A C A)

Revision
• Compute the Juke-Cantor distance between the first yeast and mouse sequences shown below.
XX X X X XX X X Xdha2_yeast 93 LRYTRHEPVGVCGEIIPWNIdhac_mouse 93 FTYTRREPIGVCGQIIPWNIdha5_yeast 92 FAYTLKVPFGVVAQIVPWNIdhal_ecoli 92 LAMIVREPVGVIAAIVPWNI
ABAB dK
3
41ln
4
3

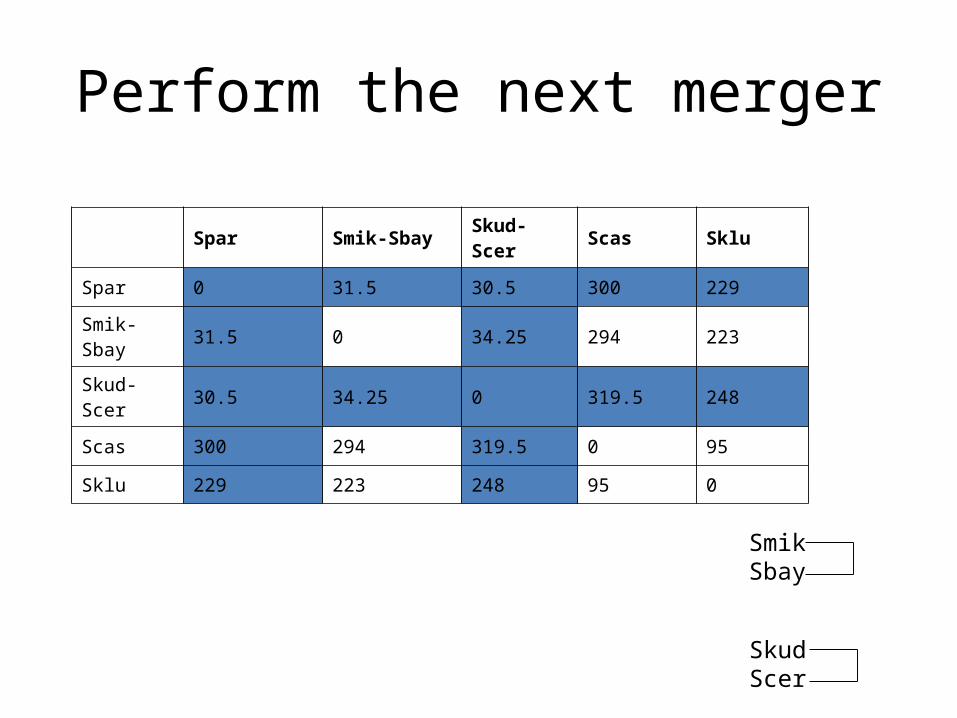
Spar Smik-Sbay Skud-Scer Scas Sklu
Spar 0 31.5 30.5 300 229
Smik-Sbay 31.5 0 34.25 294 223
Skud-Scer 30.5 34.25 0 319.5 248
Scas 300 294 319.5 0 95
Sklu 229 223 248 95 0
SmikSbay
SkudScer
Perform the next merger
Spar Smik-Sbay Skud-Scer Scas Sklu
Spar 0 31.5 30.5 300 229
Smik-Sbay 31.5 0 34.25 294 223
Skud-Scer 30.5 34.25 0 319.5 248
Scas 300 294 319.5 0 95
Sklu 229 223 248 95 0
SmikSbay
SkudScer
Perform the next merger

Skud-Scer-Spar
Smik-SbaySkud-Scer-Spar
Scas Sklu
Skud-Scer-Spar
0 32.875 0 309.75 238.5
Smik-Sbay 32.875 0 32.875 294 223
Skud-Scer-Spar
0 32.875 0 309.75 238.5
Scas 309.75 294 309.75 0 95
Sklu 238.5 223 238.5 95 0
SmikSbay
SkudScer
Perform the next merger

Smik-SbaySkud-Scer-Spar
Scas Sklu
Smik-Sbay 0 32.875 294 223
Skud-Scer-Spar
32.875 0 309.75 238.5
Scas 294 309.75 0 95
Sklu 223 2238.5 95 0
SmikSbay
SkudScer
Extend the corresponding tree
Spar
SkluScas

Maximum parsimony
for each possible treefor each column of the alignment
compute the parsimony score of the column, given the treereturn the tree with the best parsimony score

Maximum likelihood
for each possible treefor each column of the alignment
compute the likelihood of the column, given the treereturn the tree with the highest likelihood
• Similar to parsimony, but capable of using a model of evolution.
• Computationally expensive.• DNAML is the Phylip program for maximum likelihood.
FastDNAML is a fast clone.
http://evolution.genetics.washington.edu/phylip.htmlhttp://iubio.bio.indiana.edu/soft/molbio/evolve/fastdnaml/fastDNAml.html

Problem #1
• What is the probability of observing this column, given this tree and an assumed model of evolution?
ACGCGTTGGGACGCGTTGGGACGCAATGAAACACAGGGAA
T T A G
Pr(column|tree,model)+

Solution #1
• Solution: Enumerate all possible assignments to the internal nodes. Compute the probability of each tree, and sum.
T T A G T T A G T T A G
A
A
A A
C
A A
G
A

Problem #2
• What is the probability of observing this column, given this assigned tree and an assumed model of evolution?
ACGCGTTGGGACGCGTTGGGACGCAATGAAACACAGGGAA
T T A G
Pr(column|tree,model)+T
A
A

Solution #2
T T A G
T
A
A
πA, πC, πG, πT
m
The probability of the ancestral observation
being A is just πA.
The probability of observing a substitution from A to T on a branch of length m is given by
the evolutionary model.

Solution #2
T T A G
T
A
A
πA, πC, πG, πT
L0
L1 L2
L3 L4L5
L6
• The desired probability is the product of the probabilities of the branches.
• L(tree) = L0 L1 L2 L3 L4 L5 L6
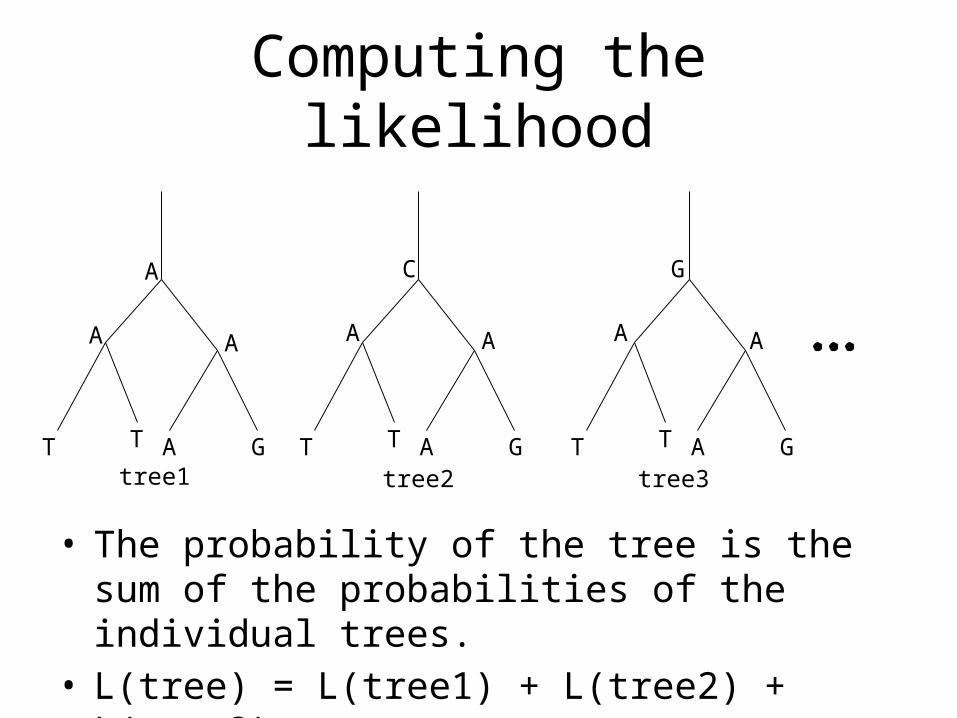
Computing the likelihood
• The probability of the tree is the sum of the probabilities of the individual trees.
• L(tree) = L(tree1) + L(tree2) + L(tree3) + …
T T A G T T A G T T A G
A
A
A A
C
A A
G
A
tree1 tree2 tree3

Maximum likelihood revisitedfor each possible tree
for each column of the alignmentfor each assignment of internal nodes
for each branch compute the probability of that branchassigned tree probability ← multiply branch probabilities
column probability ← sum assigned tree probabilitiestree probability ← multiply column probabilities
return the tree with the highest probability

Maximum likelihood revisitedfor each possible tree
for each column of the alignmentfor each assignment of internal nodes
for each branch compute the probability of that branchassigned tree probability ← multiply branch probabilities
column probability ← sum assigned tree probabilitiestree probability ← multiply column probabilities
return the tree with the highest probability
Multiply probabilities of
independent events.
Add probabilities of mutually
exclusive events.

Overview
• Parsimony• Distance methods
– Computing distances– Finding the tree
• Fitch-Margoliash• Neighbor-joining• UPGMA
• Maximum likelihood

Representing trees• ((mouse, rat), (human, chimp))
myTree = [[mouse, rat], [human, chimp]]
mouse rat human chimp

Problem #1
• Write a program to read a parenthesized tree from a file and count the number of nodes.
> cat mytree.txt(yeast, ((fly, spider), (dog, cat)))> python read-tree.py mytree.txtRead 5 species from mytree.txt.

Problem #2
• Modify the previous program to print the leaves of the tree, indenting according to the depth.
> print-tree.py mytree.txt yeast fly spider dog cat

Problem #3• Given: a three-column file in which the first two columns contain
names of species and the third column contains the distance between them.
• Print to standard output a formatted matrix in which the species names are listed in the rows and columns, and values are from the input file.– Species should be listed in alphabetical order.– The program should halt and complain if a value is missing.– The matrix is assumed to be symmetric, and each pair appears only
once.– Distances of zero along the diagonal are not included in the input.– Columns should be printed in the same width as the corresponding
species name.

./print-distance-matrix.py distances.txtRead 30 values and 6 species from distances.txt.Maximum species name width = 9. ape cat dog gerbil mouse zebrafish ape 0 0.19 0.15 0.44 0.17 0.69 cat 0.19 0 0.1 0.48 0.24 0.77 dog 0.15 0.1 0 0.43 0.25 0.78 gerbil 0.44 0.48 0.43 0 0.42 0.78 mouse 0.17 0.24 0.25 0.42 0 0.85zebrafish 0.69 0.77 0.78 0.78 0.85 0